
Submitted:
10 October 2024
Posted:
10 October 2024
You are already at the latest version
Abstract
The Cu cocatalyst supported on the surface of TiO2 photocatalysts has demonstrated unique activity and selectivity in photocatalytic CO2 reduction. The valence state of copper significantly influences the catalytic process; however, due to the inherent instability of copper's valence states, the precise role of different valence states in CO2 reduction remains inadequately understood. In this study, CuOx/TiO2 catalysts were synthesized using an in-situ growth reduction method, and we investigated the impact of various valence copper species on CO2 photocatalytic reduction. Our results indicate that Cu+ and Cu0 serve as primary active sites, with the selectivity for CH4 and CO products during CO2 photoreduction being closely related to their respective ratios on the catalyst surface. The adsorption and activation mechanisms of CO on both Cu+ and Cu0 surfaces are identified as critical factors determining product selectivity in photocatalytic processes. Furthermore, it is confirmed that Cu+ primarily facilitates CH4 production while Cu0 is responsible for generating CO. This study provides valuable insights into developing highly selective photocatalysts.
Keywords:
Photocatalytic CO2 reduction
; TiO2
; Cu valence state
; Production selectivity
; Cocatalyst
1. Introduction
With the rapid advancement of global industry and the growth of population, human consumption of fossil energy is escalating on a daily basis. On the one hand, this leads to an energy shortage[1,2], On the other hand, the substantial emissions of CO2 give rise to global warming, glacier melting, the extinction of plankton, and other issues, posing a serious threat to human health and safety[3,4,5]. In the treatment of CO2, converting CO2 directly into energy substances and chemicals is of the utmost scientific significance. Common strategies for CO2 resource utilization include: CO2 capture and storage[6,7], electrocatalytic CO2 reduction[8,9], thermal assisted catalytic CO2 reduction[10,11], and photocatalytic CO2 reduction[12,13]. Photocatalytic CO2 reduction harnesses the abundant and renewable energy of sunlight to transform CO2 into valuable chemicals, all while avoiding the consumption of precious electrical or thermal energy. This process is regarded as an optimal solution to the challenge posed by excessive CO2 emissions[14]. Since 1979, it has been reported that TiO2 can catalyze the conversion of CO2 into CH3OH, HCHO, and other chemical compounds, which has sparked a global research surge in photocatalytic CO2 conversion to energy-rich substancess[15]. Currently, a diverse array of semiconductor photocatalytic materials has been developed, with TiO2-based photocatalysts being the most extensively studied and applied in the field of photocatalysis due to their superior efficiency and stability[16,17,18,19,20,21]. suitable bandgap structure, low toxicity and other characteristics[22,23,24]. Nevertheless, the photocatalytic reduction of CO2 using TiO2 still faces challenges related to limited photocatalytic efficiency and low product selectivity[25]. In order to improve the photocatalytic performance of TiO2, surface modification can be carried out by the following common methods: doping[26,27], defect construction[28,29,30], morphology regulation[31], heterojunction construction[32,33], co-catalyst support[34,35], surface sensitization[36,37]. Among them, supported cocatalyst is an effective means to improve the photocatalytic performance of TiO2[34,35,38,39].
Transition metals are often used as cocatalysts to improve the photocatalytic activity of semiconductors, among which Cu is widely used in the field of photocatalysis, especially in the study of photocatalytic CO2 reduction, because of its abundant reserves, cheap and easy to obtain, and efficient charge separation ability[40,41,42,43,44]. The remarkable characteristic of Cu is the diversity of valence states (Cu2+, Cu+ and Cu0) and its instability. The role of different valence states of Cu in photocatalytic CO2 reduction has attracted more and more attention. Many studies have been reported the effect of copper on the selectivity of CO2 products in photocatalytic reduction[45,46,47,48,49]. Kreft et al.[46] reported a study on the control of different valence Cu components by introducing O2, and found that significantly increased product yield and complete selectivity to CO products could be observed in the presence of O2, and Cu2O was the most active species in the photocatalytic CO2 reduction process. When the proportion of Cu2O increases, the output of the corresponding product CO will also increase. Zhang et al.[50] reported a catalyst of oxygen-containing copper (Cu4O), which showed that CO on the surface of Cu4O with oxygen vacancy often continued to hydrogenate to produce high-value products, rather than desorption to produce CO. Different valence Cu components have different adsorption and activation capacities for CO and CO2, so the valence of Cu components on the catalyst surface is an important factor affecting CO2 reduction. However, due to the instability of the valence state of Cu, there is still a lack of research on the practical role of each valence Cu component in the process of CO2 reduction.
In this study, the surface of anatase TiO2 was modified with different valence Cu species. CuOx/TiO2 catalyst was prepared by in-situ growth reduction method to explore the effects of different valence Cu components on photocatalytic reduction of CO2. Combined with catalyst characterization test and photocatalytic CO2 reduction performance test, the results showed that the introduction of CuOx did not change the structure, morphology and redox potential of TiO2. Cu+ and Cu0 are the main active sites on the catalysts. The selectivity of CH4 and CO in the photocatalytic reduction of CO2 products by CuOx/TiO2 is related to the ratio of Cu+ and Cu0 content on the catalyst surface. The mechanism of the influence of different valence copper components on product selectivity was analyzed from the perspective of thermodynamics and kinetics. *CO is more likely to desorption from the Cu0 surface to produce CO, while continuing to adsorb on the Cu+ surface to produce CH4. In this study, the adsorption of CO on different valence Cu components and the relationship between Cu valence and corresponding products in the process of CO2 photocatalytic reduction were investigated, which provided a guide for the development of highly selective photocatalysts.
2. Experimental
2.1. Chemical Materials
The chemicals used in the experiment were purchased from commercial suppliers without further treatment. They are: Titanium dioxide (anatase, 99.8%, Aladdin), Copper(II) acetate monohydrate (C4H6CuO4·H2O, ≥98.0%, Kermel), Ethylene glycol (EG, (CH2OH)2 ,AR), Ethanol (CH3CH2OH, AR).
2.2. Synthesis of the CuOx/TiO2
500 mg TiO2 and 32.14 mg Cu(CH3COO)2·H2O were dispersed into a mixture of 30 mL H2O and ethylene glycol, stirred at room temperature for 1 h, then hydrothermal reaction was carried out in an oven at 200 ℃ for 2 h, cooled to room temperature, washed with H2O and ethanol three times respectively, and dried at 60 ℃ for 12 h. Solid powder catalyst was obtained by grinding. According to the addition of y mL ethylene glycol (reductant), the catalyst sample was denoted as CuOx/TiO2-y (y = 0 ~ 5).
2.3. Characterization
The crystal structures of CuOx/TiO2 were characterized by X-ray diffraction (XRD, D8-ADVANCE, Cu Kα radiation, 2 θ= 20 ~ 80°), whose operation voltage and current were set at 40 kV and 30 mA, respectively; the morphologies were analyzed by transmission electron microscopy and high resolution transmission electron microscopy equipped with FFT; (TEM, HRTEM, JEOL JEM-F200); The UV-vis diffuse reflectance spectra (DRS) absorbance spectra were obtained by a Scan UV-vis diffuse reflectance spectrophotometer (Shimadazu, UV-2600), using BaSO4 as the reflectance sample. Specific surface area, pore size distribution and CO2 physical adsorption spectrum were measured by BET surface area measurements (Quadrascorb SI-4), which was carried out by N2 adsorption-desorption isotherms. CO2-TPD was carried out on the ChemBET PULSARTMTPR/TPD chemisorption analyzer with argon (Ar) as the carrier gas. The surface chemical states of elements on different samples were characterized by X-ray photoelectron spectroscopy and auger electron spectroscopy (XPS and AES, Thermo ESCALAB 250 Xi), and the shift of the spectra owe to the relative surface charging was corrected according to the standard binding energy of C 1 s at 284.6 eV. The oxygen vacancy was measured by electron paramagnetic resonance (EPR, Bruker A300-10/12). The real loading amount of Cu in samples were measured by inductively coupled plasma optical emission spectrometer (ICP-OES, Agilent ICPOES730). The electrochemical measurement was performed on an electrochemical analyzer (CHI600E ) with a three electrodes cell at room temperature. The working electrodes were made of ITO glass and the corresponding prepared samples. The Na2SO4 aqueous solution (0.1 M) was used as electrolyte and a 300 W Xe lamp (PLS-SXE300/300UV) was used as the light source. Photoluminescence spectra (PL) were recorded on a confocal laser Raman microscope (HORIBA FLuoroMax+) using a 310 nm excitation light source; the time-resolved transient PL decay of samples were measured by transient fluorescence spectrometer (Edinburgh FLS 980).
The In-situ FTIR was tested on a Bruker Tensor II spectrometer. The samples were loaded into the in-situ reaction tank of the infrared spectrometer, and the sample was pretreated for 1 h under vacuum at 80 ℃.The photocatalytic reaction process was simulated: CO2 and 0.5mL H2O were slowly injected, and the adsorption-desorption equilibrium was reached after adsorption for 1 h, and the reaction system was illuminated. The infrared spectra under different illumination times were collected and the process of CO2 reduction catalyzed by catalysts was analyzed. FTIR spectra of CO adsorbed were measured by same pretreatment method. After the pretreatment, CO was slowly injected. Adsorb for 1 h, and then remove excess CO. The temperature of all samples was gradually increased from 30◦C to 50◦C and was measured after being stabilized at each temperature for 5 min.
2.4. Photocatalytic Reduction Reaction of CO2
The performance of photocatalytic CO2 reduction was tested in a custom-made quartz glass reactor with a volume of 0.3 L. The 20 mg catalyst powder sample was laid on the surface of the small circular table at the bottom, and 1 mL deionized water was uniformly added around the circular table. After sealing with a quartz lid, CO2 gas was poured into the reactor for 1 h to empty it, and the reactor was filled with CO2 at the same time. After ventilation, both ends of the reactor were sealed. A 300 W xenon lamp (PLS-SXE300/300UV) was used as the lamp source for analysis by gas chromatograph (Shimadzu, GC-2018). Xenon lamps are illuminated from top to bottom through the quartz glass cover of the reactor, and 1 mL of the gas in the reactor is collected every 1 h and injected into the gas chromatograph for quantitative detection of CO and CH4. The retention time and standard curve of each component gas are obtained by detecting the standard gas.
3. Results and Discussion
3.1. The Structure and Morphology
The crystal structure of the catalyst was analyzed by X-ray diffraction (XRD) and Raman spectroscopy. As shown in Figure 1(a), XRD patterns of CuOx/TiO2 show typical anatase phase TiO2 (JCPDS No.84-1286) crystal structure, indicating that the introduction of copper does not change the crystal structure of TiO2. The diffraction peaks observed at 2θ = 36.1° and 61.4° correspond to the (111) and (220) crystal faces of Cu2O (JCPDS No.78-2076), respectively. The diffraction peaks observed at 2θ = 43.3° and 50.4° correspond to the (111) and (200) crystal faces of Cu (JCPDS No.85-1326), respectively, indicating that copper species are successfully supported on the TiO2 surface in the form of Cu2O or Cu. As shown in Figure 1(b), Raman diagrams of different catalyst samples all show the Raman characteristic peaks of anatase phase TiO2, and the peak position of anatase phase TiO2 does not shift after the introduction of copper, further indicating that the crystal phase of TiO2 anatase phase remains unchanged before and after the reaction.
The surface morphology of the catalyst was analyzed by transmission electron microscopy (TEM). Results As shown in Figure 2 (a-b), there is no significant difference in the morphology of CuOx/TiO2-2 and TiO2, and the size is about 50 nm nano-sheet, indicating that the introduction of copper has no effect on the morphology of TiO2. This is further confirmed by BET surface area measurements. As shown in Figure. TiO2, CuOx/TiO2-2 and CuOx/TiO2-5 have no obvious differences in specific surface area. The results show that the addition of Cu can’t change the surface structure of TiO2, which is consistent with the results of TEM. The HRTEM image of CuOx/TiO2-2 is shown in Figure 2 (c). The lattice fringes with lattice spacing of 0.246 nm, 0.208 nm and 0.351 nm are corresponding to the Cu2O (111) crystal face, Cu (111) crystal face and anatase phase TiO2 (101) crystal face, respectively. This is consistent with the XRD results. Figure 2 (d-f) element distribution mapping shows the uniform distribution of Ti, O, and Cu elements. The loading capacity of Cu species on CuOx/TiO2-2 was 1.16 wt.% by ICP-OES. The above results show that Cu species are uniformly supported on the surface of TiO2 in the form of Cu0 and Cu2O, and are closely bound to TiO2.
3.2. Copper Valence State on the Surface of CuOx/TiO2
In the process of photocatalytic reduction of CO2 by CuOx/TiO2, Cu species is the active center of the catalytic reduction reaction, and the valence of Cu plays an important role in the selectivity of CO2 products by photocatalytic reduction. According to the XRD pattern in Figure 1 (a), it can be preliminarily concluded that the presence state of Cu changes regularly with the addition amount of glycol as a reducing agent. The valence state of Cu in catalyst was further analyzed qualitatively and quantitatively by XPS and Auger electron spectroscopy (AES). Firstly, XPS was used to characterize the peak position of each valence Cu component on the catalyst surface for qualitative analysis, and the proportion of the peak area of each valence Cu component to the total peak area of all Cu components was calculated for quantitative analysis. As shown in Figure S2 (a-e), in the XPS spectrum of Cu 2p, the characteristic peaks at the binding energies of 932.08 eV and 952.08 eV correspond to the Cu 2p3/2 and Cu 2p1/2 orbitals of Cu+/Cu0, respectively, occupying the majority of the peak area of copper components. Its proportion increased slightly with the increase of the amount of ethylene glycol. The characteristic peaks at 934 eV and 935 eV correspond to the Cu 2p3/2 and Cu 2p1/2 orbitals of Cu2+, respectively, and occupy a small part of the peak area of the copper component, which decreases slightly with the increase of the amount of ethylene glycol added. Figure S2 (f) shows that the Cu component mainly exists in the form of Cu+/Cu0, but the two components cannot be distinguished by XPS and need to be further analyzed by Auger electron spectroscopy (AES).
The AES energy spectrum of Cu LMM is shown in Figure 3 (a-e). The characteristic peak at 569.6 eV binding energy corresponds to Cu+, the characteristic peak at 567.6 eV binding energy corresponds to Cu0, and the characteristic peak at 564.5 eV is related to the Ti 2s orbital. Figure 3 (f) shows that the proportion of Cu0 on the catalyst surface increases with the addition of ethylene glycol, while the proportion of Cu+ decreases. Due to the fact that the catalyst cannot avoid contact with oxygen in the air during the test, and the copper valence state is unstable, the test results show that the copper component cannot exist completely in the form of Cu0.
3.3. Performance of Photocatalytic CO2 Reduction
The performance of the catalyst for photocatalytic CO2 reduction was tested. The yields and selectivity of CO2 products for different samples were shown in Figure 4 (a). The photocatalytic CO2 reduction performance of pure TiO2 was poor, with CO yield of 1.41 μmol·g-1·h-1 and CH4 yield of 0.28 μmol·g-1·h-1. The photocatalytic CO2 reduction performance of TiO2 was improved after loading CuOx. Comparing the photocatalytic activities of different samples, it can be found that with the increase of reduction degree, the Cu valence state decreases, and the CO yield gradually increases. The CO yield of CuOx/TiO2-5 is up to 10.68 μmol·g-1·h-1, and the CO selectivity is up to 80.12%. The CH4 yield of CuOx/TiO2-2 was up to 10.8 μmol·g-1·h-1, and the selectivity of CH4 was up to 71.9%. Comparing the selectivity of CO2 products by photocatalytic reduction of different samples, it can be found that Cu2+ has little effect on the selectivity of products. With the gradual reduction of Cu2+, the selectivity of CO first decreases and then gradually increases, and the selectivity of CH4 first increases and then decreases. According to the analysis of the proportion of copper content in different valence states in Figure 4 (b), it can be seen that copper species Cu+ and Cu0 help to improve the reduction ability of the catalyst. With the increase of Cu0 content, the selectivity of CO gradually increases, indicating that Cu0 is conducive to the selective conversion of CO, while Cu+ is conducive to the selective conversion of CH4. These results indicate that the valence state of Cu may be the key to the selectivity of CO2 products in photocatalytic reduction.
In the process of photocatalytic CO2 reduction, considering that many factors have certain effects on the activity and selectivity of the reaction, we tested the photocatalytic CO2 reduction activity of CuOx/TiO2-2 under different reaction conditions. As shown in Figure S3, a small amount of CO and CH4 were produced in the Ar atmosphere, which was generated by a small amount of carbon-containing reagents remaining in the catalyst preparation process. Compared with the results of the reaction activity and product selectivity in the CO2 atmosphere, the results were negligible, indicating that the carbon source of the photocatalytic reduction reaction products mainly came from CO2 gas. In the dark state, no CO2 reduction products were detected in the reaction system of the catalyst, indicating that light is the necessary condition for CuOx/TiO2 photocatalytic reduction of CO2 reaction.
3.4. The Selectivity of Products in Photocatalytic Reduction of CO2
The adsorption of CO2 by catalyst is the first step of photocatalytic CO2 reduction reaction. The physical and chemical adsorption capacity of different samples for CO2 is analyzed by using specific surface area and aperture analyzer and chemical absorption desorption instrument (CO2-TPD). As shown in Figure 5 (a), the physical adsorption capacity of the catalyst remained basically unchanged after the introduction of copper, which was consistent with the test results of N2 resorption desorption curve. Figure 5 (b) CO2-TPD test results show that the CO2-TPD curve of TiO2 shows the desorption of CO2 at low temperatures, and the corresponding temperatures of desorption peaks are 101 ℃ and 322 ℃, respectively, indicating that the interaction between CO2 and TiO2 is relatively low. As the desorption temperatures of CuOx/TiO2-2 and CuOx/TiO2-5 are higher, the desorption temperatures are 356 ℃, 500 ℃, 345 ℃ and 475 ℃ respectively, indicating that the interaction between CO2 and CuOx/TiO2-2 and CuOx/TiO2-5 is stronger. The results show that CuOx cocatalyst can enhance the interaction between CO2 and photocatalyst. CuOx/TiO2-2 showed a higher desorption peak than CuOx/TiO2-5, indicating that Cu+ is more conducive to the chemical adsorption of CO2 and the subsequent photocatalytic reduction of CO2 reaction process.
The UV-vis DRS spectra are shown in Figure 5 (c). Compared with pure TiO2, CuOx/TiO2 had visible light absorption, indicating that the introduction of copper increased the light absorption range of the catalyst. In addition, the absorption band edge of the catalyst remains basically unchanged (387 nm), indicating that the supported Cu does not change the band gap of TiO2, which is consistent with the results of XRD and Raman. As shown in Figure 5 (d), CuOx/TiO2 has a higher photocurrent response than pure TiO2, among which CuOx/TiO2-2 has the strongest photocurrent response, indicating that the introduction of copper improves the transport capacity of photogenerated electrons and increases the mobility of electrons. However, the selectivity of photocatalytic reduction of CO2 products cannot be directly determined by electron migration. Therefore, steady-state fluorescence spectrum and fluorescence lifetime test were used to further analyze the lifetime of photogenerated electrons. As shown in Figure 5 (e-f), the catalyst has strong characteristic peaks at wavelengths of 394 nm and 466 nm, and the fluorescence intensity decreases after the introduction of copper, which indicates that the presence of Cu can promote the migration of photogenerated electrons to the surface of the catalyst. Inhibit the recombination of photogenerated electrons and holes. The fluorescence lifetime of TiO2 is 6.00 ns, the fluorescence lifetime of CuOx/TiO2-2 is 6.40 ns, and the fluorescence lifetime of CuOx/TiO2-5 is 6.21ns. The introduction of copper can migrate electrons from TiO2 to CuOx, extending the electron lifetime. From the perspective of reaction kinetics, since the generation of CH4 is an eight-electron reaction, the catalyst with a long photo-generated electron lifetime is more likely to generate CH4 products in the photocatalytic reduction of CO2, which also explains the higher selectivity of CuOx/TiO2-2 photocatalytic reduction of CO2 to CH4.
According to the semiconductor band gap (Eg) formula: (αhν)n = k(hν−Eg), the Tauc of different samples is calculated, as shown in Figure 6 (a). The band gap of TiO2 is about 3.2 eV, and the band gap remains basically unchanged after loading CuOx. VB-XPS was used to directly test the valence band position of the catalyst, and the results were shown in Figure 6 (b). The valence band values of different samples under standard hydrogen electrodes were calculated according to the following formula: EVB, NHE = φ + EVB, XPS − 4.44, where φ is the work function of the instrument (4.5 eV). Therefore, the EVB, NHE of CuOx/TiO2-2 and CuOx/TiO2-5 are calculated to be 2.42 eV and 3.32 eV, respectively. According to the formula EVB = ECB + Eg, the conduction band (ECB) of CuOx/TiO2-2 and CuOx/TiO2-5 is −0.75 eV and −0.83 eV, respectively. As shown in Figure 6 (c), the band conduction position of TiO2 was calculated to be −0.41 eV. Based on the above results, the band gap relationship of different samples is shown in Figure 6 (d). The conduction position of CuOx/TiO2 is more negative than that of single TiO2, indicating that loaded CuOx can improve the photocatalytic reduction ability of TiO2 and enhance the photocatalytic reduction activity of CO2. The conduction positions of CuOx/TiO2-2 and CuOx/TiO2-5 are more negative than the reaction potentials of CH4 (CH4/CO2, −0.24 V vs. NHE) and CO (CO/CO2, −0.52 V vs. NHE). The results show that photocatalytic reduction of CO2 to produce CH4 and CO is thermodynamically feasible. Combined with UV-vis DRS results, the effect of different valence states of Cu on the redox potential of CuOx/TiO2 catalyst is not significant. In other words, under these conditions, the changes in product yield and selectivity of CuOx/TiO2 photocatalytic reduction of CO2 are not determined by the redox potential of the catalyst.
Carbon monoxide (CO) is not only a significant product of photocatalytic CO2 reduction but also serves as an important reaction intermediate. The key to producing high-value products lies in the continued adsorption of CO on the catalyst surface, preventing its desorption during the photocatalytic CO2 reduction process. Fourier Transform Infrared Spectroscopy (FT-IR) was employed to assess the adsorption capacity of various samples for CO. A stronger interaction between the catalyst and CO correlates with increased difficulty in desorption from the catalyst surface. As temperature rises, the rate of decrease in *CO signal intensity diminishes. The reduction rates of *CO characteristic peak intensities at 2171 cm-1 and 2100 cm-1 can be utilized to characterize the CO adsorption capacity of the photocatalyst.[51,52,53].
As shown in Figure 7 (a), when the temperature rises to 30 ℃, the *CO signal on pure TiO2 sample begins to decline rapidly, and when the temperature rises to 45 ℃, the *CO adsorption peak completely disappears, indicating that the interaction force between CO and TiO2 is weak. Compared with a pure TiO2 sample, the *CO absorption peak of catalyst CuOx/TiO2 decreased at a slower rate, indicating that the interaction force between CO and CuOx/TiO2 was strong, that is, the main adsorption site of CO was CuOx. The FT-IR spectra of CO adsorption on CuOx/TiO2-2 and CuOx/TiO2-5 are shown in Figure 7 (b) and (c), respectively. The effects of different valence states of Cu on CO adsorption are compared and analyzed. The results show that the decline rate of *CO signal intensity is as follows: RTiO2 > RCuOx/TiO2-5 > RCuOx/TiO2-2, so the adsorption capacity of CO is CuOx/TiO2-2 > CuOx/TiO2-5 > TiO2. This is consistent with the change in the ratio of copper components in the AES spectra. With the decrease of the ratio of Cu+ and the increase of the ratio of Cu0, the adsorption capacity of CO decreases, and CO is more easily resolved to form CO products. This shows that the adsorption capacity of Cu+ for CO is stronger than that of Cu0, which is conducive to further hydrogenation of CO, and the final product CH4 is formed through carbene pathway.
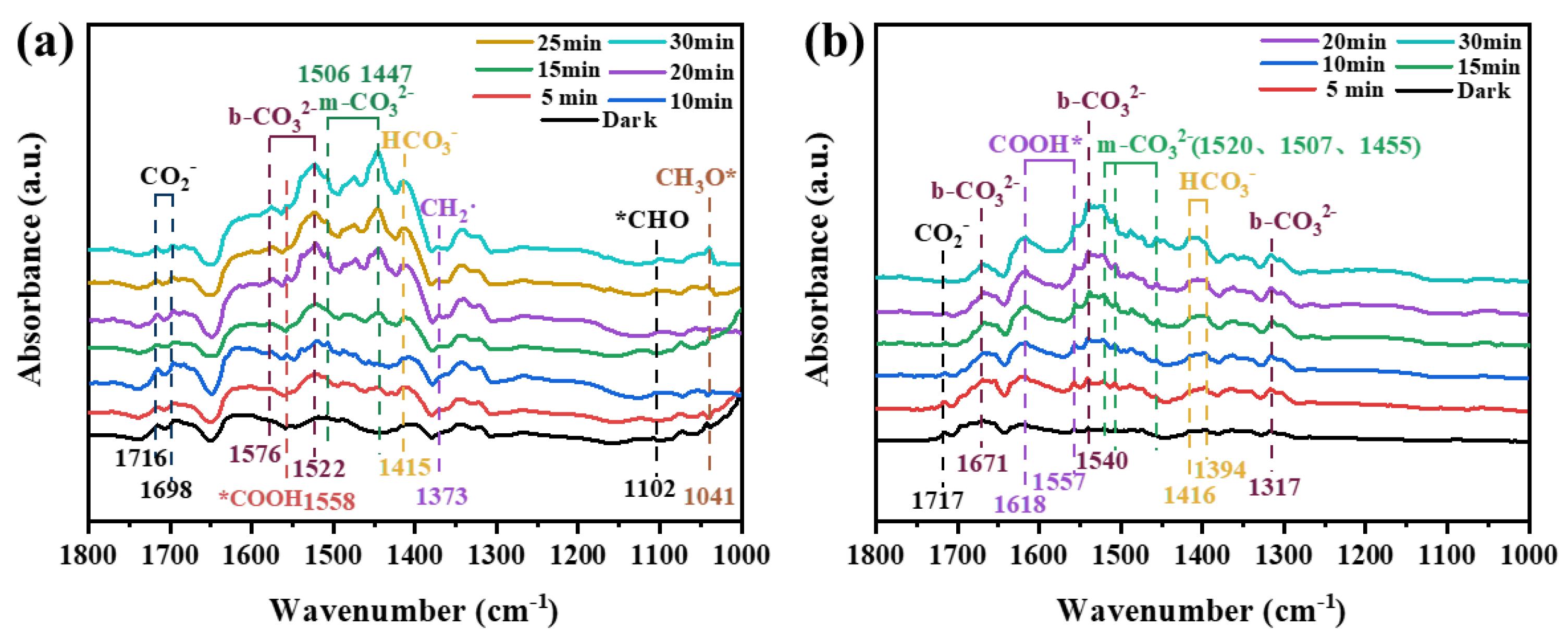
As shown in Figure 8 (a), the adsorption peaks of HCO3- (1415 cm-1), m-CO32- (1506 and 1447 cm-1) and b-CO32- (1576 and 1522 cm-1) can be observed in in-situ FT-IR spectra of CuOx/TiO2-2. The intensity of these characteristic adsorption peaks increased gradually with the extension of adsorption time, but the location did not change. COOH* (1558 cm-1), *CHO (1102 cm-1) and CH3O* (1041 cm-1) peaks appeared and increased with the increase of light time. They are all important intermediate species in the process of photocatalytic CO2 reduction. In addition, CH3O*, *CHO and CH2· (1373 cm-1)[42] participate in the reaction as important intermediates of CH4, which explains the high selectivity of CuOx/TiO2-2 catalyst for CH4 generation. It is speculated that the main conversion pathway of CH4 is the Carbene pathway: CO2 → *COOH → *CO → *CHO → C· →·CH2 → ·CH3 → CH4. As shown in Figure 8 (b), In the photocatalytic CO2 reduction process of CuOx/TiO2-5, the intermediates are mainly CO32-, HCO3- and *COOH, and no absorption peak of methane intermediates is observed. This indicates that *CO does not accumulate on the catalyst surface for further conversion, but is quickly released into the air and converted into the final product CO. This is consistent with the results of the CO adsorption FT-IR, which explains the high selectivity of CuOx/TiO2-5 photocatalytic reduction of CO2 to produce CO. It is speculated that the main conversion pathway of CO is as follows: CO2 → HCO3-/CO32- → *COOH → CO. Different catalysts produce different reaction intermediates in the light process, which directly affect the yield and selectivity of the final photocatalytic reduction of CO2 products.
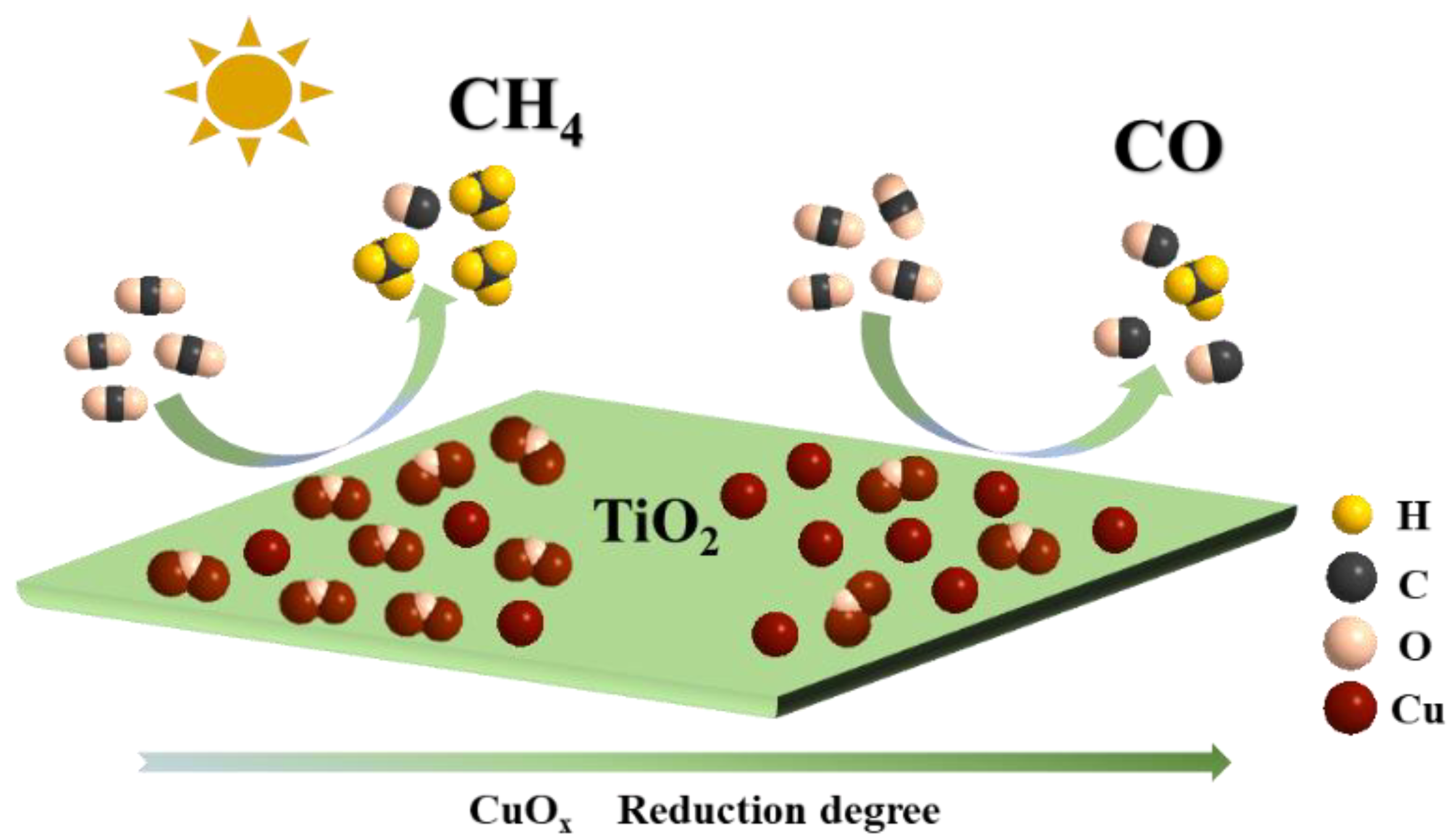
Building on the aforementioned results, a reaction mechanism for the CuOx/TiO2 photocatalytic reduction of CO2 has been proposed. The CuOx species supported on the TiO2 surface serves as a reactive site that effectively harnesses photoelectron generation, enhances charge separation efficiency, and boosts the photocatalytic activity for CO2 reduction. Results from Auger Electron Spectroscopy (AES) and evaluations of CO2 photoreduction performance indicate that the selectivity towards methane (CH4) and carbon monoxide (CO) in the products is closely linked to the relative content ratio of Cu+ and Cu0 present on the catalyst surface. Furthermore, it was observed that the adsorption and activation of *CO intermediates on this surface significantly influence final product formation. CO readily desorbs from Cu0 to yield CO, while CH4 undergoes further adsorption and hydrogenation at Cu+, thereby confirming that Cu+ acts as an active site for CH4 production, whereas Cu0 functions as an active site for CO generation
Figure 9.
Schematic image of CuOx/TiO2 photocatalytic reduction of CO2.
4. Conclusion
The CuOx/TiO2 photocatalyst was synthesized via an in-situ growth reduction method, enabling selective regulation of CO2 photoreduction products by modulating the valence state of copper. The valence state of copper is a critical determinant influencing the selectivity of CO2 reduction products. Cu+ serves as the active site for methane (CH4) formation, while Cu0 acts as the active site for carbon monoxide (CO) production. Notably, CO is not only a significant product of CO2 photoreduction but also functions as an essential reaction intermediate. Cu+ exhibits strong adsorption and activation capabilities towards CO, thereby facilitating the conversion of *CO intermediates into high-value CH4 products. This study offers valuable insights for advancing research on copper-based photocatalysts and identifying highly efficient and selective catalysts for CO2 reduction.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org
Acknowledgments
This work is financially supported by the Natural Science Foundation of Henan Province (No. 222300420406, 242300421346).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Wang J, Azam W. Natural resource scarcity, fossil fuel energy consumption, and total greenhouse gas emissions in top emitting countries. Geoscience Frontiers 2024, 15, 101757. [Google Scholar] [CrossRef]
- Yao, L. New energy utilization in environmental design and realization. Energy Reports 2022, 8, 9211–9220. [Google Scholar] [CrossRef]
- Gao W, Liang S, Wang R; et al. Industrial carbon dioxide capture and utilization: State of the art and future challenges. Chemical Society Reviews 2020, 49, 8584–8686. [Google Scholar] [CrossRef] [PubMed]
- Herndon, J. Evidence of variable earth-heat production, global non-anthropogenic climate change, and geoengineered global warming and polar melting. Journal of Geography, Environment and Earth Science International 2017, 10, 1–16. [Google Scholar] [CrossRef]
- Lowery C M, Bown P R, Fraass A J; et al. Ecological response of plankton to environmental change: Thresholds for extinction. Annual Review of Earth and Planetary Sciences 2020, 48, 403–429. [Google Scholar] [CrossRef]
- Bhanja P, Modak A, Bhaumik A. Porous organic polymers for CO2 storage and conversion reactions. ChemCatChem 2019, 11, 244–257. [Google Scholar] [CrossRef]
- Liu Y, An Y, Zhu J; et al. Integrated energy storage and CO2 conversion using an aqueous battery with tamed asymmetric reactions. Nature Communications 2024, 15, 977. [Google Scholar] [CrossRef]
- Su Y, Cheng Y, Li Z; et al. Exploring the impact of Nafion modifier on electrocatalytic CO2 reduction over Cu catalyst. Journal of Energy Chemistry 2024, 88, 543–551. [Google Scholar] [CrossRef]
- Wu Z, Gao F, Gao M. Regulating the oxidation state of nanomaterials for electrocatalytic CO2 reduction. Energy & Environmental Science 2021, 14, 1121–1139. [Google Scholar]
- Komarala E P, Alkhoori A A, Zhang X; et al. Design and synthesis of thermally stable single atom catalysts for thermochemical CO2 reduction. Journal of Energy Chemistry 2023, 86, 246–262. [Google Scholar] [CrossRef]
- Liu Y, Shang J, Zhu T. Enhanced thermal-assisted photocatalytic CO2 reduction by RGO/H-CN two-dimensional heterojunction. Journal of Materials Science & Technology 2024, 176, 36–47. [Google Scholar]
- Fu J, Jiang K, Qiu X; et al. Product selectivity of photocatalytic CO2 reduction reactions. Materials Today 2020, 32, 222–243. [Google Scholar] [CrossRef]
- Qu T, Wei S, Xiong Z; et al. Progress and prospect of CO2 photocatalytic reduction to methanol. Fuel Processing Technology 2023, 251, 107933. [Google Scholar] [CrossRef]
- Bushuyev O S, De Luna P, Dinh C T; et al. What should we make with CO2 and how can we make it? Joule 2018, 2, 825–832. [Google Scholar] [CrossRef]
- Inoue T F A, Konishi S; et al. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 1979, 277, 637–638. [Google Scholar] [CrossRef]
- Huang H, Shi R, Li Z; et al. Triphase photocatalytic CO2 reduction over silver-decorated titanium oxide at a gas-water boundary. Angewandte Chemie International Edition 2022, 61, e202200802. [Google Scholar] [CrossRef]
- Liu M, Bao X, Ma F; et al. Enhanced stability and activity towards photocatalytic CO2 reduction via supercycle ALD of Cu and TiO2. Chemical Engineering Journal 2022, 429, 132022. [Google Scholar] [CrossRef]
- Yuan S, Bao X, Chen M; et al. Unravelling the pathway determining the CO2 selectivity in photocatalytic toluene oxidation on TiO2 with different particle size. Chemical Engineering Journal 2023, 470, 144138. [Google Scholar] [CrossRef]
- Ma X, Li D, Xie J; et al. Confined space and heterojunction dual modulation of ZnO/ZnS for boosting photocatalytic CO2 reduction. Solar RRL 2023, 7, 2201093. [Google Scholar] [CrossRef]
- Wang X, Liang F, Gu H; et al. In situ synthesized α-Fe2O3/BCN heterojunction for promoting photocatalytic CO2 reduction performance. Journal of Colloid and Interface Science 2022, 621, 311–320. [Google Scholar] [CrossRef]
- Cao H, Xue J, Wang Z; et al. Construction of atomically dispersed Cu sites and S vacancies on CdS for enhanced photocatalytic CO2 reduction. Journal of Materials Chemistry A 2021, 9, 16339–16344. [Google Scholar] [CrossRef]
- Li Z, Wang S, Wu J, Zhou W. Recent progress in defective TiO2 photocatalysts for energy and environmental applications. Renewable and Sustainable Energy Reviews 2022, 156, 111980. [Google Scholar] [CrossRef]
- Collado L, Reñones P, Fermoso J; et al. The role of the surface acidic/basic centers and redox sites on TiO2 in the photocatalytic CO2 reduction. Applied Catalysis B: Environmental 2022, 303, 120931. [Google Scholar] [CrossRef]
- Rehman Z U, Bilal M, Hou J; et al. Photocatalytic CO2 reduction using TiO2-based photocatalysts and TiO2 Z-Scheme heterojunction composites: A review. Molecules 2022, 27, 2069. [Google Scholar] [CrossRef]
- Shtyka O, Shatsila V, Ciesielski R; et al. Adsorption and photocatalytic reduction of carbon dioxide on TiO2. Catalysts 2021, 11, 47. [Google Scholar]
- Reñones P, Fresno F, Oropeza F E; et al. Structural and electronic insight into the effect of indium doping on the photocatalytic performance of TiO2 for CO2 conversion. Journal of Materials Chemistry A 2022, 10, 6054–6064. [Google Scholar] [CrossRef]
- Sun X, Li F, Wang Z; et al. Efficient formic acid dehydrogenation on AuPd/N-TiO2, The role of N dopant and the effect of TiO2 crystalline phase. Chemical Engineering Journal 2023, 475, 146143. [Google Scholar] [CrossRef]
- Zhang B, Wang D, Jiao S; et al. TiO2-x mesoporous nanospheres/BiOI nanosheets S-scheme heterostructure for high efficiency, stable and unbiased photocatalytic hydrogen production. Chemical Engineering Journal 2022, 446, 137138. [Google Scholar] [CrossRef]
- Zhang Y, Li Y, Yu H; et al. Interfacial defective Ti3+ on Ti/TiO2 as visible-light responsive sites with promoted charge transfer and photocatalytic performance. Journal of Materials Science & Technology 2022, 106, 139–146. [Google Scholar]
- Lan K, Wang R, Wei Q; et al. Stable Ti3+ defects in oriented mesoporous titania frameworks for efficient photocatalysis. Angewandte Chemie International Edition 2020, 59, 17676–17683. [Google Scholar] [CrossRef]
- Wang J, Wang K, He Z; et al. Solvent-induced synthesis of hierarchical TiO2 nanoflowers with tunable morphology by monolayer self-assembly for probing the photocatalytic performance. Journal of Nanostructure in Chemistry 2022, 12, 1075–1087. [Google Scholar] [CrossRef]
- Xu Y, Wang F, Lei S; et al. In situ grown two-dimensional TiO2/Ti3CN MXene heterojunction rich in Ti3+ species for highly efficient photoelectrocatalytic CO2 reduction. Chemical Engineering Journal 2023, 452, 139392. [Google Scholar] [CrossRef]
- Yuan R, Wang M, Liao L; et al. 100% N2O inhibition in photocatalytic NOx reduction by carbon particles over Bi2WO6/TiO2 Z-scheme heterojunctions. Chemical Engineering Journal 2023, 453, 139892. [Google Scholar] [CrossRef]
- Xiao J, Chen C, Chen S; et al. Insight into the significantly enhanced photocatalytic CO2 reduction performance of Pt/MnOx dual cocatalysts on sea-urchin-like anatase TiO2 microspheres. Chemical Engineering Journal 2021, 425, 131627. [Google Scholar] [CrossRef]
- Zhang L, Hussain S, Li Q, Yang J. PdCu alloy anchored defective titania for photocatalytic conversion of carbon dioxide into methane with 100% selectivity. Journal of Energy Chemistry 2024, 91, 254–265.
- Jo M, Choi S, Jo J H; et al. Utility of squaraine dyes for dye-sensitized photocatalysis on water or carbon dioxide reduction. ACS Omega 2019, 4, 14272–14283. [Google Scholar] [CrossRef]
- Chon B, Choi S, Seo Y; et al. InP-quantum dot surface-modified TiO2 catalysts for sustainable photochemical carbon dioxide reduction. ACS Sustainable Chemistry & Engineering 2022, 10, 6033–6044. [Google Scholar]
- Liu M, Zheng L, Bao X; et al. Substrate-dependent ALD of Cux on TiO2 and its performance in photocatalytic CO2 reduction. Chemical Engineering Journal 2021, 405, 126654. [Google Scholar] [CrossRef]
- Feng X, Pan F, Tran B Z; et al. Photocatalytic CO2 reduction on porous TiO2 synergistically promoted by atomic layer deposited MgO overcoating and photodeposited silver nanoparticles. Catalysis Today 2020, 339, 328–336. [Google Scholar] [CrossRef]
- Zheng Y, Duan Z, Liang R; et al. Shape-dependent performance of Cu/Cu2O for photocatalytic reduction of CO2. ChemSusChem 2022, 15, e202200216. [Google Scholar] [CrossRef]
- Zhu Z, Yang C, Hwang Y; et al. Fuel generation through photoreduction of CO2 on novel Cu/BiVO4. Materials Research Bulletin 2020, 130, 110955. [Google Scholar] [CrossRef]
- Deng Y, Wan C, Li C; et al. Synergy effect between facet and zero-valent copper for selectivity photocatalytic methane formation from CO2. ACS Catalysis 2022, 12, 4526–4533. [Google Scholar] [CrossRef]
- Wang Z, Song H, Pang H; et al. Photo-assisted methanol synthesis via CO2 reduction under ambient pressure over plasmonic Cu/ZnO catalysts. Applied Catalysis B: Environmental 2019, 250, 10–16. [Google Scholar] [CrossRef]
- Wang T, Chen L, Chen C; et al. Engineering catalytic interfaces in Cuδ+/CeO2-TiO2 photocatalysts for synergistically boosting CO2 reduction to ethylene. ACS Nano 2022, 16, 2306–2318. [Google Scholar] [CrossRef]
- Wu Y A, McNulty I, Liu C; et al. Facet-dependent active sites of a single Cu2O particle photocatalyst for CO2 reduction to methanol. Nature Energy 2019, 4, 957–968. [Google Scholar] [CrossRef]
- Kreft S, Schoch R, Schneidewind J; et al. Improving selectivity and activity of CO2 reduction photocatalysts with oxygen. Chem 2019, 5, 1818–1833. [Google Scholar] [CrossRef]
- Yuan L, Hung S, Tang Z; et al. Dynamic evolution of atomically dispersed Cu species for CO2 photoreduction to solar fuels. ACS Catalysis 2019, 9, 4824–4833. [Google Scholar] [CrossRef]
- Jiang Z, Sun W, Miao W; et al. Living atomically dispersed Cu ultrathin TiO2 nanosheet CO2 reduction photocatalyst. Advanced Science 2019, 6, 1900289. [Google Scholar] [CrossRef]
- Lee B H, Park S, Kim M; et al. Reversible and cooperative photoactivation of single-atom Cu/TiO2 photocatalysts. Nature Materials 2019, 18, 620–626. [Google Scholar] [CrossRef]
- Zhang W, Huang C, Xiao Q; et al. Atypical oxygen-bearing copper boosts ethylene selectivity toward electrocatalytic CO2 reduction. Journal of the American Chemical Society 2020, 142, 11417–11427. [Google Scholar] [CrossRef]
- Eren B, Heine C, Bluhm H; et al. Catalyst chemical state during CO Oxidation reaction on Cu(111) studied with ambient-pressure X-ray photoelectron spectroscopy and near edge X-ray adsorption fine structure sSpectroscopy. Journal of the American Chemical Society 2015, 137, 11186–11190. [Google Scholar] [CrossRef] [PubMed]
- Chang X, Wang T, Zhao Z; et al. Tuning Cu/Cu2O interfaces for the reduction of carbon dioxide to methanol in aqueous solutions. Angewandte Chemie International Edition 2018, 57, 15415–15419. [Google Scholar] [CrossRef] [PubMed]
- Xing M, Zhou Y, Dong C; et al. Modulation of the reduction potential of TiO2–x by fluorination for efficient and selective CH4 generation from CO2 photoreduction. Nano Letters 2018, 18, 3384–3390. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(a) XRD patterns and Raman of CuOx/TiO2-y (y = 0.5.1, 2, 3, 5).
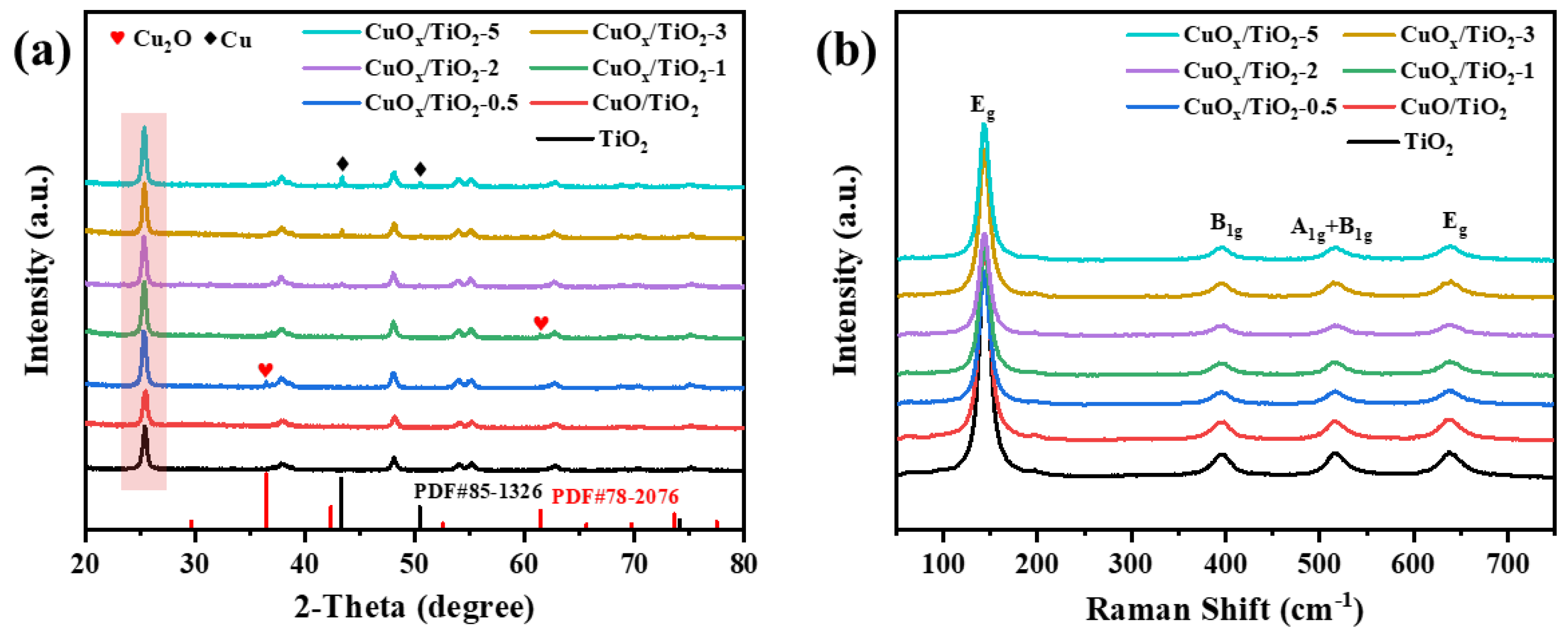
Figure 2.
(a) TEM diagram of TiO2, (b) TEM diagram, (c) HRTEM diagram, and (d-f) mapping diagram of CuOx/TiO2-2.
Figure 2.
(a) TEM diagram of TiO2, (b) TEM diagram, (c) HRTEM diagram, and (d-f) mapping diagram of CuOx/TiO2-2.
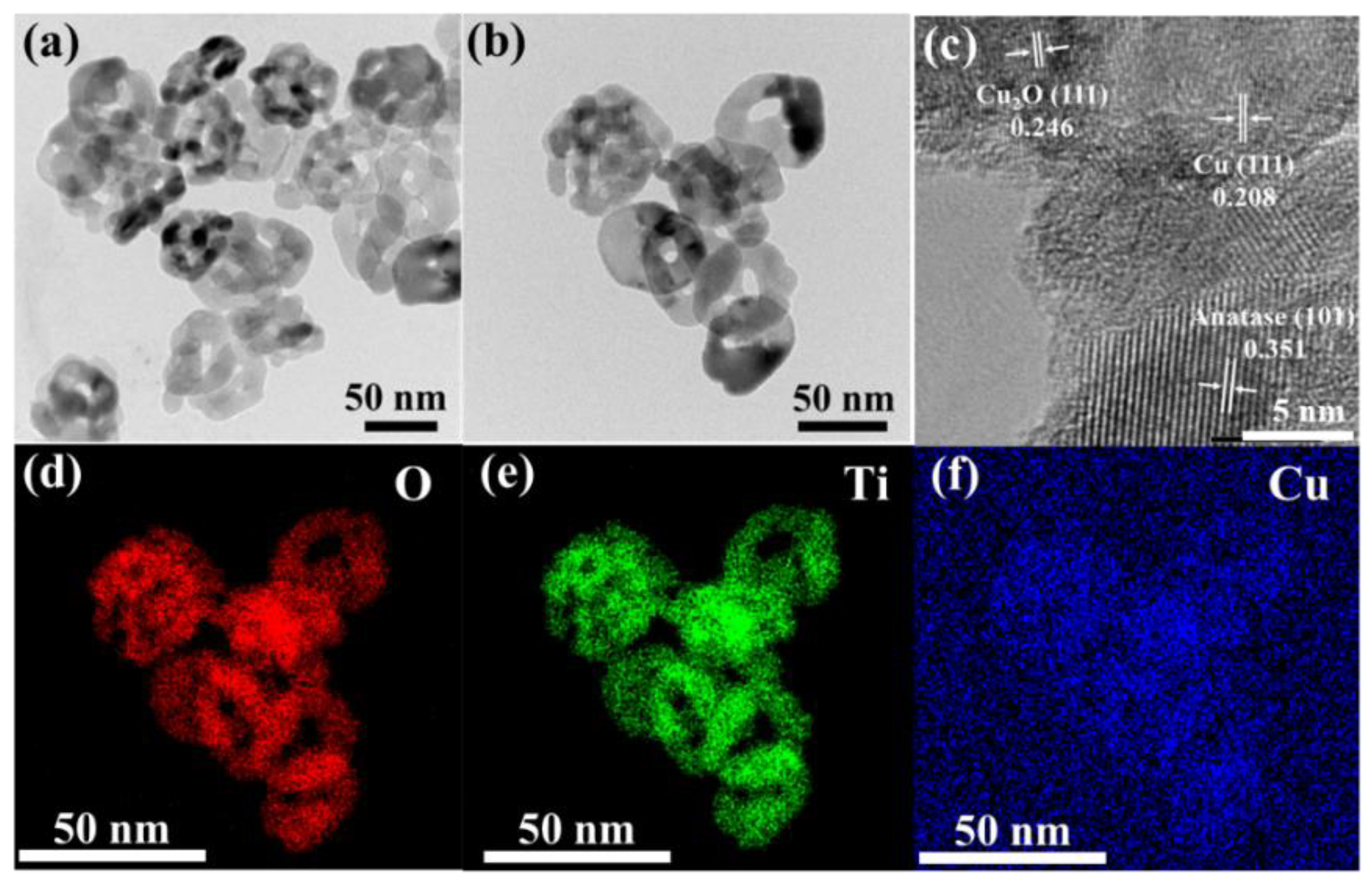
Figure 3.
Cu LMM Auger spectra of (a-e) CuOx/TiO2-y (y = 0.5, 1, 2, 3, 5), (f) Proportion of different state of Cu+ and Cu0 components in CuOx/TiO2.
Figure 3.
Cu LMM Auger spectra of (a-e) CuOx/TiO2-y (y = 0.5, 1, 2, 3, 5), (f) Proportion of different state of Cu+ and Cu0 components in CuOx/TiO2.
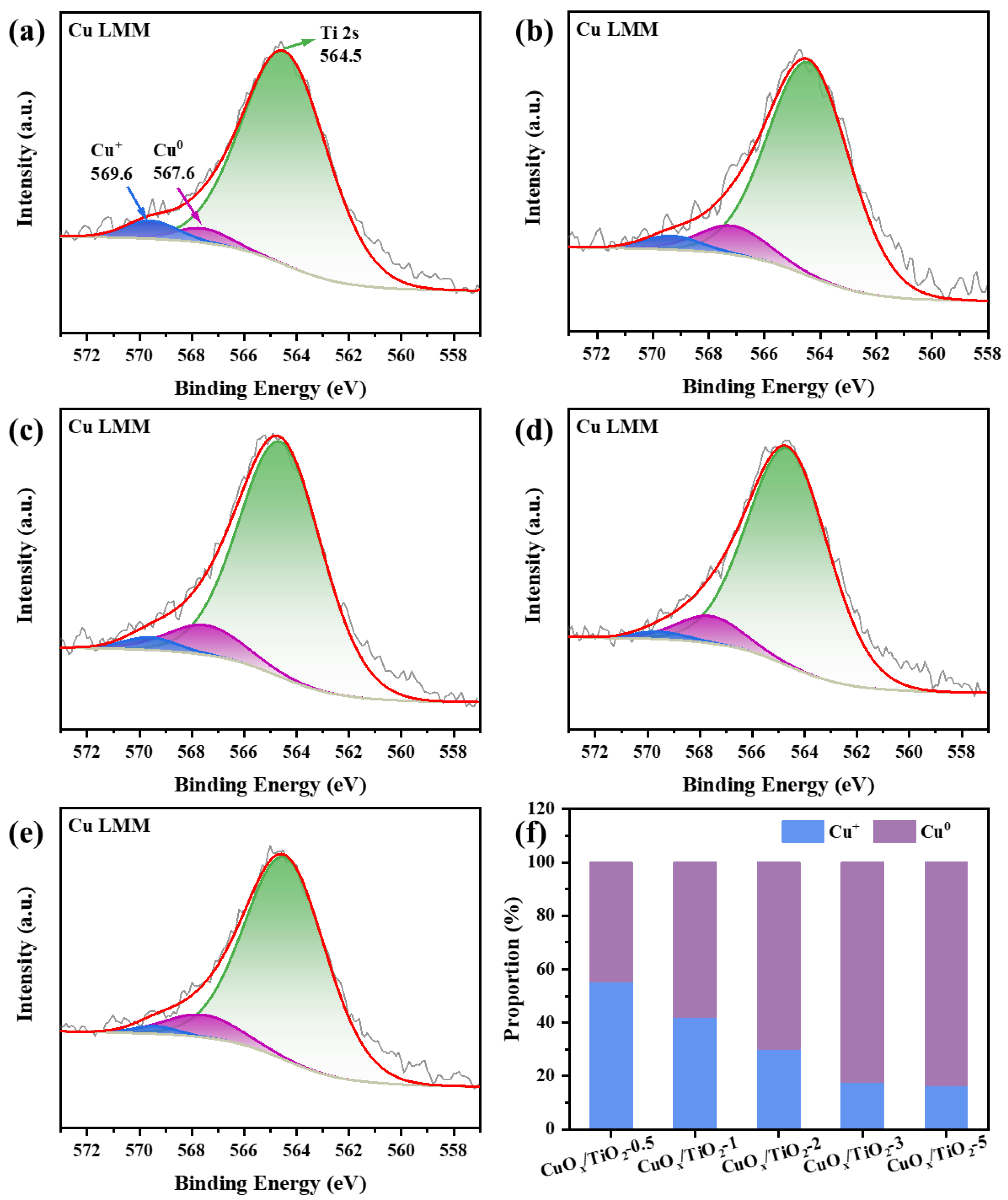
Figure 4.
(a) Photocatalytic CO2 reduction activity of CuOx/TiO2 and (b) Proportion of different state of Cu components in CuOx/TiO2.
Figure 4.
(a) Photocatalytic CO2 reduction activity of CuOx/TiO2 and (b) Proportion of different state of Cu components in CuOx/TiO2.
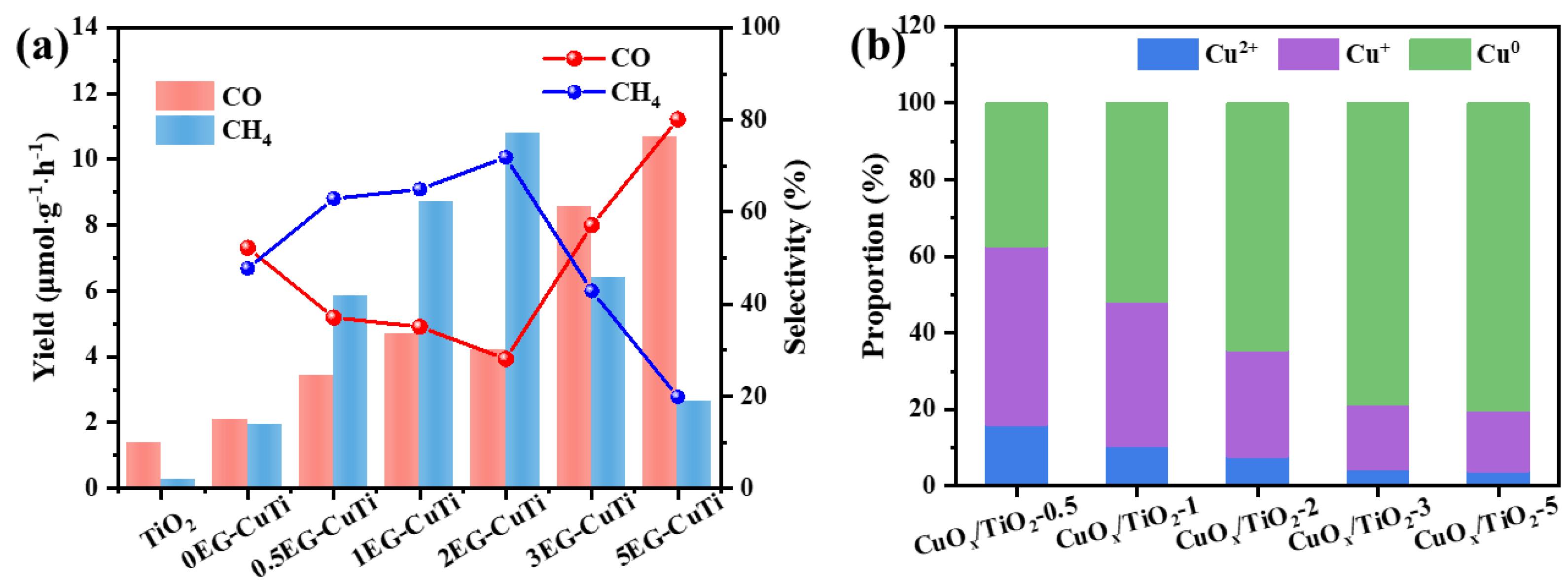
Figure 5.
(a) CO2 physical adsorption image, (b) CO2 chemisorption (CO2-TPD) image, (c) UV-vis DRS image, (d) I-t curve, (e) steady-state fluorescence spectrum image and (f) transient fluorescence lifetime image of CuOx/TiO2.
Figure 5.
(a) CO2 physical adsorption image, (b) CO2 chemisorption (CO2-TPD) image, (c) UV-vis DRS image, (d) I-t curve, (e) steady-state fluorescence spectrum image and (f) transient fluorescence lifetime image of CuOx/TiO2.
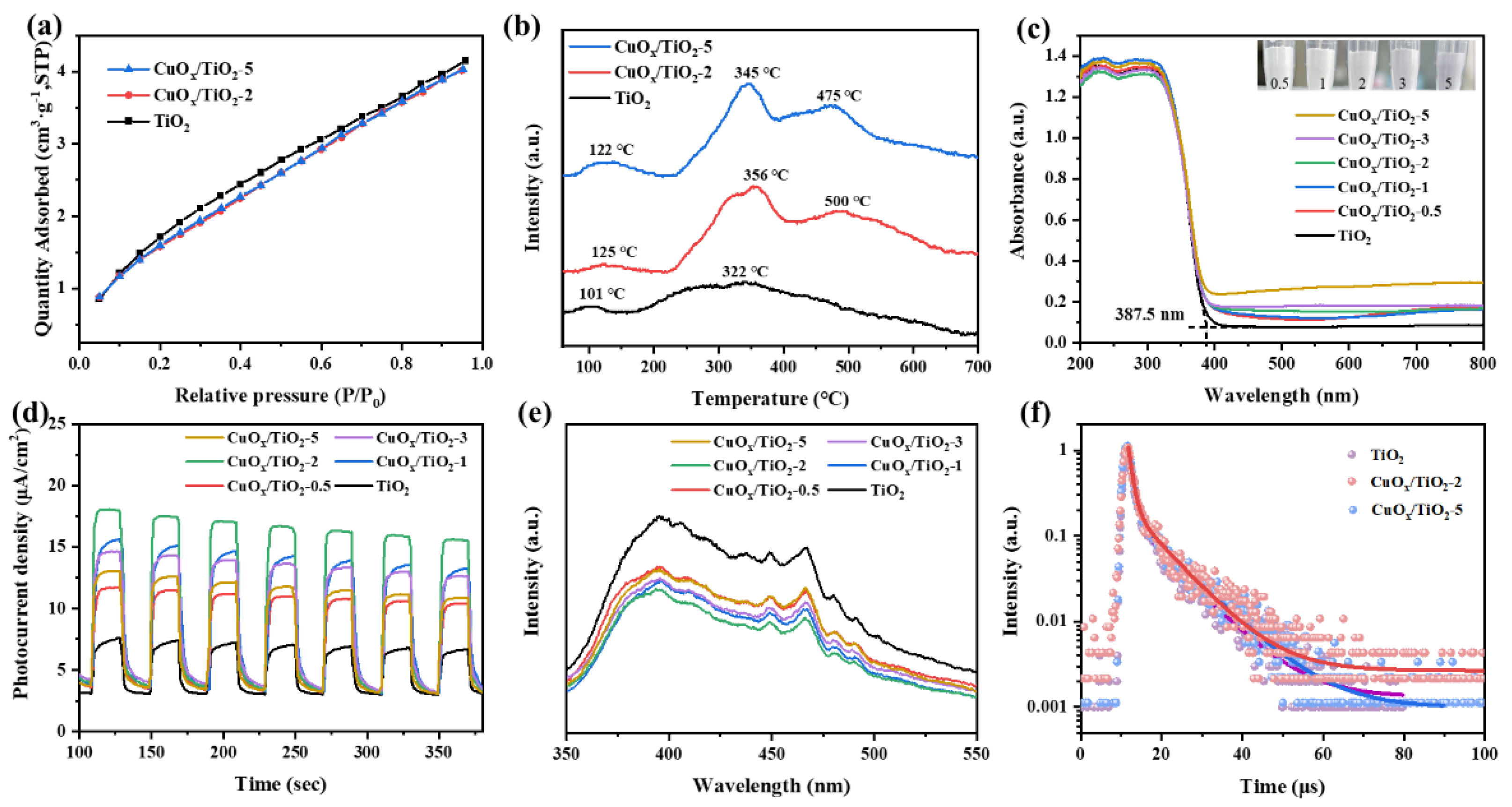
Figure 6.
(a) Tauc, (b) VB-XPS spectrum, (c) Mott-Schottky curve of TiO2 and (d) band structure of catalyst.
Figure 6.
(a) Tauc, (b) VB-XPS spectrum, (c) Mott-Schottky curve of TiO2 and (d) band structure of catalyst.
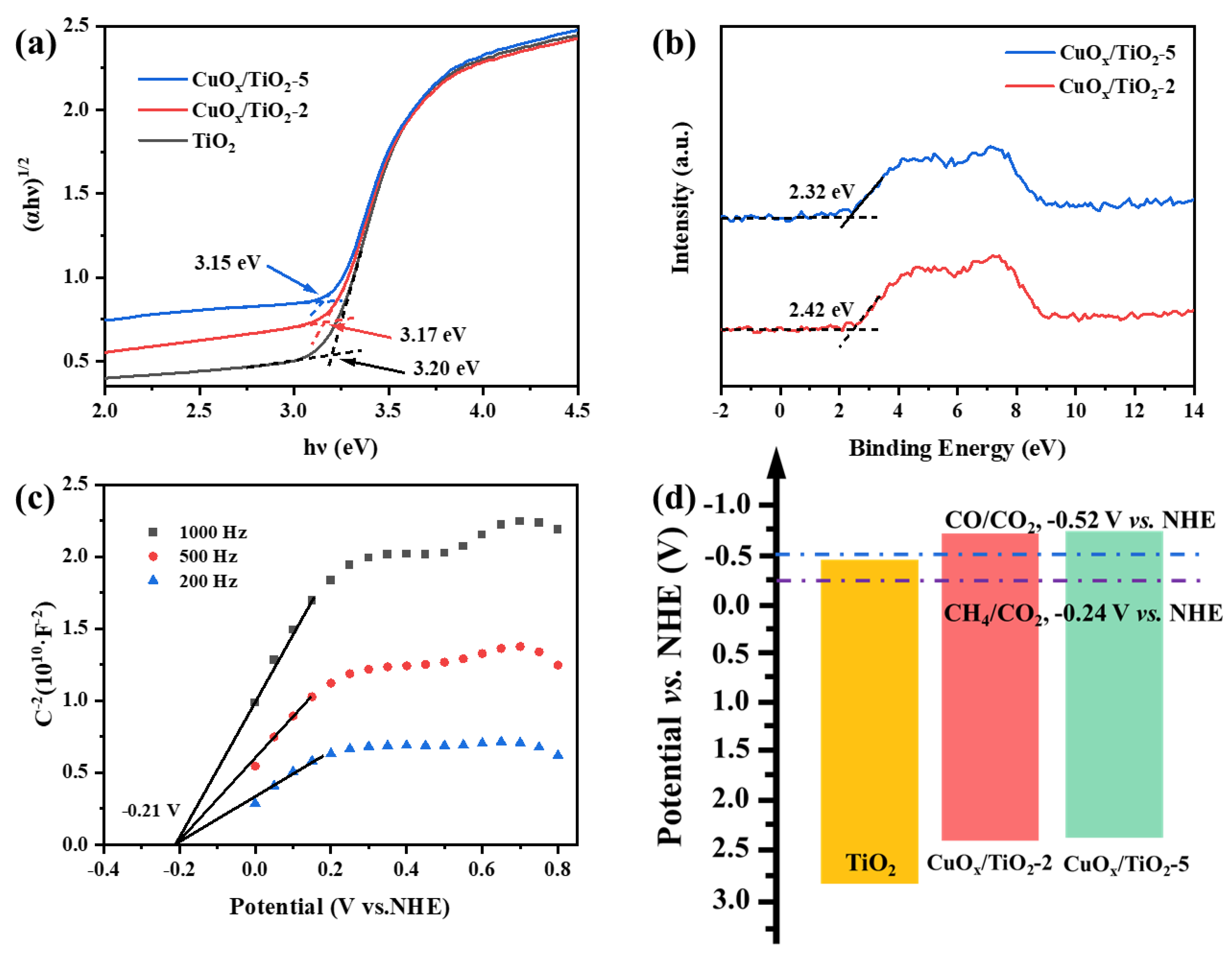
Figure 7.
FT-IR spectra of CO adsorbed on (a) TiO2, (b) CuOx/TiO2-2 and (c) CuOx/TiO2-5.
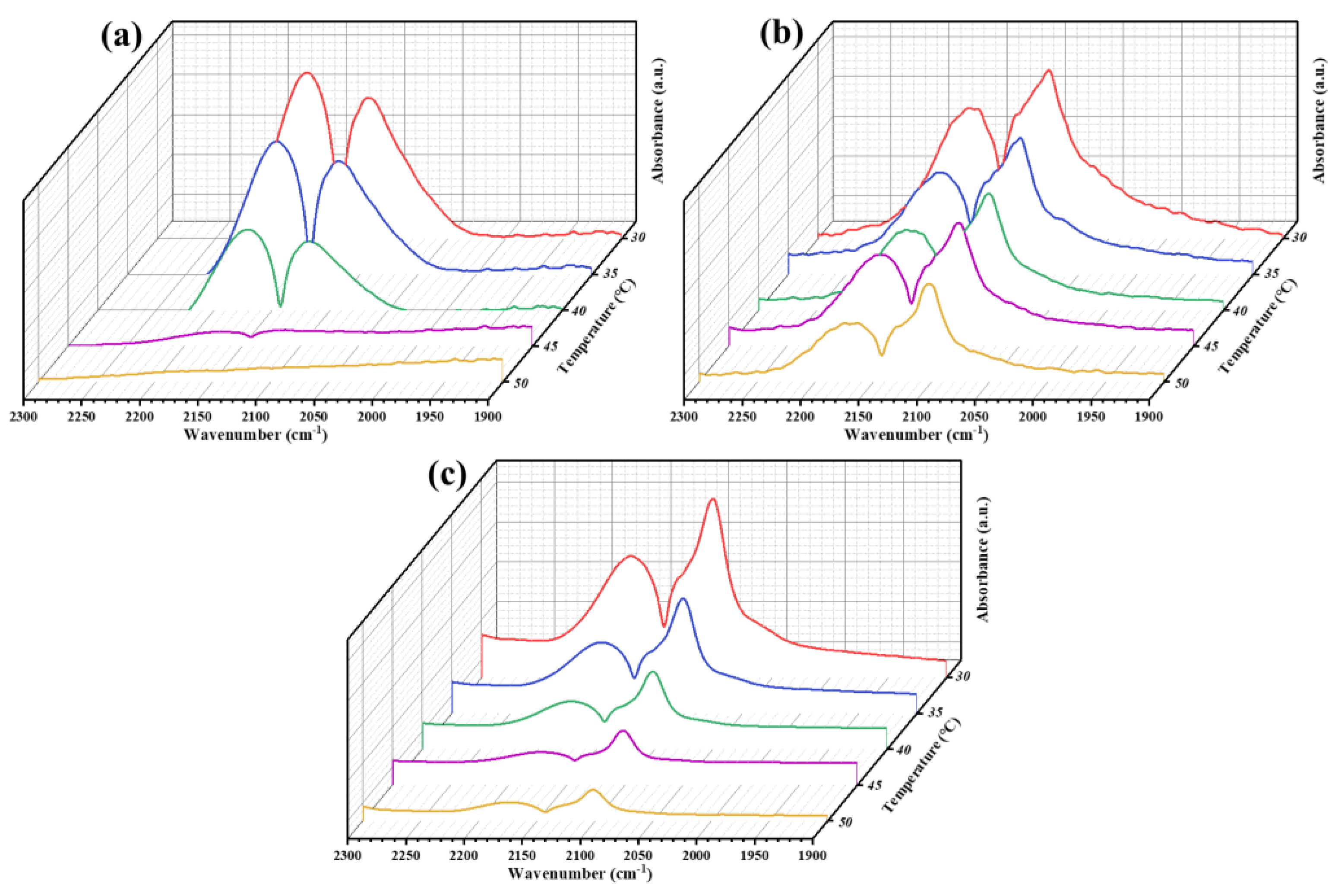
Figure 8.
In-situ FT-IR spectra of (a) CuOx/TiO2-2 and (b) CuOx/TiO2-5.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



