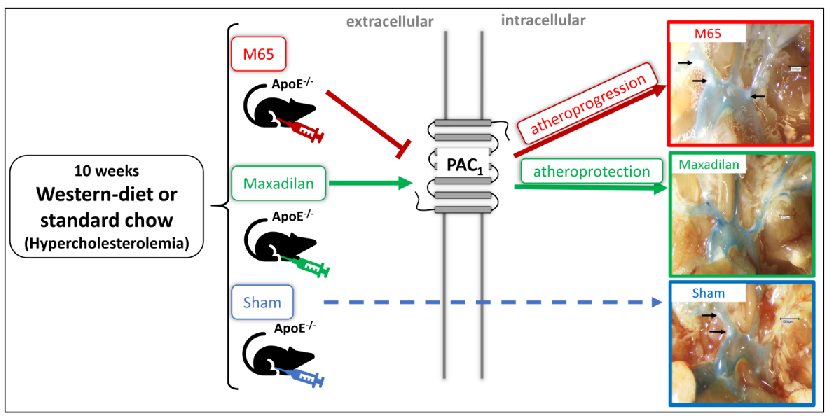
1. Introduction
Atherosclerosis is a chronic inflammatory process of arteries responsible for about 50% of deaths in the Western world [
1,
2]. Driven by low-density lipoprotein oxidation (oxLDL) and endothelial dysfunction, an inflammatory milieu arises, where inflammatory leukocytes are attracted, mainly monocytes and macrophages (MΦ). After adhesion to endothelial cells, monocytes migrate into the subendothelial space, differentiate into MΦ, internalize oxLDL via scavenger receptors, and transform into foam cells [
3,
4]. Smooth muscle cells (SMC) of the media proliferate, migrate into the intima, express scavenger receptors, internalize oxLDL, and develop into foam cells, too [
5,
6]. These processes finally lead to atherosclerotic plaque formation and calcification [
7,
8]. The presence of pituitary adenylate cyclase-activating polypeptide (PACAP) and its receptors in cardiac tissue and blood vessels was taken as evidence that this neuropeptide may play a role in cardiovascular functions [
9]. PACAP protects cultured vascular endothelial cells (EC) and SMC from lipid peroxide-induced damage, which is triggered by high lipids [
10]. These observations may suggest the involvement of PACAP in attenuating atherogenesis. In fact, using an ApoE
-/- mouse model of atherosclerosis we have recently shown that PACAP deficiency aggravates atherosclerosis under 30 weeks of standard chow [
11]. Therefore, PACAP has been suggested to act as an endogenous atheroprotectant [
11]. However, constitutive PAC1 deletion in the mouse results in atheroprotection rather than acceleration of atherogenesis [
12], a finding at odds with the loss of apparent atheroprotection elicited by PACAP knockout. Since both maneuvers result in lifelong loss of either PACAP or its receptor, and since PACAP does interact with other receptors besides PAC1, we decided to initiate experiments in which gain and loss of PAC-1 dependent PACAP function was accomplished pharmacologically, i.e. within the context of reversing atherogenesis, rather than affecting the initial factors predisposing to it.
Interestingly, Maxadilan, a selective agonist for PAC-1 isolated from the saliva of sandflies [
13], has been shown to exert anti-apoptotic effects with downregulation of cleaved caspase-3 [
14]. Furthermore, a specific PAC1 antagonist, M65, has been previously engineered based on the Maxadilan peptide backbone [
15,
16]. Therefore, we tested in the present study our hypothesis that changes in PACAP function in the adult, rather than across early development until adulthood, would separate combined developmental and adult effects of gain- and loss-of-function of PACAP and its cognate receptor during atherogenesis, providing a more therapeutically relevant assessment of its potential efficacy in the context of progressive cardiovascular disease. We show here that treatment with the PACAP agonist Maxadilan reduced the development of atherosclerosis in ApoE
-/- mice, while M65 did not recapitulate the anti-atherogenic effects of PAC1 knockout, indicating that the latter is likely to be a result of developmental dysregulation unrelated to a potential therapeutic effect of PACAP acting at its cognate receptor, in treatment of atherosclerosis.
2. Results
The essential new finding of our study is that Maxadilan reduced atherosclerosis in the aortic arch and brachiocephalic trunk of ApoE-/- mice under SC and CED with preserved or further enhanced hypercholesterolemia.
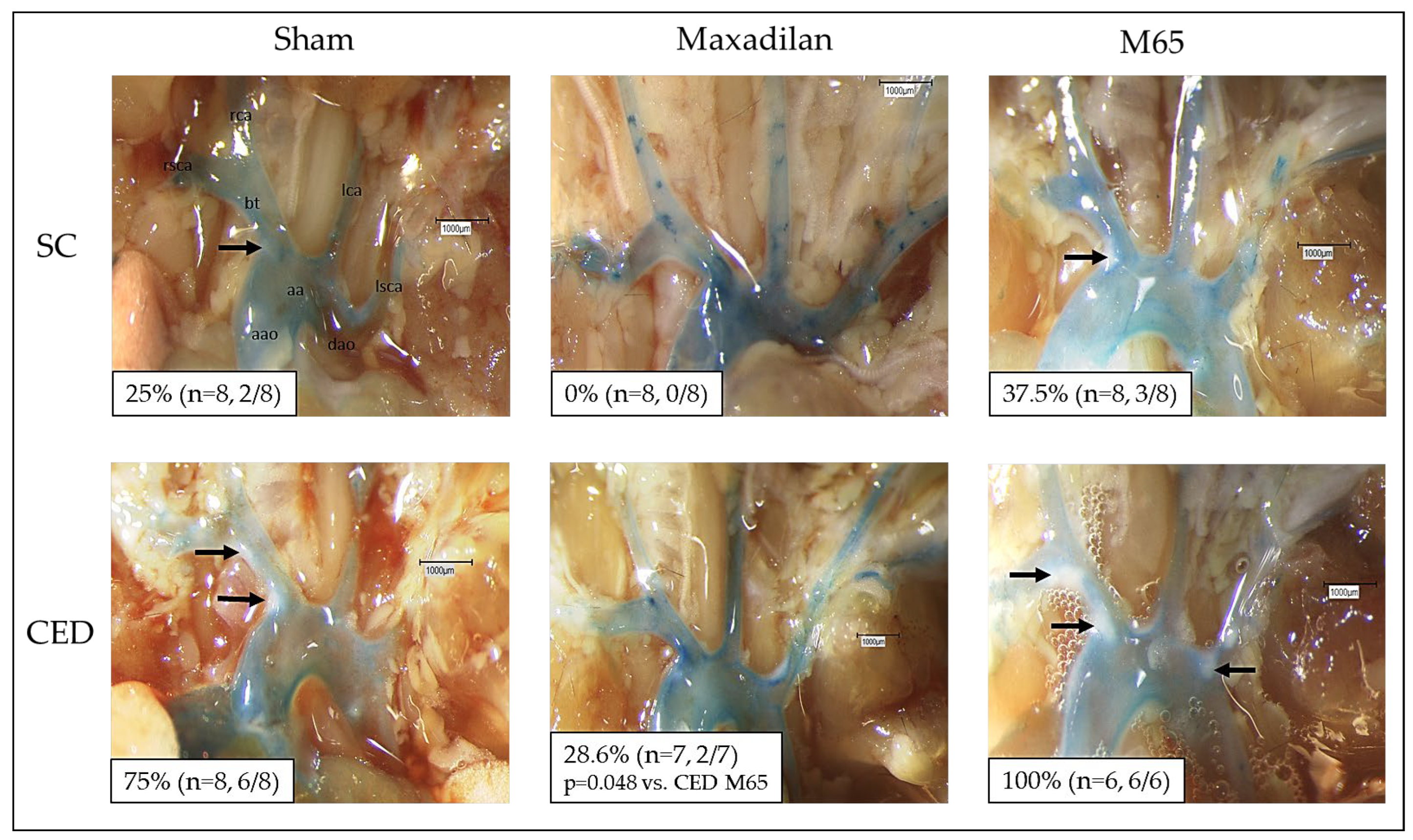
2.1. Maxadilan i.p., but Not M65 i.p., Reduced Atherosclerotic Plaque Frequency in the Aortic Arch and Its Branches
En-face dissection revealed that 25% of the ApoE
-/- mice under SC (n=8) developed macroscopically visible atherosclerotic plaques of the aortic arch and its main branches (
Figure 1). In contrast, Maxadilan-treated mice (n=8) revealed no visible plaques (
Figure 1). Under CED 75% of the Sham mice (n=8) showed distinct plaques (
Figure 1), whereas only 28.6% of the Maxadilan-treated mice (n=7) developed plaques (
Figure 1). In contrast, 37.5% of M65 treated mice under SC revealed atherosclerotic lesions and 100% of M65 treated mice under CED showed atherosclerotic plaques (
Figure 1). Thus, M65 is shown to have actions opposite to those of Maxadilan, as it enhances the percentage of mice exhibiting atherosclerotic plaques as compared to Maxadilan and Sham (
Figure 1).
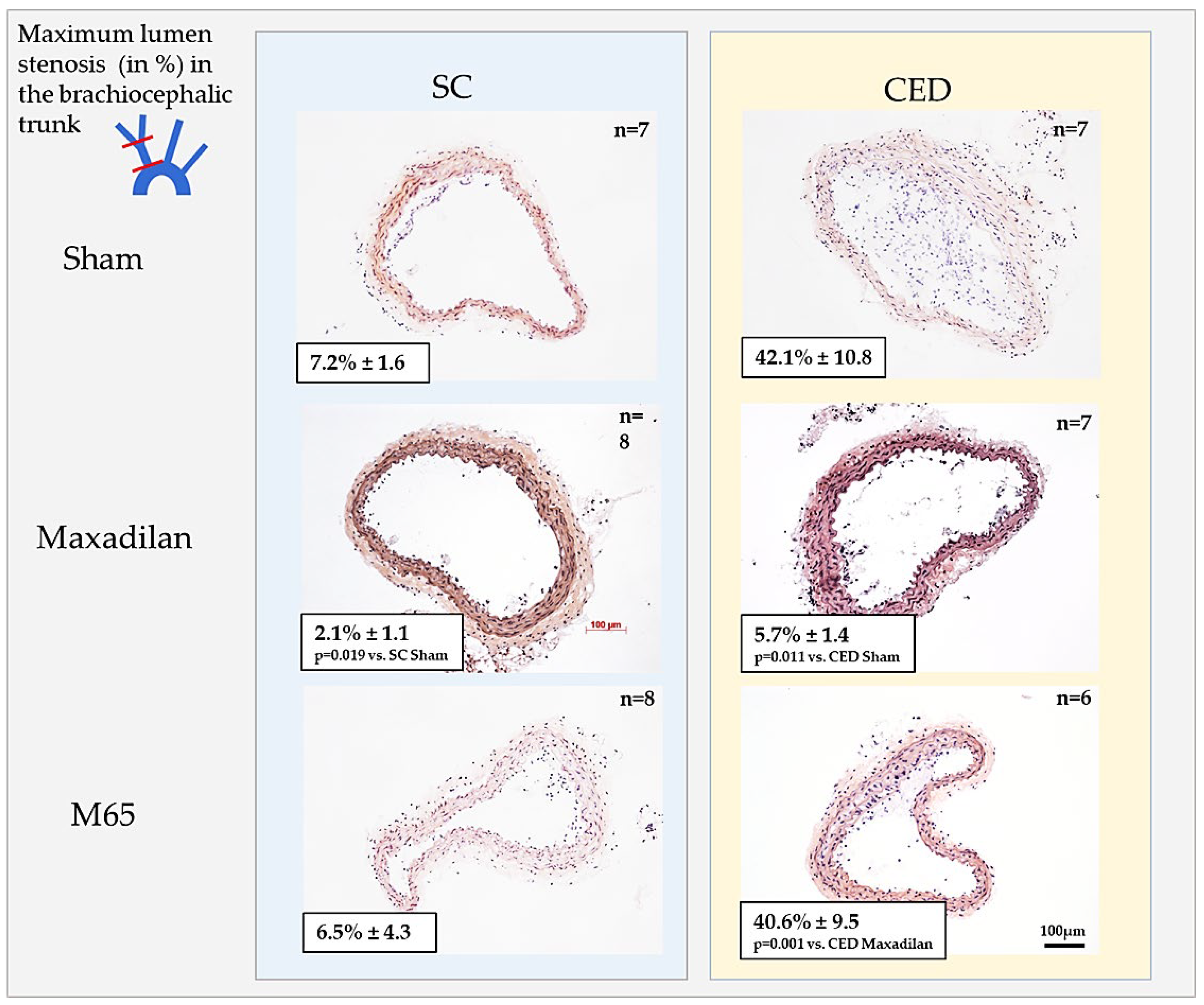
2.2. Maxadilan i.p., but Not M65 i.p., Reduced Lumen Stenosis in BT
Histomorphometric analysis of cross-sections of BT revealed that Maxadilan treatment of ApoE
-/- mice under SC significantly (p≤0.05) inhibited 3.5-fold the lumen stenosis compared to Sham (
Figure 2A,B). Under CED, Maxadilan i.p. significantly (p=0.011) reduced the lumen stenosis 7.4-fold compared to Sham (
Figure 2A,B). In spite of significantly increased cholesterol levels, lumen stenosis of Maxadilan-treated mice under CED and SC was similar (
Figure 2A,B;
Table 1). In contrast, the application of M65 to mice under SC or CED revealed neither inhibition nor progression of lumen stenosis in comparison with Sham (
Figure 2A,B).
As we have thus shown that Maxadilan, but not M65 treatment, is atheroprotective, we concentrated the following on the influence of Maxadilan treatment on distinct atherosclerosis-relevant signatures to decipher mechanisms of its atheroprotectant actions.
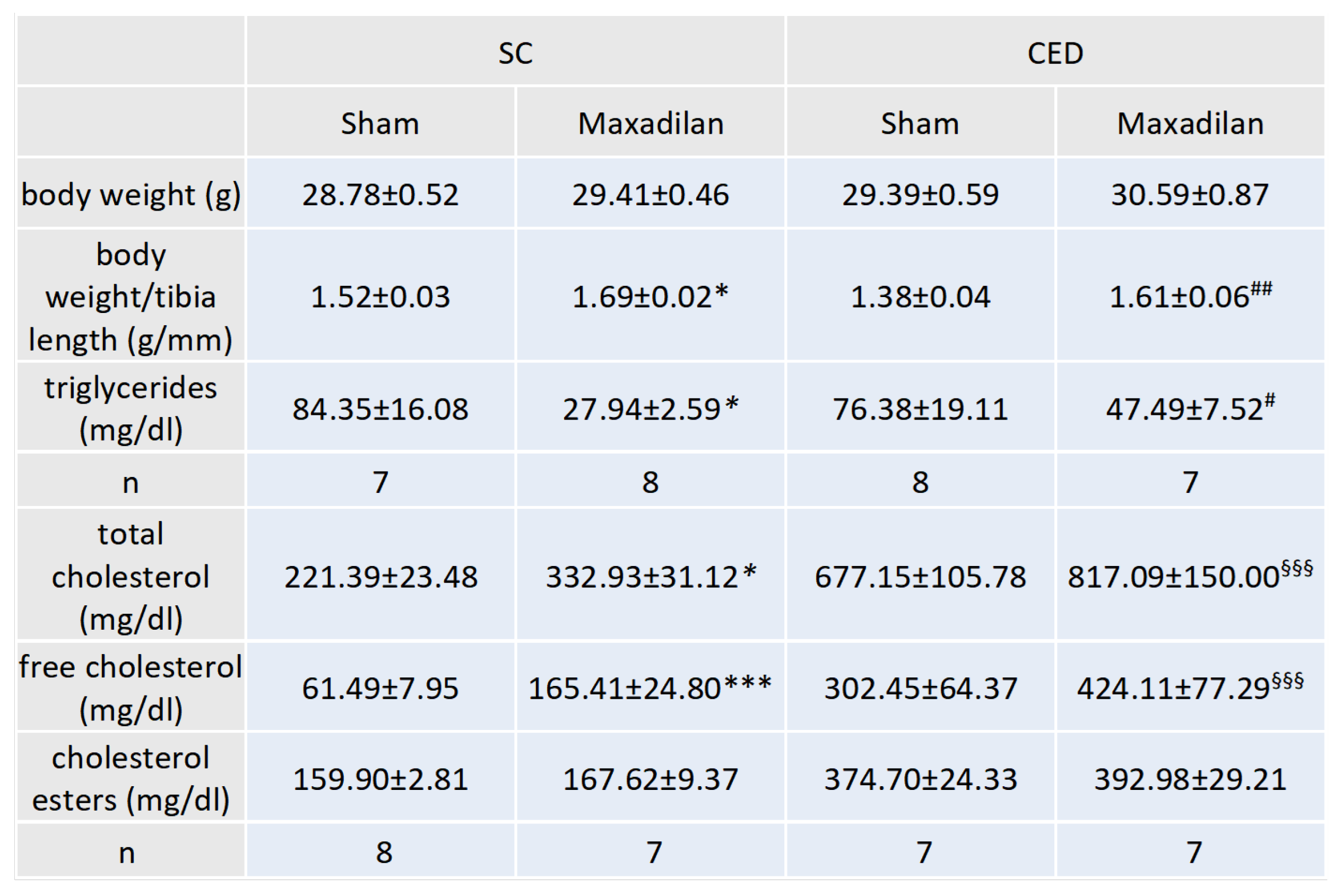
2.3. Maxadilan i.p. Decreased Plasma Triglyceride Levels and Increased Total and Free Cholesterol and Body Weight/Tibia Ratio
Under SC, Maxadilan-treated ApoE
-/- mice showed 66.9 % (p≤0.05) lower plasma triglyceride levels than Sham (
Table 1). Maxadilan-treated ApoE
-/- mice under CED exhibited 37.8% lower triglyceride levels than SC (
Table 1). Under SC, Maxadilan-treated ApoE
-/- mice showed 33.5 % (p≤0.05) higher total plasma cholesterol levels than Sham (
Table 1). After the differentiation of cholesterol into free cholesterol and cholesterol esters, Maxadilan-treated ApoE
-/- mice under SC had 169.0% (p≤0.001) higher concentrations of free cholesterol than Sham (
Table 1). Remarkably, the atheroprotective effects of Maxadilan treatment occurred in spite of increased cholesterol levels. Maxadilan-treated mice revealed significantly 11.2% (p≤0.05) or 15.9% (p≤0.01) higher body weight/ tibia length ratios than corresponding Sham mice under SC or CED (
Table 1).
Table 1.
Effect of Maxadilan application on body weight, body weight/tibia length ratio, plasma cholesterol, and triglyceride levels.
Table 1.
Effect of Maxadilan application on body weight, body weight/tibia length ratio, plasma cholesterol, and triglyceride levels.
Body weight (g), body weight/tibia length ratio (g/mm). Plasma triglyceride levels, total cholesterol, free cholesterol and cholesterol ester levels in mg/dl were evaluated via ELISA. Treated groups received PAC1 agonist maxadilan (20 nmol/kg) and sham physiological saline solution. SC, standard chow; CED, cholesterol-enriched diet; *p≤0.05, ***p≤0.001 vs. SC Sham; #p≤0.05 vs. ##p≤0.01 vs. SC maxadilan-treated; §§§p≤0.001, CED Maxadilan vs SC Maxadilan.
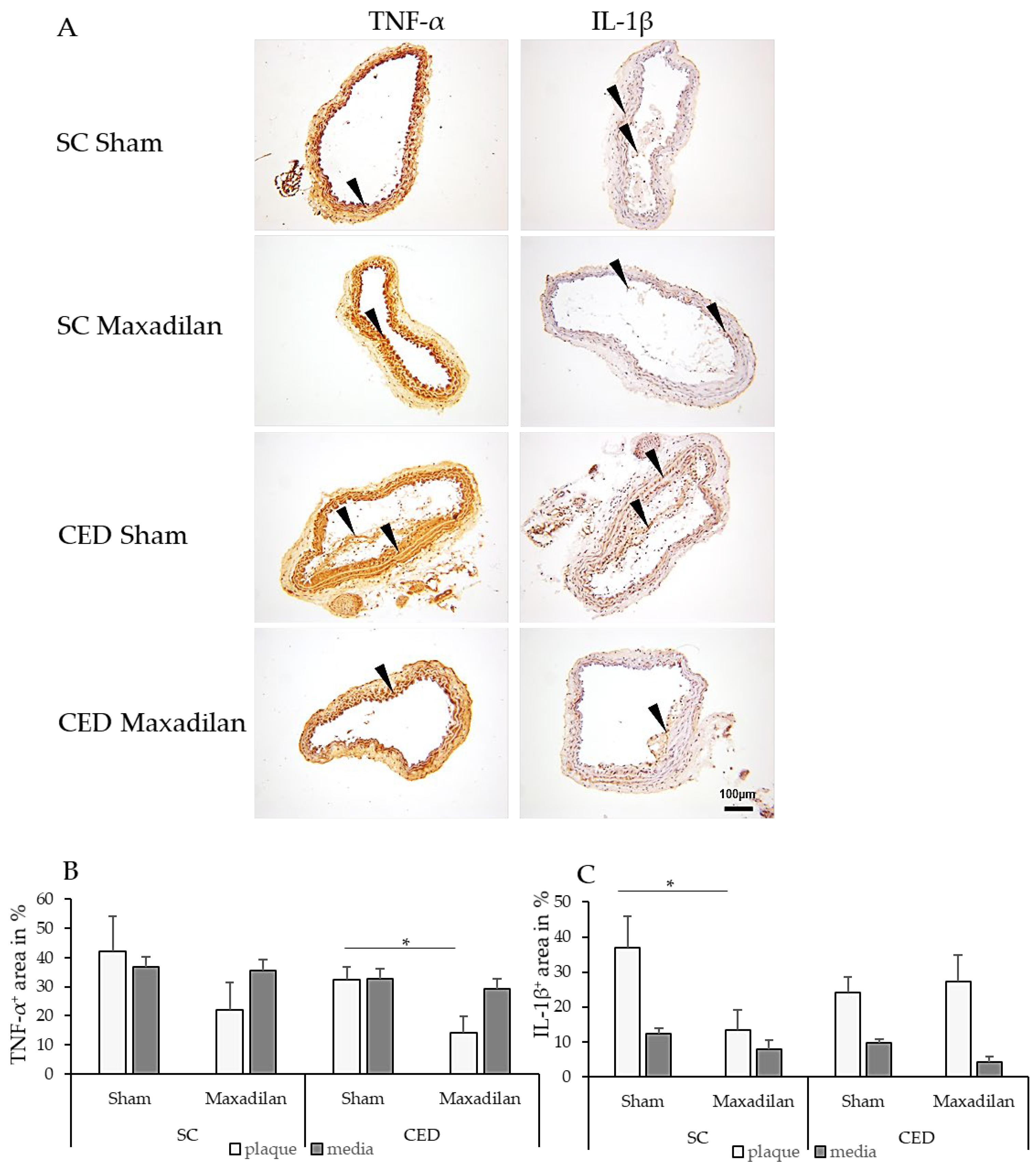
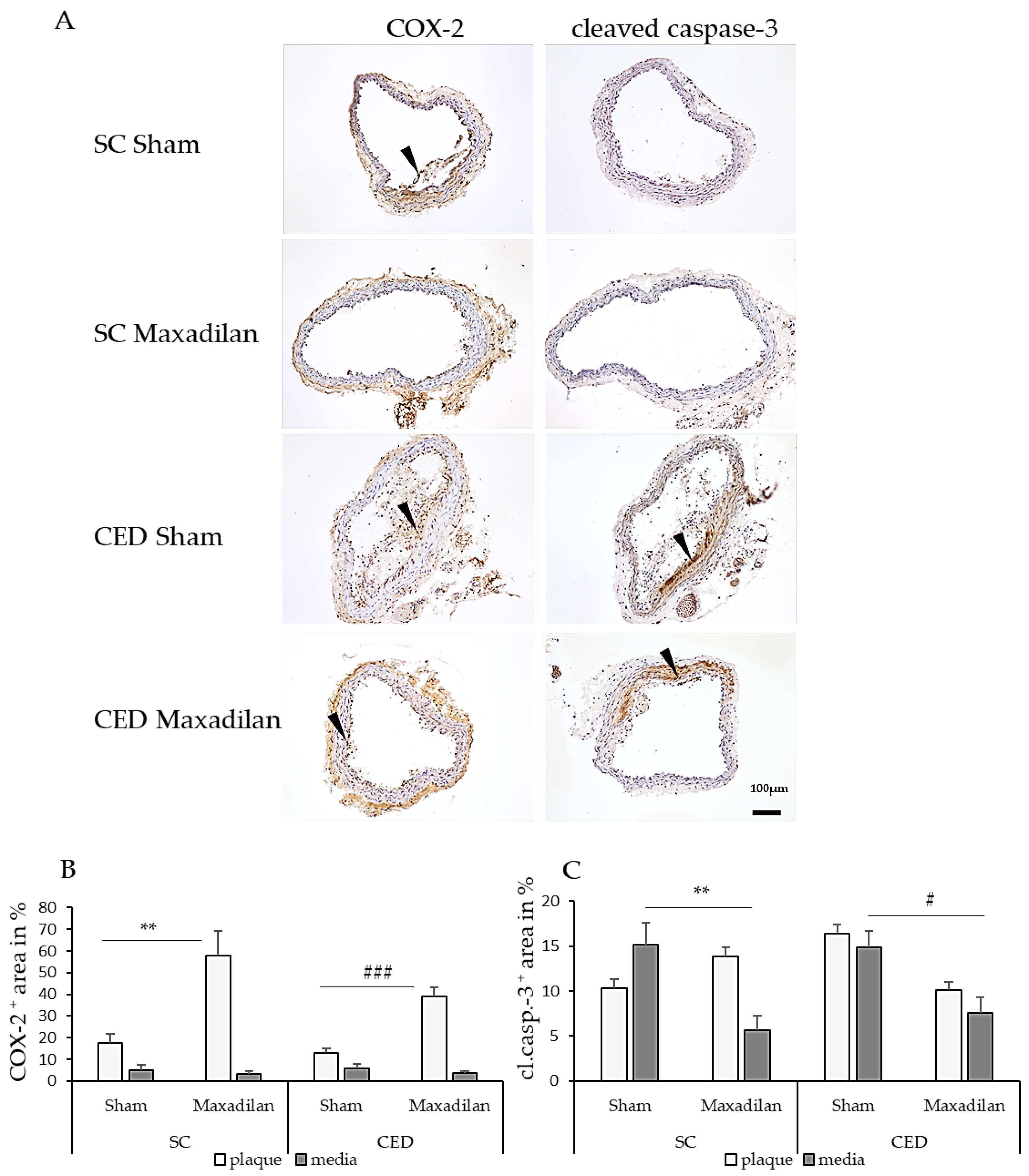
2.4. Maxadilan i.p. Reduced TNF-a+, IL-1β + and caspase-3+, and Increased COX-2+ Areas
Under CED, Maxadilan reduced TNF-α
+ area in plaques by 18.3% (p=0.04) compared to Sham (
Figure 3B). Under SC, Maxadilan-treated mice showed 23.6 % (p=0.04) lesser IL-1β
+ areas in the plaque than Sham. However, under CED, no significant changes in the percentage of IL-1β
+ areas were observed in the atherosclerotic plaques or SMC of the media. Under SC, Maxadilan treatment increased the COX-2
+ area in the lesion by 40.4% (p≤0.01) and under CED by 26.2% (p≤0.001) compared to Sham (
Figure 4B). Under SC, cleaved caspase-3
+ cells were found in plaques and media beneath the plaque in BT of Sham ApoE
-/- mice (
Figure 4C). Under SC, the percentage of cleaved caspase-3
+ area in the media of Maxadilan-treated mice was decreased by 9.5% (p≤0.01) and under CED by 7.3% (p≤0.05) compared to Sham (
Figure 4C).
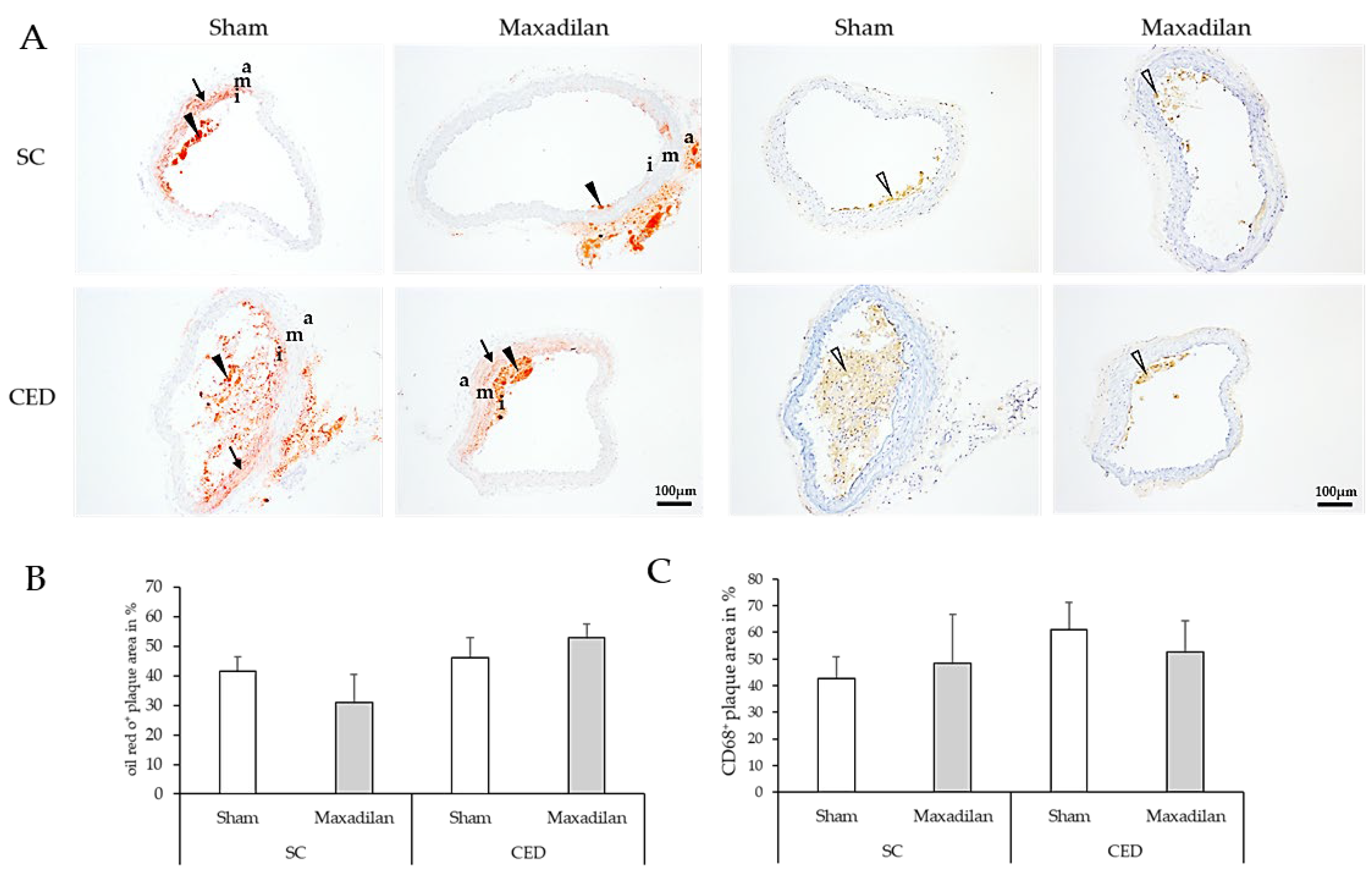
Moreover, lipid deposits were detected by ORO staining and MΦ were identified by immunohistochemistry using CD68 antibodies (
Figure 5A, C). The percentage of oil-red O plaque area and CD68
+ MΦ were similar among all groups under test (
Figure 5B,D).
3. Discussion
Here we have shown that Maxadilan reduces atherogenesis in ApoE
-/- mice both, under SC and CED confirming our hypothesis, that PACAP is atheroprotective. In contrast, M65 treatment had no significant effect on lumen stenosis under SC or CED, except that it increased the percentage of mice exhibiting macroscopically visible atherosclerotic plaques compared to Sham and especially to Maxadilan-treated mice. Our present results are in good concordance with our previous findings that PACAP deficiency in ApoE
-/- aggravates atherosclerosis [
11], suggesting that endogenous PACAP is atheroprotective. As Maxadilan is a specific PAC1 agonist, our present study indicates that the atheroprotectant effect of Maxadilan is mediated by the PAC1 receptor. It is worthy of note that a comparison of M65 and maxadilian effects shown here, resolves a discrepancy between the pro-atherogenic effects of PACAP deletion, and the amelioration of atherogenesis after CED seen in PAC1-deficient mice. It has since become clear that knockouts from conception within the PACAP system may have profound developmental effects manifest later in adulthood [
17,
18]. Thus, gain- and loss-of-function in constitutive knockout mice, at least as regards PACAP signaling, are not inevitably opposite, and in particular, depend on how long the loss (or gain) of function goes on and when it starts. Thus, pharmacological treatment with the PAC1 antagonist M65 did not copy the anti-atherogenic effect of PAC1 deficiency [
12]. In contrast, the anti-atherosclerotic effect of the PAC1 agonist Maxadilan perfectly corresponded to the loss of endogenous atheroprotection previously described in PACAP knockout mice [
11].
In the following, we consider possible mechanisms by which Maxadilan exerts its anti-atherogenic effects.
TNF-α and IL-1β are well-known pro-atherogenic, and inhibiting these cytokines attenuates atherosclerosis [19-21]. Ray and Autieri, 2019]. In fact, anti-TNF-α therapy improves microvascular endothelial dysfunction in rheumatoid arthritis patients with atherosclerosis [
22]. Therefore, it is reasonable to argue that the reduction of TNF-α and IL-1β by Maxadilan, as shown in the present study, contributes to atheroprotection by Maxadilan.
Our observation that Maxadilan is atheroprotective in spite of enhanced cholesterol levels in conjunction with the well-known anti-inflammatory effects of PAC1 activation [
23] indicates that Maxadilan acts downstream of cholesterol-induced vascular inflammation. In addition, our findings that Maxadilan reduces hypertriglyceridemia suggest that this effect may also contribute to Maxadilan’s anti-atherogenic action revealed in the present study.
Our results that Maxadilan enhances the percentage of COX-2
+ areas in atherosclerotic plaques under both, SC and CED deserves particular attention, because it is well-known that COX-2 prostanoid signaling has vasoprotective components [
24]. Previously, Maxadilan has been shown to increase the production of PGE2 by MΦ [
25] and increased expression of COX-2 reduces atherogenesis in hyperlipidemic mice [
26]. Albeit COX-2 is also known as a pro-inflammatory factor [
27] the COX-2 inhibitor Celecoxib has been reported to aggravate atherogenesis in ApoE
-/- mice [
28]. Therefore, we argue that the observed increase of COX-2
+ areas in the vascular wall seen under Maxadilan treatment could reflect the induction of COX-2-mediated vasoprotection.
Enhanced apoptosis is a characteristic feature of atherosclerosis, albeit with controversial clinical significance [23,29-31]. Clarke et al., 2008 [32.]conclude that VSMC apoptosis accelerates atherosclerosis, causes calcification and medial deterioration, and promotes stenosis. Most recently, atherosclerosis has been classified as a smooth muscle cell-driven tumor-like disease [
33]. Mice with an apoptosis defect in MΦ suggested that apoptosis is a critical self-defense mechanism in inhibiting atherogenesis [
34]. Therefore, our findings that Maxadilan reduced cleaved caspase-3
+ areas in the SMC media layer of the vascular wall suggest that part of the anti-atherogenic effect of Maxadilan is attributable to its anti-apoptotic action. Further studies will be needed to determine whether compounds such as caspase inhibitors will become clinically used to treat patients with vulnerable lesions [
35].
Taken together, we conclude that the atheroprotective effect of Maxadilan is mainly downstream of cholesterol-induced inflammation, such as induction of TNF-α and IL-1β and, in addition, may involve activation of vasoprotective signaling, reduction of apoptosis, and hypertriglyceridemia. We suggest that therapeutic approaches with PAC1 agonists under real-world clinical conditions could complement statin and PCSK-9 therapy of patients at risk of developing atherosclerosis-related pathologies.
4. Materials and Methods
4.1. Animal Experiments
The parental homozygous ApoE-/- mice were purchased from Jackson Laboratories (B6.129P2-Apoetm1Unc/J, Stock No: 002052 | ApoE KO, Jackson Laboratories, Milan). Mice were bred and genotyped in the animal facilities of the Philipps-University of Marburg. Male ApoE-/- mice at 10 weeks were fed along with standard chow (SC; LASQCdiet® Rod16 Rad; LASvendi, Soest, Germany), or 10 weeks with cholesterol-enriched diet [(CED; “Western-type diet”, 21 % fat, 0.15 % cholesterol and 19.5 % casein), Altromin GmbH, Lage, Germany]. During this period, mice were intraperitoneally (i.p.) injected three times a week with 20nmol/kg of Maxadilan or M65 (both from Bachem, Bubendorf, Switzerland) diluted in a physiological saline solution (B. Braun, Melsungen, Germany). Sham-treated animals were injected with physiological saline solution alone (B. Braun, Melsungen, Germany). The solutions were freshly generated, kept cool, and given under sterile conditions. All experiments were approved by the federal state veterinary authority, Regional Commission Gießen (V 54 - 19 c20 15 h 01 MR 20126; Nr. G 80/2019), and conducted according to the animal experiment care guidelines of GV-SOLAS. General health conditions, behavior, and body weight were monitored during all procedures, and no signs of impairment, suffering, weight loss, or abnormal weight gain were observed. After 10 weeks of treatment, animals were narcotized to Guedel stage III-IV, the thoracic cavity was opened and mice were euthanized by exsanguination from the right atrium. Blood was collected in heparinized microtubes 1.3ml LH, 25 I.U. heparin/ml blood (Sarstedt, Nümbrecht, Germany). After the right atrial incision, the vascular system was perfused with phosphate-buffered saline (PBS) solution with 0.01% heparin (B. Braun SE, Melsungen, Germany) through punctation of the left ventricle. The vascular system was injected with 200 μl PBS containing methylene blue (0.25 %; Riedel-de Haën, Seelze- Hannover, Germany). The perfused hearts and BTs were removed, frozen in liquid nitrogen-cooled isopentane, and stored at -80°C.
4.2. Determination of Cholesterol and Triglyceride Levels
Blood samples in heparinized tubes were centrifuged (10 min at 650 x g), and heparinized plasma was removed and stored at −80 °C. Using a cholesterol/ cholesteryl ester quantitation kit (ab65359, Abcam, Cambridge, UK) or triglyceride quantification assay kit (ab65336, Abcam, Cambridge, UK) according to the manufacturer's instructions, plasma cholesterol and triglyceride levels were measured using a microplate reader (Sunrise microplate reader, Tecan, Männedorf, Switzerland).
4.3. Histology and Immunohistochemistry
Serial cross sections (7µm) of BTs were transferred to superfrost plus slides (Thermo Scientific). Standard hematoxylin-eosin (Carl Roth GmbH + Co. KG, Karlsruhe, Germany) staining was performed on every 10th slide to identify maximum lumen stenosis. Morphometric analyses were performed, like lumen stenosis, neointimal area, media area, and Lillie's OilRedO (ORO, Sigma-Aldrich Chemie GmbH, Munich, Germany) staining (to detect lipids). Slides were fixed with isopropanol/1% formalin or 3% phosphate-buffered paraformaldehyde (PFA) solution for immunohistochemistry. Endogenous peroxidase was blocked with 100 U/ml glucose oxidase in PBS for 60 minutes at 37°C. Thereafter, a blocking step with 5% porcine serum for 30 minutes followed and after rinsing with PBS, sections were incubated with primary antibodies for immunohistochemical staining overnight at 4°C, by using the following antibodies, CD68 (Bio-Rad Laboratories GmbH, Feldkirchen, Germany; MCA1957, dilution 1:200), cyclooxygenase-2 (Abcam, ab15191, 1:800), cleaved caspase-3 (Cell Signaling Technology, Frankfurt, Germany, #9664, dilution 1:100), interleukin-1β (Abcam, ab9722, dilution 1:600), TNF-α (Abcam, ab6672, dilution 1:100). After application of secondary antibody matching primary host species and using ABC method (Vectastain®, Vector Laboratories, Inc. Newark, USA), DAB (Sigma, #D-5906) reaction, added with 0.3% hydrogen peroxide (Roth), was carried out. Afterward, nuclei were stained with Mayer's hemalum solution, and slides were dehydrated and mounted with Eukitt® (Sigma, #03989).
4.4. Histomorphometric Analyses
Serial cross-sections of BTs representing maximum lumen stenosis and maximum plaque formation were photographed at 100-fold magnification. The luminal area, internal elastic lamina (IEL) area, and external elastic lamina (EEL) area were measured with ImageJ/Fiji software version 1.54f (National Institute of Health, Bethesda, MD, USA). The media area is the EEL area minus the IEL area. The neointimal area is the IEL area minus the Lumen area. The area of atherosclerotic lesions, immunoreactivity and lumen stenosis were measured as described earlier [
11,
12].
4.5. Statistical Analyses
Statistical analyses were performed with Sigma Plot 12 (Systat Software Inc., San José, USA). "N" stands for the number of animals per investigated group. Double or triple samples were evaluated per animal. Statistical significance was determined by one-way analysis of variance (ANOVA) with corresponding post-hoc tests. In case the data failed the normality and/or equal variance test, Tukey's test or Dunn's method was applied. Results are presented as means ± or standard error of the mean (SEM). The results were considered statistically significant when p≤ 0.05.
Author Contributions
Conceptualization, L.E, R.K. and E.W.; methodology, G.A.B, L.M., A.S.; software, H.S.; formal analysis, J.H., L.M.; investigation, L.M.; writing—original draft preparation, L.M. and G.A.B.; writing—review and editing, L.E., R.K. and E.W.; visualization, A.S., H.S.; funding acquisition, G.A.B. and L.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the German Heart Foundation (Deutsche Herzstiftung), grant number “Projekt F/32/19”. The Open Access Publication Fund of Philipps-Universität Marburg funded the APC with the support of the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation).
Institutional Review Board Statement
All experiments were approved by the federal state veterinary authority, Regional Commission Gießen, Hessen, Germany (V 54 - 19 c20 15 h 01 MR 20126; Nr. G 80/2019), and conducted according to the animal experiment care guidelines of GV-SOLAS and to German animal welfare law and the European legislation for protection of animals used for scientific purposes (2010/63/EU) as well as to regulations for animal experiments of the Philipps University of Marburg.
Data Availability Statement
The data sets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
We thank Mr. Michael Dreher, Ms. Christa Merte-Grebe, Ms. Andrea Cordes, Ms. Anja Seiler, Ms. Katrin Lampp, Mr. Michael Schneider, and Ms. Claudia Weiss for excellent technical assistance and Ms. Vera Heine for the manuscript preparation.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analysis, or interpretation of data; in the writing of the manuscript; or in the decision to publish the result.
References
- Ross, R. Atherosclerosis-an inflammatory disease. N. Engl. J. Med. 1999, 340, 115-126. [CrossRef]
- Pahwa, R.; Jialal, I.; Pahwa, R.; Jialal, I. Atherosclerosis. 2023, In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan–. PMID: 29939576.
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653-667. [CrossRef]
- Schwartz, C.J.; Valente, A.J.; Sprague, E.A.; Kelley, J.L.; Cayatte, A.J.; Mowery, J. Atherosclerosis. Potential targets for stabilization and regression. Circulation 1992, 86(6 Suppl), III117-23.
- Allahverdian, S.; Pannu, P.S.; Francis, G.A. Contribution of monocyte-derived macrophages and smooth muscle cells to arterial foam cell formation. Cardiovasc. Res. 2012, 95, 165-172. [CrossRef]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551-9. [CrossRef]
- Alexopoulos, N.; Raggi, P. Calcification in atherosclerosis. Nat. Rev. Cardiol. 2009, 6, 681-688. [CrossRef]
- Shi, X.; Gao, J.; Lv, Q.; Cai, H.; Wang, F.; Ye, R.; Liu, X. Calcification in Atherosclerotic Plaque Vulnerability: Friend or Foe? Front. Physiol. 2020, 11, 56. [CrossRef]
- Diané, A.; Payne, G.W.; Gray, S.L. Multifaces of Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP): From Neuroprotection and Energy Homeostasis to Respiratory and Cardiovascular Systems. J. Metabolic. Synd. 2014, 3, 162. [CrossRef]
- Chang, Q. [Experimental study of the effects of pituitary adenylate cyclase-activating polypeptide (PACAP) and its mechanism on the vascular cell components--the possible relationship between PACAP and atherosclerosis]. Sheng Li Ke Xue Jin Zhan. 1997, 28, 132-135.
- Rasbach, E.; Splitthoff, P.; Bonaterra, G.A.; Schwarz, A.; Mey, L.; Schwarzbach, H.; Eiden, L.E.; Weihe, E.; Kinscherf, R. PACAP deficiency aggravates atherosclerosis in ApoE deficient mice. Immunobiology 2019, 224, 124-132. [CrossRef]
- Splitthoff, P.; Rasbach, E.; Neudert, P.; Bonaterra, G.A.; Schwarz, A.; Mey, L.; Schwarzbach, H.; Eiden, L.E., Weihe, E.; Kinscherf, R. PAC1 deficiency attenuates progression of atherosclerosis in ApoE deficient mice under cholesterol-enriched diet. Immunobiology 2020, 225, 151930. [CrossRef]
- Moro, O.; Lerner, E.A. Maxadilan, the vasodilator from sand flies, is a specific pituitary adenylate cyclase activating peptide type I receptor agonist. J. Biol. Chem. 1997, 272, 966-970. [CrossRef]
- Guo, X.; Yu, R.; Xu,Y.; Lian, R.; Yu, Y.; Cui, Z.; Ji, Q.; Chen, J.; Li, Z.; Liu, H.; Chen, J. PAC1R agonist maxadilan enhances hADSC viability and neural differentiation potential. J. Cell Mol. Med. 2016, 20, 874-890. [CrossRef]
- Moro, O.; Wakita, K.; Ohnuma, M.; Denda, S.; Lerner, E.A.; Tajima, M. Functional characterization of structural alterations in the sequence of the vasodilatory peptide maxadilan yields a pituitary adenylate cyclase-activating peptide type 1 receptor-specific antagonist. J. Biol. Chem. 1999, 274, 23103-23110. [CrossRef]
- Uchida, D.; Tatsuno, I.; Tanaka, T.; Hirai, A.; Saito, Y.; Moro, O.; Tajima, M. Maxadilan is a specific agonist and its deleted peptide (M65) is a specific antagonist for PACAP type 1 receptor. Ann. N. Y. Acad. Sci. 1998, 865, 253-258. [CrossRef]
- Bakalar, D.; Sweat, S.; Drossel, G.; Jiang, S.Z.; Samal, B.B.; Stroth, N.; Xu, W.; Zhang, L.; Zhang, H.; Eiden, L.E. Relationships between constitutive and acute gene regulation, and physiological and behavioral responses, mediated by the neuropeptide PACAP. Psychoneuroendocrinology. 2022, 135, 105447. doi: 10.1016/j.psyneuen.2021.105447. Epub 2021 Oct 16. Erratum in: Psychoneuroendocrinology 2022, 135, 105592. [CrossRef]
- Bakalar, D.; Gavrilova, O.; Jiang, S.Z.; Zhang, H.Y.; Roy, S.; Williams, S.K.; Liu, N.; Wisser, S.; Usdin, T.B.; Eiden, L.E. Constitutive and conditional deletion reveals distinct phenotypes driven by developmental versus neurotransmitter actions of the neuropeptide PACAP. J. Neuroendocrinol 2023, 35, e13286. [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685-1695. [CrossRef]
- Hansson, G.K. Inflammation and Atherosclerosis: The End of a Controversy. Circulation 2017, 136, 1875-1877. [CrossRef]
- Ray, M.; Autieri, MV. Regulation of pro- and anti-atherogenic cytokines. Cytokine 2019, 122, 154175. [CrossRef]
- Caraba, A.; Stancu, O.; Crișan, V.; Georgescu, D. Anti TNF-Alpha Treatment Improves Microvascular Endothelial Dysfunction in Rheumatoid Arthritis Patients. Int. J. Mol. Sci. 2024, 25, 9925. [CrossRef]
- Ross, J.S.; Stagliano, N.E., Donovan, M.J.; Breitbart, R.E.; Ginsburg, G.S. Atherosclerosis: a cancer of the blood vessels? Am. J. Clin. Pathol. 2001, 116 Suppl 1, S97, S107. [CrossRef]
- Ricciotti, E.; Haines, P.G.; Chai, W.; FitzGerald, G.A. Prostanoids in Cardiac and Vascular Remodeling. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 558-583. [CrossRef]
- Soares, M.B.; Titus, R.G.; Shoemaker, C.B.; David, J.R.; Bozza, M. The vasoactive peptide maxadilan from sand fly saliva inhibits TNF-alpha and induces IL-6 by mouse macrophages through interaction with the pituitary adenylate cyclase-activating polypeptide (PACAP) receptor. J. Immunol. 1998, 160, 1811-1816.
- Tang, S.Y.; Monslow, J.; Todd, L.; Lawson, J.; Puré, E.; FitzGerald, G.A. Cyclooxygenase-2 in endothelial and vascular smooth muscle cells restrains atherogenesis in hyperlipidemic mice. Circulation 2014, 129, 1761-1769. [CrossRef]
- Khalil, N.A.; Ahmed, E.M.; Tharwat, T.; Mahmoud, Z. NSAIDs between past and present; a long journey towards an ideal COX-2 inhibitor lead. RSC Adv. 2024, 14, 30647-30661. [CrossRef]
- Pang, Y.; Gan, L.; Wang, X.; Su, Q.; Liang, C.; He, P. Celecoxib aggravates atherogenesis and upregulates leukotrienes in ApoE-/- mice and lipopolysaccharide-stimulated RAW264.7 macrophages. Atherosclerosis 2019, 284, 50-58. [CrossRef]
- Kockx, M.M.; Herman, A.G. Apoptosis in atherosclerosis: beneficial or detrimental? Cardiovasc. Res. 2000, 45, 736-746. [CrossRef]
- Stoneman, V.E.; Bennett, M.R. Role of apoptosis in atherosclerosis and its therapeutic implications. Clin. Sci. (Lond) 2004, 107, 343-354. [CrossRef]
- Li, M., Wang, Z.W., Fang, L.J., Cheng, S.Q.; Wang, X.; Liu, N.F. Programmed cell death in atherosclerosis and vascular calcification. Cell Death. Dis. 2022, 13, 467. [CrossRef]
- Clarke, M.C.; Littlewood, T.D.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Bennett, M.R. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ. Res. 2008, 102, 1529-1538. [CrossRef]
- Pan, H.; Ho, S.E.; Xue, C.; Cui, J.; Johanson, Q.S.; Sachs, N.; Ross, L.S.; Li, F.; Solomon, R.A.; Connolly, E.S. Jr.; et al. Atherosclerosis Is a Smooth Muscle Cell-Driven Tumor-Like Disease. Circulation 2024, 149, 1885-1898. [CrossRef]
- Liu, J.; Thewke, D.P.; Su, Y.R.; Linton, M.F.; Fazio, S.; Sinensky, M.S. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 174-179. [CrossRef]
- Martinet, W.; Schrijvers, D.M.; De Meyer, G.R. Pharmacological modulation of cell death in atherosclerosis: a promising approach towards plaque stabilization? Br. J. Pharmacol. 2011, 164, 1-13. [CrossRef]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).