
Submitted:
11 October 2024
Posted:
12 October 2024
You are already at the latest version
Abstract
Myocardial cells and extracellular matrix fulfil their goal thanks to the energetic availability. Indeed, mechanical and electrical properties of the heart are strongly depended on the energetic production-consumption equilibrium. The produced energy is used under several forms including kinetic, dynamic, thermal energy etc. Notably, as the time goes by; aging as well as in case of heart failure, although total energy remains almost constant the contribution of each energetic form is altered. Thermal energy is increased, whereas the dynamic and kinetic energy are decreased and hence unable to satisfy adequately the cardiac work. Consequently, toxic products, unfolded /misfolded proteins, free radicals etc. are accumulated within the myocardium. Myocardial cell contraction – relaxation coupling, ion exchange, cell growth etc. function is failed, control of cell apoptosis and necrosis is lacking and cardiac micro and macro-architecture change is the final result. Energy production and consumption depends on cardiac metabolic resources and on the functional status of the cardiac silhouette including cardiomyocytes and non-cardiomyocytes cells and their metabolic - energetic behavior. Mitochondria, are intra-cellular organelles producing more than 95% of ATP and fulfill all the above prerequisites being thus very important and as such we have to better understand their anatomy, function and homeostatic properties.
Keywords:
heart failure
; mitochondria
; cardiac disease
; energy
1. Introduction
Heart is like a residence where almost everything is dependent on energetic availability in order to satisfy the everyday needs of each family’s member. Accordingly, myocardial cells and extracellular matrix fulfil their prerequisites thanks to the energetic availability. Indeed, mechanical and electrical properties of the heart are strongly depended on the energetic production-consumption equilibrium. Cardiac fuel is the master key of contraction – relaxation coupling, ion exchange, cell growth, apoptosis, necrosis etc. and serve for the maintenance of cardiac homeostatic procedure. Interestingly, although heart represent 0.5% of body weight, consumes 8% of energy, and the produced ATP supports only a small number of heart beats, forcing the entire metabolic factory to repeat the entire process within few seconds in a way to support the energetic needs [1,2].
There are several forms of produced energy including kinetic, dynamic, thermal energy etc. Notably, under different circumstances such as aging and cardiac disease, although total energy remains constant, the contribution of each energetic sub-form is different. For example, in heart failure patients’ thermal energy is increased, whereas the dynamic and kinetic energy are decreased and unable to satisfy adequately the cardiac work [3]. There is an impairment of energy production and inadequate transfer within cardiac cells proved by the decrease in cellular ATP, phosphocreatine (PCr), and the PCr/ATP ratio, observed in both heart failure with reduce [4] or preserved left ventricular ejection fraction [5]. When this bioenergetic capacity reaches its limit then a decompensate stage begins, the cardiac homeostatic disequilibrium starts, affected by the over activation of sympathetic system, renin-angiotensin-aldosterone axis, inflammation etc., and a vicious cycle leading to heart failure syndrome starts. The metabolic-energetic alteration favors the accumulation of toxic products; unfolded/misfolded proteins, free radicals etc. leading to cardiac micro and macro-architecture change; cardiac remodeling [6]. This bioenergetic capacity; energy production and consumption, depends on cardiac metabolic resources and on the functional status of both cardiomyocytes and non-cardiomyocytes cells and their metabolic - energetic behavior. A behavior that we have to better understand and to further investigate the master key of the process; mitochondria.
2. Mitochondrial Dynamics
Mitochondria are intra-cellular organelles producing more than 95% of ATP. Their normal structure, integrity, function and homeostatic properties are highly important since their unsuitable anatomy and abnormal-altered function are responsible for myocardial cell injury and death and consequently for cardiac disease genesis and progression [1,7,8]. They have their own DNA (mtDNA), circular in shape, encode 13 subunits of protein whereas the majority of the mitochondrial proteins are governed by nuclear DNA, transported within mitochondria via mitochondrial membrane. The mtDNA, because of their own low protective mechanisms, is subjected to mutations responsible for many inherited cardiomyopathies [1]. However, the amount of mutated mtDNA presented in each individual is higher than the incidence of myocardial diseases, serving thus as dormant source for subsequent emerging diseases when mitochondria mutations have reached a certain threshold [9]. On the other hand, gene–gene and gene–environment interactions do not affect proportionally cardiac mitochondria, thanks to the powerful compensatory mechanism that they possess, showing a resistance to outsider harmful events, protecting thus mitochondria malfunction and hence the manifestation of heart diseases [10]. However, when they are severely affected, they face a non-viable situation leading to detrimental effects. Usually are modestly affected, having thus the time to compensate, to rearrange their homeostatic status and change their metabolic actions. If the compensate process fails, then heart diseases emerge and within time progress [11,12,13]. When the heart failure syndrome begins the fission and fragmentation processes are involved and as the syndrome deteriorates there is a decrease in mitochondrial cristae density, areas of cluster mitochondria are observed, vacuolar degeneration and calcium overload is present [13,14,15,16] leading to myocardial cell apoptosis and necrosis [17,18]. As a response, mitochondrial defensive mechanisms are increased, especially mitophagy, trying to protect the myocardial cells and the heart as a whole. This has been reported in both preserved and reduced ejection fraction being more actively involved in the latest [14,19]. However, although the protective mechanisms are beneficial, within time as the syndrome aggravates the protective mechanisms are over whelmed unable to protect and to maintain mitochondria normal function [19,20]. Moreover, the autophago-lysosomal system is dysfunctional [21,22] making thus the mitochondria more vulnerable. The heart failure syndrome of any cause deteriorates, the fort fell, Figure 1.
There are different mitochondrial shapes across the human body depended on the tissue and the adjacent cell environment that can differentiate mitochondria structure-function-behavior. Thus, different mitochondria subpopulations are present demonstrating different response to metabolic–energetic status, underlying thus their complexity heterogeneity and diversity. In the myocardium, the most metabolically active and thus most rich organ in mitochondria, their main role is to regulate biogenesis, ion transport, and to protect themselves using their defensive mechanisms, fusion, fission, mitophagy etc. [23,24], Figure 2. This is accomplished due to the presence of three different mitochondrial subtypes within cardio myocytes, a) interfibrillar, b) subsarcolemmal and c) perinuclear mitochondria, each subtype demonstrating different shape, function – response to metabolic and pathophysiologic changes. Indeed, the interfibrillar are oval, lying in longitudinal rows within myofibrils and exhibit higher rate of oxidation. Subsarcolemmal, are responsible for electrolyte and metabolite transport and offer the higher myocardial protection. Perinuclear, are of spherical shape, control nuclear function, and regulate mostly mitochondria fusion and fission process [23]. Of interest, products of the association of mitochondrial respiratory chain complexes (I–IV) harbored in the inner mitochondria membrane create the mitochondrial super complexes. Their aim is to provide incremental capabilities in the electron transfer process. They constitute the respirasome (I+III2+IV1), responsible for mitochondrial and phospholipids (cardiolipin etc.) function, satisfying in a better way the energetic needs of the heart. Additionally, their presence and function reduce the amount of free radical production serving thus as a preventer of a possible mitochondrial dysfunction [25,26]. As mentioned before the main aim of mitochondria is the production of energy. Almost 90% of produced ATP is used to support the contraction-relaxation coupling. The separation and the assembly of actin/myosin are both highly energetic depended procedures and therefore their normal behavior is closely related to the source of energy production. Additionally, ions exchange, mainly CA2+ release and sequestration, require high amount of energy, produced at the site of mitochondria. This factory of energy; mitochondria, must act as quick as possible in order to fulfill the needs of human body. Indeed, under high energetic requirements they have to adapt accordingly and to produce enough energy to satisfy the needs. To accomplish this issue, they possess the capability to protect their self, to interconnect, to change their shape, and to move within the cell since they ‘can cross cell boundaries’ [27]. The need of change in shape and motion may occur under normal circumstances; high load exercise training or under different pathological clinical scenarios; myocardial ischemia, hypertension, cardiac hypertrophy, heart failure etc. In case of mutated or dysfunctional mitochondria etc., the production of energy is inadequate, whereas the accumulation of harmful substances; free radicals’ production, heat shock and unfolded/misfolded proteins, promote the beginning and the progress of cardiac diseases. Keeping with the above-mentioned reports it is clear that mitochondria have to have their own protective mechanisms in order to avoid their dysfunction-malfunction transformation and keep the energetic-metabolic-homeostatic status of the cell on the road. Indeed, mitochondria morphology and function adapt to the different environment, activating their self-protective actions, that is necessary for the cell survival, [23] Figure 2. These actions are under specific protein control; guanosine triphosphate hydrolase enzyme family, mitochondrial fission and fusion proteins, mitochondrial dynamics proteins 49 and 51, etc. Figure 2, facilitate a continuous adaptation of mitochondria shape and function, promote genetic material exchange between the mitochondria, ensuring their ability of optimal function [23,28]. The mechanism through which mitochondria can receive genetic material from the mitochondria of other cells, although not fully elucidated, facilitate inter-cellular molecular crosstalk that represent an adaptive mechanism trying to avoid mitochondria malfunction [27]. Three different modes of inter-cellular mitochondria transport have been proposed a) tunneling nanotubes (TNTs), b) membrane extracellular vesicles (EVs) and c) gap junctions (GJC) [29]. TNTs represent the principal way of mitochondria transport, are formed rapidly by mitochondria membrane protrusions, and are composed of F-actin and transport proteins [30]. Membrane micro-vesicles represent heterogeneous components released from intra to extra cellular environment and thus called extracellular vesicles (EVs). Smaller EVs contain exosomes, small RNAs, genomic DNA, mtDNA, while larger EVs can contain even entire mitochondria [27,31,32]. Their principal role is to eliminate abnormal proteins and can serve as an additional mode for inter cellular communication (in nervous system) [33,34]. GJCs are transport gates for several substances’ transportation including nutrients, metabolites, mitochondria [35] and it seems that play a role for the intercellular transportation of reactive oxygen substances [24,36]. Although, as it has been suggested, mitochondrial structure change is connected to several pathologies, this knowledge is not thoroughly investigated and not used in every day clinical practice [24,37]. The presence of different mitochondrial phenotypes; donut-like, ellipsoid shape etc., as well their side of action may represent defensive response to several harmful events [37,38,39]. A response that can affect the main protective mitochondrial mechanism; fission and fusion. Taken into account the presence of different mitochondrial subpopulations, their altered shape and the role of specific drivers - proteins for fission; mitochondrial fission factor, mitochondrial division 49 and 51 etc., and fusion; mitofusin1, 2 etc. Figure 2, may represent an early sign of a disease and their measurement might facilitate clinical diagnosis. Additionally, the study of mtDNA heteroplasmy (different alleles in one patient) [40] may give us further knowledge about the mitochondrial abnormal status and to let us discover earlier the upcoming consequences.
3. Mitochondria, a ‘Socialized’ Organelle
The cell’s organelles are inter-connected and they function not as a single unit but as a whole, according to the cellular needs [41]. The anatomical and functional communication between them; endoplasmic reticulum (ER), mitochondria, nucleus, plasma membrane, Golgi apparatus etc. is well known, indicating their principal role on human body homeostasis [42,43]. Mitochondria are not formed de novo, do not possess certain abilities (does not synthesize phosphatidylcholine, phosphatidylinositol, sterols, sphingolipids etc.) [43], and necessarily their activities are linked to their action to communicate with the other organelles [41,43].
Are considered as the most ‘socialized’ organelle since are interconnected with a variety of them and their defensive mechanisms; fusion, fission, mitophagy etc. are dependent on their best cross-talk. Of note, mitochondria-lysosome [44,45], mitochondria-peroxisome [46], and mitochondria -lipid droplets [47] communication serve to achieve the optimum homeostatic cell equilibrium and function. Of particular interest is the communication between ER and mitochondria, being in continuous cross-talk, since mitochondrial main function; oxidative phosphorylation and ATP production, Ca2+ exchange and buffering etc., is dependent on ER lipid and Ca2+ transport efficiency [7,43].
3.1. Mitochondria—Endoplasmic Reticulum Connection
The endoplasmic reticulum (ER) is involved in many cellular activities; secretory, protein folding, ions homeostatic process, lipids biosynthesis etc., is connected with the other cell organelles affecting thus their activities. In cardiovascular diseases several causes such as ischemia, pulmonary and arterial hypertension, metabolic disorders etc. can alter ER normal function leading to ER homeostatic imbalance characterized by the production of free radical and misfolded proteins. Consequently, the proper communication between ER and the other cardiomyocyte organelles is disrupted [48], promoting cell apoptosis, necrosis etc. Accordingly, ER–mitochondria interconnection; through mitochondria-associated membranes (MAMs), are responsible for the proper mitochondrial function; cellular metabolism, ions homeostasis, inflammation etc. Indeed, between ER and mitochondria there is an ultrastructural organization that governs various cellular life processes [42] having a crucial role on cardiovascular remodeling and hence to the progress of various cardiovascular diseases [49,50]. The interruption of ER-mitochondria communication produces redox imbalance, further perturbation of ER homeostasis, mitochondrial injury, Ca2+ homeostatic imbalance, energy depletion and programmed cell death. Consequently, myocardial contractility-relaxation coupling along with vascular smooth muscle cell differentiation are affected. Additionally, injured - abnormal mitochondria produce a huge amount of reactive oxygen species, accumulated within the cell inducing further myocardial cell injury and damage. Indeed, in patients with heart failure, the free iron is increased within mitochondria, an ion (through Fenton chemistry) necessary for the free radical production [51]. Regardless of whether mitochondrial or ER is firstly affected, the final result is the loose of homeostatic balance of both organelles as well as their capability of communication, a fact responsible for the incomplete cardiomyocyte reconstruction [52], unbalanced oxidative stress [53], Ca2+ homeostatic incapacity [54], abnormal metabolism of lipid and other substances [55], lack of adequate energy production [42], activation of MAMs, contributors of the genesis of inflammasome and of the inflammation process [56,57], and ultimately cell protection [49]. In other words, when mitochondria/ER structure and function are harmfully affected, the protective homeostatic mechanisms are altered, nuclear and mitochondrial DNA and other toxic substances are released into cytosol [58] indicating the beginning and within time the deterioration of cardiovascular diseases.
Cardiovascular diseases are characterized by the term cardiac remodeling that represent abnormal changes in the structure and function of cardiovascular system [59]. These changes are referred to abnormal response to certain stimuli and are lying on the alteration of inflammatory response, autophagy defect, lack of gene transcription normality, deficient of energy metabolism, increase of oxidative stress, ions homeostasis imbalance, cell apoptotic rhythm, cell necrosis [60,61,62,63,64,65] etc. A response that among other pathophysiological etiologies is due to the sarcoplasmic/endoplasmic reticulum–mitochondria unbalanced coupling [66,67] whereas a balanced coupling is responsible for the stability of the cellular ambient [68]. The normal function of cardiomyocytes, a highly energy demanding cells, are closely dependent on the interconnection between these two organelles, that control among others, Ca2+ buffer and transport [59]. Indeed, mitochondria represent one of the major calcium pools [69] and are part of various biochemical processes; lipid metabolism, calcium signal transduction [70] etc. Similarly, ER is the main Ca2+ homeostatic regulator [71], a site of protein, lipid biosynthesis [72] etc. Consequently, the proper communication between these two organelles is of huge importance since on their collaboration is dependent among others, 2 major properties of myocardial cell; a) the Ca2+ buffer and transport and hence contraction – relaxation coupling and b) the adequate oxidative phosphorylation that cover the myocardial energetic needs, Figure 2 and Figure 3. In this respect, various cardiovascular diseases are promoted by mitochondrial -endoplasmic reticular dysfunctional interconnection as this is shown in cardiac hypertrophy [73], heart failure, cardiomyopathy [74], ischemic heart disease [75], arrhythmogenesis [76] etc.
Calcium is the ion that regulates mitochondrial redox and energy production and during cardiomyocyte contraction, its propagation is observed from endoplasmic reticulum and cytoplasmic towards mitochondria in order to stimulate functions that are necessary for the maintenance of cardiomyocyte bioenergy [59]. However, an incontrollable Ca2+ accumulation has detrimental effect on mitochondria, provoking mitochondrial dysfunction and ultimately loss of cell homeostatic capacity, which can lead to the activation of mitochondrial apoptotic pathway, accentuation of inflammatory procedure, etc. and hence to the beginning and heart failure progress, Figure 3. Interestingly, mitochondrial dysfunction occurs also in those patients with renal insufficiency, insulin resistance etc., co-morbidities that very often coexist in heart failure patients, indicating the main role of mitochondria regarding global human homeostasis and disease progression [77].
3.1.1. Mitochondria and Quadruple Therapy in Heart Failure Patients
The pathophysiological base of heart failure syndrome of any cause and hence its therapeutic approach lies on the treatment of renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system hyper-activation. This hyper-activation leads to a metabolic disequilibrium, to a large amount of free radical production, and to the activation of the inflammatory process. Accordingly, the recommended therapy for heart failure try to block this hyperactivity, in order to reduce the myocardial oxygen consumption, to reprogram the altered metabolic remodeling and to ameliorate the energetic needs [1,78,79,80,81]. Needs that are closely related to mitochondrial normal function from which the entire myocardial ‘building’ is dependent on. Indeed, beta-blocker therapy reduce sympathetic activity, decrease serum catecholamines level, inhibit mitochondrial fatty acid uptake and increase glucose oxidation trying to compensate the energetic needs of a ‘starving’ heart [82]. While together with the use of RAS blockers reduce free radical production, and inflammation [1,83]. That’s why in patients with heart failure the use of these drugs shows substantial improvement since both drugs, among other beneficial effects, ameliorate the metabolic remodeling and thus the energetic status of myocardial cell [84]. Accordingly, SGLT2 affect metabolic and mitochondrial action [85,86,87] improve mitochondrial energetics and hence myocardial fuel needs [88]. However, even when the recommended for heart failure quadruple therapy is applied the morbidity and mortality rate remain high. It seems therefore that something is missing. Consequently, the scientific research has to try to better understand the metabolic/energetic status of the myocardial cells focused probably on mitochondria-ER homeostasis; interconnection and function.
3.2. Unusual Location of Mitochondria
Another issue that needs further investigation is the discovery of circulating in blood cell-free mitochondria and mitochondrial DNA, released from various cells triggered by stress, injury or disease [89]. Although there are controversial reports it seems that cell-free mitochondria are not energetic active [90]. Their presence in blood raises several questions that have to be addressed and further studies are required. For example, what is the meaning of their presence in healthy and in diseased individuals? Which is their cell-source of origin? Can they serve as therapeutic targets? Their presence in blood represents a non-self-recognized substances and if it is so what is the protective reaction of any single cell and tissue to diminish harmful effects? Interestingly, high levels of circulating cell-free-mtDNA have been found in several clinical scenarios including diabetes, cancer and myocardial infarction, and have been suggested as potential prognostic biomarker [27,91]. Of note, they are not related to any specific cardiovascular disease and are related to cell necrosis, acute respiratory distress syndrome, tumors, and inflammation, of any cause [92,93]. In any case this is an open issue that has to be searched in order to reach the entelechy.
4. Identify Mitochondria Dysfunction: Imaging Techniques and Biomarkers
Although mitochondria are the main player in several cardiac diseases their detection through imaging techniques and/or blood sample analysis are not well known and established. Moreover, some of them do not represent the optimum identifier, are very expensive and therefore not used in every day clinical practice. Additionally, regardless of the used technique it must be taken into account that from the calculated consumed energy only 25% is used for mechanical purposes whereas the remaining is used for non-mechanical actions; metabolism, heat production [94,95] etc. Regarding imaging techniques there are two ways, the invasive and the non-invasive technique, to calculate indirectly the capability of mitochondrial to produce the necessary energy. Using the invasive technique the input energy is measured as the coronary sinus blood flow times the arteriovenous oxygen content difference, whereas the output energy can be calculated using the pressure-volume loop. The non-invasive techniques are represented by positron emission tomography (PET), cardiovascular magnetic resonance spectroscopy [96] and identification of metabolic disturbances in plasma [97].
Regarding PET, carbon-11–labeled acetate (11C-acetate) and oxygen-15–labeled molecular oxygen (15O2) tracers have been used [95], showing however several drawbacks and therefore with limited application. Phosphorus (31P) magnetic resonance spectroscopy (MRS) can measure endogenous cardiac high-energy phosphate metabolites, creatine kinase (CK) flux [98,99,100] etc. showing the mitochondrial energetic capacity [101,102,103]. In heart failure patients’ mitochondria function is failed, biochemical sequence is altered and hence abnormal substances are utilized. This abnormality can be detected by using metabolomics; however, it is not clear the source of their production making thus this technique less accurate [104,105]. Accordingly, cardiovascular magnetic resonance spectroscopy seems to be a promising technique however due to inherent problems this technique is of limited use [106]. Several other biomarkers have been used but none of them has the potentiality to recognize and give a powerful information regarding mitochondrial function. In this respect, lactate, pyruvate, and lactate: pyruvate ratio, creatine phosphokinase etc. have been used but all of them demonstrate low specificity and sensitivity and have limited power to recognize mitochondrial deficiency. Additionally, the newer proposed biomarkers, growth differentiation factor 15 (GDF-15) and fibroblast growth factor 21 (FGF-21) although of interest show limited diagnostic power [94].
5. Strategies to Keep Mitochondrial Structural and Functional Integrity
Two strategies exist in order to protect mitochondrial integrity; the non-pharmacological and the pharmacological approach. The non-pharmacological consist on the exercise training and on the life style habits. Regarding exercise training although there are not yet conclusive results [23], it is suggested that proper exercise promote changes on mitochondrial function and metabolism [107,108] on mitochondrial fusion and fission protein activity [109] showing a cardioprotective effect [110,111]. Notably, few days of endurance exercise training are enough for mitochondrial protection against ischemic reperfusion injury [112]. Accordingly, although not totally proved, the life style habits and more specifically calorie restriction has been proposed to improve cardiac dysfunction by controlling better cardiac fibrosis, inflammation and mitochondrial defensive mechanisms [113,114].
Regarding pharmacological intervention, the proposed as the optimal medical treatment for patients with heart failure, known as quadruple therapy, contains energetic saving mechanisms for the restoration of mitochondrial function. However regardless of their use the mortality and morbidity rate remain high. Therefore, new medicines are proposed in order to find the right path, in a way to complete the real optimal medical treatment. Thus metabolism (fatty acid, glucose) antioxidants regulators etc. have been proposed [115,116]. Indeed, the control of peroxisome activated receptor-α agonists and L-Carnitine may improve left ventricular function and prevent myocardial fibrosis [117,118]. Accordingly, SGLT2 restore the use of biochemical substances (Fatty acids oxidation/glycolysis) and improve mitochondrial energetic status [119]. Also, metformin, thiazolidinediones, and statins via indirect AMPK activation, stimulate mitochondrial biogenesis [120]. The use of sacubitril/valsartan, cause an increase in natriuretic peptides, mainly αANP [121], restore internal mitochondrial membrane / outer mitochondrial membrane (IMM/OMM) ratio, decrease ROS levels, reinforce autophagy, thus expressing cardioprotective mechanism. The use of antioxidant drugs although an attractive thought, show controversial results. More specifically, the use of Coenzyme Q10 in a study showed an improvement in ejection fraction [122] whereas in another study did not [123]. Mitochondrial pyruvate carrier [124] 1-methyl-4phenyl-1,2,3,6-tetrahydropyridine (mPTP), for example cyclosporine A [125], as well other pharmacological intervention [126,127,128,129,130] have been used but none of them showed robust and conclusive results. There are studies tried to intervene on mitochondrial fusion and fission [131,132,133,134,135,136] mechanism showing an improvement of mitochondrial function whereas others did not [137,138,139,140,141,142]. Thus, based on the previous mentioned knowledge, the need for further investigation is clearly necessary.
6. Future Directions
The interest of the scientific community for mitochondria structure and function is exponential. However, till now there are not enough and robust data that can serve for the identification of malfunctioned ones. Regardless of the progress on this topic many unanswered questions are still on the table and require further elucidation. The diversity of mitochondrial phenotype may guide the determination of different diseases including the cardiovascular diseases [143]. The alteration of their side of action may indicate their different energetic status and probably the beginning of a disease. The reported multi-scale mitochondrial configuration found in different cell types is not clear and may represent a step forward [144]. Moreover, little is known about depletion or alteration of mtRNA that might affect among others the defensive mechanisms [145]. Moreover, not only mrRNA may alter the mitochondrial defensive status but also any harmful event that alter these mechanisms [146]. Again, do MAM’s play a preventive role? Do our everyday habits affect MAM’s and promote cardiovascular diseases [147]? In any case our knowledge remains limited and further effort must be made in order to better understand our factory of energy, especially regarding myocardial mitochondrial cell. Can artificial intelligence can help? This is not known yet and it remains to be answered [148].
7. Conclusions
Through time, regarding heart failure syndrome, an enormous progress is reported. A principal player in heart failure syndrome among others is mitochondria. This is an organelle of huge importance being affected by risk factors, comorbidities, etc. through multifactorial mechanisms leading to the deterioration of this syndrome. Their malfunction demonstrates a bioenergetics decline, a reduced production of energy, ion transport alteration, free radical, misfolded protein production, etc. This malfunction along with the neuro-humoral hyper-activation, leads to the homeostatic mechanisms failure and ultimately drive the whole process to the worse scenario with very severe consequences. In this respect, although many progresses have been made regarding the role of mitochondria in heart failure, there is a need for further investigation in order to understand better the role of this organelle thus finding out hopefully more effective management. The role of mitochondria as a therapeutic target in patients with heart failure is emerged [11], in a way to protect the fort not to fell.
Author Contributions
Conceptualization, I.P; methodology, IP, CK; DF; E.T; investigation, DF; CK; IP; writing—review and editing, all authors; supervision, I.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Brown, DA.; Perry, JB.; Allen, ME.; Sabbah, HN.; Stauffer, BL; Shaikh, SR.; Cleland, JGF.; Colucci, WS.; Butler, J.; Voors, AA.; et al. Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol 2017;14:238-250.
- Balaban, R. S. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J. Mol. Cell. Cardiol.2022; 34: 1259–1271.
- Neubauer, S. The Failing Heart — an Engine Out of Fuel. N Engl J Med 2007;356:1140-51.
- Beer, M.; Seyfarth, T.; Sandstede, J.; Landschutz, W.; Lipke, C.; Kostler, H.; von Kienlin, M.; Harre, K.; Hahn, D.; Neubauer, S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with 31P SLOOP magnetic resonance spectroscopy. J. Am. Coll. Cardiol.2002; 40, 1267–1274.
- Phan, T. T.; Abozguia, K.; Shivu, GN.; Mahadevan, G.; Ahmed, I.; Williams, L.; Dwivedi ,G.; Patel, K.; Steendijk, P.; Ashrafian, H.; et al. Heart failure with preserved ejection fraction is characterized by dynamic impairment of active relaxation and contraction of the left ventricle on exercise and associated with myocardial energy deficiency. J. Am. Coll. Cardiol.2009; 54, 402–409.
- Torrent-Guasp, F.; Kocica, MJ. , Corno, A., Komeda, M., Cox, J., Flotats, A., Ballester-Rodes, M., Carreras-Costa, F. Systolic ventricular filling. European Journal of Cardio-thoracic Surgery. 2004; 25:376–386.
- Lopez-Crisosto, C. , Pennanen, Ch.; Vasquez-Trincado, C.; Morales, PE.; Bravo-Sagua, R.; Quest, AFG.; Chiong, M.; Lavandero, S. Sarcoplasmic reticulum–mitochondria communication in cardiovascular pathophysiology Nat Rev Cardiol. 2017;14: 342-360.
- Dorn, GW. Mitochondrial dynamism and heart disease: changing shape and shaping change. EMBO Mol Med 2015; 7: 865–877.
- Schon E., A.; Di Mauro, S.; Hirano, M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet. 2012; 13, 878–890.
- Dorn II, GW. Mitochondrial dynamism and heart disease: changing shape and shaping change. EMBO Mol Med 2015; 7: 865–877.
- Gallo, G.; Rubattu, S.; Volpe, M. Mitochondrial Dysfunction in Heart Failure: From Pathophysiological Mechanisms to Therapeutic Opportunities. Int. J. Mol. Sci. 2024; 25:2667.
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018; 128:3716–3726.
- Chaanine, AH.; LeJemte, ThH.; Delafontaine, P. Mitochondrial Pathobiology and Metabolic Remodeling in Progression to Overt Systolic Heart Failure J. Clin. Med. 2020; 9: 3582.
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial morphology, dynamics, and function in human pressure overload or ischemic heart disease with preserved or reduced ejection fraction. Circ. Heart Fail. 2019; 12: e005131.
- Brustovetsky, N.; Brustovetsky, T.; Jemmerson, R.; Dubinsky, J.M. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J. Neurochem. 2002; 80: 207–218.
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Circ. Physiol. 2016; 311: H1540–H1559.
- Chaanine, A.H. Morphological stages of mitochondrial vacuolar degeneration in phenylephrine-stressed cardiac myocytes and in animal models and human heart failure. Medicina 2019; 55: 239.
- Sun, M.G.; Williams, J.; Munoz-Pinedo, C.; Perkins, G.A.; Brown, J.M.; Ellisman, M.H.; Green, D.R.; Frey, T.G. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat. Cell Biol. 2007; 9: 1057–1065.
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in cardiovascular diseases. J. Clin. Med. 2020; 9: 892.
- Chaanine, A.H. Autophagy and myocardial remodeling. J. Am. Coll. Cardiol. 2018; 71: 2011–2014.
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes and oxidative stress in aging and apoptosis. Biochim. Biophys. Acta (BBA) Gen. Subj. 2008; 1780: 1291–1303.
- Terman, A.; Kurz, T.; Gustafsson, B.; Brunk, U.T. The involvement of lysosomes in myocardial aging and disease. Curr. Cardiol. Rev. 2008; 4: 107–115.
- Hernandez-Resendiz, S.; Prakash, A.; Loo, SL; Semenzato, M.; Chinda, K.; Crespo-Avilan, GE.; Dam, LC.; Lu, S.; Scorrano, L.; Hausenloy DH. Targeting mitochondrial shape: at the heart of cardioprotection. Basic Research in Cardiology 2023; 118:49.
- Jenkins, BC.; Neikirk, K.; Katti, P.; Claypoo, SM.; Kirabo, A.; McReynolds, MR.; Hinton, A., Jr. Mitochondria in disease: changes in shapes and dynamics. Trends Biochem Sci. 2024; 49: 346–360.
- Lobo-Jarne, T.; Ugalde, C. Respiratory chain supercomplexes: Structures, function and biogenesis. Semin. Cell Dev. Biol. 2017; 76: 179–190.
- Nesci S.; Trombetti F.; Pagliarani A.; Ventrella V;Algieri C.; Tioli G.; et al. Molecular and supramolecular structure of the mitochondrial oxidative phosphorylation system: Implications for pathology. Life 2021; 15:242.
- Valenti, D.; Vacca, R.A.; Moro, L.; Atlante, A. Mitochondria Can Cross Cell Boundaries: An Overview of the Biological Relevance, Pathophysiological Implications and Therapeutic Perspectives of Intercellular Mitochondrial Transfer. Int. J. Mol. Sci. 2021; 22: 8312.
- Ong, SB.; Hausenloy DJ. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res 2010; 88: 16–29.
- Shanmughapriya, S.; Langford, D.; Natarajaseenivasan, K. Inter and Intracellular mitochondrial trafficking in health and disease. Ageing Res. Rev. 2020; 62: 101128.
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; Wani, M.R.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014; 33: 994–1010.
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017; 114: E9066–E9075.
- Phinney, D.; Di Giuseppe, M.; Njah, J.; Sala-Llinas, E.; Shiva, S.; Croix, C.M.S.; Stolz, D.B.; Watkins, S.; Di, Y.P.; Leikauf, G.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 2015; 6: 8472.
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Extracellular vesicles: Masters of intercellular communication and potential clinical interventions. J. Clin. Investig. 2016; 126: 1139–1143.
- Paolicelli, R.C.; Bergamini, G.; Rajendran, L. Cell-to-cell Communication by Extracellular Vesicles: Focus on Microglia. Neuroscience 2019; 405: 148–157.
- Yao, Y.; Fan, X.-L.; Jiang, D.; Zhang, Y.; Li, X.; Xu, Z.-B.; Fang, S.-B.; Chiu, S.; Tse, H.-F.; Lian, Q.; et al. Connexin 43-mediated mitochondrial transfer of iPSC-MSCs alleviates asthma inflammation. Stem Cell Rep. 2018; 11: 1120–1135.
- Lyamzaev, K.G.; Nepryakhina, O.K.; Saprunova, V.B.; Bakeeva, L.E.; Pletjushkina, O.Y.; Chernyak, B.; Skulachev, V.P. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): Formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim. Biophys. Acta (BBA) Bioenerg. 2008; 1777:, 817–825.
- Shami, GJ.; Cheng, D.; Verhaegh, Koek G. Three-dimensional ultrastructure of giant mitochondria in human nonalcoholic fatty liver disease. Sci. Rep 2021;11:3319.
- Vincent, AE.; White, K.; Daney, T.; Philips, J.; Ogden, RT.; Lawless, C.; Warren, C.; Hall, MG.; Ng, YS.; Falkous, G.; et al. (2019) Quantitative 3D mapping of the human skeletal muscle mitochondrial network. Cell Rep. 2019; 26: 996–1009.e4.
- Glancy, B.; Kim, Y.; Katti, P.; Willingham, TB. The functional impact of mitochondrial structure across subcellular scales. Front. Physiol 2020;11, 541040.
- Lewis, SC.; Uchiyama, SC.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016; 353: aaf5549.
- Bravo-Sagua, R.; Torrealba, N.; Paredes, F.; Morales, PE.; Pennanen, C.; López-Crisosto, C.; Troncoso, R.; Criollo, A.; Chiong, M.; Hill, JA.; et al. Organelle communication: Signaling crossroads between homeostasis and disease. The International Journal of Biochemistry & Cell Biology2014; 50:55-59v.
- Szymanski, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M,; Ziółkowski, W.; Duszynski, J.; Pinton, P.; Dobrzyn, A.; Wieckowski, MR. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017; 18: 1576.
- Murley, A.; Nunnari, J. The emerging network of mitochondria-organelle contacts. Mol Cell. 2016; 61: 648–653.
- Picca, A.; Guerra, F.; Calvani, R.; Romano, R.; Coelho-Junior, H.J.; Damiano, F.P.; Bucci, C.; Marzetti, E. Circulating Mitochondrial DNA and Inter-Organelle Contact Sites in Aging and Associated Conditions. Cells 2022; 11: 675.
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018; 554: 382–386.
- Mattiazzi Ušaj, M.; Brložnik, M.; Kaferle, P.; Žitnik, M.; Wolinski, H.; Leitner, F.; Kohlwein, S.D.; Zupan, B.; Petrovič, U. Genome-wide localization study of yeast Pex11 identifies peroxisome-mitochondria interactions through the ERMES complex. J. Mol. Biol.2015; 427: 2072–2087.
- Benador, I.Y.; Veliova, M.; Liesa, M.; Shirihai, O.S. Mitochondria bound to lipid droplets: Where mitochondrial dynamics regulate lipid storage and utilization. Cell Metab. 2019; 29: 827–835.
- Ren, J.; B,i Y.; Sowers, J.;, Hetz,.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol. 2021; 18: 499-521.
- Zhou, H.; Wang, S.; Hu, S.; Chen, Y.; Ren, J. ER mitochondria microdomains in cardiac ischemia-reperfusion injury: a fresh perspective. Front. Physiol. 2018a; 9:755.
- Gao, P.; Yan, Z.; Zhu, Z. Mitochondria-associated endoplasmic reticulum membranes in cardiovascular diseases. Front. Cell Dev. Biol. 2020; 8: 604240.
- Bayeva, M.; Sawicki, K. T.; Butler, J.; Gheorghiade, M.; Ardehali, H. Molecular and cellular basis of viable dysfunctional myocardium. Circ. Heart Fail. 2014; 7: 680–691.
- Chen, Q.; Thompson, J.; Hu, Y.; Lesnefsky, E. J. Chronic metformin treatment decreases cardiac injury during ischemia-reperfusion by attenuating endoplasmic reticulum stress with improved mitochondrial function. Aging 2021;13:7828–7845.
- Grings, M.; Seminotti, B.; Karunanidhi, A.; Ghaloul-Gonzalez, L; Mohsen, A.; Wipf, P.; Palmfeldt, J.; Vockley, J.; Leipnitz, G. ETHE1 and MOCS1 deficiencies: disruption of mitochondrial bioenergetics, dynamics, redox homeostasis and endoplasmic reticulum-mitochondria crosstalk in patient fibroblasts. Sci. Rep. 2019; 9:12651.
- Bagur, R.; Hajnóczky, G. Intracellular Ca sensing: its role in calcium homeostasis and signaling. Mol. Cell 217;66: 780–788.
- Biczo, G.; Vegh, E.; Shalbueva, N.; Mareninova, O.; Elperin, J.; Lotshaw, E. ; Gretler, S,; Lugea, A.; Malla, SR.; Dawsonet, D.; et al. Mitochondrial dysfunction, through impaired autophagy, leads to endoplasmic reticulum stress, deregulated lipid metabolism, and pancreatitis in animal models. Gastroenterology 2018; 154: 689–703.
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: Mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ.Heart Fail. 2013, 6, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Chen, W.; Hua, Y.; Jia, H.; Song, Y.; Wang, Y. Aerobic exercise ameliorates cardiac hypertrophy by regulatingmitochondrial quality control and endoplasmic reticulum stress through M2AChR. J. Cell. Physiol. 2021, 236, 6581–6596. [Google Scholar] [CrossRef] [PubMed]
- Paraskevaidis, I.; Farmakis, D.; Papingiotis, G.; Tsougos, E. Inflammation and Heart Failure: Searching for the Enemy—Reaching the Entelechy. J. Cardiovasc. Dev. Dis. 2023, 10, 19. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Wen, Y.; Li, S.; Lu, X.; Xu, R.; Li, C. Endoplasmic Reticulum-Mitochondria Contacts: A Potential Therapy Target for Cardiovascular, Remodeling-Associated Diseases. Front. Cell Dev. Biol. 2021;9:774989.
- Shi, Y.; Liu, L.; Deng, C.; Zhao, T.; Shi, Z.; Yan, J.; Gong, YZ.; Liao, DF.; Qin, L. Celastrol ameliorates vascular neointimal hyperplasia through Wnt5a-involved autophagy. Int. J. Biol. Sci. 2021;17, 2561–2575.
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New insights into the role of mTOR signaling in the cardiovascular system. Circ. Res. 2018a, 122, 489–505. [Google Scholar] [CrossRef]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The role of autophagy in the heart. Annu. Rev. Physiol. 2018b; 80, 1–26.
- Kimball, T.; Vondriska, T. Metabolism, epigenetics, and causal inference in heart failure. Trends Endocrinol. Metab. 2020;31, 181–191.
- Lebeau, J.; Saunders, J.; Moraes, V.; Madhavan, A.; Madrazo, N.; Anthony, M.; Wiseman, RL. The PERK arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Rep. 2018;22: 2827–2836.
- Akhmedov, A.; Rybin, V.; Marín-García, J. Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart Fail. Rev. 2015;20: 227–249.
- Nunnari, J.; Suomalainen, A. Mitochondria: in sickness and in health. Cell 2012;148: 1145–1159.
- Reddish, F. N.; Miller, C. L.; Gorkhali, R.; Yang, J. J. Calcium dynamics mediated by the endoplasmic/sarcoplasmic reticulum and related diseases. Int.J. Mol. Sci. 2017;18:1024.
- Lee, S.; Min, K. The interface between ER and mitochondria: molecular compositions and functions. Mol. Cells 2018;41:1000–1007.
- Ruan, L.; Wang, Y.; Zhang, X.; Tomaszewski, A.; McNamara, J.; Li, R. Mitochondria-associated proteostasis. Annu. Rev. Biophys. 2020; 49, 41–67.
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the trip: calcium in mitochondria back and forth. Annu. Rev. Biochem. 2016;85, 161–192.
- Raffaello, A; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci. 2016;41:1035–1049.
- Yousuf, M.S.; Maguire, A.D.; Simmen, T.; Kerr, B.J. Endoplasmic reticulum-mitochondria interplay in chronic pain: the calcium connection. Mol. Pain 2020; 16:1744806920946889.
- Tuncay, E.; Bitirim, C. V.; Olgar, Y.; Durak, A.; Rutter, G. A.; Turan, B. Zn(2+)-transporters ZIP7 and ZnT7 play important role in progression of cardiac dysfunction via affecting sarco(endo)plasmic reticulum-mitochondria coupling in yperglycemic cardiomyocytes. Mitochondrion 2019;44, 41–52.
- Liu, X.; Kwak, D.; Lu, Z.; Xu, X.; Fassett, J.; Wang, H.; et al. Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension 2014; 64, 738–744.
- Li, Y.; Wang, X.; Lou, C. Gastrodin pretreatment impact on sarcoplasmic reticulum calcium transport ATPase (SERCA) and calcium phosphate (PLB) expression in rats with myocardial ischemia reperfusion. Med. Sci. Monit. 2016;22, 3309–3315.
- Silva-Palacios, A.; Zazueta, C.; Pedraza-Chaverri, J. ER membranes associated with mitochondria: possible therapeutic targets in heartassociated diseases. Pharmacol. Res. 202;156:104758.
- Brown, DA.; Perry, JB.; Allen, ME.; Sabbah, HN.; Stauffer, BL.; Shaikh, SR.; Cleland, JGF.; Colucci, WS.; Butler, J.; Voors, AA.; et al. Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2017;14(4):238-250.
- Safari, F.; Bayat, G.; Shekarforoush, S.; Hekmatimoghaddam, S.; Anvari, Z.; Moghadam, M.F.; Hajizadeh, S. Expressional profile of cardiac uncoupling protein-2 following myocardial ischemia reperfusion in losartan- and ramiprilat-treated rats. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2014, 15, 209–217. [Google Scholar] [CrossRef]
- Kojic, Z.; Gopcevic, K.; Marinkovic, D.; Tasic, G. Effect of captopril on serum lipid levels and cardiac mitochondrial oxygen consumption in experimentally-induced hypercholesterolemia in rabbits. Physiol. Res. 2011;60 (Suppl. 1): S177–S184.
- Djanani, A.; Kaneider, N.C.; Meierhofer, C.; Sturn, D.; Dunzendorfer, S.; Allmeier, H.; Wiedermann, C.J. Inhibition of neutrophil migration and oxygen free radical release by metipranolol and timolol. Pharmacology 2003, 68, 198–203. [Google Scholar] [CrossRef]
- Betiu, A.M.; Noveanu, L.; Hâncu, I.M.; Lascu, A.; Petrescu, L.; Maack, C.; Elmér, E.; Muntean, D.M. Mitochondrial Effects of Common Cardiovascular Medications: The Good, the Bad and the Mixed. Int. J. Mol. Sci. 2022; 23:13653.
- Igarashi, N.; Fujii, N.; Suzuki, T.; Matsuki, A.; Nakadase, T.; Igawa, A.; Inoue, H. Influence of β-adrenoceptor blockade on the myocardial accumulation of fatty acid tracer and its intracellular metabolism in the heart after ischemia− reperfusion injury. Circ. J. 2006;70, 1509–1514.
- Feldman, D. S.; Carnes, C. A.; Abraham, W. T.; Bristow, M. R. Mechanism s of disease: β-adrenergic receptors — alterations in signal transduction and pharmacogenomics in heart failure. Nat. Clin. Pract. Cardiovasc. Med. 2005; 2, 475–483.
- Kourek, C.; Briasoulis, A.; Papamichail, A.; Xanthopoulos, A.; Tsougos, E.; Farmakis, D.; Paraskevaidis, I. Beyond Quadruple Therapy and Current Therapeutic Strategies in Heart Failure with Reduced Ejection Fraction: Medical Therapies with Potential to Become Part of the Therapeutic Armamentarium. Int. J. Mol. Sci. 2024, 25, 3113. [Google Scholar] [CrossRef]
- Voorrips, S.N.; Saucedo-Orozco, H.; Sánchez-Aguilera, P.I.; De Boer, R.A.; Van der Meer, P.; Westenbrink, B.D. Could SGLT2 Inhibitors Improve Exercise Intolerance in Chronic Heart Failure? Int. J. Mol. Sci. 2022; 23: 8631.
- Choi, J; Matoba, N.; Setoyama, D.; Watanabe, D.; Ohnishi, Y.; Yasui, R.; Kitai, Y.; Oomachi, A.; Kotobuki, Y.; Nishiya, Y.; et al. The SGLT2 inhibitor empagliflozin improves cardiac energy status via mitochondrial ATP production in diabetic mice. Commun. Biol 2023; 17;6(1):278.
- Cai, W.; Chong, K.; Huang, Y; Huang, C.; Yin, L. Empagliflozin improves mitochondrial dysfunction in diabetic cardiomyopathy by modulating ketone body metabolism and oxidative stress. Redox Biology 2024;69: 103010.
- Martens, P.; Mathieu, C.; Verbrugge, F.H. Promise of SGLT2 Inhibitors in Heart Failure: Diabetes and Beyond. Curr. Treat. Options Cardiovasc. Med. 2017; 19: 23.
- Miliotis, S.; Nicolalde, B.; Ortega, M.; Yepez, J.; Caicedo, A. Forms of extracellular mitochondria and their impact in health. Mitochondrion 2019, 48, 16–30. [Google Scholar] [CrossRef]
- Stier, A. Human blood contains circulating cell-free mitochondria, but are they really functional? Am. J. Physiol. Metab. 2021, 320, E859–E863. [Google Scholar] [CrossRef]
- Lindqvist, D.; Wolkowitz, O.M.; Picard, M.; Ohlsson, L.; Bersani, F.S.; Fernström, J.; Westrin, Å.; Hough, C.M.; Lin, J.; Reus, V.I.; et al. Circulating cell-free mitochondrial DNA, but not leukocyte mitochondrial DNA copy number, is elevated in major depressive disorder. Neuropsychopharmacology 2018, 43, 1557–1564. [Google Scholar] [CrossRef]
- McClintock, CR.; Mulholland, N.; Krasnodembskaya, AD. Biomarkers of mitochondrial dysfunction in acute respiratory distress syndrome: a systematic review and meta-analysis. Front Med (Lausanne) 2022;9:1.
- Rahat, B.; Ali, T.; Sapehia, D.; Mahajan, A.; Kaur, J. Circulating cell-free nucleic acids as epigenetic biomarkers in precision medicine. Front Genet 2020;11:844.
- Shayota, BJ. Biomarkers of mitochondrial disorders Neurotherapeutics 2024; 21: e00325.
- Knaapen, P.; Germans, T.; Knuuti, J.; Paulus, WJ.; Dijkmans, PA.; Allaart, CP.; Lammertsma, AA.; Visser, FC. Myocardial Energetics and Efficiency Current Status of the Noninvasive Approach. Circulation. 2007;115:918-927.
- Gabr, R. ;, El-Sharkawy, AMM.; Schär, M.; Panjrath, GS.; Gerstenblith, G.; Weiss, RG.; Bottomley PA. Cardiac work is related to creatine kinase energy supply in human heart failure: a cardiovascular magnetic resonance spectroscopy study. J Cardiovascular Magnetic Resonance 2018; 10;20(1):81.
- Cheng, ML.; Wang, CH.; Shiao, MS.; Liu, MH.; Huang, YY.; Huang, CY.; Mao, CT.; Lin, JF.; Ho, HY.; Yang, NI. Metabolic Disturbances Identified in Plasma Are Associated With Outcomes in Patients With Heart Failure. Diagnostic and Prognostic Value of Metabolomics. J Am Coll Cardiol 2015;65:1509–2084.
- Weiss, RG,; Gerstenblith, G.; Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci U S A. 2005;102:808–13.
- Tsampasian, V.; Cameron, D.; Sobhan, R.; Bazoukis, G.; Vassiliou, V.S. Phosphorus Magnetic Resonance Spectroscopy (31P MRS) and Cardiovascular Disease: The Importance of Energy. Medicina 2023, 59, 174. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, PA.; Wu, KC.; Gerstenblith.; G, Schulman, SP.; Steinberg, A.; Weiss RG. Reduced myocardial creatine kinase flux in human myocardial infarction: an in vivo phosphorus magnetic resonance spectroscopy study. Circulation. 2009;119:1918–24.
- Lamb, HJ.; Beyerbacht, HP.; van der Laarse, A.; Stoel, BC.; Doornbos, J.; van der Wall, EE.; de Roos, A. Diastolic dysfunction in hypertensive heart disease is associated with altered myocardial metabolism. Circulation 1999;99:2261–7.
- Samuel, TJ.; Lai, S.; Schär, M.; Wu, KC.; Steinberg, AM. ; Wei, A-C.; Anderson ME.; Tomaselli, GF.; Gerstenblith, G.; et al. Myocardial ATP depletion detected noninvasively predicts sudden cardiac death risk in patients with heart failure. JCI Insight 2022;7:e157557.
- Burrage, M.K.; Hundertmark, M.; Valkovic, L.; Watson, W.D.; Rayner, J.; Sabharwal, N.; Ferreira, V.M.; Neubauer, S.; Miller, J.J.; Rider, O.J.; et al. Energetic Basis for Exercise-Induced Pulmonary Congestion in Heart Failure With Preserved Ejection Fraction. Circulation 2021; 144: 1664–1678.
- Rame, JE. Chronic heart failure: a reversible metabolic syndrome? Circulation 2012;125:2809–11.
- Maekawa, K.; Hirayama, A.; Iwata, Y.; Tajima, Y.; Nishimaki-Mogami, Y.; Sugawara, S.; Ueno, N.; Abe, H.; Ishikawa, M.; Murayama, M.; et al. Global metabolomic analysis of heart tissue in a hamster model for dilated cardiomyopathy. J Mol Cell Cardiol 2013;59:76–85.
- Bakermans, AJ.; Bazil, JN.; Nederveen, AJ.; Strijkers, G.; Boekholdt, SM.; Beard, DA.; Jeneson, JAL. Human cardiac 31P-MR spectroscopy at 3 Tesla cannot detect failing myocardial energy homeostasis during exercise. Front Physiol 2017;8:93.
- Powers SK, Smuder AJ, Kavazis AN, Quindry JC. Mechanisms of exercise-induced cardioprotection. Physiology (Bethesda) 2014;29:27–38.
- Kavazis, AN. Exercise preconditioning of the myocardium. Sports Med 2009;39:923–935.
- Ding, H.; Jiang, N.; Liu, H.; Liu, X.; Liu, D.; Zhao, F.; Wen, L.; Liu, S.; Ji, LL.; Zhang, Y. Response of mitochondrial fusion and fssion protein gene expression to exercise in rat skeletal muscle. Biochim Biophys Acta 2100; 1800:250–256.
- Fulghum, K.; Hill, BG. Metabolic mechanisms of exercise induced cardiac remodeling. Front Cardiovasc Med 2018;5:127.
- Huertas, JR.; Casuso, RA.; Agustin, PH.; Cogliati, S. Stay fit, stay young: mitochondria in movement: the role of exercise in the new mitochondrial paradigm. Oxid Med Cell Longev 2019:7058350.
- Lee, Y.; Min, K.; Talbert, E.E.; Kavazis, A.N.; Smuder, A.J.; Willis, W.T.; Powers, S.K. Exercise protects cardiac mitochondria against ischemia-reperfusion injury. Med Sci Sports Exerc 2012;44:397-405.
- Niemann, B.; Chen, Y.; Issa, H.; Silber, RE.; Rohrbach, S. Caloric restriction delays cardiac ageing in rats: role of mitochondria. Cardiovasc Res 2010;88:267–276.
- Niemann, B.; Li, L.; Simm, A.; Molenda, N.; Kockskamper, J.; Boening, A.; Rohrbach, S. Caloric restriction reduces sympathetic activity similar to beta-blockers but conveys additional mitochondrio-protective efects in aged myocardium. Sci Rep 2021;11:1931].
- Wu, C.; Zhang, Z.; Zhang, W.; Liu, X. Mitochondrial dysfunction and mitochondrial therapies in heart failure. Pharmacol. Res. 2022; 175, 106038.
- Gallo, G.; Rubattu, S.; Volpe, M. Mitochondrial Dysfunction in Heart Failure: From Pathophysiological Mechanisms to Therapeutic Opportunities. Int. J. Mol. Sci. 2024; 25, 2667.
- Ogata, T.; Miyauchi, T.; Sakai, S.; Takanashi, M.; Irukayama-Tomobe, Y.; Yamaguchi, I. Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptoralpha activator fenofibrate, partly by suppressing inflammatory responses associated with the nuclear factor-kappa-B pathway. J. Am. Coll. Cardiol. 2004, 43, 1481–1488. [Google Scholar] [PubMed]
- Zhao, G.; Zhang, H.; Wang, Y.; Gao, X.; Liu, H.; Liu, W. Effects of levocarnitine on cardiac function, urinary albumin, hs-CRP, BNP, and troponin in patients with coronary heart disease and heart failure. Hellenic J. Cardiol. 2020, 61, 99–102. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors: A State-of-the-Art Review. JACC Basic. Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef]
- Kumar, A.A.; Kelly, D.P.; Chirinos, J.A. Mitochondrial Dysfunction in Heart Failure With Preserved Ejection Fraction. Circulation 2019; 139, 1435–1450.
- Raffa, S.; Forte, M.; Gallo, G.; Ranieri, D.; Marchitti, S.; Magrì, D.; Testa, M.; Stanzione, R.; Bianchi, F.; Cotugno, M.; et al. Atrial natriuretic peptide stimulates autophagy/mitophagy and improves mitochondrial function in chronic heart failure. Cell Mol. Life Sci. 2023, 80, 134]. [Google Scholar] [CrossRef]
- Fotino, A.D.; Thompson-Paul, A.M.; Bazzano, L.A. Effect of coenzyme Q10 supplementation on heart failure: A meta-analysis. Am. J. Clin. Nutr. 2013; 97, 268–275.
- Madmani, M.E.; Yusuf Solaiman, A.; Tamr Agha, K.; Madmani, Y.; Shahrour, Y.; Essali, A.; Kadro, W. Coenzyme Q10 for heart failure. Cochrane Database Syst. Rev. 2014; 6, CD008684.
- Hodges, W.T.; Jarasvaraparn, C.; Ferguson, D.; Griffett, K.; Gill, L.E.; Chen, Y.; Ilagan, M.X.G.; Hegazy, L.; Elgendy, B.; Cho, K.; et al. Mitochondrial pyruvate carrier inhibitors improve metabolic parameters in diet-induced obese mice. J. Biol. Chem. 2022, 298, 101554. [Google Scholar] [CrossRef]
- Dai, W.; Shi, J.; Gupta, R.C.; Sabbah, H.N.; Hale, S.L.; Kloner, R.A. Bendavia, a mitochondria-targeting peptide, improves postinfarction cardiac function, prevents adverse left ventricular remodeling, and restores mitochondria-related gene expression in rats. J. Cardiovasc. Pharmacol. 2014, 64, 543–553. [Google Scholar] [CrossRef]
- Koene, S.; Spaans, E.; Van Bortel, L.; Van Lancker, G.; Delafontaine, B.; Badilini, F.; Beyrath, J.; Smeitink, J. KH176 under development for rare mitochondrial disease: A first in man randomized controlled clinical trial in healthy male volunteers. Orphanet J. Rare Dis. 2017, 12, 163. [Google Scholar] [CrossRef]
- Detaille, D.; Pasdois, P.; Sémont, A.; Dos Santos, P.; Diolez, P. An old medicine as a new drug to prevent mitochondrial complex I from producing oxygen radicals. PLoS ONE 2019, 14, e0216385. [Google Scholar] [CrossRef]
- Ordog, K.; Horvath, O.; Eros, K.; Bruszt, K.; Toth, S.; Kovacs, D.; Kalman, N.; Radnai, B.; Deres, L.; Gallyas, F., Jr.; et al. Mitochondrial protective effects of PARP-inhibition in hypertension-induced myocardial remodeling and in stressed cardiomyocytes. Life Sci. 2021, 268, 118936. [Google Scholar] [CrossRef]
- Abudureyimu, M.; Yu, W.; Cao, R.Y.; Zhang, Y.; Liu, H.; Zheng, H. Berberine Promotes Cardiac Function by Upregulating PINK1/Parkin-Mediated Mitophagy in Heart Failure. Front. Physiol. 2020, 11, 565751. [Google Scholar] [CrossRef]
- Hallakou-Bozec, S.; Vial, G.; Kergoat, M.; Fouqueray, P.; Bolze, S.; Borel, A.L.; Fontaine, E.; Moller, D.E. Mechanism of action of Imeglimin: A novel therapeutic agent for type 2 diabetes. Diabetes Obes. Metab. 2021, 23, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Kitsis, RN.; Fleischer, JA.; Gavathiotis, E.; Kornfeld, OS.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, SK.; et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016;540:74–79,.
- Hernandez-Resendiz, S.; Prunier, F.; Girao, H.; Dorn, G.; Hausenloy, DJ.; Action E-CC. Targeting mitochondrial fusion and fssion proteins for cardioprotection. J Cell Mol Med 2020;24:6571– 6585.
- Maneechote, C.; Palee, S.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, SC.; Chattipakorn, N. Pharmacological inhibition of mitochondrial fssion attenuates cardiac ischemia-reperfusion injury in pre-diabetic rats. Biochem Pharmacol 202;182:114295.
- Ong, SB.; Kalkhoran, SB.; Cabrera-Fuentes, HA.; Hausenloy, DJ. Mitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular disease. Eur J Pharmacol 2015;763:104–114.
- Ong, SB.; Kalkhoran, SB.; Hernandez-Resendiz, S.; Samangouei, P.; Ong, SG.; Hausenloy, DJ. Mitochondrial-shaping proteins in cardiac health and disease—the long and the short of it! Cardiovasc Drugs Ther 2017;31:87–107.
- Rocha, AG.; Franco, A.; Krezel, AM.; Rumsey, JM.; Alberti, JM.; Knight, WC.; Biris, N.; Zacharioudakis, E.; Janetka, JW.; Baloh, RH.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018;360:336–341.
- Cao, YL.; Meng, S.; Chen. Y.; Feng, JX.; Gu, DD.; Yu, B.; Li, YJ.; Yang, JY.; Liao, S.; Chan, DC.; Gao, S. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 2017;542:372–376.
- Giacomello, M. ; Scorrano L The INs and OUTs of mitofusins. J Cell Biol 2018;217:439–440.
- Maneechote, C.; Palee, S.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, SC.; Chattipakorn, N. Diferential temporal inhibition of mitochondrial fssion by Mdivi-1 exerts efective cardioprotection in cardiac ischemia/reperfusion injury. Clin Sci (Lond) 2018;132:1669–1683.
- Bordt, EA.; Clerc, P,.;Roelofs, BA.; Saladino, AJ.; Tretter, L.; AdamVizi, V.; Cherok, E,.;Khalil, A.; Yadava, N.; Ge, SX.; et al. The putative Drp1 inhibitor mdivi-1 Is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell 2017; 40(583–594):e586.
- Bordt, EA.; Zhang, N.; Waddell, J.; Polster, BM. The non-specifc Drp1 inhibitor Mdivi-1 has modest biochemical antioxidant activity. Antioxidants (Basel) 2022;24,11:450.
- Zhang, H.; Wang, P.; Bisetto, S.; Yoon, Y.; Chen, Q.; Sheu, SS.; Wang, W. A novel fssion-independent role of dynamin-related protein 1 in cardiac mitochondrial respiration. Cardiovasc Res 2017;113:160–170.
- Dalmasso, G.; Zapata, PAM.; Brady, NR.;Hamacher-Brady, A. Agent-based modeling of mitochondria links sub-cellular dynamics to cellular homeostasis and heterogeneity. PLoS One 2017;12, e0168198.
- Glancy, B.; Kim, Y.; Katti, P.; Willingham, TB. The Functional Impact of Mitochondrial Structure Across Subcellular Scales. Front. Physiol. 2020;11:541040.
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, NG. Mitochondrial dysfunction and mitochondrial dynamics - the cancer connection. Biochim. Biophys. Acta 2017;1858, 602–614.
- Kleele, T.; Rey, T.; Winter, J. ; Zaganelli, S,.;Mahecic, D.; Perreten Lambert, H.; Ruberto, FP.; Nemir, M,.;Wai, T.; Pedrazzini, T.; et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021;593, 435–439.
- Ding, Y.; Liu, N.; Zhang, D.; Guo, L.; Shang, Q.; Liu, Y.; Ren, G.; Ma, X. Mitochondria-associated endoplasmic reticulum membranes as a therapeutic target for cardiovascular diseases. Front. Pharmacol. 2024;15:1398381.
- Neikirk, K.; Lopez, EG.; Marshall, AG.; Alghanem, A.; Krystofiak, E.; Kula, B.; Smith, N.; Shao, J.; Katti, P.; Hinton, A., Jr. Call to action to properly utilize electron microscopy to measure organelles to monitor disease. Eur. J. Cell Biol 12023;02, 151365.
Figure 1.
Changes of mitochondrial function and structure throughout the course of heart failure syndrome. As heart failure progresses a vacuolar degeneration of mitochondria is present, with membrane increased permeability, changes of biochemical substrate use, increased of free radical production etc. The defensive mechanisms, as the syndrome progresses, although increased cannot balance the upcoming decompensation. The fort fell. p: preserved, r: reduced.
Figure 1.
Changes of mitochondrial function and structure throughout the course of heart failure syndrome. As heart failure progresses a vacuolar degeneration of mitochondria is present, with membrane increased permeability, changes of biochemical substrate use, increased of free radical production etc. The defensive mechanisms, as the syndrome progresses, although increased cannot balance the upcoming decompensation. The fort fell. p: preserved, r: reduced.
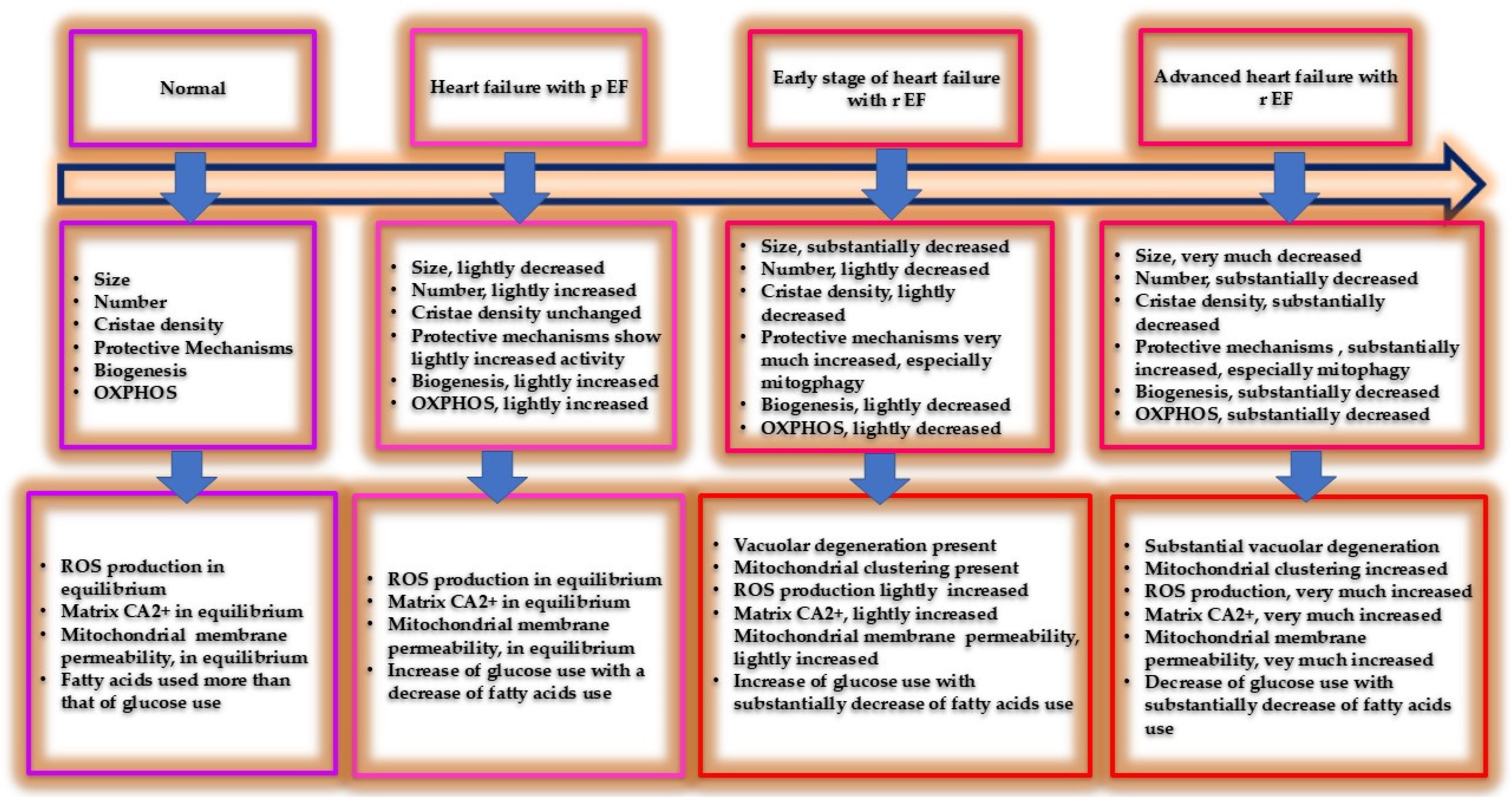
Figure 2.
The main goals of mitochondria are adenosine triphosphate (ATP) production and reactive oxygen species release (ROS). Both functions are regulated by shaping proteins that are controlled by fission and fusion systems both controlled by the relative proteins. These functioning systems are related to morphological changes (fission), and to mitophagy, apoptosis, and energy production process (fusion).
Figure 2.
The main goals of mitochondria are adenosine triphosphate (ATP) production and reactive oxygen species release (ROS). Both functions are regulated by shaping proteins that are controlled by fission and fusion systems both controlled by the relative proteins. These functioning systems are related to morphological changes (fission), and to mitophagy, apoptosis, and energy production process (fusion).
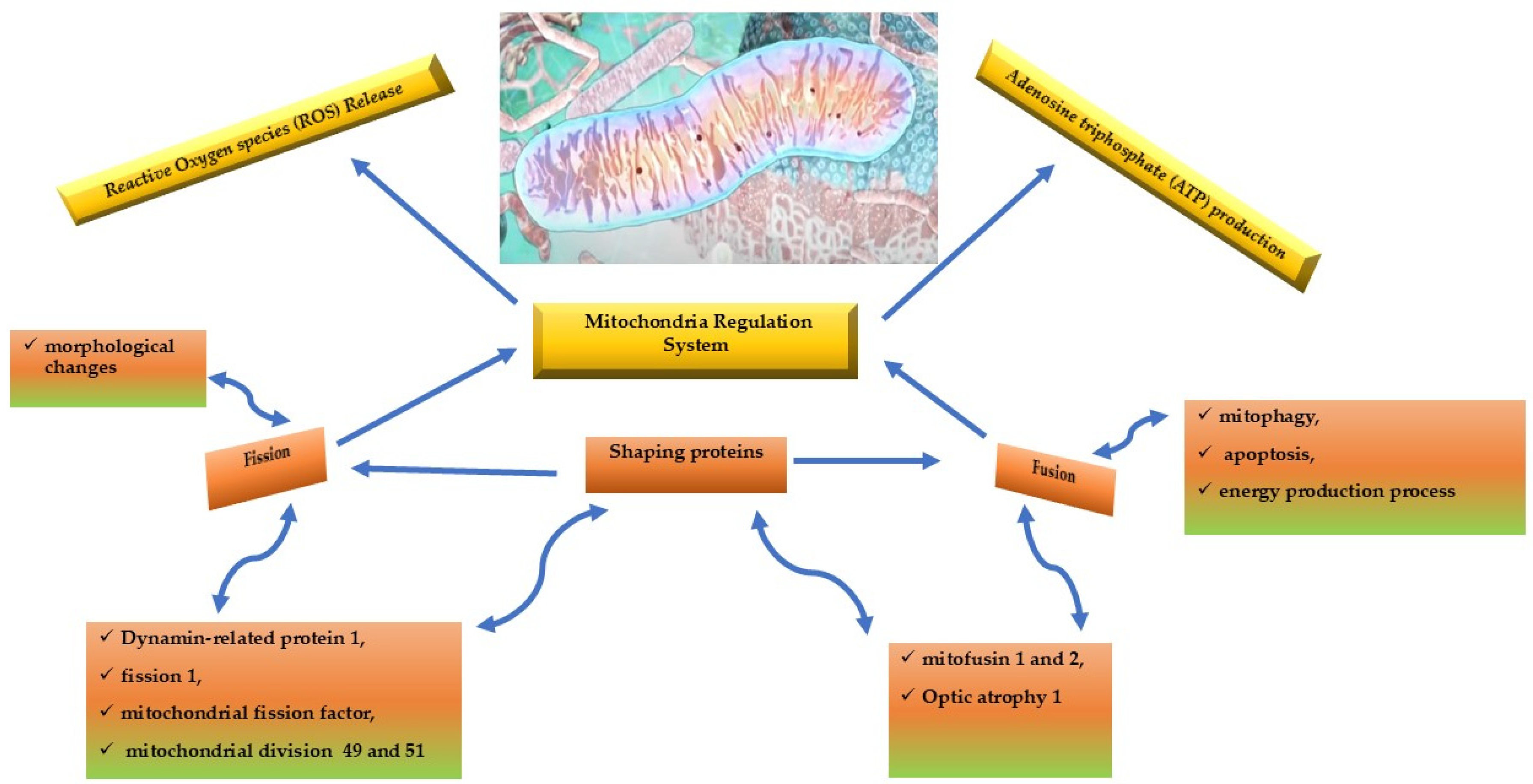
Figure 3.
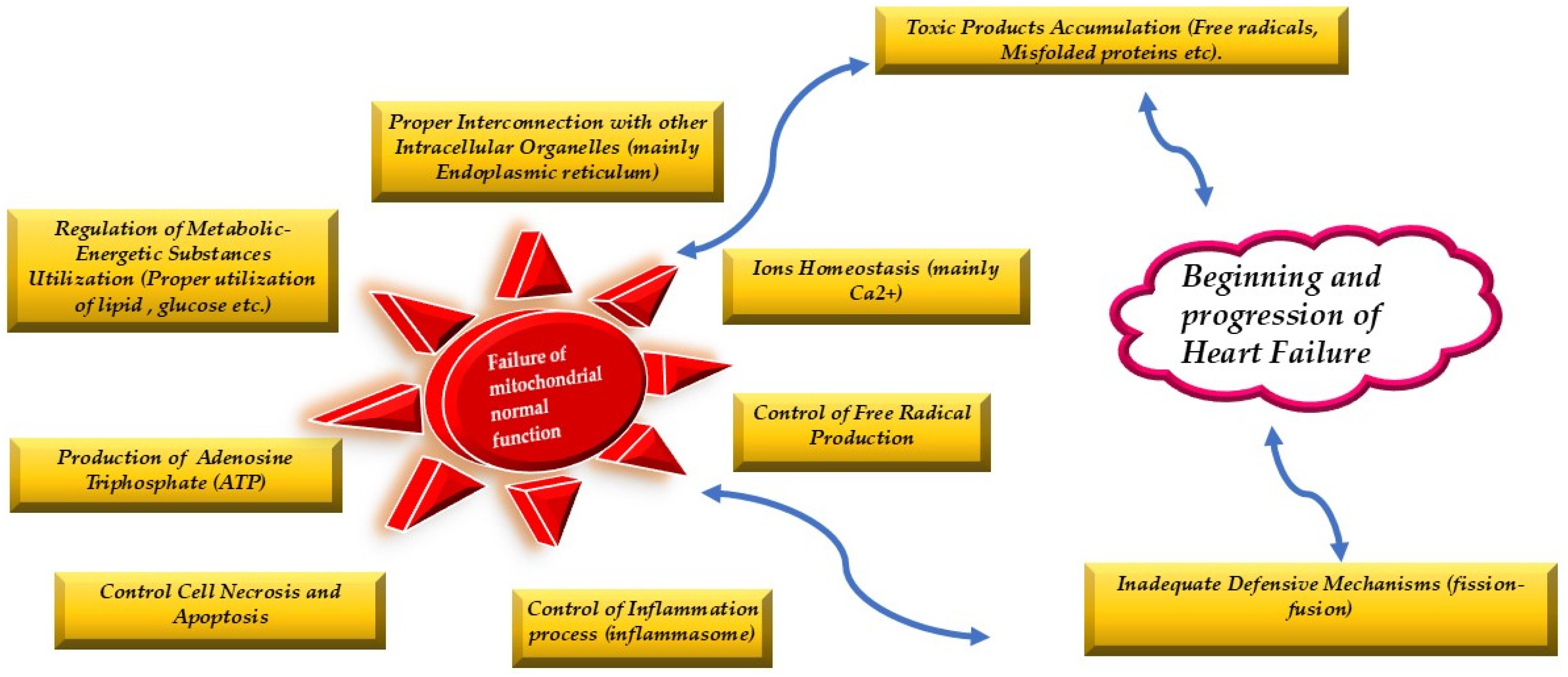
The normal mitochondrial function lies on the proper use of metabolic resources, on their interconnection with the other organelles, on the normal ions exchange, on the amount of free radical and other harmful products accumulation and on the control of cell necrosis and apoptosis. The protective mitochondrial mechanisms (fission and fusion) keep mitochondrial normality. When their protection is inadequate due both to the homeostatic failure or protective mechanisms malfunction then heart failure syndrome begins and within time progresses.
Figure 3.
The normal mitochondrial function lies on the proper use of metabolic resources, on their interconnection with the other organelles, on the normal ions exchange, on the amount of free radical and other harmful products accumulation and on the control of cell necrosis and apoptosis. The protective mitochondrial mechanisms (fission and fusion) keep mitochondrial normality. When their protection is inadequate due both to the homeostatic failure or protective mechanisms malfunction then heart failure syndrome begins and within time progresses.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



