Submitted:
17 October 2024
Posted:
18 October 2024
You are already at the latest version
Abstract
Background: Observational studies have implicated the association between gut microbiota and diverticular disease. However, the causality remains unclear. We aim to evaluate the causal association of gut microbiota alterations and circulating metabolite level with the risk of diverticular disease.
Methods: A bidirectional two-sample Mendelian randomization (MR) was conducted by using GWAS summary-level data of gut microbiota, circulating metabolites, and diverticular disease. The inverse-variance weighted (IVW) MR approach was applied as the primary analysis. The MR-Egger, weighted-median, and MR-PRESSO methods were supplemented as sensitivity analyses if applicable. The false discovery rate was adopted for multiple testing corrections.
Results: In the forward MR analysis, a genetically determined higher abundance of Ruminococcus torques group was found to be causally associated with decreased risk of diverticular disease. With each one-unit increase in log-transformed relative abundance values of the Ruminococcus torques group, there was a significant decrease in the odds ratio (OR) and 95% confidence interval (CI) for diverticular disease risk, with an OR of 0.68 (95% CI: 0.56-0.82, P=4.72ⅹ10-5). A genetically determined increased abundance of Oxalobacter (OR=0.88, 95%CI: 0.80-0.97, P=0.011) was suggestively associated with a lower risk of diverticular disease. We did not find any significant association between circulating metabolites level and diverticular disease risk. Additionally, no association between genetic liability to diverticular disease and the relative abundance of microbiota was observed in reverse MR analysis.
Conclusions: This study provides evidence for a causal association between genetically determined higher abundance of the Ruminococcus torques group and decreased risk of diverticular disease. We also suggest a potential protective role of Oxalobacter on diverticular disease.
Keywords:
Gut microbiota
; circulating metabolites
; diverticular disease
; Mendelian randomization study
Introduction
Diverticular disease, one of the most common gastrointestinal (GI) disorders, is reckoned as a heterogeneous disease, and the disease burden is increasing throughout the world.[1,2] The incidence of diverticular disease in the old has increased to 50%, while a rising trend of incidence and hospitalization rates has also been found in the young.[3] Although diverticular disease is asymptomatic in most instances, it has been one of the most burdensome GI diseases in Western countries due to its high prevalence and complex complications, such as acute diverticulitis, abscess, fistula formation, and bleeding or perforation.[4,5] Therefore, it is needful to explore the pathogenesis of diverticular disease and the mechanisms by which an asymptomatic condition shifts to a symptomatic one.
Recent studies have emphasized the role of gut microbiota imbalance in the onset of diverticular disease. A pilot study reported that the abundance of Clostridium cluster IV decreased in the feces of patients with diverticulosis and symptomatic uncomplicated diverticular disease (SUDD) when compared with controls.[6] They also showed that six urine metabolites have the potential to discriminate patients with diverticula from controls.[6] Another study also observed a reduction of Bacteroides fragilis, Collinsella aerofaciens, and Collinsella steroris species in the feces samples of diverticular disease patients.[7] In addition to the increment of Akkermansia muciniphlia in SUDD and diverticulosis, no other differences in the fecal microbial composition were identified between the patients and controls.[8] Besides, a case-control study found little evidence to support substantial associations between microbiota and diverticular disease.[9] However, the findings of conventional epidemiological studies may suffer from bias, such as residual confounding and reverse causation.
Mendelian randomization (MR) study is a widespread statistical method to evaluate causation between exposure and outcome.[10] By utilizing genetic variants as instrumental variables for exposure, the MR study can reduce residual confounding since genetic variants are randomly assigned at conception and therefore not associated with confounders.[10,11] In addition, the MR approach can diminish reverse causation because the allocation of genetic variants occurs earlier than the outcome.[11] Considering the inconclusive findings and limitations of the observational study, we conducted a two-sample bidirectional MR study to evaluate the potential causal association between gut microbiota alterations and diverticular disease risk. We also explored whether circulating metabolites level were causally associated with the risk of diverticular disease, as they may serve as diagnostic markers for diverticular disease.
Method
Study Design
The study overview and three main assumptions of MR design are displayed in Figure 1. First, a two-sample MR analysis was performed to evaluate the causal association of gut microbiota alteration and circulating metabolites level with the risk of diverticular disease, namely forward MR analysis. Then, a reverse MR analysis was conducted to explore whether genetic liability to diverticular disease has a potential effect on the relative abundance of gut microbiota.
Summary Statistics and Instrumental Variants Selection for Gut Microbiota
To derive the genetic variants for gut microbiota, summary statistics of gut microbial taxa were first obtained from a large-scale multi-ethnic GWAS meta-analysis conducted by the MiBioGen consortium which included 18,340 individuals (mostly European ancestry) from 24 cohorts.[12] Briefly, the study included subjects with different ethnic backgrounds, ages, gender, and microbiome analysis methodology and profiled microbial composition by targeting three distinct variable regions of the 16S r RNA gene (i.e., V1-V2, V3-V4, and V4).[12] The direct taxonomic binning was used to classify taxonomic and a total of 211 microbial taxa were identified at six levels (namely phylum, class, order, family, and genus). Microbial quantitative trait locus (mbQTLs) mapping analysis was performed through Spearman’s correlation analysis in which age, sex, technical covariates, and genetic principal components were employed as covariates. The details of the original GWAS have been described elsewhere.[12] The summary statistic of gut microbiota was also obtained from the MiBioGen consortium that has been described above.
Since the genus level is the lowest taxonomic level and we aim to identify each causal microbiota as specifically as possible, only the genus-level taxa were included in this study. After removing 12 unknown genera, a total of 119 genus-level taxa were retained for subsequent analysis. To obtain genetic instruments for gut microbiota, single nucleotide polymorphisms (SNPs) that are associated with each genus at a genome-wide significance (P<5×10-8) were derived as candidate instrumental variables (IVs). Then, linkage disequilibrium (LD) pruning (r2=0.001, clumping window=10,000kb) was performed to exclude the SNP which has pleiotropy with other SNPs or phenotype traits. The 1000 genomes project European samples data were used as the reference panel to estimate LD.[13] Finally, 18 SNPs associated with 17 genus-level taxa were selected as IVs for the forward MR analysis (Table S1). The microbiota variance was explained by the IVs arranged from 0.18% to 1.01%. The detailed PMID and web link of the datasets were provided in the Table 1.
Instrumental Variants Selection for Metabolites
Summary-level data of plasma metabolites was obtained from a genome-wide association study of the human metabolome, which developed an LC-MS-based platform that measures a total of 217 plasma metabolites and conducted metabolite profiling for 2076 European individuals in the Framingham Heart Study (FHS).[14] Genetic analyses were performed by leveraging normalized residuals of metabolite concentrations and adjusted for age and sex. The association between genetic variants and metabolite level was assessed through mixed effects models to adapt to pedigree data under an additive genetic model. Population stratification was adjusted in this genome-wide association analysis.[14]
We set a suggestive genome-wide significance (P < 1×10−5) to select potential IVs for circulating metabolites to acquire a higher variance explained.[15,16] This criterion has been adopted by previous studies.[17,18] We also performed LD pruning (r2=0.001, clumping window=10,000kb) to obtain independent SNPs for these metabolites. Six to fourteen SNPs were eventually employed as IVs for 12 plasma metabolites, including betaine, carnitine, carnosine, choline, indole 3 propionate, niacinamide, pantothenic acid, propionic acid, ribose, serotonin, taurine and trimethylamine N-oxide (TMAO). These IVs can explain 8.20%~54.26% genetic variance for these 12 plasma metabolites (Table S2).
Summary Statistics and Instrumental Variants Selection for Diverticular Disease
Recently, the largest-scaled GWAS analysis for diverticular disease was conducted by Wu et al., which contained 724,372 subjects coming from the UK Biobank (56,355 cases and 398,143 controls), the FinnGen Biobank (14,357 cases and 182,433 controls), and the BioVU (7,687 cases and 65,137 controls).[19] Briefly, the diagnostic criteria of diverticular disease in FinnGen and BioVU were consistent with the UK Biobank so that potential inaccuracy associated with self-report can be avoided. The GWAS analysis within each study was performed by adjusting for sex, age, and genetic principal components, and the estimates from different studies were combined by a meta-analysis.[19] The summary statistic of diverticular disease was subsequently generated.
We derived 142 SNPs that were associated with diverticular disease at the genome-wide significance (P<5×10-8) from the GWAS mentioned above.[19] We also conducted LD pruning for these SNPs with the 1000 genomes project European samples data as the reference panel. After LD pruning (r2=0.01, clumping distance=10,000 kb), 125 SNPs were finally employed to perform the reverse MR analysis (Table S3). These SNPs can explain a 10.63% genetic variance of diverticular disease.
Statistical Analysis
In the forward MR analysis, the gut microbiota and circulating metabolite were used as exposure, and diverticular disease was utilized as the outcome. The Wald ratio and random effect inverse-variance weighted (IVW) MR methods were applied as the primary analyses when there was only one IV and more than two IVs for the exposure, respectively. For metabolite, three sensitivity analyses, including MR-Egger, Weighted median, and MR pleiotropy residual sum and outlier (MR-PRESSO) methods, were also performed since there were more than three SNPs for each metabolite. Although with less statistical power, MR-Egger regression can detect horizontal pleiotropy and give an estimate with adjustment for pleiotropic effects.[20] If the weight is derived from at least half of valid IVs, the weighted median approach can provide consistent estimates even if horizontal pleiotropy exists.[21] The MR-PRESSO method can identify potential pleiotropy and outliers and provide an estimate with the removal of the outliers.[22] In the reverse MR analysis, the diverticular disease was reckoned as exposure while the gut microbiota was considered as the outcome. As mentioned above, the IVW method was leveraged as the primary analysis while MR-Egger, weighted median, and MR-PRESSO methods were employed as sensitivity analysis. In addition, we also executed Cochrane’s Q test to evaluate the heterogeneity among estimates of the used SNPs and calculated F statistics to exclude weak instruments (F<10). All statistical tests were two-sided and a false discovery rate (FDR) was performed for multiple testing corrections. The FDR P value < 0.05 indicated robust evidence of causal association. The associations did not survive after FDR correction but a P value < 0.05 was thought to be suggestive evidence. The analyses of this study were implemented through “TwoSampleMR” (0.5.6) and “MR-PRESSO” (1.0) packages in the R software 4.2.2.
Results
The Association between Gut Microbiota and Diverticular Disease Risk
As shown in Figure 2, among the 17 genus-level taxa with genetic instruments, only 13 microbiota-diverticular disease associations could be found in the summary statistic of diverticular disease. The Wald ratio MR analysis demonstrated that a genetically determined higher abundance of Ruminococcus torques group was causally associated with decreased risk of diverticular disease. For per one-unit in log-transformed relative abundance values of Ruminococcus torques group, the odd ratios (OR) and 95% confidence interval (CI) of diverticular disease risk were 0.68 (95%CI: 0.56-0.82, P=4.72ⅹ10-5). In addition, a genetically determined higher abundance of Oxalobacter (OR=0.88, 95%CI: 0.80-0.97, P=0.011) was suggestively associated with a lower risk of diverticular disease, but this association did not survive the FDR correction. F statistics illustrated there was no weak instrument (Table S1). Since all microbiota has less than three independent IVs, sensitivity analyses, such as MR-Egger, weighted median, and MR-PRESSO, were not applicable.
The Association between Circulating Metabolite Level and Diverticular Disease Risk
Among the 12 circulating metabolites, only choline level exhibited a nominal association with an increased risk of diverticular disease (OR=1.03, 95%CI: 0.99-1.06, P=0.185). No significant association was observed between genetically determined elevated metabolite concentration and the risk of diverticular disease (all P>0.05, Table 2). In addition, sensitivity analyses showed consistent estimates with the primary analysis. (Table 2) Cochrane’s Q test and F statistics indicated there was no heterogeneity and weak instrument, respectively (Table 2 and Table S2). Similarly, the MR-Egger and MR-PRESSO approaches did not detect any pleiotropy and outlier. (Table 2 and Table S4).
The Effect of Diverticular Disease on Gut Microbiome
In the reverse MR analysis, we did not find any compelling evidence to support an association between genetic susceptibility to diverticular disease and the relative abundance of taxa at the genus level (all P>0.05, Table 3). All sensitivity analyses demonstrated consistent association patterns with the IVW MR approach (Table 3). Furthermore, our calculations of F statistics and implementation of MR-Egger and MR-PRESSO analyses revealed no indications of weak instruments, pleiotropy, or outliers in these investigations (Tables S3 and S5).
Discussion
In this study, we found significant causality between a genetically determined higher abundance of Ruminococcus torques group and the decreased risk of diverticular disease, and also a suggestive association of Oxalobacter with diverticular disease. These findings might have implications for the prevention and treatment of diverticular disease. However, we did not discover any association between circulating metabolites and diverticular disease. In addition, genetic liability to diverticular disease did not affect the relative abundance of genus-level taxa.
In line with our study, a case-control study investigated the effect of colonic microbiome in the pathogenesis of acute diverticulitis (AD) and found lower abundance of Ruminococcus, Lachnospiraceae, and Faecalibacterium in the AD patients.[23] In addition, bacteremia was found to be associated with diverticular disease in two cases of bacteremia with the anaerobic bacterium Ruminococcus gnavus.[24] However, a pilot study reported that the relative abundance of Ruminococcus is positively associated with bloating severity score in 28 subjects with symptomatic uncomplicated diverticular disease.[25] These conflicting findings may be attributed to the characteristics of study populations, residual confounding and reverse causation, and the low statistical power of traditional observational studies.
The precise mechanisms by which the Ruminococcus torques group was involved in the pathogenesis of diverticular disease remain incompletely understood. However, existing research provided some insights into potential explanations. The Ruminococcus torques group is a genus reclassified as Mediterraneibater in the family Lachnospiraceae, and it is also known as Mediterraneibacter torques.[26,27] The Ruminococcus torques group is involved in carbohydrate metabolism and the production of short-chain fatty acids (SCFAs).[28] SCFAs serve as the major energy source for intestinal epithelial cells (IECs) and possess anti-inflammatory and immunomodulatory properties.[29,30] In addition, SCFAs could regulate IECs functions by modulating their proliferation, differentiation, and subpopulations, to affect gut motility and enhance the gut barrier functions as well as host metabolism.[31] Therefore, the increase in Ruminococcus torques group abundance may potentially reduce the risk of diverticular disease by regulating the gut microbial community and intestinal mucosal health through increased the level of SCFAs. Nevertheless, further research is needed to elucidate these mechanisms and evaluate the potential of Ruminococcus torques group to serve as a treatment strategy for diverticular disease.
This study also suggested a potential association between a higher abundance of Oxalobacter and a lower risk of diverticular disease, although this association did not reach statistical significance after correction for multiple testing. Oxalobacter plays a key role in oxalate metabolism, which has been deeply investigated in renal stones.[32,33] Furthermore, it has been also found to be associated with coronary artery disease, Grave’s disease, and inflammatory bowel disease.[34,35,36] However, there is no research reporting the association of Oxalobacter with diverticular disease and the underlying mechanisms so far. This might be a worthy direction to be investigated and further well-designed studies are warranted to explore this association and the potential pathogenesis pathways.
The biggest strength of this study is the MR design, which could compensate for the limitations (i.e., reverse causation and residual confounding) of traditionally observational studies and provide the first evidence supporting the involvement of gut microbiota in the development of diverticular disease. Second, we used the most recent summary statistics of diverticular disease with the largest sample size which enhances the statistical power. We applied stringent criteria to select IVs for microbiota to strengthen the reliability of the evaluation and adopt a bidirectional MR framework to exclude reverse causality bias. Besides, the study population of this work was restricted to European ancestry, which could diminish the bias caused by population structure. However, this also limits the generalizability of our findings to other ethnicities.
In addition to this limitation, we have to acknowledge other limitations of this study. First, although no weak instrument and pleiotropy were identified, the few numbers of IVs could only explain a small genetic variance of gut microbiota and make us unable to perform additional sensitivity analyses to preclude potential bias, such as MR-Egger test and MR-PRESSO. Second, we only focused on the genus level in the analysis to discover the most specific bacterial taxa which may affect the risk of diverticular disease. Therefore, this study cannot preclude potential associations of other level of microbiota with diverticular disease. Finally, the effects of environmental factors on gut microbiota and diverticular disease need to be considered in further epidemiology studies to demonstrate the gene-environment interaction effect.
In conclusion, our MR study provides evidence for a causal association between a genetically determined higher abundance of the Ruminococcus torques group and a reduced risk of diverticular disease. Additionally, we suggest a potential protective role of Oxalobacter, though further validation is needed. These findings contribute to the understanding of the complex interactions between gut microbiota and diverticular disease and highlight potential therapeutic targets for its prevention and treatment.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1. The instrumental variables of microbiota. Table S2. The instrumental variables of blood microbiota-associated metabolites. Table S3. The instrumental variables of diverticular disease. Table S4. The MR-PRESSO analysis of blood metabolites with diverticular disease. Table S5. The MR-PRESSO analysis of diverticular disease with microbiota.
Author Contributions
H.S., and K.W. designed and supervised the study. P.J., and W.Y. participated in the data curation. P.J., and W.Y. performed the data analyses and prepared the tables and figures. P.J., W.Y., X.C., Y.C., and Y.F. contributed to analysis strategy and data. P.J., and W.Y. wrote the original draft. Y.F., H.S., and K.W. contributed to project administration. All authors critically revised the content and contributed to editing the paper.
Funding
F.F.: The Natural Science Foundation Exploring Public Welfare/Healthcare of Zhejiang Province (LTGY23H030007).
Ethics Approval and Consent to Participate
Since this study was conducted based on publicly available GWASs, no additional ethical approval was required. The ethics approval and participant informed consent has been obtained by the original studies.
Consent for Publicatio
Not applicable.
Availability of Data and Materials
All data used in this study are publicly available and can be obtained from corresponding consortium and GWAS Catalog (https://www.ebi.ac.uk/gwas/). The detailed PMID and web link have been provided in the Table 1.
Acknowledgments
We extend our sincere appreciation to all participants and investigators who have made significant contributions to the MiBioGen consortium and published genome-wide association studies, for their invaluable collaboration in sharing data.
Competing Interests
All authors declare no competing interests.
List of Abbreviations
MR, Mendelian randomization. OR, odds ratio. GI, gastrointestinal. SUDD, symptomatic uncomplicated diverticular disease. mbQTLs, Microbial quantitative trait locus. LD, linkage disequilibrium. IVW, inverse-variance weighted. MR-PRESSO, MR pleiotropy residual sum and outlier. FDR, false discovery rate. AD, acute diverticulitis. SCFAs, short-chain fatty acids. IECs, intestinal epithelial cells.
References
- Fedirko V, Kopetz S, Daniel CR. Diverticular disease and cancer risk: More than a gut feeling. J Natl Cancer Inst. 2023;115(1):12-3. [CrossRef]
- Weizman AV, Nguyen GC. Diverticular disease: epidemiology and management. Can J Gastroenterol. 2011;25(7):385-9. [CrossRef]
- Etzioni DA, Mack TM, Beart RW, Jr., Kaiser AM. Diverticulitis in the United States: 1998-2005: changing patterns of disease and treatment. Ann Surg. 2009;249(2):210-7. [CrossRef]
- Sandler RS, Everhart JE, Donowitz M, Adams E, Cronin K, Goodman C, et al. The burden of selected digestive diseases in the United States. Gastroenterology. 2002;122(5):1500-11. [CrossRef]
- Tursi A, Scarpignato C, Strate LL, Lanas A, Kruis W, Lahat A, et al. Colonic diverticular disease. Nat Rev Dis Primers. 2020;6(1):20. [CrossRef]
- Barbara G, Scaioli E, Barbaro MR, Biagi E, Laghi L, Cremon C, et al. Gut microbiota, metabolome and immune signatures in patients with uncomplicated diverticular disease. Gut. 2017;66(7):1252-61. Epub 2016 Sep 12. [CrossRef]
- Lopetuso LR, Petito V, Graziani C, Schiavoni E, Paroni Sterbini F, Poscia A, et al. Gut Microbiota in Health, Diverticular Disease, Irritable Bowel Syndrome, and Inflammatory Bowel Diseases: Time for Microbial Marker of Gastrointestinal Disorders. Dig Dis. 2018;36(1):56-65. Epub 2017 Jul 7. [CrossRef]
- Tursi A, Mastromarino P, Capobianco D, Elisei W, Miccheli A, Capuani G, et al. Assessment of Fecal Microbiota and Fecal Metabolome in Symptomatic Uncomplicated Diverticular Disease of the Colon. J Clin Gastroenterol. 2016;50(Suppl 1):S9-S12. doi: 0.1097/MCG.0000000000000626.
- Jones RB, Fodor AA, Peery AF, Tsilimigras MCB, Winglee K, McCoy A, et al. An Aberrant Microbiota is not Strongly Associated with Incidental Colonic Diverticulosis. Sci Rep. 2018;8(1):4951. [CrossRef]
- Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1-22. [CrossRef]
- Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89-98. Epub 2014 Jul 4. [CrossRef]
- Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53(2):156-65. Epub 2021 Jan 18. [CrossRef]
- Clarke L, Zheng-Bradley X, Smith R, Kulesha E, Xiao C, Toneva I, et al. The 1000 Genomes Project: data management and community access. Nat Methods. 2012;9(5):459-62. [CrossRef]
- Rhee EP, Ho JE, Chen MH, Shen D, Cheng S, Larson MG, et al. A genome-wide association study of the human metabolome in a community-based cohort. Cell Metab. 2013;18(1):130-43. [CrossRef]
- Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11(3):241-7. [CrossRef]
- Sanna S, van Zuydam NR, Mahajan A, Kurilshikov A, Vich Vila A, Võsa U, et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat Genet. 2019;51(4):600-5. Epub 2019 Feb 18. [CrossRef]
- Gagnon E, Mitchell PL, Manikpurage HD, Abner E, Taba N, Esko T, et al. Impact of the gut microbiota and associated metabolites on cardiometabolic traits, chronic diseases and human longevity: a Mendelian randomization study. J Transl Med. 2023;21(1):60. [CrossRef]
- Liu X, Tong X, Zou Y, Lin X, Zhao H, Tian L, et al. Mendelian randomization analyses support causal relationships between blood metabolites and the gut microbiome. Nat Genet. 2022;54(1):52-61. Epub 2022 Jan 3. [CrossRef]
- Wu Y, Goleva SB, Breidenbach LB, Kim M, MacGregor S, Gandal MJ, et al. 150 risk variants for diverticular disease of intestine prioritize cell types and enable polygenic prediction of disease susceptibility. Cell Genom. 2023;3(7):100326. eCollection 2023 Jul 12. [CrossRef]
- Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512-25. Epub 2015 Jun 6. [CrossRef]
- Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity Analyses for Robust Causal Inference from Mendelian Randomization Analyses with Multiple Genetic Variants. Epidemiology. 2017;28(1):30-42. [CrossRef]
- Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693-8. Epub 2018 Apr 23. [CrossRef]
- Mj O, Turner GA, A S, Frizelle FA, R P. Distinct changes in the colonic microbiome associated with acute diverticulitis. Colorectal Dis. 2022;24(12):1591-601. Epub 2022 Aug 11. [CrossRef]
- Hansen SG, Skov MN, Justesen US. Two cases of Ruminococcus gnavus bacteremia associated with diverticulitis. J Clin Microbiol. 2013;51(4):1334-6. Epub 2013 Jan 30. [CrossRef]
- Kvasnovsky CL, Leong LEX, Choo JM, Abell GCJ, Papagrigoriadis S, Bruce KD, et al. Clinical and symptom scores are significantly correlated with fecal microbiota features in patients with symptomatic uncomplicated diverticular disease: a pilot study. Eur J Gastroenterol Hepatol. 2018;30(1):107-12. [CrossRef]
- Togo AH, Diop A, Bittar F, Maraninchi M, Valero R, Armstrong N, et al. Description of Mediterraneibacter massiliensis, gen. nov., sp. nov., a new genus isolated from the gut microbiota of an obese patient and reclassification of Ruminococcus faecis, Ruminococcus lactaris, Ruminococcus torques, Ruminococcus gnavus and Clostridium glycyrrhizinilyticum as Mediterraneibacter faecis comb. nov., Mediterraneibacter lactaris comb. nov., Mediterraneibacter torques comb. nov., Mediterraneibacter gnavus comb. nov. and Mediterraneibacter glycyrrhizinilyticus comb. nov. Antonie Van Leeuwenhoek. 2018;111(11):2107-28. Epub 2018 May 31. [CrossRef]
- Liu C, Finegold SM, Song Y, Lawson PA. Reclassification of Clostridium coccoides, Ruminococcus hansenii, Ruminococcus hydrogenotrophicus, Ruminococcus luti, Ruminococcus productus and Ruminococcus schinkii as Blautia coccoides gen. nov., comb. nov., Blautia hansenii comb. nov., Blautia hydrogenotrophica comb. nov., Blautia luti comb. nov., Blautia producta comb. nov., Blautia schinkii comb. nov. and description of Blautia wexlerae sp. nov., isolated from human faeces. Int J Syst Evol Microbiol. 2008;58(Pt 8):1896-902. [CrossRef]
- Ze X, Duncan SH, Louis P, Flint HJ. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012;6(8):1535-43. Epub Feb 16. [CrossRef]
- Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe. 2015;17(5):662-71. Epub Apr 9. [CrossRef]
- Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology. 2013;145(2):396-406.e1-10. Epub May 7. [CrossRef]
- Martin-Gallausiaux C, Marinelli L, Blottière HM, Larraufie P, Lapaque N. SCFA: mechanisms and functional importance in the gut. Proc Nutr Soc. 2021;80(1):37-49. Epub 2020 Apr 2. [CrossRef]
- Dawson KA, Allison MJ, Hartman PA. Isolation and some characteristics of anaerobic oxalate-degrading bacteria from the rumen. Appl Environ Microbiol. 1980;40(4):833-9. [CrossRef]
- Liu M, Zhang Y, Wu J, Gao M, Zhu Z, Chen H. Causal relationship between kidney stones and gut microbiota contributes to the gut-kidney axis: a two-sample Mendelian randomization study. Front Microbiol. 2023;14:1204311. eCollection 2023. [CrossRef]
- Zhang Y, Zhang X, Chen D, Lu J, Gong Q, Fang J, et al. Causal associations between gut microbiome and cardiovascular disease: A Mendelian randomization study. Front Cardiovasc Med. 2022;9:971376. eCollection 2022. [CrossRef]
- Cao J, Wang N, Luo Y, Ma C, Chen Z, Chenzhao C, et al. A cause-effect relationship between Graves’ disease and the gut microbiome contributes to the thyroid-gut axis: A bidirectional two-sample Mendelian randomization study. Front Immunol. 2023;14:977587. eCollection 2023. [CrossRef]
- Liu B, Ye D, Yang H, Song J, Sun X, Mao Y, et al. Two-Sample Mendelian Randomization Analysis Investigates Causal Associations Between Gut Microbial Genera and Inflammatory Bowel Disease, and Specificity Causal Associations in Ulcerative Colitis or Crohn’s Disease. Front Immunol. 2022;13:921546. eCollection 2022. [CrossRef]
Figure 1.
The study design of this study. MR, Mendelian randomization. SNP, single nucleotide polymorphism.
Figure 1.
The study design of this study. MR, Mendelian randomization. SNP, single nucleotide polymorphism.
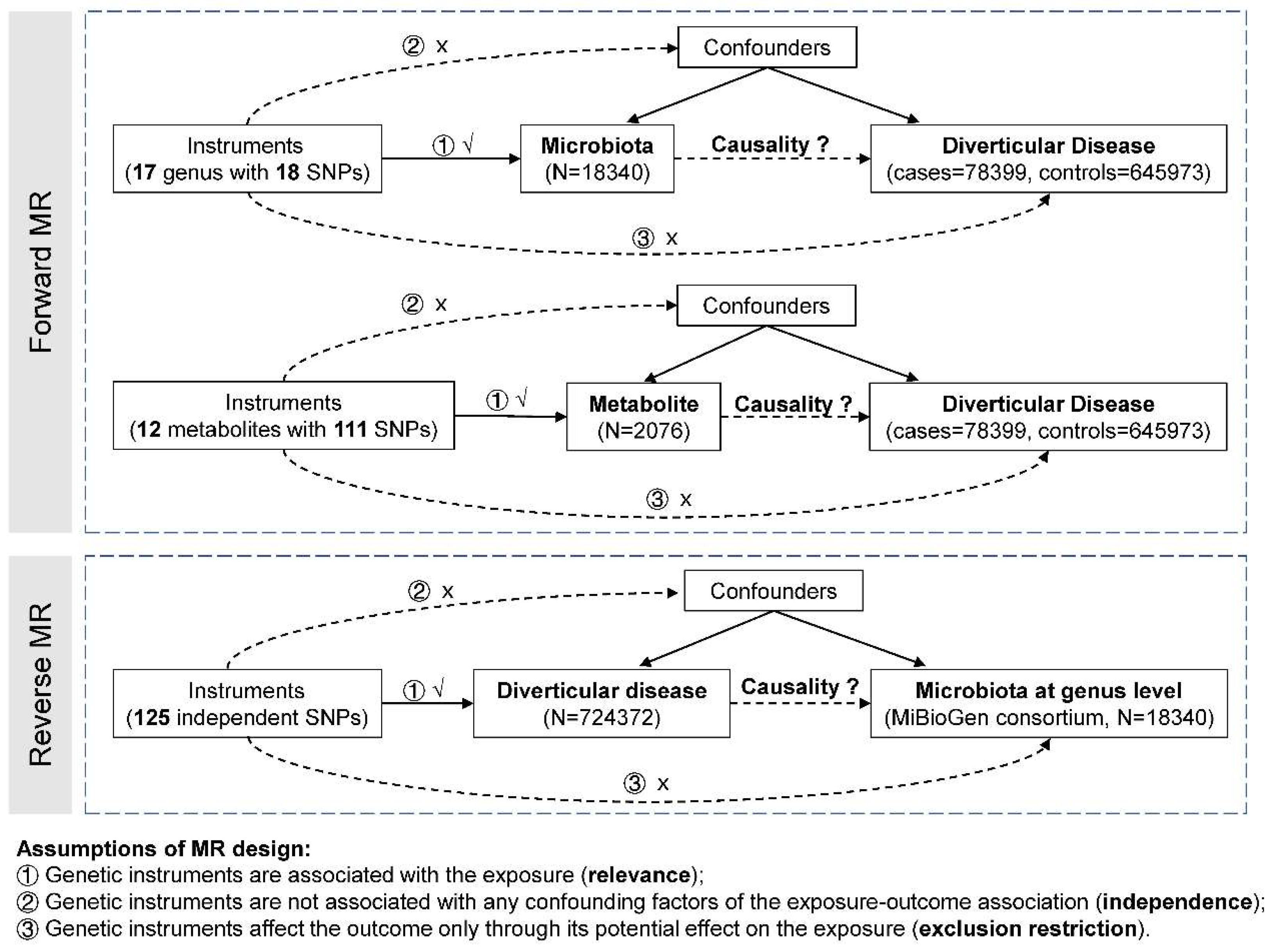
Figure 2.
The association between gut microbiota and the risk of diverticular disease. OR, odds ratio. CI, confidence interval.
Figure 2.
The association between gut microbiota and the risk of diverticular disease. OR, odds ratio. CI, confidence interval.
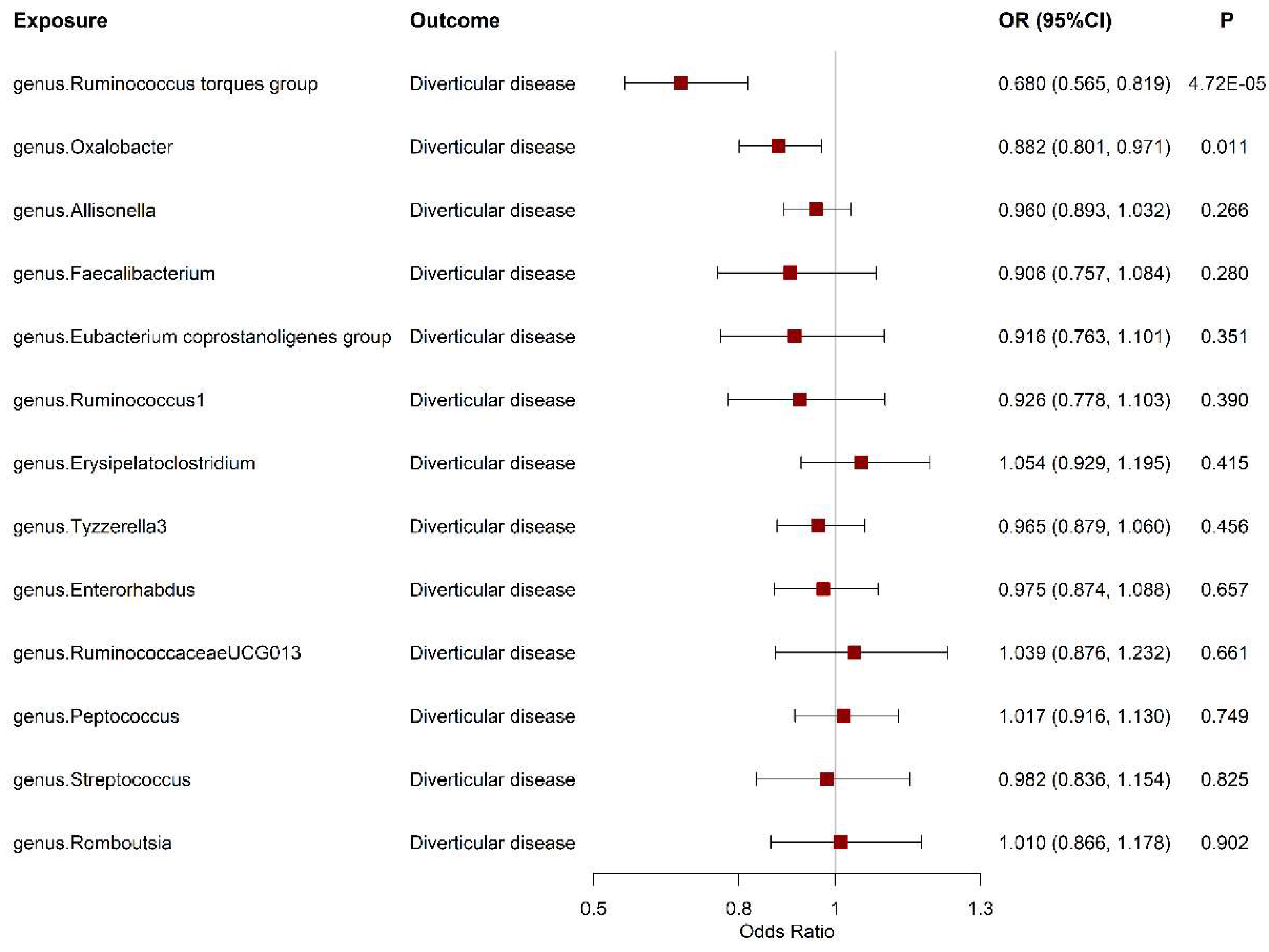

Table 1.
The summary of data sources used in this study.
| Exposure or Outcome | Participants Included in Analysis | Adjustments | Number of genetic instruments | PubMed ID and/ or URL |
|---|---|---|---|---|
| Gut microbiota taxa | 18340 European-descent individuals | Age and any necessary study-specific covariates | 18 SNPs for 17 gut microbiota taxa | 33462485 https://mibiogen.gcc.rug.nl/ |
| Gut microbial metabolites | 2076 European-descent individuals | Age, sex | 111 SNPs for 12 gut microbial metabolites | 23823483 |
| Diverticular disease | 78399 cases and 645973 controls of European ancestry | sex, age, and genetic principal components | 125 | 37492107 |
Table 2.
The association between genetically determined metabolites level and the risk of diverticular disease.
Table 2.
The association between genetically determined metabolites level and the risk of diverticular disease.
| Exposure | Outcome | Method | Nsnp | OR | LCI | UCI | pval | P_heterogeneity | P_intercept |
|---|---|---|---|---|---|---|---|---|---|
| Betaine | DD | MR Egger | 12 | 1.01 | 0.98 | 1.04 | 0.389 | 0.436 | |
| Weighted median | 12 | 1.01 | 0.99 | 1.03 | 0.310 | ||||
| Inverse variance weighted | 12 | 1.00 | 0.99 | 1.02 | 0.692 | 0.721 | |||
| Simple mode | 12 | 1.01 | 0.98 | 1.04 | 0.521 | ||||
| Weighted mode | 12 | 1.01 | 0.99 | 1.03 | 0.375 | ||||
| Choline | DD | MR Egger | 6 | 0.97 | 0.86 | 1.11 | 0.697 | 0.447 | |
| Weighted median | 6 | 1.01 | 0.97 | 1.04 | 0.655 | ||||
| Inverse variance weighted | 6 | 1.03 | 0.99 | 1.06 | 0.185 | 0.058 | |||
| Simple mode | 6 | 1.00 | 0.95 | 1.04 | 0.902 | ||||
| Weighted mode | 6 | 1.00 | 0.96 | 1.04 | 0.897 | ||||
| Serotonin | DD | MR Egger | 7 | 1.02 | 0.86 | 1.20 | 0.840 | 0.870 | |
| Weighted median | 7 | 1.00 | 0.97 | 1.04 | 0.829 | ||||
| Inverse variance weighted | 7 | 1.00 | 0.98 | 1.03 | 0.779 | 0.604 | |||
| Simple mode | 7 | 1.01 | 0.96 | 1.07 | 0.700 | ||||
| Weighted mode | 7 | 1.01 | 0.96 | 1.06 | 0.744 | ||||
| Propionic acid | DD | Inverse variance weighted | 2 | 1.01 | 0.98 | 1.05 | 0.443 | 0.425 | NA |
| Carnitine | DD | MR Egger | 11 | 1.03 | 0.99 | 1.06 | 0.168 | 0.270 | |
| Weighted median | 11 | 1.01 | 0.99 | 1.02 | 0.598 | ||||
| Inverse variance weighted | 11 | 1.01 | 0.99 | 1.02 | 0.285 | 0.862 | |||
| Simple mode | 11 | 1.00 | 0.97 | 1.03 | 0.779 | ||||
| Weighted mode | 11 | 1.00 | 0.98 | 1.03 | 0.849 | ||||
| Indole 3 propionate | DD | MR Egger | 12 | 0.99 | 0.92 | 1.07 | 0.826 | 0.803 | |
| Weighted median | 12 | 1.00 | 0.97 | 1.02 | 0.787 | ||||
| Inverse variance weighted | 12 | 1.00 | 0.98 | 1.02 | 0.919 | 0.366 | |||
| Simple mode | 12 | 0.99 | 0.94 | 1.03 | 0.574 | ||||
| Weighted mode | 12 | 0.99 | 0.95 | 1.03 | 0.641 | ||||
| Ribose 5-P and Ribulose 5-P | DD | MR Egger | 7 | 0.97 | 0.91 | 1.03 | 0.313 | 0.619 | |
| Weighted median | 7 | 0.99 | 0.96 | 1.02 | 0.506 | ||||
| Inverse variance weighted | 7 | 0.98 | 0.95 | 1.01 | 0.136 | 0.154 | |||
| Simple mode | 7 | 0.99 | 0.96 | 1.02 | 0.629 | ||||
| Weighted mode | 7 | 0.99 | 0.96 | 1.02 | 0.552 | ||||
| Taurine | DD | MR Egger | 8 | 0.99 | 0.97 | 1.01 | 0.401 | 0.110 | |
| Weighted median | 8 | 1.00 | 0.99 | 1.02 | 0.559 | ||||
| Inverse variance weighted | 8 | 1.01 | 0.99 | 1.02 | 0.358 | 0.370 | |||
| Simple mode | 8 | 1.02 | 0.98 | 1.06 | 0.291 | ||||
| Weighted mode | 8 | 1.00 | 0.99 | 1.02 | 0.708 | ||||
| Carnosine | DD | MR Egger | 13 | 1.02 | 0.99 | 1.06 | 0.218 | 0.154 | |
| Weighted median | 13 | 0.99 | 0.97 | 1.01 | 0.227 | ||||
| Inverse variance weighted | 13 | 1.00 | 0.98 | 1.01 | 0.753 | 0.084 | |||
| Simple mode | 13 | 0.99 | 0.96 | 1.02 | 0.607 | ||||
| Weighted mode | 13 | 0.99 | 0.96 | 1.01 | 0.386 | ||||
| TMAO | DD | MR Egger | 8 | 0.97 | 0.90 | 1.04 | 0.435 | 0.471 | |
| Weighted median | 8 | 0.99 | 0.96 | 1.02 | 0.384 | ||||
| Inverse variance weighted | 8 | 1.00 | 0.98 | 1.02 | 0.727 | 0.742 | |||
| Simple mode | 8 | 0.99 | 0.95 | 1.03 | 0.562 | ||||
| Weighted mode | 8 | 0.99 | 0.95 | 1.02 | 0.521 | ||||
| Niacinamide | DD | Inverse variance weighted | 2 | 1.01 | 0.97 | 1.05 | 0.682 | 0.300 | NA |
| Pantothenic acid | DD | MR Egger | 10 | 1.04 | 0.97 | 1.11 | 0.276 | 0.120 | |
| Weighted median | 10 | 0.98 | 0.96 | 1.00 | 0.102 | ||||
| Inverse variance weighted | 10 | 0.98 | 0.96 | 1.00 | 0.098 | 0.324 | |||
| Simple mode | 10 | 0.97 | 0.92 | 1.01 | 0.158 | ||||
| Weighted mode | 10 | 0.97 | 0.93 | 1.01 | 0.211 |
DD, diverticular disease. OR, odds ratio. CI, confidence interval.
Table 3.
The association between genetic liability to diverticular disease and the abundance of gut microbiota.
Table 3.
The association between genetic liability to diverticular disease and the abundance of gut microbiota.
| Exposure | Outcome | Method | nsnp | beta | se | pval | P_heterogeneity | P_intercept |
|---|---|---|---|---|---|---|---|---|
| DD | Allisonella | MR Egger | 77 | 0.237 | 0.183 | 0.200 | 0.214 | |
| Weighted median | 77 | 0.025 | 0.106 | 0.815 | ||||
| Inverse variance weighted | 77 | 0.023 | 0.067 | 0.733 | 0.556 | |||
| Simple mode | 77 | 0.096 | 0.220 | 0.665 | ||||
| Weighted mode | 77 | 0.012 | 0.159 | 0.942 | ||||
| DD | Enterorhabdus | MR Egger | 93 | -0.039 | 0.112 | 0.731 | 0.747 | |
| Weighted median | 93 | -0.033 | 0.064 | 0.605 | ||||
| Inverse variance weighted | 93 | -0.005 | 0.041 | 0.902 | 0.410 | |||
| Simple mode | 93 | -0.037 | 0.130 | 0.776 | ||||
| Weighted mode | 93 | -0.030 | 0.094 | 0.751 | ||||
| DD | Erysipelatoclostridium | MR Egger | 93 | -0.055 | 0.118 | 0.645 | 0.615 | |
| Weighted median | 93 | 0.009 | 0.057 | 0.872 | ||||
| Inverse variance weighted | 93 | 0.001 | 0.043 | 0.987 | 0.002 | |||
| Simple mode | 93 | 0.120 | 0.148 | 0.422 | ||||
| Weighted mode | 93 | 0.084 | 0.106 | 0.430 | ||||
| DD | Eubacteriumcoprostanoligenesgroup | MR Egger | 94 | -0.051 | 0.072 | 0.477 | 0.775 | |
| Weighted median | 94 | -0.055 | 0.041 | 0.184 | ||||
| Inverse variance weighted | 94 | -0.032 | 0.026 | 0.224 | 0.577 | |||
| Simple mode | 94 | -0.050 | 0.091 | 0.581 | ||||
| Weighted mode | 94 | -0.061 | 0.068 | 0.370 | ||||
| DD | Faecalibacterium | MR Egger | 94 | -0.025 | 0.071 | 0.719 | 0.937 | |
| Weighted median | 94 | -0.024 | 0.041 | 0.565 | ||||
| Inverse variance weighted | 94 | -0.020 | 0.026 | 0.435 | 0.697 | |||
| Simple mode | 94 | 0.007 | 0.094 | 0.937 | ||||
| Weighted mode | 94 | -0.008 | 0.066 | 0.904 | ||||
| DD | Oxalobacter | MR Egger | 92 | -0.023 | 0.143 | 0.870 | 0.728 | |
| Weighted median | 92 | 0.016 | 0.082 | 0.846 | ||||
| Inverse variance weighted | 92 | 0.023 | 0.052 | 0.658 | 0.407 | |||
| Simple mode | 92 | -0.019 | 0.201 | 0.923 | ||||
| Weighted mode | 92 | 0.056 | 0.161 | 0.727 | ||||
| DD | Peptococcus | MR Egger | 93 | -0.028 | 0.127 | 0.827 | 0.944 | |
| Weighted median | 93 | 0.031 | 0.073 | 0.672 | ||||
| Inverse variance weighted | 93 | -0.019 | 0.046 | 0.674 | 0.913 | |||
| Simple mode | 93 | 0.018 | 0.162 | 0.911 | ||||
| Weighted mode | 93 | 0.025 | 0.126 | 0.841 | ||||
| DD | Romboutsia | MR Egger | 94 | -0.134 | 0.085 | 0.119 | 0.247 | |
| Weighted median | 94 | -0.031 | 0.046 | 0.506 | ||||
| Inverse variance weighted | 94 | -0.042 | 0.031 | 0.185 | 0.159 | |||
| Simple mode | 94 | 0.011 | 0.100 | 0.915 | ||||
| Weighted mode | 94 | 0.001 | 0.079 | 0.991 | ||||
| DD | RuminococcaceaeUCG013 | MR Egger | 94 | -0.054 | 0.073 | 0.466 | 0.531 | |
| Weighted median | 94 | -0.007 | 0.042 | 0.872 | ||||
| Inverse variance weighted | 94 | -0.011 | 0.027 | 0.689 | 0.602 | |||
| Simple mode | 94 | -0.013 | 0.090 | 0.889 | ||||
| Weighted mode | 94 | -0.013 | 0.063 | 0.842 | ||||
| DD | Ruminococcus1 | MR Egger | 94 | -0.040 | 0.074 | 0.593 | 0.512 | |
| Weighted median | 94 | 0.029 | 0.042 | 0.480 | ||||
| Inverse variance weighted | 94 | 0.006 | 0.027 | 0.836 | 0.677 | |||
| Simple mode | 94 | 0.027 | 0.091 | 0.769 | ||||
| Weighted mode | 94 | 0.044 | 0.073 | 0.548 | ||||
| DD | Ruminococcustorquesgroup | MR Egger | 94 | -0.014 | 0.071 | 0.839 | 0.860 | |
| Weighted median | 94 | -0.015 | 0.039 | 0.711 | ||||
| Inverse variance weighted | 94 | -0.003 | 0.026 | 0.917 | 0.625 | |||
| Simple mode | 94 | -0.049 | 0.081 | 0.546 | ||||
| Weighted mode | 94 | 0.009 | 0.064 | 0.883 | ||||
| DD | Streptococcus | MR Egger | 94 | -0.016 | 0.074 | 0.834 | 0.771 | |
| Weighted median | 94 | -0.005 | 0.044 | 0.907 | ||||
| Inverse variance weighted | 94 | -0.036 | 0.027 | 0.190 | 0.612 | |||
| Simple mode | 94 | -0.011 | 0.103 | 0.917 | ||||
| Weighted mode | 94 | -0.020 | 0.064 | 0.758 | ||||
| DD | Tyzzerella3 | MR Egger | 93 | 0.315 | 0.138 | 0.025 | 0.056 | |
| Weighted median | 93 | 0.050 | 0.077 | 0.516 | ||||
| Inverse variance weighted | 93 | 0.067 | 0.051 | 0.190 | 0.146 | |||
| Simple mode | 93 | 0.113 | 0.172 | 0.514 | ||||
| Weighted mode | 93 | 0.098 | 0.119 | 0.410 |
DD, diverticular disease. OR, odds ratio. CI, confidence interval.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



