
Submitted:
16 October 2024
Posted:
16 October 2024
You are already at the latest version
Abstract
Background: Immune evasion in cancer is a multifaceted process that synchronizes pro-tumoral immune infiltration, immunosuppressive inflammation, and inhibitory immune checkpoint expression (IC). Recent developments have immunotherapies such as immune checkpoint blockade (ICB) and chimeric antigen receptor T-cell (CAR-T) therapy effectively impede this process but benefit a limited patient cohort. This investigation introduces a systemic immunotherapeutic strategy by inhibition of master-regulators of immune evasion (MR-IE). Methods: We utilized the TCGA PanCancer Atlas transcriptomic data to subset and stratify cancer samples based on IC expression levels and CIBERSORT to estimate immune cell infiltration. Differential gene expression analysis was conducted to unravel pathways associated with the immune evasion. Transcription Factor Enrichment Analysis and Survival Analysis was conducted to identify and rank candidate MR-IE per cancer type. The top-ranking candidate MR-IE of cholangiocarcinoma (CCA) was validated for its regulation of ICs using quantitative PCR and western blots. Orbitrap Proteomics was employed to assess changes in the immune-related proteome of CCA cells after MR-IE is inhibited or knocked-down. Lastly, CCA cell viability and immune-mediated cell death was assessed after MR-IE inhibition in combination with fourth-generation anti-folate receptor alpha (FRɑ) CAR-T cell therapy. Results: This workflow ranked and identified candidate MR-IE for 33 tumor types. MYC was identified as the high-ranking candidate MR-IE for CCA. MYC modulated PD-L1 expression adn key markers of inflammation and IC expression in CCA cells. Moreover, MYC inhibition was able to potentiate CAR-T mediated cell death in CCA cells. Conclusions: Cumulatively, these results offer a candidate list of MR-IE immunotherapeutic targets per cancer type that can be further explored and validated. This study also highlights the promise of MR-IE inhibition as a novel potent immunotherapeutic strategy for the treatment of CCA.
Keywords:
cancer
; transcriptomics
; immunotherapy
; cholangiocarcinoma
; Immune Checkpoints
Background
Cancer is a multi-faceted disease that confers resilience and survival advantages over normal cells. These advantages include sustained proliferative signaling, resistance to growth suppressors, invasion and metastasis, and immune evasion [1]. During cancer development, cancer cells interact intimately with the immune cells in the microenvironment to facilitate its survival and progression. These interactions promotes an immunosuppressive environment to allow cancer cells to proliferate without surveillance and elimination by immune cells. This is mediated by expression of co-inhibitory ICs, infiltration of pro-tumoral immune cells, lowering the expression of self-antigens, and the release of immunosupperssive cytokines [2].
Deciphering complex mechanisms that support immune evasion has given rise to various immunotherapies that unveil cancer cells and reinstate immune recognition and elimination of cancer cells. There are various mechanisms tackled by immunotherapies including the inhibition of co-inhibitory IC interactions by ICB, cancer-cell surface target recognition and induced cytotoxicity by CAR-T therapy, and recruiting anti-tumoral immune cell infiltration by promoting inflammation using cytokine-based therapies. Of these therapies, ICBs have shown great promise in clinical use, however, only 30-40% of the patient population are eligible for these therapies and upto 12.5% of the eligible patients respond to treatment [3]. The reported reasons for the lack of response to immunotherapies include primary and acquired resistance, low expression of intended targets amongst patient populations, and even relapses after treatment [4,5]. As the ineligible patient population expands, the exploration for alternative therapeutic strategies and targets are warranted.
Our previous review surveyed transcriptional regulators of ICs and oncoimmunology, so-called master regulators of immune evasion (MR-IE) [6]. Here we describe how transcription factors such as MYC and STAT3 play a pivotal role in the context of oncoimmunology, including the regulation of IC molecules gene expression, maturation of dendritic cells, the release of pro-tumoral cytokines, inhibiting anti-tumor immune cells, and promoting angiogenesis [7,8]. Hence, we surmise that these MR-IE, would be more effective immunotherapeutic targets for cancers.
The present investigation identified (mortality-associated) MR-IE per cancer type by a robust integration bioinformatics tools to stratify and subset The Cancer Genome Atlas (TCGA) Pan Cancer dataset based on their IC molecule expression and their predicted immune cell infiltration levels. We validated the top-ranking candidate in CCA, as it has been previously reported to have a complex immune architecture [9], and has a concerning incidence and mortality rate, with limited treatment options in Thailand [10]. This investigation not only aims to introduce a novel and effective immunotherapeutic strategy by targeting MR-IE, but also offer more potent immunotherapeutic treatment options for CCA.
Methodology
Dataset Acquisition and Classification
TCGA Pan Cancer Atlas RNA-Seq dataset was obtained from https://gdc.cancer.gov/about-data/publications/pancanatlas , along with its respective sample annotation, tumor subtype annotation and its clinical data. This dataset was used to draw immune cellular fraction estimates using CIBERSORT, as described in Thorsson et al’s investigation [11,12]. Using the datasets provided in the paper, we selected TCGA samples that were in the first and third quartile of CD8+ T-cell infiltration (less than 25% and greater than 75%) to represent low infiltration and high CD8+ T-cell infiltration, respectively. Additionally, we summed the infiltration scores of CD4+ and CD8+ T-cells of the TCGA tumors and selected samples that were in the first and third quartile to represent low and high CD4+ and CD8+ T-cell infiltration. Lastly, using the Leukocyte Fraction scores, we used the samples in the first and third quartile to represent low and high leukocyte infiltration. Thus, using these criteria, the PanCancer dataset was subset into 6 datasets (Low and High Leukocyte Infiltration, Low and High CD4+ and CD8+ T cell infiltration, and Low and High CD8+ T cell infiltration).
Hierarchical clustering of TCGA samples
The 7 datasets (6 subsets and the PanCancer dataset) were transformed using log2(x+2) to remove NA values. Using the R package pheatmap, the datasets were plotted on a heatmap that was subjected to hierarchical clustering [13]. These heatmaps were used to determine expression levels (low, intermediate and high) of co-inhibitory IC expression. Samples that belonged to each cluster were extrapolated and pooled into its respective groups for subsequent analyses.
Differential Gene Expression Analysis
Differential gene expression analysis was conducted using the R packages edgeR and limma [13]. TCGA samples within low, intermediate, and high co-inhibitory IC expression were compared in the directions: low vs. intermediate, and low vs. high co-inhibitory IC expression. P-values were adjusted using the Benjamini-Hochberg method. P-value threshold was set at below 0.001 (p<0.001)
Pathway Enrichment Analyses
To assess the pathways the differentially expressed genes (DEGs) belong to we performed enrichment analysis using the tools iLINCS Pathway Analysis [14]. The input for these tools was either the DEG list with its fold change and adjusted p-values or just the gene list, wherever appropriate.
Transcriptional Regulators Identification
DEG list was applied to STRING DB to generate a gene network. This network was used in Expression2Kinases to enrich transcription factors using known protein interactions to construct a network.
Cell Lines
Cell lines KKU-M213 and RBE were purchased from Japanese Collection of Research Bioresources Cell Bank, and were maintained in RPMI culture medium supplemented with 10% FBS (Gibco, Life technologies, Grand Island, NY, USA) and incubated at 37°C with 5% CO2.
Lipofectamine transfection for MYC Knockdown
Lipofectamine reagent was prepared using Optimem for 3 reactions. Each reaction was combined with its respective siRNA: siRNA of MYC, Negative control siRNA, reagent-control siRNA. Each of these, reactions were then applied to cells seeded on a 6-well plate, and incubated for 24 hours. After which, the treatment medium was replaced with culture medium.
Real-time PCR (qPCR)
Total RNA was extracted using the Geneaid Total RNA extraction kit, in accordance with manufacturer’s protocol. 500 ng RNA was subjected to reverse transcriptase using the Hyperscript RT mastermix kit (Geneaid) using a Bio-Rad Thermal cycler. The cDNA was subjected to qPCR using the Agilent Real-Time PCR thermal cycler.
Western Blot
8x105 cells were seeded onto a 60 mm dish for 24 hours and treated with the lipofectamine reagent carrying the siRNA of interest or inhibitor treatment. The cells (after 48 hours) were lysed according to the protocol defined in the publication [15]. Protein samples of 40 μg was loaded and separated using 12% SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked with 5% bovine serum albumin (BSA) and probed with primary antibodies at 1:1000 dilution. After incubation, the membrane was washed for 10 min in TBS/T for 3 times, incubated with secondary antibodies conjugated with horseradish-peroxidase (HRP), and immunoreactive bands was visualized by ECL Plus Western Blotting Detection system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) using G:Box ChemiXL 1.4 (Syngene; Synoptics, Cambridge, UK). Densitometry analysis was performed using Image LabTM software.
Proteomics
To investigate the protein expression profile of cancer cells upon inhibiting the candidate MR-IE, the extracted proteins were prepared according to the protocol listed in a previous investigation [16]. Cells were lysed using 1% Triton-X buffer supplemented with protease inhibitor cocktail. The resulting proteins extracted were precipitated using acetone stored at -20oC for 16 hours. After precipitation, the protein pellet was reconstituted in 0.25% RapidGest SF (Waters, UK) in 10 mM Ammonium bicarbonate. A total of 1.2 mg peptides were subjected to LC-MS/MS. The spectrum data was collected using OrbiTrap HF hybrid mass spectrometer. The raw data files were analyzed using MaxQuant to obtain protein identifications and their respective label-free quantification values using in-house standard parameters. Normalization and differential protein expression analysis was performed on the relative protein abundance matrix conducted using the Bioconductor package ‘DEP’ in R. The cutoff for candidates considered as significantly differentially expressed was LFC of log2(2) at a p-value < 0.05. Results were visualized using ‘Enhanced Volcano’ and ‘ggplot2’ packages. Differentially expressed proteins were then analyzed in iLINCS SPIA analysis using the KEGG Pathway analysis database for signaling pathway enrichment.
Cell Viability and Proliferation Assays
5 × 103 cells per well were seeded in a 96-well plate for 24 hours, and treated with either MYC inhibitor (10074-G5). Cell viability was assessed at 24-hour intervals up to 72 hours post-treatment. Vehicle control was 0.1% DMSO-containing complete medium. MTT assay was performed according to standard protocol [15]. For assays that require 96-hour treatment, 1 x 103 cells were seeded in a 96-well plate for 24 hours and then treated.
Isolation of PBMCs and activation of T-cells.
Blood from 50 ml of healthy volunteers was drawn and PBMCs were isolated by lymphocyte separating medium (Corning), centrifuged according to density. A total of 5x106 cells/well was then seeded in a 12-well plate in AIM-V cell culture medium (Gibco) and incubated for 2 hours at 37oC for monocyte cell adhesion. The resultant cell suspension are non-adherent T-cells which are cultured in AIM-V cell culture medium (Gibco) in the presence of phytohemaglutinin and cytokines IL-2 for 3 days.
Production of fourth generation FRα-CAR T-cells.
Isolated T-cells were transduced with FRα-CAR lentiviruses at a multiplicity of infection (MOI) of 50 using a reported method [17]. T-cells were cultured in AIM-V cell culture medium containing 5% human AB serum, IL-2, IL-7 and IL-15 for 4 days. Expression of FRα-CAR T-cells was assayed by flow cytometry (BD Biosciences) technique after cells stained with mouse anti-FRα antibody (1:100 in 1% BSA/PBS, Thermo Fisher Scientific), goat anti-mouse IgG (1:100, Thermo Fisher Scientific), and Alexa Fluor®488 (1:500, Thermo Fisher Scientific).
Immune-mediated Cell Death Assay
Cholangiocarcinoma cell line, KKU-M213 was stained with red fluorescence protein (mCherry), and RBE was stained with CellTrackerTM Orange (Thermo Fisher Scientific). These cancer cells were seeded onto a 96-well plate (Corning) at a cell density of 5 × 103 cells/well. After 24 hours, the cells were treated with varying concentrations of the MYC inhibitor, 10074-G5, and incubated for 48 hours. These cells were co-cultured with the FRα-CAR T-cells at a ratio of 2.5:1 for 24 hours, after which red fluorescence intensities was measured for KKU-M213. Cell viability for RBE was measured using Crystal Violet Staining. RBE cells were fixed with methanol and stained with 0.2% Crystal violet for 20 minutes. The stains were washed with PBS, and eluted with 0.5%SDS diluted in PBS.
Cancer cell death was estimated from reduced fluorescence intensity or absorbance values when compared to respective Untreated controls. These were visualized using GraphPad Prism 9.3, as a Box-and-whisker plot. Along with the fluorescent intensity and absorbance measurements, cell morphology was observed using the ECLIPSE Ti (Nikon Instruments) microscope.
Statistics and Data Analysis
Data was collected in triplicates and three independent rounds, wherever applicable. Student t-test or the One-way ANOVA with Tukey's post-hoc test was conducted to assess statistical significance at p < 0.05 (*).
Results
Analytical Pipeline
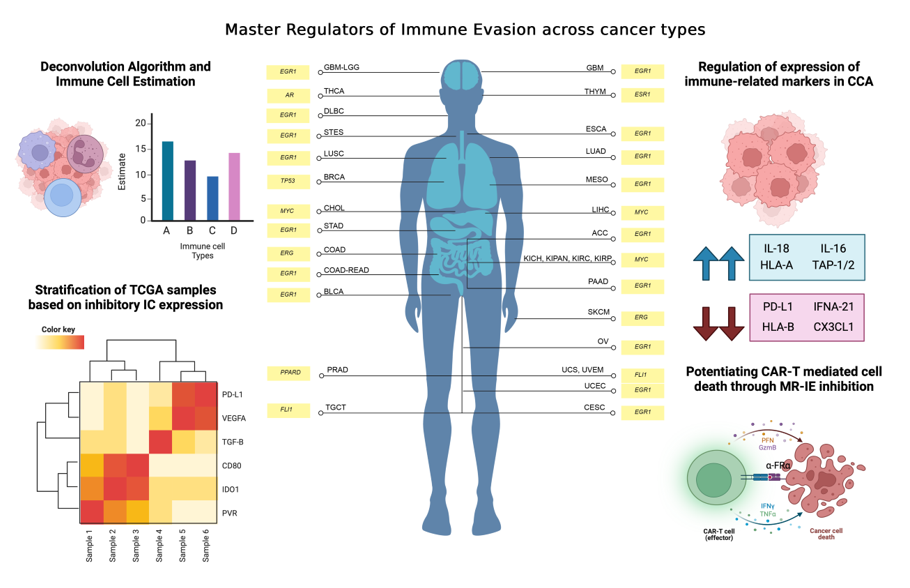
Immune evasion occurs through the synchronous process of immune cell infiltration, co-inhibitory IC expression and modulation of immune related markers. To identify transcriptional regulators that controls these processes, the PanCancer TCGA samples must be subset according to varying degrees of estimated infiltration and then stratified into varying degrees of co-inhibitory IC expression. The transcriptome of these data subsets and clusters are then compared with each other to unravel the different signaling pathways associated with the immune evasion process. The DEGs from these comparisons are then used as an input for transcription factor enrichment to identify MR-IE from varying degrees of leukocyte estimates and IC expression across cancers. These candidates are then ranked according to a composite score. This accounts for the effect of transcription factor gene expression on patient overall survival as well as statistical enrichment scores. This workflow is summarized in Figure 1.
TCGA samples can be stratified based on estimated immune cell infiltration and IC expression
In our investigation, we employed a deconvolution algorithm, CIBERSORT, to estimate the proportion of immune cells in TCGA bulk-tissue transcriptomic data. Subsequently, we utilized unsupervised hierarchical clustering to further stratify the samples in clusters of low, intermediate and high IC expression (Table 1, Figure 2, Supplementary Figures S1–S4). Interestingly, varying degrees of leukocyte estimation resulted in the formation of different clusters of samples of low, intermediate, and high IC gene expression levels (Supplementary Figure S1). Particularly, CD4+ and CD8+ T-cell estimates, resulted in more distinct clusters of mainly low and high co-inhibitory IC expression, with a few samples having intermediate expression of IC molecules (Figure 2a,b). Thus, the presence of these cell types, in particular, may influence the expression of co-inhibitory ICs in cancer cells.
Immune evasion is driven by various signaling cascades depending on different levels of IC expression and leukocyte estimation
When looking at transcriptome level changes between low and high IC expression across the varying subsets of leukocyte estimates, we unravel the signaling pathway changes that are associated with the immune evasion phenomenon. High or Intermediate IC expressing samples represent an immunologically cold (anti-tumoral immune cells do not surveil) tumor, whereas Low IC expressing samples represent an immunologically hot (anti-tumoral immune cells recognize and eliminate cancer cells) tumors.
Accordingly, our differential gene expression analysis between the differing levels of IC expression reveals an inhibition of NK-cell mediated cytotoxicity pathway and T-cell receptor signaling pathway amongst clusters of intermediate and high IC expression (Figure 2d, Supplementary Figure S8a,d–g). Notably, in subsets of data that is highly estimated with leukocytes (Supplementary Figure S8b) antigen processing and presentation pathway is seen to be inhibited, between clusters with intermediate to high IC expression. Moreover, cytokine-cytokine receptor signaling pathways are activated, and interestingly chemokine signaling pathways are inhibited in these highly CD4+ and CD8+ T-cell estimated subsets (Supplementary Figure S8d, 8f). This suggests that the levels of IC expression, and particular infiltration of CD4+ and CD8+ T-cells may trigger the secretion of cytokines to promote an immunosuppressive environment. Interestingly, amongst the low immune-cell estimated subsets (Figure 2d, Supplementary Figure S8c,e,g) the inhibition of NK-cell mediated cytotoxicity and T-cell receptor signaling pathways are maintained. However, several cancer-specific pathways are seen to be inhibited such as cAMP signaling and cGMP-PKG signaling.
Collectively, our robust workflow incorporating estimated immune infiltration of tumors and IC expression stratification of tumors confirmed and unraveled pathways related to immune evasion. This provides confidence that the DEGs would be able to identify a transcriptional regulator central to these pathways.
EGR1 and MYC are identified as top-ranking MR-IE in multiple cancers
The transcription factor enrichment analysis conducted resulted in 21 significant candidate MR-IE across the various subsets and IC expression clusters. To assess the likelihood of these candidate MR-IEs in regulating the expression of ICs, we assessed the co-expression between the transcription factors and ICs across all cancers. This revealed groups of candidate MR-IEs that are positively-correlated with specific ICs and negatively-correlated with other ICs (Figure 2e). For example, the candidate MR-IE, FOXA2, is significantly positively-correlated with the expression of ARG1, FGL1, PVR, PVRL2, CEACAM, LGALS3, HMGB1, and PD-L1 (CD276). However, it is negatively-correlated with the expression of VEGFB and TGFB1. This provides insights into the potential positive or negative regulation of these candidate MR-IE of the ICs. Nevertheless, there may be lineage-specific co-expression patterns that is not accounted for in this analysis. Thus, to rank the ideal candidate MR-IE per cancer type, we employed a cox proportional univariate analysis of each transcription factor against overall survival of each of the 33 TCGA cancer types. The hazard ratio and enrichment scores of candidate MR-IE per cancer type are tabulated in Table 2. A composite score comprising of enrichment scores, statistical significance and hazard ratios was devised to rank candidate MR-IE for each cancer type (Table 2 and Table 3). The top-ranking candidate MR-IE per cancer type is depicted in Figure 3. The cancer types are listed as per TCGA study abbreviations.
Notably, EGR1 appeared as the top-ranking candidate MR-IE in multiple cancer types, such as GBM, DLBC, ESCA, LUAD, LUSC, MESO, ACC, STAD, COAD-READ, BLCA, PAAD, OV, UCEC, and CESC, Whereas MYC appears to be the top-ranking candidate in particularly liver and renal associated cancers (i.e. LIHC, CHOL, KICH, KIPAN, KIRC, and KIRP). Nevertheless, MYC was frequently ranked second in cancer types with EGR1 as the top-ranking candidate. Thus, these two candidate MR-IE ranked based on mortality-association and statistical significance would make ideal immunotherapeutic targets. Other candidate MR-IEs include TP53 for BRCA, AR for THCA, FLI1 for TCGT, UCS and UVEM, and PPARD for PRAD.
Our investigation focuses on providing therapeutic solutions for a pressing issue in Thailand, cholangiocarcinoma (CCA; CHOL). CCA is a prevalent malignancy with a complex tumor microenvironment. Unfortunately, current therapeutic options are incapable of treating complex heterogenous patient populations,. Thus, this investigation assesses MYC as an immunotherapeutic target for CCA.
MYC perturbation in CCA results in downregulation of PD-L1
To validate MYC as an MR-IE for CCA, we assessed its role in regulating the expression of a representative IC molecule, PD-L1. In KKU-M213 cells, we observe that a therapeutic inhibition of MYC using a MYC inhibitor (10074-G5) (Supplementary Figure S9a,b) resulted in a significant downregulation of protein and gene expression of PD-L1 (Figure 4a,b). These results are maintained when the MYC gene was knocked down using siRNA interference (Figure 4d, Supplementary Figure S9e,f). Contrastingly, the RBE cells displayed a modest decrease in protein expression but no significant difference in gene expression (Figure 4e–h). Several factors could contribute towards the discrepancy in the molecular regulation of PD-L1 between these cell lines, including heterogeneity within CCA and unexplored compensatory mechanisms. Thus, in this investigation, we further explored KKU-M213 as the hypothesized ‘responsive’ cell line, and the representative cell line for a patient population with similar molecular profiles for which this novel immunotherapeutic strategy is surmised to benefit.
Inhibition of MYC potentiates CAR-T mediated cell death of CCA cells
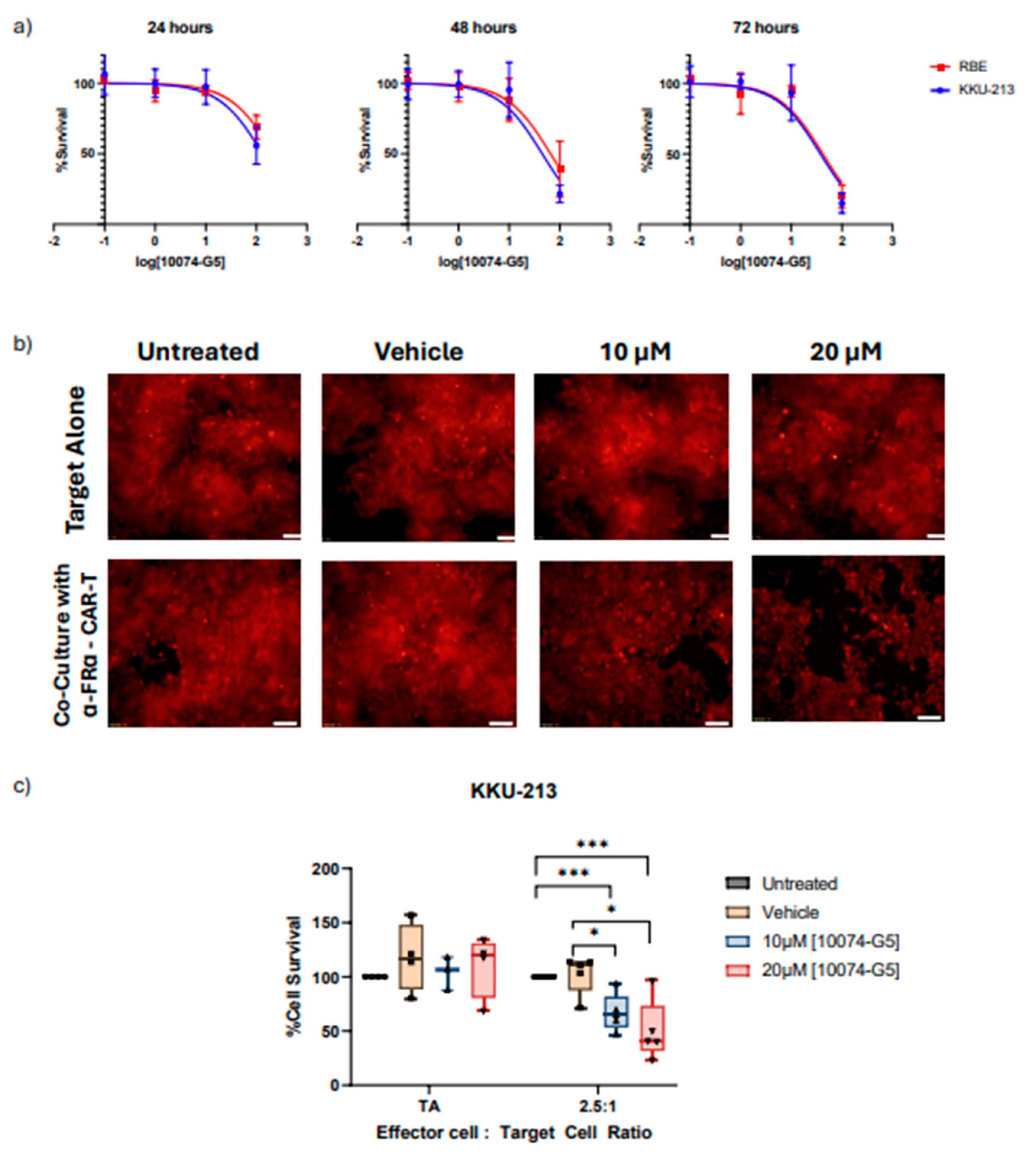
MYC, the top-ranking MR-IE candidate of CCA, modulated multiple inhibitory elements that contribute towards immune evasion (Figure 5). Thus, inhibiting MYC in cancer cells is expected to potentiate immune-mediated recognition and elimination. Here we inhibited MYC in CCA cells (KKU-M213 and RBE) using a small-molecule inhibitor before subjecting them to CAR-T cells to assess any cytotoxic effect the inhibitor may have on the cancer cells itself and to determine non-lethal concentration ranges. The cancer cell lines are responsive to MYC inhibitor treatment dose and time dependently (Figure 6a). However, the IC50 of the MYC inhibitor against CCA cell viability was higher than the reported plasma concentration (58.2µM) at 24 hours and 48 hours, ranging at 117.3µM and 50.17µM, respectively, for KKU-M213 cell line, and 260.3µM and 65.75µM for RBE cell line (Table 6). Between the two cell lines, KKU-M213 was more responsive. Nevertheless, this investigation surmises that inhibition of MYC would potentiate immune-mediated elimination and therefore, a lower concentration could potentially elicit this effect.
To functionally assess if targeting the MR-IE of CCA reinstates immune recognition and elimination of cancer cells, we used CAR-T cells as a model for cancer cell-targeted immune cell induced cytotoxicity. Acknowledging the success of fourth-generation anti-FRɑ CAR-T cell development in breast cancer [17], we sought to reposition this CAR-T cell towards CCA. To confirm if this CAR-T model can be repositioned towards CCA, we assessed the expression of FRɑ across CCA cell lines (Supplementary Figure S11). Notably, most cell lines, except HuCCA-1 and KKU-100) displayed between 20% and 70% expression, especially in the two cell lines used in this investigation, KKU-M213 (~30%) and RBE (~55%), which showed similar levels of expression. (Supplementary Figure S11a). Specifically, the relative mean fluorescent intensity (rMFI) within these cells (except HuCCA1 and KKU-100) range between 2-5. Hence, anti-FRɑ CAR-T cell has potential to be repositioned towards the treatment of CCA.
Noting that KKU-M213 is the responsive cell line and representative of the potentially responsive CCA patient population, we assessed if MYC inhibition could potentiate immune-mediated cell death. To do so, we compared the effect of MYC inhibition on target cells (CCA cells) alone versus target cells co-cultured with anti-FRɑ CAR-T cells. Interestingly, the effect of the MYC inhibitor is more pronounced when KKU-M213 cells are co-cultured with anti-FRɑ CAR-T cells, as immune-mediated cell death is potentiated (Figure 6b). Remarkably, the concentration required to elicit 50% reduction in cell viability is much lower than when KKU-M213 cells are treated alone (Figure 6c). Conversely, in RBE, the CCA cell line, deemed as ‘resistant’ because it had no significant changes in IC markers after MYC inhibition or knockdown did not potentiate immune-mediated cell death. While RBE cells were responsive to MYC inhibition induced cytotoxicity, co-culture with CAR-T cells did not increase the cell death. This suggests that the cell death observed may be a direct effect of the MYC inhibitor on the cancer cells alone (Supplementary Figure S12b).
Cumulatively, this shows that MYC inhibition exhibits immunomodulatory effects that sensitizes KKU-M213 cells to CAR-T mediated cell death. Thus, MR-IE inhibition has the potential to be an effective therapeutic strategy in CCA patients represented by KKU-M213 cells. While this result is promising, complex models that assess the infiltration of immune cells, should be utilized to further develop MYC inhibition as an immunotherapeutic strategy. Hence, this should encourage further development of MYC inhibition as an immunotherapeutic strategy for CCA, and by extension, MR-IE inhibition as an effective immunotherapeutic strategy.
Discussion
Immune evasion is a meticulous process that synchronizes several signaling cascades within tumor cells and its surrounding stromal cells to facilitate the proliferation and invasion of tumors to secondary sites [18]. These include the expression of co-inhibitory IC molecules on tumor cells and immune cells, infiltration of pro-tumoral immune cells, downregulated self-antigen presentation, and the secretion of immunosuppressive cytokines [19]. Thus, impeding any one pathway leaves room for several mechanisms to compensate for the inhibition. Existing cancer immunotherapies improved patient outcomes using ICB in lung [20], renal [21], bladder [22] and head-and-neck cancer [23]. However, only 30-40% of patients are either eligible or respond to these treatments [6]. Hence, this warrants the need for more potent and context-specific therapeutic strategies to cater to different patient populations. The present investigation offers a novel and potentially more effective therapeutic strategy by exploiting the multifunctional and multitargeted roles of transcriptional regulators of immune evasion. Our investigation identified MR-IE for each cancer type by integrating deconvolution algorithms, hierarchical clustering, transcription factor enrichment analyses, and survival analyses (Figure 1, Figure 2 and Figure 3). Therefore, this investigation provides a high-confidence list of MR-IEs as candidate immunotherapeutic biomarkers for each cancer type (Table 2, Figure 3), to encourage the development of small-molecule inhibitors against these targets.
Tumors possess a complex architecture comprising of various stromal cells, cancer associated fibroblast, and various immune cells. The infiltration of these cells is closely associated with clinical implications [24]. Hence, it is imperative to address the infiltration of immune cells in the tumors of the TCGA cohort, to fulfill our quest for the MR-IE. One caveat of using the TCGA PanCancer dataset is the information loss of tumor-infiltrated and surrounding cells due to bulk-tumor sequencing. To circumvent this issue, computational methods have been developed to estimate the proportion of various immune cells by assessing similarity of gene signatures of samples to the gene signatures of immune cells [25]. In our investigation we adopted the CIBERSORTx algorithm to estimate immune cell infiltration. Consistent with prior reviews, our investigation shows that high infiltration of CD8+ and CD4+ T-cells in the tumors influences inhibitory IC expression, and is associated with cytokine-receptor signaling pathways across cancers (Supplementary Figures S1 and S8).
CCA is a heterogenous lethal malignancy of the bile duct, with its peak incidence and mortality rate in Northeastern Thailand, and a growing incidence rate worldwide [26]. The lack of early-stage symptoms, diagnostic markers, and rapid progression contributes toward rising mortality rates. This trickles down to a lack of effective therapeutic strategies for advanced stage patients [10,27]. Recently immunotherapy including ICB, agonist antibodies and CAR-T cell therapy, has gained traction in the treatment of CCA. However, as with other cancer types, a minority of CCA patients qualify for immunotherapy based on biomarker expression and tumor mutational burden [28]. The current objective response rate for clinical trials of ICBs such as pembrolizumab (KEYNOTE-158; KEYNOTE-028), nivolumab and ipilimumab (NCT02923934) ranges between 5-20%. Thus, to amplify therapeutic options for CCA patients, this investigation proposes the therapeutic inhibition of MYC (the top-ranking MR-IE of CCA; Figure 3). Towards this, we showed that MYC inhibition potentiated immune-mediated cell death when co-cultured with anti-FRɑ CAR-T cells. Thus, this investigation highlights the potential of combining MR-IE inhibition with CAR-T cell therapy for a more potent treatment outcome (Figure 6).
Our investigation reports, for the first time, that EGR1 is the most frequently high-ranked and clinically-relevant, MR-IE in the multiple cancer types. This was followed by MYC being the second-most frequently top-ranked MR-IE (Figure 3). Currently, little is known of the role of EGR1 in anti-cancer immunity, which presents an opportunity for us to explore this as an immunotherapeutic target in the stipulated cancer types. This is supported by several investigations that report EGR1 as a “master regulator” of inflammatory enhancers in macrophages, by regulating the differentiation of monocytes to macrophages, and a myriad of inflammatory genes [29,30]. This shows merit in our workflow being able to aptly identify MR-IE in the right context. Moreover, since seldom reports thoroughly explore the therapeutic potential of targeting EGR1, prospects of the current investigation should explore this phenomenon.
MYC, on the other hand, is a well-established oncogene known to regulate several tumor-cell intrinsic mechanisms, to promote survival, growth, and proliferation of tumor cells. Recent reviews highlight the association of MYC with the expression of PD-L1 and CD-47 [19], the regulation of T-cell activation and function, and polarization of macrophages [31]. Consequently, MYC has become an ideal therapeutic target as an oncogene and as an immunotherapeutic target. However, the role of MYC is context-dependent, as the function of MYC is dependent on post-translational modifications and mutational burden of its gene targets, and the interaction of the cancer cells with tumor microenvironment [32]. For instance, MYC inactivation induced proliferative arrest in hematological cancers [33], whereas MYC inactivation in osteosarcoma resulted in terminal differentiation into bone [34]. Thus, the role of MYC and its association with patient-mortality is dependent on the cancer lineage [32]. Our workflow acknowledges the context-dependent roles of transcriptional regulators by accounting for its effect on patient overall survival when ranking the candidate MR-IEs (Table 2, Figure 3). Therefore, this ranking system yields potential MR-IEs whose higher expression results in lower patient survival, thereby making them ideal therapeutic targets in the specified cancer type. We hypothesized that targeting of MR-IEs would reinstate immune recognition and elimination of cancer cells by downregulation of inhibitory ICs and upregulation of stimulatory immune-related markers. Accordingly, our validation assays confirm the role of MYC in regulating the expression of a co-inhibitory IC molecule, PD-L1 (Figure 4). Moreover, anti-tumoral inflammatory markers such as IL-18, IL-10, and MAP2K1 were upregulated after MYC inhibition (Figure 5), in CCA cells. The modulation of these markers is associated with reinstatement of immune recognition of cancer cells. Taken together, this provides confidence in our workflow to identify and rank candidate MR-IE per cancer type, that can be developed as immunotherapeutic targets.
While our robust workflow accounted for multiple aspects of immune evasion, there are limitations to our findings. Firstly, a caveat of the TCGA PanCancer Atlas is varied sample size per cancer type, resulting in biases in clinical significance of predicted MR-IE in each cancer type. Moreover, we used bulk-tissue transcriptomics to infer immune cell estimates. Alternatively, the incorporation of single-cell RNA sequencing data may provide better resolution into the interaction between highly and lowly infiltrated tumors, context-specifically. Additionally, this data does not account for heterogeneity within cancer subtypes. This was evidenced in the discrepancy in response to MR-IE inhibition between KKU-M213 and RBE cell lines. This may be due to differences in post-translational modifications of MYC’s gene targets, or varying genetic alterations between the two chosen cell lines. Thus, our workflow may need to be revised to account for these variables. Lastly, further investigation within CCA is required to confirm the therapeutic efficacy of MR-IE inhibition, in more complex tumor models.
Conclusively, we aimed to provide insight into the regulation of immune evasion as an alternative immunotherapeutic strategy. Towards this aim, this study developed a unique workflow that identifies and ranks candidate MR-IE as novel immunotherapeutic targets for each cancer type. As a result, MYC was identified to be the candidate MR-IE for CCA, and so we encourage the development of several small-molecule inhibitors against MYC for the treatment of CCA. We hope the findings of this investigation inspires further development of these MR-IEs as therapeutic targets to benefit a wider patient population.
List of Abbreviations
| ICs | Immune Checkpoints |
| ICB | Immune Checkpoint Blockade |
| CAR-T | Chimeric Antigen T-cells |
| MR-IE | Master Regulators of Immune Evasion |
| TCGA | The Cancer Genome Atlas |
| CCA | Cholangiocarcinoma |
| X2K | Expression2Kinases |
| FRα | Folate-Receptor alpha |
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization: S.V., S.C., Data Curation: S.V., S.C., Visualization: S.V., S.C., B.B., Methodology: S.V., P.K., S.Y., S.T., B.B., N.C. S.K., M.S., S.J., C.T.,, Writing (first draft): S.V., Writing (review and revision): B.B, P.K., S.Y., N.C., S.K., S.T., M.S., K.M., S.J., C.T., J.M., S.C., R.T., T.J., Resources: S.S., R.T., C.T., S.J., Funding: S.S., R.T,, Supervision: T.J., R.T., S.C., J.M., C.T., S.J.
Ethics Approval and Consent to Participate: The use of samples and data collection protocol was approved by the Mahidol University- Institution Review Board for human ethics standard (MU-MOA COA 2022/052.1105).
Consent for Publication: All authors approve of this manuscript and have provided their consent for publication.
Availability of Data and Material: The dataset used in this investigation can be obtained from the Genomic Data Commons Portal of The Cancer Genome Atlas (TCGA) in the Pancancer Atlas publication page (https://gdc.cancer.gov/about-data/publications/pancanatlas). Software used is listed in the Key Resources Table.
Funding
This investigation was supported by Mahidol University (Fundamental Fund: fiscal year 2023 by National Science Research and Innovation Fund (NSRF)) (FF-062/2566), and by NSRF via the Program Management Unit for Human Resources and Institutional Development, Research, and Innovation grant number B36G660002 (to R.T.) and the Mahidol International Postdoctoral Fellowship grant (to S.V.).
Acknowledgments
We would like to acknowledge the generous contribution of cholangiocarcinoma cell lines by Prof.Banchop Sripa. We humbly acknowledge Prof. Pa-Thai Yenchitsomanus and his lab members for their generous contribution in the production of fourth-generation anti-FRα CAR-T cell technology. We would also like to acknowledge the contributions in methods by Ms.Pimkanya More-krong.
Competing Interest: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74.
- Gupta I, Hussein O, Sastry K, Bougarn S, Gopinath N, Chin-Smith E, et al. Deciphering the complexities of cancer cell immune evasion: Mechanisms and therapeutic implications. Advances in Cancer Biology - Metastasis. 2023.
- Haslam A, Gill J, Prasad V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for Immune Checkpoint Inhibitor Drugs. JAMA Netw Open. 2020;3(3):e200423.
- Dobosz P, Stepien M, Golke A, Dzieciatkowski T. Challenges of the Immunotherapy: Perspectives and Limitations of the Immune Checkpoint Inhibitor Treatment. Int J Mol Sci. 2022;23(5).
- Schachter J, Ribas A, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet. 2017;390(10105):1853-62.
- Venkatraman S, Meller J, Hongeng S, Tohtong R, Chutipongtanate S. Transcriptional Regulation of Cancer Immune Checkpoints: Emerging Strategies for Immunotherapy. Vaccines (Basel). 2020;8(4).
- Casey SC, Baylot V, Felsher DW. The MYC oncogene is a global regulator of the immune response. Blood. 2018;131(18):2007-15.
- Rebe C, Ghiringhelli F. STAT3, a Master Regulator of Anti-Tumor Immune Response. Cancers (Basel). 2019;11(9).
- Venkatraman S, Balasubramanian B, Pongchaikul P, Tohtong R, Chutipongtanate S. Molecularly Guided Drug Repurposing for Cholangiocarcinoma: An Integrative Bioinformatic Approach. Genes (Basel). 2022;13(2).
- Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17(9):557-88.
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity. 2019;51(2):411-2.
- Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773-82.
- R Development Core Team. R: A language and environment for statistical computing.. Vienna, Austria: R Foundation for Statistical Computing.; 2010.
- Pilarczyk M, Fazel-Najafabadi M, Kouril M, Shamsaei B, Vasiliauskas J, Niu W, et al. Connecting omics signatures and revealing biological mechanisms with iLINCS. Nat Commun. 2022;13(1):4678.
- Pongcharoen P, Jinawath A, Tohtong R. Silencing of CD44 by siRNA suppressed invasion, migration and adhesion to matrix, but not secretion of MMPs, of cholangiocarcinoma cells. Clin Exp Metastasis. 2011;28(8):827-39.
- Krobthong S, Yingchutrakul Y, Visessanguan W, Mahatnirunkul T, Samutrtai P, Chaichana C, et al. Study of the Lipolysis Effect of Nanoliposome-Encapsulated Ganoderma lucidum Protein Hydrolysates on Adipocyte Cells Using Proteomics Approach. Foods. 2021;10(9).
- Luangwattananun P, Junking M, Sujjitjoon J, Wutti-In Y, Poungvarin N, Thuwajit C, et al. Fourth-generation chimeric antigen receptor T cells targeting folate receptor alpha antigen expressed on breast cancer cells for adoptive T cell therapy. Breast Cancer Res Treat. 2021;186(1):25-36.
- Haynes NM, Chadwick TB, Parker BS. The complexity of immune evasion mechanisms throughout the metastatic cascade. Nat Immunol. 2024.
- Spranger S, Gajewski TF. Mechanisms of Tumor Cell–Intrinsic Immune Evasion. Annual Review of Cancer Biology. 2018;2(Volume 2, 2018):213-28.
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018-28.
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373(19):1803-13.
- Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558-62.
- Ferris RL, Blumenschein G, Jr., Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375(19):1856-67.
- Zhang SC, Hu ZQ, Long JH, Zhu GM, Wang Y, Jia Y, et al. Clinical Implications of Tumor-Infiltrating Immune Cells in Breast Cancer. J Cancer. 2019;10(24):6175-84.
- Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509-W14.
- Khan SA, Tavolari S, Brandi G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019;39 Suppl 1:19-31.
- Tomlinson JL, Valle JW, Ilyas SI. Immunobiology of cholangiocarcinoma. J Hepatol. 2023;79(3):867-75.
- Lo JH, Agarwal R, Goff LW, Heumann TR. Immunotherapy in Biliary Tract Cancers: Current Standard-of-Care and Emerging Strategies. Cancers (Basel). 2023;15(13).
- Trizzino M, Zucco A, Deliard S, Wang F, Barbieri E, Veglia F, et al. EGR1 is a gatekeeper of inflammatory enhancers in human macrophages. Sci Adv. 2021;7(3).
- Lee JY, Kim JH, Bang H, Cho J, Ko YH, Kim SJ, et al. EGR1 as a potential marker of prognosis in extranodal NK/T-cell lymphoma. Sci Rep. 2021;11(1):10342.
- Venkatraman S, Balasubramanian B, Thuwajit C, Meller J, Tohtong R, Chutipongtanate S. Targeting MYC at the intersection between cancer metabolism and oncoimmunology. Front Immunol. 2024;15:1324045.
- Dhanasekaran R, Deutzmann A, Mahauad-Fernandez WD, Hansen AS, Gouw AM, Felsher DW. The MYC oncogene - the grand orchestrator of cancer growth and immune evasion. Nat Rev Clin Oncol. 2022;19(1):23-36.
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4(2):199-207.
- Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A. 2007;104(32):13028-33.
Figure 1.
Schematic Workflow of Integrated in silico analysis to identify MR-IE. Blue Boxes represent analyses conducted, and yellow boxes represent details of the analyses. This Figure was made with Biorender.com.
Figure 1.
Schematic Workflow of Integrated in silico analysis to identify MR-IE. Blue Boxes represent analyses conducted, and yellow boxes represent details of the analyses. This Figure was made with Biorender.com.
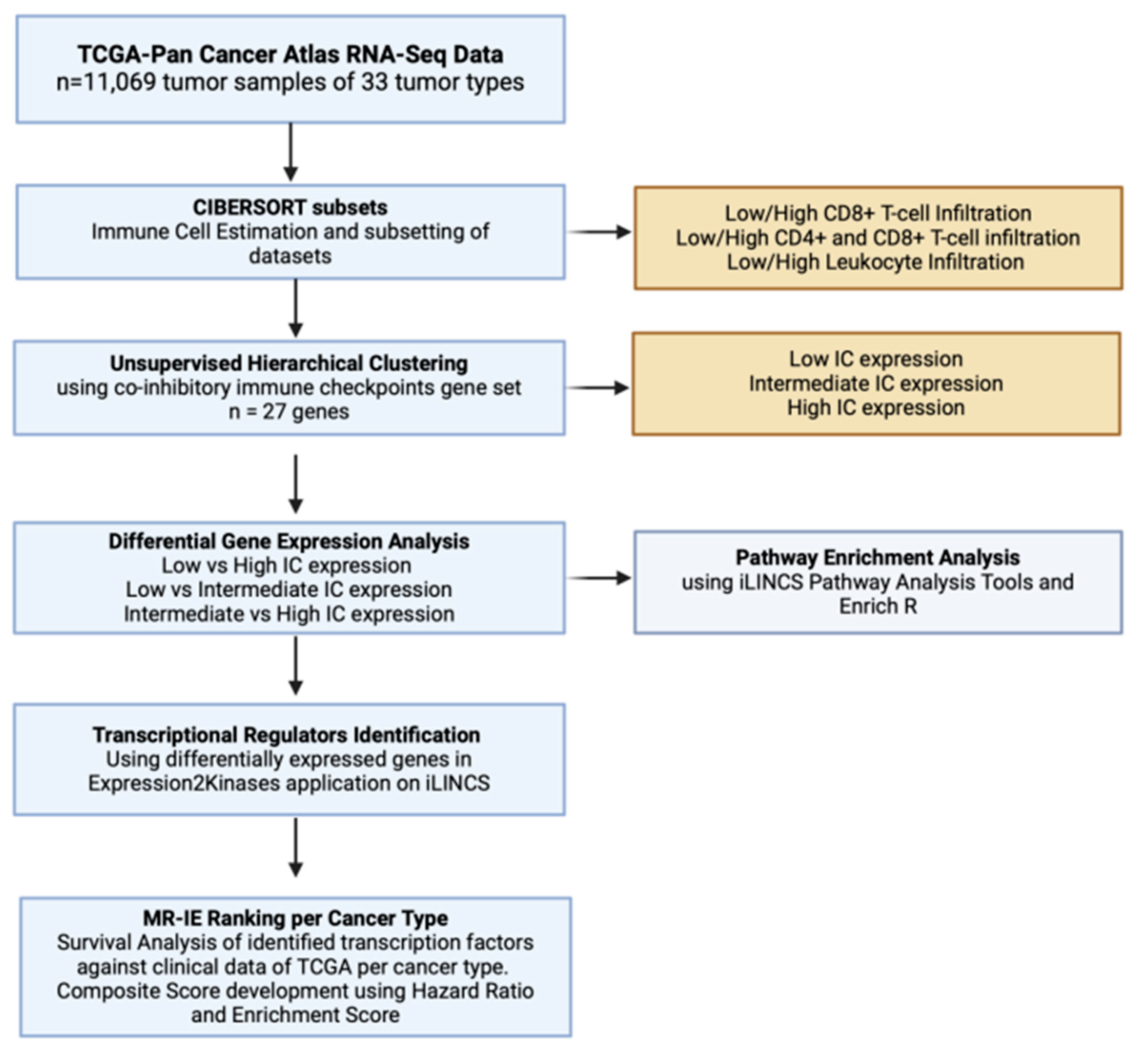
Figure 2.
Representative analysis of low CD4+ and CD8+ T-cell estimated TCGA samples. a) Unsupervised hierarchical clustering of TCGA Pan Cancer samples subset with low estimates of CD4+ and CD8+ T-cells against co-inhibitory IC gene expression. b) Multidimensional Scaling plot of clusters defined at high (red) and low (blue) IC expression. c) Volcano Plot of differentially expressed genes defined at p.
Figure 2.
Representative analysis of low CD4+ and CD8+ T-cell estimated TCGA samples. a) Unsupervised hierarchical clustering of TCGA Pan Cancer samples subset with low estimates of CD4+ and CD8+ T-cells against co-inhibitory IC gene expression. b) Multidimensional Scaling plot of clusters defined at high (red) and low (blue) IC expression. c) Volcano Plot of differentially expressed genes defined at p.
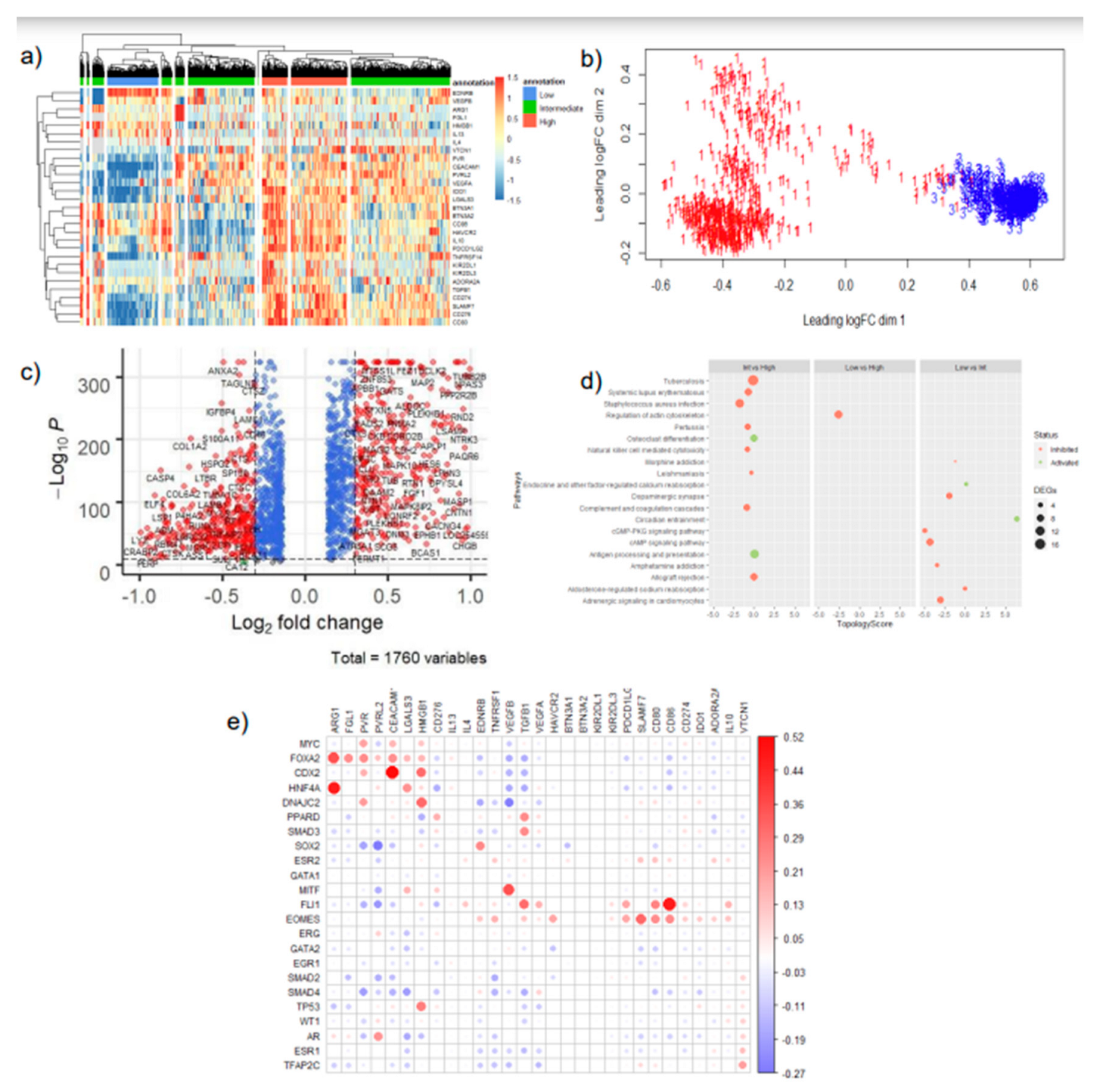
Figure 3.
Top ranking candidate MR-IE for each TCGA tumor type based on composite score. Composite Score was calculated using the Hazard Raito (effect of identified transcription factor’s effect on patients’ overall survival in each tumor type) multiplied by the statistical enrichment score obtained from the Expression2Kinases application.
Figure 3.
Top ranking candidate MR-IE for each TCGA tumor type based on composite score. Composite Score was calculated using the Hazard Raito (effect of identified transcription factor’s effect on patients’ overall survival in each tumor type) multiplied by the statistical enrichment score obtained from the Expression2Kinases application.
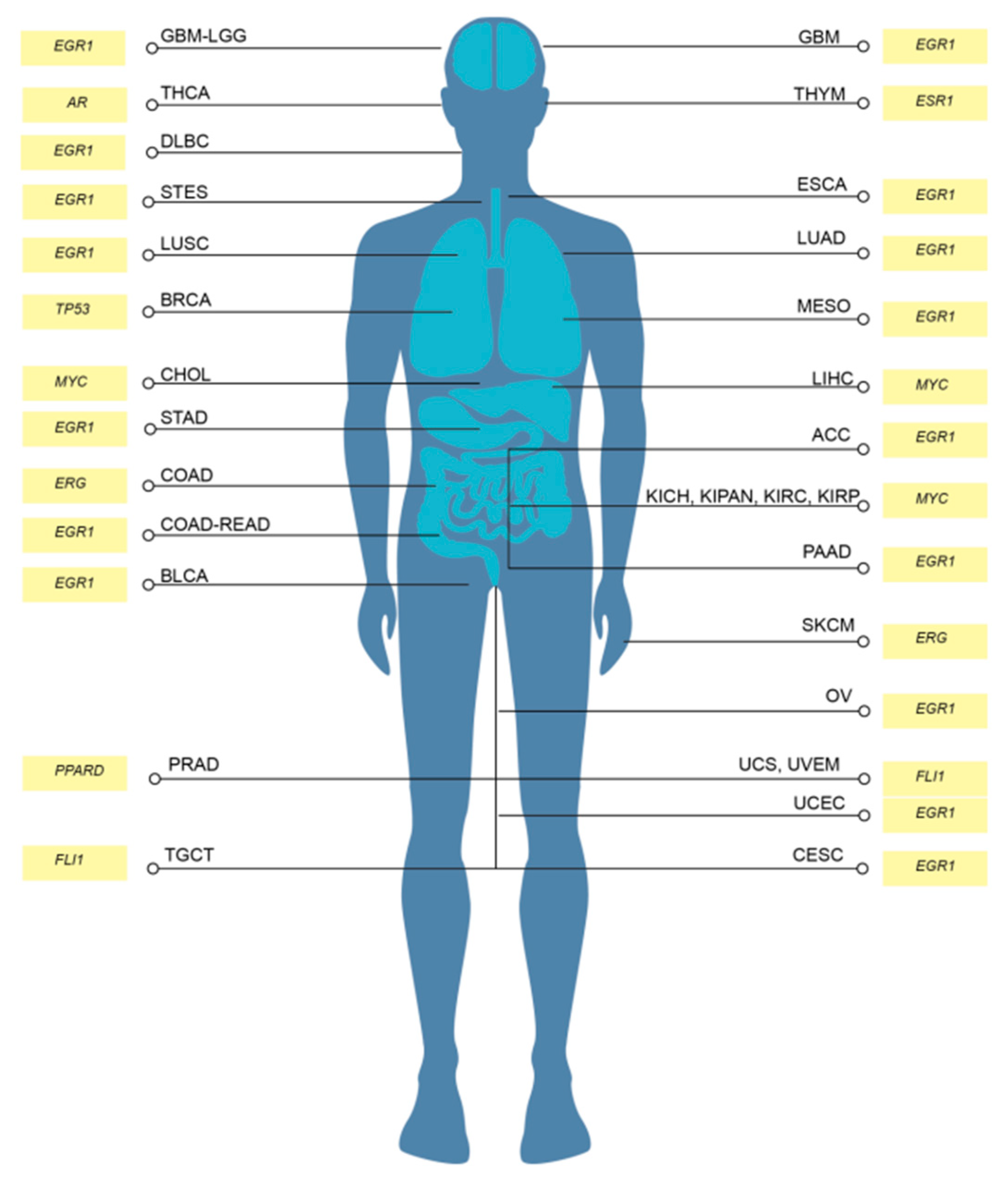
Figure 4.
Inhibition or knockdown of master regulator results in the downregulation of PD-L1 expression in CCA cell lines KKU-213 but not RBE. a) Western Blot of PD-L1 after MYC inhibitor treatment in KKU-213 cells. b) Densitometry analysis of PD-L1 bands normalized to GAPDH in KKU-213. c) mRNA expression of PD-L1 normalized to 18S expression after MYC inhibitor treatment in KKU-213 cells. d) mRNA expression of PD-L1 normalized to 18S after MYC siRNA knockdown in KKU-213 cells. e) Western Blot of PD-L1 protein after MYC inhibitor treatment in RBE cells. f) Densitometry analysis of PD-L1 bands normalized to GAPDH in RBE. g) mRNA expression of PD-L1 normalized to 18S after MYC inhibitor treatment in RBE cells. h) mRNA expression of PD-L1 normalized to 18S after MYC siRNA knockdown in RBE cells.
Figure 4.
Inhibition or knockdown of master regulator results in the downregulation of PD-L1 expression in CCA cell lines KKU-213 but not RBE. a) Western Blot of PD-L1 after MYC inhibitor treatment in KKU-213 cells. b) Densitometry analysis of PD-L1 bands normalized to GAPDH in KKU-213. c) mRNA expression of PD-L1 normalized to 18S expression after MYC inhibitor treatment in KKU-213 cells. d) mRNA expression of PD-L1 normalized to 18S after MYC siRNA knockdown in KKU-213 cells. e) Western Blot of PD-L1 protein after MYC inhibitor treatment in RBE cells. f) Densitometry analysis of PD-L1 bands normalized to GAPDH in RBE. g) mRNA expression of PD-L1 normalized to 18S after MYC inhibitor treatment in RBE cells. h) mRNA expression of PD-L1 normalized to 18S after MYC siRNA knockdown in RBE cells.
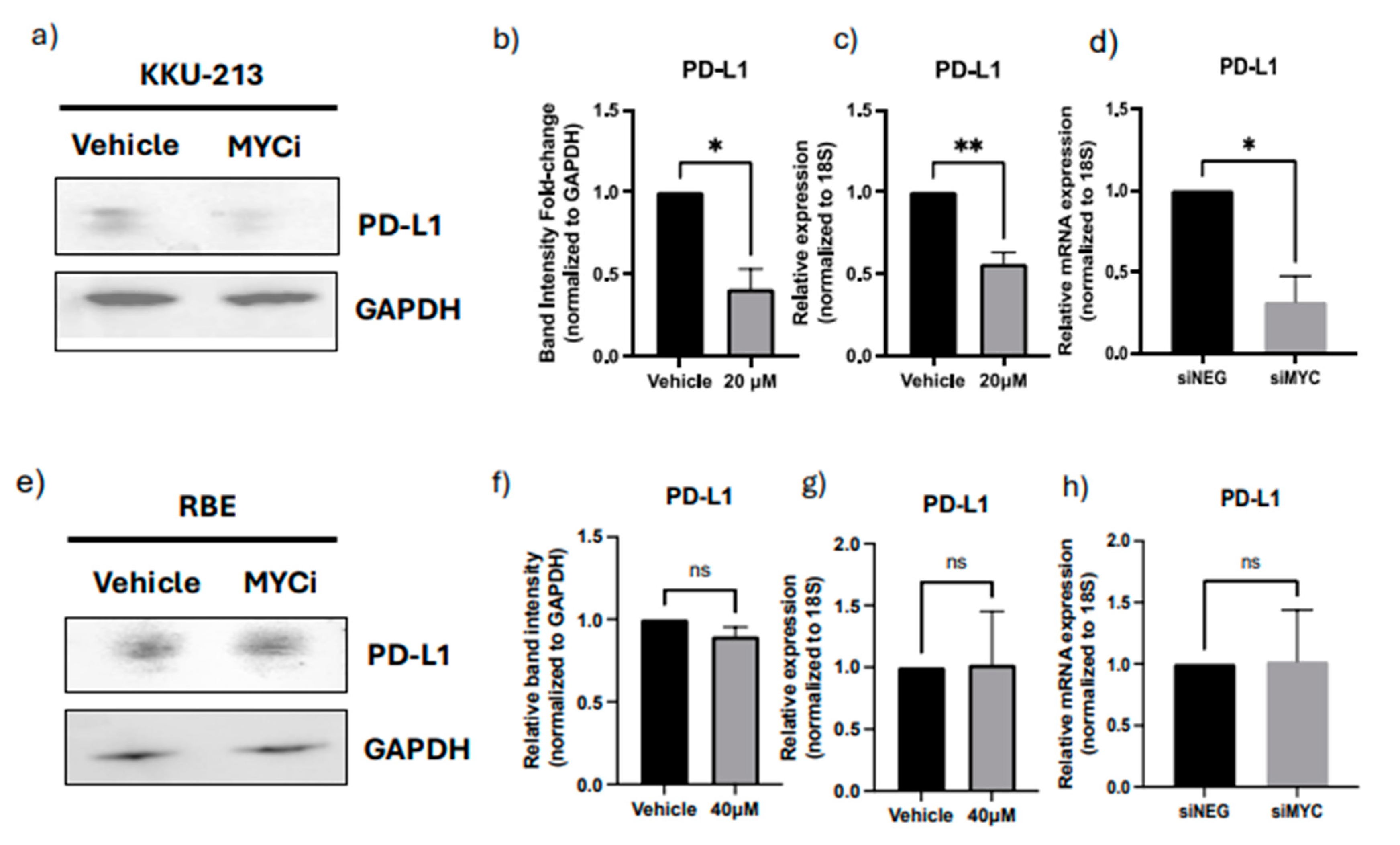
Figure 5.
Inhibition or knockdown of master regulator results in modulation of immune related markers in the cancer cell line proteome in KKU-213 cell line. a) PCA of KKU213 cell samples either treated with MYC inhibitor or Vehicle control. b) Volcano Plot of differentially expressed proteins. c) Heatmap of Differentially expressed proteins. d) Pathway Enrichment of differentially expressed proteins. e) Relative expression of Immune related proteins after MYC inhibitor treatment compared to control.
Figure 5.
Inhibition or knockdown of master regulator results in modulation of immune related markers in the cancer cell line proteome in KKU-213 cell line. a) PCA of KKU213 cell samples either treated with MYC inhibitor or Vehicle control. b) Volcano Plot of differentially expressed proteins. c) Heatmap of Differentially expressed proteins. d) Pathway Enrichment of differentially expressed proteins. e) Relative expression of Immune related proteins after MYC inhibitor treatment compared to control.
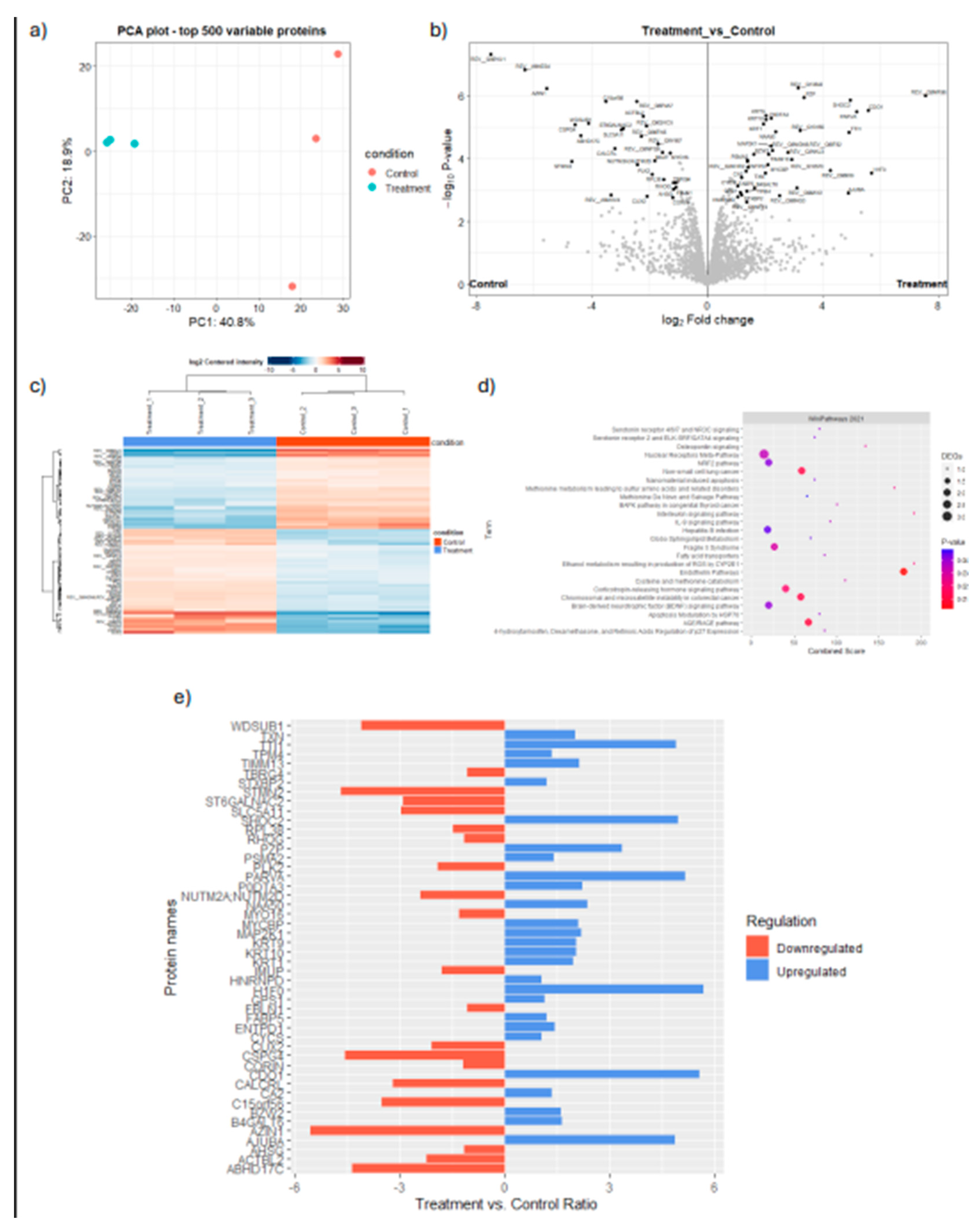
Figure 6.
Inhibition or MYC (MR-IE of CCA) results in the potentiation of immune-mediated cell death through CAR-T cell based therapy in KKU-213 cell line. a) Dose-response curves of CCA cell lines KKU-213 and RBE treated with varying doses of MYC inhibitor for 24, 48, and 72 hours. b) Fluorescent images of KKU-213 tagged with mCherry treated with MYC inhibitor in varying doses and co-cultured with anti-FR-ɑ CAR-T cells. c) Cell Survival plot compared between target cell alone (TA), and co-culture with anti-FR-ɑ at an effector to target cell ratio of 2.5:1.
Figure 6.
Inhibition or MYC (MR-IE of CCA) results in the potentiation of immune-mediated cell death through CAR-T cell based therapy in KKU-213 cell line. a) Dose-response curves of CCA cell lines KKU-213 and RBE treated with varying doses of MYC inhibitor for 24, 48, and 72 hours. b) Fluorescent images of KKU-213 tagged with mCherry treated with MYC inhibitor in varying doses and co-cultured with anti-FR-ɑ CAR-T cells. c) Cell Survival plot compared between target cell alone (TA), and co-culture with anti-FR-ɑ at an effector to target cell ratio of 2.5:1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



