Submitted:
16 October 2024
Posted:
17 October 2024
You are already at the latest version
Abstract
Objective: In recent years, the field of Essential Thrombocythemia (ET) research has seen a significant accumulation of scientific findings, but comprehensive bibliometric analyses remain lacking. This study aims to fill that gap by performing a thorough bibliometric analysis of ET research, identifying key contributors and collaboration networks, and mapping the development trends to provide insights for future research directions. Methods: A bibliometric analysis of ET-related publications from 2001 to 2024 was conducted using CiteSpace, VOSviewer, and R packages. Data were retrieved from the Web of Science Core Collection, with a focus on publication volume, citation analysis, co-authorship networks, co-citation relationships, and citation bursts. Results: A total of 4,297 studies published in 778 journals were included in the analysis. ET research has seen rapid growth, with researcher clusters in the United States and Europe driving progress through extensive regional and international collaborations. Leading researchers, such as Ayalew Tefferi from the Mayo Clinic and Alessandro M. Vannucchi from the University of Florence, have made significant advances in ET classification, molecular mechanisms, targeted therapies, and disease management. The discovery of driver mutations, such as JAK2, has revolutionized the diagnostic and therapeutic approaches to ET. Research focus has shifted from clinical morphological diagnosis to molecular diagnostics, with the field now entering the era of targeted therapies. However, the inherent heterogeneity of ET continues to present challenges for the widespread implementation of personalized precision treatment. Conclusion: Bibliometric analysis demonstrates significant advances in ET research, particularly in molecular pathology and targeted therapies. However, the heterogeneity of ET remains a major obstacle to personalized treatment. Future research should focus on further elucidating the pathogenesis of ET and improving stratified management approaches to achieve individualized precision therapy.
Keywords:
1. Introduction
2. Materials and Methods
2.1. Search Strategy and Data Acquisition
2.2. Data Analysis
3. Results
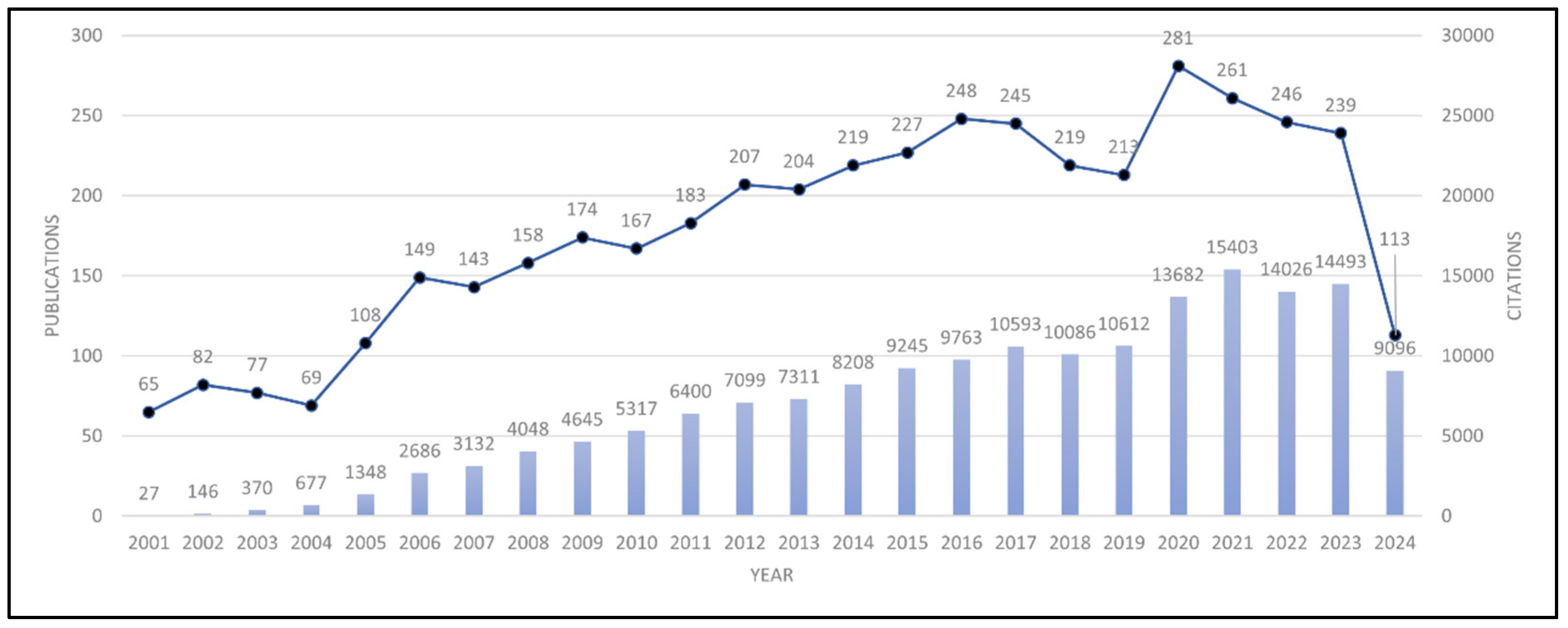
3.1. Publication and Citation Trends
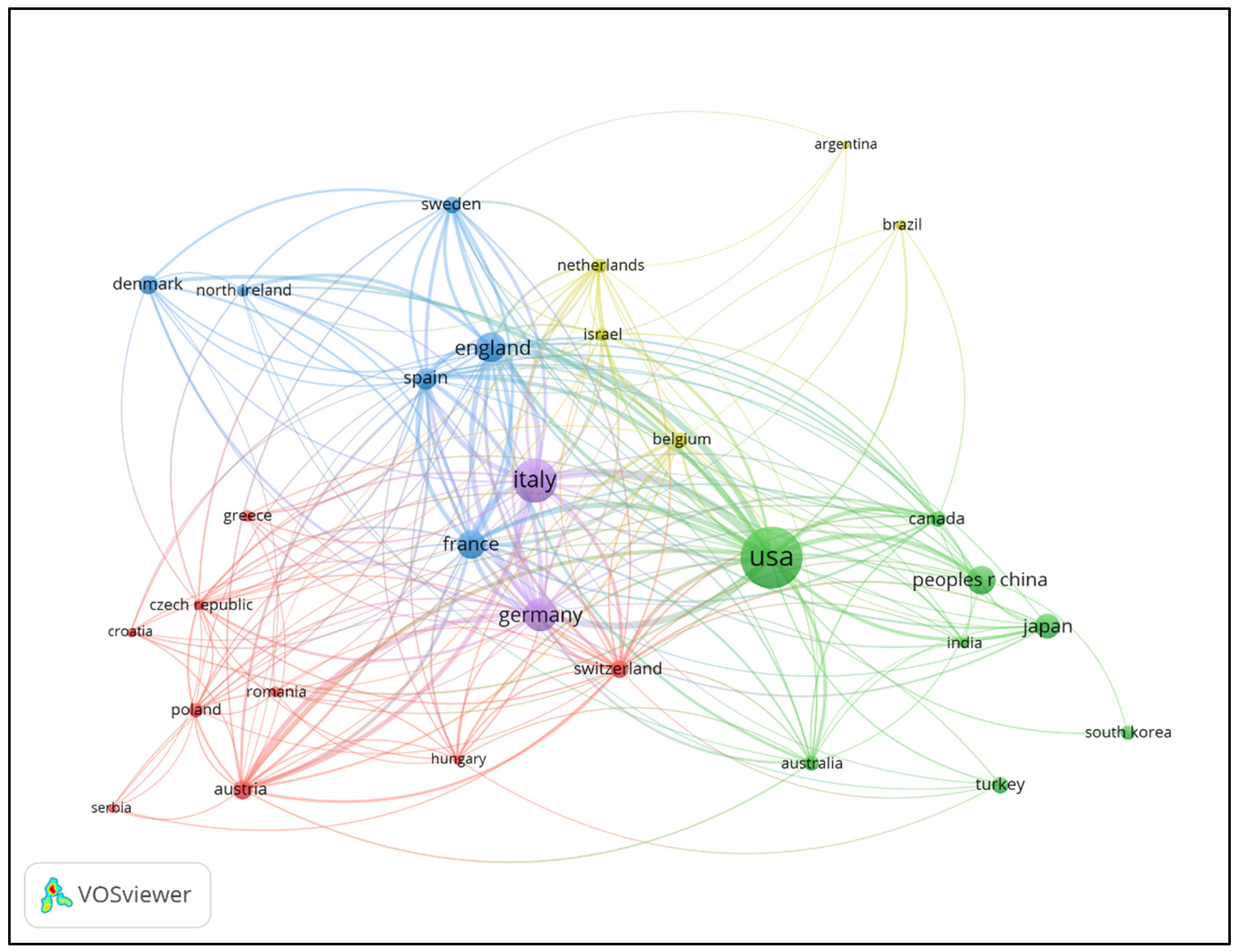
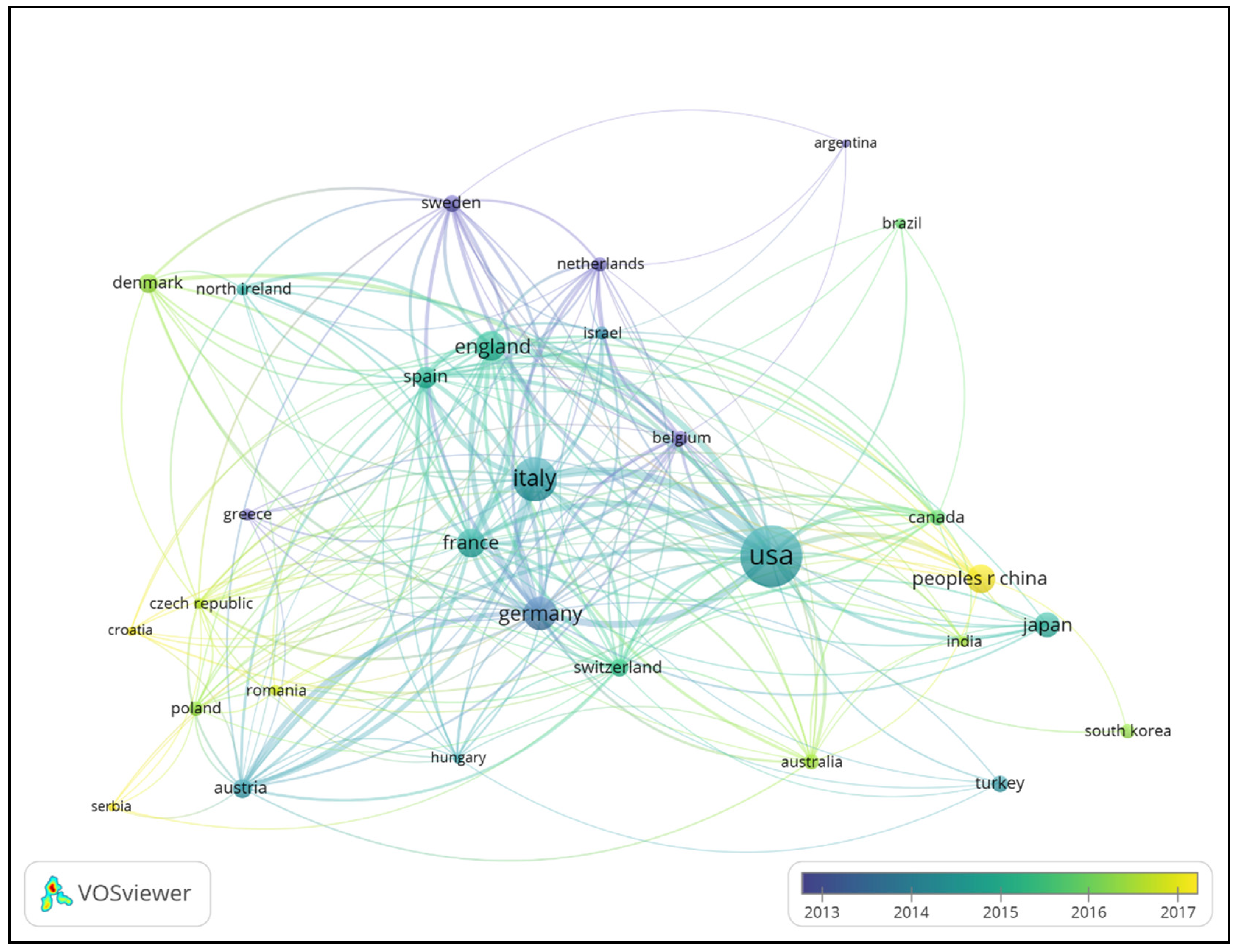
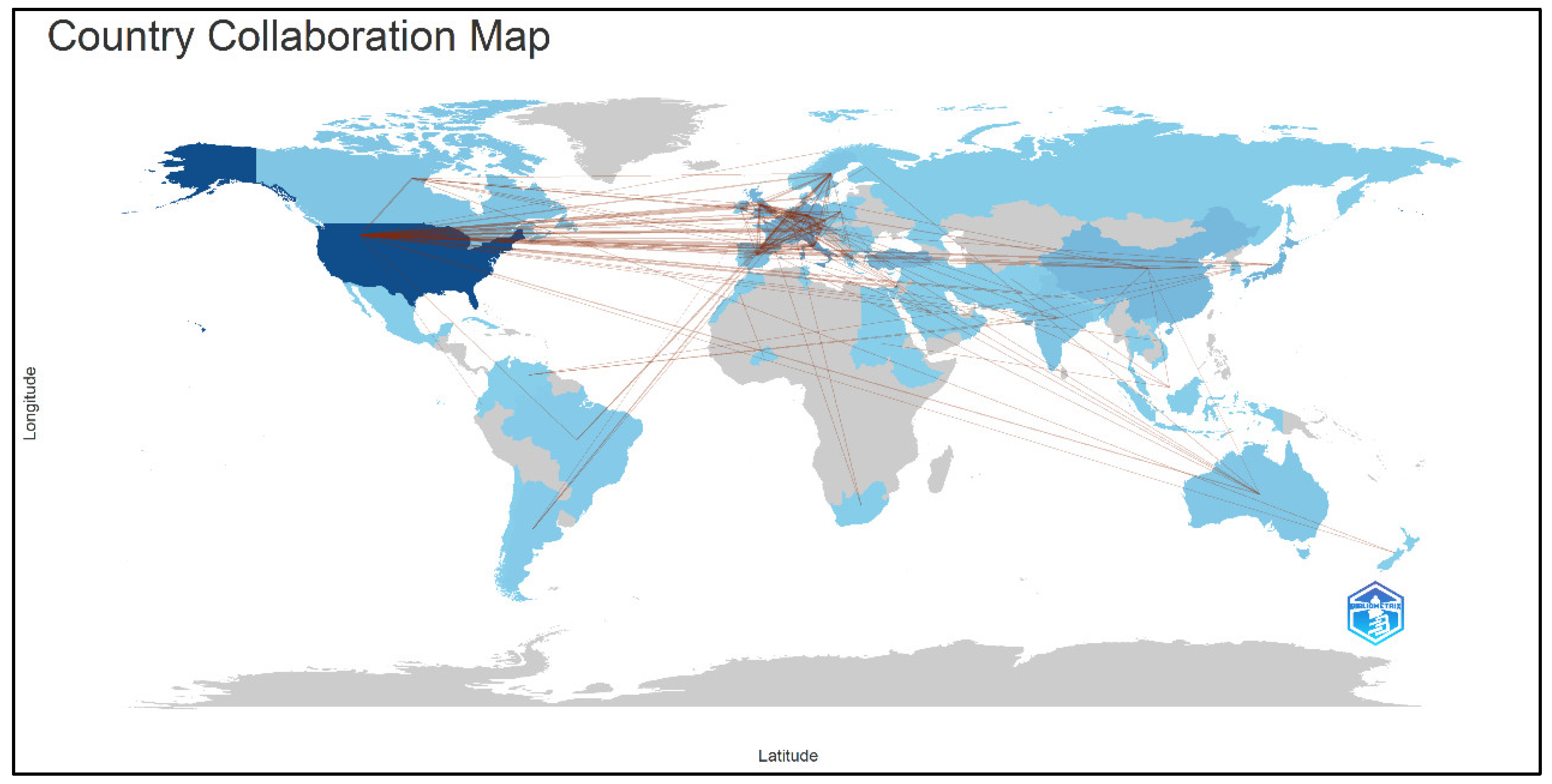
3.2. Country/Region Contributions and Collaboration Networks
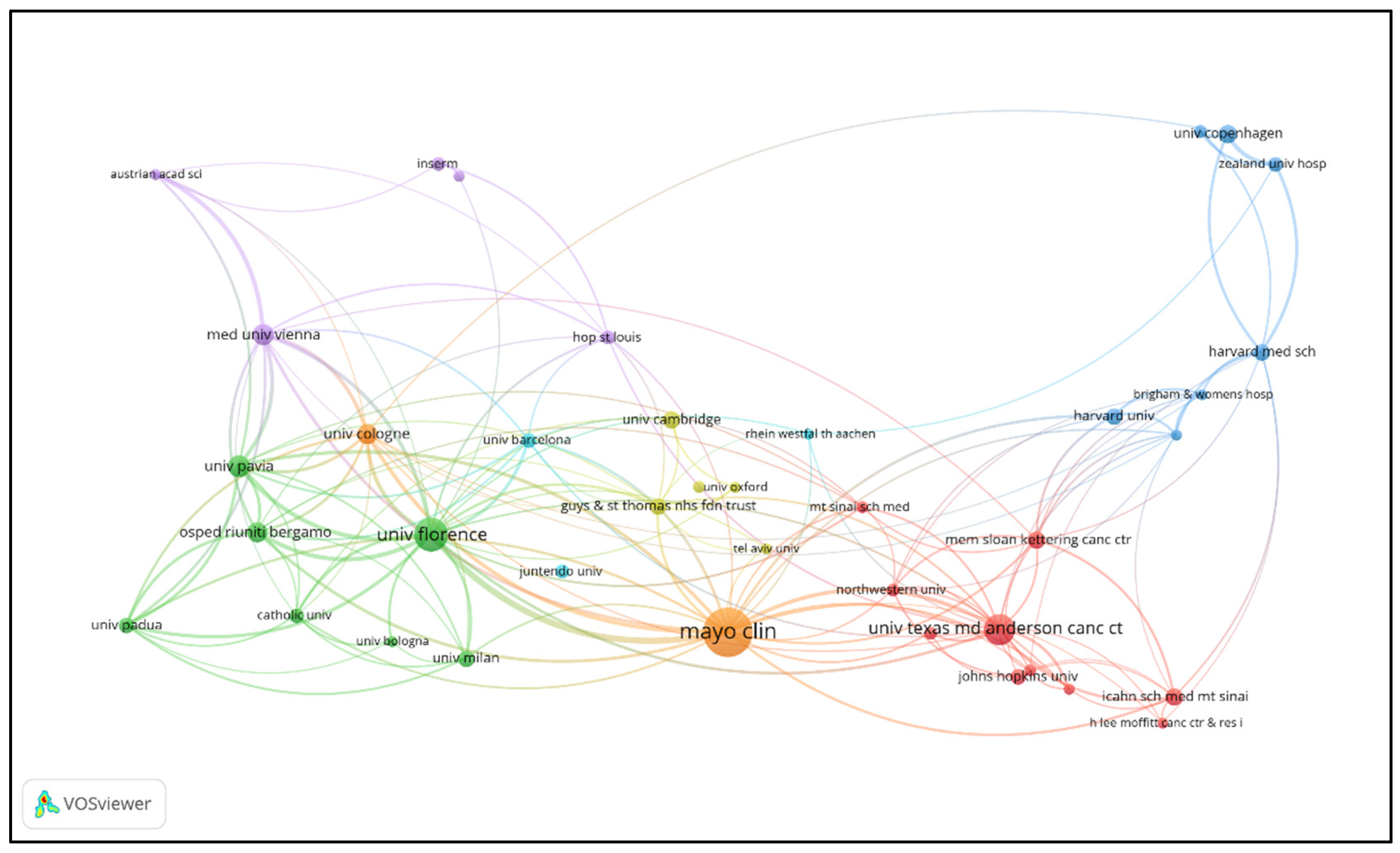
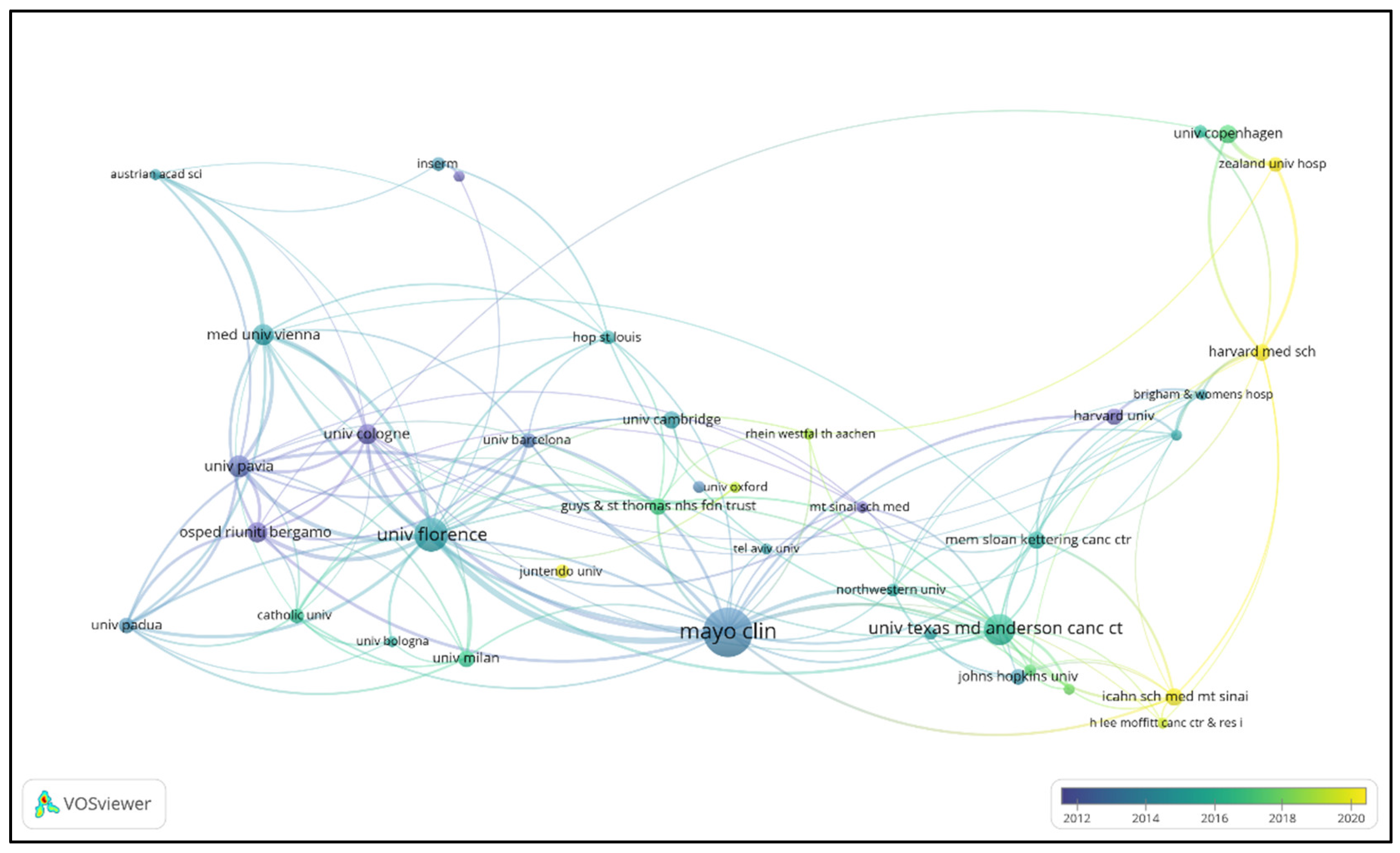
3.3. Institutional Contributions and Collaboration Networks
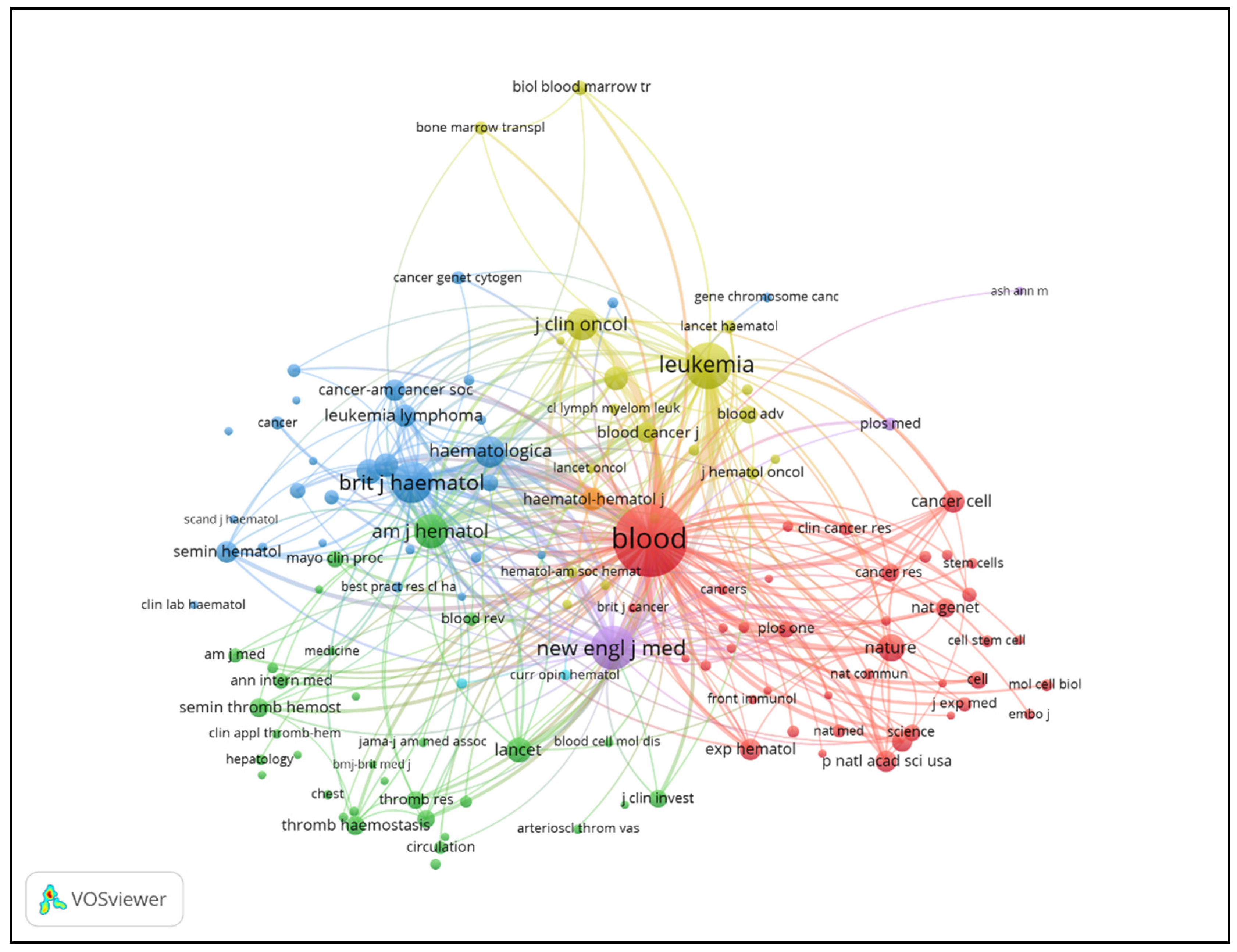
3.4. Journal Contributions and Citation Networks
3.5. Journal Contributions and Collaboration Networks
3.6. Document Contributions and Citation Analysis
3.7. Keyword Contributions and Research Trend Analysis
4. Discussion
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barbui, T.; Thiele, J.; Passamonti, F.; Rumi, E.; Boveri, E.; Ruggeri, M.; Rodeghiero, F.; D’Amore, E.S.; Randi, M.L.; Bertozzi, I.; et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol 2011, 29, 3179–3184. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vannucchi, A.M.; Barbui, T. Essential thrombocythemia treatment algorithm 2018. Blood Cancer J 2018, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Arber, D.A.; Bueso-Ramos, C.; Hasserjian, R.P.; Orazi, A. Advances in myelodysplastic/myeloproliferative neoplasms. Virchows Arch 2023, 482, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Mori, S.; Hasegawa, H.; Sasaki, D.; Mori, H.; Tsuruda, K.; Imanishi, D.; Imaizumi, Y.; Hata, T.; Kaku, N.; et al. Simultaneous screening for JAK2 and calreticulin gene mutations in myeloproliferative neoplasms with high resolution melting. Clin Chim Acta 2016, 462, 166–173. [Google Scholar] [CrossRef] [PubMed]
- McKinnell, Z.; Karel, D.; Tuerff, D.; Sh, A.M.; Nassereddine, S. Acute Myeloid Leukemia Following Myeloproliferative Neoplasms: A Review of What We Know, What We Do Not Know, and Emerging Treatment Strategies. J Hematol 2022, 11, 197–209. [Google Scholar] [CrossRef]
- Finazzi, G. Ruxolitinib in ET: not all MPN are equal. Blood 2017, 130, 1873–1874. [Google Scholar] [CrossRef]
- Harrison, C.N.; Campbell, P.J.; Buck, G.; Wheatley, K.; East, C.L.; Bareford, D.; Wilkins, B.S.; van der Walt, J.D.; Reilly, J.T.; Grigg, A.P.; et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. New Engl J Med 2005, 353, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Khosa, M.; Bhulani, N.; Ali, A.A.; Singh, J.; Khosa, F.; Nasrullah, M. Bibliometrics of Fifty Most-Cited Articles on the Mental Health of Immigrants Living in the United States. J Immigr Minor Healt 2019, 21, 414–429. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, J.; Jin, J.; Jiang, X.; Yang, L.; Fan, S.; Zhang, Q.; Chi, M. Artificial intelligence applied in cardiovascular disease: a bibliometric and visual analysis. Front Cardiovasc Med 2024, 11, 1323918. [Google Scholar] [CrossRef]
- Song, Z.; Jia, G.; Luo, G.; Han, C.; Zhang, B.; Wang, X. Global research trends of Mycoplasma pneumoniae pneumonia in children: a bibliometric analysis. Front Pediatr 2023, 11, 1306234. [Google Scholar] [CrossRef]
- Facciorusso, S.; Spina, S.; Reebye, R.; Turolla, A.; Calabro, R.S.; Fiore, P.; Santamato, A. Sensor-Based Rehabilitation in Neurological Diseases: A Bibliometric Analysis of Research Trends. Brain Sci 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jia, Z.; Xia, X.; Wang, J. Knowledge mapping of COVID-19 and autoimmune diseases: a visual and bibliometric analysis. Clin Exp Med 2023, 23, 3549–3564. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Wang, J.; Lu, C.; Zhou, Y.; Shen, L.; Ge, A.; Fan, H.; Liu, L. Updating the therapeutic role of ginsenosides in breast cancer: a bibliometrics study to an in-depth review. Front Pharmacol 2023, 14, 1226629. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Van, A.A.; Patel, T.; Mani, N.; Carnegie, A.; Corbie-Smith, G.M.; Carey, T.; Buse, J.; Dave, G. Bibliometrics approach to evaluating the research impact of CTSAs: A pilot study. J Clin Transl Sci 2020, 4, 336–344. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. New Engl J Med 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar] [CrossRef]
- Rocca, B.; Tosetto, A.; Betti, S.; Soldati, D.; Petrucci, G.; Rossi, E.; Timillero, A.; Cavalca, V.; Porro, B.; Iurlo, A.; et al. A randomized double-blind trial of 3 aspirin regimens to optimize antiplatelet therapy in essential thrombocythemia. Blood 2020, 136, 171–182. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. New Engl J Med 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. New Engl J Med 2012, 366, 787–798. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. New Engl J Med 2007, 356, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef]
- Mesa, R.A.; Camoriano, J.K.; Geyer, S.M.; Wu, W.; Kaufmann, S.H.; Rivera, C.E.; Erlichman, C.; Wright, J.; Pardanani, A.; Lasho, T.; et al. A phase II trial of tipifarnib in myelofibrosis: primary, post-polycythemia vera and post-essential thrombocythemia. Leukemia 2007, 21, 1964–1970. [Google Scholar] [CrossRef]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. New Engl J Med 2010, 363, 1117–1127. [Google Scholar] [CrossRef]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009, 113, 2895–2901. [Google Scholar] [CrossRef]
- Marchioli, R.; Finazzi, G.; Specchia, G.; Cacciola, R.; Cavazzina, R.; Cilloni, D.; De Stefano, V.; Elli, E.; Iurlo, A.; Latagliata, R.; et al. Cardiovascular events and intensity of treatment in polycythemia vera. New Engl J Med 2013, 368, 22–33. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. New Engl J Med 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. Plos Med 2006, 3, e270. [Google Scholar] [CrossRef]
- Bader, M.S.; Meyer, S.C. JAK2 in Myeloproliferative Neoplasms: Still a Protagonist. Pharmaceuticals-Base 2022, 15. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martinez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G.; et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014, 124, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef]
- Morsia, E.; Torre, E.; Poloni, A.; Olivieri, A.; Rupoli, S. Molecular Pathogenesis of Myeloproliferative Neoplasms: From Molecular Landscape to Therapeutic Implications. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells-Basel 2019, 8. [Google Scholar] [CrossRef]
- Cattaneo, D.; Croci, G.A.; Bucelli, C.; Tabano, S.; Cannone, M.G.; Gaudioso, G.; Barbanti, M.C.; Barbullushi, K.; Bianchi, P.; Fermo, E.; et al. Triple-Negative Essential Thrombocythemia: Clinical-Pathological and Molecular Features. A Single-Center Cohort Study. Front Oncol 2021, 11, 637116. [Google Scholar] [CrossRef]
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med 2018, 24, 1015–1023. [Google Scholar] [CrossRef]
- Barbui, T.; Carobbio, A.; De Stefano, V. Thrombosis in myeloproliferative neoplasms during cytoreductive and antithrombotic drug treatment. Res Pract Thromb Hae 2022, 6, e12657. [Google Scholar] [CrossRef]
- Mesa, R.A.; Niblack, J.; Wadleigh, M.; Verstovsek, S.; Camoriano, J.; Barnes, S.; Tan, A.D.; Atherton, P.J.; Sloan, J.A.; Tefferi, A. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer-Am Cancer Soc 2007, 109, 68–76. [Google Scholar] [CrossRef]
- Verstovsek, S.; Passamonti, F.; Rambaldi, A.; Barosi, G.; Rumi, E.; Gattoni, E.; Pieri, L.; Zhen, H.; Granier, M.; Assad, A.; et al. Ruxolitinib for essential thrombocythemia refractory to or intolerant of hydroxyurea: long-term phase 2 study results. Blood 2017, 130, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Michail, O.; McCallion, P.; McGimpsey, J.; Hindley, A.; Greenfield, G.; McAllister, R.; Feerick, J.; Arnold, C.; Cross, N.; Cuthbert, R.; et al. Mutational profiling in suspected triple-negative essential thrombocythaemia using targeted next-generation sequencing in a real-world cohort. J Clin Pathol 2021, 74, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Leroy, E.; Gilles, L.; Marty, C.; Plo, I.; Constantinescu, S.N. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Res 2018, 7, 82. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Passamonti, F.; Maffioli, M. The role of JAK2 inhibitors in MPNs 7 years after approval. Blood 2018, 131, 2426–2435. [Google Scholar] [CrossRef]
- Tefferi, A.; Vannucchi, A.M.; Barbui, T. Essential thrombocythemia: 2024 update on diagnosis, risk stratification, and management. Am J Hematol 2024, 99, 697–718. [Google Scholar] [CrossRef]
- Wang, R.; Tang, L.V.; Hu, Y. Genetic factors, risk prediction and AI application of thrombotic diseases. Exp Hematol Oncol 2024, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Carobbio, A.; Finazzi, G.; Antonioli, E.; Vannucchi, A.M.; Barosi, G.; Ruggeri, M.; Rodeghiero, F.; Delaini, F.; Rambaldi, A.; Barbui, T. Hydroxyurea in essential thrombocythemia: rate and clinical relevance of responses by European LeukemiaNet criteria. Blood 2010, 116, 1051–1055. [Google Scholar] [CrossRef]
- Pandey, G.; Kuykendall, A.T.; Reuther, G.W. JAK2 inhibitor persistence in MPN: uncovering a central role of ERK activation. Blood Cancer J 2022, 12, 13. [Google Scholar] [CrossRef]
- Putti, M.C.; Bertozzi, I.; Randi, M.L. Essential Thrombocythemia in Children and Adolescents. Cancers 2021, 13. [Google Scholar] [CrossRef]
- Palandri, F.; Polverelli, N.; Ottaviani, E.; Castagnetti, F.; Baccarani, M.; Vianelli, N. Long-term follow-up of essential thrombocythemia in young adults: treatment strategies, major thrombotic complications and pregnancy outcomes. A study of 76 patients. Haematologica 2010, 95, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Rimondi, E.; Melloni, E.; Gonelli, A.; Grasso, A.G.; Barbi, E.; Maximova, N. New Applications of JAK/STAT Inhibitors in Pediatrics: Current Use of Ruxolitinib. Pharmaceuticals-Base 2022, 15. [Google Scholar] [CrossRef]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov 2015, 5, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Gou, P.; Zhang, W.; Giraudier, S. Insights into the Potential Mechanisms of JAK2V617F Somatic Mutation Contributing Distinct Phenotypes in Myeloproliferative Neoplasms. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Waksal, J.A.; Harrison, C.N.; Mascarenhas, J.O. Novel therapeutics and targets in myelofibrosis. Leukemia Lymphoma 2022, 63, 1020–1033. [Google Scholar] [CrossRef] [PubMed]
- Katkenov, N.; Mukhatayev, Z.; Kozhakhmetov, S.; Sailybayeva, A.; Bekbossynova, M.; Kushugulova, A. Systematic Review on the Role of IL-6 and IL-1beta in Cardiovascular Diseases. J Cardiovasc Dev Dis 2024, 11. [Google Scholar] [CrossRef]
- Dri, E.; Lampas, E.; Lazaros, G.; Lazarou, E.; Theofilis, P.; Tsioufis, C.; Tousoulis, D. Inflammatory Mediators of Endothelial Dysfunction. Life-Basel 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Medina-Leyte, D.J.; Zepeda-Garcia, O.; Dominguez-Perez, M.; Gonzalez-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Haage, T.R.; Charakopoulos, E.; Bhuria, V.; Baldauf, C.K.; Korthals, M.; Handschuh, J.; Muller, P.; Li, J.; Harit, K.; Nishanth, G.; et al. Neutrophil-specific expression of JAK2-V617F or CALRmut induces distinct inflammatory profiles in myeloproliferative neoplasia. J Hematol Oncol 2024, 17, 43. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Antonopoulos, A.S.; Oikonomou, E.; Tsioufis, C.; Tousoulis, D. Inflammatory Mediators of Platelet Activation: Focus on Atherosclerosis and COVID-19. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Zaid, Y.; Merhi, Y. Implication of Platelets in Immuno-Thrombosis and Thrombo-Inflammation. Front Cardiovasc Med 2022, 9, 863846. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Yue, X.; Tian, X.; Liao, Z.; Meng, R.; Zou, M. Association between inflammatory biomarkers and venous thromboembolism: a systematic review and meta-analysis. Thromb J 2023, 21, 82. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leukemia Res 2013, 37, 214–220. [Google Scholar] [CrossRef]
- Koschmieder, S.; Chatain, N. Role of inflammation in the biology of myeloproliferative neoplasms. Blood Rev 2020, 42, 100711. [Google Scholar] [CrossRef]
- Cadet, J.; Davies, K. Oxidative DNA damage & repair: An introduction. Free Radical Bio Med 2017, 107, 2–12. [Google Scholar] [CrossRef]
- Pietras, E.M. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood 2017, 130, 1693–1698. [Google Scholar] [CrossRef]
- Hormaechea-Agulla, D.; Le, D.T.; King, K.Y. Common Sources of Inflammation and Their Impact on Hematopoietic Stem Cell Biology. Curr Stem Cell Rep 2020, 6, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Caiado, F.; Manz, M.G. IL-1 in aging and pathologies of hematopoietic stem cells. Blood 2024, 144, 368–377. [Google Scholar] [CrossRef]
- Caiado, F.; Pietras, E.M.; Manz, M.G. Inflammation as a regulator of hematopoietic stem cell function in disease, aging, and clonal selection. J Exp Med 2021, 218. [Google Scholar] [CrossRef]
- Marnell, C.S.; Bick, A.; Natarajan, P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J Mol Cell Cardiol 2021, 161, 98–105. [Google Scholar] [CrossRef]
- Caiado, F.; Kovtonyuk, L.V.; Gonullu, N.G.; Fullin, J.; Boettcher, S.; Manz, M.G. Aging drives Tet2+/- clonal hematopoiesis via IL-1 signaling. Blood 2023, 141, 886–903. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yalcinkaya, M.; Maestre, I.F.; Olszewska, M.; Ampomah, P.B.; Heimlich, J.B.; Wang, R.; Vela, P.S.; Xiao, T.; Bick, A.G.; et al. Blockade of IL-6 signaling alleviates atherosclerosis in Tet2-deficient clonal hematopoiesis. Nat Cardiovasc Res 2023, 2, 572–586. [Google Scholar] [CrossRef] [PubMed]
- Min, K.D.; Polizio, A.H.; Kour, A.; Thel, M.C.; Walsh, K. Experimental ASXL1-Mediated Clonal Hematopoiesis Promotes Inflammation and Accelerates Heart Failure. J Am Heart Assoc 2022, 11, e026154. [Google Scholar] [CrossRef] [PubMed]
- Gleitz, H.; Schneider, R.K. “ASXL1”-erating inflammation and bone marrow fibrosis in myeloproliferative neoplasms. Haematologica 2023, 108, 1203–1204. [Google Scholar] [CrossRef]
- Camacho, V.; Kuznetsova, V.; Welner, R.S. Inflammatory Cytokines Shape an Altered Immune Response During Myeloid Malignancies. Front Immunol 2021, 12, 772408. [Google Scholar] [CrossRef]
- Tie, Y.; Tang, F.; Wei, Y.Q.; Wei, X.W. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol 2022, 15, 61. [Google Scholar] [CrossRef]
- Rovatti, P.E.; Gambacorta, V.; Lorentino, F.; Ciceri, F.; Vago, L. Mechanisms of Leukemia Immune Evasion and Their Role in Relapse After Haploidentical Hematopoietic Cell Transplantation. Front Immunol 2020, 11, 147. [Google Scholar] [CrossRef]
- Vojdani, A.; Koksoy, S.; Vojdani, E.; Engelman, M.; Benzvi, C.; Lerner, A. Natural Killer Cells and Cytotoxic T Cells: Complementary Partners against Microorganisms and Cancer. Microorganisms 2024, 12. [Google Scholar] [CrossRef]
- Salio, M.; Cerundolo, V. Linking inflammation to natural killer T cell activation. Plos Biol 2009, 7, e1000226. [Google Scholar] [CrossRef]
- Barshidi, A.; Ardeshiri, K.; Ebrahimi, F.; Alian, F.; Shekarchi, A.A.; Hojjat-Farsangi, M.; Jadidi-Niaragh, F. The role of exhausted natural killer cells in the immunopathogenesis and treatment of leukemia. Cell Commun Signal 2024, 22, 59. [Google Scholar] [CrossRef]



| Rank | Country Or Region | Publications | Percentage(%) | Total citations | Averrage citations |
| 1 | USA (North America) | 1299 | 24.23% | 76507 | 58.90 |
| 2 | Italy (Europe) | 636 | 11.86% | 52083 | 81.89 |
| 3 | Germany (Europe) | 371 | 6.92% | 32499 | 87.60 |
| 4 | England (Europe) | 301 | 5.61% | 27032 | 89.81 |
| 5 | China (Asia) | 291 | 5.43% | 5644 | 19.40 |
| 6 | France (Europe) | 268 | 5.00% | 20004 | 74.64 |
| 7 | Japan (Asia) | 219 | 4.09% | 6059 | 27.67 |
| 8 | Spain (Europe) | 163 | 3.04% | 15267 | 93.66 |
| 9 | Denmark (Europe) | 133 | 2.48% | 5627 | 42.31 |
| 10 | Austria (Europe) | 128 | 2.39% | 12808 | 100.06 |
| 11 | Switzerland (Europe) | 112 | 2.09% | 10326 | 92.20 |
| 12 | Turkey (Asia) | 104 | 1.94% | 935 | 8.99 |
| 13 | Sweden (Europe) | 99 | 1.85% | 8109 | 81.91 |
| 14 | Canada (North America) | 89 | 1.66% | 5826 | 65.46 |
| 15 | Australia (Oceania) | 80 | 1.49% | 5733 | 71.66 |
| Rank | Insitution | Publications | Percentage(%) | Total citations | Averrage citations | total link strength |
| 1 | Mayo Clinic (USA) | 289 | 3.72% | 25780 | 89.20 | 300 |
| 2 | University of Florence (Italy) | 166 | 2.14% | 20907 | 125.95 | 325 |
| 3 | MD Anderson Cancer Center (USA) | 146 | 1.88% | 9579 | 65.61 | 138 |
| 4 | University of Pavia (Italy) | 87 | 1.12% | 21001 | 241.39 | 170 |
| 5 | Medical University of Vienna (Austria) | 82 | 1.05% | 10612 | 129.41 | 181 |
| 6 | University of Cologne (Germany) | 79 | 1.02% | 12849 | 162.65 | 140 |
| 7 | Ospedali Riuniti Bergamo (Italy) | 74 | 0.95% | 8665 | 117.09 | 138 |
| 8 | University of Copenhagen (Denmark) | 65 | 0.84% | 3194 | 49.14 | 77 |
| 9 | University of Cambridge (UK) | 63 | 0.81% | 8401 | 133.35 | 27 |
| 10 | Icahn School of Medicine at Mount Sinai (USA) | 62 | 0.80% | 1300 | 20.97 | 57 |
| 11 | Memorial Sloan Kettering Cancer Center (USA) | 61 | 0.78% | 3291 | 53.95 | 86 |
| 12 | Harvard Medical School (USA) | 57 | 0.73% | 1423 | 24.96 | 77 |
| 13 | Harvard University (USA) | 57 | 0.73% | 11086 | 194.49 | 59 |
| 14 | University of Milan (Italy) | 57 | 0.73% | 2758 | 48.39 | 59 |
| 15 | Guy’s & St Thomas’ NHS Foundation Trust (UK) | 55 | 0.71% | 3058 | 55.60 | 72 |
| 16 | Johns Hopkins University (USA) | 52 | 0.67% | 2830 | 54.42 | 30 |
| 17 | University of Padua (Italy) | 52 | 0.67% | 3781 | 72.71 | 106 |
| 18 | University of Barcelona (Spain) | 48 | 0.62% | 7702 | 160.46 | 86 |
| 19 | Zealand University Hospital (Denmark) | 48 | 0.62% | 1235 | 25.73 | 72 |
| 20 | Catholic University (Italy) | 46 | 0.59% | 2095 | 45.54 | 61 |
| Rank | Journal | Publications | Total Ciation | Average Citations | Impact Factor (2023) | JCR |
| 1 | Blood | 290 | 37349 | 128.79 | 21.0 | Q1 |
| 2 | New England Journal of Medicine | 17 | 12818 | 754.00 | 96.2 | Q1 |
| 3 | Leukemia | 126 | 9988 | 79.27 | 12.8 | Q1 |
| 4 | American Journal of Hematology | 149 | 5310 | 35.64 | 11.0 | Q1 |
| 5 | British Journal of Haematology | 107 | 5299 | 49.52 | 5.1 | Q2 |
| 6 | Journal of Clinical Oncology | 25 | 4693 | 187.72 | 45.3 | Q1 |
| 7 | Haematologica | 80 | 3396 | 42.45 | 10.3 | Q1 |
| 8 | Annals of Hematology | 132 | 2475 | 18.75 | 3.0 | Q3 |
| 9 | Haematologica-The Hematology Journal | 44 | 2416 | 54.91 | 10.3 | Q1 |
| 10 | Leukemia Research | 97 | 1881 | 19.39 | 2.1 | Q4 |
| 11 | European Journal of Haematology | 101 | 1647 | 16.31 | 4.2 | Q2 |
| 12 | Experimental Hematology | 53 | 1455 | 27.45 | 3.6 | Q3 |
| 13 | Blood Cancer Journal | 46 | 1374 | 29.87 | 12.9 | Q1 |
| 14 | Blood Advances | 43 | 1343 | 31.23 | 7.5 | Q1 |
| 15 | Leukemia & Lymphoma | 82 | 1310 | 15.98 | 2.2 | Q3 |
| 16 | Seminars In Thrombosis and Hemostasis | 47 | 1306 | 27.79 | 5.4 | Q2 |
| 17 | Cancer | 32 | 1128 | 35.25 | 8.3 | Q1 |
| 18 | International Journal of Hematology | 93 | 1029 | 11.06 | 1.8 | Q4 |
| 19 | Journal of Hematology & Oncology | 19 | 870 | 45.79 | 12.5 | Q1 |
| 20 | Journal of Thrombosis and Haemostasis | 23 | 836 | 36.35 | 6.9 | Q2 |
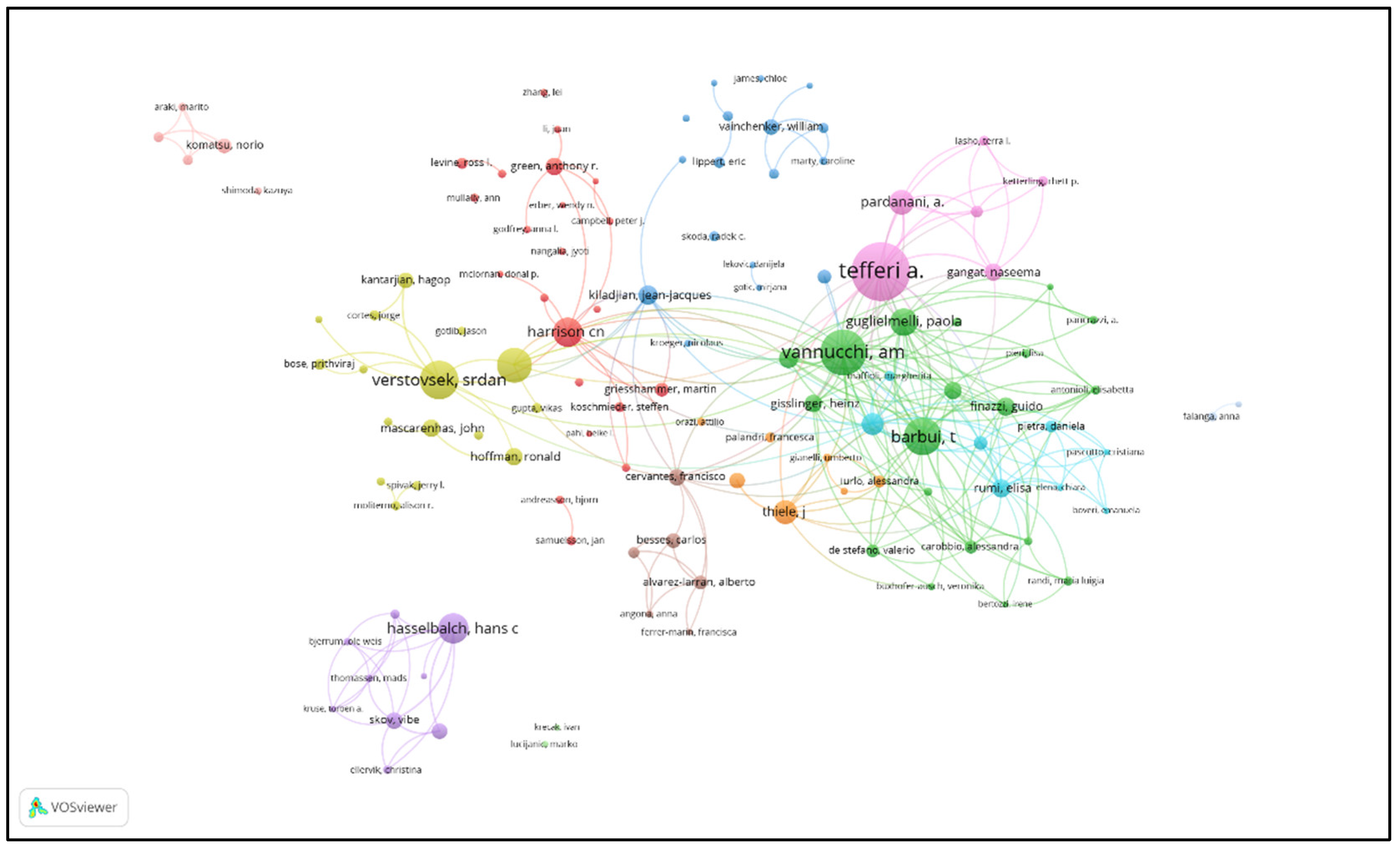
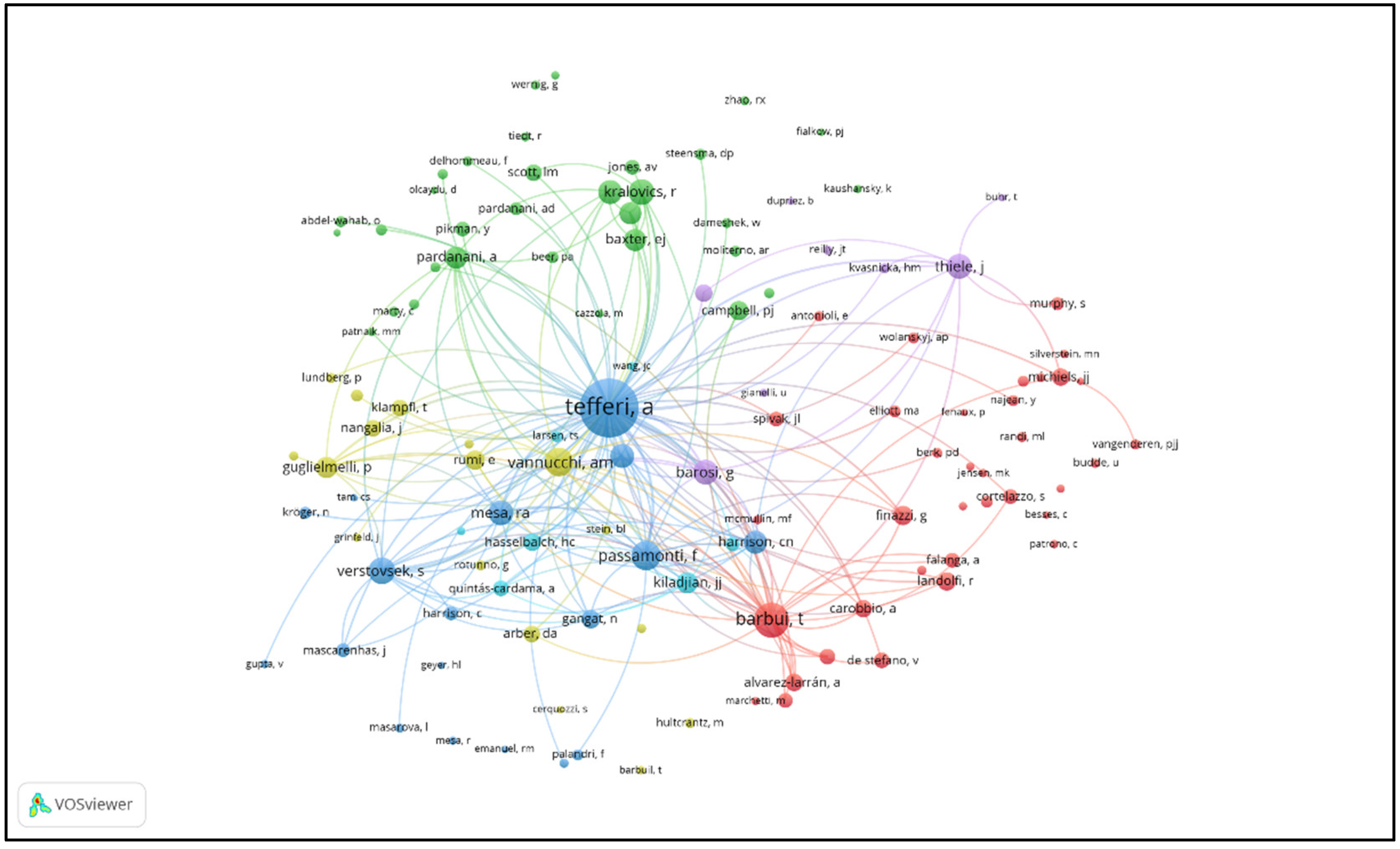
| Rank | Author | Country/Region | Institution | Documents | Citations | Total Link Strength | H-index |
| 1 | Ayalew Tefferi | USA | Mayo Clinic | 238 | 22511 | 614 | 88 |
| 2 | Alessandro M. Vannucchi | Italy | University of Florence | 177 | 18735 | 915 | 78 |
| 3 | Srdan Verstovsek | USA | MD Anderson Cancer Center | 139 | 9011 | 306 | 42 |
| 4 | Tiziano Barbui | Italy | Papa Giovanni XXIII Hospital | 138 | 13955 | 656 | 69 |
| 5 | Ruben Mesa | USA | UT Health San Antonio Cancer Center | 123 | 8738 | 277 | 42 |
| 6 | Hans Carl Hasselbalch | Denmark | Zealand University Hospital | 103 | 3462 | 241 | 35 |
| 7 | Claire N. Harrison | UK | Guy’s and St Thomas’ Hospitals | 101 | 9127 | 353 | 39 |
| 8 | Paola Guglielmelli | Italy | University of Florence | 93 | 6941 | 459 | 49 |
| 9 | Animesh Pardanani | USA | Mayo Clinic | 82 | 8242 | 304 | 43 |
| 10 | Juergen Thiele | Germany | University of Cologne | 76 | 5511 | 304 | 43 |
| 11 | Francesco Passamonti | Italy | University of Pavia | 71 | 9317 | 456 | 44 |
| 12 | Giovanni Barosi | Italy | IRCCS Policlinico San Matteo | 60 | 8348 | 327 | 36 |
| 13 | Jean-Jacques Kiladjian | France | Saint-Louis Hospital | 57 | 6115 | 256 | 33 |
| 14 | Elisa Rumi | Italy | University of Pavia | 55 | 5928 | 371 | 42 |
| 15 | Guido Finazzi | Italy | Papa Giovanni XXIII Hospital | 54 | 6230 | 358 | 47 |
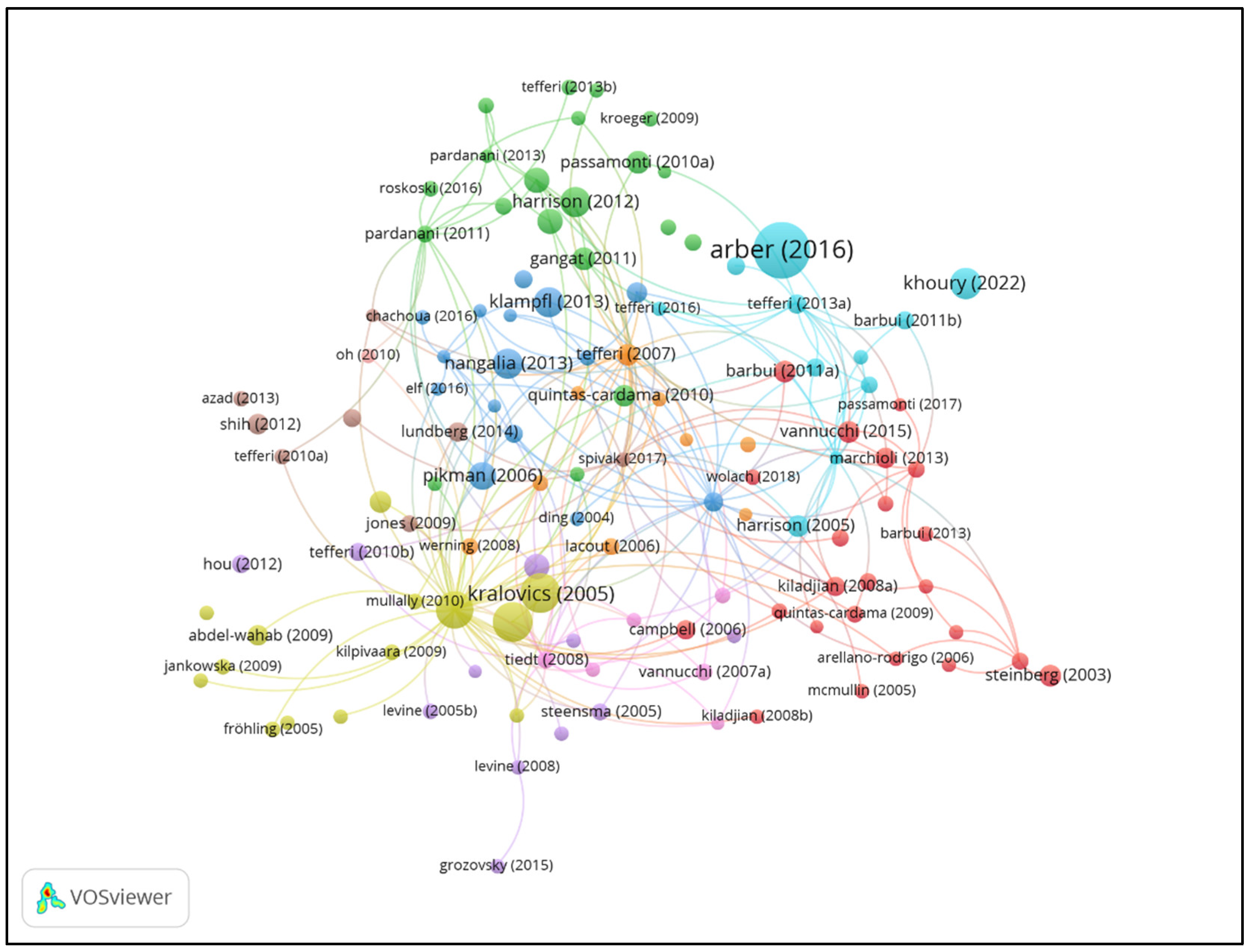
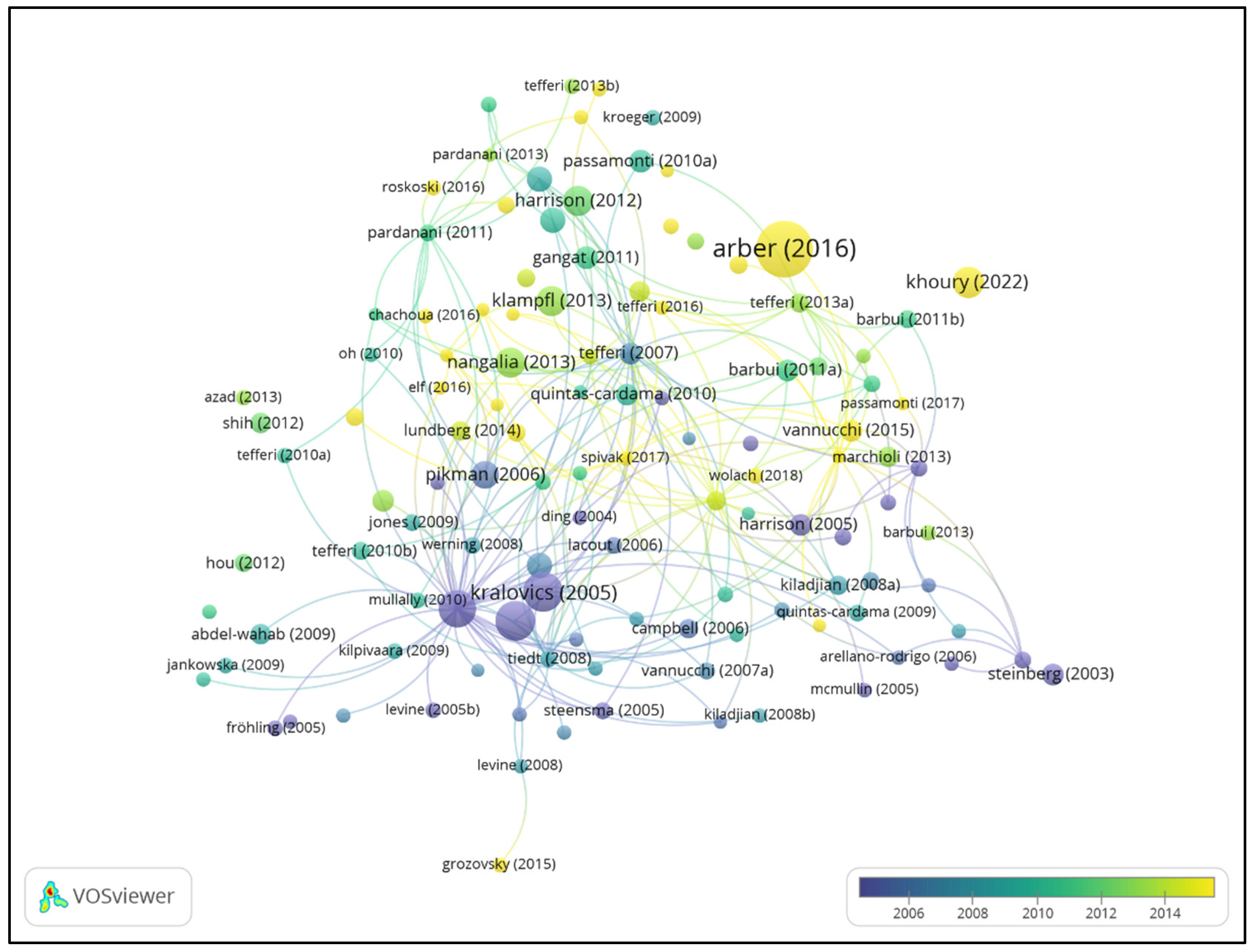
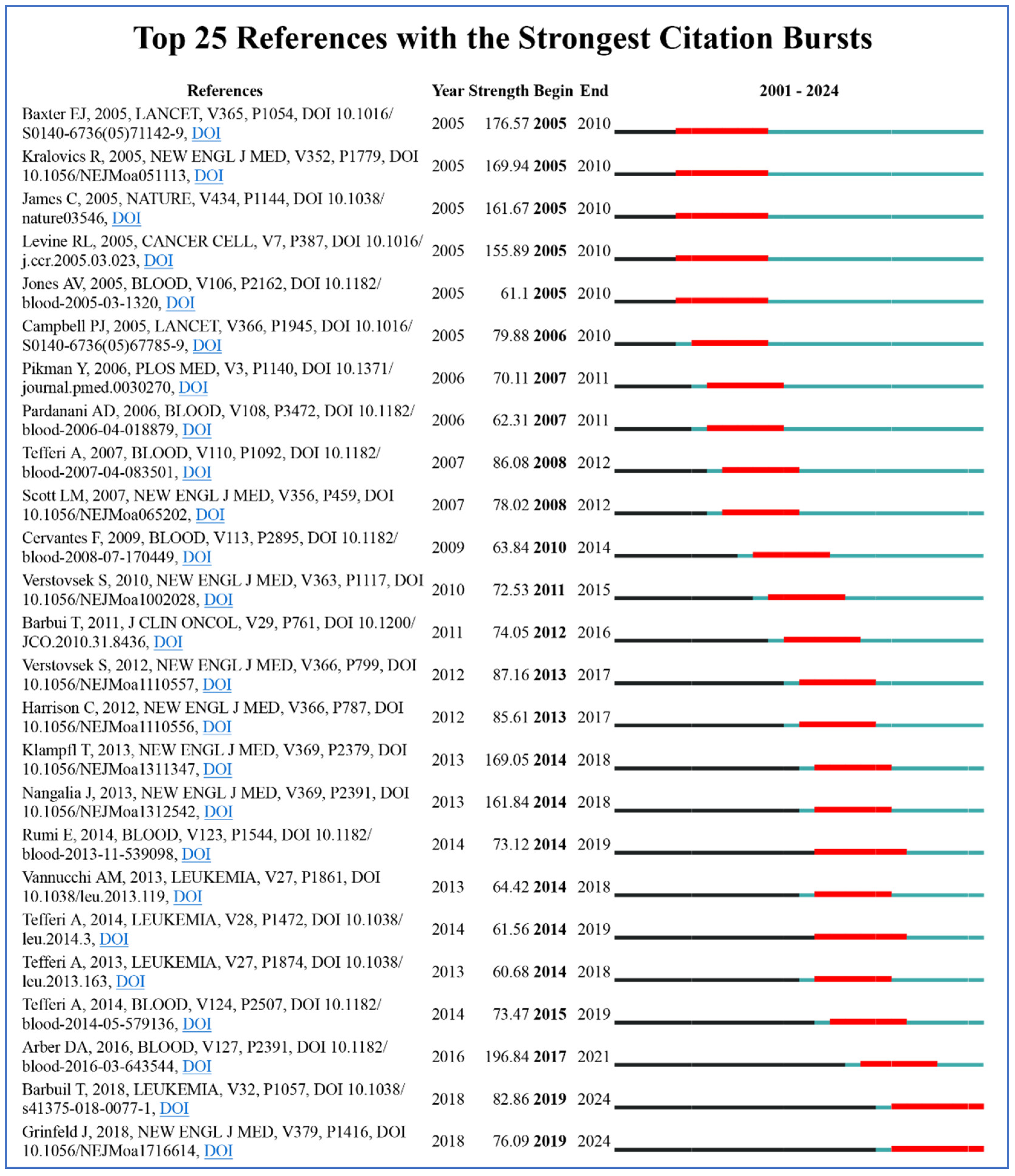
| Rank | Title | Citation | First Author | Institution | Journal |
| 1 | The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia [28] | 6674 | Daniel A. Arber | Stanford University, USA | Blood |
| 2 | A gain-of-function mutation of JAK2 in myeloproliferative disorders [19] | 2867 | Robert Kralovics | University Hospital Basel, Switzerland | The New England Journal of Medicine |
| 3 | Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders [15] | 2822 | E. Joanna Baxter | University of Cambridge, UK | The Lancet |
| 4 | Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis [23] | 2376 | Ross L. Levine | Harvard Medical School, USA | Cancer Cell |
| 5 | The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms [30] | 1515 | Joseph D. Khoury | MD Anderson Cancer Center, USA | Leukemia |
| 6 | Somatic mutations of calreticulin in myeloproliferative neoplasms [16] | 1461 | Thorsten Klampfl | CeMM, Vienna, Austria | The New England Journal of Medicine |
| 7 | JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis [20] | 1392 | Claire Harrison | Guy’s Hospital, UK | The New England Journal of Medicine |
| 8 | Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2 [29] | 1351 | Jyoti Nangalia | University of Cambridge, UK | The New England Journal of Medicine |
| 9 | MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia [31] | 1099 | Yana Pikman | Harvard Medical School, USA | PLoS Medicine |
| 10 | JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis [21] | 944 | Linda M. Scott | University of Cambridge, UK | The New England Journal of Medicine |
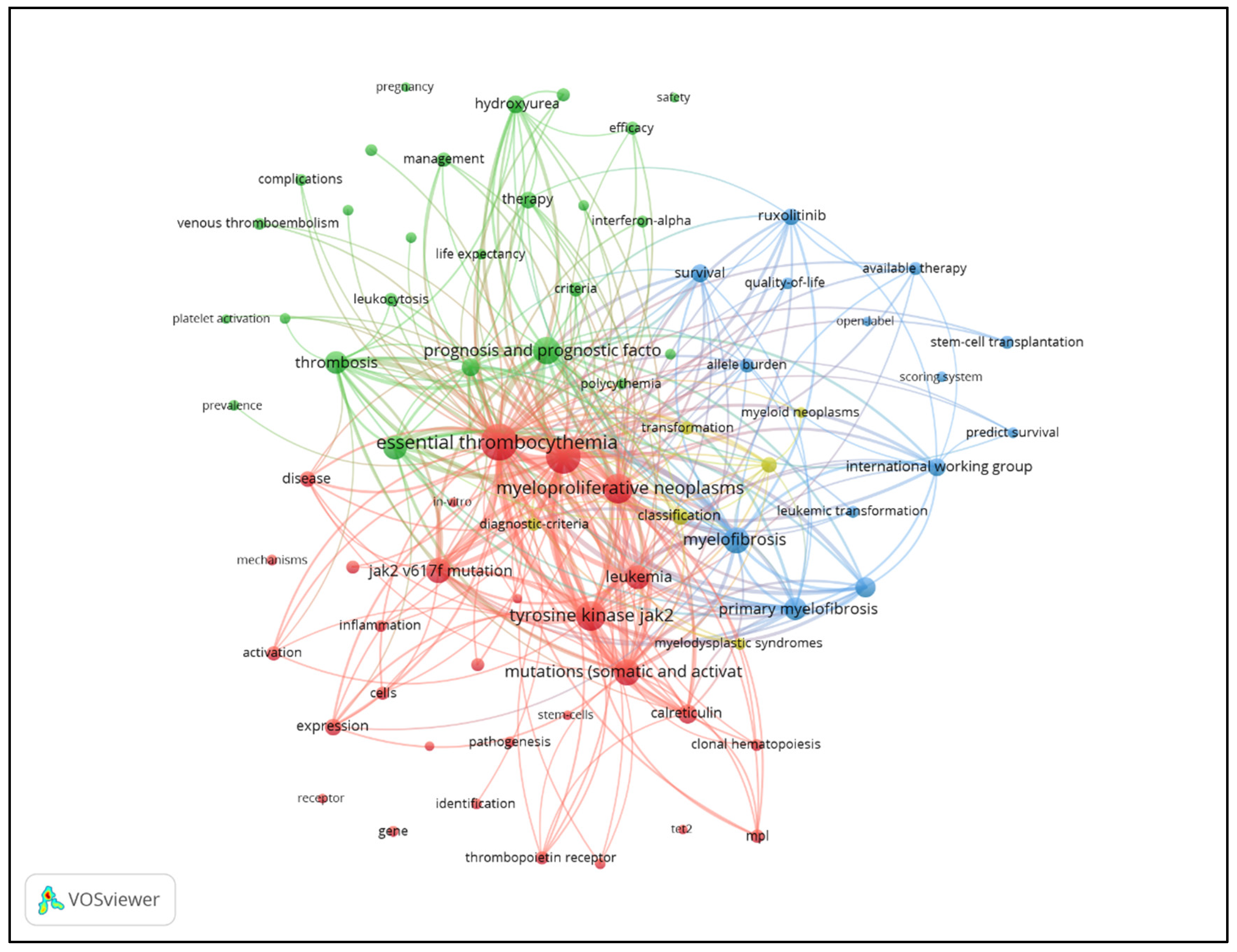
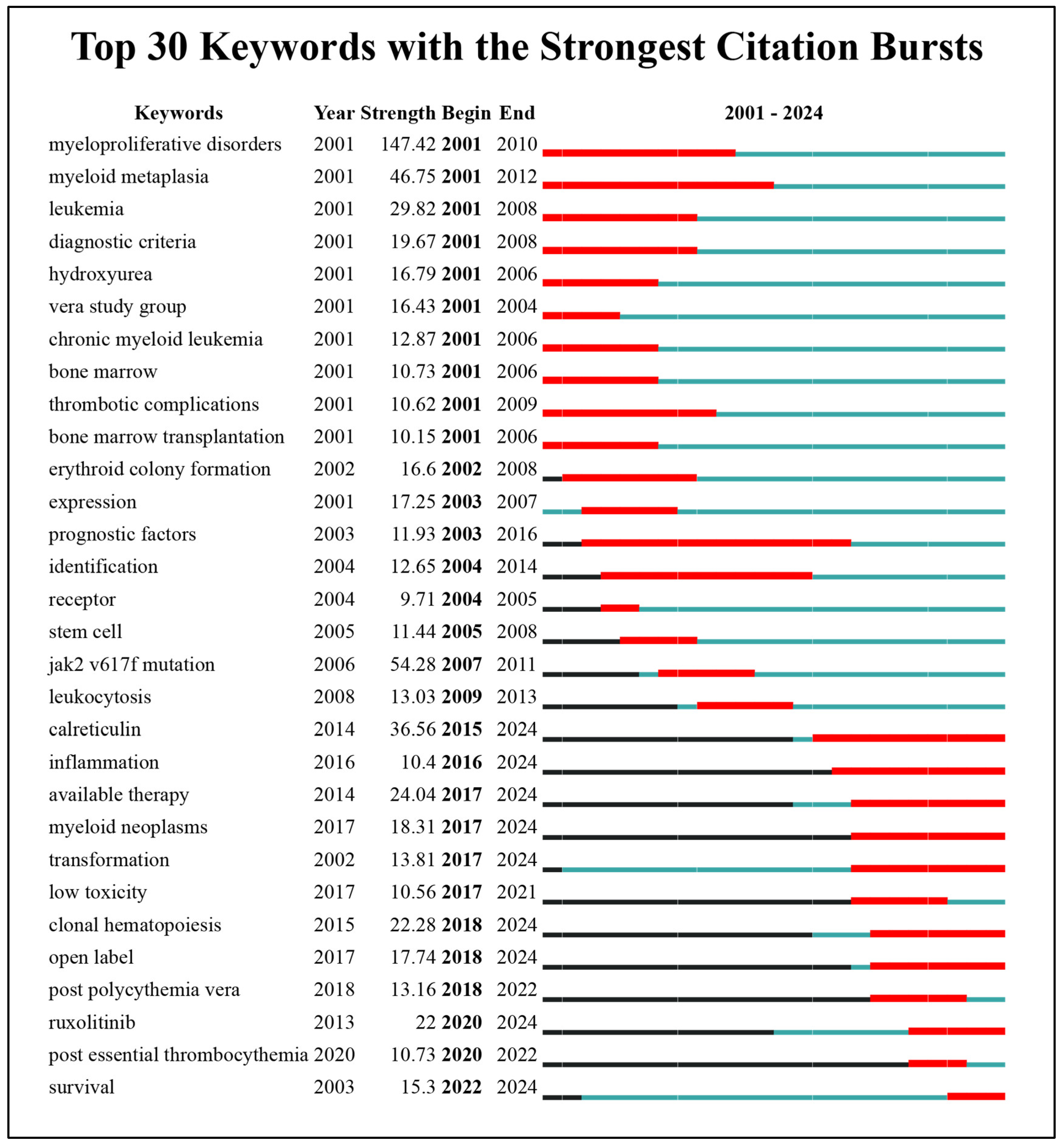
| Keyword | Freq. | Total Link Strength | Keyword | Freq. | Total Link Strength |
| Essential Thrombocythemia | 2847 | 14483 | Therapy | 233 | 1340 |
| Polycythemia Vera | 2346 | 13160 | Expression | 230 | 1089 |
| Myeloproliferative Neoplasms | 1446 | 8479 | Classification | 226 | 1362 |
| Tyrosine Kinase Jak2 | 1399 | 8245 | World-Health-Organization | 214 | 1417 |
| Prognosis And Prognostic Factors | 1044 | 6144 | Disease | 207 | 1046 |
| Mutations (Somatic and Activating) | 1020 | 5994 | Activation | 173 | 772 |
| Myelofibrosis | 906 | 5643 | Allele Burden | 157 | 1122 |
| Jak2 V617f Mutation | 804 | 4817 | Management | 156 | 862 |
| Leukemia | 743 | 3796 | Criteria | 145 | 847 |
| Myeloproliferative Disorders | 723 | 3834 | Cells | 136 | 588 |
| Thrombosis | 667 | 3644 | Available Therapy | 135 | 897 |
| Primary Myelofibrosis | 639 | 4079 | Neoplasms | 127 | 721 |
| Myeloid Metaplasia | 443 | 2804 | Diagnostic-Criteria | 118 | 718 |
| Calreticulin | 354 | 2247 | Bone-Marrow | 117 | 585 |
| Diagnosis | 333 | 1919 | Anagrelide | 117 | 672 |
| Hydroxyurea | 323 | 1921 | Efficacy | 116 | 740 |
| Survival | 302 | 1887 | Cancer | 116 | 488 |
| International Working Group | 296 | 2066 | Transformation | 111 | 728 |
| Ruxolitinib | 273 | 1813 | Stem-Cell Transplantation | 107 | 666 |
| Thrombopoietin Receptor | 248 | 1288 | Leukocytosis | 106 | 725 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



