
Submitted:
17 October 2024
Posted:
21 October 2024
You are already at the latest version
Abstract
Melanoma is a malignant, highly metastatic neoplasm showing increasing morbidity and mortality. Tumor invasion and angiogenesis are based on remodeling of the extracellular matrix (ECM). Selective inhibition of functional components of cell-ECM interaction, such as hyaluronic acid (HA), matrix metalloproteinases (MMPs), and integrins, may inhibit tumor progression and enhance the efficacy of combination treatment with checkpoint inhibitors (ICBs), chemotherapy, or immunotherapy. In this review, we have combined the results of different approaches targeting extracellular matrix elements in melanoma in preclinical and clinical studies. The identified limitations of many approaches, including side effects, low selectivity and toxicity, indicate the need for further studies to optimize therapy. Nevertheless, significant progress in expanding our understanding of tumor biology and the development of targeted therapies holds great promise for the early approaches developed several decades ago to inhibit metastasis through ECM targeting.
Keywords:
melanoma
; extracellular matrix
; MMP
; TIMP
; integrins
; CD44
; hyaluronic acid
; clinical trials
1. Introduction
Melanoma is a malignant neoplasm originating from melanocytes localized in the skin, less frequently in the mucous membranes and retina of the eye [1]. The burden of melanoma is increasing worldwide, with cases rising 170% from 1990 to 2019 [2]. According to the most recently published data, approximately 97,610 new melanoma diagnoses and 7,990 deaths were projected by the end of 2023 in the United States alone [3]. If current trends continue, the global burden is expected to reach 510,000 new cases and 96,000 deaths by 2040 [4]. These statistics underscore the weighty challenge that melanoma poses to public health worldwide, especially among light-skinned populations [5].
Although drugs with proven efficacy against melanoma have been shown previously, their mechanisms of action are predominantly focused on targeting cancer cells directly, initiating apoptosis, or exerting cytostatic effects [6]. For example, targeted therapy against BRAF, MEK and KIT protein kinases has shown improved survival rates in melanoma with a mutation in the BRAF gene [7,8,9]. However, drug resistance and unresponsiveness to therapy remains a challenge, with the tumor microenvironment (TME) having been shown to play a critical role in this process [10]. For this reason, today, combination therapy is increasingly used in cancer targeted treatment, where components of the tumor microenvironment also become an additional target [11,12]. For example, the combination of lenalidomide, sunitinib, and cyclophosphamide at low doses significantly suppressed tumor growth and angiogenesis in various cancer models, including melanoma [13]. This approach creates an unfavorable environment for tumor cells and represents a promising strategy for clinical application.
The tumor microenvironment is an altered tissue stroma localized near the tumor focus, populated by cells with both pro- and antitumor activity, with a shift in balance to one side largely determining the status of the tumor – “cold” or “hot” [14,15]. Dominant roles in the immunosuppressive status of the microenvironment have been shown for cancer-associated fibroblasts (CAFs) [16,17], tumor-associated macrophages (TAMs) [18,19], T-regulatory cells [20,21], MDSC [22,23] and other cell populations [24,25,26]. For example, TAMs interact with the collagen matrix, creating cross-links that affect tumor invasion and progression [27]. Nevertheless, for a long time, the role of the non-cellular component and especially the extracellular matrix (ECM) in the tumor-host relationship remained unknown.
In 2012, a paper by Lu et al. was published in which a group of authors put together evidence on how abnormal ECM affects cancer progression, highlighting potential therapeutic targets [28]. It is now known that ECM transformation, including changes in density and stiffness, affects different cell types in the TME, regulating tumor development and the local immune network [29]. Under altered conditions, tissue hypoxia, acidosis, and protease activity have significant effects on TME cells, which are most often associated with poor patient prognosis [30]. The cancer matrisome, or proteome of the tumor ECM, exhibits significant transformations that may serve as both valid prognostic biomarkers and potential targets for targeted drug action [31]. Understanding these complex interactions within the ECM is critical for the development of approaches to targeting cancer therapy.
2. Extracellular Matrix in Melanoma
It was long believed that the vast majority of melanomas originated from skin nevi, but current histologic data indicate that only one-third of melanomas are nevus-associated, while the remainder form de novo [32,33]. Nevertheless, characterization of the ECM of normal melanocytic nevus may serve as a starting point to investigate the pathophysiological processes of ECM restructuring during melanoma development.
The canonical ECM includes fibrillar and structural proteins such as collagen types I, III, VII, XV, XVIII, laminin, tenascin-C, fibronectin, and hydrated gel-forming macromolecules such as hyaluronan and proteoglycans, as well as integrins, which carry out adhesion signaling, and several other elements [34].
As nevi progress to melanoma, there is a loss of type IV collagen and laminin in dermal melanocytic cells, while the surrounding stroma shows increased expression of interstitial collagens, tenascin, and fibronectin, as well as close association of ECM components with intraepidermally located invading atypical melanocytes [35]. The changes shown result in a molecular pattern that increases melanocyte invasion with an altered profile of adhesive molecules [35,36]. Melanomas compared with melanocytic nevi have fewer collagen bundles, but they are significantly thickened, especially at the periphery of the neoplasm, reflecting biological differences between benign and malignant melanocytic skin lesions [37]. Excess of some ECM components, such as collagen and fibronectin, increases tissue fibrosis and hence matrix stiffness, which affects the metastatic potential of tumor cells and their invasiveness [38]. The biophysical properties of ECM also change during the progression of nevus to melanoma, as demonstrated in microrheological studies in vivo [39].
Rigid and compacted tumor ECM reduces oxygen diffusion and induces hypoxia-mediated stress, which activates associated signaling pathways [40], resulting in malignant melanocytes becoming less susceptible to drugs, including through a shift in the expression of ROR1 and ROR2 tyrosine kinase receptors [41]. In addition, growth, invasion, metastasis, and eluding the immune response of melanoma cells have been shown to be positively regulated with increasing matrix stiffness [42,43]. Increasing the rigidity and integrity of the matrisome generally reduces diffusion of small drug molecules and infiltration of immune cells into the tumor stroma [44,45].
Nevertheless, the tremendous shifts in the equilibrium of altered ECM elements and the significant involvement of some components in tumor progression make individual molecules a promising target for targeted therapy. Below we review several key ECM components and describe in detail the published preclinical approaches as well as the results of clinical trials for the targeted treatment of melanoma (Figure 1).
3. Heparanase Targeting
Heparanases are a family of endoglycosidase enzymes that degrade the glycosaminoglycan heparan sulfate (HS) in the extracellular matrix, resulting in loss of basal membrane integrity and release of heparan sulfate-associated angiogenic and growth-promoting factors that subsequently stimulate tumor blood vessel growth, cell invasion, migration, adhesion, metastasis, differentiation, and proliferation [46,47,48]. Heparanase has previously been shown to be overexpressed in 88% of metastatic melanoma specimens, with strong expression being associated with decreased survival in patients at stage IVc [49]. Another study demonstrated an almost 30-fold increase in heparanase expression in metastatic melanoma samples compared to normal tissue, highlighting its selective localization in vascularized malignant areas [50].
In preclinical models, suramin (polysulfonated naphthylurea), has been shown to have a strong inhibitory effect on heparanase activity in B16 melanoma cells and their subsequent invasiveness in reconstructed basal membranes [51]. Another compound 1,3-bis-[4-(1H-benzoimidazol-2-yl)-phenyl]-urea showed an inhibitory effect not only on the proliferative activity of B16-BL6 melanoma cells in vitro (less than 50%), but also on the metastatic potential of these cells in mouse models (reduction of about 50%) [52]. Chemically modified heparins similarly demonstrated efficacy in inhibiting heparanase and reducing B16-BL6 melanoma metastatic activity to the lung in mice [53,54]. Notably, the antimetastatic and anticoagulant activities of heparin are unrelated, allowing the development of heparanase inhibitors with minimal anticoagulant side effects [55]. All preclinical data summarized in Table 1.
3.1. PI-88
PI-88 (metformin) is a mixture of highly sulfated oligosaccharides derived from the yeast Pichia (Hanensula) holstii NRRL Y-2448, consisting primarily of phosphomannopentaose and phosphomannotetraose [56]. PI-88 inhibits heparanase, exhibits anti-angiogenic and anti-metastatic activity, and competitively inhibits the binding of heparan sulfate to growth factors such as FGF and VEGF [57]. Numerous preclinical data have shown that PI-88 inhibits angiogenesis and has antimetastatic effects in various tumor models, including melanoma [58].
The Phase I study evaluated the biological activity of PI-88, a heparanase enzyme inhibitor (250 mg/day), in combination with docetaxel (30 mg/m2 per week) in 16 patients with advanced solid malignancies, including melanoma. No partial response (PR) or complete response (CR) was observed during the study period. However, at least 2 of 5 melanoma patients (40%) evaluable for response had stable disease (SD) at the end of ≥2 cycles of therapy [59]. A similar Phase I study evaluated the pharmacokinetic and biological effects of PI-88 (80-250 mg dose) in 18 patients with advanced solid malignancies, including melanoma. Dexamethasone (20 mg) was administered additionally to prevent immune-mediated thrombocytopenia. Despite no PR or CR, 3/15 (20%) evaluable patients showed SD at 2, 4, and 10 years. One patient with melanoma (6.7%) refractory to biochemotherapy showed PR accompanied by a reduction in the size and number of pulmonary metastases [60]. Another Phase I clinical trial tested the antitumor and antiangiogenic effect of PI-88 (administered at 0.57 mg/kg for 2h - 2.28 mg/kg/day). Fourteen patients with advanced malignancies including melanoma were included in the study. Only one patient with metastatic melanoma achieved SD, but after four cycles of therapy (12 weeks) he was diagnosed with progressive disease (PD), as were the other melanoma patients in the study [61]. All clinical trials data summarized in Table 2.
Millward et al. reported a completed phase I clinical trial evaluating the efficacy of PI-88 (140 mg - 250 mg) in combination with dacarbazine (an antitumor cytostatic drug; 1000 mg/m2 every 21 days) in 19 patients with unresectable metastatic melanoma, in which the efficacy of PI-88 monotherapy was not confirmed, with dose-dependent adverse effects associated with the occurrence of grade III/IV thrombocytopenia, up to and including cerebral venous sinus thrombosis in one patient. No CR or PR were observed with PI-88 monotherapy, but one patient showed radiologic SD at 4 months. However, PR was observed in 2/5 patients (40%) initially receiving monotherapy but who later had dacarbazine added to PI-88. 3/9 patients (33%) initially receiving combination therapy had radiologic PR [62].
In a follow-up to the previous study, a Phase II trial also evaluated the efficacy of PI-88 (190 mg) with dacarbazine (1000 mg/m2 every 21 days) in 134 patients with metastatic melanoma, using the optimal drug doses identified previously. Within the study, the combination of dacarbazine and heparanase inhibitor was generally shown to be less effective than dacarbazine monotherapy. In 24 of 65 patients (36.9%) receiving the combination of PI-88 + dacarbazine was shown SD with a median duration of 117 days, while dacarbazine monotherapy was shown for 31 of 65 participants (47.7%) with a median duration of 140.6 days. However, more subjects (30.77% vs. 19.70%) experienced serious adverse effects including neutropenia (30.77%) and thrombocytopenia (27.27%) in the combination therapy option (NCT00130442).
Another Phase I trial evaluated the efficacy of the heparanase inhibitor PI-88 (80 to 250 mg/day in 2 four-day cycles over a 28-day period) in 42 patients with advanced solid tumors. Additionally, dexamethasone (10 mg per 28-day period) was also administered to potentially improve immune-mediated thrombocytopenia. Of the 17 melanoma patients in the study suitable for evaluation of antitumor activity, one (5.9%) had PR that persisted for more than 50 months, and five other patients (29.4%) had SD for 7-38 months. Three patients developed grade II-III thrombocytopenia associated with dose-limiting toxicity (NCT00073892) [63].
In a sequentially ongoing Phase II study of PI-88 in patients with advanced melanoma, the previously determined optimal dose of 250 mg/day was used. A total of 44 melanoma patients were included in the trial, with 59.1% having previously received therapy. Median time to progression and overall survival were 1.7 months and 9 months, respectively. Forty-one patients were included in the efficacy analysis. One (2.4%) patient achieved PR, six (14.6%) patients showed SD as the best response, and the remaining 30 participants (73.2%) showed PD. At the end of six cycles of treatment, three of the 41 patients studied had no disease progression (NCT00073892) [64].
3.2. PG545
Another heparanase inhibitor, PG545 (pixatimod), has also shown promising results in inhibiting tumor growth and angiogenesis. The PG500 series includes chemically engineered HS mimetics, which are fully sulfated, single component oligosaccharides attached to a lipophilic motif and optimized for drug development [65,66]. In preclinical studies, they showed heparanase inhibition and high affinity for FGF-1, FGF-2 and VEGF growth factors with minimal anticoagulant activity. PG545 was selected as a candidate molecule due to its dual functional inhibition of heparanase and angiogenesis activity [67]. Pixatimod, as an HS-mimetic, blocks the interaction of HS with growth factors by inhibiting their signaling pathways, which contributes to its anticancer action [68].
In a Phase I clinical trial, four patients with recurrent solid tumors received PG545 (25 to 50 mg per week). As a result, no RECIST responses were recorded and all patients had PD. One of the reasons may be the prescription of drug concentrations 2-4 times below the level of experimental efficacy in preclinical models. Nevertheless, levels of various target proteins over time were measured in the plasma of these patients. For example, in a melanoma patient (total of 8 doses of 25 mg), similar to most other subjects, plasma levels of VEGF and FGF-2 increased 3.5-fold and 1.5-fold, respectively, after 22 days of treatment with PG545. This was explained by the fact that blocking the interaction of growth factors and heparanase with HS in the tumor microenvironment leads to the release of free ligand into the plasma. This suppresses VEGF-induced activation of cellular signal transduction in tumor endothelial cells and stops heparanase-mediated degradation of extracellular matrix, resulting in increased levels of these proteins in plasma (NCT01252095) [69].
A Phase IIA trial investigating the effect of pixatimod in combination with nivolumab and low-dose cyclophosphamide in advanced cancer, including refractory melanoma, has recently completed, but the results are unpublished at the time of writing (NCT05061017). However, in a prior Phase Ib trial of 58 participants, three participants (5.2%) with metastatic colorectal cancer had confirmed PR and eight (13.8%) showed SD for at least 9 weeks. As demonstrated in the study, clinical benefit was associated with lower plasma levels of inflammation and IL-6, but an increased IP-10/IL-8 ratio. The participant with PR showed increased infiltration of T-lymphocytes and dendritic cells as the best response 5 weeks after treatment, making the new phase IIa study promising in terms of clinical responses [70].
Other approaches to suppress heparanase expression in melanoma stroma through genetic constructs, including adenoviral vectors carrying an antisense sequence of the heparanase gene HSPE-1, have also spread and have shown efficacy in B16-B15b / 70W melanoma lines and mouse models [71], artificial microRNA (miRNA) on A375 cell model [72] or small interfering RNA (siRNA) in B16-BL6 mouse melanoma in vivo [73]. In the described studies, genetic constructs effectively reduced heparanase enzyme expression, resulting in reduced tumor invasiveness, suppression of angiogenesis and metastasis, but approaches based on genetic suppression of heparanase haven’t been widely used in clinical trials [71,72,73].
Heparanase inhibition represents a promising area for the control of metastatic melanoma. Preclinical studies have demonstrated the efficacy of small-molecule inhibitors and gene vectors in reducing heparanase activity, suppressing angiogenesis and metastasis. However, despite promising results, clinical trials of heparanase inhibitors such as PI-88 and PG545 haven’t yet achieved significant success in melanoma monotherapy. However, in some combinations with antitumor drugs, heparanase inhibitors can improve clinical outcome, making their potential use as adjuvants in the therapy of melanoma and other malignancies possible.
4. MMP Targeting
Matrix metalloproteinases (MMPs) are a family of secreted, zinc-dependent endopeptidases that together are capable of degrading ECM components, and there is considerable evidence that they play an important role at different stages of malignancy progression [74]. MMPs play a dual role in tumor growth and metastasis: on the one hand they promote outgrowth and invasion by disrupting matrix barriers and enhancing angiogenesis, on the other hand, MMPs can also limit neovascularization [74]. Both natural tissue inhibitors of metalloproteinases (TIMPs) and artificial MMP inhibitors (MMPIs) exist to regulate the function of metalloproteinases, the latter have been developed as potential cancer therapies, including the groups of peptidomimetics, non-peptidomimetic inhibitors, tetracycline derivatives and bisphosphonates [75]. Despite promising preclinical data, early clinical trials of broad-spectrum MMP inhibitors have been unsuccessful due to serious side effects and lack of efficacy in later stage cancers [76]. A number of studies have shown that melanoma cells can express a specific pool of matrix metalloproteinases (MMP-1, MMP-2, MMP-9, MMP-13 and MT1-MMP) as well as their tissue inhibitors (TIMP-1, TIMP-2 and TIMP-3) [77].
4.1. MMP Inhibitors
As previously stated MMPIs are artificially engineered inhibitors used in experimental therapies for cancer, osteoarthritis and rheumatoid arthritis with chronic stimulation of MMP activity due to imbalance of MMP and TIMP levels in pathogenesis [78]. A number of drugs are commonly categorized as first generation synthetic non-selective MMPIs - batimastat, marimastat, cipemastat and MMI-166 [79]. In contrast, the next generation includes more selective nonpeptidomimetic inhibitors such as primostat, tanomostat, BAY12-9566, chemically modified antibiotics (e.g., COL-3), and bisphosphonates [79]. Although MMPIs have never gained traction and their clinical efficacy in cancer remains mediocre, for a holistic picture of melanoma targeting therapy, we will briefly describe the main molecules and their results.
4.1.1. First Generation MMPI
Batimastat (BB-94) is one of the first synthetic peptidomimetic MMPIs that mimics the most common MMP substrate, collagen, and has shown antitumor and antiangiogenic activity in various tumor models, including melanoma [80]. Batimastat has demonstrated broad-spectrum inhibition of virtually all types of MMPs [81]. Multiple studies have shown that this inhibitor was relatively effective in suppressing tumor growth and metastasis in mouse models of melanoma [82,83], but it was ineffective for therapy of human malignancies and clinical trials were discontinued [75,84].
Marimastat (BB2516) also belongs to synthetic peptidomimetic MMP inhibitors [85,86]. Despite improved efficacy rates compared to batimastat in preclinical settings and phase II and III advances for therapy of several types of solid tumors, a phase I study of the combination of marimastat and paclitaxel in patients with advanced malignancies, including melanoma, didn’t demonstrate greater efficacy compared to paclitaxel alone in advanced stages of disease [87]. A Phase II study also showed limited efficacy of marimastat in patients with malignant melanoma, where only 2 of 28 patients (7.1%) showed PR and 5 (17.9%) showed SD [88].
4.1.2. Second Generation MMPI
Prinomastat (AG3340) is a potent second-generation selective synthetic non-peptidomimetic inhibitor of MMP-2, -9, -13 and -14 [89]. Initial positive results from testing of prinomostat in in vivo animal models involving monotherapy of xenograft uveal melanoma in rabbits and combination therapy with carboplatin and taxol in a B16-F10 metastatic murine melanoma model have been reported [90,91]. However, phase I clinical trials in patients with advanced cancer, including melanoma, haven’t documented confirmed tumor responses to therapy with prinomastat [92].
Incyclinide (COL-3) is a non-antimicrobial chemically modified tetracycline derivative with antitumor and antimetastatic activity through inhibition of MT1-MMP and pro-MMP-2 [93]. A phase I study of oral COL-3 (36 - 98 mg/m2/d), a matrix metalloproteinase inhibitor, in 35 patients with refractory metastatic cancer, including melanoma, showed only limited efficacy in the form of SD in eight patients (22.9%) with tumors of non-epithelial origin over two months (NCT00001683) [94].
MMI270 (CGS27023A), a targeting inhibitor of MMP-2, MT1-MMP, and MMP-9 and belonging to the group of non-peptidomimetic hydroxomat inhibitors, significantly reduced the number of metastatic B16-F10 melanoma colonies in the lungs of mice without affecting colony size, in contrast to the spontaneously metastasizing melanoma line B16-BL6, due to the difference between hematogenous and lymphatic metastasis pathways [95]. Intraperitoneal injection of MMI270 after implantation of B16-BL6 melanoma cells into mice reduced the number of vessels leading to the primary tumor on the dorsal side, demonstrating a significant antiangiogenic effect of this inhibitor [96].
Rebimastat (BMS-275291) is a second-generation sulfhydryl-based matrix metalloproteinase inhibitor that binds MMP-1, MMP-2, MMP-7, MMP-9, and MMP-14. Oral treatment with rebimastat has been shown to result in dose-dependent inhibition of angiogenesis and tumor metastasis to the lung in a metastatic melanoma cell line model B16-BL6 and an in vivo Matrigel plug cell migration model [97].
Speaking of novel MMPIs, the MMP-2 targeting inhibitor JaZ-30 (C(2)-monosubstituted aziridine - aryl-1,2,3-triazole conjugate) should also be mentioned. JaZ-30 reduced melanoma cell invasion, angiogenesis, and ERK1/2 phosphorylation in a B16 4A5 melanoma cell model [98]. The authors showed that nontoxic physiologic doses of JaZ-30 reduced the invasive properties of highly metastatic melanoma cells by 40% through selective inhibition of MMP-2 catalytic activity through coordination with a zinc atom in the enzyme's active center and mediated suppression of VEGF secretion [98]. Another MMP inhibitor, SB-3CT, is a 2-[(arylsulfonyl)methyl]thiirane that selectively inhibits MMP-2/9 and enhances T-cell-mediated cytotoxicity [99,100,101]. A significant reduction of PD-L1 mRNA and protein levels in A375 and SK-MEL-28 melanoma cell lines in vitro, as well as effective suppression of B16-F10 melanoma lung metastases by combination therapy with checkpoint inhibitors (anti-PD-1 and/or anti-CTLA-4) has been shown in mouse models in vivo [99]. Marusak et al. also reported that the selective MT1-MMP/MMP-2 thiirane inhibitor ND-322 slowed melanoma tumor growth and delayed metastasis spread in a WM266-4 xenograft mouse model of melanoma [102]. Reich et al. showed that the novel selective MMP-2 inhibitor cyclopentylcarbamoylphosphonic acid similarly reduced the number of lung metastases and tumor growth in an in vivo B16-F10 mouse melanoma model [103].
4.1.3. Alternative Approaches to MMP Inhibition
Using phage display technology, Devy et al. discovered a highly selective human monoclonal MMP-14 inhibitor antibody, subsequently named DX-2400 [104]. DX-2400 has demonstrated significant anticancer effects by reducing tumor progression, decreasing the incidence of metastasis, and inhibiting angiogenesis in various murine models, including the transplanted melanoma cell line B16-F1 cells [105]. Treatment with DX-2400 reduced the number of metastatic foci in the lung in a dose-dependent manner, reaching a maximum effect (70%) at the highest dose tested (10 mg/kg) in mouse models [105]. Peptide vaccines based on synthetic immunogenic oligopeptides with MMP-2 and -9 sequences have also been described [106,107]. It was shown that depending on the source of the sequence (human/rat/mouse) the reduction in tumor size ranged from 55 to 88%, with no significant side effects in an in vivo B16-F0 mouse model of melanoma [106,107]. Short hairpin RNA (shRNA) containing a site specific for MMP-1 mRNA suppressed the expression of MMP-1 itself in a human melanoma cell line, which significantly reduced the ability of melanoma to metastasize from an orthotopic site in the dermis to the lung in an in vivo xenograft mouse model of VMM12 melanoma [108]. Tumor cells expressing MMP-1 shRNA had significantly reduced collagenase activity necessary for invasion and angiogenesis [108].
Matrix metalloproteinase inhibitors are still being studied as potential therapeutic agents for melanoma and other tumors. Despite promising results in preclinical studies, most first- and second-generation MMPIs haven’t demonstrated significant clinical efficacy in late-stage trials. However, new selective inhibitors continue to be developed that show potential in combination therapy by reducing metastasis and enhancing antitumor activity, especially when combined with checkpoint inhibitors.
4.2. Tissue Inhibitors of Metalloproteinases
Endogenous tissue inhibitors of metalloproteinases (TIMP)-like MMPIs have emerged as potential therapeutic agents for cancer treatment [109]. Normally, TIMP-1, -2, -3 and -4 regulate the activity of matrix metalloproteinases, which, as previously mentioned, are crucial for tumor invasion and metastasis [110]. TIMPs inhibit metalloproteinase activity through non-covalent binding of active zinc-binding sites of MMPs [111].
4.2.1. Recombinant TIMPs
One of the first applications of tissue inhibitors of metalloproteinases in the context of antitumor and antimetastatic effects was the work of Schultz et al. Under in vitro conditions recombinant human TIMP (rTIMP) inhibited invasion of murine melanoma cells B16-F10 through human amniotic membrane [112]. Mice injected with rTIMP showed significant inhibition of metastatic colonization of the lungs by melanoma cells from B16-F10 mice, but the size of the tumors themselves was not altered [112].
Recombinant TIMP-1 conjugated to glycosylphosphatidylinositol (TIMP-1-GPI) when combined with sub-lethal hyperthermic treatment demonstrated efficacy against melanoma cell lines 624.38-MEL, 93.04A12MEL, SK-MEL23, WM115 and WM266-4 under in vitro conditions [113]. Inhibition of proMMP-2 and proMMP-9 release from melanoma cells (WM226-4 and SK-MEL23 cell lines) as well as an overall significant increase in sensitivity to FAS-induced apoptosis was demonstrated [113].
Recombinant human TIMP-2 (r-hTIMP-2) was also shown to significantly inhibit the formation of B16-BL6 metastatic melanoma foci in mice in a dose-dependent manner regardless of route of administration [114]. In addition, a slight inhibitory effect on tumor cell growth was observed under in vitro and in vivo conditions [114]. Systemic administration of tissue metalloproteinase inhibitor 2 fused to human serum albumin (HSA-TIMP-2) at a dose of 40 mg/kg to mice inhibited the growth of B16-BL6 tumors [115]. In addition, combined treatment of HSA-TIMP-2 with 5-fluorouracil (50 mg/kg) showed a significant effect on tumor growth in this model [115]. Despite the initial view of the MMP-dependent nature of the antiangiogenic effect of TIMP-2, several studies of mutant Ala+TIMP-2 (lacking MMP-inhibitory activity) have shown that TIMP-2-mediated inhibition of tumor growth occurs at least in part independently of MMP inhibition and results from both direct effects of TIMP-2 on tumor cells themselves and modulation of the tumor microenvironment [116].
4.2.2. Genetic Vectors Encoding TIMPs
In addition to recombinant TIMP-1, a plasmid vector encoding human TIMP-1 cDNA (TIMP-1pDNA), administered intramuscularly to female mice with B16-F10 metastatic melanoma, causing a spike in serum human TIMP-1, was also used in the studies [117]. Lung metastasis was significantly reduced in mice after 4 weeks of treatment with TIMP-1 compared with controls, and further reduction of pulmonary metastases and increased overall survival were achieved by additional administration of IL-2 [117].
In another study, transfection of the melanoma cell line M24net with cDNA encoding human TIMP-2 effectively suppressed MPP-1, -2, -9 activity in a xenograft model of immunodeficient mice [118]. Induced overexpression of TIMP-2 suppressed melanoma cell growth but not metastatic activity, which was attributed to the TIMP-2 mediated ability of TIMP-2 to occluding interstitial collagen [118].
Administration of recombinant adenoviruses carrying TIMP-1, TIMP-2 and TIMP-3 genes inhibited the invasion of SK-Mel-5 and A2058 cells across the basal membrane under in vitro conditions predominantly through the expression of TIMP-3 rather than TIMP-1 and TIMP-2 [119]. In addition, overproduction of TIMP-3 reduced melanoma cell attachment to type I/IV collagen and fibronectin, ultimately leading to apoptosis in both SK-Mel-5 and A2058 cells [119]. In subsequent in vivo experiments it was shown that recombinant adenoviral vector carrying the TIMP-3 gene sequence (RAdTIMP-3) when administered to mice with xenograft melanoma line A2058 effectively inhibited gelatinase activity and suppressed tumor growth by inducing apoptosis [120].
Tissue inhibitors of metalloproteinases continue to be considered as promising therapeutic agents for melanoma, on par with synthetic inhibitors. Studies with recombinant TIMPs and various genetic vectors have shown efficacy in suppressing metastasis and tumor growth in preclinical studies. TIMPs canonically inhibit MMPs and related processes including angiogenesis and invasion. However, clinical trials using TIMPs haven’t yet been conducted. At the same time, clinical trials with synthetic MMPIs haven’t yielded the desired results because these compounds also affect other important molecules, causing serious side effects. To improve therapy, compounds with higher selectivity, low toxicity and good oral bioavailability are needed.
5. Hyaluronic Acid Targeting
Hyaluronan or hyaluronic acid (HA) is a glycosaminoglycan localized in the extracellular space of most tissues and is involved in many biological and pathophysiological processes including homeostasis, fertilization, wound healing, inflammation, angiogenesis and carcinogenesis [121]. Hyaluronic acid forms a viscous environment through inter- and intramolecular interactions, creating a dense microenvironment that limits drug delivery due to increased interstitial pressure [122]. Such changes in ECM maintain the structural integrity of the tumor, promote homeostasis, and the release of growth factors and cytokines necessary for cell proliferation [123]. HA plays a key role in cancer development by affecting signaling cascades, cell adhesion, new blood vessel formation, and metastasis [124,125]. These processes are largely related to HA/CD44 interactions, the impact of which is the subject of a separate block below.
Hyaluronic acid plays a special role in melanoma progression [126]. Melanoma cells produce large amounts of HA during early tumorigenesis, whereas at later stages HA is mainly produced by activated CAF populations [127]. Low molecular weight HA fragments may contribute to tumor invasiveness by inducing expression of cytokines and metalloproteases, in part through TLR4 signaling [128]. Summarizing, in melanoma, HA plays a key role in stimulating cell proliferation [126], adhesions [129], mobility [130], invasion and metastasis [131].
5.1. Low-Molecular Weight Inhibitors
Hymecromone or 4-Methylumbelliferone (4-MU) is a coumarin derivative that inhibits hyaluronan formation on the cell surface by suppressing hyaluronan synthase mRNA expression and by competitively binding to enzymes that precede the formation of substrate for HA synthesis [132]. This inhibition has been shown to inhibit melanoma cell adhesion and motility [133,134]. B16-F10 melanoma cells treated with 4-MU showed reduced HA formation on the cell surface [134], as well as an overall 32% reduction in the number of liver metastases after injection in mice in vivo compared to the control group [133]. Interestingly, the inhibitory effect of 4-MU was observed only in the liver, whereas no clear inhibition was found in other tissues [133]. Similarly for melanoma cell lines C8161 and MV3, exposure to 4-MU decreased hyaluronan levels in the extracellular matrix and slowed both growth and invasion of malignant cells inside collagen lattices under in vitro conditions [135].
Using a phage display method, Mummert et al. developed a synthetic Pep-1 peptide (GAHWQFNALTVR) that bound to HA and inhibited its functional activity. B16-F10 melanoma cells that constitutively expressed CD44 showed significant adhesion to HA-coated plates, and this adhesion was almost completely blocked by either neutralizing antibodies against CD44 or Pep-1. However, Pep-1 failed to inhibit in vitro proliferation of B16-F10 melanoma cells or cell growth after intravenous inoculation into mice in vivo. Importantly, a single injection of Pep-1 significantly reduced the incidence of metastasis to the lung when administered intravenously and increased the survival rate of animals with tumors [131,136]
5.2. Hyaluronidases
One of the first studies on regional chemotherapy of human melanoma transplanted into mice was the work of Spruss et al. It used a combination of hyaluronidase (an enzyme that destroys hyaluronic acid) and vinblastine (a cytostatic from the group of periwinkle alkaloids). Administration of hyaluronidase before vinblastine in three of four melanoma models (SK-Mel-2, -3, -5) demonstrated a pronounced antitumor effect, whereas separate use of these drugs didn’t lead to significant results. Eighteen weeks after treatment, tumor cells were no longer detected in the subcutaneous region of the former tumor. Interestingly, traces of the formerly injected tumor were in resident macrophages containing significant traces of melanin. This combination inhibited tumor growth and prevented metastasis, confirming its efficacy in this preclinical model. [137]
Overall, localized peritumoral administration of hyaluronidase has shown some efficacy, even though several recent studies have shown that hyaluronidase inhibition alone by delphinidin suppresses the proliferative and metastatic activity of B16-F10 melanoma cells in murine models in vivo [138].
Clinical trials with targeting agents against HA are limited to the use of recombinant enzyme when used in conjunction with drug therapy, predominantly for a simplified subcutaneous route of administration instead of longer intravenous infusions [139]. Thus, the space outside the adipocytes in the hypodermis is not a fluid but a solid extracellular matrix of collagen fibrils encased in a viscoelastic gel rich in glycosaminoglycans (including HA), which is a significant barrier to effective subcutaneous delivery of many drugs in the interstitial space, limiting injection volumes [140]. Temporary removal of HA by recombinant human hyaluronidase helps to facilitate interstitial drug administration in clinical practice [141].
The subcutaneously administered PD-1 inhibitor nivolumab (480 - 1200 mg), in combination with recombinant human hyaluronidase PH20 enzyme (rHuPH20) demonstrated good tolerability in a phase I/II clinical trial in patients with metastatic/inoperable solid tumors, including melanoma (NCT03656718) [142]. Side effects included reactions of all degrees of severity ranging from 46.4% to 75.0% in patients across the study. 11.1% had grade 3/4 TRAEs, and 5.6% reported serious TRAEs, of which one case resulted in treatment discontinuation [142]. This subcutaneous drug combination is now being actively tested in a Phase III clinical trial in patients with melanoma at Stage III A/B/C/D or IV (NCT05297565) [143], as well as in several other clinical trials (NCT03719131, NCT05625399, NCT06101134, NCT05496192, NCT06099782).
Hyaluronic acid in ECM plays a key role in processes associated with tumor growth and metastasis, creating a particularly dense environment that promotes cell proliferation and invasion. Combinations with hyaluronidase and vinblastine have demonstrated significant antitumor effects in preclinical models, and Hymecromone is currently in clinical trials for the treatment of non-tumor diseases involving pulmonary hypertension (NCT05128929). The use of recombinant enzymes in clinical trials also shows promise in terms of improving drug availability in combination treatments, but the enzyme itself is not used for melanoma monotherapy.
6. Integrins Targeting
Integrins are a large family of heterodimeric transmembrane glycoproteins that mediate cell-cell and cell-intracellular matrix interactions [144]. Integrins, especially αvβ3 [145], αvβ5 [146] and α5β1 [147], are involved in angiogenesis and metastasis of tumors, including melanoma [144,148]. For example, the vitronectin αvβ3 integrin receptor allows melanoma cells to attach to ECM components via the Arg-Gly-Asp peptide sequence [149]. Studies have shown that inhibiting the function of certain integrins can significantly reduce melanoma cell proliferation, adhesion and metastasis [148].
6.1. Disintegrins
One approach to inhibiting integrins is the use of disintegrins, small (4-16 kDa) viper snake venom proteins that contain a canonical integrin binding site (often the RGD site) [150]. These non-enzymatic proteins selectively inhibit integrin-mediated interactions, making them potential candidates as therapeutic agents for cancer and numerous other human diseases [151]. A more comprehensive review of the discovery of disintegrins, their binding sites, and their corresponding integrins has recently been published [152].
Acurhagin-C disintegrin (derived from Agkistrodon acutus venom) inhibited integrin αv/α5-dependent functions in melanoma cells by inhibiting B16-F10 cell adhesion to type VI collagen, gelatin B and fibronectin, and impaired transendothelial migration of B16-F10 cells, at higher concentrations inducing apoptosis and enhancing the effects of chemotherapy on the SK-MEL-1 cell line [153]. Tzabcanin (Crotalus simus tzabcan) was able to inhibit the adhesion of melanoma cell line A-375 to vitronectin, exhibiting weak cytotoxicity [154]. In addition, tzabkanin significantly inhibited melanoma cell migration in the scratch/wound healing assays of the scratch/wound healing tests [154]. The disintegrin contortrostatin (Agkistrodon contortrix contortrix) proved to be a potent inhibitor of M24 met melanoma cell adhesion mediated by β1 integrin, which also effectively suppressed pulmonary metastasis in mouse models [155]. Salmosin (Agkistrodon halys brevicaudus) markedly inhibited both adhesion of B16-F10 melanoma cells to extracellular matrix proteins and cell invasion through a matrigel-coated filter in vitro [156]. In vivo administration of salmosin has demonstrated the ability to suppress lung metastasis in murine models [156]. Colombistatin (Bothrops colombiensis) effectively inhibited the adhesion of SK-Mel-28 melanoma cells to fibronectin as well as their migration [157].
The recombinant disintegrins rubistatin (Crotalus ruber ruber), mujastin 1 (Crotalus scutulatus scutulatus), viridistatin 2 (Crotalus viridis viridis) originating from snake venom have similarly demonstrated the ability to initiate apoptosis in SK-Mel-28 melanoma cells, reduce their migration, adhesion, invasion and proliferation in vitro, as well as inhibit lung colonization by B16-F10 cells in mice [158,159,160].
Although the efficacy of native and recombinant disintegrins has been repeatedly demonstrated under in vitro and in vivo conditions, no clinical trials are currently underway for the therapy of melanoma and cancer in general using these types of drugs [161,162]. In turn, this is due to problems of application - instability, immunogenicity and availability of starting material [161].
6.2. Non-Disintegrins Inhibitors
At the same time, the study of non-disintegrin inhibitors can be considered a promising direction. The development of more highly selective, stable and non-immunogenic agents compared to disintegrins makes them more suitable for clinical use.
The non-peptide αvβ3 integrin inhibitor MK-0429 also demonstrated efficacy in reducing lung metastasis in mouse models of B16-F10 melanoma [145]. A novel selective αvβ3 antagonist, RGDechi-hCit, showed dose-dependent inhibition of adhesion and migration for multiple melanoma cell lines (A375, WM266-4, SK-Mel-28, Sbcl2, LB24Dagi, PR-Mel and PNP-Mel) under in vitro conditions [163]. At the same time, despite significant morphological changes in cells, proliferative activity was almost not inhibited [163].
siRNA-mediated suppression of integrin β3 expression in B16 melanoma cells significantly impaired their ability to migrate in the matrigel in vitro and metastasize in mice in vivo due to the impaired ability of the cells to bind to fibronectin [164]. The use of water-soluble plant polysaccharides extracted from Bupleurum chinense, which inhibited integrin-mediated adhesion of A375 human melanoma cells to fibronectin in vitro, may be considered an unusual approach [165]. Polysaccharides from Codonopsis lanceolata similarly inhibited B16-F10 melanoma proliferation in a mouse model of pulmonary metastasis and disrupted integrin β1-mediated cell migration under in vitro conditions [166].
Monoclonal antibodies have established themselves as excellent targeting inhibitors for a variety of targets [167]. For example, several antibodies against human integrin αvβ3 were produced in Mitjans et al. Antibody 17E6 effectively disrupted the attachment of melanoma cell line M21 melanoma cells to vitronectin and fibronectin by reversibly inhibiting their interaction with target integrins without toxic effects [168]. In experiments in mouse models, the antibody suppressed tumor growth and metastasis mediated by integrins, but not by inhibitory effects on melanoma cells themselves or antibody-mediated cellular cytotoxicity [168]. In parallel, another monoclonal mouse antibody against αvβ3 integrin, LM609, was produced, which also demonstrated efficacy in inhibiting the growth of M21 melanoma cells in vitro [169]. Notably, the mechanism of action of LM609 is based on steric hindrance: the antibody binds in the region adjacent to the RGD-binding site of the integrin, not blocking it directly but impeding the access of large ligands [170]. Two others humanized mAbs, MEDI-522 and MEDI-523, were later created based on this antibody [171].
MEDI-523 (Vitaxin) was the first humanized anti-αvβ3 mAb to be used in multiple clinical trials against cancer and despite good patient tolerability, no objective response to therapy was achieved and further trials were discontinued [172,173]. In a pilot study of Vitaxin on patients with metastatic cancer, a melanoma patient received two maximum doses of the drug, but despite this the disease continued to progress and he dropped out of the study [174]. Interestingly, in the described melanoma patient, it was possible to visualize and localize the tumor using labeled Vitaxin, which is probably due to the abundant expression of αvβ3 antigen on the surface of the tumor cells [174].
MEDI-522 (Etaracizumab) is a second-generation mAb against αvβ3 integrin and has greater stability and affinity to the target compared to its predecessor MEDI-523 [171]. Etaracizumab has been used in a number of clinical trials to treat metastatic melanoma, prostate and ovarian cancer, and other types of cancer [171]. The results of some clinical trials, despite their completed status, still remain unpublished (NCT00111696, NCT00263783).
A phase 0 study of Etaracizumab pharmacodynamics in patients with advanced melanoma showed that the drug effectively saturates tumor cells at doses of 8 mg/kg, has an acceptable safety profile with no serious toxic effects, and although no clear antitumor effects were observed, some patients could still benefit from inhibition of αvβ3 integrin-related signaling pathways [175]. In a phase I study of the monoclonal antibody MEDI-522, no CR or PR was observed in patients with advanced malignancies; however, long-term SD (34 weeks, >1 year, >2 years) was reported in patients with renal cell cancer. Two patients with melanoma and one patient with ocular melanoma showed PD after 6-8 weeks of therapy [171]. In another phase I study eteracizumab in patients with advanced solid tumors, results similar to those previously described were obtained. All patients showed absence of PR and CR, but the melanoma patient showed SD for more than 4 months [176]. The results of a randomized phase II trial of etaracizumab in combination with dacarbazine in patients with stage IV metastatic melanoma were also reported (NCT00066196). No responses were recorded in the group receiving etaracizumab alone. Therapy responses were recorded exclusively among patients given the combination of etaracizumab with dacarbazine, among whom PR was 12.7% (7 of 55). SD was observed in 45.6% (26 of 57) of patients receiving etaracizumab alone and 40% (22 of 55) in the combination group. PD was observed in 47.4% and 40%, respectively. The median time to progress was 1.8 months for monotherapy and 2.5 months for combination therapy [177].
Most adverse events in the described clinical trials were mild to moderate (grade I-II) and included fatigue, myalgia, anorexia, nausea, diarrhea and only a few patients experienced more serious side effects including hypophosphatemia, thromboembolism and neutropenia [171,175,176,177].
Integrins play a key role in angiogenesis and tumor metastasis, and particularly in melanoma. Native disintegrins from snake venom effectively inhibit adhesion, migration and invasion of melanoma cells, but remain outside the field of clinical trials due to several problems that could potentially be solved using their recombinant analogs. In addition, monoclonal antibodies, have shown potential in suppressing tumor growth and metastasis in preclinical studies. However, despite promising results, clinical trials using such inhibitors haven’t yielded significant therapeutic successes. Nevertheless, the maximum tolerated dose in clinical trials was not reached and the treatment itself demonstrated an acceptable safety profile, which creates some prospects for continued research with a bias towards combination therapy.
7. Non-Integrin Receptors Targeting
In addition to integrins, a number of other important receptors can be identified that bind ECM components and participate in its regulation, and as a consequence are actively involved in the processes of carcinogenesis, invasion, adhesion and metastasis [178]. This may include CD44 [179], DDR [180], HARE [181], LYVE-1 [182], RHAMM [183] and a number of other molecules that are promising targets for targeted therapies [178,184]. Their roles in the formation of constitutive and pathologic ECM have been described in more detail in a recent review [184]. We will summarize the major advances in the field of targeted therapy of only two molecules, CD44 and DDR.
7.1. CD44 Inhibitors
CD44 is a transmembrane glycoprotein that functions as a cell surface adhesion receptor [185]. CD44 is known to interact with a variety of ligands including hyaluronic acid, osteopontin, collagens, and matrix metalloproteinases, and these interactions are critical for its multiple cellular functions [186]. It plays an important role in cell adhesion, migration, proliferation and signaling in both normal and altered cells [187]. CD44 has been shown to be a major mediator of hyaluronic acid-mediated melanoma cell proliferation, and its high levels correlate with disease progression and poor survival in melanoma patients [129].
7.1.1. CD44 Hyaluronic Acid-Based Inhibitors
A conjugate of hyaluronan (HA) and the specific integrin ligand αvβ3 tetraiodothyroacetic acid (tetrac), was used to target docetaxel (DTX) to B16-F10 melanoma cells through localization on the surface of solid lipid nanoparticles (SLN) (TeHA-SLN/DTX). In both mice with allografted tumor and mice with in situ lung metastasis, tumor growth was significantly inhibited by the action of TeHA-SLN/DTX. Thus, TeHA-SLN demonstrated efficacy as a system for bidirectional drug delivery for melanoma treatment in vivo [188].
Coradini et al. evaluated the distribution and cytotoxic activity of hyaluronan esterified with butyric acid residues against liver metastases arising from B16-F10 (CD44+) melanoma cells in mice. Administration of the drug resulted in complete suppression of metastases in animals and significantly prolonged life expectancy compared to control groups [189].
It was demonstrated that HA conjugate with graft-poly(dimethylaminoethyl methacrylate) (HPD) could form stable complexes with siRNA and chemically cross-link through disulfide bond formation. HPD-siRNA complexes were efficiently taken up by B16-F10 melanoma cells overexpressing CD44, but not by normal fibroblasts. In vivo studies demonstrated selective accumulation of siRNA-HPD complexes at the tumor site after their systemic administration to mice, resulting in effective suppression of target gene expression. Thus, HPD conjugate can be used as an effective carrier for delivery of siRNA-based targeting drugs against melanoma [190].
In a series of papers by Peer and Margalit, it was shown how hyaluronan-coated targeted nanoliposomes (tHA-LIP) coated with doxorubicin (DXR) or mitomycin C (MMC) caused a significant reduction in the metastatic burden of melanoma cell line B16-F10.9 in the lung, compared to the use of unbound drug. Thus, tHA-LIP treatment with DXR/MMC-loaded liposomes resulted in a significant and meaningful increase in survival compared to free drug, non-targeted DXR/MMC-loaded liposomes, and Doxil, respectively [191,192].
7.1.2. CD44 Monoclonal Antibody Inhibitors
Guo et al. investigated the use of the monoclonal antibody GKW.A2 against CD44 to inhibit tumor growth and metastasis using the human melanoma cell lines SMMU-1 and SMMU-2. Administration of GKW.A3 intravenously 1 week after tumor injection to mice subcutaneously didn’t suppress local tumor development, but inhibited metastatic tumor formation and increased animal survival [193].
A monoclonal antibody against CD44 (RG7356) in a Phase I clinical trial (NCT01358903) showed low clinical efficacy for patients with a variety of solid tumors, including melanoma. Only 13 of 61 patients (21%) experienced SD lasting an average of 12 weeks. No dose-dependent changes in biological activity were reported in blood and tissue assays. Tumor targeting by positron emission tomography using 89Zr-labeled RG7356 showed that the monoclonal antibody can be used to trace tumors in the human body, providing a potential application of this agent in combination regimens [194].
7.1.3. CD44 Alternative Inhibitors
The microRNA miR-143-3p was identified as the most potent binder to the 3'-untranslated region of CD44. Overexpression of miR-143-3p was shown to inhibit CD44 translation in the melanoma cell line BLM, which was accompanied by a decrease in proliferation, migration and enhanced apoptosis of melanoma cells induced by daunorubicin in vitro. Analyses of the respective expression levels of CD44 and miR-143-3p in human melanocytic nevus and dermal melanoma samples demonstrated medium to high levels of CD44 without correlation with tumor grading or stage. Moreover, CD44 expression was inversely correlated with the infiltration of proinflammatory immune effector cells into the stroma [195].
Ahrens et al. demonstrated an approach to block HA binding to CD44 on the surface of melanoma cells using soluble CD44. It was shown that introduction of cDNA encoding a soluble form of CD44 into melanoma cells of line 1F6 inhibited their growth by competitively blocking the binding of CD44 on the cell surface to hyaluronic acid, which was confirmed experimentally under in vitro and in vivo conditions [196].
A collagen triple helix peptide mimetic, which is a triple helix “peptide-amphiphile” (α1(IV)1263-1277 PA) was designed to specifically bind to CD44. It was conjugated into liposomes of different lipid compositions loaded with the fluorophore rhodamine to create a delivery system. The presented results confirmed the efficiency of targeted liposome delivery to M14#5 melanoma cells due to the highest accumulation of rhodamine, making this peptide an attractive agent for targeted drug delivery to melanoma cells [197].
7.2. DDR1/2 Inhibitors
Discoidin domain receptors (DDR1 and DDR2), are non-integrin collagen receptors that are members of a family of receptor tyrosine kinases [198]. Both DDR receptors bind a number of different types of collagens and play important roles in key cellular processes including migration, proliferation, differentiation and cell survival. In addition, DDRs control ECM remodeling through control of matrix metalloproteinase (MMP) expression and activity and have overlapping functions with collagen-binding integrins [199].
Application of siRNA as against DDR1 [200], and DDR2 [201,202] suppressed migration, invasion and survival in human melanoma cell lines. In addition, a DDR tyrosine kinase inhibitor (DDR1-IN-1) also significantly suppressed proliferation of melanoma cell line M10 in vitro and in C8161 and SKMEL5 xenograft tumor models in vivo [200]. Berestjuk et al. in their manuscript, introduce the term matrix-mediated drug resistance (MMDR) by demonstrating that interaction with fibroblast ECM abrogates tumor antiproliferative responses to inhibition of the BRAF/MEK pathway. As part of the study, the authors demonstrate an approach to specifically target DDR1 and DDR2. In SKMEL5 cell lines and MM099 short-term melanoma cell culture and 1205Lu xenografts in mice, targeting DDR with imatinib was shown to increase the efficacy of BRAF inhibitors, counteract collagen remodeling, and delay melanoma recurrence [203].
Thus, non-integrin receptors, including CD44 and DDR, play an important role in the processes of cancer cell adhesion, migration and invasion. Inhibitors targeting CD44 show potential in targeted therapy, especially in the form of nanoparticles and conjugates in combination with chemotherapy. Collagen DDR receptors have also shown promise as targets for therapy, especially in combination approaches such as with BRAF inhibitors, which may improve melanoma treatment and reduce drug resistance of cancer cells.
8. Conclusion
The extracellular matrix remains one of the most understudied parts of tissue and the tumor microenvironment. The complex molecular network into which matrisome components are interconnected is of significant interest for targeting therapy of many diseases, including melanoma. As the experience of many described studies has shown - selective suppression of one receptor/ligand axis allows to completely inhibit tumor metastasis and invasion, however, subtle fundamental mechanisms underlying cellular interactions and adhesion may be disturbed, which will inevitably lead to disturbance of homeostasis of normal tissues. To date, enough potential targets in tumor ECM and their corresponding targeting drugs have been identified, including recombinant proteins and monoclonal antibodies. Although preclinical trials form encouraging prospects for the application of targeting therapy, clinical efficacy remains severely limited, setting the development vector towards novel highly selective and safe inhibitors.
Author Contributions
Conceptualization, A.A.R., I.Y.F. and V.V.S.; writing – original draft preparation, M.N.O., C.B.K. and A.V.G.; writing – review and editing, Y.P.M., A.I.G. and G.I.K.; visualization, Y.P.M. and K.V.K.; supervision, A.A.R. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
This paper has been supported by the Kazan Federal University Strategic Academic Leadership Program (PRIORITY-2030).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Strashilov, S.; Yordanov, A. Aetiology and Pathogenesis of Cutaneous Melanoma: Current Concepts and Advances. International Journal of Molecular Sciences 2021, 22, 6395. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Fang, Y.; Chen, H.; Zhang, T.; Yin, X.; Man, J.; Yang, X.; Lu, M. Spatiotemporal Trends of the Global Burden of Melanoma in 204 Countries and Territories from 1990 to 2019: Results from the 2019 Global Burden of Disease Study. Neoplasia 2022, 24, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Cazzato, G. Histopathological Diagnosis of Malignant Melanoma at the Dawn of 2023: Knowledge Gained and New Challenges. Dermatopathology 2023, 10, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; de Vries, E.; Whiteman, D.C.; Bray, F. Global Burden of Cutaneous Melanoma in 2020 and Projections to 2040. JAMA Dermatol 2022, 158, 495–503. [Google Scholar] [CrossRef]
- Park, S.L.; Le Marchand, L.; Wilkens, L.R.; Kolonel, L.N.; Henderson, B.E.; Zhang, Z.-F.; Setiawan, V.W. Risk Factors for Malignant Melanoma in White and Non-White/Non-African American Populations: The Multiethnic Cohort. Cancer Prev Res (Phila) 2012, 5, 423–434. [Google Scholar] [CrossRef]
- Mattia, G.; Puglisi, R.; Ascione, B.; Malorni, W.; Carè, A.; Matarrese, P. Cell Death-Based Treatments of Melanoma:Conventional Treatments and New Therapeutic Strategies. Cell Death Dis 2018, 9, 112. [Google Scholar] [CrossRef]
- Zhou, A.Y.; Johnson, D.B. Combinatorial Therapies in Melanoma: MAPK Inhibitors and Beyond. Am J Clin Dermatol 2018, 19, 181–193. [Google Scholar] [CrossRef]
- Sun, J.; Carr, M.J.; Khushalani, N.I. Principles of Targeted Therapy for Melanoma. Surg Clin North Am 2020, 100, 175–188. [Google Scholar] [CrossRef]
- Broman, K.K.; Dossett, L.A.; Sun, J.; Eroglu, Z.; Zager, J.S. Update on BRAF and MEK Inhibition for Treatment of Melanoma in Metastatic, Unresectable, and Adjuvant Settings. Expert Opin Drug Saf 2019, 18, 381–392. [Google Scholar] [CrossRef]
- Falcone, I.; Conciatori, F.; Bazzichetto, C.; Ferretti, G.; Cognetti, F.; Ciuffreda, L.; Milella, M. Tumor Microenvironment: Implications in Melanoma Resistance to Targeted Therapy and Immunotherapy. Cancers 2020, 12, 2870. [Google Scholar] [CrossRef]
- Kharouf, N.; Flanagan, T.W.; Hassan, S.-Y.; Shalaby, H.; Khabaz, M.; Hassan, S.-L.; Megahed, M.; Haikel, Y.; Santourlidis, S.; Hassan, M. Tumor Microenvironment as a Therapeutic Target in Melanoma Treatment. Cancers 2023, 15, 3147. [Google Scholar] [CrossRef] [PubMed]
- Giavina-Bianchi, M.H.; Giavina-Bianchi, P.F.; Festa, C. Melanoma: Tumor Microenvironment and New Treatments*. An. Bras. Dermatol. 2017, 92, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Blansfield, J.A.; Caragacianu, D.; Alexander, H.R.; Tangrea, M.A.; Morita, S.Y.; Lorang, D.; Schafer, P.; Muller, G.; Stirling, D.; Royal, R.E.; et al. Combining Agents That Target the Tumor Microenvironment Improves the Efficacy of Anticancer Therapy. Clin Cancer Res 2008, 14, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Swartz, M.A.; Iida, N.; Roberts, E.W.; Sangaletti, S.; Wong, M.H.; Yull, F.E.; Coussens, L.M.; DeClerck, Y.A. Tumor Microenvironment Complexity: Emerging Roles in Cancer Therapy. Cancer Res 2012, 72, 2473–2480. [Google Scholar] [CrossRef]
- Mhaidly, R.; Mechta-Grigoriou, F. Fibroblast Heterogeneity in Tumor Micro-Environment: Role in Immunosuppression and New Therapies. Semin Immunol 2020, 48, 101417. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front Immunol 2019, 10, 1835. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment. Cancers (Basel) 2014, 6, 1670–1690. [Google Scholar] [CrossRef]
- Gao, J.; Liang, Y.; Wang, L. Shaping Polarization Of Tumor-Associated Macrophages In Cancer Immunotherapy. Front Immunol 2022, 13, 888713. [Google Scholar] [CrossRef]
- Scott, E.N.; Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Regulatory T Cells: Barriers of Immune Infiltration Into the Tumor Microenvironment. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines 2016, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-Talk between Myeloid-Derived Suppressor Cells (MDSC), Macrophages, and Dendritic Cells Enhances Tumor-Induced Immune Suppression. Semin Cancer Biol 2012, 22, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front Immunol 2018, 9, 398. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, A.A.; Watkins, S.K. Immune Suppression in the Tumor Microenvironment: A Role for Dendritic Cell-Mediated Tolerization of T Cells. Cancer Immunol Immunother 2012, 61, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Shaul, M.E.; Fridlender, Z.G. Neutrophils as Active Regulators of the Immune System in the Tumor Microenvironment. J Leukoc Biol 2017, 102, 343–349. [Google Scholar] [CrossRef]
- Duong, M.N.; Geneste, A.; Fallone, F.; Li, X.; Dumontet, C.; Muller, C. The fat and the bad: Mature adipocytes, key actors in tumor progression and resistance. Oncotarget 2017, 8, 57622–57641. [Google Scholar] [CrossRef]
- Varol, C. Tumorigenic Interplay Between Macrophages and Collagenous Matrix in the Tumor Microenvironment. Methods Mol Biol 2019, 1944, 203–220. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The Extracellular Matrix: A Dynamic Niche in Cancer Progression. Journal of Cell Biology 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Vannucci, L. Stroma as an Active Player in the Development of the Tumor Microenvironment. Cancer Microenviron 2015, 8, 159–166. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular Matrix (ECM) Stiffness and Degradation as Cancer Drivers. J Cell Biochem 2019, 120, 2782–2790. [Google Scholar] [CrossRef]
- Brassart-Pasco, S.; Brézillon, S.; Brassart, B.; Ramont, L.; Oudart, J.-B.; Monboisse, J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Pampena, R.; Kyrgidis, A.; Lallas, A.; Moscarella, E.; Argenziano, G.; Longo, C. A Meta-Analysis of Nevus-Associated Melanoma: Prevalence and Practical Implications. J Am Acad Dermatol 2017, 77, 938–945e4. [Google Scholar] [CrossRef] [PubMed]
- Martín-Gorgojo, A.; Nagore, E. Melanoma Arising in a Melanocytic Nevus. Actas Dermosifiliogr (Engl Ed) 2018, 109, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Botti, G.; Cerrone, M.; Scognamiglio, G.; Anniciello, A.; Ascierto, P.A.; Cantile, M. Microenvironment and Tumor Progression of Melanoma: New Therapeutic Prospectives. J Immunotoxicol 2013, 10, 235–252. [Google Scholar] [CrossRef]
- Van Duinen, C.M.; Fleuren, G.J.; Bruijn, J.A. The Extracellular Matrix in Pigmented Skin Lesions: An Immunohistochemical Study. Histopathology 1994, 24, 33–40. [Google Scholar] [CrossRef]
- McGary, E.C.; Lev, D.C.; Bar-Eli, M. Cellular Adhesion Pathways and Metastatic Potential of Human Melanoma. Cancer Biol Ther 2002, 1, 459–465. [Google Scholar] [CrossRef]
- Smolle, J.; Fiebiger, M.; Hofmann-Wellenhof, R.; Kerl, H. Quantitative Morphology of Collagen Fibers in Cutaneous Malignant Melanoma and Melanocytic Nevus. Am J Dermatopathol 1996, 18, 358–363. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The Extracellular Matrix Modulates the Hallmarks of Cancer. EMBO Rep 2014, 15, 1243–1253. [Google Scholar] [CrossRef]
- Kavuturu, A.; Alves Constantino, M.; Sassano, A.; Merlino, G. Abstract 4148: Extracellular Matrix Biophysical Properties Change during Nevus to Melanoma Progression. Cancer Research 2024, 84, 4148–4148. [Google Scholar] [CrossRef]
- Kumar, S.M.; Yu, H.; Edwards, R.; Chen, L.; Kazianis, S.; Brafford, P.; Acs, G.; Herlyn, M.; Xu, X. Mutant V600E BRAF Increases Hypoxia Inducible Factor-1α Expression in Melanoma. Cancer Research 2007, 67, 3177–3184. [Google Scholar] [CrossRef]
- O’Connell, M.P.; Marchbank, K.; Webster, M.R.; Valiga, A.A.; Kaur, A.; Vultur, A.; Li, L.; Herlyn, M.; Villanueva, J.; Liu, Q.; et al. Hypoxia Induces Phenotypic Plasticity and Therapy Resistance in Melanoma via the Tyrosine Kinase Receptors ROR1 and ROR2. Cancer Discov 2013, 3, 1378–1393. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, M.; Zhang, L.; Shen, D.; Xu, X.; Yi, Q.; Tang, L. SNF5, a Core Subunit of SWI/SNF Complex, Regulates Melanoma Cancer Cell Growth, Metastasis, and Immune Escape in Response to Matrix Stiffness. Transl Oncol 2022, 17, 101335. [Google Scholar] [CrossRef]
- Brás, M.M.; Radmacher, M.; Sousa, S.R.; Granja, P.L. Melanoma in the Eyes of Mechanobiology. Front Cell Dev Biol 2020, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.M.; Yeyeodu, S.T.; McLaughlin, A.; Schuman, D.; Taylor, D.K. In Situ Drug Delivery to Breast Cancer-Associated Extracellular Matrix. ACS Chem. Biol. 2018, 13, 2825–2840. [Google Scholar] [CrossRef] [PubMed]
- Jalil, S.M.A.; Henry, J.C.; Cameron, A.J.M. Targets in the Tumour Matrisome to Promote Cancer Therapy Response. Cancers 2024, 16, 1847. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.-Q.; Liu, H.; Navarro, E.; Kussie, P.; Zhu, Z. Development of Heparanase Inhibitors for Anti-Cancer Therapy. Curr Med Chem 2006, 13, 2101–2111. [Google Scholar] [CrossRef]
- Yuan, F.; Yang, Y.; Zhou, H.; Quan, J.; Liu, C.; Wang, Y.; Zhang, Y.; Yu, X. Heparanase in Cancer Progression: Structure, Substrate Recognition and Therapeutic Potential. Front Chem 2022, 10, 926353. [Google Scholar] [CrossRef]
- Jayatilleke, K.M.; Hulett, M.D. Heparanase and the Hallmarks of Cancer. Journal of Translational Medicine 2020, 18, 453. [Google Scholar] [CrossRef]
- Vornicova, O.; Boyango, I.; Feld, S.; Naroditsky, I.; Kazarin, O.; Zohar, Y.; Tiram, Y.; Ilan, N.; Ben-Izhak, O.; Vlodavsky, I.; et al. The prognostic significance of heparanase expression in metastatic melanoma. Oncotarget 2016, 7, 74678–74685. [Google Scholar] [CrossRef]
- Murry, B.P.; Greiter-Wilke, A.; Paulsen, D.P.; Hiatt, K.M.; Beltrami, C.A.; Marchetti, D. Selective Heparanase Localization in Malignant Melanoma. Int J Oncol 2005, 26, 345–352. [Google Scholar] [CrossRef]
- Nakajima, M.; DeChavigny, A.; Johnson, C.E.; Hamada, J.; Stein, C.A.; Nicolson, G.L. Suramin. A Potent Inhibitor of Melanoma Heparanase and Invasion. J Biol Chem 1991, 266, 9661–9666. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Miao, H.-Q.; Xu, Y.-J.; Navarro, E.C.; Tonra, J.R.; Corcoran, E.; Lahiji, A.; Kussie, P.; Kiselyov, A.S.; Wong, W.C.; et al. 1-[4-(1H-Benzoimidazol-2-Yl)-Phenyl]-3-[4-(1H-Benzoimidazol-2-Yl)-Phenyl]-Urea Derivatives as Small Molecule Heparanase Inhibitors. Bioorg Med Chem Lett 2006, 16, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Irimura, T.; Nakajima, M.; Nicolson, G.L. Chemically Modified Heparins as Inhibitors of Heparan Sulfate Specific Endo-Beta-Glucuronidase (Heparanase) of Metastatic Melanoma Cells. Biochemistry 1986, 25, 5322–5328. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Mohsen, M.; Lider, O.; Cm, S.; Hp, E.; Vigoda, M.; Ishai-Michaeli, R.; Peretz, T. Inhibition of Tumor Metastasis by Heparanase Inhibiting Species of Heparin. Invasion & metastasis 1994.
- Bitan, M.; Mohsen, M.; Levi, E.; Wygoda, M.R.; Miao, H.Q.; Lider, O.; Svahn, C.M.; Ekre, H.P.; Ishai-Michaeli, R.; Bar-Shavit, R. Structural Requirements for Inhibition of Melanoma Lung Colonization by Heparanase Inhibiting Species of Heparin. Isr J Med Sci 1995, 31, 106–118. [Google Scholar]
- Kaur, R.; Deb, P.K.; Diwan, V.; Saini, B. Heparanase Inhibitors in Cancer Progression: Recent Advances. Current Pharmaceutical Design 27, 43–68. [CrossRef]
- Mohan, C.D.; Hari, S.; Preetham, H.D.; Rangappa, S.; Barash, U.; Ilan, N.; Nayak, S.C.; Gupta, V.K.; Basappa, null; Vlodavsky, I.; et al. Targeting Heparanase in Cancer: Inhibition by Synthetic, Chemically Modified, and Natural Compounds. iScience 2019, 15, 360–390. [CrossRef]
- Chhabra, M.; Ferro, V. PI-88 and Related Heparan Sulfate Mimetics. Adv Exp Med Biol 2020, 1221, 473–491. [Google Scholar] [CrossRef]
- Chow, L.Q.M.; Gustafson, D.L.; O’Bryant, C.L.; Gore, L.; Basche, M.; Holden, S.N.; Morrow, M.C.; Grolnic, S.; Creese, B.R.; Roberts, K.L.; et al. A Phase I Pharmacological and Biological Study of PI-88 and Docetaxel in Patients with Advanced Malignancies. Cancer Chemother Pharmacol 2008, 63, 65. [Google Scholar] [CrossRef]
- Holden, S.; Basche, M.; O’Bryant, C.; Morrow, M.; Grolnic, S.; Persky, M.; Deem, C.; Roberts, K.; Ribbons, K.; Eckhardt, S. A Phase I Study of the Heparanase Inhibitor PI-88 given Subcutaneously (Sq) in Patients (Pts) with Advanced Solid Malignancies. European Journal of Cancer 2002, 38, S74–S75. [Google Scholar] [CrossRef]
- Rosenthal, M.A.; Rischin, D.; McArthur, G.; Ribbons, K.; Chong, B.; Fareed, J.; Toner, G.; Green, M.D.; Basser, R.L. Treatment with the Novel Anti-Angiogenic Agent PI-88 Is Associated with Immune-Mediated Thrombocytopenia. Ann Oncol 2002, 13, 770–776. [Google Scholar] [CrossRef]
- Millward, M.; Hamilton, A.; Thomson, D.; Gautam, A.; Wilson, E. Final Results of a Phase I Study of Daily PI-88 as a Single Agent and in Combination with Dacarbazine (D) in Patients with Metastatic Melanoma. JCO 2007, 25, 8532–8532. [Google Scholar] [CrossRef]
- Basche, M.; Gustafson, D.L.; Holden, S.N.; O’Bryant, C.L.; Gore, L.; Witta, S.; Schultz, M.K.; Morrow, M.; Levin, A.; Creese, B.R.; et al. A Phase I Biological and Pharmacologic Study of the Heparanase Inhibitor PI-88 in Patients with Advanced Solid Tumors. Clin Cancer Res 2006, 12, 5471–5480. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.D.; Robinson, W.A.; Millward, M.J.; Powell, A.; Price, T.J.; Thomson, D.B.; Walpole, E.T.; Haydon, A.M.; Creese, B.R.; Roberts, K.L.; et al. A Phase II Study of the Heparanase Inhibitor PI-88 in Patients with Advanced Melanoma. Invest New Drugs 2008, 26, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Lanzi, C.; Cassinelli, G. Heparan Sulfate Mimetics in Cancer Therapy: The Challenge to Define Structural Determinants and the Relevance of Targets for Optimal Activity. Molecules 2018, 23, 2915. [Google Scholar] [CrossRef] [PubMed]
- Dredge, K.; Hammond, E.; Davis, K.; Li, C.P.; Liu, L.; Johnstone, K.; Handley, P.; Wimmer, N.; Gonda, T.J.; Gautam, A.; et al. The PG500 Series: Novel Heparan Sulfate Mimetics as Potent Angiogenesis and Heparanase Inhibitors for Cancer Therapy. Invest New Drugs 2010, 28, 276–283. [Google Scholar] [CrossRef]
- Mohamed, S.; Coombe, D.R. Heparin Mimetics: Their Therapeutic Potential. Pharmaceuticals 2017, 10, 78. [Google Scholar] [CrossRef]
- Johnstone, K.D.; Karoli, T.; Liu, L.; Dredge, K.; Copeman, E.; Li, C.P.; Davis, K.; Hammond, E.; Bytheway, I.; Kostewicz, E.; et al. Synthesis and Biological Evaluation of Polysulfated Oligosaccharide Glycosides as Inhibitors of Angiogenesis and Tumor Growth. J. Med. Chem. 2010, 53, 1686–1699. [Google Scholar] [CrossRef]
- Winterhoff, B.; Freyer, L.; Hammond, E.; Giri, S.; Mondal, S.; Roy, D.; Teoman, A.; Mullany, S.A.; Hoffmann, R.; von Bismarck, A.; et al. PG545 Enhances Anti-Cancer Activity of Chemotherapy in Ovarian Models and Increases Surrogate Biomarkers Such as VEGF in Preclinical and Clinical Plasma Samples. Eur J Cancer 2015, 51, 879–892. [Google Scholar] [CrossRef]
- Lemech, C.; Dredge, K.; Bampton, D.; Hammond, E.; Clouston, A.; Waterhouse, N.J.; Stanley, A.C.; Mouttie, L.L.-E.; Chojnowski, G.M.; Haydon, A.; et al. Phase Ib Open-Label, Multicenter Study of Pixatimod, an Activator of TLR9, in Combination with Nivolumab in Subjects with Microsatellite-Stable Metastatic Colorectal Cancer, Metastatic Pancreatic Ductal Adenocarcinoma and Other Solid Tumors. J Immunother Cancer 2023, 11, e006136. [Google Scholar] [CrossRef]
- Roy, M.; Reiland, J.; Murry, B.P.; Chouljenko, V.; Kousoulas, K.G.; Marchetti, D. Antisense-Mediated Suppression of Heparanase Gene Inhibits Melanoma Cell Invasion. Neoplasia 2005, 7, 253–262. [Google Scholar] [CrossRef]
- Liu, X.; Fang, H.; Chen, H.; Jiang, X.; Fang, D.; Wang, Y.; Zhu, D. An Artificial miRNA against HPSE Suppresses Melanoma Invasion Properties, Correlating with a down-Regulation of Chemokines and MAPK Phosphorylation. PLoS One 2012, 7, e38659. [Google Scholar] [CrossRef]
- Edovitsky, E.; Elkin, M.; Zcharia, E.; Peretz, T.; Vlodavsky, I. Heparanase Gene Silencing, Tumor Invasiveness, Angiogenesis, and Metastasis. J Natl Cancer Inst 2004, 96, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Reunanen, N.; Kähäri, V. Matrix Metalloproteinases in Cancer Cell Invasion. In Madame Curie Bioscience Database [Internet]; Landes Bioscience, 2013.
- Hidalgo, M.; Eckhardt, S.G. Development of Matrix Metalloproteinase Inhibitors in Cancer Therapy. JNCI: Journal of the National Cancer Institute 2001, 93, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures into Future Successes. Mol Cancer Ther 2018, 17, 1147–1155. [Google Scholar] [CrossRef]
- Hofmann, U.B.; Westphal, J.R.; Van Muijen, G.N.; Ruiter, D.J. Matrix Metalloproteinases in Human Melanoma. J Invest Dermatol 2000, 115, 337–344. [Google Scholar] [CrossRef]
- Skiles, J.W.; Gonnella, N.C.; Jeng, A.Y. The Design, Structure, and Clinical Update of Small Molecular Weight Matrix Metalloproteinase Inhibitors. Curr Med Chem 2004, 11, 2911–2977. [Google Scholar] [CrossRef]
- Lazar, A.M.; Costea, D.O.; Popp, C.G.; Mastalier, B. Skin Malignant Melanoma and Matrix Metalloproteinases: Promising Links to Efficient Therapies. Int J Mol Sci 2024, 25, 7804. [Google Scholar] [CrossRef]
- Alves, R.; Pires, A.; Jorge, J.; Balça-Silva, J.; Gonçalves, A.C.; Sarmento-Ribeiro, A.B. Batimastat Induces Cytotoxic and Cytostatic Effects in In vitro Models of Hematological Tumors. Int J Mol Sci 2024, 25, 4554. [Google Scholar] [CrossRef]
- Rasmussen, H.S.; McCann, P.P. Matrix Metalloproteinase Inhibition as a Novel Anticancer Strategy: A Review with Special Focus on Batimastat and Marimastat. Pharmacol Ther 1997, 75, 69–75. [Google Scholar] [CrossRef]
- Chirivi, R.G.; Garofalo, A.; Crimmin, M.J.; Bawden, L.J.; Stoppacciaro, A.; Brown, P.D.; Giavazzi, R. Inhibition of the Metastatic Spread and Growth of B16-BL6 Murine Melanoma by a Synthetic Matrix Metalloproteinase Inhibitor. Int J Cancer 1994, 58, 460–464. [Google Scholar] [CrossRef]
- Wylie, S.; MacDonald, I.C.; Varghese, H.J.; Schmidt, E.E.; Morris, V.L.; Groom, A.C.; Chambers, A.F. The Matrix Metalloproteinase Inhibitor Batimastat Inhibits Angiogenesis in Liver Metastases of B16F1 Melanoma Cells. Clin Exp Metastasis 1999, 17, 111–117. [Google Scholar] [CrossRef]
- Macaulay, V.M.; O’Byrne, K.J.; Saunders, M.P.; Braybrooke, J.P.; Long, L.; Gleeson, F.; Mason, C.S.; Harris, A.L.; Brown, P.; Talbot, D.C. Phase I Study of Intrapleural Batimastat (BB-94), a Matrix Metalloproteinase Inhibitor, in the Treatment of Malignant Pleural Effusions1. Clinical Cancer Research 1999, 5, 513–520. [Google Scholar] [PubMed]
- Steward, W.P. Marimastat (BB2516): Current Status of Development. Cancer Chemother Pharmacol 1999, 43 Suppl, S56–60. [Google Scholar] [CrossRef]
- Bodurtha, A.; Eisenhauer, E.; Steward, W.; Rusthoven, J.; Quirt, I.; Lohmann, R.; Wainman, N.; Rugg, T. Phase I-II Study of Marimastat (BB2516) in Patients with Metastatic Melanoma. In Proceedings of the Proc Am Soc Clin Oncol; 1997; Vol. 16, p. 493a.
- Toppmeyer, D.L.; Gounder, M.; Much, J.; Musanti, R.; Vyas, V.; Medina, M.; Orlando, T.; Pennick, M.; Lin, Y.; Shih, W.; et al. A Phase I and Pharmacologic Study of the Combination of Marimastat and Paclitaxel in Patients with Advanced Malignancy. Med Sci Monit 2003, 9, PI99–104. [Google Scholar]
- Quirt, I.; Bodurth, A.; Lohmann, R.; Rusthoven, J.; Belanger, K.; Young, V.; Wainman, N.; Stewar, W.; Eisenhauer, E. ; National Cancer Institute of Canada Clinical Trials Group Phase II Study of Marimastat (BB-2516) in Malignant Melanoma: A Clinical and Tumor Biopsy Study of the National Cancer Institute of Canada Clinical Trials Group. Invest New Drugs 2002, 20, 431–437. [Google Scholar] [CrossRef]
- Ferrario, A.; Chantrain, C.F.; von Tiehl, K.; Buckley, S.; Rucker, N.; Shalinsky, D.R.; Shimada, H.; DeClerck, Y.A.; Gomer, C.J. The Matrix Metalloproteinase Inhibitor Prinomastat Enhances Photodynamic Therapy Responsiveness in a Mouse Tumor Model. Cancer Res 2004, 64, 2328–2332. [Google Scholar] [CrossRef]
- Ozerdem, U.; Mach-Hofacre, B.; Varki, N.; Folberg, R.; Mueller, A.J.; Ochabski, R.; Pham, T.; Appelt, K.; Freeman, W.R. The Effect of Prinomastat (AG3340), a Synthetic Inhibitor of Matrix Metalloproteinases, on Uveal Melanoma Rabbit Model. Current Eye Research 2002, 24, 86–91. [Google Scholar] [CrossRef]
- Shalinsky, D.R.; Brekken, J.; Zou, H.; McDermott, C.D.; Forsyth, P.; Edwards, D.; Margosiak, S.; Bender, S.; Truitt, G.; Wood, A.; et al. Broad Antitumor and Antiangiogenic Activities of AG3340, a Potent and Selective MMP Inhibitor Undergoing Advanced Oncology Clinical Trials. Ann N Y Acad Sci 1999, 878, 236–270. [Google Scholar] [CrossRef]
- Hande, K.R.; Collier, M.; Paradiso, L.; Stuart-Smith, J.; Dixon, M.; Clendeninn, N.; Yeun, G.; Alberti, D.; Binger, K.; Wilding, G. Phase I and Pharmacokinetic Study of Prinomastat, a Matrix Metalloprotease Inhibitor. Clinical Cancer Research 2004, 10, 909–915. [Google Scholar] [CrossRef]
- Lokeshwar, B.L.; Escatel, E.; Zhu, B. Cytotoxic Activity and Inhibition of Tumor Cell Invasion by Derivatives of a Chemically Modified Tetracycline CMT-3 (COL-3). Curr Med Chem 2001, 8, 271–279. [Google Scholar] [CrossRef]
- Rudek, M.A.; Figg, W.D.; Dyer, V.; Dahut, W.; Turner, M.L.; Steinberg, S.M.; Liewehr, D.J.; Kohler, D.R.; Pluda, J.M.; Reed, E. Phase I Clinical Trial of Oral COL-3, a Matrix Metalloproteinase Inhibitor, in Patients With Refractory Metastatic Cancer. Journal of Clinical Oncology 2016. [CrossRef]
- Kasaoka, T.; Nishiyama, H.; Okada, M.; Nakajima, M. Matrix Metalloproteinase Inhibitor, MMI270 (CGS27023A) Inhibited Hematogenic Metastasis of B16 Melanoma Cells in Both Experimental and Spontaneous Metastasis Models. Clin Exp Metastasis 2008, 25, 827–834. [Google Scholar] [CrossRef]
- Nakamura, E.S.; Koizumi, K.; Yamaura, T.; Saiki, I. Anti-Tumor Angiogenic Effect of a Matrix Metalloproteinase Inhibitor MMI270. Anticancer Res 2003, 23, 411–417. [Google Scholar] [PubMed]
- Naglich, J.G.; Jure-Kunkel, M.; Gupta, E.; Fargnoli, J.; Henderson, A.J.; Lewin, A.C.; Talbott, R.; Baxter, A.; Bird, J.; Savopoulos, R.; et al. Inhibition of Angiogenesis and Metastasis in Two Murine Models by the Matrix Metalloproteinase Inhibitor, BMS-275291. Cancer Research 2001, 61, 8480–8485. [Google Scholar] [PubMed]
- Romanchikova, N.; Trapencieris, P.; Zemītis, J.; Turks, M. A Novel Matrix Metalloproteinase-2 Inhibitor Triazolylmethyl Aziridine Reduces Melanoma Cell Invasion, Angiogenesis and Targets ERK1/2 Phosphorylation. Journal of Enzyme Inhibition and Medicinal Chemistry 2014, 29, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Kuang, X.; Xie, Z.; Liang, L.; Zhang, Z.; Zhang, Y.; Ma, F.; Gao, Q.; Chang, R.; Lee, H.-H.; et al. Small-Molecule MMP2/MMP9 Inhibitor SB-3CT Modulates Tumor Immune Surveillance by Regulating PD-L1. Genome Medicine 2020, 12, 83. [Google Scholar] [CrossRef]
- Tao, P.; Fisher, J.F.; Mobashery, S.; Bernhard Schlegel, H. DFT Studies of the Ring Opening Mechanism of SB-3CT, a Potent Inhibitor of Matrix Metalloproteinase 2. Org Lett 2009, 11, 2559. [Google Scholar] [CrossRef]
- Krüger, A.; Arlt, M.J.E.; Gerg, M.; Kopitz, C.; Bernardo, M.M.; Chang, M.; Mobashery, S.; Fridman, R. Antimetastatic Activity of a Novel Mechanism-Based Gelatinase Inhibitor. Cancer Research 2005, 65, 3523–3526. [Google Scholar] [CrossRef]
- Marusak, C.; Bayles, I.; Ma, J.; Gooyit, M.; Gao, M.; Chang, M.; Bedogni, B. The Thiirane-Based Selective MT1-MMP/MMP2 Inhibitor ND-322 Reduces Melanoma Tumor Growth and Delays Metastatic Dissemination. Pharmacol Res 2016, 113, 515–520. [Google Scholar] [CrossRef]
- Reich, R.; Katz, Y.; Hadar, R.; Breuer, E. Carbamoylphosphonate Matrix Metalloproteinase Inhibitors 3: In vivo Evaluation of Cyclopentylcarbamoylphosphonic Acid in Experimental Metastasis and Angiogenesis. Clin Cancer Res 2005, 11, 3925–3929. [Google Scholar] [CrossRef]
- Devy, L.; Arulanandam, T.; Buckler, D.; Finnern, R.; Naa, L.; Rank, D.; Ladner, R.C.; Rabbani, S.; Dransfield, D.T.; Henderikx, P. 203 POSTER Antitumor Efficacy of DX-2400, a Potent and Selective Human Antibody MMP-14 Inhibitor Discovered Using Phage Display Technology. European Journal of Cancer Supplements 2006, 4, 63–64. [Google Scholar] [CrossRef]
- Devy, L.; Huang, L.; Naa, L.; Yanamandra, N.; Pieters, H.; Frans, N.; Chang, E.; Tao, Q.; Vanhove, M.; Lejeune, A.; et al. Selective Inhibition of Matrix Metalloproteinase-14 Blocks Tumor Growth, Invasion, and Angiogenesis. Cancer Research 2009, 69, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Roomi, M.; Efremov, E.; Niedzwiecki, A.; Rath, M. Matrix Metalloproteinases-9 as a Promising Target for Anti-Cancer Vaccine: Inhibition of Melanoma Tumor Growth in Mice Immunized with Syngeneic MMP-9 Peptides. World Cancer Research Journal 2019, 6. [Google Scholar] [CrossRef]
- Roomi, M.W.; Niedzwiecki, A.; Rath, A. Peptide Vaccines Directed against Human Metalloproteinases (MMPs) with Anti-Tumor Efficacy in vitro and in vivo. J Cell Med Nat Health 2018, 1–12. [Google Scholar]
- Blackburn, J.S.; Rhodes, C.H.; Coon, C.I.; Brinckerhoff, C.E. RNA Interference Inhibition of Matrix Metalloproteinase-1 Prevents Melanoma Metastasis by Reducing Tumor Collagenase Activity and Angiogenesis. Cancer Res 2007, 67, 10849–10858. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Saji, S.; Sato, F.; Noda, M.; Toi, M. Potential Clinical Applications of Matrix Metalloproteinase Inhibitors and Their Future Prospects. Int J Biol Markers 2013, 28, 117–130. [Google Scholar] [CrossRef]
- Brand, K. Cancer Gene Therapy with Tissue Inhibitors of Metalloproteinases (TIMPs). Curr Gene Ther 2002, 2, 255–271. [Google Scholar] [CrossRef]
- Gomis-Rüth, F.X.; Maskos, K.; Betz, M.; Bergner, A.; Huber, R.; Suzuki, K.; Yoshida, N.; Nagase, H.; Brew, K.; Bourenkov, G.P.; et al. Mechanism of Inhibition of the Human Matrix Metalloproteinase Stromelysin-1 by TIMP-1. Nature 1997, 389, 77–81. [Google Scholar] [CrossRef]
- Schultz, R.M.; Silberman, S.; Persky, B.; Bajkowski, A.S.; Carmichael, D.F. Inhibition by Human Recombinant Tissue Inhibitor of Metalloproteinases of Human Amnion Invasion and Lung Colonization by Murine B16-F10 Melanoma Cells. Cancer Res 1988, 48, 5539–5545. [Google Scholar]
- Djafarzadeh, R.; Milani, V.; Rieth, N.; von Luettichau, I.; Skrablin, P.S.; Hofstetter, M.; Noessner, E.; Nelson, P.J. TIMP-1-GPI in Combination with Hyperthermic Treatment of Melanoma Increases Sensitivity to FAS-Mediated Apoptosis. Cancer Immunol Immunother 2009, 58, 361–371. [Google Scholar] [CrossRef]
- Oku, T.; Ata, N.; Yonezawa, K.; Tokai, H.; Fujii, H.; Shinagawa, A.; Ohuchi, E.; Saiki, I. Antimetastatic and Antitumor Effect of a Recombinant Human Tissue Inhibitor of Metalloproteinases-2 in Murine Melanoma Models. Biol Pharm Bull 1997, 20, 843–849. [Google Scholar] [CrossRef]
- Kang, W.K.; Park, E.-K.; Lee, H.S.; Park, B.-Y.; Chang, J.-Y.; Kim, M.-Y.; Kang, H.A.; Kim, J.-Y. A Biologically Active Angiogenesis Inhibitor, Human Serum Albumin-TIMP-2 Fusion Protein, Secreted from Saccharomyces Cerevisiae. Protein Expr Purif 2007, 53, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Bourboulia, D.; Jensen-Taubman, S.; Rittler, M.R.; Han, H.Y.; Chatterjee, T.; Wei, B.; Stetler-Stevenson, W.G. Endogenous Angiogenesis Inhibitor Blocks Tumor Growth via Direct and Indirect Effects on Tumor Microenvironment. Am J Pathol 2011, 179, 2589–2600. [Google Scholar] [CrossRef]
- Shi, Y.; Parhar, R.S.; Zou, M.; Al-Mohanna, F.A.; Paterson, M.C. Gene Therapy of Melanoma Pulmonary Metastasis by Intramuscular Injection of Plasmid DNA Encoding Tissue Inhibitor of Metalloproteinases-1. Cancer Gene Ther 2002, 9, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, A.M.; Mueller, B.M.; Reisfeld, R.A.; Taylor, S.M.; DeClerck, Y.A. Effect of Tissue Inhibitor of the Matrix Metalloproteinases-2 Expression on the Growth and Spontaneous Metastasis of a Human Melanoma Cell Line. Cancer Res 1994, 54, 5467–5473. [Google Scholar]
- Ahonen, M.; Baker, A.H.; Kähäri, V.M. Adenovirus-Mediated Gene Delivery of Tissue Inhibitor of Metalloproteinases-3 Inhibits Invasion and Induces Apoptosis in Melanoma Cells. Cancer Res 1998, 58, 2310–2315. [Google Scholar]
- Ahonen, M.; Ala-Aho, R.; Baker, A.H.; George, S.J.; Grénman, R.; Saarialho-Kere, U.; Kähäri, V.-M. Antitumor Activity and Bystander Effect of Adenovirally Delivered Tissue Inhibitor of Metalloproteinases-3. Mol Ther 2002, 5, 705–715. [Google Scholar] [CrossRef]
- Takabe, P.; Siiskonen, H.; Rönkä, A.; Kainulainen, K.; Pasonen-Seppänen, S. The Impact of Hyaluronan on Tumor Progression in Cutaneous Melanoma. Front Oncol 2022, 11, 811434. [Google Scholar] [CrossRef]
- Gagneja, S.; Capalash, N.; Sharma, P. Hyaluronic Acid as a Tumor Progression Agent and a Potential Chemotherapeutic Biomolecule against Cancer: A Review on Its Dual Role. Int J Biol Macromol 2024, 275, 133744. [Google Scholar] [CrossRef]
- Turley, E.A.; Wood, D.K.; McCarthy, J.B. Carcinoma Cell Hyaluronan as a “Portable” Cancerized Prometastatic Microenvironment. Cancer Res 2016, 76, 2507–2512. [Google Scholar] [CrossRef]
- Toole, B.P. Hyaluronan-CD44 Interactions in Cancer: Paradoxes and Possibilities. Clin Cancer Res 2009, 15, 7462–7468. [Google Scholar] [CrossRef]
- Xu, Y.; Benedikt, J.; Ye, L. Hyaluronic Acid Interacting Molecules Mediated Crosstalk between Cancer Cells and Microenvironment from Primary Tumour to Distant Metastasis. Cancers 2024, 16, 1907. [Google Scholar] [CrossRef] [PubMed]
- Sapudom, J.; Nguyen, K.-T.; Martin, S.; Wippold, T.; Möller, S.; Schnabelrauch, M.; Anderegg, U.; Pompe, T. Biomimetic Tissue Models Reveal the Role of Hyaluronan in Melanoma Proliferation and Invasion. Biomater Sci 2020, 8, 1405–1417. [Google Scholar] [CrossRef]
- Edward, M.; Gillan, C.; Micha, D.; Tammi, R.H. Tumour Regulation of Fibroblast Hyaluronan Expression: A Mechanism to Facilitate Tumour Growth and Invasion. Carcinogenesis 2005, 26, 1215–1223. [Google Scholar] [CrossRef]
- Voelcker, V.; Gebhardt, C.; Averbeck, M.; Saalbach, A.; Wolf, V.; Weih, F.; Sleeman, J.; Anderegg, U.; Simon, J. Hyaluronan Fragments Induce Cytokine and Metalloprotease Upregulation in Human Melanoma Cells in Part by Signalling via TLR4. Exp Dermatol 2008, 17, 100–107. [Google Scholar] [CrossRef]
- Ahrens, T.; Assmann, V.; Fieber, C.; Termeer, C.; Herrlich, P.; Hofmann, M.; Simon, J.C. CD44 Is the Principal Mediator of Hyaluronic-Acid-Induced Melanoma Cell Proliferation. J Invest Dermatol 2001, 116, 93–101. [Google Scholar] [CrossRef]
- Ichikawa, T.; Itano, N.; Sawai, T.; Kimata, K.; Koganehira, Y.; Saida, T.; Taniguchi, S. Increased Synthesis of Hyaluronate Enhances Motility of Human Melanoma Cells. J Invest Dermatol 1999, 113, 935–939. [Google Scholar] [CrossRef]
- Mummert, M.; Mummert, D.I.; Ellinger, L.; Takashima, A. Functional Roles of Hyaluronan in B16-F10 Melanoma Growth and Experimental Metastasis in Mice. Molecular cancer therapeutics 2003.
- Kultti, A.; Pasonen-Seppänen, S.; Jauhiainen, M.; Rilla, K.J.; Kärnä, R.; Pyöriä, E.; Tammi, R.H.; Tammi, M.I. 4-Methylumbelliferone Inhibits Hyaluronan Synthesis by Depletion of Cellular UDP-Glucuronic Acid and Downregulation of Hyaluronan Synthase 2 and 3. Exp Cell Res 2009, 315, 1914–1923. [Google Scholar] [CrossRef]
- Yoshihara, S.; Kon, A.; Kudo, D.; Nakazawa, H.; Kakizaki, I.; Sasaki, M.; Endo, M.; Takagaki, K. A Hyaluronan Synthase Suppressor, 4-Methylumbelliferone, Inhibits Liver Metastasis of Melanoma Cells. FEBS Lett 2005, 579, 2722–2726. [Google Scholar] [CrossRef]
- Kudo, D.; Kon, A.; Yoshihara, S.; Kakizaki, I.; Sasaki, M.; Endo, M.; Takagaki, K. Effect of a Hyaluronan Synthase Suppressor, 4-Methylumbelliferone, on B16F-10 Melanoma Cell Adhesion and Locomotion. Biochem Biophys Res Commun 2004, 321, 783–787. [Google Scholar] [CrossRef]
- Edward, M.; Quinn, J.A.; Pasonen-Seppänen, S.M.; McCann, B.A.; Tammi, R.H. 4-Methylumbelliferone Inhibits Tumour Cell Growth and the Activation of Stromal Hyaluronan Synthesis by Melanoma Cell-Derived Factors. Br J Dermatol 2010, 162, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Mummert, M.E.; Mohamadzadeh, M.; Mummert, D.I.; Mizumoto, N.; Takashima, A. Development of a Peptide Inhibitor of Hyaluronan-Mediated Leukocyte Trafficking. J Exp Med 2000, 192, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Spruss, T.; Bernhardt, G.; Schönenberger, H.; Schiess, W. Hyaluronidase Significantly Enhances the Efficacy of Regional Vinblastine Chemotherapy of Malignant Melanoma. J Cancer Res Clin Oncol 1995, 121, 193–202. [Google Scholar] [CrossRef]
- McGuire, J.; Taguchi, T.; Tombline, G.; Paige, V.; Janelsins, M.; Gilmore, N.; Seluanov, A.; Gorbunova, V. Hyaluronidase Inhibitor Delphinidin Inhibits Cancer Metastasis. Sci Rep 2024, 14, 14958. [Google Scholar] [CrossRef]
- Locke, K.W.; Maneval, D.C.; LaBarre, M.J. ENHANZE® Drug Delivery Technology: A Novel Approach to Subcutaneous Administration Using Recombinant Human Hyaluronidase PH20. Drug Deliv 2019, 26, 98–106. [Google Scholar] [CrossRef]
- Frost, G.I. Recombinant Human Hyaluronidase (rHuPH20): An Enabling Platform for Subcutaneous Drug and Fluid Administration. Expert Opin Drug Deliv 2007, 4, 427–440. [Google Scholar] [CrossRef]
- Styles, I.K.; Feeney, O.M.; Nguyen, T.-H.; Brundel, D.H.S.; Kang, D.W.; Clift, R.; McIntosh, M.P.; Porter, C.J.H. Removal of Interstitial Hyaluronan with Recombinant Human Hyaluronidase Improves the Systemic and Lymphatic Uptake of Cetuximab in Rats. J Control Release 2019, 315, 85–96. [Google Scholar] [CrossRef]
- Lonardi, S.; Lugowska, I.; Jackson, C.G.C.A.; O’Donnell, A.; Bahleda, R.; Garrido, M.; Latten-Jansen, L.; Chacon, M.; Yimer, H.A.; Camacho, T.; et al. CheckMate 8KX: Phase 1/2 Multitumor Preliminary Analyses of a Subcutaneous Formulation of Nivolumab (± rHuPH20). JCO 2021, 39, 2575–2575. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Mohr, P.; Dronca, R.; Harris, S.; Wilson, M.; Gurm, B.; Howansky, M.; Ng, W.-T.; Ravimohan, S.; Vezina, H.; et al. 882TiP Subcutaneous vs Intravenous Nivolumab in Patients with Melanoma Following Complete Resection. Annals of Oncology 2022, 33, S951–S952. [Google Scholar] [CrossRef]
- Huang, R.; Rofstad, E.K. Integrins as Therapeutic Targets in the Organ-Specific Metastasis of Human Malignant Melanoma. J Exp Clin Cancer Res 2018, 37, 92. [Google Scholar] [CrossRef]
- Pickarski, M.; Gleason, A.; Bednar, B.; Duong, L.T. Orally Active Avβ3 Integrin Inhibitor MK-0429 Reduces Melanoma Metastasis. Oncol Rep 2015, 33, 2737–2745. [Google Scholar] [CrossRef] [PubMed]
- Ruffini, F.; Graziani, G.; Levati, L.; Tentori, L.; D’Atri, S.; Lacal, P.M. Cilengitide Downmodulates Invasiveness and Vasculogenic Mimicry of Neuropilin 1 Expressing Melanoma Cells through the Inhibition of Avβ5 Integrin. Int J Cancer 2015, 136, E545–558. [Google Scholar] [CrossRef] [PubMed]
- Mortarini, R.; Gismondi, A.; Santoni, A.; Parmiani, G.; Anichini, A. Role of the A5β1 Integrin Receptor in the Proliferative Response of Quiescent Human Melanoma Cells to Fibronectin. Cancer Research 1992.
- Arias-Mejias, S.M.; Warda, K.Y.; Quattrocchi, E.; Alonso-Quinones, H.; Sominidi-Damodaran, S.; Meves, A. The Role of Integrins in Melanoma: A Review. Int J Dermatol 2020, 59, 525–534. [Google Scholar] [CrossRef]
- Cheresh, D.A. Structure, Function and Biological Properties of Integrin Alpha v Beta 3 on Human Melanoma Cells. Cancer Metastasis Rev 1991, 10, 3–10. [Google Scholar] [CrossRef]
- Lu, X.; Lu, D.; Scully, M.F.; Kakkar, V.V. Modulation of Integrin-Binding Selectivity by Mutation within the RGD-Loop of Snake Venom Proteins: A Novel Drug Development Approach. Curr Med Chem Cardiovasc Hematol Agents 2003, 1, 189–196. [Google Scholar] [CrossRef]
- Lu, X.; Lu, D.; Scully, M.F.; Kakkar, V.V. Integrins in Drug Targeting-RGD Templates in Toxins. Curr Pharm Des 2006, 12, 2749–2769. [Google Scholar] [CrossRef]
- Almeida, G. de O.; de Oliveira, I.S.; Arantes, E.C.; Sampaio, S.V. Snake Venom Disintegrins Update: Insights about New Findings. J Venom Anim Toxins Incl Trop Dis 29, e20230039. [CrossRef]
- Shih, C.-H.; Chiang, T.-B.; Wang, W.-J. Inhibition of Integrins Av/A5-Dependent Functions in Melanoma Cells by an ECD-Disintegrin Acurhagin-C. Matrix Biol 2013, 32, 152–159. [Google Scholar] [CrossRef]
- Saviola, A.J.; Burns, P.D.; Mukherjee, A.K.; Mackessy, S.P. The Disintegrin Tzabcanin Inhibits Adhesion and Migration in Melanoma and Lung Cancer Cells. Int J Biol Macromol 2016, 88, 457–464. [Google Scholar] [CrossRef]
- Trikha, M.; De Clerck, Y.A.; Markland, F.S. Contortrostatin, a Snake Venom Disintegrin, Inhibits Beta 1 Integrin-Mediated Human Metastatic Melanoma Cell Adhesion and Blocks Experimental Metastasis. Cancer Res 1994, 54, 4993–4998. [Google Scholar]
- Kang, I.C.; Kim, D.S.; Jang, Y.; Chung, K.H. Suppressive Mechanism of Salmosin, a Novel Disintegrin in B16 Melanoma Cell Metastasis. Biochem Biophys Res Commun 2000, 275, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, E.E.; Rodríguez-Acosta, A.; Palomar, R.; Lucena, S.E.; Bashir, S.; Soto, J.G.; Pérez, J.C. Colombistatin: A Disintegrin Isolated from the Venom of the South American Snake (Bothrops Colombiensis) That Effectively Inhibits Platelet Aggregation and SK-Mel-28 Cell Adhesion. Arch Toxicol 2009, 83, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Lucena, S.; Sanchez, E.E.; Perez, J.C. Anti-Metastatic Activity of the Recombinant Disintegrin, r-Mojastin 1, from the Mohave Rattlesnake. Toxicon 2011, 57, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Lucena, S.E.; Jia, Y.; Soto, J.G.; Parral, J.; Cantu, E.; Brannon, J.; Lardner, K.; Ramos, C.J.; Seoane, A.I.; Sánchez, E.E. Anti-Invasive and Anti-Adhesive Activities of a Recombinant Disintegrin, r-Viridistatin 2, Derived from the Prairie Rattlesnake (Crotalus Viridis Viridis). Toxicon 2012, 60, 31–39. [Google Scholar] [CrossRef]
- Carey, C.M.; Bueno, R.; Gutierrez, D.A.; Petro, C.; Lucena, S.E.; Sanchez, E.E.; Soto, J.G. Recombinant Rubistatin (r-Rub), an MVD Disintegrin, Inhibits Cell Migration and Proliferation, and Is a Strong Apoptotic Inducer of the Human Melanoma Cell Line SK-Mel-28. Toxicon 2012, 59, 241–248. [Google Scholar] [CrossRef]
- Arruda Macêdo, J.K.; Fox, J.W.; de Souza Castro, M. Disintegrins from Snake Venoms and Their Applications in Cancer Research and Therapy. Curr Protein Pept Sci 2015, 16, 532–548. [Google Scholar] [CrossRef]
- Mizejewski, G. Disintegrin-Like Peptides Derived from Naturally-Occurring Proteins: A Proposed Adjunct Treatment for Cancer Therapy: A Commentary. International Journal of Cancer Research and Molecular Mechanisms 2020, 6. [Google Scholar] [CrossRef]
- Pisano, M.; Paola, I. de; Nieddu, V.; Sassu, I.; Cossu, S.; Galleri, G.; Gatto, A.D.; Budroni, M.; Cossu, A.; Saviano, M.; et al. In vitro Activity of the Avβ3 Integrin Antagonist RGDechi-hCit on Malignant Melanoma Cells. Anticancer research 2013.
- Nasulewicz-Goldeman, A.; Uszczyńska, B.; Szczaurska-Nowak, K.; Wietrzyk, J. siRNA-Mediated Silencing of Integrin Β3 Expression Inhibits the Metastatic Potential of B16 Melanoma Cells. Oncol Rep 2012, 28, 1567–1573. [Google Scholar] [CrossRef]
- Tong, H.; Jiang, G.; Qi, D.; Bi, J.; Tian, D.; Guan, X.; Zheng, S.; Sun, X. Bupleurum Chinense Polysaccharide Inhibit Adhesion of Human Melanoma Cells via Blocking Β1 Integrin Function. Carbohydr Polym 2017, 156, 244–252. [Google Scholar] [CrossRef]
- Liu, Y.; Zou, X.; Sun, G.; Bao, Y. Codonopsis Lanceolata Polysaccharide CLPS Inhibits Melanoma Metastasis via Regulating Integrin Signaling. Int J Biol Macromol 2017, 103, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Kothari, M.; Wanjari, A.; Acharya, S.; Karwa, V.; Chavhan, R.; Kumar, S.; Kadu, A.; Patil, R. A Comprehensive Review of Monoclonal Antibodies in Modern Medicine: Tracing the Evolution of a Revolutionary Therapeutic Approach. Cureus 16, e61983. [CrossRef]
- Mitjans, F.; Sander, D.; Adán, J.; Sutter, A.; Martinez, J.M.; Jäggle, C.S.; Moyano, J.M.; Kreysch, H.G.; Piulats, J.; Goodman, S.L. An Anti-Alpha v-Integrin Antibody That Blocks Integrin Function Inhibits the Development of a Human Melanoma in Nude Mice. J Cell Sci 1995, 108 ( Pt 8), 2825–2838. [Google Scholar] [CrossRef]
- Montgomery, A.M.; Reisfeld, R.A.; Cheresh, D.A. Integrin Alpha v Beta 3 Rescues Melanoma Cells from Apoptosis in Three-Dimensional Dermal Collagen. Proc Natl Acad Sci U S A 1994, 91, 8856–8860. [Google Scholar] [CrossRef] [PubMed]
- Borst, A.J.; James, Z.M.; Zagotta, W.N.; Ginsberg, M.; Rey, F.A.; DiMaio, F.; Backovic, M.; Veesler, D. The Therapeutic Antibody LM609 Selectively Inhibits Ligand Binding to Human αVβ3 Integrin via Steric Hindrance. Structure 2017, 25, 1732–1739e5. [Google Scholar] [CrossRef] [PubMed]
- McNeel, D.G.; Eickhoff, J.; Lee, F.T.; King, D.M.; Alberti, D.; Thomas, J.P.; Friedl, A.; Kolesar, J.; Marnocha, R.; Volkman, J.; et al. Phase I Trial of a Monoclonal Antibody Specific for Avβ3 Integrin (MEDI-522) in Patients with Advanced Malignancies, Including an Assessment of Effect on Tumor Perfusion. Clin Cancer Res 2005, 11, 7851–7860. [Google Scholar] [CrossRef]
- Patel, S.R.; Jenkins R.N., J.; Papadopolous, N.; Burgess, M.A.; Plager, C.; Gutterman, J.; Benjamin, R.S. Pilot Study of Vitaxin—an Angiogenesis Inhibitor—in Patients with Advanced Leiomyosarcomas. Cancer 2001, 92, 1347–1348. [Google Scholar] [CrossRef]
- Gutheil, J.C.; Campbell, T.N.; Pierce, P.R.; Watkins, J.D.; Huse, W.D.; Bodkin, D.J.; Cheresh, D.A. Targeted Antiangiogenic Therapy for Cancer Using Vitaxin: A Humanized Monoclonal Antibody to the Integrin Alphavbeta3. Clin Cancer Res 2000, 6, 3056–3061. [Google Scholar]
- Posey, J.A.; Khazaeli, M.B.; DelGrosso, A.; Saleh, M.N.; Lin, C.Y.; Huse, W.; LoBuglio, A.F. A Pilot Trial of Vitaxin, a Humanized Anti-Vitronectin Receptor (Anti Alpha v Beta 3) Antibody in Patients with Metastatic Cancer. Cancer Biother Radiopharm 2001, 16, 125–132. [Google Scholar] [CrossRef]
- Moschos, S.J.; Sander, C.A.; Wang, W.; Reppert, S.L.; Drogowski, L.M.; Jukic, D.M.; Rao, U.N.M.; Athanassiou, C.; Buzoianu, M.; Mandic, M.; et al. Pharmacodynamic (Phase 0) Study Using Etaracizumab in Advanced Melanoma. J Immunother 2010, 33, 316–325. [Google Scholar] [CrossRef]
- Delbaldo, C.; Raymond, E.; Vera, K.; Hammershaimb, L.; Kaucic, K.; Lozahic, S.; Marty, M.; Faivre, S. Phase I and Pharmacokinetic Study of Etaracizumab (AbegrinTM), a Humanized Monoclonal Antibody against Avβ3 Integrin Receptor, in Patients with Advanced Solid Tumors. Invest New Drugs 2008, 26, 35–43. [Google Scholar] [CrossRef]
- Hersey, P.; Sosman, J.; O’Day, S.; Richards, J.; Bedikian, A.; Gonzalez, R.; Sharfman, W.; Weber, R.; Logan, T.; Buzoianu, M.; et al. A Randomized Phase 2 Study of Etaracizumab, a Monoclonal Antibody against Integrin Alpha(v)Beta(3), + or - Dacarbazine in Patients with Stage IV Metastatic Melanoma. Cancer 2010, 116, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Sainio, A.; Järveläinen, H. Extracellular Matrix-Cell Interactions: Focus on Therapeutic Applications. Cell Signal 2020, 66, 109487. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, S.; Chen, S.; Ma, F. The Prognostic Value and Immunological Role of CD44 in Pan-Cancer Study. Sci Rep 2023, 13, 7011. [Google Scholar] [CrossRef]
- Wagner, D.L.; Klotzsch, E. Barring the Gates to the Battleground: DDR1 Promotes Immune Exclusion in Solid Tumors. Sig Transduct Target Ther 2022, 7, 1–2. [Google Scholar] [CrossRef]
- Harris, E.N.; Baker, E. Role of the Hyaluronan Receptor, Stabilin-2/HARE, in Health and Disease. International Journal of Molecular Sciences 2020, 21, 3504. [Google Scholar] [CrossRef]
- Karinen, S.; Juurikka, K.; Hujanen, R.; Wahbi, W.; Hadler-Olsen, E.; Svineng, G.; Eklund, K.K.; Salo, T.; Åström, P.; Salem, A. Tumour Cells Express Functional Lymphatic Endothelium-Specific Hyaluronan Receptor in vitro and in vivo: Lymphatic Mimicry Promotes Oral Oncogenesis? Oncogenesis 2021, 10, 1–11. [Google Scholar] [CrossRef]
- Hinneh, J.A.; Gillis, J.L.; Moore, N.L.; Butler, L.M.; Centenera, M.M. The Role of RHAMM in Cancer: Exposing Novel Therapeutic Vulnerabilities. Front Oncol 2022, 12, 982231. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A Guide to the Composition and Functions of the Extracellular Matrix. The FEBS Journal 2021, 288, 6850–6912. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front Cell Dev Biol 2017, 5, 18. [Google Scholar] [CrossRef]
- Goodison, S.; Urquidi, V.; Tarin, D. CD44 Cell Adhesion Molecules. Mol Pathol 1999, 52, 189–196. [Google Scholar] [CrossRef]
- Naor, D.; Nedvetzki, S.; Golan, I.; Melnik, L.; Faitelson, Y. CD44 in Cancer. Crit Rev Clin Lab Sci 2002, 39, 527–579. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Zhou, M.; Li, X.; Hu, M.; Li, C.; Li, M.; Sheng, F.; Li, Z.; Wu, G.; Luo, M.; et al. Synergistic Active Targeting of Dually Integrin Avβ3/CD44-Targeted Nanoparticles to B16F10 Tumors Located at Different Sites of Mouse Bodies. J Control Release 2016, 235, 1–13. [Google Scholar] [CrossRef]
- Coradini, D.; Zorzet, S.; Rossin, R.; Scarlata, I.; Pellizzaro, C.; Turrin, C.; Bello, M.; Cantoni, S.; Speranza, A.; Sava, G.; et al. Inhibition of Hepatocellular Carcinomas in vitro and Hepatic Metastases in vivo in Mice by the Histone Deacetylase Inhibitor HA-But. Clin Cancer Res 2004, 10, 4822–4830. [Google Scholar] [CrossRef]
- Yoon, H.Y.; Kim, H.R.; Saravanakumar, G.; Heo, R.; Chae, S.Y.; Um, W.; Kim, K.; Kwon, I.C.; Lee, J.Y.; Lee, D.S.; et al. Bioreducible Hyaluronic Acid Conjugates as siRNA Carrier for Tumor Targeting. J Control Release 2013, 172, 653–661. [Google Scholar] [CrossRef]
- Peer, D.; Margalit, R. Tumor-Targeted Hyaluronan Nanoliposomes Increase the Antitumor Activity of Liposomal Doxorubicin in Syngeneic and Human Xenograft Mouse Tumor Models. Neoplasia 2004, 6, 343–353. [Google Scholar] [CrossRef]
- Peer, D.; Margalit, R. Loading Mitomycin C inside Long Circulating Hyaluronan Targeted Nano-Liposomes Increases Its Antitumor Activity in Three Mice Tumor Models. Int J Cancer 2004, 108, 780–789. [Google Scholar] [CrossRef]
- Guo, Y.; Ma, J.; Wang, J.; Che, X.; Narula, J.; Bigby, M.; Wu, M.; Sy, M.S. Inhibition of Human Melanoma Growth and Metastasis in vivo by Anti-CD44 Monoclonal Antibody. Cancer Res 1994, 54, 1561–1565. [Google Scholar]
- Menke-van der Houven van Oordt, C.W.; Gomez-Roca, C.; van Herpen, C.; Coveler, A.L.; Mahalingam, D.; Verheul, H.M.W.; van der Graaf, W.T.A.; Christen, R.; Rüttinger, D.; Weigand, S.; et al. First-in-Human Phase I Clinical Trial of RG7356, an Anti-CD44 Humanized Antibody, in Patients with Advanced, CD44-Expressing Solid Tumors. Oncotarget 2016, 7, 80046–80058. [Google Scholar] [CrossRef]
- Fänder, J.; Kielstein, H.; Büttner, M.; Koelblinger, P.; Dummer, R.; Bauer, M.; Handke, D.; Wickenhauser, C.; Seliger, B.; Jasinski-Bergner, S. Characterizing CD44 Regulatory microRNAs as Putative Therapeutic Agents in Human Melanoma. Oncotarget 2019, 10, 6509–6525. [Google Scholar] [CrossRef]
- Ahrens, T.; Sleeman, J.P.; Schempp, C.M.; Howells, N.; Hofmann, M.; Ponta, H.; Herrlich, P.; Simon, J.C. Soluble CD44 Inhibits Melanoma Tumor Growth by Blocking Cell Surface CD44 Binding to Hyaluronic Acid. Oncogene 2001, 20, 3399–3408. [Google Scholar] [CrossRef]
- Rezler, E.M.; Khan, D.R.; Lauer-Fields, J.; Cudic, M.; Baronas-Lowell, D.; Fields, G.B. Targeted Drug Delivery Utilizing Protein-like Molecular Architecture. J Am Chem Soc 2007, 129, 4961–4972. [Google Scholar] [CrossRef] [PubMed]
- Leitinger, B. Discoidin Domain Receptor Functions in Physiological and Pathological Conditions. Int Rev Cell Mol Biol 2014, 310, 39–87. [Google Scholar] [CrossRef]
- Cario, M. DDR1 and DDR2 in Skin. Cell Adhesion & Migration 2018, 12, 386–393. [Google Scholar] [CrossRef]
- Reger de Moura, C.; Battistella, M.; Sohail, A.; Caudron, A.; Feugeas, J.P.; Podgorniak, M.-P.; Pages, C.; Mazouz Dorval, S.; Marco, O.; Menashi, S.; et al. Discoidin Domain Receptors: A Promising Target in Melanoma. Pigment Cell Melanoma Res 2019, 32, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Poudel, B.; Lee, Y.-M.; Kim, D.-K. DDR2 Inhibition Reduces Migration and Invasion of Murine Metastatic Melanoma Cells by Suppressing MMP2/9 Expression through ERK/NF-κB Pathway. Acta Biochim Biophys Sin (Shanghai) 2015, 47, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Badiola, I.; Villacé, P.; Basaldua, I.; Olaso, E. Downregulation of Discoidin Domain Receptor 2 in A375 Human Melanoma Cells Reduces Its Experimental Liver Metastasis Ability. Oncol Rep 2011, 26, 971–978. [Google Scholar] [CrossRef]
- Berestjuk, I.; Lecacheur, M.; Carminati, A.; Diazzi, S.; Rovera, C.; Prod’homme, V.; Ohanna, M.; Popovic, A.; Mallavialle, A.; Larbret, F.; et al. Targeting Discoidin Domain Receptors DDR1 and DDR2 Overcomes Matrix-mediated Tumor Cell Adaptation and Tolerance to BRAF-targeted Therapy in Melanoma. EMBO Mol Med 2022, 14, e11814. [Google Scholar] [CrossRef]
Figure 1.
Various approaches to inhibit functional components of the ECM. Abbreviations: MMPs – matrix metalloproteinases, MT1-MMP – membrane type 1-matrix metalloproteinase, MMP-1, -2, -9, -13 – metalloproteinase-1, -2, -9, -13, shRNA – short hairpin RNA, MMPIs – matrix metalloproteinase inhibitors, TIMPs – tissue inhibitors of metalloproteinases, AV – adenovirus, siRNA – small interfering RNA, miRNA – microRNA, mABs – monoclonal antibodies, HA – hyaluronic acid, CAF – cancer-associated fibroblasts, TAM – tumor-associated macrophages, ECM – extracellular matrix. Created in BioRender. Mayasin, Y. (2024) BioRender.com/h80w259.
Figure 1.
Various approaches to inhibit functional components of the ECM. Abbreviations: MMPs – matrix metalloproteinases, MT1-MMP – membrane type 1-matrix metalloproteinase, MMP-1, -2, -9, -13 – metalloproteinase-1, -2, -9, -13, shRNA – short hairpin RNA, MMPIs – matrix metalloproteinase inhibitors, TIMPs – tissue inhibitors of metalloproteinases, AV – adenovirus, siRNA – small interfering RNA, miRNA – microRNA, mABs – monoclonal antibodies, HA – hyaluronic acid, CAF – cancer-associated fibroblasts, TAM – tumor-associated macrophages, ECM – extracellular matrix. Created in BioRender. Mayasin, Y. (2024) BioRender.com/h80w259.
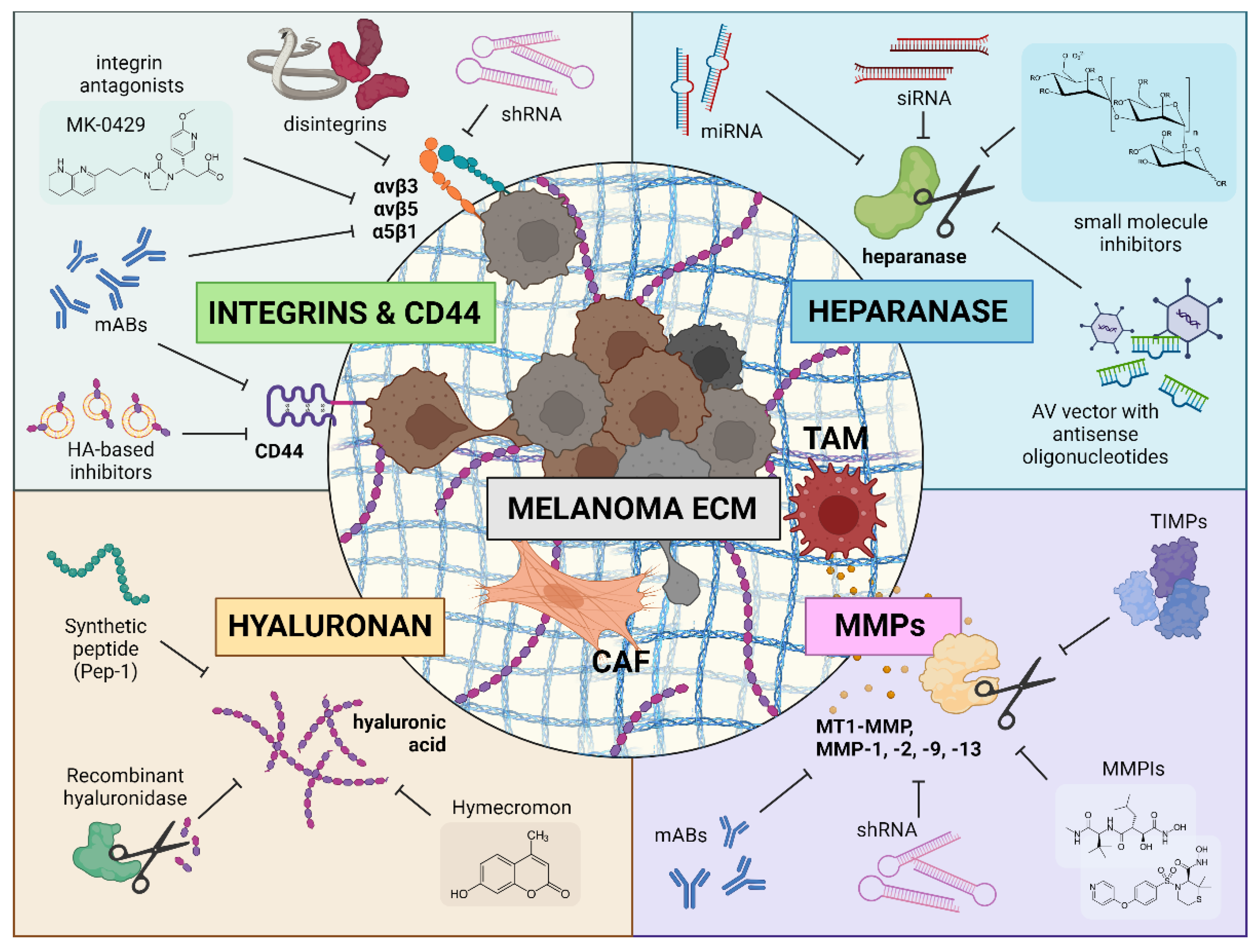

Table 1.
Preclinical researches of ECM-targeted melanoma therapy.
| Target | Type of drug | Additional Terms | Research object | Results | Reference |
|---|---|---|---|---|---|
| Heparanase | Suramin (polysulfonated naphthylurea) | _ | allograft B16-F10 in mice in vivo | Strong inhibitory effect on heparanase activity in melanoma cells; demonstration of reduced invasiveness in reconstructed basal membranes | [51] |
| 1,3-bis-[4-(1H-benzoimidazol-2-yl)-phenyl]-urea | _ | B16-BL6 cell line in vitro; syngeneic B16 in C57 mice in vivo | Inhibitory effect observed on the proliferative activity of melanoma cells in vitro (less than 50%); reduction in metastatic potential of these cells in mouse models (about 50% reduction) | [52] | |
| Chemically modified heparins | _ | syngeneic B16-BL6 in C57BL/6 mice in vivo | Significant reductions in the numbers of experimental melanoma lung metastases occurred | [53] | |
| Modified species of heparin and size-homogeneous oligosaccharides derived from depolymerized heparins | _ | syngeneic B16-BL6 in C57BL/6 mice in vivo | Effective inhibition of heparanase-mediated degradation of heparan sulfate in the ECM and reduction of lung colonization by melanoma cells | [54] | |
| Adenoviral vector carrying a cDNA with an antisense sequence of the heparanase gene HSPE-1 | _ | B16-B15b and 70W in nude mouse models in vivo | Significant reduction of HPSE-1 content in melanoma cells after adenoviral vector infection; significant decrease in melanoma invasiveness | [71] | |
| Artificial microRNA (miRNA) | _ | A375 cell line in vitro | Effective inhibition of HPSE protein expression and mRNA synthesis; reduction of invasive properties of melanoma cells in vitro and in vivo | [72] | |
| Plasmid vector carrying a small interfering RNA (siRNA) construct | _ | syngeneic B16-BL6 in C57BL/6 mice in vivo | Less vascularization of tumors and formation of fewer metastases; longer lifespan of mice injected with modified cells compared to mice injected with control cells without the genetic constructs | [73] | |
| MMPs | Prinomastat (AG3340) | Carboplatin; Taxol | syngeneic B16-F10 in C57BL/6 mice in vivo | Reduction of tumor growth, angiogenesis, and proliferation with increased necrosis and apoptosis; enhanced efficacy of carboplatin and taxol; decreased metastasis in melanoma; improved therapeutic index over cytotoxic drugs | [91] |
| MMI270 (CGS27023A) | _ | syngeneic B16-F10 and B16-BL6 in BDF1 mice in vivo | Significant reduction of the metastatic colonies in the lungs; no effect on colony size | [95] | |
| _ | syngeneic B16-BL6 in mice in vivo | Reduction of vessels leading to the primary tumor | [96] | ||
| Rebimastat (BMS-275291) | _ | syngeneic B16-BL6 in C57BL/6 mice in vivo | Dose-dependent inhibition of tumor metastasis to the lungs; dose-dependent anti-angiogenic effect | [97] | |
| JaZ-30 (C(2)-monosubstituted aziridine - aryl-1,2,3-triazole conjugate) | _ | B16 4A5 cell line in vitro | Reduction of VEGF secretion and ERK1/2 phosphorylation; inhibition of invasion through Matrigel and angiogenesis reduction in HUVEC cells; moderate decreasion of cell viability | [98] | |
| Small-molecule MMP2/MMP9 inhibitor SB-3CT | ICB (anti-PD-1/anti-CTLA-4) | A375 and SK-Mel-28 cell lines in vitro; syngeneic B16-F10 in C57/BL6 mice in vivo | Significant reduction in mRNA and protein levels of PD-L1 in melanoma cell lines; suppression of lung metastases when combined with ICB therapy | [99] | |
| ND-322 | _ | xenograft WM266-4 in mice in vivo | Effective inhibition of MT1-MMP and MMP2 activity resulting in reduction of melanoma cells growth, migration and invasion in vitro | [102] | |
| CPCPA (cyclopentylcarbamoylphosphonic acid) | _ | syngeneic B16-F10 in C57BL mice in vivo | Effective inhibition of tumor cell invasion through Matrigel without affecting cell proliferation; reduction of metastasis, inhibition of MMP expression and angiogenesis in mice | [103] | |
| Anti-MT1-MMP antibody DX-2400 | _ | allograft B16-F10 in mice in vivo | Blockade of proMMP-2 activation, reduction of MMP-9 expression, reduction of endothelial cell invasion, inhibition of tumor progression, reduction of metastasis rate and angiogenesis | [105] | |
| Peptide vaccines based on synthetic immunogenic oligopeptides with MMP sequences | Human MMP-2 and MMP-9 | syngeneic B16-F0 in C57BL/6 mice in vivo | Up to 88% reduction in tumor volume with human MMP-2 oligopeptide; 80% reduction with one of the human MMP-9 oligopeptides; no pronounced side effects | [106] | |
| MMP-9 of mice and rats | syngeneic B16-F0 in C57BL/6J mice in vivo | Reduction in tumor size (55 to 77% depending on the oligopeptide); no differences in clinical serum analyses, haematological parameters and histopathology of major organs compared to controls | [107] | ||
| MMP-1 inhibitory shRNA | _ | VMM12 cell line in vitro; xenograft VMM12 in immunodeficient nu/nu mice in vivo | Suppression of MMP-1 expression in vitro; reduction of metastatic activity in the lungs; reduction of collagenase activity and mediated suppression of invasion and angiogenesis | [108] | |
| Recombinant human TIMP | _ | B16-F10 cell line with amniotic membrane in vitro; syngeneic B16-F10 in C57BL/6 mice in vivo | Inhibition of metastasis; no effect on tumor growth | [112] | |
| Recombinant TIMP-1 conjugated to glycosylphosphatidylinositol | Sub-lethal hyperthermic treatment | 624.38-MEL, 93.04A12MEL, SK-MEL23, WM115, WM266-4 cell lines in vitro | Inhibition of proMMP-2 and proMMP-9 release from melanoma cells; significant increase in sensitivity to FAS-induced apoptosis | [113] | |
| Recombinant human TIMP-2 (r-hTIMP-2) | _ | syngeneic B16-BL6 in C57BL/6 mice in vivo | Inhibition of metastatic foci formation and limited inhibitory effect on tumor cell growth under in vitro and in vivo | [114] | |
| Recombinant human TIMP-2 fused to human serum albumin | Fluorouracil | syngeneic B16-BL6 in C57BL/6 mice in vivo | Inhibition of tumor growth | [115] | |
| Plasmid vector encoding TIMP-1 cDNA | Intraperitoneal injection of IL-2 | syngeneic B16-F10 in C57BL/6 mice in vivo | Significant reduction of lung metastasis; Further reduction of pulmonary metastases and increased survival were achieved by IL-2 administration combined with TIMP-1 treatment | [117] | |
| cDNA encoding human TIMP-2 | _ | xenograft M24 net in immunodeficient mice in vivo | Suppression of melanoma cell growth due to TIMP-2-mediated occlusion of interstitial collagen; no effect on metastatic activity | [118] | |
| Recombinant adenoviruses encoding TIMP-3 | _ | SK-Mel-5 and A2058 cell lines in Matrigel in vitro | Inhibition of invasion through the basal membrane; reduction of cell attachment to collagen types I and IV and fibronectin; induction of apoptosis | [119] | |
| Recombinant adenovirus encoding TIMP-3 | _ | xenograft A2058 in SCID/SCID mice in vivo | Inhibition of gelatinase activity and xenograft growth; Induction of apoptosis | [120] | |
| Integrins | 4-Methylumbelliferone (4-MU) | _ | syngeneic B16-F10 in C57BL/6 mice in vivo | Enhancement of melanoma cell adhesion and motility due to the presence of HA; inhibition of HA formation on the cell surface by 4-MU; decrease in the number of metastatic nodules by 32% in liver tissue | [133] |
| _ | B16-F10 cell line in vitro | Promotion of melanoma cell adhesion and locomotion by HA; dose-dependent reduction of cell adhesion (up to 49%) and locomotion (up to 37%) by 4-MU | [134] | ||
| _ | C8161 and MV3 cell lines in vitro | Reduction of hyaluronan levels in the matrix by 4-MU; inhibition of both growth and invasion in collagen lattices of melanoma cells; reversible growth suppression without induction of apoptosis | [135] | ||
| Synthetic peptide Pep-1 | _ | B16-F10 cell line in vitro; allograft B16-F10 in mice in vivo | Blocking of CD44-mediated adhesion to HA by Pep-1; no reduction in melanoma cell proliferation in vitro or growth in vivo; significant reduction in lung metastasis incidence and increased survival observed following a single intravenous injection of Pep-1 | [136] | |
| Hyaluronidase | Vinblastin | xenograft SK-Mel-2, -3, -5, -24 in nu/nu mice in vivo | Pronounced antitumor effect of combination therapy; ineffectiveness of individual drugs; prevention of inflammatory reactions with prior hyaluronidase; disappearance of tumor cells after 18 weeks, with no lymph node metastases | [137] | |
| Delphinidin | _ | syngeneic B16-F10 in C57BL/6 mice in vivo | Inhibition of cell proliferation, migration, and invasion; reduction of melanoma cell growth by 50% and over 90%; decrease in migration by approximately 45%; reduction of metastasis to sentinel lymph nodes from 80% in control mice to 25% | [138] | |
| Integrin inhibitor MK-0429 | Cyclophosphamide | allograft B16-F10 in B6D2F1 mice in vivo | Reduction of metastatic tumor colonies by 64%, decrease in tumor area by 60%, inhibition of tumor progression, and a 40% reduction in lung tumor burden | [145] | |
| Acurhagin-C | Methotrexate | B16-F10 and SK-Mel-1 cell lines in vitro | Reducing cell adhesion and transendothelial migration; induction of apoptosis via caspase-8 and -9 activation; enhancement of methotrexate's anti-proliferative effects in melanoma cells, sparing human epidermal melanocytes | [153] | |
| Tzabcanin | _ | A375 cell line in vitro | Reduction of melanoma cell adhesion to vitronectin with an IC50 of 747 nM; inhibition of melanoma cell migration by approximately 45% | [154] | |
| Contortrostatin | _ | M24 met cell line in vitro; xenograft M24 met in SCID mice in vivo | Reduction of adhesion to type I collagen (IC50 = 20 nM), vitronectin (IC50 = 75 nM), and fibronectin (IC50 = 220 nM); reduction of lung tumor foci by 51% at 20 µg and by 73% at 100 µg in vivo. | [155] | |
| Recombinant Salmosin | _ | B16-F10 cell line in vitro; syngeneic B16-F10 in C57BL/6 mice in vivo | Reduction of adhesion and invasion in vitro by blocking αvβ3 integrin; inhibition of cell proliferation on collagen I-coated plates; inhibition of lung colonization by melanoma cells in vivo | [156] | |
| Recombinant Colombistatin | _ | SK-Mel-28 cell line in vitro | Inhibition of adhesion of melanoma cells to fibronectin; reduction of migration activity | [157] | |
| Recombinant Mujastin 1 | _ | SK-Mel-28 and B16-F10 cell lines in vitro | Inhibition of SK-Mel-28 cell adhesion to fibronectin; reduction of lung tumor colonization in mouse models | [158] | |
| Recombinant Viridistatin 2 | _ | xenograft SK-Mel-28 and syngeneic B16-F10 in C57BL/6 and BALB/c mice in vivo | Inhibition of SK-Mel-28 cell adhesion, migration, and invasion; reduction of SK-Mel-28 migration by 96% and invasion of various cell lines by up to 85%; significant reduction of lung colonization of murine melanoma cells by 71% in vivo | [159] | |
| Recombinant Rubistatin | _ | SK-Mel-28 cell line in vitro | Inhibition of cell migration, proliferation, and adhesion to fibronectin | [160] | |
| Selective antagonist of αvβ3 RGDechi-hCit | Cisplatinum; Etoposide | A375, WM266-4, SK-Mel-28, Sbcl2, LB24Dagi, PR-Mel и PNP-Mel cell lines in vitro | Partial inhibition of adhesion and migration was observed, particularly in WM266 cells with the highest αvβ3 levels; no direct correlation between inhibition and αvβ3 expression | [163] | |
| siRNA against β3 integrin | _ | B16 cell line in matrigel in vitro; syngeneic B16 in C57BL/6/IiW mice in vivo | Over 90% reduction in β3 expression; significant impairment of fibronectin binding and migration through Matrigel; lung metastases decreation | [164] | |
| Bupleurum chinense Polysaccharides | _ | A375 cell line in vitro | Reduction of F-actin stress fibers by 54% to 28% compared to control; reduction of melanoma adhesion to fibronectin by 35% to 64%; reduction of phosphorylation of FAK by 50% to 65% and paxillin by 55% to 70% at various concentrations; reduction of focal adhesions per cell by 36% | [165] | |
| Codonopsis lanceolata Polysaccharides | _ | B16-F10 cell line in vitro; syngeneic B16-F10 in C57BL/6 mice in vivo | Inhibition of cell proliferation and pulmonary metastasis; disruption of integrin β1-mediated cell migration under in vitro conditions | [166] | |
| Anti-αv-integrin 17E6 antibody | _ | xenograft M21 cell line in Balb/c nu/nu mice in vivo | Inhibition of tumor growth and metastasis mediated by integrins; lack of inhibitory effects on melanoma cells themselves or antibody-mediated cellular cytotoxicity | [168] | |
| Anti-αvβ3 integrin monoclonal antibody LM609 | _ | xenograft M21 in Balb/c nu/nu mice in vivo | Elimination of the survival advantage from αvβ3 ligation in melanoma cells; significant reduction of melanoma cell viability in collagen matrices; no significant impact on cell adhesion or migration in cells with low αvβ3 expression | [169] | |
| CD44 | Hyaluronan (HA) + tetraiodothyroacetic acid (tetrac) conjugate (TeHA-SLN) | Docetaxel (DTX) | syngeneic B16-F10 in mice in vivo; melanoma metastasis in situ | Tumor growth inhibition was significant due to the action of TeHA-SLN/DTX; efficacy of TeHA-SLN as a bidirectional drug delivery system was demonstrated | [188] |
| Hyaluronan esterified with butyric acid residues | _ | syngeneic B16-F10 in C57BL/6 mice in vivo | Complete suppression of metastases in animals and significantly prolonged life expectancy compared to control groups | [189] | |
| HPD-siRNA complexes | _ | syngeneic B16-F10 in Balb/c nude mice in vivo | Selective accumulation of siRNA-HPD complexes at the tumor site after systemic administration to mice resulted in effective suppression of target gene expression; significant impact on tumor growth and progression was observed | [190] | |
| Nano-sized hyaluronan-liposomes (tHA-LIP) | Doxorubicin (DXR); Doxil | syngeneic B16-F10.9 in C57BL/6 mice in vivo | Selective accumulation of DXR in tumors; enhanced therapeutic effects observed; reduced tumor progression and metastatic burden; improved survival rates in syngeneic models compared to control | [191] | |
| Mitomycin C (MMC) | Increased potency of MMC-loaded tHA-LIP in receptor-overexpressing cells; prolonged circulation and enhanced accumulation in tumor-bearing lungs; improved delivery of MMC; significant improvements in tumor progression, metastasis, and survival outcomes | [192] | |||
| Anti-CD44 monoclonal antibody GKW.A2 | _ | xenograft SMMU-1 and SMMU-2 in mice in vivo | Local tumor development was not suppressed one week after subcutaneous injection in mice; however, the formation of metastatic tumors was inhibited, and animal survival was prolonged | [193] | |
| miR-143-3p | _ | BLM cell line in vitro | Decrease in melanoma cell proliferation; reduction in cell migration; increase in apoptosis of melanoma cells | [195] | |
| cDNA encoding the soluble form of CD44 | _ | 1F6 cell lines in vitro; xenograft 1F6 in MF1 nu/nu mice in vivo | Ihibition of cell growth by competitively blocking cell surface binding of CD44 to hyaluronic acid | [196] | |
| A peptide mimetic of collagen triple helix peptide (α1(IV)1263-1277 PA) | Liposomes loaded with rhodamine | М14#5 cell line in vitro | PA-associated improvement in targeting specificity; promotion of greater accumulation of therapeutic agents in tumor cells within melanoma models compared to non-targeting liposomes | [197] | |
| DDR | siRNA against DDR2 | _ | B16-BL6 cell line in vitro | Suppression of migration, invasion, and survival in human melanoma cell lines | [201] |
| DDR tyrosine kinase inhibitor (DDR1-IN-1) | siRNA against DDR1 | M10 cell line in vitro; xenograft C8161 and SK-Mel-5 in nude/c mice in vivo | Significant inhibition of melanoma cell proliferation in vitro and in vivo | [200] | |
| Imatinib | BRAF inhibitors | SK-Mel-5 and MM099 cell lines in vitro; xenograft 1205Lu in nude mice in vivo | Increase in the efficacy of BRAF inhibitors; counteraction of collagen remodeling; delay in melanoma recurrence | [203] | |
| siRNA against DDR2 | _ | A375 cell line in vitro | Reduction in gelatinase activity and JNK phosphorylation in melanoma cells; decrease in proliferation and migration rates compared to mock-transfected cells | [202] |
miRNA – micro RNA, HPSE – heparanase, mRNA – messenger RNA, VEGF – vascular endothelial growth factor, ERK1/2 - extracellular-signal regulated kinases 1/2, CPCPA - cyclopentylcarbamoylphosphonic acid, TIMPs – tissue inhibitors of metalloproteinases, IL-2 – interleukin-2, 4-MU – methylumbelliferone, FAK – focal adhesion kinase, SCID – severe combined immunodeficiency, HA – hyaluronic acid, TeHA-SLN – Hyaluronan and tetraiodothyroacetic acid conjugate, DTX – Docetaxel, HPD – hyaluronic acid-graft-poly(dimethylaminoethyl methacrylate), MMC – Mitomycin C, DXR – Doxorubicin, tHA-LIP – nano-sized hyaluronan-liposomes, siRNA – small interfering RNA, cDNA – complementary DNA, PA – peptide mimetic of collagen triple helix peptide, DDR – discoidin domain receptors, JNK – c-Jun N-terminal kinases.
Table 2.
Clinical trials of ECM-targeted melanoma therapy.
| Target | Type of drug | Additional Terms | Clinical Trials ID | Phase | Disease | Status/Results | Reference |
|---|---|---|---|---|---|---|---|
| Heparanase | PI-88 (metformin) | Docetaxel | _ | I | Advanced Malignancies (including melanoma) | Completed. No PR or CR was observed during the study period. However, at least 2 of 5 melanoma patients (40%) evaluable for response had SD at the end of ≥2 cycles of therapy | [59] |
| Dexamethasone | _ | I | Advanced Solid Malignancies (including melanoma) | Completed. Despite no PR or CR, 3/15 (20%) evaluable patients showed SD at 2, 4, and 10 years. One patient with melanoma (6.7%) refractory to biochemotherapy showed PR accompanied by a reduction in the size and number of pulmonary metastase | [60] | ||
| _ | _ | I | Advanced Malignancies (including melanoma) | Completed. 14 patients with advanced malignancies, including melanoma, were included in the study, where only one patient (7.1%) with metastatic melanoma achieved SD, but after four cycles of therapy (12 weeks), he was diagnosed with PD, as were the other melanoma patients in the study | [61] | ||
| Dacarbazin | _ | I | Unresectable Metastatic Melanoma | Completed. No CR or PR were observed with PI-88 monotherapy, but one patient showed radiologic SD at 4 months. However, PR was observed in 2/5 patients (40%) initially receiving monotherapy but who later had dacarbazine added to PI-88. 3/9 patients (33%) initially receiving combination therapy had radiologic PR | [62] | ||
| NCT00130442 | II | Metastatic Melanoma | Completed. Twenty-four of 65 patients (36.9%) showed SD with a median duration of 117 days. However, in the combination therapy option, more subjects (30.77% vs. 19.70%) experienced serious adverse effects including neutropenia (30.77%) and thrombocytopenia (27.27%) | _ | |||
| _ | NCT00073892 | II | Progressive Melanoma | Completed. One (2.4%) patient achieved PR, six (14.6%) patients showed SD as the best response, and the remaining 30 participants (73.2%) showed PD. At the end of six cycles of treatment, 3 of the 41 patients studied had no disease progression | [64] | ||
| PG545 (pixatimod) | _ | NCT01252095 | I | Melanoma | Terminated (Unexpected injection site reactions). The results are unpublished, but there is additional clinical data (summarised below). As a result, no RECIST responses were recorded and all patients had PD. Plasma levels of VEGF and FGF-2 increased 3.5-fold and 1.5-fold, respectively, after 22 days of treatment with PG545 | [69] | |
| Nivolumab | NCT05061017 | Ib | Solid Tumors (not including melanoma) | Completed. Of the 58 participants, three people (5.2%) with metastatic colorectal cancer had confirmed PR and eight (13.8%) had SD for at least 9 weeks | [70] | ||
| Nivolumab; Cyclophosphamide | IIA | Refractory Metastatic Melanoma | Completed. Results not published | _ | |||
| MMPs | BB-94 (Batimastat) | _ | _ | I | Malignant Pleural Effusion (including melanoma) | Completed. The melanoma patient treated with an intrapleural dose of 60 mg/m² showed a PR with a reduced need for pleural aspirations and some improvement in dyspnoea scores one month after treatment. Although BB-94 did not induce systemic tumor regression, the patient experienced symptomatic relief | [84] |
| BB2516 (Marimastat) | Paclitaxel | _ | I | Advanced Malignancies (including melanoma) | Completed. Two melanoma patients were included in the study. While no CR or PR was observed, seven patients achieved SD. One melanoma patient showed symptomatic relief, but the disease progressed to PD. The combination was well tolerated with no effect on the pharmacokinetics of paclitaxel, suggesting safe co-administration at single agent doses | [87] | |
| _ | _ | II | Malignant Melanoma | Completed. No CR were observed among the 28 eligible patients. Two patients (7.1%) achieved confirmed PR, lasting approximately 3 months. Five patients (17.9%) experienced SD for a median duration of 1.8 months, while 16 patients showed PD | [88] | ||
| AG3340 (Prinomastat) | _ | _ | I | Advanced Cancer (including melanoma) | Completed. No confirmed tumor responses to therapy. The primary toxicities identified were joint and muscle pain, generally reversible with rest and/or dose reduction | [92] | |
| COL-3 (Incyclinide) | _ | NCT00001683 | I | Refractory Metastatic Cancer (including melanoma) | Completed. Demonstrated limited efficacy in the form of SD in eight patients (22.9%) with tumors of non-epithelial origin over two months | [94] | |
| Hyaluronic acid | Recombinant human hyaluronidase PH20 (rHuPH20) | Nivolumab | NCT03656718 | I/II | Unresectable Melanoma; Metastatic Melanoma | Completed. The SC Nivolumab + rHuPH20 dose-related exposures were well tolerated | [142] |
| NCT05297565 | III | Stage III A/B/C/D or Stage IV Melanoma | Completed. Results not published | [143] | |||
| Rituximab | NCT03719131 | II | Stage III A/B/C/D or Stage IV Cutaneous melanoma; Unresectable Melanoma | Active, not recruiting. Results not published | _ | ||
| Relatlimab; Nivolumab | NCT05625399 | III | Stage III or Stage IV Melanoma | Recruiting. Results not published | _ | ||
| Relatlimumab; Nivolumab | NCT06101134 | II | Melanoma | Recruiting. Results not published | _ | ||
| Nivolumab | NCT05496192 | II | Stage III A/B/C/D or Stage IV Melanoma | Withdrawn (Replaced it with another clinical trial) | _ | ||
| Hyaluronidase | Pembrolizumab | NCT06099782 | II | Stage II B/C or Stage III Melanoma | Recruiting. Results not published | _ | |
| Integrins | MEDI-523 (Vitaxin) | _ | _ | Metastatic Cancer (including melanoma) | Completed. One melanoma patient received two maximum doses of the drug, but continued to have PD, leading to withdrawal from the study. Notably, in this patient, the labelled Vitaxin successfully visualised and localised the tumor, likely due to the high expression of the αvβ3 integrins | [174] | |
| MEDI-522 (Etaracizumab) | _ | _ | I | Advanced Malignancies (including melanoma) | Completed. No CR or PR was observed in patients with advanced malignancies; however, long-term SD (34 weeks, >1 year, >2 years) was reported in patients with renal cell cancer. Two patients with melanoma and one patient with ocular melanoma showed PD after 6-8 weeks of therapy | [171] | |
| _ | _ | 0 | Advanced Melanoma | Completed. Pharmacodynamics in patients with advanced melanoma showed that the drug effectively saturates tumor cells at a dose of 8 mg/kg. Demonstrated an acceptable safety profile with no serious toxic effects and although no clear antitumor effects were observed, some patients may still benefit from inhibition of αvβ3 integrin-related signalling pathways | [175] | ||
| _ | _ | I | Advanced Solid Tumors (including melanoma) | Completed. All patients showed absence of PR and CR, but the melanoma patient showed SD for more than 4 months | [176] | ||
| Dacarbazin | NCT00066196 | II | Stage IV Melanoma | Completed. Responses were seen in the etoracizumab plus dacarbazine group, with 7 of 55 patients (12.7%) achieving a PR. There were no responses in the monotherapy group. SD was observed in 26 of 57 (45.6%) patients receiving etoracizumab alone and 22 of 55 (40%) in the combination group. PD was observed in 47.4% and 40%, respectively | [177] | ||
| _ | NCT00111696 | I | Stage IV Melanoma; Recurrent Malignant Melanoma | Completed. Results not published | _ | ||
| _ | NCT00263783 | I | Melanoma | Completed. Results not published | _ | ||
| _ | NCT00111696 | I | Advanced Malignant Melanoma | Completed. Results not published | _ | ||
| CD44 | Anti-CD44 Antibody RG7356 | _ | NCT01358903 | I | Melanoma | Completed. Has shown low clinical efficacy for patients with a variety of solid tumors, including melanoma. Only 13 out of 61 patients (21%) experienced SD lasting an average of 12 weeks. Labelled antibody showed efficacy in tumor tracing | [194] |
CR – complete response, PR – partial response, PD – progressive disease, SD – stable disease, RECIST – response evaluation criteria in solid tumors, VEGF – vascular endothelial growth factor, FGF-2 – fibroblast growth factor 2, rHuPH20 – recombinant human hyaluronidase PH20 enzyme, SC – subcutaneous.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



