Submitted:
13 October 2024
Posted:
14 October 2024
You are already at the latest version
Abstract
Artificial intelligence (AI) shows great potential in medical diagnosis and drug analysis. Through deep learning technology, AI can automatically extract features from large amounts of complex medical data, significantly improving diagnostic accuracy and drug development efficiency. Especially in drug target discovery and antiviral peptide classification, AI technology can accelerate data processing and prediction, helping researchers identify potential therapeutic molecules more quickly and optimize the drug development process. This study proposes and validates a Deep learn-based model, deep-Avpiden, to improve the classification and discovery efficiency of antiviral peptides (AVPs). By using sequential convolutional networks (TCNs), the model outperforms traditional recurrent neural networks (RNNs) and long short-term memory networks (LSTMs) in capturing long-term dependencies and parallel computing capabilities. In terms of dataset, we used AVPs and non-AVPS samples from multiple databases, totaling 5,414 cleaned and de-weighted peptide sequences for model training after data preprocessing and embedding. The Deep-AVPiden model has been shown to outperform existing advanced classifiers in experiments, and its effectiveness has been verified by accuracy, precision, recall rate, and area under the ROC curve (OC-ROC). In addition, to accommodate computing resource constraints, we propose an optimized version of Deep-AvPIDen (DS), which utilizes Deep separation convolution technology to significantly reduce computing resource consumption. Through the online application platform, researchers can efficiently classify antiviral proteins and discover new AVPs. Future research could further optimize the model's computational efficiency, handle larger data sets, and expand its potential for biomedical problems such as drug combination prediction and new drug discovery.
Keywords:
Deep learning
; Drug target discovery
; Medical diagnosis
; Antiviral peptide
; Artificial intelligence
1. Introduction
Since the 20th century, deep learning theory has undergone extensive research and development. With the progress of science and technology and the significant improvement of computing power, deep learning technology has gradually been widely used in the past decade, including machine translation, target tracking, automatic driving, and other fields. These successful applications not only promote the progress of deep learning technology itself but also provide a new way to solve bioinformatics problems such as drug-target interaction prediction. [1]Deep learning techniques learn higher-dimensional, more abstract models from large-scale biomedical data by making multiple nonlinear transformations of the original input features. This capability enables the application of deep learning in the prediction of drug-target interactions. Drug target interaction prediction is an important task, which involves extracting discriminative features from the original characterization of existing drugs and targets and predicting the interactions between drugs and targets through these features.
In recent years, drug target interaction prediction methods based on deep learning technology have gradually attracted the attention of researchers. These methods have made significant improvements in the prediction of drug-target association problems, but they also face new challenges. Specifically, drug target interaction prediction is often viewed as a binary classification problem that requires extracting valid molecular fingerprint descriptors or features from complex and diverse raw data.
Figure 1.
Enzymes as Drug Targets.
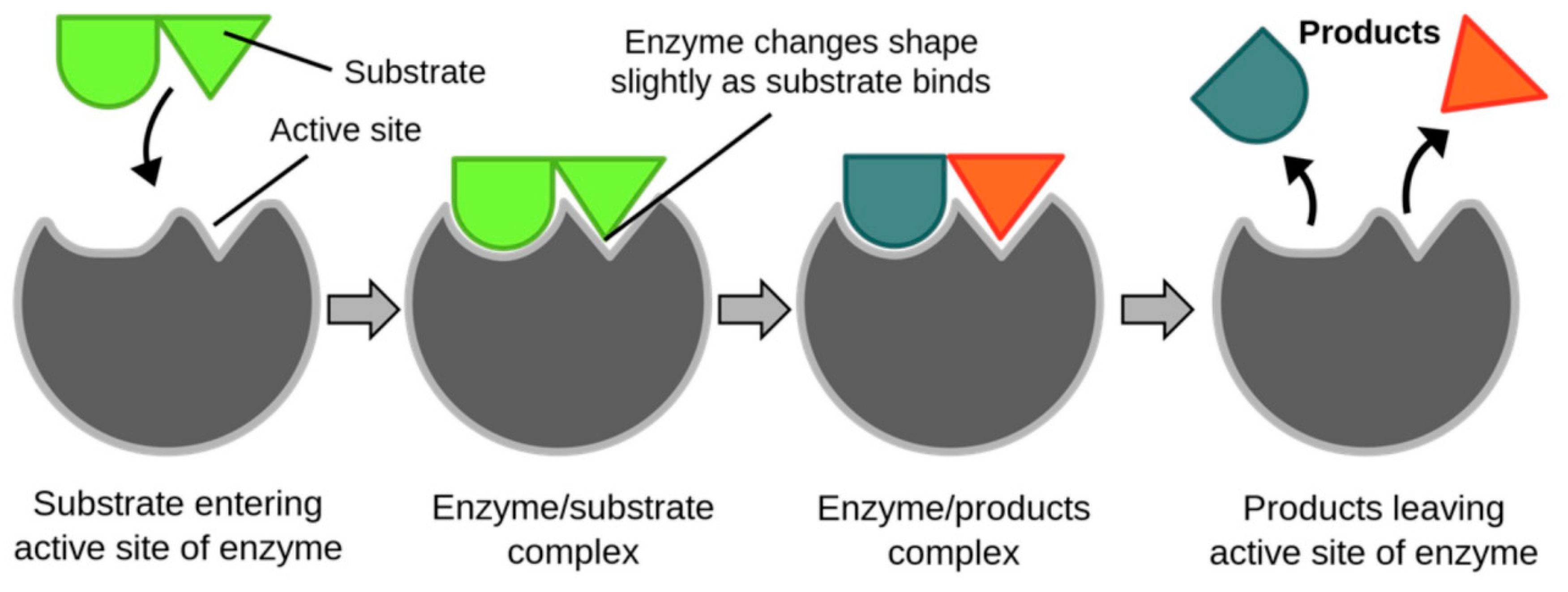
Enzymes are a type of protein and essential biomolecules within the human body that act as biological catalysts to facilitate specific chemical reactions. [2]They speed up these reactions while remaining unchanged. Substrates are the molecules that enzymes interact with, and when they bind to the enzyme’s active site, they undergo a reaction to form new molecules known as products. The enzyme secures the substrate in its active site through various interactions, including hydrogen bonding, ionic bonding, and van der Waals forces, enabling a successful reaction to occur.
The other function of the enzyme is to provide functional groups that will react with the substrates to carry out the necessary chemical reactions that support all our life processes. This puts forward high requirements on how to design effective deep learning structures. In addition, compared to traditional machine learning methods (such as support vector machines, logistic regression, etc.) with better interpretability, deep learning methods are often referred to as "black boxes" and their predictive results are less interpretable. Although deep learning models excel in predictive performance, their lack of interpretability may limit their widespread adoption in industrial applications, particularly in the improvement and design of new drugs.
Based on deep learning technology, this paper aims to mine and integrate known biological experimental data from the perspective of bioinformatics to predict potential drug target interactions by constructing reasonable models. This paper will focus on solving the problems existing in the prediction of drug target interactions, and in-depth analysis of the interpretability of deep learning models to improve the accuracy of drug screening. This research will provide a scientific basis for drug development and is expected to reduce research and development costs and promote the development of biomedicine.
2. Related Work
2.1. Prediction of Drug-Target Interactions
Drug-target interaction prediction task is a typical binary problem. Specifically, if there is an actual interaction between the drug and the target, the sample is labeled 1;[3] If it does not exist, it is marked as 0. With the rapid development of computer technology and algorithms, drug-target interaction screening based on computational biology has been paid more and more attention. The high concurrent processing capacity and powerful computing power of the computer significantly shorten the screening time of drug-target interaction, and the computational method provides a more efficient alternative to the full biomedical experimental verification of traditional methods.
At present, the research results of drug-target interaction prediction can be mainly divided into three categories: ligand-similarity-based methods, molecular docking-based methods, and chemical genome-based methods. Ligand-similarity-based methods make predictions by comparing chemical structural similarities between drugs and targets. [4]The molecule-docking approach evaluates the likelihood of binding by simulating the binding process between the drug and the target. Chemogenome-based approaches involve analyzing the effects of drugs on the genome to predict their targets.
Recent advances in cancer treatment indicate that drug combination therapy has become the standard clinical strategy for the treatment of complex diseases such as cancer. The rationale for drug combination is that multiple drugs target multiple targets, pathways, and cellular metabolic processes in the disease, thereby reducing the toxicity and development of resistance to single-drug therapy. For example, anastrozole combined with fulvestrant in the treatment of metastatic breast cancer, amiloride combined with hydrochlorothiazide in the treatment of hypertension, glibenclamide combined with metformin in the treatment of type 2 diabetes, etc.[5,6,7], all demonstrate the effectiveness of combination therapy. However, although traditional clinical trial methods can provide intuitive drug combination effects, the trial design is complex and inefficient due to the possible side effects of new drugs and the unpredictability of therapeutic effects.
Figure 2.
A deep learning-based method for predicting novel drug-target interactions.
To accelerate the discovery of drug combinations, large-scale screening techniques such as multiple screening and high-throughput screening were introduced. These methods detect activity through models at the cellular or molecular level and accumulate a large amount of data for drug combinations. However, the exponential growth of the search space for large amounts of data makes the screening process difficult and time-consuming. Therefore, many computational methods and predictive models have been proposed to shorten the search time.
In recent years, computational methods have been widely used in drug combination prediction. Machine learning methods have become the focus of research, but there are also systems biology methods, dynamic models, and random search methods. [8]The systems biology approach focuses on analyzing biological networks, and Yu et al. use network embedding to predict the feasibility of drug combinations. The dynamic model uses equations to simulate the dynamic changes of network nodes, and Sun et al. analyze the efficacy of signal transduction networks through pharmacological perturbations. Random search algorithms such as MACS[9] screen drug combinations by search algorithm and fitness function. Although each of these methods has its advantages and disadvantages, the emergence of machine learning, especially deep learning methods, has provided a new direction for drug-target interaction prediction and has richer application potential.
2.2. Prediction Method of Drug-Target Interaction based on Traditional Machine Learning
In recent years, drug combination prediction has seen the application of various computational methods, with machine learning being the most widely used. Additional approaches, such as systems biology, dynamic models, and stochastic search methods, have their niche applications while also facing certain limitations; many of these methods often incorporate machine learning for enhanced predictions. Systems biology, for instance, centers on the control and analysis of biological networks—Yu et al. harnessed these networks for predicting drug-target interactions, illustrating the potential of network embedding in drug combination prediction. [10,11]Cheng and colleagues introduced classic network proximity calculations, which quantify the relationships between drug targets and disease proteins in human protein-protein interaction networks by assessing the topological connections between drug modules (subnetworks of drug targets) and cancer modules (subnetworks of cancer proteins), identifying six unique topological categories for all potential drug-drug-disease combinations.Dynamic models use differential equations to simulate the dynamic changes of network nodes. Sun et al. analyzed the impact of pharmacological perturbations on signal transduction networks, examining the role of crosstalk intensity in switching between drug sensitivity and resistance. However, most dynamic definitions of biological networks lack reliable explanations.
The accuracy of mathematical methods relies on underlying model assumptions. Li et al. proposed the Combination Drug Assembler (CDA), which employs hypergeometric tests for pathway gene set enrichment analysis to assess signal pathway expression patterns and drug set patterns. Stochastic search algorithms explore iterative drug combinations and measurements within the possibility space. Zinner et al. introduced the Medical Algorithm Combination Screening (MACS)[12], which integrates search algorithms with a novel fitness function based on inhibition levels and drug quantity. However, due to computational time and space costs, these methods are typically applicable only to small datasets. Compared to these algorithms, machine learning and the recently emerging deep learning methods offer more advanced capabilities. Traditional machine learning methods in drug-target interaction prediction mainly rely on supervised learning to predict the interaction between drugs and targets by training their molecular descriptors (features). [13]These methods compute large amounts of biomedical data and utilize statistical models to make predictions about potential drug-target interactions. Commonly used traditional machine learning methods include support vector machine (SVM), logistic regression (LR), random forest (RF), etc., each of which has its characteristics and has been applied to drug screening and target identification.
- PredAntiCoV: PredAntiCoV is a two-stage classifier tool designed to solve the prediction problem of antiviral drugs (AVPs). The first stage uses the amino acid composition, dipeptide, physicochemical properties, and other characteristics of the drug to predict whether the drug is an antiviral drug through the random forest (RF) [14]model. The second phase further predicts whether these drugs have the potential to fight specific viruses. The tool uses a variety of undersampling methods to deal with data imbalances and analyzes the importance of features with p-values to help optimize predictive performance.
- AVPIden: AVPIden is a two-stage predictive model focused on identifying antiviral drugs and their targets. The first stage predicts whether the drug is an antiviral drug (AVP)[15], and the second stage predicts the targeting effect of the drug against different viruses through multi-task learning. The model not only supports the prediction of multiple viruses but also explains the influence of biometrics on the prediction results of the model through the Shapley value, which has important significance for understanding the prediction mechanism of the model and improving drug design.
- CIAntiCoV: The CIAntiCoV tool analyzes existing antiviral drug prediction methods and integrates multiple prediction models to improve the prediction accuracy of drug-target interactions. By comparing and analyzing the advantages and disadvantages of different approaches, CIAntiCoV helps to identify effective drug combinations and optimize the drug screening process.
Traditional machine learning methods achieve efficient analysis of complex biological data through feature engineering and model training in drug-target interaction prediction. However, these methods still face challenges when dealing with large-scale data and high-dimensional features, such as the complexity of feature selection, the interpretability issues of the model, and the difficulty of dealing with data imbalances. Nevertheless, these approaches have shown significant advantages in drug development, such as shortening screening cycles, reducing experimental costs, and providing valuable information for the discovery of new drugs. [16]However, in order to further improve the accuracy and efficiency of prediction, the combination of advanced technologies such as deep learning is still an important direction for future development.
2.3. Application of Deep Learning-Based Methods to the Prediction of Drug-Target Interactions
In the field of drug-target interaction prediction, deep learning methods have significant advantages. First, deep learning models can automatically extract complex features through their hierarchical structure, thus reducing the need for manual feature engineering. Whereas traditional shallow learning methods rely on manual selection and extraction of features, deep learning automatically learns and identifies high-level features from raw data through architectures such as convolutional neural networks (CNNS), recurrent neural networks (RNN)[17], and Transformer. This ability is critical for processing complex drug and target data, which often have complex patterns and relationships.
Second, deep learning models often provide better predictive accuracy. Compared to shallow learning algorithms, deep learning is able to capture more subtle patterns when processing large-scale data, thus improving prediction performance. This high accuracy is particularly applicable in the prediction of drug-target interactions, where small differences in characteristics can affect the final prediction results. Deep learning models also excel in multimodal data processing, integrating data from different sources, such as the chemical structure of a drug and sequence information of a target, to improve the comprehensiveness and accuracy of predictions.
However, deep learning approaches also face some challenges. One of the main problems is the high data requirements. The training of deep learning models typically requires large amounts of data, and drug-target interaction datasets tend to be small, which can lead to undertraining of the models, which affects predictive performance. In addition, the training process of deep learning models often requires a lot of computing resources and time, especially with high-performance servers and graphics processing units (Gpus), which can be a bottleneck in resource-limited environments.
In a study applying deep learning to ACVP [18](antiviral peptide) prediction, ENNAVIA and iACVP demonstrated practical applications of these techniques. ENNAVIA uses a transfer learning approach to improve model performance through pre-training and fine-tuning steps and optimizes parameters using grid search strategies. Despite its superior performance on independent test sets, it has strict limits on the input data length, limiting its ability to identify some potential ACVPs. In contrast, iACVP integrates traditional machine learning and deep learning methods, including Transformer, CNN, and BiLSTM, and combines different encodings and k-mer values to optimize the model. This integrated approach makes up for the shortcomings of a single model and can handle data of different lengths, improving the accuracy and robustness of the prediction.
Overall, deep learning provides powerful feature learning and data processing capabilities in drug-target interaction prediction. [19]Despite the challenges of data volume and computational resources, techniques such as ensemble learning and model generation can further improve the performance of models, providing more accurate predictive tools for drug development and disease treatment.
3. Methodology
With the rapid development of deep learning and artificial intelligence technology, drug research and development, especially in the field of drug-target discovery, has ushered in unprecedented changes. While traditional drug development often relies on experience and laboratory screening, deep learning technology can significantly improve the efficiency and accuracy of new drug discovery by processing large amounts of data and complex pattern recognition. In particular, antimicrobial peptides (AMPs)[20] and antiviral peptides (AVPs), as new therapeutic agents, have attracted wide attention in recent years because of their effective ability against traditional antibiotic-resistant pathogens.
This research focuses on developing and validating a novel Deep learning-based model, deep-Avpiden, which aims to improve the classification and discovery efficiency of AVPs. We use temporal convolutional networks (TCNs) for sequence modeling. This network architecture has significant advantages over traditional recurrent neural networks (RNNs) and long short-term memory networks (LSTMs) in capturing long-term dependencies and parallel computing power in sequence data. TCNs can optimize the drug-target identification process by reducing computational resource consumption and improving prediction accuracy when processing complex biological sequence data.
In addition, in order to meet the requirements of the traditional model in computing resources, we also propose an optimized version of the model, Deep-AVPiden (DS)[21]. The model utilizes deep separation convolution technology to significantly reduce computing resource consumption during training and deployment. By building an online application platform that connects the functions of these models to the needs of laboratory researchers, we have achieved rapid classification of anti-viral proteins and discovery of AVPs. The platform not only improves the efficiency of data processing, but also provides a convenient tool for researchers to conduct auxiliary medical research.
In our experiments, we compared Deep-AVPiden with existing state-of-the-art classifiers, and the results show that our method outperforms other models in performance. In addition, through statistical analysis and performance evaluation of the model, we verified its effectiveness in predicting antiviral proteins in different organisms. This study not only provides new methods and tools for drug discovery, but also demonstrates the potential for deep learning and artificial intelligence to be widely used in the field of assisted medicine.
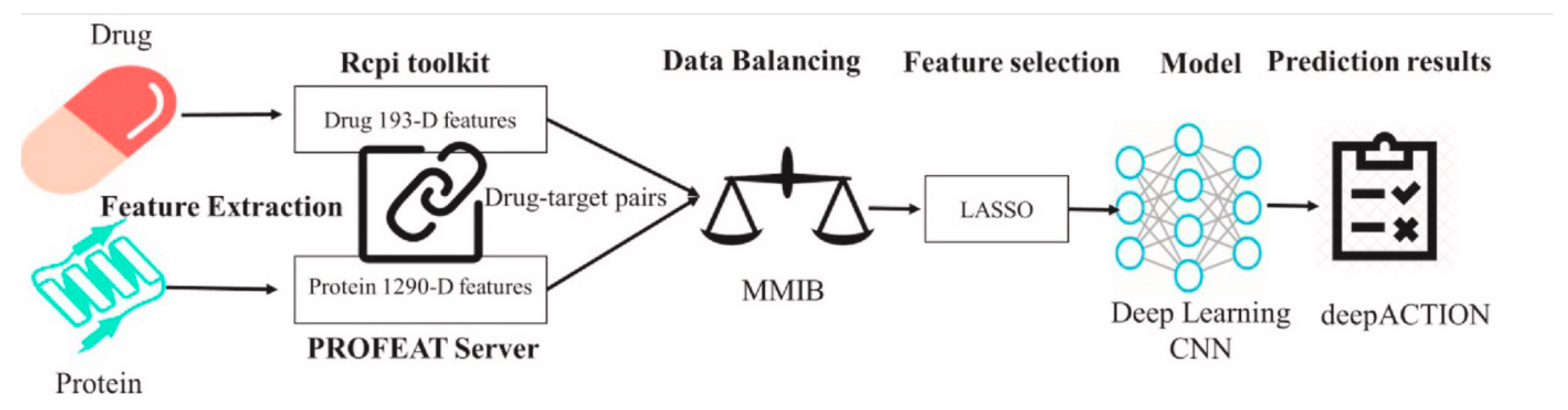
3.1. Dataset
The dataset used in this study contains samples of both antiviral peptides (AVPs) and non-AVPs, where each sample is a string of letters made up of standard amino acids. Data on antiviral peptides came from multiple databases, including AVPdb, HIPdb, starPep Database, DRAMP, and SATPdb. Non-antiviral peptides were obtained from Swiss-Prot database and AVPdb. The initial numbers of AVPs and non-AVPs collected were 10,500 and 9,000, respectively. To ensure data quality, we cleaned the data to exclude non-standard amino acids (e.g. B, J, O, U, X, Z) and peptide sequences less than 5 or greater than 50 amino acids in length. Subsequently, a CD-HIT program was used to de-duplicate both AVPs and non-AVPs at a threshold of 0.9 to filter out similar sequences. In order to eliminate the influence of inter-class sample quantity imbalance on model performance, 699 non-AVPs were randomly removed. The final dataset contained 5,414 peptide sequences, including 2,707 AVPs and 2,707 non-AVPs. This data is further divided into training sets (70%), test sets (15%), and validation sets (15%).
Figure 3.
Te layout of the proposed work.
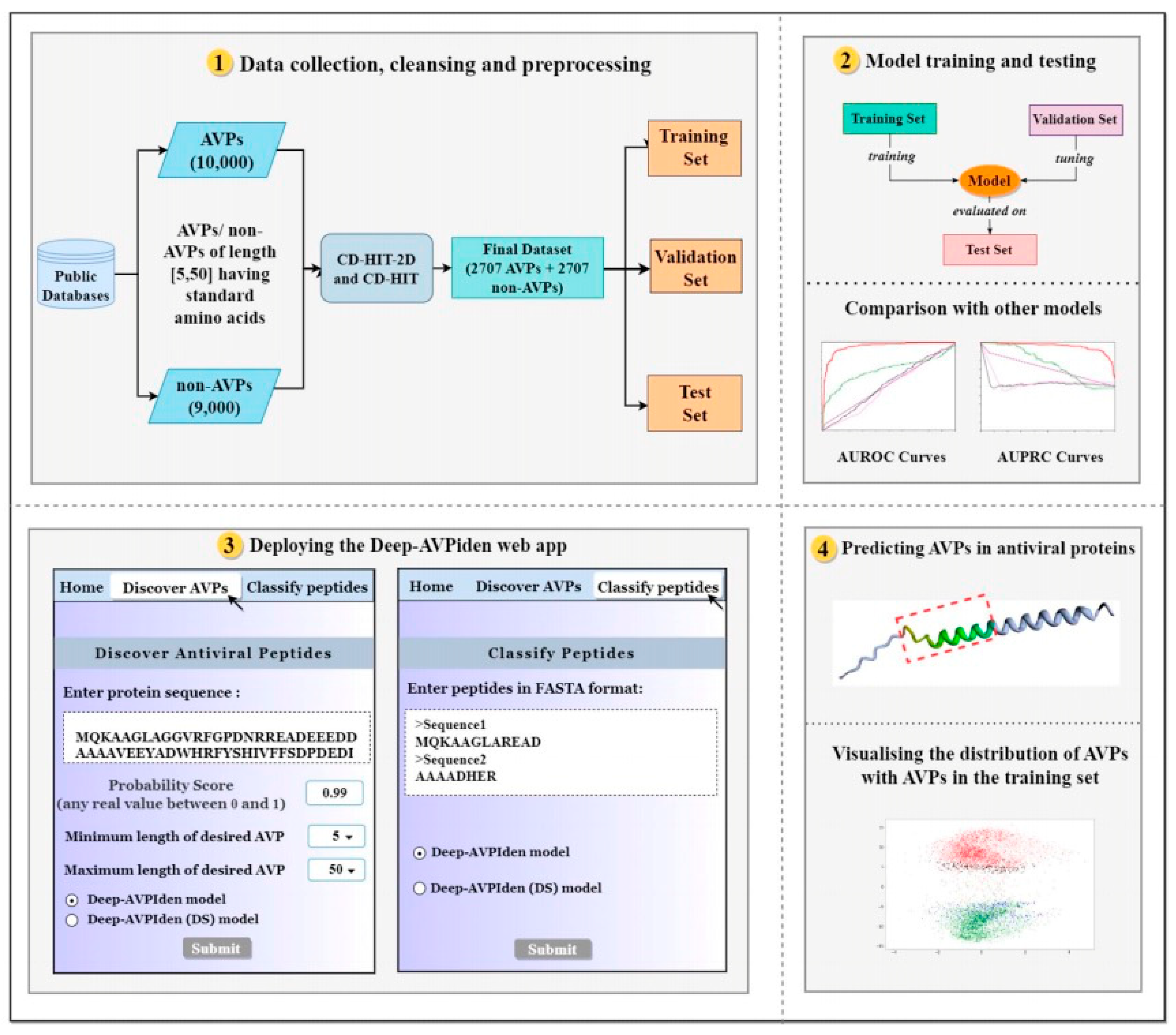
3.2. Data Pre-Processing
In order to convert the data into a format understandable by the computer, we first convert the amino acid sequence into a string of numbers, achieved through a one-to-one character-to-integer mapping. Since the length of strings in the data set varies, strings in the length [5,49] are normalized by padding from zero to length 50 in order to unify the format. The set of numerical strings after this processing is divided into training set, verification set and test set. The training set is then used to generate the word embedding matrix, a detailed process described in the "Proposed Model" section.
In TCN, the input x is processed by a convolution operation ∗d and a one-dimensional filter f of size k. skip connections can be used in TCN blocks, which prevents gradient disappearance/explosion problems and helps avoid degradation issues and overfitting. Each residual block consists of two one-dimensional convolution layers and introduces a jump connection by adding the input and output of the block. In this way, an ordinary TCN block is transformed into a residual TCN block whose output (y) can be calculated from the given equation.
In the TCN block, F(x) represents the output from the final layer, followed by an activation function such as ReLU to introduce non-linearity. Skip connections are utilized to enable the residual block to learn the identity function of the input, which can contribute to stabilizing the learning process in deep neural networks.Depth-wise Separable Convolutions (DwSCs) emerged from the research community's interest in creating smaller and more efficient models. Before this, pre-trained models were either compressed or had shallow networks. DwSCs, introduced in studies and later used to train deep ConvNets, factorize a standard convolution operation into two stages. First, depth-wise convolutions apply a single filter to each input channel separately, reducing the computational load. Second, point-wise convolutions use a 1x1 convolutional layer with multiple filters to combine the outputs of the depth-wise convolutions. This factorization significantly reduces the number of training parameters and computations, leading to faster training times and reduced space usage, making models suitable for resource-constrained platforms like mobile devices.
3.3. Proposed model
As depicted in Figure 4, the Deep-AVPiden model includes several integral layers, starting with the embedding layer that employs the skip-gram algorithm to create a word embedding matrix for the 20 standard amino acids. This process converts numerical strings into a feature matrix of dimensions (50,512), with 50 indicating the length of the string and 512 being the fixed-length vector that represents each amino acid.
Next, the Spatial Dropout layer performs regularization by dropping entire columns from the feature matrix, rather than individual elements, to handle high correlations between frames. This is followed by dilated causal Temporal Convolutional Network (TCN) blocks with two parallel 1D-CONV layers, where filters vary in size, and ReLU activation is applied. Batch normalization layers are used for stability, with dilation factors increasing by powers of 2.
The final model components include a Global Average Pooling (GAP) layer that averages the features from each TCN block, a concatenation layer that merges these outputs, and a dense layer with 64 units and dropout for further processing. The output layer uses a sigmoid function to produce a probability score between 0 and 1, classifying peptides as AVPs if the score is 0.5 or higher. The model also includes an alternative design using depth-wise separable convolutions for efficiency, comparing performance against the standard convolution-based approach.
4. Results and Discussions
4.1. Experimental Design
In this study, deep learning models are trained and evaluated in detail. The training used high-performance computing nodes equipped with a 2.4GHz Intel Xeon Skylake 6148 CPU processor, 192 GB RAM, and an NVIDIA V100 GPU. We used Python for programming and utilized the Keras framework (with TensorFlow as the back end) and the Keras-TCN library to build and train the model.
To verify the effect of the model, we compared it to several advanced classifiers, including DeepAVP, AVPIden, IMP-CA2L, ENNAVIA, Meta-iAVP, PreTP-Stack, and iACVP. These models are treated specifically to ensure the fairness of the test set. For example, these models are compared only after removing duplicate and homologous sequences to avoid bias in performance evaluations. In particular, the test sets of the ENNAVIA and AVPIden models are restricted to specific length intervals; for example, ENNAVIA classifies only sequences with lengths in between, while AVPIden classifies sequences with lengths in between. The iACVP model requires the sequence to contain more than five amino acid residues. Therefore, in order to accurately evaluate the performance of these models, we adjusted the test set accordingly to meet these requirements.
In addition, when using the IPAMP-CA2L model, we found that the model sometimes failed to accurately label the type of function of AMP (such as antibacterial or antiviral). To address this issue, we removed instances from the test set when reporting results that could not clearly label the type of function to avoid uncertainty in the results. In this way, we ensure the accuracy of experimental results and a fair comparison of model performance.
The models’ performance was evaluated using key metrics such as accuracy, precision, and the area under the receiver operating characteristic curve (AUC-ROC), which are computed using four values: true positives (TP, correctly identified AVPs), false positives (FP, non-AVPs incorrectly identified as AVPs), true negatives (TN, correctly identified non-AVPs), and false negatives (FN, AVPs incorrectly identified as non-AVPs). A thorough analysis revealed that the Deep-AVPiden model outperforms other models significantly across these performance indicators. The formula is as follows:
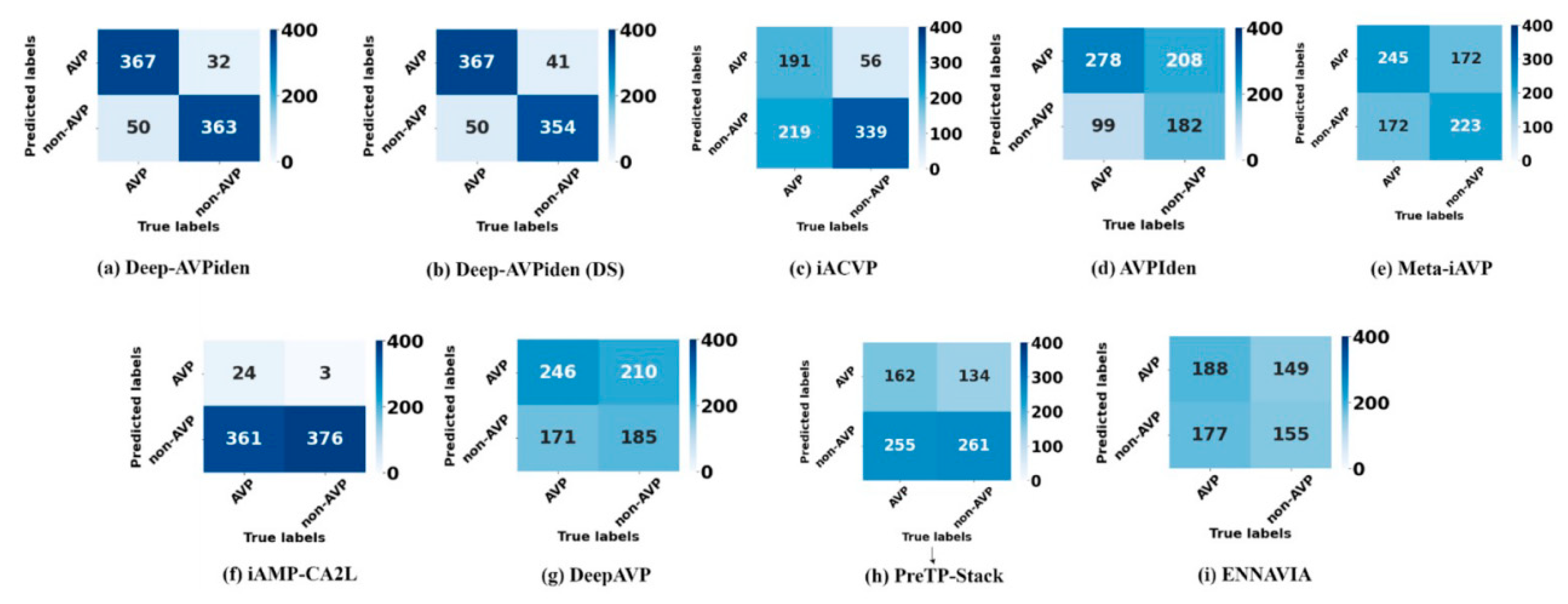
We consider causal and non-causal temporal convolutional networks (TCNs) during model construction. Although the performance difference between the two is small, as Table 1 shows, the model using causal convolution outperforms the non-causal convolution model in terms of average accuracy, recall, and OC-ROC. Therefore, we chose causal TCNs to construct the Deep-AVPiden model. Table 2 shows the performance results of various advanced models, including Deep-AVPiden and Deep-AVPiden (DS). It is evident that these two models significantly outperform other models on all performance indicators. The confusion matrix in Figure 3 shows that the proposed model provides more true cases (TPs) and true negative cases (TNs) and fewer false positive cases (FPs) and false negative cases (FNs) than other models.
Figure 3.
Confusion matrices obtained for various models including Deep-AVPiden on the test set.
The Deep-AVPiden web app, accessible at https://deep-avpiden.anvil.app, facilitates the prediction of antiviral peptides (AVPs) across various organisms such as mammals, plants, and fish. Utilizing this tool, we have identified several AVPs within proteins from distinct families, including ribosome-inactivating proteins (RIPs), which can halt protein synthesis, RNA-binding proteins (RBPs) that inhibit RNA virus replication, and Dicer-like proteins (DCLs) that engage in RNA silencing through the cleavage of double-stranded RNA. Notably, plant-derived AVPs like pokeweed antiviral protein (PAP) and trichosanthin have demonstrated efficacy against viruses such as Potato virus Y (PVY) and Cucumber mosaic virus (CMV).Interferons (IFNs), classified into type I, II, and III based on receptor structure, are known antiviral proteins used in treating infections like hepatitis C and HIV. We also analyzed these proteins using the online tool https://heliquest.ipmc.cnrs.fr, which visualizes hydrophobic moments in alpha-helical peptides. A higher hydrophobic moment indicates better antiviral potential. Our findings suggest that the discovered AVPs possess a high hydrophobic moment, implying strong antiviral activity.
Furthermore, the AVPs identified are significantly shorter than their parent proteins, highlighting that the tool effectively pinpoints the core antiviral regions. We performed clustering analysis with CD-HIT and used isometric mapping to compare the discovered AVPs with those in our training set. The 2D visualization shows that the predicted AVPs closely resemble the training set's AVPs, suggesting that the identified peptides are likely to have substantial antiviral activity. Future laboratory synthesis and validation will confirm their effectiveness.
5. Conclusions
1. Summarize the application of deep learning in the prediction of drug-target interaction
Deep learning techniques show significant advantages in the prediction of drug-target interactions. By automatically extracting and learning complex features, these methods reduce the need for manual feature engineering and provide highly accurate prediction results. Despite the challenges of large data volumes and high computational resource requirements, Deep learning models, such as Deep-Avpiden, significantly improve the accuracy and efficiency of predictions through their powerful feature learning and data processing capabilities, thus providing a more reliable tool for drug development and biomedical research.
2. Compare the effects of traditional and deep learning methods
Compared with traditional machine learning methods, deep learning methods show better performance in drug-target interaction prediction. Although traditional methods have certain advantages when dealing with small-scale data, deep learning's automatic feature extraction ability and high prediction accuracy make it a better choice when facing high-dimensional features and large-scale data. The experimental results show that the Deep-AVPIDen model exceeds other existing advanced classifiers in many performance indicators, which proves the practical application value of Deep learning in this field.
3. Future research direction and application potential
Although the current study shows that deep learning has achieved remarkable results in drug-target interaction prediction, there is still room for further improvement. Future research could focus on optimizing the model's computational efficiency, processing larger data sets, and improving the model's interpretability. At the same time, applying deep learning methods to more biomedical problems, such as drug combination prediction and new drug discovery, will further expand its application potential and promote progress in the field of biomedicine.
References
- Katsila, T., Spyroulias, G. A., Patrinos, G. P., & Matsoukas, M. T. (2016). Computational approaches in target identification and drug discovery. Computational and structural biotechnology journal, 14, 177-184.
- Colburn, W. A. (2003). Biomarkers in drug discovery and development: from target identification through drug marketing. The Journal of Clinical Pharmacology, 43(4), 329-341.
- Yu, K., Bao, Q., Xu, H., Cao, G., & Xia, S. (2024). An Extreme Learning Machine Stock Price Prediction Algorithm Based on the Optimisation of the Crown Porcupine Optimisation Algorithm with an Adaptive Bandwidth Kernel Function Density Estimation Algorithm.
- Li A, Zhuang S, Yang T, Lu W, Xu J. Optimization of logistics cargo tracking and transportation efficiency based on data science deep learning models. Applied and Computational Engineering. 2024 Jul 8;69:71-7.
- Xu, J., Yang, T., Zhuang, S., Li, H. and Lu, W., 2024. AI-based financial transaction monitoring and fraud prevention with behaviour prediction. Applied and Computational Engineering, 77, pp.218-224.
- Ling, Z., Xin, Q., Lin, Y., Su, G. and Shui, Z., 2024. Optimization of autonomous driving image detection based on RFAConv and triplet attention. Applied and Computational Engineering, 77, pp.210-217.
- Yang, T., Li, A., Xu, J., Su, G. and Wang, J., 2024. Deep learning model-driven financial risk prediction and analysis. Applied and Computational Engineering, 77, pp.196-202.
- Zhou, Y., Zhan, T., Wu, Y., Song, B., & Shi, C. (2024). RNA Secondary Structure Prediction Using Transformer-Based Deep Learning Models. arXiv preprint arXiv:2405.06655.
- Liu, B., Cai, G., Ling, Z., Qian, J., & Zhang, Q. (2024). Precise Positioning and Prediction System for Autonomous Driving Based on Generative Artificial Intelligence. Applied and Computational Engineering, 64, 42-49.
- Cui, Z., Lin, L., Zong, Y., Chen, Y., & Wang, S. (2024). Precision Gene Editing Using Deep Learning: A Case Study of the CRISPR-Cas9 Editor. Applied and Computational Engineering, 64, 134-141.
- Xiao, J., Wang, J., Bao, W., Deng, T., & Bi, S. (2024). Application progress of natural language processing technology in financial research. Financial Engineering and Risk Management, 7(3), 155-161.
- Li, J., Wang, Y., Xu, C., Liu, S., Dai, J., & Lan, K. (2024). Bioplastic derived from corn stover: Life cycle assessment and artificial intelligence-based analysis of uncertainty and variability. Science of The Total Environment, 174349.
- Zhang, X., 2024. Machine learning insights into digital payment behaviors and fraud prediction. Applied and Computational Engineering, 67, pp.61-67.
- Zhang, X. (2024). Analyzing Financial Market Trends in Cryptocurrency and Stock Prices Using CNN-LSTM Models.
- Li, J., Wang, Y., Xu, C., Liu, S., Dai, J., & Lan, K. (2024). Bioplastic derived from corn stover: Life cycle assessment and artificial intelligence-based analysis of uncertainty and variability. Science of The Total Environment, 174349.
- Xiao, J., Wang, J., Bao, W., Deng, T., & Bi, S. (2024). Application progress of natural language processing technology in financial research. Financial Engineering and Risk Management, 7(3), 155-161.
- Liu, S., Yan, K., Qin, F., Wang, C., Ge, R., Zhang, K., Huang, J., Peng, Y. and Cao, J., 2024. Infrared Image Super-Resolution via Lightweight Information Split Network. arXiv preprint arXiv:2405.10561.
- Zhu, Y., Yu, K., Wei, M., Pu, Y., & Wang, Z. (2024). AI-Enhanced Administrative Prosecutorial Supervision in Financial Big Data: New Concepts and Functions for the Digital Era. Social Science Journal for Advanced Research, 4(5), 40-54.
- Zhao, Fanyi, et al. "Application of Deep Reinforcement Learning for Cryptocurrency Market Trend Forecasting and Risk Management." Journal of Industrial Engineering and Applied Science 2.5 (2024): 48-55.
- Zhang, M., Yuan, B., Li, H., & Xu, K. (2024). LLM-Cloud Complete: Leveraging Cloud Computing for Efficient Large Language Model-based Code Completion. Journal of Artificial Intelligence General science (JAIGS) ISSN: 3006-4023, 5(1), 295-326.
- Li, P., Hua, Y., Cao, Q., & Zhang, M. (2020, December). Improving the Restore Performance via Physical-Locality Middleware for Backup Systems. In Proceedings of the 21st International Middleware Conference (pp. 341-355).
- Zhou, S., Yuan, B., Xu, K., Zhang, M., & Zheng, W. (2024). THE IMPACT OF PRICING SCHEMES ON CLOUD COMPUTING AND DISTRIBUTED SYSTEMS. Journal of Knowledge Learning and Science Technology ISSN: 2959-6386 (online), 3(3), 193-205.
- Shang, F., Zhao, F., Zhang, M., Sun, J., & Shi, J. (2024). Personalized Recommendation Systems Powered By Large Language Models: Integrating Semantic Understanding and User Preferences. International Journal of Innovative Research in Engineering and Management, 11(4), 39-49.
- Ju, Chengru, and Yida Zhu. "Reinforcement Learning Based Model for Enterprise Financial Asset Risk Assessment and Intelligent Decision Making." (2024).
- Yu, Keke, et al. "Loan Approval Prediction Improved by XGBoost Model Based on Four-Vector Optimization Algorithm." (2024).
- Zhou, S., Sun, J., & Xu, K. (2024). AI-Driven Data Processing and Decision Optimization in IoT through Edge Computing and Cloud Architecture.
- Sun, J., Zhou, S., Zhan, X., & Wu, J. (2024). Enhancing Supply Chain Efficiency with Time Series Analysis and Deep Learning Techniques.
- Wang, S., Zheng, H., Wen, X., Xu, K., & Tan, H. (2024). Enhancing chip design verification through AI-powered bug detection in RTL code. Applied and Computational Engineering, 92, 27-33.
- He, Z., Shen, X., Zhou, Y., & Wang, Y. (2024, January). Application of K-means clustering based on artificial intelligence in gene statistics of biological information engineering. In Proceedings of the 2024 4th International Conference on Bioinformatics and Intelligent Computing (pp. 468-473).
- Gong, Y., Zhu, M., Huo, S., Xiang, Y., & Yu, H. (2024, March). Utilizing Deep Learning for Enhancing Network Resilience in Finance. In 2024 7th International Conference on Advanced Algorithms and Control Engineering (ICAACE) (pp. 987-991). IEEE.
- Strachan, Ryan T., Gina Ferrara, and Bryan L. Roth. "Screening the receptorome: an efficient approach for drug discovery and target validation." Drug discovery today 11.15-16 (2006): 708-716.
- You, Y., Lai, X., Pan, Y., Zheng, H., Vera, J., Liu, S., ... & Zhang, L. (2022). Artificial intelligence in cancer target identification and drug discovery. Signal Transduction and Targeted Therapy, 7(1), 156.
- Xin, Q., Xu, Z., Guo, L., Zhao, F., & Wu, B. (2024). IoT traffic classification and anomaly detection method based on deep autoencoders. Applied and Computational Engineering, 69, 64-70.
- Phoebe Chen, Y. P., & Chen, F. (2008). Identifying targets for drug discovery using bioinformatics. Expert opinion on therapeutic targets, 12(4), 383-389.
- Ou-Yang, S. S., Lu, J. Y., Kong, X. Q., Liang, Z. J., Luo, C., & Jiang, H. (2012). Computational drug discovery. Acta Pharmacologica Sinica, 33(9), 1131-1140.
- Yu, K., Bao, Q., Xu, H., Cao, G., & Xia, S. (2024). An Extreme Learning Machine Stock Price Prediction Algorithm Based on the Optimisation of the Crown Porcupine Optimisation Algorithm with an Adaptive Bandwidth Kernel Function Density Estimation Algorithm.
- Ling, Z., Xin, Q., Lin, Y., Su, G. and Shui, Z., 2024. Optimization of autonomous driving image detection based on RFAConv and triplet attention. Applied and Computational Engineering, 77, pp.210-217.
Figure 4.
Te deep-AVPiden architecture.
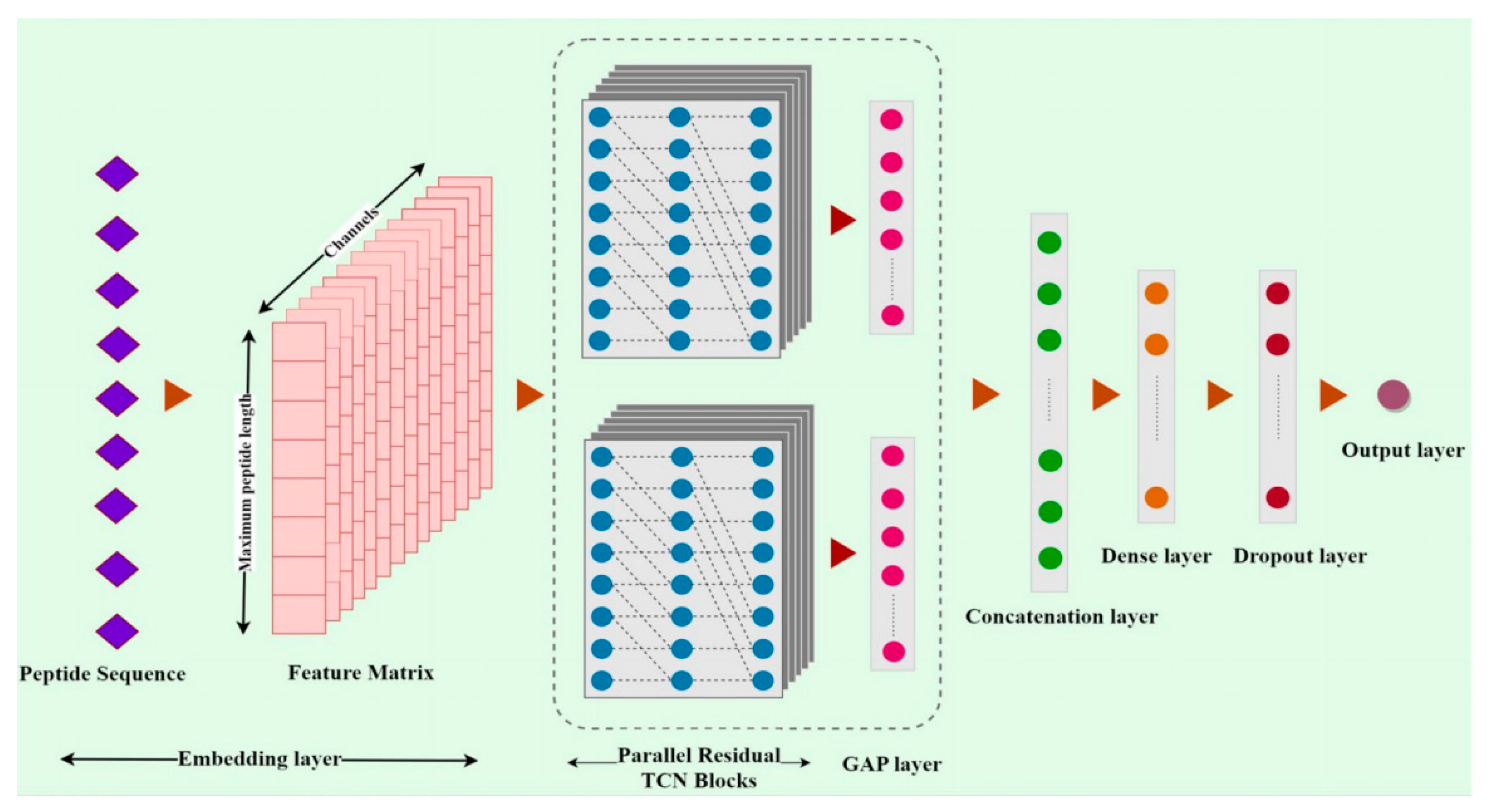

Table 1.
Comparison between acausal and causal TCNs considered while building the model.
| Model | Accuracy (%) | Precision (%) | Recall (%) | AUROC (%) |
|---|---|---|---|---|
| Deep-AVPiden (causal) | 89.88 ± 0.01 | 90.29 ± 1.74 | 90.09 ± 1.72 | 95.99 ± 0.01 |
| Deep-AVPiden (acausal) | 89.77 ± 0.38 | 90.55 ± 1.32 | 88.73 ± 1.89 | 95.89 ± 0.31 |
Table 2.
Comparison ofdeep-AVPiden with existing models on test set.
| Model | Accuracy (%) | Precision (%) | Recall (%) | AUROC (%) |
|---|---|---|---|---|
| Deep-AVPiden | 89.88 ± 0.00 | 90.29 ± 1.74 | 90.09 ± 1.72 | 95.99 ± 0.01 |
| Deep-AVPiden (DS) | 88.47 ± 0.13 | 88.49 ± 0.40 | 88.98 ± 0.38 | 94.90 ± 0.05 |
| iACVP | 65.83 | 77.33 | 46.59 | 75.49 |
| AVPIden | 59.98 | 57.2 | 73.74 | 68.81 |
| Meta-iAVP | 57.63 | 58.75 | 58.75 | 58.29 |
| DeepAVP | 53.08 | 53.94 | 58.99 | 52.77 |
| iAMP-CA2L | 52.36 | 88.89 | 6.23 | 52.72 |
| PreTp-Stack | 52.09 | 54.73 | 38.85 | 52.46 |
| ENNAVIA | 51.27 | 55.79 | 51.51 | 48.99 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



