
Submitted:
11 October 2024
Posted:
11 October 2024
You are already at the latest version
Abstract
The present review is related to the novel approach for improvement of the optical properties of wide bandgap metal oxides, in particular TiO2, based on the formation of the inorganic-organic hybrids that display absorption in the visible spectral range due to the formation of interfacial charge transfer (ICT) complexes. We outlined the property requirements of TiO2-based ICT complexes for efficient photo-induced catalytic reactions, emphasizing the simplicity of the synthetic procedure, the possibility of the fine-tuning of the optical properties supported by the density functional theory (DFT) calculations, and the formation of a covalent linkage between inorganic and organic components of hybrids, i.e., the nature of the interface. In addition, this study provides a comprehensive insight into potential applications of TiO2-based ICT complexes in photo-driven catalytic reactions (water splitting and degradation of organic molecules), including the identification of reactive species that participate in photocatalytic reactions by the spin-trapping electron paramagnetic resonance (EPR) technique. Considering the practically limitless number of combinations between inorganic and organic components capable of forming oxide-based ICT complexes and with the knowledge that this research area is unexplored, we are confident it is worth studying, and we emphasized some further perspectives.
Keywords:
Interfacial charge transfer complexes
; Titanium dioxide
; Solar energy conversion
; Optical properties
; Water splitting reaction
; Photocatalytic oxidation reactions
; Interface properties
; EPR spin-trapping
1. General Background
The scientific and engineering interest in the photocatalytic reactions, initiated by the pioneering work by Fujishima and Honda in the early seventies of the last century on water splitting using a TiO2 electrode [1], has grown tremendously, addressing a variety of environmental problems such as water and air purification and inactivation of bacteria and viruses, as well as hydrogen production. Considering the importance of photocatalysis, over the years, many review articles have appeared, so we refer readers to reviews by Hoffman et al. [2] and Kudo et al. [3] for additional background information and relevant literature that covers an initial period of the development of this field.
Among the variety of semiconductors, TiO2 is the most studied photocatalyst since it is chemically stable and affordable. The anatase has a bandgap energy of 3.2 eV, and the pH-dependent position of valence band maximum (VBmax) and conduction band minimum (CBmin) is presented in Figure 1 [4]. Upon TiO2 excitation with photons with an energy exceeding the bandgap energy, electrons from the VB are promoted to the CB, leaving holes behind.
Photogenerated charge carriers can recombine non-radiatively and dissipate the absorbed energy as heat, recombine radiatively, or get trapped at surface states and react with electron donors and electron acceptors adsorbed on its surface. In most photocatalytic experiments, oxygen is present and serves as the primary electron acceptor.
Further, in water, two protonated superoxide radical anions disproportionate into oxygen and hydrogen peroxide in a bimolecular reaction.
On the other hand, hydroxyl radicals (HO∙) are formed in the reaction of holes with adsorbed water or hydroxyl ions.
So, the excitation of aerated aqueous dispersions of semiconductors leads to generating reactive oxygen species (ROS).
The adsorption of reactants, either electron donors or electron acceptors, to metal oxides is governed by their surface chemistry, i.e., the charge of surface hydroxyl groups. Based on the literature survey by Professor Kosmulski [5], it is well-known that the average and median pH values of zero point charge (pHZPC) for anatase are 5.9 and 6, respectively, while for the rutile, they are 5.4 and 5.5, respectively.
Considering the position of VBmax and CBmin in TiO2, the photogenerated holes are powerful oxidants, while photogenerated electrons are good reductants. Besides the limitless use of solar energy, the advantage of photocatalysis is the possibility to carry on photo-driven heterogeneous reactions under mild experimental conditions (room temperature and atmospheric pressure), including non-spontaneous reactions with selective product synthesis [6,7]. However, two obstacles must be overcome to achieve the high yield of photocatalytic reactions over TiO2. First, based on the laser flash photolysis experiments, it is well known that the photogenerated charge carriers recombination is a fast process occurring on the nanosecond time scale [8,9]. Second, as a wide bandgap semiconductor, TiO2 absorbs only the UV photons, i.e., 5% of solar radiance. So, efficient separation of photogenerated electrons and holes and improved optical properties are prerequisites to achieve efficient photocatalytic reactions over TiO2.
Three different approaches are distinguished to address the previously mentioned issues. First, the deposition of co-catalysts (noble metal particles) on the photocatalysts’ surface provides enhanced electron transfer reactions and improved optical properties by plasmon resonance absorption [10,11]. Second, doping with light and heavy elements promotes less energetic excitations of electrons from mid-gap dopant levels to the conduction band of TiO2 [12,13,14]. And finally, third, surface modification of TiO2 with organic and organometallic molecules absorbing in the visible spectral range [15], that letter led to the discovery of dye-sensitized solar cells, the so-called Grätzel solar cells [16].
Despite tremendous efforts to use other semiconductor materials for photocatalytic purposes under visible light excitation, improving their optical properties by doping [17], forming heterojunctions to enhance their oxidation or reduction power [18], or synthesizing Z-scheme photocatalysts to improve charge separation [19], TiO2 remains as a photocatalyst of choice.
The most recent approach to bring the absorption of TiO2 and other wide bandgap metal oxides in the visible spectral range is interfacial charge transfer (ICT) formation [20]. Colorless aromatic molecules are recognized as suitable ligands to facilitate the formation of ICT complexes. During this time, from fundamental studies, the research in this area evolved to potential applications of TiO2, including the photocatalytic production of hydrogen and degradation of organic dyes [21,22,23,24], the light-to-current conversion [25,26,27], and photo-induced antimicrobial activity [28]. However, this review will be limited to photocatalytic reactions over TiO2-based ICT complexes.
2. Interfacial Charge Transfer Complexes
2.1. Synthesis
The formation of interfacial charge transfer (ICT) complexes is facilitated by a polycondensation reaction between hydroxyl groups originating from the surface of metal oxide (Me‒OH) and the colorless aromatic molecules, inducing the formation of a covalent linkage between surface metal, oxygen, and carbon. A schematic presentation for TiO2-based ICT complex formation is presented in Scheme 1. The successful formation of the ICT complexes is accompanied by the red absorption shift, providing a simple way to bring the optical properties of materials to the more practical spectral range for photo-driven catalytic reactions. As an instructive example, the absorption spectra of pristine 45-Å TiO2 colloid and TiO2-based ICT complexes, displaying tunable red absorption shifts induced by different surface-active ligands, are shown in Figure 2 [29].
The appearance of absorption in the visible spectral range in TiO2-based hybrids is due to the electronic coupling of localized orbitals of surface-attached ligands with the delocalized electron levels from the conduction band of TiO2 [30]. Consequently, absorption of light by the ICT complex yields direct, single-step promotion of electrons from the ligand directly into the CB of TiO2 without energy loss [31], resulting in a red absorption shift compared to unmodified TiO2. It should be emphasized that there is a fundamental difference in the photogeneration of charge carriers in ICT complexes and metal oxides sensitized with dye molecules; a graphical presentation is shown in Scheme 2. In the case of dye-sensitized metal oxides, photogeneration of charge carriers involves two steps: first, excitation of the dye molecules and subsequent electron transfer from the excited state into the semiconductor conduction band. In addition, photogenerated electrons and holes in ICT complexes are separated instantaneously into two phases, so holes are localized on the donating organic modifier, and the electrons are delocalized in the CB of TiO2 [32].
In the early stages of development of this field, when revealing the basic properties of the ICT complexes, most of the studies were done on small-sized colloidal TiO2 nanoparticles due to their unique surface structure. It is well-known, based on the X-ray absorption near-edge spectroscopy (XANES), that the bond length and coordination of the surface Ti atoms change from octahedral (six-coordinate) to square-pyramidal (penta-coordinate) when the size of TiO2 particles is sufficiently small, i.e., when particles have large curvature [20,33]. Later, for practical purposes, instead of using small colloidal nanoparticles, TiO2-based ICT complexes were prepared using submicronic TiO2 particles prepared by ultrasonic spray pyrolysis [34,35], TiO2 nanopowders prepared by sol-gel method [22], and commercial TiO2 photocatalysts such as Degussa P25 and Aerosil P90 [21,36,37,38,39,40,41].
Having in mind that on one side, the efficiency of the photocatalytic reactions is higher for small-sized particles, while, on the other side, powders are easier to handle, the visible-light-responsive TiO2 nanoparticles were in-situ prepared on polymer support [24]. Macroporous copolymer, based on glycidyl methacrylate and ethylene glycol dimethacrylate (poly(GMA-co-EGDMA)), with a large specific surface area (36 m2/g) and average pore size (130 nm) was used as a starting material since its functionalization is easy due to the presence of the reactive epoxy group. A two-step synthetic procedure is presented schematically in Figure 3A. The first step includes dopamine modification of poly(GMA-co-EGDMA) copolymer, indicated by the complete disappearance of vibrations that belong to the epoxy ring. In the second step, the hydrolysis of titanium(IV) isopropoxide (TTIP) in the presence of functionalized poly(GMA-co-EGDMA) copolymer dispersed in organic solvent results in the simultaneous formation of the TiO2 nanoparticles and the appearance of the ICT complex via two adjacent phenolic groups from dopamine coordinated to poly(GMA-co-EGDMA) copolymer over the amino groups. The microscopy data showed that the polymer support is decorated with a large number of well-separated small-in-size TiO2 nanoparticles (<10 nm) with a reasonably narrow size distribution (Figure 3B). In addition, the spectroscopy measurements (Figure 3C) revealed that the absorption spectrum of the resulting composite displays a significant redshift.
Until recently, the ICT complex formation has been almost exclusively studied using various morphological forms of TiO2 [20,21,22,23,24,25,26,27,28,29,31,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70]. In the last several years, the successful formation of the ICT complexes for other wide-band-gap oxides, including titanates [71,72,73,74,75,76], CeO2 [77,78], ZrO2 [79,80], ZnO [81,82,83,84,85], Al2O3 [86,87,88], and hydroxyapatite [89,90], has been reported. However, the number of inorganic components of the ICT complexes is quite limited, while the number of organic components is practically limitless. A literature overview of the ICT complexes between various wide-band-gap oxides and different types of ligands is summarized in Table 1. For clarity reasons, we classified ligands into four groups: ligands forming two neighboring Me‒O‒C linkages (catechol (CAT), salicylic acid (SA), and their derivatives), ligands forming single Me‒O‒C linkage (phenol and its derivatives), ligands forming Me‒S‒C linkage/linkages (thiols), and miscellaneous, refereeing to all other ligand types. It should be emphasized that the formation of the ICT complex between silver nanoparticles and aromatic amino acids was recently predicted by the DFT calculations, providing new insight into peculiar optical properties and reactivity of surface-modified silver nanoparticles [91], indicating that the ICT complex formation is not exclusive for metal-oxides.
2.2. Composition and Surface Structure
Infrared spectroscopy is a method of choice to reveal the coordination of ligand molecules to the surface of wide bandgap oxides. The differences in infrared (IR) spectra between free and bound ligands (appearance or disappearance, as well as the shift of vibrational bands) provide the possibility to understand the surface structure of organic-inorganic hybrids. So far, besides infrared spectroscopy, there is only one attempt, combining solid-state NMR with density functional theory (DFT), to understand the geometry of attached catechol (CAT) to the TiO2 surface [92].
Knowing that the ICT complex formation occurs by polycondensation reaction between hydroxyl groups from inorganic and organic parts of hybrid, the coordination of phenol (Ph) and its derivatives to the TiO2 surface is almost intuitive. An instructive example is a study by Fujisawa et al. [37]. The Kubelka-Munk function spectra of pristine TiO2 and TiO2‒Ph samples and the IR spectra of free Ph and attached Ph to the TiO2 surface are shown in Figure 4. The absorption onset in the TiO2‒Ph sample is red-shifted (around 550 nm), compared to unmodified TiO2, due to the ICT complex formation. The IR spectrum of Ph shows pronounced peaks at 1472 and 1593 cm−1 that belong to in-plane starching C=C vibration mixed with C‒H and O‒H bending vibrations and two relatively broad peaks at 1223 and 1369 cm−1 that belong to C‒H and O‒H bending vibrations. While the peaks at 1472 and 1593 cm−1 are practically intact upon adsorption of Ph on TiO2, the lower-energy peaks at 1223 and 1369 cm−1 are drastically changed, indicating a significant structural change around the hydroxy group of Ph and suggesting the formation of a Ti‒O‒C linkage.
However, in the case of ligands with two neighboring hydroxyl groups (CAT and SA, and their derivatives), the IR spectroscopy can’t provide an answer about the mode of their binding to Ti surface atom (Tisurf), is it bidentate binuclear (bridging) coordination or bidentate mononuclear (chelating) coordination; for clarity reasons chelating and bridging coordination of CAT to Tisurf is presented in Scheme 3. In other words, the question is, what is the molar ratio between Tisurf and these types of ligands? Since the ICT complexes exhibit optical properties distinct from their constituents, Job’s method of continuous variation [93] was applied to determine their composition, assuming that a single-type complex is present in the solution. The molar concentration of surface Ti atoms ([Tisurf]) can be calculated from the following equation [94], knowing the molar concentration of TiO2 ([TiO2]) and the diameter of particles in angstroms (D):
Of course, the prerequisite for using equation 1 to calculate the molar concentration of Tisurf is the narrow size distribution of nanometer in size TiO2 particles. The stoichiometric ratio (n) between Tisurf and the ligand (L) can be determined by plotting the absorbance of the ICT complex versus the mole fraction (x) of the metal or ligand. An instructive example, the Job’s plot for the surface-modified 45-Å TiO2 colloids with CAT and SA [29], is presented in Figure 5. Mole fraction (x=[Tisurf]/([Tisurf]+[L])) at the absorbance maximum corresponds to the stoichiometric ratio between Tsurf and L. The stoichiometric ratio between Tisurf and various catecholate- and salicylate-type ligands was found to be 2:1, indicating bridging coordination of these ligand types to the surface Ti atoms [22,29,34,35,42,43,44,45,46,47].
Besides composition, the simple spectrophotometric method, based on Benesi-Hildebrand analysis [95], is suitable for the determination of stability constants in transparent heterogeneous colloidal systems, i.e., when the inorganic-organic hybrid particles are sufficiently small, and the ICT complex exhibits optical properties distinct from its constituents. The stability constant of the ICT complex (Kb) can be expressed as follows:
Since the absorption in the visible spectral range solely originates from the ICT complex, the concentration of the ICT complex can be expressed by the Lambert-Beer equation ([ICTcomplex]=A/εl), so equation 2 can be rearranged in the following way:
where A and Amax are absorbances for a given and saturation concentration of ligand ([L]), respectively.
The stability constants of the TiO2-based ICT complexes, obtained using this simple spectrophotometric method, are similar in the order 103 M-1 [22,29,34,35,42,43,44,45,46,47]. Only when thiosalicylic acid was used as a ligand a slightly smaller stability constant was found [57]. However, the stability constants for surface-modified TiO2 with SA and CAT, obtained by different methodologies, the adsorption approach after filtration [96,97,98,99,100] or IR measurements [101,102,103,104], are one order of magnitude larger (~104 M-1). This discrepancy may be due to different applied methodologies and the TiO2’s different particle sizes. In addition, for spectroscopic determination of the stability constants of the TiO2-based ICT complexes, only adsorption curves were used, and since the Benesi-Hildebrand analysis requires equilibrium conditions when the adsorption and desorption rates are equal, they might be underestimated [105].
Density functional theory (DFT) calculations, with a time lag of more than a decade, supported the experimental efforts to understand the composition and coordination of ligands to TiO2 surface. Even initial studies on small model systems provided theoretical results with the same trend as experimental ones [43,44]. For example, the calculated IR spectrum of CAT bound to the TiO2 surface, assuming bridging coordination and using molecular complex Ti2O(OH)4‒L, is quite similar to the measured one, while the calculated IR using chelating model (Ti(OH)2‒L) does not have resemblance [43] (Figure 6A). Later on, the use of model clusters with increasing complexity, such as Ti8O14(OH)3−L [38] and Ti21O40H(OH)4−L [68], provided a more realistic description of the geometry of the ICT complexes, including the bond length. An instructive example is a study by Fujisawa et al. [38], showing that the calculated IR spectra on model complexes mimicking TiO2‒aminophenol system (Figure 6C) follow in the same manner the influence of an electron-donating amino group at different positions (o-, m-, and p-) on the Ti‒O‒C vibration observed experimentally in the 1200-1300 cm-1 spectral range. Optimized structures of model systems are presented in Figure 6B. It is fair to say that during the years, the DFT calculation evolved at the predictability level, providing an opportunity to minimize a trial-and-error approach.
2.3. Fine-Tuning of Optical Properties – The Role of Free Functional Group
The visible-light response of the TiO2-based ICT complexes originates from the excitation from the donor levels created in the TiO2 midband. The presence of different ligands attached to the TiO2 surface leads to different spectral responses. Higashimoto et al. [21], probing the photocatalytic ability of surface-modified TiO2 with CAT and its derivatives in water splitting reaction, were the first to establish the influence of phenyl-ring-substituted groups on the electronic structures of the ICT complexes, i.e., the possibility to tune optical properties of inorganic-organic hybrids without trial-and-error approach. Simply speaking, the ligands’ free electron-donating groups (EDG) induce the decrease of the energy gap of the TiO2-based ICT complex compared to the ligand without substituent group. On the opposite, the presence of electron-withdrawing groups (EWG) leads to the enlargement of the energy gap. For clarity, the energy-level diagram displaying the red/blue absorption shift, i.e., the decrease/increase of energy gap in the presence of ligands with EDG/EWG, is presented in Scheme 4.
A recent study by Fujisawa et al. [53] provides an illustrative example of the substituent effect on the optical properties of the TiO2-based ICT complexes. In this study, the optical properties of the ICT complexes with phenol derivatives, having EDG groups (4-tert-butylphenol and 4-methoxyphenol) and EWG group (pentafluorophenol), were compared with the optical properties of the anatase TiO2‒phenol complex; the Kubelka-Munk spectra are shown in Figure 7. The absorption of the surface-modified TiO2 with phenol derivatives having EDGs (‒C(CH3)3 and ‒OCH3) is red-shifted compared to the TiO2‒phenol complex. Also, a phenol derivative with a stronger electron-donating –OCH3 group induced a larger absorption shift toward the infrared spectral region. On the other hand, pentafluorophenol, a phenol derivative with five electron-withdrawing fluorine atoms, induced the blue absorption shift compared to TiO2‒phenol complex. In addition, the experimental findings are well-supported with DFT and time-dependent DFT (TD-DFT) calculations on large model systems, mimicking anatase TiO2 (Ti34O66(OH)4) and TiO2-phenol complexes (TiO2‒O‒Ph‒R, where R is H, C(CH3)3, OCH3, or F5).
Although the spectroscopy measurements combined with the DFT calculations provide information concerning relevant electronic levels in the ICT complexes, ionization potential (IP) measurements by photoelectron spectroscopy (PS) offer more precise information. The pinned position of IP in CAT-functionalized TiO2, SrTiO3, and BaTiO3 powders, corresponding to the highest occupied molecular orbital (HOMO) of the chemisorbed CAT to metal oxide surfaces, indicates an alternative way, although limited, to tune optical properties of hybrids by changing their inorganic component while using the same ligand [75].
Step forward in IP determination provided the vacuum-ultraviolet electron imaging angle-resolved photoelectron spectroscopy (VUV ARPES). This unique technique allows the measurements of IP in separated colloidal nanoparticles instead of measuring isolated powders. The absorption spectra of 50-Å TiO2 colloid and surface-modified TiO2 colloids with SA and 5-aminosalicylic acid (5-ASA) are shown in Figure 8, together with corresponding photoelectron spectra [49]. The amino group is EDG, and the TiO2‒5-ASA complex is red-shifted compared to the TiO2‒SA complex. The ionization energies for TiO2, TiO2‒SA, and TiO2‒5-ASA, determined by VUV ARPES, are 7.2, 6.5, and 5.9 eV, respectively. Differences in ionization energies between functionalized TiO2 with SA and 5-ASA and pristine TiO2 (0.7. and 1.3 eV, respectively) correspond to the red absorption shift observed in absorption spectra.
Besides the direct tuning of the optical properties of the ICT complexes, the proper introduction of substituent functional groups provides the possibility to prepare higher hierarchical structures. For example, the amino group introduction with a strong reducing capacity [106,107] offers a simple way for in-situ reduction of silver ions to metallic silver particles linked to metal oxide over ligand [76,80]. So, the presence of Ag nanoparticles, conjugated to the ICT complex, has a twofold effect, simultaneously improving the optical properties of the hybrid due to surface plasmon absorption and separation of photo-generated charge carriers. As an example, the TEM image of the titanate nanotube surface modified by 5-ASA and decorated with Ag nanoparticles, prepared using the free functional amino group to reduce Ag+ ions, is presented in Figure 9, together with Kubelka-Munk spectra [76]. This simple synthetic approach leads to the formation of well-separated, nearly spherical nanometer in-size Ag particles linked to the titanate nanotube surfaces, as indicated by microscopy and spectroscopy data. Of course, Ag nanoparticles display antimicrobial activity, and the above-mentioned synthetic procedure makes it possible to support them with a biocompatible material prepared from waste, like hydroxyapatite [89], or by magnetite, simplifying from a technological point of view separation process [108]. In addition, the introduction of desired functionality is pivotal in other potential applications non-related to photo-driven processes, such as enhanced sorption capacity and selectivity of sorbents [90] and visualization/recognition of drug molecules [109,110].
3. Photocatalytic Degradation of Organic Pollutants
Although the photocatalytic degradation of organic pollutants, in particular phenol and its derivatives, has been in this field one of the main topics for decades, the formation of ICT complexes with this class of organic compounds and the possibility to use the visible light to drive photocatalytic reaction was recognized with considerable delay. The reason for that is the most probable use of low concentration of phenol-based compounds (micromolar), typical for photocatalytic experiments, and the low extinction coefficient of the ICT complexes with this type of ligands. The 4-chlorophenol (4-CP) is a frequent representative of phenols to optimize the efficiency of their photocatalytic degradation since degradation of 4-CP leads to complete mineralization and generation of chlorides and CO2. Because of that, we will use, as an example, the photocatalytic degradation of 4-CP to emphasize the difference in the reaction mechanism under visible light excitation.
The detailed influence of the TiO2 loading and excitation wavelength in the UV spectral range from 300 to 400 nm on the mineralization rate and the quantum yield of 4-CP degradation is in-depth analyzed in a study by Stafford et al. [111]. The postulated mechanism of photocatalytic degradation of 4-CP under UV light excitation of TiO2 is complex, occurring in three parallel reaction pathways, including reactive oxygen species, hydroxyl radicals (HO•), and superoxide radical anion (O2•‒), formed from photogenerated electrons and holes, respectively [112,113]. Photomineralization kinetics of 4-CP follow a Langmuir-Hinshelwood kinetics, typical of many similar systems.
However, it is possible to degrade 4-CP using exclusively visible light (hν > 420 nm) by exciting TiO2‒4-CP‒O2 system, generating as final products chlorides and CO2, as pointed out by Agrios et al. [114,115] and Kim et al. [116]. Of course, direct ligand-to-metal electron transfer without involving the excited state of 4-CP is responsible for the visible light reactivity. Then, the injected electron into the CB of TiO2 reduces oxygen, electron acceptor, forming superoxide radical anion (O2•‒). In deaerated suspensions saturated with nitrogen, the photocatalytic degradation of 4-CP does not occur (Figure 10) since photogenerated electrons recombine with the surface complex, making a null cycle. However, when an alternative electron acceptor, ferric ions, is present in the anoxic suspension, the photocatalytic degradation of 4-CP is as fast as in the presence of oxygen. It is important to point out that photooxidation of 4-CP is not affected by the presence of radical scavengers, such as tert-butyl alcohol or enzyme superoxide dismutase.
Over the years, the above-mentioned approach for photocatalytic degradation of organic pollutants, preferably phenol derivatives, was widely exploited [117,118,119,120,121,122,123,124,125]. Since these are examples of self-sensitized degradation of pollutants under visible irradiation, and TiO2-based ICT complexes are unstable when exposed to light and not characterized in detail, we did not include the corresponding references in the literature overview presented in Table 1.
The prerequisite to consider any material as a photocatalyst is its stability. Higashimoto et al. [21] were the first to report that the visible-light-responsive TiO2-based ICT complexes with catechol and its derivatives retain their optical properties when exposed to light and display the photocatalytic activities for H2 evolution, which we will discuss in the following chapter of this review. Simultaneously, Milićević et al. [22] noticed that the same class of hybrid materials is capable of inducing photocatalytic degradation of an organic dye crystal violet under exclusive illumination with photons whose energy is smaller than 2.75 eV, i.e., smaller than the band gap energy of TiO2. The organic dyes, such as methylene blue (MB), crystal violet (CV), and methyl orange (MO), are frequently used to test the photocatalytic performance of semiconductors because the mechanism of their degradation kinetics is well-established, it is easy to follow their degradation kinetics experimentally, and they do not undergo to direct photolysis, i.e., their degradation in the absence of photocatalyst is negligible [126,127].
An illustrative example of enhanced photooxidative capability is the degradation of CV over ICT complex between TiO2 and dopamine linked to poly(GMA-co-EGDMA) copolymer with extended absorption in the visible spectral range [24], whose synthesis is described in the previous chapter (see Figure 3). Before photocatalytic experiments, sorption equilibrium was established in the dark, and from absorption spectra, the sorption capacity of the hybrid towards CV was estimated. The exclusive visible light excitation ensured the use of the low-energy band-pass 450 nm cutoff filter. The absorption spectra of CV as a function of time, before (in dark) and under visible light illumination, are presented in Figure 11. The results indicate that the ICT complex between TiO2 and dopamine linked to poly(GMA-co-EGDMA) copolymer can induce photocatalytic degradation of CV by exclusive visible light excitation. Of course, the degradation kinetics of CV is faster when the light source mimics the solar spectrum (compare kinetic curves given in the inset to Figure 11).
So far, photocatalytic degradation of different organic dyes, taking advantage of enhanced optical properties due to ICT complex formation, has been reported for TiO2 functionalized with tiron [23,41] and rhodizonic acid [65]. Also, other surface-modified metal oxides, such as titanates [71,72], ZrO2 [80], and Al2O3 [86,88], display photooxidative ability. Among the mentioned studies, the most striking example is photocatalytic degradation of organic dyes (MB and CV) over surface-modified Al2O3 with catechol [86] and 5-aminosalicylic acid [88], knowing that Al2O3 is an insulator with the band gap of about 8.7 eV [128]. We can say that the ICT complex formation transforms the insulator into a hybrid semiconductor-like material capable of harvesting a large portion of the solar spectrum.
Silver and silver compounds are powerful biocides, and the increased resistance of microbial species towards antibiotics renewed the interest in using Ag nanoparticles as disinfectant agents, either free-standing [129] or deposited onto different supports, inorganic [89] and organic [130,131], or within matrices for water treatment [132] and food packaging applications [133,134]. As an alternative to avoid the undesired impact of silver on the environment, the photocatalytic inactivation of microbial species by TiO2 thin films [135,136] or deposited TiO2 nanoparticles onto fibers [137], including textiles [138,139] has arisen. The TiO2, doped with light [140] and heavy elements [141], with extended absorption in the visible spectral range, was applied to avoid the use of harmful UV light sources and replace them with less expensive, harmless visible light sources. Concerning the use of TiO2-based ICT complexes, to the best of our knowledge, the biocidal effect against E. coli and S. aureus upon exclusive visible light excitation was reported only for TiO2 nanofibers surface-modified with rhodizonic acid [28]. Of course, the reaction mechanism includes reactive oxygen species (hydroxyl radical and superoxide radical anion), the same ones participating in the photocatalytic degradation of organic dyes.
4. Photocatalytic Hydrogen Generation
Hydrogen is the ultimate clean energy source that can replace fossil fuels, considerably solving energy and environmental issues. Semiconductors must have the proper energy alignment of the conduction and valence band and the band gap energy to perform as photocatalysts in a water-splitting reaction. Consequently, the potentials of CBmin and VBmax have to be negative compared to the redox potential of H+/H2 (0 V versus NHE) and more positive than the potential of O2/H2O (1.23 V), respectively. Therefore, the theoretical minimum band gap for water splitting is 1.23 eV. Like electrolysis, photogenerated electrons reduce water molecules to form H2 and oxidize water by the holes to form O2.
Another significant issue is the stability of photocatalysts. For example, CdS has suitable band positions and visible light response but is inactive for water splitting into H2 and O2. Instead of oxidizing water, photogenerated holes oxidize S2‒ in CdS, followed by the release of Cd2+ ions [142].
This reaction is called photocorrosion and is characteristic of an entire class of metal sulfides.
In strict terminology, water splitting means splitting water molecules into H2 and O2 in a stoichiometric ratio without the sacrificial agents. However, the sacrificial agents, i.e., electron donors, are frequently used, particularly alcohols, since H2 yield might be low. For example, 2-propanol (isopropyl alcohol) reacts with holes or their successors, hydroxyl radicals, forming alkoxy ((CH3)2CHO∙) or α-hydroxyalkyl ((CH3)2C∙(OH)) radicals (Equation 9).
The redox potential of α-hydroxyalkyl radical ((CH3)2C∙(OH)) is sufficiently negative (-1.23 V versus NHE [143]) to transfer electrons into the semiconductor conduction band. So, the absorption of one photon leads to the generation of two electrons, and alcohols are frequently termed current doubling agents. But, in this case, compared to the water-splitting reaction, photocatalytic generation of hydrogen is a half-reaction.
Despite the low overlap of its absorption with the solar spectrum, chemically inert and cost-effective TiO2 remains the most attractive photocatalytic material, and the use of TiO2-based ICT complexes is the most recent attempt to enhance its photocatalytic performance in water-splitting reactions. However, the number of studies in this research area is still small.
Pioneering work by Ikeda et al. [144] is an instructive example of improved photocatalytic hydrogen evolution due to visible-light absorption of surface-modified TiO2 with 1,1’-binaphtalene-2,2’-diol. Before modifying TiO2 with binaphthol, a small amount of co-catalyst, Pt particles, was deposited on TiO2 to make active sites for hydrogen generation. The photocatalytic experiments were carried out in the presence of sacrificial electron donor triethanolamine (TEOA) under exclusive visible light excitations, filtering out high-energy photons by cut-off filters. The kinetics of hydrogen evolution over surface-modified TiO2 with binaphthol as a function of excitation wavelength is shown in Figure 12. We can draw the following conclusions from these results: First, hydrogen production does not occur if excitation wavelengths are longer than 580 nm since this wavelength corresponds to the absorption onset of the TiO2-based ICT complex with binaphthol. Second, hydrogen evolution is becoming more efficient by broadening the visible excitation range. Finally, the hydrogen generation over unmodified TiO2 is non-existent under visible light excitation (>430 nm).
Another instructive example is a study by Higashimoto et al. [21] concerning the influence of functional groups in catechol derivatives on the photocatalytic hydrogen production of functionalized TiO2 under visible light excitation. It should be mentioned that the TiO2 was platinized, and triethanolamine (TEOA) was used as the hole scavenger, similar to the work of Ikeda et al. [144]. Figure 13A shows time-dependent hydrogen production over surface-modified TiO2 with various catechol derivatives. The photocatalytic activity of TiO2-based ICT complexes with catechol derivatives having electron-donating groups (4-tert-butylcatechol and 3-methoxycatechol) is lower, almost non-existent, than that of the surface-modified TiO2 with catechol. On the other hand, hybrids with catechol derivatives having electron-withdrawing groups (2,3-dihydroxybenzoic acid, 3,4-dihydroxybenzonitrile, and tiron) display better photocatalytic performance. The observed photoactivity trend is unexpected since electron-donating groups are decreasing the energy gap of the TiO2-based ICT complex, while the presence of electron-withdrawing groups leads to the enlargement of the energy gap compared to the ligand without substituent group (Scheme 4). Simple speaking, the better photoactivity displayed photocatalysts absorbing less in the visible spectral region. The explanation for the observed effect is the necessity of using a sacrificial electron donor to facilitate the efficient separation of photogenerated charge carriers. Figure 13B correlates the photocatalytic activities of TiO2-based ICT complexes with catechol derivatives with oxidative potentials of corresponding catechol derivatives (the oxidative potential of TEOA is marked by an arrow). The catechol derivatives with electron-withdrawing groups have more anodic oxidative potential than the sacrificial hole scavenger (TEOA), and consequently, the larger the difference, the better the photoactivity. On the other hand, the catechol derivatives with electron-donating groups have more cathodic oxidative potential than TEOA, and, of course, TEOA does not perform as a hole scavenger. So, recombination between photogenerated charge carriers prevails, lowering the efficiency of the water-splitting reaction.
Oxidative potentials of small alcohols (methanol, ethanol, and isopropyl alcohol) are significantly more negative (0.016, 0.084, and 0.105 V versus NHE [145], respectively) than the oxidative potential of TEOA (1.14 V versus Ag/AgCl electrode [21]). So, on one side, their ability to scavenge photogenerated holes and hydroxyl radicals is better than TEOA. On the other side, the generated alcohol radicals can transfer electrons to the conduction band of TiO2, leading to two electrons in the conduction band per one absorbed photon. The time-dependent rate of photocatalytic hydrogen generation, using coordinated over dopamine TiO2 nanoparticles to polymer support as a photocatalyst, is shown in Figure 14. The schematized synthetic procedure of the photocatalyst, its morphology, and the optical properties are presented in Figure 3, accompanied by comments in section B1 [24]). The photocatalytic hydrogen generation rate over TiO2-based ICT complex supported by polymer increases and, after approximately one hour, reaches the steady value, which is about two times higher compared to the rate obtained under the same experimental conditions using the most studied commercial TiO2 photocatalyst Degussa P25. It is worth mentioning that the photocatalytic experiments were performed without a co-catalyst (Pt nanoparticles), unlike the above-described studies by Ikeda et al. [144] and Higashimoto et al. [21].
5. Identification of Reactive Species
Reactive species induced by photoexcitation, primary (electron-hole pair) and secondary (radicals), can be followed either by “fast” time-resolved spectroscopic techniques (pulse radiolysis, pump-probe flash photolysis, and time-resolved EPR techniques) or “static” continuous-wave EPR techniques based on the nature of reactive species, i.e., the presence of paramagnetic centers in photogenerated reactive species. The spectroscopic fingerprint of the hydrated electron [146] and hydroxyl radical [147], discovered by Professor Hart in the early sixties by pulse radiolysis technique, provided a burst in studying the radical reactions. Later on, Professor Grätzel [148] showed in simple experiments that stationary illumination of the deaerated TiO2 colloid led to the appearance of a blue color with absorption maximum peaking at 700 nm, which is identical to the absorption spectrum of hydrated electron observed in pulse radiolysis experiment. However, time-resolved spectroscopic techniques have never been used to provide in-depth insight into the photocatalytic reaction mechanism of any oxide-based ICT complexes. On the other hand, just a few recent studies employed the EPR technique to identify radical species that participate in photocatalytic reactions, following initial works by Howe et al. [149] and Micic et al. [150] on pristine TiO2.
Photogenerated charge carriers and radical intermediates are paramagnetic species, so EPR spectroscopy is the technique of choice for their detection and identification. It provides valuable insight concerning the origin of the photocatalytic activity in specific structures. Photogenerated electrons and holes are highly reactive, so a photocatalytic system of interest has to be frozen to suppress their disappearance. The low-temperature EPR spectra of pristine TiO2 and surface-modified TiO2 with 4-chlorophenol (4-CP) in dark and under illumination with light sources having different spectral profiles (UV and Vis) are shown in Figure 15 [40]. EPR signals reflecting the effective photoinduced electron transfer were observed upon in situ UV exposure of pristine TiO2 powder (Degussa P25) [40,151]. An axially symmetric EPR signal with the g-tensor components g⊥=1.988 and unresolved g||=1.945 can be attributed to the Ti(III) bulk centers in anatase lattice representing the trapped photogenerated electrons., while the signals with g > 2.00 are compatible with the formation of the trapped holes [152,153]. Since the commercial TiO2 powder (Degussa P25) contains about 30% rutile (Eg=3.0 eV) [154]), the Vis light exposure also evokes the generation of analogous paramagnetic signals, however, of lower intensity [40] (Figure 15A).
On the other hand, the EPR spectra of the TiO2/4-CP preserved the axially symmetric signal of Ti(III) centers, displaying an additional single-line EPR signal with the g-value of 2.005 even in the dark (Figure 15B) due to the formation of persistent free radicals and reduced metal ion via interaction of chemisorbed phenol with metal oxide surfaces [40,155,156]. The intensity of the single-line signal with the g-value of 2.005, characteristic of the oxygen-centered organic radicals [157] increasing under UV excitation, and becomes even more pronounced under Vis light illumination, reflecting the effective photoinduced electron transfer from the organic moiety of the TiO2/4-CP complex to the conduction band of TiO2, producing phenoxy radical and Ti(III)surf characterized with the unresolved broad EPR signals [40]. These experimental data correspond well with the previously published results by Kim et al. [116] and described in Section B, concerning visible-light-induced photocatalytic degradation of 4-CP and phenolic compounds in aqueous suspensions of TiO2, mediated by a surface complex, where the formation of O-centered phenol radical in the course of the photodegradation process was proposed [158]. To briefly conclude, the low-temperature EPR spectra obtained for unmodified and surface-modified TiO2 powder with 4-CP [40] strongly supported the visible-light-induced photocatalytic oxidation mechanism of phenolic compounds proposed in the literature [114,115,116].
An alternative, or better to say complementary techniques, to the low-temperature EPR technique, suitable for studying intermediate radicals, are the spin-trapping and spin-scavenging EPR techniques. The modus operandi of spin-trapping relies on the formation of persistent radical species, e.g., nitroxide radicals, via the reaction of diamagnetic spin trap with non-persistent photogenerated radicals. On the other hand, the spin-scavenging approach enables monitoring the disappearance of persistent radical species added to the experimental system that efficiently scavenge the photogenerated reactive radicals. So far, the spin-trapping EPR technique has been used to get a deeper insight into the mechanism of photocatalytic processes over TiO2-based ICT complexes with squaric acid [67] and polyphenol taxifolin [159], as well as surface-modified titanate nanotubes by 5-aminosalicylic acid decorated with silver nanoparticles [76].
The semi-stable 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) radical cation (ABTS•+) with a characteristic highly-resolved EPR signal is known to undergo reduction to ABTS. Consequently, the decrease of the overmodulated EPR signal upon the excitation in the presence of a photocatalyst reflects the reactions involving the photogenerated electrons [160]. Figure 16 shows the time-dependent relative ABTS•+ concentration evaluated from the EPR spectra, monitored in the aerated aqueous suspensions of the pristine TiO2 powder and TiO2-based ICT complex with squaric acid (SqA) upon UV (λmax = 365 nm) and Vis excitation (λ > 420 nm) [67]. Control experiments, in the absence of photocatalysts, showed that the intensity of the ABTS•+ signal does not change upon exposure to UV or Vis light (see Figure 16). However, different behavior displayed pristine TiO2 and TiO2-based ICT complex with SqA upon excitation with light sources emitting photons in the UV and Vis spectral range. Upon UV excitation in the presence of both photocatalysts (TiO2 and TiO2/SqA), the EPR signal of ABTS•+ decreases immediately after the beginning of exposure, quickly reaching the zero relative concentration of radical cation (Figure 16A). On the other hand, the ABTS•+ EPR signal remains intact upon exposure of pristine TiO2 to Vis light, while on the other hand, in an analogous experiment with the TiO2/SqA, a continuous decrease of the ABTS•+ concentration can be noticed (Figure 16B). Of course, these results are a consequence of the optical properties of the TiO2/SqA, absorbing in the Vis spectral range, which provides the possibility for photoinduced electron transfer from the organic moiety of TiO2/SqA to the conduction band of TiO2 upon exclusive Vis light excitation. Of course, the application of spin trap, reactive towards hydroxyl radicals, such as 5,5-dimethy-1-pyrroline N-oxide (DMPO) [160,161], or stable radicals capable of scavenging all reactive paramagnetic species and measuring the photo-induced radical-producing capacity of the system of interest, such 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical and Tempo derivatives, such as 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (Tempol) [162] can provide complementary data to previously described.
6. Perspectives for Future Studies
In light of the preceding overview, the development of oxide-based ICT complexes is still at the early stage, including the most studied inorganic-organic hybrid using TiO2 as an inorganic component, and studies of their potential applications based on photo-induced catalytic reactions are seldom. However, the advantages of using oxide-based ICT complexes, in particular TiO2-based ICT complexes, are easily recognizable and may be summarized as follows:
The simple synthetic procedure with cost-effective components.
Covalent linkage between inorganic and organic components of ICT complexes.
Enhanced optical properties with absorption in a more practical visible spectral range.
The simple way of fine-tuning optical properties.
Excitation without energy loss.
Efficient separation of photo-induced charge carriers.
The possibility of preparing composite materials with a higher hierarchical structure.
Considering the current status in this field and the emphasized advantages of oxide-based ICT complexes, we believe these novel materials are competitive with other well-developed hybrid materials intended to bring absorption in the desired spectral range. So, in our opinion, oxide-based ICT complexes are worth further studies, and we humbly suggest the following directions:
Diversification of prepared ICT complexes, using other oxides besides TiO2 with bioactive organic components as ligands.
Extensive use of the DFT calculations at the predictability level is needed to avoid a trial-and-error approach as much as possible.
Photo-induced catalytic reactions (water splitting reaction and degradation of organic molecules) should be carried out on a long timescale to estimate the stability of oxide-based ICT complexes.
The antimicrobial activity of oxide-based ICT complexes should be tested to avoid toxic agents and harmful UV light sources; the literature data are almost non-existent.
Besides photo-induced catalytic reactions, other potential applications of oxide-based ICT complexes should be explored, such as the recognition of organic molecules, particularly drugs, and their sorption abilities towards heavy metal ions, taking advantage of functionalization that can provide selectivity and increase the sorption capacity.
Acknowledgments
We thank Professor Vlasta Brezová for critically reading the manuscript. This study was supported by the Ministry of Science, Technological Development, and Innovation of the Republic of Serbia (grant 451-03-66/2024-03/200017).
References
- A. Fujishima, K. Honda, Electrochemical Photolysis of Water at a Semiconductor Electrode, Nature, 238, 37-38 (1972).
- M.R. Hoffmann, S.T. Martin, W. Choi, D.W. Bahnemann, Environmental applications of semiconductor photocatalysis, Chem. Rev., 95, 69-96 (1995).
- A. Kudo, Y. Miseki, Heterogeneous photocatalyst materials for water splitting, Chem. Soc. Rev., 38, 253-278 (2009).
- T. Bak, J. Nowotny, M. Rekas, C.C. Sorrell, Photo-electrochemical hydrogen generation from water using solar energy. Materials-related aspects, Int. J. Hydrogen Energ., 27, 991-1022 (2002).
- M. Kosmulski, The significance of the difference in the point of zero charge between rutile and anatase, Adv. Colloid Interface Sci., 99, 255-264 (2002).
- A. Kraeutler, A.J. Bard, Photoelectrosynthesis of Ethane from Acetate Ion at an n-Type TiO2 Electrode. The Photo-Kolbe Reaction, J. Am. Chem. Soc., 99, 7729-7731 (1977).
- A. Kraeutler, A.J. Bard, Heterogeneous Photocatalytic Decomposition of Saturated Carboxylic Acids on TiO2 Powder. Decarboxylative Route to Alkanes, J. Am. Chem. Soc., 100, 5985-5992 (1978).
- S.T. Martin, H. Herrmann, W. Choi, M.R. Hoffmann, Time-resolved microwave conductivity. Part 1.‒TiO2 photoreactivity and size quantization. Trans. Faraday Soc., 90, 3315-3323 (1994).
- S.T. Martin, H. Herrmann, M.R. Hoffmann, Time-resolved microwave conductivity. Part 2.‒Quantum-sized TiO2 and the effect of adsorbates and light intensity on charge-carrier dynamics, Faraday Soc., 90, 3323-3330 (1994).
- C.G. Silva, R. Juárez, T. Marino, R. Molinari, H. García, Influence of Excitation Wavelength (UV or Visible Light) on the Photocatalytic Activity of Titania Containing Gold Nanoparticles for the Generation of Hydrogen or Oxygen from Water, J. Am. Chem. Soc., 133, 595-602 (2011).
- A. Meng, L. Zhang, B. Cheng, J. Yu, Dual Cocatalysts in TiO2 Photocatalysis, Adv. Mater., 31, 1807660 (2019).
- R. Asahi, T. Morikawa, K. Aoki, Y. Taga, Visible-light photocatalysis in nitrogen doped titanium oxides, Science, 293, 269-271 (2001).
- S. Sakthivel, H. Kisch, Angew. Daylight photocatalysis by carbon-modified titanium dioxide, Chem. Int. Ed., 42, 4908-4911 (2003).
- X. Chen, C. Burda, The Electronic Origin of the Visible-Light Absorption Properties of C-, N- and S-Doped TiO2 Nanomaterials, J. Am. Chem. Soc., 130, 5018-5019 (2008).
- J. Kiwi, E. Pelizzetti, M. Visca, M. Grätzel, Sustained Water Cleavage by Visible Light, J. Am. Chem. Soc., 103, 6324-6329 (1981).
- B. O’Regan, M. Grätzel, A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films, Nature, 353, 737-740 (1991).
- S. Shen, L. Zhao, Z. Zhou, L. Guo, Enhanced Photocatalytic Hydrogen Evolution over Cu-Doped ZnIn2S4 under Visible Light Irradiation, J. Phys. Chem. C, 112, 16148-16155 (2008).
- J. Low, J. Yu, M. Jaroniec, S. Wageh, A.A. Al-Ghamdi, Heterojunction Photocatalysts, Adv. Mater., 29, 1601694 (2017).
- Q. Xu, L. Zhang, J. Yu, S. Wageh, A.A. Al-Ghamdi, M. Jaroniec, Direct Z-scheme photocatalysts: Principles, synthesis, and applications, Mater. Today, 21, 1042-1063 (2018).
- T. Rajh, J.M. Nedeljković, L.X. Chen, O. Poluektov, M.C. Thurnauer, Improving optical and charge separation properties of nanocrystalline TiO2 by surface modification with vitamin C, J. Phys. Chem. B, 103, 3515-3519 (1999).
- S. Higashimoto, T. Nishi, M. Yasukawa, M. Azuma, Y. Sakata, H. Kobayashi, Photocatalysis of titanium dioxide modified by catechol-type interfacial surface complexes (ISC) with different substituted groups, J. Catal., 329, 286-291 (2015).
- B. Milićević, V. Ðorđević, D. Lončarević, S.P. Ahrenkiel, M.D. Dramićanin, J.M. Nedeljković, Visible light absorption of surface modified TiO2 powders with bidentate benzene derivatives, Micropor. Mesopor. Mat., 217, 184-189 (2015).
- Yao, C. Peng, P. Lu, Y. He, W. Zhang, Q. Zhang, Fabrication of Tiron-TiO2 charge-transfer complex with excellent visible-light photocatalytic performance, Mat. Chem. Phys., 184, 298-305 (2016).
- Vukoje, T. Kovač, J. Džunuzović, E. Džunuzović, D. Lončarević, S.P. Ahrenkiel, J.M. Nedeljković, Photocatalytic ability of visible-light-responsive TiO2 nanoparticles, J. Phys. Chem. C, 120, 18560-18569 (2016).
- S. Günes, N. Marjanović, J.M. Nedeljković, N.S. Sariciftci, Photovoltaic characterization of hybrid solar cells using surface modified TiO2 nanoparticles and poly(3-hexyl)thiophene, Nanotechnology, 19, 424009 (2008).
- J. Fujisawa, M. Nagata, Efficient light-to-current conversion by organic-inorganic interfacial charge-transfer transitions in TiO2 chemically adsorbed with 2-anthroic acid, Chem. Phys. Lett., 619, 180-184 (2015).
- S.-B. Ko, T.I. Ryu, A.-N. Cho, J. Fujisawa, H. Segawa, N.-G. Park, Visible light absorption and photoelectrochemical activity of colorless molecular 1,3-bis (dicyanomethylidene)indane (BDMI) by surface complexation on TiO2, Phys. Chem. Chem. Phys., 17, 18541-18546 (2015).
- M. Shahriari-Khalaji, F. Zabihi, A. Bahi, D. Sredojević, J.M. Nedeljković, D.K. Macharia, M. Ciprian, S. Yang, F. Ko, Photon-driven bactericidal performance of surface-modified TiO2 nanofibers, J. Mater. Chem. C, 11, 5796-5805 (2023).
- I.A. Janković, Z.V. Šaponjić, M.I. Čomor, J.M. Nedeljković, Surface modification of colloidal TiO2 nanoparticles with bidentate benzene derivatives, J. Phys. Chem. C, 113, 12645-12652 (2009).
- P. Persson, R. Bergström, S. Lunell, Quantum Chemical Study of Photoinjection Processes in Dye-Sensitized TiO2 Nanoparticles. J. Phys. Chem. B, 104, 10348-10351 (2000).
- J. Fujisawa, M. Hanaya, Light harvesting and direct electron injection by interfacial charge-transfer transitions between TiO2 and carboxy-anchor dye LEG4 in dye-sensitized solar cells, J. Phys. Chem. C, 122, 8-15 (2018).
- T. Rajh, Z. Šaponjić, J. Liu, N.M. Dimitrijević, N.F. Scherer, M. Vega-Arroyo, P. Zapol, L.A. Curtiss, M.C. Thurnauer, Charge transfer across the nanocrystalline-DNA interface: probing DNA recognition, Nano Lett., 4, 1017-1023 (2004).
- L.X. Chen, T. Rajh, W. Jäger, J. Nedeljković, M.C. Thurnauer, X-ray absorption reveals surface structure of titanium dioxide nanoparticles, J. Synchrotron Rad., 6, 445-447 (1999).
- I.M. Dugandžić, D.J. Jovanović, L.T. Mančić, N. Zheng, S.P. Ahrenkiel, O.B. Milošević, Z.V. Šaponjić, J.M. Nedeljković, Surface modification of submicronic TiO2 particles prepared by ultrasonic spray pyrolysis for visible light absorption, J. Nanopart. Res., 14, 1157-1167 (2012).
- I.M. Dugandžić, D.J. Jovanović, L.T. Mančić, O.B. Milošević, S.P. Ahrenkiel, Z.V. Šaponjić, J.M. Nedeljković, Ultrasonic spray pyrolysis of surface modified TiO2 nanoparticles with dopamine, Mater. Chem. Phys., 143, 233-239 (2013).
- J. Fujisawa, R. Muroga, M. Hanaya, Interfacial charge-transfer transitions in a TiO2-benzenedithiol complex with Ti–S–C linkages, Phys. Chem. Chem. Phys., 17, 29867-29873 (2015).
- J. Fujisawa, S. Matsumura, M. Hanaya, A single Ti‒O‒C linkage induces interfacial charge-transfer transitions between TiO2 and a π-conjugated molecule, Chem. Phys. Lett., 657, 172-176 (2016).
- J. Fujisawa, T. Eda, G. Giorgi, M. Hanaya, Visible-to-Near-IR Wide-Range Light Harvesting by Interfacial Charge-Transfer Transitions between TiO2 and p-Aminophenol and Evidence of Direct Electron Injection to the Conduction Band of TiO2, J. Phys. Chem. C, 121, 18710-18716 (2017).
- D.N. Sredojević, T. Kovač, E. Džunuzović, V. Ðorđević, B.N. Grgur, J.M. Nedeljković, Surface-modified TiO2 powders with phenol derivatives: A comparative DFT and experimental study, Chem. Phys. Lett., 686, 167-172 (2017).
- Z. Barbieriková, D. Dvoranová, V. Brezová, E. Džunuzović, D.N. Sredojević, V. Lazić, J.M. Nedeljković, Visible-light-responsive surface-modified TiO2 powder with 4-chlorophenol: A combined experimental and DFT study, Opt. Mater., 89, 237-242 (2019).
- V. Lazić, D. Sredojević, A. Ćirić, J.M. Nedeljković, G. Zelenková, M. Férová, T. Zelenka, M.P. Chavhan, V. Slovák, The photocatalytic ability of visible-light-responsive interfacial charge transfer complex between TiO2 and Tiron, J. Photoch. Photobio. A, 449, 115394 (2024).
- I.A. Janković, Z.V. Šaponjić, E.S. Džunuzović, J.M. Nedeljković, New hybrid properties of TiO2 nanoparticles surface modified with catecholate type ligands, Nanoscale Res. Lett., 5, 81-88 (2010).
- T.D. Savić, I.A. Janković, Z.V. Šaponjić, M.I. Čomor, D.Ž. Veljković, S.D. Zarić, J.M. Nedeljković, Surface modification of anatase nanoparticles with fused catecholate type ligands: a combined DFT and experimental study of optical properties, Nanoscale, 4, 1612-1619 (2012).
- T.D. Savić, Z.V. Šaponjić, M.I. Čomor, J.M. Nedeljković, M.D. Dramićanin, M.G. Nikolić, D.Ž. Veljković, S.D. Zarić, I.A. Janković, Surface modification of anatase nanoparticles with fused ring salicylate-type ligands (3-hydroxy-2-naphtoic acids): a combined DFT and experimental study of optical properties, Nanoscale, 5, 7601-7612 (2013).
- T.D. Savić, M.I. Čomor, J.M. Nedeljković, D.Ž. Veljković, S.D. Zarić, V.M. Rakić, I.A. Janković, The effect of substituents on the surface modification of anatase nanoparticles with catecholate-type ligands: a combined DFT and experimental study, Phys. Chem. Chem. Phys., 16, 20796-20805 (2014).
- T.D. Savić, M.I. Čomor, N.D. Abazović, Z.V. Šaponjić, M.T. Marinović-Cincović, D.Ž. Veljković, S.D. Zarić, I.A. Janković, Anatase nanoparticles surface modified with fused ring salicylate-type ligands (1-hydroxy-2-naphthoic acids): a combined DFT and experimental study, J. Alloy Compd., 630, 226-235 (2015).
- V. Bajić, B. Spremo-Potparević, L. Živković, A. Čabarkapa, J. Kotur-Stevuljević, E. Isenović, D. Sredojević, I. Vukoje, V. Lazić, S.P. Ahrenkiel, J.M. Nedeljković, Surface-modified TiO2 nanoparticles with ascorbic acid: Antioxidant properties and efficiency against DNA damage in vitro, Colloid Surface B, 155, 323-331 (2017).
- D. Dekanski, B. Spremo-Potparević, V. Bajić, L. Živković, D. Topalović, D.N. Sredojević, V. Lazić, J.M. Nedeljković, Acute toxicity study in mice of orally administrated TiO2 nanoparticles functionalized with caffeic acid, Food Chem. Toxicol., 115, 42-48 (2018).
- D.K. Božanić, G.A. Garcia, L. Nahon, D. Sredojević, V. Lazić, I. Vukoje, S.P. Ahrenkiel, V. Djoković, Ž. Šljivančanin, J.M. Nedeljković, Interfacial Charge Transfer Transitions in Colloidal TiO2 Nanoparticles Functionalized with Salicylic acid and 5-Aminosalicylic acid: A Comparative Photoelectron Spectroscopy and DFT Study, J. Phys. Chem. C, 123, 29057-29066 (2019).
- A. Todorović, K. Bobić, F. Veljković, S. Pejić, S. Glumac, S. Stanković, T. Milovanović, I. Vukoje, J.M. Nedeljković, S. Radojević Škodrić, S.B. Pajović, D. Drakulić, Comparable Toxicity of Surface-Modified TiO2 Nanoparticles: An In Vivo Experimental Study on Reproductive Toxicity in Rats, Antioxidants, 13, 231 (2024).
- T.S. Kovač, E.S. Džunuzović, J.V. Džunuzović, B. Milićević, D.N. Sredojević, E.N. Brothers, J.M. Nedeljković, Visible light absorption of TiO2 nanoparticles surface-modified with vitamin B6: A comparative experimental and DFT study, J. Serb. Chem. Soc., 83, 899-909 (2018).
- J. Fujisawa, S. Kato, M. Hanaya, Detailed study of a TiO2-phenol complex using deuterated phenol, Chem. Phys. Lett., 803, 139833 (2022).
- J. Fujisawa, S. Kato, M. Hanaya, Substituent effects on interfacial charge-transfer transitions in a TiO2-phenol complex, Chem. Phys. Lett., 827, 140688 (2023).
- T. Rajh, D.M. Tiede, M.C. Thurnauer, Surface modification of TiO2 nanoparticles with bidentate ligands studied by EPR spectroscopy J. Non-Cryst. Solids, 205-207, 815-820 (1996).
- M.C. Thurnauer, T. Rajh, D.M. Tiede, Surface Modification of TiO2: Correlation between Structure, Charge Separation and Reduction Properties, Acta Chem. Scand., 51, 610-618 (1997).
- J. Fujisawa, R. Muroga, M. Hanaya, Interfacial charge-transfer transitions and reorganization energies in sulfur-bridged TiO2-x-benzenedithiol complexes (x: o, m, p), Phys. Chem. Chem. Phys., 18, 22286‒22292 (2016).
- B. Milićević, V. Ðorđević, D. Lončarević, J.M. Dostanić, S.P. Ahrenkiel, M.D. Dramićanin, D. Sredojević, N.M. Švrakić, J.M. Nedeljković, Charge-transfer complex formation between TiO2 nanoparticles and thiosalicylic acid: A comprehensive experimental and DFT study, Opt. Mater., 73, 163-171 (2017).
- J. Fujisawa, N. Kaneko, M. Hanaya, Interfacial Charge-Transfer Transitions in TiO2 Nanoparticles Adsorbed with 4-Mercaptobenzenoic Acid: Carboxy versus Thiol Anchor and Adsorbate-to-TiO2 versus TiO2-to-Adsorbate Charge Transfer, J. Phys. Chem. C, 125, 13534-13541 (2021).
- J. Fujisawa, S. Kato, M. Hanaya, Interfacial Charge-Transfer Transitions in Weakly Coupled TiO2 Complexes with Aromatic Monothiols for Visible-Light Photocatalytic Reactions, J. Phys. Chem. C, 126, 11602-11610 (2022).
- S. Manzhos, R. Jono, K. Yamashita, J. Fujisawa, M. Nagata, H. Segawa, Study of Interfacial Charge Transfer Bands and Electron Recombination in the Surface Complexes of TCNE, TCNQ, and TCNAQ with TiO2, J. Phys. Chem. C, 115, 21487-21493 (2011).
- J. Fujisawa, M. Nagata, M. Hanaya, Charge-transfer complex versus σ-complex formed between TiO2 and bis(dicyanomethylene) electron acceptors, Phys. Chem. Chem. Phys., 17, 27343‒27356 (2015).
- B. Moongraksathum, P.T. Hsu, Y.W. Chen, Photocatalytic activity of ascorbic acidmodified.
- TiO2 sol prepared by the peroxo sol‒gel method. J. Sol-Gel Sci. Technol., 78, 647-659 (2016).
- J. Fujisawa, N. Kikuchi, M. Hanaya, Coloration of tyrosine by organic-semiconductor interfacial charge-transfer transitions, Chem. Phys. Lett., 664, 178-183 (2016).
- L. Tian, J. Xu, A. Alnafisah, R. Wang, X. Tan, N.A. Oyler, L. Liu, X. Chen, A novel green TiO2 photocatalyst with surface charge-transfer complex of Ti and hydrazine groups, Chem. Eur. J., 23, 5345-5351 (2017).
- Y. Sun, J.B. Mwandeje, L.M. Wangatia, F. Zabihi, J. Nedeljkovic, S. Yang, Enhanced Photocatalytic Performance of Surface-Modified TiO2 Nanofibers with Rhodizonic Acid, Adv. Fiber Mater., 2, 118-122 (2020).
- J. Fujisawa, Interfacial charge-transfer transitions between TiO2 and indole, Chem. Phys. Lett., 739, 136974 (2020).
- Z. Barbieriková, M. Šimunková, V. Brezová, D. Sredojević, V. Lazić, D. Lončarević, J.M. Nedeljković, Interfacial charge transfer complex between TiO2 and non-aromatic ligand squaric acid, Opt. Mater., 123, 111918 (2022).
- J. Fujisawa, S. Kato, M. Hanaya, Interfacial Charge-Transfer Transitions between TiO2 Nanoparticles and Benzoic Acid Derivatives J. Phys. Chem. C, 125, 25075-25086 (2021).
- J. Fujisawa, S. Kato, M. Hanaya, Aromatic-ring dependence of interfacial charge-transfer transitions between TiO2 nanoparticles and aromatic carboxylic acids, Chem. Phys. Lett., 788, 139274 (2022).
- J. Fujisawa, S. Kato, M. Hanaya, Adsorption, Electronic, and Optical Properties of TiO2–Pyridine Complexes: General Principles for Interfacial Charge-Transfer Transitions, J. Phys. Chem. C, 127, 17888-17895 (2023).
- D. Jaušovec, M. Božić, J. Kovač, J. Štrancar, V. Kokol, Synergies of phenolic-acids’ surface-modified titanate nanotubes (TiNT) for enhanced photo-catalytic activities, J. Colloid Interf. Sci., 38, 277-290 (2015).
- M.M. Medić, M. Vasić, A.R. Zarubica, L.V. Trandafilović, G. Dražić, M.D. Dramićanin, J.M. Nedeljković, Enhanced photoredox chemistry in surface-modified Mg2TiO4 nano-powders with bidentate benzene derivatives, RSC Adv., 6, 94780-94786 (2016).
- J. Fujisawa, T. Eda, M. Hanaya, Interfacial Charge-Transfer Transitions in BaTiO3 Nanoparticles Adsorbed with Catechol, J. Phys. Chem. C, 120, 21162-21168 (2016).
- J. Fujisawa, T. Eda, M. Hanaya, Comparative study of conduction-band and valence-band edges of TiO2, SrTiO3, and BaTiO3 by ionization potential measurements, Chem. Phys. Lett., 685, 23-26 (2017).
- T. Eda, J. Fujisawa, M. Hanaya, Inorganic Control of Interfacial Charge-Transfer Transitions in Catechol-Functionalized Titanium Oxides Using SrTiO3, BaTiO3, and TiO2, J. Phys. Chem. C, 122, 16216-16220 (2018).
- Z. Barbieriková, D. Lončarević, J. Papan, I.D. Vukoje, M. Stoiljković, S.P. Ahrenkiel, J.M. Nedeljković, Photocatalytic hydrogen evolution over surface-modified titanate nanotubes by 5-aminosalicylic acid decorated with silver nanoparticles, Adv. Powder Technol., 31, 4683-4690 (2020).
- M. Prekajski-Ðordević, I. Vukoje, V. Lazić, V. Ðordević, D. Sredojević, J. Dostanić, D. Lončarević, S.P. Ahrenkiel, M.R. Belić, J.M. Nedeljković, Electronic structure of surface complexes between CeO2 and benzene derivatives: A comparative experimental and DFT study, Mater. Chem. Phys., 236, 121816 (2019).
- V. Lazić, L.S. Živković, D. Sredojević, M.M. Fernandes, S. Lanceros-Mendez, S.P. Ahrenkiel, J.M. Nedeljković, Tuning Properties of Cerium Dioxide Nanoparticles by Surface Modification with Catecholate-type of Ligands, Langmuir, 36, 9738-9746 (2020).
- V. Lazić, A. Pirković, D. Sredojević, J. Marković, J. Papan, S.P. Ahrenkiel, I. Janković-Častvan, D. Dekanski, M. Jovanović-Krivokuća, J.M. Nedeljković, Surface-modified ZrO2 nanoparticles with caffeic acid: Characterization and in vitro evaluation of biosafety for placental cells, Chem.-Biol. Interact., 347, 109618 (2021).
- A. Zarubica, D. Sredojević, R. Ljupković, M. Randjelović, N. Murafa, M. Stoiljković, V. Lazić, J.M. Nedeljković, Photocatalytic ability of visible-light-responsive hybrid ZrO2 particles, Sustain. Energ. Fuels, 7, 2279-2287 (2023).
- M. Dukić, D. Sredojević, M. Férová, V. Slovak, D. Lončarević, J. Dostanić, H. Šalipur, V. Lazić, J.M. Nedeljković, Interfacial charge transfer complexes between ZnO and benzene derivatives: Characterization and photocatalytic hydrogen production, Int. J. Hydrogen Energ., 62, 628-636 (2024).
- J. Fujisawa, N. Kaneko, M. Hanaya, Interfacial charge-transfer transitions in ZnO induced exclusively by adsorption of aromatic thiols, Chem. Comm., 56, 4090-4093 (2020).
- J. Fujisawa, M. Hanaya, Linkage Dependence of Interfacial Charge-Transfer Transitions in ZnO: Carboxylate versus Sulfur Linker, J. Phys. Chem. A, 125, 5903-5910 (2021).
- J. Fujisawa, Definitive assignment and mechanistic study of interfacial charge-transfer transitions between ZnO and benzenethiol, Chem. Phys. Lett., 778, 138774 (2021).
- J. Fujisawa, S. Kato, M. Hanaya, Interfacial Charge-Transfer Transitions between ZnO Nanoparticles and Aliphatic Thiols, J. Phys. Chem. C, 127, 15300-15306 (2023).
- V. Ðordević, J. Dostanić, D. Lončarević, S.P. Ahrenkiel, D.N. Sredojević, N. Švrakić, M. Belić, J.M. Nedeljković, Hybrid visible-light responsive Al2O3 particles, Chem. Phys. Lett., 685, 416-421 (2017).
- V. Ðorđević, D.N. Sredojević, J. Dostanić, D. Lončarević, S.P. Ahrenkiel, N. Švrakić, E. Brothers, M. Belić, J.M. Nedeljković, Visible light absorption of surface-modified Al2O3 powders: A comparative DFT and experimental study, Micropor. Mesopor. Mat., 273, 41-49 (2019).
- A. Zarubica, R. Ljupković, J. Papan, I. Vukoje, S. Porobic, S.P. Ahrenkiel, J.M. Nedeljković, Visible-light-responsive Al2O3 powder: Photocatalytic study, Opt. Mater., 106, 110013 (2020).
- V. Lazić, I. Smičiklas, J. Marković, D. Lončarević, J. Dostanić, S.P. Ahrenkiel, J.M. Nedeljković, Antibacterial ability of supported silver nanoparticles by functionalized hydroxyapatite with 5-aminosalicylic acid, Vacuum, 148, 62-68 (2018).
- I.D. Smičiklas, V.M. Lazić, L.S. Živković, S.J. Porobić, S.P. Ahrenkiel, J.M. Nedeljković, Sorption of divalent heavy metal ions onto functionalized biogenic hydroxyapatite with caffeic acid and 3,4-dihydroxybenzoic acid, J. Environ. Sci. Heal. A, 54, 899-905 (2019).
- D. Sredojević, S. Stavrić, V. Lazić, S.P. Ahrenkiel, J.M. Nedeljković, Interfacial charge transfer complex formation between silver nanoparticles and aromatic amino acids, Phys. Chem. Chem. Phys., 24, 16493-16500 (2022).
- D. Finkelstein-Shapiro, S.K. Davidowski, P.B. Lee, C. Guo, G.P. Holland, T. Rajh, K.A. Gray, J.L. Yarger, M. Calatayud, Direct evidence of chelated geometry of catechol on TiO2 by a combined solid-state NMR and DFT study, J. Phys. Chem. C, 120, 23625-23630 (2016).
- P. Job, Formation and Stability of Inorganic Complexes in Solution, Ann. Chim., 9, 113-203 (1928).
- L.X. Chen, T. Rajh, Z. Wang, M.C. Thurnauer, XAFS studies of surface structures of TiO2 nanoparticles and photocatalytic reduction of metal ions, J. Phys. Chem. B, 101, 10688-10697 (1997).
- H.A. Benesi, J.H. Hildebrand, A spectrophotometric investigation of the interaction of iodine with aromatic hydrocarbons, J. Am. Chem. Soc., 71, 2703-2707 (1949).
- J. Moser, S. Punchihewa, P.P. Infelta, M. Grätzel, Surface Complexation of Colloidal Semiconductors Strongly Enhances Interfacial Electron-Transfer Rates, Langmuir, 7, 3012-3018 (1991).
- S.T. Martin, J.M. Kesselman, D.S. Park, N.S. Lewis, M.R. Hoffman, Surface Structures of 4-Chlorocatechol Adsorbed on Titanium Dioxide, Environ. Sci. Technol., 30, 2535-2542 (1996).
- P. Rodriguez, M.A. Blesa, A.E. Regazzoni, Surface Complexation at the TiO2 (anatase)/AqueousSolution Interface: Chemisorption of Catechol, J. Colloid Interface Sci., 177, 122-131 (1996).
- A.E. Regazzoni, P. Mandelbaum, M. Matsuyoshi, S. Schiller, S.A. Bilmes, M.A. Blesa, Adsorption and Photooxidation of Salicylic Acid on Titanium Dioxide: A Surface Complexation Description, Langmuir, 14, 868-874 (1998).
- D.V. Heyd, B. Au, Fluorescence development during 514 nm irradiation of catechol adsorbed on nanocyrstalline titanium dioxide, J. Photochem. Photobiol. A: Chem., 174, 62-70 (2005).
- A.D. Weisz, A.E. Regazzoni, M.A. Blesa, ATR–FTIR study of the stability trends of carboxylate complexes formed on the surface of titanium dioxide particles immersed in water, Solid State Ionics, 143, 125-130 (2001).
- A.D. Weisz, L. García Rodenas, P.J. Morando, A.E. Regazzoni, M.A. Blesa, FTIR study of the adsorption of single pollutants and mixtures of pollutants onto titanium dioxide in water: oxalic and salicylic acids, Catal. Today, 76, 103-112 (2002).
- P.Z. Araujo, C.B. Mendive, L.A. García Rodenas, P.J. Morando, A.E. Regazzoni, M.A. Blesa, D. Bahnemann, FT-IR–ATR as a tool to probe photocatalytic interfaces, Colloid Surface A, 265, 73-80 (2005).
- P.Z. Araujo, P.J. Morando, M.A. Blesa, Interaction of Catechol and Gallic Acid with Titanium Dioxide in Aqueous Suspensions. 1. Equilibrium Studies, Langmuir, 21, 3470-3474 (2005).
- T. Rajh, L.X. Chen, K. Lukas, T. Liu, M.C. Thurnauer, D.M. Tiede, Surface Restructuring of Nanoparticles: An Efficient Route for Ligand-Metal Oxide Crosstalk, J. Phys. Chem. B, 106, 10543-10552 (2002).
- M. Chen, Y.G. Feng, X. Wang, T.C. Li, J.Y. Zhang, D.J. Qian, Silver nanoparticles capped by oleylamine: formation, growth, and self-organization, Langmuir, 23, 5296-5304 (2007).
- S. Mourdikoudis, L.M. Liz-Marzan, Oleylamine in nanoparticle synthesis, Chem. Mater., 25, 1465-1476 (2013).
- V. Lazić, K. Mihajlovski, A. Mraković, E. Illés, M. Stoiljković, S.P. Ahrenkiel, J.M. Nedeljković, Antimicrobial activity of silver nanoparticles supported by magnetite, ChemistrySelect, 4, 4018-4024 (2019).
- J. Fujisawa, S. Matsumura, M. Hanaya, Facile and rapid visualization of colorless endocrine disruptor bisphenol A by interfacial charge-transfer transitions with TiO2 nanoparticles, ChemistrySelect, 2, 6097-6099 (2017).
- J. Fujisawa, T. Eda, M. Hanaya, Facile, quick and selective visible-light sensing of phenol-containing drug molecules acetaminophen and biosol by use of interfacial charge transfer transitions with TiO2 nanoparticles, Chem. Phys. Lett., 684, 328-332 (2017).
- U. Stafford, K.A. Gray, P.V. Kamat, Photocatalytic Degradation of 4-Chlorophenol: The Effects of Varying TiO2 Concentration and Light Wavelength, J. Catal., 167, 25-32 (1997).
- A. Mills, R.H. Davies, D. Worsley, Water purification by semiconductor photocatalysis, Chem. Soc. Rev., 22, 417-425 (1993).
- A. Mills, S. Morris, Photomineralization of 4-chlorophenol sensitized by titanium dioxide: a study of the initial kinetics of carbon dioxide photogeneration, J. Photochem. Photobiol. A: Chem., 71, 75-83 (1993).
- A.G. Agrios, K.A. Gray, E. Weitz, Photocatalytic Transformation of 2,4,5-Trichlorophenol on TiO2 under Sub-Band-Gap Illumination, Langmuir, 19, 1402-1409 (2003).
- A.G. Agrios, K.A. Gray, E. Weitz, Narrow-Band Irradiation of a Homologous Series of Chlorophenols on TiO2: Charge-Transfer Complex Formation and Reactivity, Langmuir, 20, 5911-5917 (2004).
- S. Kim, W. Choi, Visible-Light-Induced Photocatalytic Degradation of 4-Chlorophenol and Phenolic Compounds in Aqueous Suspension of Pure Titania: Demonstrating the Existence of a Surface-Complex-Mediated Path, J. Phys. Chem. B, 109, 5143-5149 (2005).
- T. Paul, P. Miller, T.J. Strathmann, Visible-Light-Mediated TiO2 Photocatalysis of Fluoroquinolone Antibacterial Agents, Environ. Sci. Technol., 41, 4720-4727 (2007).
- M. Li, P. Tang, Z. Hong, M. Wang, High efficient surface-complex-assisted photodegradation of phenolic compounds in single anatase titania under visible-light, Colloid Surface A, 318, 285-290 (2008).
- N. Wang, L. Zhu, Y. Huang, Y. She, Y. Yu, H. Tanga, Drastically enhanced visible-light photocatalytic degradation of colorless aromatic pollutants over TiO2 via a charge-transfer complex path: A correlation between chemical structure and degradation rate of the pollutants, J. Catal., 266, 199-206 (2009).
- S. Higashimoto, N. Kitao, N. Yoshida, T. Sakura, M. Azuma, H. Ohue, Y. Sakata, Selective photocatalytic oxidation of benzyl alcohol and its derivatives into corresponding aldehydes by molecular oxygen on titanium dioxide under visible light irradiation, J. Catal., 266, 279-285 (2009).
- S. Higashimoto, N. Suetsugu, M. Azuma, H. Ohue, Y. Sakata, Efficient and selective oxidation of benzylic alcohol by O2 into corresponding aldehydes on a TiO2 photocatalyst under visible light irradiation: Effect of phenyl-ring substitution on the photocatalytic activity, J. Catal., 274, 76-83 (2010).
- S. Higashimoto, K. Okada, T. Morisugi. M. Azuma, H. Ohue, T.-H. Kim, M. Matsuoka, M. Anpo, Effect of Surface Treatment on the Selective Photocatalytic Oxidation of Benzyl Alcohol into Benzaldehyde by O2 on TiO2 Under Visible Light, Top. Catal., 53, 578-583 (2010).
- S. Girish Kumar, L. Gomathi Devi, Review on Modified TiO2 Photocatalysis under UV/Visible Light: Selected results and Related Mechanism on Interfacial Charge Transfer Carrier Transfer Dynamics, J. Phys. Chem. A, 115, 13211-13241 (2011).
- S. Higashimoto, R. Shirai, Y. Osano, M. Azuma, H. Ohue, Y. Sakata, H. Kobayashi, Influence of metal ions on the photocatalytic activity: Selective oxidation of benzyl alcohol on iron (III) ion-modified TiO2 using visible light, J. Catal., 311, 137-143 (2014).
- G. Kim, S.-H. Lee, W. Choi, Glucose–TiO2 charge transfer complex-mediated photocatalysis under visible light, Appl. Catal. B: Environ., 162, 463-469 (2015).
- A. Houas, H. Lachheb, M. Ksibi, E. Elaloui, C. Guillard, J.-M. Herrmann, Photocatalytic degradation pathway of methylene blue in water, Appl. Catal. B: Environ., 31, 145-157 (2001).
- R. Djellabi, M. Ghorab, G. Cerrato, S. Morandi, S. Gatto, V. Oldani, A. Di Michele, C. Bianchi, Photoactive TiO2−Montmorillonite Composite for Degradation of Organic Dyes in Water, J. Photochem. Photobio. A, 295, 57-63 (2014).
- B. Ealet, M.H. Elyakhloufi, E. Gillet, M. Ricci, Electronic and crystallographic structure of γ-alumina thin films, Thin Solid Films, 250, 92-100 (1994).
- A. Panáček, L. Kvítek, R. Prucek, M. Kolář, R. Večeřová, N. Pizúrová,V.K. Sharma, T. Nevĕčná, R. Zbořil, Silver colloid nanoparticles: synthesis, characterization, and their antibacterial activity, J. Phys. Chem. B, 110, 16248-16253 (2006).
- T. Yuranova, A.G. Rincon, A. Bozzi, S. Parra, C. Pulgarin, P. Albers, J. Kiwi, Antibacterial textiles prepared by RF-plasma and vacuum-UV mediated deposition of silver, J. Photochem. Photobiol. A, 161, 27-34 (2003).
- V. Ilić, Z. Šaponjić, V. Vodnik, B. Potkonjak, P. Jovančić, J. Nedeljković, M. Radetić, The influence of silver content on antimicrobial activity and color of cotton fabrics functionalized with Ag nanoparticles, Carbohyd. Polym., 78, 564-569 (2009).
- V.A. Oyanedel-Craver, J.A. Smith, Sustainable colloidal-silver-impregnated ceramic filter for point-of-use water treatment, Environ. Sci. Technol., 42, 927-933 (2008).
- S. Davidović, V. Lazić, M. Miljković, M. Gordić, M. Sekulić, M. Marinović-Cincović, I.S. Ratnayake, S.P. Ahrenkiel, J.M. Nedeljković, Antibacterial ability of immobilized silver nanoparticles in agar-agar films co-doped with magnesium ions, Carbohyd. Polym., 224, 115187 (2019).
- V. Lazić, V. Vivod, Z. Peršin, M. Stoiljković, I.S. Ratnayake, P.S. Ahrenkiel, J.M. Nedeljković, V. Kokol, Dextran-coated silver nanoparticles for improved barrier and controlled antimicrobial properties of nanocellulose films used in food packaging, Food Packaging Shelf, 26, 100575 (2020).
- W.A. Daoud, J.H. Xin, Low temperature sol-gel processed photocatalytic titania coating, J. Sol-Gel Sci. Technol., 29, 25-29 (2004).
- C. Pulgarin, J. Kiwi, V. Nadtochenko, Mechanism of photocatalytic bacterial inactivation on TiO2 films involving cell-wall damage and lysis, Appl. Catal. B-Environ., 128, 179-183 (2012).
- W.A. Daoud, J.H. Xin, Y.H. Zhang, Surface functionalization of cellulose fibers with titanium dioxide nanoparticles and their combined bactericidal activities, Surf. Sci., 599: 69-75 (2005).
- D. Mihailović, Z. Šaponjić, M. Radoičić, T. Radetić, P. Jovančić, J Nedeljković, M. Radetić, Functionalization of polyester fabrics with alginates and TiO2 nanoparticles, Carbohyd. Polym., 79, 526-532 (2010).
- D. Mihailović, Z. Šaponjić, R. Molina, N. Pauč, P. Jovančić, J. Nedeljković, M. Radetić, Improved properties of oxygen and argon RF plasma-activated polyester fabrics loaded with TiO2 nanoparticles, ACS Appl. Mater. Inter., 2, 1700-1706 (2010).
- J.A. Rengifo-Herrera, E. Mielczarski, J. Mielczarski, N.C. Castillo, J. Kiwi, C. Pulgarin, Escherichia coli inactivation by N, S co-doped commercial TiO2 powders under UV and visible light, Appl. Catal. B-Environ., 84, 448-456 (2008).
- D. Meng, X. Liu, Y. Xie, Y. Du, Y. Yang, C. Xiao, Antibacterial Activity of Visible Light-Activated TiO2 Thin Films with Low Level of Fe Doping, Adv. Mater. Sci. Eng., 5819805 (2019).
- Y.V. Pleskov, Y.Y. Gurevich, in Semiconductor Photoelectrochemistry, ed. P.N. Bartlett, Plenum, New York, 1986.
- H. Schwarz, R. Dodson, Reduction Potentials of CO2-and the Alcohol Radicals, J. Phys. Chem., 93, 409-414 (1989).
- S. Ikeda, C. Abe, T. Torimoto, B. Ohtani, Photochemical hydrogen evolution from aqueous triethanolamine solutions sensitized by binaphthol-modified titanium(IV) oxide under visible-light irradiation, J. Photoch. Photobio. A, 160, 61-67 (2003).
- Z.H.N. Al-Azri, W.-T. Chen, A. Chan, V. Jovic, T. Ina, H. Idriss, G.I.N. Waterhouse, The roles of metal co-catalysts and reaction media in photocatalytic hydrogen production: Performance evaluation of M/TiO2 photocatalysts (M = Pd, Pt, Au) in different alcohol–water mixtures, J. Catal., 329, 355-367 (2015).
- E.J. Hart, J.W. Boag, Absorption Spectrum of the Hydrated Electron in Water and in Aqueous Solutions, J. Am Chem. Soc., 84, 4090-4095 (1962).
- J.K. Thomas, J. Rabani, M.S. Matheson, E.J. Hart, S. Gordon, Absorption Spectrum of the Hydroxyl Radical, J. Phys. Chem., 70, 2409-2410 (1966).
- D. Duonghong, J. Ramsden, Michael Grätzel, Dynamics of Interfacial Electron-Transfer Processes in Colloidal Semiconductor Systems, J. Am. Chem. Soc., 104, 2977-2985 (1982).
- R.F. Howe, M. Grӓtzel, EPR Observation of Trapped Electrons in Colloidal TiO2, J. Phys. Chem., 89, 4495-4499 (1985).
- O.I. Micic, Y. Zhang, K.R. Cromack, A.D. Trifunac, M.C. Thurnauer, Trapped Holes on TiO2 Colloids Studied by Electron Paramagnetic Resonance, J. Phys. Chem., 97, 7277-7283 (1993).
- D. Dvoranová, V. Brezová, M. Mazúr, M.A. Malati, Investigations of metal-doped titanium dioxide photocatalysts, Appl. Catal. B Env., 37, 91-105 (2002).
- M. Chiesa, M.C. Paganini, S. Livraghi, E. Giamello, Charge trapping in TiO2 polymorphs as seen by Electron Paramagnetic Resonance spectroscopy, Phys. Chem. Chem. Phys., 15, 9435-9447 (2013).
- E.G. Panarelli, S. Livraghi, S. Maurelli, V. Polliotto, M. Chiesa, E. Giamello, Role of surface water molecules in stabilizing trapped hole centres in titanium dioxide (anatase) as monitored by electron paramagnetic resonance, J. Photoch. Photobio. A, 322-323, 27-34 (2016).
- B. Ohtani, O. Prieto-Mahaney, D. Li, R. Abe, What is Degussa (Evonik) P25? Crystalline composition analysis, reconstruction from isolated pure particles and photocatalytic activity test, J. Photoch. Photobio. A, 216, 179-182 (2010).
- M.C. Patterson, N.D. Keibart, L.W. Kiruri, C.A. Thibodeaux, S. Lomnicki, R.L. Kurtz, E.D. Poliakoff, B. Dellinger, P.T. Sprunger, EPFR formation from phenol adsorption on Al2O3 and TiO2: EPR and EELS studies, Chem. Phys., 422, 277-282 (2013).
- M.C. Patterson, C.A. Thibodeaux, O. Kizilkaya, R.L. Kurtz, E.D. Poliakoff, P.T. Sprunger, Electronic signatures of a model pollutant-particle system: chemisorbed phenol on TiO2 (110), Langmuir, 31, 3869-3875 (2015).
- Landolt-Börnstein, Numerical Data and Functional Relationships in Science and Technology. Vol. 17 Magnetic Properties of Free radicals. Subvolume e, Radicals centered on heteroatoms with Z>7 and selected anion radicals. pp. 35-249.
- G. Zhang, G. Kim, W. Choi, Visible light driven photocatalysis mediated via ligand-to- metal charge transfer (LMCT): an alternative approach to solar activation of titania, Energy Environ. Sci., 7, 954-966 (2014).
- V. Nikšić, M. Malček Šimunková, Z. Dyrčíková, D. Dvoranová, V. Brezová, D. Sredojević, J.M. Nedeljković, V. Lazić, Photoinduced reactive species in interfacial charge transfer complex between TiO2 and taxifolin: DFT and EPR study, Opt. Mater., 152, 115454 (2024).
- V. Brezová, D. Dvoranová, A. Staško, Characterization of titanium dioxide photoactivity following the formation of radicals by EPR spectroscopy, Res. Chem. Intermed., 33, 251-268 (2007).
- K. Jomová, L. Hudecova, P. Lauro, M. Simunkova, S.H. Alwasel, I.M. Alhazza, M. Valko, A switch between antioxidant and prooxidant properties of the phenolic compounds myricetin, morin, 3′,4′-dihydroxyflavone, taxifolin and 4-hydroxy-coumarin in the presence of copper(II) ions: a spectroscopic, absorption titration and dna damage study, Molecules, 24, 4335 (2019).
- A. Staško, V. Brezová, S. Biskupič, V. Misík, The potential pitfalls of using 1,1- dipehnylpicrylhydrazyl to characterize antioxidant in mixed water solvent, Free Radic. Res., 41, 379-390 (2007).
Figure 1.
The pH-dependent energy level position of VBmax and CBmin in anatase toward the vacuum level and the normal hydrogen electrode (reprinted from T. Bak, J. Nowotny, M. Rekas, C.C. Sorrell, Int. J. Hydrogen Energ., 27, 991-1022 (2002); copyright 2002 Elsevier).
Figure 1.
The pH-dependent energy level position of VBmax and CBmin in anatase toward the vacuum level and the normal hydrogen electrode (reprinted from T. Bak, J. Nowotny, M. Rekas, C.C. Sorrell, Int. J. Hydrogen Energ., 27, 991-1022 (2002); copyright 2002 Elsevier).
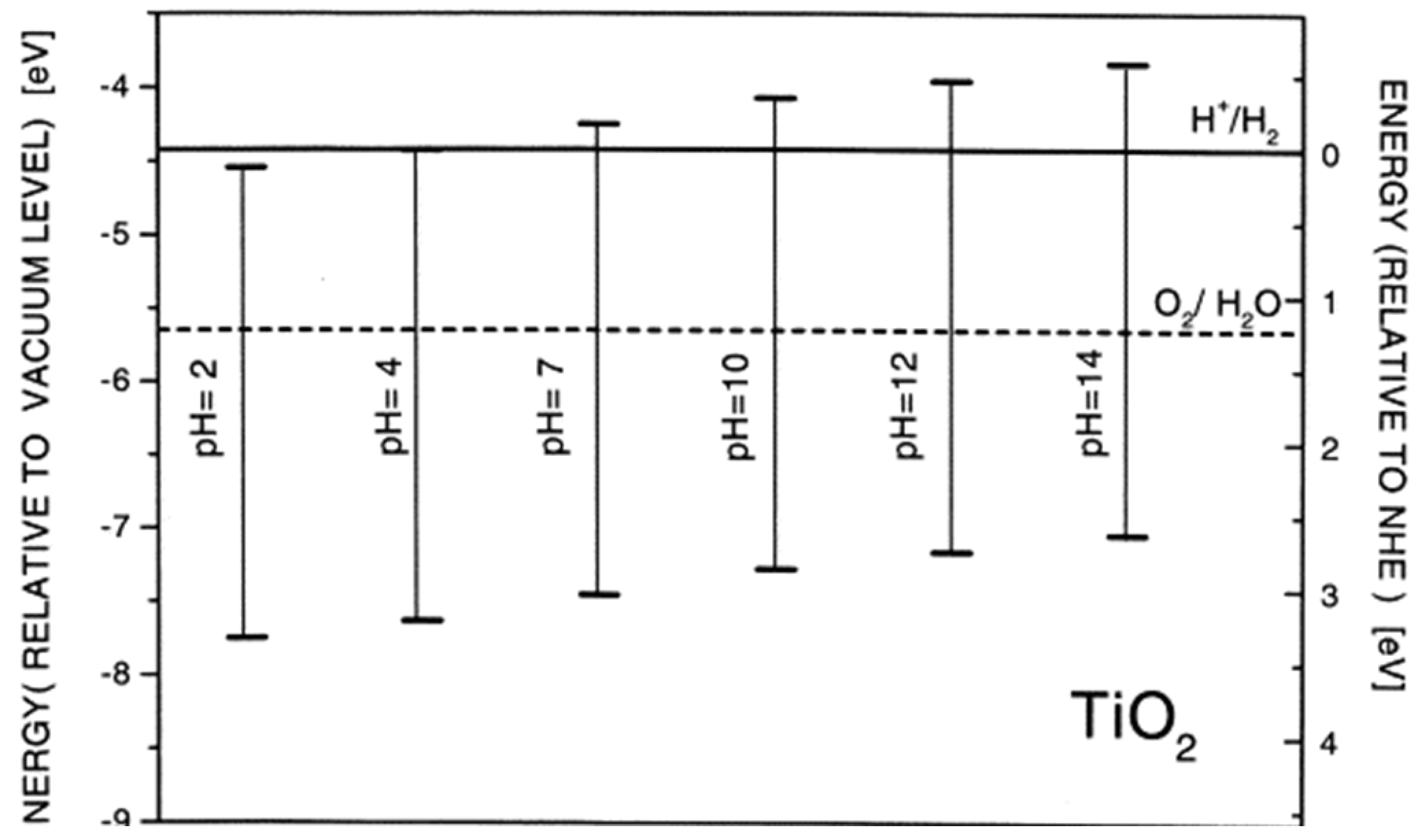
Scheme 1.
A schematic presentation for the formation of the TiO2-based ICT complexes.
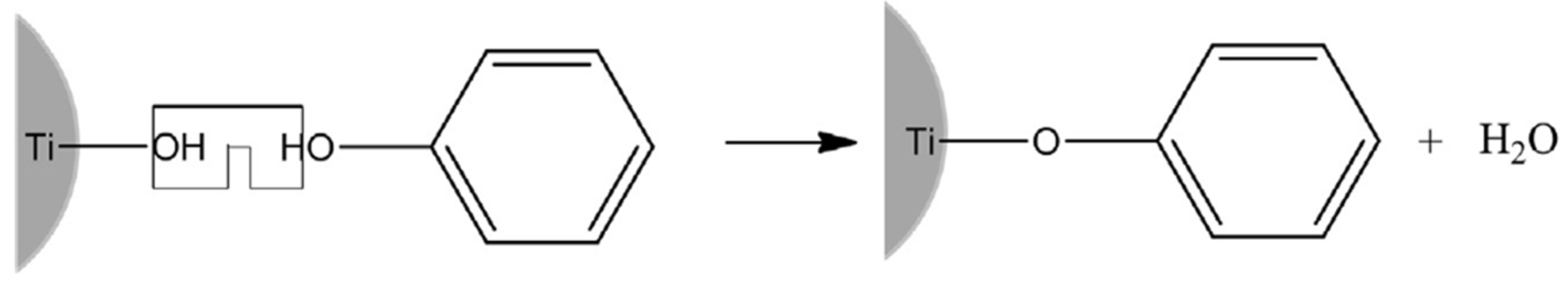
Figure 2.
Absorption spectra and photo images of colloidal solutions of 45-Å TiO2 nanoparticles surface modified with different ligands: (A) bare TiO2, (B) 2-hydroxybenzoic acid, (C) 2,5-dihydroxybenzoic acid, (D) 2,3-dihydroxybenzoic acid, (E) 3,4-dihydroxybenzoic acid, and (F) catechol (reprinted from I.A. Janković, Z.V. Šaponjić, M.I. Čomor, J.M. Nedeljković, J. Phys. Chem. C, 113, 12645-12652 (2009); copyright 2009 American Chemical Society).
Figure 2.
Absorption spectra and photo images of colloidal solutions of 45-Å TiO2 nanoparticles surface modified with different ligands: (A) bare TiO2, (B) 2-hydroxybenzoic acid, (C) 2,5-dihydroxybenzoic acid, (D) 2,3-dihydroxybenzoic acid, (E) 3,4-dihydroxybenzoic acid, and (F) catechol (reprinted from I.A. Janković, Z.V. Šaponjić, M.I. Čomor, J.M. Nedeljković, J. Phys. Chem. C, 113, 12645-12652 (2009); copyright 2009 American Chemical Society).
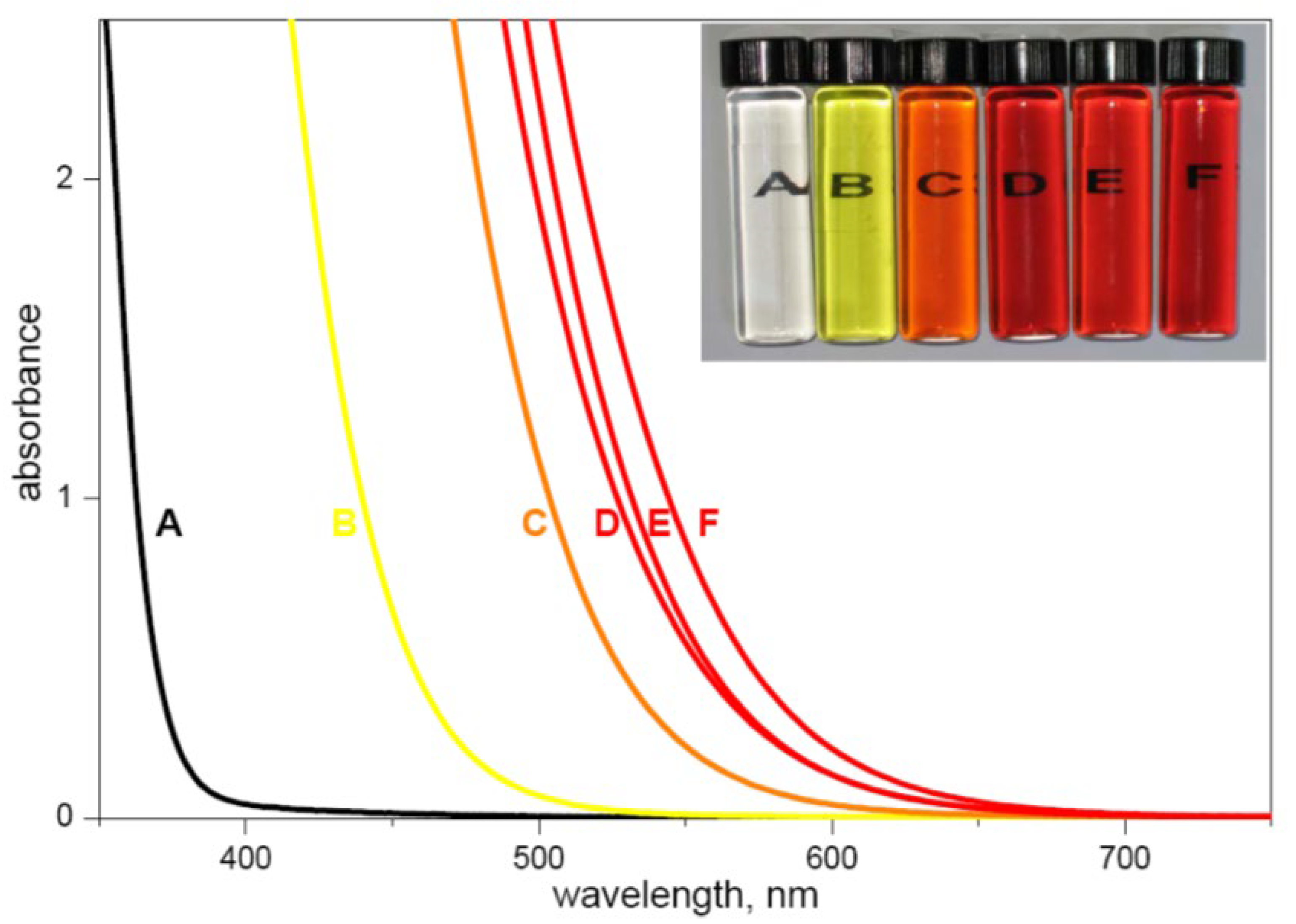
Scheme 2.
Energy-diagram of organic-to-inorganic ICT transition (A) and photoexcitation of dye-sensitized semiconductor (B).
Scheme 2.
Energy-diagram of organic-to-inorganic ICT transition (A) and photoexcitation of dye-sensitized semiconductor (B).
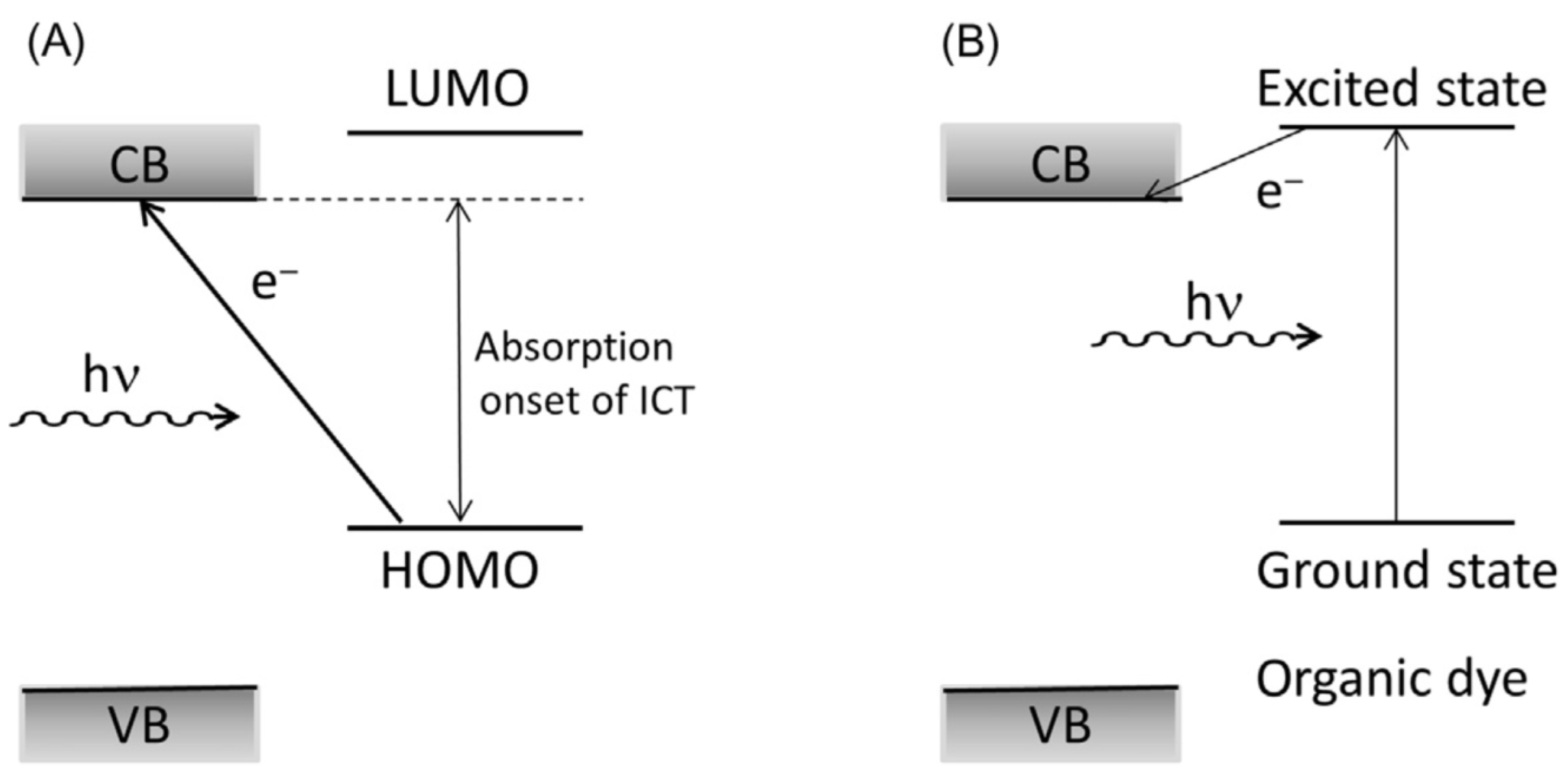
Figure 3.
(A) Reaction mechanism for the in-situ formation of surface-modified TiO2 NPs attached to polymer support, and (B) TEM image of obtained composite. (C) Kubelka-Munk transformations of diffuse reflection data for surface-modified TiO2 NPs supported by polymer, including photo image (reprinted from I. Vukoje, T. Kovač, J. Džunuzović, E. Džunuzović, D. Lončarević, S.P. Ahrenkiel, J.M. Nedeljković, J. Phys. Chem. C, 120, 18560-18569 (2016); copyright 2016 American Chemical Society).
Figure 3.
(A) Reaction mechanism for the in-situ formation of surface-modified TiO2 NPs attached to polymer support, and (B) TEM image of obtained composite. (C) Kubelka-Munk transformations of diffuse reflection data for surface-modified TiO2 NPs supported by polymer, including photo image (reprinted from I. Vukoje, T. Kovač, J. Džunuzović, E. Džunuzović, D. Lončarević, S.P. Ahrenkiel, J.M. Nedeljković, J. Phys. Chem. C, 120, 18560-18569 (2016); copyright 2016 American Chemical Society).
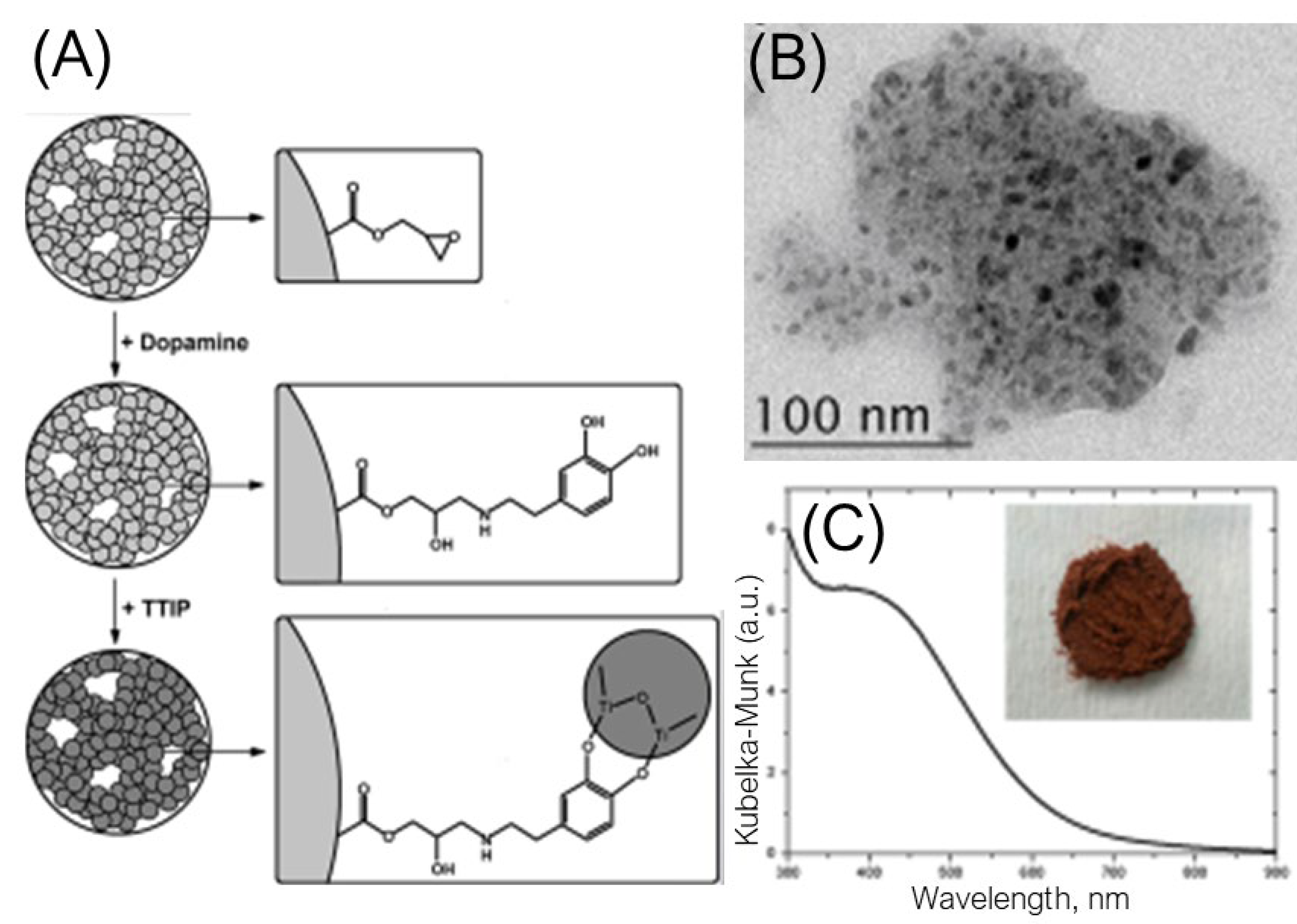
Figure 4.
(A) Kubelka–Munk function spectra of TiO2 (blank) and the TiO2‒phenol sample together with photographs of these samples, and (B) FT-IR spectra of phenol (above) and the TiO2–phenol sample (below) together with that of TiO2 (blank; dashed curve) (reprinted from J. Fujisawa, S. Matsumura, M. Hanaya, Chem. Phys. Lett., 657, 172-176 (2016); copyright 2016 Elsevier).
Figure 4.
(A) Kubelka–Munk function spectra of TiO2 (blank) and the TiO2‒phenol sample together with photographs of these samples, and (B) FT-IR spectra of phenol (above) and the TiO2–phenol sample (below) together with that of TiO2 (blank; dashed curve) (reprinted from J. Fujisawa, S. Matsumura, M. Hanaya, Chem. Phys. Lett., 657, 172-176 (2016); copyright 2016 Elsevier).
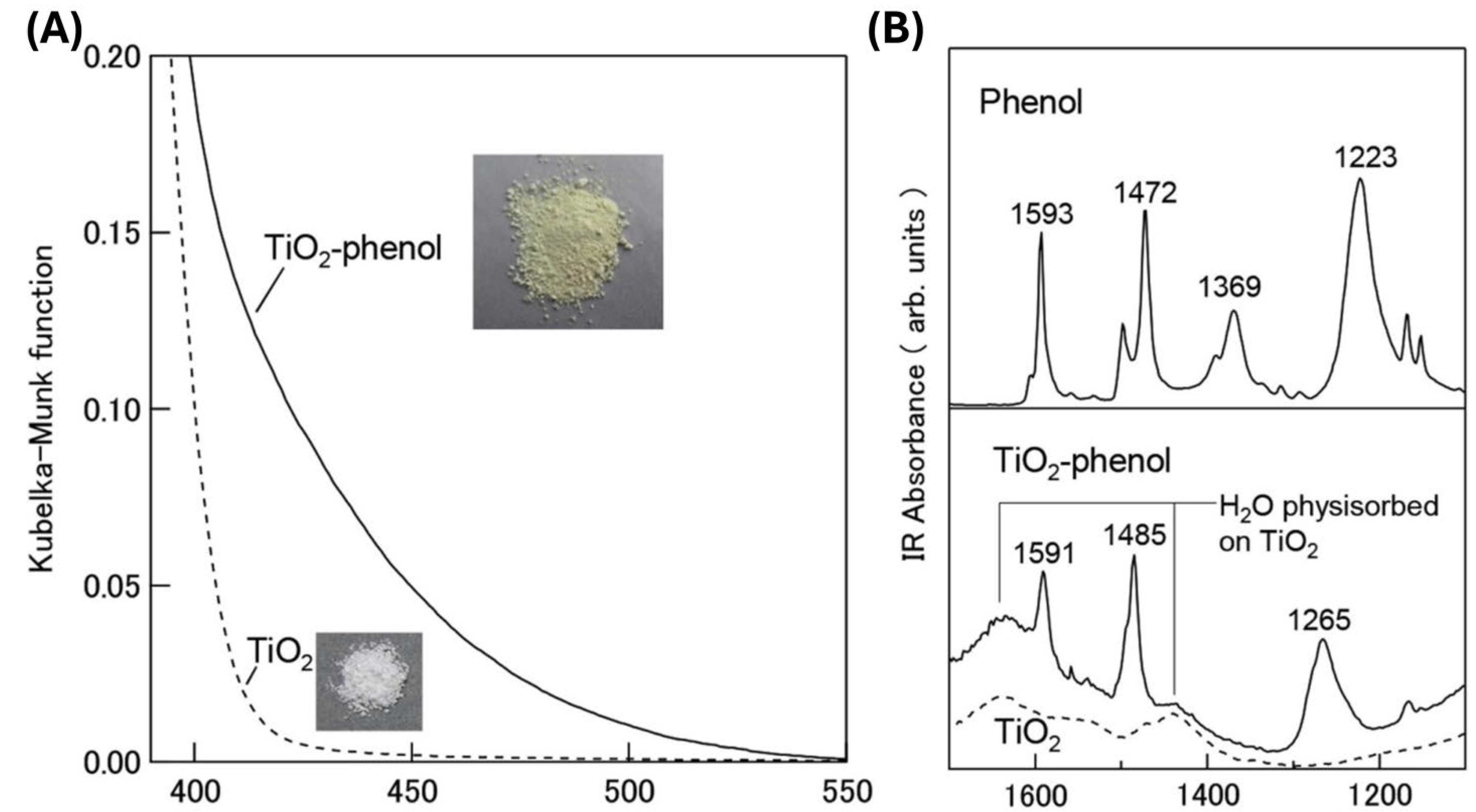
Scheme 3.
(A) Chelating and (B) bridging coordination of CAT to TiO2 surface.
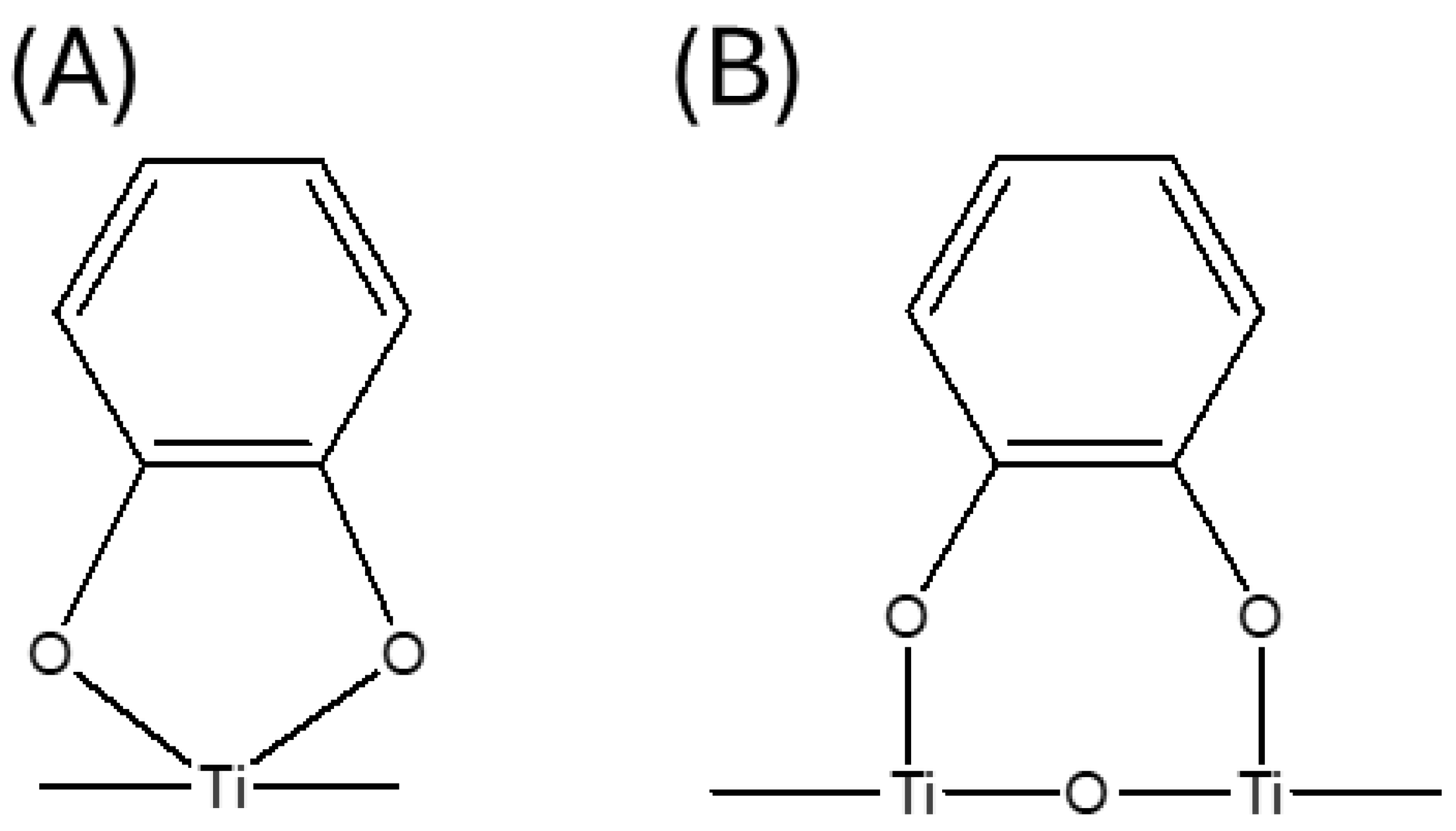
Figure 5.
Job’s curves for ligand‒Tsurf complexes (ligands are CAT and SA; [Tisurf]+[L]=2.0 mM) (reprinted from I.A. Janković, Z.V. Šaponjić, M.I. Čomor, J.M. Nedeljković, J. Phys. Chem. C, 113, 12645-12652 (2009); copyright 2009 American Chemical Society).
Figure 5.
Job’s curves for ligand‒Tsurf complexes (ligands are CAT and SA; [Tisurf]+[L]=2.0 mM) (reprinted from I.A. Janković, Z.V. Šaponjić, M.I. Čomor, J.M. Nedeljković, J. Phys. Chem. C, 113, 12645-12652 (2009); copyright 2009 American Chemical Society).
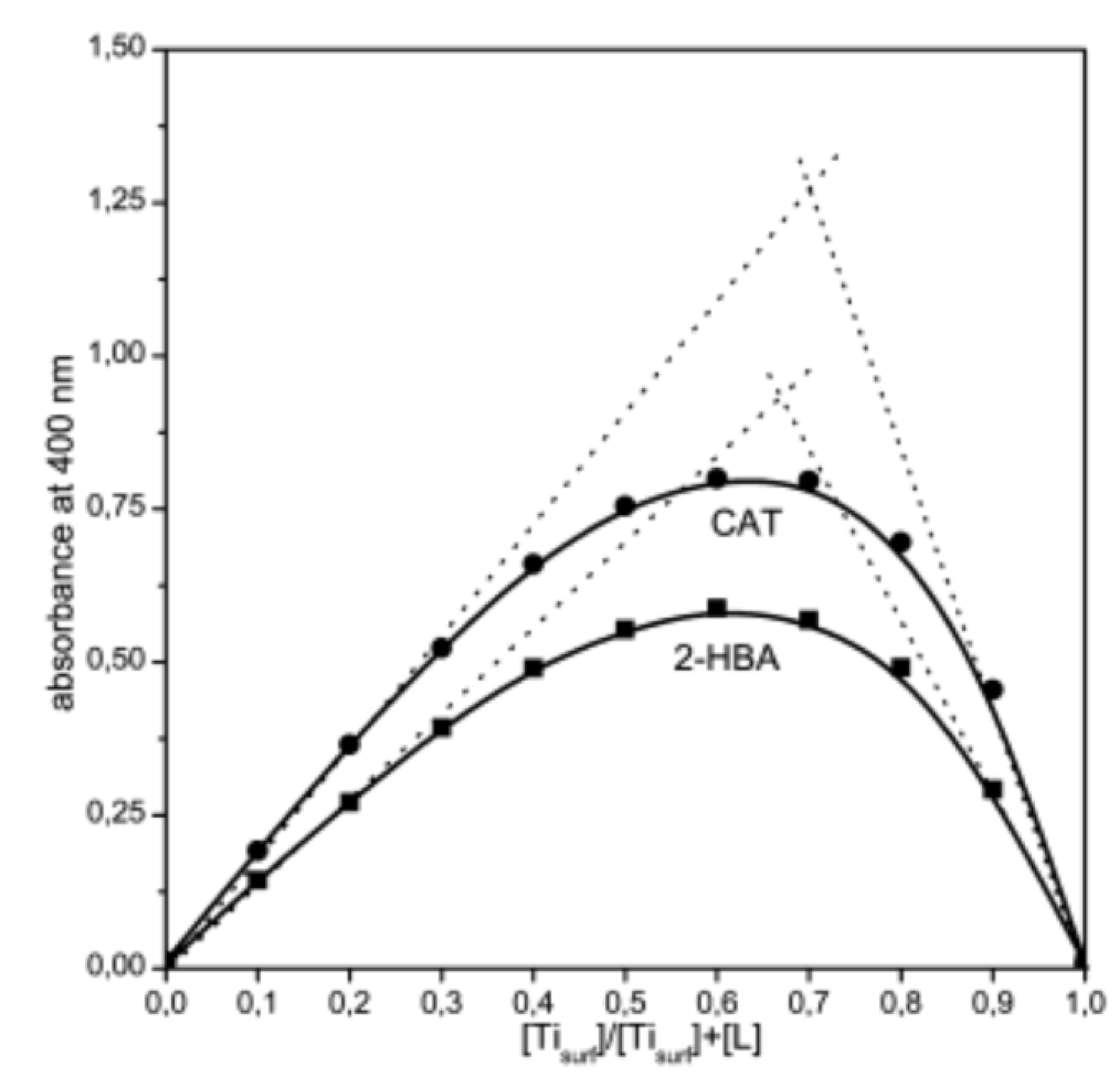
Figure 6.
(A) IR spectra of free and adsorbed CAT to TiO2 nanoparticles and calculated IR spectra for bridging and chelating coordination (top-dawn) (reprinted from T.D. Savić, I.A. Janković, Z.V. Šaponjić, M.I. Čomor, D.Ž. Veljković, S.D. Zarić, J.M. Nedeljković, Nanoscale, 4, 1612-1619 (2012); copyright 2012 Royal Society of Chemistry). (B) Optimized structures and (C) calculated vibrational spectra of [Ti8O14(OH)3−o-AP] (green), [Ti8O14(OH)3−m-AP] (orange), and [Ti8O14(OH)3−p-AP] (red). Large white: titanium; small white: hydrogen; gray: carbon; blue: nitrogen; red: oxygen atom (reprinted from J. Fujisawa, T. Eda, G. Giorgi, M. Hanaya, J. Phys. Chem. C, 121, 18710-18716 (2017); copyright 2017 American Chemical Society).
Figure 6.
(A) IR spectra of free and adsorbed CAT to TiO2 nanoparticles and calculated IR spectra for bridging and chelating coordination (top-dawn) (reprinted from T.D. Savić, I.A. Janković, Z.V. Šaponjić, M.I. Čomor, D.Ž. Veljković, S.D. Zarić, J.M. Nedeljković, Nanoscale, 4, 1612-1619 (2012); copyright 2012 Royal Society of Chemistry). (B) Optimized structures and (C) calculated vibrational spectra of [Ti8O14(OH)3−o-AP] (green), [Ti8O14(OH)3−m-AP] (orange), and [Ti8O14(OH)3−p-AP] (red). Large white: titanium; small white: hydrogen; gray: carbon; blue: nitrogen; red: oxygen atom (reprinted from J. Fujisawa, T. Eda, G. Giorgi, M. Hanaya, J. Phys. Chem. C, 121, 18710-18716 (2017); copyright 2017 American Chemical Society).
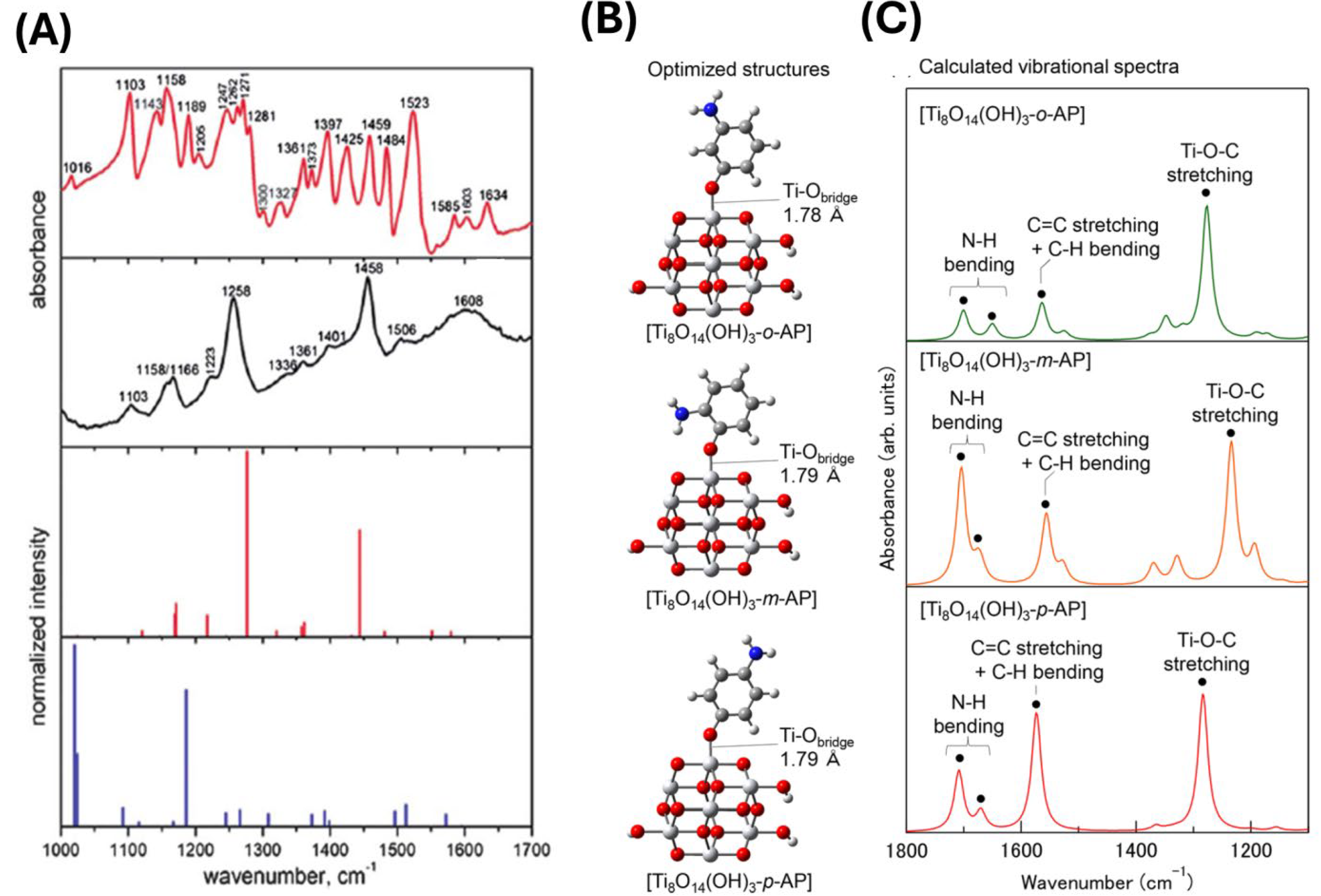
Scheme 4.
The influence of the EDG/EWG on the alignment of energy levels in the ICT complexes.
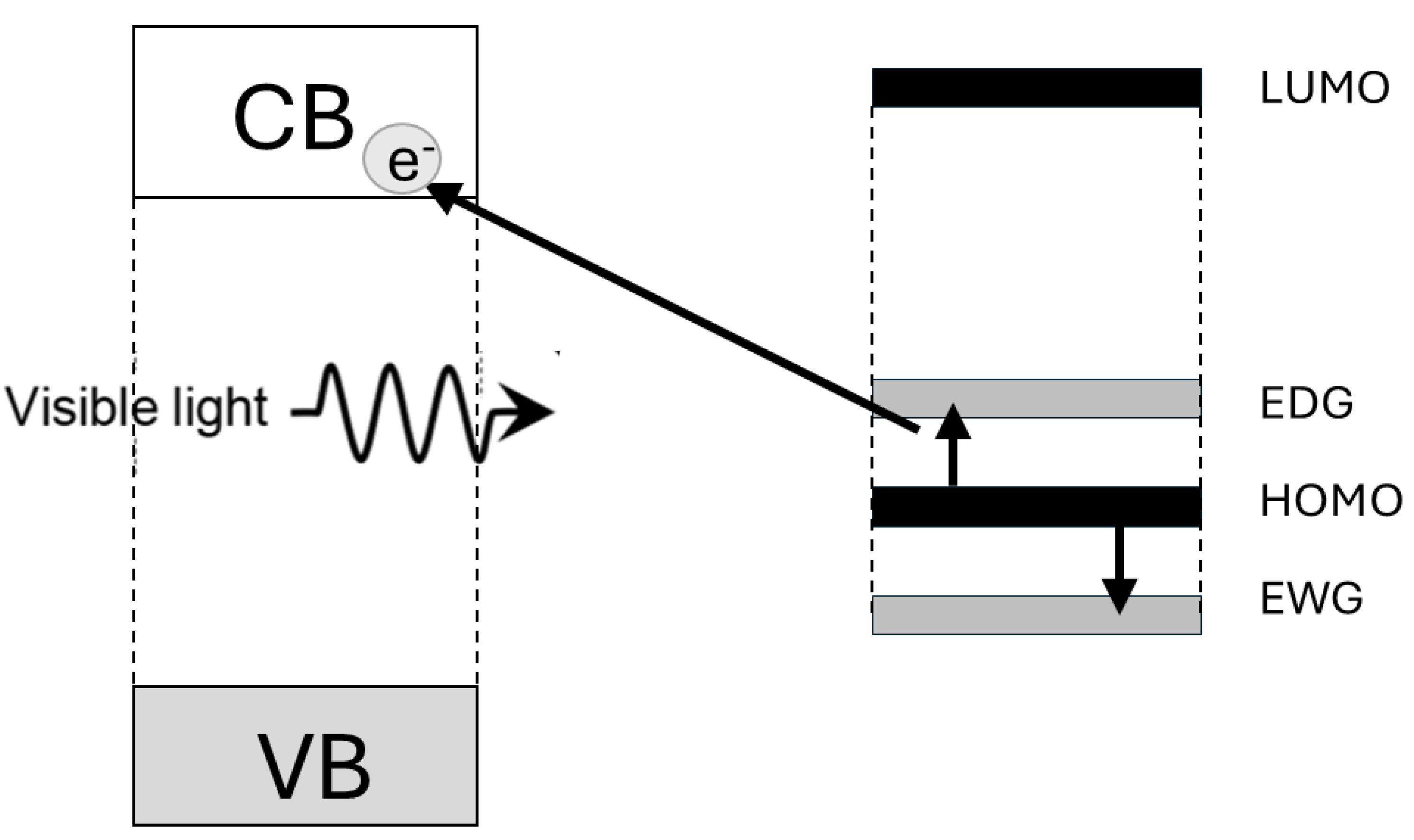
Figure 7.
Kubelka-Munk spectra (solid curves) of TiO2 and TiO2‒O‒Ph‒R (R: H, C(CH3)3, OCH3, and F5) together with absorption spectra (dashed curves) of HO‒Ph‒R in CH3CN solution (reprinted from J. Fujisawa, S. Kato, M. Hanaya, Chem. Phys. Lett., 827, 140688 (2023); copyright 2023 Elsevier).
Figure 7.
Kubelka-Munk spectra (solid curves) of TiO2 and TiO2‒O‒Ph‒R (R: H, C(CH3)3, OCH3, and F5) together with absorption spectra (dashed curves) of HO‒Ph‒R in CH3CN solution (reprinted from J. Fujisawa, S. Kato, M. Hanaya, Chem. Phys. Lett., 827, 140688 (2023); copyright 2023 Elsevier).
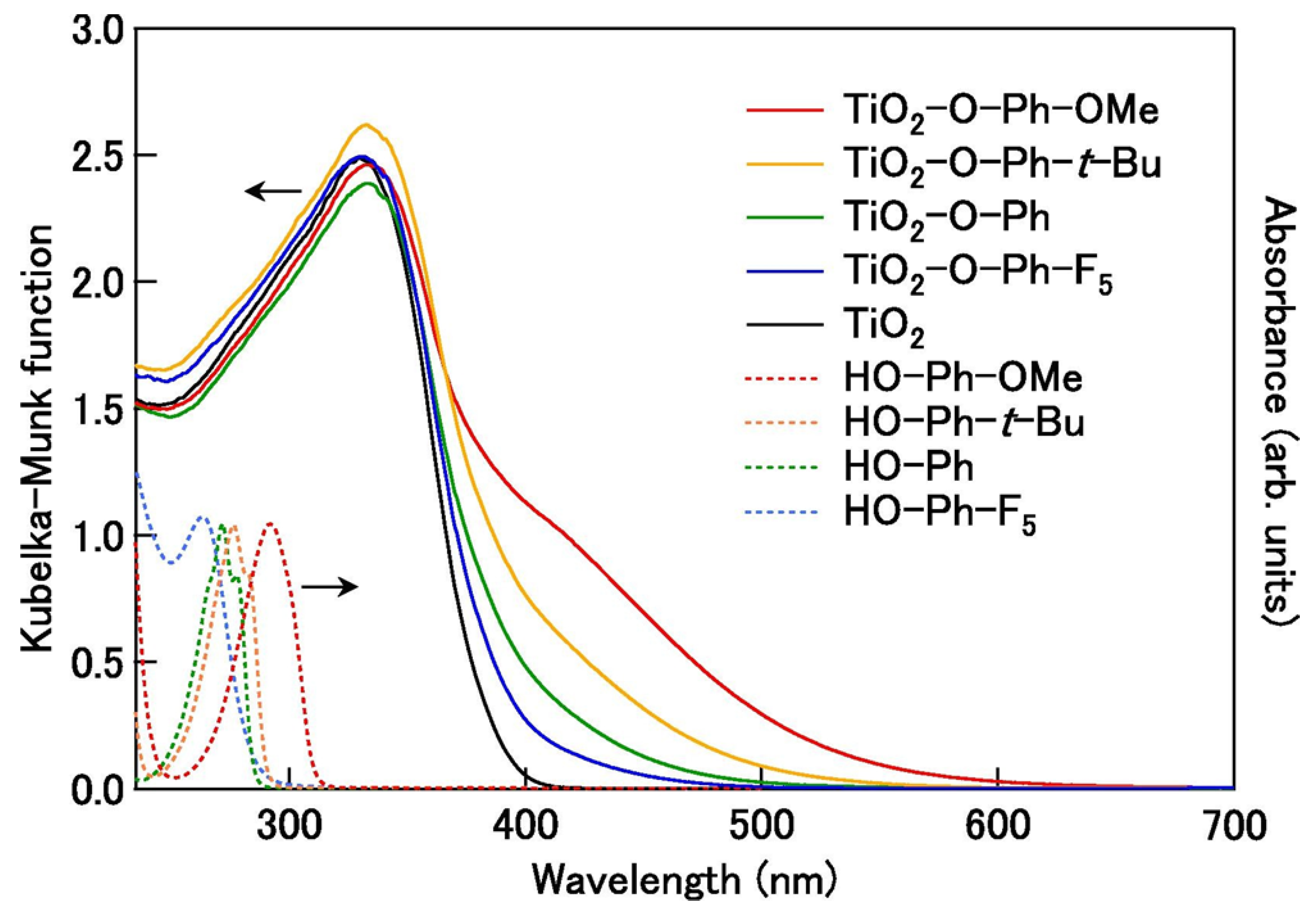
Figure 8.
(A) Absorption spectra of pristine 50-Å TiO2 colloid and surface-modified TiO2 colloids with SA and 5-ASA and corresponding photoimages. (B) Photoelectron spectra of TiO2, TiO2‒SA, and TiO2‒5-ASA recorded at 11 eV photon energy (reprinted from D.K. Božanić, G.A. Garcia, L. Nahon, D. Sredojević, V. Lazić, I. Vukoje, S.P. Ahrenkiel, V. Djoković, Ž. Šljivančanin, J.M. Nedeljković, J. Phys. Chem. C, 123, 29057-29066 (2019); copyright 2019 American Chemical Society).
Figure 8.
(A) Absorption spectra of pristine 50-Å TiO2 colloid and surface-modified TiO2 colloids with SA and 5-ASA and corresponding photoimages. (B) Photoelectron spectra of TiO2, TiO2‒SA, and TiO2‒5-ASA recorded at 11 eV photon energy (reprinted from D.K. Božanić, G.A. Garcia, L. Nahon, D. Sredojević, V. Lazić, I. Vukoje, S.P. Ahrenkiel, V. Djoković, Ž. Šljivančanin, J.M. Nedeljković, J. Phys. Chem. C, 123, 29057-29066 (2019); copyright 2019 American Chemical Society).
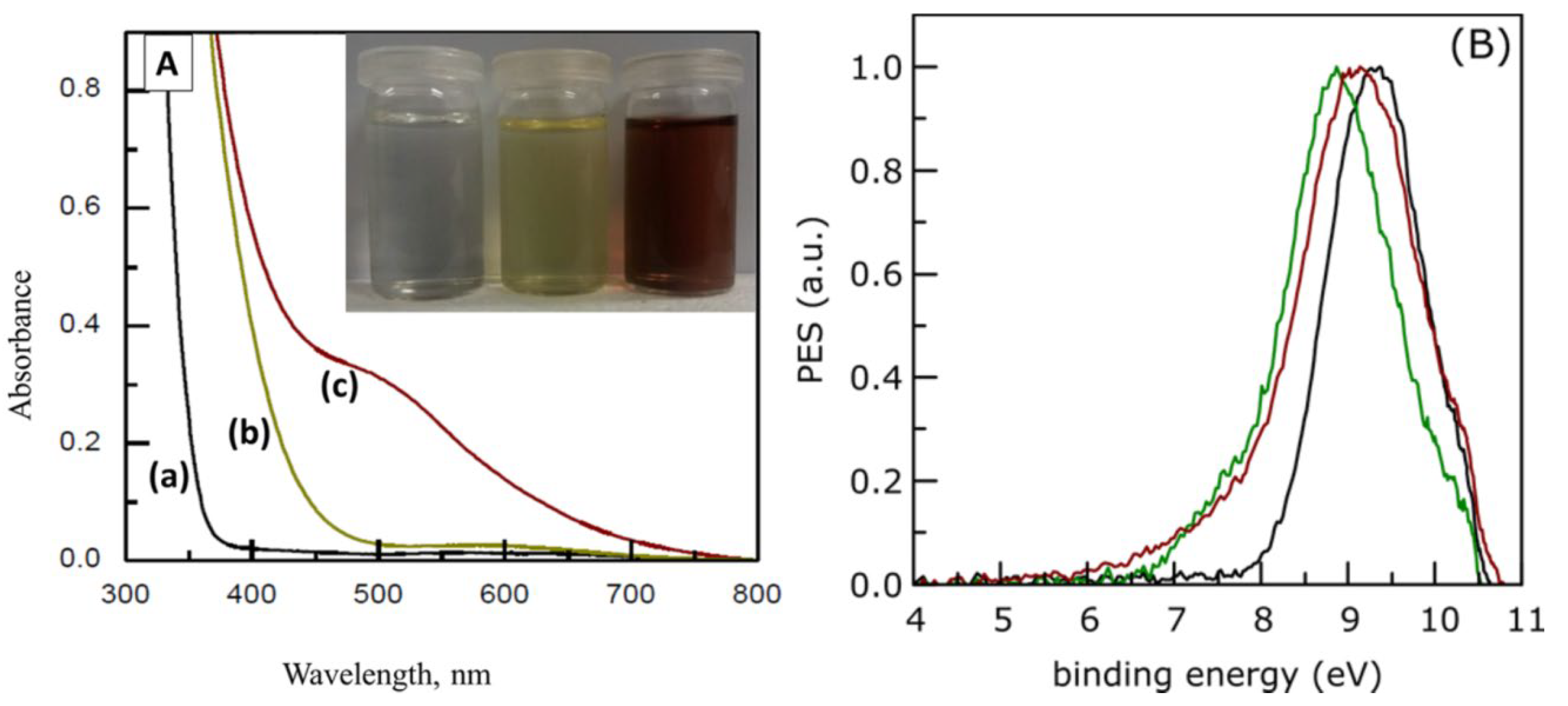
Figure 9.
(A) Kubelka-Munk spectra of pristine titanate nanotubes, surface-modified titanate nanotubes with 5-ASA, and surface-modified titanate nanotubes with 5-ASA decorated with Ag nanoparticles. (B) TEM image of surface-modified titanate nanotubes with 5-ASA decorated with Ag nanoparticles (reprinted from Z. Barbieriková, D. Lončarević, J. Papan, I.D. Vukoje, M. Stoiljković, S.P. Ahrenkiel, J.M. Nedeljković, Adv. Powder Technol., 31, 4683-4690 (2020); copyright 2020 Elsevier).
Figure 9.
(A) Kubelka-Munk spectra of pristine titanate nanotubes, surface-modified titanate nanotubes with 5-ASA, and surface-modified titanate nanotubes with 5-ASA decorated with Ag nanoparticles. (B) TEM image of surface-modified titanate nanotubes with 5-ASA decorated with Ag nanoparticles (reprinted from Z. Barbieriková, D. Lončarević, J. Papan, I.D. Vukoje, M. Stoiljković, S.P. Ahrenkiel, J.M. Nedeljković, Adv. Powder Technol., 31, 4683-4690 (2020); copyright 2020 Elsevier).
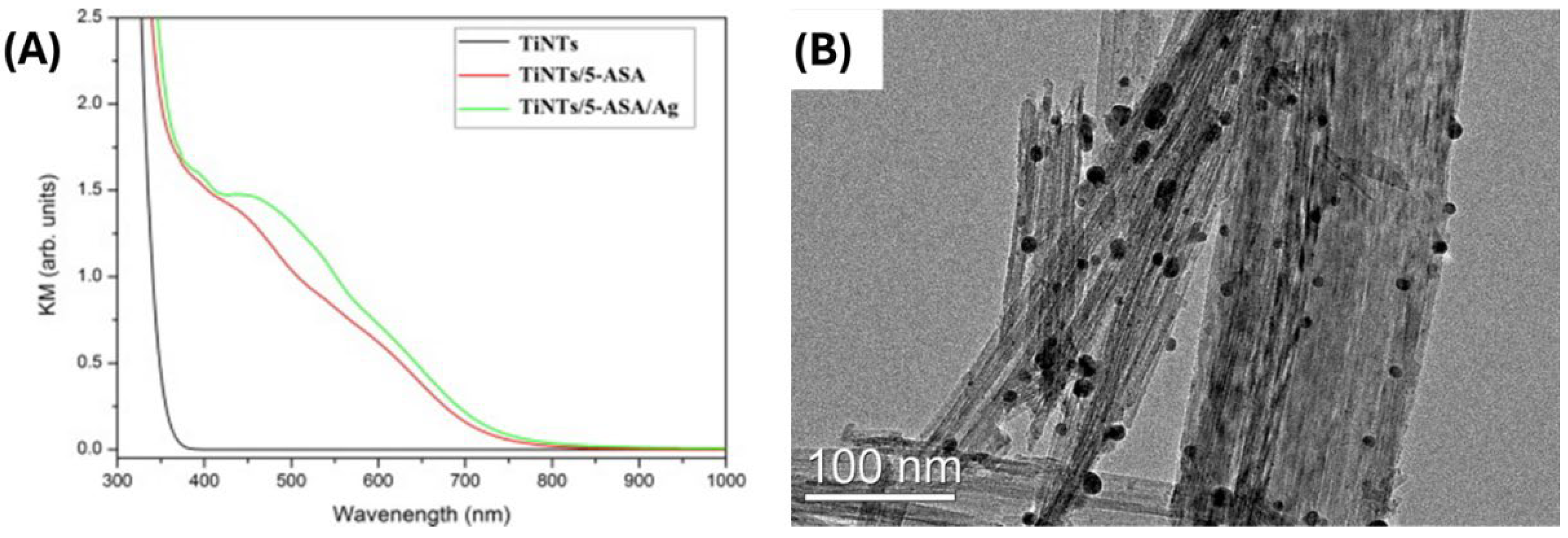
Figure 10.
Effects of electron acceptor on visible-light-induced photocatalytic degradation of 4-CP (reprinted from S. Kim, W. Choi, J. Phys. Chem. B, 109, 5143-5149 (2005); copyright 2005 American Chemical Society).
Figure 10.
Effects of electron acceptor on visible-light-induced photocatalytic degradation of 4-CP (reprinted from S. Kim, W. Choi, J. Phys. Chem. B, 109, 5143-5149 (2005); copyright 2005 American Chemical Society).
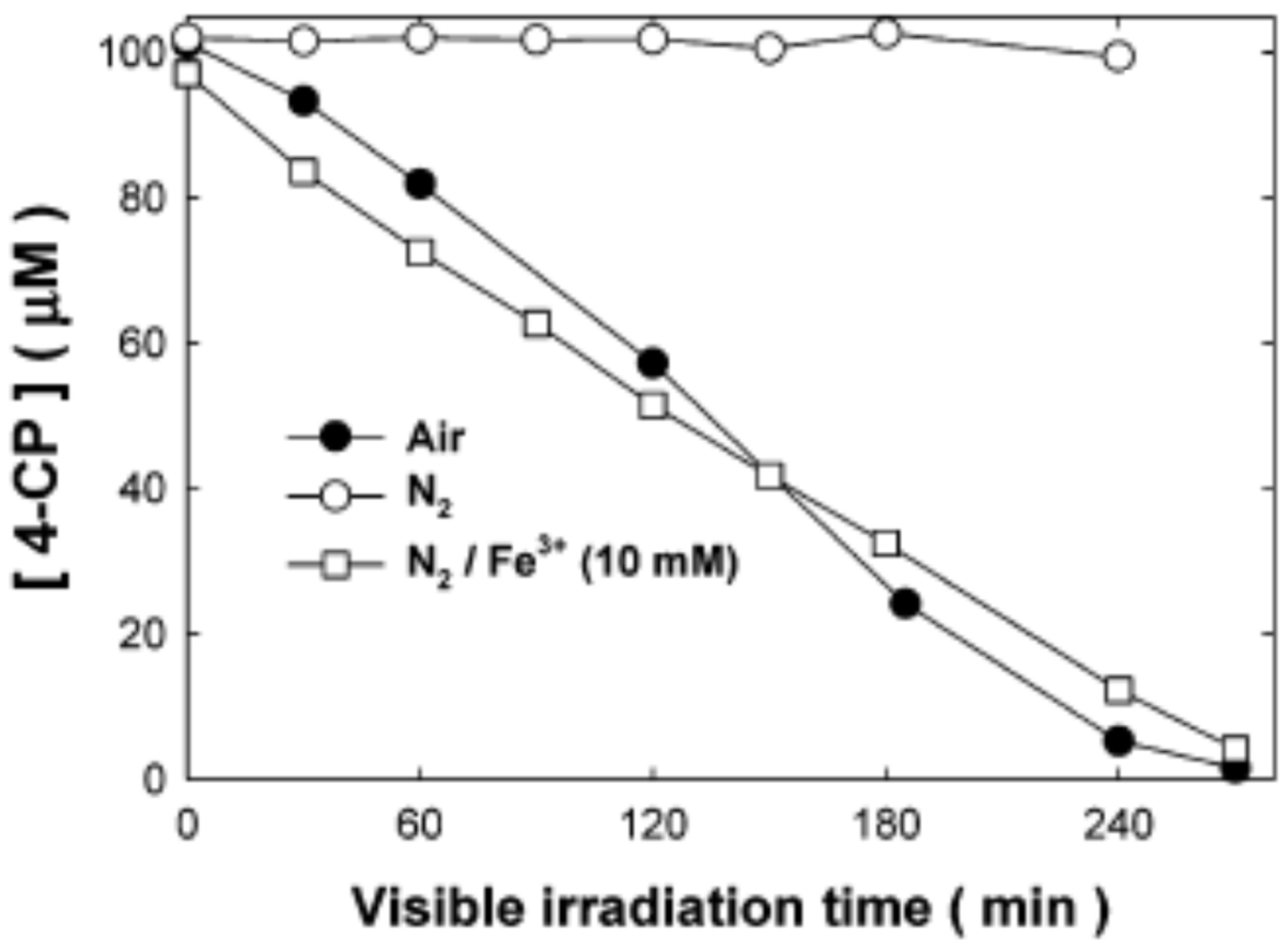
Figure 11.
Adsorption in dark and photocatalytic degradation of crystal violet (CV) over TiO2-based ICT complex with dopamine supported by macroporous polymer upon illumination with visible light (hν<2.75 eV) followed with absorption spectroscopy; inset: adsorption in dark and photocatalytic degradation kinetic curves of the CV using for excitation light with or without UV part of the spectrum (reprinted from I. Vukoje, T. Kovač, J. Džunuzović, E. Džunuzović, D. Lončarević, S.P. Ahrenkiel, J.M. Nedeljković, J. Phys. Chem. C, 120, 18560-18569 (2016); copyright 2016 American Chemical Society).
Figure 11.
Adsorption in dark and photocatalytic degradation of crystal violet (CV) over TiO2-based ICT complex with dopamine supported by macroporous polymer upon illumination with visible light (hν<2.75 eV) followed with absorption spectroscopy; inset: adsorption in dark and photocatalytic degradation kinetic curves of the CV using for excitation light with or without UV part of the spectrum (reprinted from I. Vukoje, T. Kovač, J. Džunuzović, E. Džunuzović, D. Lončarević, S.P. Ahrenkiel, J.M. Nedeljković, J. Phys. Chem. C, 120, 18560-18569 (2016); copyright 2016 American Chemical Society).
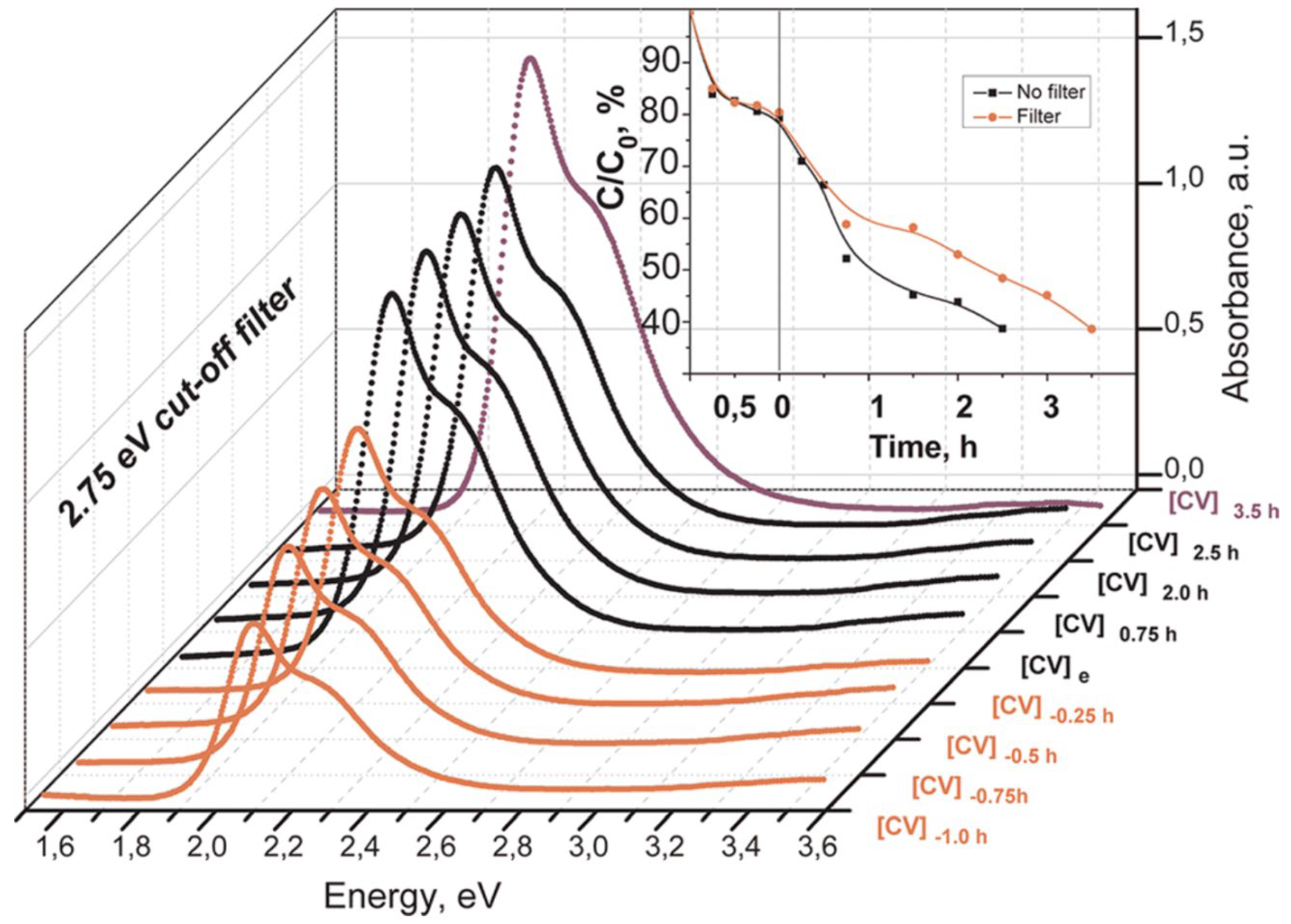
Figure 12.
Dependence of rate of H2 evolution by 1,1’-binaphtalene-2,2’-diol‒modified TiO2 loaded with 0.5 wt.-% of Pt on the wavelength of photoirradiated light of (A) >430, (B) >490, (C) >540, and (D) >580 nm (reprinted from H. Schwarz, R. Dodson, J. Phys. Chem., 93, 409-414 (1989); copyright 1989 American Chemical Society).
Figure 12.
Dependence of rate of H2 evolution by 1,1’-binaphtalene-2,2’-diol‒modified TiO2 loaded with 0.5 wt.-% of Pt on the wavelength of photoirradiated light of (A) >430, (B) >490, (C) >540, and (D) >580 nm (reprinted from H. Schwarz, R. Dodson, J. Phys. Chem., 93, 409-414 (1989); copyright 1989 American Chemical Society).
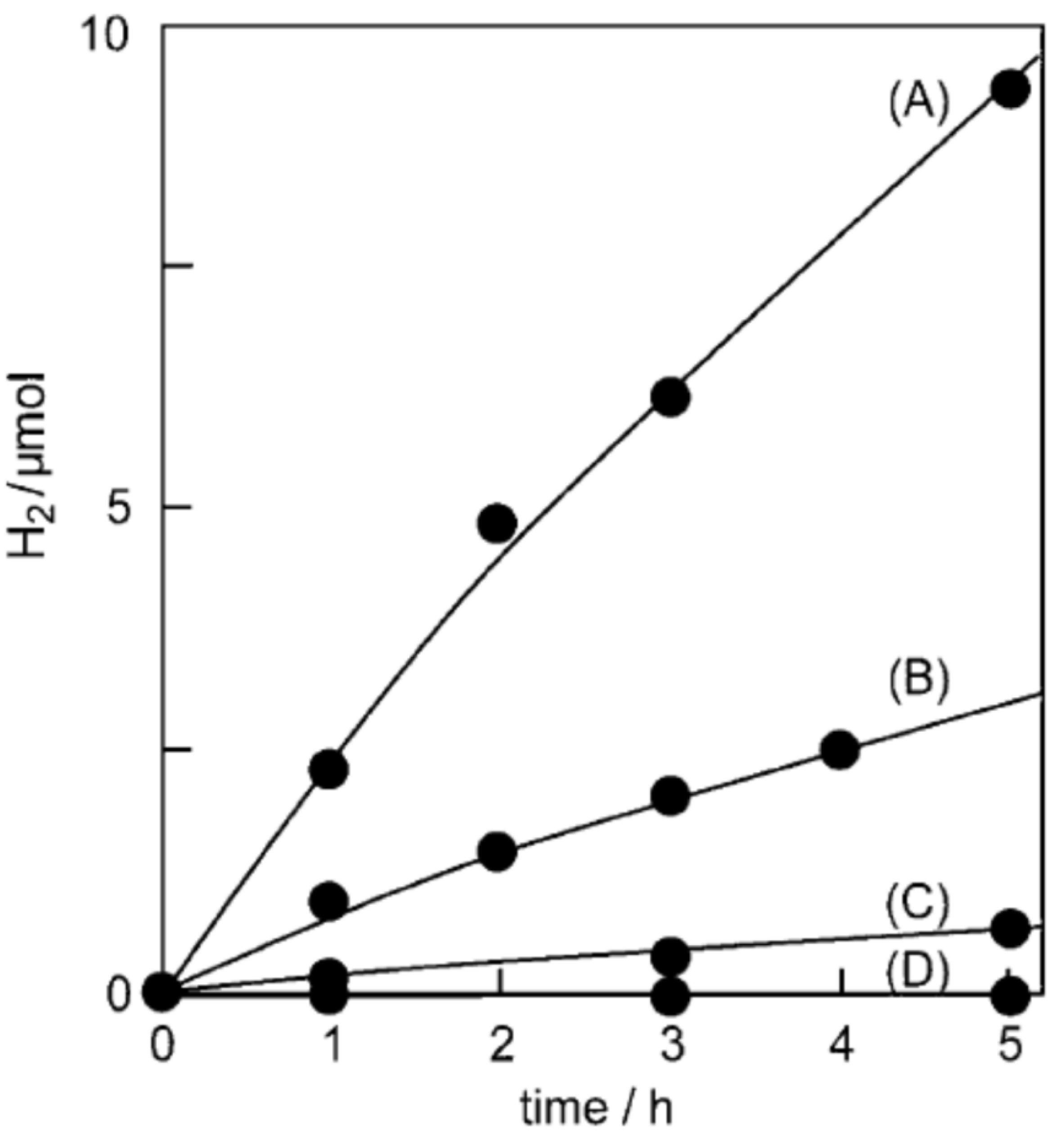
Figure 13.
(A) Photocatalytic hydrogen evolution from aqueous TEOA (10 vol.-%) over surface-modified TiO2 with (a) 4-tert-butylcatechol, (b) 3-methoxycatechol, (c) catechol, (d) 2,3-dihydroxybenzoic acid, (e) 3,4-dihydroxybenzonitrile, and (f) tiron. (B) Photocatalytic activity of surface-modified TiO2 with catechol derivatives as a function of the corresponding oxidative modifier potentials (EA). The arrow represents the EA of TEOA (reprinted from S. Higashimoto, T. Nishi, M. Yasukawa, M. Azuma, Y. Sakata, H. Kobayashi, J. Catal., 329, 286-291 (2015); copyright 2015 Elsevier).
Figure 13.
(A) Photocatalytic hydrogen evolution from aqueous TEOA (10 vol.-%) over surface-modified TiO2 with (a) 4-tert-butylcatechol, (b) 3-methoxycatechol, (c) catechol, (d) 2,3-dihydroxybenzoic acid, (e) 3,4-dihydroxybenzonitrile, and (f) tiron. (B) Photocatalytic activity of surface-modified TiO2 with catechol derivatives as a function of the corresponding oxidative modifier potentials (EA). The arrow represents the EA of TEOA (reprinted from S. Higashimoto, T. Nishi, M. Yasukawa, M. Azuma, Y. Sakata, H. Kobayashi, J. Catal., 329, 286-291 (2015); copyright 2015 Elsevier).
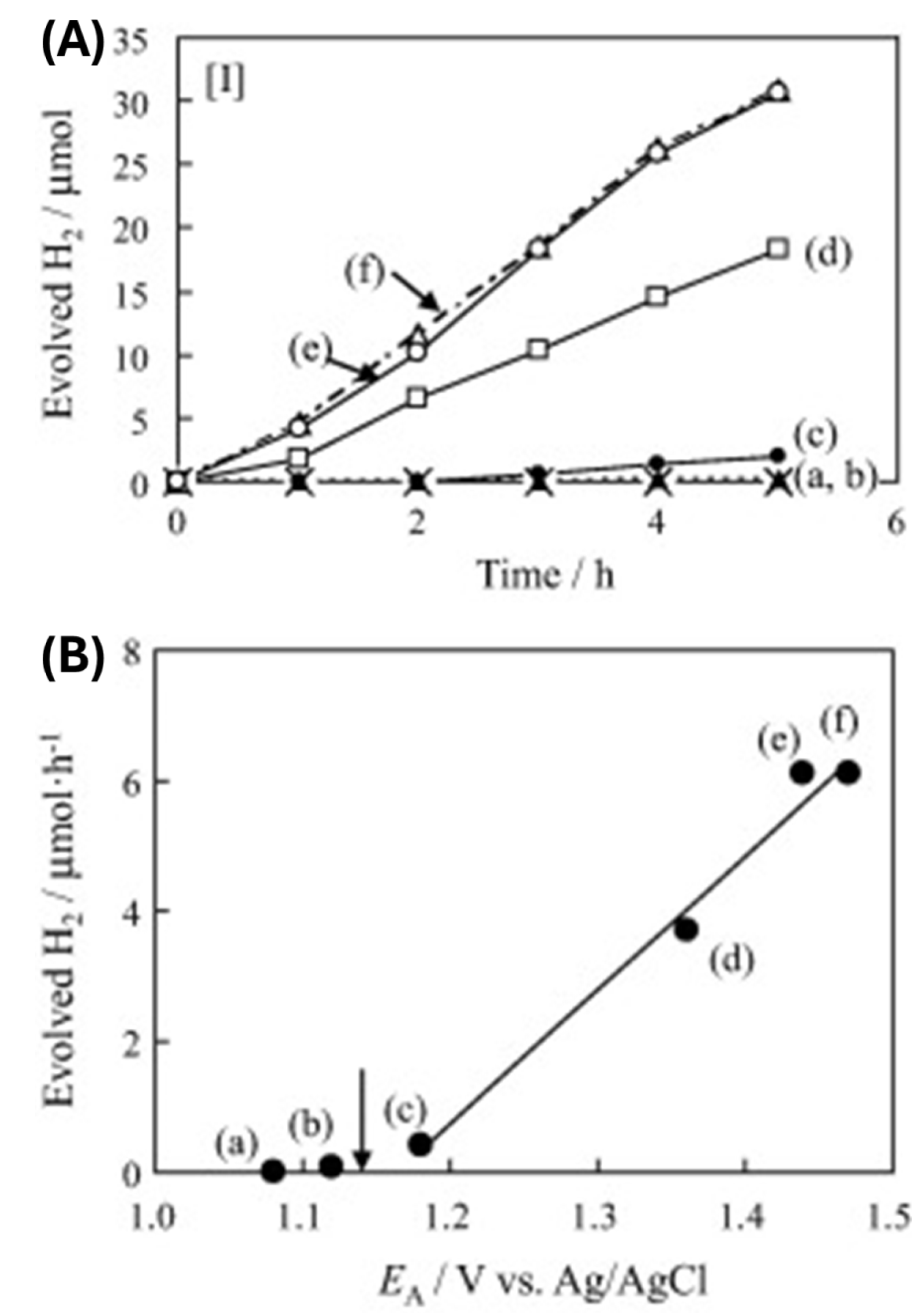
Figure 14.
Rates of photocatalytic generation of hydrogen as a function of illumination time (medium pressure Hg lamp) over TiO2 nanoparticles coordinated over dopamine to polymer support (a) and TiO2 Degussa P25 powder (b) (reprinted from I. Vukoje, T. Kovač, J. Džunuzović, E. Džunuzović, D. Lončarević, S.P. Ahrenkiel, J.M. Nedeljković, J. Phys. Chem. C, 120, 18560-18569 (2016); copyright 2016 American Chemical Society).
Figure 14.
Rates of photocatalytic generation of hydrogen as a function of illumination time (medium pressure Hg lamp) over TiO2 nanoparticles coordinated over dopamine to polymer support (a) and TiO2 Degussa P25 powder (b) (reprinted from I. Vukoje, T. Kovač, J. Džunuzović, E. Džunuzović, D. Lončarević, S.P. Ahrenkiel, J.M. Nedeljković, J. Phys. Chem. C, 120, 18560-18569 (2016); copyright 2016 American Chemical Society).
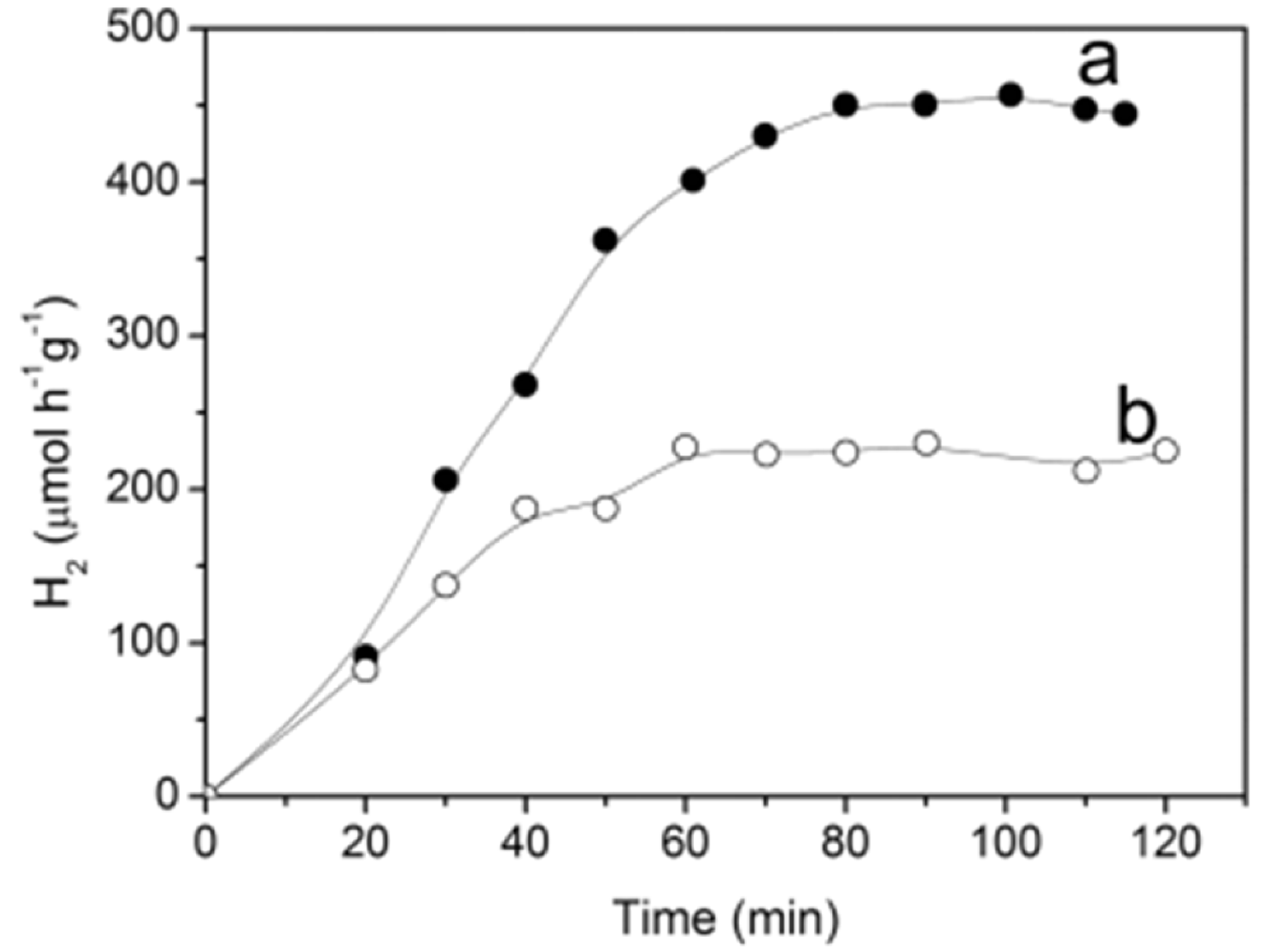
Figure 15.
(A) EPR spectra of pristine TiO2 and (B) surface-modified TiO2 with 4-CP in the dark and upon UV or Vis excitation at 100 K (reprinted from Z. Barbieriková, D. Dvoranová, V. Brezová, E. Džunuzović, D.N. Sredojević, V. Lazić, J.M. Nedeljković, Opt. Mater., 89, 237-242 (2019); copyright 2019 Elsevier).
Figure 15.
(A) EPR spectra of pristine TiO2 and (B) surface-modified TiO2 with 4-CP in the dark and upon UV or Vis excitation at 100 K (reprinted from Z. Barbieriková, D. Dvoranová, V. Brezová, E. Džunuzović, D.N. Sredojević, V. Lazić, J.M. Nedeljković, Opt. Mater., 89, 237-242 (2019); copyright 2019 Elsevier).
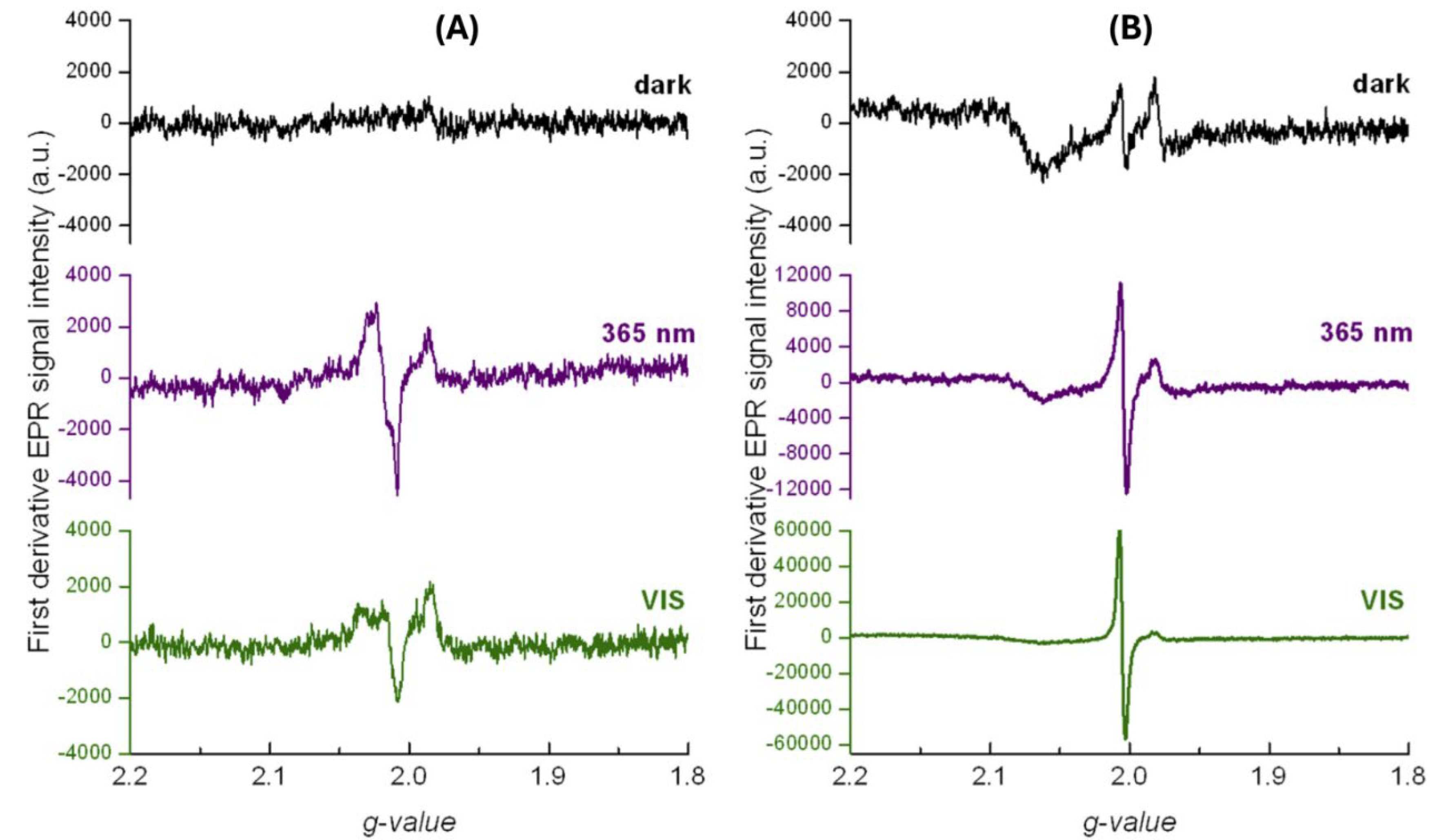
Figure 16.
Time dependence of ABTS•+ relative concentration evaluated from double-integrated EPR spectra monitored in the aqueous aerated suspensions of TiO2, surface-modified TiO2 with squaric acid, and ABTS•+ reference (photocatalyst-free) solution upon excitation with: (A) UVA (λ = 365 nm) and (B) VIS light (λ > 420 nm) (reprinted from Z. Barbieriková, M. Šimunková, V. Brezová, D. Sredojević, V. Lazić, D. Lončarević, J.M. Nedeljković, Opt. Mater., 123, 111918 (2022); copyright 2022 Elsevier).
Figure 16.
Time dependence of ABTS•+ relative concentration evaluated from double-integrated EPR spectra monitored in the aqueous aerated suspensions of TiO2, surface-modified TiO2 with squaric acid, and ABTS•+ reference (photocatalyst-free) solution upon excitation with: (A) UVA (λ = 365 nm) and (B) VIS light (λ > 420 nm) (reprinted from Z. Barbieriková, M. Šimunková, V. Brezová, D. Sredojević, V. Lazić, D. Lončarević, J.M. Nedeljković, Opt. Mater., 123, 111918 (2022); copyright 2022 Elsevier).
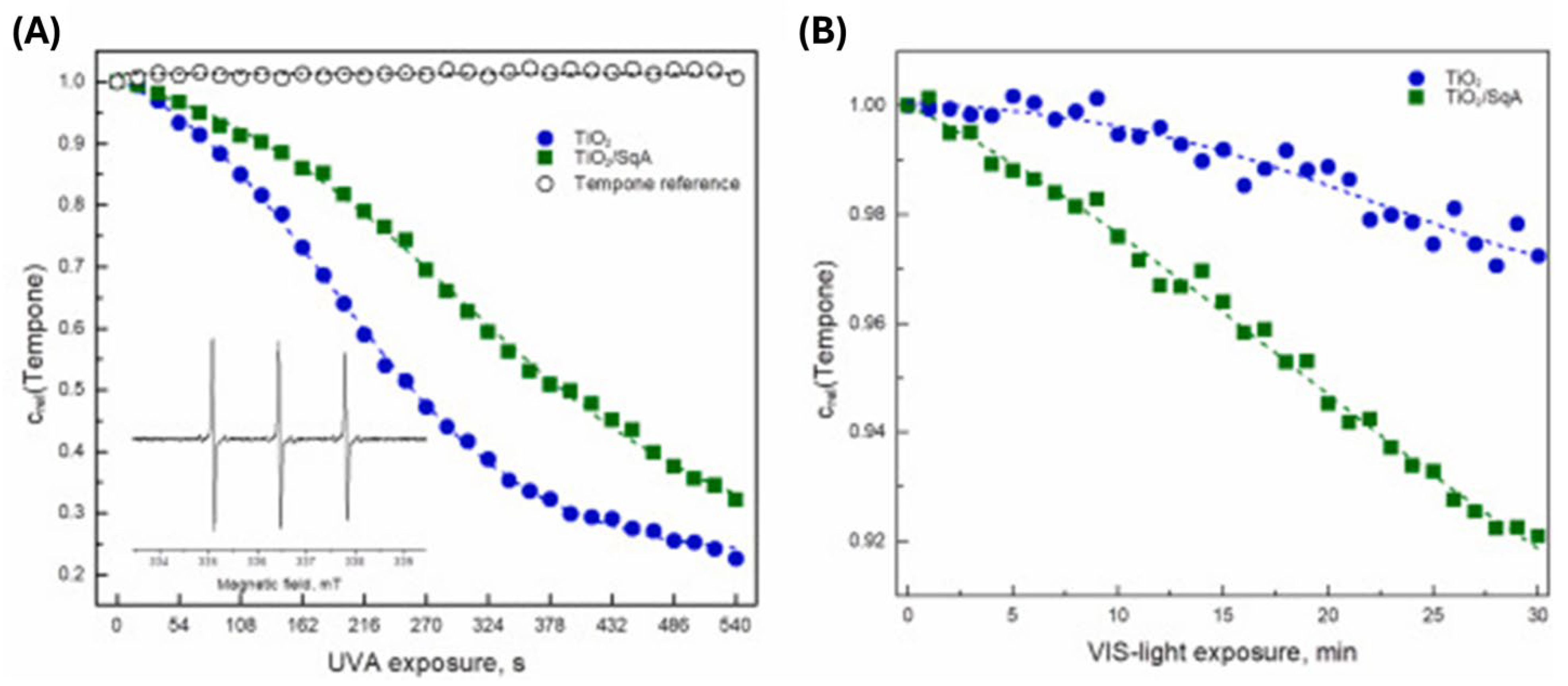

Table 1.
Literature overview ‒ the ICT complexes between various wide-band-gap oxides (Me‒Ox) and different types of ligands.
Table 1.
Literature overview ‒ the ICT complexes between various wide-band-gap oxides (Me‒Ox) and different types of ligands.
| Me‒Ox | Type of ligand | |||
|---|---|---|---|---|
| Two Me‒O‒C linkages | Single Me‒O‒C linkage | Me‒S‒C linkage or linkages | Miscellaneous | |
| TiO2 | 21-25, 29, 33-35, 41-50 | 26, 37-40, 51-53 | 36, 54-59 | 20, 27, 28, 31, 60-70 |
| Titanates | 72-76 | |||
| CeO2 | 77, 78 | |||
| ZrO2 | 79-80 | |||
| ZnO | 81 | 82-85 | ||
| Al2O3 | 86-88 | |||
| HAP* | 89, 90 | |||
* Hydroxyapatite.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



