
Submitted:
11 October 2024
Posted:
14 October 2024
You are already at the latest version
Abstract
Background: Ovarian clear cell carcinoma (OCCC) is a type of ovarian cancer with a poor prognosis if detected in the progressive stage since there is less effective chemotherapy. Recent advancements in molecular-targeted drugs have not substantially affected OCCC treatment. Therefore, we explored the potential of targeting MDM2 in OCCC cells. Methods: We used TOV-21G and KOC7c cells as the ARID1A mutant-type, and RMG-I and ES2 cells as the ARID1A wild-type. Then, we performed small interfering library screening, Western blotting, real-time polymerase chain reaction analysis, cell proliferation assay, cell cycle analysis, time-lapse cell proliferation assessment, and DNA damage assessment. Next, to generate murine intraperitoneal tumors, 7.5 × 106 TOV-21G cells in 200 μL of phosphate-buffered saline were injected subcutaneously into the intraperitoneum in 5–6-week-old athymic nude mice. Results: Using various cell lines with ARID1A mutations or without mutation, the results showed that the interference of MDM2 effectively reduced cell proliferation in ARID1A-mutant cells but not in ARID1A wild-type cells. Additionally, interference with ARID1A against ARID1A wild strains reproduced susceptibility to MDM2 interference. In vivo experiments demonstrated that nutlin-3, an MDM2 inhibitor, significantly suppressed tumor growth in the ovarian cancer mouse model.
Conclusion: These findings suggest that targeting MDM2 may be a viable strategy for the treat-ment of ARID1A-mutated OCCC, offering a new therapeutic approach for this challenging type of cancer.
Keywords:
clear cell carcinoma
; synthetic lethality
; MDM2
; nutlin-3
; ARID1A
1. Introduction
Ovarian cancer is the fifth most common cause of cancer-related mortality in females, and it has the lowest 5-year survival rate among gynecological cancers [1,2]. The major pathological subtypes of ovarian cancer include high-grade serous carcinoma, endometrial carcinoma, clear cell carcinoma, and mucinous carcinoma. Clear cell carcinoma is resistant to platinum chemotherapy and has a poor prognosis when detected in the progressed stage or recurrence [3,4,5,6,7]. Currently, molecular-targeted drugs have been developed consecutively for the treatment of ovarian cancer, and angiogenesis inhibitors and poly ADP-ribose polymerase (PARP) inhibitors are covered by insurance [8]. Although the high efficacy of PARP inhibitors has been reported in serous carcinomas with a high mutation rate of BRCA1/2, there are no highly effective molecular-targeted drugs because their genetic background is very different from that of clear cell carcinomas [9].
Molecular features of ovarian clear cell carcinoma (OCCC) include TGF-βII (66%), ARID1A (46–57%), PIK3CA (50%), PTEN (20%), and KRAS (5–16%) mutations [9,10]. We focused on the ARID1A mutation, known as the SWI-SNF complex. ARID1A is considered a tumor-suppressor gene with important functions in gene transcription regulation and in replication, repair, and cell cycle arrest [10] in the same manner as BRCA1/2 mutations responsible for homologous recombination repair in serous carcinomas. ARID1A mutations have been reported in various carcinomas: 27% in gastric cancer, 13% in hepatocellular carcinoma, 13% in bladder cancer, 15% in esophageal cancer, and 17% in Burkitt lymphoma. Recent advancements in molecular-targeted drugs have not substantially affected OCCC treatment. In our previous study, cycline E1 (CCNE1) was one of the synthetic lethal candidates with ARID1A mutation in OCCC [11]; however, in the present study, we aimed to investigate the synthetic lethal mechanism of MDM2 as a novel candidate for ARID1A mutation in OCCC.
2. Results
2.1. Screening of Candidate Genes Harboring a Synthetic Lethal Effect with ARID1A Downregulation in OCCC
We confirmed the effective interference of ARID1A to RMG-I (ARID1A wild-type line) at 96 hours by 5 nM of siRNA (87.2 ± 0.3 versus [vs.] 100.0 ± 0.4, p < 0.001) (Figure 1B). Figure 1C shows the result of the first siRNA screening. The MTT assay was used to identify candidate genes whose interference was significantly reduced (p < 0.05) the cell viability of the ARID1A-knockdown group compared with that of the control group. Seven candidate genes were identified, but we focused on MDM2 as one such candidate.
The RMG-I cell line (ARID1A wild type) was grown in a six-well plate at a concentration of 4.0×105 cells per well. Si-ARID1A or si-control was rapidly reverse-transfected at a concentration of 5nM. Forty-eight hours after transfection, ARID1A-knockdown and control cells were plated in three wells of a 96-well plate at a concentration of 5000 cells per well, and screening siRNA transfection was conducted (A). Figure 1B illustrates the effective interference of ARID1A to RMG-I at 96 h using 5 nM of the siRNA. The volcano plot indicates that MDM2 (mouse double minute protein 2) exhibited the most significant reduction in proliferation compared to the control (C). MDM2, mouse double minute protein 2; CDC6, cell division cycle 6; CCNE1, cyclin E1; **, p < 0.01.
2.2. MDM2 Has a Synthetic Lethal Effect Only in ARID1A-Mutated Cell Lines
In TOV-21G (ARID1A mutation type), the MDM2-knockdown group showed significantly reduced proliferation compared with the control group in a time-dependent manner at 24, 48, and 72 hours (104.7 ± 4.3 vs. 100.0 ± 5.5, p = 0.171; 82.7 ± 4.6 vs. 100.0 ± 9.9, p = 0.014; and 83.5 ± 2.3 vs. 100.0 ± 7.9, p = 0.002, respectively). In KOC7c cells (ARID1A mutation type), the MDM2-knockdown group showed reduced proliferation compared with the control group in a time-dependent manner at 24, 48, and 72 hours (81.2 ± 8.5 vs. 100.0 ± 8.7, p = 0.009; 85.1 ± 6.0 vs. 100.0 ± 6.3, p = 0.005; and 81.0 ± 11.9 vs. 100.0 ± 9.2, p = 0.023, respectively).
In contrast, in RMG-I cells (ARID1A wild-type), the MDM2-knockdown group did not show a significant reduction in cell proliferation compared to the control group. In ES2 cells (ARID1A wild type), the MDM2-knockdown group did not show significantly reduced proliferation (24 hours: 101.33 ± 7.5 and 100.00 ± 12.2, p = 0.804; 48 hours: 102.85 ± 3.2 and 100.00 ± 11.5, p = 0.527; 72 hours: 108.65 ± 16.7 and 100.00 ± 12.4, p = 0.282, respectively) (Figure 2A). To confirm the interference of MDM2, we further assessed the relative MDM2 messenger RNA (mRNA) expression levels in the MDM2-knockdown and control groups using RT-PCR. si-MDM2 sufficiently suppressed the mRNA levels of MDM2 (Figure 2B). Next, we assessed MDM2 protein expression. In all cell types, the MDM2-knockdown group showed reduced protein expression compared with the control group at 72 hours (Figure 2A).
2.3. Interference of MDM2 Expression Affects the Cell Cycle
We assessed the effect of MDM2 expression on cell cycle progression in TOV-21G cells. Knockdown of MDM2 showed a significant increase in the proportion of the G1 phase compared with that in the control group at 48 and 72 hours (59.9 ± 2.3 vs. 48.6 ± 1.0, p = 0.002 and 63.3 ± 2.2 vs. 50.2 ± 3.7, p = 0.007, respectively), and a decrease in the S phase (22.2 ± 1.4 vs. 29.4 ± 3.6, p = 0.034 and 19.8 ± 0.4 vs. 25.1 ± 1.8, p = 0.009, respectively) (Figure 2C). The apoptosis assay did not reveal any significant difference between the si-MDM2 and si-control groups (data not shown). These results suggest that interference of MDM2 with TOV-21G affects the cell cycle in the G1 to S phase and does not induce apoptosis in tumor cells.
In Figure 2A, MDM2 protein expression was lower in the MDM2-knockdown group compared to the control group at 72 hours. Figure 2B shows that si-MDM2 effectively suppressed MDM2 mRNA levels. Knockdown of MDM2 increased the proportion of the G1 phase and decreased the S phase in TOV-21G cells at 48 and 72 hours (2C). MDM2, mouse double minute protein 2; RT-PCR, Reserve transcription polymerase chain reaction; mRNA, messenger ribonucleic acid; PBS, phosphate-buffered saline; *, p < 0.05, **, p < 0.01.
2.4. Time-Lapse Cell Proliferation Assessment
The results of time-lapse cell proliferation are shown in Figure 3. TOV-21G, which is an ARID1A mutated cell line, showed a slight proliferative tendency at 4.44 µM, whereas RMG-I which is ARID1A wild shows a marked proliferative tendency at 13.33 µM. Although the concentrations at which the effect was observed were similar for KOC7c and RMG-I, there was a difference in time course and cell proliferation between these two cell lines. RMG-I increased even after decreasing once, suggesting that the inhibitory effect on cell proliferation was insufficient. A different tendency was seen in ES2 which is ARID1A wild. ES2 has a TP53 mutation, suggesting that nutlin-3 may have anti-tumor effects in a TP53 pathway-dependent manner.
TOV-21G, KOC-7c, ES2, and RMG-I were added with a concentration gradient (40.0μM, 13.33μM, 4.44μM, 1.48μM, 0.49μM, and 0μM). IncuCyte ZOOM™ apparatus and incubation continued over 120 h. OCCC, ovarian clear cell carcinoma. The ARID1A mutation cell lines decreased or plateau at 13.33µM compared to the ARID1A wild-type cell lines.
2.5. Knockdown of MDM2 Inhibits Cell Proliferation of ARID1A-Interfered Cell Lines
To confirm that the interference of MDM2 showed selective effects on the ARID1A deficient status, we interfered with ES2 and RMG-I (ARID1A wild-type OCCC lines) using si-ARID1A (10 nM) and si-control, and 48 hours after the first knockdown, these cells were transfected with 10-nM si-MDM2. Setting this secondary transfection time as 0 hours, we measured cell proliferation chronologically by IncuCyte ZOOM™ for 100 hours. At the endpoint of these assays, under interference of 10-nM MDM2, the RMG-I with si-ARID1A (10 nM) group showed significant cell proliferative suppression compared with the si-control group (63.0 ± 8.5 vs. 94.8 ± 4.5, p < 0.001) (Figure 4A). However, under interference of 10-nM MDM2, the ES2 with si-ARID1A (10 nM) group did not show significant cell proliferative suppression compared to the si-control group (98.8 ± 0.4 and 99.5 ± 0.8, respectively; p = 0.001) (Figure 4A). To clarify the mechanism underlying the synthetic lethality of MDM2 in the ARID1A mutation, the basal expression level of MDM2 is shown in Figure 4B. TOV-21G, KOC7c, and ES2 cells showed lower protein expression levels than RMG-I cells (28.0, 22.8, and 53.5 vs. 100%). Interference of ARID1A by 10-nM siRNA showed decreased protein expression of MDM2 at 96 hours (55.6 ± 5.0 vs. 100.0 ± 11.2, p = 0.003) (Figure 4B). These results suggest that MDM2 interference affected only ARID1A mutative cells because temporary downregulation of ARID1A was sensitive to MDM2 interference.
We hypothesized that interference of MDM2, which is known as a suppressor of p53, would increase DNA damage, but there were no significant differentiations between the si-MDM2 and si-control groups at 24 and 48 hours (1847.1 ± 6902.8 and 1716.5 ± 7088.2, p = 0.925 and 2378.0 ± 2849.4 and 1622.2 ± 2481.6, p = 0.088, respectively) (Supplementary Figure 2). Since ARID1A supports the expression level of MDM2, it is likely that MDM2 interference is ineffective when the ARID1A gene is wild-type, whereas the ARID1A mutation effectively reduces the amount of MDM2 in cells only by MDM2 interference, resulting in the suppression of cell growth.
In Figure 4A, the time-lapse cell proliferation assay of ES2 and RMG-I cells was conducted. After seeding, si-ARID1A (10 nM) was transfected, followed by a secondary transfection using si-MDM2 (10 nM). Images were captured every three hours for around 100 hours. Figure 4B shows the protein expression level of MDM2 for each cell line. Interference of ARID1A by 10 nM siRNA resulted in decreased MDM2 protein expression at 96 hours. MDM2, mouse double minute protein 2; siRNA, small interfering ribonucleic acid. **, p < 0.01.
2.6. Nutlin-3 Inhibits Tumor Growth in a Xenograft Mouse Model
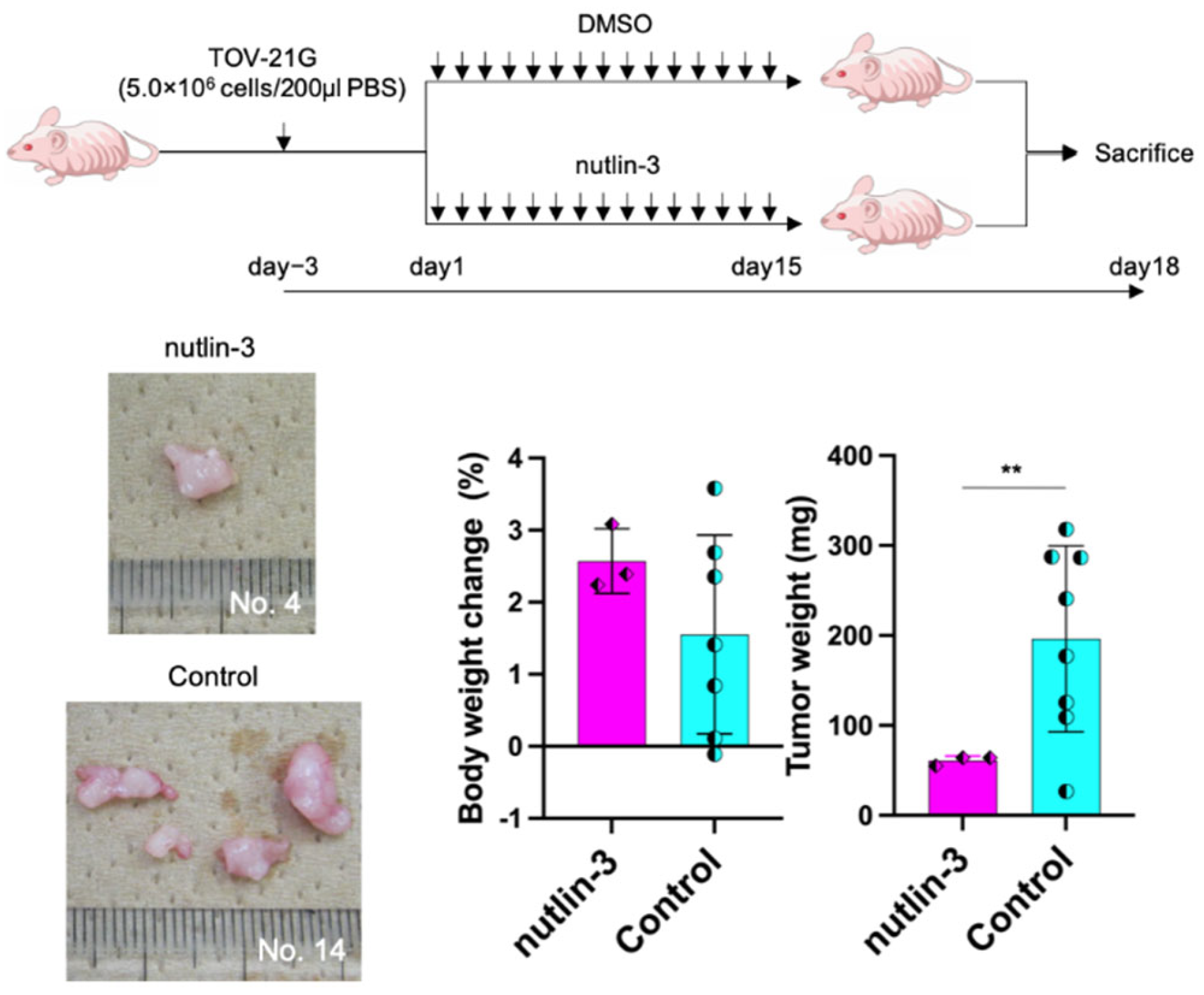
To determine whether the interference of MDM2 shows a suppressive effect on tumor growth, we conducted an in vivo assay using a xenograft mouse model. We used nutlin-3 as a molecule that suppresses MDM2. Cases with confirmed tumor viability at the time of sacrifice were included in this analysis: nutlin-3 group, n=3 and control group, n=7. Although body weight did not show significant differences between the groups, tumor weight determination indicated that the target group showed significantly decreased tumor growth than the control group (61.1 ± 4.2 vs. 196.4 ± 96.8, p = 0.008) (Figure 5).
7.5 × 106 of TOV-21G cells were injected into intraperitoneal in five-to-six-week-old athymic nude mice to generate murine intraperitoneal tumors. We used 40mg/kg of nutin-3 and the same Dimethyl sulfoxide as control. One week after the injection, we separated the mice into two groups based on their weight: the nutlin-3 group (n = 7) and control group (n = 10). Reagents were injected once a day for 15 days. Three days after the last injection, the mice were sacrificed. Although body weight did not show significant differences between the two groups, tumor weight determination indicated that the target group showed significantly decreased tumor growth compared to the control group. MDM2, mouse double minute protein 2; **, p < 0.01.
3. Discussion
MDM2 is a protein that inhibits regulation of the activity the cancer suppressor p53. This study showed that MDM2 could be a synthetic partner of ARID1A mutations. In a small number of cell lines, ARID1A accelerated the expression level of MDM2 and played an essential role in proliferation. The ARID1A mutation effectively reduced the necessary amount of MDM2 in the cells only by MDM2 interference, which suppressed cell growth.
ARID1A mutations exist in more than 50% of OCCC cases and approximately 45% of endometriosis-associated ovarian carcinoma (EAOC) cases [14,15,16,17]. EAOC is a group of malignant tumors that arise from ovarian endometrioma, characterized by repeated hemorrhages in the ovaries and a reactive oxygen species (ROS)-rich environment due to iron accumulation [18]. The ARID1A gene encodes ARID1A/BAF250A, a key subunit of the SWI-SNF chromatin remodeling complex [16,19]. Thus, the ARID1A gene is considered a tumor-suppressor gene, and ARID1A pathogenic mutations are generally loss-of-function mutations (nonsense, frame-shifts, and large deletions) that lead to the loss of ARID1A protein expression [20,21,22]. The importance of ARID1A mutation in the malignant transformation of ovarian endometriosis remains unclear, although the possibility of a two-hit hypothesis has been suggested [23]. Interestingly, some studies have revealed that ROS decreases ARID1A expression by promoter methylation in ovarian cancers [24,25], and ARID1A loss sensitizes ovarian cancer cells to ROS-inducing agents [26]. Because EAOC, including OCCC, arise from a ROS-rich environment, our study’s results are consistent with those in the aforementioned studies.
MDM2 binds to the p53 tumor-suppressor protein with high affinity and negatively modulates its transcriptional activity and stability [27,28]. It also affects the nuclear export of p53 and serves as a ubiquitin ligase that promotes p53 degradation [28,29]. These functions contribute to its oncogenic effects, e.g., pro-angiogenic activity, chromosomal instability, and degradation of cell cycle regulators [29]. Nutlin-3 is the first small molecule to inhibit MDM2/p53 binding, as reported by Vassilev et al. [28,29]. It can induce apoptosis in cancer cells by activating or stabilizing p53 [27,30]. Moreover, nutlin-3 induces increase MDM2 and inhibits DNA double-strand break repair [27], and it is effective against chemo resistant tumors through activation of the p53 pathway [31]. This mechanism suggests that nutlin-3 is effective in treating wild-type p53 wild ovarian cancer [27,31]. In particular, OCCC, which is the therapeutic target of this report, comprises only 10% of p53 mutated type cancer [32]. The use of nutlin-3, which is mostly a wild-type p53, makes sense as a therapeutic strategy. Until now, the effect of nutlin-3 has focused only on its association with p53 mutations; however, we believe that the presence or absence of ARID1A mutations, in addition to p53 mutations, can be considered to more efficiently identify cancers in which nutlin-3 exerts its effect. There have been no clinical trials on nutlin-3. However, similar compounds, e.g., RG7112, idasanutlin, and AMG-232, have been used in clinical trials for sarcoma, leukemia, lymphoma, and melanoma [29,33].
This study has a limitation. In ovarian clear cell carcinomas arising from high ROS levels, mutation or loss of the ARID1A gene is likely to contribute to cell survival by decreasing MDM2 expression and increasing p53 stability, although it seems to conflict with the original role of p53 in terms of tumor suppression. Nevertheless, ARID1A gene mutation and tumorigenesis are still unclear.
4. Materials and Methods
4.1. Ethics Statements
All animal experiments were conducted in accordance with the Guidelines for Proper Conduct of Animal Experiments (June 1, 2006, Science Council of Japan), and this study was approved by the Animal Ethics Committee of Nara Medical University (number: 13491).
4.2. Cell Lines
We used TOV-21G and KOC7c cells as the ARID1A mutant-type, and RMG-I and ES2 cells as the ARID1A wild-type. All cells were maintained in a humidified incubator at 37°C with 5% carbon dioxide (CO2). TOV-21G and ES2 cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA), whereas KOC7c and RMG-I cells were provided by Itamochi (Tottori University School of Medicine, Yonago, Japan).
These cells were maintained in the Dulbecco Modified Eagle Medium/Ham F-12 with L-glutamine and phenol red containing 10% fetal bovine serum and 100-U/mL penicillin and streptomycin and used at a sub-confluent status.
4.3. Small Interfering RNA Library Screening
We performed small interfering RNA (siRNA) library screening of human cell cycle regulation-related genes, deubiquitinating enzymes, and DNA damage response genes (G-003205, G-006005, and G-004705; Dharmacon™, Cambridge, UK). The RMG-I cell line was grown in six-well plate at a concentration of 4.0 × 105 cells per well, and si-ARID1A (SI03051461; Qiagen, Hilden, Germany) or si-control (D-001210-02; Dharmacon™) was reverse transfected rapidly at 5 nM according to the manufacturer’s recommended protocol (Figure 1A). At 48 hours after transfection, ARID1A-knockdown and control cells were plated in three wells of a 96-well plate at a concentration of 5000 cells per well (Figure 1A). In each of the three wells of ARID1A- knockdown and control cells, 5 nM of the respective siRNA was transfected for screening. After 48 hours, cell viability was measured using the MTT assay (Cell Proliferation Kit I; Roche, Salzburg, Austria) according to the recommended protocol. For each 96-well plate, we transfected the si-control as the negative control and si-PLK1 (M-003290-01; Dharmacon™) as the positive control [12]. Candidates were extracted as follows. First, the difference in cell viability between cells transfected with si-control and si-ARID1A was considered to be an effect of ARID1A downregulation. Second, to avoid cell population error between the two cell groups at the start of the assay, given that the cell viability of the negative control group showed a normal distribution, we corrected the test results on the basis of the difference from the negative control. Lastly, for further assessment, the most effective sequence was determined by MTT assay using TOV-21G cells.
4.4. Western Blotting
TOV-21G, KOC7c, and ES2 cell lines were grown in a six-well dish (2.0 × 105 cells per well for 48 hours and 1.0 × 105 cells per well for 72 hours), and RMG-I cell lines were grown in a six-well dish (4.0 × 105 cells per well for 48 hours and 3.0 × 105 cells per well for 72 hours), as determined by the growing speed of cell lines. After placing the cell lines in dishes, si-MDM2 and si-control were reverse transfected at 5 nM using the manufacturer’s recommended protocol. We extracted proteins at 48 and 72 hours after transfection. Samples were applied to Mini-PROTEAN®TGX™ Gels 4–15% and transferred by Trans-Blot®Turbo™ Transfer Pack (Bio-Rad, Hercules, CA, USA). The following antibodies were used for Western blotting: primary antibodies against MDM2 (#86934; Cell Signaling Technology, San Diego, CA, USA; 1:1,000 dilution) and ß-actin (#4970; Cell Signaling Technology; 1:10,000 dilution). Horseradish peroxidase-conjugated secondary antibodies against rabbit immunoglobulin G (sc-2004; Santa Cruz Biotechnology, Dallas, TX, USA; 1:10,000 dilution) were used.
4.5. Real-Time Polymerase Chain Reaction
RNA extraction to four cell lines (TOV-21G, KOC7c, ES2, and RMG-I) was performed at 24 and 48 hours after transfection using a TaqMan Gene Expression Cells-to-CT™ Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Real-time polymerase chain reaction (RT-PCR) was performed on a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with 4 μL of complementary DNA, 10 μL of the TaqMan Gene Expression Master Mix (4369016; Applied Biosystems), 1 μL of MDM2 or GAPDH TaqMan Gene Expression Assay (Hs01066938_m1 or Hs99999905_m1; Applied Biosystems), and 5 μL of nuclease-free water (B-003000-WB-100; Dharmacon™), and the results were analyzed using the relative quantitative method.
4.6. Cell Proliferation Assay
The viabilities of the four cell lines (TOV-21G, KOC7c, ES2, and RMG-I) were assessed after reverse transfection with si-MDM2 or si-control. After placing 150 µL of cell lines on dishes (5.0×103 cells/μL of TOV-21G, KOC7c and ES2 and 1.0×104 cells/μL of RMG-I), si-MDM2 and si-control were reverse transfected at 5 nM using the manufacturer’s recommended protocol. We then performed cell proliferation assays at 24, 48, and 72 hours after transfection. Next, we pipetted 40 μL of CellTiter 96® AQueous One Solution Reagent into each well of the 96-well assay plate containing the samples in 200 μL of culture medium. Finally, we incubated the plate at 37℃ for 30 minutes in a humidified, 5% CO2 atmosphere, and the absorbance was recorded at 492 nm using a 96-well plate reader (Supplementary Figure 1).
4.7. Cell Cycle Analysis
TOV-21G cells were grown in six-well dishes (2.0 × 105 cells per well), and si-MDM2 and si-control were reverse transfected at 5 nM according to the manufacturer’s recommended protocol. The cells were harvested and washed in phosphate-buffered saline (PBS) before fixation in cold 70% ethanol, which was added dropwise to the pellet while vortexing. Cells were fixed for 30 minutes at 4℃, and the fixed cells were washed twice with PBS and centrifuged at 250 × g for 5 minutes. Then, cells were incubated with 50 μL of a 100-μg/mL stock of RNase and 200-μL propidium iodide (from 50-μg/mL stock solution). A FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA) was used to analyze the cell population for cell cycle changes (Figure 2C).
4.8. Time-Lapse Cell Proliferation Assessment
Cell proliferation was studied using the IncuCyte ZOOM™ Live Cell Imaging system (Essen BioScience, Ann Arbor, MI, USA), as previously described for kinetic monitoring of proliferation and cytotoxicity of cultured cells [13]. The IncuCyte image assay quantifies how rapidly the proportion of area covered by cells increases with time as a function of the cell proliferation rate.
First, TOV-21G, KOC-7c, ES2, and RMG-I cells were seeded into 96-well plates at a concentration of 5000 cells each per well, and nutlin-3 was added at a concentration gradient (40.0, 13.33, 4.44, 1.48, 0.49, and 0 μM). The IncuCyte ZOOM™ apparatus was used, and incubation continued over 120 hours (Figure 3).
ES2 and RMG-I cells were seeded into six-well plates at a concentration of 2.0 × 105 cells and 4.0 × 105 cells each per well, and si-ARID1A (10 nM) was transfected. Forty-eight hours later, each cell line was seeded into a 96-well plate, and a secondary transfection was performed using si-MDM2 (10 nM) (0 hours). The cells were transferred to the IncuCyte ZOOM™ apparatus, and incubation continued for approximately 100 hours. During the incubation period, the IncuCyte captured images every 3 hours. After defining the area of the cells, all images were analyzed chronologically, focusing on confluence (%) (Figure 4A).
4.9. DNA Damage Assessment
The cells were seeded into six-well plates at a concentration of 2.0 × 105 cells per well and then transfected with 15-nM si-MDM2 and si-control (0 hours). After 24 or 48 hours, cells were fixed with 4% paraformaldehyde containing 0.1% Triton-X and 250-mM HEPES for 15 minutes. For permeabilization, 1% was used for permeabilization. The blocking solution, which contained the antibody of phosphorylated histone H2AX (γH2AX) and the secondary antibody, was included in the DNA damage detection kit (G265, Dojindo Laboratories, Kumamoto, Japan) and used according to the manufacturer’s instructions. Images were captured using a fluorescence microscope BZ-X710 (Keyence, Osaka, Japan). All images were captured at 20× magnification (Supplementary Figure 2).
4.10. In Vivo Assay
To generate murine intraperitoneal tumors, 7.5 × 106 TOV-21G cells in 200 μL of PBS were injected subcutaneously into the intraperitoneum in 20 of 5–6-week-old female athymic nude mice (SLC, Hamamatsu, Japan). We administered 40-mg/kg nutin-3 (Cat. No. S1061; Selleckchem, Houston, USA) or the same amount of dimethyl sulfoxide (control) subcutaneously once daily. One week after the injection, we separated the mice into two groups based on their body weight: the nutlin-3 group (n = 10) and the control group (n = 10). The reagents were injected once daily for 15 days. Three days after the last injection, we sacrificed the mice (Figure 5). Cases in which the tumor tissue could not be identified and cases that resulted in death over the course of the study were excluded.
4.11. Statistical Analysis
Data are presented as mean ± standard deviation. The Student t-test was used to assess the difference between the target and control groups, and the Mann–Whitney U test was applied for variables that did not present a normal distribution. For multiple comparisons, one-way analysis of variance (ANOVA) was conducted, followed by the Grams–Howell test. The synergistic effect of concomitant cisplatin use was assessed using two-way ANOVA. A two-sided p-value <0.05 was considered statistically significant, and all statistical analyses were performed using SPSS (version 29.0; IBM Corp., Armonk, NY, USA).
5. Conclusions
This study demonstrates that MDM2 could be a synthetic lethal target for ARID1A mutative OCCC, offering a new therapeutic approach for this challenging type of cancer.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: DNA damage assessment of MDM2 interference on TOV-21G. There was no significant differentiation between the si-MDM2 and the si-control group at 24h or 48h. MDM2, mouse double minute protein 2; HEPES, 4-(2-hydroxyethy)-piperazine-1-ethanesulfonic acid. Figure S2: Cell Proliferation Assay of OCCC cells under MDM2 interference. The ARID1A mutated cells showed a significant difference between si-MDM2 and si-control. MDM2, mouse double minute protein 2; *, p < 0.05, **, p < 0.01.
Author Contributions
Conceptualization, T.M., N.K. and H.K.; methodology, T.M., N.K. and Y.Y.; validation, T.M. and N.K.; formal analysis, T.M. and N.K.; investigation, T.M. and N.K.; resources, T.M., N.K., R.K., and F.K.; data curation, T.M., N.K., M.M., S.Y. and Y.Y.; writing—original draft preparation, T.M., N.K., and K.N.; writing—review and editing, T.M., N.K., R.K., and F.K.; visualization, T.M., M.M., and S.Y.; supervision, F.K.; project administration, F.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by JSPS KAKENHI (grant no. 22K09644).
Institutional Review Board Statement
All animal experiments were conducted according to the Guidelines for the Proper Conduct of Animal Experiments (1 June 2006, Science Council of Japan) and this study was approved by the animal ethics committee of Nara Medical University (No. 13491).
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare that no competing financial interests.
References
- Dinkelspiel HE, Champer M, Hou J, Tergas A, Burke WM, Huang Y, Neught AI, Ananth CV, Hershman DL and Wright JD: Long-Term Mortality Among Women with Epithelial Ovarian Cancer. Gynecol Oncol 138: 421-428, 2015.
- Siegel RL, Miller KD and Jemal A: Cancer statistics, 2019. CA Cancer J Clin 69: 7-34, 2019.
- Cunningham JM, Winham SJ, Wang C, Weiglt B, Fu Z, Armasu SM, McCauley BM, Brand AH, Chiew YE, Elishaev E, Gourley C, Kennedy CJ, Laslavic A, Lester J, Piskorz A, Sekowska M, Brenton JD, Churchman M, DeFazio A, Drapkin R, Elias KM, Huntsman DG, Karlan BY, Köbel M, Konner J, Lawrenson K, Papaemmanuil E, Bolton KL, Modugno F, Goode EL. DNA Methylation Profiles of Ovarian Clear Cell Carcinoma. Cancer Epidemiol Biomarkers Prev 31: 132-141, 2022.
- Makii C, Ikeda Y, Oda K, Uehara Y, Nishijima A, Koso T, Kawata Y, Kashiyama T, Miyasaka A, Sone K, Tanikawa M, Tsuruga T, Mori-Uchino M, Nagasaka K, Matsumoto Y, Wada-Hiraike O, Kawana K, Hasegawa K, Fujiwara K, Aburatani H, Osuga Y, Fujii T. Anti-tumor activity of dual inhibition of phosphatidylinositol 3-kinase and MDM2 against clear cell ovarian carcinoma. Gynecol Oncol 155: 331-339, 2019.
- Adams M, Cookson VJ, Higgins J, Martin HL, Tomlinson DC, Bond J, Morrison EE, Bell SM. A high-throughput assay to identify modifiers of premature chromosome condensation. J Biomol Screen 19: 176–183, 2013.
- Kurman RJ, Shih IeM. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer--shifting the paradigm. Hum Pathol. 42:918-31, 2011.
- Kawahara N, Yamada Y, Kobayashi H. CCNE1 Is a Putative Therapeutic Target for ARID1A-Mutated Ovarian Clear Cell Carcinoma. Int J Mol Sci. 22:5869, 2021.
- Johnston ST, Shah ET, Chopin LK, Sean McElwain DL, Simpson MJ. Estimating cell diffusivity and cell proliferation rate by interpreting IncuCyte ZOOM™ assay data using the Fisher-Kolmogorov model. BMC Syst Biol. 9:38, 2015.
- Kobayashi Y, Masuda K, Hiraswa A, Takehara K, Tsuda H, Watanabe Y, Oda K, Nagase S, Mandai M, Okamoto A, Yaegashi N, Mikami M, Enomoto T, Aoki D, Katabuchi H; Working Group on Clinical Practice for Cancer Genomic Medicine and HBOC, Japan Society of Gynecologic Oncology. Current status of hereditary breast and ovarian cancer practice among gynecologic oncologists in Japan: a nationwide survey by the Japan Society of Gynecologic Oncology (JSGO). J Gynecol Oncol. 33:e61, 2022.
- Gadducci A, Multinu F, Cosio S, Carinelli S, Ghioni M, Aletti GD. Clear cell carcinoma of the ovary: Epidemiology, pathological and biological features, treatment options and clinical outcomes. Gynecol Oncol. 162:741-750, 2021.
- Kawahara N, Ogawa K, Nagayasu M, Kimura M, Sasaki Y, Kobayashi H, Candidate synthetic lethality partners to PARP inhibitors in the treatment of ovarian clear cell cancer. Biomed. Rep. 7: 391–399, 2017.
- Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 363:1532–1543, 2010.
- Jones S, Wang TL, Shih IM, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA, Vogelstein B, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 330:228–231, 2010.
- Guan B, Gao M, Wu CH, Wang TL, Shih IM. Functional analysis of in-frame indel ARID1A mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia. 14:986–993, 2012.
- Köbel M, Kalloger SE, Huntsman DG, Santos JL, Swenerton KD, Seidman JD, Gilks CB. Differences in tumor type in low-stage versus high-stage ovarian carcinomas. Int. J. Gynecol. Pathol. 29:203–211, 2010.
- Chan JK, Teoh D, Hu JM, Shin JY, Osann K, Kapp DS. Do clear cell ovarian carcinomas have poorer prognosis compared to other epithelial cell types? A study of 1411 clear cell ovarian cancers. Gynecol. Oncol. 109:370–376, 2008.
- Mackay HJ, Brady MF, Oza AM, Reuss A, Pujade-Lauraine E, Swart AM, Siddiqui N, Colombo N, Bookman MA, Pfisterer J, et al. Prognostic relevance of uncommon ovarian histology in women with stage III/IV epithelial ovarian cancer. Int. J. Gynecol. Cancer. 20:945–952, 2010.
- Takahashi K, Takenaka M, Okamoto A, Bowtell DDL, Kohno T. Treatment Strategies for ARID1A-Deficient Ovarian Clear Cell Carcinoma. Cancers (Basel)13:1769, 2021.
- Carrillo AM, Hicks M, Khabele D, Eischen CM. Pharmacologically Increasing Mdm2 Inhibits DNA Repair and Cooperates with Genotoxic Agents to Kill p53-Inactivated Ovarian Cancer Cells. Mol Cancer Res13:1197-205, 2015.
- Mir R, Tortosa A, Martinez-Soler F, Vidal A, Condom E, Pérez-Perarnau A, Ruiz-Larroya T, Gil J, Giménez-Bonafé P. Mdm2 antagonists induce apoptosis and synergize with cisplatin overcoming chemoresistance in TP53 wild-type ovarian cancer cells. Int J Cancer132:1525-36, 2013.
- Son DS, Kabir SM, Dong YL, Lee E, Adunyah SE. Inhibitory effect of tumor suppressor p53 on proinflammatory chemokine expression in ovarian cancer cells by reducing proteasomal degradation of IκB. PLoS One7:e51116, 2012.
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science303:844-8, 2004.
- Haronikova L, Bonczek O, Zatloukalova P, Kokas-Zavadil F, Kucerikova M, Coates PJ, Fahraeus R, Vojtesek B. Resistance mechanisms to inhibitors of p53-MDM2 interactions in cancer therapy: can we overcome them? Cell Mol Biol Lett26:53, 2021.
- Makii C, Oda K, Ikeda Y, Sone K, Hasegawa K, Uehara Y, Nishijima A, Asada K, Koso T, Fukuda T, Inaba K, Oki S, Machino H, Kojima M, Kashiyama T, Mori-Uchino M, Arimoto T, Wada-Hiraike O, Kawana K, Yano T, Fujiwara K, Aburatani H, Osuga Y, Fujii T. MDM2 is a potential therapeutic target and prognostic factor for ovarian clear cell carcinomas with wild type TP53. Oncotarget7:75328-75338, 2016.
- Traweek RS, Cope BM, Roland CL, Keung EZ, Nassif EF, Erstad DJ. Targeting the MDM2-p53 pathway in dedifferentiated liposarcoma. Front Oncol2:1006959, 2022.
- Doi, T. , Yoshino T., Shitara K., Matsubara N., Fuse N., Naito Y., Uenaka K., Nakamura T., Hynes S.M., Lin A.B. Phase I study of LY2603618, a CHK1 inhibitor, in combination with gemcitabine in Japanese patients with solid tumors. Anticancer Drugs26:1043–1053, 2015.
- Farmer, H. , McCabe N., Lord C.J., Tutt A.N.J., Johnson D.A., Richardson T.B., Santarosa M., Dillon K.J., Hickson I.D., Knights C., et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature434:917–921, 2005.
- Pejovic T, Cathcart AM, Alwaqfi R, Brooks MN, Kelsall R, Nezhat FR. Genetic Links between Endometriosis and Endometriosis-Associated Ovarian Cancer-A Narrative Review (Endometriosis-Associated Cancer). Life (Basel)14:704, 2024.
- Samartzis, E.P. , Labidi-Galy S.I., Moschetta M., Uccello M., Kalaitzopoulos D.R., Perez-Fidalgo J.A., Boussios S. Endometriosis-associated ovarian carcinomas: Insights into pathogenesis, diagnostics, and therapeutic targets—A narrative review. Ann. Transl. Med8:1712, 2020.
- Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer11:481-92, 2011.
- Calvo, E. , Chen V.J., Marshall M., Ohnmacht U., Hynes S.M., Kumm E., Diaz H.B., Barnard D., Merzoug F.F., Huber L., et al. Preclinical analyses and phase I evaluation of LY2603618 administered in combination with pemetrexed and cisplatin in patients with advanced cancer. Investig. New Drugs32:955–968, 2014.
- Italiano, A. , Infante J., Shapiro G., Moore K., LoRusso P., Hamilton E., Cousin S., Toulmonde M., Postel-Vinay S., Tolaney S., et al. Phase I study of the checkpoint kinase 1 inhibitor GDC-0575 in combination with gemcitabine in patients with refractory solid tumors. Ann. Oncol29:1304–1311, 2018.
- Iglehart, J.D. , Silver D.P. Synthetic lethality: A new direction in cancer-drug development. N Engl J Med361:189–191, 2009.
- Yachida N, Yoshihara K, Suda K, et al. ARID1A protein expression is retained in ovarian endometriosis with ARID1A loss-of-function mutations: implication for the two-hit hypothesis. Sci Rep10:14260, 2020.
- Zhou W, Liu H, Yuan Z, et al. Targeting the mevalonate pathway suppresses ARID1A-inactivated cancers by promoting pyroptosis. Cancer Cell41:740-756, 2023.
- Chan JK, Teoh D, Hu JM, Shin JY, Osann K, and Kapp DS. Do clear cell ovarian carcinomas have poorer prognosis compared to other epithelial cell types? A study of 1411 clear cell ovarian cancersGynecologic oncology. 109: 370–376, 2008.
- Mackay HJ, Brady MF, Oza AM, Reuss A, Pujade-Lauraine E, Swart AM, Siddiqui N, Colombo N, Bookman MA, Pfisterer J, du Bois A; Gynecologic Cancer InterGroup. Prognostic relevance of uncommon ovarian histology in women with stage III/IV epithelial ovarian cancer. Int J Gynecol Cancer. 9: 20, 2010; -52.
- Yamaguchi K, Mandai M, Toyokuni S, Hamanishi J, Higuchi T, Takakura K, Fujii S. Contents of endometriotic cysts, especially the high concentration of free iron, are a possible cause of carcinogenesis in the cysts through the iron-induced persistent oxidative stress. Clin Cancer Res14:32-40, 2008.
- Xie H, Chen P, Huang HW, Liu LP, Zhao F. Reactive oxygen species downregulate ARID1A expression via its promoter methylation during the pathogenesis of endometriosis. Eur Rev Med Pharmacol Sci21:4509-4515, 2017.
- Winarto H, Tan MI, Sadikin M, Wanandi SI. ARID1A Expression is Down-Regulated by Oxidative Stress in Endometriosis and Endometriosis-Associated Ovarian Cancer. Transl Oncogenomics. 9:1177272716689818, 2017.
- Kwan SY, Cheng X, Tsang YT, Choi JS, Kwan SY, Izaguirre DI, Kwan HS, Gershenson DM, Wong KK. Loss of ARID1A expression leads to sensitivity to ROS-inducing agent elesclomol in gynecologic cancer cells. Oncotarget7:56933-56943, 2016.
Figure 1.
The synthetic lethal elucidation for ARID1A mutative ovarian clear cell carcinoma.
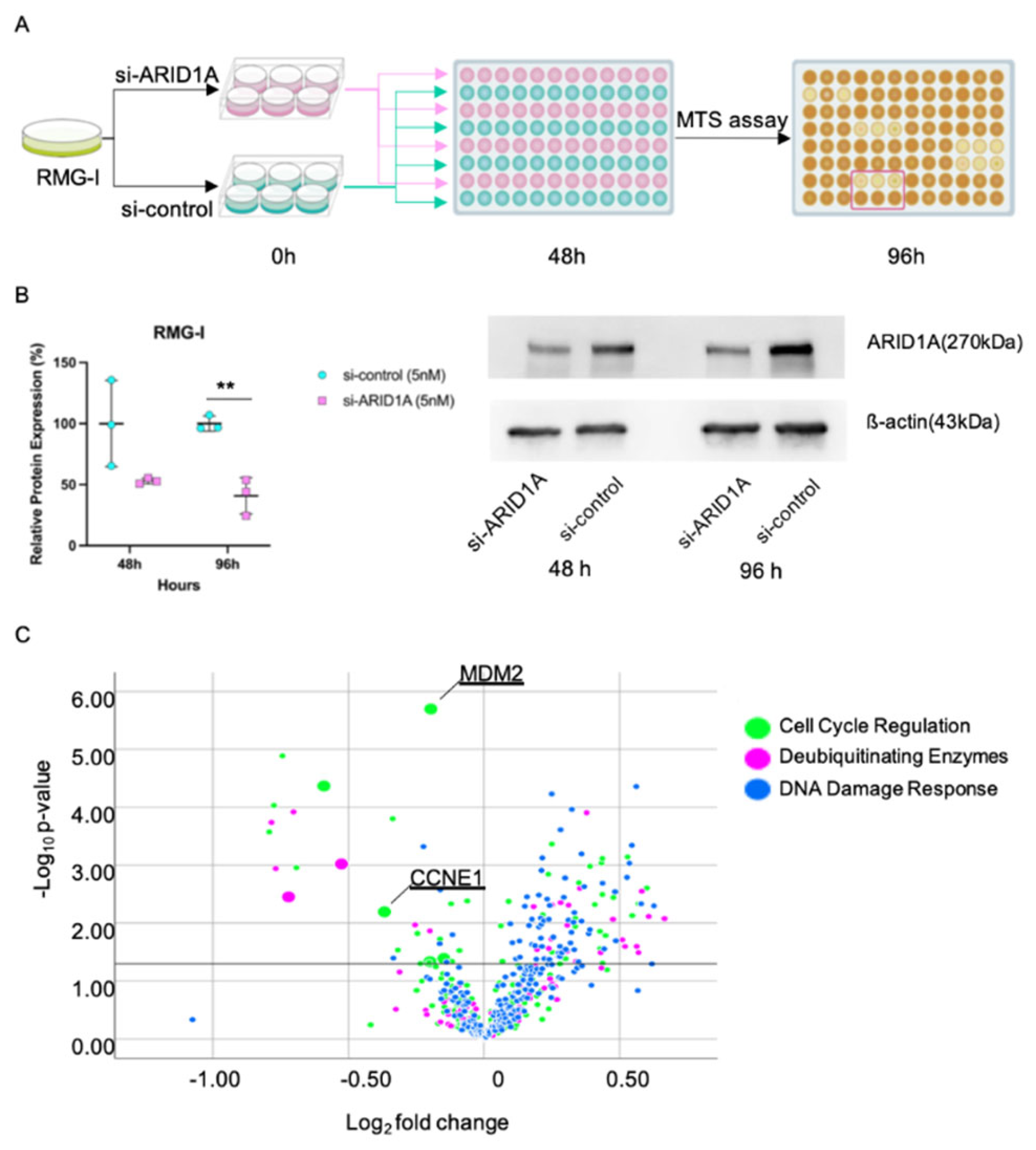
Figure 2.
The effects of si-MDM2 on gene, cell cycle, and protein expression.
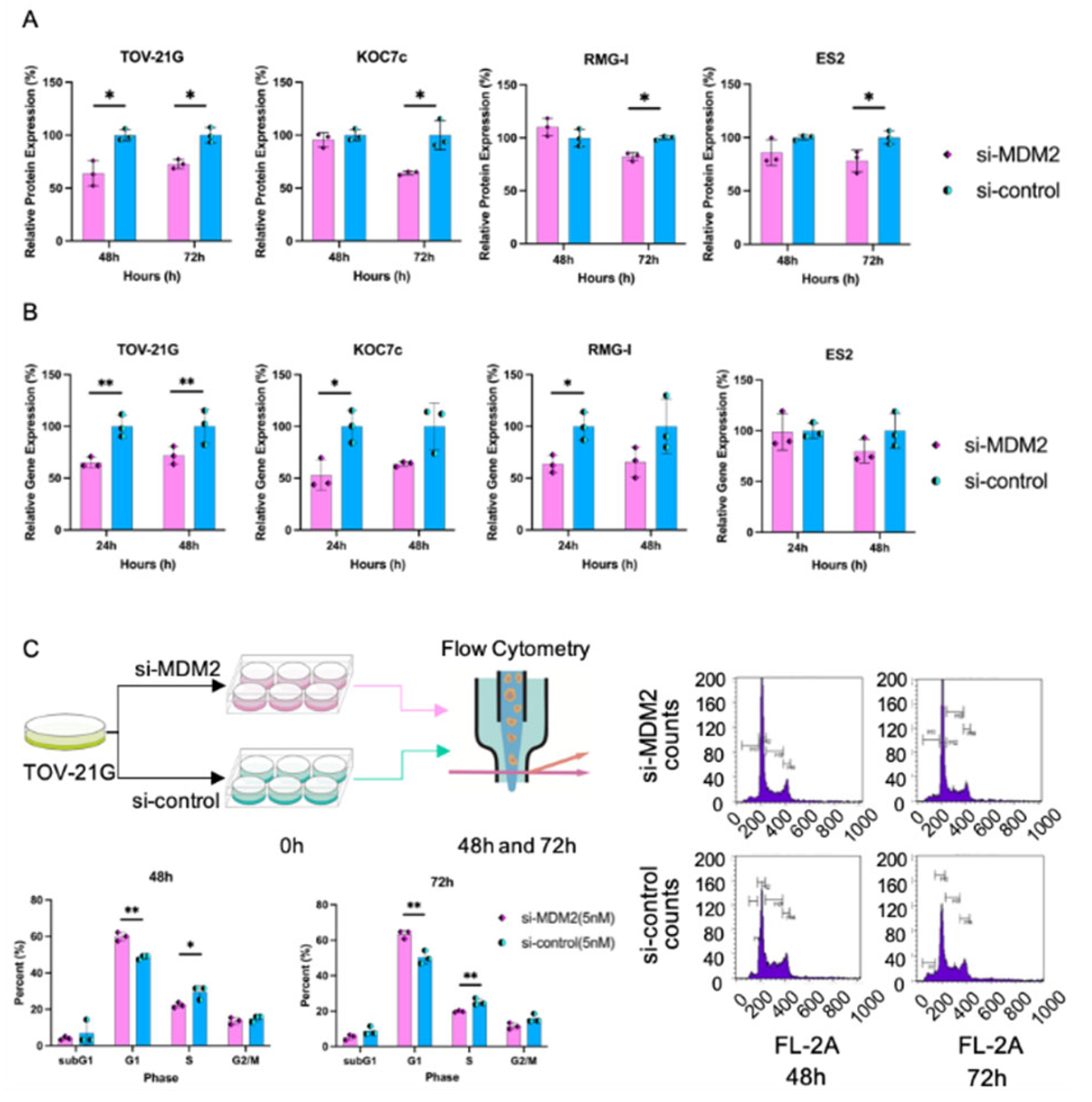
Figure 3.
Growth pattern of each OCCC cell upon nutlin-3 administration.
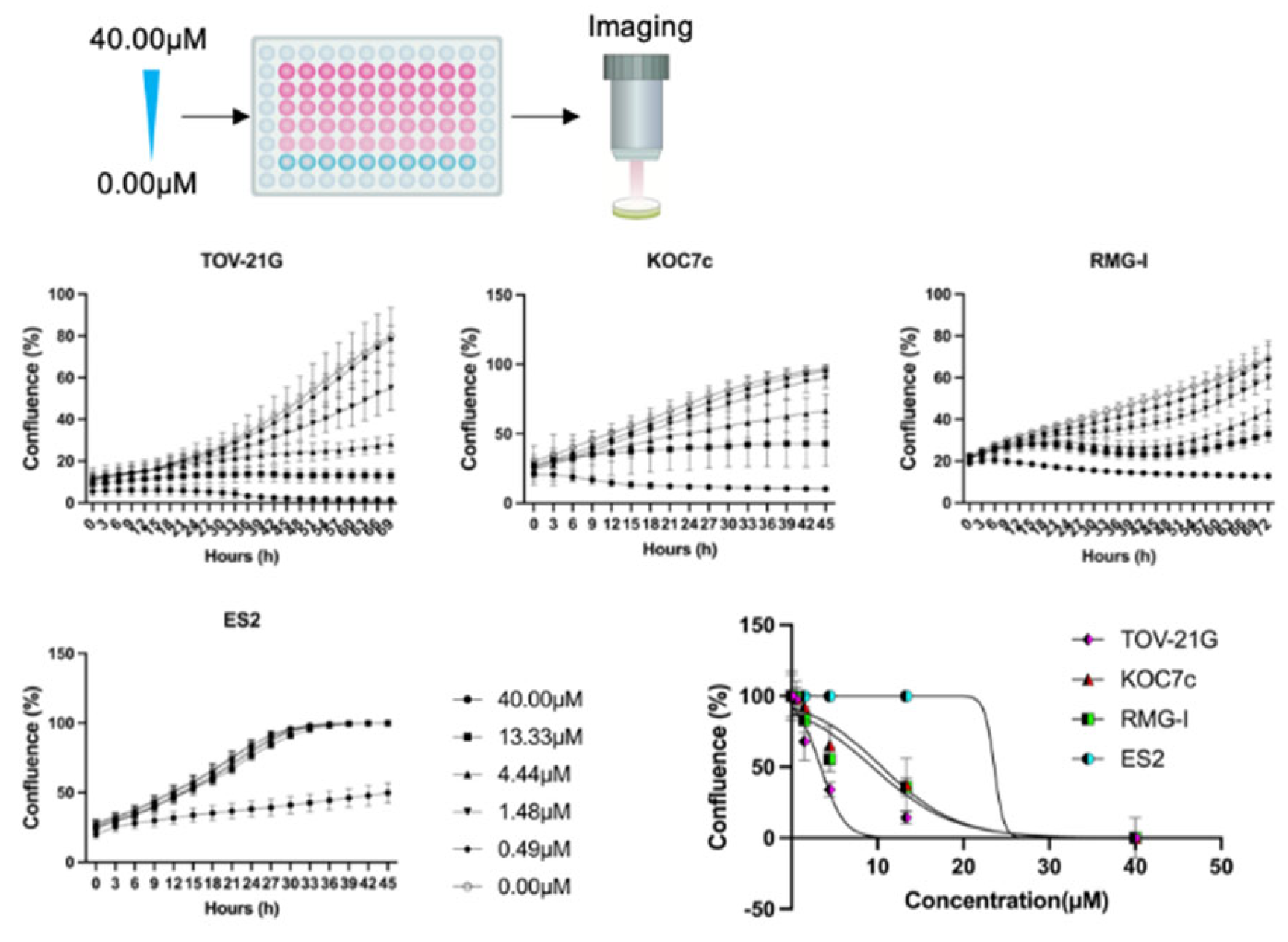
Figure 4.
Synthetic lethality of MDM2 interference under ARID1A interference.
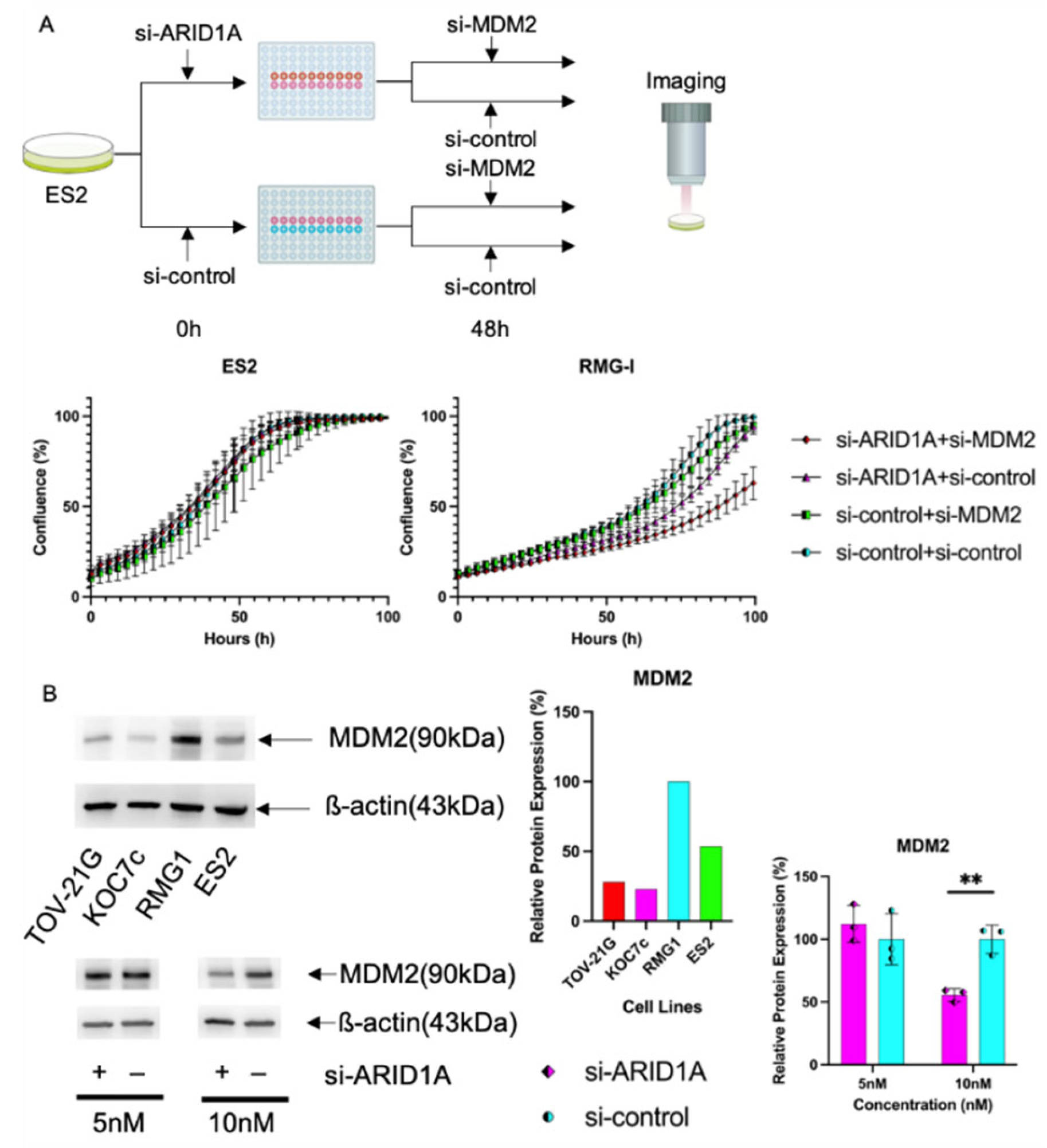
Figure 5.
The nutlin-3 administration in TOV-21G xenograft mouse model.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



