
Submitted:
17 October 2024
Posted:
18 October 2024
You are already at the latest version
Abstract
The endocannabinoid signalling system (ECS) plays a critical role from the very beginning of embryogenesis. Accordingly, the ECS is engaged early-on in nervous system development, starting from neurulation, supported by the identification of ECS components - both receptors and enzymes controlling endocannabinoid metabolism – at these early stages. In particular, regarding the brain, the ECS is involved in the tightly regulated sequence of events that comprise brain development, from neurogenesis to neuronal migration, morphological guidance for neuronal connectivity, and synaptic circuitry refinement. The importance of this broad role of the ECS across various brain development processes is further underscored by the growing understanding of the consequences of cannabis exposure at different developmental stages. Despite the considerable knowledge we have on the role of the ECS in brain development, significant gaps in our understanding remain, particularly regarding the long-term impact and underlying mechanisms of cannabis exposure at different developmental stages. This review provides an overview of the current state of knowledge on the role of the ECS throughout brain development, from embryogenesis to adulthood, and discusses the impact of cannabis exposure, especially during adolescence—a critical period of circuitry maturation and refinement coinciding with an increased risk of cannabis use.
Keywords:
CB1 receptor
; CB2 receptor
; TRPV1 receptor
; GPR55
; brain development
; neurogenesis
; neuronal migration
; axon pathfinding
; synaptogenesis
; Cannabis
1. Introduction to Cannabis and the Endocannabinoid System
The endocannabinoid system (ECS) was discovered during efforts to understand how marijuana produces its recreational and medicinal effects on the human body. Cannabis research gained momentum during the era of the hippies, when Gaoni and Mechoulam elucidated the chemical structure of the two principal phytocannabinoids, cannabidiol (CBD) and Δ9-tetrahydrocannabinol (Δ9-THC) [1] (Figure 1). Over the last 30 years, it became evident that Δ9-THC is primarily responsible for the recreational (psychotomimetic) effects of marijuana, despite the plant producing over 120 additional phytocannabinoids [2]. Δ9-THC interacts with various receptors in the human body, though only the canonical cannabinoid receptors, CB1 and CB2 (CB1Rs and CB2Rs) (Figure 1), are part of the sensu stricto ECS. Through CB1R activation, Δ9-THC elicits effects such as hypolocomotion, catalepsy, hypothermia, and analgesia, collectively known as the tetrad model in drug-naïve subjects [1,2]. Importantly, other hemp variants with low levels of Δ9-THC acid are neither illicit nor psychotomimetic. In contrast, CBD, the other principal phytocannabinoid, is not only devoid of psychoactivity but also antagonizes the effects of Δ9-THC in most biological assays [2,3,4].
1.1. The Endocannabinoid System
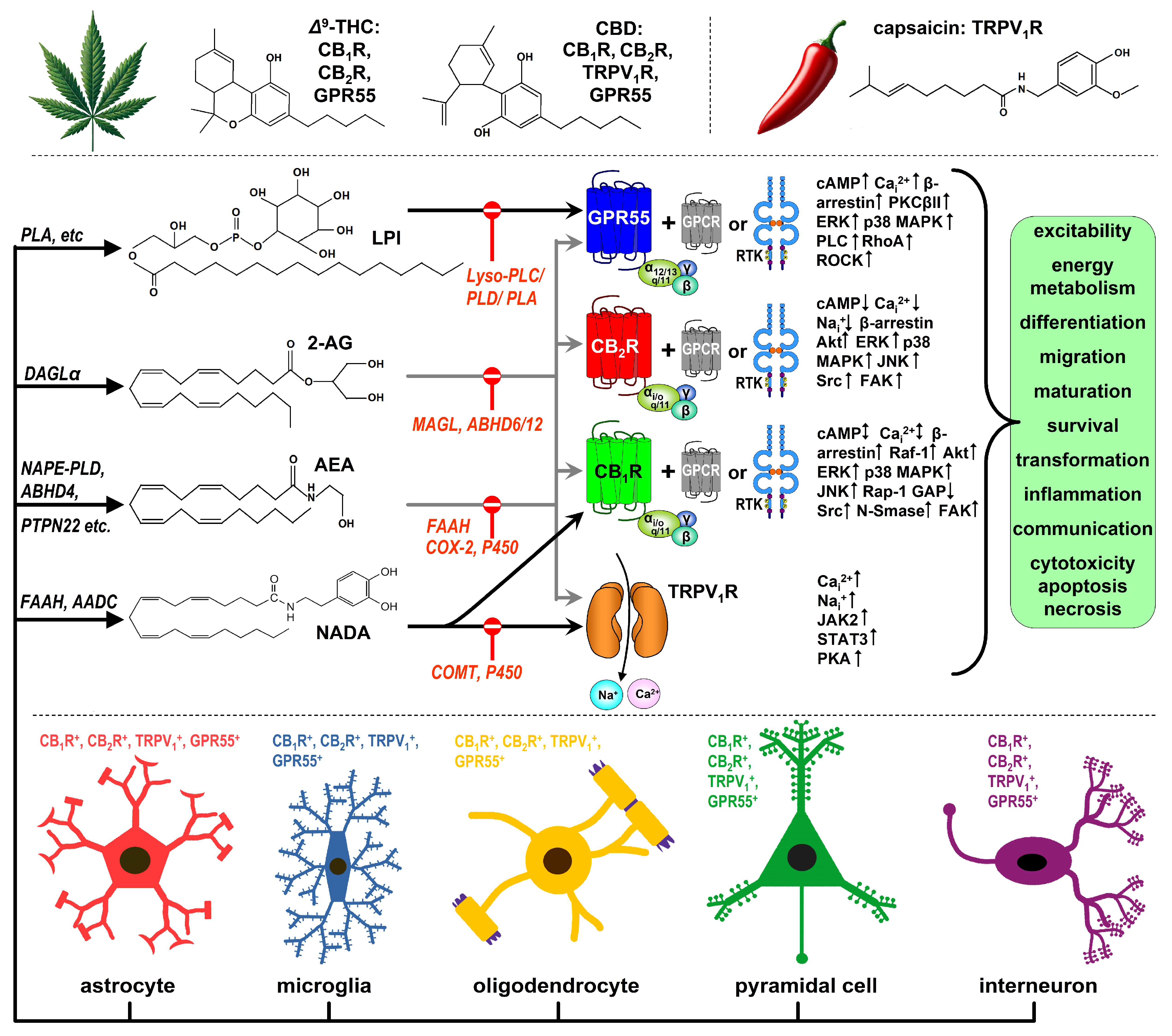
The term “endocannabinoid” was coined 30 years ago to distinguish cannabinoids produced by the body from synthetic and phytocannabinoids [5]. The most studied endocannabinoid messengers are the lipophilic N-arachidonoyl-ethanolamine (anandamide or AEA) and 2-arachidonoyl glycerol (2-AG) (Figure 1). Both can activate CB1R and CB2R, which are located in various subcellular compartments, including intracellularly [6,7,8,9,10,11,12,13].
In addition to CB1R and CB2R, several other receptors, both on the cell surface and intracellularly, are influenced by cannabinoids. One such receptor is the G protein-coupled receptor 55 (GPR55), a L-α-lysophosphatidyl-inositol (LPI) receptor (Figure 1) that shares a modest (13-14%) sequence homology with CB1R and CB2R [2,14]. Another key receptor is TRPV1, a polymodal sensor that responds to heat, toxins including chilli pepper’s capsaicin, protons, and voltage, and functions as a Na+/Ca2+ channel (Figure 1). Both GPR55 and TRPV1 interact with eCBs, synthetic cannabinoids, and phytocannabinoids, making them important targets for medical cannabis formulations such as Epidiolex, an antiepileptic medication based on CBD [15]. Some argue that GPR55 and TRPV1 should be considered bona fide endocannabinoid receptors, even though they also play key roles in other signalling systems (Figure 1).
1.2. Cannabinoid Receptors
CB1R was the first identified and remains the most significant cannabinoid receptor, with high expression levels in the brain. Initially, CB1R was detected in cholecystokinin+ GABAergic interneurons in the rodent and human brain [16,17], but later studies identified CB1R in various other cell types, including VGLUT1+ glutamatergic cells, monoaminergic neurons, certain cholinergic neurons, astrocytes, and microglia [2,6,7,18] (Figure 1). In contrast, CB2R was long regarded as “the peripheral cannabinoid receptor,” absent from the healthy brain. However, in the past two decades, its presence and function in neurons have been increasingly accepted [11,12,19,20,21] (Figure 1).
Both CB1R and CB2R engage with various intracellular signalling pathways, depending on the cellular context. In their homodimeric forms, these receptors predominantly couple with inhibitory Gi/o proteins. Activation of CB1Rs and CB2Rs typically inhibits adenylyl cyclase, and activates pathways such as focal adhesion kinase (FAK), extracellular signal-regulated kinase 1/2 (ERK1/2), p38 mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), c-Src kinase (Src), neutral sphingomyelinase (N-SMase), ceramide synthesis, and phosphoinositide 3-kinase (PI3K)/Akt. These pathways are crucial for cytoskeletal reorganization, proliferation, migration, and cell survival or apoptosis. Additionally, via the Gi/o βγ subunit, CB1R and CB2R can inhibit voltage-gated Ca2+ channels and activate inwardly rectifying K+ channels, leading to membrane hyperpolarization in neurons [2,22,23,24] (Figure 1).
CB1R and CB2R often form heteromeric complexes with other G protein-coupled receptors (GPCRs), resulting in novel functional entities with unique responses to cannabinoids, which play an essential role in brain development [25]. One example is the CB1R-CB2R heteromer, where the unilateral activation of either receptor stimulates Akt/PKB phosphorylation, ERK1/2 activation, and neurite outgrowth in transfected neurons and globus pallidus slices [26]. CB1R can also form heteromers with receptor tyrosine kinases (RTKs) that are critical for growth and development [27]. One example involves the transactivation of the TrkB receptor of brain-derived neurotrophic factor (BDNF) via Src kinase in cholecystokinin+ GABAergic interneurons of the developing hippocampus and cortex [28] (see below) (Figure 1).
GPR55 was first discovered in humans in 1999 and soon emerged as a potential third metabotropic endocannabinoid receptor [25,29]. GPR55 activation by Δ9-THC, AEA, 2-AG, and other endogenous LPI-like ligands triggers coupling with Gα12, Gα13, or Gαq/11, leading to increased intracellular calcium levels or the activation of β-arrestin, PKCβII, ERK, p38 MAPK, PLC, RhoA and ROCK [2,25,29,30,31] (Figure 1). GPR55’s involvement in regulating cell proliferation, growth, migration, metabolism, and survival has garnered significant interest in cancer research [32,33]. These functions suggest that GPR55 could play a role in brain development, although GPR55 knockout (KO) mice show no macroscopic brain abnormalities [34] (see below).
Among the many members of the “transient receptor potential” (TRP) superfamily of ligand-gated ion channels, TRPV1 serves as an ionotropic receptor for several cannabinoid ligands. It is activated by AEA, 2-AG, and their close relatives, including N-arachidonoyl dopamine (NADA) and N-oleoyl dopamine (OLDA)—both belonging to the so-called endovanilloid class—as well as botanical substances such as capsaicin, CBD, and resiniferatoxin [2,35,36]. The TRPV1 channel is composed of four subunits that form a central pore, which is permeable to Na+ and Ca2+ (Figure 1). These six-transmembrane-domain subunits are prone to alternative splicing, often resulting in functionally distinct TRPV1 receptors [37,38]. Notably, the TRPV1b splice variant is strongly expressed in the human fetal brain, suggesting a role in development [39]. Both the presynaptic density and functional role of TRPV1 receptors decline in the first weeks of postnatal life [40], further supporting the hypothesis that TRPV1 may play a developmental role.
1.3. Endocannabinoids
1.3.1. 2-arachidonoyl glycerol (2-AG)
In the brain, 2-AG synthesis primarily involves the action of diacylglycerol lipases α and β (DAGLα and DAGLβ) [41,42]. Consistent with the mechanism of retrograde 2-AG signalling, DAGLα is postsynaptic and colocalizes with dendritic markers in both rodent and human brains [43,44]. Typically, 2-AG synthesis is triggered by postsynaptic Ca2+ entry and activation of Gq/11-coupled metabotropic receptors such as the mGluR5, which in turn activates phospholipase Cβ1 (PLCβ1), releasing sn-2-arachidonoyl-DAG, the precursor of 2-AG [41,45,46]. Postsynaptic Ca2+ elevation also activates DAGLα, cleaving 2-AG from its precursor. Although this describes “on-demand” synthesis, evidence supports the existence of a basal synaptic pool of pre-synthesized 2-AG, stored in adiposomes, that is readily releasable [41,47] (Figure 1).
In brain homogenates, monoacylglycerol lipases (MAGL 1 and 2) are responsible for 85% of 2-AG degradation, with the remaining 15% hydrolytic activity attributed to α/β hydrolase domain 6 (ABHD6; 4%) and α/β hydrolase domain 12 (ABHD12; 9%) [42,46,48] (Figure 1).
1.3.2. Anandamide (AEA)
Anandamide is synthesized through several pathways, most notably from N-acylphosphatidylethanolamine by NAPE-specific phospholipase D (NAPE-PLD) [5], as well as by other enzymes such as protein tyrosine phosphatase non-receptor type 22, and through a multi-step process involving α/β hydrolase domain 4 (ABHD4) and glycerophosphodiesterase GDE1 [2,47,49] (Figure 1).
While several enzymes can degrade anandamide, the bulk of its metabolism is carried out by fatty acid aminohydrolase-1 (FAAH-1), which hydrolyzes anandamide into arachidonic acid and ethanolamine [2,50]. Humans also possess FAAH-2, an enzyme functionally similar to FAAH-1 but with only 20% sequence similarity [51]. Additional enzymes such as COX-2 and cytochrome P450 are involved in anandamide degradation [2,50] (Figure 1).
Figure 1.
Overview of the endocannabinoid system (ECS) in the brain. The endocannabinoid system (ECS) was uncovered through research investigating the molecular targets of key phytocannabinoids found in Cannabis sativa, particularly Δ9-tetrahydrocannabinol (Δ9-THC), the psychoactive component, and cannabidiol (CBD), a non-psychoactive compound. Both Δ9-THC and CBD interact with numerous targets within the brain, and here we focus on four key receptors: the cannabinoid receptors CB1 and CB2 (CB1R and CB2R), GPR55, and the transient receptor potential vanilloid type 1 (TRPV1) receptor. CB1R, CB2R, and GPR55 are G protein-coupled receptors (GPCRs) with seven transmembrane-spanning domains. While Δ9-THC acts as a partial agonist at these GPCRs, CBD’s pharmacological actions are more complex, often resembling negative allosteric modulation and weak partial agonism. CBD also activates and rapidly desensitizes the ionotropic TRPV1R, similar to capsaicin from chili peppers, but without the associated pungency. TRPV1R agonists are referred to as vanilloid ligands. These receptors are expressed across various brain cell types, including astrocytes, microglia, oligodendrocytes [52], glutamatergic neurons, GABAergic interneurons, and projection neurons (GABAergic, monoaminergic, and cholinergic), depending on factors like brain region, age, and neuropsychiatric conditions. While all four receptors are typically found in the cytoplasm—primarily in nerve terminals, dendrites, and cell bodies—there is substantial evidence for their intracellular localization. In addition to receptors, the ECS includes enzymes responsible for synthesizing lipid ligands that activate these receptors. One of the most well-studied eCBs, anandamide (N-arachidonoyl-ethanolamine or AEA), is synthesized from N-acylphosphatidylethanolamine (NAPE) via NAPE-specific phospholipase D (PLD). Several alternative pathways also contribute to AEA production. Diacylglycerol lipase α (DAGLα) is the primary enzyme that synthesizes 2-arachidonoyl-glycerol (2-AG), another major eCB. Both AEA and 2-AG activate all four receptors, though other ligands exhibit more receptor-selective actions. For example, N-arachidonoyl-dopamine (NADA), likely produced by fatty acid amide hydrolase (FAAH) in dopaminergic cells, acts as a hybrid agonist for CB1R and TRPV1R [53]. Similarly, L-α-lysophosphatidyl-inositol (LPI) and its congeners resemble classical eCBs but selectively activate GPR55. The activation of these receptors can influence virtually all functions of the brain cells expressing them, but their actions are highly context-dependent. The effects depend on factors such as receptor splice variants, heteromeric interactions with other receptors (e.g., TrkB, insulin receptor, or EGF receptor), the cell’s metabolic state and age, and the ontogenetic stage of the organism. Many receptor-mediated effects are tied to brain cell processes such as differentiation, maturation, migration, circuit formation, and plasticity, which are key topics in this review. Finally, after eCBs activate their receptors, they are primarily metabolized intracellularly by a variety of enzymes. The key enzymes for this review are FAAH and cyclooxygenase-2 (COX-2), which degrade anandamide, and monoacylglycerol lipase (MAGL), which metabolizes 2-AG. Cytochrome P450 (P450) enzymes may also contribute to eCB metabolism. LPI is broken down by various lysophospholipases (A, C, and D).
Figure 1.
Overview of the endocannabinoid system (ECS) in the brain. The endocannabinoid system (ECS) was uncovered through research investigating the molecular targets of key phytocannabinoids found in Cannabis sativa, particularly Δ9-tetrahydrocannabinol (Δ9-THC), the psychoactive component, and cannabidiol (CBD), a non-psychoactive compound. Both Δ9-THC and CBD interact with numerous targets within the brain, and here we focus on four key receptors: the cannabinoid receptors CB1 and CB2 (CB1R and CB2R), GPR55, and the transient receptor potential vanilloid type 1 (TRPV1) receptor. CB1R, CB2R, and GPR55 are G protein-coupled receptors (GPCRs) with seven transmembrane-spanning domains. While Δ9-THC acts as a partial agonist at these GPCRs, CBD’s pharmacological actions are more complex, often resembling negative allosteric modulation and weak partial agonism. CBD also activates and rapidly desensitizes the ionotropic TRPV1R, similar to capsaicin from chili peppers, but without the associated pungency. TRPV1R agonists are referred to as vanilloid ligands. These receptors are expressed across various brain cell types, including astrocytes, microglia, oligodendrocytes [52], glutamatergic neurons, GABAergic interneurons, and projection neurons (GABAergic, monoaminergic, and cholinergic), depending on factors like brain region, age, and neuropsychiatric conditions. While all four receptors are typically found in the cytoplasm—primarily in nerve terminals, dendrites, and cell bodies—there is substantial evidence for their intracellular localization. In addition to receptors, the ECS includes enzymes responsible for synthesizing lipid ligands that activate these receptors. One of the most well-studied eCBs, anandamide (N-arachidonoyl-ethanolamine or AEA), is synthesized from N-acylphosphatidylethanolamine (NAPE) via NAPE-specific phospholipase D (PLD). Several alternative pathways also contribute to AEA production. Diacylglycerol lipase α (DAGLα) is the primary enzyme that synthesizes 2-arachidonoyl-glycerol (2-AG), another major eCB. Both AEA and 2-AG activate all four receptors, though other ligands exhibit more receptor-selective actions. For example, N-arachidonoyl-dopamine (NADA), likely produced by fatty acid amide hydrolase (FAAH) in dopaminergic cells, acts as a hybrid agonist for CB1R and TRPV1R [53]. Similarly, L-α-lysophosphatidyl-inositol (LPI) and its congeners resemble classical eCBs but selectively activate GPR55. The activation of these receptors can influence virtually all functions of the brain cells expressing them, but their actions are highly context-dependent. The effects depend on factors such as receptor splice variants, heteromeric interactions with other receptors (e.g., TrkB, insulin receptor, or EGF receptor), the cell’s metabolic state and age, and the ontogenetic stage of the organism. Many receptor-mediated effects are tied to brain cell processes such as differentiation, maturation, migration, circuit formation, and plasticity, which are key topics in this review. Finally, after eCBs activate their receptors, they are primarily metabolized intracellularly by a variety of enzymes. The key enzymes for this review are FAAH and cyclooxygenase-2 (COX-2), which degrade anandamide, and monoacylglycerol lipase (MAGL), which metabolizes 2-AG. Cytochrome P450 (P450) enzymes may also contribute to eCB metabolism. LPI is broken down by various lysophospholipases (A, C, and D).
2. Cannabinoid Receptors and Brain Development
The involvement of ECS in embryogenesis starts from the very beginning, controlling gametogenesis, fertilization, oviductal transport, blastocysts development and implantation, entailing a fine-tuned regulation of CB1R and CB2R activity tightly controlled mainly by precise AEA levels at this early stage [54,55,56,57,58,59]. A precise tone of ECS was also shown to be required in normal trophoblast stem cells proliferation and differentiation [60,61,62,63], being involved in placentation via CB1R [63]. In the inner cell mass, embryonic stem cells express both CB1R and CB2R [64,65,66], significantly up-regulated with differentiation and associated with cell survival [64,67,68]. Mouse embryonic stem cells also express TRPV1R, but its role, if any, remains to be defined [66]. This increased expression of CB1R and CB2R along with differentiation is reflected in their involvement in cell lineage commitment and the development of the germinal layers [64,69]. Accordingly, it was shown in chick embryos that the exposure to Δ9-THC analogue, O-2545, at gastrulation impaired the formation of brain, heart, somite, and spinal cord primordia [70], corroborated by recent studies in zebrafish also showing that the exposure to Δ9-THC and/or CBD during gastrulation induces several later developmental defects including in nervous system development [71,72,73,74]. Such exposure induced alterations in neural plate formation and patterning indicating a most likely involvement of ECS in the neurulation process [70]. Interestingly, it was shown an interaction between cannabinoid signalling and morphogenetic factors [75,76], critical to nervous system partnering.
Such involvement of ECS from the earliest stages of nervous system development is supported by the identification of ECS components, both receptors and enzymes controlling the endocannabinoid metabolism, as well as the endocannabinoids 2-AG and AEA in the earliest stages of nervous system development. Both CB1R transcripts and protein were identified in the neural plate, during neurulation and onwards in chick embryos [77,78], as well as 2-AG and AEA, and the enzymes involved in their metabolism [78]. CB1R expression in such early stages of nervous system development was observed also in zebrafish [79,80] and rodents (from E7.5) [81]. GPR55 mRNA expression was also recently found at such early stages in zebrafish [80].
Particularly concerning the brain, CB1R mRNA can be detected in the mice telencephalon both in the pallium and subpalium from E11.5 [81,82,83,84], increasing their expression along with neuronal differentiation [80], as observed also in chick embryos [77,85], peaking at E16.5 [82,86]. Embryonic-derived neural progenitors in vitro display functional CB1R [87,88,89] and there is some evidence of mRNA expression in proliferative ventricular regions [84], but there is a consistent body evidence pointing to an absence or very low levels of CB1R protein in both ventricular and subventricular zones (VZ/SVZ) in the developing brain [82,83,90,91,92]. It has been identified CB1R immunoreactivity in intermediate precursor cells exiting subventricular zone [82], but there is clearly a robust increase in CB1R expression in post-mitotic neurons in the developing brain [82,83,90,92,93,94]. Similar pattern of CB1R expression in more differentiated cellular stages has been observed in human developing brain, detected as early as gestational week (GW) 9 [95], and more recently in the monkey, where it was observed a more intense immunoreactivity for CB1R in comparison with mice, but completely absent in the VZ/SVZ [92]. Accordingly, in developing cortex, CB1R immunoreactivity has been detected in mice at E12.5-E13.5 in the preplate in reelin-expressing Cajal-Retzius cells and newly differentiated glutamatergic neurons [83,90,94], also observed in developing human brain [95], and later on in postmitotic radial migrating principal neurons [92,94,96] and migrating interneurons [90,94,97,98]. A similar increase in CB1R expression with neural differentiation was also observed in human inducible pluripotent stem cells (IPSC)-derived organoids [99]. From E13.5, CB1R expression becomes transiently prominent in developing axons of pyramidal neurons in the intermediate zone (IZ; [94]), in particular in long-range corticofugal axonal tracts such as cortico-thalamic and cortico-spinal tracts [82,83,93,98,100], and perinatally in the afferent fibres cruising the brainstem and cerebellum [101,102]. Such transient prominent subcellular expression in developing axons is also observed in embryonic chicken [77,85], zebrafish [85] and rats [103]. A similar pattern of expression of CB1R in neuronal fibre tracts is observed in developing human brain [95,104,105,106]. Such transient cellular and subcellular distribution at cortical projection neurons fades in early postnatal life coincident with synaptic contact formation/stabilization [83,84,100]. CB1R is also present in developing cholinergic neurons [107].
The spatial-temporal dynamics in the cellular and subcellular expression of CB1R is accompanied by a precise spatial-temporal tone of endocannabinoids (eCB) controlling the activity of CB1R tightly regulated by a concomitant dynamic cellular and subcellular distribution of the enzymes controlling the metabolism of eCBs. While in early embryogenesis AEA seems to take a prominent role [58,108,109], at mid-late embryogenesis, in brain development, 2-AG gains relevance [93,108]. For instance, it has been elegantly shown the existence of a precise and concerted cellular and subcellular expression of DAGL and MAGL supporting a spatially restricted bioavailability of 2-AG necessary for the correct axonal guidance and growth of corticofugal axons [93,98,100,110] and development of cholinergic afferents [107,111].
CB2R has been also identified in embryonic-derived neural progenitors in vitro [112,113], supported by the observation of an increase in cell proliferation in E14.5 mice-derived cortical slices upon a selective activation of CB2R [88]. Interestingly, in opposition to the observed for CB1R, its expression decreases with differentiation [112]. Additionally, it has been provided evidence for its expression in retinal ganglion cells that project to the thalamus and midbrain [114] and functional evidence for CB2R expression in oligodendrocytes and their progenitors [115].
Regarding the other receptors able to sense eCBs, TRPV1 can be transiently expressed during the embryonic development in some brain regions [116] and prenatal capsaicin exposure in mice (E7-E13) has a behavioural outcome [117]. Yet, its eventual expression in developing brain remains elusive. In relation to GPR55, as aforementioned, it has been shown mRNA expression throughout the developing brain in zebrafish [80]. Functional evidence suggests its expression in retinal projections [118]. Yet, its presence in the developing brain also remains poorly defined.
2.1. Cannabinoid Receptors and the Development of Brain Cytoarchitecture
The development of brain cytoarchitecture encompasses the proliferation and differentiation of neurons and their migration to their final positions in an tightly-regulated manner, in order to attain a subsequent and proper brain wiring. Pharmacological or genetic manipulation of ECS interferes with brain cytoarchitecture in both the number and final position of different neuronal populations from glutamatergic [82,84,92,94,96,99,119] to GABAergic [28,120,121] or cholinergic neurons [107,111]. This may arise from a control of proliferation and/or neuronal migration and differentiation by ECS, for which has been provided evidence.
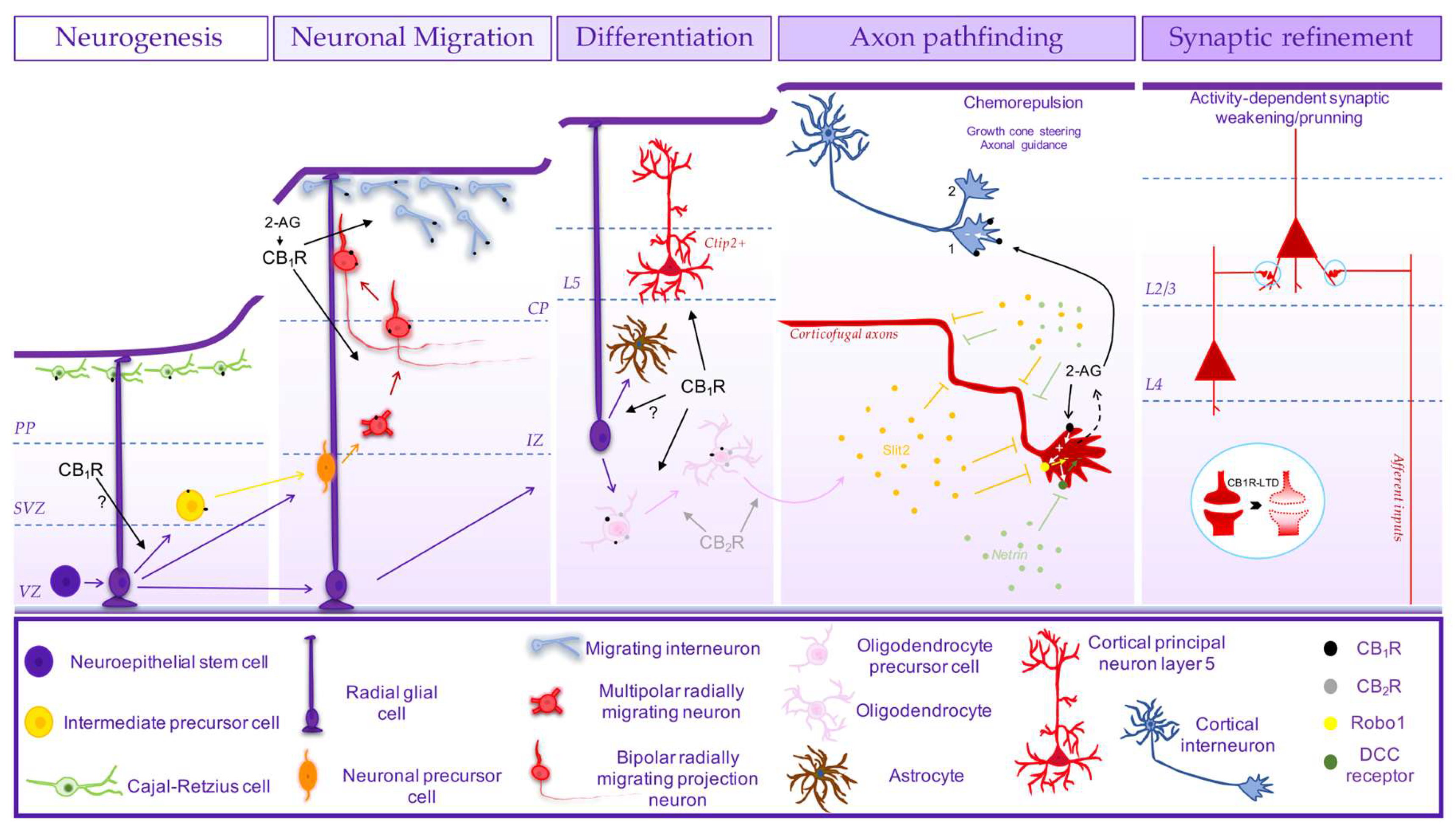
In vitro studies in cultured embryonic-derived neural progenitor cells (NPC) indicate that NPCs produce and release the two major eCB species, namely AEA and 2-AG [87], and pharmacological and genetic manipulation (KO mice) of both CB1R and CB2R showed that the activity of either CB1R or CB2R promotes proliferation of cultured NPCs derived from different embryonic brain regions [87,88,112,113,122,123]. Accordingly, the increase in the tonic activity of ECS by inhibition or deletion of FAAH induce an increase in NPC proliferation [87]. A CB2R-induced cell proliferation has been also observed in organotypic E14.5 mice-derived cortical slices [88]. In vivo, it has been shown that CB1R-KO mice display a reduced proliferation in the developing cortex [82,84,124] (Figure 2), hippocampus [125] and cerebellum [123]. Further evidence indicated that activation of CB1R promoted proliferation, inhibiting neuronal differentiation, as observed in vitro both in human neural stem cells [126] or cultured embryonic NPC [127]. In vivo, CB1R was also shown to control the generation of Tbr2+ intermediate precursor cells and its absence (CB1R-KO) leads to premature cell cycle exit [91]. This promotion of cell proliferation during development by CB1R may entail a bidirectional cross-talk with TNFα[122]. In contrast, WIN55,212-2 exposure during embryogenesis had no effect on cell proliferation [94]. Also, exposure of murine NPCs to AEA has also been shown to decrease proliferation [128] and in mouse neural stem cells, activation of CB1R favoured differentiation into neurons [89]. In fact, the evidence pointing for an absence or very low levels of CB1R in proliferative regions both in the ganglionic eminences in the subpallium [98] and in the developing cortex [92] led to question if the observed CB1R-mediated promotion of cell proliferation in vivo may be due to a direct action [92]. In addition, it should be noticed that GPR55 activation promotes both proliferation and differentiation of human neural stem cells [129], which needs to be further addressed to better understand the eventual contribution of GPR55 to the role of ECS in neurogenesis.
More consistent body of evidence supports the involvement of ECS in neuronal migration and differentiation of post-mitotic neurons, in line with the increased expression of CB1R along differentiation [77,82], which contributes to the development of cytoarchitecture. Interference with the ECS by prenatal exposure to cannabinoids or genetic manipulation of CB1R affects brain cytoarchitecture (e.g., [130,131]), both excitatory [82,84,92,94,96,99,119,132] and inhibitory [28,120,121]. CB1R-KO mice display at P2.5 a different distribution pattern of cortical projection neurons labelled with BrdU at E14.5, presenting a higher number of cells at deeper layers and lower at superficial layers [82]. Accordingly, while pharmacological activation of CB1R accelerate radial migration, overexpression of the FAAH enzyme inhibits radial migration [82]. Such tonic action of ECS through CB1R in radial migration was later reinforced by the observation that the knockdown of CB1R at E14.5 in mice by in utero electroporation of plasmids encoding siRNAs induced an accumulation of migrating neurons in the IZ and consequently a lower number of cells reaching the cortical plate (CP) at E17.5 [96] (Figure 2). This resulted in an increase in the number of cells at the deeper layers and a decrease in the upper layers at P2 and P10 [96], similar to the observed in the CB1R-KO mice [82], indicating for a delay in radial migration in the absence or reduced levels of CB1R in post-mitotic neurons [96]. Morphological analysis of radially migrating neurons in CB1R-KO mice revealed that these neurons at the IZ display deviations in their vertical orientation with misoriented processes, suggesting for a role of CB1R in correct cell movement from the IZ to the CP [92]. In this regard, it should be mentioned the reported ability of CB1R, endogenously activated by 2-AG, to increase neuronal motility of E14.5 mice-derived NPC, increasing the frequency of bursts of movement, while reducing their turning frequency [133]. Besides an eventual control of movement, at the IZ, migrating neurons need to polarize, undergoing a multipolar-bipolar transition [134,135], forming a leading process (future apical dendrite) oriented towards the CP and a trailing process (future axon) growing orthogonally to the radial migration direction, in the transition from the lower to the upper IZ [136], necessary for subsequent radial migration towards the CP [137,138], through glial fibre–dependent guidance. While radial glial scaffold seems not to be affected by CB1R activation [94], CB1R-KO mice at the IZ display a considerable low percentage of cells with a bipolar morphology in comparison with wild-type mice embryos [92]. This indicates that CB1R may be affecting radial migration at the IZ-CP transition eventually by controlling neuronal polarization and/or through the well-established control of axon formation/outgrowth (see next section; Figure 2). In addition, by controlling neuronal differentiation of glutamatergic neurons [82], CB1R is also involved in cortical projection neurons distribution across the different cortical layers, in particular by controlling the differentiation/maturation of deep cortical layer 5 pyramidal neurons [84,99,119]. While genetic ablation of CB1R in post-mitotic cortical projection neurons reduced the number of sub-cerebral projection neurons of layer 5 (Ctip2+) and a consequent decrease in cortical thickness, FAAH-KO mice displayed a higher number of Ctip2+ cells [84]. Interestingly, interfering with this tonic action induced by 2-AG via CB1R by prenatal exposure to Δ9-THC in mice between E12.5-E16.5 also reduced the number of neurons in layer 5 [119]. Such balanced CB1R activity drives the generation of deep layer Ctip2+-neurons by preventing Satb2-mediated repression, increasing Ctip2 expression [84] (Figure 2). These observations performed in mice were more recently recapitulated in human IPSC-derived brain organoids [99]. The dysregulation of CB1R-mediated generation of sub-cerebral projection neurons leads to long-term impairments in corticospinal motor function [84,119]. ECS may also affect early born cortical projection neurons placement through CB1R expressed in Cajal-Retzius cells [83,90,94], which contribute to guide early-born post-mitotic glutamatergic neurons through the expression of reelin [136,139,140], since CB1R controls the number of Cajal-Retzius cells [94] (Figure 2).
As already mentioned, ECS also controls the development of inhibitory cytoarchitecture, since prenatal Δ9-THC exposure or genetic deletion of CB1R (KO mice) affect the number of different types of interneurons [28,120,121]. This seems to reflect on one hand a CB1R-mediated control of tangential migration of interneurons, since WIN55,212,2 exposure from E5 in rats induced and increase in the number of GABA cells tangentially migrating in the marginal zone [94]. This should entail a chemoattract action of ECS through CB1R activation on migrating interneurons, as chemotaxis of cholecystokinin+-interneurons by CB1R was observed in vitro through the transactivation of TrkB receptors [28], previously shown to be involved in the tangential migration of medial ganglionic eminence-derived cells [141] (Figure 2). Such interplay between ECS and BDNF may also be involved in radial migration (see [142]). Moreover, there is also functional interplay between neuregulin-1, which is a major chemoattractant of cortical tangentially migrating interneurons [143] and ECS. Neuregulin-1 downregulates MAGL expression leading to enhanced 2-AG signalling [144] and it was observed a cross-talk between neuregulin-1 and ECS in the control of movement of cortical embryonic neuroblasts [133]. This opens the possibility of ECS to be also controlling the guidance/movement in tangential migration through an interaction with neuregulin-1. Moreover, while principal neurons are endowed with the capacity of eCB synthesis during their development, self-sustaining ECS [82], GABAergic interneurons seem to lack synthetic enzymes until the switch to radial intracortical migration [98], being most likely attracted by paracrine guidance by target-derived eCBs. This suggests that ECS may play a role in the integration of the excitatory and inhibitory cytoarchitecture. Furthermore, ECS is also involved in the differentiation/maturation of GABAergic neurons, also shown to involve the activity of TrkB receptors [28].
In addition to the control of differentiation of glutamatergic and GABAergic neurons, ECS also contributes for the differentiation of cholinergic neurons [107,111]. Cell autonomous DAGLα-derived 2-AG signalling via CB1R controls spatial organization and morphogenesis of cholinergic neurons with an impact in cholinergic basal forebrain projections. This is under the control of nerve growth factor (NGF) trough TrkA receptors by regulating 2-AG spatial availability through the control of MAGL subcellular levels [107,111].
ECS is also involved in gliogenesis. In cultured neuronal progenitor cells derived from P2 rat cortices, the pharmacological activation of CB1R increased the generation of GFAP+-cells [125]. In vivo, CB1R-KO mice displayed a decrease in astrogliogenesis and an increase in neurogenesis in rat developing hippocampus postnatally (P15), in contrast to a CB1R-induced neuronal commitment observed prenatally (e.g., [82,84,89]). More consistent body of evidence supports a role of ECS in oligodendrogenesis. 2-AG produced by cultured rat-derived oligodendrocyte precursors (OPC) expressing DAGLα and DAGL, and CB1R and CB2R [146], promoted both OPC survival [146], proliferation [147] and oligodendrocytes differentiation [145] via CB1R or CB2R, through PI3K/Akt and mTOR signalling [146,147,148] (Figure 2). In vivo, while the postnatal (P1-P15) activation of CB1R in rats induced an increase in oligodendrocyte cell commitment, CB2R was more associated with migrating OPCs [149]. Yet, only the activation of both CB1R and CB2R increased the expression of myelin basic protein in subcortical white matter [149]. Accordingly, postnatal Δ9-THC exposure (P6-P9) in mice increases the density of mature myelinating oligodendrocytes in subcortical white matter decreasing OPC by inducing OPC cell cycle exit, while promoting oligodendrocyte differentiation, effects prevented by selective antagonists of CB1R or CB2R [150]. Likewise, in mice, at late embryogenesis, ECS also promotes oligodendrocyte differentiation since the inhibition of MAGL in vivo lead to premature differentiation of oligodendrocytes, although only via CB2R and not CB1R [115] (Figure 2).
2.2. Cannabinoid Receptors and the Development of Brain Circuitry
ECS is involved in the development of brain circuitry, not only by governing the development of cytoarchitecture, but also through an involvement in the axonal pathfinding for the formation of synaptic connectivity and their maturation/refinement.
As mentioned, during brain development CB1Rs display a predominant expression in developing axons [151], in particular in distal segments and growth cones as observed in diverse neuronal types such as glutamatergic [82,83,84,93,98,100,152], GABAergic [98] or cholinergic neurons [107,111]. Activated by target-derived 2-AG or produced by DAGL located in the axonal tips [82,93,98,100,111,114] and spatially limited to the motile growth cones by MAGL located at proximal axonal segments [93,107,111], CB1R promotes axonal development by controlling their directional growth [85,93,100,107,110,111,115,153]. This achieved by a chemorepulsion action of CB1R at the actin-rich growth cone, including motile filopodial extensions, driving growth cone steering [93,98,107,115,152]. Accordingly, the genetic or pharmacological manipulation of CB1R or DAGL or MAGL has been shown to have an impact in the development of axons and correct axon pathfinding from diverse neuronal populations.
In developing chick embryos or zebrafish, the genetic knockdown or pharmacological blockade of CB1R impairs axonal growth, guidance and fasciculation [85,154]. In mammals, in agreement with the observed transient expression of CB1R in white matter tracts in long-range corticofugal developing axons at mid-late embryogenesis [82,83,90,98,103,105,106,155], the genetic deletion of CB1R selectively in post-mitotic cortical projection neurons impaired axon fasciculation of corticofugal axons due to impaired axon pathfinding [82], both corticothalamic [100] or corticofugal tracts [84] (Figure 2). Interestingly, CB1R-KO mice display aberrant fasciculation and misrouting not only of corticothalamic axons (CTA), but also of thalamocortical axons (TCA)[100]. Taking into account that CTAs express CB1R, whereas TCAs do not, but express MAGL and DAGL [93,100], these findings indicate that CB1R signalling in CTA, triggered by tightly spatially-regulated availability of 2-AG, is involved not only in the development of CTA, but also in the partnering of TCA, mediating the reciprocal fasciculation of afferent and efferent cortico-thalamic projections [93,100]. Indeed, CB1R-KO mice display a significant increase in the innervation by thalamocortical axons of cortical layers 2/3 [156], although it may entail also activity-dependent mechanisms (see below). In agreement with a role of CB1R in the development of long-range axonal projections, in utero exposure to Δ9-THC also impairs corticofugal tracts [119,132]. In contrast, MAGL inhibition triggered corpus callosum enlargement due to corticofugal axon spreading [115]. Moreover, it was elegantly shown that ECS guides corticofugal axons by a concomitant CB1R-induced Robo1 positioning at the growth cones and a CB2R-induced production of Slit2 by oligodendrocytes inducing a chemorepellent signal [115] (Figure 2). Concomitantly, it may be involved in myelination of these fibres, as Δ9-THC exposure enhanced subcortical white matter myelination in a CB1R and CB2R dependent manner [150].
The development of retinal projections in mice also entails CB1R-driven guidance by controlling growth cone steering [152]. This was show to be mediated by the regulation of the trafficking of deleted in colorectal cancer (DCC) receptor, which tethers netrin-induced growth cone steering, in a PKA-dependent manner [152]. A similar mechanism was observed in cultured cortical neurons [152]. In vivo, while the activation of CB1R reduced retinal projection growth, its blockade promoted growth and caused aberrant projections [152]. Later on, it was shown that CB2R is also expressed in retinal ganglion cells growth cones and engaged in the development of their axons also by controlling growth cone morphological changes through a similar mechanism [114]. Likewise, genetic deletion or pharmacological blockade of CB2R increased retinal axonal length, aberrant projections, affecting retino-thalamic projections [114]. GPR55 was also shown to regulate retinal axon growth and guidance. Pharmacological activation of GPR55 increases the surface area and filopodia in growth cones, inducing retinal axon growth [118]. In vivo, GPR55 activation leads to aberrant retinal ganglion cells projections affecting target selection [118].
ECS also controls axon pathfinding of cortical GABAergic interneurons through CB1R [98]. While eCBs were show to be chemoatractants in interneuron migration [28], they control their axonal guidance by inducing growth cone collapse through CB1R activation, most likely by a target-derived 2-AG, as suggested by a downregulation of DGAL with GABAergic differentiation [157] and by the observed dendritic redistribution of DAGL in glutamatergic pyramidal cells at late embryogenesis [98]. CB1R also controls cholinergic innervation of the hippocampus. CB1R activated by cell-autonomous 2-AG signalling produced by DAGL, co-located with CB1R at the growth cones, and spatially restricted to the motile segments by MAGL selectively located at the proximal axonal stems, facilitates outgrowth of cholinergic afferents, inhibiting growth cone differentiation, while controlling their guidance, eventually by 2-AG paracrine signalling [107,111]. This role of CB1R signalling in cholinergic axon pathfinding was shown to be regulated by NGF [111]. More recently, the observation that CB1R-KO-mice display an impaired striatonigral connectivity suggests for a role of CB1R also in axonal pathfinding of striatal neurons onto dopaminergic neurons in the substantia nigra [158]. Concerning TRPV1 receptor, the observation that temperature-induced axonal repulsion in rat cortical neurons is mediated by TRPV1 [159] suggest that it may also be involved in axonal pathfinding, yet its role remains ill defined.
The chemorepellent signalling induced by CB1R and controlling axon guidance was first shown to involve RhoA activation and subsequent ROCK activation in GABAergic interneurons [98] (Figure 2). In cultured rat hippocampal neurons and organotypic slices, this was show to induce non-muscle myosin II dependent contraction of the actomyosin cytoskeleton, leading to actin-rich growth cone retraction, a mechanism shown to be required for the correct pathfinding of corticofugal neurons [160]. Moreover, in mice developing cortical neurons, CB1R-induced growth cone collapse was shown to entail a deactivation of Rac1 leading to F-actin disassembly, being proposed that CB1R induces the retraction of filopodia by Rac1 deactivation and of lammelipodia by RhoA activation [161]. In fact, both DCC trafficking and Slit-Robo pathway shown to be involved in CB1R-mediated growth cone repulsion in retinal ganglion cells and cortical neurons [115,152] have been associated with RhoA [162] or Rac [163]. Hence, similar intracellular mechanisms seem to be engaged by CB1R to induce growth cone collapse in different neuronal populations. Concomitantly, CB1R may be also able to control microtubule stability by regulating superior cervical ganglion 10 (SCG10)/stathmin-2 protein [132], involved in microtubule disassembly [164]. Furthermore, the targeting of CB1R to axonal growth cones, namely in corticofugal axons, was recently shown to be mediated by kinesin-1 [165]. The genetic deletion of kinesin-1 leads to abnormal fasciculation and pathfinding defects of corticofugal axons with a reduction in CB1R levels [165]. When the axon reaches their post-synaptic target, there is a cellular and subcellular redistribution of the ECS components. Essentially, MAGL accumulates in growth cones, limiting 2-AG signalling, most likely decreasing the growth cone motility, allowing presynapse differentiation, keeping a presynaptic location [93], whereas DAGL is targeted to postsynaptic dendritic spines [82,98,110] for retrograde signalling.
Regarding synaptogenesis per se, in cultured rat hippocampal neurons, the pharmacological activation of CB1R inhibited synapse formation [166]. The inhibition of tonic activity of CB1R by DAGL inhibition induced an increase in synaptogenesis in cultured cortical neurons [82]. An increase in synaptogenesis was also observed in a cortical spheroid model of human brain development [167]. In vivo, the genetic deletion of CB1R in cortical interneurons led not only to an increase of inhibitory synaptic contacts at cortical pyramidal cells, but also to an altered synaptic distribution [98]. Likewise, genetic deletion of DAGLα impairs cholinergic afferents in the hippocampus, but mainly their targeting and not their density [111]. Hence, ECS and CB1R seems to contribute to the development of synaptic contacts mainly through the morphological guidance towards their postsynaptic target, rather than a direct role in structural formation of synapses. In spite of this, recent evidence indicates that CB1R can contribute to synapse formation and stabilization, but in an activity-dependent manner [168]. In mice organotypic slices, it was shown that exogenous activation of CB1R induces the formation and stabilization of inhibitory boutons at principal neurons, independently of neuronal activity [168]. However, physiologically, this is triggered in locations of strong excitatory input, entailing postsynaptic 2-AG production and activation of CB1R at inhibitory axons, most likely to tune excitation/inhibition balance [168,169]. Furthermore, ECS through CB1R is also involved in synaptic circuitry refinement in an activity-dependent manner. CB1R-KO mice display altered circuitry in primary somatosensory cortex [131,170] and visual cortex [130], most likely due to the deletion CB1R at glutamatergic neurons [131]. This may reflect in part the role of CB1R in the development of cytoarchitecture and axon pathfinding. However, it seems also to rely on the control of synaptic pruning by CB1R through the induction of long-term depression (LTD) (Figure 2). In mouse visual cortex, the blockade of CB1R during brief monocular deprivation prevented experience-dependent synaptic weakening selectively at L2/3, by blocking CB1R-induced LTD [171]. Likewise, in rodent primary somatosensory cortex, CB1R-LTD is also required not only for weakening of deprived sensory inputs in L2/3, but also of L4-L2/3 synapses [172], previously shown to display a CB1R-dependent LTD [173,174] (Figure 2). This may contribute to normal circuit development, since CB1R blockade disturbed whisker map formation [172]. CB1R expressed in TCA-L2/3 synapses controls their synaptic pruning through the ability to induce LTD [156]. In rat prefrontal cortex (PFC), while the blockade of CB1R during adolescence of female rats seems to prevent the occurrence of pruning of glutamatergic synapses [175], Δ9-THC exposure induced a decrease in spine density at L2/3 pyramidal neurons [175,176], as well as impairment of eCB-mediated LTD [175]. Accordingly, early onset consumers of marijuana during adolescence display thicker cortex, possible due to disrupted synaptic pruning [177]. CB1R may also contribute to synaptic pruning by mediating hetero-LTD as observed in L2/3 of mice visual cortex [178] and developing hippocampal CA1 area in rats [179] in the first two postnatal weeks. CB1R may also interfere in circuitry development and maturation by controlling the excitatory-inhibitory switch of GABAergic signalling, since Δ9-THC exposure in postnatal days 1-10 caused a delay in this switch via CB1R [180].
In addition to glutamatergic, GABAergic or cholinergic signalling, ECS may also be involved in the development of other neurotransmitter signalling systems, as suggested by studies showing that the exposure to cannabinoids perinatally can also affect for instance dopaminergic (e.g., [181,182,183]) or serotoninergic [184,185] systems.
Figure 2.
Schematic representation of the involvement of endocannabinoid signalling system (ECS) in corticogenesis. ECS through CB1R may be involved in cortical cell proliferation [82,84,124] and intermediate precursor cell generation [91]. CB1R is expressed in Cajal-Retzius cells and may control early born cortical projection neurons positioning [83,90,94]. CB1R is involved in radial migration [82] in the transition from the intermediate zone (IZ) towards the cortical plate [96] by controlling the neuronal polarization [92] and eventually through the control of cell movement [92,133], controlling the distribution of neurons across the different cortical layers [82,96]. CB1R also regulates tangential migration of cortical interneurons [28,94]. ECS, via CB1R, is also engaged in the development of cortical excitatory cytoarchitecture by controlling the differentiation of cortical projection neurons of layer 5 (Ctip2+) [84,99,119]. Next, ECS controls the guidance of corticofugal axons [82,84,93,100,115] by regulating growth cone steering through autocrine signalling by 2-AG via CB1R at the growth cones, through the regulation of Robo1 receptor and the concomitant CB2R-induced release of Slit2 by oligodendrocytes [115], whose differentiation was shown to entail CB1R and CB2R [115,146,147,148,149]. ECS may also control axon pathfinding through the regulation of the trafficking of deleted in colorectal cancer (DCC) receptor, which tethers the action of the guidance cue netrin [152]. 2-AG signalling through CB1R also controls growth cone steering of cortical interneurons via RhoA activation [98]. ECS is later involved in cortical synaptic refinement by controlling synaptic weakening/pruning through CB1R-mediated long-term depression (LTD), observed in afferent inputs at layer 2/3 and layer 4-layer 2/3 synapses [156,171,172,173,174,175,176].
Figure 2.
Schematic representation of the involvement of endocannabinoid signalling system (ECS) in corticogenesis. ECS through CB1R may be involved in cortical cell proliferation [82,84,124] and intermediate precursor cell generation [91]. CB1R is expressed in Cajal-Retzius cells and may control early born cortical projection neurons positioning [83,90,94]. CB1R is involved in radial migration [82] in the transition from the intermediate zone (IZ) towards the cortical plate [96] by controlling the neuronal polarization [92] and eventually through the control of cell movement [92,133], controlling the distribution of neurons across the different cortical layers [82,96]. CB1R also regulates tangential migration of cortical interneurons [28,94]. ECS, via CB1R, is also engaged in the development of cortical excitatory cytoarchitecture by controlling the differentiation of cortical projection neurons of layer 5 (Ctip2+) [84,99,119]. Next, ECS controls the guidance of corticofugal axons [82,84,93,100,115] by regulating growth cone steering through autocrine signalling by 2-AG via CB1R at the growth cones, through the regulation of Robo1 receptor and the concomitant CB2R-induced release of Slit2 by oligodendrocytes [115], whose differentiation was shown to entail CB1R and CB2R [115,146,147,148,149]. ECS may also control axon pathfinding through the regulation of the trafficking of deleted in colorectal cancer (DCC) receptor, which tethers the action of the guidance cue netrin [152]. 2-AG signalling through CB1R also controls growth cone steering of cortical interneurons via RhoA activation [98]. ECS is later involved in cortical synaptic refinement by controlling synaptic weakening/pruning through CB1R-mediated long-term depression (LTD), observed in afferent inputs at layer 2/3 and layer 4-layer 2/3 synapses [156,171,172,173,174,175,176].
3. Cannabinoids and the Adolescent Brain
Adolescence represents a period of profound neurodevelopment, marked by structural and functional changes within the brain’s cytoarchitecture and synaptic circuitry, particularly in PFC, a region critical for executive functions, decision-making, and impulse control [186,187]. This transition from childhood to adulthood involves the maturation of several brain regions, particularly the PFC, amygdala, and hippocampus, which regulate executive functions, emotions, and learning. These areas undergo extensive synaptic pruning, myelination and circuit refinement, making adolescence a sensitive window for both adaptive and maladaptive plasticity [188,189]. These fine-tuning processes eliminate redundant or weak synapses and facilitate signal transmission across brain circuits, especially those related to cognitive and emotional regulation [190]. Human imaging studies reveal significant reductions in gray matter volume in the PFC and temporal lobes during adolescence, consistent with synaptic pruning observed in animal models [191,192]. White matter increases, attributed to enhanced myelination, have also been documented in regions such as the corpus callosum and other subcortical areas [193,194]. These structural changes reflect a shift toward more efficient neural processing and enhanced cognitive control, with notable improvements in functions such as working memory, impulse control, and decision-making [195,196]. However, this ongoing synaptic and circuit refinement opens a critical window during which external factors such as substance use can significantly influence brain development [190].
Adolescence also coincides with increased risk-taking behaviours, emotional instability, and heightened social influence, potentially leading to drug experimentation, including cannabis use [187,197]. The developing brain is particularly vulnerable to cannabis exposure, which has been associated with various negative outcomes, including impaired cognitive function, increased risk of psychiatric disorders, and long-lasting changes in brain structure [198,199]. Adolescence is a critical period when both the dopaminergic system and the ECS take centre stage in PFC development [200]. The susceptibility of the adolescent brain to such effects is thought to stem from the intricate roles of the ECS in the ongoing brain maturation. During adolescence, the ECS undergoes dynamic changes, with peaks in CB1R expression and endocannabinoid ligand levels observed in the PFC and hippocampus [201,202,203,204] (Table 1). These fluctuations make the adolescent brain highly sensitive to perturbations in ECS, including those induced by exogenous cannabinoids such as Δ9-THC [199]. Both human and animal research demonstrate that adolescent cannabis exposure results in persistent changes to brain structure, function, and behaviour. These changes increase the risk of psychiatric disorders, including anxiety, depression, and schizophrenia, and result from the disruption of normal ECS during a critical period of brain development. Understanding how cannabis use during adolescence affects the maturation of the ECS and related neural circuits is critical for developing interventions to mitigate its long-term consequences [205,206].
3.1. Animal Studies on the Role of the Endocannabinoid System in the Adolescent Brain
Adolescence is also crucial period for the rodent brain development, characterized by dynamic changes in corticolimbic structures [207]. These regions, including the PFC, amygdala, and hippocampus, are involved in regulating emotional behaviours such as fear, anxiety, and executive function. The ECS plays a central role in controlling the orchestration and the function of these circuits, primarily through the CB1R [208]. Rodent studies have revealed that the ECS undergoes significant developmental changes during adolescence. The ECS regulates the balance between excitatory and inhibitory neurotransmission, which is crucial for the maturation of synaptic connections and the refinement of corticolimbic circuits [208,209]. The expression of CB1Rs peaks at the onset of adolescence, especially in the PFC and striatum, before declining into adulthood [210]. In adolescent rats, Molla et al. (2024) found that the ECS was not yet fully engaged to regulate afferent transmission from these brain regions [211]. By late adolescence, however, both 2-AG and anandamide could be recruited to limit hippocampal drive, although only 2-AG inhibited basolateral amygdalar inputs. The protracted development of the ECS in the PFC and its fluctuating developmental trajectory in other corticolimbic regions may leave the adolescent brain particularly vulnerable to disruptions by cannabis exposure during this critical window of development [201,211].
These vulnerabilities can be assessed in adolescent rodents exposed to cannabinoids, as this experimental paradigm recapitulates key behavioural and structural alterations that are often found in regular cannabis consumer adolescents [205,206,208]. The following animal studies unanimously indicate that perturbations in ECS signalling during adolescence, whether through stress or exogenous cannabinoid exposure, can result in long-lasting effects on emotional regulation and cognitive processing [212]. In the rodent brain, significant cellular and molecular alterations can be found after cannabinoid exposure, particularly in the PFC, hippocampus, and other corticolimbic areas. Importantly, these are brain areas critical for memory and cognition. Chronic exposure of adolescent rodents to Δ9-THC or synthetic CB1R agonists has been shown several times to cause long-term impairments in tasks such as short term memory, object recognition, spatial working memory, social interaction memory, and affective functions [205,213]. These effects are associated with changes in proteins involved in synaptic plasticity (e.g., PSD95, NMDA receptors), abnormal firing patterns of pyramidal neurons, reduced dendritic complexity especially of the pyramidal neurons in layer 2/3 in the medial PFC (mPFC) and reduced hippocampal connectivity, together with the downregulation and desensitization of CB1Rs in various brain regions, with a more pronounced effect in females. This is likely due to dynamic and sexually dimorphic changes in the expression and molecular pharmacology of CB1Rs during adolescence, especially in regions involved in cognition and emotional regulation [206,208].
Indeed, Bernabeu et al. (2023) reported how synaptic plasticity, particularly eCB-LTD, exhibits sex-specific differences during adolescence [214]. While other forms of plasticity like long-term potentiation (LTP) and mGluR-LTD are already mature in both sexes by adolescence, eCB-LTD is expressed early in females, but only appears at puberty in males. This study also found greater synaptic levels of CRIP1a (a CB1R-interacting protein that reduces CB1R signalling via G proteins) and ABHD6 in juvenile males, which likely contributed to the repressed eCB signalling as compared to juvenile females. Additionally, this milestone study systematically analysed the expression of other elements of the eCB system across both sexes of juvenile, pubescent and adult rats, and they found significant and likely meaningful age- and sex-dependent changes in the expression of the CB1R, CB2R, TRPV1R, DAGLα, MAGL, NAPE-PLD, FAAH and mGluR5 (the activity of the latter is associated with retrograde 2-AG release - see above). These findings highlight that synaptic plasticity in the PFC is not uniform across sexes or developmental stages. The differences were specific to the PFC and were not observed in other brain regions like the nucleus accumbens, supporting the notion that the PFC is one of the last regions to mature (Table 1).
In conclusion, the findings of Bernabeu et al. (2023) underscore the critical role of the ECS in adolescent brain development and the long-term impacts of early cannabinoid exposure [214]. Adolescence is a period of heightened vulnerability to changes in synaptic plasticity, and sex-specific differences in ECS function may shape how the brain responds to cannabinoid agonists during this crucial developmental window. In line with this affirmation, adolescent rodents exposed to cannabinoids showed impaired maturation of the glutamatergic and GABAergic systems, in particular, abnormal glutamate receptor distribution and altered inhibitory/excitatory balance. At the ultrastructural level, disrupted normal patterns of synaptic pruning, reduced dendritic spine density and alterations in dendritic length and remodelling were observed in the hippocampus and PFC of adolescent rodents subject to cannabinoid agonist exposure [206,208]. Synaptic maturation is critically dependent on intact glial cell functioning, however, adolescent cannabinoid agonists exposure can also modulate the function of diverse glial cell types. There are several studies reporting changes in astrocytic markers (GFAP) and microglial morphology, contributing to neuroinflammation and abnormal synaptic pruning during brain maturation. These alterations lead to worsened working memory, cognitive flexibility and spatial recognition tasks, which is translated into persistent impairments in executive functions and decision-making [205,206] (Table 1).
The role of microglia in adolescent brain development is far from fully appreciated. Lee et al. (2022) examined the effects of adolescent low-dose Δ9-THC exposure on microglial function and the broader ECS, particularly focusing on how Δ9-THC disrupts microglia’s homeostasis and impairs their responses to microbial infection and social stress into young adulthood [215]. Repeated low-dose Δ9-THC exposure during adolescence induced a state of dyshomeostasis in microglia isolated from the brains of male and female mice. This was evident from broad alterations in the expression of genes critical to microglial homeostasis, such as those related to innate immunity (e.g., Il-1β, Il-6, Tlr2-9). The observed dysfunction persisted into early adulthood (postnatal day 70), but returned to baseline at full maturity (postnatal day 120), thus revealing a critical period in adolescence where Δ9-THC can significantly disrupt microglial function, which in turn could influence brain health during crucial developmental windows. The study of Lee et al. (2022) also showed alterations in the ECS upon repeated Δ9-THC exposure, particularly in microglial cells [215] (Table 1). This includes increased in FAAH and a decrease in NAPE-PLD and MAGL expressions. These perturbations imply an enduring change in anandamide and 2-AG signalling, contributing toward the altered immune response and microglial dysregulation. In addition to immune dysregulation, adolescent Δ9-THC exposure caused impairments in the response to psychosocial stress (social defeat paradigm). Normally, social stress would induce anxiety-like behaviours and an immune response, but Δ9-THC-exposed mice showed a blunted response, suggesting a diminished capacity to handle stress. This further points to long-term effects on the brain’s neuroimmune interface and stress-processing pathways. As already expected from the above studies, sex differences were also observed, because male mice showed more pronounced changes in microglial morphology, while both sexes exhibited reduced cytokine responses post-Δ9-THC exposure. Surprisingly, these pathological changes were fully abolished by peripheral CB1R blockade, suggesting that peripheral CB1Rs, potentially on circulating monocytes, may play a key role in mediating Δ9-THC’s impact on microglia, highlighting a potential cross-talk between the central and peripheral immune systems [215] (Table 1).
However, the impact of cannabinoid agonists on microglia, especially those that are selective for the CB2R, can be positive too. For instance, it is known that chronic alcohol exposure (CAE) during late adolescence increases anxiety-like behaviours, especially during withdrawal, which may persist into adulthood. These effects are linked to neuroinflammation in the PFC. Li et al. (2023) found that CAE triggers the activation of microglia which displayed deramification (retraction of their processes) and cell body enlargement [216]. These changes are often linked to a transition from a homeostatic (M2-like) to a pro-inflammatory (M1-like) state, which is characterized by the secretion of pro-inflammatory cytokines like IL-1β and TNF-α. These cytokines are involved in synaptic pruning and may damage neuronal circuitry. The authors also found that CAE increased CB2R density in PFC microglia, and CB2R activation by its selective agonist AM1241 that does not bind CB1R, prevented CAE-induced anxiety-like behaviours, mitigated microglial activation by reducing their pro-inflammatory M1-like phenotype, restored normal microglial morphology and reduced the secretion of inflammatory cytokines [216]. It suppressed NLRP3 inflammasome activation, which is critical in promoting inflammation through the caspase-1/IL-1β pathway. Altogether, these findings suggest that CB2R activation offers a potential therapeutic strategy for treating alcohol-induced neuroinflammation and related mood disorders such as anxiety in late adolescence (Table 1).
Exposure to alcohol and stress is increased during adolescence in many human societies, and often negatively impact brain development in synergism [187]. A recent investigation shed light on the role of hippocampal CB1R in impulsivity and alcohol abuse during adolescence [217]. This report demonstrate that adolescent rats exhibit more impulsive choices and consume more alcohol than adults – behaviours that are associated with elevated CB1R expression in the CA3 and dentate gyrus (DG) regions of the adolescent hippocampus. These findings support the notion that CB1Rs in the this brain area plays a significant role in mediating impulsive behaviours and substance-seeking tendencies, further emphasizing the involvement of ECS in adolescent brain maturation. Besides the CB1R, the role of TRPV1Rs in mediating stress responses is also implicated in adolescence, suggesting that ECS dysregulation during this critical period may lead to long-term vulnerability to stress-related disorders [214]. In concert with this, another study in adolescent mice found that CAE impairs CB1R-dependent synaptic plasticity (eCB-LTD) in the DG medial perforant pathway (MPP-LTD) [218]. Furthermore, environmental enrichment (EE) rescued eCB-LTD, and additionally, in the control mice, EE reverted the eCB-LTD into a novel form of TRPV1R-dependent LTP (MPP-LTD to MPP-LTP switch). In conclusion, the study provides evidence that EE influences different synaptic plasticity pathways involving the CB1R and the TRPV1R in the hippocampus, potentially offering therapeutic strategies to counteract the cognitive deficits induced by adolescent alcohol exposure [218] (Table 1).
Actually, the CB1R and the TRPV1R have been demonstrated to exert opposing effects on anxiety, the former being anxiolytic, the latter anxiogenic [219]. Hence, simultaneous blockade of FAAH and TRPV1R blockade may be an interesting tool to be explored in anxiety disorder in adolescents. Nevertheless, stress, fear and anxiety-related behaviours are difficult to dissociate from one another in animal models, where they have been shown particularly sensitive to CB1R modulation, during adolescence [220,221]. In animal models, cannabinoid exposure produces mixed outcomes regarding anxiety, with some studies reporting anxiolytic effects while others show increased anxiety. CB1R activation has been shown to reduce fear and anxiety responses by dampening excitatory inputs in the PFC and amygdala, thereby promoting emotional regulation [222]. Others shown enduring increases in anxiety-like behaviours and dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis in adulthood [208,223] (Table 1).
Disruption of ECS during adolescence also impairs the maturation of fear extinction circuits, leading to persistent deficits in the ability to regulate anxiety and fear responses in adulthood [212,224]. Such findings underscore the importance of the ECS in modulating brain plasticity and emotional development during this critical period. Chronic Δ9-THC exposure in adolescent rats reduced dendritic complexity and synaptic density, especially in regions associated with executive function and emotional regulation [206]. This reduction in synaptic strength is accompanied by behavioural deficits, such as increased impulsivity and impaired decision-making [211]. Δ9-THC exposure during adolescence has also been associated with depressive-like behaviours, including passive coping strategies and anhedonia. Additionally, adolescent exposure to natural and synthetic cannabinoids affects the mesolimbic dopamine system, probably due to the presence of cannabinoid receptors in both dopaminergic cells and their input terminals [2], further exacerbating decision-making impairments [200,213,225].
One might wonder not only whether chronic alterations in ECS signalling during adolescence shape stress- and anxiety-related behaviours later in life, but also whether stress itself influences the ECS in the adolescent brain, creating a reciprocal relationship between stress exposure and ECS modulation during this critical developmental period. Indeed, Demaili and colleagues (2023) recently reported that early life stress (ELS) and adolescent stress independently or in combination influence the ECS of young female rats, particularly the expression of CB1R and FAAH in the mPFC [226]. These changes were driven by epigenetic mechanisms, specifically DNA methylation, which led to long-term modulation of stress responses. The findings offer insights into how ELS can reprogram the ECS to either buffer or exacerbate responses to subsequent stress in adolescence, with implications for mental health outcomes later in life. Curiously, both ELS and adolescent stress independently led to CB1R upregulation in the mPFC, suggesting that ECS changes persist into adulthood. However, when ELS was followed by adolescent stress, CB1R expression returned to control levels, indicating a “buffering” effect. In contrast, only adolescent stress (forced swimming) caused an upregulation of FAAH, while ELS alone did not have this effect. Nevertheless, ELS exposure buffered the upregulation of FAAH by adolescent stress. These changes in gene expression were paralleled by decreased DNA methylation across specific CpG sites at the promoter regions of the CB1R and FAAH genes. Overall, the study supports the two-hit hypothesis, where ELS reprograms the response to later (adolescent) stressors [226] (Table 1).
Altogether, prolonged exposure to Δ9-THC or synthetic cannabinoids during adolescence is associated with persistent behavioural abnormalities, such as deficits in social interaction and various types of memory, increased anxiety, anhedonia, cognitive filtering, which all persist into adulthood. At the neurophysiological level, GABAergic hypofunction is found in the PFC that contributes to overactivation of the mesolimbic dopamine system. Furthermore, dysregulation of cortical pyramidal neurons, the reduction in gamma oscillations and sensorimotor gating deficits (prepulse inhibition) are consistently observed in these animal models. At the molecular level, reduced expression of GAD67 and GAT-1 is found, together with dampened signalling pathways such as Akt1/GSK-3, and mTOR, which are associated with the regulation of dopamine and GABAergic neurotransmission [227,228]. Importantly, these alterations strongly resemble schizophrenia-related psychopathology and recapitulate psychosis-related behaviours in man, which is often associated with precedent marijuana use during adolescence (see below) [208,229,230,231] (Table 1).
Recently, a ground-breaking study recapitulated on how chronic adolescent Δ9-THC exposure leads to severe behavioural, anatomical, and molecular impairments in animals, resembling neuropsychiatric disorders like schizophrenia [213]. The authors used a Δ9-THC dosing range that mimics the effects of a moderate to heavy use regimen of marijuana on a human adolescent, and it was previously shown to cause a profound and enduring neuropsychiatric phenotype [227]. As many times seen before and discussed above, these rats display cognitive deficits, affective abnormalities, impaired sensorimotor filtering, aberrant pyramidal cell firing patterns and a hyperactive mesocorticolimbic dopaminergic system. Intriguingly, this study found that L-theanine, a neuroprotective compound, counteracts these effects by normalizing brain activity and signalling pathways, preserving cognitive and emotional functions, and preventing long-term brain dysregulation [213]. In detail, L-theanine effectively blocked Δ9-THC-induced cognitive and affective abnormalities, restoring normal memory functions, reducing anxiety, and preventing anhedonia. L-theanine also normalized dopaminergic signalling in both the PFC and ventral tegmental area and prevented the downregulation of the Akt/GSK-3 pathway in the PFC. Finally, L-theanine prevented the Δ9-THC-induced disruptions in gamma oscillations, which are essential for proper cognitive and sensorimotor gating functions. In summary, L-theanine offers hope to mitigate the detrimental effects of marijuana abuse by adolescents.
However, not only chronic CB1R activation can be a concern, but also, long-term treatment with CBD. CBD is a negative allosteric modulator of CB1R, CB2R and GPR55, while it activates (and likely desensitizes) TRPV1R and inhibits eCB reuptake, among other pharmacological actions [2,232,233]. The number of phytocannabinoid-based medications is steadily growing, and these formulations often contain Δ9-THC, CBD or both. The anticonvulsant Epidiolex is a purified CBD solution, which is taken twice daily during several weeks or months by children with intractable epilepsy [15]. Even though their benefit clearly outweighs their influence on brain development if administered to children and adolescents, the possible neurodevelopmental effects nevertheless remain a valid concern. This concern was thoroughly allayed by Aguiar et al. (2024), who evaluated the consequences of long-term oral treatment of adolescent and young adult rats with CBD [234]. Treatment with a CBD-enriched cannabis extract (low Δ9-THC, high CBD) for 15 days did not result in any changes in body weight, locomotor activity, memory consolidation, or cognitive behaviour in healthy rats. The study showed no detrimental impact on short-term memory or locomotor behaviour, indicating the absence of adverse behavioural effects even during a sensitive period like adolescence to early adulthood (Table 1). However, the chronic treatment with the extract did induce notable changes in the glutamatergic synapses in the hippocampus. There was a reduction in the GluA1 subunit of AMPA receptors, coupled with an increase in PSD95 protein levels. That is, CBD just like other cannabinoids, is able to interfere with the dynamic rearrangement and maturation of glutamatergic synapses. This however may contribute to neuroprotective adaptations against excitotoxicity, potentially benefiting developmentally acquired neurological disorders of excitatory synaptic transmission, such as epilepsy and autism spectrum disorder [15,235]. Additionally, the expression of GFAP (a marker of astrocytic activation) was reduced in treated animals, suggesting that the CBD-enriched extract may prevent reactive astrogliosis, which is associated with neuroinflammation and excitotoxicity. Moreover, microglial arborization in the CA1 and CA3 hippocampal regions was reduced, indicating changes in microglial morphology, although their phagocytic activity was not significantly altered. Altogether, the study of Aguiar et al. (2023) underscores the potential safety of CBD-enriched cannabis extracts for therapeutic use in adolescents. The absence of behavioural detriments, coupled with neuroprotective changes in synaptic and glial components, suggests that such treatments may be well-tolerated, although further studies are needed, particularly regarding long-term effects [234] (Table 1).
3.2. The Maturating Human Brain Is Vulnerable to Cannabinoids
The human ECS undergoes significant changes during adolescence, a period marked by critical neurodevelopmental processes that affect emotional regulation, cognitive function, and vulnerability to psychiatric disorders. Emerging research suggests that the ECS is particularly sensitive to genetic polymorphisms and environmental influences, such as marijuana consumption, during this time, which can have long-term consequences on brain maturation [212,220] (Table 1). Adolescent exposure to Δ9-THC has been linked to persistent changes in the PFC, hippocampus and amygdala, regions critical for decision-making, memory, and impulse control. Human and rodent studies both have invariably demonstrated that Δ9-THC disrupts the balance of excitatory and inhibitory neurotransmission, which is essential for the refinement of synaptic connections during adolescence [231,236,237]. A recent study exploring the acute effects of cannabis on brain network connectivity have shown that cannabis disrupts multiple resting-state networks, particularly affecting the default mode, executive control, salience, hippocampal, and limbic striatal networks [238]. The authors tested the hypothesis that acute cannabis use could interfere with the undergoing significant structural changes of the PFC and hippocampus in the immature brain, thus contributing to impaired cognition and emotional processing. Using fMRI, Ertl and colleagues compared adolescents (16–17 years) and young adults (26–29 years) and found that cannabis significantly reduced within-network connectivity across these brain networks, with no significant difference between the age groups. Contrary to expectations, CBD did not attenuate the effects of Δ9-THC, and in some cases exacerbated the disruptions in connectivity, further challenging the assumption that CBD can counteract the negative effects of Δ9-THC. These disruptions in brain network connectivity are closely tied to cognitive functions, particularly decision-making, memory, and emotional regulation, which are especially vulnerable during adolescence due to ongoing brain maturation [238] (Table 1).
As for psychiatric outcomes, cannabis use during adolescence doubles the risk of developing anxiety disorders in adulthood [236]. This risk is particularly pronounced in individuals who begin using cannabis before age 15, and it is more prevalent in females. Depressive disorders are also more common in adolescent cannabis users, and this is linked to reduced hippocampal and white matter volumes, probably because of a lesser connectivity among brain regions regulating mood and emotions [239], but more direct effects on glutamate and monoamine turnovers can also be considered. Genetic variations in the ECS can too influence mental health outcomes during adolescence. Desai et al. (2024) examined how the FAAH C385A variant affect anandamide metabolism, modulates anxiety, depression, and brain activity related to threat and reward processing [240]. They found that youth with the FAAH AA genotype showed lower depressive symptoms compared to those with the AC or CC genotypes. This nonsynonymous FAAH C385A polymorphism is found in one quarter of humans with Caucasian ancestry, and it reduces FAAH activity and thus elevates anandamide levels. The 385A allele has been associated with lower anxiety and more efficient amygdala regulation in response to stress, but also with a greater index of impulsivity, stronger reward-related activity in the ventral striatum, street drug use, problem drug/alcohol abuse, as well as obesity [241] (Table 1). The impact of FAAH polymorphism can be particularly pronounced during adolescence, when corticolimbic circuits involved in emotional regulation, such as the PFC and amygdala, are still maturing [242] (Table 1).
In addition to genetic vulnerabilities, marijuana consumption during adolescence exerts significant effects on brain development, particularly through the disruption of CB1R-mediated signalling. The major culprit is very likely Δ9-THC, the psychoactive component of drug-type cannabis preparation, which, during adolescence, has been shown to alter the trajectory of synaptic pruning and neuroplasticity in corticolimbic circuits, leading to long-term impairments in cognitive function and emotional regulation [212,236]. Clearly, early cannabis use, particularly before age 17, is linked to lasting deficits in cognitive functions such as working memory, attention, decision-making, attention, and executive functions and verbal IQ. Higher Δ9-THC concentrations in modern cannabis strains amplify the potential for psychiatric disorders [229,243] (Table 1). Neuroimaging studies have shown structural abnormalities, including reduced gray matter volume in the PFC, altered white matter integrity, and reduced hippocampal volume and functioning, which correlate with cognitive impairments [239,244,245]. An important and rare longitudinal study enrolling almost 800 young subjects, examined how cannabis use during adolescence affects brain development, focusing on cortical thickness changes over time. Results show that greater cannabis use is associated with increased thinning in the left and right PFC, 5 years after the establishment of baseline cortical thickness. However, baseline cortical thickness was not associated with experimentation with cannabis. The extent of PFC atrophy was dose-dependent and linked to attentional impulsiveness at follow-up [246].
Notwithstanding, it is still largely debated to which extent adolescence marijuana use affects brain development. A recent systematic review and meta-analysis of voxel-based morphometry studies investigated the overall effects of adolescent cannabis use on brain morphology, with a focus on age, sex, and gray matter volume (GMV) differences [247]. Curiously, when combining all six included studies, no significant GMV differences were found between cannabis-using youth and typically developing youth. The study identified age-related GMV changes in the left superior temporal gyrus (L-STG). The L-STG is involved in auditory, speech, language, and emotional processing. Structural abnormalities in this region could contribute to impairments in social cognition and increase the risk of psychotic or affective disorders, particularly since cannabis use is associated with higher risks for these conditions in adolescence. Supplemental analyses found that a longer duration of cannabis use was associated with decreased GMV in the L-STG, supporting the idea that cumulative cannabis exposure may contribute to structural brain changes [247]. Older cannabis user youth showed decreased GMV compared to age-matched cannabis-naïve youth, while younger cannabis user youth showed increased GMV. This suggests a developmental gradient, with cannabis exposure potentially affecting GMV differently, depending on the age at which cannabis use occurs. A meta-regression revealed that studies with a higher proportion of female participants showed increased GMV in the right middle occipital gyrus in cannabis user youth compared to typically developing youth. Conversely, in studies with a higher proportion of males, cannabis user youth showed decreased GMV in this region. This indicates that sex may moderate the relationship between cannabis use and brain morphology, with females showing different neuroanatomical effects of cannabis compared to males. These differences may be accounted for hormonal influences or differences in cannabis-related behaviour between the sexes. These findings can be best explained assuming that cannabis-related GMV increases in younger adolescents may be due to disrupted synaptic pruning, while in older adolescents or young adults, a reduced GMV may be a result of neurotoxic processes [247].
All in all, these studies emphasize that more longitudinal research is needed to disentangle the complex relationships between age, sex, cannabis exposure, and brain development, especially during the critical period of adolescence, and additional confounding factors also need to be considered, including alcohol and tobacco use, socioeconomic status, the strength of marijuana strains consumed and the mode of ingestion. Another longitudinal study assessed the cognitive performance of over 1,000 individuals born after 1972 [244]. Initial neuropsychological testing was conducted at age 13, before any cannabis use had begun. The participants had varying histories of cannabis use, ranging from non-use to cannabis dependence. Follow-up assessments were completed when the participants reached age 38. Persistent cannabis users exhibited significant impairments in memory function, including challenges with both short-term memory (working memory) and long-term memory retention. This decline was observed across multiple domains of neuropsychological testing and was particularly severe in individuals who started using cannabis in adolescence (Table 1).
Cannabis users, especially those who started young, also showed marked deficits in executive functioning such as problem-solving, decision-making, planning, and the ability to inhibit impulsive behaviour. One of the notable declines was in processing speed, the cognitive ability to quickly and efficiently perform mental tasks. Slow processing speed can make it difficult for individuals to follow instructions, keep up with conversations, or respond quickly in demanding environments. Persistent cannabis users, particularly those with adolescent-onset use, showed slower processing speeds over time. Cannabis users also experienced significant problems with sustained attention and focus. This manifested as distractibility, difficulty concentrating for long periods, and an inability to stay engaged with tasks. These issues were noticeable not just in test results but also in daily life, as reported by friends and family members of the participants. Finally, the study found a clear association between persistent cannabis use and a measurable decline in IQ. Those with the most severe decline in IQ were individuals who started using cannabis during adolescence and continued using it persistently. This study of Meier et al. (2012) thus clearly confirms that cannabis use during brain development may have a neurotoxic effect, leading to long-lasting cognitive impairments, with IQ drops as significant as 6 to 8 points over the span of the study [244].
Marijuana use, particularly during adolescence, is also strongly associated with an increased risk of psychosis. It is easy to understand why, since the ECS controls the development of all domains and systems which are affected in schizophrenia, including certain brain areas (PFC, hippocampus, amygdala, striatum, L-STG), GABAergic and glutamatergic signalling, monoaminergic neuromodulation and even brain metabolism [230]. This risk can manifest as temporary psychotic episodes or symptoms, but in some cases, it may persist and contribute to the development of more chronic conditions, such as schizophrenia. While marijuana use during adolescence may elevate the risk of schizophrenia in some individuals, the association between marijuana and schizophrenia is more complex and less direct than its link to psychosis. Schizophrenia is a chronic mental disorder that typically emerges in late adolescence or early adulthood, characterized not only by psychotic symptoms but also by cognitive impairments and negative symptoms like social withdrawal [229,230,237]. Longitudinal studies indicate that adolescent marijuana users, especially those who use it frequently or consume high-potency strains, are at a higher risk of developing schizophrenia later in life. However, marijuana use alone is unlikely to cause schizophrenia; rather, it may act as a trigger in individuals who are genetically predisposed, e.g., those carrying variants in their catecholamine-O-methyltransferase (COMT) gene or in their CB1R gene CNR1. This is supported by the observation that while adolescent marijuana use is a growing problem, the incidence of new schizophrenia cases has not shown a corresponding increase. Additionally, it is possible that individuals with a genetic predisposition to schizophrenia are more likely to experiment with marijuana during adolescence, further complicating the relationship between marijuana use and schizophrenia risk [230].
The following ground-breaking study of Tao et al. (2020) shed new light on how genetic predispositions, environmental influences, and marijuana use converge in the development of schizophrenia [203]. They found that in the PFC and the hippocampus, CB1R mRNA expression is highest in the foetal period, followed by a sharp decline post-natally, which stabilizes throughout adulthood. This strongly implies that CB1R activity is critical during human brain development. Notably, carriers of the COMT Val158 allele showed a stronger negative correlation between CNR1 expression in the dorsolateral PFC (DLPFC) and age, potentially linking cannabis exposure during adolescence to dysregulated brain development. Furthermore, CNR1 expression was significantly decreased in the DLPFC of patients with schizophrenia and major depressive disorder, suggesting that ECS dysregulation is involved in the pathology of these psychiatric conditions. Interestingly, Δ9-THC or ethanol exposure upregulated CNR1 expression in patients with affective disorders, and CNR1 expression was also increased in schizophrenia patients who completed suicide, pointing to the complex interaction between cannabis use, mental health, and suicide risk. DNA methylation at specific loci (e.g., cg02498983) correlated with age and COMT genotype in the PFC. Carriers of the Val158 allele showed the steepest increase in methylation over time, and this negatively correlated with CNR1 expression. This well correlates with the above animal studies, suggesting that epigenetic modulation induced by environmental factors including marijuana abuse can reprogram brain circuits during adolescence, increasing the risk of psychosis. Additionally, the study identified a novel CNR1 transcript, whose expression was associated with a single nucleotide polymorphism rs806368, a genetic variant previously linked to substance dependence. This transcript might regulate CB1R expression in response to cannabis exposure, contributing to the development of addiction and psychiatric disorders in genetically predisposed individuals [203].
Although the level of expression (mRNA) and protein density are not interchangeable terms, most studies reported in this review agree upon that both peak at early stages of brain development. We reported a steady decline in rat hippocampal CB1R density during the post-natal life [248]. However, we also found much higher CB1R density in the embryonic hippocampus, with a steep decline until birth (unpublished). A post-mortem study also found that CB1R mRNA expression in the human DLPFC decreases significantly over time, peaking during neonatal life and declining steadily into adulthood [249] (Table 1). This pattern was particularly evident in cortical layer 2, suggesting that eCB-mediated regulation of neurotransmission is robust in early life but diminishes with age. DAGLα expression followed a bell-shaped curve, with low levels in infancy and adulthood but peaking during school age to young adulthood. This suggests that the production of 2-AG is particularly important during cognitive development in childhood. While the typically presynaptic expression of MAGL declined after infancy, the expression of the post-synaptic 2-AG-metabolizing enzyme, ABHD6, showed a steady increase across development. This may reflect a developmental switch from retrograde inhibition to dendritic self-inhibition [8]. In contrast, both NAPE-PLD and FAAH steadily increased from infancy to adulthood, indicating that AEA becomes increasingly important after adolescence. CB1R mRNA was highly expressed in cortical layer 2 during early life (neonates and toddlers), while the deep cortical layers 5 and 6 showed weaker but still significant CB1R mRNA expression. CB1R expression decreased significantly with age, particularly in superficial layers like 2 and 3, and the intensity of expression in the deeper layers (5 and 6) also declined by adulthood. Notably, CB1R mRNA showed clear association with GABAergic interneuron markers, supporting the notion about the role of CB1R in early-life regulation of cortical interneuron development [249] (Table 1).
Additional post-mortem studies in patients with schizophrenia reveal a strong GABAergic dysfunction in the corticolimbic areas, particularly of the parvalbumin+ GABAergic neurons, leading to impaired inhibitory control of pyramidal neurons and disrupted gamma oscillations, which are essential for cognitive processing, together with the hyperactivity of the mesolimbic dopaminergic system [229,236,250]. The negative symptoms (alogia, anhedonia, affective flattening, avolition, memory problems, social withdrawal) are mostly linked with hypofrontality, more closely, disturbances in GABAergic and glutamatergic activities of the PFC. The positive symptoms of schizophrenia (hallucinations, paranoia, disorganized thinking, abnormal motor behaviour) are closely linked with a hyperdopaminergic state, particularly in the mesolimbic pathway [250]. As the animal studies made very clear, chronic exposure to CB1R agonists during adolescence indeed causes hypofrontality and hyperdopaminergic state via multiple mechanisms, consistent with lasting developmental, neurochemical and neurophysiological changes in the corticolimbic system and beyond [237]. While acutely, Δ9-THC administration in humans induces several schizophrenia-like symptoms, including paranoia, hallucinations and cognitive impairments, on the long run, Δ9-THC exposure can exacerbate psychotic symptoms in individuals already diagnosed with schizophrenia, or it can facilitate the onset of schizophrenia in individuals with genetic predisposition [229,230,251]. These effects have a strong neurodevelopmental component when marijuana abuse occurs during adolescence [237].
Importantly, CBD has been proposed as a possible antipsychotic medicine [237,252], with proven therapeutic potential against a multitude of complications in schizophrenia, including:
- Positive Symptoms: CBD has been shown to ameliorate hyperlocomotion and stereotypies, which are proxies for positive symptoms like psychomotor agitation and hallucinations in schizophrenia. CBD may exert antipsychotic effects by normalizing dopamine signalling and counteracting Δ9-THC’s psychotomimetic effects.
- Negative Symptoms: There is evidence that CBD can improve social interaction deficits and reduce immobility in animal models of schizophrenia, suggesting it could treat negative symptoms such as social withdrawal, anhedonia, and lack of motivation.
- Cognitive Symptoms: CBD has shown promise in reversing cognitive deficits in preclinical models, particularly in memory and attention tasks. It has been shown to restore object recognition memory and working memory, likely by modulating PFC and hippocampal circuits.
CBD’s antipsychotic effects may stem from its ability to modulate CB1Rs, CB2Rs and TRPV1Rs, by affecting AEA turnover, by acting as a partial D2R/D3R agonist and as a partial 5-HT1AR agonist, thus normalizing monoaminergic signalling and conferring antidepressant and antipsychotic effects [237,252]. Nevertheless, an increasing body of studies fail to provide direct evidence that CBD can counteract the psychotomimetic effects of Δ9-THC [238,253], thus adding to the complexity of the role of cannabis preparations in psychosis.
4. Conclusions
The intricate role of the ECS in brain development has been well-documented, with its influence beginning as early as embryogenesis and continuing through key developmental stages, including adolescence. Despite extensive research highlighting both its regulatory functions and its vulnerabilities, significant gaps in our understanding remain. For instance, the impact of cannabis exposure, both in utero and during adolescence, presents a multifaceted challenge. While studies consistently show that early exposure to Δ9-THC can disrupt brain development and maturation, particularly in the PFC and hippocampus, the exact mechanisms remain incompletely understood. While much of the focus has been on the immediate effects of other substances, such as alcohol and tobacco, the long-term implications of prenatal cannabis exposure should not be overlooked. As cannabis becomes increasingly legalized and socially accepted in many regions, the potential for underestimating its risks to foetal development grows. This calls for heightened awareness, education and caution among healthcare providers and the general public, ensuring that expectant mothers are fully informed about the potential consequences of cannabis use during pregnancy.
One of the key takeaways from this review is the need for more longitudinal studies that track the effects of cannabis exposure across the lifespan. From one hand, there are undeniable discrepancies in findings related to cannabis-induced neurodevelopmental damage. While animal models provide robust evidence of Δ9-THC’s negative impact on synaptic pruning, memory, and emotional regulation, human studies yield mixed results, particularly regarding the role of genetic predispositions. Studies like those investigating FAAH and CNR1 polymorphisms suggest that genetic vulnerabilities may modulate the effects of cannabis, underscoring the importance of personalized approaches in future research and potential interventions. On the other hand, existing research provides compelling evidence that adolescence is a critical window for ECS modulation with possible or putative long-term consequences, yet most studies focus on short-term outcomes. Longitudinal research is necessary to determine whether cannabis-induced changes observed in the adolescent brain persist into adulthood and how they manifest in long-term cognitive, emotional, and psychiatric health outcomes.
The role of CBD, often proposed as a counterbalance to Δ9-THC’s detrimental effects, remains controversial, with some studies suggesting it may exacerbate rather than mitigate the disruptions caused by Δ9-THC. Thus, the field needs to address the ongoing debate about CBD’s protective or harmful effects in the context of adolescent brain development. Although CBD is touted for its neuroprotective properties and potential therapeutic applications, conflicting data point to the need for caution in using CBD-based therapies in adolescents. More detailed mechanistic studies are needed to clarify how CBD interacts with the ECS during this vulnerable period and whether it truly mitigates the risks posed by Δ9-THC or other cannabinoids.
In conclusion, while significant progress has been made in understanding the ECS’s role in brain development and its disruption by exogenous cannabinoids or by ECS polymorphism, more comprehensive research is needed. Specifically, longitudinal human studies, attention to genetic variability, and careful examination of therapeutic cannabinoids like CBD are crucial for filling the current knowledge gaps. Only through such efforts can we fully appreciate the complex relationship between cannabis and neurodevelopment, ensuring that both public health policies and clinical practices are informed by the latest, most reliable data.

Table 1.
The involvement of the endocannabinoid system in adolescent brain development.
| ReceptorEnzyme Ligand |
Function in the Adolescent Brain | Effects of External Cannabinoids | Consequences if Perturbed | Sex-Dependent Effects | References |
|---|---|---|---|---|---|
| CB1R | Regulates excitatory/inhibitory neurotransmission, synaptic pruning, and maturation of corticolimbic circuits (e.g., PFC, hippocampus). Peaks during adolescence, declines in adulthood. | Δ9-THC acts as a CB1R agonist. Chronic exposure downregulates CB1R, desensitizes receptors, impairs synaptic plasticity, and reduces dendritic complexity. | Persistent changes in PFC and hippocampal structure. Increased risk of psychiatric disorders (e.g., anxiety, schizophrenia). Impaired executive function, memory, and emotional regulation. | Greater CB1R density in males, more efficient CB1R coupling in females. More pronounced cognitive and emotional impairments in female rodents. Males show delayed onset of CB1R-mediated synaptic plasticity. | [206,214,254] |
| CB2R | Involved in immune regulation and neuroinflammation. Low neuronal expression in the brain but increases in microglia with neuroinflammation. | Chronic Δ9-THC exposure reduces CB2R density in adolescent brains. Selective acute CB2R activation (e.g., by AM1241) can reduce neuroinflammation, prevent anxiety-like behaviors during adolescence. | Chronic Δ9-THC exposure unequivocally downregulates CB2R expression, which may exacerbate anxiety and neuroinflammation caused by substance abuse or stress. | Two-fold greater CB2R expression in adolescent but not adult females. | [214,215,216] |
| TRPV1R | Involved in modulating stress and anxiety responses during adolescence. Opposes CB1R effects on anxiety regulation. | TRPV1R activation by CBD or stress can exacerbate anxiety responses. TRPV1R-dependent LTP in hippocampus may be linked to cognitive deficits caused by alcohol exposure. | Increased anxiety and cognitive deficits when activated by cannabinoids or stress. TRPV1 blockade may provide therapeutic potential for treating anxiety disorders. | Females show earlier onset of TRPV1-mediated synaptic plasticity. Male rodents show stronger anxiety-related responses to TRPV1 activation. | [214,218,219] |
| Δ9-THC | Partial agonist of the CB1R and the CB2R. Interferes with the maturation of corticolimbic circuits, synaptic pruning, and neuroplasticity during adolescence. | May lead to downregulation and desensitization of its receptors with chronic use. Disrupts synaptic plasticity, reduces dendritic complexity, and impairs signaling in the PFC and hippocampus. Triggers hypoGABAergic and hyperdopaminergic state. | Affects cortical thickness and wiring. Long-lasting cognitive impairments (e.g., memory, decision-making) and emotional dysregulation. Increases the risk of psychiatric disorders like anxiety, depression, and schizophrenia. | Females are more susceptible to Δ9-THC-induced emotional and cognitive impairments, showing greater downregulation of CB1R. Males tend to exhibit delayed onset of Δ9-THC-induced synaptic plasticity changes. | [208,229,230,243,244,254] |
| CBD | Negative allosteric modulator of CB1R and CB2R. Activates TRPV1R and inhibits eCB reuptake. Potential neuroprotective role during brain development. | Long-term CBD exposure can affect glutamatergic synapses and synaptic plasticity. May have neuroprotective effects but can also exacerbate disruptions in brain network connectivity when co-administered with Δ9-THC. | Reduction in GluA1 AMPA subunit and increased PSD95. Alters brain connectivity, especially when combined with Δ9-THC. No adverse effects on cognitive or motor functions in healthy adolescents. | Males may experience greater cognitive protection from CBD. Females show increased susceptibility to CBD’s effects on synaptic plasticity when combined with Δ9-THC. | [234,238,245] |
| FAAH | Breaks down anandamide. Controls anandamide levels and regulates emotional responses, stress, and cognitive functions. | Polymorphism FAAH C385A reduces enzyme activity, leading to elevated anandamide levels and altered stress responses. Chronic Δ9-Δ9-THC exposure interferes with FAAH activity, but reports are conflicting. | Reduced FAAH activity is associated with heightened emotional regulation and impulsivity, but can increase susceptibility to substance abuse and psychiatric disorders. | Females generally have lower FAAH expression during adolescence, leading to prolonged anandamide signaling. Males with FAAH C385A polymorphism show stronger reward-related acivity, impulsivity and risk-taking behaviors. | [214,215,240,241,242,255,256,257] |
| MAGL | Breaks down 2-AG. Regulates synaptic plasticity, excitatory/inhibitory balance, and emotional regulation. | Decrease in function during adolescence. Chronic Δ9-THC exposure reduces microglial but increases overall MAGL expression, leading to altered synaptic transmission. | Dysregulation of synaptic connections and plasticity in corticolimbic circuits. Long-term emotional and cognitive deficits. | Inhibiting MAGL uncover LTD in juvenile males. | [214,215,249,257] |
| DAGLα | Synthesizes 2-AG, essential for synaptic plasticity and connectivity in brain maturation. Peaks during adolescence. | Its expression peaks during adolescence. Δ9-THC can alter DAGL activity, affecting 2-AG synthesis and overall cannabinoid signaling during brain development. | Disruption in the production of 2-AG, leading to impaired synaptic connectivity, memory, and emotional regulation. | Females show earlier DAGL maturation and heightened synaptic plasticity, whereas males exhibit more delayed effects on synaptic development. | [201,249,257] |
| NAPE-PLD | Synthesizes anandamide, plays a role in regulating emotional and cognitive functions. | Gain of function during adolescence. Chronic Δ9-THC exposure can reduce NAPE-PLD expression in microglia, leading to altered anandamide production. | Impaired emotional regulation and stress response. Increased risk of psychiatric disorders due to disrupted anandamide signaling. | Females show higher baseline NAPE-PLD activity, contributing to sex differences in emotional regulation under stress or cannabinoid exposure. | [214,215,249] |
| ABHD6 | Degrades 2-AG, plays a role in regulating synaptic plasticity and emotional responses. | Chronic Δ9-THC exposure can increase ABHD6 expression in the placenta but not in the brain. ABHD6 expression is increased in the PFC of schizophrenic adolescents. | Dysregulated synaptic transmission and plasticity in corticolimbic circuits, leading to cognitive and emotional deficits. | Males show higher ABHD6 levels during adolescence, leading to stronger inhibition of 2-AG signaling compared to females. | [214,258,259] |
Author Contributions
All authors contributed to conceptualization, writing and editing. All authors have read and agreed to the published version of the manuscript.
Funding
The research activity of the authors has been supported by FCT – Fundação para a Ciência e a Tecnologia under the project 2022.03263.PTDC (to R.J.R.) and through the COMPETE 2020 - Operational Programme for Competitiveness and Internationalisation and Portuguese national funds via FCT – Fundação para a Ciência e a Tecnologia, under project[s] PTDC/MED-NEU/28160/2017 (to J.M.M.) and UIDB/04539/2020, UIDP/04539/2020 and LA/P/0058/2020.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
2-AG, 2-arachidonoylglycerol; ABHD, Alpha/Beta Hydrolase Domain-Containing; AEA, N-arachidonoyl-ethanolamine (Anandamide); Akt, Protein Kinase B (PKB); BDNF, Brain-Derived Neurotrophic Factor; CAE, Chronic Alcohol Exposure; CB1R, Cannabinoid Receptor 1; CB2R, Cannabinoid Receptor 2; CBD, Cannabidiol; COMT - Catechol-O-Methyltransferase; CP, Cortical Plate; CRIP1a, Cannabinoid Receptor-Interacting Protein 1a; CTA, Cortico-Thalamic Axons; DAGLα, Diacylglycerol Lipase alpha; DAGLβ, Diacylglycerol Lipase beta; DCC, Deleted in Colorectal Cancer; DLPFC, Dorsolateral Prefrontal Cortex; DG, Dentate Gyrus; EE, Environmental Enrichment; ERK1/2, Extracellular Signal-Regulated Kinase 1/2; eCB, endocannabinoids; ECS, Endocannabinoid Signalling System; FAAH, Fatty Acid Amide Hydrolase; FAK, Focal Adhesion Kinase; GABA, Gamma-Aminobutyric Acid; GAD67, Glutamate Decarboxylase 67; GAT-1, GABA Transporter 1; GFAP, Glial Fibrillary Acidic Protein; GMV, Gray Matter Volume; GPCR, G-Protein Coupled Receptor; GPR55, G-protein Coupled Receptor 55; GSK-3, Glycogen Synthase Kinase-3; GW, Gestational Week; HPA, Hypothalamic-Pituitary-Adrenal (axis); IPSC, Inducible Pluripotent Stem Cell; IZ, Intermediate Zone; JNK, c-Jun N-terminal Kinase; KO, Knockout; L-STG, Left Superior Temporal Gyrus; LPI, Lysophosphatidyl-inositol; LTD, Long-Term Depression; LTP, Long-Term Potentiation; MAPK, Mitogen-Activated Protein Kinase; MAGL, Monoacylglycerol Lipase; mGluR5, Metabotropic Glutamate Receptor 5; mPFC, medial Prefrontal Cortex; MPP-LTD, Medial Perforant Pathway-Long Term Depression; mTOR, Mechanistic Target of Rapamycin; N-SMase, Neutral Sphingomyelinase; NAPE-PLD, N-acyl phosphatidylethanolamine-specific Phospholipase D; NGF, Nerve Growth Factor; NLRP3, NOD-, LRR- and Pyrin domain-containing protein 3; NMDA,N-methyl-D-aspartate (NMDA receptor); NPC, Neuronal progenitor cells; OPC, Oligodendrocyte Precursor Cell; PFC, Prefrontal Cortex; PI3K, Phosphoinositide 3-kinase; PKA, Protein Kinase A; PKCβII, Protein Kinase C beta II; PLC, Phospholipase C; PLCβ1, Phospholipase C beta 1; PSD95, Postsynaptic Density Protein 95; Rac1, Ras-related C3 botulinum toxin substrate 1; RGC, Retinal Ganglion Cells; RhoA, Ras homolog family member A; ROCK, Rho-associated protein kinase; SCG10, Superior Cervical Ganglion 10 (Stathmin-2); Src, Proto-Oncogene Tyrosine-Protein Kinase Src; TCA, Thalamocortical Axons; TrkB, Tropomyosin receptor kinase B; TRPV1, Transient Receptor Potential Vanilloid 1 (receptor); VGLUT1, Vesicular Glutamate Transporter 1; VZ/SVZ, Ventricular/Subventricular Zones; Δ9-THC, Δ9-tetrahydrocannabinol.
References
- Mechoulam, R.; Hanuš, L.O.; Pertwee, R.; Howlett, A.C. Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat. Rev. Neurosci. 2014, 15(11), 757–764. [Google Scholar] [CrossRef] [PubMed]
- Solymosi, K.; Köfalvi, A. Cannabis: A treasure trove or Pandora’s box? Mini Rev. Med. Chem. 2017, 17(13), 1223–1291. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Borrelli, F.; Capasso, R.; Di Marzo, V.; Mechoulam, R. Non-psychotropic plant cannabinoids: New therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci. 2009, 30(10), 515–527. [Google Scholar] [CrossRef]
- Sideris, A.; Doan, L.V. An Overview of Cannabidiol. Anesth. Analg. 2024, 138(1), 54–68. [Google Scholar] [CrossRef]
- Di Marzo, V.; Fontana, A.; Cadas, H.; Schinelli, S.; Cimino, G.; Schwartz, J.C.; Piomelli, D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 1994, 372(6507), 686–691. [Google Scholar] [CrossRef]
- Katona, I.; Freund, T.F. Multiple functions of endocannabinoid signaling in the brain. Annu. Rev. Neurosci. 2012, 35, 529–558. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Castillo, P.E.; Manzoni, O.J.; Tonini, R. Synaptic functions of endocannabinoid signaling in health and disease. Neuropharmacology 2017, 124, 13–24. [Google Scholar] [CrossRef]
- Bacci, A.; Huguenard, J.R.; Prince, D.A. Long-lasting self-inhibition of neocortical interneurons mediated by endocannabinoids. Nature 2004, 431(7006), 312–316. [Google Scholar] [CrossRef]
- Marinelli, S.; Pacioni, S.; Cannich, A.; Marsicano, G.; Bacci, A. Self-modulation of neocortical pyramidal neurons by endocannabinoids. Nat. Neurosci. 2009, 12(12), 1488–1490. [Google Scholar] [CrossRef]
- Den Boon, F.S.; Chameau, P.; Schaafsma-Zhao, Q.; van Aken, W.; Bari, M.; Oddi, S.; Kruse, C.G.; Maccarrone, M.; Wadman, W.J.; Werkman, T.R. Excitability of prefrontal cortical pyramidal neurons is modulated by activation of intracellular type-2 cannabinoid receptors. Proc. Natl. Acad. Sci. USA 2012, 109(9), 3534–3539. [Google Scholar] [CrossRef]
- Stempel, A.V.; Stumpf, A.; Zhang, H.Y.; Özdoğan, T.; Pannasch, U.; Theis, A.K.; Otte, D.M.; Wojtalla, A.; Rácz, I.; Ponomarenko, A.; Xi, Z.X.; Zimmer, A.; Schmitz, D. Cannabinoid type 2 receptors mediate a cell type-specific plasticity in the hippocampus. Neuron 2016, 90(4), 795–809. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, A.; Parthier, D.; Sammons, R.P.; Stempel, A.V.; Breustedt, J.; Rost, B.R.; Schmitz, D. Cannabinoid type 2 receptors mediate a cell type-specific self-inhibition in cortical neurons. Neuropharmacology 2018, 139, 217–225. [Google Scholar] [CrossRef]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19(3), 833. [Google Scholar] [CrossRef]
- Alhouayek, M.; Masquelier, J.; Muccioli, G.G. Lysophosphatidylinositols, from cell membrane constituents to GPR55 ligands. Trends Pharmacol. Sci. 2018, 39(6), 586–604. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.A.; Whalley, B.J. The proposed mechanisms of action of CBD in epilepsy. Epileptic Disord 2020, 22(S1), 10–15. [Google Scholar] [CrossRef]
- Katona, I.; Sperlágh, B.; Sík, A.; Köfalvi, A.; Vizi, E.S.; Mackie, K.; Freund, T.F. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J. Neurosci. 1999, 19(11), 4544–4558. [Google Scholar] [CrossRef]
- Katona, I.; Sperlágh, B.; Maglóczky, Z.; Sántha, E.; Köfalvi, A.; Czirják, S.; Mackie, K.; Vizi, E.S.; Freund, T.F. GABAergic interneurons are the targets of cannabinoid actions in the human hippocampus. Neuroscience 2000, 100(4), 797–804. [Google Scholar] [CrossRef]
- Lutz, B. Neurobiology of cannabinoid receptor signaling. Dialogues Clin. Neurosci. 2020, 22(3), 207–222. [Google Scholar] [CrossRef]
- Callén, L.; Moreno, E.; Barroso-Chinea, P.; Moreno-Delgado, D.; Cortés, A.; Mallol, J.; Casadó, V.; Lanciego, J.L.; Franco, R.; Lluis, C.; Canela, E.I.; McCormick, P.J. Cannabinoid receptors CB1 and CB2 form functional heteromers in brain. J. Biol. Chem. 2012, 287(25), 20851–20865. [Google Scholar] [CrossRef]
- Ishiguro, H.; Kibret, B.G.; Horiuchi, Y.; Onaivi, E.S. Potential role of cannabinoid type 2 receptors in neuropsychiatric and neurodegenerative disorders. Front. Psychiatry 2022, 13, 828895. [Google Scholar] [CrossRef]
- Grabon, W.; Rheims, S.; Smith, J.; Bodennec, J.; Belmeguenai, A.; Bezin, L. CB2 receptor in the CNS: From immune and neuronal modulation to behavior. Neurosci. Biobehav. Rev. 2023, 150, 105226. [Google Scholar] [CrossRef] [PubMed]
- Harkany, T.; Guzmán, M.; Galve-Roperh, I.; Berghuis, P.; Devi, L.A.; Mackie, K. The emerging functions of endocannabinoid signaling during CNS development. Trends Pharmacol. Sci. 2007, 28(2), 83–92. [Google Scholar] [CrossRef] [PubMed]
- Dalton, G.D.; Carney, S.T.; Marshburn, J.D.; Norford, D.C.; Howlett, A.C. CB1 Cannabinoid receptors stimulate Gβγ-GRK2-mediated FAK phosphorylation at tyrosine 925 to regulate ERK activation involving neuronal focal adhesions. Front. Cell. Neurosci. 2020, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Oyagawa, C.R.M.; Grimsey, N.L. Cannabinoid receptor CB1 and CB2 interacting proteins: Techniques, progress and perspectives. Methods Cell Biol. 2021, 166, 83–132. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Reggio, P.H. An update on non-CB1, non-CB2 cannabinoid related G-protein-coupled receptors. Cannabis Cannabinoid Res. 2017, 2(1), 265–273. [Google Scholar] [CrossRef]
- Navarro, G.; Varani, K.; Lillo, A.; Vincenzi, F.; Rivas-Santisteban, R.; Raïch, I.; Reyes-Resina, I.; Ferreiro-Vera, C.; Borea, P.A.; Sánchez de Medina, V.; Nadal, X.; Franco, R. Pharmacological data of cannabidiol- and cannabigerol-type phytocannabinoids acting on cannabinoid CB1, CB2 and CB1/CB2 heteromer receptors. Pharmacol. Res. 2020, 159, 104940. [Google Scholar] [CrossRef]
- Dalton, G.D.; Howlett, A.C. Cannabinoid CB1 receptors transactivate multiple receptor tyrosine kinases and regulate serine/threonine kinases to activate ERK in neuronal cells. Br. J. Pharmacol. 2012, 165(8), 2497–2511. [Google Scholar] [CrossRef]
- Berghuis, P.; Dobszay, M.B.; Wang, X.; Spano, S.; Ledda, F.; Sousa, K.M.; Schulte, G.; Ernfors, P.; Mackie, K.; Paratcha, G.; Hurd, Y.L.; Harkany, T. Endocannabinoids regulate interneuron migration and morphogenesis by transactivating the TrkB receptor. Proc. Natl. Acad. Sci. USA 2005, 102(52), 19115–19120. [Google Scholar] [CrossRef]
- Baker, D.; Pryce, G.; Davies, W.L.; Hiley, C.R. In silico patent searching reveals a new cannabinoid receptor. Trends Pharmacol. Sci. 2006, 27(1), 1–4. [Google Scholar] [CrossRef]
- Kapur, A.; Zhao, P.; Sharir, H.; Bai, Y.; Caron, M.G.; Barak, L.S.; Abood, M.E. Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J. Biol. Chem. 2009, 284(43), 29817–29827. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Balenga, N.A.; Schröder, R.; Kargl, J.K.; Platzer, W.; Martini, L.; Arthur, S.; Penman, J.; Whistler, J.L.; Kostenis, E.; Waldhoer, M.; Irving, A.J. GPR55 ligands promote receptor coupling to multiple signalling pathways. Br. J. Pharmacol. 2010, 160(3), 604–614. [Google Scholar] [CrossRef]
- Ross, R.A. L-α-lysophosphatidylinositol meets GPR55: A deadly relationship. Trends Pharmacol. Sci. 2011, 32(5), 265–269. [Google Scholar] [CrossRef] [PubMed]
- Calvillo-Robledo, A.; Cervantes-Villagrana, R.D.; Morales, P.; Marichal-Cancino, B.A. The oncogenic lysophosphatidylinositol (LPI)/GPR55 signaling. Life Sci. 2022, 301, 120596. [Google Scholar] [CrossRef]
- He, Y.; Shen, H.; Bi, G.H.; Zhang, H.Y.; Soler-Cedeño, O.; Alton, H.; Yang, Y.; Xi, Z.X. GPR55 is expressed in glutamate neurons and functionally modulates drug taking and seeking in rats and mice. Transl. Psychiatry 2024, 14(1), 101. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Szállási, Á. Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacol. Rev. 2014, 66(3), 676–814. [Google Scholar] [CrossRef]
- Muller, C.; Reggio, P.H. An analysis of the putative CBD binding site in the ionotropic cannabinoid receptors. Front. Cell. Neurosci. 2020, 14, 615811. [Google Scholar] [CrossRef]
- Nagy, I.; White, J.P.M.; Paule, C.C.; Maze, M.; Urban, L. Functional molecular biology of the TRPV1 ion channel. In Cannabinoids and the Brain; Köfalvi, A., Ed.; Springer: New York, USA, 2007; pp. 101–130. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Eilers, H. TRPV1 splice variants: Structure and function. Front. Biosci. 2010, 15(3), 872–882. [Google Scholar] [CrossRef]
- Vos, M.H.; Neelands, T.R.; McDonald, H.A.; Choi, W.; Kroeger, P.E.; Puttfarcken, P.S.; Faltynek, C.R.; Moreland, R.B.; Han, P. TRPV1b overexpression negatively regulates TRPV1 responsiveness to capsaicin, heat and low pH in HEK293 cells. J. Neurochem. 2006, 99(4), 1088–1102. [Google Scholar] [CrossRef] [PubMed]
- Köles, L.; Garção, P.; Zádori, Z.S.; Ferreira, S.G.; Pinheiro, B.S.; da Silva-Santos, C.S.; Ledent, C.; Köfalvi, A. Presynaptic TRPV1 vanilloid receptor function is age- but not CB1 cannabinoid receptor-dependent in the rodent forebrain. Brain Res. Bull. 2013, 97, 126–135. [Google Scholar] [CrossRef]
- Alger, B.E. Endocannabinoids at the synapse a decade after the dies mirabilis (29 March 2001): What we still do not know. J. Physiol. 2012, 590(10), 2203–2212. [Google Scholar] [CrossRef]
- Murataeva, N.; Straiker, A.; Mackie, K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br. J. Pharmacol. 2014, 171(6), 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Fukaya, M.; Uchigashima, M.; Miura, E.; Kamiya, H.; Kano, M.; Watanabe, M. Localization of diacylglycerol lipase-alpha around postsynaptic spine suggests close proximity between production site of an endocannabinoid, 2-arachidonoyl-glycerol, and presynaptic cannabinoid CB1 receptor. J. Neurosci. 2006, 26(18), 4740–4751. [Google Scholar] [CrossRef] [PubMed]
- Mulder, J.; Zilberter, M.; Pasquaré, S.J.; Alpár, A.; Schulte, G.; Ferreira, S.G.; Köfalvi, A.; Martín-Moreno, A.M.; Keimpema, E.; Tanila, H.; Watanabe, M.; Mackie, K.; Hortobágyi, T.; de Ceballos, M.L.; Harkany, T. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 2011, 134(4), 1041–1060. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 70-81, 70–81. [Google Scholar] [CrossRef]
- Savinainen, J.R.; Saario, S.M.; Laitinen, J.T. The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol. (Oxf.) 2012, 204(2), 267–276. [Google Scholar] [CrossRef]
- Maccarrone, M.; Di Marzo, V.; Gertsch, J.; Grether, U.; Howlett, A.C.; Hua, T.; Makriyannis, A.; Piomelli, D.; Ueda, N.; van der Stelt, M. Goods and bads of the endocannabinoid system as a therapeutic target: Lessons learned after 30 years. Pharmacol. Rev. 2023, 75(5), 885–958. [Google Scholar] [CrossRef]
- Blankman, J.L.; Cravatt, B.F. Chemical probes of endocannabinoid metabolism. Pharmacol. Rev. 2013, 65(2), 849–871. [Google Scholar] [CrossRef]
- Simon, G.M.; Cravatt, B.F. Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J. Biol. Chem. 2008, 283(14), 9341–9349. [Google Scholar] [CrossRef]
- Maccarrone, M. Metabolism of the endocannabinoid anandamide: Open questions after 25 years. Front. Mol. Neurosci. 2017, 10, 166. [Google Scholar] [CrossRef]
- Wei, B.Q.; Mikkelsen, T.S.; McKinney, M.K.; Lander, E.S.; Cravatt, B.F. A second fatty acid amide hydrolase with variable distribution among placental mammals. J. Biol. Chem. 2006, 281(48), 36569–36578. [Google Scholar] [CrossRef]
- Moreno-Luna, R.; Esteban, P.F.; Paniagua-Torija, B.; Arevalo-Martin, A.; Garcia-Ovejero, D.; Molina-Holgado, E. Heterogeneity of the endocannabinoid system between cerebral cortex and spinal cord oligodendrocytes. Mol. Neurobiol. 2021, 58(2), 689–702. [Google Scholar] [CrossRef] [PubMed]
- Grabiec, U.; Dehghani, F. N-Arachidonoyl dopamine: A novel endocannabinoid and endovanilloid with widespread physiological and pharmacological activities. Cannabis Cannabinoid Res. 2017, 2(1), 183–196. [Google Scholar] [CrossRef] [PubMed]
- Paria, B.C.; Wangm, H.; Dey, S.K. Endocannabinoid signaling in synchronizing embryo development and uterine receptivity for implantation. Chem. Phys. Lipids 2002, 121, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dey, S.K. Lipid signaling in embryo implantation. Prostaglandins Other Lipid. Mediat. 2004, 77, 84–102. [Google Scholar] [CrossRef]
- Sun, X. & Dey, K. Aspects of encocannabinoid signaling in periimplantation biology. Mol. Cell. Endocrinol. 2008, 286S, S3–S11. [Google Scholar] [CrossRef]
- Macarrone, M. CB2 receptors in reproduction. Br. J. Pharmacol. 2008, 153, 189–198. [Google Scholar] [CrossRef]
- Correa, F.; Wolfson, M.L.; Valchi, P.; Aisemberg, J.; Franchi, A.M. Endocannabinoid system and pregnancy. Reprod. 2016, 152, R191–R200. [Google Scholar] [CrossRef]
- Innocenzi, E.; De Domenico, E.; Ciccarone, F.; Zampieri, M.; Rossi, G.; Cicconi, R.; Bernardini, R.; Mattei, M.; Grimaldi, P. Paternal activation of CB2 cannabinoid receptor impairs placental and embryonic growth via an epigenetic mechanism. Sci. Rep. 2019, 9, 17034. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Paria, B.C.; Dey, S.K.; Armant, D.R. Stage-specific excitation of cannabinoid receptor exhibits differential effects on mouse embryonic development. Biol. Reprod. 1999, 60, 839–844. [Google Scholar] [CrossRef]
- Taylor, A.H.; Ang, C.; Bell, S.C.; Konje, J.C. The role of the endocannabinoid system in gametogenesis implantation and early pregnancy. Hum. Reprod. Update 2007, 13(5), 501–513. [Google Scholar] [CrossRef]
- Habayeb, O.M.H.; Taylor, A.H.; Bell, S.C.; Taylor, D.J.; Konje, J.C. Expression of the endocannabinoid system in human first trimester placenta and its role in trophoblast proliferation. Endocrinol. 2008, 149(10), 5052–5060. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xie, H.; Yang, J.; Wang, H.; Bradshaw, H.B.; Dey, S.K. Endocannabinoid signaling directs differentiation of trophoblast cell lineages and placentation. Proc. Natl. Acad. Sci. U S A 2010, 107(39), 16887–16892. [Google Scholar] [CrossRef]
- Jiang, S.; Fu, Y.; Williams, J.; Wood, J.; Pandarinathan, L.; Avraham, S.; Makriyannis, A.; Avraham, S.; Avraham, H.K. Expression and function of cannabinoid receptors CB1 and CB2 and their cognate cannabinoid ligands in murine embryonic stem cells. PLoS ONE 2007, 2(7), e641. [Google Scholar] [CrossRef]
- Sharov, A.; Piao, Y.; Matoba, R.; Dudekula, D.B.; Qian, Y.; VanBuren, V.; Falco, G.; Martin, P.R.; Stagg, C.A.; Bassey, U. C.; Wang, Y.; Carter, M. G.; Hamatani, T.; Aiba, K.; Akutsu, H.; Sharova, L.; Tanaka, T. S.; Kimber, W. L.; Yoshikawa, T.; Jaradat, S. A.; Pantano, S.; Nagaraja, R.; Boheler, K. R.; Taub, D.; Hodes, R. J.; Longo, D. L.; Schlessinger, D.; Keller, J.; Klotz, E.; Kelsoe, G.; Umezawa, A.; Vescovi, A. L.; Rossant, J.; Kunath, T.; Hogan, B.L.M.; Curci, A.; D’Urso, M.; Kelso, J.; Hide, W.; Ko, M.S.H. Transcriptome analysis of mouse stem cells and early embryos. PLoS Biol. 2003, 1(3), e74. [Google Scholar] [CrossRef] [PubMed]
- Bari, M.; Tedesco, M.; Battista, N.; Pasquariello, N.; Pucci, M; Gasperi, V.; Scaldaferri, M.L.; Farini, D.; De Felici, M.; Maccarrone, M. Characterization of the endocannabinoid system in mouse embryonic stem cells. Stem Cells Dev 2011, 20, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Nones, J.; Spohr, T.C.; Furtado, D.R.; Sartore, R.C.; Paulsen, B.S.; Guimarães, M.Z.; Rehen, S.K. Cannabinoids modulate cell survival in embryoid bodies. Cell Biol. Int. 2010, 34(4), 399–408. [Google Scholar] [CrossRef]
- Oh, H.A.; Kwon, S.; Choi, S.; Shin, H.; Yoon, K.H.; Kim, W.J.; Lim, H.J. Uncovering a role for endocannabinoid signaling in autophagy in preimplantation mouse embryos. Mol. Hum. Reprod. 2013, 19(2), 93–101. [Google Scholar] [CrossRef]
- Galve-Roperh, I.; Chiurchiù, V.; Díaz-Alonso, J.; Bari, M.; Guzmán, M.; Maccarrone, M. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog. Lip. Res. 2013, 52, 633–650. [Google Scholar] [CrossRef]
- Psychoyos, D.; Hungund, B.; Cooper, T.; Finneli, R. A cannabinoid analogue of Δ9-tetrahydrocannabinol disrupts neural development in chick. Birth Defects Res. 2008, 83, 477–488. [Google Scholar] [CrossRef]
- Ahmed, K.T.; Amin, M.R.; Shah, P.; Ali, D.W. Motor neuron development in zebrafish is altered by brief (5-hr) exposures to THC (∆9-tetrahydrocannabinol) or CBD (cannabidiol) during gastrulation. Sci. Rep. 2018, 8(1), 10518. [Google Scholar] [CrossRef]
- Amin, M.R.; Ahmed, K.T.; Ali, D.W. Early exposure to THC alters M-cell development in zebrafish embryos. Biomedicines 2020, 8(1), 5. [Google Scholar] [CrossRef] [PubMed]
- Kanyo, R.; Amin, M.R.; Locskai, L.F.; Bouvier, D.; Olthuis, A.M.; Allison, W.T.; Ali, D.W. Medium-throughput zebrafish optogenetic platform identifies deficits in subsequent neural activity following brief early exposure to cannabidiol and Δ9-tetrahydrocannabinol. Sci. Rep. 2021, 11, 11515. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.R.; Ahmed, K.T.; Ali, D.W. Cannabinoid receptor 2(Cb2r) mediates cannabinol (CBN) induced developmental defects in zebrafish. Sci. Rep. 2022, 12, 20251. [Google Scholar] [CrossRef] [PubMed]
- Khaliullina, H.; Bilgin, M.; Sampaio, J.L.; Shevchenko, A.; Eaton, S. Endocannabinoids are conserved inhibitors of the Hedgehog pathway. Proc. Natl. Acad. Sci. U S A 2015, 112(11), 3415–3420. [Google Scholar] [CrossRef] [PubMed]
- Boa-Amponsem, O.; Zhang, C.; Burton, D.; Williams, K.P.; Cole, G.J. Ethanol and cannabinoids regulate zebrafish GABAergic neuron development and behavior in a Sonic Hedgehog and Fibroblast Growth Factor-dependent mechanism. Alcohol Clin. Exp. Res. 2020, 44(7), 1366–1377. [Google Scholar] [CrossRef]
- Begbie, J.; Doherty, P.; Graham, A. Cannabinoid receptor, CB1, expression follows neuronal differentiation in the early chick embryo. J. Anat. 2004, 205(3), 213–218. [Google Scholar] [CrossRef]
- Psychoyos, D.; Vinod, K.Y.; Cao, J.; Xie, S.; Hyson, R.L.; Wlodarczyk, B.; He, W.; Cooper, T.B.; Hungund, B.L.; Finnell, R.H. Cannabinoid receptor 1 signaling in embryo neurodevelopment. Birth Defects Res. 2012, 95(2), 137–50. [Google Scholar] [CrossRef]
- Migliarini, B.; Carnevali, O. A novel role for the endocannabinoid system during zebrafish development. Mol. Cell Endocrinol. 2009, 299(2), 172–177. [Google Scholar] [CrossRef]
- Son, H.W.; Ali, D.W. Endocannabinoid receptor expression in early zebrafish development. Dev. Neurosci. 2022, 44(3), 142–152. [Google Scholar] [CrossRef]
- Buckley, N.E.; Hansson, S.; Harta, G.; Mezey, E. Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neuroscience 1998, 82(4), 1131–49. [Google Scholar] [CrossRef]
- Mulder, J.; Aguado, T.; Keimpema, E.; Barabás, K.; Ballester-Rosado, C.J.; Nguyen, L.; Monory, K.; Marsicano, G.; Di Marzo, V.; Hurd, Y.L.; Guillemot, F.; Mackie, K.; Lutz, B.; Guzmán, M.; Lu, H.C.; Galve-Roperh, I.; Harkany, T. Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc. Natl. Acad. Sci. U S A 2008, 105(25), 8760–8765. [Google Scholar] [CrossRef] [PubMed]
- Vitalis, T.; Lainé, J.; Simon, A.; Roland, A.; Leterrier, C.; Lenkei, Z. The type 1 cannabinoid receptor is highly expressed in embryonic cortical projection neurons and negatively regulates neurite growth in vitro. Eur. J. Neurosci. 2008, 28(9), 1705–1718. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Alonso, J.; Aguado, T.; Wu, CS.; Palazuelos, J.; Hofmann, C.; Garcez, P.; Guillemot, F.; Lu, H.C.; Lutz, B.; Guzmán, M.; Galve-Roperh, I. The CB(1) cannabinoid receptor drives corticospinal motor neuron differentiation through the Ctip2/Satb2 transcriptional regulation axis. J. Neurosci. 2012, 32(47), 16651–16665. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.; Chambers, D.; Hobbs, C.; Doherty, P.; Graham, A. The endocannabinoid receptor, CB1, is required for normal axonal growth and fasciculation. Mol. Cell. Neurosci. 2008, 38(1), 89–97. [Google Scholar] [CrossRef]
- Berrendero, F.; Sepe, N.; Ramos, J.A.; Di Marzo, V.; Fernández-Ruiz, J.J. Analysis of cannabinoid receptor binding and mRNA expression and endogenous cannabinoid contents in the developing rat brain during late gestation and early postnatal period. Synapse 1999, 33(3), 181–191. [Google Scholar] [CrossRef]
- Aguado, T.; Monory, K.; Palazuelos, J.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzmán, M.; Galve-Roperh, I. The endocannabinoid system drives neural progenitor proliferation. FASEB J. 2005, 19(12), 1704–1706. [Google Scholar] [CrossRef]
- Palazuelos, J.; Ortega, Z.; Díaz-Alonso, J.; Guzmán, M.; Galve-Roperh, I. CB2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J. Biol. Chem. 2012, 287(2), 1198–209. [Google Scholar] [CrossRef]
- Compagnucci, C.; Di Siena, S.; Bustamante, M.B.; Di Giacomo, D.; Di Tommaso, M.; Maccarrone, M.; Grimaldi, P.; Sette, C. Type-1 (CB1) cannabinoid receptor promotes neuronal differentiation and maturation of neural stem cells. PLoS ONE 2013, 8(1), e54271. [Google Scholar] [CrossRef]
- Morozov, Y.M.; Torii, M.; Rakic, P. Origin, early commitment, migratory routes, and destination of cannabinoid type 1 receptor-containing interneurons. Cereb. Cortex 2009, 19 (Suppl 1), i78–89. [Google Scholar] [CrossRef]
- Díaz-Alonso, J.; Aguado, T.; de Salas-Quiroga, A.; Ortega, Z.; Guzmán, M.; Galve-Roperh, I. CB1 cannabinoid receptor-dependent activation of mTORC1/Pax6 signaling drives Tbr2 expression and basal progenitor expansion in the developing mouse cortex. Cereb. Cortex 2015, 25(9), 2395–2408. [Google Scholar] [CrossRef]
- Morozov, Y.M.; Mackie, K.; Rakic, P. Cannabinoid type 1 receptor is undetectable in rodent and primate cerebral neural stem cells but participates in radial neuronal migration. Int. J. Mol. Sci. 2020, 21(22), 8657. [Google Scholar] [CrossRef] [PubMed]
- Keimpema, E.; Barabas, K.; Morozov, Y.M.; Tortoriello, G.; Torii, M.; Cameron, G.; Yanagawa, Y.; Watanabe, M.; Mackie, K.; Harkany, T. Differential subcellular recruitment of monoacylglycerol lipase generates spatial specificity of 2-arachidonoyl glycerol signaling during axonal pathfinding. J. Neurosci. 2010, 30(42), 13992–134007. [Google Scholar] [CrossRef] [PubMed]
- Saez, T.M.; Aronne, M.P.; Caltana, L.; Brusco, A.H. Prenatal exposure to the CB1 and CB2 cannabinoid receptor agonist WIN 55,212-2 alters migration of early-born glutamatergic neurons and GABAergic interneurons in the rat cerebral cortex. J Neurochem. 2014, 129(4), 637–648. [Google Scholar] [CrossRef] [PubMed]
- Zurolo, E.; Iyer, A.M.; Spliet, W.G.; Van Rijen, P.C.; Troost, D.; Gorter, J.A.; Aronica, E. CB1 and CB2 cannabinoid receptor expression during development and in epileptogenic developmental pathologies. Neuroscience 2010, 170(1), 28–41. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Alonso, J.; de Salas-Quiroga, A.; Paraíso-Luna, J.; García-Rincón, D.; Garcez, P.P.; Parsons, M.; Andradas, C.; Sánchez, C.; Guillemot, F.; Guzmán, M.; Galve-Roperh, I. Loss of cannabinoid CB1 receptors induces cortical migration malformations and increases seizure susceptibility. Cereb. Cortex 2017, 27(11), 5303–5317. [Google Scholar] [CrossRef]
- Morozov, Y.M.; Freund, T.F. Post-natal development of type 1 cannabinoid receptor immunoreactivity in the rat hippocampus. Eur. J. Neurosci. 2003, 18(5), 1213–1222. [Google Scholar] [CrossRef]
- Berghuis, P.; Rajnicek, A.M.; Morozov, Y.M.; Ross, R.A.; Mulder, J.; Urbán, G.M.; Monory, K.; Marsicano, G.; Matteoli, M.; Canty, A.; Irving, A.J.; Katona, I.; Yanagawa, Y.; Rakic, P.; Lutz, B.; Mackie, K.; Harkany, T. Hardwiring the brain: Endocannabinoids shape neuronal connectivity. Science 2007, 316(5828), 1212–1216. [Google Scholar] [CrossRef]
- Paraíso-Luna, J.; Aguareles, J.; Martín, R.; Ayo-Martín, A.C.; Simón-Sánchez, S.; García-Rincón, D.; Costas-Insua, C.; García-Taboada, E.; de Salas-Quiroga, A.; Díaz-Alonso, J.; Liste, I.; Sánchez-Prieto, J.; Cappello, S.; Guzmán, M.; Galve-Roperh, I. Endocannabinoid signalling in stem cells and cerebral organoids drives differentiation to deep layer projection neurons via CB1 receptors. Development 2020, 147(24), dev192161. [Google Scholar] [CrossRef]
- Wu, C.S.; Zhu, J.; Wager-Miller, J.; Wang, S.; O’Leary, D.; Monory, K.; Lutz, B.; Mackie, K.; Lu, H.C. Requirement of cannabinoid CB(1) receptors in cortical pyramidal neurons for appropriate development of corticothalamic and thalamocortical projections. Eur. J. Neurosci. 2010, 32(5), 693–706. [Google Scholar] [CrossRef]
- Barnes, J.L.; Mohr, C.; Ritchey, C.R.; Erikson, C.M.; Shiina, H.; Rossi, D.J. Developmentally transient CB1Rs on cerebellar afferents suppress afferent input, dowstream synaptic excitation, and signaling to migrating neurons. J. Neurosci. 2020, 40(32), 6133–6145. [Google Scholar] [CrossRef]
- Martinez, L.R.; Black, K.C.; Webb, B.T.; Bell, A.; Baygani, S.K.; Mier, T.J.; Dominguez, L.; Mackie, K.; Kalinovsky, A. Components of endocannabinoid signaling system are expressed in the perinatal mouse cerebellum and required for its normal development. eNeuro 2020, 7(2), 0471–0519. [Google Scholar] [CrossRef] [PubMed]
- Gómez, M.; Hernández, M.L.; Pazos, M.R.; Tolón, R.M.; Romero, J.; Fernández-Ruiz, J. Colocalization of CB1 receptors with L1 and GAP-43 in forebrain white matter regions during fetal rat brain development: Evidence for a role of these receptors in axonal growth and guidance. Neuroscience 2008, 153(3), 687–699. [Google Scholar] [CrossRef]
- Biegon, A.; Kerman, I.A. Autoradiographic study of pre- and postnatal distribution of cannabinoid receptors in human brain. Neuroimage 2001, 14(6), 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Mato, S.; Del Olmo, E.; Pazos, A. Ontogenetic development of cannabinoid receptor expression and signal transduction functionality in the human brain. Eur. J. Neurosci. 2003, 17(9), 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dow-Edwards, D.; Keller, E.; Hurd, Y.L. Preferential limbic expression of the cannabinoid receptor mRNA in the human fetal brain. Neuroscience 2003, 118(3), 681–694. [Google Scholar] [CrossRef] [PubMed]
- Keimpema, E.; Tortoriello, G.; Alpár, A.; Capsoni, S.; Arisi, I.; Calvigioni, D.; Hu, S.S.; Cattaneo, A.; Doherty, P.; Mackie, K.; Harkany, T. Nerve growth factor scales endocannabinoid signaling by regulating monoacylglycerol lipase turnover in developing cholinergic neurons. Proc. Natl. Acad. Sci. USA 2013, 110(5), 1935–1940. [Google Scholar] [CrossRef] [PubMed]
- Berrendero, F.; Sepe, N.; Ramos, J.A.; Di Marzo, V.; Fernández-Ruiz, J.J. Analysis of cannabinoid receptor binding and mRNA expression and endogenous cannabinoid contents in the developing rat brain during late gestation and early postnatal period. Synapse 1999, 33(3), 181–191. [Google Scholar] [CrossRef]
- Paria, B.C.; Song, H.; Wang, X.; Schmid, P.C.; Krebsbach, R.J.; Schmid, H.H.; Bonner, T.I.; Zimmer, A.; Dey, S.K. Dysregulated cannabinoid signaling disrupts uterine receptivity for embryo implantation. J. Biol. Chem. 2001, 276(23), 20523–20528. [Google Scholar] [CrossRef]
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A.; Matias, I.; Schiano-Moriello, A.; Paul, P.; Williams, E.J.; Gangadharan, U.; Hobbs, C.; Di Marzo, V.; Doherty, P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell. Biol. 2003, 163(3), 463–468. [Google Scholar] [CrossRef]
- Keimpema, E.; Alpár, A.; Howell, F.; Malenczyk, K.; Hobbs, C.; Hurd, Y.L.; Watanabe, M.; Sakimura, K.; Kano, M.; Doherty, P.; Harkany, T. Diacylglycerol lipase α manipulation reveals developmental roles for intercellular endocannabinoid signaling. Sci. Rep. 2013, 3, 2093. [Google Scholar] [CrossRef]
- Palazuelos, J.; Aguado, T.; Egia, A.; Mechoulam, R.; Guzmán, M.; Galve-Roperh, I. Non-psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J. 2006, 20(13), 2405–2407. [Google Scholar] [CrossRef] [PubMed]
- Molina-Holgado, F.; Rubio-Araiz, A.; García-Ovejero, D.; Williams, R.J.; Moore, J.D.; Arévalo-Martín, A.; Gómez-Torres, O.; Molina-Holgado, E. CB2 cannabinoid receptors promote mouse neural stem cell proliferation. Eur. J. Neurosci. 2007, 25(3), 629–634. [Google Scholar] [CrossRef]
- Duff, G.; Argaw, A.; Cecyre, B.; Cherif, H.; Tea, N.; Zabouri, N.; Casanova, C.; Ptito, M.; Bouchard, J.F. Cannabinoid receptor CB2 modulates axon guidance. PLoS ONE 2013, 8(8), e70849. [Google Scholar] [CrossRef] [PubMed]
- Alpár, A.; Tortoriello, G.; Calvigioni, D.; Niphakis, M.J.; Milenkovic, I.; Bakker, J.; Cameron, G.A.; Hanics, J.; Morris, C.V.; Fuzik, J.; Kovacs, G.G.; Cravatt, B.F.; Parnavelas, J.G.; Andrews, W.D.; Hurd, Y.L; Keimpema, E.; Harkany, T. Endocannabinoids modulate cortical development by configuring Slit2/Robo1 signalling. Nat. Commun. 2014, 5, 4421. [Google Scholar] [CrossRef] [PubMed]
- Cavanaugh, D.J.; Chesler, A.T.; Jackson, A.C.; Sigal, Y.M.; Yamanaka, H.; Grant, R.; O’Donnell, D.; Nicoll, R.A.; Shah, N.M.; Julius, D.; Basbaum, A.I. Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J. Neurosci. 2011, 31(13), 5067–5077. [Google Scholar] [CrossRef]
- Perchuk, A.; Bierbower, S.M.; Canseco-Alba, A.; Mora, Z.; Tyrell, L.; Joshi, N.; Schanz, N.; Gould, G.G.; Onaivi, E.S. Developmental and behavioral effects in neonatal and adult mice following prenatal activation of endocannabinoid receptors by capsaicin. Acta Pharmacol. Sin. 2019, 40(3), 418–424. [Google Scholar] [CrossRef]
- Cherif, H.; Argaw, A.; Cécyre, B.; Bouchard, A.; Gagnon, J.; Javadi, P.; Desgent, S.; Mackie, K.; Bouchard, J.F. Role of GPR55 during axon growth and target innervation. eNeuro 2015, 2, ENEURO.0011-15.2015. [Google Scholar] [CrossRef]
- de Salas-Quiroga, A.; Díaz-Alonso, J.; García-Rincón, D.; Remmers, F.; Vega, D.; Gómez-Cañas, M.; Lutz, B.; Guzmán, M.; Galve-Roperh, I. Prenatal exposure to cannabinoids evokes long-lasting functional alterations by targeting CB1 receptors on developing cortical neurons. Proc. Natl. Acad. Sci. U S A 2015, 112(44), 13693–13698. [Google Scholar] [CrossRef]
- Rogers, S.A.; Kempen, T.A.; Pickel, V.M.; Milner, T.A. Enkephalin levels and the number of neuropeptide Y-containing interneurons in the hippocampus are decreased in female cannabinoid-receptor 1 knock-out mice. Neurosci. Lett. 2016, 620, 97–103. [Google Scholar] [CrossRef]
- de Salas-Quiroga, A.; García-Rincón, D.; Gómez-Domínguez, D.; Valero, M.; Simón-Sánchez, S.; Paraíso-Luna, J.; Aguareles, J.; Pujadas, M.; Muguruza, C.; Callado, L.F.; Lutz, B.; Guzmán, M.; de la Prida, L.M.; Galve-Roperh, I. Long-term hippocampal interneuronopathy drives sex-dimorphic spatial memory impairment induced by prenatal THC exposure. Neuropsychopharmacology 2020, 45(5), 877–886. [Google Scholar] [CrossRef]
- Rubio-Araiz, A.; Arévalo-Martín, A.; Gómez-Torres, O.; Navarro-Galve, B.; García-Ovejero, D.; Suetterlin, P.; Sánchez-Heras, E.; Molina-Holgado, E.; Molina-Holgado, F. The endocannabinoid system modulates a transient TNF pathway that induces neural stem cell proliferation. Mol. Cell Neurosci. 2008, 38, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Trazzi, S.; Steger, M.; Mitrugno, V.M.; Bartesaghi, R.; Ciani, E. CB1 cannabinoid receptors increase neuronal precursor proliferation through AKT/glycogen synthase kinase-3beta/beta-catenin signaling. J. Biol. Chem. 2010, 285(13), 10098–10109. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Xie, L.; Kim, S.H.; Parmentier-Batteur, S.; Sun, Y.; Mao, X.O.; Childs, J.; Greenberg, D.A. Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol. Pharmacol. 2004, 66(2), 204–208. [Google Scholar] [CrossRef] [PubMed]
- Aguado, T.; Palazuelos, J.; Monory, K.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzmán, M.; Galve-Roperh, I. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J. Neurosci. 2006, 26(5), 1551–1561. [Google Scholar] [CrossRef]
- Rueda, D.; Navarro, B.; Martinez-Serrano, A.; Guzman, M.; Galve-Roperh, I. The endocannabinoid anandamide inhibits neuronal progenitor cell differentiation through attenuation of the Rap1/B-Raf/ERK pathway. J. Biol. Chem. 2002, 277(48), 46645–46650. [Google Scholar] [CrossRef]
- Jiang, W.; Zhang, Y.; Xiao, L.; Van Cleemput, J.; Ji, S.P.; Bai, G.; Zhang, X. Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic- and antidepressant-like effects. J. Clin. Invest. 2005, 115, 3104–3116. [Google Scholar] [CrossRef]
- Soltys, J.; Yushak, M.; Mao-Draayer, Y. Regulation of neural progenitor cell fate by anandamide. Biochem. Biophys. Res. Commun. 2010, 400(1), 21–26. [Google Scholar] [CrossRef]
- Hill, J.D.; Zuluaga-Ramirez, V.; Gajghate, S.; Winfield, M.; Persidsky, Y. Activation of GPR55 increases neural stem cell proliferation and promotes early adult hippocampal neurogenesis. Br. J. Pharmacol. 2018, 175(16), 3407–3421. [Google Scholar] [CrossRef]
- Abbas-Farishta, R.; Robert, C.; Turcot, O.; Thomas, S.; Vanni, M.P.; Bouchard, J.F.; Casanova, C. Impact of CB1 receptor deletion on visual responses and organization of primary visual cortex in adult mice. Invest. Ophthalmol. Vis. Sci. 2015, 56(13), 7697–7707. [Google Scholar] [CrossRef]
- Hedrich, J.; Angamo, E.A.; Conrad, A.; Lutz, B.; Luhmann, H.J. Cell type specific impact of cannabinoid receptor signaling in somatosensory barrel map formation in mice. J. Comp. Neurol. 2020, 528(1), 3–13. [Google Scholar] [CrossRef]
- Tortoriello, G.; Morris, C.V.; Alpar, A.; Fuzik, J.; Shirran, S.L.; Calvigioni, D.; Keimpema, E.; Botting, C.H.; Reinecke, K.; Herdegen, T.; Courtney, M.; Hurd, Y.L.; Harkany, T. Miswiring the brain: Δ9-tetrahydrocannabinol disrupts cortical development by inducing an SCG10/stathmin-2 degradation pathway. EMBO J. 2014, 33(7), 668–685. [Google Scholar] [CrossRef] [PubMed]
- Turunen, P.M.; Louhivuori, L.M.; Louhivuori, V.; Kukkonen, J.P.; Åkerman, K.E. Endocannabinoid signaling in embryonic neuronal motility and cell-cell contact - Role of mGluR5 and TRPC3 Channels. Neuroscience 2018, 375, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, Y.; Hisanaga, S.; Heizmann, C.W.; Murakami, F. Distinct migratory behavior of early- and late-born neurons derived from the cortical ventricular zone. J. Comp. Neurol. 2004, 479(1), 1–14. [Google Scholar] [CrossRef]
- Calderon de Anda, F.; Gärtner, A.; Tsai, L.H.; Dotti, C.G. Pyramidal neuron polarity axis is defined at the bipolar stage. J. Cell Sci. 2008, 121(Pt 2) Pt 2, 178–185. [Google Scholar] [CrossRef]
- Jossin, Y.; Cooper, J.A. Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat. Neurosci. 2011, 14(6), 697–703. [Google Scholar] [CrossRef]
- Nadarajah, B.; Brunstrom, J.E.; Grutzendler, J.; Wong, R.O.; Pearlman, A.L. Two modes of radial migration in early development of the cerebral cortex. Nat. Neurosci. 2001, 4(2), 143–50. [Google Scholar] [CrossRef] [PubMed]
- Noctor, S.C.; Martínez-Cerdeño, V.; Ivic, L.; Kriegstein, A.R. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci. 2004, 7(2), 136–44. [Google Scholar] [CrossRef]
- Franco, S.J.; Martinez-Garay, I.; Gil-Sanz, C.; Harkins-Perry, S.R.; Müller, U. Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron. 2011, 69(3), 482–97. [Google Scholar] [CrossRef]
- Gil-Sanz, C.; Franco, S.J.; Martinez-Garay, I.; Espinosa, A.; Harkins-Perry, S.; Müller, U. Cajal-Retzius cells instruct neuronal migration by coincidence signaling between secreted and contact-dependent guidance cues. Neuron. 2013, 79, 461–477. [Google Scholar] [CrossRef]
- Polleux, F.; Whitford, K.L.; Dijkhuizen, P.A.; Vitalis, T.; Ghosh, A. Control of cortical interneuron migration by neurotrophins and PI3-kinase signaling. Development. 2002, 129, 3147–3160. [Google Scholar] [CrossRef]
- Fukumitsu, H.; Ohtsuka, M.; Murai, R.; Nakamura, H.; Itoh, K.; Furukawa, S. Brain-derived neurotrophic factor participates in determination of neuronal laminar fate in the developing mouse cerebral cortex. J. Neurosci. 2006, 26(51), 13218–30. [Google Scholar] [CrossRef] [PubMed]
- Flames, N.; Long, J.E.; Garratt, A.N.; Fischer, T.M.; Gassmann, M.; Birchmeier, C.; Lai, C.; Rubenstein, J.L.; Marín, O. Short- and long-range attraction of cortical GABAergic interneurons by neuregulin-1. Neuron. 2004, 44(2), 251–61. [Google Scholar] [CrossRef]
- Du, H.; Kwon, I.K.; Kim, J. Neuregulin-1 impairs the long-term depression of hippocampal inhibitory synapses by facilitating the degradation of endocannabinoid 2-AG. J. Neurosci. 2013, 33(38), 15022–31. [Google Scholar] [CrossRef] [PubMed]
- Gomez, O.; Arevalo-Martin, A.; Garcia-Ovejero, D.; Ortega-Gutierrez, S.; Cisneros, J.A.; Almazan, G.; Sánchez-Rodriguez, M.A.; Molina-Holgado, F.; Molina-Holgado, E. The constitutive production of the endocannabinoid 2-arachidonoylglycerol participates in oligodendrocyte differentiation. Glia 2010, 58, 1913–1927. [Google Scholar] [CrossRef]
- Molina-Holgado, E.; Vela, J.M.; Arévalo-Martín, A.; Almazán, G.; Molina-Holgado, F.; Borrell, J.; Guaza, C. Cannabinoids promote oligodendrocyte progenitor survival: Involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J. Neurosci. 2002, 22(22), 9742–53. [Google Scholar] [CrossRef]
- Gomez, O.; Sanchez-Rodriguez, M.A.; Ortega-Gutierrez, S.; Vazquez-Villa, H.; Guaza, C.; Molina-Holgado, F.; Molina-Holgado, E. A basal tone of 2-arachidonoylglycerol contributes to early oligodendrocyte progenitor proliferation by activating phosphatidylinositol 3-kinase (PI3K)/akt and the mammalian target of rapamycin (mTOR) pathways. J. Neuroimmune Pharmacol. 2015, 10(2), 309–317. [Google Scholar] [CrossRef] [PubMed]
- Gomez, O.; Sanchez-Rodriguez, A.; Le, M.; Sanchez-Caro, C.; Molina-Holgado, F.; Molina-Holgado, E. Cannabinoid receptor agonists modulate oligodendrocyte differentiation by activating PI3K/Akt and the mammalian target of rapamycin (mTOR) pathways. Br. J. Pharmacol. 2011, 163(7), 1520–32. [Google Scholar] [CrossRef]
- Arévalo-Martín, A.; García-Ovejero, D.; Rubio-Araiz, A.; Gómez, O.; Molina-Holgado, F.; Molina-Holgado, E. Cannabinoids modulate Olig2 and polysialylated neural cell adhesion molecule expression in the subventricular zone of post-natal rats through cannabinoid receptor 1 and cannabinoid receptor 2. Eur. J. Neurosci. 2007, 26(6), 1548–59. [Google Scholar] [CrossRef]
- Huerga-Gómez A, Aguado T, Sánchez-de la Torre A, Bernal-Chico A, Matute C, Mato S, Guzmán M, Galve-Roperh I, Palazuelos J. Δ9-Tetrahydrocannabinol promotes oligodendrocyte development and CNS myelination in vivo. Glia. 2021, 69, 532–545. [CrossRef]
- Leterrier, C. ; Lainé J, Darmon, M.; Boudin, H.; Rossier, J.; Lenkei, Z. Constitutive activation drives compartment-selective endocytosis and axonal targeting of type 1 cannabinoid receptors. J. Neurosci. 2006, 26, 3143–3153. [Google Scholar] [CrossRef]
- Argaw, A.; Duff, G.; Zabouri, N.; Cécyre, B.; Chainé, N.; Cherif, H.; Tea, N.; Lutz, B.; Ptito, M.; Bouchard, J.F. Concerted action of CB1 cannabinoid receptor and deleted in colorectal cancer in axon guidance. J. Neurosci. 2011, 31(4), 1489–99. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.J.; Walsh, F.S.; Doherty, P. The FGF receptor uses the endocannabinoid signaling system to couple to an axonal growth response. J. Cell Biol. 2003, 160(4), 481–6. [Google Scholar] [CrossRef]
- Zuccarini, G.; D’Atri, I.; Cottone, E.; Mackie, K.; Shainer, I.; Gothilf, Y.; Provero, P.; Bovolin, P.; Merlo, G.R. Interference with the Cannabinoid Receptor CB1R Results in Miswiring of GnRH3 and AgRP1 Axons in Zebrafish Embryos. Int. J. Mol Sci. 2019, 21(1), 168. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; Berrendero, F.; Hernández, M.L.; Ramos, J.A. The endogenous cannabinoid system and brain development. Trends Neurosci. 2000, 23(1), 14–20. [Google Scholar] [CrossRef]
- Itami, C.; Huang, J.Y.; Yamasaki, M.; Watanabe, M.; Lu, H.C.; Kimura, F. Developmental switch in spike timing-dependent plasticity and cannabinoid-dependent reorganization of the thalamocortical projection in the barrel cortex. J. Neurosci. 2016, 36(26), 7039–54. [Google Scholar] [CrossRef]
- Walker, D.J.; Suetterlin, P.; Reisenberg, M.; Williams, G.; Doherty, P. Down-regulation of diacylglycerol lipase-alpha during neural stem cell differentiation: Identification of elements that regulate transcription. J. Neurosci. Res. 2010, 88(4), 735–745. [Google Scholar] [CrossRef]
- Crittenden, J.R.; Yoshida, T.; Venu, S.; Mahar, A.; Graybiel, A.M. Cannabinoid receptor 1 is required for neurodevelopment of striosome-dendron bouquets. eNeuro. 2022, 9, ENEURO.0318-21.2022. [Google Scholar] [CrossRef] [PubMed]
- Black, B.; Vishwakarma, V.; Dhakal, K.; Bhattarai, S.; Pradhan, P.; Jain, A.; Kim, Y.T.; Mohanty, S. Spatial temperature gradients guide axonal outgrowth. Sci. Rep. 2016, 6, 29876. [Google Scholar] [CrossRef]
- Roland, A.B.; Ricobaraza, A.; Carrel, D.; Jordan, B.M.; Rico, F.; Simon, A.; Humbert-Claude, M.; Ferrier, J.; McFadden, M.H.; Scheuring, S.; Lenkei, Z. Cannabinoid-induced actomyosin contractility shapes neuronal morphology and growth. Elife. 2014, 3, e03159. [Google Scholar] [CrossRef]
- Njoo, C.; Agarwal, N.; Lutz, B.; Kuner, R. The Cannabinoid receptor CB1 interacts with the WAVE1 complex and plays a role in actin dynamics and structural plasticity in neurons. PLoS Biol. 2015, 13(10), e1002286. [Google Scholar] [CrossRef]
- Moore, S.W.; Correia, J.P.; Lai Wing Sun, K.; Pool, M.; Fournier, A.E.; Kennedy, T.E. Rho inhibition recruits DCC to the neuronal plasma membrane and enhances axon chemoattraction to netrin 1. Development. 2008, 135(17), 2855–64. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bashaw, G.J. Son of sevenless directly links the Robo receptor to rac activation to control axon repulsion at the midline. Neuron. 2006, 52(4), 595–607. [Google Scholar] [CrossRef] [PubMed]
- Manna, T.; Grenningloh, G.; Miller, H.P.; Wilson, L. Stathmin family protein SCG10 differentially regulates the plus and minus end dynamics of microtubules at steady state in vitro: Implications for its role in neurite outgrowth. Biochemistry. 2007, 46(11), 3543–52. [Google Scholar] [CrossRef] [PubMed]
- Saez, T.M.M.; Fernandez-Bessone, I.; Rodriguez, M.S.; Alloatti, M.; Otero, M.G.; Cromberg, L.E.; Pozo-Devoto, V.M.; Oubiña, G.; Sosa, L.; Buffone, M.G.; Gelman, D.M.; Falzone, T.L. Kinesin-1-mediated axonal transport of CB1 receptors is required for cannabinoid-dependent axonal growth and guidance. Development 2020, 147(8), dev184069. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Thayer, S.A. Cannabinoids inhibit the formation of new synapses between hippocampal neurons in culture. J. Neurosci. 2001, 21(10), RC146. [Google Scholar] [CrossRef]
- Papariello, A.; Taylor, D.; Soderstrom, K.; Litwa, K. CB1 antagonism increases excitatory synaptogenesis in a cortical spheroid model of fetal brain development. Sci. Rep. 2021, 11(1), 9356. [Google Scholar] [CrossRef]
- Hu, H.Y.; Kruijssen, D.L.H.; Frias, C.P.; Rózsa, B.; Hoogenraad, C.C.; Wierenga, C.J. Endocannabinoid Signaling Mediates Local Dendritic Coordination between Excitatory and Inhibitory Synapses. Cell Rep. 2019, 27, 666–675.e5. [Google Scholar] [CrossRef]
- Liang, J.; Kruijssen, D.L.H.; Verschuuren, A.C.J.; Voesenek, B.J.B.; Benavides, F.F.W.; Sáez Gonzalez, M.; Ruiter, M.; Wierenga, C.J. Axonal CB1 Receptors Mediate Inhibitory Bouton Formation via cAMP Increase and PKA. J. Neurosci. 2021, 41(40), 8279–8296. [Google Scholar] [CrossRef]
- Deshmukh, S.; Onozuka, K.; Bender, K.J.; Bender, V.A.; Lutz, B.; Mackie, K.; Feldman, D.E. Postnatal development of cannabinoid receptor type 1 expression in rodent somatosensory cortex. Neuroscience. 2007, 145(1), 279–87. [Google Scholar] [CrossRef]
- Liu, C.H.; Heynen, A.J.; Shuler, M.G.; Bear, M.F. Cannabinoid receptor blockade reveals parallel plasticity mechanisms in different layers of mouse visual cortex. Neuron. 2008, 58(3), 340–5. [Google Scholar] [CrossRef]
- Li, L.; Bender, K.J.; Drew, P.J.; Jadhav, S.P.; Sylwestrak, E.; Feldman, D.E. Endocannabinoid signaling is required for development and critical period plasticity of the whisker map in somatosensory cortex. Neuron. 2009, 64(4), 537–49. [Google Scholar] [CrossRef] [PubMed]
- Bender, K.J.; Allen, C.B.; Bender, V.A.; Feldman, D.E. Synaptic basis for whisker deprivation-induced synaptic depression in rat somatosensory cortex. J. Neurosci. 2006, 26(16), 4155–65. [Google Scholar] [CrossRef] [PubMed]
- Nevian, T.; Sakmann, B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J. Neurosci. 2006, 26(43), 11001–13. [Google Scholar] [CrossRef] [PubMed]
- Rubino, T.; Prini, P.; Piscitelli, F.; Zamberletti, E.; Trusel, M.; Melis, M.; Sagheddu, C.; Ligresti, A.; Tonini, R.; Di Marzo, V.; Parolaro, D. Adolescent exposure to THC in female rats disrupts developmental changes in the prefrontal cortex. Neurobiol. Dis. 2015, 73, 60–9. [Google Scholar] [CrossRef]
- Miller, M.L.; Chadwick, B.; Dickstein, D.L.; Purushothaman, I.; Egervari, G.; Rahman, T.; Tessereau, C.; Hof, P.R.; Roussos, P.; Shen, L.; Baxter, M.G.; Hurd, Y.L. Adolescent exposure to Δ9-tetrahydrocannabinol alters the transcriptional trajectory and dendritic architecture of prefrontal pyramidal neurons. Mol. Psychiatry. 2019, 24(4), 588–600. [Google Scholar] [CrossRef]
- Filbey, F.M.; McQueeny, T.; DeWitt, S.J.; Mishra, V. Preliminary findings demonstrating latent effects of early adolescent marijuana use onset on cortical architecture. Dev. Cogn. Neurosci. 2015, 16, 16–22. [Google Scholar] [CrossRef]
- Huang, Y.; Yasuda, H.; Sarihi, A.; Tsumoto, T. Roles of endocannabinoids in heterosynaptic long-term depression of excitatory synaptic transmission in visual cortex of young mice. J Neurosci. 2008, 28(28), 7074–83. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Huang, Y.; Tsumoto, T. Regulation of excitability and plasticity by endocannabinoids and PKA in developing hippocampus. Proc. Natl. Acad. Sci. U S A. 2008, 105(8), 3106–11. [Google Scholar] [CrossRef]
- Scheyer, A.F.; Borsoi, M.; Wager-Miller, J.; Pelissier-Alicot, A.L.; Murphy, M.N.; Mackie, K.; Manzoni, O.J.J. Cannabinoid exposure via lactation in rats disrupts perinatal programming of the gamma-aminobutyric acid trajectory and select early-life behaviors. Biol. Psychiatry. 2020, 87(7), 666–677. [Google Scholar] [CrossRef]
- García-Gil, L.; De Miguel, R.; Muñoz, R.M.; Cebeira, M.; Villanua, M.A.; Ramos, J.A.; Fernández-Ruiz, J.J. Perinatal delta(9)-tetrahydrocannabinol exposure alters the responsiveness of hypothalamic dopaminergic neurons to dopamine-acting drugs in adult rats. Neurotoxicol. Teratol. 1997, 19(6), 477–87. [Google Scholar] [CrossRef]
- Wang, X.; Dow-Edwards, D.; Anderson, V.; Minkoff, H.; Hurd, Y.L. In utero marijuana exposure associated with abnormal amygdala dopamine D2 gene expression in the human fetus. Biol. Psychiatry. 2004, 56(12), 909–15. [Google Scholar] [CrossRef] [PubMed]
- DiNieri, J.A.; Wang, X.; Szutorisz, H.; Spano, S.M.; Kaur, J.; Casaccia, P.; Dow-Edwards, D.; Hurd, Y.L. Maternal cannabis use alters ventral striatal dopamine D2 gene regulation in the offspring. Biol. Psychiatry. 2011, 70(8), 763–769. [Google Scholar] [CrossRef] [PubMed]
- Molina-Holgado, F.; Amaro, A.; González, M.I.; Alvarez, F.J.; Leret, M.L. Effect of maternal delta 9-tetrahydrocannabinol on developing serotonergic system. Eur. J. Pharmacol. 1996, 316(1), 39–42. [Google Scholar] [CrossRef]
- Molina-Holgado, F.; Alvarez, F.J.; Gonzalez, I.; Antonio, M.T.; Leret, M.L. Maternal exposure to delta 9-tetrahydrocannabinol (Δ9-THC) alters indolamine levels and turnover in adult male and female rat brain regions. Brain Res. Bull. 1997, 43(2), 173–8. [Google Scholar] [CrossRef]
- Giedd, J.N.; Blumenthal, J.; Jeffries, N.O.; Castellanos, F.X.; Liu, H.; Zijdenbos, A.; Paus, T.; Evans, A.C.; Rapoport, J.L. Brain development during childhood and adolescence: A longitudinal MRI study. Nat. Neurosci. 1999, 2(10), 861–863. [Google Scholar] [CrossRef]
- Sicher, A.R.; Duerr, A.; Starnes, W.D.; Crowley, N.A. Adolescent alcohol and stress exposure rewires key cortical neurocircuitry. Front. Neurosci. 2022, 16, 896880. [Google Scholar] [CrossRef]
- Huttenlocher, P.R.; Dabholkar, A.S. Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 1997, 387(2), 167–178. [Google Scholar] [CrossRef]
- Sturman, D.A.; Moghaddam, B. The neurobiology of adolescence: Changes in brain architecture, functional dynamics, and behavioral tendencies. Neurosci. Biobehav. Rev. 2011, 35(8), 1704–1712. [Google Scholar] [CrossRef] [PubMed]
- Paus, T.; Keshavan, M.; Giedd, J.N. Why do many psychiatric disorders emerge during adolescence? Rev. Neurosci. 2008, 9(12), 947–957. [Google Scholar] [CrossRef]
- Gogtay, N.; Giedd, J.N.; Lusk, L.; Hayashi, K.M.; Greenstein, D.; Vaituzis, A.C.; Nugent, T.F. 3rd; Herman, D.H.; Clasen, L.S.; Toga, A.W.; Rapoport, J.L.; Thompson, P.M. Dynamic mapping of human cortical development during childhood through early adulthood. Proc. Natl. Acad. Sci. USA 2004, 101(21), 8174–8179. [Google Scholar] [CrossRef]
- Sowell, E.R.; Peterson, B.S.; Thompson, P.M.; Welcome, S.E.; Henkenius, A.L.; Toga, A.W. Mapping cortical change across the human life span. Nat. Neurosci. 2003, 6(3), 309–315. [Google Scholar] [CrossRef] [PubMed]
- Paus, T.; Zijdenbos, A.; Worsley, K.; Collins, D.L.; Blumenthal, J.; Giedd, J.N.; Rapoport, J.L.; Evans, A.C. Structural maturation of neural pathways in children and adolescents: In vivo study. Science 1999, 283(5409), 1908–1911. [Google Scholar] [CrossRef] [PubMed]
- Paus, T.; Collins, D.L.; Evans, A.C.; Leonard, G.; Pike, B.; Zijdenbos, A. Maturation of white matter in the human brain: A review of magnetic resonance studies. Brain Res. Bull. 2001, 54(3), 255–266. [Google Scholar] [CrossRef]
- Hwang, K.; Velanova, K.; Luna, B. Strengthening of top-down frontal cognitive control networks underlying the development of inhibitory control: A functional magnetic resonance imaging effective connectivity study. J. Neurosci. 2010, 30(46), 15535–15545. [Google Scholar] [CrossRef]
- Luna, B.; Padmanabhan, A.; O’Hearn, K. What has fMRI told us about the development of cognitive control through adolescence? Brain Cogn. 2010, 72(1), 101–113. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, L.; Albert, D.; Cauffman, E.; Banich, M.; Graham, S.; Woolard, J. Age differences in sensation seeking and impulsivity as indexed by behavior and self-report: Evidence for a dual systems model. Dev. Psychol. 2008, 44(6), 1764–1778. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Baler, R.D.; Compton, W.M.; Weiss, S.R. Adverse health effects of marijuana use. N. Engl. J. Med. 2014, 370(23), 2219–2227. [Google Scholar] [CrossRef]
- Lubman, D.I.; Cheetham, A.; Yücel, M. Cannabis and adolescent brain development. Pharmacol. Ther. 2015, 148, 1–16. [Google Scholar] [CrossRef]
- Peters, K.Z.; Naneix, F. The role of dopamine and endocannabinoid systems in prefrontal cortex development: Adolescence as a critical period. Front. Neural Circuits 2022, 16, 939235. [Google Scholar] [CrossRef]
- Ellgren, M.; Artmann, A.; Tkalych, O.; Gupta, A.; Hansen, H.S.; Hansen, S.H.; Devi, L.A.; Hurd, Y.L. Dynamic changes of the endogenous cannabinoid and opioid mesocorticolimbic systems during adolescence: THC effects. Eur. Neuropsychopharmacol. 2008, 18(11), 826–834. [Google Scholar] [CrossRef]
- Marco, E.M.; Echeverry-Alzate, V.; López-Moreno, J.A.; Giné, E.; Peñasco, S.; Viveros, M.P. Consequences of early life stress on the expression of endocannabinoid-related genes in the rat brain. Behav. Pharmacol. 2014, 25, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Li, C.; Jaffe, A.E.; Shin, J.H.; Deep-Soboslay, A.; Yamin, R.; Weinberger, D.R.; Hyde, T.M.; Kleinman, J.E. Cannabinoid receptor CNR1 expression and DNA methylation in human prefrontal cortex, hippocampus and caudate in brain development and schizophrenia. Transl. Psychiatry 2020, 10(1), 158. [Google Scholar] [CrossRef]
- Dallabrida, K.G.; de Oliveira Bender, J.M.; Chade, E.S.; Rodrigues, N.; Sampaio, T.B. Endocannabinoid system changes throughout life: Implications and therapeutic potential for autism, ADHD, and Alzheimer’s disease. Brain Sci. 2024, 14(6), 592. [Google Scholar] [CrossRef] [PubMed]
- Renard, J.; Krebs, M.O.; Jay, T.M.; Le Pen, G. Long-term cognitive impairments induced by chronic cannabinoid exposure during adolescence in rats: A strain comparison. Psychopharmacology (Berl.) 2013, 225(4), 781–790. [Google Scholar] [CrossRef] [PubMed]
- Renard, J.; Vitalis, T.; Rame, M.; Krebs, M.O.; Lenkei, Z.; Le Pen, G.; Jay, T.M. Chronic cannabinoid exposure during adolescence leads to long-term structural and functional changes in the prefrontal cortex. Eur. Neuropsychopharmacol. 2016, 26(1), 55–64. [Google Scholar] [CrossRef]
- Schneider, M. Adolescence as a vulnerable period to alter rodent behavior. Cell Tissue Res. 2013, 354(1), 99–106. [Google Scholar] [CrossRef]
- Rubino, T.; Parolaro, D. The impact of exposure to cannabinoids in adolescence: Insights from animal models. Biol. Psychiatry 2016, 79(7), 578–585. [Google Scholar] [CrossRef]
- Katona, I.; Freund, T.F. Multiple functions of endocannabinoid signaling in the brain. Annu. Rev. Neurosci. 2012, 35, 529–558. [Google Scholar] [CrossRef]
- Rodríguez de Fonseca, F.; Ramos, J.A.; Bonnin, A.; Fernández-Ruiz, J.J. Presence of cannabinoid binding sites in the brain from early postnatal ages. Neuroreport 1993, 4(2), 135–138. [Google Scholar] [CrossRef]
- Molla, H.M.; Miguelez Fernández, A.M.M.; Tseng, K.Y. Late-adolescent onset of prefrontal endocannabinoid control of hippocampal and amygdalar inputs and its impact on trace-fear conditioning behavior. Neuropsychopharmacology 2024, 49(9), 1417–1424. [Google Scholar] [CrossRef]
- Meyer, H.C.; Lee, F.S.; Gee, D.G. The role of the endocannabinoid system and genetic variation in adolescent brain development. Neuropsychopharmacology 2018, 43(1), 21–33. [Google Scholar] [CrossRef] [PubMed]
- De Felice, M.; Renard, J.; Hudson, R.; Szkudlarek, H.J.; Pereira, B.J.; Schmid, S.; Rushlow, W.J.; Laviolette, S.R. l-Theanine prevents long-term affective and cognitive side effects of adolescent Δ-9-tetrahydrocannabinol exposure and blocks associated molecular and neuronal abnormalities in the mesocorticolimbic circuitry. J. Neurosci. 2021, 41(4), 739–750. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu, A.; Bara, A.; Murphy Green, M.N.; Manduca, A.; Wager-Miller, J.; Borsoi, M.; Lassalle, O.; Pelissier-Alicot, A.L.; Chavis, P.; Mackie, K.; Manzoni, O.J.J. Sexually dimorphic adolescent trajectories of prefrontal endocannabinoid synaptic plasticity equalize in adulthood, reflected by endocannabinoid system gene expression. Cannabis Cannabinoid Res. 2023, 8(5), 749–767. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Jung, K.M.; Fotio, Y.; Squire, E.; Palese, F.; Lin, L.; Torrens, A.; Ahmed, F.; Mabou Tagne, A.; Ramirez, J.; Su, S.; Wong, C.R.; Jung, D.H.; Scarfone, V.M.; Nguyen, P.U.; Wood, M.; Green, K.; Piomelli, D. Frequent low-dose Δ9-tetrahydrocannabinol in adolescence disrupts microglia homeostasis and disables responses to microbial infection and social stress in young adulthood. Biol. Psychiatry 2022, 92(11), 845–860. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, H.; Liu, D.; Li, X.; He, L.; Pan, J.; Shen, Q.; Peng, Y. CB2R activation ameliorates late adolescent chronic alcohol exposure-induced anxiety-like behaviors during withdrawal by preventing morphological changes and suppressing NLRP3 inflammasome activation in prefrontal cortex microglia in mice. Brain Behav. Immun. 2023, 110, 60–79. [Google Scholar] [CrossRef]
- Romero-Torres, B.M.; Alvarado-Ramírez, Y.A.; Duran-Alonzo, S.R.; Ruiz-Contreras, A.E.; Herrera-Solis, A.; Amancio-Belmont, O.; Prospéro-García, O.E.; Méndez-Díaz, M. A potential role of hippocampus on impulsivity and alcohol consumption through CB1R. Pharmacol. Biochem. Behav. 2023, 225, 173558. [Google Scholar] [CrossRef]
- Rico-Barrio, I.; Peñasco, S.; Lekunberri, L.; Serrano, M.; Egaña-Huguet, J.; Mimenza, A.; Soria-Gómez, E.; Ramos, A.; Buceta, I.; Gerrikagoitia, I.; Mendizabal-Zubiaga, J.; Elezgarai, I.; Puente, N.; Grandes, P. Environmental enrichment rescues endocannabinoid-dependent synaptic plasticity lost in young adult male mice after ethanol exposure during adolescence. Biomedicines 2021, 9(7), 825. [Google Scholar] [CrossRef]
- Micale, V.; Cristino, L.; Tamburella, A.; Petrosino, S.; Leggio, G.M.; Drago, F.; Di Marzo, V. Anxiolytic effects in mice of a dual blocker of fatty acid amide hydrolase and transient receptor potential vanilloid type-1 channels. Neuropsychopharmacology 2009, 34(3), 593–606. [Google Scholar] [CrossRef]
- Renard, J.; Krebs, M.O.; Le Pen, G.; Jay, T.M. Long-term consequences of adolescent cannabinoid exposure in adult psychopathology. Front. Neurosci. 2014, 8, 361. [Google Scholar] [CrossRef]
- Saravia, R.; Ten-Blanco, M.; Julià-Hernández, M.; Gagliano, H.; Andero, R.; Armario, A.; Maldonado, R.; Berrendero, F. Concomitant THC and stress adolescent exposure induces impaired fear extinction and related neurobiological changes in adulthood. Neuropharmacology 2019, 144, 345–357. [Google Scholar] [CrossRef]
- Akirav, I. The role of cannabinoids in modulating emotional and non-emotional memory processes in the hippocampus. Front. Behav. Neurosci. 2011, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Rubino, T.; Viganò, D.; Realini, N.; Guidali, C.; Braida, D.; Capurro, V.; Castiglioni, C.; Cherubino, F.; Romualdi, P.; Candeletti, S.; Sala, M.; Parolaro, D. Chronic delta-9-tetrahydrocannabinol during adolescence provokes sex-dependent changes in the emotional profile in adult rats: Behavioral and biochemical correlates. Neuropsychopharmacology 2008, 33(11), 2760–2771. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.T.; Hill, M.N.; Lee, F.S. Developmental regulation of fear learning and anxiety behavior by endocannabinoids. Genes Brain Behav. 2016, 15(1), 108–124. [Google Scholar] [CrossRef] [PubMed]
- Abboussi, O.; Andaloussi, Z.L.; Chris, A.D.; Taghzouti, K. Chronic exposure to WIN55,212-2 during adolescence alters prefrontal dopamine turnover and induces sensorimotor deficits in adult rats. Neurotox. Res. 2020, 38(3), 682–690. [Google Scholar] [CrossRef]
- Demaili, A.; Portugalov, A.; Dudai, M.; Maroun, M.; Akirav, I.; Braun, K.; Bock, J. Epigenetic (re)programming of gene expression changes of CB1R and FAAH in the medial prefrontal cortex in response to early life and adolescence stress exposure. Front. Cell. Neurosci. 2023, 17, 1129946. [Google Scholar] [CrossRef]
- Renard, J.; Rosen, L.G.; Loureiro, M.; De Oliveira, C.; Schmid, S.; Rushlow, W.J.; Laviolette, S.R. Adolescent cannabinoid exposure induces a persistent sub-cortical hyper-dopaminergic state and associated molecular adaptations in the prefrontal cortex. Cereb. Cortex 27(2), 1297–1310. [CrossRef]
- Renard, J.; Szkudlarek, H.J.; Kramar, C.P.; Jobson, C.E.L.; Moura, K.; Rushlow, W.J.; Laviolette, S.R. Adolescent THC exposure causes enduring prefrontal cortical disruption of GABAergic inhibition and dysregulation of sub-cortical dopamine function. Sci. Rep. 7(1), 11420. [CrossRef]
- Renard, J.; Rushlow, W.J.; Laviolette, S.R. Effects of adolescent THC exposure on the prefrontal GABAergic system: Implications for schizophrenia-related psychopathology. Front. Psychiatry 2018, 9, 281. [Google Scholar] [CrossRef]
- Köfalvi, A.; Fritzsche, M. The endocannabinoid system is a major player in schizophrenia. In Köfalvi, A., Ed.; Cannabinoids and the Brain; Springer: New York, USA, 2008; pp. 485–528. [Google Scholar] [CrossRef]
- Bossong, M.G.; Niesink, R.J. Adolescent brain maturation, the endogenous cannabinoid system and the neurobiology of cannabis-induced schizophrenia. Prog. Neurobiol. 2010, 92(3), 370–385. [Google Scholar] [CrossRef]
- Vitale, R.M.; Iannotti, F.A.; Amodeo, P. The (poly)pharmacology of cannabidiol in neurological and neuropsychiatric disorders: Molecular mechanisms and targets. Int. J. Mol. Sci. 2021, 22(9), 4876. [Google Scholar] [CrossRef]
- Castillo-Arellano, J.; Canseco-Alba, A.; Cutler, S.J.; León, F. The polypharmacological effects of cannabidiol. Molecules 2023, 28(7), 3271. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, A.F.L.; Campos, R.M.P.; Isaac, A.R.; Paes-Colli, Y.; Carvalho, V.M.; Sampaio, L.S.; de Melo Reis, R.A. Long-term treatment with cannabidiol-enriched cannabis extract induces synaptic changes in the adolescent rat hippocampus. Int. J. Mol. Sci. 2023, 24(14), 11775. [Google Scholar] [CrossRef] [PubMed]
- Poleg, S.; Golubchik, P.; Offen, D.; Weizman, A. Cannabidiol as a suggested candidate for treatment of autism spectrum disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 89, 90–96. [Google Scholar] [CrossRef]
- Dow-Edwards, D.; Silva, L. Endocannabinoids in brain plasticity: Cortical maturation, HPA axis function and behavior. Brain Res. 2017, 1654(Pt B), 157–164. [Google Scholar] [CrossRef]
- Stark, T.; Di Martino, S.; Drago, F.; Wotjak, C.T.; Micale, V. Phytocannabinoids and schizophrenia: Focus on adolescence as a critical window of enhanced vulnerability and opportunity for treatment. Pharmacol. Res. 2021, 174, 105938. [Google Scholar] [CrossRef]
- Ertl, N.; Freeman, T.P.; Mokrysz, C.; Ofori, S.; Borissova, A.; Petrilli, K.; Curran, H.V.; Lawn, W.; Wall, M.B. Acute effects of different types of cannabis on young adult and adolescent resting-state brain networks. Neuropsychopharmacology 2024, 49(10), 1640–1651. [Google Scholar] [CrossRef] [PubMed]
- Medina, K.L.; Nagel, B.J.; Park, A.; McQueeny, T.; Tapert, S.F. Depressive symptoms in adolescents: Associations with white matter volume and marijuana use. J. Child Psychol. Psychiatry 2007, 48(6), 592–600. [Google Scholar] [CrossRef]
- Desai, S.; Zundel, C.G.; Evanski, J.M.; Gowatch, L.C.; Bhogal, A.; Ely, S.; Carpenter, C.; Shampine, M.; O’Mara, E.; Rabinak, C.A.; Marusak, H.A. Genetic variation in endocannabinoid signaling: Anxiety, depression, and threat- and reward-related brain functioning during the transition into adolescence. Behav. Brain Res. 2024, 463, 114925. [Google Scholar] [CrossRef]
- Hariri, A.R.; Gorka, A.; Hyde, L.W.; Kimak, M.; Halder, I.; Ducci, F.; Ferrell, R.E.; Goldman, D.; Manuck, S.B. Divergent effects of genetic variation in endocannabinoid signaling on human threat- and reward-related brain function. Biol. Psychiatry 2009, 66(1), 9–16. [Google Scholar] [CrossRef]
- Kovner, R.; Oler, J.A.; Kalin, N.H. Cortico-limbic interactions mediate adaptive and maladaptive responses relevant to psychopathology. Am. J. Psychiatry 2019, 176(12), 987–999. [Google Scholar] [CrossRef]
- Di Forti, M.; Marconi, A.; Carra, E.; Fraietta, S.; Trotta, A.; Bonomo, M.; Bianconi, F.; Gardner-Sood, P.; O’Connor, J.; Russo, M.; Stilo, S.A.; Marques, T.R.; Mondelli, V.; Dazzan, P.; Pariante, C.; David, A.S.; Gaughran, F.; Atakan, Z.; Iyegbe, C.; Powell, J.; Morgan, C.; Lynskey, M.; Murray, R.M. Proportion of patients in south London with first-episode psychosis attributable to use of high-potency cannabis: A case-control study. Lancet Psychiatry 2015, 2(3), 233–238. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.H.; Caspi, A.; Ambler, A.; Harrington, H.; Houts, R.; Keefe, R.S.; McDonald, K.; Ward, A.; Poulton, R.; Moffitt, T.E. Persistent cannabis users show neuropsychological decline from childhood to midlife. Proc. Natl. Acad. Sci. USA 2012, 109(40), E2657–2664. [Google Scholar] [CrossRef] [PubMed]
- Hermann, D.; Schneider, M. Potential protective effects of cannabidiol on neuroanatomical alterations in cannabis users and psychosis: A critical review. Curr. Pharm. Des. 2012, 18(32), 4897–4905. [Google Scholar] [CrossRef]
- Albaugh, M.D.; Ottino-Gonzalez, J.; Sidwell, A.; Lepage, C.; Juliano, A.; Owens, M.M.; Chaarani, B.; Spechler, P.; Fontaine, N.; Rioux, P.; Lewis, L.; Jeon, S.; Evans, A.; D’Souza, D.; Radhakrishnan, R.; Banaschewski, T.; Bokde, A.L.W.; Quinlan, E.B.; Conrod, P.; Desrivières, S.; Flor, H.; Grigis, A.; Gowland, P.; Heinz, A.; Ittermann, B.; Martinot, J.L.; Paillère Martinot, M.L.; Nees, F.; Papadopoulos Orfanos, D.; Paus, T.; Poustka, L.; Millenet, S.; Fröhner, J.H.; Smolka, M.N.; Walter, H.; Whelan, R.; Schumann, G.; Potter, A.; Garavan, H.; IMAGEN Consortium. Association of cannabis use during adolescence with neurodevelopment. Association of cannabis use during adolescence with neurodevelopment. JAMA Psychiatry 2021, 78(9), 1–11. [Google Scholar] [CrossRef]
- Allick, A.; Park, G.; Kim, K.; Vintimilla, M.; Rathod, K.; Lebo, R.; Nanavati, J.; Hammond, C.J. Age- and sex-related cortical gray matter volume differences in adolescent cannabis users: A systematic review and meta-analysis of voxel-based morphometry studies. Front. Psychiatry 2021, 12, 745193. [Google Scholar] [CrossRef]
- Canas, P.M.; Duarte, J.M.; Rodrigues, R.J.; Köfalvi, A.; Cunha, R.A. Modification upon aging of the density of presynaptic modulation systems in the hippocampus. Neurobiol. Aging 2009, 30(11), 1877–1884. [Google Scholar] [CrossRef]
- Long, L.E.; Lind, J.; Webster, M.; Weickert, C.S. Developmental trajectory of the endocannabinoid system in human dorsolateral prefrontal cortex. BMC Neurosci. 2012, 13, 87. [Google Scholar] [CrossRef]
- Kinon, B.J.; Leucht, S.; Tamminga, C.; Breier, A.; Marcus, R.; Paul, S.M. Rationale for adjunctive treatment targeting multiple mechanisms in schizophrenia. J. Clin. Psychiatry 2024, 85(3), 23nr15240. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.C.; Sewell, R.A.; Ranganathan, M. Cannabis and psychosis/schizophrenia: Human studies. Eur. Arch. Psychiatry Clin. Neurosci. 2009, 259(7), 413–431. [Google Scholar] [CrossRef]
- Zuardi, A.W.; Crippa, J.A.; Hallak, J.E.; Bhattacharyya, S.; Atakan, Z.; Martin-Santos, R.; McGuire, P.K.; Guimarães, F.S. A critical review of the antipsychotic effects of cannabidiol: 30 years of a translational investigation. Curr. Pharm. Des. 2012, 18(32), 5131–5140. [Google Scholar] [CrossRef]
- Gorbenko, A.A.; Heuberger, J.A.A.C.; Klumpers, L.E.; de Kam, M.L.; Strugala, P.K.; de Visser, S.J.; Groeneveld, G.J. Cannabidiol increases psychotropic effects and plasma concentrations of Δ9-tetrahydrocannabinol without improving its analgesic properties. Clin. Pharmacol. Ther. 2024. [CrossRef] [PubMed]
- Rubino, T.; Parolaro, D. Sexually dimorphic effects of cannabinoid compounds on emotion and cognition. Front. Behav. Neurosci. 2011, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Elmes, M.W.; Kaczocha, M.; Berger, W.T.; Leung, K.; Ralph, B.P.; Wang, L.; Sweeney, J.M.; Miyauchi, J.T.; Tsirka, S.E.; Ojima, I.; Deutsch, D.G. Fatty acid-binding proteins (FABPs) are intracellular carriers for Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD). J. Biol. Chem. 2015, 290(14), 8711–8721. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.R.; Watts, J.J.; Da Silva, T.; Tyndale, R.F.; Rusjan, P.M.; Houle, S.; Wilson, A.A.; Ross, R.A.; Boileau, I.; Mizrahi, R. Fatty acid amide hydrolase is lower in young cannabis users. Addict. Biol. 2021, 26(1), e12872. [Google Scholar] [CrossRef]
- Ostlund, I.; Von Gunten, M.; Smith, C.; Edwards, J.G. Chronic Δ9-tetrahydrocannabinol impact on plasticity, and differential activation requirement for CB1-dependent long-term depression in ventral tegmental area GABA neurons in adult versus young mice. Front. Neurosci. 2023, 16, 1067493. [Google Scholar] [CrossRef] [PubMed]
- Volk, D.W.; Siegel, B.I.; Verrico, C.D.; Lewis, D.A. Endocannabinoid metabolism in the prefrontal cortex in schizophrenia. Schizophr. Res. 2013, 147(1), 53–57. [Google Scholar] [CrossRef]
- Maia, J.; Fonseca, B.M.; Cunha, S.C.; Braga, J.; Gonçalves, D.; Teixeira, N.; Correia-da-Silva, G. Impact of tetrahydrocannabinol on the endocannabinoid 2-arachidonoylglycerol metabolism: ABHD6 and ABHD12 as novel players in human placenta. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865(12), 158807. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



