
Submitted:
13 October 2024
Posted:
14 October 2024
You are already at the latest version
Abstract
Anti-oxidative and anti-inflammatory properties of high-density lipoprotein (HDL) are thought to be mediated by paraoxonase 1 (PON1), a calcium-dependent hydrolytic enzyme carried on a subfraction of HDL that also carries other anti-oxidative and anti-inflammatory proteins. In humans and mice, low PON1 activity is associated with elevated oxidized lipids, homocysteine (Hcy)-thiolactone, and proteins modified by these metabolites, which can cause oxidative stress and inflammation. PON1-dependent metabolic changes can lead to atherothrombotic cardiovascular disease, Alzheimer’s disease, and cancer. The molecular bases underlying these associations are not fully understood. Biochemical, proteomic, and metabolic studies have significantly expanded our understanding of mechanisms by which low PON1 leads to disease and high PON1 is protective. Studies discussed in this review highlight the changes in gene expression affecting proteostasis as a cause of the pro-oxidative and pro-inflammatory phenotypes associated with attenuated PON1 activity. Accumulating evidence supports the conclusion that PON1 regulates the expression of anti-oxidative and anti-inflammatory proteins, and that disruption of these processes leads to disease.
Keywords:
PON1 physiological substrates
; homocysteine thiolactone
; 5-(3’
; 4’-dihydroxyphenyl)-?-valerolactone
; PON1 proteomics
; antioxidant proteins
; anti-inflammatory proteins
; cardiovascular disease
; Alzheimer’s disease
1. Introduction
Atherosclerosis is the main cause of morbidity and mortality in the Western world. It is a multifactorial chronic inflammatory disease that involves a complex interaction of circulating cells and blood factors with the blood vessels. The disease starts with endothelial dysfunction that leads to accumulation of oxidized lipids in the artery wall [1,2]. Lipid oxidation plays a central role in atherogenesis [3] by inducing a pro-inflammatory phenotype in the arterial wall that underlies the development and progression of atherosclerosis [4].
High-density lipoprotein (HDL) is an established protective factor against atherosclerosis due to its ability to mediate reverse cholesterol transport as well as anti-oxidative, anti-inflammatory, and endothelial protective functions [4,5,6]. HDL can inhibit endothelial cell adhesion molecules, such as vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and E-selectin that enable monocytes to bind at the sites of developing atherosclerosis [7]. HDL can remove peroxidized lipids from LDL and subsequently reduce them in a reaction with methionine residues of apolipoprotein A1 (APOA1) [8]. Lipid-free APOA1 can also remove lipid peroxide molecules from low density lipoprotein (LDL) [9]. Reconstituted HDL containing only APOA1 and phospholipids, inhibits LDL oxidation like the native HDL3b and HDL3c particles do [8]. Other HDL-associated lipoproteins and enzymes, including paraoxonase 1 (PON1), have also been implicated in HDL’s anti-oxidative, anti-inflammatory, and endothelial protective functions [10,11].
Studies over the last decade have significantly expanded our knowledge regarding the natural substrates of PON1 and their role in human disease. Other studies have shown that the protective function of PON1 in human health is due to the ability of PON1 to affect the expression of genes involved in anti-oxidative and anti-inflammatory processes. These studies are discussed in the present review, highlighting the involvement of reduced PON1 expression/activity in the pro-oxidative, pro-atherogenic, pro-amyloidogenic, and pro-cancerogenic phenotypes.
2. Hydrolytic activities of the PON1 enzyme
PON1, a hydrolytic enzyme that requires calcium for activity, is expressed in the liver, kidney, colon [12], brain [13,14,15], and circulates attached to HDL in the blood. It is a minor HDL protein with potential clinical significance. Proteomic studies revealed that HDL particles carrying PON1, are enriched in several other important proteins such as A2M, ALB, CLU, IGHG1, IGLC2, PROS1, and TF [16]. The PON1 gene is located on the long arm of chromosome 3 in the PON cluster together with PON2 and PON3 genes. Its polymorphic variants include PON1-Q192R [17] that involves the glutamine (Q) to arginine (R) change at position 192 of the amino acid sequence of the PON1 protein and affects its hydrolytic activity. Historically, the hydrolytic activity of PON1 has been assayed with non-natural substrates such as the organophosphate paraoxon (for which the PON1 enzyme has been named) and phenyl acetate [18] (Figure 1).
Studies of homocysteine (Hcy) metabolism have led to the discovery that PON1 is responsible for enzymatic hydrolysis of Hcy-thiolactone to Hcy (Figure 1) in human serum, thus identifying the first natural substrate of PON1 [19]. Hcy-thiolactone, a cyclic chemically reactive thioester, is a product of Hcy editing by methionyl-tRNA synthetase during protein biosynthesis [20,21,22].
PON1 is the only Hcy-thiolactone hydrolyzing enzyme in the human blood [19,39]. The Hcy-thiolactonase activity of the PON1 enzyme shows an interindividual variability of over 10-fold [23,24], similar to the interindividual variability in the paraoxonase activity [17]. This variability is mostly due to polymorphisms in the human PON1 gene [17]. For example, PON1-192RR variant exhibits high activity while PON1-192QQ variant has low activity towards Hcy-thiolactone [23] and paraoxon [17]. In contrast, PON1-Q192R polymorphism has an opposite effect on the arylesterase activity: the PON1-192RR variant exhibits low arylesterase activity while PON1-192QQ variant has high arylesterase activity [16,25,26,27,28]. Individuals who have the low activity PON1-192QQ polymorphic variant produce significantly more Hcy-thiolactone than those who have the high activity PON1-192RR polymorphic variant [25].
Low PON1 expression/activity is accompanied by increased oxidative stress and predicts adverse outcomes in cardiovascular disease (CVD) [27,29], diabetes [30,31], neurological disease [32], and cancer [33]. This has been suggested to be due to the mediation by PON1 of anti-oxidative and anti-inflammatory effects of HDL [34,35]. Many other HDL components have also been shown to mediate the anti-oxidative activity of HDL [10,11,36], including APOA1, which accounts for 70% of HDL protein mass [36] and anti-apoptotic activity [37] and for most of the HDL anti-oxidative activity [8]. Accumulating evidence suggests that influence of PON1 on oxidative stress and inflammation is indirect rather than direct [38].
Hcy-thiolactone is harmful because it reacts with ε-amino group of protein lysine residues, forming N-homocysteinylated-proteins, which impairs protein’s structure and function [22]. Hydrolytic detoxification of Hcy-thiolactone by PON1 is beneficial because it prevents protein damage by N-homocysteinylation [19,23,39]. For example, serum from donors with PON1-LL55/RR192 genotype hydrolyzed Hcy-thiolactone (Figure 2A) to Hcy (Figure 2B) faster and afforded better protection from protein N-homocysteinylation than serum from donors with the PON1-MM55/QQ192 genotype (Figure 2C). Notably, PON1 in rabbit serum hydrolyzed Hcy-thiolactone (Figure 2A) even faster and afforded much better protection from protein N-homocysteinylation than any human serum (Figure 2C).
In a large randomized clinical trial, urinary Hcy-thiolactone was associated with myocardial infarction in coronary artery disease patients [40]. In a mouse and cellular models of Alzheimer’s disease (AD), Hcy-thiolactone promoted the accumulation of amyloid beta (Aβ) by inhibiting autophagy [15]. Hcy-thiolactone can promote the progression to AD by upregulating amyloid precursor protein (APP), which results in increased generation of Aβ [15].
The involvement of Hcy-thiolactone in disease can also be explained by its ability to impair protein structure/function via N-homocystinylation of protein lysine residues [22]. For example, N-homocysteinylation of fibrinogen by Hcy-thiolactone, which impairs lysis of fibrin clots in vitro [41], explains the association of Hcy-thiolactone with impaired lysis of fibrin clots in vivo in humans (manifested by longer time of fibrinolysis) as we have recently shown in a large randomized controlled trial [42].
Enzymological studies in vitro led to a contention that the lactonase activity is a native physiological activity of PON1, but no physiological evidence was provided [43,44]. Other studies repeated this contention by stating that the lactonase activity is “the established native physiological activity of PONs” [45] even though no physiological evidence supporting such statement has been reported either. A study that attempted to identify endogenous lipophilic lactones as possible in vivo substrates for PON1 in human serum, found none [46]. Possible involvement of PON1 in metabolism of endogenous lipophilic lactones in vivo as proposed in refs [43,44] remains to be proven.
Nevertheless, recent findings showed that some phenyl-γ-valerolactones (PVLs), phase 2 metabolites derived from dietary flavan-3-ols, are substrates for PON1 and PON3 in vivo [47]. Flavan-3-ols constitute the main class of polyphenolic bioactive compounds present in the food and beverages such as tea, pome fruits, cocoa products, and berries. Large-scale randomized clinical trials show that flavan-3-ol intake was associated with beneficial cardiovascular effects [48] but had no effect on cognition [49]. After intake, flavan-3-ols are catabolized by gut microbiota to PVLs and phenyl-γ-valeric acids (PVAs), which enter the circulation and are distributed throughout the human body [50]. After intraperitoneal administration of 5-(3′,4′-dihydroxyphenyl)-γ-valerolactone (γVL), the sulfated form of γVL was detected in the brain while γVL aglycon was not detected [51]. In TNF-α stimulated human brain primary microvascular endothelial cells, 5-(4′-hydroxyphenyl)-γ-valerolactone-3′-sulfate and 5-(4′-hydroxyphenyl)-γ-valerolactone-3′-O-glucuronide were biologically active at low nanomolar concentrations and influenced the expression of genes involved in biological pathways such as cell adhesion, cytoskeleton organization, focal adhesion, signaling pathways, pathways regulating endothelial permeability, and interaction with immune cells [52]. However, it is not known whether corresponding PVAs (i.e., products of PVLs hydrolysis by PON1) can also influence gene expression.
In human serum, γVL was rapidly hydrolyzed to the corresponding γ-substituted valeric acid (γVA) by PON1 and PON3 (t1/2 = 9.8 min) (Figure 3B) [47]. The hydrolysis was prevented by treatments with EGTA (calcium chelator and an inhibitor of PON1) or with heat (Figure 3A). Km was 269 μM (Figure 3C), way above sub micromolar γVL concentrations in humans [50]. Some but not all, phase II metabolites (sulfated or glucuronidated γVL) were also hydrolyzed by PON1/PON3 in serum. In general, conjugated γVLs were worse substrates of PON than unconjugated γVL. Additional conjugations of γVL significantly reduced or prevented the hydrolysis of γVL metabolites by PON. Another polyphenol-derived lactone, enterolactone, was not a substrate [47].
In contrast to the established influence of PON1 genotype on the hydrolysis of Hcy-thiolactone [23,39], the EPIC-Norfolk sub-cohort study found that the sum of urinary conjugated γVLs was not influenced by PON1 genotype (Q192R, rs662 in the coding region; -162A/G rs705381 in the promoter region) in males but there was a small effect in females [47]. These findings suggest that PON1 genotype has a minor sex-dependent effect on the hydrolysis of conjugated γVLs; this, however, remains to be examined in future studies.
3. Association of PON1 with Human Disease
PON1 has been studied in the fields of toxicology, CVD, renal disease, liver disease, Alzheimer’s disease, and cancer. Initial studies have shown that Pon1-/- mice are highly susceptible to the toxicity of organophosphate insecticides [53] and to atherosclerosis induced by the metabolic stress of a high-fat diet [54], or by ApoE depletion [55]. Molecular basis of PON1’s protective role in organophosphate poisoning is well understood [53]. In contrast, the nature of PON1 targets in atherosclerosis and other human diseases is not fully understood. Elevated oxidative stress and inflammation, associated with CVD [5], are also observed in Pon1-deficient mice [54,55] and humans with attenuated PON1 activity [27].
Reduced PON1 activity accompanied by increased oxidative stress and inflammation is a common finding in patients with CVD [27,29,56], diseases of kidney [30,31] and liver [57], Alzheimer’s disease [32,58,59], and cancer [33,60] (Table 1). This has been suggested to be due to the mediation by PON1 of anti-oxidative and anti-inflammatory effects of HDL [34,35]. Many other HDL components have also been shown to mediate the anti-oxidative and anti-inflammatory activity of HDL [10,11,36], including APOA1, which accounts for 70% of HDL protein mass [36] and anti-apoptotic activity [37], and for most of the HDL anti-oxidative activity [8]. Although how attenuated PON1 expression or activity can induce oxidative stress and inflammation is not clear, accumulating evidence, discussed below, suggests that the influence of PON1 on oxidative stress and inflammation is indirect rather than direct [38].
3.1. PON1, Oxidative Stress, Inflammation, and CVD
In Pon1-/- mice fed with a high fat diet, the enhancement of atherosclerosis is accompanied by upregulation of oxidative stress, which is manifested by elevated levels of lipid peroxides in purified HDL [54]. In Pon1-/-ApoE-/- mice, elevated oxidized phospholipid epitopes in plasma, bioactive oxidized phospholipids in purified endogenous intermediate density lipoprotein/LDL, and upregulated expression of oxidative stress-responsive genes such as heme oxygenase-1 (HO1), peroxisome proliferator-activated receptor gamma (PPARγ), and oxLDL receptor (oxLDL-R) in the liver were observed [55]. Although no lipid hydroperoxides were found in fresh purified LDL from any Pon1 genotype, LDL from Pon1-/- mice, the LDL from Pon1-/- nevertheless stimulated lipid hydroperoxide generation and monocyte transmigration better than did LDL from Pon1+/+ mice in a coculture model. These results suggested that LDL from Pon1-/- mice was changed somehow to become prone to oxidation. Lipid hydroperoxide formation in LDL was inhibited by the pretreatment with purified human PON1 [54].
Overexpression in mice of human PON1 using bacterial artificial chromosome genomic clones increased plasma PON1 levels 2- to 4- fold and significantly reduced aortic lesions in dietary as well as ApoE-/- models [61]. Overexpression of human PON1 in LDL-/- mouse model of metabolic syndrome via adenovirus-mediated PON1 gene transfer increased paraoxonase activity of PON1 4.4-fold, significantly reduced plaque-associated oxLDL, titer of auto-antibodies against LDL modified by malondialdehyde (MDA) (a proxy for oxLDL), and plaque volume by 80% [62]. Unfortunately, how Pon1 overexpression influences the expression of genes involved in inflammation and oxidative stress has not been studied in these two mouse models.
Overexpression of the human PON gene cluster (PC Tg) in ApoE-/- mice using bacterial artificial chromosome increased the enzymatic activity towards the paraoxon substrate in isolated HDL by 60%, stabilized atherosclerotic lesions, and significantly attenuated (by 20-30%) plaque area [63]. PC Tg HDL significantly inhibited oxLDL production (by 40-67%) compared to wild type HDL. Inflammation markers ICAM-1 and MCP-1 were significantly downregulated (by 31-51%) in PC Tg/ApoE-/- mice compared to ApoE-/- mice. The inflammatory response of isolated PC Tg macrophages (due to human PON2 overexpression) was significantly attenuated compared to macrophages isolated from ApoE-/- mice as shown by quantification of TNF-α and IL-6. Human PON2 inhibited also macrophage MMP-9 expression and foam cell formation from PC Tg macrophages compared to macrophages from ApoE-/- mice [63]. These findings show that PON gene cluster overexpression protects from atherosclerosis by ameliorating oxidative stress and downregulating the expression of genes involved in inflammation.
The first large-scale prospective study that evaluated the relationship between oxidative stress and PON1 genotype and activity and their prognostic value as predictors of future CVD, involved 1339 patients (65-year-old, 72% male) and 283 controls without CVD (57-year-old, 48% male) who underwent coronary angiography [27]. The study found at baseline that low paraoxonase or arylesterase activity of PON1 as well as PON1-192QQ polymorphic variant were associated with elevated various oxidized fatty acids (5-, 8-, 9-, 11-, 12-, 15-hydroxyeicosatetraenoic acids (HETEs), 9-, 13-hydroxyoctadecadienoic acids (HODEs), 8-isoprostane prostaglandin F2α (8-isoPGF2α)) in patients (n = 150) (Table 1). Participants carrying PON1-192QQ alleles had significantly increased risk of adverse CVD outcomes such as death, myocardial infarction (MI), and stroke, compared with PON1-192RR and PON1-192QR carriers over a 3-year-followup (18.0% vs. 13.6%, adjusted hazard ratio 1.48, P = 0.01) and all-cause mortality (11.1% vs 6.75%, adjusted hazard ratio 2.05, P = 0.001). Although PON1-192QQ genotype was not associated with nonfatal stroke or MI, low activity of PON1predicated increased frequency of stroke and MI, all-cause mortality, and the sum of adverse CVD outcomes. Specifically, participants with the lowest paraoxonase or arylesterase activity of PON1 (1st quartile) showed increased frequency of adverse CVD outcomes (paraoxonase: 25.1%; arylesterase 23.5%) compared to participants with the highest activities of PON1 (4th quartile) (7.3% or 7.7%, respectively). The adjusted hazard ratios for nonfatal MI and stroke, all-cause mortality, and the sum of in the lowest vs. highest PON1 activity quartiles were 4.4, 2.4, and 3.4, respectively, for paraoxonase and 4.5, 2.2, and 2.9 for arylesterase, respectively, and were independent in multivariate analysis in models (separate for paraoxonase and arylesterase) adjusted for all traditional cardiac risk factors and medications). These findings demonstrate that PON1 genotype/activity affect oxidative stress and predict future CVD risk.
However, it should be noted that the PON1-Q192R polymorphism, associated with indices of oxidative stress in vivo [27], has opposite effects on paraoxonase and arylesterase activities of PON1: the 192Q allele associates with low paraoxonase and high arylesterase activity while the 192R allele associates with high paraoxonase and low arylesterase activities were [25,28,64,65]. In this context it is not clear why low paraoxonase and arylesterase activities of PON1 were associated with nonfatal stroke and MI, but the PON1-192QQ genotype was not [27].
A population-based cross-sectional study of 1,895 participants (32-year-old, 46% male) examined a relationship between rs669 SNP (PON1-Q192R), PON1 activity, and conjugated dienes in lipoprotein lipids [66] as a measure of oxLDL lipids [67] that is known to correlate with lipid hydroperoxides and MDA [68]. In multiple regression models, paraoxonase activity of PON1was inversely correlated with oxLDL lipids (P = 0.0001) but not with oxHDL lipids, and tended to be associated with oxLDL protein (P = 0.08). A stronger association between PON1 activity and oxLDL lipids was seen in the PON1-192RR carriers than in the PON1-192QQ carriers. Although PON1 rs662 SNP was strongly associated with paraoxonase activity of PON1, it was not associated with oxHDL lipids or protein.
A case-control study found that CAD patients confirmed by angiography (n = 105) had significantly increased plasma 8-isoprostane F2 (8-iso-PGF2α, produced by the non-enzymatic peroxidation of arachidonic acid in membrane phospholipids) and reduced paraoxonase and arylesterase activities of PON1 compared to healthy controls (n = 45) [69]. Paraoxonase and arylesterase activities of PON1 were significantly negatively associated with the severity of CAD (Gensini score) in univariate analyses, while 8-iso-PGF2α was associated positively. Such associations were also seen in multiple regression models adjusted for traditional risk factors. These findings suggest that PON1 may protect phospholipids from oxidation [69]. However, the mechanism underlying these findings remains to be elucidated.
One study examined how PON1 SNPs and PON1 arylesterase activity are related to oxidation susceptibility of LDL isolated from male CAD patients and healthy control participants [70]. The susceptibility of LDL to oxidation was measured using an assay, inwhich LDL is oxidized by copper and the resulting conjugated dienes are monitored by absorbance at 234 nm. During LDL preparation from each participant, HDL/PON1 was removed. The study involved CAD patients (n = 205, 70-year-old, > 80% stenosis) and control participants (n = 232, 66-year-old, < 15% stenosis). It was found that the susceptibility of LDL to oxidation, was not correlated with arylesterase activity of PON1, although correlated with CAD. In contrast, PON1 promoter SNP, PON1 -108C/T, which affects PON1 expression, and other PON1 SNPs, were associated with the susceptibility of LDL to copper induced . The absence of congruency in the relationships between the susceptibility of LDL to oxidation and CAD, PON1 arylesterase activity, and PON1 -108C/T SNP raises doubts regarding validity of the experimental approach used in this study.
3.2. PON1, Lipid Oxidation, Hcy-thiolactone, and Alzheimer’s Disease
OxLDL is pro-inflammatory and induces oxidative stress and cytotoxicity in neurons, astrocytes, and microglia [52,53]. Importantly, amyloid beta binds to oxLDL and accelerates the formation of macrophage foam cells [54], suggesting that oxLDL can directly participate in the development of AD.
PON1 plays an important role in the detoxification of neurotoxins including organophosphate pesticides, potent inhibitors of acetylcholinesterase; people with low PON1 activity show increased sensitivity to neurotoxins while those with high PON1 activity are less susceptible [54]. Notably, exposure to organophosphates increases the risk of developing AD [71,72]. Treatments with doses of chlorpyrifos oxone that do not affect wild type Pon1+/+ mice are known to induce seizures and death in Pon1 knockout mice [54]. Importantly, Pon1 is also known to detoxify Hcy-thiolactone in mice. For example, treatments with Hcy-thiolactone induce seizures significantly faster (Figure 4A) and with increased incidence (Figure 4B) in Pon1-/- mice compared to wild type Pon1+/+ mice. Death incidence was also higher in Pon1-/- mice (Figure 4C) [39].
3.2.1. Mice
Proteomic analyzes of Pon1-/- and Pon1+/+ mice have shown that, Pon1 plays an important role in maintaining cellular proteostasis [58], in addition to controlling Hcy-thiolactone and N-Hcy-protein levels [39]. Pon1 gene deletion affects the expression of cellular proteins in an organ-specific way, with the patterns of expression modulated by hyperhomocysteinemia (HHcy). In brains of Pon1-/- mice, proteins involved in anti-oxidant defenses (Sod1, DJ-1), brain-specific function (Nrgn), and the assembly of cytoskeleton (Tbcb) were significantly downregulated, while CapZa2 protein involved in the assembly of cytoskeleton was significantly upregulated compared to wild type Pon1+/+ mouse brain [58].
In brains of HHcy Pon1-/- mice that were fed with high methionine diet, Prdx2 and DJ-1proteins participating in anti-oxidant defense; Ncald, Nrgn, and Stmn1 proteins involved in brain-specific function; energy metabolism protein Ak1; cell cycle GDI1 and Ran proteins; cytoskeleton assembly Tbcb protein; and Hdhd2 protein of unknown function were all upregulated (Table 2). Notably, Pon1 gene deletion affected the expression of DJ-1 (Park7), Sod1, and Prdx2 proteins involved in the oxidative stress response that are also known to be associated with AD [58].
Clusterin (CLU or APOJ), involved in the transport of amyloid beta (Aβ) from plasma to brain in humans (reviewed in [73]), is carried on a minor HDL subspecies, that contains two other proteins: APOA1 and PON1 [74]. Importantly, Clu (ApoJ) levels are significantly elevated in plasma of Pon1-/- mice compared to wild type Pon1+/+ animals [65].
PON1 involvement in AD was examined using Pon1-/-5xFAD mice and in Aβ-overexpressing mouse neuroblastoma N2a-APPswe cells [15]. 5xFAD mice overexpress the K670N/M671L (Swedish), I716V (Florida), and V717I (London) mutations in human APP (695), and M146L and L286V mutations in human PS1 and start to accumulate high levels of Aβ42 at about 2-month-old [75]. The dysregulation of mTOR signaling and autophagy are linked to Aβ accumulation in AD patients [76,77], while histone H4K20me1 demethylation by the histone demethylase PHF8 maintains homeostasis of mTOR signaling [78].
The study revealed that Pon1 plays an important role in protecting from the amyloidogenic processing of APP to Aβ in brains of mice and identified mechanism of this new function of Pon1 in the central nervous system (Figure 5). Specifically, Pon1 depletion in Pon1-/-5xFAD mice significantly downregulated Phf8 and upregulated methylated histone H4K20me1 mark. This led to upregulation of mTOR expression and increased its active form, phospho-mTOR, which impaired autophagy by downregulating Bcln1, Atg5, and Atg7 proteins in Pon1-/-5xFAD mouse brains compared to Pon1+/+5xFAD brains. Silencing of the Pon1 gene in N2a-APPswe cells by RNA interference using siRNA targeting Pon1 gene led to Phf8 downregulation, which increased histone H4K20me1 binding at the mTOR promoter thereby upregulating mTOR expression and signaling. Upregulated mTOR signaling impaired autophagy and significantly elevated APP expression and Aβ levels. Hcy-thiolactone or N-Hcy-protein (metabolites known to accumulated Pon1-/- mice), or Phf8 depletion by RNA interference elevated Aβ levels in N2a-APPswe cells [15]. Notably, Phf8 gene silencing did not influence the expression of APP, indicating that Aβ levels increased independently of APP [79].
Pon1 depletion induced changes in the Phf8->H4K20me1->mTOR->autophagy pathway (Figure 5) that were similar to the changes induced by HHcy [15], suggesting that the same Hcy metabolites were involved. This suggestion is supported by our previous findings showing that a common primary physiological outcome of Pon1 depletion and of HHcy was essentially the same: Pon1 depletion [39,81] and HHcy [82] each increased Hcy-thiolactone and N-Hcy-protein levels. Taken together, our findings show that Aβ generation in the Pon1-/- brain is mediated by the influence of Hcy-related metabolites on mTOR signaling/autophagy [15]. These findings provide mechanistic explanation for the link between attenuated PON1 activity [35] or elevated Hcy [83] and AD.
3.2.2. Humans
A few studies examined PON1 activity in relation to oxidative stress in AD patients. In one study, PON1 activities were found to be significantly decreased (Table 1) while PAF-AH activity and oxLDL levels were significantly increased in forty-nine AD patients (74-year-old, 59% female, MMSE score = 21 ± 5) compared to thirty-four age/sex-matched control individuals [84]. The study found a significant inverse correlation of oxLDL with PON1 activities (but not with activity of PAF-AH) in AD patients and non-AD control individuals. Further, activities of PON1 and levels of oxLDL were associated with the severity of AD (assessed by using the MMSE test, which quantifies global cognition). Patients with moderate (MMSE score of 11 to 24) and severe AD (MMSE score < 10) had significantly decreased activities of PON1 and increased oxLDL levels compared to patients with mild AD (MMSE score > 24). Even though PAF and oxidized phospholipids hydrolysis by PAF-AH generates free oxidized fatty acids, which have potent biological activity, PAF-AH activity was not correlated with oxLDL nor with severity of AD. Although these findings suggest that PON1 may participate in oxLDL metabolism in AD, the nature of this participation is not clear.
Another study evaluated oxLDL in fifty-four late-onset AD patients (aged 77 years, 81% female, MMSE score = 18) and 51 healthy elderly individuals (aged 77 years, 73% male, MMSE score = 29) and a relationship between oxLDL and PON1-107C/T polymorphism and APOE genotype [85]. Patients with AD and control individuals with the PON1-107TT genotype had significantly elevated levels of plasma oxLDL compared to PON1-107CC/CT genotype. The distribution of lipoprotein cholesterol in patients with AD was shifted toward a greater prevalence of smaller, denser LDL. In AD patients, the smaller, denser LDL levels were significantly associated with the levels of oxLDL. Lipoprotein distribution was not influenced by APOE genotype. These findings suggest that plasma oxLDL levels could modulate the association of PON1-107TT polymorphism with AD [85].
A study that examined relationships between PON1, lipid peroxidation, and dementia with AD patients (n = 63), vascular dementia patients (n = 40), and mixed dementia patients (n = 33) found that MDA/thiobarbituric acid-reactive substances were elevated to a greater extent in vascular dementia than AD [59]. In patients with vascular involvement, increase in MDA/TBARS reflected the extent of global cortical atrophy. Arylesterase activity of PON1 was significantly attenuated in patients with dementia, more so in patients with severe cognitive deficits. In patients with vascular dementia, low arylesterase activity of PON1 was associated with increased brain ischemia and medial temporal lobe atrophy. These findings show that reduced activity of PON1 and increased levels of the MDA/TBARS oxidative stress marker are associated with brain atrophy and vascular dementia rather than with cognitive decline. However, it is not clear how reduced PON1 activity can lead to oxidative stress and impaired cognition.
3.3. PON1 Depletion, Dysregulation of Signaling Pathways, and Cancer
Hepatocellular carcinoma (HCC) is one of the most common neoplasms, the third leading cause of cancer death, and a leading cause of death among patients with cirrhosis [86]. Liver cirrhosis is widely prevalent worldwide and can be a consequence of different causes, such as obesity, non-alcoholic fatty liver disease, high alcohol consumption, hepatitis B or C infection, autoimmune diseases, cholestatic diseases, and iron or copper overload [57].
Recent studies show that PON1 activity and expression are compromised in HCC patients. Specifically, serum PON1 activity, measured with 4-nitrophenylacetate as a substrate, was significantly reduced in HCC patients [60]. Transcriptomic analysis showed that the expression of PON1was significantly downregulated in HCC tissues compared to normal tissues [33]. However, there was also a significant variation in PON1 expression between HCC patients. Patients with low PON1 expression manifested significant differences in pathology severity, tumor size and grade. Female HCC patients with low PON1 expression had a higher degree of tumor malignancy.
Differences in PON1 expression influenced the clinical manifestations, biological processes, immune infiltration, and expression of immune checkpoints in HCC, suggesting that PON1 plays an important role in modulating tumor progression and immune cell infiltration, thus establishing PON1 as a new biomarker important for prognosis, targeted therapy, and immunotherapy in HCC patients [33].
Bioinformatic analysis of pathway enrichment in the high and low PON1 mRNA expression groups showed that PON1 gene inhibits key signaling pathways, such as the PI3K/Akt/mTOR signaling, the cell cycle G2 checkpoint, the TGF-β signaling, and the Wnt/β-catenin signaling, which play a crucial role in pathogenesis and progression of HCC [33]. Interestingly, we found that Pon1 depletion upregulated mTOR signaling and inhibited autophagy via Pcft/H4K20me1 in mouse brain and neuroblastoma cells [15], suggesting that effects of Pon1 depletion on gene expression are disease/tissue-specific.
4. PON1 Has no Intrinsic Anti-oxidant Activity: Don’t Waste Clean Thoughts on Dirty Enzymes
PON1 has been stated to hydrolyze oxidized lipids and thus to promote atheroprotective effects, e.g., refs [27,87], which incorrectly implies that PON1 has an intrinsic anti-oxidant function. That PON1 has the ability to hydrolyze oxidized lipids was originally proposed by a study that reported the ability of purified native human PON1 to inhibit copper-induced oxidation of LDL in an in vitro assay that quantified lipo-peroxides and TBARS [88].
The availability of an assay for a biological event in a cell-free system usually facilitates studies of its molecular mechanism. Indeed, this assay has been used in many in vitro studies using purified native (e.g., refs. [89,90,91,92]) and recombinant [93,94] PON1 preparations. Unfortunately, these and other studies in the PON1 field did not follow the maxim “don’t waste clean thoughts on dirty enzymes” attributed by Arthur Kornberg in his ‘ten commandments of enzymology’ [95] to Efraim Racker, a pioneer in the enzymology of oxidative phosphorylation [96].
Some labs did not replicate the finding that purified PON1 protects LDL from oxidation [44,97] while those that did [98,99,100] went on to corrected themselves by showing in a more rigorous and well-controlled studies that their earlier findings were due to PAF-AH contamination in PON1 preparations and that PAF-AH-free that PON1 does not protect lipoproteins from oxidation nor hydrolyze oxidized lipids [44,101,102]. Rigorously purified PON1, or plasma from an individual with a mutation in the PAF-AH gene, did not hydrolyze PAF nor the oxidized phospholipids from oxLDL [97].
One study purported to show that purified PON1 was capable of hydrolyzing PAF. In that study [103], purified PON1 preparations were tested by western blotting (20 μg) and amino acid sequencing (50 μg, or about 1 nmol PON1) and found not to have any detectable PAF-AH contamination. However, such evidence does not exclude PAF-AH contamination, considering that as little as 5 to 10 ng of PAF-AH (undetectable by western blotting and not sufficient for sequencing) is sufficient to account for all the phospholipase activity in purified PON1 preparations [97]. In fact, other labs have shown that purified PON1 has no phospholipase A2-like activity toward PAF or pro-atherogenic oxidized phospholipids and that PAF-AH is the sole phospholipase A2 of HDL [97,101]. Rigorously purified PON1, or plasma from an individual with a mutation in the PAF-AH gene, did not hydrolyze PAF nor the oxidized phospholipids from oxLDL [97].
Although Aviram et al. [93] and Liu et al. [94] reported that PON1, PON2, and PON3 protect LDL from oxidative modification, Draganov et al. found no protection [44]. Recombinant human PON1 was expressed from a baculovirus vector in insect cells and purified. When PON1 hydrolytic activity and a putative anti-oxidant activity were monitored during PON1 purification, the two activities did not co-purify at any stage and in any of the preparations. The putative anti-oxidant activity was shown to be associated with a low mass contaminant and the detergent used in PON1 purification [44]. That putative anti-oxidant activity in PON1 preparations was associated with the detergent present in these preparations was confirmed by another lab that also showed that anti-oxidant activity was not associated with hydrolytic PON1 activities such as arylesterase and lactonase, or with phospholipase activity [101]. Unfortunately, it appears that many other laboratories didn’t seem bother to control their PON1 preparations for contaminants.
Studies that examined contribution of individual protein components to the ability of HDL to inhibit LDL oxidation showed that APOA1 is the major anti-oxidant protein in HDL [10]. APOA1 is also the major factor responsible for the protection of human endothelial cells from oxLDL-induced apoptosis, accounting for 70% of HDL antiapoptotic activity [37]. APOA1 is one of the two phosphatidylcholine peroxide reducing enzymes isolated from human plasma (the other is glutathione peroxidase) [104]. APOA1 is essential for HDL structure and for activation of the HDL-associated enzymes PON1 and LCAT [105]. Two methionine residues in APOA1 (Met112, Met148) are oxidized to sulfoxides during reduction of lipid peroxides to redox-inactive hydroxides [106,107]. Reconstituted HDL containing only purified APOA1, and phospholipids (palmitoyloleoyl phosphatidylcholine at a molar ratio of 1.0/77.1) has the capacity to inhibit LDL oxidation like that of native normolipidemic small, dense HDL3b and 3c isolated from normal human plasma. Oxidation of APOA1 Met residues in HDL3 incubated with oxLDL is accompanied by concomitant reduction of lipid peroxides to lipid hydroxides [8].
To assess a role of HDL-associated enzymes, such as PON1, PAF-AH, and LCAT, in oxLDL inactivation, HDL3 was pretreated with inhibitors such as DFP that inhibits the 3 enzymes, Pefabloc that inhibits only PAF-AH, or EDTA that inhibits only PON1, and then incubated with oxLDL. As expected, pretreatment significantly reduced the activities of LCAT (by 50%), PAF-AH (by 90%), and PON1 (by 99%). In contrast, the capacity of HDL3 to inactivate lipid peroxides in oxLDL or to delay LDL oxidation was not affected. None of the inhibitors impaired the capacity of HDL3 to delay the accumulation of conjugated dienes in LDL [8]. Two earlier studies have also reported that inactivation of PON1 activity by EDTA did not affect the anti-oxidant activity of HDL3 [108] or PON1 preparations [91] in the copper-induced LDL oxidation assay. These findings do not support the contention that paraoxonase activity inhibits the formation of 'minimally oxidized' LDL by hydrolyzing biologically active oxidized phospholipids [9,89].
5. Mechanistic Bases of PON1 Involvement in Human Disease
The findings that PON1 is associated with oxidative stress, inflammation, and disease in humans and in mouse models (discussed in section 2) but cannot be directly linked to these processes (discussed in section 3). This suggests that the influence of PON1 on oxidative stress, inflammation, and disease is indirect.
Studies of proteomic and transcriptomic changes in mice and humans in relation to changes in PON1 expression and activity (listed in Table 3 and discussed below) support this suggestion and provide insights into the biological function of PON1. Specifically, these studies suggest that vascular inflammation and oxidative stress that are associated with PON1 depletion are caused by the dysregulation of genes involved in these processes. Majority of studies discussed in this section are related to CVD with two related to kidney disease and one to brain disease. Studies related to AD and cancer were discussed in section 2.
5.1. Low PON1 Activity in Dysfunctional HDL Is Associated with Impaired Nitric Oxide Production in Endothelial Cells
Native HDL possesses anti-inflammatory and anti-oxidative properties [34] and directly influences the vascular endothelium by activation of nitric oxide (NO) synthesis by eNOS [116,117] thus promoting endothelial repair [118]. Such cardio-protective processes are, at least in part, mediated by binding of HDL to endothelial scavenger receptor B, type I (SR-BI), and by PON1 [6], carried in the circulation on a minor fraction of HDL [16].
Patients with stable coronary artery disease (CAD) or an acute coronary syndrome, carry dysfunctional HDLCAD which does not promote endothelial NO synthesis, anti-inflammatory effects, and repair that is characteristic of normal HDL isolated from healthy individuals [6]. This has been shown to be due to activation by HDLCAD of the endothelial lectin-like oxLDL receptor 1 (LOX-1), which induces endothelial PKCβII activation, thereby inhibiting eNOS-dependent NO generation. These newly acquired properties were conferred by elevated MDA, a product of lipid peroxidation, that chemically modified PON1 thereby reducing its activity and generating dysfunctional HDLCAD, which actvates PKCβII and lacks anti-oxidative, anti-inflammatory properties of normal HDL. Moreover, HDL from Pon1-/- mice failed to enhance endothelial NO production while addition of pure PON1 or HDL from healthy individuals partially ameliorated stimulating effects of HDL on NO production. Even though PON1 activity in HDLCAD was decreased, PON1 protein content in HDLCAD was elevated compared to HDL from healthy controls, suggesting that enzymatic activity of PON1 was inactivated in HDLCAD. These findings show that HDL-associated PON1 activity has an important function in maintaining the ability to stimulate endothelial-atheroprotective effects of HDL, i.e., NO production. Impairment of this fundamental role of PON1/HDL by oxidative stress can account for the increased risk of adverse cardiovascular events in CAD patients [27,28]. These findings also show that PON1 regulates the expression of genes involved in endothelial homeostasis and that dysregulation of these processes leads to CAD or acute coronary syndrome.
5.2. Pon1 Depletion Affects Expression of Genes Involved in Inflammation, Oxidative Stress, and Blood Clotting
Although Pon1 depletion in mice in the absence of hyperlipidemia does not induce atherosclerosis [54], Pon1-/- mice fed with a standard normolipidemic chow diet show altered expression of proteins involved in vascular inflammation, oxidative stress, and thrombogenicity [119]. Specifically, there was a significant 2-fold increase in leukocyte adhesion revealed by intravital microscopy, but no significant change in leukocyte rolling in Pon1-/- mice compared to Pon1+/+ control animals. The increase in adhesion was correlated with significant increases in aortic P-selectin and Icam mRNA levels (P = 0.016) and an 1.3-fold increase in Vcam1 mRNA (P =0.096). Aortic Tnf-α mRNA expression was not affected. The rate of aortic superoxide production was significantly increased in Pon1-/- vs. Pon1+/+ mice (3-fold, P = 0.04). Pon1-/- mice were also predisposed to thrombosis as shown by a significant 57% reduction in time to occlusion in a carotid thrombosis assay (P < 0.001). Notably, these vascular changes mimic those seen in severely hyperlipidemic ApoE-/- mice [119]. These findings also show that Pon1 interacts with genes involved in inflammation, oxidative stress, and blood clotting.
5.3. Pon1 Depletion Increases Expression of Liver Oxidative Stress Genes and Accelerates Atherosclerosis
In mice fed with a high fat diet, Pon1 gene deletion led to increased atherosclerosis and increased lipid peroxides levels in isolated HDL compared to Pon+/+ animals [54]. In ApoE-/- mice, Pon1 gene deletion also increased atherosclerosis and oxidative stress, manifested by elevated epitopes in plasma oxidized phospholipid, biologically active oxidized phospholipids in isolated endogenous intermediate density lipoprotein/LDL. RT-qPCR analyses showed that these changes were accompanied by upregulated expression of genes involved in oxidative stress-responsive genes in the liver ) HO-1, PPARγ, and oxidized LDL-R) [55] (Table 3). These findings show that Pon1 deficiency promotes oxidative stress and atherogenesis. These results also suggest that Pon1 interacts with oxidative stress-responsive genes and that disruption of these interactions induces oxidative stress and causes atherosclerosis.
5.4. Pon1 Depletion Increases Expression of Oxidative Stress Genes in Liver, Kidney, and Brain
Proteomic analyses of Pon1-/- mice show that, in addition to controlling Hcy-thiolactone and N-Hcy-protein levels [39], Pon1 is important in maintaining cellular proteostasis. Specifically, in brains of Pon1-/- mice fed with a normal chow diet, levels of proteins involved in anti-oxidant defenses (Sod1, DJ-1) were significantly reduced compared to Pon+/+ animals. In the presence of hyperhomocysteinemia (HHcy) induced by feeding with a high-methionine diet, DJ-1 and Prdx2 proteins were significantly upregulated in HHcy Pon1-/- mice compared to HHcy Pon+/+ animals [58]. In kidneys [111] and livers [110] of Pon1-/- mice, levels of the anti-oxidant protein Prdx2 were significantly elevated compared to Pon+/+ animals. These findings suggest that Pon1 interacts with oxidative stress-responsive proteins in an organ-specific way and that HHcy modulates these interactions.
5.5. Pon1 Depletion in Scarb1-/- Mice Is Associated with Upregulated Expression of Oxidative Stress Genes
Scavenger receptor BI (SR-BI) plays a central role in reverse cholesterol transport (RCT) as the major receptor for HDL cholesterol (HDL-C) [120]. Although elevated plasma HDL-C levels are associated with a lower risk of CVD in humans, a rate mutation in the human SCARB1 gene encoding SR-BI increases the risk of CVD, suggesting that high concentrations of HDL-C are not causally protective against CVD and that cholesterol flux and HDL function are more important than the steady-state levels [121]. The SR-BI knockout mice (Scarb1-/- mice) have dysfunctional HDL characterized by impaired macrophage reverse cholesterol transport (RCT) [122], high plasma HDL-C levels, impaired anti-oxidative and anti-inflammatory properties, and are susceptible to atherosclerosis [123].
Notably, the dysfunctional HDL is also characterized by reduced PON1 arylesterase and paraoxonase activities and is associated in a tissue-dependent way with indices of oxidative stress such as isoprostane F2α-VI (iPF2α-VI) and protein carbonyls in Scarb1-/- mice. Levels of monocyte chemoattractant protein-1 (MCP1) were similar in Scarb1-/- and wild type mice indicating that SR-BI deletion has no effect on inflammation. Western diet did not affect MCP1 levels in Scarb1-/- mice but increased serum PON1 paraoxonase activity and urinary iPF2α-VI in both Scarb1-/- and wild-type mice [124].
The dysfunctional HDL and reduced PON1 activity in Scarb1-/- mice were associated with upregulated expression of genes encoding oxidative stress proteins. Specifically, mRNAs for glutathione peroxidases GPx1 and GPx4, superoxide dismutase SOD1 and SOD2, glutathione S-transferases GSTA2 and GSTA4, which reduce lipid peroxidation products, and HO-1, which removes free prooxidant heme and generates of the anti-oxidant bilirubin, were upregulated in Scarb1-/- mice compared to wild-type animals. PAF-AH activity and catalase expression were not affected by Scarb1 depletion and reduced Pon1 activity [124].
5.6. Pon1 Depletion in Scarb1-/- Mice Affects Expression of Oxidative Stress and Inflammation-Related Liver Genes
Lipo-proteomics analysis showed that protein content of dysfunctional HDL from SR-BI-/- mice was decreased by 25%, compared to wild type SR-BI+/+ animals [113]. Out of seventy-eight proteins identified in SR-BI−/− HDL, twenty-six were upregulated and ten were downregulated compared to SR-BI+/+ HDL. Specifically, ApoA1, ApoA2, ApoC1, ApoC2, ApoM and PON1 were downregulated, while ApoE, ApoH, Lcat, acute phase proteins ApoA4, Saa, complement C3, proteinase inhibitors such as A1AT, inter alpha-trypsin inhibitor (3 of Itih1-4) and α-2-macroglobulin were upregulated in SR-BI−/− HDL compared to SR-BI+/+ HDL. Interestingly, plasma proteomics showed that these proteins were also affected in plasma of Pon1-/- mice compared to wild-type Pon1+/+ animals (Table 3) and in PON1-Q192R polymorphism in humans [65]. Proteins involved in lipid metabolism were significantly decreased (37.34% vs. 57.98%) as were anti-oxidant proteins (1.94% vs. 7.09%). In contrast, proteins involved in inflammatory/immune response were significantly increased (22.14% vs. 9.19%) as were proteinase-inhibiting proteins (15.67% vs. 8.49%). In in vitro experiments, SR-BI+/+ HDL significantly reduced Mcp1 and Tnf-α levels in oxLDL-treated macrophages while SR-BI−/− HDL had no effect [113].
Probucol is a cholesterol-lowering and anti-oxidant drug [125] that rescues female infertility in SR-BI−/−mice [126]. Treatments with probucol lowered plasma total and free cholesterol mainly in the HDL-C fraction, upregulated Pon1 and ApoA1, and downregulated ApoA4, Saa, A1AT, and myeloperoxidase (MPO) activity in SR-BI−/−mice. These findings indicate that anti-oxidant properties of HDL were improved by the probucol treatment [113].
5.7. PON1 Regulates the Expression of Hepatic Genes Involved in HDL Metabolism, Oxidative Stress, and Inflammation.
Lentivirus-mediated Pon1 overexpression resulted in a significant elevation of PON1 levels in liver and plasma by 63% in SR-BI-/- mice [115]. Pon 1 overexpression improved the anti-oxidative and anti-inflammatory properties of dysfunctional HDL and reduced hepatic steatosis and aortic atherosclerosis through its effects on the expression of genes involved in these processes. Specifically, cholesterol transporter Scarb1, inflammatory cytokines Il-6, Tnf-α, and Nox1 mRNAs were significantly downregulated, while Abca1 mRNA and the anti-inflammatory cytokines Il-4 and Il-10 were upregulated in macrophages treated with plasma from Pon1+SR-BI-/- mice. Levels of plasma MPO activity, an oxidative enzyme secreted by activated neutrophils, monocytes, and macrophages were significantly reduced in Pon1+SR-BI-/- mice. Lecithin-cholesterol acyltransferase (Lcat) (which removes cholesterol from the blood and tissues), ApoA1, and ApoE were significantly upregulated in Pon1+SR-BI-/- plasma. Histological examinations showed that aortic lesions and hepatic lipid depositions were significantly reduced in Pon1+SR-BI-/- mice. Hepatic ApoA1, ApoE, LDLR, LXRα, Abca1, Abcg5/8, and Acat were significantly upregulated while inflammatory cytokines Il-6 and Tnf-α were downregulated in Pon1+SR-BI-/- mice. These findings indicate that Pon1 can regulate proteins important for HDL function and ameliorate atherosclerosis and hepatosteatosis [115].
5.8. Pon1 1 Ameliorates Renal Lipotoxicity by Regulating Genes Involved in Activating Lipophagy and Inhibiting Pyroptosis
Excessive lipid accumulation can lead to lipotoxicity due to the generation of toxic lipid intermediates. In the kidney, one of the more vulnerable organs, lipotoxicity causes tissue damage and dysfunction via oxidative stress, inflammation, and autophagy impairment, which lead to renal disease [127]. Pon1 is expressed in the glomerular clusters and the epithelial cells of the proximal tubules in the kidney [128]. Pon1-/- mice show increased oxidative stress in the kidney manifested by elevated expression of renal Prdx2 [111]. Renal disease patients show significantly reduced plasma HDL-C, APOA-I, serum PON1 protein concentration, PON1 arylesterase/ paraoxonase activity, and LCAT activity [129], indicating impaired interactions of PON1 with APOAI and LCAT in dysfunctional HDL in these patients.
Mice deficient in the scavenger receptor class B type I (SR-BI-/-) fed with a normal diet showed significantly reduced serum PON1 activity and renal PON1 expression, which was accompanied by renal pathology involving oxidative stress, inflammation, and fibrosis [114]. Western blot analysis and qRT-PCR showed that the expression of p47phox protein, a key regulator of NADPH oxidase, was significantly higher in the kidneys of SR-BI-/- mice compared with wild type SR-BI+/+ mice. mRNA levels of NADPH oxidase genes Nox 1 and Nox4 were also significantly increased. Levels of inflammation related Il1b and Il6 mRNAs were significantly increased, while anti-inflammatory cytokine Il10 mRNA tended to decrease. These findings show that the renal oxidative stress and inflammation were significantly upregulated in SR-BI-/- mice compared to wild type SR-BI+/+ animals.
Overexpression of PON1 (mRNA, 2.03-fold; protein, 3.36-fold) using a lentivirus vector significantly attenuated the pathologic changes in the kidneys of SR-BI-/- mice fed with a high-fat diet. Specifically, PON1-overexpressing (Pon1+) SR-BI-/- mice showed a significant decrease in the fluorescence intensity of dihydroethidium bromide staining, a significant decrease of immunohistochemical renal staining for lipid peroxidation indicator 4-hydroxynonenal, a significant decrease in the oxidative stress-related indicators such as renal p47phox protein and Nox1, Nox2, Nox4 mRNAs, as well as a significant increase in the activity of the anti-oxidant enzyme SOD in the kidney. In addition, PON1-overexpressing mice showed reduced levels of Tnf-α and Il6 mRNAs and elevated levels of anti-inflammatory cytokine Il10. These findings show that PON1 overexpression has beneficial effects on the kidney function in SR-BI-/- mice by reducing renal ROS production, improving anti-oxidant status, and ameliorating renal inflammation. PON1 overexpression also attenuated renal lipid accumulation by upregulating cholesterol ester hydrolysis-related genes (Nceh1, Lipa) and cholesterol efflux related receptors (Abca1, Abcag1). Western blot and qRT-PCR analyses showed that fibrosis-related proteins (fibronectin, collagen, type I, a1, and actin a2 in smooth muscle, aorta) were significantly downregulated in PON1-overexpressing mice compared to the control lentivirus-GFP-injected mice; mRNA levels for fibronectin, Col1a1, Ccn2, and Lcn2, a marker of kidney injury, were also significantly reduced, indicating that PON1 overexpression ameliorated fibrosis. Moreover, Pon1 overexpression inhibited mTOR signaling and restored autophagy flux in the mouse kidney [114]. That Pon1 can regulate mTOR signaling and autophagy was also reported in another study that found upregulated mTOR signaling and downregulated autophagy in Pon1−/− mice brains and in Pon1-silenced mouse neural cells [15].
5.9. PON1-Q192R Polymorphism Influences Oxidative Stress and Inflammation Proteins in Human Plasma
To ascertain how changes in PON1 expression/activity affect the expression of other proteins, plasma proteomes were analyzed in healthy participants recruited from the population of Poznań, Poland [65]. PON1-Q192R polymorphism affected serum paraoxonase activity (7-fold reduction in PON1-192QQ vs. PON1-192RR) and protein (40% reduction PON1-192QQ vs. PON1-192RR) (Table 4). Label-free nanoLC-MS/MS mass spectrometry analyses showed that there was an overlap in the principal component analysis (PCA) profiles between low activity PON1-192QQ, intermediate activity PON1-192QR, and high activity PON1-192RR genotypes (Figure 6B).
The PON1-Q192R polymorphism affected the expression of twenty-one plasma proteins, including six oxidative stress-related proteins (APOA1↓, PON1↓, APOD↑, APOM↑, haptoglobin (HP)↓, glutathione peroxidase 3 (GPX3)↑), four immune response proteins: CFP↑, IGHG3↑, ↑PGLYRP2, ↑V2-6 (IGL), four lipoprotein metabolism proteins (APOA1↓, APOB↑, APOC1↓, PON1↓), two acute phase response protein (TTR ↑, AMBP↑), and seven blood coagulation protein (F13B↓, PLG↓, SERPINA10↓, VTN↓, ↑C9, ↑V2-17 (IGL), FETUB↑). Six of those proteins (APOA1, APOB, APOC1, APOD, APOM, PON1), representing 29% of total number of proteins affected by the PON1-Q192R polymorphism, are components of HDL [10,130]. These findings suggest that PON1 regulates the expression of other components of HDL as well as HDL proteins involved in oxidative stress, inflammation, and complement/coagulation in humans. Dysregulation of these processes may account for the pro-oxidant and pro-atherogenic phenotypes associated with attenuated PON1 levels in humans [27] and mice [54].
5.10. Pon1−/− Genotype Influences Oxidative Stress and Inflammation Proteins in Mouse Plasma
Pon1 activity and protein are absent in Pon1-/- mice [54] (Table 5). To determine how Pon1 depletion affects the expression of other proteins, plasma proteomes were analyzed in Pon1−/− mice (n = 17) and their Pon1+/+ littermates (n = 8) using label-free nanoLC-MS/MS mass spectrometry [65]. PCA profiles differed between Pon1-/- vs. Pon1+/+ siblings (Figure 6A). Pon1 depletion in mice affected the expression of fifty plasma proteins, including seven redox proteins (↓Alb, ↓Blvrb, ↑ Ambp, ↑Hp, ↑Hpx, ↑ApoD, ↑ApoM), seven lipoprotein metabolism proteins (↓ApoA1, ↑ApoB, ↓ApoC1, ↑ApoD, ↑ApoM, ↓Pon1, ↑Lcat), four acute phase response proteins (↑Ambp, ↑Hp, ↑Hpx, ↑Ttr), and eleven complement/coagulation proteins (↑Al182371, ↑Cfh, ↑Clu, ↑F2 (prothrombin), ↓Klkb1, ↓Mbl1; ↓Serpinc1, ↑Fetub, ↓F13B, ↑Hrg, ↓Itih1) (arrows indicate direction of change). Nine of those proteins (↓Alb, ↓ApoA1, ↓ApoC1, ↑ApoD, ↑ApoM ,↑Clu, ↑Saa1, ↑Saa2, ↑Lcat), representing 18% of total number of proteins affected by Pon1−/− genotype, are components of HDL [10,16] . Clu (ApoJ) is also involved in amyloid beta (Aβ) transport from plasma to brain [73].
Nine proteins that were affected by Pon1 genotype in mice (↓ApoA1↓, ↑ApoD↑, ↑ApoM↑, ↓Pon1↓, ↑haptoglobin (Hp)↓, ↓Ighg3↑, ↑Ttr↓, ↓F13B↓, ↑Fetub↓), were also affected by PON1-Q192R polymorphism in humans (representing 22% and 43% of the total number of differentiating proteins in mice and humans, respectively) (right and left arrows refer to the change in humans and mice, respectively). These findings show that changes in the mouse plasma proteome associated with the Pon1−/− genotype were like those in the human plasma proteome associated with PON1-Q192R polymorphism. These findings also suggest that Pon1 regulates the expression of other protein components of HDL in addition to proteins involved in oxidative stress, inflammation, and complement/coagulation in mice.
Ingenuity Pathway Analysis identified the “Lipid Metabolism, Molecular Transport, Small Molecule Biochemistry” network, affected by PON1 activity in mice (Figure 7A) and humans (Figure 7B). Proteins in the human network are involved in acute phase/immune response, lipid metabolism, and exhibit strong interactions focusing on LDL and HDL, the cytokine IL6, and the transforming growth factor beta 1 (TGFB1). In mice, this network contains oxidative stress proteins such as Alb, Hpx, Hp, Blvrd, Ambp, ApoD, and ApoM in mice.
These findings suggest that PON1 interacts with molecular pathways involved in oxidative stress, inflammatory response, and complement/blood coagulation, lipoprotein metabolism, processes that are essential for blood homeostasis. Dysregulation of these processes by attenuated PON1 protein/activity levels can account for PON1’s association with cardiovascular and neurological diseases and cancer.
5.11. Metabolic Stress Amplifies Pro-Inflammatory, Pro-Oxidative, and Pro-Atherogenic Changes in Mouse Plasma Proteome Induced by Pon1 Depletion
To determine effects of Pon1 depletion under condition of metabolic stress on gene expression, plasma proteomes were examined in Pon1-/- mice and their wild type Pon1+/+ littermates fed with a high methionine HHcy diet [112]. There was a clear difference in PCA profiles of LFQ intensities for Pon1-/- mice compared to wild type Pon1+/+ animals, with a partial overlap between them (7 out of 44, 16%; Pon1-/-, blue squares □ vs. Pon1+/+, green triangles ∆) (Figure 8). In mice fed with a standard chow diet, the overlap was greater (28 out of 48, 58%; Pon1-/-, blue crosses + vs. Pon1+/+, purple circles ○), indicating that the Pon1-/- genotype exerts a stronger influence under the conditions of metabolic stress of HHcy (Figure 8).
Pon1 depletion in HHcy mice changed the expression of 89 proteins, 1.8-fold more than in chow diet mice, including 18 redox proteins, 24 immune response proteins, 6 acute phase proteins, 15 complement/blood coagulation proteins, 9 lipoprotein/lipid metabolism proteins, protein turnover proteins, and 10 other proteins (Table 6). Eight of those proteins (↓Alb, ↓ApoA1, ↓ApoA2, ↑ApoB, ↓ApoC1, ↓ApoC2, ↑Clu, ↓Pon1), representing 8% of total number of proteins affected by Pon1−/− genotype, are components of HDL [10,130].
The largest changes in the number of Pon1−/− genotype-dependent proteins in HHcy vs. chow diet mice were observed for oxidative stress-related proteins (18 proteins in HHcy diet vs. 4 proteins in chow diet mice), acute phase proteins (7 vs. 2), and protein turnover proteins (6 vs. 2) (Table 6). Smaller changes between the diets were observed in the number of proteins involved in immune response (24 vs. 19), complement/coagulation (8 vs. 7), blood coagulation (6 vs. 3), lipoprotein/lipid metabolism (9 vs. 8), and other proteins (10 vs. 5) (Table 6). These findings clearly show that the metabolic stress of HHcy greatly amplifies effects of Pon1-/- genotype on oxidative stress and inflammation.
Eleven of the proteins (12%) affected by Pon1−/− genotype in HHcy mice (ApoA1, ApoA2, ApoC1, ApoC2, Pltp, Saa1, Saa2, Alb, A2m, Pros1, Pon1) are the components of HDL [10,130], some of which were found to be enriched in the PON1-containg HDL subfraction (Alb, Clu, A2m, and Pros1) [16]. Phospholipid transfer protein (Pltp), found to be upregulated in Pon1−/− HHcy mice (Table 6), regulates the size/composition of HDL in the circulation and controls plasma HDL levels [131]. These findings show that Pon1 affects the expression of plasma proteins involved in oxidative stress, inflammation, and other processes linked to CVD. Dysregulation of these processes may account for the pro-oxidant and pro-atherogenic phenotypes associated with attenuated PON1 levels in humans [27] and mice [54].
Nineteen oxidative stress-related proteins such as Parkinson disease protein 7 (Park7, DJ-1), peroxiredoxin-2 (Prdx2), peroxiredoxin-6 (Prdx6), thioredoxin (Txn) were significantly downregulated, while seven inflammatory response proteins were upregulated in HHcy Pon1-depleted mice (Table 6). Impairment of anti-oxidant and anti-inflammatory defenses caused by changes in the expression of oxidative stress- and inflammation-related proteins can account for increased oxidative stress and inflammation observed in Pon1-/- mice [54] and in humans with low activity of PON1 [27].
6. Conclusions
Transcriptomic and proteomic analyses provided new insights regarding PON1 function by identifying proteins and molecular pathways influenced by PON1 depletion or PON1 overexpression. Accumulating evidence shows that changes in PON1 expression/activity influence both extracellular and cellular proteostasis by impairing epigenetic regulation, upregulating mTOR signaling and inhibiting autophagy. Pon1 depletion induces oxidative stress and inflammation by influences expression of genes involved in these processes. The changes in gene expression caused by low PON1 expression/activity levels are exacerbated by metabolic stress of hyperlipidemia or hyperhomocysteinemia. Although these changes are linked to CVD, Alzheimer’s disease, and cancer, molecular mechanisms by which PON1 affects gene expression remain to be elucidated in future studies.
Data Availability Statement
The data that support the findings of this study are available in the methods and/or supplementary material of this article.
Acknowledgments
Supported in part by grants 2018/29/B/NZ4/00771, 2019/33/B/NZ4/01760, and 2021/43/B/NZ4/00339 from the National Science Center, Poland, and Grant 17GRNT32910002 from the American Heart Association.
Disclosures
No conflicts of interest, financial or otherwise, are declared by the author.
References
- Libby, P., Inflammation in atherosclerosis. Nature 2002, 420, (6917), 868-74.
- Hopkins, P. N., Molecular biology of atherosclerosis. Physiol Rev 2013, 93, (3), 1317-542. [CrossRef]
- Witztum, J. L.; Steinberg, D., The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends Cardiovasc Med 2001, 11, (3-4), 93-102.
- Navab, M.; Ananthramaiah, G. M.; Reddy, S. T.; Van Lenten, B. J.; Ansell, B. J.; Fonarow, G. C.; Vahabzadeh, K.; Hama, S.; Hough, G.; Kamranpour, N.; Berliner, J. A.; Lusis, A. J.; Fogelman, A. M., The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J Lipid Res 2004, 45, (6), 993-1007. [CrossRef]
- Libby, P.; Ridker, P. M.; Hansson, G. K., Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, (7347), 317-25. [CrossRef]
- Besler, C.; Heinrich, K.; Rohrer, L.; Doerries, C.; Riwanto, M.; Shih, D. M.; Chroni, A.; Yonekawa, K.; Stein, S.; Schaefer, N.; Mueller, M.; Akhmedov, A.; Daniil, G.; Manes, C.; Templin, C.; Wyss, C.; Maier, W.; Tanner, F. C.; Matter, C. M.; Corti, R.; Furlong, C.; Lusis, A. J.; von Eckardstein, A.; Fogelman, A. M.; Luscher, T. F.; Landmesser, U., Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest 2011, 121, (7), 2693-708.
- Barter, P. J.; Nicholls, S.; Rye, K. A.; Anantharamaiah, G. M.; Navab, M.; Fogelman, A. M., Antiinflammatory properties of HDL. Circ Res 2004, 95, (8), 764-72. [CrossRef]
- Zerrad-Saadi, A.; Therond, P.; Chantepie, S.; Couturier, M.; Rye, K. A.; Chapman, M. J.; Kontush, A., HDL3-mediated inactivation of LDL-associated phospholipid hydroperoxides is determined by the redox status of apolipoprotein A-I and HDL particle surface lipid rigidity: relevance to inflammation and atherogenesis. Arterioscler Thromb Vasc Biol 2009, 29, (12), 2169-75.
- Navab, M.; Hama, S. Y.; Anantharamaiah, G. M.; Hassan, K.; Hough, G. P.; Watson, A. D.; Reddy, S. T.; Sevanian, A.; Fonarow, G. C.; Fogelman, A. M., Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: steps 2 and 3. J Lipid Res 2000, 41, (9), 1495-508. [CrossRef]
- Brites, F.; Martin, M.; Guillas, I.; Kontush, A., Antioxidative activity of high-density lipoprotein (HDL): Mechanistic insights into potential clinical benefit. BBA Clin 2017, 8, 66-77. [CrossRef]
- Soran, H.; Schofield, J. D.; Liu, Y.; Durrington, P. N., How HDL protects LDL against atherogenic modification: paraoxonase 1 and other dramatis personae. Curr Opin Lipidol 2015, 26, (4), 247-56.
- Mackness, B.; Beltran-Debon, R.; Aragones, G.; Joven, J.; Camps, J.; Mackness, M., Human tissue distribution of paraoxonases 1 and 2 mRNA. IUBMB Life 2010, 62, (6), 480-2. [CrossRef]
- Marsillach, J.; Mackness, B.; Mackness, M.; Riu, F.; Beltran, R.; Joven, J.; Camps, J., Immunohistochemical analysis of paraoxonases-1, 2, and 3 expression in normal mouse tissues. Free radical biology & medicine 2008, 45, (2), 146-57.
- Leduc, V.; Legault, V.; Dea, D.; Poirier, J., Normalization of gene expression using SYBR green qPCR: a case for paraoxonase 1 and 2 in Alzheimer's disease brains. J Neurosci Methods 2011, 200, (1), 14-9.
- Witucki, L.; Jakubowski, H., Depletion of Paraoxonase 1 (Pon1) Dysregulates mTOR, Autophagy, and Accelerates Amyloid Beta Accumulation in Mice. Cells 2023, 12, (5). [CrossRef]
- Moren, X.; Lhomme, M.; Bulla, A.; Sanchez, J. C.; Kontush, A.; James, R. W., Proteomic and lipidomic analyses of paraoxonase defined high density lipoprotein particles: Association of paraoxonase with the anti-coagulant, protein S. Proteomics Clin Appl 2016, 10, (3), 230-8. [CrossRef]
- Humbert, R.; Adler, D. A.; Disteche, C. M.; Hassett, C.; Omiecinski, C. J.; Furlong, C. E., The molecular basis of the human serum paraoxonase activity polymorphism. Nat Genet 1993, 3, (1), 73-6. [CrossRef]
- Gan, K. N.; Smolen, A.; Eckerson, H. W.; La Du, B. N., Purification of human serum paraoxonase/arylesterase. Evidence for one esterase catalyzing both activities. Drug Metab Dispos 1991, 19, (1), 100-6.
- Jakubowski, H., Calcium-dependent human serum homocysteine thiolactone hydrolase. A protective mechanism against protein N-homocysteinylation. J Biol Chem 2000, 275, (6), 3957-62. [CrossRef]
- Jakubowski, H.; Goldman, E., Synthesis of homocysteine thiolactone by methionyl-tRNA synthetase in cultured mammalian cells. FEBS Lett 1993, 317, (3), 237-40.
- Jakubowski, H., Quality control in tRNA charging. Wiley Interdiscip Rev RNA 2012, 3, (3), 295-310. [CrossRef]
- Jakubowski, H., Homocysteine Modification in Protein Structure/Function and Human Disease. Physiol Rev 2019, 99, (1), 555-604.
- Jakubowski, H.; Ambrosius, W. T.; Pratt, J. H., Genetic determinants of homocysteine thiolactonase activity in humans: implications for atherosclerosis. FEBS Lett 2001, 491, (1-2), 35-9. [CrossRef]
- Jakubowski, H., Homocysteine thiolactone: metabolic origin and protein homocysteinylation in humans. J Nutr 2000, 130, (2S Suppl), 377S-381S. [CrossRef]
- Perla-Kajan, J.; Borowczyk, K.; Glowacki, R.; Nygard, O.; Jakubowski, H., Paraoxonase 1 Q192R genotype and activity affect homocysteine thiolactone levels in humans. FASEB J 2018, fj201800346R. [CrossRef]
- Costa, L. G.; Cole, T. B.; Jarvik, G. P.; Furlong, C. E., Functional genomic of the paraoxonase (PON1) polymorphisms: effects on pesticide sensitivity, cardiovascular disease, and drug metabolism. Annu Rev Med 2003, 54, 371-92.
- Bhattacharyya, T.; Nicholls, S. J.; Topol, E. J.; Zhang, R.; Yang, X.; Schmitt, D.; Fu, X.; Shao, M.; Brennan, D. M.; Ellis, S. G.; Brennan, M. L.; Allayee, H.; Lusis, A. J.; Hazen, S. L., Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 2008, 299, (11), 1265-76. [CrossRef]
- Tang, W. H.; Hartiala, J.; Fan, Y.; Wu, Y.; Stewart, A. F.; Erdmann, J.; Kathiresan, S.; Consortium, C. A.; Roberts, R.; McPherson, R.; Allayee, H.; Hazen, S. L., Clinical and genetic association of serum paraoxonase and arylesterase activities with cardiovascular risk. Arterioscler Thromb Vasc Biol 2012, 32, (11), 2803-12. [CrossRef]
- Mackness, B.; Durrington, P.; McElduff, P.; Yarnell, J.; Azam, N.; Watt, M.; Mackness, M., Low paraoxonase activity predicts coronary events in the Caerphilly Prospective Study. Circulation 2003, 107, (22), 2775-9. [CrossRef]
- Dube, P.; Khalaf, F. K.; DeRiso, A.; Mohammed, C. J.; Connolly, J. A.; Battepati, D.; Lad, A.; Breidenbach, J. D.; Kleinhenz, A. L.; Khatib-Shahidi, B.; Patel, M.; Tassavvor, I.; Gohara, A. F.; Malhotra, D.; Morgan, E. E.; Haller, S. T.; Kennedy, D. J., Cardioprotective Role for Paraoxonase-1 in Chronic Kidney Disease. Biomedicines 2022, 10, (9). [CrossRef]
- Meneses, M. J.; Silvestre, R.; Sousa-Lima, I.; Macedo, M. P., Paraoxonase-1 as a Regulator of Glucose and Lipid Homeostasis: Impact on the Onset and Progression of Metabolic Disorders. Int J Mol Sci 2019, 20, (16). [CrossRef]
- Cervellati, C.; Trentini, A.; Romani, A.; Bellini, T.; Bosi, C.; Ortolani, B.; Zurlo, A.; Passaro, A.; Seripa, D.; Zuliani, G., Serum paraoxonase and arylesterase activities of paraoxonase-1 (PON-1), mild cognitive impairment, and 2-year conversion to dementia: A pilot study. J Neurochem 2015, 135, (2), 395-401.
- Dong, L.; Dong, C.; Yu, Y.; Jiao, X.; Zhang, X.; Zhang, X.; Li, Z., Transcriptomic analysis of Paraoxonase 1 expression in hepatocellular carcinoma and its potential impact on tumor immunity. Clin Transl Oncol 2024. [CrossRef]
- Zhang, Q.; Jiang, Z.; Xu, Y., HDL and Oxidation. Adv Exp Med Biol 2022, 1377, 63-77.
- Marsillach, J.; Adorni, M. P.; Zimetti, F.; Papotti, B.; Zuliani, G.; Cervellati, C., HDL Proteome and Alzheimer's Disease: Evidence of a Link. Antioxidants (Basel) 2020, 9, (12). [CrossRef]
- Davidson, W. S.; Silva, R. A.; Chantepie, S.; Lagor, W. R.; Chapman, M. J.; Kontush, A., Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol 2009, 29, (6), 870-6. [CrossRef]
- de Souza, J. A.; Vindis, C.; Negre-Salvayre, A.; Rye, K. A.; Couturier, M.; Therond, P.; Chantepie, S.; Salvayre, R.; Chapman, M. J.; Kontush, A., Small, dense HDL 3 particles attenuate apoptosis in endothelial cells: pivotal role of apolipoprotein A-I. J Cell Mol Med 2010, 14, (3), 608-20. [CrossRef]
- Jakubowski, H., Proteomic Exploration of Paraoxonase 1 Function in Health and Disease. Int J Mol Sci 2023, 24, (9). [CrossRef]
- Borowczyk, K.; Shih, D. M.; Jakubowski, H., Metabolism and neurotoxicity of homocysteine thiolactone in mice: evidence for a protective role of paraoxonase 1. Journal of Alzheimer's disease : JAD 2012, 30, (2), 225-31. [CrossRef]
- Borowczyk, K.; Piechocka, J.; Glowacki, R.; Dhar, I.; Midtun, O.; Tell, G. S.; Ueland, P. M.; Nygard, O.; Jakubowski, H., Urinary excretion of homocysteine thiolactone and the risk of acute myocardial infarction in coronary artery disease patients: the WENBIT trial. J Intern Med 2019, 285, (2), 232-244. [CrossRef]
- Sauls, D. L.; Lockhart, E.; Warren, M. E.; Lenkowski, A.; Wilhelm, S. E.; Hoffman, M., Modification of fibrinogen by homocysteine thiolactone increases resistance to fibrinolysis: a potential mechanism of the thrombotic tendency in hyperhomocysteinemia. Biochemistry 2006, 45, (8), 2480-7. [CrossRef]
- Sikora, M.; Skrzydlewski, P.; Perla-Kajan, J.; Jakubowski, H., Homocysteine thiolactone contributes to the prognostic value of fibrin clot structure/function in coronary artery disease. PLoS One 2022, 17, (10), e0275956. [CrossRef]
- Khersonsky, O.; Tawfik, D. S., Structure-reactivity studies of serum paraoxonase PON1 suggest that its native activity is lactonase. Biochemistry 2005, 44, (16), 6371-82.
- Draganov, D. I.; Teiber, J. F.; Speelman, A.; Osawa, Y.; Sunahara, R.; La Du, B. N., Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities. J Lipid Res 2005, 46, (6), 1239-47. [CrossRef]
- Mohammed, C. J.; Lamichhane, S.; Connolly, J. A.; Soehnlen, S. M.; Khalaf, F. K.; Malhotra, D.; Haller, S. T.; Isailovic, D.; Kennedy, D. J., A PON for All Seasons: Comparing Paraoxonase Enzyme Substrates, Activity and Action including the Role of PON3 in Health and Disease. Antioxidants (Basel) 2022, 11, (3). [CrossRef]
- Slutsky Smith, E. A.; Khatib, S.; Szuchman-Sapir, A., Fishing for lipid lactones using selective reaction and characteristic fragmentation pattern. J Chromatogr B Analyt Technol Biomed Life Sci 2022, 1197, 123201. [CrossRef]
- Momma, T. Y.; Kuhnle, G. G. C.; Fong, R. Y.; Ensunsa, J. L.; Crozier, A.; Schroeter, H.; Ottaviani, J. I., 5-(3',4'-Dihydroxyphenyl)-gamma-Valerolactone Is a Substrate for Human Paraoxonase: A Novel Pathway in Flavan-3-ol Metabolism. Mol Nutr Food Res 2023, 67, (17), e2300281.
- Sesso, H. D.; Manson, J. E.; Aragaki, A. K.; Rist, P. M.; Johnson, L. G.; Friedenberg, G.; Copeland, T.; Clar, A.; Mora, S.; Moorthy, M. V.; Sarkissian, A.; Carrick, W. R.; Anderson, G. L.; Group, C. R., Effect of cocoa flavanol supplementation for the prevention of cardiovascular disease events: the COcoa Supplement and Multivitamin Outcomes Study (COSMOS) randomized clinical trial. Am J Clin Nutr 2022, 115, (6), 1490-1500. [CrossRef]
- Baker, L. D.; Manson, J. E.; Rapp, S. R.; Sesso, H. D.; Gaussoin, S. A.; Shumaker, S. A.; Espeland, M. A., Effects of cocoa extract and a multivitamin on cognitive function: A randomized clinical trial. Alzheimers Dement 2023, 19, (4), 1308-1319. [CrossRef]
- Mena, P.; Bresciani, L.; Brindani, N.; Ludwig, I. A.; Pereira-Caro, G.; Angelino, D.; Llorach, R.; Calani, L.; Brighenti, F.; Clifford, M. N.; Gill, C. I. R.; Crozier, A.; Curti, C.; Del Rio, D., Phenyl-gamma-valerolactones and phenylvaleric acids, the main colonic metabolites of flavan-3-ols: synthesis, analysis, bioavailability, and bioactivity. Nat Prod Rep 2019, 36, (5), 714-752. [CrossRef]
- Angelino, D.; Carregosa, D.; Domenech-Coca, C.; Savi, M.; Figueira, I.; Brindani, N.; Jang, S.; Lakshman, S.; Molokin, A.; Urban, J. F., Jr.; Davis, C. D.; Brito, M. A.; Kim, K. S.; Brighenti, F.; Curti, C.; Blade, C.; Del Bas, J. M.; Stilli, D.; Solano-Aguilar, G. I.; Santos, C. N. D.; Del Rio, D.; Mena, P., 5-(Hydroxyphenyl)-gamma-Valerolactone-Sulfate, a Key Microbial Metabolite of Flavan-3-ols, Is Able to Reach the Brain: Evidence from Different in Silico, In Vitro and In Vivo Experimental Models. Nutrients 2019, 11, (11). [CrossRef]
- Corral-Jara, K. F.; Nuthikattu, S.; Rutledge, J.; Villablanca, A.; Morand, C.; Schroeter, H.; Milenkovic, D., Integrated Multi-Omic Analyses of the Genomic Modifications by Gut Microbiome-Derived Metabolites of Epicatechin, 5-(4'-Hydroxyphenyl)-gamma-Valerolactone, in TNFalpha-Stimulated Primary Human Brain Microvascular Endothelial Cells. Front Neurosci 2021, 15, 622640. [CrossRef]
- Costa, L. G.; Giordano, G.; Cole, T. B.; Marsillach, J.; Furlong, C. E., Paraoxonase 1 (PON1) as a genetic determinant of susceptibility to organophosphate toxicity. Toxicology 2013, 307, 115-22. [CrossRef]
- Shih, D. M.; Gu, L.; Xia, Y. R.; Navab, M.; Li, W. F.; Hama, S.; Castellani, L. W.; Furlong, C. E.; Costa, L. G.; Fogelman, A. M.; Lusis, A. J., Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature 1998, 394, (6690), 284-7.
- Shih, D. M.; Xia, Y. R.; Wang, X. P.; Miller, E.; Castellani, L. W.; Subbanagounder, G.; Cheroutre, H.; Faull, K. F.; Berliner, J. A.; Witztum, J. L.; Lusis, A. J., Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit increased lipoprotein oxidation and atherosclerosis. J Biol Chem 2000, 275, (23), 17527-35. [CrossRef]
- Hong, C. G.; Florida, E.; Li, H.; Parel, P. M.; Mehta, N. N.; Sorokin, A. V., Oxidized low-density lipoprotein associates with cardiovascular disease by a vicious cycle of atherosclerosis and inflammation: A systematic review and meta-analysis. Front Cardiovasc Med 2022, 9, 1023651. [CrossRef]
- Kotani, K.; Watanabe, Y.; Miura, K.; Gugliucci, A., Paraoxonase 1 and Non-Alcoholic Fatty Liver Disease: A Meta-Analysis. Biomolecules 2021, 26(8), 2323. [CrossRef]
- Suszynska-Zajczyk, J.; Luczak, M.; Marczak, L.; Jakubowski, H., Inactivation of the paraoxonase 1 gene affects the expression of mouse brain proteins involved in neurodegeneration. J Alzheimers Dis 2014, 42, (1), 247-60. [CrossRef]
- Bednarz-Misa, I.; Berdowska, I.; Zboch, M.; Misiak, B.; Zielinski, B.; Placzkowska, S.; Fleszar, M.; Wisniewski, J.; Gamian, A.; Krzystek-Korpacka, M., Paraoxonase 1 decline and lipid peroxidation rise reflect a degree of brain atrophy and vascular impairment in dementia. Adv Clin Exp Med 2020, 29, (1), 71-78. [CrossRef]
- Bade, J. D.; Veeramalla, V.; Naidu, M. B. R.; Lalitha, D. L.; Ponnada, S. C.; Kandi, V., Serum Activities of Paraoxonase 1 (PON1) in Predicting Liver Damage Among Patients Diagnosed With Hepatocellular Carcinoma: A Case-Control Study. Cureus 2023, 15, (9), e46234. [CrossRef]
- Tward, A.; Xia, Y. R.; Wang, X. P.; Shi, Y. S.; Park, C.; Castellani, L. W.; Lusis, A. J.; Shih, D. M., Decreased atherosclerotic lesion formation in human serum paraoxonase transgenic mice. Circulation 2002, 106, (4), 484-90. [CrossRef]
- Mackness, B.; Quarck, R.; Verreth, W.; Mackness, M.; Holvoet, P., Human paraoxonase-1 overexpression inhibits atherosclerosis in a mouse model of metabolic syndrome. Arterioscler Thromb Vasc Biol 2006, 26, (7), 1545-50. [CrossRef]
- She, Z. G.; Zheng, W.; Wei, Y. S.; Chen, H. Z.; Wang, A. B.; Li, H. L.; Liu, G.; Zhang, R.; Liu, J. J.; Stallcup, W. B.; Zhou, Z.; Liu, D. P.; Liang, C. C., Human paraoxonase gene cluster transgenic overexpression represses atherogenesis and promotes atherosclerotic plaque stability in ApoE-null mice. Circ Res 2009, 104, (10), 1160-8. [CrossRef]
- Brophy, V. H.; Jampsa, R. L.; Clendenning, J. B.; McKinstry, L. A.; Jarvik, G. P.; Furlong, C. E., Effects of 5' regulatory-region polymorphisms on paraoxonase-gene (PON1) expression. American journal of human genetics 2001, 68, (6), 1428-36. [CrossRef]
- Sikora, M.; Bretes, E.; Perla-Kajan, J.; Lewandowska, I.; Marczak, L.; Jakubowski, H., Genetic Attenuation of Paraoxonase 1 Activity Induces Proatherogenic Changes in Plasma Proteomes of Mice and Humans. Antioxidants (Basel) 2020, 9, (12). [CrossRef]
- Kresanov, P.; Vasankari, T.; Ahotupa, M.; Kaikkonen, J.; Hutri-Kahonen, N.; Juonala, M.; Kahonen, M.; Lehtimaki, T.; Viikari, J.; Raitakari, O. T., Paraoxonase-1 and oxidized lipoprotein lipids. The Cardiovascular Risk in Young Finns Study. Atherosclerosis 2015, 241, (2), 502-6. [CrossRef]
- Ahotupa, M.; Marniemi, J.; Lehtimaki, T.; Talvinen, K.; Raitakari, O. T.; Vasankari, T.; Viikari, J.; Luoma, J.; Yla-Herttuala, S., Baseline diene conjugation in LDL lipids as a direct measure of in vivo LDL oxidation. Clin Biochem 1998, 31, (4), 257-61. [CrossRef]
- Esterbauer, H.; Striegl, G.; Puhl, H.; Rotheneder, M., Continuous monitoring of in vitro oxidation of human low density lipoprotein. Free Radic Res Commun 1989, 6, (1), 67-75.
- Kuchta, A.; Strzelecki, A.; Cwiklinska, A.; Toton, M.; Gruchala, M.; Zdrojewski, Z.; Kortas-Stempak, B.; Gliwinska, A.; Dabkowski, K.; Jankowski, M., PON-1 Activity and Plasma 8-Isoprostane Concentration in Patients with Angiographically Proven Coronary Artery Disease. Oxid Med Cell Longev 2015, 2015, 5136937. [CrossRef]
- Carlson, C. S.; Heagerty, P. J.; Hatsukami, T. S.; Richter, R. J.; Ranchalis, J.; Lewis, J.; Bacus, T. J.; McKinstry, L. A.; Schellenberg, G. D.; Rieder, M.; Nickerson, D.; Furlong, C. E.; Chait, A.; Jarvik, G. P., TagSNP analyses of the PON gene cluster: effects on PON1 activity, LDL oxidative susceptibility, and vascular disease. J Lipid Res 2006, 47, (5), 1014-24. [CrossRef]
- Hayden, K. M.; Norton, M. C.; Darcey, D.; Ostbye, T.; Zandi, P. P.; Breitner, J. C.; Welsh-Bohmer, K. A.; Cache County Study, I., Occupational exposure to pesticides increases the risk of incident AD: the Cache County study. Neurology 2010, 74, (19), 1524-30.
- Jones, N., Alzheimer disease: Risk of dementia and Alzheimer disease increases with occupational pesticide exposure. Nat Rev Neurol 2010, 6, (7), 353.
- Tanzi, R. E., The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2012, 2, (10).
- Blatter, M. C.; James, R. W.; Messmer, S.; Barja, F.; Pometta, D., Identification of a distinct human high-density lipoprotein subspecies defined by a lipoprotein-associated protein, K-45. Identity of K-45 with paraoxonase. Eur J Biochem 1993, 211, (3), 871-9.
- Oakley, H.; Cole, S. L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; Berry, R.; Vassar, R., Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 2006, 26, (40), 10129-40.
- Khayati, K.; Antikainen, H.; Bonder, E. M.; Weber, G. F.; Kruger, W. D.; Jakubowski, H.; Dobrowolski, R., The amino acid metabolite homocysteine activates mTORC1 to inhibit autophagy and form abnormal proteins in human neurons and mice. FASEB J 2017, 31, (2), 598-609. [CrossRef]
- Yates, S. C.; Zafar, A.; Hubbard, P.; Nagy, S.; Durant, S.; Bicknell, R.; Wilcock, G.; Christie, S.; Esiri, M. M.; Smith, A. D.; Nagy, Z., Dysfunction of the mTOR pathway is a risk factor for Alzheimer's disease. Acta Neuropathol Commun 2013, 1, 3. [CrossRef]
- Chen, X.; Wang, S.; Zhou, Y.; Han, Y.; Li, S.; Xu, Q.; Xu, L.; Zhu, Z.; Deng, Y.; Yu, L.; Song, L.; Chen, A. P.; Song, J.; Takahashi, E.; He, G.; He, L.; Li, W.; Chen, C. D., Phf8 histone demethylase deficiency causes cognitive impairments through the mTOR pathway. Nat Commun 2018, 9, (1), 114. [CrossRef]
- Witucki, L., Jakubowski, H.,, Homocysteine Metabolites Inhibit Autophagy and Elevate Amyloid Beta by Impairing Phf8/H4K20me1-dependent Epigenetic Regulation of mTOR in Cystathionine β-Synthase-Deficient Mice. 2023 bioRxiv. [CrossRef]
- Jakubowski, H., Homocysteine Thiolactone Detoxifying Enzymes and Alzheimer's Disease. Int J Mol Sci 2024, 25, (15). [CrossRef]
- Perla-Kajan, J.; Jakubowski, H., Paraoxonase 1 protects against protein N-homocysteinylation in humans. FASEB J 2010, 24, (3), 931-6.
- Jakubowski, H.; Perla-Kajan, J.; Finnell, R. H.; Cabrera, R. M.; Wang, H.; Gupta, S.; Kruger, W. D.; Kraus, J. P.; Shih, D. M., Genetic or nutritional disorders in homocysteine or folate metabolism increase protein N-homocysteinylation in mice. Faseb J 2009, 23, (6), 1721-7. [CrossRef]
- Smith, A. D.; Refsum, H., Homocysteine - from disease biomarker to disease prevention. J Intern Med 2021, 290, (4), 826-854. [CrossRef]
- Bacchetti, T.; Vignini, A.; Giulietti, A.; Nanetti, L.; Provinciali, L.; Luzzi, S.; Mazzanti, L.; Ferretti, G., Higher Levels of Oxidized Low Density Lipoproteins in Alzheimer's Disease Patients: Roles for Platelet Activating Factor Acetyl Hydrolase and Paraoxonase-1. J Alzheimers Dis 2015, 46, (1), 179-86.
- Cagnin, A.; Leon, A.; Vianello, D.; Colavito, D.; Favaretto, S.; Zarantonello, G.; Stecca, A.; Ermani, M.; Zambon, A., LDL density and oxidation are modulated by PON1 promoter genotype in patients with Alzheimer's disease. J Alzheimers Dis 2013, 34, (2), 377-85.
- Forner, A.; Reig, M.; Bruix, J., Hepatocellular carcinoma. Lancet 2018, 391, (10127), 1301-1314.
- Mackness, M. I.; Durrington, P. N.; Mackness, B., The role of paraoxonase 1 activity in cardiovascular disease: potential for therapeutic intervention. Am J Cardiovasc Drugs 2004, 4, (4), 211-7.
- Mackness, M. I.; Arrol, S.; Durrington, P. N., Paraoxonase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett 1991, 286, (1-2), 152-4. [CrossRef]
- Watson, A. D.; Berliner, J. A.; Hama, S. Y.; La Du, B. N.; Faull, K. F.; Fogelman, A. M.; Navab, M., Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest 1995, 96, (6), 2882-91.
- Aviram, M.; Rosenblat, M.; Bisgaier, C. L.; Newton, R. S.; Primo-Parmo, S. L.; La Du, B. N., Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxonase. J Clin Invest 1998, 101, (8), 1581-90. [CrossRef]
- Aviram, M.; Billecke, S.; Sorenson, R.; Bisgaier, C.; Newton, R.; Rosenblat, M.; Erogul, J.; Hsu, C.; Dunlop, C.; La Du, B., Paraoxonase active site required for protection against LDL oxidation involves its free sulfhydryl group and is different from that required for its arylesterase/paraoxonase activities: selective action of human paraoxonase allozymes Q and R. Arterioscler Thromb Vasc Biol 1998, 18, (10), 1617-24.
- Bayrak, A.; Bayrak, T.; Bodur, E.; Kilinc, K.; Demirpence, E., The effect of HDL-bound and free PON1 on copper-induced LDL oxidation. Chem Biol Interact 2016, 257, 141-6. [CrossRef]
- Aviram, M.; Rosenblat, M., Paraoxonases 1, 2, and 3, oxidative stress, and macrophage foam cell formation during atherosclerosis development. Free Radic Biol Med 2004, 37, (9), 1304-16. [CrossRef]
- Liu, Y.; Mackness, B.; Mackness, M., Comparison of the ability of paraoxonases 1 and 3 to attenuate the in vitro oxidation of low-density lipoprotein and reduce macrophage oxidative stress. Free Radic Biol Med 2008, 45, (6), 743-8. [CrossRef]
- Kornberg, A., Ten commandments of enzymology, amended. Trends Biochem Sci 2003, 28, (10), 515-7. [CrossRef]
- Kresge, N.; Simoni, R. D.; Hill, R. L., Unraveling the Enzymology of Oxidative Phosphorylation: the Work of Efraim Racker. Journal of Biological Chemistry 2006, 281, (4), e4-e6. [CrossRef]
- Marathe, G. K.; Zimmerman, G. A.; McIntyre, T. M., Platelet-activating factor acetylhydrolase, and not paraoxonase-1, is the oxidized phospholipid hydrolase of high density lipoprotein particles. J Biol Chem 2003, 278, (6), 3937-47. [CrossRef]
- Ahmed, Z.; Ravandi, A.; Maguire, G. F.; Emili, A.; Draganov, D.; La Du, B. N.; Kuksis, A.; Connelly, P. W., Apolipoprotein A-I promotes the formation of phosphatidylcholine core aldehydes that are hydrolyzed by paraoxonase (PON-1) during high density lipoprotein oxidation with a peroxynitrite donor. J Biol Chem 2001, 276, (27), 24473-81. [CrossRef]
- Ahmed, Z.; Ravandi, A.; Maguire, G. F.; Emili, A.; Draganov, D.; La Du, B. N.; Kuksis, A.; Connelly, P. W., Multiple substrates for paraoxonase-1 during oxidation of phosphatidylcholine by peroxynitrite. Biochem Biophys Res Commun 2002, 290, (1), 391-6. [CrossRef]
- Ahmed, Z.; Babaei, S.; Maguire, G. F.; Draganov, D.; Kuksis, A.; La Du, B. N.; Connelly, P. W., Paraoxonase-1 reduces monocyte chemotaxis and adhesion to endothelial cells due to oxidation of palmitoyl, linoleoyl glycerophosphorylcholine. Cardiovasc Res 2003, 57, (1), 225-31. [CrossRef]
- Teiber, J. F.; Draganov, D. I.; La Du, B. N., Purified human serum PON1 does not protect LDL against oxidation in the in vitro assays initiated with copper or AAPH. J Lipid Res 2004, 45, (12), 2260-8. [CrossRef]
- Connelly, P. W.; Draganov, D.; Maguire, G. F., Paraoxonase-1 does not reduce or modify oxidation of phospholipids by peroxynitrite. Free Radic Biol Med 2005, 38, (2), 164-74. [CrossRef]
- Rodrigo, L.; Mackness, B.; Durrington, P. N.; Hernandez, A.; Mackness, M. I., Hydrolysis of platelet-activating factor by human serum paraoxonase. Biochem J 2001, 354, (Pt 1), 1-7.
- Mashima, R.; Yamamoto, Y.; Yoshimura, S., Reduction of phosphatidylcholine hydroperoxide by apolipoprotein A-I: purification of the hydroperoxide-reducing proteins from human blood plasma. J Lipid Res 1998, 39, (6), 1133-40. [CrossRef]
- Rye, K. A.; Barter, P. J., Cardioprotective functions of HDLs. J Lipid Res 2014, 55, (2), 168-79. [CrossRef]
- Garner, B.; Waldeck, A. R.; Witting, P. K.; Rye, K. A.; Stocker, R., Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. J Biol Chem 1998, 273, (11), 6088-95.
- Panzenbock, U.; Stocker, R., Formation of methionine sulfoxide-containing specific forms of oxidized high-density lipoproteins. Biochim Biophys Acta 2005, 1703, (2), 171-81. [CrossRef]
- Graham, A.; Hassall, D. G.; Rafique, S.; Owen, J. S., Evidence for a paraoxonase-independent inhibition of low-density lipoprotein oxidation by high-density lipoprotein. Atherosclerosis 1997, 135, (2), 193-204. [CrossRef]
- Suszynska-Zajczyk, J.; Luczak, M.; Marczak, L.; Jakubowski, H., Hyperhomocysteinemia and bleomycin hydrolase modulate the expression of mouse brain proteins involved in neurodegeneration. J Alzheimers Dis 2014, 40, (3), 713-26. [CrossRef]
- Suszynska-Zajczyk, J.; Jakubowski, H., Paraoxonase 1 and dietary hyperhomocysteinemia modulate the expression of mouse proteins involved in liver homeostasis. Acta Biochim Pol 2014, 61, (4), 815-23. [CrossRef]
- Suszynska-Zajczyk, J.; Sikora, M.; Jakubowski, H., Paraoxonase 1 deficiency and hyperhomocysteinemia alter the expression of mouse kidney proteins involved in renal disease. Mol Genet Metab 2014, 113, (3), 200-6. [CrossRef]
- Sikora, M.; Jakubowski, H., Changes in redox plasma proteome of Pon1-/- mice are exacerbated by a hyperhomocysteinemic diet. Free Radic Biol Med 2021, 169, 169-180.
- Cao, J.; Xu, Y.; Li, F.; Shang, L.; Fan, D.; Yu, H., Protein markers of dysfunctional HDL in scavenger receptor class B type I deficient mice. J Transl Med 2018, 16, (1), 155. [CrossRef]
- Liu, Q.; Xiao, J. J.; Wang, S.; Li, Y.; Yang, L. J.; Lu, Q. Y.; Wu, X. Y.; Cao, J.; Yu, H.; Zhang, B. F., Paraoxonase 1 Ameliorates Renal Lipotoxicity by Activating Lipophagy and Inhibiting Pyroptosis. Am J Pathol 2022, 192, (11), 1531-1545. [CrossRef]
- Zhao, X. J.; Liu, L. C.; Guo, C.; Shen, W. W.; Cao, J.; Du, F.; Wu, D. F.; Yu, H., Hepatic paraoxonase 1 ameliorates dysfunctional high-density lipoprotein and atherosclerosis in scavenger receptor class B type I deficient mice. Ann Transl Med 2021, 9, (13), 1063. [CrossRef]
- Yuhanna, I. S.; Zhu, Y.; Cox, B. E.; Hahner, L. D.; Osborne-Lawrence, S.; Lu, P.; Marcel, Y. L.; Anderson, R. G.; Mendelsohn, M. E.; Hobbs, H. H.; Shaul, P. W., High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med 2001, 7, (7), 853-7.
- Trigatti, B. L.; Fuller, M., HDL signaling and protection against coronary artery atherosclerosis in mice. J Biomed Res 2016, 30, (2), 94-100.
- Seetharam, D.; Mineo, C.; Gormley, A. K.; Gibson, L. L.; Vongpatanasin, W.; Chambliss, K. L.; Hahner, L. D.; Cummings, M. L.; Kitchens, R. L.; Marcel, Y. L.; Rader, D. J.; Shaul, P. W., High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ Res 2006, 98, (1), 63-72.
- Ng, D. S.; Chu, T.; Esposito, B.; Hui, P.; Connelly, P. W.; Gross, P. L., Paraoxonase-1 deficiency in mice predisposes to vascular inflammation, oxidative stress, and thrombogenicity in the absence of hyperlipidemia. Cardiovasc Pathol 2008, 17, (4), 226-32. [CrossRef]
- Acton, S.; Rigotti, A.; Landschulz, K. T.; Xu, S.; Hobbs, H. H.; Krieger, M., Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, (5248), 518-20. [CrossRef]
- Zanoni, P.; Khetarpal, S. A.; Larach, D. B.; Hancock-Cerutti, W. F.; Millar, J. S.; Cuchel, M.; DerOhannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; Trompet, S.; Jukema, J. W.; De Craen, A.; Deloukas, P.; Sattar, N.; Ford, I.; Packard, C.; Majumder, A.; Alam, D. S.; Di Angelantonio, E.; Abecasis, G.; Chowdhury, R.; Erdmann, J.; Nordestgaard, B. G.; Nielsen, S. F.; Tybjaerg-Hansen, A.; Schmidt, R. F.; Kuulasmaa, K.; Liu, D. J.; Perola, M.; Blankenberg, S.; Salomaa, V.; Mannisto, S.; Amouyel, P.; Arveiler, D.; Ferrieres, J.; Muller-Nurasyid, M.; Ferrario, M.; Kee, F.; Willer, C. J.; Samani, N.; Schunkert, H.; Butterworth, A. S.; Howson, J. M.; Peloso, G. M.; Stitziel, N. O.; Danesh, J.; Kathiresan, S.; Rader, D. J.; Consortium, C. H. D. E.; Consortium, C. A. E.; Global Lipids Genetics, C., Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016, 351, (6278), 1166-71.
- Zhang, Y.; Da Silva, J. R.; Reilly, M.; Billheimer, J. T.; Rothblat, G. H.; Rader, D. J., Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest 2005, 115, (10), 2870-4. [CrossRef]
- Leiva, A.; Verdejo, H.; Benitez, M. L.; Martinez, A.; Busso, D.; Rigotti, A., Mechanisms regulating hepatic SR-BI expression and their impact on HDL metabolism. Atherosclerosis 2011, 217, (2), 299-307. [CrossRef]
- Van Eck, M.; Hoekstra, M.; Hildebrand, R. B.; Yaong, Y.; Stengel, D.; Kruijt, J. K.; Sattler, W.; Tietge, U. J.; Ninio, E.; Van Berkel, T. J.; Pratico, D., Increased oxidative stress in scavenger receptor BI knockout mice with dysfunctional HDL. Arterioscler Thromb Vasc Biol 2007, 27, (11), 2413-9. [CrossRef]
- Chester, J.; Johnston, E.; Walker, D.; Jones, M.; Ionescu, C. M.; Wagle, S. R.; Kovacevic, B.; Brown, D.; Mikov, M.; Mooranian, A.; Al-Salami, H., A Review on Recent Advancement on Age-Related Hearing Loss: The Applications of Nanotechnology, Drug Pharmacology, and Biotechnology. Pharmaceutics 2021, 13, (7). [CrossRef]
- Miettinen, H. E.; Rayburn, H.; Krieger, M., Abnormal lipoprotein metabolism and reversible female infertility in HDL receptor (SR-BI)-deficient mice. J Clin Invest 2001, 108, (11), 1717-22.
- Ruan, X. Z.; Varghese, Z.; Moorhead, J. F., An update on the lipid nephrotoxicity hypothesis. Nat Rev Nephrol 2009, 5, (12), 713-21. [CrossRef]
- Rodrigo, L.; Hernandez, A. F.; Lopez-Caballero, J. J.; Gil, F.; Pla, A., Immunohistochemical evidence for the expression and induction of paraoxonase in rat liver, kidney, lung and brain tissue. Implications for its physiological role. Chem Biol Interact 2001, 137, (2), 123-37. [CrossRef]
- Miljkovic, M.; Stefanovic, A.; Vekic, J.; Zeljkovic, A.; Gojkovic, T.; Simic-Ogrizovic, S.; Bogavac-Stanojevic, N.; Cerne, D.; Ilic, J.; Stefanovic, I.; Jelic-Ivanovic, Z.; Spasojevic-Kalimanovska, V.; Kotur-Stevuljevic, J., Activity of paraoxonase 1 (PON1) on HDL(2) and HDL(3) subclasses in renal disease. Clin Biochem 2018, 60, 52-58. [CrossRef]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M. J.; Davidson, W. S., Structure of HDL: particle subclasses and molecular components. Handb Exp Pharmacol 2015, 224, 3-51.
- Huuskonen, J.; Olkkonen, V. M.; Jauhiainen, M.; Ehnholm, C., The impact of phospholipid transfer protein (PLTP) on HDL metabolism. Atherosclerosis 2001, 155, (2), 269-81. [CrossRef]
Figure 1.
Hydrolytic activities of the PON1 enzyme. Hcy-thiolactone [19,23,24,25] and 5-(3′,4′-dihydroxyphenyl)-γ-valerolactone [47] are biological substrates of PON1; paraoxon and phenylacetate are nonbiological substrates of PON1 [17,18].
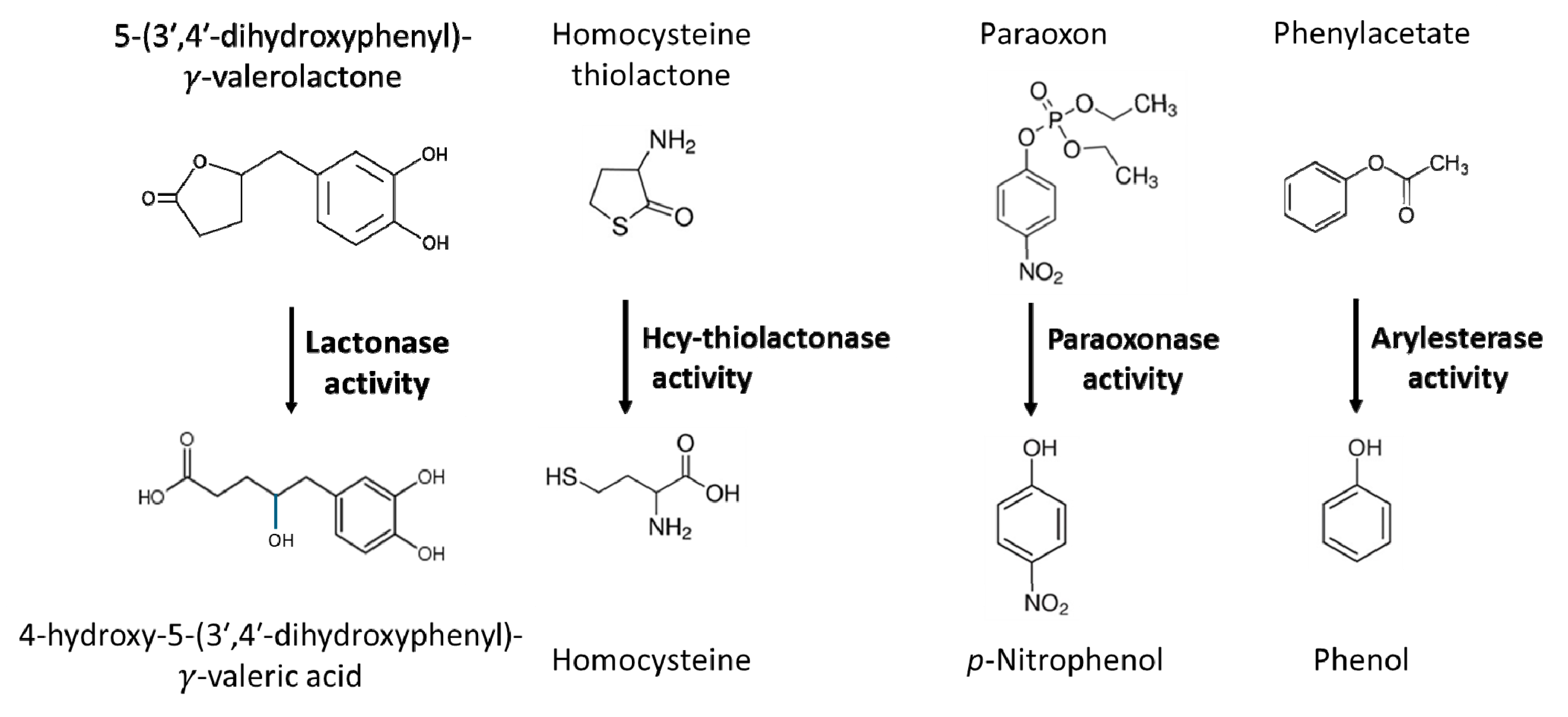
Figure 2.
Turnover of Hcy-thiolactone and accumulation of N-Hcy-protein in serum at 37˚C. Changes in Hcy thiolactone (3.5 µM at time zero) (A), Hcy (B), and N-Hcy-protein (C) levels in human serum from individuals with PON1-LL55/RR192 genotype (■, t1/2 = 0.5 h), PON1-MM55/QQ192 genotype (♦, t1/2 = 1 h), human sera with the PON1 enzyme inactivated by 5 mM EDTA/2 mM D-penicillamine (●, 1.5 h), and rabbit serum (□, 0.25 h). Reproduced with permission from Ref. [23]. Copyright 2001 by John Wiley and Sons.
Figure 2.
Turnover of Hcy-thiolactone and accumulation of N-Hcy-protein in serum at 37˚C. Changes in Hcy thiolactone (3.5 µM at time zero) (A), Hcy (B), and N-Hcy-protein (C) levels in human serum from individuals with PON1-LL55/RR192 genotype (■, t1/2 = 0.5 h), PON1-MM55/QQ192 genotype (♦, t1/2 = 1 h), human sera with the PON1 enzyme inactivated by 5 mM EDTA/2 mM D-penicillamine (●, 1.5 h), and rabbit serum (□, 0.25 h). Reproduced with permission from Ref. [23]. Copyright 2001 by John Wiley and Sons.
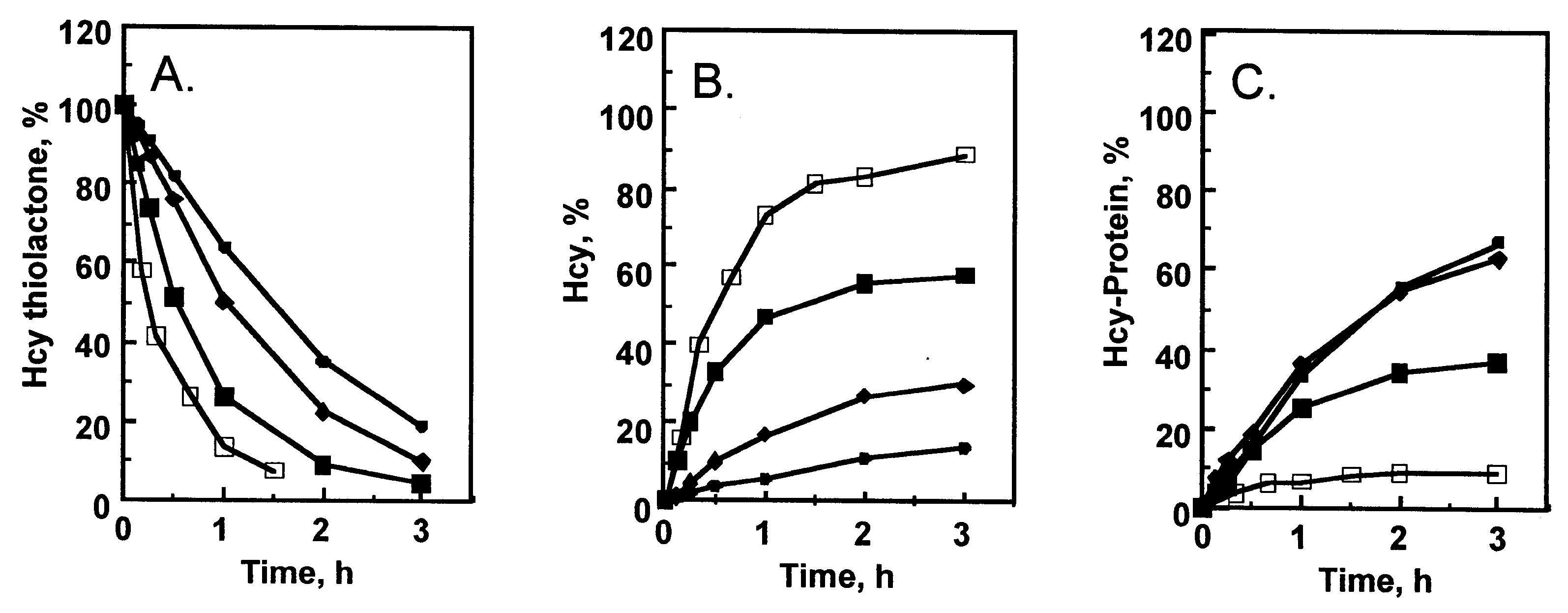
Figure 3.
Turnover of 5-(3′,4′-dihydroxyphenyl)-γ-valerolactone (γVL) and accumulation of 5-(3′,4′-dihydroxyphenyl)-γ-valeric acid (γVA) in human serum at 37˚C. (A) Accumulation of γVA in control serum supplemented with 1 μM γVL after 1 h, serum + 5 mM EGTA , and heat-inactivated serum (57˚C, 30 min; HI). Data represents mean ± standard deviation (n = 6). (B) Kinetics of γVL disappearance and γVA generation in serum. (C) Serum γVL-hydrolyzing activity as a function of γVL concentration. Data represents mean ± standard deviation (n = 4 per time point or concentration). Reproduced from Ref. [47].
Figure 3.
Turnover of 5-(3′,4′-dihydroxyphenyl)-γ-valerolactone (γVL) and accumulation of 5-(3′,4′-dihydroxyphenyl)-γ-valeric acid (γVA) in human serum at 37˚C. (A) Accumulation of γVA in control serum supplemented with 1 μM γVL after 1 h, serum + 5 mM EGTA , and heat-inactivated serum (57˚C, 30 min; HI). Data represents mean ± standard deviation (n = 6). (B) Kinetics of γVL disappearance and γVA generation in serum. (C) Serum γVL-hydrolyzing activity as a function of γVL concentration. Data represents mean ± standard deviation (n = 4 per time point or concentration). Reproduced from Ref. [47].
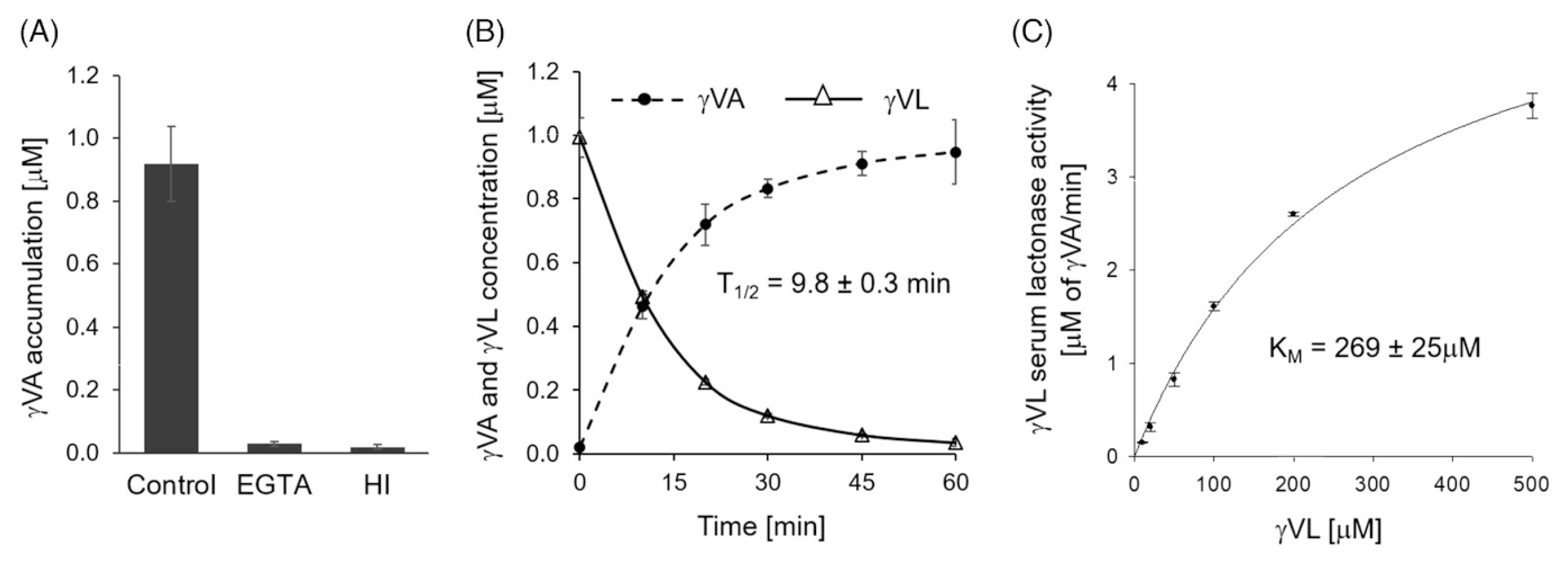
Figure 4.
Latency (A), incidence of seizures (B) and death (C) in L-Hcy-thiolactone-injected (intraperitoneally, 3.7 μmol/g body weight) Pon1-/- mice (n = 19) and their Pon1+/+ siblings (WT, n =13) monitored for 90 min after injection. The figure was drawn based on data from Borowczyk et al. [39].
Figure 4.
Latency (A), incidence of seizures (B) and death (C) in L-Hcy-thiolactone-injected (intraperitoneally, 3.7 μmol/g body weight) Pon1-/- mice (n = 19) and their Pon1+/+ siblings (WT, n =13) monitored for 90 min after injection. The figure was drawn based on data from Borowczyk et al. [39].
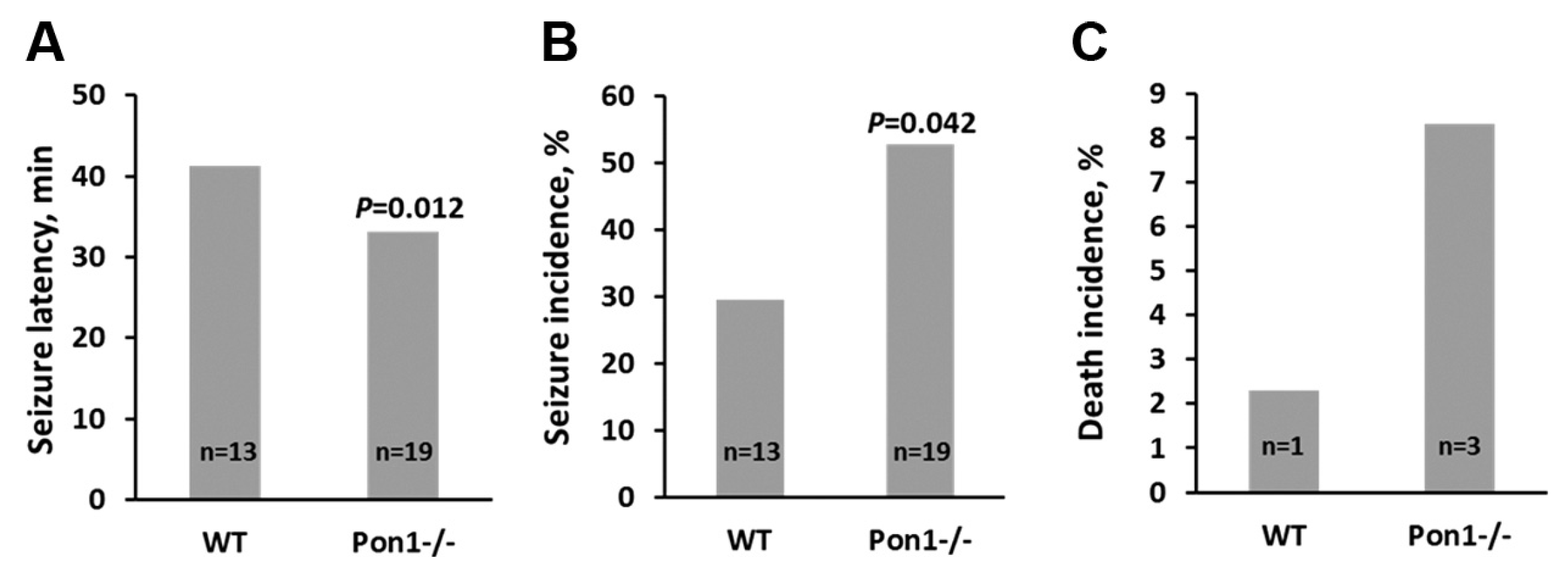
Figure 5.
Proposed mechanism of Aβ generation in Pon1-/-5xFAD mice. Up and down arrows show the direction of changes. Pon1, paraoxonase 1; Hcy, homocysteine; HTL, Hcy-thiolactone; APP, amyloid β precursor protein; mTOR, mammalian target of rapamycin; pmTOR, phospho-mTOR; Phf8, Plant Homeodomain Finger protein 8. [H4K20me1-mTOR] represents H4K20me1 bound at the mTOR promoter. Reprinted from Ref. [80].
Figure 5.
Proposed mechanism of Aβ generation in Pon1-/-5xFAD mice. Up and down arrows show the direction of changes. Pon1, paraoxonase 1; Hcy, homocysteine; HTL, Hcy-thiolactone; APP, amyloid β precursor protein; mTOR, mammalian target of rapamycin; pmTOR, phospho-mTOR; Phf8, Plant Homeodomain Finger protein 8. [H4K20me1-mTOR] represents H4K20me1 bound at the mTOR promoter. Reprinted from Ref. [80].
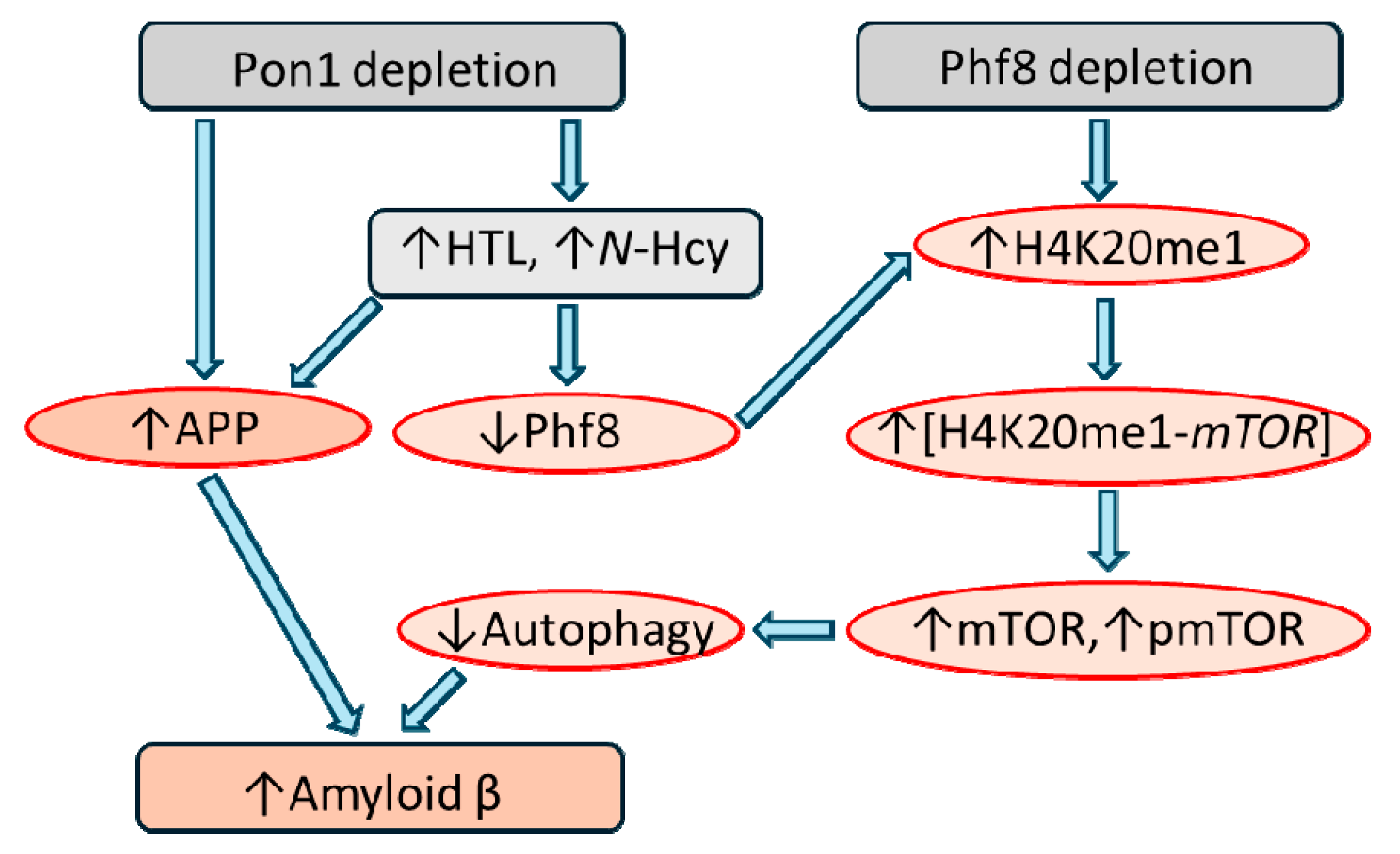
Figure 6.
Principal component analysis of the LFQ intensities for plasma proteins. (A) Pon1-/- mice (n = 17; blue cross) and Pon1+/+ siblings (n = 8; red square). B. PON1-/--192QQ (n = 50; blue cross) and PON1-/--192QQ healthy humans (n = 50; red square). Calculations were performed with Perseus. Adapted from Ref. [65].
Figure 6.
Principal component analysis of the LFQ intensities for plasma proteins. (A) Pon1-/- mice (n = 17; blue cross) and Pon1+/+ siblings (n = 8; red square). B. PON1-/--192QQ (n = 50; blue cross) and PON1-/--192QQ healthy humans (n = 50; red square). Calculations were performed with Perseus. Adapted from Ref. [65].
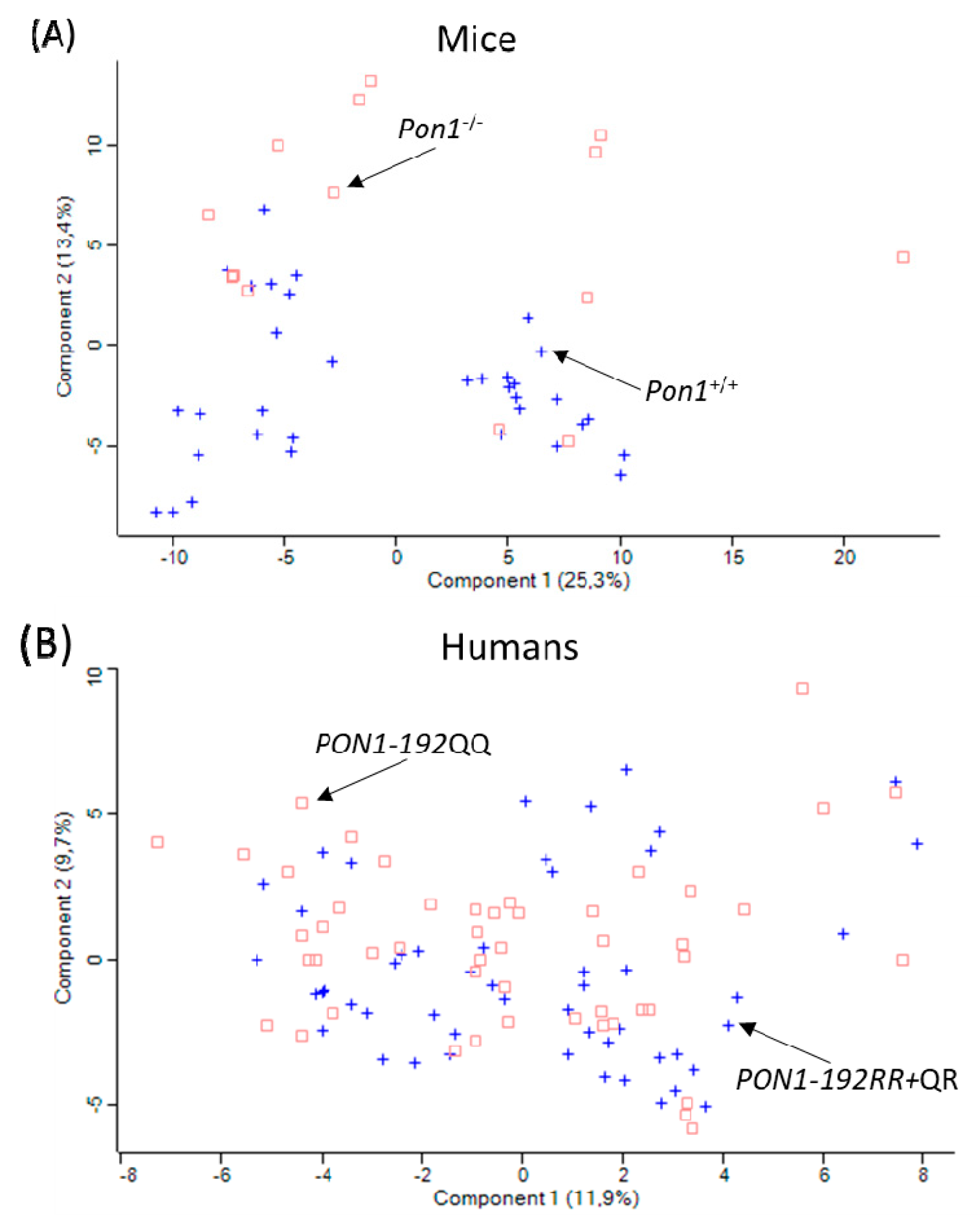
Figure 7.
Top molecular network ‘Lipid Metabolism, Molecular Transport, Small Molecule Biochemistry’ associated with (A) Pon1-/- genotype in mice and (B) PON1-Q192R polymorphism in humans. The mouse network (A) includes redox-related proteins (Alb, Ambp, Hp, Hpx, ApoD, ApoM) and inflammation-related proteins (Ambp and Ttr). The human network (B) contains inflammation-related AMBP and TTR proteins. Reprinted with permission from Ref. [65].
Figure 7.
Top molecular network ‘Lipid Metabolism, Molecular Transport, Small Molecule Biochemistry’ associated with (A) Pon1-/- genotype in mice and (B) PON1-Q192R polymorphism in humans. The mouse network (A) includes redox-related proteins (Alb, Ambp, Hp, Hpx, ApoD, ApoM) and inflammation-related proteins (Ambp and Ttr). The human network (B) contains inflammation-related AMBP and TTR proteins. Reprinted with permission from Ref. [65].
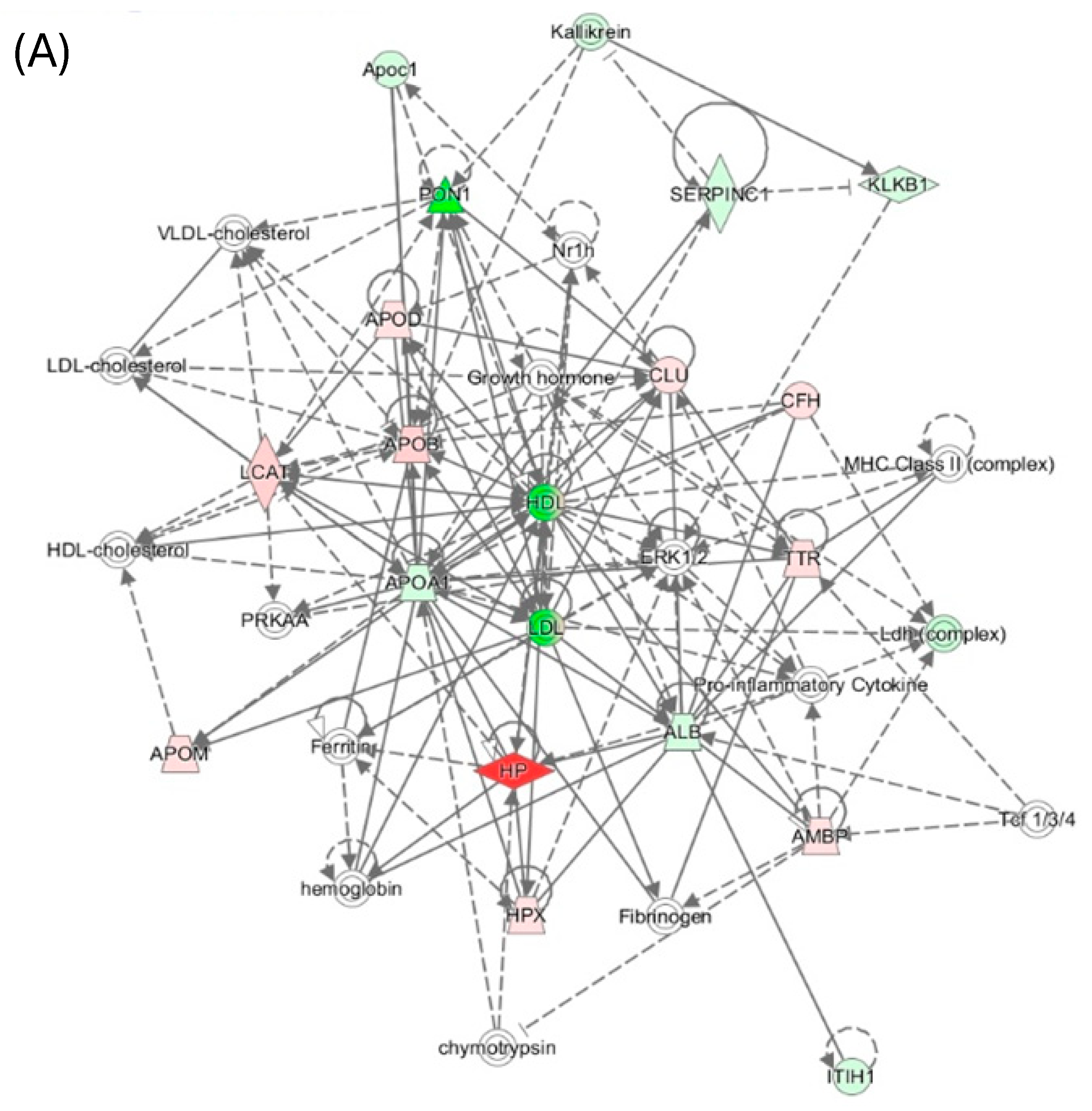
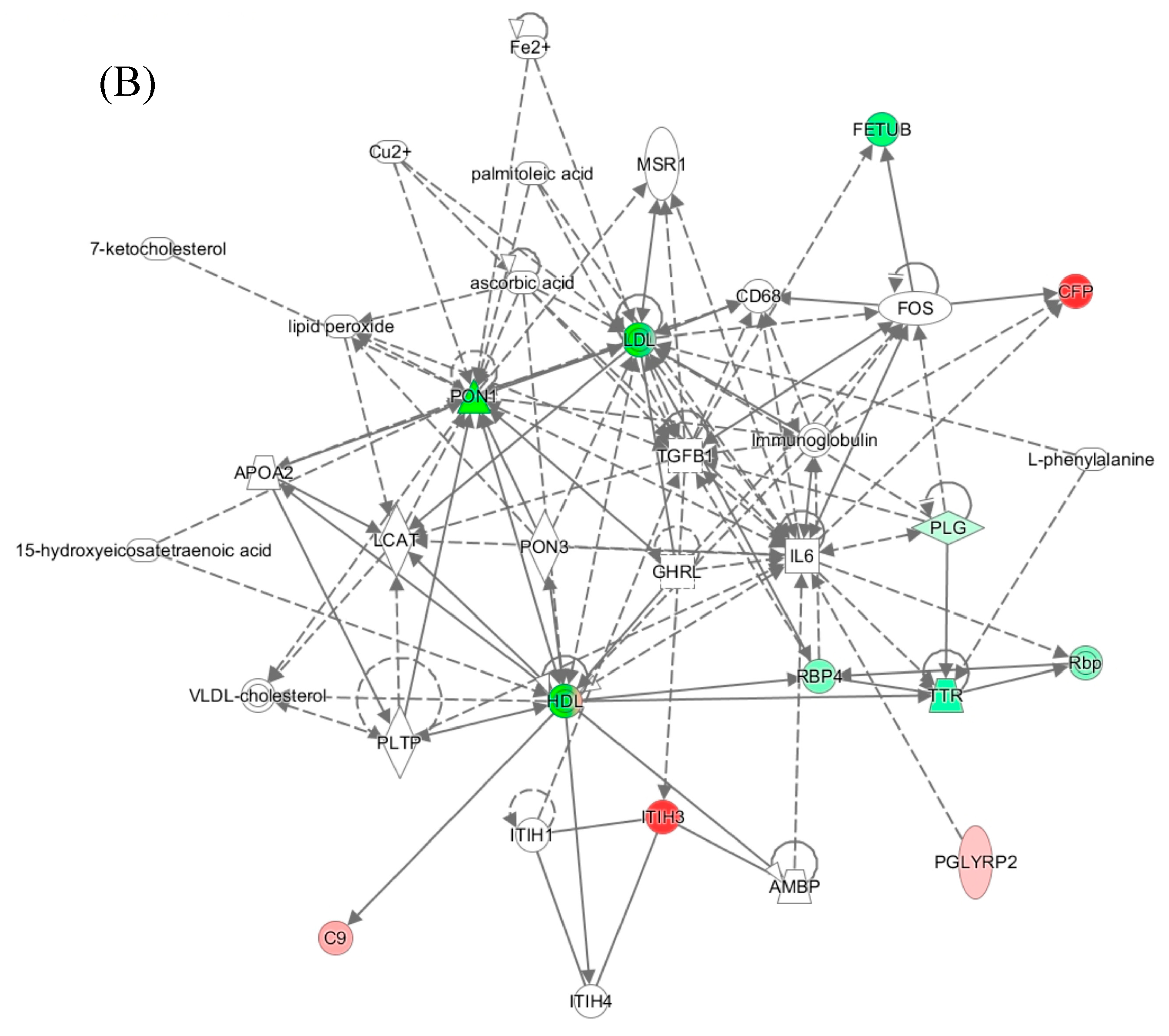
Figure 8.
Principal component analysis (PCA) of label-free quantification (LFQ) intensities of mouse plasma proteins: effects of Pon1 genotype and diet. Pon1-/- mice, control diet (n = 17, blue cross), Pon1-/- mice, HHcy diet (n = 15, blue square), Pon1+/+ siblings, control diet (n = 8, purple circle), Pon1+/+ siblings, HHcy diet (n = 7, green triangle). Calculations were carried out using Perseus. Ovals illustrate a smaller overlap between data points for Pon1-/- and Pon1+/+ mice fed with high-Met (A.) than for mice fed with control diet (B.). Reprinted with permission from Ref. [112]. Copyright 2021 by Elsevier, Inc.
Figure 8.
Principal component analysis (PCA) of label-free quantification (LFQ) intensities of mouse plasma proteins: effects of Pon1 genotype and diet. Pon1-/- mice, control diet (n = 17, blue cross), Pon1-/- mice, HHcy diet (n = 15, blue square), Pon1+/+ siblings, control diet (n = 8, purple circle), Pon1+/+ siblings, HHcy diet (n = 7, green triangle). Calculations were carried out using Perseus. Ovals illustrate a smaller overlap between data points for Pon1-/- and Pon1+/+ mice fed with high-Met (A.) than for mice fed with control diet (B.). Reprinted with permission from Ref. [112]. Copyright 2021 by Elsevier, Inc.
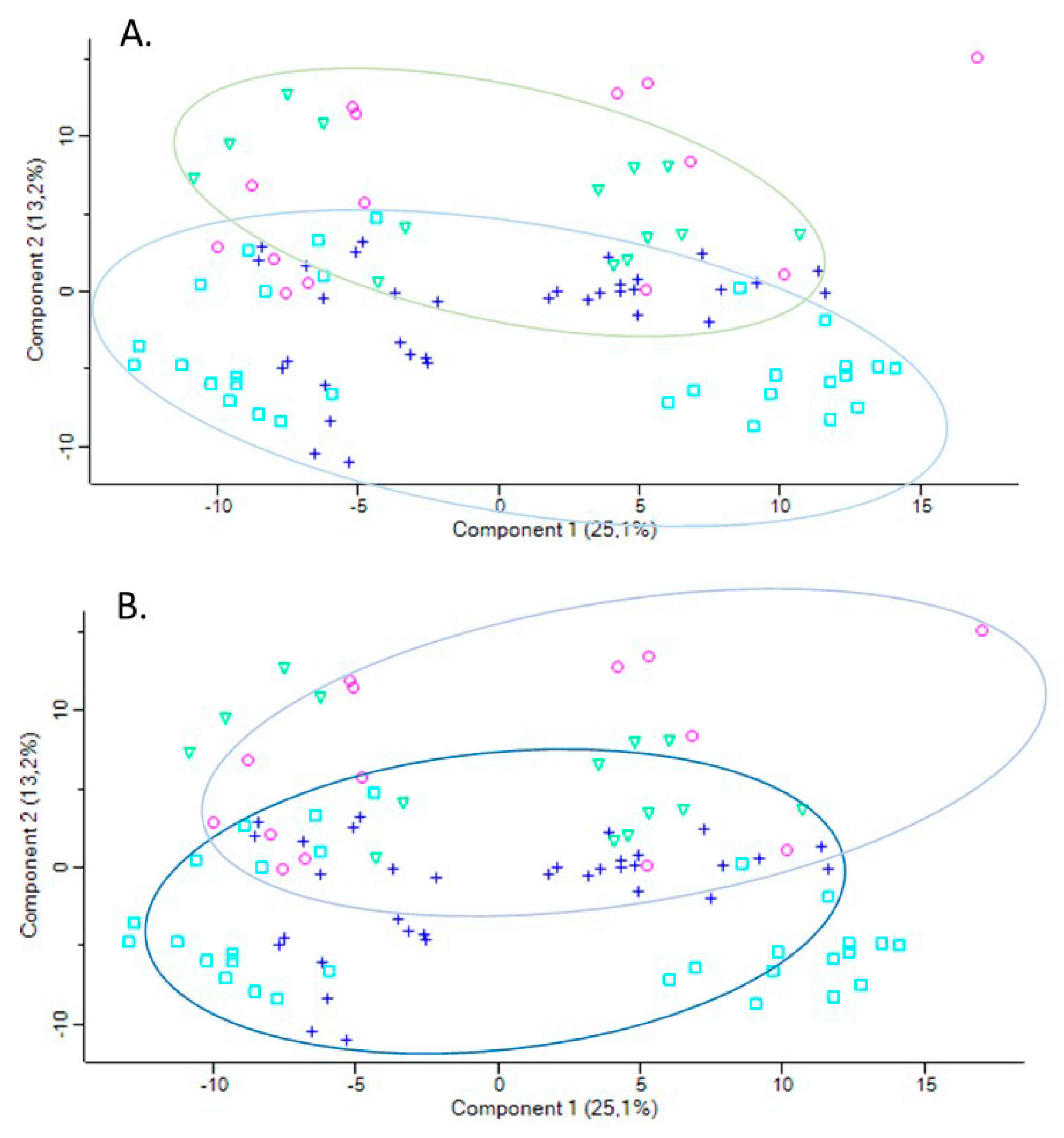

Table 1.
Association of PON1 with oxidative stress, inflammation, and human disease*.
| Disease | PON1 activity | PON1 genotype | Oxidative stress | Inflammation | References |
| Cardiovascular disease | ↓ | PON1-192QQ | ↑ | ↑ | [27,29,56,69] |
| Kidney disease | ↓ | ND | ND | ↑ | [30,31] |
| Fatty liver disease | ↓ | ND | ↑ | ↑ | [57] |
| Alzheimer’s disease | ↓ | PON1-107TT | ↑ | ND | [32,58,59,84] |
| Hepatocellular carcinoma | ↓ | ND | ↑ | ↑ | [33,60] |
| *ND, not determined. Up and down arrows indicate the direction of change. | |||||
Table 2.
Brain proteins affected by Pon1 depletion and/or HHcy in mice are also affected in AD and other neuropathies.
Table 2.
Brain proteins affected by Pon1 depletion and/or HHcy in mice are also affected in AD and other neuropathies.
| Protein name | Change in Pon1-/- vs. Pon1+/+ brain* |
Change in human AD brain (another neuropathy or animal model) ** | |
|---|---|---|---|
| Std. diet | 1%-Met diet | ||
| Brain-specific | |||
| Ncald | – | ↑ | ↓, (↓ Gls-/- mouse) |
| Nrgn | ↓ | ↑ | ↓ |
| Stmn1 | – | ↑ | ↓, (↑ MS, TLE, SMA, schizophrenia), (↑ HD4 mouse model) |
| Anti-oxidant defense | |||
| Sod1 | ↓ | – | (↑ ALS) |
| Prdx2 | – | ↑ | ↑ |
| DJ-1 (Park7) | ↓ | ↑ | ↑ |
| Energy metabolism | |||
| Ak1 | – | ↑ | ↑ |
| Cell cycle | |||
| GDI1 | – | ↑ | (↑ rat ischemic brain) |
| Ran | – | ↑ | ↑ |
| Cytoskeleton assembly | |||
| Tbcb | ↓ | ↑ | (↑ GAN) |
| CapZa2 | ↑ | – | ↑ CapZb2# |
| Other proteins | |||
| Hdhd2 | – | ↑ | |
* The up ‘↑’ and down ‘↓’ arrows indicate up-regulated and down-regulated proteins, respectively. No significant change is indicated by dash ‘–‘. ** AD, Alzheimer’s disease; MS, multiple sclerosis; HD, Huntington disease; GAN, giant axon neuropathy; TLE, temporal lobe epilepsy; SMA, spinal muscular atrophy. # The b2 subunit of the CapZ heterodimer. Adapted from Ref. [58].
Table 3.
PON1 influences oxidative stress- and inflammation-related gene expression.
| Model | Treatment | Gene expression assessment | Outcome | Referencee | ||
|---|---|---|---|---|---|---|
| Methods | Tissue | In vivo | In vitro | |||
| Pon1-/- vs Pon1+/+mice, high fat diet | Pon1 deletion in WT mice | Enzymatic activity assay, northern blot, RT-qPCR, western blot |
Liver, plasma | Pon1 protein absent in Pon1-/- mice, ↑lipid peroxides in HDL, no lipid peroxides in LDL, ↑atherosclerosis; no atherosclerosis in Pon1-/- mice on a chow diet | ↑Lipid peroxides in hLDL, ↑MCP1, ↑monocyte migration ameliorated by Pon1+/+ HDL in a cell co-culture model of the arterial wall; Pon1-/- HDL does not prevent hLDL oxidation | Shih DM et al. [54] |
| Pon1-/-ApoE-/- vs. Pon1+/+ApoE-/- mice, std chow diet | Pon1 deletion in ApoE-/- mice | Enzymatic activity assay, northern blot, RT-qPCR |
Liver, plasma | ↑Lipid peroxides in LDL, ↑oxidized phospholipid epitopes in plasma; ↑HO1, PPARγ, ↑oxLDL receptors (SRA, CD36, macrosialin), but not HDL receptor SR-BI, in the liver; no change in anti-oxLDL and anti-MDA-LDL autoantibody levels; ↑atherosclerosis | LDL from Pon1-/- mice elevated lipid hydroperoxide and monocyte transmigration | Shih DM et al. [55] |
| Human PON cluster transgenic mice | PON1, PON2, PON3 overexpression | Enzymatic activity assay, RT-qPCR, western blot, ELISA |
↑PON1 expression, ↓atherosclerosis ↓Icam-1, ↓Mcp-1, ↓TNF-α, ↓IL-6, ↓Mmp-9 |
PON Tg HDL ameliorated Cu-induced LDL oxidation | She ZG et al. [63] | |
| Pon1-/- vs. Pon1+/+mice, std chow diet | Pon1 deletion | 2D-SDS-PAGE gel electrophoresis, MALDI-TOF mass spectrometry | Brain | ↓Sod1, ↓DJ-1 | [109] Brain | |
| Liver | ↑Prdx2, ↑Ftl, ↑ApoE | [110] Liver | ||||
| Kidney | ↑Prdx2, ↓ApoA1 | [111] Kidney | ||||
| Enzymatic activity assay, Label-free nanoLC-MS/MS mass spectrometry |
Plasma | ↓Alb, ↓Blvrb, ↑Ambp, ↑(Hpx, ↑ApoD, ↑ApoM, ↑Hp), ↑Ttr | Sikora M et al. [65] | |||
| Pon1-/- vs. Pon1+/+mice, high methionine diet | Pon1 deletion | Enzymatic activity assay, Label-free nanoLC-MS/MS mass spectrometry |
Plasma | ↓Alb$, ↑Hp, ↑Hpx ,↓Alad, ↑Cp, ↓Gclm, ↓Cat, ↑Ctsb, ↓Gsn, ↑Grn, ↓Prdx2#, ↓Prdx6, ↓Txn, ↓Igfbp3, ↓Park7#, ↓Pebp1#, ↓Ppia, ↓Serpina3k | Sikora M et al. [112] | |
| PON1-192QQ vs. PON1-192RR+QR humans | none | Enzymatic activity assay, Label-free nanoLC-MS/MS mass spectrometry |
plasma | APOA1↓, PON1↓, APOD↑, APOM↑, HP↓, GPX3↑ | Sikora M et al. [65] | |
| SR-BI-/- vs SR-BI-/- mice | SR-BI deletion, probucol treatment | Enzymatic activity assay, 2D-SDS-PAGE gel electrophoresis, LC-MS/MS mass spectrometry, western blot, RT-qPCR | Plasma, HDL | ↓HDL protein, ↓ApoA1, ↓Pon1 protein and activity, ↑Saa, ↑ApoA4, ↑A1AT, ↑Mpo Probucol upregulated Pon1, downregulated Saa, ApoA4, A1AT, and Mpo thereby improving HDL function |
↑Mcp1, Tnf-α in oxLDL treated macrophages; SR-BI+/+ HDL reduced Mcp1, Tnf-α while SR-BI−/− HDL had no effect |
Cao J et al. [113] |
| Pon1+SR-BI-/- mice | Pon1 overexpression SR-BI-/- mice using lentivirus vector | Enzymatic activity assay, RT-qPCR, western blot |
Kidney |
Pon1+SR-BI-/- mice: ↓renal Pon1 expression & plasma activity, ↑ expression of redox (p47phox, Nox1, Nox4) and inflammation related (Il1b, Il6) genes, ↓anti-inflammatory cytokine Il10. Pon1+SR-BI-/- mice fed with a high-fat diet : ↑plasma & renal Pon1 expression & activity, ↓renal redox (p47phox, Nox1, Nox4, Sod) and inflammation related (Tnfα, Il6) genes, ↑anti-inflammatory cytokine Il10 |
Liu Q et al. [114] | |
| Liver | Pon1+SR-BI-/- mice: ↑hepatic/plasma Pon1, ↑ApoE, ↑Lcat, ↓plasma ALT activity, ↓ROS levels & MPO activity, ↓acute-phase & pro-inflammatory plasma proteins (ApoA4, A1AT, Saa), ↑hepatic ApoA1, Ldlr, Lxrα, Abca, Abcg5, Abcg8, ↓Tnf-α, ↓Il6, ↓ atherosclerosis | Macrophages treated with Pon1+SR-BI-/- HDL: ↓mRNA for inflammatory cytokines IL-6, TNFα, NOX1, ↑mRNA for anti-inflammatory cytokines IL-4, IL-10; mRNA for cholesterol transport ↓Scarb1, ↑Abca1 | Zhao X J et al. [115] | |||
HO1, heme oxidase 1; PPARγ, peroxisome proliferator-activated receptor gamma; MCP1, monocyte chemotactic protein; MPO, myeloperoxidase; Pon1+SR-BI-/- mice, Pon1 overexpressing SR-BI-/- mice; Up and down arrows indicate the direction of change in gene expression.
Table 4.
PON1 genotype, activity, and protein levels in humans.
| Human PON1 | ||
|---|---|---|
| Genotype (n) | Activity a | Protein b |
| PON1-192RR (19) | 100 | 100 |
| PON1-192QR (30) | 20.6 | 63.0 |
| PON1-192QQ (51) | 14.3 | 60.0 |
a Relative mean values, determined in serum using paraoxon as a substrate. b PON1 protein quantified by label-free mass spectrometry. Adapted with permission from Ref. [65]. Copyright 2020 by the Authors.
Table 5.
Pon1 genotype, activity, and protein levels in mice.
| Mouse Pon1 | ||
|---|---|---|
| Genotype (n) | Activity a | Protein b |
| Pon1+/+ (17) | 100 | 100 |
| Pon1-/- (8) | 0.0 | 2.0 |
a Relative mean values, determined in serum using paraoxon as a substrate. b PON1 protein quantified by label-free mass spectrometry Adapted with permission from Ref. [65].
Table 6.
Plasma proteins affected by Pon1-/- genotype in mice fed with HHcy or control diet*.
| Unique to HHcy diet mice (n = 66) | Unique to control diet mice (n = 27) |
Proteins affected both in HHcy and control diet mice (n = 23) |
|---|---|---|
| Oxidative stress (n = 15): ↓Alad, ↑Cp, ↓Gclm, ↓Cat, ↑Ctsb, ↓Gsn, ↑Grn, ↓Prdx2#, ↓Prdx6, ↓Txn, ↓Igfbp3, ↓Park7#, ↓Pebp1#, ↓Ppia, ↓Serpina3k |
Oxida tive stress (n = 1): ↓Blvrb |
Oxidative stress (n = 3): ↓Alb$, ↑Hp, ↑Hpx |
| Immune response (n = 15): ↓Il1rap, Igh (n=10↑, 1↓), ↑Igk (n = 3), | Immune response (n = 10): ↑Clu$, ↑Igh (n=3↑, 1↓), ↑Igk (n=3), ↑Igl, ↑Igm | Immune response (n = 9): ↑Igh (n = 4), ↑Igj, ↑Igk (n = 3), ↑Igl |
| Acute phase response (n = 6): ↑A2m$, ↑Ahsg, ↑Orm1, ↑Orm2, ↑Saa1, ↑Saa2 |
Acute phase response (n = 1): ↑Ttr |
Acute phase response (n = 1): ↑Ambp |
| Complement/coagulation (n = 5): ↑Apcs, ↓F13a1, ↑C3, ↑Cfb, ↑Cfhr1 |
Complement/coagulation (n = 4): ↑AI182371, ↑F2, ↓F13b, ↓Mbl1 | Complement/coagulation (n = 3): ↑Cfh, ↓Klkb1, ↓Serpinc1 |
| Blood coagulation (n = 6): ↑Serpina10, ↓Gp1ba, ↑Gp5, ↑Itih3, ↑Pros1$, ↓Proz | Blood coagulation (n = 3): ↓Hgfac, ↑Hrg‡, ↓Itih1 | |
| Lipoprotein/lipid metabolism (n = 5): ↓ApoA2, ↓ApoC2, ↓Azgp1, ↓Pgp, ↑Pltp |
Lipoprotein metabolism (n=4): ↓Afm, ↑ApoD, ↑ApoM, ↑Lcat |
Lipoprotein metabolism (n = 4): ↓ApoA1, ↑ApoB, ↓ApoC1, ↓Pon1 |
| Protein turnover (n = 5): ↓Apeh, ↓Mug2, ↓Serpina3m, ↓Uba1, ↓Uba52 | Protein turnover (n = 1): ↑Fetub | Protein turnover (n=1): ↓Mug1; |
| Other proteins (n = 8): ↓Atic, ↓Nme1, ↓Pnp (purine metabolism), ↓Tpi, ↓Tkt (glucose metabolism), ↓Ran# (nucleoplasmic transport), ↓Rbp4 (retinol transport), ↓Spp2 (bone remodeling) |
Other proteins (n = 3): ↓Aldoa, ↓Ldha (glucose metabolism), ↓Lifr (tissue regeneration) |
Other proteins (n = 2): ↓Bpgm (glucose metabolism), ↓Ica (carbonic anhydrase inhibitor) |
* Up and down arrows indicate the direction of change in protein levels. # Proteins affected also in mouse brain [58]. $ Enriched in, or ‡ largely excluded from, PON1-containg HDL subfraction of normal human HDL [16]. Bold font indicates proteins identified as components of HDL. Adapted with permission from Ref. [112]. Copyright 2021 by Elsevier, Inc.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



