
Submitted:
15 October 2024
Posted:
15 October 2024
You are already at the latest version
Abstract
Compounds of natural origin found in varying quantities in plant-based products constitute a highly significant category, possessing structural significance as well as the capacity to regulate oxidative processes. The activity of these compounds may be modulated by the composition of the biological milieu in which they operate, the pH of the environment, or the presence of metal cations in plants or plant extracts. This work shows the synthetic methodology and the optimization process that took place to synthesize a catechin-copper complex which demonstrated antioxidant activity tested for iron chelation ability, hydroxyl radical test and inhibition of lipoxygenase (15-LOX) activity. Antidiabetic assay was performed by determination of the alpha-amylase and alpha-glucosidase inhibition assays and the antibacterial activity showed results against Staphylococcus aureus and Candida albicans. The complex was characterized by FT-IR, UV-VIS and EDX techniques and the biological activity assays were tested in triplicate to fully confirm the activity.
Keywords:
copper-catechin-complex
; antioxidant activity
; antibacterial
1. Introduction
Metal chelation is an important property of flavonoids, and it holds considerable importance due to its profound influence on the magnitude of the pharmacological effects that flavonoids can exert by regulating the bioavailability of diverse minerals and ions. Catechins are a class of natural polyphenolic compounds known as flavanols, which are members of the flavonoid family. Abundant concentrations of these compounds can be found in a diverse range of fruits, vegetables, and plant-based beverages. The term “catechin” is etymologically derived from the scientific name of the Cutch tree, known as Acacia catechu L.f. Fresh tea leaves, rock-rose leaves, broad beans, red wine, black grapes, strawberries, and apricots are known to contain elevated levels of catechin [1,2,3].
Certain flavonoids have been identified to possess the ability to chelate plurivalent metals such as: iron, zinc and copper. According to [4] catechin has the ability to mitigate the harmful consequences of oxidation on the erythrocyte membrane. This is achieved by counteracting the impact of various oxidizing agents that trigger the liberation of iron in its free, redox-active state. The binding sites of metals to flavonoids are located in the B ring, at the 3-hydroxyl and 4-oxo groups in the heterocyclic ring C, as well as at the 4-oxo and 5-hydroxyl groups between the C and A groups [5,6,7].
Chelates, which are complexes formed by flavonoids with metal ions, can be observed in the exhibit significant effects in terms of their antioxidative, antibacterial, and antidiabetic activities. Multiple studies have demonstrated that the anti-radical properties of flavonoid-metal complexes are more potent compared to flavonoid itself. Kostyuk et al. (2001) [8] conducted a study that provided evidence for the enhanced efficacy of rutin, quercetin, and catechin when complexed with Fe (II), Fe (III), Cu (II), or Zn (II) compared to their free forms. This increased effectiveness can be attributed to the incorporation of an additional dismutation center [9,10,11].
The human body possesses a significant array of defensive mechanisms to protect itself from the detrimental effects of oxidative stress. Enzymes, namely superoxide dismutase (SOD), catalases (CAT), and glutathione peroxidases (Gpx), are integral components of the cellular defense mechanism against reactive oxygen species (ROS). These enzymes are further aided by the activities of glutathione reductases (GR) and glucose-6-phosphate dehydrogenases. Copper (Cu) is a constituent of cytosolic superoxide dismutase (SODs), while manganese (Mn) is a component of mitochondrial SODs. Additionally, there exist extracellular superoxide dismutase (SODs). The Cu/Zn plasma ratio holds significance in relation to SOD activity, as a reduction in enzymatic cofactors results in a decline in SOD activity [12]. The antioxidant capacities of flavonoids exhibit a notable superior activity in comparison to vitamins. Flavonoids possess the capability to directly counteract free radicals through the process of hydrogen atom donation. The biological activities of flavonoids in an in vitro setting are contingent upon the arrangement of functional groups within their molecular structure. According to [8], the antioxidant activity is consistently influenced by the arrangement and quantity of hydroxyl groups.
The present study has contributed to an enhanced comprehension of the factors that dictate the associations between the molecular structure of flavonoids and their capacity to engage in metal complexation and it is focused on understanding the complex synthesis and to fully understand the antibacterial, antioxidant and antidiabetic activity of the synthesized copper-catechin complex[13] .
2. Materials and Methods
2.1. Starting Materials
Commercial starting materials were used as provided and the all the solvents used for the reactions were spectrophotometric grade. (+)-catechin, copper sulphate, alpha-glucosidase, alpha-amylase, p-nitrophenyl-alpha-D-glucopyranoside (pNFG), sodium carbonate, starch, dinitrosalicilic acid, sodium hydroxide, iron sulphate, hydrogen peroxide, boric acid, linoleic acid, soy lipoxygenase, sodium acetate, ferrozine ethanol, methanol, DMSO, chloroform, acetone, were all provided by Sigma-Aldrich and used without further purifications. Ciprofloxacin 10 µg (Oxoid) and nystatin 100 µg (HiMedia Laboratory).
2.2. Synthesis of Cat-Cu Complex
One of the methods we used in order to obtain the catechin-copper complex provides for the use of catechin and copper sulphate in a 1:1 molar ratio. The catechin is dissolved in methanol under continuous stirring, then the copper salt is added gradually over 30 minutes, after which the reaction is maintained for 3 hours at 40℃ under continuous stirring. Various systems were tried to adjust the pH, but the best results were obtained with 1N NaOH at pH=8.5. The reaction was stopped and allowed to cool to room temperature. The solvent was removed by filtration, and the precipitate was washed several times with acetone and dried in an oven for 48h at 40℃ to make sure all the residual solvent is removed, and the precipitate is dried to constant mass. The yielded complex thus obtained was further used for structural confirmation determinations, UV-VIS absorption spectroscopy, morphological analysis and EDX.
2.3. Characterization of the Complex
- Instrumentation
Structural characterization of the individual reagents and reaction products was carried out by Fourier-transform infrared spectroscopy (FTIR). The FTIR spectra were registered on an FT-IR Bruker Vertex 70 Spectrophotometer by ATR technique using samples as powders. The solubility of the synthesized complex was qualitatively evaluated by the dissolution of 5 mg of complex in 1ml of several solvents (chloroform, acetone, ethanol, methanol, DMSO). The UV-VIS spectra were recorded on a Shimadzu UV-1280 spectrophotometer using diluted polymer solution approx.10 ֿ5 M) in methanol. The morphology of the polymer coatings was evaluated by SEM using Verios G4 UC Scanning Electron Microscope from Thermo Fisher Scientific (Waltham, MA, USA).
- b. In vitro biological activity assay
For the newly synthetized complex, the investigation regarding the biological activity was assessed by the in vitro antioxidant, antidiabetic and antibacterial activity.
Antioxidant assay
Antioxidant potential was evaluated by three different methods: iron chelation test, hydroxyl radical test and inhibition of lipoxygenase (15-LOX) activity [13,14,15].
For the iron (II) chelation test, ferrozine was used, due to its ability to form complexes with ferrous iron in a quantitative manner, resulting in the production of a pink color. Nevertheless, the introduction of chelating agents results in the disturbance of complex formation, which leads to a decreasing in the intensity of coloration. The monitoring of the ferrous ion was conducted through the measurement of the formation of a pink ferrous ion-ferrozine complex at a wavelength of 562 nm. The methodology employed in this study closely resembled the approach outlined by Burlec et al. [16]. Using 0.2 mL of sample solution in ultrapure water, 0.74 mL of 0.1 M acetate buffer solution (pH 5.25), 0.02 mL of 2 mM ferrous sulphate solution, 0.2 M hydrochloric acid were added, and after stirring for 10-15 seconds, 0.04 mL of 5 mM ferrozine solution was slowly poured. After 10 minutes of resting in the dark, the absorbance of the solution at 562 nm was determined using a standard solution, prepared under the same conditions as the sample (the ferrous sulphate solution was replaced by ultrapure distilled water). In parallel, the control solution and its blank were prepared, the control containing 0.2 mL of ultrapure water, 0.74 mL of 0.1 M acetate buffer solution (pH 5.25), 0.02 mL of 2 mM ferrous sulphate solution in 0.2 M hydrochloric acid, and after stirring for 10-15 seconds, 0.04 mL of 5 mM ferrozine solution [16,17].
The chelating capacity of the ferrous ion was calculated according to the formula:
where:
activity % = 100 x (Ac – Ap)/(Ac)
Ac - absorbance of the control solution,
Ap - absorbance of the sample solution
The hydroxyl radical is generated through the reaction between the ferrous ion and hydrogen peroxide. This hydroxyl radical then reacts with salicylic acid, resulting in the formation of a pink-purple compound that exhibits its highest absorbance at a wavelength of 562 nm [5,13]. The absorbance of the control sample was measured at a wavelength of 562 nm, relative to the control sample where the ferrous sulfate solution was substituted with distilled water. In this experiment, a volume of 0.225 mL of sample solution dissolved in dimethyl sulfoxide (DMSO) was combined with 0.750 mL of a 1.5 mM iron (II) sulfate solution, 0.9 mL of a 20 mM sodium salicylate solution, and 0.525 mL of a 6 mM hydrogen peroxide solution. The mixture was incubated for 30 minutes at 37℃. Subsequently, the mixture was allowed to cool down to the ambient room temperature. The absorbance of the sample (referred to as the control) was then measured at a wavelength of 562 nm. This measurement was compared to the control sample, where the ferrous sulfate solution was substituted with bi-distilled water. The positive control underwent the same processing conditions as the samples, with the exception that dimethyl sulfoxide (DMSO) was utilized in place of the sample solution [12,18,19].
The determination of lipoxygenase inhibition involved assessing the activity of 15-sLOX by monitoring spectrophotometrically the formation of reaction products at a wavelength of 234 nm. The experimental procedures involved conducting all reactions with a final volume of 2 ml and stirring the mixture using a magnetic bar at ambient temperature. The experimental setup involved the utilization of a reaction medium comprising a HEPES buffer with a concentration of 0.1 M and a pH value of 7.4. The experimental procedure involved the addition of the inhibitor (complex) in methanol to the cuvette containing the substrate buffer, followed by the subsequent addition of the enzyme. [1,9].
0.05 mL of 15-lipoxygenase solution in borate buffer pH 9 was treated with 0.05 mL of analyte solution diluted in DMSO and the mixture was left to rest for 10 minutes at room temperature, after which 2 mL of 0.16 mM linoleic acid solution in 0.1M borate buffer pH 9 was added. The absorbance of the solution was recorded at 234 nm, in the range of 0-120 seconds. In parallel, the positive control was processed in which the solution to be analyzed was replaced by DMSO. Gallic acid was used as a reference substance, and solutions of gallic acid in DMSO were processed under the same conditions as the methanolic extract.
All determinations were performed in triplicate, the results being expressed as the mean of three determinations ± standard deviation.
The lipoxygenase inhibition capacity was calculated according to the formula:
where:
activity% = (AEFI – AECI) x 100/AEFI
AEFI - represents the difference between the absorbance of the enzyme solution without inhibitor at 90 seconds and the absorbance of the same solution at 30 seconds;
AECI - represents the difference between the absorbance of the inhibitor-treated enzyme solution (sample) at 90 seconds and the absorbance of the same solution at 30 seconds.
Antidiabetic activity
Antidiabetic assay was performed by determining the alpha-amylase and alpha-glucosidase assays.
Alpha-amylase catalyzes the hydrolysis of starch with the release of glucose which reacts with dinitrosalicylic acid and forms a yellow-orange colored compound with maximum absorbance at 540 nm. In the presence of enzyme inhibitors, enzyme activity is blocked or reduced with a reduction in the absorbance of the solution at 540 nm. A 0.4 mL sample solution dissolved in DMSO was combined with 0.08 mL of an enzyme solution with a concentration of 2 IU/mL, 0.2 mL of a starch solution with a concentration of 0.5%, and 0.16 mL of a phosphate buffer with a concentration of 20 mM and a pH of 6.7. The resulting mixture was incubated at 37°C 10 minutes. Following a time interval of 10 minutes, a volume of 0.32 mL of dinitrosalicylic reagent solution was introduced into the reaction mixture, which was subsequently subjected to a temperature of 100℃ for 15 minutes. The solution underwent a cooling process, following which the absorbance of the sample was measured relative to the sample control that lacked the addition of any enzyme. The positive control was acquired using the same procedure, employing DMSO instead of the complex [9,20,21,22].
The enzyme inhibition capacity was calculated according to the formula:
where:
activity % = 100 x (Ac – Ap)/(Ac)
Ac - represents the absorbance of the control solution,
Ap - represents the absorbance of the sample solution.
Alpha-glucosidase catalyzes the hydrolysis of pNFG to p-nitrophenylphosphate, a yellow compound with maximum absorbance at 405 nm. In the presence of inhibitors, the enzyme activity decreases or is blocked with the reduction of absorbance of the solution at 405 nm [23,24].
We prepared the solutions following this protocol: 0.08 mL of 2 IU/mL enzyme solution, 0.2 mL of 0.5% starch and 0.16 mL of 20 mM phosphate buffer solution pH 6.7 were added to 0.4 mL of sample solution in dimethyl sulfoxide and the solution was maintained for 10 minutes at 37oC. After 10 minutes, 0.32 m dinitrosalicylic reagent solution was added, and the reaction was maintained for 15 minutes at 100℃. The solution was cooled, and the absorbance of the sample was read against the sample control in which no enzyme was added. The positive control was processed under the same conditions as the samples, but dimethyl sulfoxide was used instead of the sample solution.
The enzyme inhibition capacity was calculated according to the formula:
where:
activity % = 100 x (Ac – Ap)/(Ac)
Ac - absorbance of the control solution,
Ap - absorbance banks of the sample solution
The IC50 value, expressed in µg sample/mL final solution, was calculated for samples exhibiting an enzyme inhibition capacity exceeding 50%. The IC50 value was determined by considering the concentration of the antioxidant agent solution that corresponds to an activity of 50%, using linear interpolation based on the first lower and higher values of 50%. The experiments were conducted in triplicate, and the outcomes were reported as t the mean of three determinations ± standard deviation. In order to establish accurate correlations and determine statistical significance, a t-student analysis was conducted.
Antibacterial activity
The antibacterial activity was determined using the disk diffusion test against Staphylococcus aureus ATCC 25923, Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853, Klebsiella and Candida albicans ATCC 90028.
We tested the antimicrobial activity of the synthesized complexes both by applying qualitative methods, through the diffusometric method, and by quantitative methods, applying the method of microdilutions in broth (CLSI 2023, CLSI 2009). For the tests, the microorganisms were incubated in inclined tubes, with nutrient agar for bacteria, respectively Sabouraud agar for fungi.
A standardized inoculum of 5x104UFC/ml of the test bacterial strain was seeded in a discontinuous concentration gradient of the test products, made in Mueller-Hinton broth/Liquid Sabouraud medium. After overnight incubation at 37℃, we determined the minimum inhibitory concentration, as the lowest antibiotic dilution that inhibited visible bacterial growth.
The limits of the concentrations tested for each sample were between 0.01-10 mg/mL. CMB/CMF values were determined by transferring 0.1 µl of each well that showed complete inhibition of visible growth onto the surface of a solid medium plate. The subcultures were incubated for 24 hours at 37°C (for bacteria) and 30°C (for fungi). The CMB/CMF value was considered as the lowest compound concentration that kills 99.9% of the tested microorganisms.
The inoculum was obtained from the 24-hour culture of each test microorganism and 48 hours for Candida spp. The suspension in isotonic saline solution (0.9% NaCl) was adjusted to the density of the 0.5 McFarland standard, with the final density of ~108 Colony Forming Units/ml (CFU/ml) for bacteria, respectively ~107 CFU/ml for Candida (CLSI, 2023). Mueller-Hinton agar medium was inoculated with this suspension, melted and brought to a temperature of 44-45°C. The inoculated medium was distributed in Petri plates with a diameter of 9 cm, in a volume of 10 ml/plate. For Candida spp. we used Sabouraud agar medium [10,25,26,38].
In stainless steel cylinders, with a diameter of 7 mm, placed on the surface of the medium inoculated with each test microorganism, we deposited with a micropipette a volume of 100 µl of each sample, after which the plates were incubated at 37℃ for bacteria, respectively 30℃ for Candida spp. After 24-48 hours of incubation, the antimicrobial activity was evaluated by measuring the diameter of the zone of inhibition of the growth of the test microorganisms compared to the control (reference standard) - disks impregnated with precise concentrations of antibiotics: ciprofloxacin 10 µg (Oxoid) and nystatin 100 µg (HiMedia Laboratory) [27,36,37].
Each sample was tested 3 times, and the final result represents the average of the values of three diameters of the zones of inhibition of the growth of test microorganisms.
3. Results and Discussions
3.1. Synthesis and Complexation Yield
The primary parameters evaluated in this study included adjusting the molar ratio of the reactants, the choice of solvents for achieving optimal yield, control of reaction temperature, and adjustment of pH solution in order to encourage precipitate formation and improve the yield of the reaction. The resulting precipitate was confirmed to be the desired complex, after which we determined the yield of the reaction by weighing the dry precipitate.
Furthermore, the stirring velocity and duration were modified to promote enhanced interaction between the main reactants (catechin and copper salt) and the reaction solvent. Table I presents the reaction yields achieved by varying these parameters. Despite our attempts to modulate the reaction medium through pH adjustments, the most intriguing outcomes were observed in the system where the pH was adjusted to 8.5 using a 1N NaOH solution.

Table 1.
Yields obtained changing the pH and solvent.
3.2. UV-VIS
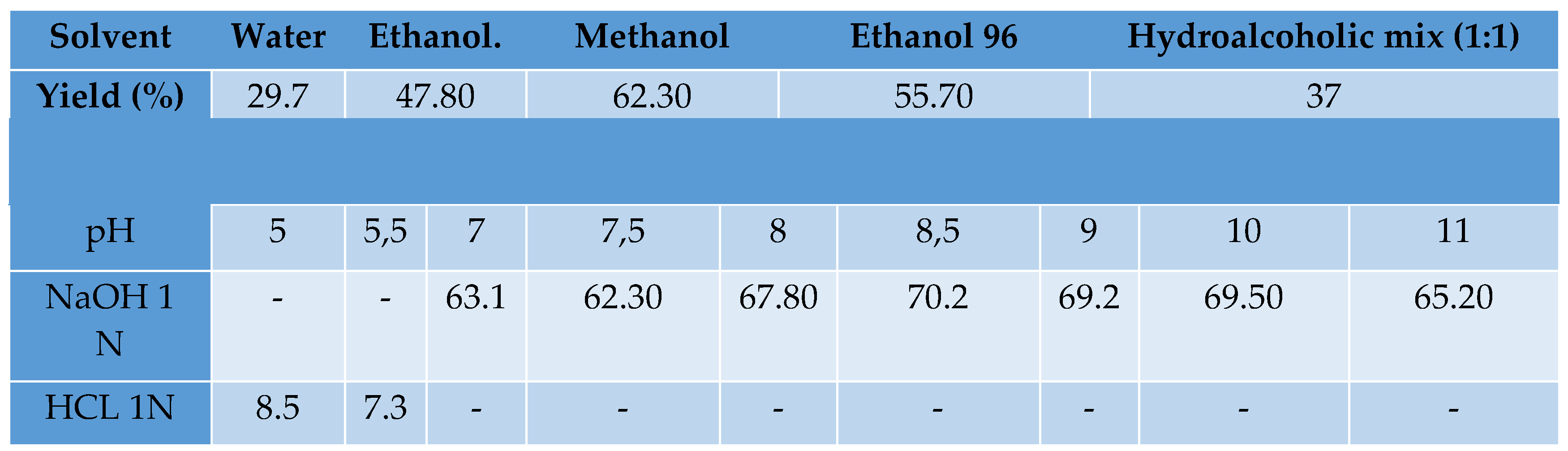
The absorption spectra of the synthesized catechin-copper complex were measured using UV-VIS spectroscopy, as shown in Figure 1. The results indicate a significant shift in the absorption maximum of the complex when compared to that of catechin. This shift might serve as evidence that the complex has been formed. The UV-VIS spectra provided insights into the mechanism underlying the formation of the catechin-copper bond. The presence of the copper ion facilitated a transition of the π → π∗ nature, leading to the formation of a condensed ring structure with catechin. This interaction involved the participation of the 3-OH and 4-oxo functional groups. The lack of the hydroxyl (OH) group in the complex can be ascribed to its reduced protonic acidity and the presence of steric hindrance.
3.3. FT-IR
Upon binding with copper ion, the v(C=O) band of free catechin in the infrared spectrum exhibited a shift from 1657 cm−1 to 1598 cm−1 (Figure 2). This phenomenon can be attributed to the establishment of a coordination bond between the oxygen atom of the carbonyl group and the metal ions.
The formation of a novel ring within the complex resulted in the amplification of the conjugative effect, leading to a noticeable shift in the characteristic infrared absorption band ν(C=C) from 1619 cm−1 to 1509 cm−1. The identification of vibrations at a wavenumber of 634 cm−1 in the complex indicated the existence of an O-Cu bond, which was absent in the unbound catechin molecule.
Figure 3 depicts the proposed structure for the catechin-Cu(II) complex based on the findings obtained from FT-IR analysis.
3.4. Morphological Analysis
Scanning electron microscopy (SEM) was employed to capture images of the cat-Cu complex at various resolutions such as: 20, 5, 2, and 1 μm. The obtained images exhibit a fibrous morphology characterized by an acicular structure and an irregular fracture pattern (Figure 4).
The present study documents the observation of the formation of minute clusters of crystals measuring 1 μm in diameter. These clusters exhibit adhesion at certain larger segments, potentially suggesting the presence of aggregation centers with varying degrees of complexity.
3.5. EDX
The chemical composition and elemental presence within the chemical structure of the synthesized catechin-copper complex were confirmed through EDX analysis (Figure 5). Therefore, the presence of a copper content of 39.9% by weight is detected in the already purified and spectroscopically analyzed precipitate, thereby confirming the formation of the complex. These observations align with the data acquired through the utilization of FT-IR spectroscopy and UV-VIS spectroscopy.
The peaks registered by EDX analysis indicate a high concentration of copper in the sample, Figure 5 shows the analyzed area on a micron scale as well as the different concentration of atoms by weight and by their concentration. The EDX technique focuses mainly on the analysis of electrons found in the K layer of atoms.
3.6. In Vitro Antioxidant Activity
Copper, copper salt and the cat-Cu complex have a higher ferrous ion chelation capacity compared to that of catechin, but comparing the data there is the possibility of copper ions interfering with ferrozine that forms the colored complex with the Fe2+ ion. Maintaining the chelating capacity of catechin even after complexation with copper ions has antioxidant benefits through the catechin itself, but also through the copper ions that enter the body can be used for the synthesis of superoxide dismutase, an enzyme with a role in the decomposition of the superoxide radical anion. Figure 6 shows the similar activity of the catechin and cat-Cu complex in terms of iron chelation capacity [7,13,28,29].
3.7. Lipoxygenase Activity
The test to evaluate the ability to inhibit lipoxygenase highlighted the fact that by complexing catechin with copper ions, the inhibitory effect is greatly reduced both by comparison with catechin, but also by comparison with copper ions. Studies in which catechin and copper were used showed that the antioxidant effects of catechin are better maintained when synthesizing nanoparticles with zinc oxide, compared to the catechin-zinc ion complex [3].
Both catechin and cat-Cu complex reduce lipoxygenase activity, reducing the amount of linoleic acid hydroperoxides (the substrate used for this determination). The inhibitory activity of cat-Cu complex is compared to that of catechin is shown graphically in Figure 7.
3.8. Determination of the Scavenger Capacity of the Hydroxyl Radical
The hydroxyl radicals are capable of altering the spatial structure of functional groups within protein structures, thereby impacting their biological function. This modification can lead to the development of pathological conditions such as atherosclerosis, neurological disorders, and cancer [18].
In both the lipoxygenase inhibition test and the hydroxyl radical scavenger test, it was observed that the cat-Cu complex exhibited similar activity in comparison to catechin. This outcome can be attributed to the partial obstruction of the hydroxyl groups within the catechin structure. Hydroxyl groups, particularly those classified as phenolic, exhibit proton donor properties and have the capability to counteract the effects of free radicals. In contrast, the process of complexation demonstrates a more pronounced effect on the scavenging activity of zinc ions, resulting in a significant increase (p < 0.001). The results we obtained are displayed in Figure 8.
3.9. In Vitro Antidiabetic Activity
3.9.1. Inhibition of Alpha-Amylase
The digestion of food starch involves the participation of pancreatic alpha-amylase and alpha-glucosidase which will transform the starch into glucose molecules that are absorbed in the intestine, pass into the blood and cause postprandial hyperglycemia. Its value is dependent on the available starch for digestion, the activity of the enzymes involved in digestion and the capacity of the intestinal absorption systems. The increase in blood sugar, above the physiological levels, determines the accentuation of the phenomenon of oxidative stress and the uncontrolled glycosylation of proteins with the worsening of symptoms in diabetes and the risk of organic complications [32,33,34].
Metal ions reduce or block the activity of the enzyme by changing its secondary structure, with an increase in the frequency of areas with a beta-folded or linear structure at the expense of those with an alpha-helix structure [35]. Changing the secondary structure will affect the structure of the active center of the enzyme and reduce the interaction with the substrate. Figure 9 depicts the comparative alpha-amylase inhibition potential for the cat-Cu complex.
3.9.2. Inhibition of Alpha-Glucosidase
According to Choudhary et al. (2020), the activity of alpha-glucosidase is diminished by catechin through the formation of hydrogen bonds with the functional groups of the amino acids present in the enzyme’s structure [7].
Several studies have demonstrated the capacity of catechin, and other polyphenols derived from vegetables to effectively inhibit or decrease the activity of alpha-amylase or alpha-glucosidase enzymes. In certain cases, their efficacy in this regard surpasses that of acarbose, a pharmaceutical compound commonly employed to hinder carbohydrate digestion. The inhibitory effect of copper on alpha-amylase or alpha-glucosidase remains consistent when administered in the form of nanoparticles, yielding results similar to those obtained in the current study. Notably, a more pronounced impact on alpha-glucosidase is observed compared to alpha-amylase [14]. The comparative assay of catechin and cat-Cu complex can be seen in Figure 10.
3.10. Antibacterial Activity
The evaluation of the antimicrobial activity expressed by the diameter of the inhibition zone was completed with the determination of the minimum inhibitory concentrations (MIC) and the minimum bactericidal concentrations (CMB), respectively fungicidal (CMF) on Staphylococcus aureus ATCC 25923, Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853 and Candida albicans ATCC 90028. Minimum inhibitory concentration (MIC) values are associated with the values of the lowest concentration of the synthesized and tested complexes that inhibited the growth of microbial cultures, compared to the positive control. We determined the minimum inhibitory concentrations (MIC) by the microdilution method in the broth, for products with definite antimicrobial activity in the diffusometric method.
Synthesized cat-Cu complex exhibited modest antibacterial activity against S. aureus and modest antifungal activity tested for Candida albicans, and no antibacterial activity against E. coli and P. aeruginosa as can be seen in Table 2.
The evaluation of the antimicrobial activity, expressed by the diameter of the inhibition zone, was supplemented by determining the minimum inhibitory concentrations (MIC) and the minimum bactericidal concentrations (MBC), as well as the minimum fungicidal concentrations (MFC) against S. aureus, Escherichia coli, and Candida albicans.
The minimum inhibitory concentration (MIC) values represent the lowest concentration of the synthesized and tested complexes that inhibited the growth of microbial cultures, compared to the positive control. We determined the minimum inhibitory concentrations (MIC) using the broth microdilution method, for the products with confirmed antimicrobial activity in the diffusion method.
The MBC/MFC values were determined by transferring 0.1 µl from each well that showed complete inhibition of visible growth onto the surface of a solid medium plate. The subcultures were incubated for 24 hours at 37°C (for bacteria) and 30°C (for fungi). The MBC/MFC value was considered the lowest concentration of the compound that destroyed 99.9% of the tested microorganisms.
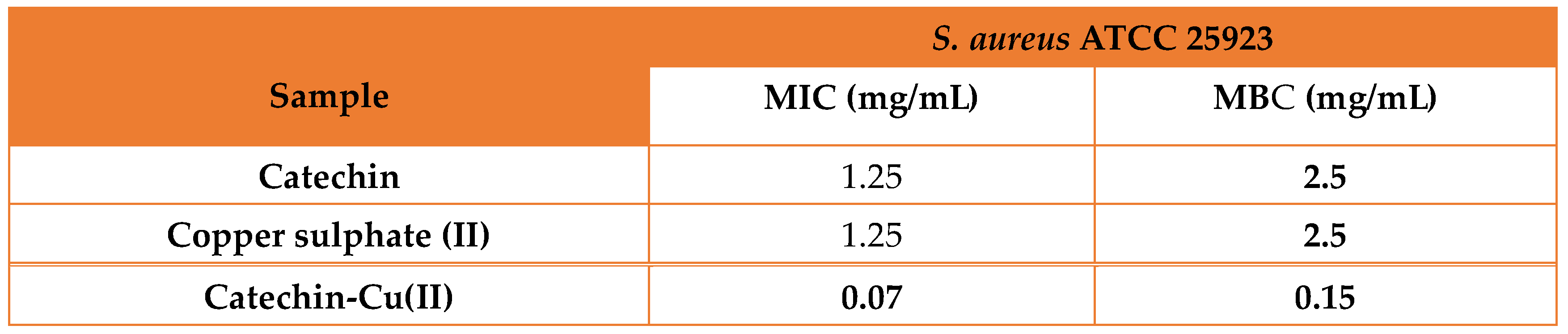
The MIC and MBC against S. aureus are presented in Table 3.
Many metals are known for their biocidal properties through different mechanism of actions, making them ideal for sterilizing surface or even as antibiotics. Copper is a potent antimicrobial agent, second to silver cations. The mechanism in which copper causes apoptosis of bacteria is not perfectly defined but there are many theories to it [39,40,41].
Generally, copper cations enter bacteria by rupturing the membrane and being further carried by different proteins. Contact sterilization theory states that copper cations (I) and (II) accumulate on the outside of the pathogen, this accumulation causes a high-density charge which creates an imbalance in the outer bacterial space. This imbalance leads to the deformation of the cell causing the cell membrane to rupture and for the contents to leak out [42,43].
(1) Cu2+ + H2O2 → Cu+ + 2H+ + 2O•-
Cu2+ + 2O- → Cu+ + O2
Cu2+ + H2O2 → Cu+ + HO• + HO-
The Fenton reaction (1) is another way in which copper ions induce apoptosis, copper participates in redox reactions leading to the formation of ROS which in turn lead to oxidative stress and cellular death [44].
4. Conclusions
Maintaining or improving the antioxidant or enzymatic inhibitory or antibacterial properties of catechin, after complexation with metal ions, may represent a benefit in the case of the use of such compounds in preclinical studies.
Given the significance of copper ions in keeping insulin stability and the advantageous impact of the complex in decreasing carbohydrate digestion, these complexes have potential utility in the treatment of individuals with diabetes. The maintenance of physiological parameters related to oxidative processes in living organisms, such as the human body, frequently necessitates the assistance of endogenous antioxidant mechanisms, which can be supplemented by the intake of compounds known for their protective effects.
The assessment of the antioxidant potential of the cat-Cu complex synthesized revealed a similar activity in the antioxidant and antidiabetic capacity of catechin. Therefore, in order to preserve the antioxidative activity, it is imperative to optimize the metal-catechin complex extraction process or explore the potential utilization of catechin derivatives.
Consequently, it becomes imperative to conduct in vivo toxicological and pharmacokinetic assessments to evaluate their suitability for therapeutic use.
Author Contributions
Conceptualization, I.I.L. and M.H.; methodology, O.C.; software, A.S.; validation, C.M., C.T. and R.H.; formal analysis, I.I.L.; investigation, M.H.; resources, A.S.; data curation, C.T.; writing—original draft preparation, C.M.; writing—review and editing, O.C.; visualization, R.H.; supervision, M.H.; project administration, C.M.; funding acquisition, R.H. All authors have read and agreed to the published version of the manuscript. Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Humulescu, I.; Flutur, M.-M.; Cioanca, O.; Mircea, C.; Robu, S.; Marin-Batir, D.; Spac, A.; Corciova, A.; Hancianu, M. Comparative Chemical and Biological Activity of Selective Herbal Extracts. Farmacia 2021, 69, 861–866. [Google Scholar] [CrossRef]
- Tan, Z.; Deng, J.; Ye, Q.; Zhang, Z. The Antibacterial Activity of Natural-Derived Flavonoids. Curr Top Med Chem 2022, 22, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Finaud, J.; Lac, G.; Filaire, E. Oxidative Stress : Relationship with Exercise and Training. Sports Med 2006, 36, 327–358. [Google Scholar] [CrossRef] [PubMed]
- Ferrali, M.; Signorini, C.; Caciotti, B.; Sugherini, L.; Ciccoli, L.; Giachetti, D.; Comporti, M. Protection against Oxidative Damage of Erythrocyte Membrane by the Flavonoid Quercetin and Its Relation to Iron Chelating Activity. FEBS Lett 1997, 416, 123–129. [Google Scholar] [CrossRef]
- Lungu, I.; Huzum, B.; Humulescu, I.A.; Cioancă, O.; Morariu, D.; Șerban, I.-L.; Hăncianu, M. Flavonoids as promising therapeutic and dietary agents. Med.-Surg. J. 2020, 124, 151–156. [Google Scholar]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An Overview. J Nutr Sci 2016, 5, e47. [Google Scholar] [CrossRef]
- Chen, C.-M.; Wu, C.-T.; Yang, T.-H.; Chang, Y.-A.; Sheu, M.-L.; Liu, S.H. Green Tea Catechin Prevents Hypoxia/Reperfusion-Evoked Oxidative Stress-Regulated Autophagy-Activated Apoptosis and Cell Death in Microglial Cells. J Agric Food Chem 2016, 64, 4078–4085. [Google Scholar] [CrossRef]
- Kostyuk, V.A.; Potapovich, A.I.; Vladykovskaya, E.N.; Korkina, L.G.; Afanas’ev, I.B. Influence of Metal Ions on Flavonoid Protection against Asbestos-Induced Cell Injury. Arch Biochem Biophys 2001, 385, 129–137. [Google Scholar] [CrossRef]
- Iancu C, Mircea C, Petrariu FL, Cioancă O, Stan C, Corciovă A, Murărașu A, Filip N, Hăncianu M, The evaluation of normo-glycemic and cyto-regenerative effects of Pelargonium species extracts. Farmacia 2020, 68, 135–141. [CrossRef]
- Tan, Z.; Deng, J.; Ye, Q.; Zhang, Z. The Antibacterial Activity of Natural-Derived Flavonoids. Curr Top Med Chem 2022, 22, 1009–1019. [Google Scholar] [CrossRef]
- Lungu, I.; Panainte, A.; Mircea, C.; Tuchiluș, C.; Ștefanache, A.; Szasz, F.; Grigorie, D.; Robu, S; Cioancă, O. Catechin-Zinc-Complex: Synthesis, Characterization and Biological Activity Assessment. Farmacia 2023, 71. [Google Scholar] [CrossRef]
- Kumamoto, M.; Sonda, T.; Nagayama, K.; Tabata, M. Effects of pH and Metal Ions on Antioxidative Activities of Catechins. Biosci Biotechnol Biochem 2001, 65, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Bors, W.; Heller, W.; Michel, C.; Saran, M. Flavonoids as Antioxidants: Determination of Radical-Scavenging Efficiencies. Methods Enzymol 1990, 186, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Ameena, S.; Rajesh, N.; Anjum, S.M.; Khadri, H.; Riazunnisa, K.; Mohammed, A.; Kari, Z.A. Antioxidant, Antibacterial, and Anti-Diabetic Activity of Green Synthesized Copper Nanoparticles of Cocculus Hirsutus (Menispermaceae). Appl Biochem Biotechnol 2022, 194, 4424–4438. [Google Scholar] [CrossRef]
- Souza, R.F.V.; De Giovani, W.F. Antioxidant Properties of Complexes of Flavonoids with Metal Ions. Redox Report 2004, 9, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Sarker, U.; Oba, S. Nutraceuticals, Antioxidant Pigments, and Phytochemicals in the Leaves of Amaranthus spinosus and Amaranthus viridis Weedy Species. Sci Rep. 2019, 9, 20413. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.B.; Memon, S.; Mahroof-Tahir, M.; Bhanger, M.I. Synthesis, Characterization and Antioxidant Activity Copper–Quercetin Complex. Spectrochim Acta A Mol Biomol Spectrosc 2009, 71, 1901–1906. [Google Scholar] [CrossRef]
- Sharma, N.; Phan, H.T.; Chikae, M.; Takamura, Y.; Azo-Oussou, A.F.; Vestergaard, M.C. Black Tea Polyphenol Theaflavin as Promising Antioxidant and Potential Copper Chelator. J Sci Food Agric 2020, 100, 3126–3135. [Google Scholar] [CrossRef]
- Szczepanik, J.; Podgórski, T.; Domaszewska, K. The Level of Zinc, Copper and Antioxidant Status in the Blood Serum of Women with Hashimoto’s Thyroiditis. Int J Environ Res Public Health 2021, 18, 7805. [Google Scholar] [CrossRef]
- Ali, H.; Houghton, P.J.; Soumyanath, A. Alpha-Amylase Inhibitory Activity of Some Malaysian Plants Used to Treat Diabetes; with Particular Reference to Phyllanthus Amarus. J Ethnopharmacol 2006, 107, 449–455. [Google Scholar] [CrossRef]
- Apostolidis, E.; Lee, C.M. In Vitro Potential of Ascophyllum Nodosum Phenolic Antioxidant-Mediated Alpha-Glucosidase and Alpha-Amylase Inhibition. J Food Sci 2010, 75, H97–102. [Google Scholar] [CrossRef]
- Choudhary, D.K.; Chaturvedi, N.; Singh, A.; Mishra, A. Characterization, Inhibitory Activity and Mechanism of Polyphenols from Faba Bean (Gallic-Acid and Catechin) on α-Glucosidase: Insights from Molecular Docking and Simulation Study. Prep Biochem Biotechnol 2020, 50, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Rajeshkumar, S.; Menon, S.; Venkat Kumar, S.; Tambuwala, M.M.; Bakshi, H.A.; Mehta, M.; Satija, S.; Gupta, G.; Chellappan, D.K.; Thangavelu, L.; et al. Antibacterial and Antioxidant Potential of Biosynthesized Copper Nanoparticles Mediated through Cissus Arnotiana Plant Extract. J Photochem Photobiol B 2019, 197, 111531. [Google Scholar] [CrossRef] [PubMed]
- El-Fattah, A.A.; Azzam, M.; Elkashef, H.; Elhadydy, A. Antioxidant Properties of Milk: Effect of Milk Species, Milk Fractions and Heat Treatments. Int. J. Dairy Sci. 2019, 15, 1–9. [Google Scholar] [CrossRef]
- Hasanin, M.; Al Abboud, M.A.; Alawlaqi, M.M.; Abdelghany, T.M.; Hashem, A.H. Ecofriendly Synthesis of Biosynthesized Copper Nanoparticles with Starch-Based Nanocomposite: Antimicrobial, Antioxidant, and Anticancer Activities. Biol Trace Elem Res 2022, 200, 2099–2112. [Google Scholar] [CrossRef]
- Shahrajabian, M.H.; Sun, W.; Cheng, Q. Importance of Epigallocatechin and Its Health Benefits. Free Radicals and Antioxidants 2021, 10, 47–51. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.; He, Y.; Jia, L.; Yang, C.S.; Reiter, R.J.; Zhang, J. Antioxidant and Pro-Oxidant Activities of Melatonin in the Presence of Copper and Polyphenols In Vitro and In Vivo. Cells 2019, 8, 903. [Google Scholar] [CrossRef]
- Ferrali, M.; Signorini, C.; Caciotti, B.; Sugherini, L.; Ciccoli, L.; Giachetti, D.; Comporti, M. Protection against Oxidative Damage of Erythrocyte Membrane by the Flavonoid Quercetin and Its Relation to Iron Chelating Activity. FEBS Lett 1997, 416, 123–129. [Google Scholar] [CrossRef]
- Serpen, A.; Gökmen, V.; Fogliano, V. Total antioxidant capacities of raw and cooked meats. Meat Sci. 2012, 90, 60–65. [Google Scholar] [CrossRef]
- Jhong, C.-H.; Riyaphan, J.; Lin, S.-H.; Chia, Y.-C.; Weng, C.-F. Screening Alpha-Glucosidase and Alpha-Amylase Inhibitors from Natural Compounds by Molecular Docking in Silico. Biofactors 2015, 41, 242–251. [Google Scholar] [CrossRef]
- Kim, K.T.; Rioux, L.E.; Turgeon, S.L. Alpha-Amylase and Alpha-Glucosidase Inhibition Is Differentially Modulated by Fucoidan Obtained from Fucus Vesiculosus and Ascophyllum Nodosum. Phytochemistry 2014, 98, 27–33. [Google Scholar] [CrossRef]
- Etxeberria, U.; De La Garza, A.L.; Campin, J.; Martnez, J.A.; Milagro, F.I. Antidiabetic Effects of Natural Plant Extracts via Inhibition of Carbohydrate Hydrolysis Enzymes with Emphasis on Pancreatic Alpha Amylase. Expert Opin Ther Targets 2012, 16, 269–297. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Chen, Y.; Ye, X.; Wang, L.; Shao, J.; Jing, H.; Jiang, C.; Wang, H.; Ma, C. Three Flavanols Delay Starch Digestion by Inhibiting α-Amylase and Binding with Starch. Int J Biol Macromol 2021, 172, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Ademiluyi, A.O.; Oboh, G. Phenolic-Rich Extracts from Selected Tropical Underutilized Legumes Inhibit α-Amylase, α-Glucosidase, and Angiotensin I Converting Enzyme in Vitro. J Basic Clin Physiol Pharmacol 2012, 23, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, K.; Cymbaluk-Płoska, A. The Role of Zinc and Copper in Gynecological Malignancies. Nutrients 2020, 12, 3732. [Google Scholar] [CrossRef]
- Lisa EL, Carac G, Barbu V, Robu SI. The synergistic antioxidant effect and antimicrobial efficacity of propolis, myrrh and chlorhexidine as beneficial toothpaste components. Rev. Chim.-Buchar 2017, 68, 2060–2065. [CrossRef]
- Marin DB, Cioanca OA, Apostu MI, Tuchilus CG, Mircea C, Robu SI, Tutunaru D, Corciova AN, Hancianu M. The Comparative Study of Equisetum pratense, E. Sylvaticum, E. Telmateia: Accumulation of silicon, antioxidant and antimicrobial screening. Revista de Chimie 2019, 70, 2519–2523. [CrossRef]
- Maftei NM, Bogdan RE, Boev M, Marin DB, Ramos-Villarroel AY, Iancu AV. Innovative Fermented Soy Drink with the Sea Buckthorn Syrup and the Probiotics Co-Culture of Lactobacillus Paracasei ssp. Paracasei (L. Casei® 431) and Bifidobacterium Animalis ssp. Lactis (Bb-12®).
- Yu, J.; Huang, X.; Ren, F.; Cao, H.; Yuan, M.; Ye, T.; Xu, F. Application of antimicrobial properties of copper. Appl Organomet Chem 2024, 38, e7506. [Google Scholar] [CrossRef]
- Sadani, K. , Nag, P., Pisharody, L., Thian, X. Y., Bajaj, G., Natu, G.,... & Mukherji, S. Polyphenol stabilize copper nanoparticle formulations for rapid disinfection of bacteria and virus on diverse surfaces. Nanotechnology 2021, 33(3), 035701. [Google Scholar]
- Wang, Z. , & An, P. Characterization of copper complex nanoparticles synthesized by plant polyphenols. BioRxiv 2017, 134940. [Google Scholar]
- Salah, I.; Parkin, I. P.; Allan, E. Copper as an antimicrobial agent: Recent advances. RSC advances 2021, 11, 18179–18186. [Google Scholar] [CrossRef]
- Rajalakshmi, S. , Fathima, A., Rao, J. R., & Nair, B. U. Antibacterial activity of copper (II) complexes against Staphylococcus aureus. RSC advances 2014, 4, 32004–32012. [Google Scholar]
- Scattareggia Marchese, A. , Destro, E., Boselli, C., Barbero, F., Malandrino, M., Cardeti, G.,... & Lanni, L. Inhibitory Effect against Listeria monocytogenes of Carbon Nanoparticles Loaded with Copper as Precursors of Food Active Packaging. Foods 2022, 11. [Google Scholar]
Figure 1.
comparative UV-VIS spectra of catechin and cat-Cu complex.
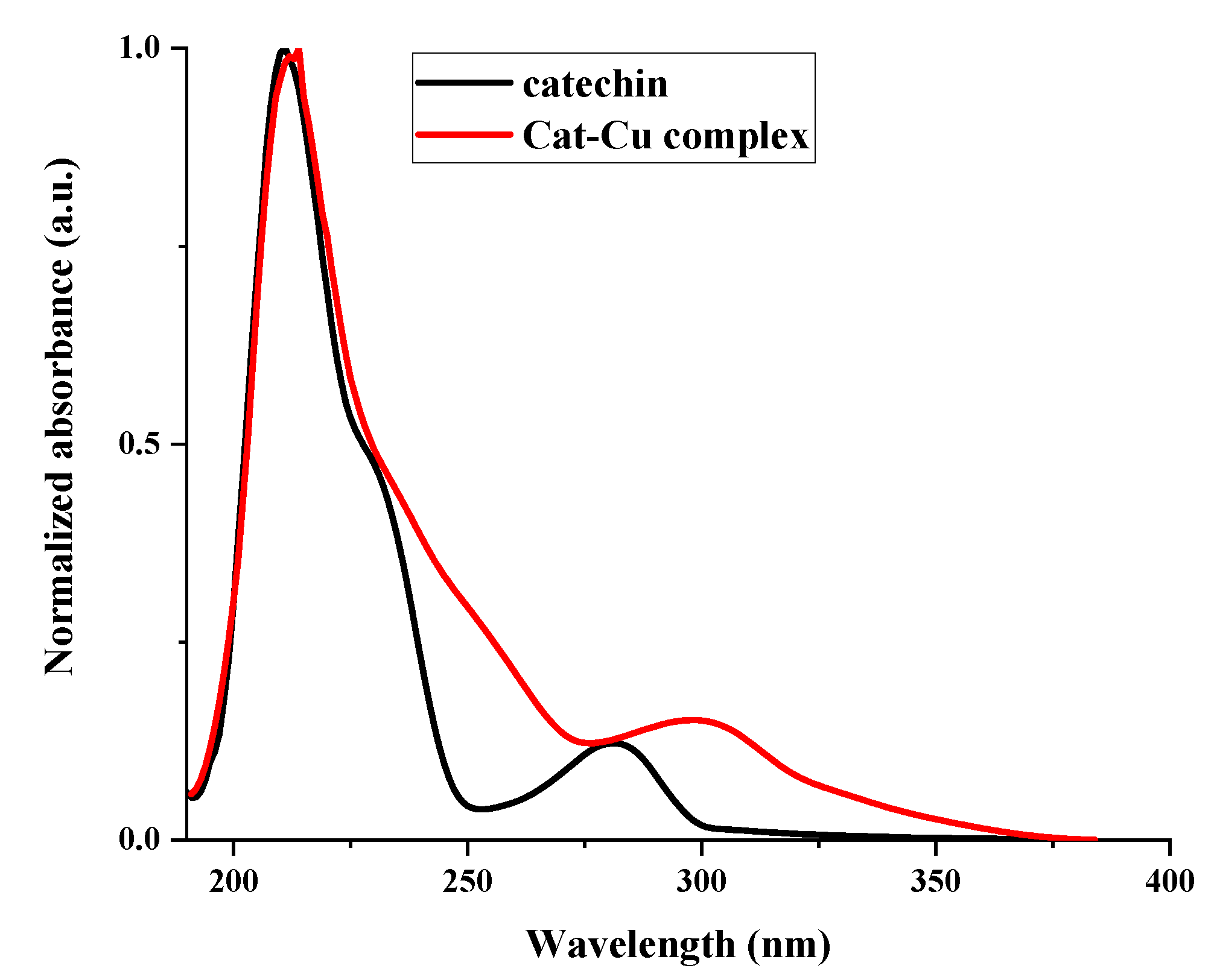
Figure 2.
Comparative FT-IR spectra of catechin and cat-Cu complex.
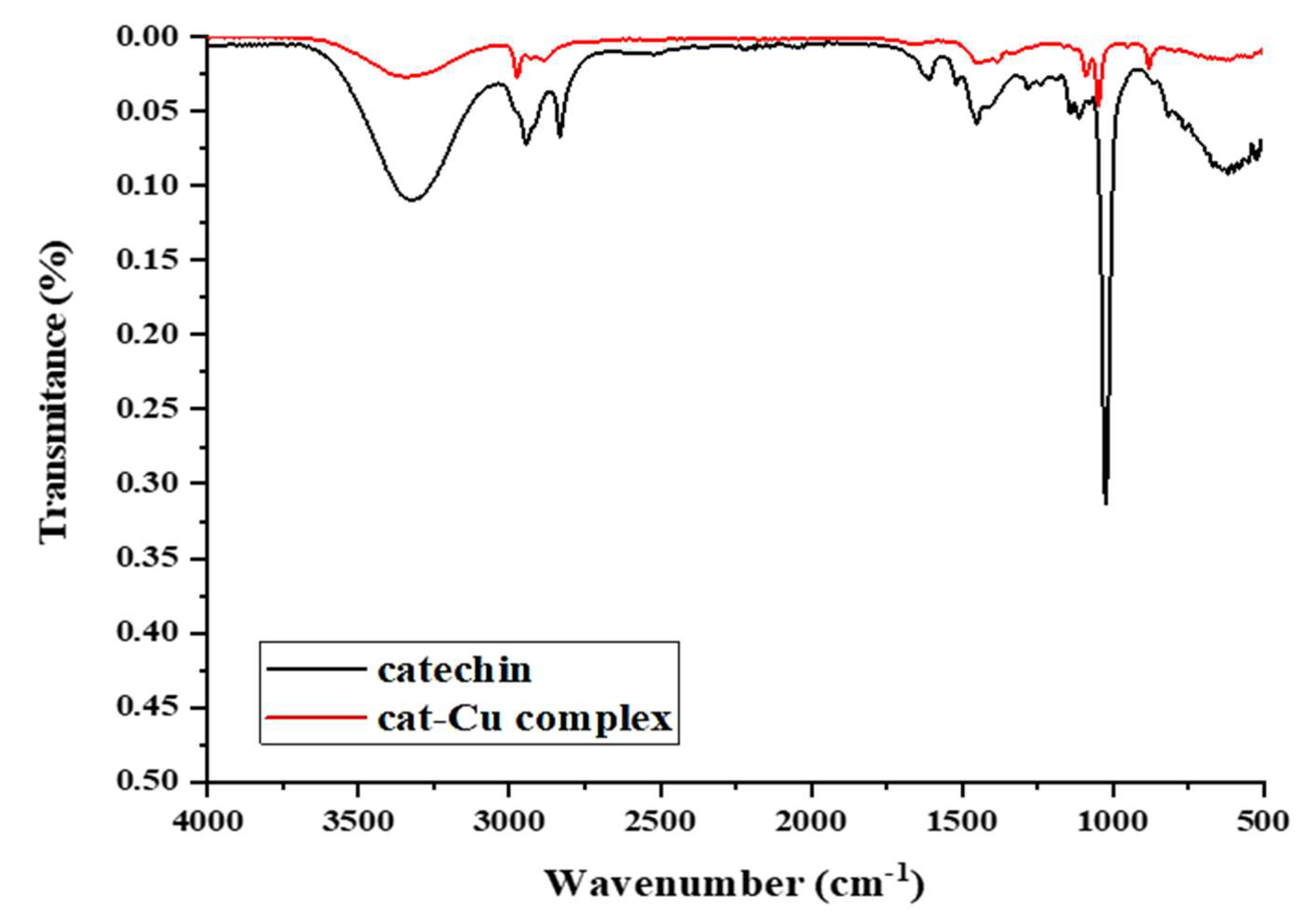
Figure 3.
Proposed structure for the synthesized catechin-Cu(II) complex.
Figure 4.
SEM images for cat-Cu complex, A) x 1000; B) x 5000; C) x 19 998; D) x 19 997.

Figure 5.
EDX of cat-Cu complex synthesized complex.
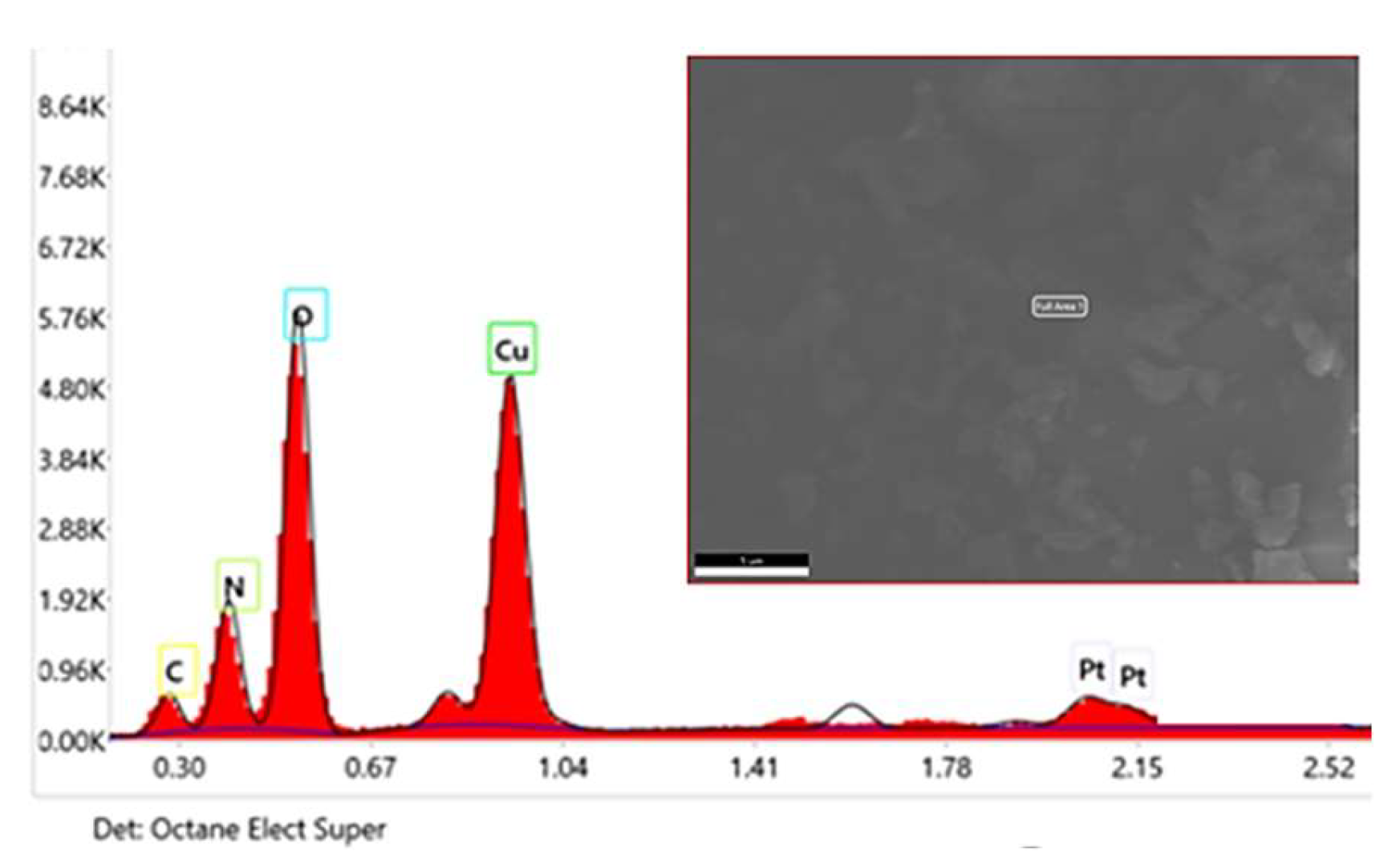
Figure 6.
Graphical representation of the iron chelation capacity of cat-Cu complexes compared to catechin.
Figure 6.
Graphical representation of the iron chelation capacity of cat-Cu complexes compared to catechin.
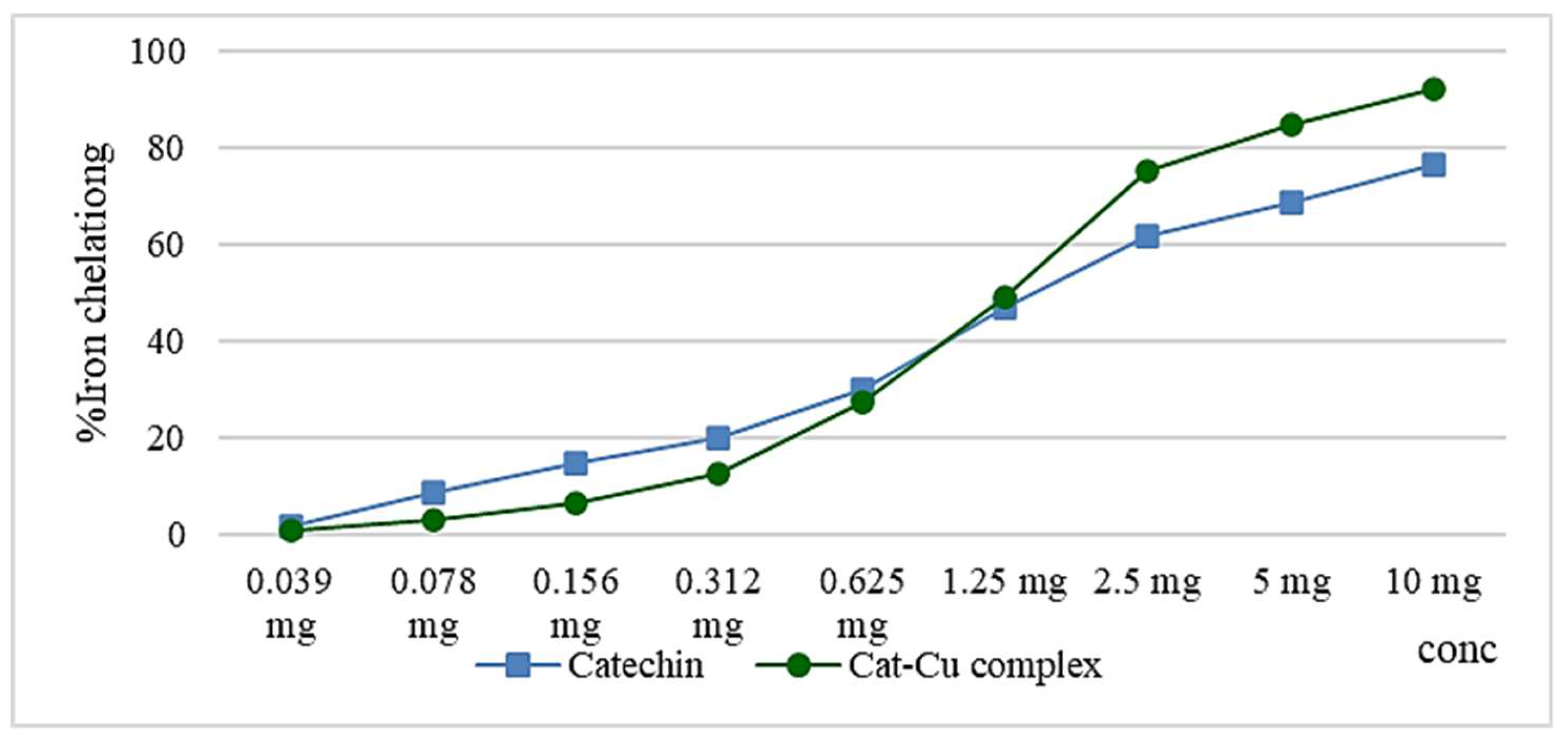
Figure 7.
Graphic representation of the activity of the cat-Cu complex that reduces lipoxygenase activity.
Figure 7.
Graphic representation of the activity of the cat-Cu complex that reduces lipoxygenase activity.
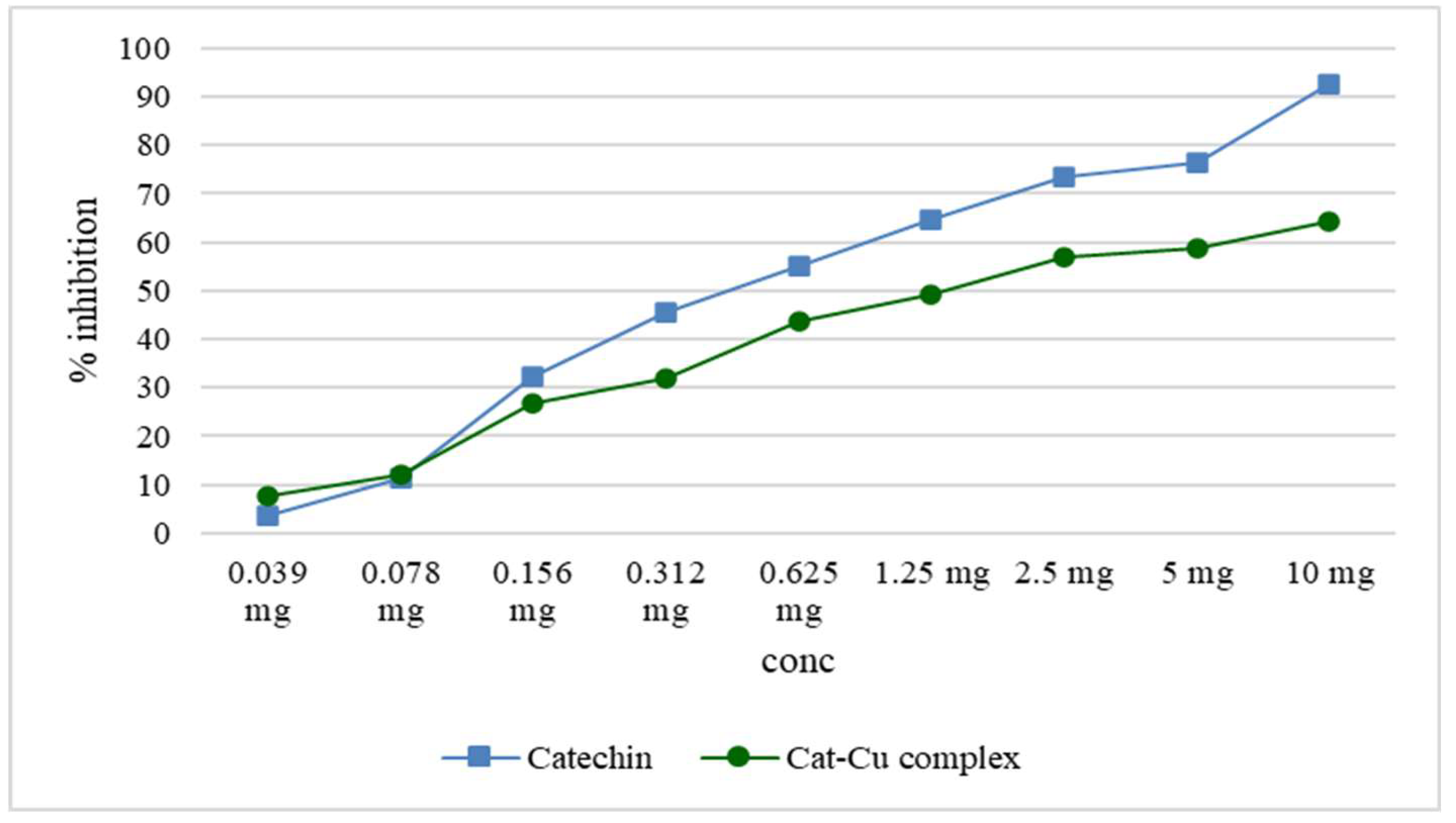
Figure 8.
Hydroxyl radical scavenger activity of the cat-Cu complex.
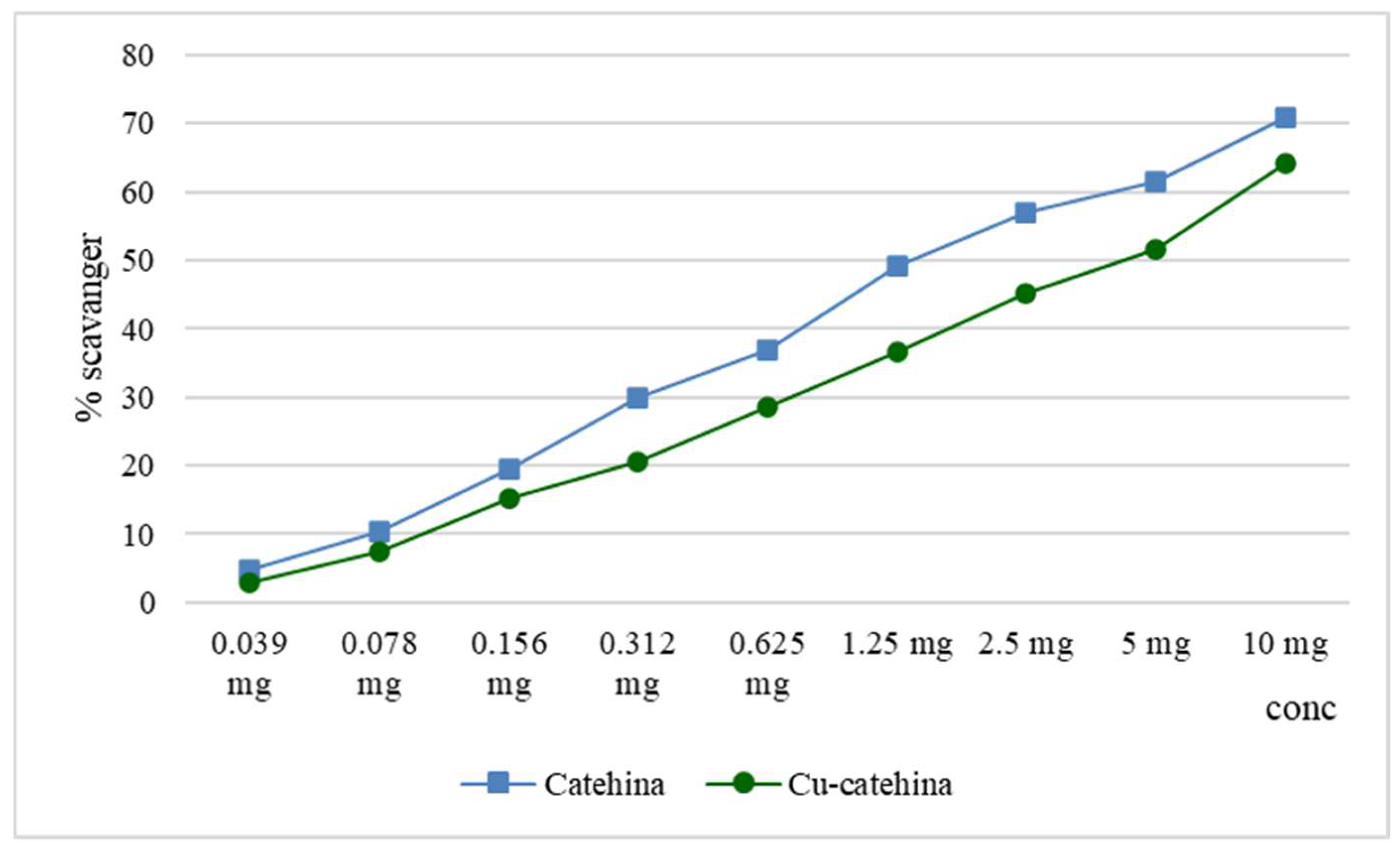
Figure 9.
Alpha-amylase inhibition potential for the cat-Cu complex.
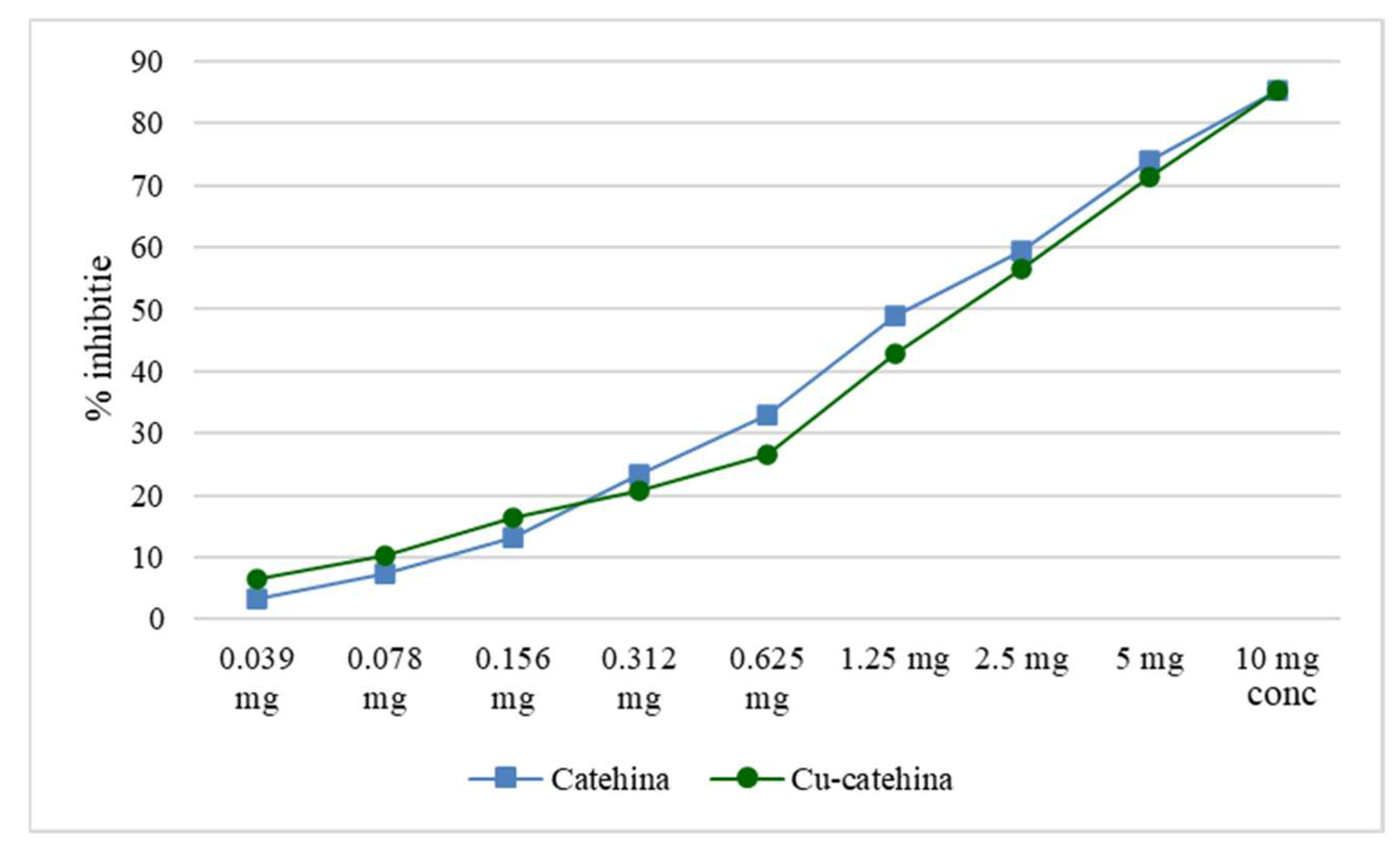
Figure 10.
Graphic representation of alpha-glucosidase inhibition capacity of cat-Cu complex compared to catechin.
Figure 10.
Graphic representation of alpha-glucosidase inhibition capacity of cat-Cu complex compared to catechin.
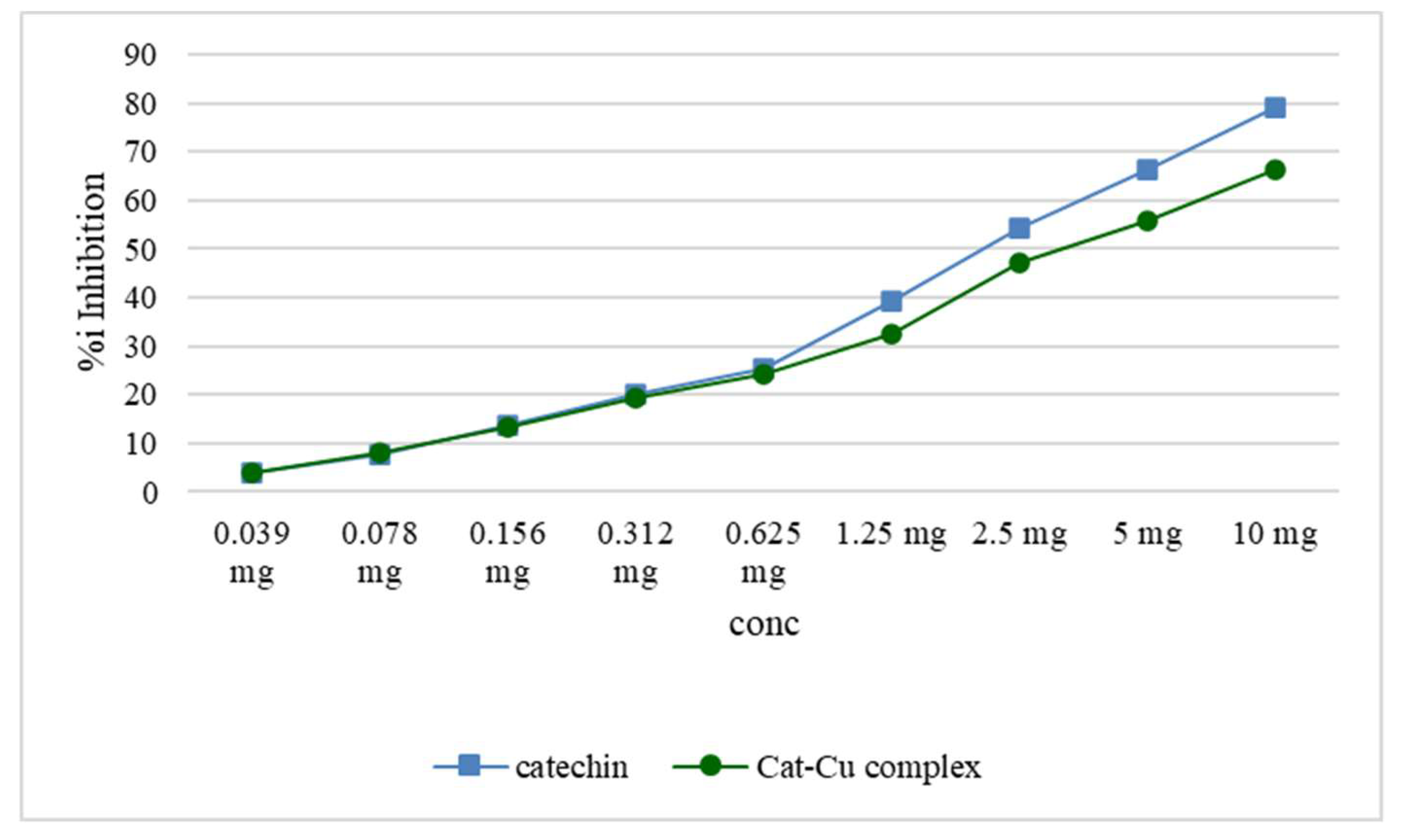
Table 2.
Comparing study of diameter of inhibition for catechin, copper sulphate and cat-Cu complex for S. aureus, E. coli, P. aeruginosa and Candida albicans.
Table 2.
Comparing study of diameter of inhibition for catechin, copper sulphate and cat-Cu complex for S. aureus, E. coli, P. aeruginosa and Candida albicans.
| Tested substance | S. aureus ATCC 25923 | E. coli ATCC 25922 | P. aeruginosa ATCC 27853 | C. albicansATCC 90028 |
|---|---|---|---|---|
| Catechin | 20.0±0.00 | 0 | 16.0±0.00 | 0 |
| Copper sulphate | 14.0±0.00 | 0 | 0 | 0 |
| cat-Cu complex | 17.3±0.57 | 0 | 0 | 17.0±0.00 |
| blank (DMSO) | 0 | 0 | 0 | 0 |
| I. Ciprofloxacin (5 µg/disc) | 30.0±0.00 | 34.0±0.00 | 31.3±0.57 | Not tested |
| I.Fluconazol (25 µg/disc) | Not tested | Not tested | Not tested NT* | 30.0±0.00 |
Table 3.
The MIC and MBC of the synthesized complex and individual reagents against S. aureus.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



