
Submitted:
11 October 2024
Posted:
12 October 2024
You are already at the latest version
Abstract
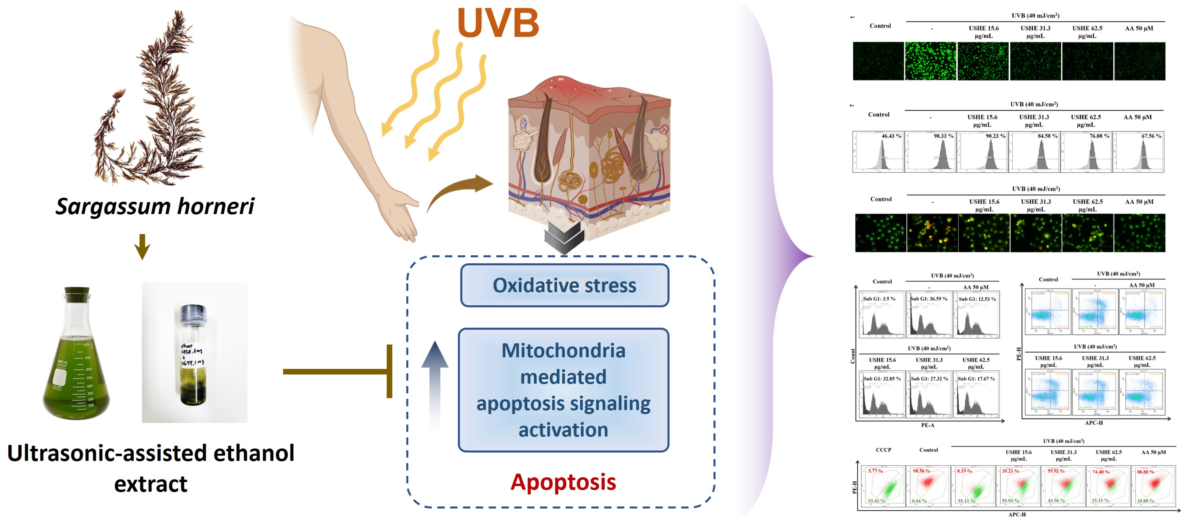
The present study investigated the photoprotective effect of the ultrasonic-assisted ethanol extract (USHE) from Sargassum horneri, a brown seaweed containing fucosterol (6.22 ± 0.06 mg/g) and sulfoquinovosyl glycerolipids (C23H43O11S, C25H45O11S, C25H47O11S, C27H49O11S), against oxidative damage in ultraviolet B (UVB)-exposed HaCaT keratinocytes. USHE indicated the antioxidant activity with increasing ferric-reducing antioxidant power (FRAP) and 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging. Moreover, USHE increased cell viability by markedly reducing the production of intracellular reactive oxygen species (ROS) in UVB-exposed HaCaT keratinocytes. Additionally, USHE reduced the apoptosis, and sub-G1 cell population, and increased the mitochondrial membrane potential. Moreover, USHE modulated the protein expression levels of anti-apoptotic molecules (Bcl-xL, Bcl-2, and PARP), and pro-apoptotic molecules (Bax, cleaved caspase-3, p53, cleaved PARP, and cytochrome C). This modulation accorded with the upregulation of cytosolic heme oxygenase (HO)-1, NAD(P)H quinone oxidoreductase 1 (NQO 1), and nuclear factor erythroid-2-related factor 2 (Nrf2), collectively known as components of the antioxidant system. These findings suggest that USHE has a photoprotective effect in UVB-exposed HaCaT keratinocytes and can be utilized to develop cosmeceuticals for UVB protection.
Keywords:
Sargassum horneri
; Photoprotective effect
; Apoptosis
; Oxidative stress
; Antioxidant capacity
1. Introduction
Ultraviolet radiation ranges from 290 to 400 nm in wavelength and reaches the earth surface due to the ozone layer being destroyed increasingly [1,2]. Ultraviolet B (UVB) radiation with wavelengths ranging from 290 to 320 nm [3], serves as a crucial catalyst for vitamin D synthesis in the skin [4]. The UVB leads to reactive oxygen species (ROS) production within cells in the epidermis and dermis of the skin [1,5]. ROS, such as superoxide radicals and hydrogen peroxide, act as signaling molecules and contribute to various cellular processes by modifying the activity of metabolic enzymes and transcription factors [6]. However, the production of ROS due to prolonged or intense exposure to UVB rays can overpower the natural defense mechanisms of antioxidants in skin cells. The oxidative stress affects the cellular elements including lipids, proteins, and DNA, and ultimately induces apoptosis known as programmed cell death [6,7]. Disrupted DNA leads to degenerative skin disorders for instance sunburns, suntans, blisters, erythema (redness), and melanomas [6].
Since UVB exposure is inevitable, it is essential to comprehend the delicate balance between potential benefits and health risks. Understanding this balance emphasizes the significance of implementing comprehensive sun protection strategies to preserve optimal skin health. These strategies may include antioxidant-rich skincare, protective clothing, and judicious use of sunscreen to mitigate the adverse effects of UVB radiation. Furthermore, many studies have been conducted recently to refine skin protective materials from safe, less toxic, and readily available bioresources [8]. Yet, it is crucial to clarify the molecular mechanism behind the biological function of natural products when searching for potential therapeutic remedies [8]. Moreover, recent studies have identified that marine resources consist of various bioactive components including polysaccharides, phenols, sterols, proteins, and a variety of pigments indicating photoprotective effects [9,10,11,12].
Sargassum is a genus of brown macroalgae in the order Fucales of the Phaeophyceae class. Sargassum spp. has gained attention as a resource for extracting bio functional components such as meroterpenoids, phlorotannins, sulfated polysaccharides, sterols, and glycolipids [13,14]. Sargassum horneri is an edible brown alga that is highly abundant along the coasts of Jeju Island in South Korea. As reported, due to its high content of bioactive compounds, S. horneri has drawn more interest in studies of anti-inflammatory, antioxidant, anti-allergy, and anticancer agents [14,15,16,17]. The current trend, which prioritizes synergistic pharmacological benefits over the 'one-target, one-drug' strategy in both cosmeceutical and pharmaceutical industries, serves as the basis for the present study.
Ethanol extracts and sterols refined from ethanol extracts of S. horneri have been widely studied in the past few years [14,17,18,19]. Nevertheless, the ethanol extract of S. horneri indicated a comparatively low extraction yield [14,20]. Therefore, this study was designed to reveal the photoprotective effects of the ultrasonic-assisted ethanol extract (USHE) from S. horneri on UVB-exposed HaCaT keratinocytes. The hypothesis is that USHE regulates intracellular ROS production and thereby recovers the cells from mitochondria-mediated apoptosis and downregulates oxidative stress through the nuclear factor erythroid-2-related factor 2(Nrf2)/heme oxygenase (HO)-1 signaling pathway.
2. Materials and Methods
2.1. Materials
Dulbecco’s modified eagle medium (DMEM), penicillin/streptomycin mixture, and fetal bovine serum (FBS) were purchased from GibcoBRL (Grand Island, NY, USA). Folin and Ciocalteu’s phenol reagent, 2,2-diphenyl-1-picrylhydrazyl (DPPH), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 2’,7’-dichlorodihydrofluorescein diacetate (DCF-DA), dimethyl sulfoxide (DMSO), bovine serum albumin (BSA), propidium iodide (PI), acridine orange (AO), ethidium bromide (EB), ascorbic acid (AA), agarose, and gallic acid were bought from Sigma-Aldrich (St. Louis, MO, USA). D-glucose was purchased from Junsei Chemical Co., Ltd. (Tokyo, Japan). Skim milk powder was obtained from BD DifcoTM (Sparks, MD, USA). BCA protein assay kit, NE-PER® nuclear and cytoplasmic extraction kit, 1-Step transfer buffer, Pierce™ RIPA buffer, and protein ladder were purchased from Thermo Fisher Scientific (Rockford, IL, USA). Primary and secondary antibodies needed for the western blot analysis were bought from Cell Signaling Technology (Beverly, MA, USA). The remaining chemicals and reagents used in the study were analytical grade.
2.2. Ultrasonic-Assisted Ethanol Extraction
S. horneri was obtained from the Jeju Island, South Korea. The sample was identified by the Biodiversity Research Institute in Jeju, South Korea. The sample was cleaned with running water at first. Afterward, samples were dried in the ventilated room at room temperature (27.5 ± 2.0°C) and stored at room temperature for three weeks until extraction began. After soaking the 200 g of dried sample for 1 h with distilled water, 30 L of 50% ethanol was filled into the ultrasonic apparatus (MD-1200PG, Mirae ultrasonic, KR) along with the sample. The ultrasonic strength used in this extraction was 28 kHz with output power at 1200W, the stirring speed was 60 rpm, and the initial temperature of the extract was 4°C. The average temperature during the extraction process was maintained within 4 ± 2.0°C using an ice bath. After 5 h of extraction, the supernatant was collected by centrifugation at 4800 rpm for 10 mins. Ethanol was evaporated from the filtered extract and stored at -20°C. The sample was dissolved in DMSO and diluted with Phosphate-Buffered Saline (PBS) for the experiments.
2.3. Compositional Analysis of USHE
The crude compositions of USHE were determined by following the methods mentioned in the previous study [21]. The phenolic content was quantified by employing a gradient concentration of gallic acid as the calibration standard. To determine the total protein content, the Lowry method was utilized with BSA serving as the benchmark. Carbohydrate content was determined using the phenol–sulfuric acid method, with d-glucose as the reference standard.
2.4. High-Performance Liquid Chromatography (HPLC) and Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Analysis
HPLC analysis was conducted to determine the fucosterol content in the USHE. For this analysis, a Waters Alliance e2695 Separations Module (USA) coupled with a Waters 2489 UV vis Detector (USA) and a YMC Pack-Pro C18 column (4.6 × 250 mm, 5 μm) was used. The solvent system comprised 35% ethanol and 65% acetonitrile. Peaks observed in each sample at 210 nm and 35°C were compared to a standard fucosterol sample dissolved in methanol. The other prominent compounds were identified by using a Waters Arc HPLC system (Waters Corp., USA) coupled with a Quattro Premier XE (Waters Corp., USA) mass spectrometer. A chromatogram was generated using an XBridge® C18 Column (4.6 x 150 mm, 3.5 μm, Waters Corp., USA). The mobile phase consisted of water (A) and acetonitrile (B), both with 0.1% formic acid. A linear gradient started from 10% B to 90% B over the first 15 min, followed by an increase from 90% to 100% B over the next 5 min. The flow rate through the column was 0.6 mL/min. The injection volume for the analysis was 10 μL.
2.5. Antioxidant Capacity of USHE
The DPPH radical scavenging activity of USHE was assessed using a method outlined in a previous study [21]. In brief, 100 μL of USHE hydrolysates was mixed with 100 μL of DPPH solution (150 μM). The absorbance of the mixture was measured utilizing a SpectraMax M2 microplate (Molecular Devices, Silicon Valley, CA, USA) reader at 517 nm after it had been kept in the dark at 25 °C for 30 mins. In addition, the reducing capacity of the sample is directly measured by the ferric reducing antioxidant power (FRAP) assay, which is an essential indicator for assessing its effectiveness as a strong antioxidant [22]. The 10% ferric chloride solution was used to measure the FRAP of USHE. In brief, a mixture was prepared with 0.1 M phosphate buffer (pH 6.6–7.0), 1% potassium ferricyanide, and either the sample or distilled water (as a control) in a 3:5:2 ratio. This mixture was vortexed and incubated at 50 °C for 20 mins. Following this, 10% trichloroacetic acid was added, and the mixture was centrifuged at 3000 rpm for 10 mins. The supernatant was then mixed with distilled water and 10% ferric chloride in a 5:5:1 ratio, and the absorbance was measured at 700 nm using a microplate reader.
2.6. Cell Culture
HaCaT keratinocytes were cultured in DMEM supplemented with 10% heat-inactivated FBS and a 1% penicillin/streptomycin antibiotic mixture. The cell culture was maintained in a humidified atmosphere with 5% CO2 at 37°C. The cells were regularly sub-cultured until they reached exponential growth. During cell subculturing, cells were washed with PBS after the culture medium was removed. Following, cells are allowed to detach with the aid of trypsin-EDTA (Gibco, Grand Island, NY, USA) for a brief period at 37°C. Subsequently, the DMEM medium is used to neutralize the trypsin activity. The cells are gently moved, and the suspension is centrifuged. Next, the pellet is reconstituted in a new DMEM medium, and the concentration of cells is ascertained. The cells are seeded into new culture flasks for further growth. Cells in the exponential growth phase, specifically between passages 3 and 10, were seeded and utilized for the respective experiments.
2.7. Cell Viability Analysis
Cytotoxicity of the USHE and its cytoprotective effect on HaCaT keratinocytes against UVB stimulation was analyzed by following the MTT colorimetric cell viability analysis method [23]. The HaCaT keratinocytes were seeded at 1 × 105 cells/well density in 96-well plates. The cell seeded-well plates were incubated for 24 h at the cell culture conditions mentioned in section 2.5. Following the incubation, cells were subjected to USHE at concentrations of 15.6, 31.3, 62.5, 125, and 250 µg/mL treatment to evaluate the cytotoxic effect. Then, DMSO was added to the wells after being incubated with MTT and the mixture was gently shaken in the dark to dissolve the formazan crystals. After 30 mins of shaking in the dark, absorbance was measured at 570 nm. The stimulated cells were subjected to the MTT cell survival analysis by following the same procedure after being stimulated with 40 mJ/cm2 of UVB exposure for 3.0 s at room temperature using an ultraviolet cross-linker (UVP CL-1000L, Upland, CA, USA).
2.8. Analysis of Intracellular ROS Production
Intracellular ROS production in stimulated cells was assessed using a DCF-DA assay. Briefly, 24 h after seeding, cells were treated with USHE at concentrations of 15.6, 31.3, and 62.5 µg/mL and incubated for 2 h. Following this, UVB stimulation was applied to enhance intracellular ROS production. After a 1 h stimulation period, DCF-DA was added to the cells, and fluorometric analysis was performed using a microplate reader with excitation and emission wavelengths of 485 and 528 nm, respectively. Additionally, DCF-DA treated cells were examined using a Thermo Fisher Scientific EVOS M5000 Imaging fluorescence microscope (Rockford, IL, USA) and a CytoFLEX flow cytometer (Beckman Coulter, Brea, CA, USA) as well as CytoFLEX flow cytometer (Beckman Coulter, Brea, CA, USA).
2.9. Nuclear Double Staining
Cells were seeded in 24 well plates and treated with selected concentrations of USHE for 2 h. After 24 h of UVB stimulation, cells were treated with an AO and EB 1:1 mixture (10 μg/mL). Stained cells were evaluated by using a fluorescence microscope following 10 mins of incubation.
2.10. Cell Cycle Analysis
Changes in the cell cycle were analyzed using the procedures outlined in a previous study [24]. Briefly, harvested cells were centrifuged at 1700 rpm, for 5 mins, and resulted cell pellet was permeabilized in 70% ethanol at 4°C overnight. Following a 5 mins centrifugation at 1700 rpm, ethanol was removed by using suction. Then, cells were washed with PBS containing 2 mM EDTA and centrifuged at 1500 rpm, for 5 mins. Subsequently, cells were resuspended in PBS containing EDTA, RNase, and propidium iodide, and incubated at 37°C for 30 mins before being analyzed using a flow cytometer.
2.11. Annexin V and JC-1 Assays
The stimulated cells were harvested after a 6 h incubation period for the flow cytometry-based Annexin V assay to assess apoptosis by using Annexin V-conjugated fluorescein isothiocyanate and propidium iodide kit (Invitrogen, CAT #V13242, Carlsbad, CA, USA). The cellular mitochondrial health status was evaluated using the MitoProbeTM JC-1 assay kit (Invitrogen, Carlsbad, CA, USA) after harvesting stimulated cells following a 4 h incubation period. The manufacturer's instructions were followed while conducting the analyses.
2.12. Western Blot Analysis
The harvested cells were lysed using NE-PER® nuclear and cytoplasmic extraction kits and separately collected the cytoplasmic and nuclear proteins. Afterward, protein levels in the lysates were determined using the BCA protein assay kit, and 30 µg of protein from each lysate was electrophoresed. Tris-glycine SDS-PAGE was done with 10% resolving gels and 5% stacking gels [15]. The resolved proteins in the gels were then transferred onto nitrocellulose membranes (Merck Millipore, Ireland), and blocked. Following this, the bands were sequentially incubated with primary (1:1000) and HRP-conjugated secondary (1:3000) antibodies. Images were taken by using the Core Bio Davinch ChemiTM imaging system (Seoul, Korea)
2.13. Statistical Analysis
The data are shown as mean ± standard error (SE), and the experiments were carried out in triplicate (n = 3). Significant differences among the data sets were evaluated using a one-way analysis of variance (ANOVA) followed by Duncan’s multiple range test, performed using IBM SPSS software (Version 24.0, Chicago, IL, USA). p ˂ 0.05 was considered statistically significant in this study.
3. Results
3.1. Extraction Yield and Proximate Composition of USHE, and Identification of Active Compounds
The extraction yield of USHE was 29.50 ± 1.32% and the proximate compositional analysis of USHE indicated a higher crude protein content (15.01 ± 1.06%) than carbohydrate (9.83 ± 0.87%) and total phenolic compounds (6.97 ± 0.36%) content (Table 1). HPLC analysis results revealed that the fucosterol content in USHE was 6.22 ± 0.06 mg/g (Figure S1) (Table 1). Furthermore, the main peaks in LC-MS/MS chromatography were identified as 1-O-Hexadecanoyl-3-O-(6′-sulfo-α-D-quinovopyranosyl) glycerol (Figure S2). The tandem mass analysis predicted other peaks to be related to isomer compounds. The high-resolution mass spectrum displayed m/z values of 527.2718, 553.3187, 555.3054, and 581.3199 ([M-H]), tentatively identified as 1-O-Tetradecanoyl-3-O-(6′-sulfo-α-D-quinovopyranosyl) glycerol, 1-O-(11-Hexadecenoyl)-3-O-(6′-sulfo-α-D-quinovopyranosyl) glycerol, 1-O-Hexadecanoyl-3-O-(6′-sulfo-α-D-quinovopyranosyl) glycerol, and 1-O-(9-Octadecenoyl)-3-O-(6′-sulfo-α-D-quinovopyranosyl) glycerol, respectively, based on high-resolution mass and AntiBase search (Table 2).
3.2. Antioxidant Capacity of USHE
According to the findings, USHE indicated DPPH radical scavenging activity with a respective IC50 value of 49 ± 0.24 µg/mL (Figure 1A). The FRAP of the USHE was increased dose-dependently with a maximum absorbance of 0.2927 at 62.5 μg/mL (Figure 1B). The reference standard (10 mM ascorbic acid) was used to compare the sample activities [21,25].
3.3. Effect of USHE on Cell Viability and Intracellular ROS Production in UVB-Exposed HaCaT Keratinocytes
Cytotoxicity of USHE concentrations and the effect on cell viability of the UVB-exposed HaCaT keratinocytes were evaluated by MTT assay. As illustrated in Figure 2A, a significant cytotoxic effect on HaCaT keratinocytes was not observed for any of the tested USHE concentrations. The cell viability was decreased upon UVB stimulation, however, markedly increased with USHE in a dose-dependently (Figure 2B). Nonetheless, 15.6, 31.3, and 62.5 µg/mL concentrations of USHE were selected to be used throughout the investigation due to the significant upsurge in cell viability without cytotoxic effect. Figure 2C shows that intracellular ROS production in UVB-exposed HaCaT keratinocytes was dramatically raised, while decreased with USHE. The ROS scavenging ability of USHE was further confirmed by DCF-DA fluorescence imaging and DCF-DA flow cytometric analysis. As depicted in Figure 2D, the intensity of green fluorescence elevated with UVB stimulation while dose-dependently reduced with USHE. The DCF-DA flow cytometry revealed that UVB radiation increased the number of cells with a higher fluorescence (x-axis), which is correlated with a higher ROS production (Figure 2E). However, the number of cells with a higher fluorescence was diminished with USHE in a dose-dependent manner. A 50 µM concentration of AA was used as a positive control for the cell experiments [21].
3.4. Effect of USHE on Proportion of Apoptotic Cells in UVB-Exposed HaCaT Keratinocytes
Apoptotic cell death is triggered by UVB-exposed oxidative stress in HaCaT keratinocytes [10]. Damaged membranes of late apoptotic and dead cells can be identified with EB staining. Figure 3A demonstrated an increase in the number of cells with orange-colored nuclei, which was subsequently decreased by USHE in a dose-dependent manner, indicating the attenuation of late apoptosis in UVB-exposed cells. In addition, cell cycle analysis was conducted to detect apoptosis and DNA damage after PI staining [24]. According to Figure 3B, the sub-G1 apoptotic cell population (36.59 ± 1.1%) was increased after UVB stimulation compared to the control group (3.5 ± 0.05%). However, the sub-G1 apoptotic cell population was dose-dependently lowered with USHE. Annexin V flow cytometry analysis can be used to investigate the cellular early apoptosis. As shown in Figure 3C, the number of viable cells declined with UVB stimulation compared to the control group, increasing early apoptotic cells. Nevertheless, USHE notably reduced the early apoptotic cell population in a dose-dependent manner. The early apoptotic cell population was lowest at 62.5 µg/mL of USHE concentration.
3.5. Effect of USHE against UVB-Exposed Oxidative Stress by Inhibiting Mitochondria-Mediated Apoptotic Signaling Pathway in HaCaT Keratinocytes
The JC-1 assay is used to evaluate the membrane potential and health of mitochondria in cells. The ratio of red to green fluorescence is frequently utilized in JC-1 analysis as a measure of mitochondrial membrane potential. A healthy, polarized mitochondria with stable membrane potential has a higher red/green ratio. Reduced red/green ratios are indicative of mitochondrial depolarization, which may be a marker of early apoptosis or mitochondrial dysfunction. According to the findings of the study (Figure 4A, 4B), UVB-exposed cells exhibited a lower red/green ratio, indicating a significant presence of unhealthy/depolarized mitochondria. USHE effectively increased the red/green ratio, suggesting the presence of healthy, polarized mitochondria with a stable membrane potential. Moreover, the release of chemicals from the mitochondria, including cytochrome c, into the cytoplasm occurs when cells are stimulated by UVB. This release leads to a series of reactions in the cell that subsequently cause apoptosis by activating several proteins and enzymes. The protein levels of Bcl-2-associated X (Bax), cleaved caspase-3, p53, cleaved PARP, and cytochrome c were upregulated by UVB exposure, while inhibiting anti-apoptotic mediators, including B-cell lymphoma (Bcl)-xL, Bcl-2, and PARP (Figure 4C). Interestingly, USHE improved the levels of Bcl-xL, Bcl-2, and PARP in a dose-dependent manner while downregulating levels of Bax, cleaved caspase-3, p53, cleaved PARP, and cytochrome c (Figure 4C). Relative folds of the above proteins are indicated in Figure 4D.
3.6. Effect of USHE on Nrf2/HO-1 Activation in UVB-Exposed HaCaT Keratinocytes
The Nrf2/HO-1 signaling pathway includes key factors that regulate the cellular oxidative stress response via inhibiting ROS generation. The findings from the western blot analysis of protein expressions of cytosolic NQO1, HO-1, and nuclear translocated Nrf2 revealed the protective effect of USHE. According to Figure 5, the pretreatment with USHE increased the levels of cytosolic NQO1, HO-1, and nuclear translocated Nrf2 in UVB-exposed HaCaT keratinocytes in a dose-dependent manner.
4. Discussion
The present study revealed the photoprotective effect of USHE against oxidative stress and apoptosis in HaCaT keratinocytes which is caused by UVB exposure. Commonly, many researchers have used the organic solvent extraction technique to prepare extracts from marine resources [14,17,18,20]. The ultrasonic-assisted seaweed extraction method represents a cutting-edge approach to the extraction of bioactive compounds from seaweed [26,27]. The advantages of this method include reduced extraction time, lower energy consumption, and improved yields of bioactive components [27]. The results of the current study indicated that USHE showed 29.50 ± 1.32% yield (w/w), confirming ultrasonic-assisted ethanol extraction method has the potential to obtain higher yields compared to the ethanol extracts of previous studies [14,20]. It suggests the potential of USHE for the development of natural industrial materials. Previous studies have indicated that the ethanol extract of S. horneri contains various active components [14,19]. One of our previous reports indicated that fucosterol from S. horneri 70% ethanol extract exhibited potential antioxidant and anti-inflammatory effects against TNF-α/IFN-γ-stimulated HDF cells by regulating the Nrf2/HO-1, NF-κB, MAPK signaling pathways [14]. Besides, the intracellular ROS scavenging effect of fucosterol isolated from S. crassifolium was reported [28]. Interestingly, the HPLC analysis in the current study indicated the fucosterol content in USHE was 6.22 ± 0.06 mg/g. In addition, total phenolic compounds were 6.97 ± 0.36% in USHE on a dry basis (%). As reported, phenols can reduce oxidative stress through several mechanisms, such as Nrf2 activation and scavenging of ROS [29]. Additionally, four sulfoquinovosyl glycerolipids (SQGLs) in USHE sample were identified by using MS analysis. The unique structure of SQGLs allows them to integrate into cellular membranes, contributing to membrane stability and protecting against lipid peroxidation, a common result of oxidative stress [30]. These compounds have potential applications in cosmeceuticals, where they can combat skin aging and enhance skin integrity due to their antioxidant effects.
The antioxidant capacity of the USHE was determined by DPPH radical scavenging activity and FRAP assay. The DPPH assay has been widely utilized as a pre-screening technique for novel antioxidants derived from natural resources [31]. The findings of the study showed that USHE indicated DPPH radical scavenging activity with a respective IC50 value of 49 ± 0.24 µg/mL. Besides, the FRAP assay is used to measure antioxidant power based on the reduction of ferric to ferrous ions at low pH [32]. According to the FRAP assay, there was a significant and dose-dependent increase with a maximum absorbance of 0.2927 at 62.5 μg/mL. Therefore, further analyses were carried out to assess the protective effect of USHE against oxidative stress in UVB-exposed HaCaT keratinocytes based on the promising radical scavenging capacity and antioxidant power of USHE.
As social trends such as training or outdoor sports are becoming popular, skin cancer in melanoma as well as carcinoma has strikingly increased. Nonetheless, precancerous mutations in the keratinocyte genome have occurred even in the absence of burning while exposed to sublethal chronic UV irradiation [33]. UVB-exposed skin apoptosis is a complex process involving various molecular mechanisms, such as DNA damage, activation of pro-apoptotic proteins, and inflammation [34]. Even though chromophores in human skin can absorb UV radiation [35], UVB increases intracellular ROS production, either through direct changes in cells or via photosensitization mechanisms [36]. In this study, different concentrations of USHE were used to examine the effect on the cell viability of UVB-exposed HaCaT keratinocytes. The findings of the study revealed that there is a significant increment in the cell viability with USHE dose-dependently. Dysregulated ROS accumulation in cells causes oxidative stress and subsequently initiates DNA damage and cellular apoptosis [37]. One of our previous studies indicated that fucosterol from S. horneri 70% ethanol extract decreased the cellular ROS levels in TNF-α/IFN-γ-stimulated HDF cells [14]. According to the outcomes of the DCF-DA analysis, UVB stimulation markedly increased the intracellular ROS production in HaCaT keratinocytes in contrast to the control group. However, intracellular ROS production in UVB-exposed HaCaT keratinocytes was reduced with the fucosterol-containing USHE, in line with the indications of radical scavenging and reducing power antioxidant capability of USHE. Furthermore, the nuclear double staining experiment along with flow cytometric analysis with Annexin V and PI staining showed the dose-dependent reduction of early and late apoptotic cell population with the sample.
Previous studies have extensively revealed that DNA damage is caused by UV radiation in human keratinocytes. Specifically, an early fraction of apoptotic cells in the Sub G1 phase of the cell cycle has been detected [33]. As reported, sub-G1 apoptotic cell populations increased with UVB exposure compared to the control [38]. In like manner, upon cell cycle investigation of the present study, UVB stimulation increased the cell accumulation in the Sub G1 phase while significantly decreased by USHE doses in contrast to the stimulation group of HaCaT keratinocytes. The findings strongly supported the recovery of physiological differentiation of the UVB-exposed HaCaT keratinocytes by the USHE. Besides, prior studies have shown that tumor suppressor p53 mediates UVB-exposed apoptosis in epidermal cells [39]. For more clarifications, the underlying apoptosis inhibitory mechanism was investigated by using flow cytometric and western blot analysis. Mitochondrial alterations and their role in apoptosis have been known for several years. In apoptotic experimental models, a loss of mitochondrial membrane potential (ΔΨm) is a hallmark of apoptosis [40]. As far as ΔΨm plays an important role, the JC-1 analysis technique is frequently used to assess cells with high or low ΔΨm. Previous research explored that UVB-exposed HaCaT keratinocytes showed a large proportion of unhealthy mitochondria, similar to the CCCP group [10]. Results obtained from JC-1 analysis in the present study allowed us to identify two identical populations with different ΔΨm. A first population with high ΔΨm and the other one with low ΔΨm. USHE gradually increases the ΔΨm in contrast to the CCCP group in a dose-dependent manner. Moreover, mitochondrial fission is fundamental for apoptosis which facilitates the release of pro-apoptotic factors, such as cytochrome c, from the mitochondrial intermembrane space. This release triggers a cascade of events leading to cell death. The increase of cytochrome c, Bax, cleaved caspase-3, cleaved PARP, and p53 levels in UVB-exposed HaCaT keratinocytes, with a decrease of Bcl-2 and Bcl-xL levels, suggests the initiation of apoptosis in this study. Moreover, PARP cleavage took place with the UVB stimulation. Similarly, apoptogenic protein expressions have been shown due to UVB stimulation in one of the prior studies [11]. The results of the present study showed a dose-dependent reduction in apoptotic molecular mediators after treatment with USHE, demonstrating its protective properties against UVB-exposed oxidative stress in HaCaT keratinocytes.
Activation of Nrf2 to promote the expression of HO-1 and NQO1 is considered a promising therapeutic strategy to mitigate harmful cellular responses [21]. Nrf2 remains bound to the cytoplasmic inhibitor Keap1. Upon stimulation, Nrf2 undergoes translocation to the nucleus, initiating the transcription of various antioxidant genes, such as HO-1, and NQO1. Induction of HO-1 and NQO1 in UVB-exposed cells suppresses increased intracellular ROS levels, indicating an antioxidative impact [10]. HO-1 has been widely recognized for its antioxidative, anti-inflammatory, cytoprotective, and anti-apoptotic effects across various cell types [10,14,25]. The findings indicate that USHE enhances Nrf2 activation and its nuclear translocation. This is supported by the increased levels of HO-1 and NQO1, which further highlight the antioxidant potential of USHE. Collectively, these results show that USHE boosts Nrf2 activation and regulates the expression of antioxidant enzymes such as HO-1 and NQO1 in UVB-exposed HaCaT keratinocytes. Given its antioxidant properties and the presence of beneficial phytochemicals, USHE holds promise as a valuable ingredient for skincare products and cosmeceuticals. Future studies should explore its broader bioactivity through both in vitro and in vivo evaluations for various diseases.
5. Conclusions
Based on the present study, USHE containing fucosterol and SQGLs effectively inhibits oxidative stress by reducing ROS production in UVB-exposed HaCaT keratinocytes. Furthermore, USHE indicates a protective effect by downregulating mitochondria-mediated apoptotic signaling and nuclear damage. Therefore, USHE possesses antioxidant potential against photodamage of the skin and can be utilized for the development of nutraceuticals/cosmeceuticals for UVB protection. Furthermore, this study suggests that USHE could be further investigated through in vitro and in vivo studies to evaluate its application as an ingredient in cosmeceuticals with antioxidant capabilities.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, K.G.I.S.K. and A.M.K.J.; methodology, K.G.I.S.K., G.A., A.M.K.J., C.I.K., Y.S.A., S.J.H. and E.A.K.; validation, K.G.I.S.K., A.M.K.J. and Y.S.A.; formal analysis, K.G.I.S.K., A.M.K.J. and C.I.K.; investigation, K.G.I.S.K., N.C., and A.M.K.J.; resources, G.A., C.I.K., Y.S.A., S.J.H. and E.A.K.; data curation, K.G.I.S.K., N.C., and A.M.K.J.; writing—original draft preparation, K.G.I.S.K.; writing—review and editing, G.A. and A.M.K.J.; visualization, K.G.I.S.K. and A.M.K.J.; supervision, G.A.; project administration, G.A., funding acquisition, G.A., C.I.K., Y.S.A. and E.A.K.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Korea Institute of Marine Science & Technology Promotion (KIMST) funded by the Ministry of Oceans and Fisheries (20220128) and the Technology Development Program (S3266086) funded by the Ministry of SMEs and Startups (MSS, Korea).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Taylor, S.C.; Alexis, A.F.; Armstrong, A.W.; Fuxench, Z.C.C.; Lim, H.W. Misconceptions of photoprotection in skin of color. J. Am. Acad. Dermatol. 2022, 86, S9–S17. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Yazar, S.; Young, A.; Norval, M.; De Gruijl, F.; Takizawa, Y.; Rhodes, L.; Sinclair, C.; Neale, R. Human health in relation to exposure to solar ultraviolet radiation under changing stratospheric ozone and climate. Photochemical & Photobiological Sciences 2019, 18, 641–680. [Google Scholar]
- Mapoung, S.; Arjsri, P.; Thippraphan, P.; Semmarath, W.; Yodkeeree, S.; Chiewchanvit, S.; Piyamongkol, W.; Limtrakul, P. Photochemoprotective effects of Spirulina platensis extract against UVB irradiated human skin fibroblasts. S. Afr. J. Bot. 2020, 130, 198–207. [Google Scholar] [CrossRef]
- Leal, A.C.; Corrêa, M.P.; Holick, M.F.; Melo, E.V.; Lazaretti-Castro, M. Sun-induced production of vitamin D3 throughout 1 year in tropical and subtropical regions: Relationship with latitude, cloudiness, UV-B exposure and solar zenith angle. Photochemical & Photobiological Sciences 2021, 20, 265–274. [Google Scholar]
- Xiao, Z.; Yang, S.; Liu, Y.; Zhou, C.; Hong, P.; Sun, S.; Qian, Z.-J. A novel glyceroglycolipid from brown algae Ishige okamurae improve photoaging and counteract inflammation in UVB-induced HaCaT cells. Chemico-Biological Interactions 2022, 351, 109737. [Google Scholar] [CrossRef]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Molecular Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. European journal of medicinal chemistry 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Cavinato, M.; Waltenberger, B.; Baraldo, G.; Grade, C.V.; Stuppner, H.; Jansen-Dürr, P. Plant extracts and natural compounds used against UVB-induced photoaging. Biogerontology 2017, 18, 499–516. [Google Scholar] [CrossRef]
- Priyan Shanura Fernando, I.; Kim, K.-N.; Kim, D.; Jeon, Y.-J. Algal polysaccharides: Potential bioactive substances for cosmeceutical applications. Crit. Rev. Biotechnol. 2019, 39, 99–113. [Google Scholar] [CrossRef]
- Fernando, I.P.S.; Dias, M.K.H.M.; Madusanka, D.M.D.; Han, E.J.; Kim, M.J.; Jeon, Y.-J.; Ahn, G. Fucoidan refined by Sargassum confusum indicate protective effects suppressing photo-oxidative stress and skin barrier perturbation in UVB-induced human keratinocytes. International Journal of Biological Macromolecules 2020, 164, 149–161. [Google Scholar] [CrossRef]
- Fernando, I.P.S.; Heo, S.-J.; Dias, M.K.H.M.; Madusanka, D.M.D.; Han, E.-J.; Kim, M.-J.; Sanjeewa, K.K.A.; Lee, K.; Ahn, G. (−)-loliolide isolated from sargassum horneri abate uvb-induced oxidative damage in human dermal fibroblasts and subside ecm degradation. Marine Drugs 2021, 19, 435. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.-J.; Ko, S.-C.; Cha, S.-H.; Kang, D.-H.; Park, H.-S.; Choi, Y.-U.; Kim, D.; Jung, W.-K.; Jeon, Y.-J. Effect of phlorotannins isolated from Ecklonia cava on melanogenesis and their protective effect against photo-oxidative stress induced by UV-B radiation. Toxicology in vitro 2009, 23, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Heinrich, M.; Myers, S.; Dworjanyn, S.A. Towards a better understanding of medicinal uses of the brown seaweed Sargassum in Traditional Chinese Medicine: A phytochemical and pharmacological review. J. Ethnopharmacol. 2012, 142, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Kirindage, K.G.I.S.; Jayasinghe, A.M.K.; Han, E.-J.; Jee, Y.; Kim, H.-J.; Do, S.G.; Fernando, I.P.S.; Ahn, G. Fucosterol isolated from dietary brown alga Sargassum horneri protects TNF-α/IFN-γ-stimulated human dermal fibroblasts via regulating Nrf2/HO-1 and NF-κB/MAPK pathways. Antioxidants 2022, 11, 1429. [Google Scholar] [CrossRef] [PubMed]
- Sanjeewa, K.K.A.; Fernando, I.P.S.; Kim, E.-A.; Ahn, G.; Jee, Y.; Jeon, Y.-J. Anti-inflammatory activity of a sulfated polysaccharide isolated from an enzymatic digest of brown seaweed Sargassum horneri in RAW 264.7 cells. Nutr. Res. Pract. 2017, 11, 3–10. [Google Scholar] [CrossRef]
- Han, E.J.; Kim, H.-S.; Sanjeewa, K.K.A.; Jung, K.; Jee, Y.; Jeon, Y.-J.; Fernando, I.P.S.; Ahn, G. Sargassum horneri as a functional food ameliorated IgE/BSA-induced mast cell activation and passive cutaneous anaphylaxis in mice. Marine drugs 2020, 18, 594. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, H.J.; Jee, Y.; Jeon, Y.-J.; Kim, H.J. Antioxidant potential of Sargassum horneri extract against urban particulate matter-induced oxidation. Food Sci. Biotechnol. 2020, 29, 855–865. [Google Scholar] [CrossRef]
- Herath, K.H.I.N.M.; Cho, J.; Kim, A.; Kim, H.-S.; Han, E.J.; Kim, H.J.; Kim, M.S.; Ahn, G.; Jeon, Y.-J.; Jee, Y. Differential modulation of immune response and cytokine profiles of Sargassum horneri ethanol extract in murine spleen with or without Concanavalin A stimulation. Biomed. Pharmacother. 2019, 110, 930–942. [Google Scholar] [CrossRef]
- Zhao, D.; Zheng, L.; Qi, L.; Wang, S.; Guan, L.; Xia, Y.; Cai, J. Structural features and potent antidepressant effects of total sterols and β-sitosterol extracted from Sargassum horneri. Marine drugs 2016, 14, 123. [Google Scholar] [CrossRef]
- Dias, M.K.H.M.; Madusanka, D.M.D.; Han, E.J.; Kim, H.-S.; Jeon, Y.-J.; Jee, Y.; Kim, K.-N.; Lee, K.; Fernando, I.P.S.; Ahn, G. Sargassum horneri (Turner) C. Agardh ethanol extract attenuates fine dust-induced inflammatory responses and impaired skin barrier functions in HaCaT keratinocytes. J. Ethnopharmacol. 2021, 273, 114003. [Google Scholar] [CrossRef]
- Kirindage, K.G.I.S.; Fernando, I.P.S.; Jayasinghe, A.M.K.; Han, E.-J.; Dias, M.K.H.M.; Kang, K.-P.; Moon, S.-I.; Shin, T.-S.; Ma, A.; Ahn, G. Moringa oleifera hot water extract protects Vero cells from hydrogen peroxide-induced oxidative stress by regulating mitochondria-mediated apoptotic pathway and Nrf2/HO-1 signaling. Foods 2022, 11, 420. [Google Scholar] [CrossRef]
- Firuzi, O.; Lacanna, A.; Petrucci, R.; Marrosu, G.; Saso, L. Evaluation of the antioxidant activity of flavonoids by “ferric reducing antioxidant power” assay and cyclic voltammetry. Biochimica et Biophysica Acta (BBA)-General Subjects 2005, 1721, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Kirindage, K.G.I.S.; Jayasinghe, A.M.K.; Han, E.-J.; Han, H.-J.; Kim, K.-N.; Wang, L.; Heo, S.-J.; Jung, K.-S.; Ahn, G. Phlorofucofuroeckol-A refined by edible brown algae Ecklonia cava indicates anti-inflammatory effects on TNF-α/IFN-γ-stimulated HaCaT keratinocytes and 12-O-tetradecanoylphorbol 13-acetate-induced ear edema in BALB/c mice. J. Funct. Foods 2023, 109, 105786. [Google Scholar] [CrossRef]
- Kirindage, K.G.I.S.; Jayasinghe, A.M.K.; Cho, N.; Cho, S.H.; Yoo, H.M.; Fernando, I.P.S.; Ahn, G. Fine-dust-induced skin inflammation: Low-molecular-weight fucoidan protects keratinocytes and underlying fibroblasts in an integrated culture model. Marine Drugs 2022, 21, 12. [Google Scholar] [CrossRef]
- Fernando, I.P.S.; Kirindage, K.G.I.S.; Jayasinghe, A.M.K.; Han, E.J.; Dias, M.K.H.M.; Kang, K.P.; Moon, S.I.; Shin, T.S.; Ma, A.; Jung, K. Hot Water Extract of Sasa borealis (Hack.) Makino & Shibata Abate Hydrogen Peroxide-Induced Oxidative Stress and Apoptosis in Kidney Epithelial Cells. Antioxidants 2022, 11, 1013. [Google Scholar] [CrossRef]
- Balicki, S.; Pawlaczyk-Graja, I.; Gancarz, R.; Capek, P.; Wilk, K.A. Optimization of Ultrasound-Assisted Extraction of Functional Food Fiber from Canadian Horseweed (Erigeron canadensis L.). ACS omega 2020, 5, 20854–20862. [Google Scholar] [CrossRef]
- Kirindage, K.G.I.S.; Jayasinghe, A.M.K.; Ko, C.-I.; Ahn, Y.-S.; Heo, S.-J.; Oh, J.-Y.; Kim, E.-A.; Cha, S.-H.; Ahn, G. Unveiling the Potential of Ultrasonic-Assisted Ethanol Extract from Sargassum horneri in Inhibiting Tyrosinase Activity and Melanin Production in B16F10 Murine Melanocytes. Frontiers in Bioscience-Landmark 2024, 29, 194. [Google Scholar] [CrossRef]
- Chi, H.K.; Van, N.T.H.; Hang, T.T.N.; Duc, T.M.; Ha, T.T.H. Intracellular reactive oxygen species scavenging effect of fucosterol isolated from the brown alga Sargassum crassifolium in Vietnam. Vietnam Journal of Marine Science and Technology 2020, 20, 308–316. [Google Scholar] [CrossRef]
- Pawlowska, E.; Szczepanska, J.; Koskela, A.; Kaarniranta, K.; Blasiak, J. Dietary polyphenols in age-related macular degeneration: protection against oxidative stress and beyond. Oxidative medicine and cellular longevity 2019, 2019. [Google Scholar] [CrossRef]
- Plouguerné, E.; da Gama, B.A.; Pereira, R.C.; Barreto-Bergter, E. Glycolipids from seaweeds and their potential biotechnological applications. Frontiers in cellular and infection microbiology 2014, 4, 174. [Google Scholar] [CrossRef]
- Wang, T.; Jonsdottir, R.; Ólafsdóttir, G. Total phenolic compounds, radical scavenging and metal chelation of extracts from Icelandic seaweeds. Food chemistry 2009, 116, 240–248. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Analytical biochemistry 1996, 239, 70–76. [Google Scholar] [CrossRef]
- de Pedro, I.; Alonso-Lecue, P.; Sanz-Gómez, N.; Freije, A.; Gandarillas, A. Sublethal UV irradiation induces squamous differentiation via a p53-independent, DNA damage-mitosis checkpoint. Cell Death Dis. 2018, 9, 1094. [Google Scholar] [CrossRef]
- Garg, C.; Sharma, H.; Garg, M. Skin photo-protection with phytochemicals against photo-oxidative stress, photo-carcinogenesis, signal transduction pathways and extracellular matrix remodeling—An overview. Ageing Research Reviews 2020, 62, 101127. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hu, J.Y.; Wang, S.Q. The role of antioxidants in photoprotection: a critical review. J. Am. Acad. Dermatol. 2012, 67, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- De Jager, T.; Cockrell, A.; Du Plessis, S. Ultraviolet light induced generation of reactive oxygen species. Ultraviolet Light in Human Health, Diseases and Environment 2017, 15-23.
- Fernando, I.P.S.; Dias, M.K.H.M.; Madusanka, D.M.D.; Han, E.J.; Kim, M.J.; Heo, S.-J.; Ahn, G. Fucoidan Fractionated from Sargassum coreanum via Step-Gradient Ethanol Precipitation Indicate Promising UVB-Protective Effects in Human Keratinocytes. Antioxidants 2021, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Fernando, I.P.S.; Dias, M.K.H.M.; Madusanka, D.M.D.; Han, E.J.; Kim, M.J.; Jeon, Y.-J.; Ahn, G. Step gradient alcohol precipitation for the purification of low molecular weight fucoidan from Sargassum siliquastrum and its UVB protective effects. International journal of biological macromolecules 2020, 163, 26–35. [Google Scholar] [CrossRef]
- Ziegler, A.; Jonason, A.S.; Leffellt, D.J.; Simon, J.A.; Sharma, H.W.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D.E. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar] [CrossRef]
- Lugli, E.; Troiano, L.; Ferraresi, R.; Roat, E.; Prada, N.; Nasi, M.; Pinti, M.; Cooper, E.L.; Cossarizza, A. Characterization of cells with different mitochondrial membrane potential during apoptosis. Cytometry Part A 2005, 68, 28–35. [Google Scholar] [CrossRef]
Figure 1.
Antioxidant activities of USHE: (A) DPPH radical scavenging activity, and (B) reducing power antioxidant activity. Values are expressed as the mean ± SE from triplicate experiments. Bars with different letters within the same graph represent significant differences (p < 0.05).
Figure 1.
Antioxidant activities of USHE: (A) DPPH radical scavenging activity, and (B) reducing power antioxidant activity. Values are expressed as the mean ± SE from triplicate experiments. Bars with different letters within the same graph represent significant differences (p < 0.05).
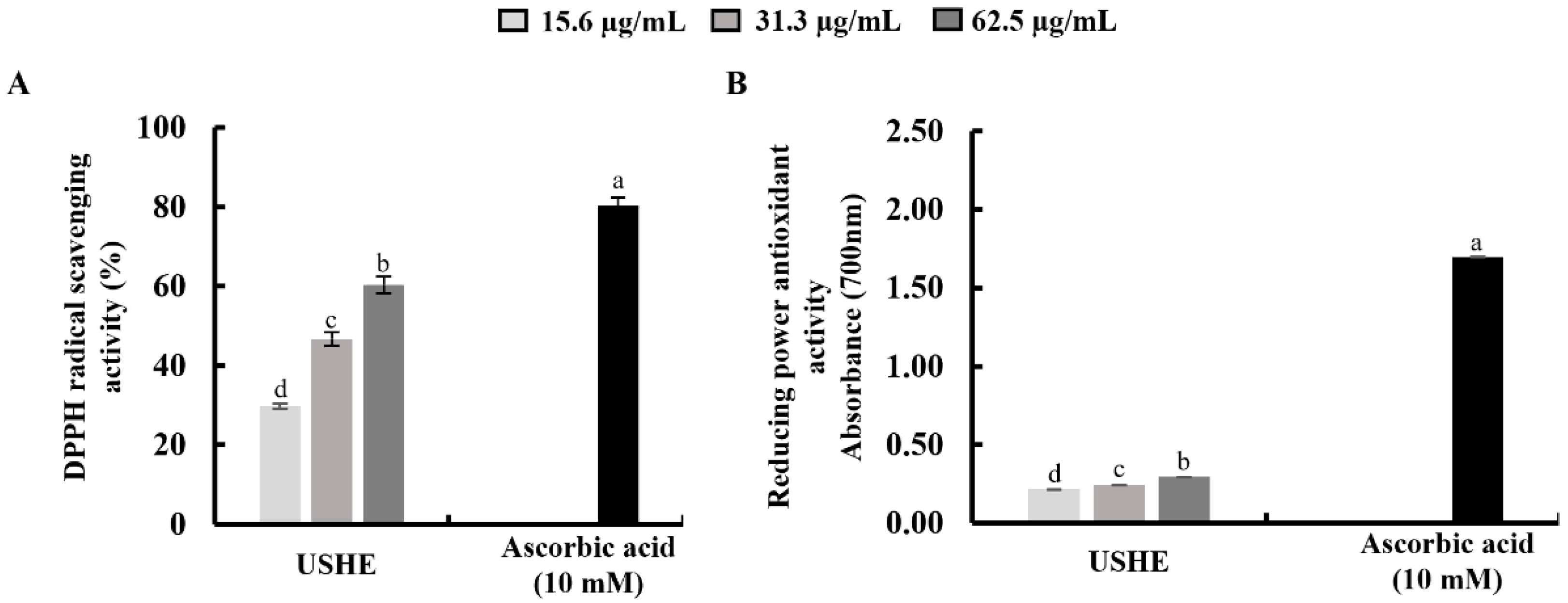
Figure 2.
Analysis of the protective effects of USHE on UVB-exposed HaCaT keratinocytes. Evaluation of (A) cytotoxicity, (B) cell viability, and intracellular ROS production using (C) fluorometry, (D) fluorescence microscopy, and (E) flow cytometry. Values are expressed as the mean ± SE from triplicate experiments. Bars with different letters within the same graph represent significant differences (p < 0.05).
Figure 2.
Analysis of the protective effects of USHE on UVB-exposed HaCaT keratinocytes. Evaluation of (A) cytotoxicity, (B) cell viability, and intracellular ROS production using (C) fluorometry, (D) fluorescence microscopy, and (E) flow cytometry. Values are expressed as the mean ± SE from triplicate experiments. Bars with different letters within the same graph represent significant differences (p < 0.05).
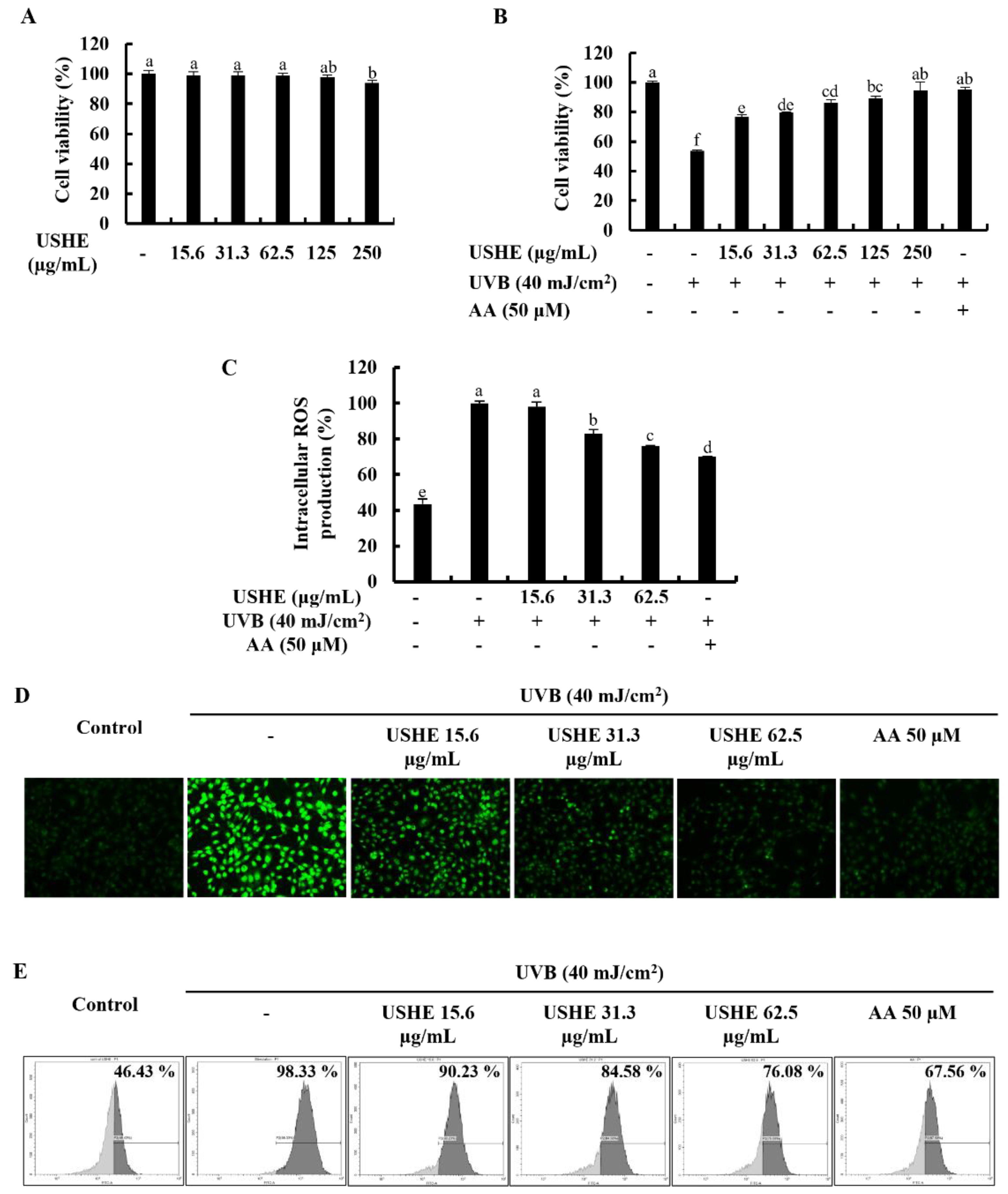
Figure 3.
Effect of USHE on the apoptotic cell population in UVB-exposed HaCaT keratinocytes. (A) Nuclear morphology analysis using EB and AO double staining, (B) sub-G1 apoptotic population analysis by flow cytometry using PI, and (C) early apoptosis investigation using flow cytometry Annexin V analysis. Values are expressed as the mean ± SE from triplicate experiments. Bars with different letters within the same graph represent significant differences (p < 0.05).
Figure 3.
Effect of USHE on the apoptotic cell population in UVB-exposed HaCaT keratinocytes. (A) Nuclear morphology analysis using EB and AO double staining, (B) sub-G1 apoptotic population analysis by flow cytometry using PI, and (C) early apoptosis investigation using flow cytometry Annexin V analysis. Values are expressed as the mean ± SE from triplicate experiments. Bars with different letters within the same graph represent significant differences (p < 0.05).
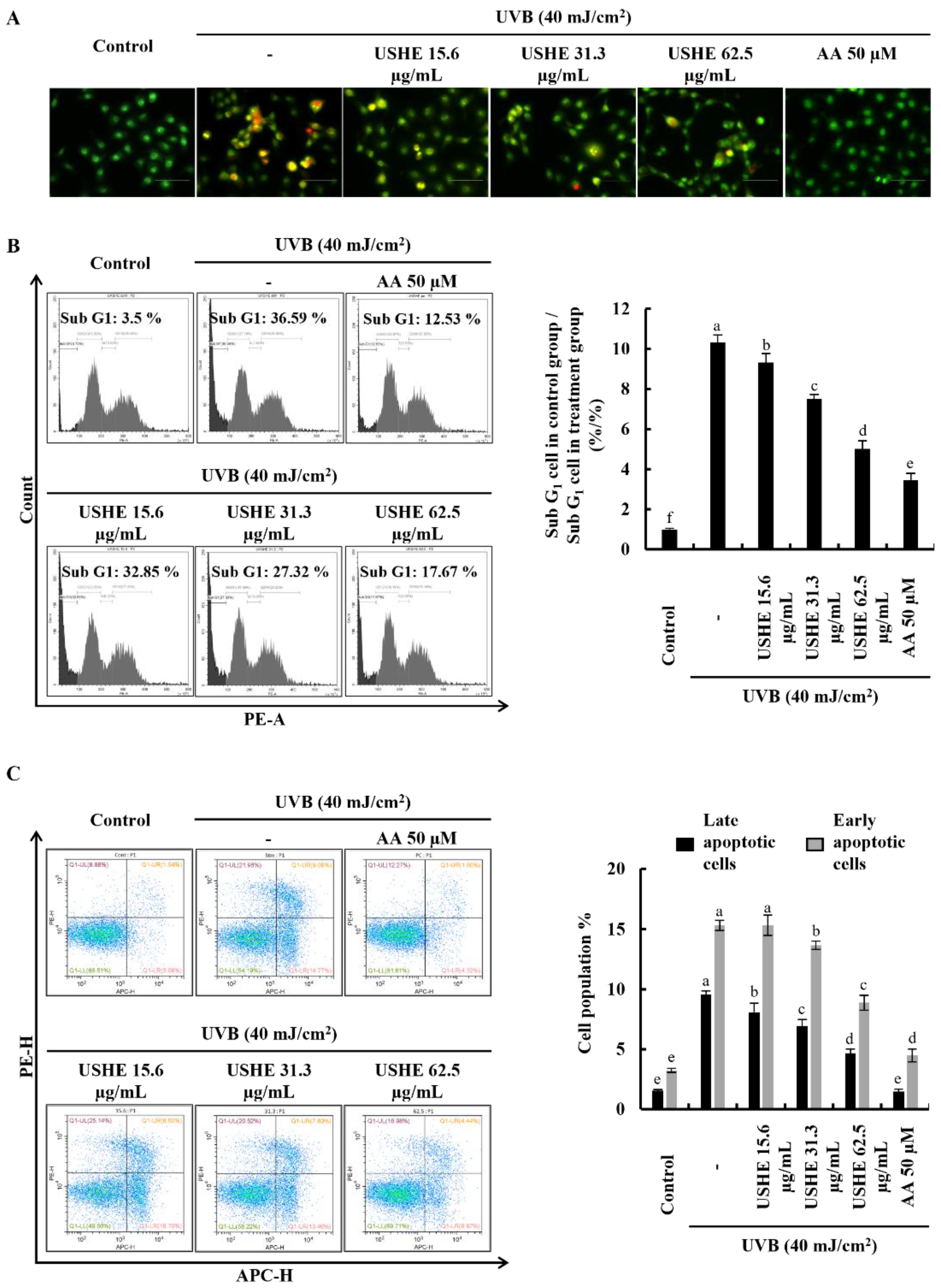
Figure 4.
Effect of USHE on mitochondria-mediated apoptosis in UVB-exposed HaCaT keratinocytes. (A, B) Mitochondrial membrane potential analysis by JC-1 assays, with numbers indicating the percentage of cells. Western blot analysis of (C) mitochondria-mediated apoptotic pathway protein expression levels and (D) their relative fold changes. Values are expressed as the mean ± SE from triplicate experiments. Bars with different letters within the same graph represent significant differences (p < 0.05).
Figure 4.
Effect of USHE on mitochondria-mediated apoptosis in UVB-exposed HaCaT keratinocytes. (A, B) Mitochondrial membrane potential analysis by JC-1 assays, with numbers indicating the percentage of cells. Western blot analysis of (C) mitochondria-mediated apoptotic pathway protein expression levels and (D) their relative fold changes. Values are expressed as the mean ± SE from triplicate experiments. Bars with different letters within the same graph represent significant differences (p < 0.05).
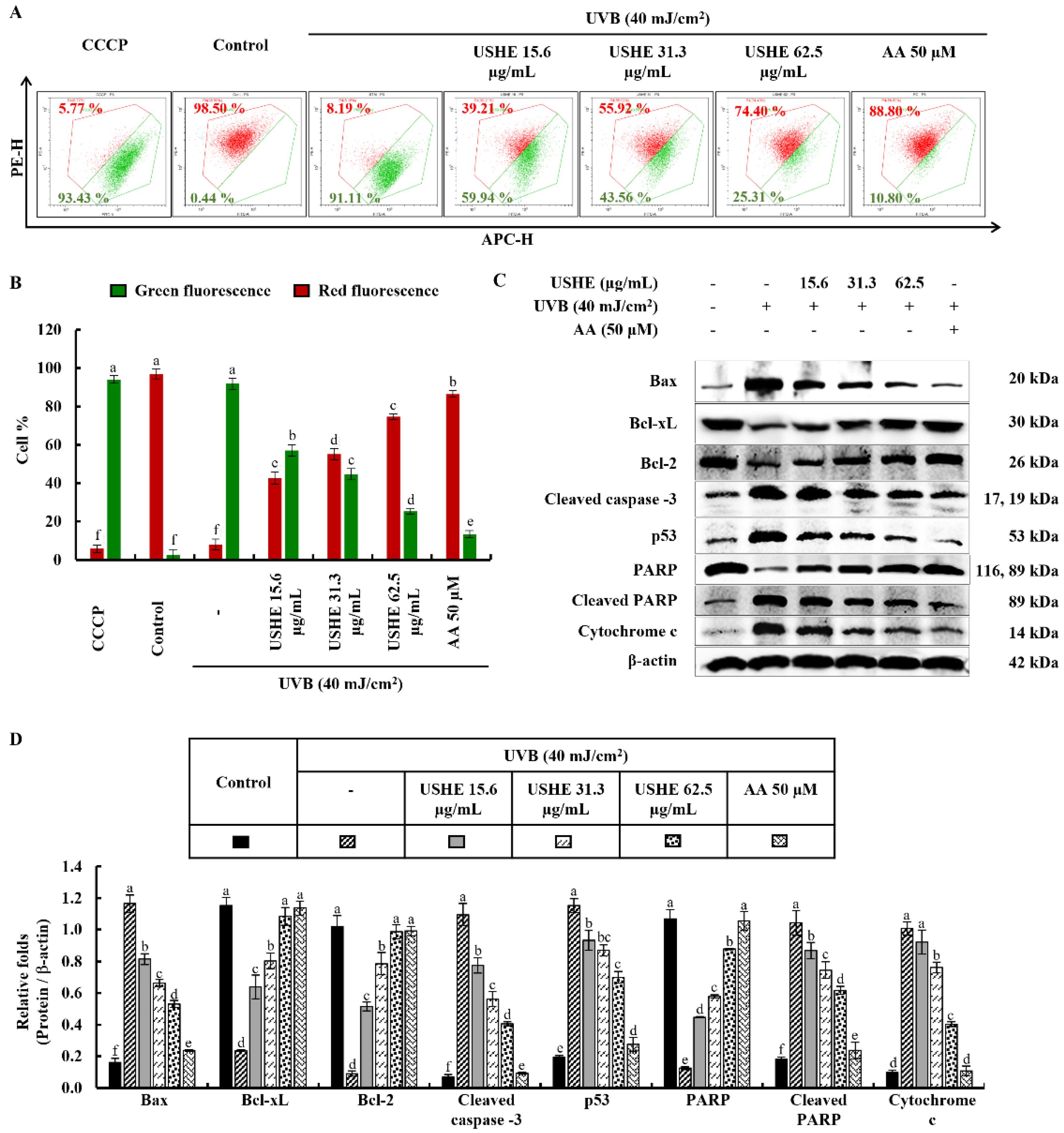
Figure 5.
Cytoprotective effect of USHE on the activation of the Nrf2/HO-1 signaling pathway. Western blot analysis of (A) cytosolic HO-1, NQO1, and (B) nuclear translocated Nrf2 protein expression in UVB-exposed HaCaT keratinocytes. Values are expressed as the mean ± SE from triplicate experiments. Bars with different letters within the same graph represent significant differences (p < 0.05).
Figure 5.
Cytoprotective effect of USHE on the activation of the Nrf2/HO-1 signaling pathway. Western blot analysis of (A) cytosolic HO-1, NQO1, and (B) nuclear translocated Nrf2 protein expression in UVB-exposed HaCaT keratinocytes. Values are expressed as the mean ± SE from triplicate experiments. Bars with different letters within the same graph represent significant differences (p < 0.05).
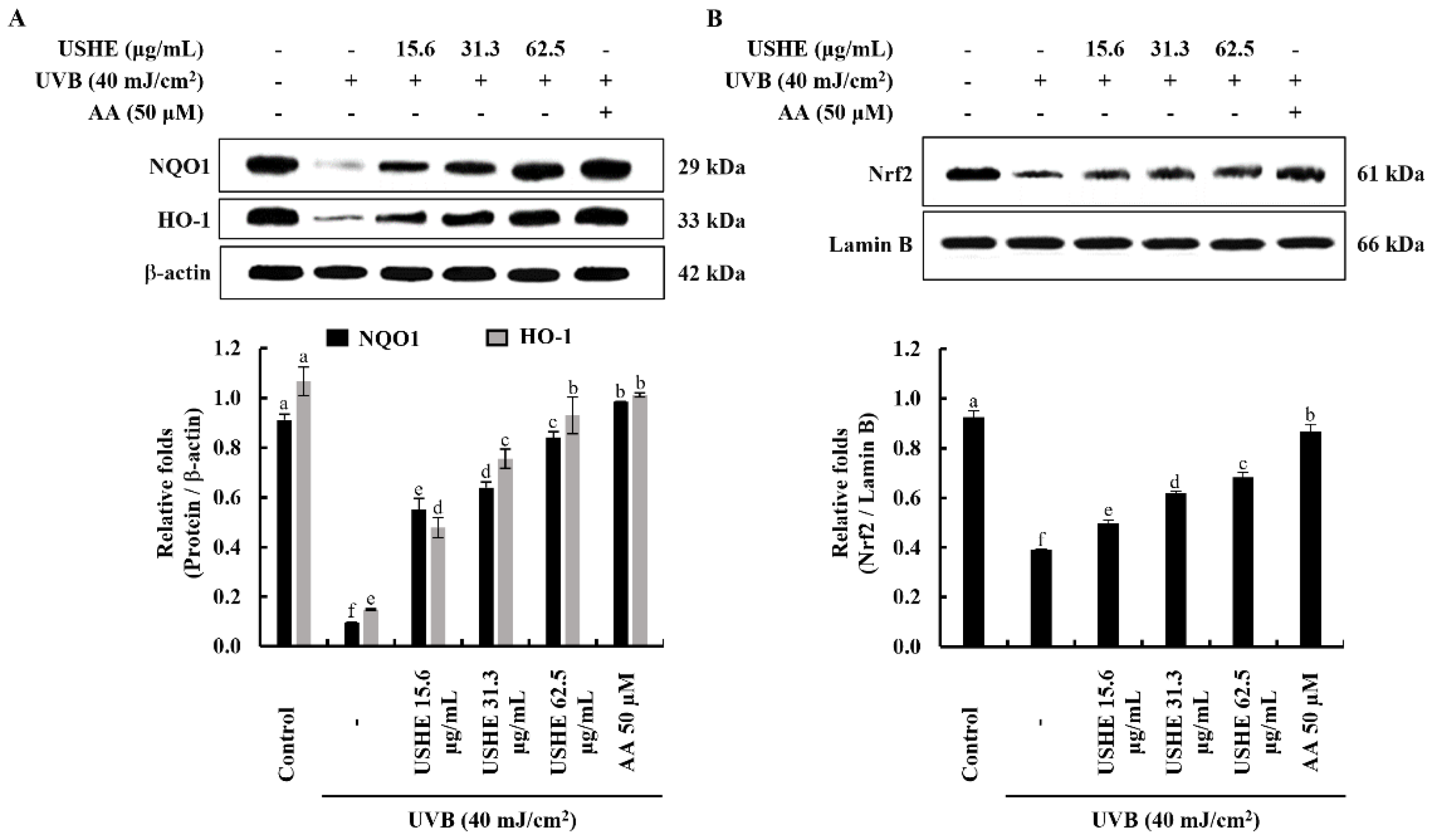

Table 1.
Yield and proximate composition of USHE.
| Sample | Yield (%) 1 | Carbohydrate (%)2 | Crude protein (%)2 | Total phenolic compounds (%)2 |
|---|---|---|---|---|
| USHE | 29.50 ± 1.32 | 9.83 ± 0.87 | 15.01 ± 1.06 | 6.97 ± 0.36 |
1 Average value was present on a dry basis and, 2 average value was present as a proportion of resulted in yield ± SEM (all experiments were performed in triplicate (n = 3) to determine the repeatability).
Table 2.
Metabolite profiling of USHE.
| RT (min) |
m/z ([M-H]) |
Formula | Δppm | Compound |
|---|---|---|---|---|
| 16.12 | 527.2518 | C23H43O11S | -0.0192 | 1-O-Tetradecanoyl-3-O-(6′-sulfo-a-D-quinovopyranosyl) glycerol |
| 17.17 | 553.3187 | C25H45O11S | -0.0504 | 1-O-(11-Hexadecenoyl)-3-O-(6′-sulfo-a-D-quinovopyranosyl) glycerol |
| 19.02 | 555.3054 | C25H47O11S | -0.0215 | 1-O-Hexadecanoyl-3-O-(6′-sulfo-a-D-quinovopyranosyl) glycerol |
| 20.13 | 581.3199 | C27H49O11S | -0.0007 | 1-O-(9-Octadecenoyl)-3-O-(6′-sulfo-a-D-quinovopyranosyl) glycerol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



