
Submitted:
10 October 2024
Posted:
11 October 2024
You are already at the latest version
Abstract
Type 2 diabetes mellitus (T2DM) is a multifactorial disease, influenced by dietary and environmental factors that can modify the intestinal microbiota. The aim was to evaluate changes in composition and diversity of the intestinal microbiota, associated with carbohydrate (CH) consumption in T2DM patients. Forty patients participated, with and without T2DM. Feces samples were collected for the characterization of microbial diversity from the massive sequencing of the 16S rRNA gene. Carbohydrate consumption was quantified from the FCF, the groups were categorized according to BMI and BMI + CH consumption. The group without T2DM showed normal biochemical and anthropometric parameters, although it had high carbohydrate consumption compared to the group with T2DM. At the Phylum level, there were differences in relative abundance, Control overweight group (CL–OW>CH) and T2DM-Normal Weight > CH patients, had increased Bacteroides and decreased Firmicutes. In contrast, CL–OW>CH and T2DM-OW<CH, showed reduced Bacteroidetes and elevated Firmicutes amount. At the genus level, the differences were in the relative abundance of Roseburia, Clostridium_IV, Prevotella, and Sporobacter, associated with the consumption of carbohydrate. The groups that consumed high amounts of carbohydrates, regardless of whether they had diabetes mellitus and overweight, had a significantly reduced proportion of Faecalibacterium, an altered proportion of Bacteroides. The high consumption of carbohydrate showed considerable modifications in the composition and diversity of the bacterial communities.
Keywords:
Carbohydrate
; Body Mass Index
; Type 2 Diabetes Mellitus
; Microbiota
1. Introduction
Type 2 diabetes mellitus (T2DM) occurs when the pancreas does not produce enough insulin or the body does not use it effectively, which causes glucose intolerance [1]. It has been estimated that in 2015 there were 415 million people with diabetes mellitus; by 2040, the number will be 642 million [2]. In Mexico, diabetes mellitus is the main cause of morbidity and mortality in those aged between 45 and 64 years [3]. It is a multifactorial disease involving genetic, dietary, lifestyle (such as sedentarism) [4], and environmental factors (such as the intestinal microbiota) [5]. The intestinal microbial composition is considered a factor of environmental origin because it differs between patients according to their environments [6]. In this sense, the type, quantity, and quality of the diet modifies patients’ microbiota, which causes dysbiosis that influences the pathophysiology of T2DM [7]. The intestinal microbiota is defined as a complex and diverse community dominated by bacteria living in the intestine [8]. In adults, the most abundant bacterial phyla are Bacteroidetes (Gram-negative bacteria) and Firmicutes (Gram-positive bacteria) [9]; Actinobacteria, Fusobacterium, and Verrucomicrobia are variable between individuals [10]. Each nutrient consumed in the diet alters the composition of the intestinal microbiota, promoting the expansion of microorganisms in a particular way [11]. Studies describe the effect of high-fat diets [12], but no studies involve the influence of consumption of different types of carbohydrates on the intestinal microbiota. For example, most individuals, include sucrose in different proportions as a dietary additive [13]. Hydrolyzed sucrose is retained in the distal intestine, exposing the microbiota to significant amounts of fructose and glucose for a longer period [14]. Sucrose has been found to decrease the abundance of Bacteroides thetaiotaomicron, which are important regulators of intestinal colonization [15]. For this reason, the objective of this work was to evaluate the changes in the composition and diversity of the intestinal microbiota associated with carbohydrate consumption in type 2 diabetes mellitus patients.
2. Results
2.1. Socio-Demographic Data
The median age in the T2DM group was higher (49 years) than the median age in the control group (42 years). In the T2DM subgroups, the T2DM-OW subgroup was younger (median 47 years), compared to the T2DM-NW subgroup (median 50 years). In the CL-OW and CL-NW subgroups, the median ages were 36 and 45 years, respectively. All the patients were healthy, without pathologies at the time of the study for the control group and the T2DM patients didn’t have aggregate pathologies recorded. In each group we found 18 women and 2 men. In the diabetes mellitus group, a total of 12 patients had a 6-10-year evolution of T2DM, while only 8 had less than 5 years of diagnosis. The treatment most frequently taken by patients was metformin (40%), followed by insulin (25%), the combination of metformin plus glibenclamide (20%), the combination of insulin plus metformin (10%), and the combination of sitagliptin plus metformin (5%).
2.2. Anthropometric Evaluation
The BMI was of 24.75 kg / m2 for the control group and a BMI of 24.8 kg / m2 for the T2DM group. The BMI was categorized, with the following findings in the control group: 12 patients with normal weight and 8 with overweight. In the T2DM group, 12 had normal weight, and 8 were overweight.
2.3. Biochemical Evaluation
The biochemical parameters of glycaemia (144 ±9.1 mg / dL) and HbA1c (8.95 ±2.2 mg / dL), were found to be significantly elevated (P < 0.001, in both cases) in the T2DM group, compared to the control group (glycaemia: 82 ±3.9 mg / dL and HbA1c: 5.25±0.356 mg / dL). The glucose and HbA1c results, the control group patients of CL-OW subgroup showed elevation in both parameters compared with the patients of CL-NW subgroup. In contrast, in the T2DM group, the T2DM-NW subgroup of patients showed elevated glycaemia and HbA1c, compared to the T2DM-OW subgroup (Table 1). Regarding triglycerides, a difference was found between the control and the T2DM group (P <0.024); the differences between subgroups was observed in T2DM-OW subgroup showed high triglycerides, compared the T2DM-NW, CL-OW, and CL-NW subgroups (Table 1). On the other hand, total cholesterol was lower in the control group compared to the T2DM group (P<0.029), however, there are no significant differences between the subgroups (P<0.519), as shown in Table 1.
2.4. Dietary Evaluation
The dietary evaluation between the groups showed significant differences (P <0.009). The group with T2DM consumed less energy (1,266 Kcal / day) with an adequate proportion of carbohydrates (113 g / day), fiber (13 g / day), protein (60 g / day), and lipids (34 g / day); in comparison with the patients without diabetes, who consumed higher energy (2,284 Kcal / day), carbohydrates (249 g / day), fibber (25 g / day), proteins (86 g / day), and lipids (66 g / day). The control group consumed more CH such us sucrose (22 g / day), starch (18 g / day), fructose (8 g / day) except glucose (3.5 g / day); compared to the T2DM group who consumed 30 g / day of starch, 25 g / day of sucrose, the same proportion of fructose (8.6 g / day), with a higher amount of glucose (6 g / day), they consumed more CH than the T2DM group.
2.5. Microbiota Analysis
The richness and diversity of OTUs were estimated using the Chao 1 index as an indicator of richness, the Shannon and Inversa indices of Simpson as indicators of diversity, and the Pielou index as an indicator of equity (Table 2a and Table 2b). Under these approaches, the fecal bacterial communities of the T2DM-NW subgroup showed lower values compared to those obtained in the T2DM-OW subgroup. This was contrary to what was observed in the CL-OW subgroup, which had richness and diversity greater than that of the T2DM-OW subgroup. The percentage of coverage was very similar in all samples of the different subgroups (Table 2a and Table 2b). The results of the massive sequencing directed to the V4-V5 region of the 16S rRNA gene of the fecal samples were used for taxonomic characterization at different levels. For the analysis of relative abundance, the levels of Phylum and gender were considered in the subgroups categorized by BMI and BMI + CH. The analysis of the relative abundance at the Phylum level showed that the taxonomic distribution of the subgroups BMI and BMI + CH had a higher abundance of Firmicutes and Bacteroidetes, followed by Proteobacteria, Actinobacteria, and Verrucomicrobia. In groups categorized according to BMI, Firmicutes were represented mostly in the CL-OW subgroup (70.62%) and Bacteroidetes in the T2DM-OW subgroup (54%). Proteobacteria had a higher representation in the CL-NW subgroup (0.95%), while Actinobacteria did not reach a representativeness of greater than 0.5% in all subgroups. However, the subgroups with the highest representation were T2DM-NW (0.27%) and CL-NW (0.26%); Verrucomicrobia (1.20%) was represented mostly in the T2DM-NW subgroup (Figure S2). In the BMI + CH subgroups, the Firmicutes were represented mostly in the CL-OW < CH subgroup (75.30%). This was contrary to Bacteroidetes, which had a higher representation in the T2DM-OW < CH subgroup (54%), followed by T2DM-NW > CH (49%). Proteobacteria showed greater representation in the T2DM-NW > CH (1.16%) and CL-NW < CH (1.04%) subgroups. Actinobacteria had a majority in the CL-NW > CH subgroup (0.65%) and was so low in the other subgroups, it did not reach 0.5%, while Verrucomibrobia (1.43%) was represented mostly in the T2DM-NW < CH subgroup. The results of the evaluation of relative abundance at the genus level showed that the taxonomic distribution according to BMI and BMI + CH had a greater abundance in 25 taxa, with the results of the CL-NW subgroup taken as a reference.

Table 2.
a. Number of qualified readings, estimators of richness, species, diversity and uniformity of the bacterial communities present in samples of patients without T2DM categorized by BMI.
Table 2.
a. Number of qualified readings, estimators of richness, species, diversity and uniformity of the bacterial communities present in samples of patients without T2DM categorized by BMI.
|
Control Group |
Nseqsa | Coverageb (%) | OTUc |
Chao1 (lcid-hcie) |
Inv-Simpson (lcid-hcie) |
Shannon (lcid-hcie) |
Pielou |
| CL-NW (n = 12) | |||||||
| M15 | 25575 | 99.75 | 278 | 412 (357-520) | 6 (5.94-6.12) | 2.81 (2.8-2.83) | 0.993 |
| M45 | 55419 | 99.82 | 437 | 594 (572-638) | 6.27 (6.21-6.34) | 4.75 (4.72-4.79) | 0.991 |
| M13 | 58542 | 99.86 | 612 | 857 (795-97) | 4.74 (4.70-4.78) | 3.45 (3.44-3.47) | 0.994 |
| M23 | 57984 | 99.86 | 1029 | 1297 (1245-1391) | 6.44 (6.41-6.47) | 4.44 (4.43-4.45) | 0.998 |
| M29 | 98637 | 99.86 | 906 | 1264 (1162-1374) | 5.29 (5.26-5.31) | 4.12 (4.11-4.13) | 0.997 |
| M32 | 56648 | 99.86 | 1125 | 1468 (1436-1666) | 4.39 (4.35-4.44) | 3.10 (3.09-3.11) | 0.998 |
| M33 | 45032 | 99.88 | 832 | 1160 (1059-1363) | 5.73 (5.71-5.75) | 4.60 (4.59-4.61) | 0.997 |
| M34 | 64036 | 99.83 | 1081 | 1396 (1344-1508) | 6.55 (6.5-6.6) | 4.99 (4.98-5) | 0.996 |
| M37 | 50042 | 99.86 | 458 | 597 (505-641) | 4.66 (4.62-4.71) | 2.46 (2.45-2.47) | 0.996 |
| M38 | 5021 | 99.66 | 1191 | 1490 (1488-1688) | 6.52 (6.38-6.67) | 4.86 (4.82-4.87) | 0.998 |
| M43 | 46370 | 99.87 | 883 | 1239 (1217-1288) | 6.03 (5.98-6.09) | 4.60 (4.58-4.61) | 0.997 |
| M46 | 39738 | 99.83 | 950 | 1290 (1188-1334) | 6.32 (6.23-6.40) | 4.55 (4.53-4.56) | 0.998 |
| CL-OW (n = 8) | |||||||
| M48 | 6333 | 99.82 | 1146 | 1503 (1471-1704) | 5.59 (5.51-5.68) | 4.75 (4.73-4.76) | 0.997 |
| M49 | 14430 | 99.83 | 998 | 1330 (1318-1434) | 5.41 (5.36-5.45) | 4.85 (4.84-4.88) | 0.994 |
| M6 | 8343 | 99.75 | 1037 | 1345 (1253-1410) | 5.71 (5.63-5.8) | 4.84 (4.82-4.86) | 0.997 |
| M16 | 59311 | 99.85 | 681 | 908 (849-1009) | 5.99 (5.95-6) | 4.9 (4.8-5) | 0.979 |
| M17 | 52056 | 99.84 | 1015 | 1332 (1267-1450) | 4.87 (4.82-4.92) | 3.43 (3.42-3.44) | 0.997 |
| M19 | 40529 | 99.81 | 861 | 1128 (1072-1229) | 6.14 (6-6.2) | 4.75 (4.74-4.76) | 0.999 |
| M25 | 64872 | 99.88 | 822 | 1091 (1080-1202) | 5.43 (5.41-5.45) | 4.59 (4.58-4.61) | 0.778 |
| M30 | 21765 | 99.74 | 625 | 884 (872-919) | 4.94 (4.86-5) | 2.14 (2.13-2.16) | 0.992 |
The parameters were estimated using MOTHUR. Samples from patients without T2DM (M). a Nseqs = Sequence numbers were obtained after removing low quality sequences (N ≥ 2; homopolymers ≥8) and short sequences (<200 bp). b The smallest library (5021 sequences) was used for normalization of the data. The results presented are the average of 1000 repetitions. c OTU = number of operational taxonomic units defined over the maximum distance of 3%. dlci = lower confidence interval. ehci = upper confidence interval.
Table 2.
b. Number of qualified readings, estimators of richness, species, diversity and uniformity of the bacterial communities present in stool samples of patients with T2DM categorized by BMI.
Table 2.
b. Number of qualified readings, estimators of richness, species, diversity and uniformity of the bacterial communities present in stool samples of patients with T2DM categorized by BMI.
|
T2DM Group |
Nseqsa | Coverageb (%) | OTUc |
Chao1 (lcid-hcie) |
Inv-Simpson (lcid-hcie) |
Shannon (lcid-hcie) |
Pielou |
| T2DM-NW (n = 12) | |||||||
| DM5 | 44662 | 99.86 | 715 | 944 (897-1037) | 5.65 (5.6-5.7) | 4.3 (4.29-4.32) | 0.995 |
| DM38 | 28103 | 99.84 | 1130 | 1471 (1431-1559) | 5.62 (5.54-5.7) | 4 (4.05-4.08) | 0.995 |
| DM1 | 42462 | 99.81 | 703 | 890 (830-998) | 6.24 (6.17-6.32) | 3.47 (3.46-3.48) | 0.997 |
| DM2 | 63930 | 99.84 | 1047 | 1346 (1276-1454) | 5.98 (5.92-6) | 3.53 (3.52-3.54) | 0.997 |
| DM6 | 51875 | 99.86 | 879 | 1172 (1125-1257) | 3.77 (3.74-3.8) | 3 (3-3.03) | 0.995 |
| DM8 | 53503 | 99.85 | 790 | 1057 (993-1175) | 4.27 (4.22-4.31) | 3.89 (3.88-3.9) | 0.997 |
| DM9 | 63090 | 99.82 | 908 | 1168 (1090-1299) | 4.62 (4.56-4.68) | 4.53 (4.52-4.54) | 0.999 |
| DM17 | 37353 | 99.83 | 1187 | 1517 (1460-1629) | 6.53 (6.46-6.61) | 4.87 (4.86-4.88) | 0.998 |
| DM19 | 10255 | 99.74 | 738 | 1097 (1073-1161) | 4.72 (4.6-4.86) | 3.95 (3.92-3.97) | 0.994 |
| DM27 | 33597 | 99.79 | 680 | 943 (866-1093) | 7.1 (7-7.19) | 5 (5.01-5.03) | 0.998 |
| DM50 | 40129 | 99.82 | 931 | 1278 (1203-1425) | 6.95 (6.88-7) | 4 (4.06-4.08) | 0.998 |
| DM51 | 38123 | 99.81 | 799 | 1056 (981-1198) | 6.64 (6.59-6.7) | 4.48 (4.47-5) | 0.894 |
| T2DM-OW (n = 8) | |||||||
| DM16 | 46862 | 99.84 | 1009 | 1328 (1275-1425) | 5.37 (5.32-5.43) | 4.82 (4.81-4.83) | 0.999 |
| DM20 | 67321 | 99.86 | 799 | 1125 (1052-1253) | 4.87 (4.84-4.9) | 3.33 (3.32-3.34) | 0.996 |
| DM21 | 58523 | 99.85 | 1250 | 1609 (1531-1749) | 6.23 (6.18-6.28) | 5.23 (5.22-5.24) | 0.997 |
| DM22 | 58044 | 99.87 | 1011 | 1390 (1333-1495) | 4.85 (4.81-4.9) | 4 (4.06-4.08) | 0.997 |
| DM23 | 39135 | 99.84 | 981 | 1270 (1220-1368) | 5.57 (5.52-5.63) | 4.75 (4.74-4.77) | 0.996 |
| DM28 | 37674 | 99.85 | 922 | 1366 (1327-1443) | 4.75 (4.69-4.82) | 2.92 (2.9-2.93) | 0.996 |
| DM42 | 18130 | 99.74 | 933 | 1228 (1166-1367) | 5.14 (5-5.25) | 3.84 (3.82-3.86) | 0.994 |
| DM42 | 71836 | 99.86 | 646 | 879 (809-1000) | 5.96 (5.92-6) | 4 (4.04-4.06) | 0.998 |
The parameters were estimated using Mothur. Samples from patients with T2DM (DM). a Nseqs = Sequence numbers were obtained after removing low quality sequences (N ≥ 2; homopolymers ≥8) and short sequences (<200 bp). b The smallest library (5021 sequences) was used for normalization of the data. The results presented are the average of 1000 repetitions. c OTU = number of operational taxonomic units defined over the maximum distance of 3%. dlci = lower confidence interval. ehci = upper confidence interval.
In the categorization based on BMI (Figure S2) the following were found: CL-NW subgroup: Faecalibacterium and Bacteroides; CL-OW subgroup: Megamones and Catenibacterium; T2DM-NW subgroup: Lachnospiracea_incertae_sedis and Ruminococcus; and T2DM-OW subgroup: Prevotella and Clostridium_IV. On the other hand, in the subgroups categorized according to BMI + CH, the following were found: CL-NW > CH: Faecalibacterium and Roseburia; CL-NW < CH: Streptococcus and Dorea; CL-OW > CH: Blautia and Ruminococcus; CL-OW < CH: Megamones and Catenibacterium; T2DM-NW > CH: Bacteroides and Coprococcus; T2DM-NW < CH: Lachnospiracea_incertae_sedis and Lactobacillus; and T2DM-OW < CH: Prevotella and Clostridium_IV, as seen in Table 3.
Different taxonomic groups were identified at the gender level with statistical significance according to the condition of BMI + CH. The results showed 4 taxa with statistical significance: a) Roseburia (P = 0.025), b) Clostridium_IV (P = 0.017), c) Prevotella (P = 0.017), and d) Sporobacter (P = 0.020), these correlations by level between the BMI + CH. Furthermore, to test for differences in bacterial composition between samples, a principal component analysis (PCA) was performed for all genus-level 16S rRNA gene reads in STAMP. In this analysis, the difference that exists between the dominances of the bacterial communities and the characteristics of each subgroup based on BMI and BMI + CH was observed. In the BMI group, the T2DM-OW subgroup showed a position clearly differentiated from that of the other subgroups. In contrast, in the BMI + CH group, the T2DM-OW < CH subgroup is separated from the other subgroups.
The data represent the Means ± Standard Deviations of the biochemical values of patients with and without diabetes mellitus. One-way ANOVA* was performed to analyze differences between groups, with Tukey’s post hoc** test to compare intragroup differences. Significance was considered with a p > 0.05. Control Normal Weight (CL-NW), Control Overweight (CL-OW), Type 2 Diabetes mellitus Normal Weight (T2DM-NW), Type 2 Diabetes mellitus Overweight (T2DM-OW).
The table shows the percentage and type of bacteria at the genus level classified by group of patients who consumed more than 200 g of CH or less than this amount. The percentage of less than 5% represents genera of bacteria with little representation in the bacterial microenvironment.
Study groups: Control and Type 2 Diabetes Mellitus, classified by Body Mass Index (BMI) and subcategorized by BMI + HCO consumption.
Figure S2a and S2b.
Structure bacterial communities of the faeces samples of the CL and T2DM groups and their respective subgroups according to BMI. Average relative abundance at the Phylum level of the BMI subgroups. Only taxa with mean relative abundances > 0.05% were graphed. Control Normal Weight (CL-NW), Control Overweight (CL-OW), Type 2 Diabetes Mellitus Normal Weight (T2DM-NW), Type 2 Diabetes Mellitus Overweight (T2DM-OW).
Figure S3ad S3b.
Structure of the bacterial communities of faeces samples for CL and T2DM groups, with the subgroups categorized by BMI. Average relative abundance at the gender level of the BMI subgroups. Taxa with mean relative abundance P > 0.05 % were plotted. Control Normal Weight (CL-NW), Control Overweight (CL-OW), Type 2 Diabetes Mellitus Normal Weight (T2DM-NW), Type 2 Diabetes Mellitus Overweight (T2DM-OW).
3. Discussion
In this study, the dietary evaluation showed higher carbohydrate consumption in the healthy control group than in the patients with T2DM. This phenomenon acts as a risk factor for the healthy group but is protective for the diabetic group. For example, the dietary recommendation in patients with T2DM establishes a carbohydrate consumption less than 45%, and sugar consumption less than 10% / day of the total energy consumed. [22]; a sucrose consumption below 5% has been proposed [23]. In this study, both groups consumed more than 50% carbohydrates and 10% sucrose in their diet, situation that can be associated with the presence of overweight in these patients [24]. The richness and diversity of the bacterial communities of the feces samples of the subgroups categorized by BMI and BMI + CH (Table 2a and Table 2b, and Figure S2) allowed us to observe the modifications in the structure of the bacterial communities of these subjects. In the T2DM-NW > CH subgroup, richness was much lower compared to that of the CL-NW > CH subgroup, while in the CL-OW < CH subgroup, there was greater richness than in the T2DM-OW < CH subgroup (Table 2a and Table 2b). These results show the changes in the diversity of the intestinal microbiota in patients with overweight, T2DM, and high carbohydrate consumption. Similar behavior has been reported in overweight patients, who tend toward obesity and who present changes in the diversity of the intestinal microbiota, which leads to low-grade inflammation [25] and, consequently, in the long term, may be the cause of T2DM [26]. Currently, that information is unclear. It is known that the type of intestinal microbiota will depend on the quality and quantity of the nutrients consumed, particularly carbohydrates. Consumption of metformin in patients with T2DM for glycemic control has been reported to cause changes in the intestinal microbiota [27]. With the results that this study obtained, it can be sustained that the consumption of carbohydrates and the status of being overweight alter the structure of the intestinal microbiota of patients with T2DM [28].
From the characterization of bacterial communities of feces, it was possible to detect that the relative abundance of Firmicutes in the T2DM-OW subgroup decreased by almost 50% of the total (Firmicutes / Bacteroidetes ratio) compared to in the CL-OW group, where the percentage of Firmicutes reached 70.62% (Figure S2). These findings contrast with the results of the study by Chávez-Carbajal et al. [18], who found that the group with T2DM + metformin presented an increase in Bacteroidetes and a decrease in Firmicutes, allowing for the association of metformin as a modifier of the microbiota. In addition, an interesting datum is that the abundance percentages of the T2DM-OW group (Bacteroidetes 54% and Firmicutes 45.23%) of this study (Figure S2) are like those of the T2DM group without metformin (Bacteroidetes 53.18% / Firmicutes 43.11%) from the work of Chávez-Carbajal et al. [18]. The foregoing makes it possible to clarify that there is a close relationship between overweight, T2DM, metformin consumption, and changes in the intestinal microbiota. Similarly, other studies show a clear relationship between overweight/obesity, T2DM [29], and the diversity of Bacteroidetes vs Firmicutes [7], where patients with T2DM and obesity have a lower percentage of Bacteroidetes (19.5%) and a higher percentage of Firmicutes (55.7%) compared to patients without T2DM (Bacteroidetes: 32.1% / Firmicutes: 36.9%) [30]. The importance of maintaining a normal weight in patients suffering from T2DM is clear, as being overweight/obese could affect the therapeutic effects of metformin [29,30]. When the subgroups of BMI + CH are combined, it can be inferred that suffering from T2DM combined with high consumption of carbohydrate could be the main cause of the changes in the abundance of Firmicutes. This was observed in the T2DM-NW > CH subgroup (49.72%), in which there was less Firmicutes in comparison to the CL-OW > CH subgroup (56.59%). On the other hand, this same Phylum was higher in the CL-OW < CH subgroup (75.30%), a combination of overweight and low carbohydrate consumption without DMT2, was lower in the T2DM-NW < CH subgroup (67.25%), in which patients are suffering from T2DM, with low carbohydrate consumption and normal weight; overweight plus T2DM and low carbohydrate consumption (T2DM-OW < CH, 45.23%) also showed a lower percentage of Firmicutes (Figure S2). This is consistent with published data regarding dysbiosis attributed to Firmicutes of the intestinal microbiota in women [31]; 90% of patients in this study were women. Additionally, it was reported that Firmicutes could contribute to the development of obesity [32], which agrees with the results of this study, as there is an abundance of Firmicutes, associated with overweight/obesity, in Mexican women. On the other hand, the prevalence of Bacteroidetes was more represented in the T2DM-OW < CH subgroup (54%), with a lower proportion of Proteobacteria, Actinobacteria, and Verrucomicrobia found (Figure S2). In relation to Bacteroidetes and Actinobacteria, it is believed that they could be of importance to the growth of Firmicutes, as they could be inhibiting thier growth [33]. The present study found that the Bacteroides (29.75%) had a higher relative abundance than the other genera of the T2DM-NW > CH subgroup. It has been reported that changes in the abundance of Bacteroides could be attributed to the amount consumed vs. absorption; for example, sucrose, which is not absorbed in the small intestine, is absorbed in the colon, where it also interacts with the colonic microbiota [34]. This has also been reported in Japanese patients with T2DM [50]. These results are controversial because the ingestion of sucrose produces a reduction in Bacteroides by inhibiting the expression of the BT3172 gene, which is essential for its survival [15,35,36].
Another genus associated with a high prevalence of prediabetes [37] and childhood obesity [38] is the Megamones, which, in this study, showed a high prevalence in the CL-OW < CH subgroup. In addition, Megamones was found to be highly enriched in a group with normal glucose tolerance [39], revealing that the different species of Megamones have some particular functions; some are capable of fermenting glucose into acetate and propionate, short-chain fatty acids (SCFA), which are beneficial to health [40].
The genus Prevotella appeared with higher relative abundance in the T2DM-OW < CH subgroup. Prevotella is a bacterium associated with chronic intestinal inflammation that has been found to be not very abundant in the Chinese population with T2DM [41]. In this study, when the abundance analysis was performed between the subgroups, Prevotella was identified within the group of most abundant intestinal bacteria. The abundance of these bacteria increases in subgroups with T2DM, making dysbiosis evident in these patients [6,7,42]. In addition, the inflammation of the intestinal mucosa mediated by this bacterium promotes systemic inflammation, with increased intestinal permeability and translocation of bacterial products [43,44]. The genus Faecalibacterium presented a higher relative prevalence in the CL-NW > CH subgroup and a lower relative prevalence in the T2DM-NW > CH subgroup. Faecalibacterium is one of the most abundant bacterial species in the healthy human intestine [45]. Currently, F. prausnitzii is the only known species of the genus Faecalibacterium that, in recent years, has been of great interest as a biomarker of intestinal health [46]. The decrease in the abundance of this bacterial genus is associated with inflammatory bowel syndrome (IBS), inflammatory bowel disease (IBD), colorectal cancer, and T2DM [47]. However, the effects derived from the interaction between F. prausnitzii and the consumption of carbohydrate in patients with T2DM are not yet known. There are variations in the microbial composition of the subgroups categorized by BMI and BMI + CH. At the genus level, a clear bacterial profile was observed within the T2DM-OW and T2DM-OW < CH subgroups. This finding would help to confirm the existence of a microbial signature in T2DM, reported in other studies [34]. Therefore, this study could serve as a basis for identifying the microbial molecular fingerprint of the colon in patients with T2DM, overweight, and carbohydrate consumption. The fact that the intestinal microbiota is a dynamic ecosystem that changes with the type of diet [11] could explain the modifications found with the consumption of carbohydrate in this study. The carbohydrate consumption significantly marked the abundance of certain bacteria in the different subgroups of BMI + CH. At the gender level, there was a significant increase in Roseburia and Clostridium IV (CL-NW > CH), as well as Prevotella (T2DM-OW < CH) and Sporobacter (T2DM-NW > CH). These findings provide insight into the effect and interaction between sucrose consumption and the changes it causes in the host’s intestinal microbiota [10, 14, and 48]. In the colon, the interaction between the microbiota and the products of sucrose metabolism [34] can influence the inflammatory state, the function of the intestinal barrier, and, therefore, the development of complications of DM2. [49]. With this study, new nutritional education strategies can be proposed, which favor the reduction of carbohydrate in the diet and, with this, modulate various bacterial groups associated with the development of TDM2.
4. Materials and Methods
4.1. Ethics Statement
The present study protocol was reviewed and approved by the ethics committee of the hospital and the Faculty of Medicine (project 015/2018) of the Universidad Autónoma del Estado de México (UAEMéx). All participants included in the study gave their informed consent in accordance with the Declaration of Helsinki revised in 2013 [16].
4.2. Study Design
This was a prospective, cross-sectional, and comparative study. One hundred subjects with and without a diagnosis of T2DM were invited to attend the hospital’s outpatient clinic an informative talk. From this, forty subjects of both sexes attended the outpatient clinic of the General Hospital “Dr. Nicolás San Juan” in Toluca, State of Mexico A brief medical history was given to assess whether the patients met the inclusion criteria.
4.3. Study Subjects
Of the 100 subjects invited to participate, 40 met the inclusion criteria. These were divided into 2 groups: 1) Type 2 Diabetes mellitus group, T2DM (n = 20); and 2) Control group of healthy subjects without Diabetes mellitus, CL (n = 20). The inclusion criteria for both groups were 25 to 65 years old, without any other pathology at the time of the study and less than 10 years of suffering the disease, regardless of chronological age. The following exclusion criteria were used in both cases: pregnancy; chronic alcohol, drug, or tobacco use; and acute or chronic autoimmune, bacterial, or viral diseases.
4.4. Anthropometric Evaluation
To calculate the body mass index (BMI), each participant’s body weight in kilograms was determined using a Tanita-brand scale model Bf-682 (Monterrey, Mexico) after an 8-hour fast. The participants were placed standing on the scale, with the soles of their feet on the scale’s surface. Height was measured while standing, without shoes, with a stadiometer (Seca® Model 240; Accuracy ± 2 mm, Seca GmbH & Co., Hamburg, Germany). Once these data were obtained, BMI was calculated using the following formula: weight (Kg) / height (m2). Once the BMI was obtained, it was categorized as follows: a) Normal weight (NW = 18.5 - 24.99 Kg / m2), b) Overweight (OW = 25.0 - 29.99 Kg / m2), and c) Obese > 30 Kg / m2.
4.5. Determination of Biochemical Profile
To obtain the blood samples, the patients were asked to fast for 8 hours prior to the collection of the sample. Whole venous blood was collected in tubes with heparin (BD, Vacutainer, Franklin Lakes, NJ, USA). Glycaemia, glycosylated haemoglobin, total cholesterol and triglycerides were determined from whole blood, using immunoturbidimetric tests with reinforcing particles (Innovastar, Diasys Diagnostic Systems, Holzheim, Germany).
4.6. Dietary Evaluation and Patient Categorization
A Food Consumption Frequency (FCF) questionnaire was used to evaluate the food consumed during the last month. From this information, the energy intake of the macronutrients (carbohydrates, lipids, and proteins) was calculated for each patient, with the help of two instruments: The Mexican List of Composition of Food Equivalents “Mexican System of Equivalents” [17] and the FoodData Central, U.S. Department of Agriculture (https://fdc.nal.usda.gov/) [18]. From the intake in grams per day of CH, a consumption classification was made in more than 200 g / day (> 200) or less than 200 g / day (< 200), considering the proportion of sucrose, fructose, glucose and maltose. With these data, the groups were categorized by BMI and sub-categorized according to BMI + carbohydrate consumption as shown in Figure 1.
4.7. Collection of Feces
To collect feces samples, a sterile bottle was provided with instructions for correct collection and storage. Once the samples were collected, they were processed in a sterile microbiological hood. Four aliquots with 1 g of feces were stored in sterile 1.5 mL micro tubes at -70ºC for subsequent batch analysis [19].
4.8. Analysis of the Fecal Microbiota
1). DNA extraction from faeces
The 40 samples from the study participants were processed, using 150 mg of feces from each patient, for DNA extraction, following the instructions provided in the protocol of the Quick-DNA™ Fecal / Soil Microbe Miniprep Kit from the Zymo Research brand. The times of mechanical lysis with disruptor were modified from 10 to 15 minutes; cooling cycles were at the same times.
1). Gel electrophoresis of DNA extraction and Metagenomics’ Material Amplification Test (PCR)
Electrophoresis was performed at 110 Volts for 35 minutes on samples of 3 µL of DNA loaded on 1% agarose gels (100 mL of 1X Buffer TAE per 1 g of agarose). A total of 1 mL of DNA from each sample was used to amplify by PCR targeting the 16S rRNA gene, to verify that they were capable of being amplified [20].
3). Massive sequencing
4.10. Illumina Sequencing by Amplicons of the V4-V5 Region of the 16s rRNA Gene
The 40 samples resulted in metagenomics DNA that met the quality of purity and concentration (260/280 = A1.8-2.0). They were sent to the sequencing service of the Center for Comparative Genomics and Evolutionary Bioinformatics (CGBE) of Dalhousie University, Canada. Massive sequencing of the Illumina MiSeq type (300 + 300 bp PE) was requested [21].
4.11. Bioinformatic Analysis of the Sequences
The 16S rRNA data were processed with Mothur software version 1.39.5, following the pipeline recommended by the developer of the MiSeq SOP. The selected reads met the following criteria: no ambiguous bases, a minimum length of 200 bp, and homopolymers with a maximum length of 8 bp. Similarly, removal of the chimeric sequences and the lineages of eukaryotes, mitochondria, and chloroplasts, unknown and unclassified, was carried out. Subsequently, the diversity of the sequences by operational taxonomic units (OTU) was examined considering quality readings with a dissimilarity of 3%, while the rarefaction curves were calculated in a similarity of 97% with the Mothur alpha diversity flow. Different metrics were calculated to evaluate the bacterial communities, including the number of OTUs observed, the Shannon diversity indices and Pielou uniformity, and the Chao 1 and Inv-Simpson estimators for species richness. The calculation of these parameters was carried out by normalizing all the libraries. Finally, the composition of the bacterial communities was determined by beta diversity. The abundance was expressed as a percentage with respect to the total number of sequences in each sample (relative abundance) [20,21]. Subsequently, the file generated by Mothur was analyzed using the Statistical Analysis of metagenomics Profiles Test (STAMP) software, and the results were represented by graphs. The obtained sequences were registered in the NCBI BioSample database, at the following link: https://www.ncbi.nlm.nih.gov/sra/PRJNA807457, with the number to access and cite these SRA data: PRJNA807457.
4.12. Statistical analysis
The anthropometric, biochemical, and diet profiles were analyzed using SPSS v22 software, employing the non-parametric Kruskal-Wallis test. To identify the differences between the subgroups of the intestinal microbiota in the taxonomic levels of Phylum, Class, Order, and Genus, the relative abundance (%) of bacterial communities and the principal component analysis (PCA) we used the ANOVA test and the Kruskal-Wallis test to calculate p values < 0.05 in STAMP [18,21].
5. Conclusions
The composition and diversity of the gut microbiota in patients with and without T2DM were significantly different. The habitual consumption of carbohydrate is closely related to differences in the taxonomic levels of Phylum and genus. Although more studies are necessary, it can be concluded that, with a reduction in the consumption of 10% per day, it is possible to modulate the bacterial communities in healthy patients and, through this, perhaps help to prevent dysbiosis and the development of T2DM. But, with an increase in the consumption (more than 15%), the modification in the proportion and diversity of the microbiota was altered. This was observed in the groups that consumed high amounts of carbohydrates (CL-NW > CH, CL OW > CH, T2DM-NW > CH), regardless of whether they had diabetes mellitus and overweight or not, which significantly reduced the proportion of Faecalobacterium, altered the proportion of Bacteroides, with important modifications in other bacterial genera. The high consumption of carbohydrate considerably modified the composition and diversity of the bacterial communities. If comorbidities such as T2DM and overweight are added to this, the changes in the intestinal microbiota will be more evident. In this study, carbohydrate consumption showed, at the genus level, an abundance of bacteria, such as Faecalobacterium, Bacteroides, Blautia, Roseburia, Prevotella, and Sporobacter.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, BEMC and ADSM; methodology, JFAG and BEMC; validation, JACC and RVR; FDMCE; ADSM and BEMC writing—original draft preparation, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. This project was funded by Universidad Autónoma del Estado de México.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Acknowledgments
To Consejo Nacional de Ciencia y Tecnología, for the scholarship granted to the master’s student.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- NORMA Oficial Mexicana 015-SSA2-2010, Para la prevención, tratamiento y control de la diabetes mellitus. Secretaria de Gobernación. Diario Oficial de la Federación. Diabetes Tipo 2. Available from: http://www.salud.gob.mx/unidades/cdi/nom/m015ssa24.html (Update of January 20, 2022).
- Zimmet, P.; Alberti, K.G.; Magliano, D.J.; Bennett, P.H. Diabetes mellitus statistics on prevalence and mortality: facts and fallacies. Nat. Rev. Endocrinol. 2016, 12, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Instituto Nacional de Estadística y Geografía (INEGI) (2015). Estadísticas de mortalidad 2015. Consulta interactiva de datos. Available from: https://www.inegi.org.mx/contenidos/programas/enasem/2018/doc/enasem_2018_presentacion.pdf. (Update of January 20, 2022).
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef]
- Candela, M.; Biagi, E.; Soverini, M.; Consolandi, C.; Quercia, S.; Severgnini, M.; Peano, C.; Turroni, S.; Rampelli, S.; Pozzilli, P.; et al. Modulation of gut microbiota dysbioses in type 2 diabetic patients by macrobiotic Ma-Pi 2 diet. Br. J. Nutr. 2016, 116, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Wen, L.; Duffy, A. Factors Influencing the Gut Microbiota, Inflammation, and Type 2 Diabetes. J. Nutr. 2017, 147, 1468S–1475S. [Google Scholar] [CrossRef]
- Harsch, I.A.; Konturek, P.C. The Role of Gut Microbiota in Obesity and Type 2 and Type 1 Diabetes Mellitus: New Insights into “Old” Diseases. Med Sci. 2018, 6, 32. [Google Scholar] [CrossRef]
- Bridgewater, L.C.; Zhang, C.; Wu, Y.; Hu, W.; Zhang, Q.; Wang, J.; Li, S.; Zhao, L. Gender-based differences in host behavior and gut microbiota composition in response to high fat diet and stress in a mouse model. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Rinninella, E.; Cintoni, M.; Raoul, P.; Lopetuso, L.R.; Scaldaferri, F.; Pulcini, G.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. Food Components and Dietary Habits: Keys for a Healthy Gut Microbiota Composition. Nutrients 2019, 11, 2393. [Google Scholar] [CrossRef]
- Muralidharan, J.; Galiè, S.; Hernández-Alonso, P.; Bulló, M.; Salas-Salvadó, J. Plant-Based Fat, Dietary Patterns Rich in Vegetable Fat and Gut Microbiota Modulation. Front. Nutr. 2019, 6, 157. [Google Scholar] [CrossRef]
- US Department of Health and Human Services and US Department of Agriculture. 2015–2020 Dietary Guidelines for Americans. 8th ed. December 2015. Available online: http://health.gov/dietaryguidelines/2015/guidelines/ (accessed on 21 March 2019).
- Townsend GE 2nd, Han W ND 3rd, V Raghavan, NA Barry, AL Goodman, EA Groisman. Dietary sugar silences a colonization factor in a mammalian gut symbiont. Proc Natl Acad Sci 2019; 116 (1):233-238.
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef] [PubMed]
- World Medical Association [WMA], Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects. Clin Rev Ed. 2013;310(20):2191-2194. Available from: https://www.wma.net/wp-content/uploads/2016/11/DoH-Oct2013-JAMA.pdf (Update of , 2022). 20 January.
- AB Pérez-Lizaur, B Palacios-González, AL Castro-Becerra, I Flores-Galicia, Sistema Mexicano de Alimentos Equivalentes, 4th ed. México: Porrúa; 2014. P168.
- Chávez-Carbajal, A.; Pizano-Zárate, M.L.; Hernández-Quiroz, F.; Ortiz-Luna, G.F.; Morales-Hernández, R.M.; De Sales-Millán, A.; Hernández-Trejo, M.; García-Vite, A.; Beltrán-Lagunes, L.; Hoyo-Vadillo, C.; et al. Characterization of the Gut Microbiota of Individuals at Different T2D Stages Reveals a Complex Relationship with the Host. Microorganisms 2020, 8, 94. [Google Scholar] [CrossRef]
- Akkermans ADL, van Elsas JD, de Bruijn F. Manual molecular microbial ecology. 1rst ed. eBook: Springer, Kluwer Academic Publishers, Dordrecht; 1996. Chapter 1.4.4, Ramírez-Saad H, Akkermans WM, Akkermans ADL. DNA extraction from actinorhizal nodules; p1-11.
- Aguirre-Garrido, J.F.; Ramírez-Saad, H.C.; Toro, N.; Martínez-Abarca, F. Bacterial Diversity in the Soda Saline Crater Lake from Isabel Island, Mexico. Microb. Ecol. 2016, 71, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Marañón, M.; Miralles, I.; Aguirre-Garrido, J.F.; Anguita-Maeso, M.; Millán, V.; Ortega, R.; García-Salcedo, J.A.; Martínez-Abarca, F.; Soriano, M. Changes in the soil bacterial community along a pedogenic gradient. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.A. Sugar-Sweetened and Artificially-Sweetened Beverages in Relation to Obesity Risk. Adv. Nutr. Int. Rev. J. 2014, 5, 797–808. [Google Scholar] [CrossRef]
- American Diabetes Association (ADA) (2019) Making sense of food labels. Available from:https://www.diabetes.org/nutrition/understanding-food-labels/making-sense-of-food-labels (Update of January 20, 2022).
- World Health Organization (WHO). Sugar intake for adults and children. 2015 WHO reference number: WHO/NMH/NHD/15.2. Available from: https://www.who.int/publications/i/item/9789241549028 (update of January 20, 2022).
- Murugesan, S.; Ulloa-Martínez, M.; Martínez-Rojano, H.; Galván-Rodríguez, F.M.; Miranda-Brito, C.; Romano, M.C.; Piña-Escobedo, A.; Pizano-Zárate, M.L.; Hoyo-Vadillo, C.; García-Mena, J. Study of the diversity and short-chain fatty acids production by the bacterial community in overweight and obese Mexican children. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1337–1346. [Google Scholar] [CrossRef]
- Leite, A.Z.; Rodrigues, N.d.C.; Gonzaga, M.I.; Paiolo, J.C.C.; de Souza, C.A.; Stefanutto, N.A.V.; Omori, W.P.; Pinheiro, D.G.; Brisotti, J.L.; Junior, E.M.; et al. Detection of Increased Plasma Interleukin-6 Levels and Prevalence of Prevotella copri and Bacteroides vulgatus in the Feces of Type 2 Diabetes Patients. Front. Immunol. 2017, 8, 1107. [Google Scholar] [CrossRef]
- MetaHIT consortium; Forslund, K. ; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; et al. Erratum: Corrigendum: Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2017, 545, 116–116. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Ahmad, A.; Yang, W.; Chen, G.; Shafiq, M.; Javed, S.; Zaidi, S.S.A.; Shahid, R.; Liu, C.; Bokhari, H. Analysis of gut microbiota of obese individuals with type 2 diabetes and healthy individuals. PLOS ONE 2019, 14, e0226372. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.; Vogensen, F.K.; Van Den Berg, F.W.J.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut Microbiota in Human Adults with Type 2 Diabetes Differs from Non-Diabetic Adults. PLoS ONE 2019, 5, e9085. [Google Scholar] [CrossRef] [PubMed]
- Lippert, K.; Kedenko, L.; Antonielli, L.; Kedenko, I.; Gemeier, C.; Leitner, M.; Kautzky-Willer, A.; Paulweber, B.; Hackl, E. Gut microbiota dysbiosis associated with glucose metabolism disorders and the metabolic syndrome in older adults. Benef. Microbes 2017, 8, 545–556. [Google Scholar] [CrossRef]
- Chávez-Carbajal, A.; Nirmalkar, K.; Pérez-Lizaur, A.; Hernández-Quiroz, F.; Ramírez-Del-Alto, S.; García-Mena, J.; Hernández-Guerrero, C. Gut Microbiota and Predicted Metabolic Pathways in a Sample of Mexican Women Affected by Obesity and Obesity Plus Metabolic Syndrome. Int. J. Mol. Sci. 2019, 20, 438. [Google Scholar] [CrossRef]
- Barczynska, R.; Kapusniak, J.; Litwin, M.; Slizewska, K.; Szalecki, M. Dextrins from Maize Starch as Substances Activating the Growth of Bacteroidetes and Actinobacteria Simultaneously Inhibiting the Growth of Firmicutes, Responsible for the Occurrence of Obesity. Plant Foods Hum. Nutr. 2016, 71, 190–196. [Google Scholar] [CrossRef]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Hamaguchi, M.; Kaji, A.; Sakai, R.; Osaka, T.; Inoue, R.; Kashiwagi, S.; Mizushima, K.; Uchiyama, K.; Takagi, T.; et al. Intake of sucrose affects gut dysbiosis in patients with type 2 diabetes. J. Diabetes Investig. 2020, 11, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Maya-Lucas, O.; Murugesan, S.; Nirmalkar, K.; Alcaraz, L.D.; Hoyo-Vadillo, C.; Pizano-Zárate, M.L.; García-Mena, J. The gut microbiome of Mexican children affected by obesity. Anaerobe 2019, 55, 11–23. [Google Scholar] [CrossRef]
- Dong, T.S.; Mayer, E.A.; Osadchiy, V.; Chang, C.; Katzka, W.; Lagishetty, V.; Gonzalez, K.; Kalani, A.; Stains, J.; Jacobs, J.P.; et al. A Distinct Brain-Gut-Microbiome Profile Exists for Females with Obesity and Food Addiction. Obesity 2020, 28, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Chevrot R, Carlotti A, Sopena V, Marchand P, Rosenfeld E. Megamonas rupellensis sp. nov., an anaerobe isolated from the caecum of a duck. Int J Syst Evol Microbiol 2008; 58(Pt 12): 2921–2914.
- Sakon H, Nagai F, Morotomi M, Tanaka R Sutterella parvirubra sp. nov. and Megamonas funiformis sp. nov., isolated from human faeces. Int J Syst Evol Microbiol 2008; 58(Pt4):970–975.
- Wu X, Mac Han L, Nawaz M, Gao F, Zhang X, et.al. Molecular characterization of the fecal microbiota in patients with type II diabetes. Curr Microbiol 2010; 61(1):69–78.
- Anhê FF, Jensen BAH, Varin TV, Servant F, Van Blerk S, Richard D, & Schertzer JD. Type 2 diabetes influences bacterial tissue compartmentalization in human obesity. Nat Metab 2020; 2(3):233-242.
- Larsen, J.M. The immune response toPrevotellabacteria in chronic inflammatory disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef]
- Leite, A.Z.; Rodrigues, N.d.C.; Gonzaga, M.I.; Paiolo, J.C.C.; de Souza, C.A.; Stefanutto, N.A.V.; Omori, W.P.; Pinheiro, D.G.; Brisotti, J.L.; Junior, E.M.; et al. Detection of Increased Plasma Interleukin-6 Levels and Prevalence of Prevotella copri and Bacteroides vulgatus in the Feces of Type 2 Diabetes Patients. Front. Immunol. 2017, 8, 1107. [Google Scholar] [CrossRef]
- Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, et.al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018; 555(7695):210-215.
- Leylabadlo, H.E.; Ghotaslou, R.; Feizabadi, M.M.; Farajnia, S.; Moaddab, S.Y.; Ganbarov, K.; Khodadadi, E.; Tanomand, A.; Sheykhsaran, E.; Yousefi, B.; et al. The critical role of Faecalibacterium prausnitzii in human health: An overview. Microb. Pathog. 2020, 149, 104344. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Halder, C.V.; de Sousa Faria, A.V.; Andrade, S.S. Action and function of Faecalibacterium prausnitzii in health and disease. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 643–648. [Google Scholar] [CrossRef]
- Lopez-Siles, M.; Duncan, S.H.; Garcia-Gil, L.J.; Martinez-Medina, M. Faecalibacterium prausnitzii: from microbiology to diagnostics and prognostics. ISME J. 2017, 11, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tapia, M.; Martínez-Medina, J.; Tovar, A.R.; Torres, N. Natural and Artificial Sweeteners and High Fat Diet Modify Differential Taste Receptors, Insulin, and TLR4-Mediated Inflammatory Pathways in Adipose Tissues of Rats. Nutrients 2019, 11, 880. [Google Scholar] [CrossRef] [PubMed]
- Könner, A.C.; Brüning, J.C. Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol. Metab. 2011, 22, 16–23. [Google Scholar] [CrossRef]
- Kasprzak-Drozd, K.; Oniszczuk, T.; Stasiak, M.; Oniszczuk, A. Beneficial Effects of Phenolic Compounds on Gut Microbiota and Metabolic Syndrome. Int. J. Mol. Sci. 2021, 22, 3715. [Google Scholar] [CrossRef]
Figure 1.
Classification of groups by Body Mass Index and subgroups categorized by Body Mass Index plus carbohydrate consumption. Carbohydrate (CH), Control Normal Weight (CL-NW), Control Overweight (CL-OW), Type 2 Diabetes mellitus Normal Weight (T2DM-NW), Type 2 Diabetes mellitus Overweight (T2DM-OW).
Figure 1.
Classification of groups by Body Mass Index and subgroups categorized by Body Mass Index plus carbohydrate consumption. Carbohydrate (CH), Control Normal Weight (CL-NW), Control Overweight (CL-OW), Type 2 Diabetes mellitus Normal Weight (T2DM-NW), Type 2 Diabetes mellitus Overweight (T2DM-OW).
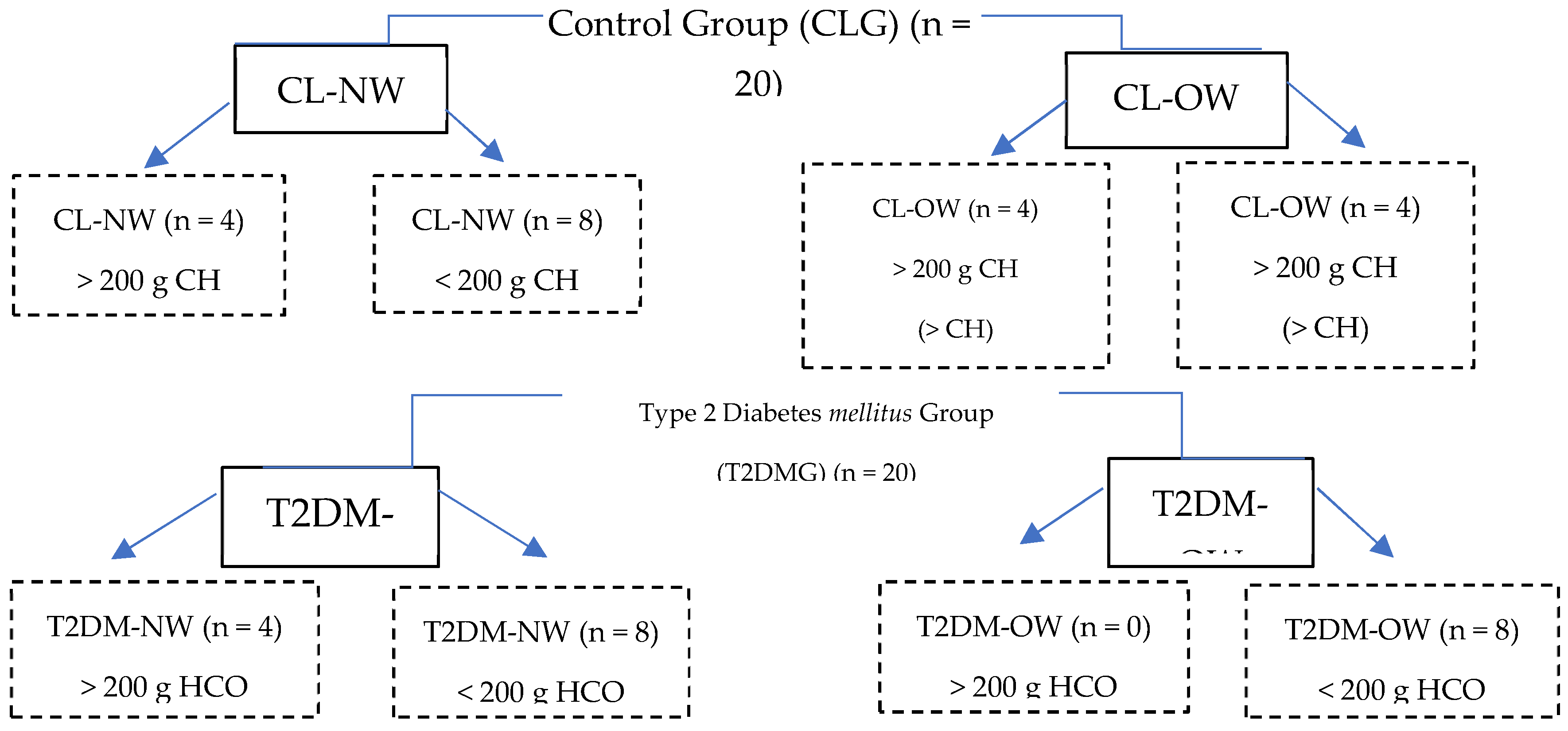
Table 1.
Biochemical data of patients with and without Diabetes mellitus.
|
CL-NW n = 12 |
CL-OW n = 8 |
T2DM-NW n = 12 |
T2DM-OW n = 8 |
||
| Mean ±SD | Mean ±SD | Mean ±SD | Mean ±SD | P value | |
| Serum glycaemia | 81.1 ±1.7 | 83.3 ±5.9 | 150 ± 7.1** | 135 ±4.3** | 0.001* |
| Hb1Ac | 5.1 ±0.206 | 5.4 ±0.471 | 9.2 ±1.5** | 8.4 ±3.3** | 0.001* |
| TG | 128 ±3.6 | 155 ±8.3 | 208 ± 1.5 | 251 ±1.5 | 0.119 |
| CT | 175.2 ±2.3 | 174 ±2.4 | 195 ±6 | 192 ±3.9 | 0.519 |
Table 3.
Average relative abundance at the gender level, categorized for consumption of Carbohydrates per group.
Table 3.
Average relative abundance at the gender level, categorized for consumption of Carbohydrates per group.
|
CLNW n = 12 |
% |
CLOW n = 8 |
% |
T2DMNW n = 12 |
% |
T2DMOW n = 8 |
||
|
>200 g HCO |
Faecalibacterium Prevotella Roseburia Blautia Roseburia Bacteroides Less than 5% |
23.92 18.48 8.47 8.11 8.47 5.85 26.7 |
Faecalibacterium Bacteroides Prevotella Blautia Ruminococcus Parabacteroides Less than 5% |
23.3 21.23 15.23 11.09 7.5 4.83 16.82 |
Bacteroides Prevotella Faecalibacterium Roseburia Blautia Less than 5% |
29.75 14.19 10.13 8.47 6.82 30.64 |
No patient consumed more than 200 g of CHO per day, then we don’t have samples in this group. |
-- |
| n = 4 | n = 4 | n = 4 | n = 0 | -- | ||||
|
< 200 g HCO |
Faecalibacterium Bacteroides Prevotella Blautia Roseburia Streptococcus Less than 5% |
20.49 16.79 10.87 9.32 8.36 4.10 15.67 |
Faecalibacterium Roseburia Prevotella Megamones Bacteroides Blautia Less than 5% |
16.3 15.52 11.41 10.89 10.3 10.28 25.3 |
Faecalibacterium Prevotella Roseburia Bacteroides, Blautia Lachnospiracea_ incertae_sedis Less than 5% |
16.63 12.15 12.1 9.85 8.22 5.71 35.34 |
Prevotella Faecalobacterium Blautia Roseburia Less than 5% |
52.13 13.57 5.94 5.03 23.33 |
| n = 8 | n = 4 | n = 8 | n = 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



