
Submitted:
14 October 2024
Posted:
15 October 2024
You are already at the latest version
Abstract
Most of our bone are formed thought a process called endochondral ossification, which is a tiny, regulated process leaded by the chondrocytes of the GP and which require continuous cross talk between these cells and chondrocytes and invading cell populations, including osteoblasts, chondroclasts and vascular cells. The better understanding of these signaling pathways is not only important in the normal growth and maturation of the skeleton but might be highly relevant to pathophysiological processes in bones and joints. Since there is poor information about communication between chondrocytes and other cell types in the developing bones, this review examines the current knowledge of how interactions between chondrocytes and bone forming cells modulate bone growth.
Keywords:
chondrocytes
; osteocyte
; osteogenesis
; growth plate
; x linked hypophosphatemic rickets
1. Introduction
Longitudinal growth is a continuous and multifactorial process genetically determined and modulated by multiple influences such as nutritional, environmental, and hormonal factors. Normal growth reflects the general health of a child and proceeds in a predictable fashion: growth is rapid during the 2 first years of life and gradually decelerates with age until puberty. During puberty, occurs a second growth spurt and, at the end, growth eventually ceases when growth plates close and the person achieves its final size [1,2,3].
Short stature is defined, at least, as two standard deviations (SD) below the mean height of a specific population adjusted for age, sex, and pubertal stage. The severity of longitudinal growth failure indicates the likelihood of pathology. Thus, it is very low (below 2%) with a height standard deviation score (SDS) above -2 (around the third percentile) and increases to around 50% between -2 and -3 SDS and to 80% beyond -3 SDS (0.1 percentile) [4,5].
According to the International Classification of Pediatric Endocrine Diagnosis, short stature may be idiopathic (when no cause is identified), secondary to organ system disease (e.g., chronic kidney disease) or to environmental factors, or may arise from genetic disorders (primary short stature)[5].
The development of bone mineral mass during the entire growth period, including during pubertal maturation relies on a harmonious interplay of environmental, dietary, hormonal, and genetic factors. Integral to this process, is endochondral ossification within the growth plate (GP), which facilitates bone elongation until adulthood. Chondrocytes, along with osteocytes, are pivotal in orchestrating this growth length and development. Their intricate communication entails sophisticated signaling pathways and interactions, all of them crucial for proper bone formation. In this review, we have attempted to present the current knowledge concerning the cellular and molecular basis of the communication between growth plate, bone and surrounding tissues on longitudinal bone growth. This article also provides information about GP alterations observed in certain renal diseases associating growth failure.
2. Physiological Process of Longitudinal Growth
As previously pointed, longitudinal bone growth occurs at the growth plate and bone by a process called endochondral ossification. Bone formation begins in utero in the six or seven weeks of age and started as a cartilage model template [6]. This cartilage ossifies at the diaphyseal side and grows on the epiphyseal side. In the late teens or early twenties, a person reaches skeletal maturity [7]. By then, all the cartilage has been replaced by bone, so no further growth in bone length is possible. This mechanism is highly regulated and requires continuous signal interchange between cartilage and bone cells. Two types of bone ossification characterize the human skeleton: intramembranous and endochondral (Figure 1, Figure 2).
2.1. The Growth Plate
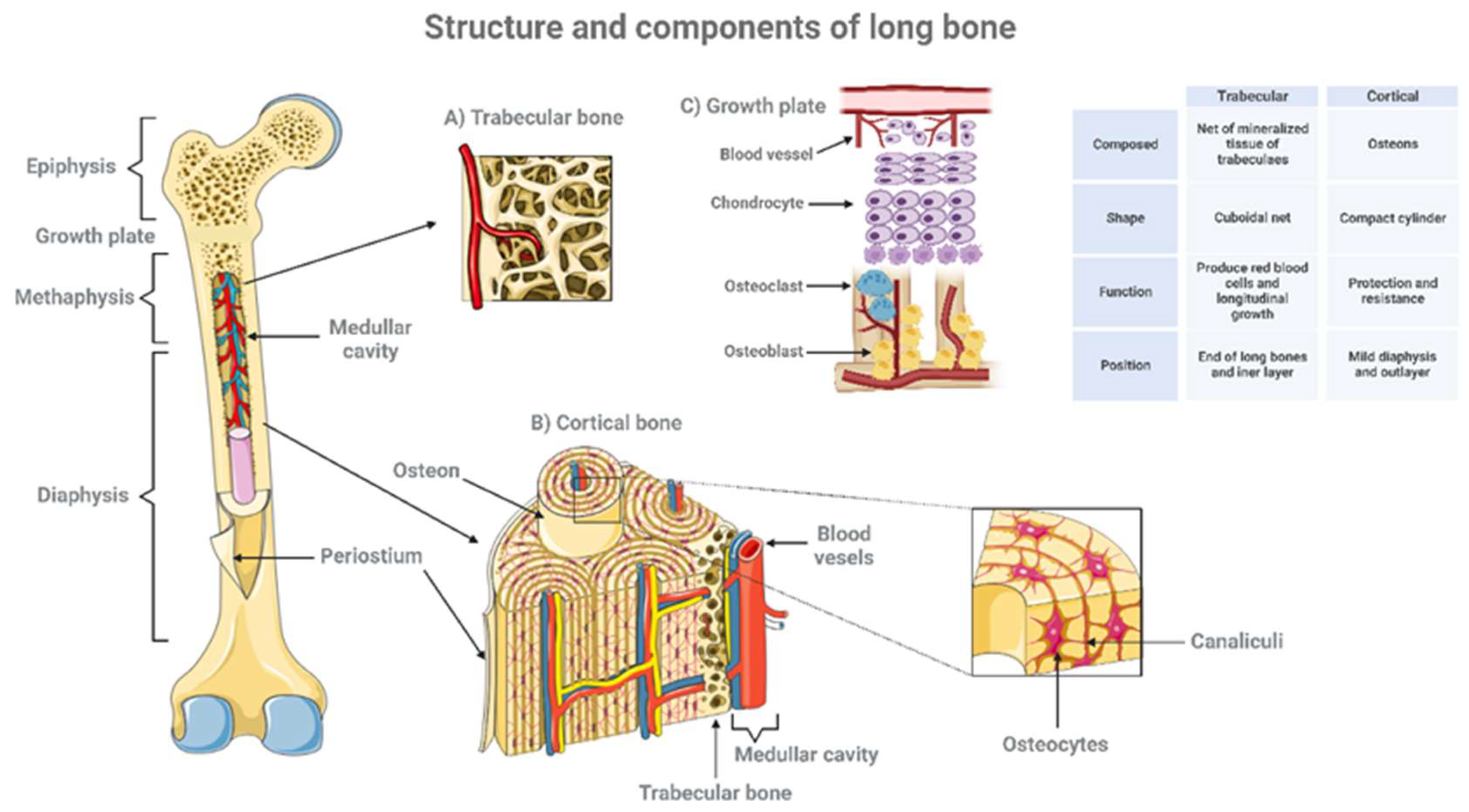
GP also called epiphyseal plate, physis, or growth cartilage, is the organ responsible for longitudinal growth and is the place where new bone apposition takes place [8]. GP is only found in children and adolescents; in adults, who have stopped growing, the plate disappears, and it is replaced by an epiphyseal line. This replacement is known as epiphyseal closure. GP is functionally and cytologically specialized region. It is made up of three tissue types: the cartilage component divided into four distinct zones, the bony tissue of the metaphysis and the vascular supply that surrounds the GP and the close interaction between them is critical for a normal bone formation and growth (Figure 3, Table 1).
GP chondrocytes are a major player in the process of endochondral ossification that contributes to the longitudinal growth of the skeleton, the determinants of chondrocyte shape and the coordination of GP function with the development bone is currently under study.
Chondrocytes of the growth plate are settled into vertical columns and in which chondrocytes display a continuous structural variation that reflects successive maturation stages of a cell-differentiation cycle. Maturation occurs synchronously for cells located at the same horizontal level in different columns and this gives rise to horizontal strata. Based on cell morphology and phenotype this soft tissue is organized in four distinct zones that contain chondrocytes in resting, proliferative, prehypertrophic or hypertrophic states. The replacement of cartilage by mineralized bone (endochondral ossification) is a complex process, not completely understood. It is triggered by the differentiation of proliferating chondrocytes in the center of the cartilage into a non-proliferative hypertrophic state. Hypertrophic chondrocytes (HC) interact with the surrounded environment and secrete cartilage matrix and other molecules; as a result, the tissue is subsequently invaded by blood vessels and osteogenic cells, that remodel the cartilage into bone tissue.
2.2. Bone Structure
Bone formation begins in utero in the six or seven weeks of age and started as a cartilage model template [6]. This cartilage ossifies at the diaphyseal side and grows on the epiphyseal side. In the late teens or early twenties, a person reaches skeletal maturity [7]. By then, all the cartilage has been replaced by bone, so no further growth in bone length is possible. This mechanism is highly regulated and requires continuous signal interchange between cartilage and bone cells (Figure 1).
Bone is a metabolically active organ that provides structural support, facilitates movement, and protects vital organs. It is also an endocrine organ that plays an important role in regulating mineral and acid-base homeostasis and in blood cell production (hematopoiesis). Therefore, although bones are hard structures, they continually replenish themselves and regenerate, consistently making new bone. In fact, the human skeleton is replaced every 7-10 years [9]. During childhood, that process allows bones to growth in length and change the shape. This process requires the interaction of 3 types of cells: osteoclasts, osteocytes and osteoblasts. There is also a 4th type of cell (Figure 1) that are the osteoprogenitor population.
Osteoblasts: Bone Forming Cells [10]. They are cuboidal cells that are located along the bone surface comprising 4–6% of the total and are largely known for their bone forming function [11]. They arise from a common plenipotentiary multipotent stromal cell (MSC). The synthesis of bone matrix by osteoblasts occurs in two main steps: deposition of organic matrix and its subsequent mineralization.
Osteocytes: Mature Bone Cells. They are the most numerous cells in the bone, representing 90–95% of the total amount, as well as the most long-lived with a life span of up to 25 years. Osteocytes are spider-shaped cells buried in the mineralized bone matrix, descended from MSC lineage through osteoblast differentiation [12]. Each osteocyte has up to 50 long and 450 branched cellular processes that extend along the bone within a network of interconnected canaliculi. These facilitate the cell-cell communication trough inter-cellular transport signaling molecules, such as prostaglandins and nitric oxide. This network is essential in coordinating the response of bone to mechanical and biological signals. Through mechanical transduction processes, osteocytes can transduce extracellular signals to elicit cellular responses to regulate bone formation and resorption.
Osteoclasts: Bone Resorbing Cells. They are terminally differentiated multinucleated cells originate from mononuclear cells of the hematopoietic stem cell lineage. They are polarized cells that breaks down bone and cartilage tissue. This function is critical in growth, maintenance, repair, and remodeling of bones.
Osteoprogenitors: They are stem cells located in the bone that play a prodigal role in bone repair and growth. These cells developed from MSC, and they are the precursors to the more specialized bone cells (osteocytes and osteoblasts). They reside in the bone marrow, and they reduce with age. Dysfunction of osteoprogenitor cells may delay ossification and lead to a spectrum of diseases such as dwarfism and Kashin-Beck disease [13].
The architecture performs its essential mechanical functions (Figure 4). Bone is covered by an external surface called periosteum that consists of two layers: an outer fibrous layer and an inner more cellular and vascular layer that can form bone. The cortical bone or compact bone is the dense outer surface of bone that forms a protective layer around the internal cavity. It is located in the diaphysis, and it has double blood supply that allows loss of one source of circulation without adversely affecting the viability of the tissue. Cortical bone forms the external layer of all bones but is found principally in the diaphysis of long bones. Cortical bone is made up of osteons, osteocytes are present within the osseous tissue surrounded by an extracellular matrix composed of calcium and phosphorous-rich hydroxyapatite. Collagen fibers present within the extracellular matrix provide limited flexibility to cortical bone. Trabecular or cancellous bone is present in the extreme of long bones or metaphysis. It is porous and trabeculated tissue and its spatial complexity contributes the maximal strength with minimum mass. The epiphysis is the enlarged end of the long bones produced by trabecular bone. Any disorganization of this net structure implicates not only bone deformities but also a smaller amount of adaptation to stress forces and more chances to bone fractures [14].
Figure 4.
Structure and components of long bone. Long bones consisting of a long shaft (the diaphysis) plus two articular (joint) surfaces, called epiphyses. They are comprised mostly of compact bone, but they also contain spongy or trabecular bone and marrow in the hollow center (the medullary cavity). The diagram shows the main structural features of bone as well as a magnified view showing some of the finer detail of trabecular (A) cortical bone (B) and growth plate (C). Created with BioRender.com.
Figure 4.
Structure and components of long bone. Long bones consisting of a long shaft (the diaphysis) plus two articular (joint) surfaces, called epiphyses. They are comprised mostly of compact bone, but they also contain spongy or trabecular bone and marrow in the hollow center (the medullary cavity). The diagram shows the main structural features of bone as well as a magnified view showing some of the finer detail of trabecular (A) cortical bone (B) and growth plate (C). Created with BioRender.com.
2.3. Endochondral Ossification, by Which the Long Bones Are Form
Endochondral ossification starts with a cartilage matrix, GP, aggregated in cluster in continuous cell division of chondrocytes [17,18]. Chondrocytes form a column-like structure with an orientation that directs the lengthening of the bone along a particular axis. Then, cartilage is eroded by osteoclasts and replaced by bone. Consequently, when a bone has reached its full size, its GP is converted into bone [19]. Endochondral ossification is a dynamic process that requires coordinated cellular activities among all cells. Therefore, the nature of growing entails tightly regulated mechanisms of intercellular recognition and communication that permit the cells to sort and migrate, synchronize activity, differentiate, equalize hormonal responses, and diffuse locally generated signals. Consequently, any miss regulation in any of the process could produce a growth defect.
Given the gradual progress of skeletal evolution over millions of years, it is not surprising that different cell lineages contribute to different parts of the mammalian skeleton. Skeletal tissue may be broadly classified as into two different types, bone and cartilage and the basic cell type which produces these mineralized tissues are: osteoblast (bone producing cell) and chondroblast (cartilage producing cell)[20]. According to previous dogma, chondrocytes and osteoblasts came from independent lineages derived from a common osteochondro-progenitor (Figure 1). The proposal for the existence of a population of MSC was first postulated by Friendenstein and colleagues [21] who reported a population of bone marrow stromal cells capable of generating bone following heterotopic transplantation. This cell has the ability to differentiate into osteoblasts, adipocytes and chondrocytes.
2.4. Skeleton Development and Growth Rely on the Coordinated Interaction of Cartilage and Bone Cells
Development and growth of the skeleton occur through the coordinated interaction of cartilage and bone cells. HC in the GP are the greatest contributors to lengthening of the cartilage template, therefore better understanding of how hypertrophic chondrocyte interacts with bone tissue will be critical to comprehend growth defects.
During bone growth, changes in both the cartilage and the bone compartments occurs, and for that, exist a close interaction between chondrocytes an osteoblast cells lineage. The idea that chondrocytes and bone cells talk to each other in the GP exists for decades [22]. However, until recently there has been little functional evidence that chondrocytes can directly control the differentiation and activity of either osteoclasts or osteoblasts. Nowadays we know that cell enlargement is not the only important function of hypertrophic chondrocytes. In vivo and ex vivo studies has shown that small molecules can transit between these tissues. In fact, several studies pointed out that osteocytes produce cytokines, as receptor activator of nuclear factor kappa-β ligand (RANKl), essential for the osteoclast differentiation and function and survival in mature bone [23,24,25]. Now we also know that the whole joint is involved in the development of articular cartilage (AC). In particular, the interaction (crosstalk) between cartilage and subchondral bone is a central feature for osteoarthritis (OA) progression.
To understand the crosstalk between cartilage and bone cells, it is necessary to know the GP development. Both AC and the GP arise in part from the embryonic growth zone and are regulated by the axis involving Indian Hedgehog and Parathyroid hormone related protein (IHH-PTHrP) [26]. The proliferative chondrocytes express PTHrP and colonize both the forming articular cartilage and GP structures. Once the secondary ossification center (SOC) is formed, PTHrP is detected in the resting zone of the GP and in the superficial region of the AC. It is believed that PTHrP and its receptor retards chondrocyte hypertrophy preventing their mineralization. In the other size, Ihh produced by prehypertrophic chondrocytes has an opposite function, and blocks proliferation promoting final differentiation and hypertrophy.
After birth and during normal growth, the epiphyseal plate will expand in length by continuous cell division of chondrocytes in the proliferative zone, which is accompanied by further secretion of extracellular matrix. Then chondrocytes in the hypertrophic zone begin to increase in size (hypertrophy), stop secreting collagen and other proteoglycans, and begin secreting alkaline phosphatase, an enzyme essential for mineral deposition. Final step is the replacement of hypertrophic cartilage by bone. This requires vascular invasion, a step initiated and controlled by the interaction of angiogenic and anti-angiogenic factors [27,28]. Hypertrophic chondrocytes attract blood vessels into the center of the cartilage template, leading to the formation of a highly vascularized endosteum within the nascent marrow space, stimulated primarily by the secretion of vascular endothelial growth factor (VEFG)[29].
Osteoblasts, accompanying vascular invasion, lay down endochondral bone to replace cartilage. Bone tissue formation takes place through a series of phases, osteoblast proliferation, followed by extracellular matrix maturation and matrix mineralization [30]. A range of transcription factors are known to be involved in the regulation of osteogenesis but two of the most broadly studied are Runt-related transcription factor 2 (Runx2) and Osterix (Osx) [31,32]. Runx2 is considered the major transcription factor controlling osteoblast commitment and differentiation. Runx2 expression is upregulated in several murine OA models, suggesting a role in disease pathogenesis [33]. In fact, overexpression of Runx2 can induce osteogenesis in vitro and in vivo, demonstrated by increased osteoblastic markers, Osteopontin and Osteocalcin [34,35]. Contrary, Runx2 null mice show a complete absence of ossification, owing to the maturational arrest of osteoblasts [36]. Osx is another important transcription factor, since Osx-deficient mice show an absence of osteoblasts and defective bone formation [37]. Other transcription factors of interest in relation to osteogenesis are the distal less gene family. Overexpression of Dlx5 can accelerate osteoblast differentiation in vitro. Osteocytes derive from osteoblasts, being essentially osteoblasts surrounded by the products they have previously secreted. Osteocytes surrounded by matrix have an extensive canalicular network connecting them to each other and to bone surface (Figure1).
Sclerostin is another regulator factor controlling osteocyte differentiation. The loss of sclerostin, derived from the inactivation of the SOST gene, leads to sclerosteosis, a skeletal disorder characterized by high bone mass due to increased osteoblast activity. Several studies have suggested that sclerostin may be an osteocyte-derived factor that is transported to osteoblasts at the bone surface and inhibits bone formation [38,39]. Osteoclasts are multinucleated cells that resorb the bone matrix and degrade the cartilage during the endochondral ossification. They arise from the monocyte-macrophage lineage present in the hematopoietic marrow, under the stimulation of two pivotal cytokines: macrophage-colony stimulating factor (M-CSF) and RANKL, both mainly produced by osteoblasts [35] (Figure 1). Osteoblasts also produce osteoprotegerin (OPG) that acts as a decoy receptor for RANKL, inhibiting its binding to RANK expressed and negative regulation osteoclast maturation [40]. Studies have established those HC also express RANKL/OPG and support osteoclast formation [40,41]. Osteoclasts break down the newly formed bone to open up the medullary cavity and trabecular formation.
For years, we thought that death is the ultimate fate of terminally differentiated HCs. However, now we know that HC can become into osteoblasts and contribute to the full osteogenic lineage [38]. Chondrocytes transdifferentiate into osteoblasts that make new bone matrix during development [39]. This process occurs in a region it is known as “the transition zone or ossification zone” that is located adjacent to the hypertrophic zone. HC located in the transition have two fates. Some of them die, while others re-enter the cell cycle, switch off the chondrogenic program, activate the molecular program associated with pluripotency, and with osteogenesis, and they become osteoblasts able to synthesize new bone matrix. Support comes from imaging, morphological, and ultrastructural studies in vivo as well as linear tracing studies. Yang et al. [30] genetically labeled either hypertrophic chondrocytes (HC) by Col10a1-Cre or chondrocytes by tamoxifen-induced Agc1-CreERT2 using Enhanced green fluorescent protein (EGFP), LacZ or Tomato expression. Both Cre drivers were specifically active in chondrocytic cells and not in perichondrium, in periosteum or in any of the osteoblast lineage cells. After labeling, these cells were distributed throughout trabeculae surfaces and, in the endosteum, embedded within the bone matrix. In vitro studies demonstrated that a proportion of the non-chondrocytic cells derived from chondrocytes labeled by Col10a1-Cre or by Agc1-CreERT2 were functional osteoblasts [42]. HC can also instruct adjacent perichondrial cells to differentiate into osteoblasts [43]. Cell chondrocyte transdifferentiation has also been described in the literature in multiple times. Rat chondrocytes have been shown, when stimulated by Fibroblastic growth factor 23 (FGF-23), Neurobasal-A, epidermal growth factor (EGF), and insulin-like growth factor-1 (IGF-1) they can transdifferentiate into stellate neuronal cells [44]. Another example is what happens in atherosclerotic lesions. It was described in mice that they presented what seems to be the transdifferentiation of vascular smooth muscle to chondrogenic tissue via increased expression of tissue non-specific alkaline phosphatase and bone morphogenetic protein (BMP-2) activation [45]. Beyond these examples is important to know that transdifferentiation is not limited to artificial cell culture settings, or to a pathology but is also a natural phenomenon.
Besides transdifferentiation into bone-forming cells, HC induce bone formation at their adjacent regions by secreting matrix proteins, including type X collagen, and critical paracrine factors. As previously described, HC secrete VEGF that induce invasion of blood vessels from the perichondrium, and IHH hat regulate proliferation and differentiation and directs perichondrial cells to become osteoblasts. In addition, IHH acts further on periarticular chondrocytes at the end of the cartilage and therefore promotes the production of parathyroid hormone–related peptide (PTHrP)[46]. PTHrP acts opposite as Ihh and delays the chondrocyte differentiation into hypertrophic chondrocytes. These series of interactions establish the PTHrP-Ihh feedback loop that is essential to maintaining the GP structure and AC health (Figure 3). Actually, deletion of either the PTHrP gene or the PTH/PTHrP receptor gene leads to acceleration of differentiation of GP chondrocytes, bones exhibit a striking increase in osteoblast number and matrix accumulation and a reduction of vascular invasion [47,48]. Wnt signaling also plays an important role, studies have shown that misexpression of Wnt-4 accelerates the nonhypertrophic to hypertrophic transition and results in slightly advanced ossification [49]. Matrix metalloproteinase (MMP13) is expressed by hypertrophic chondrocytes and osteoblasts. We demonstrate that MMP13 is required for proper resorption of hypertrophic cartilage. Another pathway that has been recently implicated in the communication between cartilage and other bone cells is the epidermal growth factor receptor (EGFR) pathway. EGFR on chondrocytes activates the induction of matrix metalloproteinases (MMPs) as well as RANKL (Figure 4).
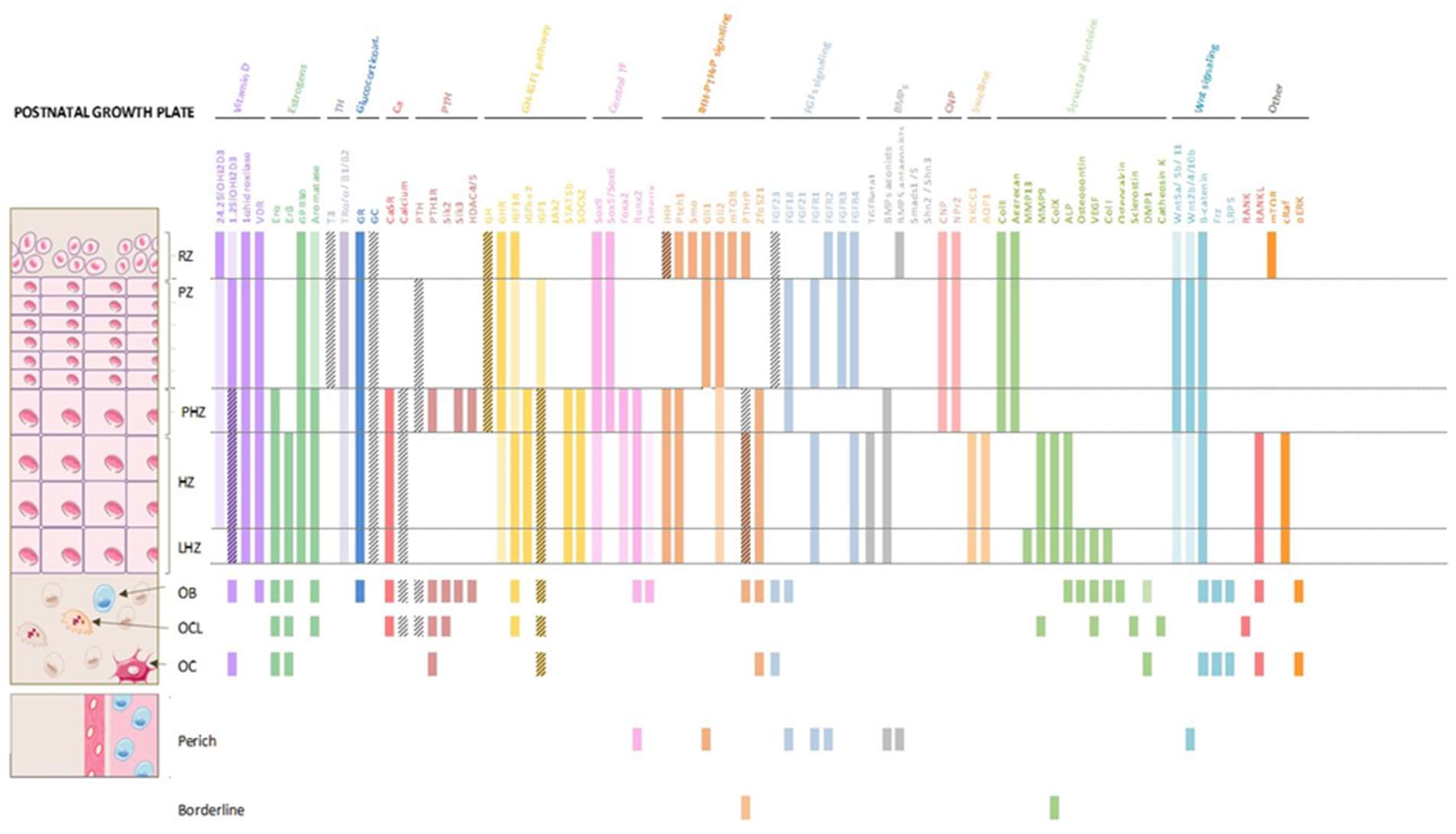
Figure 4.
Molecular expression of the main factors involved in the dynamics of the postnatal growth plate. Colored bars mean that the protein is expressing in that region. If the bar does not have color and is only with oblique stripes, means that it is that it is not expressed in that area, but it is where it performs its function. When the bar has color and stripes means that it is expressed there, and the cell also has receptors for the protein.
Figure 4.
Molecular expression of the main factors involved in the dynamics of the postnatal growth plate. Colored bars mean that the protein is expressing in that region. If the bar does not have color and is only with oblique stripes, means that it is that it is not expressed in that area, but it is where it performs its function. When the bar has color and stripes means that it is expressed there, and the cell also has receptors for the protein.
In this process, α-parvin, an integrin-associated focal adhesion protein, plays a significant role, regulating α-parvin the rotation of chondrocytes, which is an essential process for chondrocytes to form a columnar structure. The loss of this protein in animal models increases binucleation, raises cell death, and causes dilation of the resting zones of mature GP. Single-cell RNA-seq analyses revealed alterations in the transcriptome in all three zones (i.e., resting, proliferative, and hypertrophic zones) of the GP [50]. In conclusion, HC can recruit osteoclasts and osteoblasts to sites of cartilage remodeling. Thus, they can be considered as master regulators of endochondral bone formation.
In the other hand, articular cartilage, molecular crosstalk between chondrocytes and osteoblasts/osteocytes are likely of primary importance during bone growth. Osteocytes form an interconnected network throughout the cortical and trabecular bone, and these cells act as mechanosensory. Cytokine-sized and larger molecules can traverse between bone and cartilage in either healthy or diseased joints. These molecules are able to traverse osteocyte canaliculi, and this transport is increased by bone loading [51,52]. During the osteoarthritic process marked alterations in the composition of the cartilage matrix occurs, resulting in swelling of the matrix, and increased metabolic activity of chondrocytes and inducing hypertrophy and apoptosis. Production of matrix modifying factors such as MMPs by chondrocytes induce the response of load-sensing osteocytes. IGF-1 is an important factor in GP development, it is transported into cartilage mainly from osteocytes into the circulation. This transport occurs partly by diffusion across a concentration gradient but, in addition, mechanical loading has been shown to enhance the transport and promote growth [50]. Osteocytes also secrete FGF-23. FGF-23 suppressed chondrocyte proliferation and maturation via Indian hedgehog. FGF-23 is well established to play crucial roles in X-linked hypophophatemia (XLH) and growth retardation [53], actually inhibition of FGF23 partially rescued XLH phenotype [54] promoting growth. Inhibiting FGF-23 sems to be a crucial factor in OA disorders however this phenomenon is not completely explored.
GH interacts with growth plate through GH receptors (GHRs) located in chondrocytes and, indirectly, through systemic and local stimulation of IGF-1. Upon binding to GHR at the growth plate’s chondrocytes, it induces dimerization of the receptor, resulting in tyrosine phosphorylation of Janus-associated kinase 2 (JAK2), a tyrosine kinase associated with the intracellular domain of the receptor. Phosphorylation induces the kinase activity of JAK2, which in turn phosphorylates/activates a group of molecules known as signal transducers and activators of transcription (STATs). Upon activation, the STATs dimerize and translocate to the nucleus, where they regulate the expression of target genes responsible for GH action, including IGF-1 [55].
3. Defects in Interaction Lead to Growth Disorders
Many human skeletal growth disorders are due to abnormalities in the endocrine regulation system. One example can be Jansen Metaphyseal Chondrodysplasia (JMC) [56], an autosomal dominant disease caused by activating PTH/PTHrP receptor (PTH1R) in the GP (Figure 1). GP chondrocytes are a major player in the process of endochondral ossification that contributes to the longitudinal growth of the skeleton, the determinants of chondrocyte shape and the coordination of GP function with the development bone is currently under study.
An elevated number of inherited human conditions presents by defects in osteoclasts (e.g., osteopetrosis, pycnodysostosis) [56]. However, the phenotype in many osteoclast diseases is a combination of osteosclerosis with osteolytic lesions. In such conditions, the primary defect is hyperactivity of osteoclasts.
Several skeletal dysplasias are associated with defects in differentiation or function of chondrocytes, osteoblasts and osteoclasts, which will cause defects in production or functionality of cartilage and/or bone matrix. Any alteration of one of these three cell types always results in compromised skeletal growth and mineralization defect. Bone growth disorders in children can result from injury, chronic diseases and sometimes by specific genetic disorders affecting GP or bone development led to abnormalities in the endochondral ossification and growth impairment.
Although diagnoses of a skeletal dysplasia can be made frequently in the newborn period, there actually may be no short stature at that time, but by 1 year of age the growth differential between the skeletally dysplastic child and a normal child becomes apparent.
An imbalance between the different cellular actors leads to poor plate function, which leads to growth failure that is reflected as a structural alteration of the GP. Many pathologies that occur during childhood can have an incidence and affect final height in an evident way.
The GP can have different thickness, and some abnormalities associated with a defect of mineralization are associated with thicker GPs with persistence of cartilage in the hypertrophic zone and adjacent metaphysis [57]. Interference with the hypertrophy progression of GP chondrocytes, leads to abnormal cartilage formation, ossification, and modifications in the rates of bone formation, leading in the end to growth problems, as for example rickets and dwarfism [58]. In the other hand, GH/IGF-1 pathway is the key regulator of hypertrophy. The identification of IGF1 as a key regulator of hypertrophy is not a surprise, given the GP phenotype of IGF-1 KO mice where they have a markedly reduced chondrocyte proliferation, increased apoptosis, and delayed maturation and differentiation of chondrocytes, resulting in a severely shortened and under mineralized skeleton [59]. Some children have growth failure, despite normal or even high levels of GH. In some of these children has abnormally low levels of IGF-1, this condition is called Primary Insulin-like Growth Factor Deficiency (PIGFD). Others have shown, in both that nutritional deficiency inhibits IGF-1 signaling in the GP and reduces final adult height [60]. While a degree of circulating IGF-1 is necessary [61], local action of IGF-1 may be more critical for longitudinal bone growth. IGF-1 expression it has been observed in the HZ of epiphyseal GPs and in bone [55]. In mice carrying liver-specific IGF-1 deletion, which display a reduction in serum IGF-1, a decreased cortical bone is demonstrated [62]. The systemic IGF-1 contributes to cortical bone integrity, while the bone trabecular integrity is sustained by the locally produced skeletal IGF-1. This points out that local regulation of growth, is crucial for final length [48].
4. What We Already Know about Clinical Conditions That Associate Growth Retardation
Disorders associated with a defect in the GP and bone are numerous. Among these, we can find congenital/hereditary disorders (complex syndromes, dysplasias, rickets) or acquired conditions (Chronic Kidney Disease (CKD), malnourished) [63]. Genetic disorders can affect different phases of the maturational cycle of chondrocytes and then, alterations may occur in the growth plate at the various zones, reserve, proliferative, hypertrophic, and at the border of the growth plate. These disorders influence chondrocyte activity and extracellular matrix content which subsequently disrupts growth plate performance and decreases its ability to properly develop and lengthen the bones. There are similarities in growth plate anomalies observed in clinical conditions associated with growth impairment. These disruptions may arise from various factors, including genetic abnormalities, hormonal imbalances, nutritional deficiencies or other medical conditions (Supplemental tables: Table S1, S2 and S3). By studying these similarities, researchers aim to better understand the underlying mechanisms of growth impairment and develop more effective treatments.
The precise mechanism behind growth delay in children with chronic inflammation, remains intangible. While inflammation is a normal immune response to protect the body from harmful stimuli, chronic inflammation occurs when the immune system remains activated for prolonged periods, leading to tissue damage and long-term health issues. Chronic inflammation in children are present in several conditions such as, allergies, infections, environmental triggers or autoimmune disorders, where there exist elevated serum levels of main pro-inflammatory cytokines such as interleukin 6 (IL-6), tumor necrosis factor-α (TNF-α), and interleukin 1β (IL-1β) [64]. Thus, besides malnutrition, chronic stress, and prolonged use of glucocorticoids, are known conductors to longitudinal growth retardation, it has been observed that these cytokines also lead to abnormal bone development by impairment the activity of the GH/IGF-1 axis [65,66,67,68,69,70,71]. It should be considered that IGF-1 as a valuable laboratory indicator of nutritional status could be decreased in states of poor nutrition due to limited nutrient intake and malabsorption, which often accompany chronic inflammation [69,72,73]. Serum levels of IGFBP-3 do not seem to be as dependent on nutritional deprivation as IGF-1, and they better reflect GH signaling [74]. Experimental studies have also confirmed that IL-6 could act locally at the growth plate chondrocytes [75,76,77,78]. Moreover, IL-1β, IL-6, and TNF-α could affect the function of the growth plate by suppressing IGF-1 intracellular signaling and by inhibiting the effect of IGF-1 on chondrocytes proliferation and differentiation at the growth plate [67,68,79].
In a model of cultured fetal rat metatarsal bones, it was seen that TNF-α and IL-1β could act in synergy at the growth plate chondrocytes to inhibit longitudinal growth due to a decrease in chondrocyte proliferation and an increase in its apoptosis [80]. However, it is worth noting that both TNF-α and IL-1β, produced endogenously in growth plate chondrocytes, appear to play a role in normal growth, while negative effects before mentioned of these cytokines on chondrocytes are observed at supraphysiological levels [81,82]. Current data indicate that IL-6, TNF-α, and IL-1β could affect the GH/IGF-1 axis through both systemic and local mechanisms, acting individually or in combination. Systemic action of pro-inflammatory cytokines leads mainly to hepatic GH resistance and suppression of IGF-1 action in target tissues, while their local action directly affects the growth plate [(65,68,83–85). It has been confirmed that IL-6 could suppress GH action leading to reduction in JAK/STAT signaling by induction of suppressor of cytokine signaling (SOCS)-3 protein (68,84]. Based on recent research, it’s important to highlight the role of microRNAs as potential additional factors connecting chronic inflammation with growth retardation seen in childhood-onset chronic inflammatory diseases[71]. Multiple studies suggest that miRNAs, acting as post-transcriptional regulators of gene expression, may influence various proteins and cytokines involved in controlling the GH/IGF-1 axis [86,87,88].
CKD-Metabolic bone disease (MBD) is assumed as a systemic disorder defined by at least one of these features: (i) abnormal calcium, phosphorus, PTH, FGF23, and/or vitamin D metabolisms; (ii) impaired bone formation and maintenance translated into short stature, reduced mineralization and increased risk of fractures and (iii) extra skeletal calcifications [89]. While impairment of body growth is a main manifestation in pediatric patients with CKD-MBD, the achievement of a normal final height is a major challenge in their management [90]. Experimental young murine models with renal insufficiency, present a reduction in length gain and growth velocity due to a delay of the GP calcification-vector known as osseous front advance (OFA) [55,91]. That OFA formation correlates directly to final size of the hypertrophic chondrocytes at the growth cartilage, is well known. Thus, the delay in cartilage calcification in uremia is associated to a lower gain in volume and density by terminal chondrocytes. There are three main molecules, directly involved in improving the proportions of chondrocytes: Aquaporin (Aqp1) and Na–K–Cl cotransporter 1 (Nkcc1), involved in track water and solutes through the plasmatic membrane for volume and density gain at first hypertrophic steps. The third factor, IGF-1, leads the real hypertrophy at final phase depending on the JACK-STATS signal transducers and activators of transcription of GH pathway [92]. In uremia, probably as consequence of abnormal modulation of IHH or WNT signals, a poor expression of Aqp1 impairs normal volume increment at the first hypertrophic levels. Curiously, and maybe in a try to compensate this scenario, Nkcc1 maintains its expression along he whole HZ and facilitating the cellular growth by augmenting cytoplasm density. However, the reduced IGF-1 local production for inducing the real hypertrophy at last steps, results irremediably in the inappropriate final chondrocyte size observed in uremic growth plates.
But it is also necessary to think that CKD associates a miss-balance of systemic factors such as GH, vitamin D, glucocorticoids, estrogens, thyroid hormones, cytokines… Thus, as in juvenile idiopathic arthritis (JIA) other inflammatory conditions, uremia could be favoring an inflammatory scenario supporting the consequences associated to the disease. Thus, besides the abnormal final chondrocytes size, uremia must create an inflammatory scenario in the hypertrophic zone of the growth plate, dysregulating local transcription factors (FGF23 and receptors, PTHrP, MEPE) and a modifying the expression patterns of matrix proteins (Col10a1, MMP13/9) that, combined with altered behaviors of SOX9 and RUNX2/3 [93,94], may orchestrate the dysfunction of local factors directly involved in new bone progression (VEGF, BMPs or HMGBP1).
Hereditary hypophosphatemias, are a group of genetic disorders involving renal phosphate loss, characterized by hypophosphatemia, rickets, and normal serum calcium levels. The characteristic clinical features include rickets, slow growth/stature shortening, bone pain, and bone deformities. Hypophosphatemic rickets typically presents in infancy or early childhood with skeletal deformities and GP abnormalities. The most common is X-linked hypophosphatemia (XLH) [63]. The genetic basis for XLH is the loss of function of the PHEX gene, located in chromosome Xp22; this loss results in an elevation of the serum levels of fibroblast growth factor 23 (FGF23), and impaired renal production of 1,25-dihydroxyvitamin D (1,25(OH)2D3). FGF-23 and its growth plate local receptors are increased in hypophosphatemic animal models and has been reported to be responsible for hypophosphatemia and reduced 1,25-(OH)2D3 levels [64,65], while in vitro analyses of primary murine chondrocytes, have demonstrated that phosphate mediates hypertrophic chondrocyte apoptosis by activating the caspase-9-dependent mitochondrial pathway [67].
Children with XLH show an over-activation of both extracellular signal-regulated kinase (ERK) and pERK1/2, leading to an expansion of the hypertrophic chondrocyte layer and a decrease in type I collagen in vitro, as well as an upregulation of the mitogen activated protein kinase (MAPK) signaling pathway in the GP, while serum calcium levels remain normal. In this regard, pERK1/2 inhibition activity in Hyp mice relates a partial recovery of cartilage deformities and skeletal abnormalities [66]. It has been observed that excess FGF23, which is characteristic of rXLH, can influence the signaling of pathways involved in terminal hypertrophy of chondrocytes. FGF23 may interact with other signaling pathways, such as the IGF signaling pathway, altering chondrocyte terminal hypertrophy and repercuting over the expression of other factors at the osteochondral junction (MMP9, MMP13, VEFG, OCN, ALP, TRAP…).
As we have seen, in various clinical conditions characterized by growth retardation, whether originating from genetic factors or acquired conditions, there is a convergence of molecular changes occurring in the hypertrophic zone of the growth plate and the osteochondral junction. Abnormal expression of IGF1 is likely one of the contributors.
The coupling between osteoblast, osteoclast and chondrocytes functions is critical for skeletal growth, remodeling, and repair. However, although the general principles and mechanisms of endochondral bone formation are well established, many details remain to be well defined, such as the mechanisms by which chondrocytes send signals to adjacent tissue to trigger bone growth.
In conclusion, while significant advances have been made in understanding the intricate communication between chondrocytes, osteoblasts, and osteoclasts during skeletal growth, there are still gaps in our knowledge. The precise mechanisms by which chondrocytes signal to adjacent tissues to stimulate bone growth remain an active area of research. Addressing these unknowns will not only enhance our fundamental understanding of skeletal development but also open avenues for more effective treatments for growth-related disorders. Continued exploration into the molecular interactions at the growth plate will be key to unlocking new therapeutic strategies.
5. Conclusions and Future Directions
In conclusion, while significant advances have been made in understanding the intricate communication between chondrocytes, osteoblasts, and osteoclasts during skeletal growth, there are still gaps in our knowledge. The precise mechanisms by which chondrocytes signal to adjacent tissues to stimulate bone growth remain an active area of research. Addressing these unknowns will not only enhance our fundamental understanding of skeletal development but also open avenues for more effective treatments for growth-related disorders. Continued exploration into the molecular interactions at the growth plate will be key to unlocking new therapeutic strategies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Classification of primary disorders affecting the growth plate and bone during the growth stage.; Table S2: Classification of secondary disorders affecting the growth plate and bone during the growth stage; Table S3: Other disorders affecting the growth plate and bone during the growth stage.
Author Contributions
Author Contributions: Conceptualization, R.F. and F.H.G; Methodology, F.H.G and R.F.; Software, F.H.G; Validation, J.R.S., H.G.P and J.M.L.; Formal Analysis, J.R.S., H.G.P., J.M.L. and R.F.; Investigation, F.H.G.; Resources, F.H.G and R.F.; Data Curation, F.H.G., R.F. and H.G.P.; Writing – Original Draft Preparation, F.H.G and R.F.; Writing – Review & Editing, J.R.S., H.G.P., J.M.L. and R.F.; Visualization, F.H.G., A.F.I. and J.M.L.; Supervision, R.F., J.R.S., H.G.P. and J.M.L.; Project Administration, H.G.P.; Funding Acquisition, H.G.P. All the authors have read and agreed to the published version of the manuscript.
Funding
This review has been funded by the Carlos III Health Institute (ISCIII) -projects PI20/00922, PI23/00174- and co-funded by the European Union.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank the University of Oviedo for the grant from the Vice-Rectorate for Research (reference PAPI-22-OF-4) and the Fundación Nutrición y Crecimiento (FUNDNYC) for its support.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Nichols J. UpToDate. 2022. p. 1–50 Normal growth patterns in infants and prepubertal children.
- Polidori, N.; Castorani, V.; Mohn, A.; Chiarelli, F. Deciphering short stature in children. Ann. Pediatr. Endocrinol. Metab. 2020, 25, 69–79. [CrossRef]
- Graber EG. MSD Manual, Professional Version. 2023. Physical Growth of Infants and Children.
- Bogarín, R.; Richmond, E.; Rogol, A.D. A new approach to the diagnosis of short stature. Minerva Pediatr. 2020, 72, 250–262. [CrossRef]
- Chiarelli, F.; Primavera, M.; Mastromauro, C. Evaluation and management of a child with short stature. Minerva Pediatr. 2021, 72, 452–461. [CrossRef]
- Blumer, M.J. Bone tissue and histological and molecular events during development of the long bones. Ann. Anat. - Anat. Anz. 2021, 235, 151704. [CrossRef]
- Furdock, R.J.; Sanders, J.O.; Cooperman, D.R.; Liu, R.W. Using Skeletal Maturity in Pediatric Orthopaedics: A Primer. J. Pediatr. Orthop. 2022, 42, e793–e800. [CrossRef]
- Ağırdil, Y. The growth plate: a physiologic overview. EFORT Open Rev. 2020, 5, 498–507. [CrossRef]
- Csukasi, F.; Bosakova, M.; Barta, T.; Martin, J.H.; Arcedo, J.; Barad, M.; Rico-Llanos, G.A.; Zieba, J.; Becerra, J.; Krejci, P.; et al. Skeletal diseases caused by mutations in PTH1R show aberrant differentiation of skeletal progenitors due to dysregulation of DEPTOR. Front. Cell Dev. Biol. 2023, 10, 963389. [CrossRef]
- Coates P. Bone turnover markers. Aust Fam Physician. 2013 May;42(5):285–7.
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [CrossRef]
- Florencio-Silva, R.; da Silva Sasso, G.R.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. BioMed Res. Int. 2015, 2015, 421746. [CrossRef]
- Dallas, S.L.; Bonewald, L.F. Dynamics of the transition from osteoblast to osteocyte. Ann. N. Y. Acad. Sci. 2010, 1192, 437–443. [CrossRef]
- Helfrich MH. Osteoclast diseases. Microsc Res Tech. 2003 Aug 15;61(6):514–32.
- Ren, X.; Zhou, Q.; Foulad, D.; Tiffany, A.S.; Dewey, M.J.; Bischoff, D.; Miller, T.A.; Reid, R.R.; He, T.-C.; Yamaguchi, D.T.; et al. Osteoprotegerin reduces osteoclast resorption activity without affecting osteogenesis on nanoparticulate mineralized collagen scaffolds. Sci. Adv. 2019, 5, eaaw4991. [CrossRef]
- Šromová, V.; Sobola, D.; Kaspar, P. A Brief Review of Bone Cell Function and Importance. Cells 2023, 12, 2576. [CrossRef]
- Morgan, E.F.; Unnikrisnan, G.U.; Hussein, A.I. Bone Mechanical Properties in Healthy and Diseased States. Annu. Rev. Biomed. Eng. 2018, 20, 119–143. [CrossRef]
- Hannink G, Arts JJC. Bioresorbability, porosity and mechanical strength of bone substitutes: What is optimal for bone regeneration? Injury. 2011 Sep;42:S22–5.
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.-S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [CrossRef]
- Deng D, Liu X, Huang W, Yuan S, Liu G, Ai S, et al. Osteoclasts control endochondral ossification via regulating acetyl-CoA availability. Bone Res. 2024 Aug 28;12(1):49.
- Mackie, E.J.; Tatarczuch, L.; Mirams, M. The skeleton: a multi-functional complex organ. The growth plate chondrocyte and endochondral ossification. J. Endocrinol. 2011, 211, 109–121. [CrossRef]
- Hall, B.K. Evolutionary Consequences of Skeletal Differentiation. Am. Zoöl. 1975, 15, 329–350. [CrossRef]
- Friedenstein AJ, Piatetzky-Shapiro II, Petrakova KV. Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol. 1966;16(3):381–90.
- Bonewald LF. The amazing osteocyte. Journal of Bone and Mineral Research. 2011 Feb 1;26(2):229–38.
- Xu, J.; Yu, L.; Liu, F.; Wan, L.; Deng, Z. The effect of cytokines on osteoblasts and osteoclasts in bone remodeling in osteoporosis: a review. Front. Immunol. 2023, 14, 1222129. [CrossRef]
- Takegahara N, Kim H, Choi Y. RANKL biology. Bone. 2022 Jun;159:116353.
- Zhou, P.; Zheng, T.; Zhao, B. Cytokine-mediated immunomodulation of osteoclastogenesis. Bone 2022, 164, 116540–116540. [CrossRef]
- Chen, X.; Macica, C.M.; Nasiri, A.; Broadus, A.E. Regulation of articular chondrocyte proliferation and differentiation by indian hedgehog and parathyroid hormone–related protein in mice. Arthritis Rheum. 2008, 58, 3788–3797. [CrossRef]
- Dreyer, C.H.; Kjaergaard, K.; Ding, M.; Qin, L. Vascular endothelial growth factor for in vivo bone formation: A systematic review. J. Orthop. Transl. 2020, 24, 46–57. [CrossRef]
- Grosso, A.; Lunger, A.; Burger, M.G.; Briquez, P.S.; Mai, F.; Hubbell, J.A.; Schaefer, D.J.; Banfi, A.; Di Maggio, N. VEGF dose controls the coupling of angiogenesis and osteogenesis in engineered bone. npj Regen. Med. 2023, 8, 1–15. [CrossRef]
- Street, J.; Bao, M.; DeGuzman, L.; Bunting, S.; Peale, F.V., Jr.; Ferrara, N.; Steinmetz, H.; Hoeffel, J.; Cleland, J.L.; Daugherty, A.; et al. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc. Natl. Acad. Sci. USA 2002, 99, 9656–9661. [CrossRef]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S.E. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. 2014, 111, 12097–12102. [CrossRef]
- Zhu, S.; Chen, W.; Masson, A.; Li, Y.-P. Cell signaling and transcriptional regulation of osteoblast lineage commitment, differentiation, bone formation, and homeostasis. Cell Discov. 2024, 10, 1–39. [CrossRef]
- Chan, W.C.W.; Tan, Z.; To, M.K.T.; Chan, D. Regulation and Role of Transcription Factors in Osteogenesis. Int. J. Mol. Sci. 2021, 22, 5445. [CrossRef]
- Nagata, K.; Hojo, H.; Chang, S.H.; Okada, H.; Yano, F.; Chijimatsu, R.; Omata, Y.; Mori, D.; Makii, Y.; Kawata, M.; et al. Runx2 and Runx3 differentially regulate articular chondrocytes during surgically induced osteoarthritis development. Nat. Commun. 2022, 13, 1–17. [CrossRef]
- Mollentze, J.; Durandt, C.; Pepper, M.S. An In Vitro and In Vivo Comparison of Osteogenic Differentiation of Human Mesenchymal Stromal/Stem Cells. Stem Cells Int. 2021, 2021, 1–23. [CrossRef]
- Xu J, Li Z, Hou Y, Fang W. Review Article Potential mechanisms underlying the Runx2 induced osteogenesis of bone marrow mesenchymal stem cells [Internet]. Vol. 7, Am J Transl Res. 2015. Available from: www.ajtr.org.
- Qin, X.; Jiang, Q.; Komori, H.; Sakane, C.; Fukuyama, R.; Matsuo, Y.; Ito, K.; Miyazaki, T.; Komori, T. Runt-related transcription factor-2 (Runx2) is required for bone matrix protein gene expression in committed osteoblasts in mice. J. Bone Miner. Res. 2021, 36, 2081–2095. [CrossRef]
- Liu, Q.; Li, M.; Wang, S.; Xiao, Z.; Xiong, Y.; Wang, G. Recent Advances of Osterix Transcription Factor in Osteoblast Differentiation and Bone Formation. Front. Cell Dev. Biol. 2020, 8, 601224. [CrossRef]
- van Bezooijen, R.L.; Roelen, B.A.; Visser, A.; van der Wee-Pals, L.; de Wilt, E.; Karperien, M.; Hamersma, H.; Papapoulos, S.E.; Dijke, P.T.; Löwik, C.W. Sclerostin Is an Osteocyte-expressed Negative Regulator of Bone Formation, But Not a Classical BMP Antagonist. J. Exp. Med. 2004, 199, 805–814. [CrossRef]
- Carpenter, K.A.; Alkhatib, D.O.; Dulion, B.A.; Guirado, E.; Patel, S.; Chen, Y.; George, A.; Ross, R.D. Sclerostin antibody improves alveolar bone quality in the Hyp mouse model of X-linked hypophosphatemia (XLH). Int. J. Oral Sci. 2023, 15, 1–10. [CrossRef]
- Maurizi, A.; Rucci, N. The Osteoclast in Bone Metastasis: Player and Target. Cancers 2018, 10, 218. [CrossRef]
- Marcadet, L.; Bouredji, Z.; Argaw, A.; Frenette, J. The Roles of RANK/RANKL/OPG in Cardiac, Skeletal, and Smooth Muscles in Health and Disease. Front. Cell Dev. Biol. 2022, 10, 903657. [CrossRef]
- Zhou, X.; von der Mark, K.; Henry, S.; Norton, W.; Adams, H.; de Crombrugghe, B. Chondrocytes Transdifferentiate into Osteoblasts in Endochondral Bone during Development, Postnatal Growth and Fracture Healing in Mice. PLOS Genet. 2014, 10, e1004820. [CrossRef]
- Long, F.; Chung, U.-I.; Ohba, S.; McMahon, J.; Kronenberg, H.M.; McMahon, A.P. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 2004, 131, 1309–1318. [CrossRef]
- Greene, C.A.; Green, C.R.; Sherwin, T. Transdifferentiation of chondrocytes into neuron-like cells induced by neuronal lineage specifying growth factors. Cell Biol. Int. 2014, 39, 185–191. [CrossRef]
- Fakhry, M.; Roszkowska, M.; Briolay, A.; Bougault, C.; Guignandon, A.; Diaz-Hernandez, J.I.; Diaz-Hernandez, M.; Pikula, S.; Buchet, R.; Hamade, E.; et al. TNAP stimulates vascular smooth muscle cell trans-differentiation into chondrocytes through calcium deposition and BMP-2 activation: Possible implication in atherosclerotic plaque stability. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2017, 1863, 643–653. [CrossRef]
- Kobayashi, T.; Soegiarto, D.W.; Yang, Y.; Lanske, B.; Schipani, E.; McMahon, A.P.; Kronenberg, H.M. Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J. Clin. Investig. 2005, 115, 1734–1742. [CrossRef]
- Esbrit, P. The role of parathyroid hormone related protein (PTHrP) in bone metabolism: from basic to clinical research. Rev. de Osteoporos. y Metab. Miner. 2023, 15, 1–5. [CrossRef]
- Lanske, B.; Amling, M.; Neff, L.; Guiducci, J.; Baron, R.; Kronenberg, H.M. Ablation of the PTHrP gene or the PTH/PTHrP receptor gene leads to distinct abnormalities in bone development. J. Clin. Investig. 1999, 104, 399–407. [CrossRef]
- Li, X.; Han, Y.; Li, G.; Zhang, Y.; Wang, J.; Feng, C. Role of Wnt signaling pathway in joint development and cartilage degeneration. Front. Cell Dev. Biol. 2023, 11, 1181619. [CrossRef]
- Yuan, J.; Guo, L.; Wang, J.; Zhou, Z.; Wu, C. α-parvin controls chondrocyte column formation and regulates long bone development. Bone Res. 2023, 11, 1–12. [CrossRef]
- Wang, B.; Zhou, X.; Price, C.; Li, W.; Pan, J.; Wang, L. Quantifying load-induced solute transport and solute-matrix interaction within the osteocyte lacunar-canalicular system. J. Bone Miner. Res. 2013, 28, 1075–1086. [CrossRef]
- Hopkins, T.; Wright, K.T.; Kuiper, N.J.; Roberts, S.; Jermin, P.; Gallacher, P.; Kuiper, J.H. An In Vitro System to Study the Effect of Subchondral Bone Health on Articular Cartilage Repair in Humans. Cells 2021, 10, 1903. [CrossRef]
- Fuente R, Gil-Peña H, Claramunt-Taberner D, Hernández O, Fernández-Iglesias A, Alonso-Durán L, et al. X-linked hypophosphatemia and growth. Vol. 18, Reviews in Endocrine and Metabolic Disorders. Springer New York LLC; 2017. p. 107–15.
- Wöhrle, S.; Henninger, C.; Bonny, O.; Thuery, A.; Beluch, N.; E Hynes, N.; Guagnano, V.; Sellers, W.R.; Hofmann, F.; Kneissel, M.; et al. Pharmacological inhibition of fibroblast growth factor (FGF) receptor signaling ameliorates FGF23-mediated hypophosphatemic rickets. J. Bone Miner. Res. 2013, 28, 899–911. [CrossRef]
- Fernández-Iglesias, .; López, J.M.; Santos, F. Growth plate alterations in chronic kidney disease. Pediatr. Nephrol. 2020, 35, 367–374. [CrossRef]
- Voller, T.; Cameron, P.; Watson, J.; Phadnis, J. The growth plate: anatomy and disorders. Orthop. Trauma 2020, 34, 135–140. [CrossRef]
- Kornak, U.; Mundlos, S. Genetic Disorders of the Skeleton: A Developmental Approach. Am. J. Hum. Genet. 2003, 73, 447–474. [CrossRef]
- Wang, Y.; Cheng, Z.; ElAlieh, H.Z.; Nakamura, E.; Nguyen, M.-T.; Mackem, S.; Clemens, T.L.; Bikle, D.D.; Chang, W. IGF-1R signaling in chondrocytes modulates growth plate development by interacting with the PTHrP/Ihh pathway. J. Bone Miner. Res. 2011, 26, 1437–1446. [CrossRef]
- Savage MO. Insulin-Like Growth Factors, Nutrition and Growth. In: R. Shamir, D. Turck, M. Phillip, editors. World Review of Nutrition and Dietetics [Internet]. 2013 [cited 2023 Nov 27]. p. 52–9. Available from: . [CrossRef]
- Kim, S.-M.; Sultana, F.; Korkmaz, F.; Rojekar, S.; Pallapati, A.; Ryu, V.; Lizneva, D.; Yuen, T.; Rosen, C.J.; Zaidi, M. Neuroendocrinology of bone. Pituitary 2024, 1–17. [CrossRef]
- Zhang, M.; Xuan, S.; Bouxsein, M.L.; von Stechow, D.; Akeno, N.; Faugere, M.C.; Malluche, H.; Zhao, G.; Rosen, C.J.; Efstratiadis, A.; et al. Osteoblast-specific Knockout of the Insulin-like Growth Factor (IGF) Receptor Gene Reveals an Essential Role of IGF Signaling in Bone Matrix Mineralization. J. Biol. Chem. 2002, 277, 44005–44012. [CrossRef]
- Saltarelli MA, Quarta A, Chiarelli F. Growth plate extracellular matrix defects and short stature in children. Ann Pediatr Endocrinol Metab. 2022 Dec 31;27(4):247–55.
- Witkowska-Sędek, E.; Pyrżak, B. Chronic inflammation and the growth hormone/insulin-like growth factor-1 axis. Central Eur. J. Immunol. 2020, 45, 469–475. [CrossRef]
- MacRae, V.; Wong, S.; Farquharson, C.; Ahmed, S. Cytokine actions in growth disorders associated with pediatric chronic inflammatory diseases (review).. Int. J. Mol. Med. 2006, 18, 1011–1018. [CrossRef]
- Bechtold, S.; Simon, D. Growth abnormalities in children and adolescents with juvenile idiopathic arthritis. Rheumatol. Int. 2014, 34, 1483–1488. [CrossRef]
- Choukair D, Hügel U, Sander A, Uhlmann L, Tönshoff B. Inhibition of IGF-I–related intracellular signaling pathways by proinflammatory cytokines in growth plate chondrocytes. Pediatr Res. 2014 Sep 18;76(3):245–51.
- Witkowska-Sędek, E.; Pyrżak, B. Chronic inflammation and the growth hormone/insulin-like growth factor-1 axis. Central Eur. J. Immunol. 2020, 45, 469–475. [CrossRef]
- Fazeli, P.K.; Klibanski, A. Determinants of GH resistance in malnutrition. J. Endocrinol. 2013, 220, R57–R65. [CrossRef]
- Soendergaard, C.; Young, J.A.; Kopchick, J.J. Growth Hormone Resistance—Special Focus on Inflammatory Bowel Disease. Int. J. Mol. Sci. 2017, 18, 1019. [CrossRef]
- Cirillo, F.; Lazzeroni, P.; Sartori, C.; Street, M.E. Inflammatory Diseases and Growth: Effects on the GH–IGF Axis and on Growth Plate. Int. J. Mol. Sci. 2017, 18, 1878. [CrossRef]
- Patel, L. Growth and Chronic Disease. 2007, 65, 129–136. [CrossRef]
- Amaro, F.; Chiarelli, F. Growth and Puberty in Children with Inflammatory Bowel Diseases. Biomedicines 2020, 8, 458. [CrossRef]
- Gupta, N.; Lustig, R.H.; Kohn, M.A.; McCracken, M.; Vittinghoff, E. Sex differences in statural growth impairment in Crohnʼs disease: Role of IGF-1. Inflamm. Bowel Dis. 2011, 17, 2318–2325. [CrossRef]
- Gaspari, S.; Marcovecchio, M.L.; Breda, L.; Chiarelli, F. Growth in juvenile idiopathic arthritis: the role of inflammation.. 2011, 29, 104–10.
- Fernandez-Vojvodich, P.; Palmblad, K.; Karimian, E.; Andersson, U.; Sävendahl, L. Pro-Inflammatory Cytokines Produced by Growth Plate Chondrocytes May Act Locally to Modulate Longitudinal Bone Growth. Horm. Res. Paediatr. 2012, 77, 180–187. [CrossRef]
- Fernandez-Vojvodich, P.; Zaman, F.; Sävendahl, L. Interleukin-6 acts locally on the growth plate to impair bone growth. Ann. Rheum. Dis. 2013, 72, e24–e24. [CrossRef]
- Nakajima, S.; Naruto, T.; Miyamae, T.; Imagawa, T.; Mori, M.; Nishimaki, S.; Yokota, S. Interleukin-6 inhibits early differentiation of ATDC5 chondrogenic progenitor cells. Cytokine 2009, 47, 91–97. [CrossRef]
- D’angelo, D.M.; Di Donato, G.; Breda, L.; Chiarelli, F. Growth and puberty in children with juvenile idiopathic arthritis. Pediatr. Rheumatol. 2021, 19, 1–13. [CrossRef]
- Mårtensson, K.; Chrysis, D.; Sävendahl, L. Interleukin-1β and TNF-α Act in Synergy to Inhibit Longitudinal Growth in Fetal Rat Metatarsal Bones. J. Bone Miner. Res. 2004, 19, 1805–1812. [CrossRef]
- Fernandez-Vojvodich, P.; Palmblad, K.; Karimian, E.; Andersson, U.; Sävendahl, L. Pro-Inflammatory Cytokines Produced by Growth Plate Chondrocytes May Act Locally to Modulate Longitudinal Bone Growth. Horm. Res. Paediatr. 2012, 77, 180–187. [CrossRef]
- Sederquist, B.; Fernandez-Vojvodich, P.; Zaman, F.; Sävendahl, L. RECENT RESEARCH ON THE GROWTH PLATE: Impact of inflammatory cytokines on longitudinal bone growth. J. Mol. Endocrinol. 2014, 53, T35–T44. [CrossRef]
- Zhao Y, Meng C, Wang Y, Huang H, Liu W, Zhang JF, et al. IL-1β inhibits β-Klotho expression and FGF19 signaling in hepatocytes. American Journal of Physiology-Endocrinology and Metabolism. 2016 Feb 15;310(4):E289–300.
- Zhao, Y.; Xiao, X.; Frank, S.J.; Lin, H.Y.; Xia, Y. Distinct mechanisms of induction of hepatic growth hormone resistance by endogenous IL-6, TNF-α, and IL-1β. Am. J. Physiol. Metab. 2014, 307, E186–E198. [CrossRef]
- E MacRae, V.; Farquharson, C.; Ahmed, S.F. The restricted potential for recovery of growth plate chondrogenesis and longitudinal bone growth following exposure to pro-inflammatory cytokines. J. Endocrinol. 2006, 189, 319–328. [CrossRef]
- Cirillo, F.; Catellani, C.; Lazzeroni, P.; Sartori, C.; Street, M.E. The Role of MicroRNAs in Influencing Body Growth and Development. Horm. Res. Paediatr. 2020, 93, 7–15. [CrossRef]
- Fedorczak, A.; Lewiński, A.; Stawerska, R. Involvement of Sirtuin 1 in the Growth Hormone/Insulin-like Growth Factor 1 Signal Transduction and Its Impact on Growth Processes in Children. Int. J. Mol. Sci. 2023, 24, 15406. [CrossRef]
- Catellani, C.; Ravegnini, G.; Sartori, C.; Angelini, S.; Street, M.E. GH and IGF System: The Regulatory Role of miRNAs and lncRNAs in Cancer. Front. Endocrinol. 2021, 12. [CrossRef]
- Shah A, Hashmi MF, Aeddula NR. StatPearls [Internet]. 2024. Chronic Kidney Disease-Mineral Bone Disorder (CKD-MBD).
- Cirillo, L.; De Chiara, L.; Innocenti, S.; Errichiello, C.; Romagnani, P.; Becherucci, F. Chronic kidney disease in children: an update. Clin. Kidney J. 2023, 16, 1600–1611. [CrossRef]
- Molinos, I.; Santos, F.; Carbajo-Perez, E.; Garcia, E.; Rodriguez, J.; Garcia-Alvarez, O.; Gil, H.; Ordoñez, F.; Loredo, V.; Mallada, L. Catch-up growth follows an abnormal pattern in experimental renal insufficiency and growth hormone treatment normalizes it. Kidney Int. 2006, 70, 1955–1961. [CrossRef]
- Hu, B.; Li, H.; Zhang, X. A Balanced Act: The Effects of GH–GHR–IGF1 Axis on Mitochondrial Function. Front. Cell Dev. Biol. 2021, 9. [CrossRef]
- Chen, N.; Wu, R.W.; Lam, Y.; Chan, W.C.; Chan, D. Hypertrophic chondrocytes at the junction of musculoskeletal structures. Bone Rep. 2023, 19, 101698. [CrossRef]
- Hallett, S.A.; Ono, W.; Ono, N. Growth Plate Chondrocytes: Skeletal Development, Growth and Beyond. Int. J. Mol. Sci. 2019, 20, 6009. [CrossRef]
- Ikegawa, K.; Hasegawa, Y. Fracture risk, underlying pathophysiology, and bone quality assessment in patients with Turner syndrome. Front. Endocrinol. 2022, 13, 967857. [CrossRef]
- Papadopoulou, A.; Bountouvi, E. Skeletal defects and bone metabolism in Noonan, Costello and cardio-facio-cutaneous syndromes. Front. Endocrinol. 2023, 14, 1231828. [CrossRef]
- LaCombe, J.M.; Roper, R.J. Skeletal dynamics of Down syndrome: A developing perspective. Bone 2019, 133, 115215–115215. [CrossRef]
- Homans, J.F.; Tromp, I.N.; Colo, D.; Schlösser, T.P.C.; Kruyt, M.C.; Deeney, V.F.X.; Crowley, T.B.; McDonald-McGinn, D.M.; Castelein, R.M. Orthopaedic manifestations within the 22q11.2 Deletion syndrome: A systematic review. Am. J. Med Genet. Part A 2017, 176, 2104–2120. [CrossRef]
- Funato, N. Craniofacial Phenotypes and Genetics of DiGeorge Syndrome. J. Dev. Biol. 2022, 10, 18. [CrossRef]
- Piché J, Van Vliet PP, Pucéat M, Andelfinger G. The expanding phenotypes of cohesinopathies: one ring to rule them all! Cell Cycle. 2019 Nov 2;18(21):2828–48.
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.-M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat. Rev. Genet. 2018, 19, 649–666. [CrossRef]
- Ishida, M. New developments in Silver-Russell syndrome and implications for clinical practice. Epigenomics 2016, 8, 563–580. [CrossRef]
- M Saal H, D Harbison M, Netchine I. GeneReviews® [Internet]. 2024. Silver-Russell Syndrome. [Cited 2024 Sep 10]. https://www.ncbi.nlm.nih.gov/books/NBK1324/.
- Pauli, R.M. Achondroplasia: a comprehensive clinical review. Orphanet J. Rare Dis. 2019, 14, 1–49. [CrossRef]
- Hypochondroplasia. In: Atlas of Genetic Diagnosis and Counseling. New York, NY: Springer US; 2012. p. 1105–12.
- B Bober M, A Bellus G, M Nikkel S, E Tiller G. GeneReviews® [Internet]. 2020. Hypochondroplasia. [Cited 2024 Sep 10]. https://www.ncbi.nlm.nih.gov/sites/books/NBK1477/.
- Fukami, M.; Seki, A.; Ogata, T. SHOX Haploinsufficiency as a Cause of Syndromic and Nonsyndromic Short Stature. Mol. Syndr. 2016, 7, 3–11. [CrossRef]
- Charoenngam, N.; Nasr, A.; Shirvani, A.; Holick, M.F. Hereditary Metabolic Bone Diseases: A Review of Pathogenesis, Diagnosis and Management. Genes 2022, 13, 1880. [CrossRef]
- Besio, R.; Chow, C.; Tonelli, F.; Marini, J.C.; Forlino, A. Bone biology: insights from osteogenesis imperfecta and related rare fragility syndromes. FEBS J. 2019, 286, 3033–3056. [CrossRef]
- Subramanian S., Anastasopoulou C., Krishnan Viswanathan V. StatPearls [Internet]. 2023. Osteogenesis Imperfecta.
- Nakamura-Utsunomiya, A. Bone Biomarkers in Mucopolysaccharidoses. Int. J. Mol. Sci. 2021, 22, 12651. [CrossRef]
- Melbouci, M.; Mason, R.W.; Suzuki, Y.; Fukao, T.; Orii, T.; Tomatsu, S. Growth impairment in mucopolysaccharidoses. Mol. Genet. Metab. 2018, 124, 1–10. [CrossRef]
- Jiang, Z.; Byers, S.; Casal, M.L.; Smith, L.J. Failures of Endochondral Ossification in the Mucopolysaccharidoses. Curr. Osteoporos. Rep. 2020, 18, 759–773. [CrossRef]
- Fuente, R.; García-Bengoa, M.; Fernández-Iglesias, .; Gil-Peña, H.; Santos, F.; López, J.M. Cellular and Molecular Alterations Underlying Abnormal Bone Growth in X-Linked Hypophosphatemia. Int. J. Mol. Sci. 2022, 23, 934. [CrossRef]
- Faienza, M.F.; D'Amato, E.; Natale, M.P.; Grano, M.; Chiarito, M.; Brunetti, G.; D'Amato, G. Metabolic Bone Disease of Prematurity: Diagnosis and Management. Front. Pediatr. 2019, 7, 143. [CrossRef]
- Cho, W.K.; Suh, B.-K. Catch-up growth and catch-up fat in children born small for gestational age. Korean J. Pediatr. 2016, 59, 1–7. [CrossRef]
- Bandeira, F.; de Oliveira, L.B.; Caldeira, R.B.; Toscano, L.S. Skeletal consequences of heart failure. Women's Heal. 2022, 18. [CrossRef]
- Trost, S.; Tesfaye, N.; Harindhanavudhi, T. The interplay between bone and heart health as reflected in medication effects: A narrative review. Women's Heal. 2023, 19. [CrossRef]
- Alonso-Gonzalez R, Massarella D, Swan L. Skeletal system in adult congenital heart disease. International Journal of Cardiology Congenital Heart Disease. 2023 Sep;13:100460.
- Ma, Y.; Qiu, S.; Zhou, R. Osteoporosis in Patients With Respiratory Diseases. Front. Physiol. 2022, 13, 939253. [CrossRef]
- Zhang, L.; Sun, Y. Muscle-Bone Crosstalk in Chronic Obstructive Pulmonary Disease. Front. Endocrinol. 2021, 12. [CrossRef]
- Danford, C.J.; Trivedi, H.D.; Bonder, A. Bone Health in Patients With Liver Diseases. J. Clin. Densitom. 2020, 23, 212–222. [CrossRef]
- Xia, S.; Qin, X.; Wang, J.; Ren, H. Advancements in the pathogenesis of hepatic osteodystrophy and the potential therapeutic of mesenchymal stromal cells. Stem Cell Res. Ther. 2023, 14, 1–12. [CrossRef]
- Saeki, C.; Saito, M.; Tsubota, A. Association of chronic liver disease with bone diseases and muscle weakness. J. Bone Miner. Metab. 2024, 42, 399–412. [CrossRef]
- Harris, L.; Senagore, P.; Young, V.B.; McCabe, L.R. Inflammatory bowel disease causes reversible suppression of osteoblast and chondrocyte function in mice. Am. J. Physiol. Liver Physiol. 2009, 296, G1020–G1029. [CrossRef]
- Gordon, H.; Burisch, J.; Ellul, P.; Katsanos, K.; Allocca, M.; Bamias, G.; Acosta, M.B.-D.; Braithwaite, T.; Greuter, T.; Harwood, C.; et al. ECCO Guidelines on Extraintestinal Manifestations in Inflammatory Bowel Disease. J. Crohn’s Colitis 2023, 18, 1–37. [CrossRef]
- Yang, H.R. Updates on bone health in children with gastrointestinal diseases. Ann. Pediatr. Endocrinol. Metab. 2020, 25, 10–14. [CrossRef]
- Braga CBM, Bizari L, Suen VMM, Marchini JS, Paula FJA de, Cunha SF de C da. Bone mineral density in short bowel syndrome: correlation with BMI and serum vitamins C, E and K. Arch Endocrinol Metab. 2015 Jun;59(3):252–8.
- Santos, F.; Díaz-Anadón, L.; A Ordóñez, F.; Haffner, D. Bone Disease in CKD in Children. Calcif. Tissue Int. 2021, 108, 423–438. [CrossRef]
- Lalayiannis, A.D.; Soeiro, E.M.D.; Moysés, R.M.A.; Shroff, R. Chronic kidney disease mineral bone disorder in childhood and young adulthood: a ‘growing’ understanding. Pediatr. Nephrol. 2023, 39, 723–739. [CrossRef]
- Zhang, H.; Yang, F.; Cao, Z.; Xu, Y.; Wang, M. The influence of iron on bone metabolism disorders. Osteoporos. Int. 2023, 35, 243–253. [CrossRef]
- Toxqui, L.; Vaquero, M.P. Chronic Iron Deficiency as an Emerging Risk Factor for Osteoporosis: A Hypothesis. Nutrients 2015, 7, 2324–2344. [CrossRef]
- Kaltsas, G.; Makras, P. Skeletal Diseases in Cushing’s Syndrome: Osteoporosis versus Arthropathy. Neuroendocrinology 2010, 92, 60–64. [CrossRef]
- Leszczyńska, D.; Szatko, A.; Papierska, L.; Zgliczyński, W.; Glinicki, P. Musculoskeletal complications of Cushing syndrome. Rheumatology 2023, 61, 197–208. [CrossRef]
- Wang, D.; Dang, C.-X.; Hao, Y.-X.; Yu, X.; Liu, P.-F.; Li, J.-S. Relationship between osteoporosis and Cushing syndrome based on bioinformatics. Medicine 2022, 101, e31283. [CrossRef]
- Loxton P, Narayan K, Munns CF, Craig ME. Bone Mineral Density and Type 1 Diabetes in Children and Adolescents: A Meta-analysis. Diabetes Care. 2021 Aug 1;44(8):1898–905.
- Hofbauer, L.C.; Busse, B.; Eastell, R.; Ferrari, S.; Frost, M.; Müller, R.; Burden, A.M.; Rivadeneira, F.; Napoli, N.; Rauner, M. Bone fragility in diabetes: novel concepts and clinical implications. Lancet Diabetes Endocrinol. 2022, 10, 207–220. [CrossRef]
- Parthasarathy, L.; Khadilkar, V.; Chiplonkar, S.; Khadilkar, A. Longitudinal growth in children and adolescents with type 1 diabetes. Indian Pediatr. 2016, 53, 990–992. [CrossRef]
- Csonka, V.; Varjú, C.; Lendvay, M. Diabetes mellitus-related musculoskeletal disorders: Unveiling the cluster of diseases. Prim. Care Diabetes 2023, 17, 548–553. [CrossRef]
- Zhu, S.; Pang, Y.; Xu, J.; Chen, X.; Zhang, C.; Wu, B.; Gao, J. Endocrine Regulation on Bone by Thyroid. Front. Endocrinol. 2022, 13, 873820. [CrossRef]
- Williams, G.R.; Bassett, J.H.D. Thyroid diseases and bone health. J. Endocrinol. Investig. 2017, 41, 99–109. [CrossRef]
- Sidhu, K.; Ali, B.; Burt, L.A.; Boyd, S.K.; Khan, A. Spectrum of microarchitectural bone disease in inborn errors of metabolism: a cross-sectional, observational study. Orphanet J. Rare Dis. 2020, 15, 1–13. [CrossRef]
- Langeveld, M.; Hollak, C.E.M. Bone health in patients with inborn errors of metabolism. Rev. Endocr. Metab. Disord. 2018, 19, 81–92. [CrossRef]
- Kalra, S.; Grimer, R.J.; Spooner, D.; Carter, S.R.; Tillman, R.M.; Abudu, A. Radiation-induced sarcomas of bone. J. Bone Jt. Surgery. Br. Vol. 2007, 89, 808–813. [CrossRef]
- Hua, J.; Huang, J.; Li, G.; Lin, S.; Cui, L. Glucocorticoid induced bone disorders in children: Research progress in treatment mechanisms. Front. Endocrinol. 2023, 14. [CrossRef]
- A Alfaedi, S.; Kubbara, M.F.; A Alaithan, A.; Alhudhaif, H.M.; A Al Abdullah, A.; Sahool, H.M.; AL Jawad, M.S.; A Almatar, M.; Alnakhli, I.R.; A Altawili, M. Beneath the Surface: Exploring Hidden Threats of Long-Term Corticosteroid Therapy to Bone Density. Cureus 2024, 16, e55109. [CrossRef]
- Cooper, M.S. Glucocorticoids in bone and joint disease: the good, the bad and the uncertain. Clin. Med. 2012, 12, 261–265. [CrossRef]
- Dixit, M.; Poudel, S.B.; Yakar, S. Effects of GH/IGF axis on bone and cartilage. Mol. Cell. Endocrinol. 2020, 519, 111052–111052. [CrossRef]
- Lindsey, R.C.; Mohan, S. Skeletal effects of growth hormone and insulin-like growth factor-I therapy. Mol. Cell. Endocrinol. 2015, 432, 44–55. [CrossRef]
- Oichi T, Kodama J, Wilson K, Tian H, Imamura Kawasawa Y, Usami Y, et al. Nutrient-regulated dynamics of chondroprogenitors in the postnatal murine growth plate. Bone Res. 2023 Apr 21;11(1):20.
- Prentice A, Schoenmakers I, Ann Laskey M, de Bono S, Ginty F, Goldberg GR. Symposium on ‘Nutrition and health in children and adolescents’ Session 1: Nutrition in growth and development Nutrition and bone growth and development. Proceedings of the Nutrition Society. 2006 Nov 21;65(04):348–60.
- Kelly, R.R.; McDonald, L.T.; Jensen, N.R.; Sidles, S.J.; LaRue, A.C. Impacts of Psychological Stress on Osteoporosis: Clinical Implications and Treatment Interactions. Front. Psychiatry 2019, 10, 200. [CrossRef]
- Legroux-Gerot, I.; Vignau, J.; Collier, F.; Cortet, B. Bone loss associated with anorexia nervosa. Jt. Bone Spine 2005, 72, 489–495. [CrossRef]
- Mollard, E.; Bilek, L.; Waltman, N. Emerging evidence on the link between depressive symptoms and bone loss in postmenopausal women. Int. J. Women's Heal. 2017, ume 10, 1–9. [CrossRef]
- Wang, S.-T.; Ni, G.-X. Depression in Osteoarthritis: Current Understanding. Neuropsychiatr. Dis. Treat. 2022, ume 18, 375–389. [CrossRef]
- Rajkumar V, Waseem M. StatPearls [Internet]. . 2023. Familial Short Stature. [Cited 2024 September 10]. https://www.ncbi.nlm.nih.gov/books/NBK559123/.
Figure 1.
Membranous ossification vs. Endochondral ossification. Mesenchymal stem cells (MSCs) present in the bone marrow can differentiate to become either chondrocytes or osteoblasts. Osteoblasts buildup bone directly through a process called intramembranous ossification (in red), while chondrocytes proliferate, hypertrophy, mineralize, and then new bone is deposited onto the cartilaginous matrix through a process called endochondral ossification (in blue). Endochondral ossification forms the bones of the limbs and long bones, while membranous ossification forms the bones of the axial skeleton. In this process, various factors and precursors are involved from mesenchymal cell differentiation to hypertrophic chondrocyte. Created with BioRender.com.
Figure 1.
Membranous ossification vs. Endochondral ossification. Mesenchymal stem cells (MSCs) present in the bone marrow can differentiate to become either chondrocytes or osteoblasts. Osteoblasts buildup bone directly through a process called intramembranous ossification (in red), while chondrocytes proliferate, hypertrophy, mineralize, and then new bone is deposited onto the cartilaginous matrix through a process called endochondral ossification (in blue). Endochondral ossification forms the bones of the limbs and long bones, while membranous ossification forms the bones of the axial skeleton. In this process, various factors and precursors are involved from mesenchymal cell differentiation to hypertrophic chondrocyte. Created with BioRender.com.
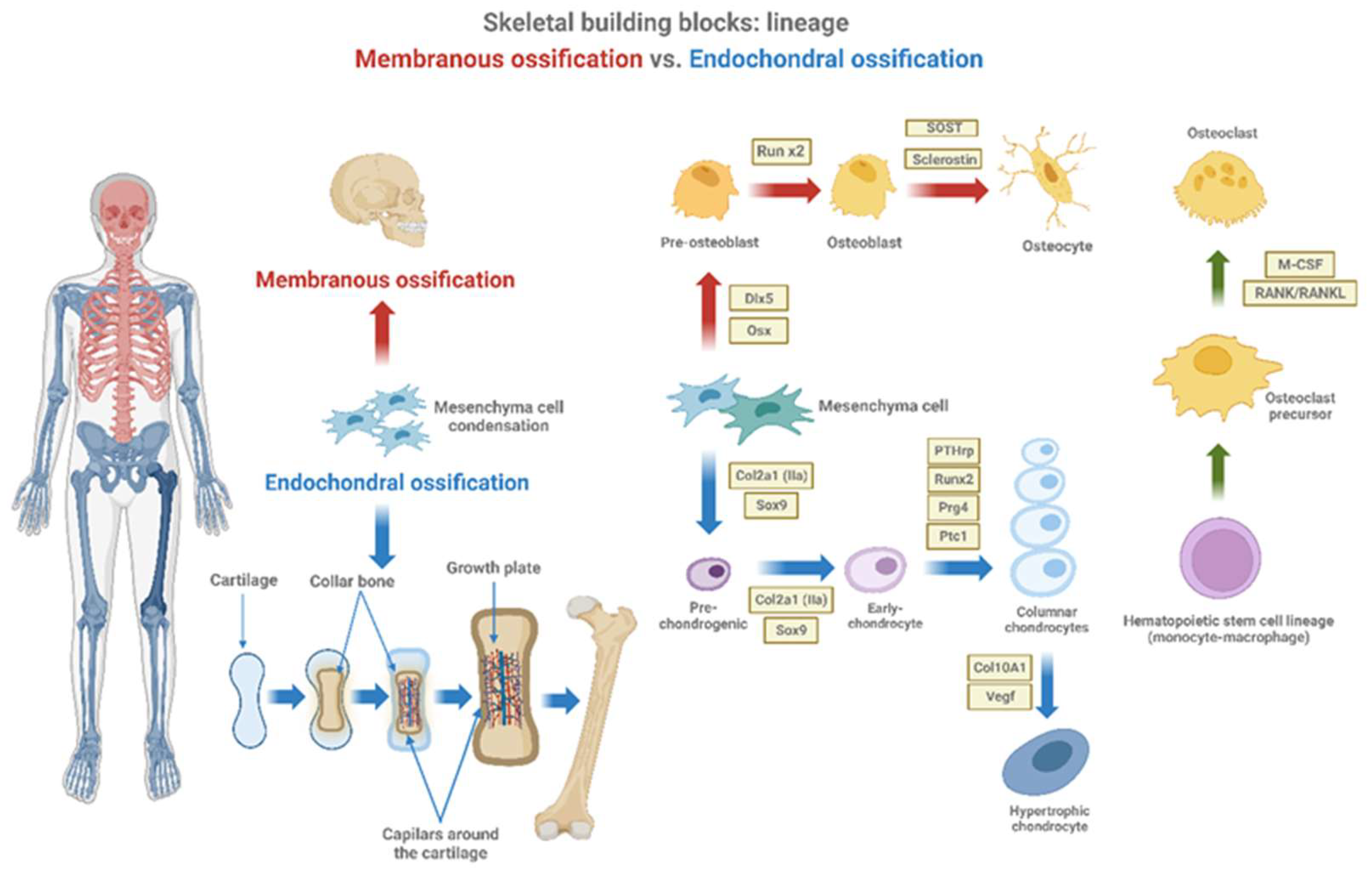
Figure 2.
Histological sections of the tibial epiphysis of a rat of 35 days of age. Fig. is a Paraffin section stained with Alcian blue/acid fuchsin showing the cartilage of the growth plate (stained in blue) and the bone tissue (stained in fuchsia) in a typical endochondral ossification process where the cartilage is progressively replaced by bone. Fig. B is a magnification of figure A showing that longitudinal septa of the growth plate serve as a scaffold upon which osteoblasts deposite mineralized osseous matrix. Fig. C is a semithin section of a rat tibia showing a group of osteoblasts secreting bone matrix on a bone trabecula in a representative membranous ossification process.
Figure 2.
Histological sections of the tibial epiphysis of a rat of 35 days of age. Fig. is a Paraffin section stained with Alcian blue/acid fuchsin showing the cartilage of the growth plate (stained in blue) and the bone tissue (stained in fuchsia) in a typical endochondral ossification process where the cartilage is progressively replaced by bone. Fig. B is a magnification of figure A showing that longitudinal septa of the growth plate serve as a scaffold upon which osteoblasts deposite mineralized osseous matrix. Fig. C is a semithin section of a rat tibia showing a group of osteoblasts secreting bone matrix on a bone trabecula in a representative membranous ossification process.
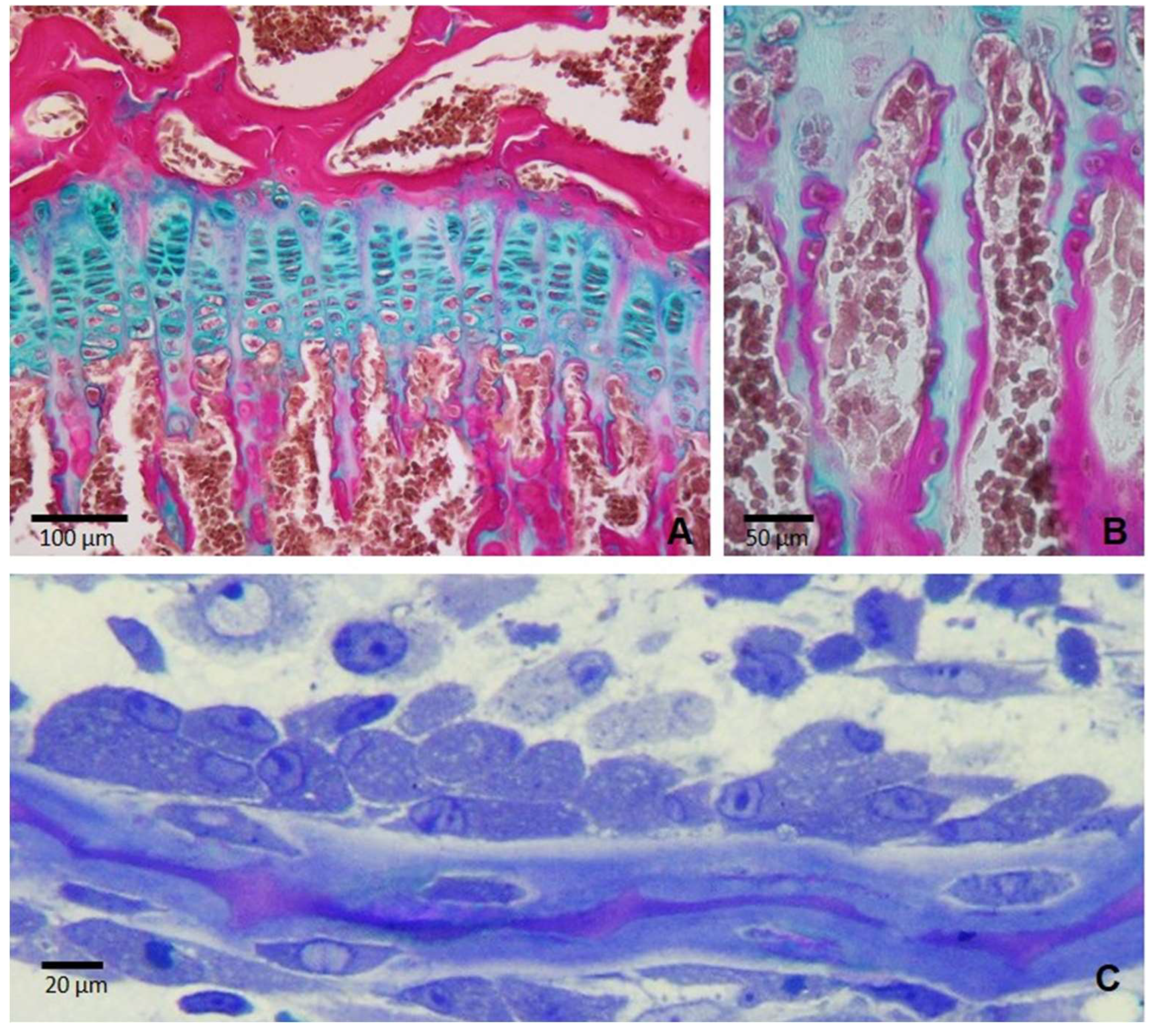
Figure 3.
Schematic diagram of a longitudinal section of the epiphyseal growth plate. Growth plate is a cartilage-like structure situated between the metaphysis and the diaphysis of all long bones, as for example the tibia. It is hyaline cartilage. Growth plate is histological formed by three zones. The epiphysis is above the reserve zone, is followed by the proliferative and the prehypertrophic zone, finally the metaphysis is below called as the hypertrophic zone. Created with BioRender.com.
Figure 3.
Schematic diagram of a longitudinal section of the epiphyseal growth plate. Growth plate is a cartilage-like structure situated between the metaphysis and the diaphysis of all long bones, as for example the tibia. It is hyaline cartilage. Growth plate is histological formed by three zones. The epiphysis is above the reserve zone, is followed by the proliferative and the prehypertrophic zone, finally the metaphysis is below called as the hypertrophic zone. Created with BioRender.com.
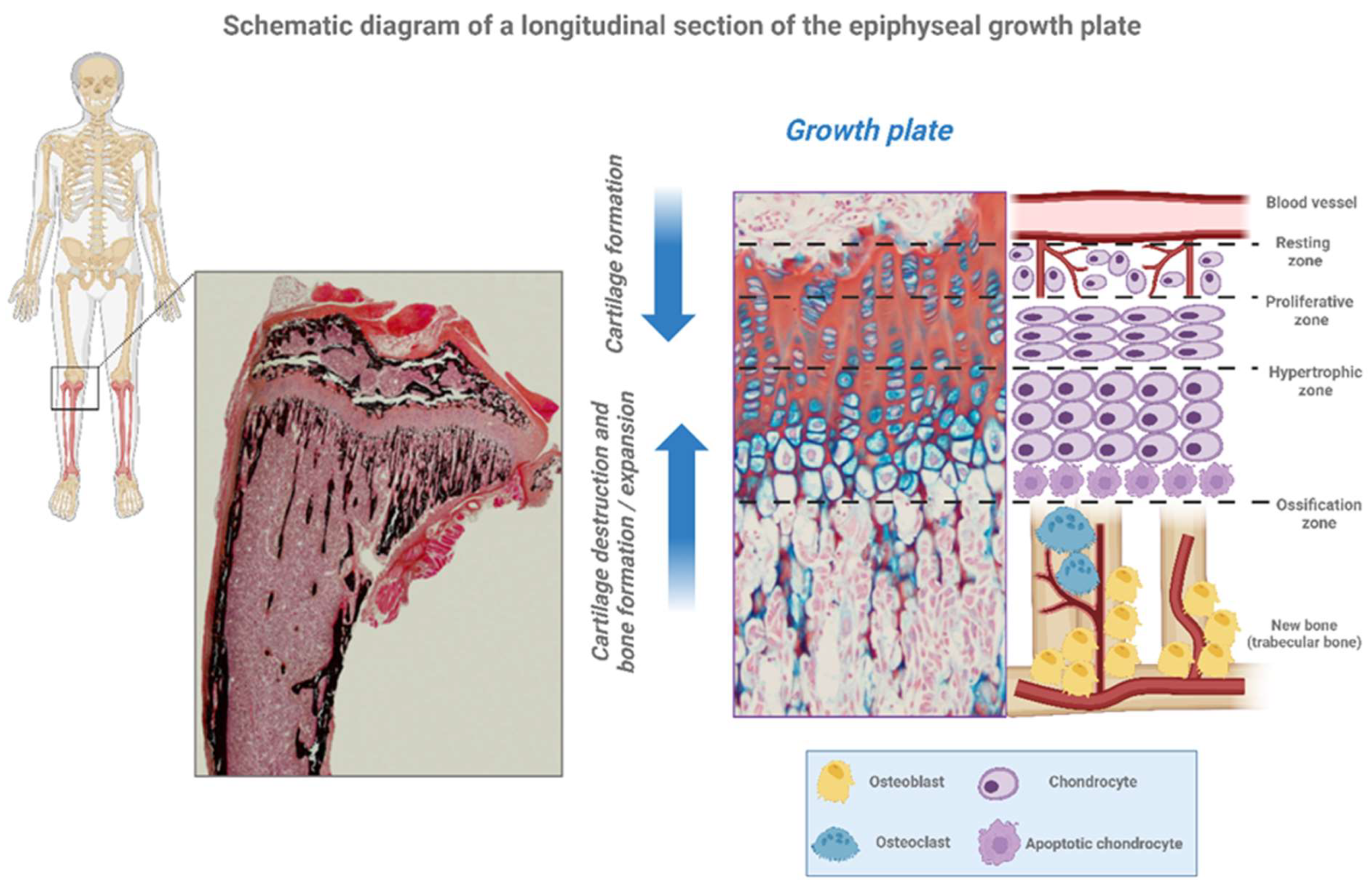

Table 1.
Description of the growth plate parts.
| Epiphyseal Growth Plate | Description |
|---|---|
| Resting Zone (RZ) | Quiescent chondrocytes. These resting cells can be moved to the zone of proliferation. |
| Proliferative Zone (PZ) | Chondrocytes are arranged in columns and multiply. These cells, undergo rapid mitosis by growth hormone (GH) effect. |
| Pre-Hypertrophic Zone (PHZ) | Matrix synthesis is started |
| Hypertrophic Zone (HZ) | Chondrocytes stop proliferation and change their phenotype to hypertrophic by synthesis of proteins and swelling. |
|
Calcification Zone (CZ) |
The terminally hypertrophic chondrocytes undergo apoptosis and differentiation, and the cartilaginous mold welcomes the osteoblasts so that they become osteoclasts and thus induce the formation of new bone tissue. Simultaneously, vascular invasion occurs. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



