
Submitted:
13 October 2024
Posted:
15 October 2024
You are already at the latest version
Abstract
Oxyria (Polygonaceae) is a small genus only comprising two species, Oxyria digyna and O. sinensis. Both species are well-known Chinese herbal medicines with a long history of use. We sequenced and assembled the complete mitogenomes of these two species and conducted a comparative analysis of the mitogenomes within Polygonaceae. Both O. digyna and O. sinensis displayed distinctive multi-branched conformations, consisting of one linear and one circular molecule. These two species shared similar gene compositions and exhibited distinct codon preferences, with mononucleotides as the most abundant type of simple sequence repeats. In the mitogenome of O. sinensis, a pair of long forward repeat sequences can mediate the division of molecule 1 into two sub-genomic circular molecules. Homologous sequence analysis revealed the occurrence of gene transfer between the chloroplast and mitochondrial genomes within Oxyria species. Additionally, a substantial number of homologous collinear blocks with varied arrangements were observed across different Polygonaceae species. Phylogenetic analysis suggested that mitogenome genes can serve as reliable markers for constructing phylogenetic relationships within Polygonaceae. Comparative analysis of eight species revealed Polygonaceae mitogenomes exhibited variability in gene presence, and most protein-coding genes (PCGs) have undergone negative selection. Overall, our study provided a comprehensive overview of the structural, functional, and evolutionary characteristics of the Polygonaceae mitogenomes.
Keywords:
Oxyria
; Polygonaceae
; Mitogenomes
; Phylogenetic analysis
; Repeat-mediated recombination
1. Introduction
Oxyria is a small genus in the Polygonaceae, consisting of two species, Oxyria digyna (L.) Hill and O. sinensis Hemsley. Oxyria digyna thrives in various moderately humid tundra habitats and is widely distributed across the Northern Hemisphere. It is highly adapted to high-altitude environments, reaching elevations up to ca. 4,900 m [1,2]. In contrast, O. sinensis is native to China, predominantly grows in the middle to high altitude regions of southwestern mountains of China like Yunnan, Sichuan and Xizang (Tibet), ranging from 1,600 to 3,800 m [3,4]. Oxyria sinensis is dioecious that typically grows in resource-poor habitats, such as slopes and valleys. They are often found in copper-zinc mining areas and serves as indicator plants for copper deposits, earning it the nickname “copper-weed” [1,5]. All wild records showed Oxyria species have demonstrated remarkable adaptation to harsh environment conditions, such as cold temperatures, high altitudes, and nutrient-poor soils. Both O. digyna and O. sinensis have been used as medicinal herbs for their anti-inflammatory, cathartic, antimicrobial, and antidiarrheal properties [1,6]. The strong ecological adaptative ability and diverse natural compounds made O. digyna and O. sinensis excellent candidates for investigating the genetic information, which, is associated with genome evolution and natural compound synthesis within the Polygonaceae. While the chloroplast genomes of Oxyria have been published [7], investigation on their mitogenomes remains blank.
The Polygonaceae is globally distributed and particularly abundant in the temperate regions of the Northern Hemisphere [8]. The family comprises approx. 50 genera and more than 1,000 species [7]. Approximately 13 genera and more than 200 species are distributed in China. Many species within the family have important economic value. For example, buckwheat (Fagopyrum esculentum) is a crucial grain crop [9], Polygonum orientale and P. capitata are introduced as ornamental plants. Also, some species, such as Pleuropterus multiflorus, Reynoutria japonica, Rheum palmatum, and Fallopia aubertii, are widely used in traditional Chinese medicine [10,11,12,13]. The chloroplast genomes of multiple species within the Polygonoideae have been assembled and reported for phylogenetic, evolutionary, and horticultural studies [7,14]. The entire nuclear genomes of some species with high commercial and ecological value, such as F. esculentum, Reynoutria japonica, Rheum officinale, and Rheum nobile have been studied [15,16,17,18].
The mitogenomes, typically consisting of circular or linear DNA, are crucial for metabolic processes like respiration and photosynthesis, which significantly impact the growth and development of organisms [19,20,21,22]. Mitochondria exhibited considerable genetic diversity, largely attributed to their high frequency of recombination. Investigating the diverse and complex nature of plant mitogenomes meets significant challenges but offers opportunities for improving our understanding in the field of evolutionary biology [21]. Many phylogenetic and molecular identification studies on Polygonaceae have been conducted using transcriptomes or chloroplast genomes [7,14]. However, to date, only six mitogenomes in the Polygonaceae have been reported. They are, Rheum rhabarbarum, Rheum palmatum, Pleuropterus multiflorus, Reynoutria japonica, Fallopia aubertii and Fagopyrum esculentum [9,10,11,12,13]. Previous studies have shown that plant mitogenomes exhibit rich diversity in size and structure, even among closely related species [23,24]. In the six published species of Polygonaceae, mitogenome sizes ranged from 302,229 bp in Reynoutria japonica to 404,063 bp in Fagopyrum esculentum [9,11]. These mitogenomes are exclusively composed of circular structures, with variations in the number of circular chromosomes: one in Fallopia aubertii, two in Rheum palmatum, three in Rheum rhabarbarum, and ten in Fagopyrum esculentum [9,10,13]. Despite these findings, comparative analyses among the mitogenomes of Polygonaceae remain limited, requiring the necessity for further investigation into mitochondrial diversity within this family.
In this study, we focus on assembling and comparatively analyzing the mitogenomes of O. digyna and O. sinensis to examine their features like codon usage, sequence repeats, and gene migration events from the chloroplast to the mitogenomes. Furthermore, we compared the reported mitogenomes of the Polygonaceae to identify their general characters. This study also explored the effectiveness of utilizing mitogenome data for phylogenetic inference within Polygonaceae. Our aim is to provide comprehensive insights into the mitogenome evolution within Polygonaceae.
2. Results
2.1. Basic Features of the Oxyria Mitogenomes
The mitogenome of O. digyna had a total length of 384,115 bp and consisted of two main structures: a linear molecule (Molecule 1: 271,961 bp) and a circular molecule (Molecule 2: 112,154 bp), with GC contents of 45.0% and 44.4%, respectively (Figure 1A). In comparison, the mitogenome of O. sinensis measured 411,437 bp in total length and also exhibited a multi-branched structure, consisting of a circular molecule (Molecule 1, 374,651 bp) and a linear molecule (236,786 bp), with GC contents of 44.5% and 42.0%, respectively (Figure 1B). The functional categorization and physical locations of the annotated genes were displayed (Figure 1).
We annotated 34 and 36 PCGs in the mitogenomes of O. digyna and O. sinensis, respectively (Figure 1; Table 1). Both species shared the same types of PCGs (34 types) and three rRNA genes. In O. digyna, all PCGs present as single copies, whereas in O. sinensis, the cox2 and matR genes were in duplicates (Figure 1; Table 1). Additionally, 25 tRNA genes were annotated in O. digyna (with trnL-CAA and trnV-GAC absent), while 28 ones in O. sinensis. Both species had multiple copies of trnM-CAU and trnS-GGA. O. digyna had five copies of trnM-CAU and three copies of trnS-GGA, while O. sinensis had six copies of trnM-CAU and three copies of trnS-GGA (Figure 1; Table 1). Additionally, O. digyna had three copies of trnP-UGG, whereas O. sinensis had three copies of trnE-UUC. The overall gene composition of the two mitogenomes appears to be highly conserved.
2.2. Mitochondrial Repetitive Sequences
In the O. digyna mitogenome, we identified 16 simple sequence repeats (SSRs) in molecule 1 (linear) and 3 SSRs in molecule 2 (circular) (Figure 2A; Table S2-S3). In molecule 1, the majority of SSRs (93.75%, 15 out of 16) were monomeric. Among these monomeric SSRs, thymine (T) repeats were the most prevalent (7 repeats), followed by adenine (A) repeats (6 repeats), constituting 46.67% and 40% of the monomeric SSRs, respectively (Table S2). Additionally, molecule 1 contained one trinucleotide SSR of the GAA type. In molecule 2, all SSRs were monomeric, comprising two thymine (T) repeats and one adenine (A) repeat (Table S3). For the O. sinensis mitogenome, 28 SSRs were found in molecule 1 (circular) and 3 SSRs in molecule 2 (linear) (Figure 2B; Table S4-S5). In molecule 1, most SSRs were also monomeric, accounting for 92.86% (26 out of 28). These monomeric SSRs consisted solely of adenine (A) repeats (15 repeats) and thymine (T) repeats (11 repeats), which accounted for 57.69% and 42.31% of the monomeric SSRs, respectively. Additionally, two SSRs in molecule 1 were dimeric, with AT and TC repeats, and one of each type (Table S4). In molecule 2, all SSRs were monomeric, consisting of two cytosine (C) repeats and one adenine (A) repeat (Table S5).
The analysis identified abundant tandem repeats in both O. digyna and O. sinensis mitogenomes. In O. digyna, six tandem repeats were found in both Molecule 1 and Molecule 2, with repeat lengths ranging from 16 to 42 bp and 16 to 41 bp, respectively (Figure 2; Table S6-S7). In O. sinensis, 12 tandem repeats ranging from 12 to 39 bp were identified in chromosome 1, while chromosome 2 contained only one tandem repeat with a consensus size of 17 bp (Figure 2; Table S8-S9).
We identified 181 pairs of dispersed repeats in O. digyna (156 in molecule 1 and 25 in molecule 2) and 267 pairs in O. sinensis (262 in molecule 1 and 5 in molecule 2), all longer than 30 bp (Table S10-S13). The repeats were predominantly forward and palindromic (Figure 2). In chromosome 1 of O. digyna, we identified 63 pairs of palindromic repeats with the longest 156 bp, and 93 pairs of forward repeats with the longest 745 bp. In chromosome 2 of O. digyna, six pairs of palindromic repeats (up to 136 bp) and 19 pairs of forward repeats (up to 71 bp) were found. For O. sinensis, chromosome 1 contained 113 pairs of palindromic repeats, with the longest 390 bp, and 149 pairs of forward repeats, with the longest extending up to 17,960 bp. In chromosome 2 of O. sinensis, three pairs of palindromic repeats (up to 51 bp) and 2 pairs of forward repeats (up to 32 bp) were identified. Our study further identified that the longest forward repeat sequences in O. sinensis could mediate the formation of two circular molecules in molecule 1 of the mitogenome (Figure 2C). PCR experiments validated these findings, as the length of the PCR products matched the expected results (Figure 2D).
2.3. Codon Usage Preference
The codon usage analysis was performed on the 34 PCGs in the mitogenomes of O. digyna and O. sinensis, respectively (Figure 3). The RSCU values for the start codon AUG (Met) and UGG (Trp) were consistently 1, reflecting no preference. However, a general trend of codon preference was observed in the mitochondrial PCGs of both O. digyna (A) and O. sinensis (B). In both species, there were 30 codons with RSCU values greater than 1, in which 27 ended in A or U, accounting for 90%. Conversely, there were 32 codons with RSCU values less than 1, with 28 of them ending in G or C bases, making up 87.5%. Among all amino acid codons, those encoding Leucine (Leu), Serine (Ser), and Arginine (Arg) were the most abundant in both O. digyna and O. sinensis. In O. digyna, the RSCU values exceeding 1.5 included GCU (Ala), CAU (His), CAA (Gln), AGA (Arg), and ACY (Thr), with GCU (Ala) having the highest RSCU value of 1.58 in mitochondrial PCGs. Conversely, in O. sinensis, only CAA (Gln), AGA (Arg), and GCU (Ala) had RSCU values above 1.5, with CAA (Gln) showing the highest RSCU value of 1.54 in mitochondrial PCGs (Figure 3).
2.4. Characterization of Chloroplast Genome Transfer into the Mitogenome
We identified 27 and 33 homologous fragments between the mitochondrial and chloroplast genomes in O. digyna and O. sinensis, respectively (Figure 4). In O. digyna, the total length of homologous fragments was 10,585 bp, constituting 2.76% of the mitogenome length. Notably, five of the 27 homologous fragments exceeded 1,000 bp, with fragments 1 and 2 being the longest at 1,439 bp (Table S14). In O. sinensis, the total length of the homologous fragments was 34,819 bp, comprising 8.46% of the mitogenome length. Ten of the 33 homologous fragments exceeded 1,000 bp, with fragments 1 and 2 the longest at 6,104 bp (Table S15). Annotation of these homologous sequences found 13 complete genes within the 27 homologous fragments of the O. digyna mitogenome, including ten complete tRNA genes (trnD-GUC, two trnH-GUG, three trnM-CAU, two trnN-GUU, trnS-GGA, trnW-CCA) and three PCGs (petG and two rpl23). In the O. sinensis mitogenome, 17 complete genes were annotated in 33 homologous fragments, including eight PCGs (two ndhB, two rps7, rpoC1, rbcL and two rpl23) and nine tRNA genes (two trnA-UGC, two trnI-CAU, trnL-CAA, trnM-CAU, two trnV-GAC and trnW-CCA).
2.5. Phylogeny and Synteny
The phylogenetic tree was constructed using 20 conserved mitochondrial PCGs (atp8, atp9, ccmB, ccmC, ccmFC, ccmFN, cox1, cox2, cox3, matR, mttB, nad3, nad4L, nad6, nad9, rps12, rps13, rps3, rps4, rps7) from the ten species. The phylogenetic tree showed strong support for all clades and nodes, particularly for the sister relationship between O. digyna and O. sinensis. The Oxyria clade was sister to Rheum (Figure 5A), congruent with previous study [48]. Pairwise synteny analysis revealed a large number of homologous collinear blocks that were not arranged in the same order across the species (Table S16; Figure 5B). The highest abundance of short homologous sequences was observed between the mitogenomes of Fallopia aubertii and Fagopyrum esculentum, despite these species belonging to different genera. The mitogenomes of O. digyna and O. sinensis contained a significantly higher number of homologous sequences compared to those of other species. Conversely, the fewest homologous sequences were identified between Pleuropterus multiflora and Reynoutria japonica. However, they contained much longer homologous fragments.
2.6. Variation in Gene Composition in Mitogenomes of Polygonaceae
We compared the distribution of mitochondrial genes among species in Polygonaceae (Figure 5C), which revealed a high diversity in their gene composition. The trnM-CAU gene was present in multiple copies across all species within the Polygonaceae. In contrast, the rps10, trnfM-CAU (present only in Rheum rhabarbarum), and trnT-GUA genes (found exclusively in Fagopyrum esculentum) were mostly absent in other species of the Polygonaceae. Among the ten species examined, Rheum rhabarbarum possessed the highest number of mitochondrial genes (68), whereas Reynoutria japonica had the fewest (41). Both O. digyna and O. sinensis exhibited the loss of the same PCGs (rps10 and sdh3). Additionally, Reynoutria japonica showed the highest gene loss, missing genes including atp1, atp4, atp6, cob, rpl5, rps1, rps10, rps14, and sdh3.
2.7. Variation in Substitution Rates of Mitochondrial PCGs
We estimated the pairwise nucleotide substitution rates, including the nonsynonymous substitution rate (dN), synonymous substitution rate (dS), and the dN/dS ratio (Figure 6; Table S17). The dN/dS values for most genes were less than 1.0, accounting for 50% of the 20 PCGs analyzed (Figure 6), including atp9, ccmB, ccmC, cox1, cox3, matR, nad3, nad4L, rps12, and rps7. Among these genes, atp9 and cox1 had the lowest dN/dS values (dN/dS < 0.25), suggesting that they have undergone strong purifying selection. The dN/dS values were consistently conserved across the studied species. However, some genes, such as atp8, ccmFC, ccmFN, cox2, mttB, nad6, rps13, rps3, and rps4, showed dN/dS values greater than 1, indicating that these genes may have been subject to positive selection during evolution. Notably, the atp8 gene exhibited an exceptionally high dN/dS value (e.g., O. sinensis vs. Fallopia aubertii: 1.92), suggesting strong positive selection pressure.
3. Discussion
3.1. General Features of the Oxyria Mitogenomes
In our study, the mitogenomes of the Oxyria were sequenced and assembled for the first time. We discovered that both O. digyna and O. sinensis exhibit notable structural polymorphisms, characterized by the presence of both linear and circular molecules. This is distinct from the mitochondrial structures reported in other reported Polygonaceae species, which typically have only circular molecules (one circular chromosome in Fallopia aubertii, two in Rheum palmatum, three in Rheum rhabarbarum, and ten in Fagopyrum esculentum) [9,10,13]. This structural variation may be considered as a phylogenetic signature in the independent evolution of Oxyria in Polygonaceae. A similar genome conformation has also been reported in other plants. For example, the mitogenome of Mentha spicata consists of a linear chromosome and two circular chromosomes [25]. Such structural variability is not rare across various plant species and is likely influenced by the presence of repetitive sequences. Generally, a higher level of structural variability corresponds to a greater number of repetitive sequences. The gene composition in Oxyria species is generally similar, although O. digyna has lost the trnL-CAA and trnV-GAC. This suggests that while the PCGs are highly conserved, tRNA genes demonstrated greater variability within the mitogenomes of the genus Oxyria. These findings may contribute to our understanding to the diversity of mitogenome structures within Polygonaceae.
Small repeat sequences are crucial determinants of the size of plant mitogenomes [26,27]. The size of the O. digyna or O. sinensis mitogenome is comparable to that of other Polygonaceae species with reported mitogenomes, but larger than that of Fagopyrum esculentum [9]. Notably, the mitogenome of O. sinensis is the longest among them, featuring a pair of long repetitive sequences spanning 17,960 bp, which resulted in the duplication of the cox2 and matR genes. A similar phenomenon is observed in Taraxacum mongolicum, which contains a pair of 21,809 bp repeat sequences [28]. As active sites of recombination, these repeat sequences may have significantly influenced the structure of plant mitogenomes, leading to diverse forms such as circular, linear, or branched molecules. In this study, we identified 19 and 31 SSRs in the mitogenomes of O. digyna and O. sinensis, respectively. Notably, mononucleotide repeats were the most prevalent type of SSRs in both O. digyna and O. sinensis. Most monomeric SSRs consisted of A and T bases rather than G and C, consistent with the observations in Rheum palmatum [10] and Cymbidium ensifolium [29]. This bias may be attributed to the lower energy requirement to break A-T bonds compared to G-C bonds. Long repeat sequences also play a critical role in mediating homologous recombination in plant mitogenomes [19]. The repeat sequences analysis indicated a considerable diversity of tandem repeats and dispersed repeats between O. digyna and O. sinensis. Repetitive sequences in mitogenomes can serve as recombination sites, contributing to the formation of multiple molecular conformations. In our study, a long pair of repeat sequences can mediate the formation of two independent circular molecules. Our results provided additional evidence for the presence of multiple conformations within the mitogenome of plants.
Codon bias analysis can provide inferences into horizontal gene transfers and evolutionary relationships among organisms, as closely related species often display similar codon usage patterns [30,31]. The codon usage analysis of PCGs in the mitogenomes of O. digyna and O. sinensis revealed a preference for A/U-ending codons, which is consistent with the patterns observed in Rheum palmatum [10] and Reynoutria japonica [11]. This preference may reflect an evolutionary adaptation for maintaining cellular equilibrium. Both Oxyria species showed a higher usage of codons encoding leucine (Leu), serine (Ser), and arginine (Arg), with specific codons like GCU (Ala), CAU (His), AGA (Arg), and CAA (Gln) notably prevalent. This bias towards certain codons and amino acids may influence mitochondrial gene expression and the efficiency of protein synthesis.
3.2. Intergenomic Sequence Transfers
Chloroplasts and mitochondria are semi-autonomous organelles of endosymbiotic origin [32,33]. Horizontal gene transfer frequently occurs between plastids and mitochondria, resulting in DNA exchange from plastid to mitochondria (MTPT) [34,35,36,37,38]. In this study, we identified MTPT fragments 10,585 bp in O. digyna and 34,819 bp in O. sinensis, which constituted 2.76% and 8.46% of their respective mitogenome lengths. Comparatively, the MTPT homologous sequence lengths in other Polygonaceae species, such as Fallopia aubertii and Reynoutria japonica, were found to be 47,757 bp and 26,123 bp, accounting for 13.6% and 8.64% of their mitogenomes, respectively. Our results revealed that MTPTs are particularly common, and their abundance varies significantly among different species in Polygonaceae. In the mitogenomes of O. digyna and O. sinensi, ten and nine complete homologous tRNA gene fragments were found, a similar phenomenon also presented in Fallopia aubertii and Reynoutria japonica [11,13]. The three complete MTPT genes (trnA-UGC, trnL-CAA, trnV-GAC) detected in O. sinensis were also found in Fallopia aubertii and Reynoutria japonica [11,13]. The six genes, including trnH-GUG, trnM-CAU, trnN-GUU, trnW-CCA, trnP-UGG, and trnS-GGA are commonly recognized as MTPT genes in angiosperms [39], and our study reaffirms this observation. MTPTs played a crucial role in the precise assembly of chloroplast and mitochondrial genomes. The sequence transfers analysis enhanced our understanding to the evolution of organelle genomes.
3.3. Phylogenetic Inference and Synteny Analysis
The mitogenome, with its advantages such as maternal inheritance, rapid evolution, low recombination rates, has become a valuable tool for studies in taxonomy, phylogeny, evolution, and population genetics [19,40]. The phylogenetic relationships revealed by our analysis were consistent with previous studies [9,10,11,12,13,41,42]. This consistency may have underscored the reliability of mitochondrial genes in constructing phylogenetic relationships within Polygonaceae. The mitogenomes of O. digyna and O. sinensis can serve as informative resources for further phylogenetic studies in Polygonaceae. With the rapid advancements in sequencing technology, an increasing number of complete plant mitogenomes have been assembled and reported, facilitating comparative analyses of mitogenome characteristics across various species [21,40]. Pairwise synteny analysis suggested that mitogenomes within Polygonaceae have undergone extensive rearrangements and exhibited a high degree of structural non-conservation, which may play a crucial role in the evolution and diversification of plant mitogenomes.
3.4. Comparative Analysis of Gene Composition and Selective Pressure Analysis
The mitogenome composition in Polygonaceae exhibited a mix of conservation and variability among PCGs, rRNA, and tRNA, reflecting evolutionary adaptations and functional requirements. Common PCGs are highly conserved and are essential for mitochondrial functions. However, O. digyna and O. sinensis have lost some PCGs, specifically rps10 and sdh3, which are involved in the biosynthesis of ribosomal proteins and succinate dehydrogenase, respectively. Similarly, Reynoutria japonica has lost the most genes among the species studied, though it retains all NADH dehydrogenase-related genes [11]. Previous research has proposed that some mitochondrial genes appear to have lost or transferred to the nuclear genome, with chloroplast genes potentially compensating for these losses [34,35,36]. The extensive intergenomic sequence transfers observed may have contributed to the evolutionary dynamics and functional adaptations of mitogenomes within Polygonaceae [11,43]. The universal presence of rRNA genes (rrn18, rrn26, rrn5) emphasized their critical role in mitochondrial ribosome function. While tRNA genes are broadly represented, there are notable variations. For example, high copies of trnM-CUA were detected across all species in the Polygonaceae. Conversely, trnfM-CAU (present only in Rheum rhabarbarum) and trnT-GUA (present only in Fagopyrum esculentum) were nearly absent in other species within the family, indicating possible gene loss or reduced functional necessity.
Substitutions at synonymous and nonsynonymous sites help differentiate between neutral and selective forces acting on genes [44,45,46]. The analysis of nucleotide substitution rates among mitochondrial PCGs showed most genes had a dN/dS ratio less than 1.0, indicating purifying selection. This result was consistent with previous studies [11,13,34,47], revealing that most mitochondrial genes are conserved. Genes such as atp9 and cox1 exhibited particularly low dN/dS values, suggesting strong conservation across the mitogenomes of Polygonaceae. This conservation reflects the essential roles of these genes in mitochondrial function and the selective pressures to maintain their integrity. The dN/dS values of protein-coding genes such as atp8, ccmFC, ccmFN, and cox2 were found to be larger than 1. These three genes may have experienced positive selection due to environmental stress, leading to the development of new functions to adapt to the changing environment.
4. Materials and Methods
4.1. Sample Collection and DNA Sequencing
The sample of O. sinensis was collected from a plant growing in the Wuhan Botanical Garden (Wuhan, China), which was originally introduced from Shade Town, Kangding City, Sichuan Province, southwestern China (Longitude: 101.3334, Latitude: 29.5552). The sample of O. digyna was collected from the Changbai Mountain in Antu County, Yanbian Korean Autonomous Prefecture, Jilin Province, northeastern China (Longitude: 128.0575, Latitude: 42.0453). Fresh leaves of the two species were collected, immediately frozen in liquid nitrogen, and stored at -80 °C. High-quality genomic DNA was extracted using the CTAB method [48]. We utilized both Illumina short-read sequencing and Oxford Nanopore long-read sequencing. The short-paired reads were sequenced by Illumina HiSeq X ten (Illumina, Inc.; San Diego, CA, USA). For Oxford Nanopore sequencing, a 10 kb insert size library was prepared and sequenced on the Nanopore PromethION platform (Oxford Nanopore Technologies, Oxford, UK). We employed NanoPack to assess and ensure the quality of the raw reads [49].
4.2. Mitogenome Assembly and Annotation
We assembled the mitogenomes of the two Oxyria species using a hybrid approach that combined Illumina short reads with long reads from Oxford Nanopore sequencing. Initially, a de-novo assembly was performed using SMARTdenovo software v3.0 with Nanopore long reads [50]. Predicted mitochondrial contigs were then filtered against the mitogenome reference of Rheum palmatum (Accession number: OR148905). The selected contigs were polished for three iterations using miniasm v0.3 and Racon v1.5.0 [51,52]. Mitochondrial sequences from Illumina short reads were filtered by mapping the clean short reads to the predicted mitochondrial contigs using Bowtie2 and SAMtools [53,54]. The final assembly of the mitogenome was conducted by combining the filtered Illumina short reads and Nanopore long reads with Unicycler v0.5.0 [55]. The assemblies were visualized using Bandage [56], with GFA format files generated by Unicycler [55]. Additionally, the chloroplast genomes of Oxyria were assembled using GetOrganelle v1.7.7.0 [57] with the parameters “-R 10 -t 1 -k 21,45,65,85,105”.
We annotated the mitogenomes of two Oxyria species using the GeSeq web server (https://chlorobox.mpimp-golm.mpg.de/geseq.html) [58], with the mitogenome of R. palmatum mentioned above as the reference. Transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs) were identified using tRNAscan-SE [59]. The circular maps of the mitogenomes were generated with OGDraw [60]. For the chloroplast genomes, annotations were performed using Geneious v9.0.2 [61] and GeSeq (https://chlorobox.mpimp-golm.mpg.de/geseq.html). All annotations of both mitochondrial and chloroplast genomes were thoroughly reviewed and manually corrected using Apollo software [62].
4.3. Analysis of Repetitive Sequence and Repeat-Mediated Homologous Recombination
We identified three types of repeats: simple sequence repeats (SSRs), tandem repeats, and dispersed repeats. SSRs were identified using MISA-web (https://webblast.ipk-gatersleben.de/misa/) with the following parameters: 1–10, 2–5, 3–4, 4–3, 5–3, 6–3 [63]. Tandem repeats were detected using Tandem Repeats Finder v4.09 (http://tandem.bu.edu/trf/trf.submit.options.html) with the parameters: match +2, mismatch –7, and indel –7 [64]. Dispersed repeats, including forward (F), reverse (R), palindromic (P), and complement (C) repeats, were identified using the online version of REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer/). The search criteria included a minimum repeat size of 30 bp and a repeat identity greater than 90%, with the following parameters: Hamming distance of 3, a maximum of 50 computed repeats, and a minimum repeat size of 8 [65]. The results of the repeat analysis of the mitogenomes were visualized using the Circos package, which was implemented in TBtools [66].
We identified potential structures of circular molecules in the mitogenome using long-read data. Specifically, we extracted the repetitive sequences and their flanking regions (1,000 bp) from the mitogenome, representing sequences that support the master structure. We then swapped the flanking regions to represent sequences that might support possible recombination. These major and recombinant sequences were mapped to the long reads using BLASTN software [67] to determine the presence of recombination sequences. To verify the reliability, we performed a PCR experiment, designing primers based on the repeat sequences and their upstream and downstream regions. The PCR reaction mixture consisted of 1 μL DNA, 1 μL of 10 μM forward and reverse primers each, 13 μL of 2× Taq PCR Master Mix, and 10 μL of ddH2O. The PCR conditions were as follows: initial denaturation at 94 °C for 3 minutes; 30 cycles of 94 °C for 30 seconds, 55 °C for 30 seconds, and 72 °C for 1 minute, followed by a final extension at 72 °C for 10 minutes.
4.4. Analysis of Codon Preference and Chloroplast to Mitochondrion DNA Transformation
PhyloSuite software was used to extract protein-coding genes (PCGs) from the mitogenomes [68]. Codon preference analysis of the mitochondrial PCGs was then performed using MEGA v7.0, and relative synonymous codon usage (RSCU) values were calculated [69]. To identify homologous sequences between the chloroplast and mitogenomes, we assembled the chloroplast genomes of O. digyna and O. sinensis using short reads with GetOrganelle [57]. Homologous sequences were identified using BLASTN software [67] with an e-value cut-off of 1e-5. The distribution of mitochondrial plastid sequences (MTPTs) across all genomes was visualized using the Circos package [70] implemented in TBtools [66].
4.5. Phylogenetic and Syntenic Analysis
To determine the phylogenetic position of Oxyria species within Polygonaceae, we integrated the mitogenomes of six published Polygonaceae species from five genera (Rheum, Pleuropterus, Reynoutria, Fallopia and Fagopyrumn). Myricaria laxiflora and Myricaria elegans, belonging to the Tamaricaceae family within the order Caryophyllales, were used as outgroups. The mitogenome sequences were downloaded from NCBI (Table S1). A maximum likelihood (ML) phylogenetic tree was constructed based on shared PCGs using MEGA v7.0 software [71], applying the GTR+G+I nucleotide substitution model with 1,000 bootstrap replicates. Additionally, we conducted a comparative analysis of the gene composition of the mitogenomes. The results were visualized using heatmap plots generated with the ggplot2 package in R-package [72]. Pairwise synteny analysis of mitogenomes among Oxyria and other six species was performed using BLASTN software [67]. This analysis involved identifying conserved collinear segments, specifically focusing on homologous sequences longer than 500 bp [73]. The visualization of these homologous segments was accomplished using the Circos package [70].
4.6. Nucleotide Substitution Rate Estimation
Phylosuite software [40] was used to locate and extract shared mitochondrial genes of species. We used MAFFT v7.4 [74] and PhyloSuite [68] for the alignment and concatenation of the nucleotide sequences. Pairwise nucleotide substitution rates, including nonsynonymous (dN) and synonymous (dS) substitution rates, as well as the dN/dS ratio, were calculated using the yn00 module in PAML v4.9 [75]. Visualization of the pairwise dN/dS values was achieved through box plots generated with the ggplot2 package in R-package [72].
5. Conclusion
In this study, the mitogenomes of O. digyna and O. sinensis were sequenced, assembled, annotated, and compared. Both O. digyna and O. sinensis exhibited notable structural polymorphism, characterized with both linear and circular configurations. A long repeat sequence was identified and validated to mediate alternative conformations in the mitogenome of O. sinensis, enhancing our understanding of structural diversity of mitogenome within Polygonaceae. Meanwhile, O. digyna and O. sinensis mitogenomes shared similar gene contents. The comparison of the mitochondrial and chloroplast genomes of Oxyria showed homologous regions and genes, suggesting potential gene transfer events between these organellar genomes. Phylogenetic analysis based on mitochondrial genes across eight Polygonaceae taxa confirmed that the dataset is reliable resource for phylogenetic purpose. The analysis of mitochondrial gene composition across eight Polygonaceae species showed a mix of conservation and variability among PCGs, rRNA, and tRNA. This study provides potential genomic background for better understanding and utilizing medicinal herbs.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
L.J.L. performed data analysis and drafted the manuscript. Z.J., Y.X., and Y.S.W. participated in the acquisition and interpretation of data. C.O.A., O.M.T., and H.Z. contributed to the review and editing of the manuscript. Y.X.S. and H.C.W. provided critical revisions to the manuscript. H.J.Z. conducted the overall planning and supervision of the study.
Funding
This study was supported by grants from the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502), the Project funded by China Postdoctoral Science Foundation (2022M713333).
Data availability
The chloroplast genome accession numbers for O. digyna and O. sinensis in GenBank are PP151304 and PP151305, respectively. The mitogenome accession numbers of O. digyna are PQ301199 and PQ301200. The mitogenome accession numbers of O. sinensis are PQ320132 and PQ320133. The sequencing reads used for mitogenomes assembly in this study have been released on the NCBI with those accession numbers PRJNA1157214 (BioProject), SAMN43508666 and SAMN43508667. (BioSample), SRR30599217 and SRR30599218 (SRA). The sample was deposited in the Wuhan Botanical Garden with the accession number zhsl-2023 and sl-2023.
Acknowledgments
We would like to express our sincere gratitude to the Horticultural Conservation Center of Wuhan Botanical Garden for their invaluable support throughout this research. Special thanks go to Wenbin Xu for his assistance and provision of essential materials.
Conflicts of Interest
The authors declare no competing interests.
References
- Ahmad, I.; Milella, L.; Alotaibi, G. Oxyria digyna: A review on the nutritional value, phytochemistry and ethnopharmacology. PHYTONutrients 2022, 02–16. [Google Scholar] [CrossRef]
- Allen, G.A.; Marr, K.L.; McCormick, L.J.; Hebda, R.J. The impact of Pleistocene climate change on an ancient arctic–alpine plant: Multiple lineages of disparate history in Oxyria digyna. Ecology and Evolution 2012, 2(3), 649–665. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Chen, G.; Li, Z.; Yang, Y.; Wang, Z.; Wang, L. Refugial isolation and range expansions drive the genetic structure of Oxyria sinensis (Polygonaceae) in the Himalaya-Hengduan Mountains. Scientific Reports 2015, 5(1), 10396. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Yong-Ping, Y. Reproductive allocation in a dioecious perennial Oxyria sinensis (Polygonaceae) along altitudinal gradients. Journal of Systematics and Evolution 2008, 46(6), 830. [Google Scholar]
- Yang, J.; Hu, L.; Wang, Z.; Zhu, W.; Meng, L. Responses to drought stress among sex morphs of O xyria sinensis (Polygonaceae), a subdioecious perennial herb native to the East Himalayas. Ecology and Evolution 2014, 4(21), 4033–4040. [Google Scholar] [CrossRef]
- Xing-yu, L.; Chun-lin, L.; Yue-hu, W.; Rong, G. Chemical Constituents of Oxyria digyna. Natural Product Research & Development 2008, 20(5). [Google Scholar]
- Zhang, H.; Zhang, X.; Sun, Y.; Landis, J.B.; Li, L.; Hu, G.; Sun, J.; Tiamiyu, B.B.; Kuang, T.; Deng, T. Plastome phylogenomics and biogeography of the subfam. Polygonoideae (Polygonaceae). Frontiers in Plant Science 2022, 13, 893201. [Google Scholar] [CrossRef]
- Cao, D.L.; Zhang, X.J.; Qu, X.J.; Fan, S.J. Phylogenomics, divergence time estimation, and adaptive evolution in the Polygonoideae (Polygonaceae). Journal of Systematics and Evolution 2023, 61(6), 1004–1019. [Google Scholar] [CrossRef]
- Logacheva, M.D.; Schelkunov, M.I.; Fesenko, A.N.; Kasianov, A.S.; Penin, A.A. Mitochondrial genome of Fagopyrum esculentum and the genetic diversity of extranuclear genomes in buckwheat. Plants 2020, 9(5), 618. [Google Scholar] [CrossRef]
- Gao, L.; Hao, L.; Xu, W.; Xin, T.; Song, C.; Lin, Y.; Zhu, L.; Song, J. Characterization of Rheum palmatum mitochondrial genome and comparative analysis among Caryophyllales species. Acupuncture and Herbal Medicine 2023, 3(4), 323–332. [Google Scholar] [CrossRef]
- Chen, J.; Ma, H.; Fan, H.; Lin, F.; Chai, T.; Wang, H. De novo assembly and comparative analysis of the mitochondrial genome of Reynoutria japonica. Frontiers in Genetics 2023, 14, 1289811. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-K.; Kim, Y.-K. The multipartite mitochondrial genome of Fallopia multiflora (Caryophyllales: Polygonaceae). Mitochondrial DNA Part B 2018, 3(1), 155–156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, Y.-C.; Wang, J.-L. Assembly and Characterization of the Mitochondrial Genome of Fallopia aubertii (L. Henry) Holub. Frontiers in Bioscience-Landmark 2023, 28(10), 233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Zhang, X.; Landis, J.B.; Sun, Y.X.; Sun, J.; Kuang, T.H.; Li, L.J.; Tiamiyu, B.B.; Deng, T.; Sun, H. Phylogenomic and comparative analyses of Rheum (Polygonaceae, Polygonoideae). Journal of Systematics and Evolution 2022, 60(6), 1229–1240. [Google Scholar] [CrossRef]
- He, Q.; Ma, D.; Li, W.; Xing, L.; Zhang, H.; Wang, Y.; Du, C.; Li, X.; Jia, Z.; Li, X. High-quality Fagopyrum esculentum genome provides insights into the flavonoid accumulation among different tissues and self-incompatibility. Journal of Integrative Plant Biology 2023, 65(6), 1423–1441. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, L.; Zheng, Y.; Zhou, C.; Huang, P.; Xiao, X.; Zhao, Y.; Hao, X.; Hu, Z.; Chen, Q. Assembly and annotation of a draft genome of the medicinal plant Polygonum cuspidatum. Frontiers in plant science 2019, 10, 1274. [Google Scholar] [CrossRef]
- Zhang, H.; He, Q.; Xing, L.; Wang, R.; Wang, Y.; Liu, Y.; Zhou, Q.; Li, X.; Jia, Z.; Liu, Z. The haplotype-resolved genome assembly of autotetraploid rhubarb Rheum officinale provides insights into its genome evolution and massive accumulation of anthraquinones. Plant Communications 2024, 5(1). [Google Scholar] [CrossRef]
- Feng, T.; Pucker, B.; Kuang, T.; Song, B.; Yang, Y.; Lin, N.; Zhang, H.; Moore, M.J.; Brockington, S.F.; Wang, Q. The genome of the glasshouse plant noble rhubarb (Rheum nobile) provides a window into alpine adaptation. Communications biology 2023, 6(1), 706. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Smith, D.R.; Keeling, P.J. Mitochondrial and plastid genome architecture: Reoccurring themes, but significant differences at the extremes. Proceedings of the National Academy of Sciences 2015, 112(33), 10177–10184. [Google Scholar] [CrossRef]
- Wang, J.; Kan, S.; Liao, X.; Zhou, J.; Tembrock, L.R.; Daniell, H.; Jin, S.; Wu, Z. Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 2024. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, X.; Xu, Y.; Zhang, Z.; Wei, Y.; Hu, Y.; Zheng, C.; Qu, X. Assembly and comparative analysis of the first complete mitochondrial genome of a traditional Chinese medicine Angelica biserrata (Shan et Yuan) Yuan et Shan. International Journal of Biological Macromolecules 2024, 257, 128571. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’model of plant mitochondrial DNA structure. New Phytol. 2013, 200(4), 978–985. [Google Scholar] [CrossRef] [PubMed]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15(8), e1008373. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Ni, Y.; Zhang, J.; Li, J.; Liu, C. Complete mitochondrial genome of Mentha spicata L. reveals multiple chromosomal configurations and RNA editing events. International Journal of Biological Macromolecules 2023, 251, 126257. [Google Scholar] [CrossRef]
- Andre, C.; Levy, A.; Walbot, V. Small repeated sequences and the structure of plant mitochondrial genomes. Trends Genet. 1992, 8(4), 128–132. [Google Scholar] [CrossRef]
- Liu, J.; Feng, Y.; Chen, C.; Yan, J.; Bai, X.; Li, H.; Lin, C.; Xiang, Y.; Tian, W.; Qi, Z. Genomic insights into the clonal reproductive Opuntia cochenillifera: Mitochondrial and chloroplast genomes of the cochineal cactus for enhanced understanding of structural dynamics and evolutionary implications. Frontiers in Plant Science 2024, 15, 1347945. [Google Scholar] [CrossRef]
- Jiang, M.; Ni, Y.; Li, J.; Liu, C. Characterisation of the complete mitochondrial genome of Taraxacum mongolicum revealed five repeat-mediated recombinations. Plant Cell Rep. 2023, 42(4), 775–789. [Google Scholar] [CrossRef]
- Shen, B.; Shen, A.; Liu, L.; Tan, Y.; Li, S.; Tan, Z. Assembly and comparative analysis of the complete multichromosomal mitochondrial genome of Cymbidium ensifolium, an orchid of high economic and ornamental value. BMC Plant Biol. 2024, 24(1), 255. [Google Scholar] [CrossRef]
- Athey, J.; Alexaki, A.; Osipova, E.; Rostovtsev, A.; Santana-Quintero, L.V.; Katneni, U.; Simonyan, V.; Kimchi-Sarfaty, C. A new and updated resource for codon usage tables. BMC Bioinformatics 2017, 18, 1–10. [Google Scholar] [CrossRef]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49(1), 539–565. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J. The endosymbiotic origin, diversification and fate of plastids. Philosophical Transactions of the Royal Society B: Biological Sciences 2010, 365(1541), 729–748. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.F.; Gray, M.W.; Burger, G. Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 1999, 33(1), 351–397. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; An, B.; Park, S. Dynamic changes in the plastid and mitochondrial genomes of the angiosperm Corydalis pauciovulata (Papaveraceae). BMC Plant Biol. 2024, 24(1), 303. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Grewe, F.; Zhu, A.; Ruhlman, T.A.; Sabir, J.; Mower, J.P.; Jansen, R.K. Dynamic evolution of Geranium mitochondrial genomes through multiple horizontal and intracellular gene transfers. New Phytol. 2015, 208(2), 570–583. [Google Scholar] [CrossRef]
- Zhao, N.; Wang, Y.; Hua, J. The roles of mitochondrion in intergenomic gene transfer in plants: A source and a pool. International Journal of Molecular Sciences 2018, 19(2), 547. [Google Scholar] [CrossRef]
- Sloan, D.B.; Wu, Z. History of plastid DNA insertions reveals weak deletion and at mutation biases in angiosperm mitochondrial genomes. Genome biology and evolution 2014, 6(12), 3210–3221. [Google Scholar] [CrossRef]
- Guo, S.; Li, Z.; Li, C.; Liu, Y.; Liang, X.; Qin, Y. Assembly and characterization of the complete mitochondrial genome of Ventilago leiocarpa. Plant Cell Rep. 2024, 43(3), 77. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Watase, Y.; Nagase, M.; Makita, N.; Yagura, S.; Hirai, A.; Sugiura, M. The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: Comparative analysis of mitochondrial genomes in higher plants. Mol. Genet. Genomics 2005, 272, 603–615. [Google Scholar] [CrossRef]
- Møller, I.M.; Rasmusson, A.G.; Van Aken, O. Plant mitochondria–past, present and future. The Plant Journal 2021, 108(4), 912–959. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Sun, Y.; Landis, J.; Li, L.; Hu, G.; Sun, J.; Tiamiyu, B.; Kuang, T.; Deng, T. Plastome phylogenomics and biogeography of the subfam. Polygonoideae (Polygonaceae). Frontiers in plant science 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.L.; Zhang, X.J.; Qu, X.J.; Fan, S.J. Phylogenomics, divergence time estimation, and adaptive evolution in the Polygonoideae (Polygonaceae). Journal of Systematics and Evolution 2022. [Google Scholar] [CrossRef]
- Adams, K.L.; Qiu, Y.-L.; Stoutemyer, M.; Palmer, J.D. Punctuated evolution of mitochondrial gene content: High and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proceedings of the National Academy of Sciences 2002, 99(15), 9905–9912. [Google Scholar] [CrossRef] [PubMed]
- Buschiazzo, E.; Ritland, C.; Bohlmann, J.; Ritland, K. Slow but not low: Genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol. Biol. 2012, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 2002, 19(6), 908–917. [Google Scholar] [CrossRef]
- Hurst, L.D. Genetics and the understanding of selection. Nature Reviews Genetics 2009, 10(2), 83–93. [Google Scholar] [CrossRef]
- Xia, C.; Li, J.; Zuo, Y.; He, P.; Zhang, H.; Zhang, X.; Wang, B.; Zhang, J.; Yu, J.; Deng, H. Complete mitochondrial genome of Thuja sutchuenensis and its implications on evolutionary analysis of complex mitogenome architecture in Cupressaceae. BMC Plant Biol. 2023, 23(1), 84. [Google Scholar] [CrossRef]
- Arseneau, J.R.; Steeves, R.; Laflamme, M. Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Molecular ecology resources 2017, 17(4), 686–693. [Google Scholar] [CrossRef]
- De Coster, W.; D’hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34(15), 2666–2669. [Google Scholar] [CrossRef]
- Liu, H.; Wu, S.; Li, A.; Ruan, J. SMARTdenovo: A de novo assembler using long noisy reads. Gigabyte 2021, 2021. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27(5), 737–746. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016, 32(14), 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9(4), 357–359. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10(2), giab008. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comp. Biol. 2017, 13(6), e1005595. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31(20), 3350–3352. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; DePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome biology 2020, 21, 1–31. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45(W1), W6–W11. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25(5), 955–964. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47(W1), W59–W64. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28(12), 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.E.; Searle, S.; Harris, N.; Gibson, M.; Iyer, V.; Richter, J.; Wiel, C.; Bayraktaroglu, L.; Birney, E.; Crosby, M. Apollo: A sequence annotation editor. Genome biology 2002, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33(16), 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27(2), 573–580. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29(22), 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Molecular plant 2020, 13(8), 1194–1202. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43(16), 7762–7768. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Molecular ecology resources 2020, 20(1), 348–355. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33(7), 1870–1874. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19(9), 1639–1645. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30(4), 772–780. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, L. ggplot2: Elegant graphics for data analysis by WICKHAM, H. Oxford University Press, 2011. [Google Scholar]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40(7), e49–e49. [Google Scholar] [CrossRef] [PubMed]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47(W1), W5–W10. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24(8), 1586–1591. [Google Scholar] [CrossRef]
Figure 1.
O. digyna and O. sinensis mitogenomes gene map. Genes shown on the outside and inside of the circle are transcribed clockwise and counterclockwise, respectively. The dark grey region within the inner circle represents the GC content.
Figure 1.
O. digyna and O. sinensis mitogenomes gene map. Genes shown on the outside and inside of the circle are transcribed clockwise and counterclockwise, respectively. The dark grey region within the inner circle represents the GC content.
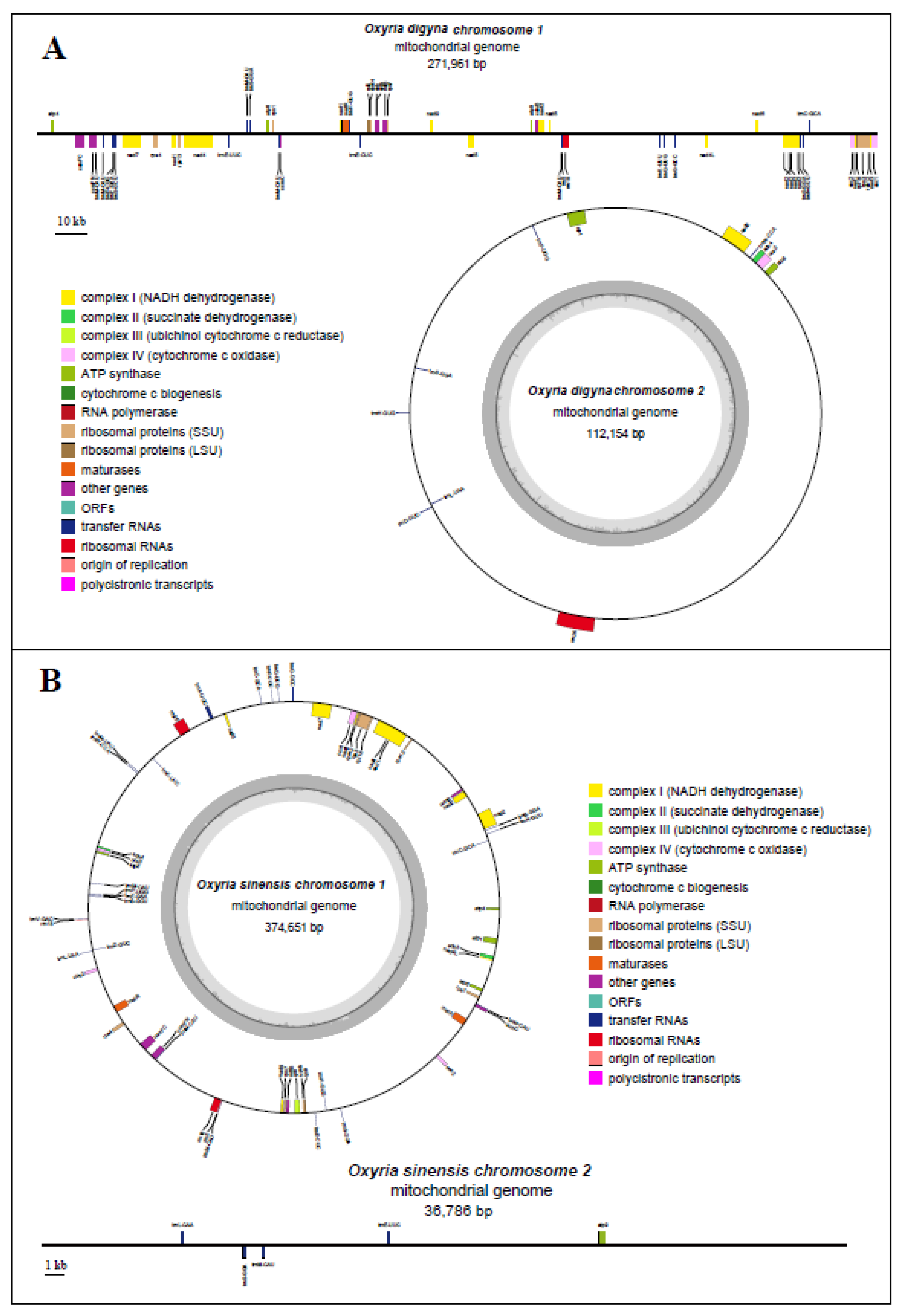
Figure 2.
O. digyna (A) and O. sinensis (B) mitogenome repeated sequence diagram. The inner circle shows the dispersed repeats with a length greater than or equal to 50 bp, in which blue represents forward repeats and yellow represents palindromic repeats. The two outer circles show tandem repeats and simple sequence repeats as short bars, respectively. C. The long repeat sequence in the mitogenome of O.sinensis molecule 1 mediates the potential conformations generated from the recombination. D. The gel electrophoresis results of PCR products were amplified using primers. The amplified sequences correspond to the four sites of the recombination conformation. For repeat sequences that were too long to be fully amplified by PCR, we selected portions of the repeat sequences along with 200 bp of their flanking regions for the PCR experiments.
Figure 2.
O. digyna (A) and O. sinensis (B) mitogenome repeated sequence diagram. The inner circle shows the dispersed repeats with a length greater than or equal to 50 bp, in which blue represents forward repeats and yellow represents palindromic repeats. The two outer circles show tandem repeats and simple sequence repeats as short bars, respectively. C. The long repeat sequence in the mitogenome of O.sinensis molecule 1 mediates the potential conformations generated from the recombination. D. The gel electrophoresis results of PCR products were amplified using primers. The amplified sequences correspond to the four sites of the recombination conformation. For repeat sequences that were too long to be fully amplified by PCR, we selected portions of the repeat sequences along with 200 bp of their flanking regions for the PCR experiments.
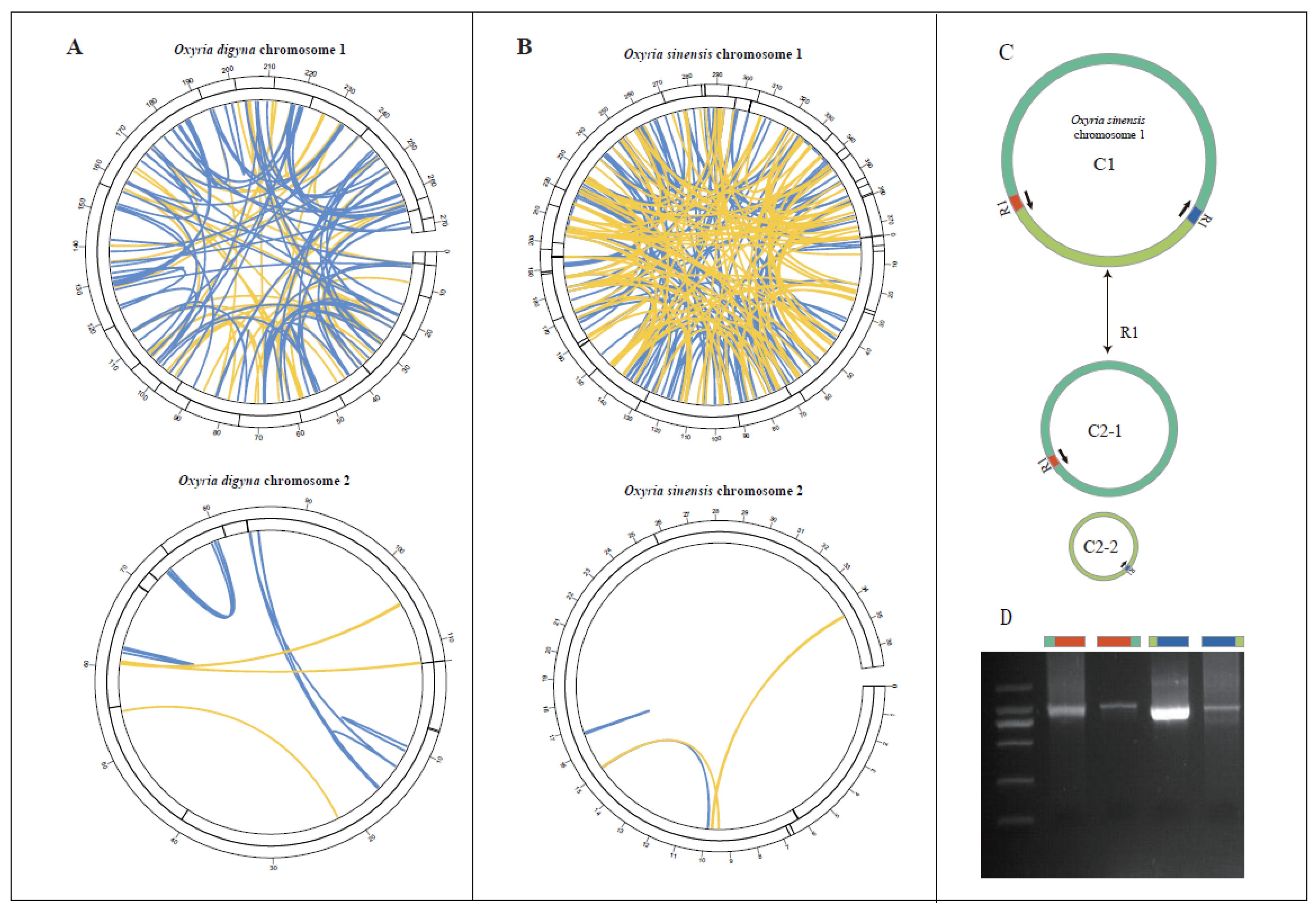
Figure 3.
O. digyna (A) and O. sinensis (B) mitogenome relative synonymous codon usage. The codon families are shown on the X-axis. The RSCU values indicate how frequently a specific codon is observed compared to its expected frequency under uniform synonymous codon usage. RSCU values greater than 1 suggest a preference for specific amino acids in codon usage.
Figure 3.
O. digyna (A) and O. sinensis (B) mitogenome relative synonymous codon usage. The codon families are shown on the X-axis. The RSCU values indicate how frequently a specific codon is observed compared to its expected frequency under uniform synonymous codon usage. RSCU values greater than 1 suggest a preference for specific amino acids in codon usage.
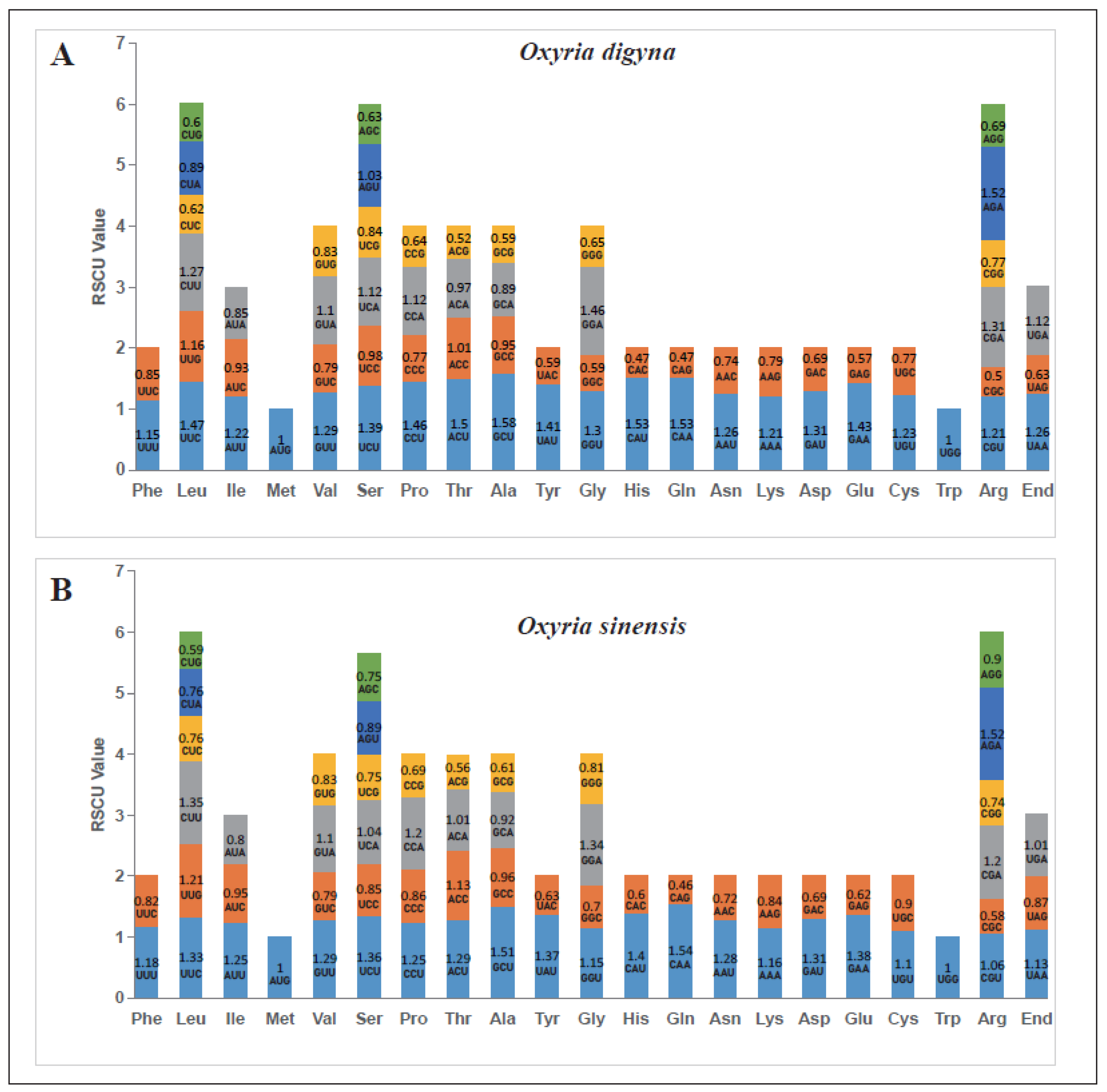
Figure 4.
Schematic diagram of gene transfer between chloroplast and mitogenomes in O. digyna (A) and O. sinensis (B). The orange and green arcs represent the mitogenome and chloroplast genomes, respectively, with the yellow lines between the arcs corresponding to homologous genomic fragments.
Figure 4.
Schematic diagram of gene transfer between chloroplast and mitogenomes in O. digyna (A) and O. sinensis (B). The orange and green arcs represent the mitogenome and chloroplast genomes, respectively, with the yellow lines between the arcs corresponding to homologous genomic fragments.
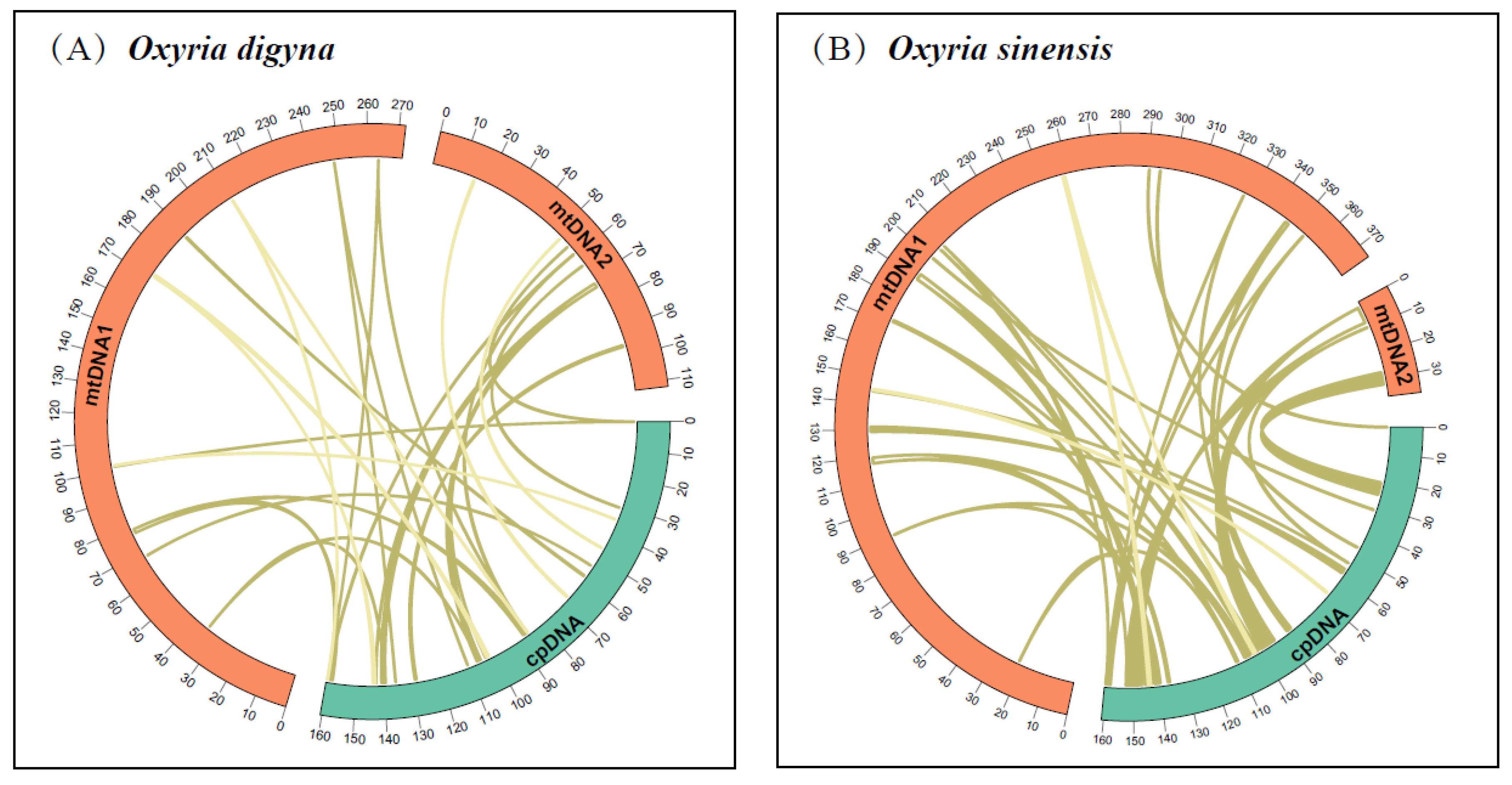
Figure 5.
The phylogenetic relationships (A) and mitogenomes synteny (B) and mitochondrial genes distribution (C) of O. digyna and O. sinensis with the 8 closely related species. The Maximum Likelihood tree was constructed based on the sequences of 20 conserved PCGs. Regarding mitogenome synteny, bars indicate the mitogenomes, and the ribbons display the homologous sequences between the adjacent species. The red areas indicate where the reversal occurred; the grey areas indicate regions of good homology. Common blocks less than 500bp in length are not retained, and regions that fail to have a common block indicate that they are peculiar to the species. Regarding mitochondrial gene distribution, the colors of the boxes indicate the number of copies that exist in the mitogenomes.
Figure 5.
The phylogenetic relationships (A) and mitogenomes synteny (B) and mitochondrial genes distribution (C) of O. digyna and O. sinensis with the 8 closely related species. The Maximum Likelihood tree was constructed based on the sequences of 20 conserved PCGs. Regarding mitogenome synteny, bars indicate the mitogenomes, and the ribbons display the homologous sequences between the adjacent species. The red areas indicate where the reversal occurred; the grey areas indicate regions of good homology. Common blocks less than 500bp in length are not retained, and regions that fail to have a common block indicate that they are peculiar to the species. Regarding mitochondrial gene distribution, the colors of the boxes indicate the number of copies that exist in the mitogenomes.
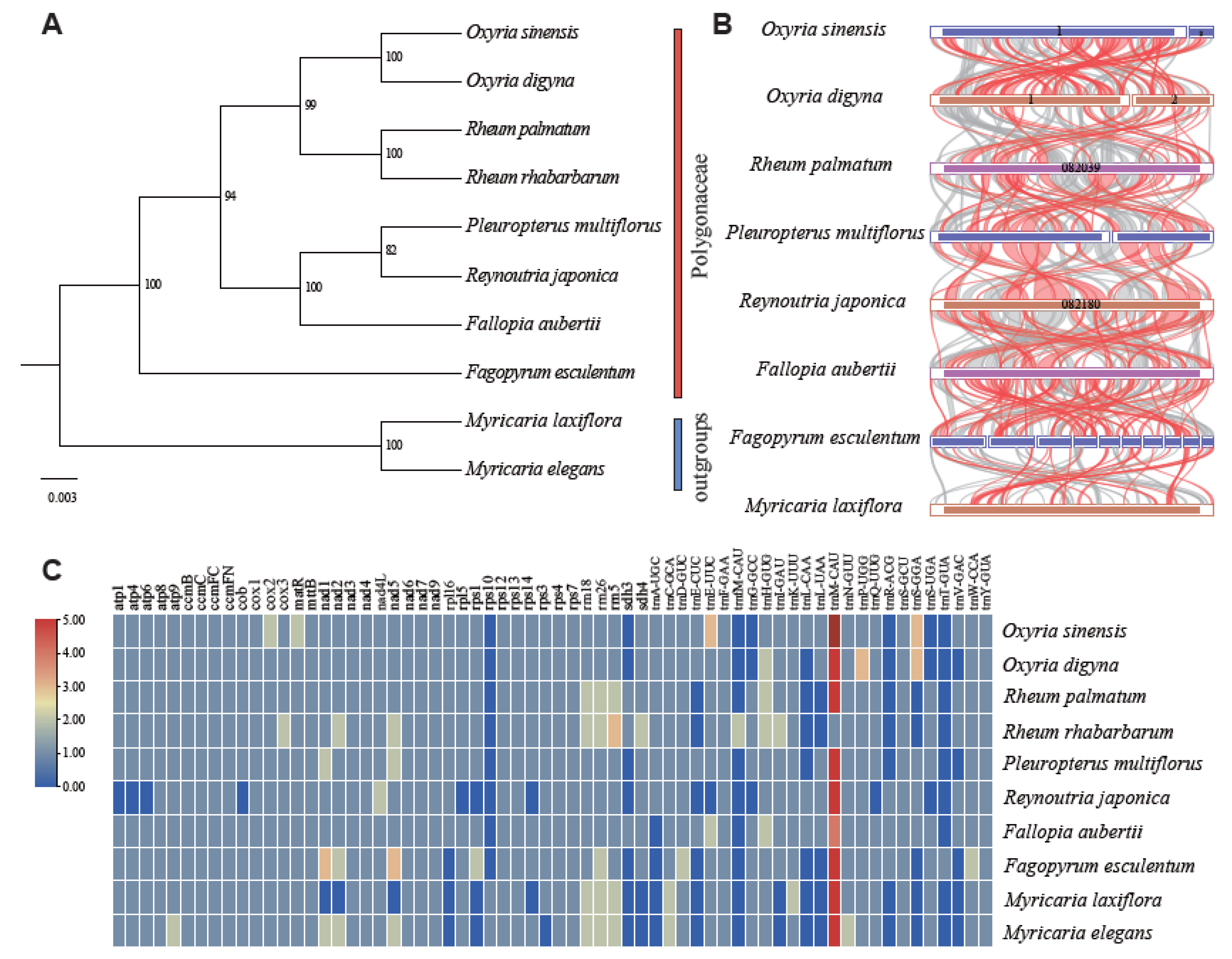
Figure 6.
Boxplots of pairwise dN, dS values and their ratio among 20 mitochondrial genes in O. digyna and O. sinensis and 8 closely related species mitogenomes.
Figure 6.
Boxplots of pairwise dN, dS values and their ratio among 20 mitochondrial genes in O. digyna and O. sinensis and 8 closely related species mitogenomes.
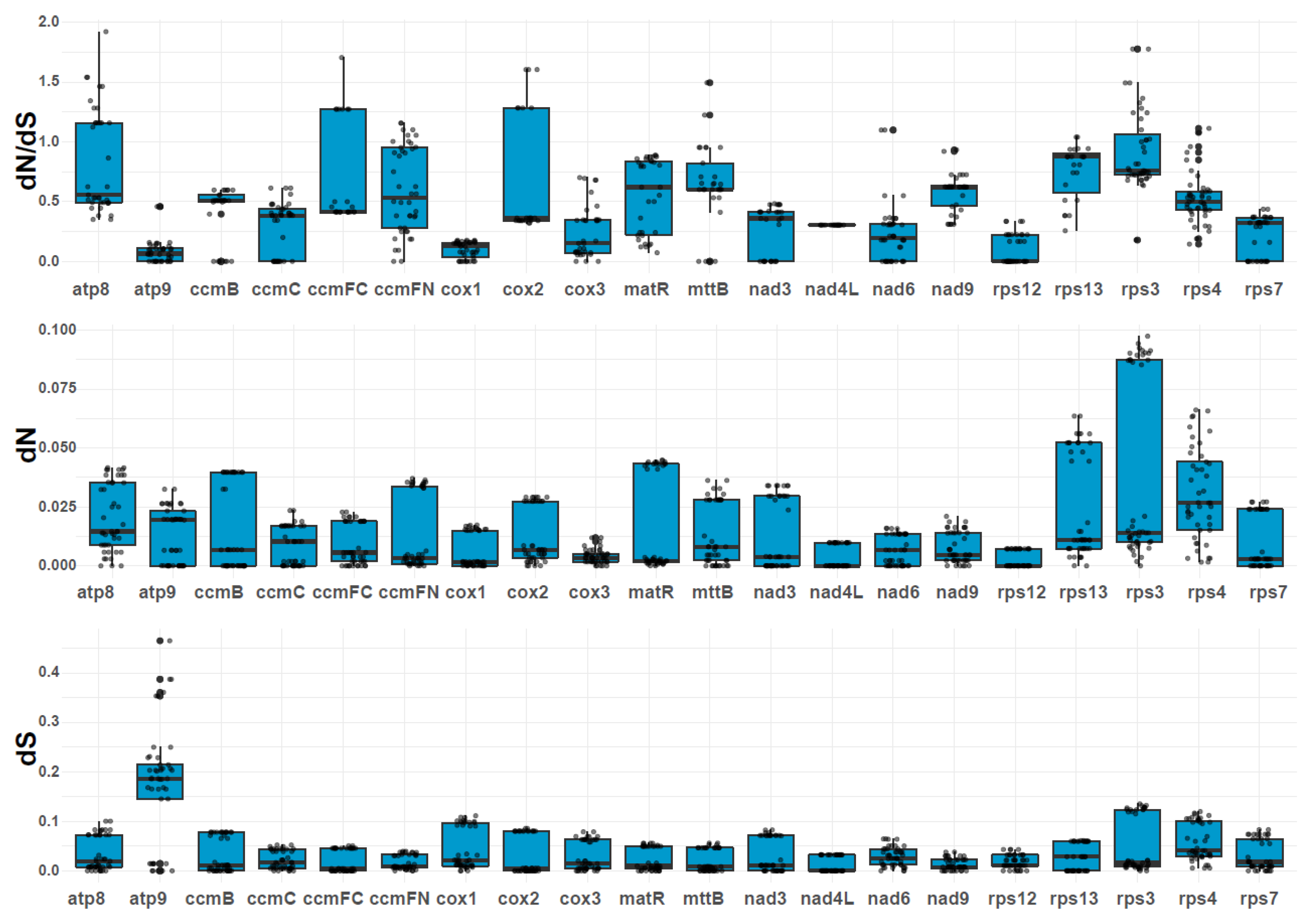

Table 1.
Gene composition of the O. digyna and O. sinensis mitogenomes.
| Group of genes | O. digyna mitogenome | O. sinensis mitogenome | |||
|---|---|---|---|---|---|
| chromosome 1 | chromosome 2 | chromosome 1 | chromosome 2 | ||
| Core genes | Complex I (NADH dehydrogenase) | nad1, nad2, nad3, nad4*, nad5, nad4L, nad6, nad7*, nad9 | nad1, nad2, nad3, nad4*, nad4L, nad5, nad6, nad7*, nad9 | ||
| Cytochrome c biogenesis | cob | cob | |||
| Complex IV (cytochrome c oxidase) | cox1, cox2 | cox3 | cox1, cox2 (2), cox3 | ||
| ATP synthase | atp4, atp6, atp9 | atp1, atp8 | atp1, atp4, atp6, atp8, | atp9 | |
| Maturases | matR | matR (2) | |||
| Cytochrome c biogenesis | ccmB, ccmC, ccmFC*, ccmFN | ccmB, ccmC, ccmFC*, ccmFN | |||
| Transport membrane protein | mttB | mttB | |||
| Variable genes | rpl5, rpl16 | rpl5, rpl16 | |||
| Ribosomal protein | rps1, rps3*, rps4, rps7, rps12, rps13 rps14 | rps1, rps3*, rps4, rps7, rps12, rps13, rps14 | |||
| Complex II (succinate dehydrogenase) | sdh4 | sdh4 | |||
| rRNA genes | Ribosomal RNAs | rrn5, rrn18 | rrn26 | rrn18, rrn26, rrn5 | |
| tRNA | Transfer RNAs | trnC-GCA, trnE-CUC, trnE-UUC, trnF-GAA, trnG-GCC, trnH-GUG, trnK-UUU, trnM-CAU(5), trnN-GUU, trnP-UGG, trnQ-UUG, trnS-GCU, trnS-GGA(2), trnY-GUA | trnA-UGC, trnD-GUC, trnH-GUG, trnL-UAA, trnP-UGG, trnS-GGA, trnW-CCA | trnA-UGC, trnC-GCA, trnD-GUC, trnE-CUC, trnE-UUC, trnF-GAA, trnG-GCC, trnH-GUG, trnK-UUU, trnL-UAA, trnM-CAU(5), trnN-GUU, trnP-UGG, trnQ-UUG, trnS-GCU, trnS-GGA(2), trnV-GAC, trnW-CCA | trnE-UUC(2), trnL-CAA, trnM-CAU, trnS-GAA |
* Labeled intron containing genes, and bracketed numbers represent copy number of each gene.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



