
Submitted:
08 October 2024
Posted:
09 October 2024
You are already at the latest version
Abstract
N-methyl-D-aspartate receptors (NMDARs) are critical components of the mammalian central nervous system, involved in synaptic transmission, plasticity, and neurodevelopment. This review focuses on the structural and functional characteristics of NMDARs, with a particular emphasis on the GRIN2 subunits (GluN2A-D). The diversity of GRIN2 subunits, driven by alternative splicing and genetic variants, significantly impacts receptor function, synaptic localization, and disease manifestation. We explore the physiological properties and developmental regulation of these subunits, highlighting their roles in the pathophysiology of various NDDs, including ASD, epilepsy, and schizophrenia. By reviewing current knowledge and experimental models, including mouse models and human-induced pluripotent stem cells (hiPSCs), this article aims to elucidate different approaches through which the intricacies of NMDAR dysfunction in NDDs are currently being explored. The comprehensive understanding of NMDAR subunit composition and their mutations provides a foundation for developing targeted therapeutic strategies to address these complex disorders.
Keywords:
NDD – neurodevelopmental disorder
; EPSCs – Excitatory postsynaptic currents
; mEPSCs – Miniature Excitatory postsynaptic currents
; ATD – Amino-terminal domain
; LBD – Ligand binding domain
; TMD – Transmembrane domain
; CTD – C-terminal domain
; CNS – Central nervous system
; SCZ – Schizophrenia
; ASD - autism spectrum disorder
1. Introduction
The most dominant excitatory neurotransmitter in the CNS is the non-essential amino acid, glutamate. It acts as a fast transmitter and plays a crucial role in modulating cellular excitability and synaptic transmission between neuronal networks [1]. Glutamate receptors (GluRs) are distributed widely across the mammalian brain, serving as the principal excitatory transmitter system. Based on the preference of these receptors towards different excitatory small molecule agonists, initially the glutamate receptors were thought to be divided into NMDA and non-NMDA receptors, that were considered to mediate fast postsynaptic potentials by activating the ion channels directly [2]. Following the successful expression and functional studies of a glutamate receptor in Xenopus oocyte [3], a large family of glutamate receptor subunits was discovered, and a strong correlation between molecular subtype and pharmacological properties was established among the receptor subunits [4,5]. Based on the structural and functional aspects, GluRs in the CNS are majorly categorized into two families namely, metabotropic glutamate receptors (mGluRs), and ionotropic glutamate receptors (iGluRs) [6,7]. The mGluRs are a family of G-protein coupled receptors (GPCRs), that are activated by extracellular ligands such as neurotransmitters and mediate downstream signaling through the G-proteins [8,9]. The most classical neurotransmitter-GPCRs are part of family A. mGluRs are class-C GPCRs with a unique structure characterized by a large extracellular N-terminal domain that houses the endogenous ligand-binding site [10]. Ionotropic glutamate receptors, on the other hand, are cation-permeable ligand-gated ion channels including, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), Kainate receptors (KARs), N-methyl-D-aspartate receptors (NMDARs), and delta receptors, also known as orphan receptors (Figure 1A–D) [11].
AMPA receptors are the primary members of the ionotropic glutamate receptor family that mediate fast excitatory synaptic transmission in the CNS [11,13]. Upon glutamate binding, a conformational change ensues the opening of AMPAR ion channels, allowing the influx of sodium ions (Na⁺) into the postsynaptic neuron, generating EPSCs [14]. These EPSCs are essential for initiating action potentials and propagating excitatory signals within neural circuits, which consequently leads to the activation of NMDA receptors [15]. Delta receptors however, are different from the other members of the iGluR family with respect to their lack of typical glutamate-induced ion channel currents [16]. Both the subtypes of delta receptors (GluD1 and GluD2) are characterized by their unique ligand-binding domains (LBDs), contributing to their divergent electrophysiological and gating properties compared to AMPA, Kainate, and NMDA receptors [17]. This member of the iGluR family was named as an “orphan receptor” for many years due to the absence of identified endogenous interacting ligands. However, recent studies have shed light on the interaction of GluD subunits with glycine and D-serine in vitro as well as in situ (Figure: 1D) [12,18]. The roles of these receptors in synaptic plasticity are particularly prominent, with GluD2 being essential for cerebellar long-term depression (LTD) and proper cerebellar function, and GluD1 implicated in synapse formation and maintenance in regions such as the prefrontal cortex and hippocampus [19,20,21]. Collectively, ionotropic glutamate receptors play a crucial role in healthy functioning of the mammalian central nervous system. Dysfunction of the iGluRs is associated with a variety of neurological disorders, including autism spectrum disorder, epilepsy, Alzheimer’s disease, schizophrenia, and Huntington’s disease [22,23,24,25].
According to the genome aggregation database (gnomAD), there are around 700 variants of the GRIN family of proteins in humans. Evidently, GRIN2 protein subunits account for most variants including frame shift, nonsense, missense, splice-site, deletion, inversion, or translocation variations and mutations that are observed in NDDs [26]. Therefore, understanding the pathophysiology of ionotropic glutamate receptors is crucial for elucidating their involvement in NDDs for developing targeted therapeutic interventions. In line with this, for characterizing NMDAR pathophysiology in different NDDs, several disease models have been developed over the years including, drosophila models, mouse models, non-human primate models, zebrafish, and human-derived induced pluripotent stem cells (hiPSCs) [27,28,29,30,31]. These models offer critical insights into the molecular and cellular mechanisms underlying these conditions by allowing researchers to manipulate specific genes and observe resultant effects on brain development and function. They further help elucidate how genetic and environmental factors interact to contribute to disease pathophysiology, facilitating the identification of risk factors and potential therapeutic targets. Additionally, they serve as essential platforms for preclinical drug testing, enabling the assessment of safety and efficacy of new treatments. Overall, these models are invaluable in advancing our understanding and developing new treatment for NDDs. This review centers on exploring the structural aspects, functional mechanisms, and pathophysiological implications, focusing on the NMDARs, and disease models pertinent to characterizing NMDA receptors in NDDs.
2. Structure and Function of NMDA Receptors
Characteristic structural features that are conserved among different subtypes of iGluRs include, the extracellular amino-terminal domain (ATD) which contains an agonist-binding site, ligand binding domain (LBD) that is proximal to the plasma membrane, a transmembrane domain (TMD) for enabling ion permeation into the cells, gating elements that control opening and closing of the permeation pore, and an intracellular C-terminal domain (CTD) for downstream signaling [11]. NMDA receptors (NMDARs) are cation-specific ligand-gated ion channels that mediate glutamatergic synaptic transmission throughout the CNS. The NMDARs are localized on presynaptic, postsynaptic, as well as extrasynaptic membranes and play pivotal roles in excitatory neurotransmission and synaptic plasticity [32,33]. Functional NMDARs generally exhibit a heterotetrameric structure that is formed by combination of distinct subunits known as, GluN1 (NR1), GluN2A-D (NR2A-D), and GluN3A-B (NR3A-B) [34]. NMDA receptors necessitate the binding of two co-agonists glutamate and glycine simultaneously for channel activation, exhibit high permeability to Ca2+ ions, and are subject to voltage-dependent inhibition by extracellular Mg2+ ions [11]. Initial depolarization of NMDA receptors is stimulated by its interaction with the presynaptically released glutamate and extracellularly abundant glycine, which is followed by an influx of Na+ ions, generating the EPSCs [11]. Another temporally distinct component contributing to these EPSCs arises from the APMA receptors. The AMPA receptors mediate a synaptic current with rapid rise and decay kinetics, NMDA receptors on the other hand exhibit rather slow kinetics with activation time of approximately 10 milliseconds (rise time) and deactivation time (decay time) in the range of tens to thousands of milliseconds [35]. This difference in the kinetics enables depolarization of NMDAR and voltage-dependent release of Mg2+ ions. Therefore, NMDA receptors are considered as “co-incidence detectors” due to their dependence on the frequency of synaptic release of agonists glutamate and glycine, and the depolarization of the membrane potential. The slow synaptic currents enable prolonged influx of Ca2+ ions into the postsynaptic neurons (Figure 2). This substantial increase in Ca2+ concentration stimulates intracellular signaling which modulate gene regulation to induce various processes and behaviors in postsynaptic neurons, including synaptic plasticity [36], maintenance of dendrite arborization for memory formation [37], neuroprotection [38], and regulation of synapse formation [39].
2.1. Glycine-Binding GluN1 Subunit
In the CNS, GRIN1 gene encodes the GluN1 subunits of the NMDA receptor, which harbor the glycine-binding sites, and act as a fundamental component of the functional receptors. During developmental stages, GluN1 subunits exhibit constitutive expression and are notably prevalent across various regions of the brain when compared to GluN2 subunits of the NMDA receptors [41]. The GRIN1 gene consists of 22 exons, out of which, 3 exons undergo alternative RNA splicing and generate 8 functional isoforms of the GluN1 protein (Figure 3) [42]. The GluN1 variants are referred to as N1, C1, C2, and C2’. These polypeptide cassettes are encoded through different splicing combinations of exons 5, 21, and 22 or 22’. Exons 5 and 21 encode for 21 amino acids (N1) in the ATD and 37 amino acids (C1) in the CTD, respectively. The C2 cassette within the CTD, encoded by Exon 22, comprises 38 amino acids. Deletion or splicing out of C2 segment leads to the elimination of the initial termination signal, leading to a modified open reading frame (ORF) that integrates alternative 22-amino-acid cassette (denoted as C2’) into the C-terminal of the final protein product [43]. The CTD of GluN1 subunits interact with various intracellular proteins such as postsynaptic density protein 95 (PSD95), which is involved in regulating the trafficking and localization of NMDA receptors [44]. Additionally, GluN1 CTD interaction with calmodulin and neurofilaments plays a role in activation of regulatory proteins (ex. CaMKII) and maintenance of structural integrity of neuronal axons, respectively [45,46].
The splice variants of GRIN1 can form homomeric ion channels that exhibit ligand gated ion-channel properties, like the NMDA receptors. However, the homomeric receptors differ in physiological and pharmacological properties such as affinity towards exogenous and endogenous agonists, potentiation by polyamines, and regulation by protein kinase C [47,48]. Apart from the homomeric receptors, identity of GluN1 variants in the di-heteromeric and tri-heteromeric receptors also plays a crucial role in defining the kinetic and pharmacological properties of NMDARs. Exon 5 undergoes alternative splicing, giving rise to two distinct isoforms of the GluN1 protein, denoted GluN1-1a and GluN1-1b. GluN1-1a is distinguished by the exclusion of residues encoded by exon 5, whereas GluN1-1b contains the peptide sequence encoded by exon 5 [43]. Rumbaugh et al. have shown that deactivation kinetics of GluN2B subunits are influenced by specific residues encoded by exon 5 within the GluN1 subunit. Specifically, GluN2B receptors deactivate more rapidly when paired with GluN1-1b [49]. Similar observations were made by Vance et al. in Xenopus oocytes, where GluN1 splice variants affected the agonist potency, deactivation time course, and single channel properties of GluN2D containing NMDA receptors [50]. This study examined the effect of exon-5 encoded residues of GRIN1 subunit on the overall functionality of GluN1-GluN2D containing NMDA receptors. Inclusion of exon-5 encoded residues resulted in prolonged deactivation and reduced interaction with agonists L-glutamate and glycine [50]. Moreover, agonist stereochemistry also acts as a contributing factor in regulating receptor deactivation time course. For instance, linear agonists such as D-glutamate, L-aspartate, and D-aspartate resulted in faster deactivation of Glun1/Glun2D containing NMDARs compared to L-glutamate [51]. These findings underscore the importance of GluN1 splice variants and agonist characteristics in determining the functional properties of NMDA receptors, highlighting their potential impact on receptor behavior and pharmacological responses.
2.2. The GluN2 Diversity and Its Role in the NMDA Receptors
The incorporation of two GluN1 subunits is indispensable for the constitution of operational NMDAR complexes, however the assortment of GluN2/3 subunits govern the receptor’s functional versatility. In contrast to the GluN1 subunit, GluN2 expression fluctuates dynamically during embryonic brain development, reflecting an intricate regulation of stage-specific, region-specific, and cell-specific expression patterns [52]. Initially, during embryogenesis in rodents, a widespread distribution characterizes both GluN2B and GluN2D subunits. As the developmental period progresses, GluN2B maintains a high level of expression restricted to the forebrain, while GluN2D diminishes notably, primarily localizing to midbrain structures like the diencephalon and mesencephalon in adulthood [53,54]. Conversely, GluN2A emerges at birth, progressively amplifying its presence across the CNS, and is particularly enriched in regions of high cognitive function, such as the cortex and hippocampus [54,55]. GluN2C-containing receptors emerge during the first postnatal week, maintaining a localized expression within the olfactory bulb, thalamus, and vestibular nuclei. Eventually, during the second week a developmental transition from GluN2B- to GluN2C-containing NMDARs is observed in cerebellar granule cells, which remains the primary cite of GluN2C expression in adult rats [41,54,56]. The intricacies of GluN2 subunit distribution in mammalian brain further entails the cell-specific expression of GluN2-containing NMDARs. For instance, despite their low expression in the cortex and hippocampus, GluN2C and GluN2D exhibit selective expression in interneurons and glial cells [57,58]. Similarly, GluN2B and GluN2D localize in cerebellar Golgi cells, despite their overall low expression in the cerebellum [59,60]. Within the mature forebrain, GluN2A-containing receptors are predominantly localized at the synaptic terminals. In contrast, GluN2B receptors exhibit a broader spatial distribution, spanning peri- or extra-synaptic regions. Remarkably, synaptic NMDAR subunit composition displays dynamic adaptability in response to neuronal activity and environmental stimuli, affording precise modulation of receptor subtypes during synaptic plasticity [61,62].
The temporospatial distribution of the GluN2 proteins is regulated by alternative splicing, which serves as a pivotal mechanism influencing the expression patterns of GluN2 proteins, shaping their functional diversity and regulatory dynamics within the CNS. Based on the human-brain RNA-seq data, a Glun2A isoform (GluN2A-short) was identified, which was characterized by the excision of 343 nucleotides from the final canonical exon of GRIN2A gene, thereby yielding a C-terminally truncated GluN2A with 1281 amino acids (aa) [63]. Warming et al. demonstrated that the GluN2A-short subunit is abundantly expressed (up to 25%) in the human brain and forms functional receptors upon co-expression with the GluN1 subunit [64]. Its prevalence extends to chimpanzee and macaque datasets but is notably absent in rat and mouse, indicating its primate-specific nature [63,65,66]. The truncation of the GluN2A CTD bears significant functional implications, notably resulting in the loss of critical interaction motifs that act as the binding sites for other regulatory proteins such as CaMKII and PSD-95 [52]. A previous study by Tabish et al., investigating GluN2B transcript variants in a mouse model reported two transcripts of the NR2B gene, arising from alternate splicing in the noncoding exons of the 5′ untranslated region (UTR) [67]. Interestingly, GluN2C, predominantly expressed in the cerebellum, exhibits alternative 5′-UTR exons, particularly GRIN2C-a, which appears to be more abundant than the canonical exon 1, suggesting a primary UTR isoform. Additionally, a less common isoform (GRIN2C-b) with alternative splice cite at 5′-exon junction, was observed along with elongation of exon 2, indicating variability in the 5′-UTR of the GRIN2C gene [63]. However, the RNA-seq data suggests that no splice site consensus sequences are observed in rodents or primates for the GRIN2D gene [63,68]. These findings underscore the complexity of alternative splicing mechanisms and their implications for the diversity of GluN2 subunits in NMDA receptors. Understanding this intricate regulation is crucial for elucidating the roles of NMDARs in synaptic plasticity, neural function, and neurodevelopmental processes.
3. NMDA Receptors in Neurodevelopmental Disorders: Variants of GluN2 Subunits
The GluN2 subunits of NMDA receptors play pivotal roles in brain function and are critically involved in various neurological disorders. Each GluN2 subtype—GluN2A, GluN2B, GluN2C, and GluN2D—contributes to synaptic transmission, plasticity, and excitability, and their diverse combinations and interactions with other receptor subunits may influence pathological mechanisms underlying conditions such as Epilepsy, ASD, developmental delay/intellectual disability (DD/ID), schizophrenia, and other NDDs [25,69,70,71,72,73]. Identification of GluN2 variants associated with these disorders plays a crucial role in understanding the intricate dynamics of NMDAR pathophysiology, and is essential for developing effective therapeutic strategies. The genome aggregation database (gnomAD) comprises over 700 variants of the GRIN genes that exhibit no correlation with the healthy population, out of which GRIN2A accounts for 44% of all known disease associated variants of the GRIN family [26]. Disease-relevant variants are scattered across the domains of the mature protein, where mutations in the transmembrane domain and linker regions are known to be particularly associated with more severe phenotypes and poor prognosis [74,75].
Most common phenotypes associated with GRIN2A mutations are epilepsy/seizures, intellectual disability, and schizophrenia (SCZ) [76,77]. GRIN2A variants majorly exhibit truncation and missense mutations in schizophrenia resulting in haploinsufficiency and loss of function. The SCZ-linked GRIN2A variants predominantly result in loss of function in NMDARs. Whereas, M653I and S809R mutations linked to epilepsy and DD/ID may lead to gain-of-function or loss-of-function of NMDA receptors depending upon the location of the mutation [78]. For example, in a SCZ cohort, a patient was observed to bear a de novo missense mutation (c.2902G>A; p.A968T) in the intracellular region of GRIN2A, which did not have significant implications on the NMDAR function [79]. Evidently, Individuals with a severe developmental disorder and intellectual disabilities are more prone to the missense mutations in GRIN2A rather than the protein-truncation variants [80]. Given the diversity of subunit composition in NMDA receptors, GRIN2B might compensate the loss of GRIN2A. However, electrophysiological data from the rats suggests that Grin2a+/− and Grin2a−/− knockouts exhibit reduced NMDA-evoked currents. Furthermore, percentage inhibition of NMDA-evoked currents in Grin2a−/− neurons after ifenprodil treatment is significantly greater than that of Grin2a+/− or Grin2a+/+ neurons; suggesting that subunit compensation does not occur in case of allelic deletions [74,81]. Another study explored the impact of GluN2B variants (G689C and G689S) on NMDA receptor function across different receptor configurations (di- and tri- hetermomers). These variants drastically reduced glutamate potency but exhibited partial restoration in mixed receptors (GluN2A and GluN2B) due to positive cooperativity. The neurosteroid Pregnenolone Sulphate was observed to effectively potentiate the receptors, indicating therapeutic potential for GRINopathies despite concerns about non-specific effects. [82].
GRIN2B variants account for around 37% of all variants observed in NMDA receptors in NDDs [26]. GRIN2B variants arising from missense, nonsense, frameshift, or splice site mutations are frequently associated with the NDDs such as autism, schizophrenia, DD/ID, and epileptic encephalopathy. Mutations affecting the NMDAR function are distributed throughout the structural domains of the GRIN2B subunit [83]. Whole exome sequencing data from patient cohorts suggests that rare de novo mutations in the ABD and TMD are observed exclusively in the patient population, and have significant contributions in altering NMADR functional properties [69,70,84]. For example, gene screening and variation analysis on an ASD cohort, identified a missense mutation (c.2473T>G; p.L825 V) in a highly conserved region of the transmembrane region of GRIN2B, which was predicted to be relatively damaging in an in-silico pathogenicity analysis [79]. In another study, a trio-based exome sequencing of individuals with sporadic ASD was performed, 21 de novo mutations were identified, 11 of which were found to be protein-altering mutations. A single-base substitution was discovered in a proband at the canonical 3’ splice site of GRIN2B (exon 10), which resulted in possible regression and co-morbidity for mild ID [85]. Recently, it was established that the truncated GRIN2B resultant of the point mutation in splice site mentioned above affects the cellular phenotype of neurons in ASD. The mutant subunit (GRIN2B724t), which is truncated in the second extracellular loop (S2), upon expression with WT-GRIN1 subunit hinders the trafficking of NMDAR to the cell surface or dendrites. Additionally, GRIN2B724t disrupts the dendrite morphogenesis by decreasing outgrowth rates while increasing retraction and subsequent pruning [86,87].
While GRIN2-A/B variants are most frequently observed in the NDDs, GRIN2C and GRIN2D variants account for 2.8% and 3.8% of total GRIN variants identified in NMDARs, respectively [26]. Molecular characterization of postmortem brain from SCZ patients, established significant reduction in GRIN2C mRNA levels in the dorsolateral prefrontal cortex (DLPFC) tissue, which may lead to altered NMDAR stoichiometry and endogenous NMDAR deficit in schizophrenia [88,89,90,91]. Li et al. identified of rare de novo GRIN2D variants in two independent probands having epileptic encephalopathy. A missense variant (c.1999G>A; p.Val667Ile), in the GRIN2D gene resulting in a single amino acid substitution in the M3 transmembrane domain, was considered the most likely disease-causing variant [92]. Additionally, a GRIN2D mutation (c.1412G > A), potentially affecting the splicing site, which may be a possible risk factor in SCZ [93].
4. Insights into NMDA Receptor Pathophysiology from Mouse Models of Schizophrenia and ASD
Rodents are extensively utilized in modeling human disorders owing to several key factors. Their genetic similarity to humans provides a strong foundation for research. The ability to modify their genomes, combined with their rapid reproductive cycle, facilitates efficient study. Their small size and social behavior make them easy to maintain in laboratory settings. Researchers have developed a variety of neurological, behavioral, pharmacological, and other tests to assess features of neurodevelopmental disorders in these models, broadening our understanding of these conditions [94,95,96,97]. Additionally, mouse models have been instrumental in elucidating the underlying biological and pathophysiological mechanisms of the NMDA receptor-related neurodevelopmental disorders [28,98,99,100]. For instance, mouse models with targeted genetic modifications in the NMDA receptor subunits to explore the complex neurobiology underlying schizophrenia (SCZ) have been previously developed, that are instrumental in identifying abnormal neural circuits, synaptic dysfunctions, and behavioral abnormalities of the disorder [28,101,102]. Mouse models used to study schizophrenia often focus on NMDA receptor dysfunction. Over the years various strategies including pharmacological models, genetic models, and neurodevelopmental models, have been developed to study the NMDA receptor hypofunction in SCZ [103].
One of the approaches in pharmacological models entails the usage of NMDAR antagonists that act as an open-channel blockers. Phencyclidine (PCP), ketamine, and MK-801, AKA the “trapping blockers”, that gets trapped inside NMDAR ion channel pore, which renders the receptor to a closed inactive state [104,105,106,107]. MK-801 (dizocilpine) is a highly potent noncompetitive NMDA receptor antagonist, more effective than ketamine and PCP, and is therefore widely used in animal studies to induce schizophrenia-like behavioral and neurochemical changes [108,109]. Genetic analyses reveal that polymorphisms in the coding and promoter regions of GRIN subunits can affect NMDA receptor transcript levels and functionality. While various mouse models with targeted deletions of NMDA receptor subunits have been created, those with homozygous deletion of GRIN1 or GRIN2B subunits fail to survive past the perinatal period [110,111,112,113]. To address lethality, NR1 knockdown (KD) mice were created by expressing the NR1-1a splice variant in NR1 knockout mice. These NR1 KD mice exhibit altered dendritic differentiation, increased axonal arborizations, and faster development of corpus callosum projection neurons. However, their average lifespan depends on the transgene expression levels, limiting their use for further behavioral, molecular, and structural studies in adults [114,115]. Another NR1 KD mouse line, created by inserting a neomycin cassette into the GRIN1 gene, shows significantly reduced NR1 expression. Nonetheless, these mice exhibit a range of schizophrenia-like behaviors, including impaired social and sexual interactions, cognitive inflexibility, abnormal ERPs, spatial cognition, and sensorimotor gating deficits, hyperlocomotion, stereotypy, self-injury, decreased anxiety, and reduced nest building [98,116,117,118,119,120].
Comparable strategies are employed in developing mouse models for ASD, using genetic modifications and behavioral assessments to investigate NMDA receptor dysfunction and its role in ASD. For instance, in 16p11+/− mice, which exhibit deficits in spatial memory and social motivation, NMDA receptor activation in the medial prefrontal cortex (mPFC) layer 5 pyramidal neurons is notably reduced. This impairment in NMDA receptor function is attributed to decreased phosphorylation of the GluN2B subunit at the S1303 site, rather than a decrease in the overall expression of the subunit. To counter this, Wang et al. used a chemogenetic strategy involving the “designer receptors exclusively activated by designer drugs (DREADDs)” and delivered via viral vectors to the mPFC. The activation of these receptors was done with clozapine-N-oxide (CNO) facilitated CaMKII-mediated phosphorylation of the S1303 site on the GluN2B subunit. This led to successful restoration of NMDA receptor-mediated EPSCs to normal levels and alleviated the behavioral deficits observed in the mice [121]. Interestingly, zinc, which is co-released with glutamate and accumulates in postsynaptic spines, significantly modulates NMDAR function. Zinc deficiency is one of the major factor in etiology of behavioral defects associated with ASDs [122,123], and studies have shown that dietary supplementation of zinc can help alleviate the phenotypes of ASD [124]. Dietary supplementation of zinc was observed to significantly improve the auditory fear memory and social interaction in the Tbr1+/- mice [125]. In a Grin2b mice with ASD-risk mutation (Grin2b+/C456Y), reduced GluN2B levels led to decreased hippocampal NMDAR currents, enhanced LTD, and normal LTP, suggesting protein degradation and LTD sensitivity. These mice had normal social behavior but showed increased anxiolytic-like behavior. Early D-cycloserine treatment rescued NMDAR function and improved adult anxiolytic behavior [126].
Other than NMDAR-associated ASDs, several other risk genes have been identified, out of which the SHANK family genes are particularly implicated in severe behavioral deficits [127,128]. SHANK variants play a critical role in determining the specific type of NMDA receptor functional deficits observed in ASD mouse models [129]. For example, mice with either a haploinsufficiency (Shank3e4–9+/−) or complete deficiency (Shank3e4–9−/−) in the ankyrin repeat (ANK) domain of SHANK3 exhibited a significant decrease in the NMDA/AMPA excitatory postsynaptic current (EPSC) ratio at cortical excitatory synapses onto striatal medium spiny neurons. Contrastingly, in a Shank313–16−/− mice, which lacks the PSD95/DlgA/Zo-1 (PDZ) domain-coding exons 13–16 of Shank3, NMDAR-mediated synaptic transmission at striatal synapses remained normal; however, the decay kinetics of NMDAR-mediated EPSCs were altered [130,131].
Overall, mouse models serve as significant research tools for studying NMDAR associated NDDs by providing a platform to investigate the effects of NMDA receptor dysfunction on brain function and behavior. Through targeted genetic modifications and detailed behavioral assessments, these models have been crucial in uncovering the underlying mechanisms and elucidating the complex interactions involved in these disorders. Despite their utility, mouse models of neurodevelopmental disorders have considerable limitations; as they do not fully replicate the complexity of human disease condition, provide limited understanding of the etiopathology of the disease. Moreover, special care is required in certain cases, as pharmacologic agents such as PCP, used for inducing the disease phenotype can render mice hyperexcitable and aggressive [132,133,134]. Primate models overcome some limitations of rodent models, such as brain anatomy and drug response differences, but they only replicate basic behaviors like simple social interactions and repetitive actions [29,135] and their use is restricted by stringent ethics limitations. These limitations hinder the study of complex behavioral changes in NDDs and the development of effective therapies. This warrants the need of an alternative model system which can replicate the phenotypes relevant to the human NDDs.
5. Human iPSC-Derived Neurons in the Research of NMDA Receptor Pathophysiology
The emergence of human-induced pluripotent stem cell (hiPSC) technology stands as a pivotal milestone in biomedical research, offering a potent tool to explore human biology and diseases in unprecedented detail. Since its inception following Yamanaka’s groundbreaking work in 2006-2007, hiPSCs have provided researchers with an unparalleled opportunity to unlock the secrets of human development and pathophysiology in vitro [136,137]. By reprogramming somatic cells into a pluripotent state, hiPSCs enable the generation of patient-specific cellular models, circumventing ethical concerns and offering a unique window into the molecular underpinnings of various disorders [138]. The neurons derived from patient cells, faithfully recapitulate key aspects of human brain development and pathology, allowing researchers to explore the nuanced interactions between genetic predispositions, environmental factors, and neuronal phenotypes (Figure 4) [139].
With their ability to model complex synaptic processes, aberrant connectivity, and neurotransmitter imbalances, hiPSC-derived neurons serve as invaluable tools for dissecting the molecular mechanisms underlying neuropsychiatric disorders such as ASD, schizophrenia, bipolar disorder, and intellectual disability [140,141,142,143,144]. Integration-free methods for iPSC generation avoids genomic integration of vectors, thereby preserving genetic integrity and reducing tumorigenic risks [145]. Consequently, several groups have successfully adopted this method for modeling different neurological disorders. For instance, iPSCs were generated by reprogramming fibroblasts derived from a Phelan-McDermid syndrome (PMS) patient, harboring an insertion mutation in SHANK3 (C.3679insG) [146]. The iPSCs were observed to express the pluripotency markers, differentiate into the three germ layers, retain the disease-causing mutation, and display normal karyotypes. Therefore, this technology allows researchers to explore the functional properties of cellular factors involved in the pathology of NDDs, which can be translated into a patient specific therapeutic intervention.
In the CNS, the diversity among NMDAR subunits shapes receptor properties and functions, impacting both physiological and pathological conditions. GluN2 subunits demonstrate unique expression patterns across various brain regions and neuron types. Notably, GluN2A and GluN2B are prominently present in complex brain structures like the cortex, suggesting their pivotal roles in neural processes [11,148,149]. Expression of functional NMDA receptors as well as other ion channels in hiPSCs, which exhibit normal electrophysiological characteristics, serves as a bona-fide system for studying human brain in dish [150,151,152]. For instance, whole-cell patch-clamp experiments suggest that hiPSC-derived neurons have spontaneously occurring mEPSCs, indicating the presence of functional synapses in the cells [153]. Studies have shown mRNA expression of NMDAR subunits, such as GRIN1, GRIN2(A-D), and GRIN3(A-B) in neurons derived from human induced pluripotent stem cells (hiPSCs) [154,155]. Additionally, electrophysiology experiments indicate that while GluN2A subunits mediate 10-30% of NMDA-induced currents, GluN2B subunits account for over 70% in neurons differentiated via an NPC stage and NGN2 neurons, highlighting the predominance of functional GluN2B subunits in these cells [153]. The decay time constant of NMDAR-mediated synaptic currents provides further insight into the subunit composition of NMDARs in hiPSC-derived neurons. Prolonged decay time, exceeding 250 milliseconds, indicates that the synaptic currents are dominated by the GluN2b containing receptors [153,156]. Moreover, loss-of-function mutation in GRIN2B results in impairment of differentiation and increased proliferation in hiPSC-derived neural progenitor cells (NPCs) [157]. The distinct expression patterns and functional roles of NMDAR subunits, especially GluN2A and GluN2B, highlight their significance in brain physiology. In line with this, researchers have utilized iPSC-derived neurons to study the molecular mechanisms underlying NDDs, including ASD.
5.1. iPSC Models for Studying NMDARs in ASD
Recent studies emphasize the effective use of iPSC-derived neurons to model ASD in the investigation of intricate neuronal phenotypes [158,159,160,161]. For example, a de novo heterozygous point mutation in Down Syndrome Cell Adhesion Molecule (DSCAM) gene was associated with ASD. iPSCs derived from the 12-year-old patient was used for generating neuronal cells. The DSCAM mutation resulted in impairment of the NMDAR function, which was significantly restored by the exogenous expression of DSCAM [31]. Dysregulation of SHANK family of proteins is also implicated in ASD. Located at the core of the postsynaptic density (PSD) in glutamatergic synapses, SHANK proteins bind to structural proteins, glutamate receptors, and the actin cytoskeleton, thereby modulating the structure, plasticity, and maturation of excitatory synapses [162,163]. SHANK3 plays an important role in regulating the functional properties of NMDA and AMPA receptors. It enhances synaptic transmission mediated by these receptors and boosts glutamate release by forming trans-synaptic signaling complexes with neurexin and neuroligin [164]. Recently, it was established that SHANK3 mutation is associated with S-nitrosylation (SNO), a nitric oxide-mediated posttranslational modification (PTM), which leads to neuronal pathology [165]. Consequently, the effect of SNO was studied parallelly in ASD mouse model and hiPSC-derived neurons. The study demonstrated elevated levels of nitric oxide, which induced oxidative and nitrosative stress leading to the enrichment of SNO-proteome. Post neuronal nitric oxide synthase (nNOS)-inhibitor treatment, the ASD deficits were significantly restored in both the models [160].
Advanced techniques like transcriptome screening and whole-exome sequencing have expanded the list of ASD-associated genes and helped integrate these genes into relevant biological pathways, deepening our understanding of the underlying mechanisms [166,167]. Hussein et al. investigated the physiological properties of ASD-patient derived neurons harboring mutations in different candidate genes including, SHANK3, UBTF, and GRIN2B. Interestingly, electrophysiological recordings of cortical neurons corresponding to all the mutants exhibited a similar phenotype of early maturation and hyperexcitability, suggesting common pathological pathways despite different genetic alterations [168]. Furthermore, hiPSCs provide a platform to decipher intrinsic differences in the pathology of NDDs among patients containing similar genetic makeup, indicating a role of environmental factors and epigenetic modifications in the manifestation of the neuropsychiatric diseases [169,170]. These findings suggest that human-derived iPSC models can perfectly recapitulate the pathophysiological behaviors observed in ASD patients. Moreover, reprogrammed cells can be valuable for pinpointing shared pathway disruptions in the development of different NDDs such as ASD and SCZ. Studies on neurons derived from patients with ASD and SCZ using induced pluripotent stem cells (iPSCs) reveal distinct yet converging developmental patterns. Initially, neurons from ASD patients mature rapidly, showing increased excitability, enhanced sodium and potassium currents, and more extensive branching and synaptic connections compared to control neurons. However, as they continue to develop, these neurons lose their early advantages, becoming less excitable, with reduced synaptic activity and fewer branched neurites. In contrast, neurons derived from SCZ patients begin with lower functionality, characterized by less branching, reduced excitability, and decreased synaptic activity. Although SCZ neurons consistently develop more slowly than control neurons, they eventually exhibit a phenotype like mature ASD neurons, suggesting that while the developmental trajectories differ, both disorders result in comparable neurodevelopmental disruptions [171].
5.2. iPSC Models for Studying NMDARs in Schizophrenia
Schizophrenia is a complex psychiatric disorder with a global prevalence of about 1%, typically manifesting in late adolescence or early adulthood. SCZ is a highly heterogeneous disorder with variable clinical presentations, complicating the ability to draw generalized conclusions from postmortem data. For instance, postmortem studies on schizophrenia patients have reported inconsistent expression of GRIN1 subunit in the frontal cortex, due to the differences in examined regions, and variations in tissue handling and dissections [172,173,174,175]. Therefore, Patient derived iPSCs serve as a great alternative model system to overcome these limitations [176]. In a pioneering study, Chiang et al. derived integration-free iPSCs from schizophrenia patients with the DISC1 mutation, a genetic variant linked to the disorder’s pathology. These iPSCs displayed the characteristic morphology of human embryonic stem cells (hESCs), exhibited normal karyotypes, and expressed a range of pluripotency markers such as Nanog, Oct4, Sox2, SSEA3, SSEA4, TRA-1-60, TRA-1-81, and TRA-2-49 [177]. A subsequent in-depth study on iPSC-derived neurons from a schizophrenia patient revealed key functional deficits, including reduced neurite length and decreased levels of the synaptic protein PSD-95 [178]. Several studies have explored iPSC-derived neurons from schizophrenia patients, consistently finding abnormalities in neuronal and synaptic development, as well as in various intracellular signaling mechanisms [179,180,181,182,183]. Schizophrenia concordance rate in monozygotic twins ranges between 41-79%, which is much higher compared to that of dizygotic twins (10-19%) [184,185,186]. Despite the high concordance in monozygotic twins, the almost 50% of cases in these subjects exhibit discordance for SCZ [187]. The iPSC-derived neurons serve as a great model to study SCZ pathology in monozygotic twins, as it can measure disease associated changes in a similar genetic background. In line with this, a recent study has shown that iPSC-derived dentate gyrus (DG) neurons of affected individuals are less arborized and exhibit delayed maturation in comparison to the unaffected and control twins. Consequently, the excitatory postsynaptic currents are drastically reduced in the DG neurons of affected twins. Moreover, genetic alterations were observed in pathways related to neuronal development, synapse-related, and Wnt signaling in the affected individuals [169].
In iPSC-derived neurons from patients with schizophrenia, researchers have identified changes in the expression of various glutamate receptor subunits, including GRIN2A, GRIN2B, GRIK1, GRIK2, and GRM1, GRM7, as well as glutamate transporter genes [176]. The glutamate hypothesis, supported by findings that NMDA receptor antagonists can induce psychosis in healthy individuals, suggests that impairments in glutamatergic neurotransmission, involving NMDA receptors, play a crucial role in SCZ [188]. This hypothesis is further supported by reduced mismatch negativity (MMN), an event that indicates early dysfunctions in auditory processing and may signal the onset of schizophrenia-related alterations in brain function from a young age [189,190]. The PDE4 inhibitor rolipram was recently used to restore synaptic function and boost excitatory synaptic activity in iPSC-derived neurons with DISC1 mutations [191]. Additionally, PDE4 inhibitors have demonstrated clinical benefits in improving mismatch negativity (MMN) and correcting working memory-related theta activity changes [192,193]. Moreover, Recent research highlights the involvement of ADCYAP1, the gene responsible for encoding the PACAP protein, in several disrupted pathways in neurons derived from schizophrenia patients [194]. PACAP plays a critical role in enhancing NMDA receptor function by activating the cAMP/PKA pathway, which results in the release of RACK1 from GRIN2B subunits [195]. A deficiency in PACAP, commonly seen in schizophrenia, likely reduces NMDA receptor activity, particularly in SST interneurons, which are highly enriched with PACAP receptors and GRIN2B subunits. This impairment may contribute to the underlying neural dysfunction associated with the disorder.
Numerous strategies have been devised for therapeutic targeting of NMDA receptors in SCZ. NMDARs are targeted in schizophrenia treatment to address symptoms that current antipsychotics fail to manage effectively, such as negative symptoms and cognitive dysfunction. Research focuses on various strategies to modulate NMDAR function. These include targeting the glycine modulatory site (GMS) on the NMDA receptors with compounds like GlyT-1 inhibitors [196], D-serine [197], DAAO inhibitors [198], and D-cycloserine (DCS) [199]. Additionally, positive allosteric modulators (PAMs) for metabotropic glutamate receptors, such as mGluR5 and Group II mGlu receptors, are explored to indirectly influence NMDAR function. Antioxidants like N-acetylcysteine (NAC) and sulforaphane are also investigated for their potential to mitigate NMDAR hypofunction. Despite mixed clinical outcomes—where some drugs show promise while others fail—this approach highlights the ongoing effort to refine therapeutic strategies and improve the efficacy of treatments targeting NMDAR dysfunction in SCZ [200].
5.3. iPSC Models for Studying NMDARs in Epilepsy
Induced pluripotent stem cell (iPSC) models have emerged as powerful tools in epilepsy research, offering unique opportunities to study the cellular and molecular mechanisms underlying the disorder. Patients with mutations in the same gene can exhibit widely varying seizure types, severity, and responses to treatment. iPSC models derived from these patients have been instrumental in studying several neurodevelopmental disorders associated with epilepsy. These include conditions such as Dravet Syndrome [201], Angelman Syndrome [202], CDKL5-related neurodevelopmental delay [203], STXBP1-associated epileptic encephalopathy [204], and Timothy Syndrome [205]. Rett syndrome is one of the most thoroughly investigated epileptic disorders using iPSC technology, caused by mutations in the MeCP2 gene on the X chromosome, with 50% to 90% of patients experiencing seizures [206]. iPSC models have shown reductions in neuronal soma size, neurite outgrowth, synapse formation, and spontaneous activity compared to controls [138,207]. Furthermore, wild-type neurons display similar morphological abnormalities when co-cultured with astrocytes from Rett syndrome patients, highlighting the significant role of astrocytes in epileptogenesis [208].
Recent research has highlighted the significant role of NMDA receptors (NMDARs) in the pathophysiology of epilepsy, particularly through genetic mutations and excitotoxic signaling pathways [209]. Mutations in genes encoding NMDAR subunits, such as GRIN1, GRIN2A, GRIN2B, GRIN2C, and GRIN2D, have been linked to various epileptic phenotypes, ranging from focal and generalized seizures to severe epileptic encephalopathies [209]. These mutations often disrupt NMDAR function, leading to increased neuronal excitability, which is central to epileptogenesis. For example, mutations in GRIN1 are associated with early infantile encephalopathy and developmental delay [210], while GRIN2A and GRIN2B mutations contribute to epilepsy syndromes like Landau-Kleffner syndrome and epileptic encephalopathy, respectively [211,212]. In addition to genetic factors, NMDAR-mediated excitotoxicity, triggered by excessive glutamate release, plays a pivotal role in neuronal death. This mechanism, characterized by calcium overload and activation of destructive cellular pathways, is not only relevant to epilepsy but also to neurodegenerative diseases like Alzheimer’s and Parkinson’s [209]. Targeting NMDAR dysfunction and excitotoxicity thus represents a promising therapeutic avenue for addressing refractory forms of epilepsy. In line with this, researchers have developed patient derived iPSC models for studying NMDAR dysfunction in epilepsy. For instance, iPSCs were generated from peripheral blood mononuclear cells (PBMCs) from a seven-year-old child with epilepsy, who carried a heterozygous mutation of GRIN2A gene (c.1832 A>T) [213]. Another group generated iPSCs from fibroblasts of a developmental and epileptic encephalopathy (DEE) patient carrying a de novo heterozygous pathogenic variant of GRIN2D [214]. Similarly, Shi et al. developed an iPSC line from a patient with autosomal dominant neurodevelopmental disorder carrying the GRIN1 c.389A > G (p.Asp130Gly) mutation. This cell line serves as a valuable model for investigating GRIN1-related disorders, including epilepsy [215]. Overall, the integration of iPSC models in epilepsy research holds promise for enhancing our understanding of the disease’s heterogeneity and developing personalized therapeutic approaches for patients with various genetic backgrounds.
6. Discussion
This review delves into the multifaceted roles of NMDA receptors (NMDARs) within the central nervous system (CNS) and their critical involvement in various neurodevelopmental disorders. The intricate interplay of GluN2 subunits in shaping receptor functionality and synaptic plasticity underscores the essential contributions of these receptors to brain physiology. The literature suggests that the variability in NMDAR subunit composition is a fundamental factor influencing receptor kinetics and pharmacological properties. Particularly noteworthy are the differential expression patterns of GluN2 subunits across various developmental stages and brain regions, highlighting their distinct contributions to neurodevelopmental processes. The discovery of primate-specific isoforms, like the truncated GluN2A-short, adds another layer of complexity, suggesting unique evolutionary adaptations that may influence cognitive functions. The impact of NMDAR dysfunction becomes strikingly apparent when we consider the disease-associated GRIN gene variants linked to a spectrum of neurological disorders, including epilepsy, schizophrenia, intellectual disabilities, and ASD. The prevalence of GRIN2A and GRIN2B variants in these conditions correlates with their critical roles in synaptic signaling. The complexity of NDDs is further compounded by the variable effects of different types of mutations on NMDA receptor function. For instance, while certain missense mutations may subtly alter receptor kinetics, others might drastically impair receptor function or localization [216,217]. Similarly, truncation mutations often lead to a complete loss of function, but their effects can vary depending on the specific region of the gene that is affected. Studies have shown that missense mutations in GRIN2A are frequently observed in patients with severe developmental disorders and intellectual disabilities, whereas protein-truncating variants tend to be less common. On the other hand, truncation mutations can have more pronounced effects, such as the GRIN2B truncation mutation that disrupts NMDAR trafficking and dendritic development, particularly in ASD [87,218]. This heterogeneity in mutation effects highlights the importance of developing sophisticated approaches to unraveling genotype-phenotype correlations.
The use of mouse models and patient-derived induced pluripotent stem cell (iPSC) neurons has significantly enriched our understanding of NMDAR-related pathophysiology. These models offer a window into the functional consequences of GRIN mutations in a controlled environment. Mouse models, engineered to carry human-relevant mutations, provide vital insights into the in vivo effects of NMDAR dysfunction. For instance, mouse models of autism spectrum disorders (ASD) with genetic modifications have shed light on specific types of NMDA receptor functional deficits. In 16p11+/− mice, which exhibit deficits in spatial memory and social motivation, NMDA receptor activation in the medial prefrontal cortex (mPFC) layer 5 pyramidal neurons is notably reduced due to decreased phosphorylation of the GluN2B subunit at the S1303 site; similarly, the Shank3e4–9+/− and Shank3e4–9−/− mice, which have haploinsufficiency or complete deficiency in the SHANK3 gene, show a significant decrease in the NMDA/AMPA excitatory postsynaptic current (EPSC) ratio at cortical excitatory synapses onto striatal medium spiny neurons. These findings Suggest that mouse models are essential for understanding the pathophysiology of NMDA receptors in neurodevelopmental disorders by elucidating the specific genetic and molecular mechanisms regulating receptor function. Despite their utility, mouse models of neurodevelopmental disorders have limitations; they do not fully replicate the complexity of human disease conditions and provide a limited understanding of the etiopathology of the disease. This limitation hinders the study of complex behavioral changes in NDDs and the development of effective therapies. Complementing the insights gained from animal models, patient-derived iPSC neurons offer a human-specific perspective. The emergence of human-induced pluripotent stem cell (hiPSC) technology stands as a pivotal milestone in biomedical research, offering a potent tool to explore human biology and diseases in unprecedented detail. Neurons derived from patients faithfully recapitulate key aspects of human brain development and pathology, allowing researchers to explore the nuanced interactions between genetic predispositions, environmental factors, and neuronal phenotypes. The study on GRIN-related pathologies enhances our understanding of the phenotypic spectrum associated with NMDAR-subunit variants. For instance, GRIN2B variants that are linked to NDDs like intellectual disability, autism, and epilepsy. Functional analyses classify these variants as gain- or loss-of-function based on assays that assess key receptor properties such as agonist sensitivity and ion channel function. This framework for categorizing variants plays a critical role in precision medicine, helping to guide targeted therapies by determining whether a variant enhances or impairs NMDAR function, ultimately aiding in better diagnosis and treatment strategies for ion channel-related disorders [219,220].
Looking ahead, several avenues for future research stand out. There is a pressing need for more detailed studies on the less-explored GluN2C and GluN2D subunits, whose distinct expression profiles suggest unique functions in the CNS. Additionally, the role of alternative splicing in generating NMDAR diversity warrants further investigation, particularly in the context of disease. Understanding these mechanisms could unlock new therapeutic targets, offering hope for more effective interventions. As we continue to advance our genomic and neuropharmacological tools, the potential for precision medicine in treating NMDAR-related disorders becomes increasingly tangible, promising a future where therapies are as unique as the patients they are designed to help.
In conclusion, NMDARs are pivotal to both normal brain function and the pathogenesis of a wide range of neurodevelopmental disorders. By unraveling the complex genetic, structural, and functional aspects of these receptors, we can better understand their roles in health and disease, ultimately guiding the development of innovative therapeutic strategies. The ongoing progress in this field holds great promise for improving clinical outcomes and enhancing the quality of life for individuals affected by these challenging conditions.
Author Contributions
Conceptualization, S.S.; literature search and original draft preparation, R.T.; review of the original draft and editing, S.S.; review of the subsequent draft and editing, T.G.S.; All authors have read and agreed to the published version of the manuscript.
Funding
The authors would like to thank the Israel Science Foundation (ISF grant 1994/21 and 3252/21), 2bcured (Improving treatment options for GRIN2B Related Neurodevelopmental Disorder), and Zuckerman (Zuckerman STEM leadership program) for funding and support to Shani Stern. The article processing charge (APC) was funded by the University of Haifa.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Crupi R, Impellizzeri D, Cuzzocrea S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front Mol Neurosci. 2019; 12: 20.
- Watkins JC, Jane DE. The glutamate story. Br J Pharmacol. 2006; 147: 100-8.
- Michael Hollmann AOS-G, Scott W. Rogers & Stephan Heinemann. Cloning by functional expression of a member of the glutamte receptor family. Nature. 1989; 342: 643-8.
- Seeburg, PH. The TiPS/TINS Lecture: The molecular biology of mammalian glutamate receptors. TiPS Reviews. 1993; 14: 297-303.
- Heinemann MHaS. Cloned Glutamate Receptors. Annu Rev Neurosci. 1994; 17: 31-108.
- SEIJI OZAWA. Glutamate receptors in the mammalian central nervous system. Prog Neurobiol. 1998; 54: 581-618.
- Hiroyuki Sugiyama II, Chikara Hirono. A new type of glutamate receptor linked to inositol phospholipid metabolism. Nature. 1987; 325: 531-3.
- Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010; 50: 295-322.
- Petronella Kettunen PK, Dietmar Hess, Abdeljabbar El Manira. Signaling Mechanisms of Metabotropic Glutamate Receptor 5 Subtype and Its Endogenous Role in a Locomotor Network. J Neurosci. 2002; 22: 1868–73.
- Pin JP, Galvez T, Prezeau L. Evolution, structure, and activation mechanism of family 3/C G-protein-coupled receptors. Pharmacol Ther. 2003; 98: 325-54.
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010; 62: 405-96.
- Elisa Carrillo1 CUG, 2, Vladimir Berka1, Vasanthi Jayaraman1,2*. Delta glutamate receptors are functional glycine- and d-serine–gated cation channels in situ. Sci Adv. 2021; 7: 1-9.
- Jane, D. AMPA glutatmate receptors. xPharm: The Comprehensive Pharmacology Reference. 2007:1-19.
- Yang Y, Calakos N. Presynaptic long-term plasticity. Front Synaptic Neurosci. 2013; 5: 8.
- Gan Q, Salussolia CL, Wollmuth LP. Assembly of AMPA receptors: mechanisms and regulation. J Physiol. 2015; 593: 39-48.
- Sabine, M. Schmid MH. To Gate or not to Gate: Are the Delta Subunits in the Glutamate Receptor Family Functional Ion Channels?. Mol Neurobiol. 2008; 37: 126-41.
- Orth A, Tapken D, Hollmann M. The delta subfamily of glutamate receptors: characterization of receptor chimeras and mutants. Eur J Neurosci. 2013; 37: 1620-30.
- Peter Naur* KBH, Anders S. Kristensen‡, Shashank M. Dravid‡, Darryl S. Pickering§, Lars Olsen*, Bente Vestergaard*, Jan Egebjerg†, Michael Gajhede*, Stephen F. Traynelis‡, and Jette S. Kastrup*¶. Ionotropic glutamate-like receptor δ2 binds D-serine and glycine. PNAS. 2007; 104: 14116–21.
- Gao J, Maison SF, Wu X, Hirose K, Jones SM, Bayazitov I, et al. Orphan glutamate receptor delta1 subunit required for high-frequency hearing. Mol Cell Biol. 2007; 27: 4500-12.
- Yuzaki, M. The delta2 glutamate receptor: a key molecule controlling synaptic plasticity and structure in Purkinje cells. Cerebellum. 2004; 3: 89-93.
- Yadav R, Gupta SC, Hillman BG, Bhatt JM, Stairs DJ, Dravid SM. Deletion of glutamate delta-1 receptor in mouse leads to aberrant emotional and social behaviors. PLoS One. 2012; 7: e32969.
- Bowie, D. Ionotropic Glutamate Receptors & CNS Disorders. CNS Neurol Disord Drug Targets. 2008; 7: 129-143.
- Balazs R, Bridges RJ, Cotman CW, Cotman CA. Glutamate and Glutamate Receptors in Neurological Diseases. Excitatory Amino Acid Transmission in Health and Disease, 2005.
- Ragnarsson L, Dodd PR, Hynd MR. Role of Ionotropic Glutamate Receptors in Neurodegenerative and Other Disorders. In: Kostrzewa RM, editor. Handbook of Neurotoxicity. New York, NY: Springer New York; 2014. p. 1039-70.
- Chen TS, Huang TH, Lai MC, Huang CW. The Role of Glutamate Receptors in Epilepsy. Biomedicines. 2023; 11: 738.
- Hansen KB, Wollmuth LP, Bowie D, Furukawa H, Menniti FS, Sobolevsky AI, et al. Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol Rev. 2021; 73: 298-487.
- Xia S, Miyashita T, Fu TF, Lin WY, Wu CL, Pyzocha L, et al. NMDA receptors mediate olfactory learning and memory in Drosophila. Curr Biol. 2005; 15: 603-15.
- Douglas Forrest M, * Holly D. Soares,*, Lily Ng DCL, * Morgan Sheng,+ Colin 1. Stewart,* James I. Morgan,* John A. Connor,* and Tom Curran*. Targeted Disruption of NMDA Receptor 1 Gene Abolishes NMDA Response and Results in Neonatal Death. Neuron. 1994; 13: 325-38.
- Gil-da-Costa R, Stoner GR, Fung R, Albright TD. Nonhuman primate model of schizophrenia using a noninvasive EEG method. PNAS. 2013; 110: 15425-30.
- Philip D Campbell, MG. Zebrafish as a tool to study schizophrenia-associated copy number variants. Disease Models & Mechanisms. 2020; 13: 1-24.
- Lim CS, Kim MJ, Choi JE, Islam MA, Lee YK, Xiong Y, et al. Dysfunction of NMDA receptors in neuronal models of an autism spectrum disorder patient with a DSCAM mutation and in Dscam-knockout mice. Mol Psychiatry. 2021; 26: 7538-7549.
- Petralia RS, Wang YX, Hua F, Yi Z, Zhou A, Ge L, et al. Organization of NMDA receptors at extrasynaptic locations. NeuroScience. 2010; 167: 68-87.
- Ralf Mohrmann HHaKG. Developmental regulation of subunit composition of extrasynaptic NMDA receptors in neocortical neurones. Dev Neurosci. 2000; 11: 1203-1208.
- Chang HR, Kuo CC. The activation gate and gating mechanism of the NMDA receptor. J Neurosci. 2008; 28: 1546-1556.
- Jeffrey, A. Dzubay CEJ. Kinetics of NMDA Channel Opening. The Journal of NeuroScience. 1996; 16: 4129–4134.
- Kawamoto EM, Vivar C, Camandola S. Physiology and pathology of calcium signaling in the brain. Front Pharmacol. 2012; 3: 61.
- Mauceri D, Freitag HE, Oliveira AM, Bengtson CP, Bading H. Nuclear calcium-VEGFD signaling controls maintenance of dendrite arborization necessary for memory formation. Neuron. 2011; 71: 117-30.
- Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J Neurosci. 2005; 25: 4279-87.
- Saneyoshi T, Fortin DA, Soderling TR. Regulation of spine and synapse formation by activity-dependent intracellular signaling pathways. Curr Opin Neurobiol. 2010; 20: 108-15.
- Sprengel R, Eltokhi A. Ionotropic Glutamate Receptors (and Their Role in Health and Disease). NeuroScience in the 21st Century. 2022. p. 57-86.
- Suzuki Y, Nakamoto C, Watanabe-Iida I, Watanabe M, Takeuchi T, Sasaoka T, et al. Quantitative analysis of NMDA receptor subunits proteins in mouse brain. Neurochem Int. 2023; 165: 105517.
- Li H, Rajani V, Han L, Chung D, Cooke JE, Sengar AS, et al. Alternative splicing of GluN1 gates glycine site-dependent nonionotropic signaling by NMDAR receptors. PNAS. 2021; 118: e2026411118.
- R. Suzanne Zukin MVLB. Alternatively spliced isoforms of the NMDAR I receptor subunit. Trends in NeuroSciences. 1995; 18: 306-13.
- Lin Y, Skeberdis VA, Francesconi A, Bennett MV, Zukin RS. Postsynaptic density protein-95 regulates NMDA channel gating and surface expression. J Neurosci. 2004; 24: 10138-10148.
- Incontro S, Diaz-Alonso J, Iafrati J, Vieira M, Asensio CS, Sohal VS, et al. The CaMKII/NMDA receptor complex controls hippocampal synaptic transmission by kinase-dependent and independent mechanisms. Nat Commun. 2018; 9: 2069.
- Michael, D. Ehlers ETF, Richard J. O’Brien, Richard L. Huganir. Splice Variant-Specific Interaction of the NMDA Receptor Subunit NR1 with Neuronal Intermediate Filaments. J Neurosci. 1998; 18: 720-30.
- Guylaine, M. Durand mvlb, R. Suzanne zukin. Splice variants of the N-methyl-D-aspartate receptor NR1 identify domains involved in regulation by polyamines and protein kinase C. PNAS. 1993; 90: 6731-5.
- Ling zhang xz, Marie-christine paupard, Alice p. Wang, Linda santchi, Linda k. Friedman, r. Suzanne zukin, Michael v. L. Bennett. Spermine potentiation of recombinant N-methyl-D-aspartate receptors is affected by subunit composition. PNAS. 1994; 91: 10883-10887.
- Gavin rumbaugh kp, Jian feng wang, Stefano vicini. Exon 5 and Spermine Regulate Deactivation of NMDA Receptor Subtypes. J Neurophysiol. 2000; 83: 1300-1306.
- Vance KM, Hansen KB, Traynelis SF. GluN1 splice variant control of GluN1/GluN2D NMDA receptors. J Physiol. 2012; 590: 3857-3875.
- Vance KM, Simorowski N, Traynelis SF, Furukawa H. Ligand-specific deactivation time course of GluN1/GluN2D NMDA receptors. Nat Commun. 2011; 2: 294.
- Vieira M, Yong XLH, Roche KW, Anggono V. Regulation of NMDA glutamate receptor functions by the GluN2 subunits. J Neurochem. 2020; 154: 121-143.
- Hannah Monyer NB, David J. Laurie, Bert Sakmann, Peter H. Seeburg. Developmental and Regional Expression in the Rat Brain and Functional Properties of Four NMDA Receptors. Neuron. 1994; 12: 529-540.
- Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol. 1994; 347: 150-160.
- Sheng M, Cummings, J., Roldan, L. A., Jan, Y. N., Jan, L. Y. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994; 368: 144–147.
- Wenzel A, Fritschy JM, Mohler H, Benke D. NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J Neurochem. 1997; 68: 469-478.
- Xi D, Keeler B, Zhang W, Houle JD, Gao WJ. NMDA receptor subunit expression in GABAergic interneurons in the prefrontal cortex: application of laser microdissection technique. J Neurosci Methods. 2009; 176: 172-181.
- Perszyk RE, DiRaddo JO, Strong KL, Low CM, Ogden KK, Khatri A, et al. GluN2D-Containing N-methyl-d-Aspartate Receptors Mediate Synaptic Transmission in Hippocampal Interneurons and Regulate Interneuron Activity. Mol Pharmacol. 2016; 90: 689-702.
- Stephen, G. Brickley CM, M. H. Selina Mok, Masayoshi Mishina, Stuart G. Cull-Candy. NR2B and NR2D Subunits Coassemble in Cerebellar Golgi Cells to Form a Distinct NMDA Receptor Subtype Restricted to Extrasynaptic Sites. J Neurosci. 2003; 23: 4958–4966.
- Charu Misra SGB, Mark Farrant, Stuart G. Cull-Candy. Identification of subunits contributing to synaptic and extrasynaptic NMDA receptors in Golgi cells of the rat cerebellum. J Physiol. 2000; 524: 147-162.
- Andres Barria, RM. Subunit-Specific NMDA Receptor Trafficking to Synapses. Neuron. 2002; 35: 345-353.
- Kenneth, R. Tovar GLW. The Incorporation of NMDA Receptors with a Distinct Subunit Composition at Nascent Hippocampal Synapses In Vitro. J Neurosci. 1999; 19: 4180–4188.
- Herbrechter R, Hube N, Buchholz R, Reiner A. Splicing and editing of ionotropic glutamate receptors: a comprehensive analysis based on human RNA-Seq data. Cell Mol Life Sci. 2021; 78: 5605-5630.
- Warming H, Pegasiou CM, Pitera AP, Kariis H, Houghton SD, Kurbatskaya K, et al. A primate-specific short GluN2A-NMDA receptor isoform is expressed in the human brain. Mol Brain. 2019; 12: 64.
- Hiroyuki Meguro HM, Kazuaki Araki, Etsuko Kushiya, Tatsuya Kutsuwada, Makoto Yamazaki, Toshiro Kumanishi, Masaaki Arakawa, Kenji Sakimura, Masayoshi Mishina Functional Characterization of heteromeric NMDA receptor channel expressed from cloned cDNAs. Nature 1992; 357: 70–74.
- Hannah Monyer RS, Ralf Schoepfer, Anne Herb, Miyoko Higuchi, Hilda Lomeli, Nail Burnashev, Bert Sakmann, Peter H. Seeburg. Heteromeric NMDA Receptors: Molecular and Functional Distinction of Subtypes. Science. 1992; 256: 1217-1221.
- Tabish M, Ticku MK. Alternate splice variants of mouse NR2B gene. Neurochem Int. 2004; 44: 339-343.
- Takahiro Ishii KM, Hidemitsu Sugihara, Kazuhiro Sakurada, Hiroshi Kadotani, Mineto Yokoi, Chihiro Akazawa, Ryuichi Shigemoto, Noboru Mizuno, Masayuki Masu, Shigetada Nakanishi. Molecular Characterization of the Family of the N-Methyl-D-Aspartate Receptor Subunits. JBC. 1993; 268: 2836-2843.
- Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. 2010; 42: 1021-1026.
- Brian, J. O’Roak LV, Wenqing Fu, Jarrett D. Egertson, Ian B. Stanaway, Ian G. Phelps, Gemma Carvill, Akash Kumar, Choli Lee, Katy Ankenman, Jeff Munson, Joseph B. Hiatt, Emily H. Turner, Roie Levy, Diana R. O’Day, Niklas Krumm, Bradley P. Coe, Beth K. Martin, Elhanan Borenstein, Deborah A. Nickerson, Heather C. Mefford, Dan Doherty, Joshua M. Akey, Raphael Bernier, Evan E. Eichler, Jay Shendure. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science. 2012; 338: 1619-1622.
- EM Kenny PC, S Furlong, E Heron, G Kenny, C Fahey, E Kelleher, S Ennis, D Tropea, R Anney, AP Corvin, G Donohoe, L Gallagher, M Gill, DW Morris. Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol Psychiatry. 2014; 19: 872–879.
- Myers RA, Casals F, Gauthier J, Hamdan FF, Keebler J, Boyko AR, et al. A population genetic approach to mapping neurological disorder genes using deep resequencing. PLoS Genet. 2011; 7(2): e1001318.
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012; 367: 1921-1929.
- Strehlow V, Heyne HO, Vlaskamp DRM, Marwick KFM, Rudolf G, de Bellescize J, et al. GRIN2A-related disorders: genotype and functional consequence predict phenotype. Brain. 2019; 142: 80-92.
- Garcia-Recio A, Santos-Gomez A, Soto D, Julia-Palacios N, Garcia-Cazorla A, Altafaj X, et al. GRIN database: A unified and manually curated repertoire of GRIN variants. Hum Mutat. 2021; 42: 8-18.
- Liu XR, Xu XX, Lin SM, Fan CY, Ye TT, Tang B, et al. GRIN2A Variants Associated With Idiopathic Generalized Epilepsies. Front Mol Neurosci. 2021; 14: 720984.
- Harrison PJ, Bannerman DM. GRIN2A (NR2A): a gene contributing to glutamatergic involvement in schizophrenia. Mol Psychiatry. 2023; 28: 3568-3572.
- Shepard N, Baez-Nieto D, Iqbal S, Kurganov E, Budnik N, Campbell AJ, et al. Differential functional consequences of GRIN2A mutations associated with schizophrenia and neurodevelopmental disorders. Sci Rep. 2024; 14: 2798.
- Tarabeux J, Kebir O, Gauthier J, Hamdan FF, Xiong L, Piton A, et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry. 2011; 1: e55.
- Singh T, Poterba T, Curtis D, Akil H, Al Eissa M, Barchas JD, et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature. 2022; 604: 509-16.
- Camp CR, Vlachos A, Klockner C, Krey I, Banke TG, Shariatzadeh N, et al. Loss of Grin2a causes a transient delay in the electrophysiological maturation of hippocampal parvalbumin interneurons. Commun Biol. 2023; 6: 952.
- Kellner S, Berlin S. Rescuing tri-heteromeric NMDA receptor function: the potential of pregnenolone-sulfate in loss-of-function GRIN2B variants. Cell Mol Life Sci. 2024; 81: 235.
- Hu C, Chen W, Myers SJ, Yuan H, Traynelis SF. Human GRIN2B variants in neurodevelopmental disorders. J Pharmacol Sci. 2016; 132: 115-121.
- Epi KC, Epilepsy Phenome/Genome P, Allen AS, Berkovic SF, Cossette P, Delanty N, et al. De novo mutations in epileptic encephalopathies. Nature. 2013; 501: 217-221.
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011; 43: 585-589.
- Bahry JA, Fedder-Semmes KN, Sceniak MP, Sabo SL. An Autism-Associated de novo Mutation in GluN2B Destabilizes Growing Dendrites by Promoting Retraction and Pruning. Front Cell Neurosci. 2021; 15: 692232.
- Sceniak MP, Fedder KN, Wang Q, Droubi S, Babcock K, Patwardhan S, et al. An autism-associated mutation in GluN2B prevents NMDA receptor trafficking and interferes with dendrite growth. J Cell Sci. 2019; 132: jcs232892.
- Weickert CS, Fung SJ, Catts VS, Schofield PR, Allen KM, Moore LT, et al. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol Psychiatry. 2013; 18: 1185-1192.
- Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology. 2008; 33(9):2175-86.
- S. Akbarian NJS, D. Bradley,’ A. Tafazzoli, l D. Trinh, l W. P. Hetrick, S. G. Potkin,, C. A. Sandman WEB, Jr., E. G. Jones. Selective Alterations in Gene Expression for NMDA Receptor Subunits in Prefrontal Cortex of Schizophrenics The Journal of NeuroScience. 1996; 16: 19-30.
- Catts VS, Lai YL, Weickert CS, Weickert TW, Catts SV. A quantitative review of the postmortem evidence for decreased cortical N-methyl-D-aspartate receptor expression levels in schizophrenia: How can we link molecular abnormalities to mismatch negativity deficits? Biol Psychol. 2016; 116: 57-67.
- Li D, Yuan H, Ortiz-Gonzalez XR, Marsh ED, Tian L, McCormick EM, et al. GRIN2D Recurrent De Novo Dominant Mutation Causes a Severe Epileptic Encephalopathy Treatable with NMDA Receptor Channel Blockers. Am J Hum Genet. 2016; 99: 802-816.
- Yu Y, Lin Y, Takasaki Y, Wang C, Kimura H, Xing J, et al. Rare loss of function mutations in N-methyl-D-aspartate glutamate receptors and their contributions to schizophrenia susceptibility. Transl Psychiatry. 2018; 8: 12.
- Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, et al. Molecular Architecture of the Mouse Nervous System. Cell. 2018; 174: 999-1014.
- Matthaei, KI. Genetically manipulated mice: a powerful tool with unsuspected caveats. J Physiol. 2007; 582:481-488.
- Dali Li ZQ, Yanjiao Shao, Yuting Chen, Yuting Guan, Meizhen Liu, Yongmei Li, Na Gao, Liren Wang, Xiaoling Lu, Yongxiang Zhao, Mingyao Liu. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol. 2013; 31: 681-683.
- Wohr M, Scattoni ML. Behavioural methods used in rodent models of autism spectrum disorders: current standards and new developments. Behav Brain Res. 2013; 251: 5-17.
- Amy, R. Mohn RRG, Marc G. Caron, Beverly H. Koller. Mice with Reduced NMDA Receptor Expression Display Behaviors Related to Schizophrenia. Cell. 1999; 98: 427-36.
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010; 13: 76-83.
- Xu W, Morishita W, Buckmaster PS, Pang ZP, Malenka RC, Sudhof TC. Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron. 2012; 73: 990-1001.
- Yuqing Li RSE, Chong Chen, Sonal Jhaveri, Susumu Tonegawa. Whisker-Related Neuronal Patterns Fail to Develop in the Trigeminal Brainstem Nuclei of NMDARI Knockout Mice. Cell. 1994; 76: 427-37.
- Tatsuya Kutsuwada KS, Toshiya Manabe, Chitoshi Takayama,, Nobuo Katakura EK, Rie Natsume, Masahiko Watanabe, Yoshiro Inoue, Takeshi Yagi, Shinichi Aizawa, Masaaki Arakawa, Tomoyuki Takahashi, Yoshio Nakamura, Hisashi Mori, Masayoshi Mishina. Impairment of Suckling Response, Trigeminal Neuronal Pattern Formation, and Hippocampal LTD in NMDA Receptor E2 Subunit Mutant Mice. Neuron. 1996; 16: 333–344.
- Bialon M, Wasik A. Advantages and Limitations of Animal Schizophrenia Models. Int J Mol Sci. 2022; 23(11).
- Yavas E, Young AMJ. Repeated phencyclidine disrupts nicotinic acetylcholine regulation of dopamine release in nucleus accumbens: Implications for models of schizophrenia. Neurochem Int. 2020; 140: 104836.
- Cao T, Tang M, Jiang P, Zhang B, Wu X, Chen Q, et al. A Potential Mechanism Underlying the Therapeutic Effects of Progesterone and Allopregnanolone on Ketamine-Induced Cognitive Deficits. Front Pharmacol. 2021; 12: 612083.
- Chen G, Lin X, Li G, Jiang D, Lib Z, Jiang R, et al. Risperidone reverses the spatial object recognition impairment and hippocampal BDNF-TrkB signalling system alterations induced by acute MK-801 treatment. Biomed Rep. 2017; 6: 285-290.
- Sobolevsky AI, Yelshansky MV. The trapping block of NMDA receptor channels in acutely isolated rat hippocampal neurones. J Physiol. 2000; 526: 493-506.
- Wallach J, Kang H, Colestock T, Morris H, Bortolotto ZA, Collingridge GL, et al. Pharmacological Investigations of the Dissociative ‘Legal Highs’ Diphenidine, Methoxphenidine and Analogues. PLoS One. 2016; 11: e0157021.
- Vales K, Holubova K. Minireview: Animal model of schizophrenia from the perspective of behavioral pharmacology: Effect of treatment on cognitive functions. Neurosci Lett. 2021; 761: 136098.
- Salmi M, Bolbos R, Bauer S, Minlebaev M, Burnashev N, Szepetowski P. Transient microstructural brain anomalies and epileptiform discharges in mice defective for epilepsy and language-related NMDA receptor subunit gene Grin2a. Epilepsia. 2018; 59: 1919-1930.
- Kazutaka Ikeda KA, Chitoshi Takayama, Yoshiro Inoue, Takeshi Yagi, Shinichi Aizawa, Masayoshi Mishina. Reduced spontaneous activity of mice defective in the e4 subunit of the NMDA receptor channel. Brain Res Mol Brain Res. 1995; 33: 61-71.
- Alexander, K. Ebralidze DJR, 2 Susumu Tonegawa,1 and N. Traverse Slater2. Modification of NMDA Receptor Channels and Synaptic Transmission by Targeted Disruption of the NR2C Gene. J Neurosci. 1996; 16: 5014-5025.
- Saumya Das* YFS, Thomas Rothe†‡, Louis S. Premkumar§, Mari Takasu*, James E. Crandallk¶, Pieter Dikkes#, David A. Conner$, Posina V. Rayudu‡, Wing Cheung‡, H.-S. Vincent Chen‡, Stuart A. Lipton‡¶ & Nobuki Nakanishi*¶. Increased NMDA current and spine density in mice lacking the NMDA receptor subunit NR3A. Nature 1998; 393: 377-381.
- Takuji Iwasato RSE, † Patricio T. Huerta,* Dong Feng Chen,* Toshikuni Sasaoka,* Emel Ulupinar,† and Susumu Tonegawa*. NMDA Receptor-Dependent Refinement of Somatotopic Maps. Neuron. 1997; 19: 1201–1210.
- Lee LJ, Lo FS, Erzurumlu RS. NMDA receptor-dependent regulation of axonal and dendritic branching. J Neurosci. 2005; 25: 2304-2311.
- Duncan GE, Moy SS, Perez A, Eddy DM, Zinzow WM, Lieberman JA, et al. Deficits in sensorimotor gating and tests of social behavior in a genetic model of reduced NMDA receptor function. Behav Brain Res. 2004; 153: 507-519.
- Moy SS, Perez A, Koller BH, Duncan GE. Amphetamine-induced disruption of prepulse inhibition in mice with reduced NMDA receptor function. Brain Res. 2006; 1089: 186-194.
- Bickel S, Lipp HP, Umbricht D. Impaired attentional modulation of auditory evoked potentials in N-methyl-D-aspartate NR1 hypomorphic mice. Genes Brain Behav. 2007; 6: 558-568.
- Dzirasa K, Ramsey AJ, Takahashi DY, Stapleton J, Potes JM, Williams JK, et al. Hyperdopaminergia and NMDA receptor hypofunction disrupt neural phase signaling. J Neurosci. 2009; 29: 8215-8224.
- Halene TB, Ehrlichman RS, Liang Y, Christian EP, Jonak GJ, Gur TL, et al. Assessment of NMDA receptor NR1 subunit hypofunction in mice as a model for schizophrenia. Genes Brain Behav. 2009; 8: 661-675.
- Wang W, Rein B, Zhang F, Tan T, Zhong P, Qin L, et al. Chemogenetic Activation of Prefrontal Cortex Rescues Synaptic and Behavioral Deficits in a Mouse Model of 16p11.2 Deletion Syndrome. J Neurosci. 2018; 38: 5939-5948.
- do Nascimento P, Oliveira Silva DF, de Morais T, de Rezende AA. Zinc Status and Autism Spectrum Disorder in Children and Adolescents: A Systematic Review. Nutrients. 2023; 15: 3663.
- Sauer AK, Hagmeyer S, Grabrucker AM. Prenatal Zinc Deficient Mice as a Model for Autism Spectrum Disorders. Int J Mol Sci. 2022; 23: 6082.
- Faber S, Zinn GM, Kern JC, 2nd, Kingston HM. The plasma zinc/serum copper ratio as a biomarker in children with autism spectrum disorders. Biomarkers. 2009; 14: 171-180.
- Lee K, Jung Y, Vyas Y, Skelton I, Abraham WC, Hsueh YP, et al. Dietary zinc supplementation rescues fear-based learning and synaptic function in the Tbr1(+/-) mouse model of autism spectrum disorders. Mol Autism. 2022; 13:13.
- Shin W, Kim K, Serraz B, Cho YS, Kim D, Kang M, et al. Early correction of synaptic long-term depression improves abnormal anxiety-like behavior in adult GluN2B-C456Y-mutant mice. PLoS Biol. 2020; 18: e3000717.
- Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet. 2010; 42: 489-91.
- Leblond CS, Heinrich J, Delorme R, Proepper C, Betancur C, Huguet G, et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012; 8: e1002521.
- Delling JP, Boeckers TM. Comparison of SHANK3 deficiency in animal models: phenotypes, treatment strategies, and translational implications. J Neurodev Disord. 2021; 13: 55.
- Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011; 472: 437-442.
- Fourie C, Vyas Y, Lee K, Jung Y, Garner CC, Montgomery JM. Dietary Zinc Supplementation Prevents Autism Related Behaviors and Striatal Synaptic Dysfunction in Shank3 Exon 13-16 Mutant Mice. Front Cell Neurosci. 2018; 12: 374.
- Ey E, Leblond CS, Bourgeron T. Behavioral profiles of mouse models for autism spectrum disorders. Autism Res. 2011; 4: 5-16.
- Tordjman S, Drapier D, Bonnot O, Graignic R, Fortes S, Cohen D, et al. Animal models relevant to schizophrenia and autism: validity and limitations. Behav Genet. 2007; 37: 61-78.
- Tania M, Khan MA, Xia K. Recent advances in animal model experimentation in autism research. Acta Neuropsychiatr. 2014; 26: 264-271.
- Feng G, Jensen FE, Greely HT, Okano H, Treue S, Roberts AC, et al. Opportunities and limitations of genetically modified nonhuman primate models for neuroscience research. PNAS. 2020; 117: 24022-24031.
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126: 663-676.
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131: 861-872.
- Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010; 143: 527-539.
- Vahsen BF, Gray E, Candalija A, Cramb KML, Scaber J, Dafinca R, et al. Human iPSC co-culture model to investigate the interaction between microglia and motor neurons. Sci Rep. 2022; 12: 12606.
- Nascimento JM, Saia-Cereda VM, Zuccoli GS, Reis-de-Oliveira G, Carregari VC, Smith BJ, et al. Proteomic signatures of schizophrenia-sourced iPSC-derived neural cells and brain organoids are similar to patients’ postmortem brains. Cell Biosci. 2022; 12: 189.
- Fan Y, Li Y, Yang X, Zhang H, Wang B, Guan J, et al. Generation and characterization of PBMCs-derived human induced pluripotent stem cell (iPSC) line SDQLCHi051-A from an autism spectrum disorder patient with compound CHD8 gene mutations. Stem Cell Res. 2023; 69: 103114.
- Longobardi E, Miceli F, Secondo A, Cicatiello R, Izzo A, Tinto N, et al. Generation of an iPSC line (UNINAi001-A) from a girl with neonatal-onset epilepsy and non-syndromic intellectual disability carrying the homozygous KCNQ3 p.PHE534ILEfs*15 variant and of an iPSC line (UNINAi002-A) from a non-carrier, unaffected brother. Stem Cell Res. 2021; 53: 102311.
- Stern S, Sarkar A, Stern T, Mei A, Mendes APD, Stern Y, et al. Mechanisms Underlying the Hyperexcitability of CA3 and Dentate Gyrus Hippocampal Neurons Derived from Patients with Bipolar Disorder. Biol Psychiatry. 2020; 88: 139-149.
- Zhou YY, Zeng F. Integration-free methods for generating induced pluripotent stem cells. Genomics Proteomics Bioinformatics. 2013; 11: 284-287.
- Nayak R, Rosh I, Rabinski T, Falik D, Mendel Percia M, Stern S. Generation and characterization of iPSC lines (UOHi003-A, UOHi002-A) from a patient with SHANK3 mutation and her healthy mother. Stem Cell Res. 2022; 64: 102899.
- Liu C, Oikonomopoulos A, Sayed N, Wu JC. Modeling human diseases with induced pluripotent stem cells: from 2D to 3D and beyond. Development. 2018; 145: dev156166.
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013; 14: 383-400.
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994; 12: 529-540.
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013; 78: 785-798.
- Stein JL, de la Torre-Ubieta L, Tian Y, Parikshak NN, Hernandez IA, Marchetto MC, et al. A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron. 2014; 83: 69-86.
- Nehme R, Zuccaro E, Ghosh SD, Li C, Sherwood JL, Pietilainen O, et al. Combining NGN2 Programming with Developmental Patterning Generates Human Excitatory Neurons with NMDAR-Mediated Synaptic Transmission. Cell Rep. 2018; 23: 2509-2523.
- Zhang WB, Ross PJ, Tu Y, Wang Y, Beggs S, Sengar AS, et al. Fyn Kinase regulates GluN2B subunit-dominant NMDA receptors in human induced pluripotent stem cell-derived neurons. Sci Rep. 2016; 6: 23837.
- Frega M, Linda K, Keller JM, Gumus-Akay G, Mossink B, van Rhijn JR, et al. Neuronal network dysfunction in a model for Kleefstra syndrome mediated by enhanced NMDAR signaling. Nat Commun. 2019; 10: 4928.
- Neagoe I, Liu C, Stumpf A, Lu Y, He D, Francis R, et al. The GluN2B subunit represents a major functional determinant of NMDA receptors in human induced pluripotent stem cell-derived cortical neurons. Stem Cell Res. 2018; 28: 105-114.
- Ruden JB, Dixit M, Zepeda JC, Grueter BA, Dugan LL. Robust Expression of Functional NMDA Receptors in Human Induced Pluripotent Stem Cell-Derived Neuronal Cultures Using an Accelerated Protocol. Front Mol Neurosci. 2021; 14: 777049.
- Bell S, Maussion G, Jefri M, Peng H, Theroux JF, Silveira H, et al. Disruption of GRIN2B Impairs Differentiation in Human Neurons. Stem Cell Reports. 2018; 11: 183-196.
- Ben-Reuven L, Reiner O. Modeling the autistic cell: iPSCs recapitulate developmental principles of syndromic and nonsyndromic ASD. Dev Growth Differ. 2016; 58: 481-491.
- Lim CS, Yang JE, Lee YK, Lee K, Lee JA, Kaang BK. Understanding the molecular basis of autism in a dish using hiPSCs-derived neurons from ASD patients. Mol Brain. 2015; 8: 57.
- Tripathi MK, Ojha SK, Kartawy M, Hamoudi W, Choudhary A, Stern S, et al. The NO Answer for Autism Spectrum Disorder. Adv Sci. 2023; 10: e2205783.
- Brant B, Stern T, Shekhidem HA, Mizrahi L, Rosh I, Stern Y, et al. IQSEC2 mutation associated with epilepsy, intellectual disability, and autism results in hyperexcitability of patient-derived neurons and deficient synaptic transmission. Mol Psychiatry. 2021; 26: 7498-7508.
- Boeckers TM, Bockmann J, Kreutz MR, Gundelfinger ED. ProSAP/Shank proteins - a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J Neurochem. 2002; 81: 903-910.
- Kreienkamp, HJ. Scaffolding proteins at the postsynaptic density: shank as the architectural framework. Handb Exp Pharmacol. 2008; 186: 365-380.
- Arons MH, Thynne CJ, Grabrucker AM, Li D, Schoen M, Cheyne JE, et al. Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. J Neurosci. 2012; 32: 14966-14978.
- Amal H, Barak B, Bhat V, Gong G, Joughin BA, Wang X, et al. Shank3 mutation in a mouse model of autism leads to changes in the S-nitroso-proteome and affects key proteins involved in vesicle release and synaptic function. Mol Psychiatry. 2020; 25: 1835-1848.
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012; 74: 285-299.
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012; 485: 237-241.
- Hussein Y, Tripathi U, Choudhary A, Nayak R, Peles D, Rosh I, et al. Early maturation and hyperexcitability is a shared phenotype of cortical neurons derived from different ASD-associated mutations. Transl Psychiatry. 2023; 13: 246.
- Stern S, Zhang L, Wang M, Wright R, Rosh I, Hussein Y, et al. Monozygotic twins discordant for schizophrenia differ in maturation and synaptic transmission. Mol Psychiatry. 2024; 29: 3208-3222.
- Vilain J, Galliot AM, Durand-Roger J, Leboyer M, Llorca PM, Schurhoff F, et al. Environmental risk factors for schizophrenia: a review. Encephale. 2013; 39: 19-28.
- Romanovsky E, Choudhary A, Peles D, Abu-Akel A, Stern S. Uncovering convergence and divergence between autism and schizophrenia using genomic tools and patients’ neurons. Mol Psychiatry 2024. (ahead of print). [CrossRef]
- Dracheva S, Marras SA, Elhakem SL, Kramer FR, Davis KL, Haroutunian V. N-methyl-D-aspartic acid receptor expression in the dorsolateral prefrontal cortex of elderly patients with schizophrenia. Am J Psychiatry. 2001; 158: 1400-1410.
- Le Corre S, Harper CG, Lopez P, Ward P, Catts S. Increased levels of expression of an NMDARI splice variant in the superior temporal gyrus in schizophrenia. Neuroreport. 2000; 11: 983-986.
- Sokolov, BP. Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: evidence on reversible up-regulation by typical neuroleptics. J Neurochem. 1998; 71: 2454-2464.
- Akbarian S, Sucher NJ, Bradley D, Tafazzoli A, Trinh D, Hetrick WP, et al. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J Neurosci. 1996; 16: 19-30.
- Rasanen N, Tiihonen J, Koskuvi M, Lehtonen S, Koistinaho J. The iPSC perspective on schizophrenia. Trends Neurosci. 2022; 45: 8-26.
- Chiang CH, Su Y, Wen Z, Yoritomo N, Ross CA, Margolis RL, et al. Integration-free induced pluripotent stem cells derived from schizophrenia patients with a DISC1 mutation. Mol Psychiatry. 2011; 16: 358-360.
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011; 473: 221-225.
- Balan S, Toyoshima M, Yoshikawa T. Contribution of induced pluripotent stem cell technologies to the understanding of cellular phenotypes in schizophrenia. Neurobiol Dis. 2019; 131: 104162.
- Nakazawa T, Hashimoto R, Takuma K, Hashimoto H. Modeling of psychiatric disorders using induced pluripotent stem cell-related technologies. J Pharmacol Sci. 2019; 140: 321-324.
- Powell SK, O’Shea CP, Shannon SR, Akbarian S, Brennand KJ. Investigation of Schizophrenia with Human Induced Pluripotent Stem Cells. Adv Neurobiol. 2020; 25: 155-206.
- Wen Z, Christian KM, Song H, Ming GL. Modeling psychiatric disorders with patient-derived iPSCs. Curr Opin Neurobiol. 2016; 36: 118-127.
- Choudhary A, Peles D, Nayak R, Mizrahi L, Stern S. Current progress in understanding schizophrenia using genomics and pluripotent stem cells: A meta-analytical overview. Schizophr Res. 2022.
- Fischer, M. Psychoses in the offspring of schizophrenic monozygotic twins and their normal co-twins. Br J Psychiatry. 1971; 118: 43-52.
- Narayan CL, Shikha D, Shekhar S. Schizophrenia in identical twins. Indian J Psychiatry. 2015; 57: 323-324.
- Hilker R, Helenius D, Fagerlund B, Skytthe A, Christensen K, Werge TM, et al. Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register. Biol Psychiatry. 2018; 830: 492-498.
- Castellani CA, Laufer BI, Melka MG, Diehl EJ, O’Reilly RL, Singh SM. DNA methylation differences in monozygotic twin pairs discordant for schizophrenia identifies psychosis related genes and networks. BMC Med Genomics. 2015; 8: 17.
- Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012; 37: 4-15.
- Rosburg T, Kreitschmann-Andermahr I. The effects of ketamine on the mismatch negativity (MMN) in humans - A meta-analysis. Clin Neurophysiol. 2016; 127: 1387-1394.
- Laurens KR, Murphy J, Dickson H, Roberts RE, Gutteridge TP. Trajectories of Mismatch Negativity and P3a Amplitude Development from Ages 9 to 16 Years in Children with Risk Factors for Schizophrenia. Biol Psychiatry Cogn Neurosci Neuroimaging. 2020; 5: 1085-1094.
- Kim NS, Wen Z, Liu J, Zhou Y, Guo Z, Xu C, et al. Pharmacological rescue in patient iPSC and mouse models with a rare DISC1 mutation. Nat Commun. 2021; 12:1398.
- Burrack N, Yitzhaky A, Mizrahi L, Wang M, Stern S, Hertzberg L. Altered Expression of PDE4 Genes in Schizophrenia: Insights from a Brain and Blood Sample Meta-Analysis and iPSC-Derived Neurons. Genes. 2024; 15: 609.
- Gilleen J, Nottage J, Yakub F, Kerins S, Valdearenas L, Uz T, et al. The effects of roflumilast, a phosphodiesterase type-4 inhibitor, on EEG biomarkers in schizophrenia: A randomised controlled trial. J Psychopharmacol. 2021; 35: 15-22.
- Tiihonen J, Koskuvi M, Lahteenvuo M, Trontti K, Ojansuu I, Vaurio O, et al. Molecular signaling pathways underlying schizophrenia. Schizophr Res. 2021; 232: 33-41.
- Yaka R, He DY, Phamluong K, Ron D. Pituitary adenylate cyclase-activating polypeptide (PACAP(1-38)) enhances N-methyl-D-aspartate receptor function and brain-derived neurotrophic factor expression via RACK1. J Biol Chem. 2003; 278: 9630-8.
- Rosenbrock H, Desch M, Wunderlich G. Development of the novel GlyT1 inhibitor, iclepertin (BI 425809), for the treatment of cognitive impairment associated with schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2023; 273: 1557-1566.
- Panizzutti R, Fisher M, Garrett C, Man WH, Sena W, Madeira C, et al. Association between increased serum d-serine and cognitive gains induced by intensive cognitive training in schizophrenia. Schizophr Res. 2019; 207: 63-69.
- O’Donnell P, Dong C, Murthy V, Asgharnejad M, Du X, Summerfelt A, et al. The D-amino acid oxidase inhibitor luvadaxistat improves mismatch negativity in patients with schizophrenia in a randomized trial. Neuropsychopharmacology. 2023; 48: 1052-1059.
- Goff, D. The Therapeutic Role of d-Cycloserine in Schizophrenia. Adv Pharmacol. 2016; 76: 39-66.
- Wu Q, Huang J, Wu R. Drugs Based on NMDAR Hypofunction Hypothesis in Schizophrenia. Front Neurosci. 2021; 15: 641047.
- Chen W, Liu J, Zhang L, Xu H, Guo X, Deng S, et al. Generation of the SCN1A epilepsy mutation in hiPS cells using the TALEN technique. Sci Rep. 2014; 4: 5404.
- Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, et al. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. PNAS. 2010; 107: 17668-17673.
- Ricciardi S, Ungaro F, Hambrock M, Rademacher N, Stefanelli G, Brambilla D, et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat Cell Biol. 2012; 14: 911-923.
- Yamashita S, Chiyonobu T, Yoshida M, Maeda H, Zuiki M, Kidowaki S, et al. Mislocalization of syntaxin-1 and impaired neurite growth observed in a human iPSC model for STXBP1-related epileptic encephalopathy. Epilepsia. 2016; 57: e81-86.
- Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011; 17: 1657-1662.
- Dolce A, Ben-Zeev B, Naidu S, Kossoff EH. Rett syndrome and epilepsy: an update for child neurologists. Pediatr Neurol. 2013; 48: 337-345.
- Cheung AY, Horvath LM, Grafodatskaya D, Pasceri P, Weksberg R, Hotta A, et al. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum Mol Genet. 2011; 20: 2103-2105.
- Maezawa I, Swanberg S, Harvey D, LaSalle JM, Jin LW. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J Neurosci. 2009; 29: 5051-5061.
- Chen S, Xu D, Fan L, Fang Z, Wang X, Li M. Roles of N-Methyl-D-Aspartate Receptors (NMDARs) in Epilepsy. Front Mol Neurosci. 2021; 14: 797253.
- Zehavi Y, Mandel H, Zehavi A, Rashid MA, Straussberg R, Jabur B, et al. De novo GRIN1 mutations: An emerging cause of severe early infantile encephalopathy. Eur J Med Genet. 2017; 60: 317-320.
- Hanada, T. Ionotropic Glutamate Receptors in Epilepsy: A Review Focusing on AMPA and NMDA Receptors. Biomolecules. 2020; 10: 464.
- Sharawat IK, Yadav J, Saini L. Novel GRIN2B mutation: A rare cause of severe epileptic encephalopathy. Neurol India. 2019; 67: 562-563.
- Sun C, Yang M, Qin F, Guo R, Liang S, Hu H. Generation of an induced pluripotent stem cell line SYSUi-003-A from a child with epilepsy carrying GRIN2A mutation. Stem Cell Res. 2020; 43: 101706.
- Rabinski T, Sagiv ST, Hausman-Kedem M, Fattal-Valevski A, Rubinstein M, Avraham KB, et al. Reprogramming of two induced pluripotent stem cell lines from a heterozygous GRIN2D developmental and epileptic encephalopathy (DEE) patient (BGUi011-A) and from a healthy family relative (BGUi012-A). Stem Cell Res. 2021; 51: 102178.
- Shi Z, Liu H, Feng F, Huang Z, Chen WX. Generation of an induced pluripotent stem cell line GWCMCi006-A from a patient with autosomal dominant neurodevelopmental disorder with or without hyperkinetic movements and seizures harboring GRIN1 c.389A > G mutation. Stem Cell Res. 2024; 76: 103371.
- Fedele L, Newcombe J, Topf M, Gibb A, Harvey RJ, Smart TG. Disease-associated missense mutations in GluN2B subunit alter NMDA receptor ligand binding and ion channel properties. Nat Commun. 2018; 9: 957.
- Addis L, Virdee JK, Vidler LR, Collier DA, Pal DK, Ursu D. Epilepsy-associated GRIN2A mutations reduce NMDA receptor trafficking and agonist potency - molecular profiling and functional rescue. Sci Rep. 2017; 7: 66.
- Liu S, Zhou L, Yuan H, Vieira M, Sanz-Clemente A, Badger JD, 2nd, et al. A Rare Variant Identified Within the GluN2B C-Terminus in a Patient with Autism Affects NMDA Receptor Surface Expression and Spine Density. J Neurosci. 2017; 37: 4093-4102.
- Platzer K, Yuan H, Schutz H, Winschel A, Chen W, Hu C, et al. GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J Med Genet. 2017; 54: 460-470.
- Myers SJ, Yuan H, Perszyk RE, Zhang J, Kim S, Nocilla KA, et al. Classification of missense variants in the N-methyl-d-aspartate receptor GRIN gene family as gain- or loss-of-function. Hum Mol Genet. 2023; 32: 2857-2871.
Figure 1.
Structural composition of the NMDA receptors; A) Graphical representation of the subunit composition of NMDA receptors, each monomer of the receptor contains four functional domains, Amino-terminal domain (ATD), Ligand-binding domain (LBD), Transmembrane domain (TMD), and C-terminal domain. B) Ion channel activity of AMPA and Kainate receptors (members of iGluR family). C) Crystal structure displaying 3D conformation of the heterotetramer NMDA receptor containing GluN1 and GluN2B subunits (PDB: 6WHX); GluN3 subunits also form functional receptors with GluN1 subunit. D) Proposed mechanism of GluD2 ion channel activity through interaction with pre-synaptic linker proteins; the representation is adapted from Carillo et al. SciAdv, 2021 [12].
Figure 1.
Structural composition of the NMDA receptors; A) Graphical representation of the subunit composition of NMDA receptors, each monomer of the receptor contains four functional domains, Amino-terminal domain (ATD), Ligand-binding domain (LBD), Transmembrane domain (TMD), and C-terminal domain. B) Ion channel activity of AMPA and Kainate receptors (members of iGluR family). C) Crystal structure displaying 3D conformation of the heterotetramer NMDA receptor containing GluN1 and GluN2B subunits (PDB: 6WHX); GluN3 subunits also form functional receptors with GluN1 subunit. D) Proposed mechanism of GluD2 ion channel activity through interaction with pre-synaptic linker proteins; the representation is adapted from Carillo et al. SciAdv, 2021 [12].
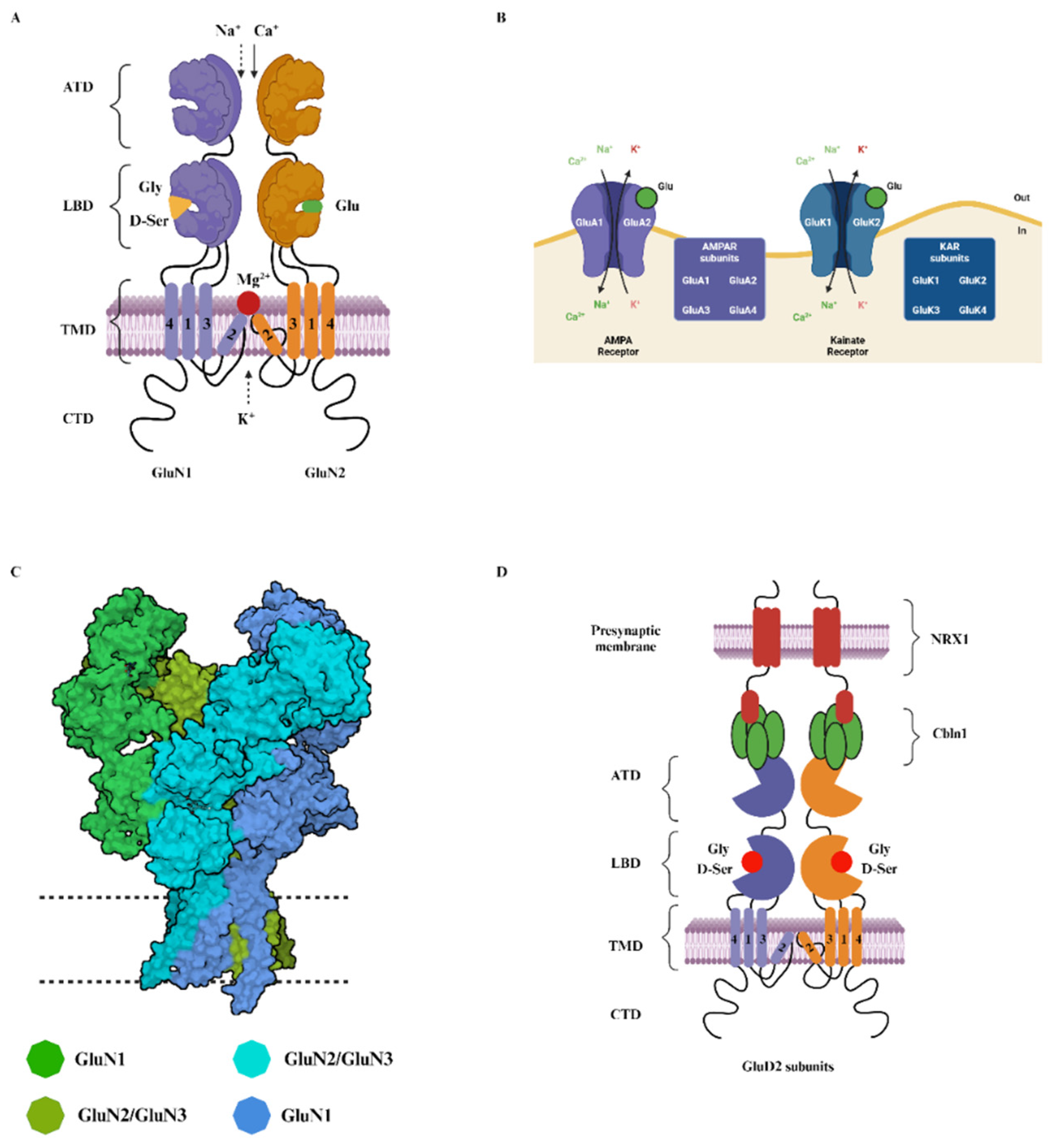
Figure 2.
Activation of the “coincidence receptor”. A) Initial interaction of AMPAR )purple) and NMDAR (blue) with pre-synaptic glutamate; the receptors are inactive at the resting membrane potential (-70 mV), upon interaction with the presynaptic glutamate, NMDAR remains inactive still since Mg2+ is blocking the ion channel pore, while AMPA receptor allows in-flow of Na+ ions initiating the depolarization of the post-synaptic membrane. B) Depolarization of the postsynaptic membrane facilitates the release of Mg2+ ions from the NMAR ion channel pore leading to the activation of receptor. C) Activation of NMDAR and repetitive presynaptic glutamate release leads to increased in-flow of Ca2+ and Na+ ions into the postsynaptic neurons. Resultant calcium signaling induces various cellular processes. Symbols for ions and glutamate are given below the image. The representation is adapted from Sprengel et al. 2022 [40].
Figure 2.
Activation of the “coincidence receptor”. A) Initial interaction of AMPAR )purple) and NMDAR (blue) with pre-synaptic glutamate; the receptors are inactive at the resting membrane potential (-70 mV), upon interaction with the presynaptic glutamate, NMDAR remains inactive still since Mg2+ is blocking the ion channel pore, while AMPA receptor allows in-flow of Na+ ions initiating the depolarization of the post-synaptic membrane. B) Depolarization of the postsynaptic membrane facilitates the release of Mg2+ ions from the NMAR ion channel pore leading to the activation of receptor. C) Activation of NMDAR and repetitive presynaptic glutamate release leads to increased in-flow of Ca2+ and Na+ ions into the postsynaptic neurons. Resultant calcium signaling induces various cellular processes. Symbols for ions and glutamate are given below the image. The representation is adapted from Sprengel et al. 2022 [40].
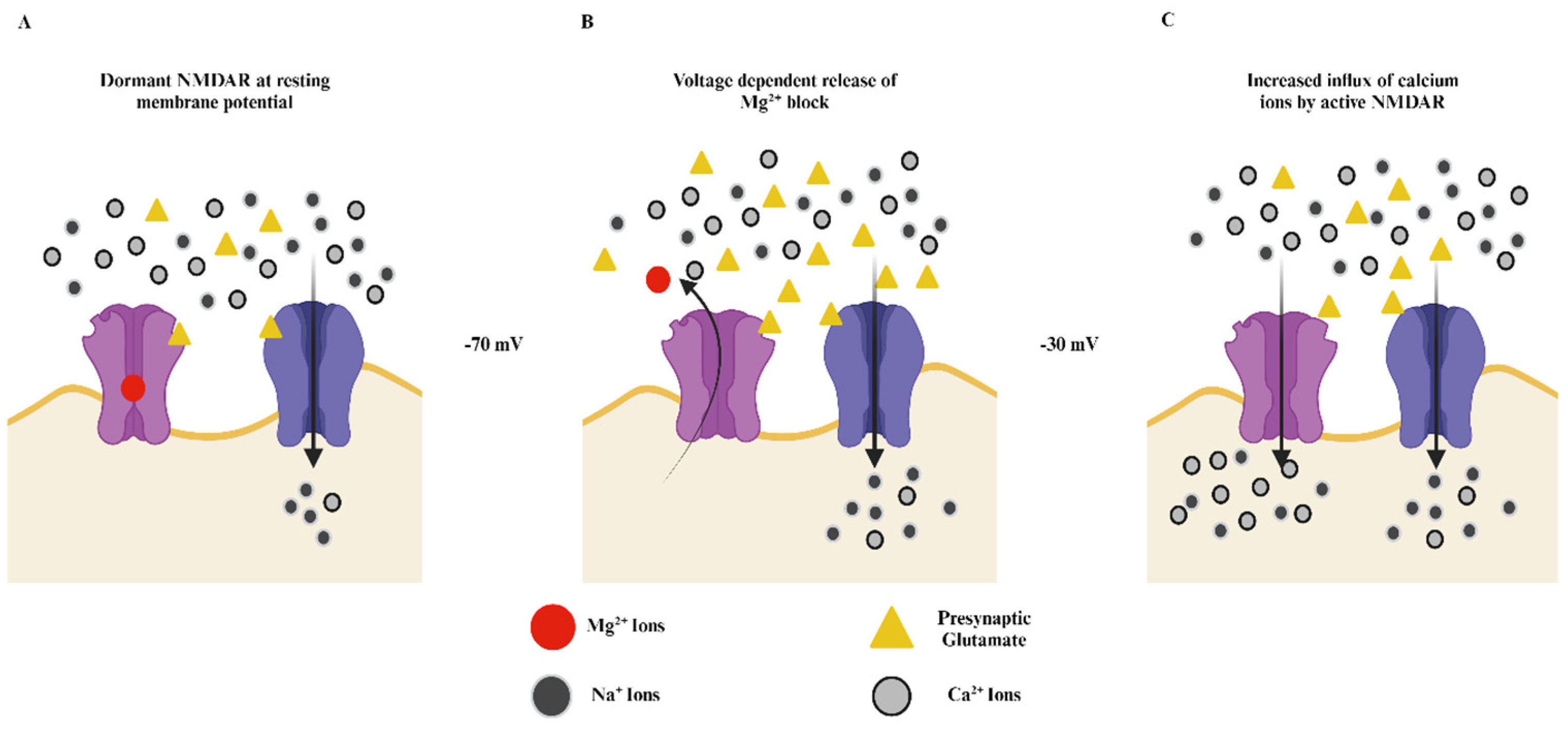
Figure 3.
Schematics of the eight functional isoforms of GluN1 subunit resulting from alternative RNA splicing. Color coding represents the splicing sites at exons 5, 21, and 22 or 22’, denoted as cassettes N1, C1, C2, and C2’, respectively. N1 cassette site is in the amino-terminal domain, whereas C1, C2, and C2’ sites are in the C-terminal domain. The schematics are adapted from Li et al. PNAS, 2021 [42].
Figure 3.
Schematics of the eight functional isoforms of GluN1 subunit resulting from alternative RNA splicing. Color coding represents the splicing sites at exons 5, 21, and 22 or 22’, denoted as cassettes N1, C1, C2, and C2’, respectively. N1 cassette site is in the amino-terminal domain, whereas C1, C2, and C2’ sites are in the C-terminal domain. The schematics are adapted from Li et al. PNAS, 2021 [42].
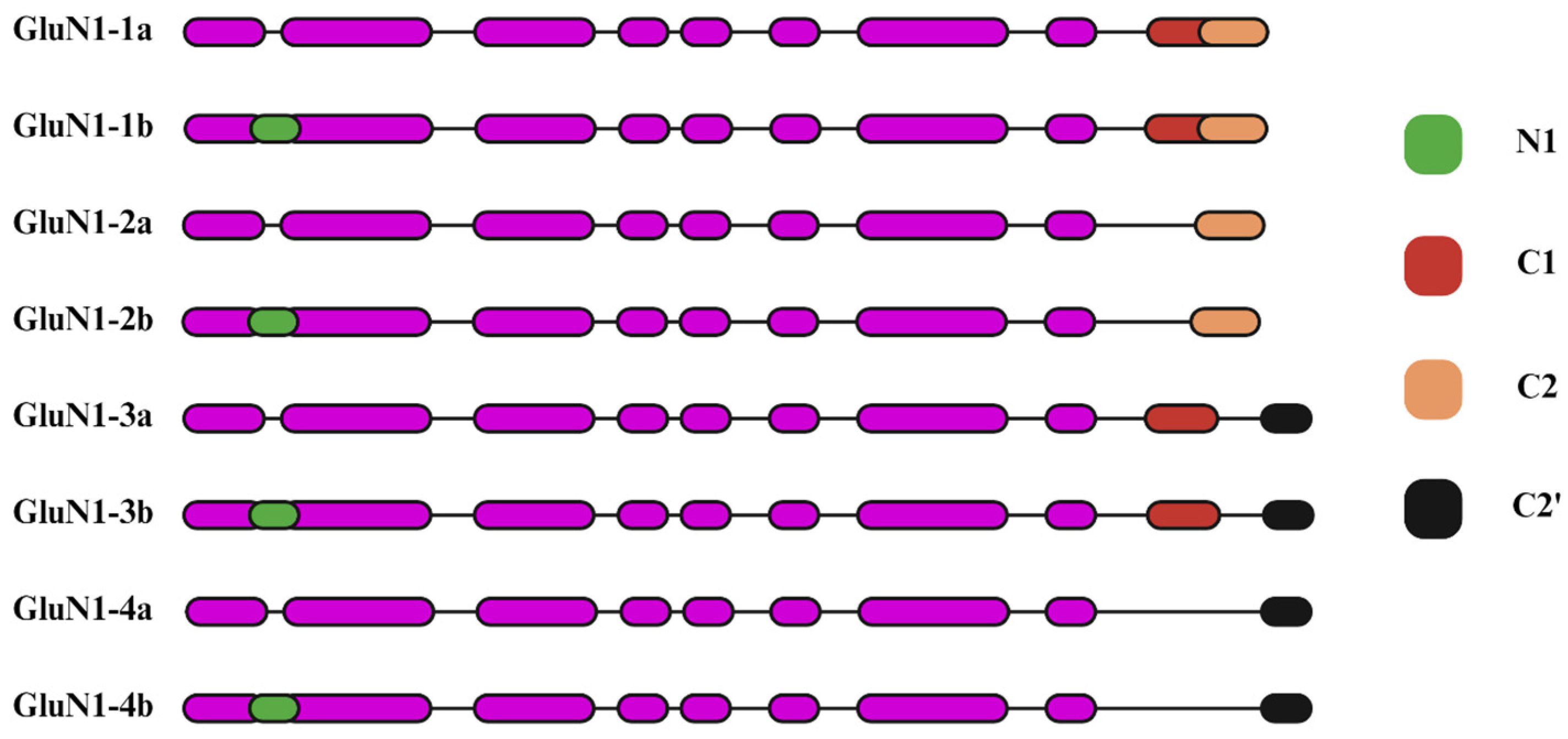
Figure 4.
Modeling neurodevelopmental disorders using patient derived iPSCs. The illustration depicts the general protocol for modelling human neurodevelopmental disorders (NDDs) using patient derived induced pluripotent stem cells (iPSCs). The iPSCs can be derived from the fibroblasts, PBMCs, or lymphoblastoid cell lines (LCLs) of the patients as well as the healthy subjects (Isogenic controls). These iPSCs provide a platform to study the disease biology and explore novel therapeutic strategies through various techniques that can lead to the development of personalized and more effective treatment options for patients suffering with different NDDs. The schematics of the figure are adapted from Liu et al, Development, 2018 [147].
Figure 4.
Modeling neurodevelopmental disorders using patient derived iPSCs. The illustration depicts the general protocol for modelling human neurodevelopmental disorders (NDDs) using patient derived induced pluripotent stem cells (iPSCs). The iPSCs can be derived from the fibroblasts, PBMCs, or lymphoblastoid cell lines (LCLs) of the patients as well as the healthy subjects (Isogenic controls). These iPSCs provide a platform to study the disease biology and explore novel therapeutic strategies through various techniques that can lead to the development of personalized and more effective treatment options for patients suffering with different NDDs. The schematics of the figure are adapted from Liu et al, Development, 2018 [147].
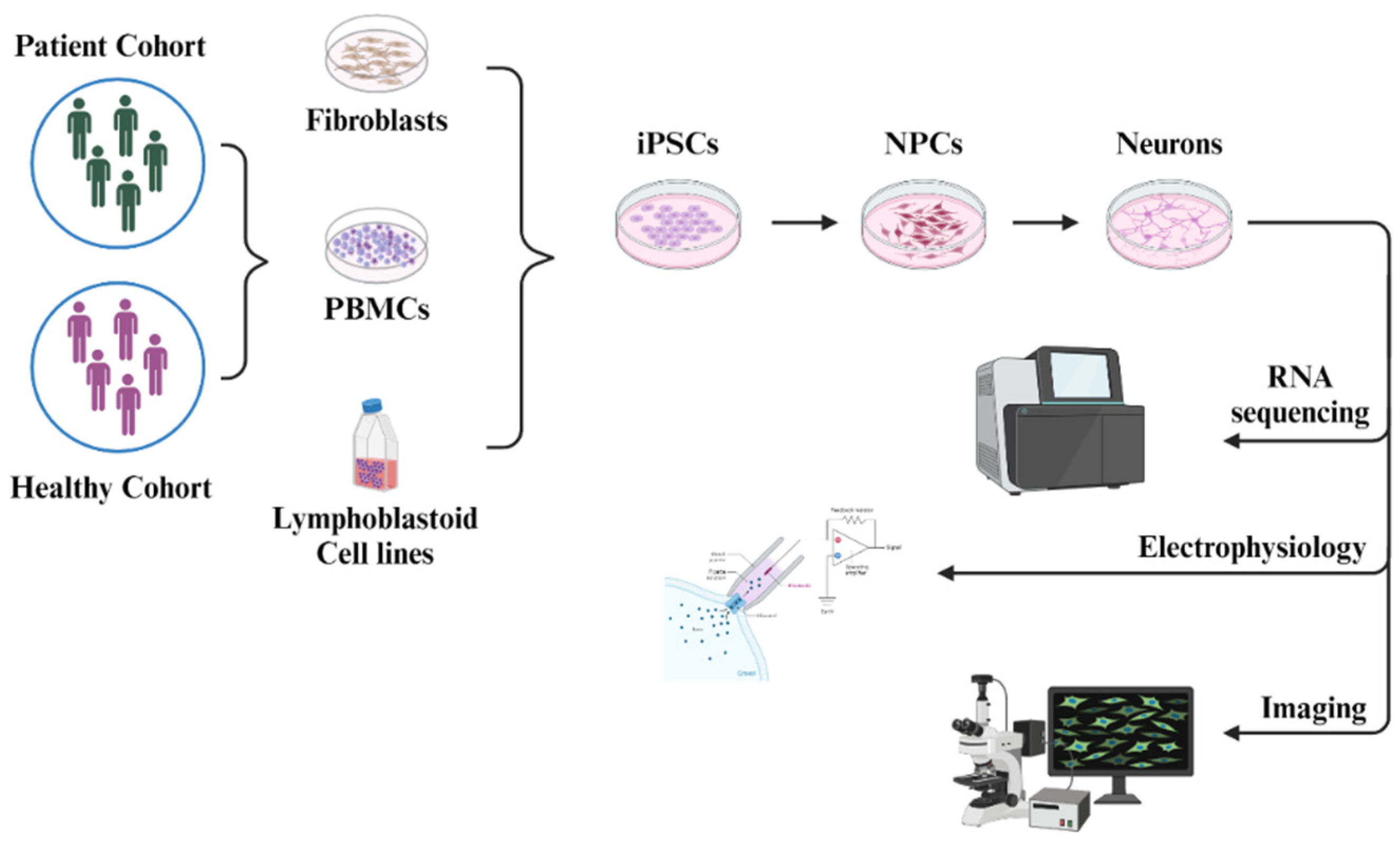
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



