
Submitted:
20 October 2024
Posted:
21 October 2024
You are already at the latest version
Abstract
Background/Objectives: The rise of virtual bioequivalence studies has transformed the pharmaceutical landscape, enabling more efficient drug development processes. This systematic review aims to explore advancements in Physiologically-Based Pharmacokinetic (PBPK) modeling, its regulatory implications, and its role in achieving virtual bioequivalence, particularly for complex drug formulations. Methods: We conducted a systematic review of clinical trials using computational methods, particularly PBPK modeling, for bioequivalence assessments. Eligibility criteria are emphasized in silico modeling and pharmacokinetic simulations. Comprehensive literature searches were performed across databases such as PubMed, Scopus, and the Cochrane Library. A search strategy using key terms and Boolean operators ensured extensive coverage. We adhered to PRISMA guidelines for study selection, data extraction, and quality assessment, focusing on key characteristics, methodologies, outcomes, and regulatory perspectives from the FDA and EMA. Results: Our findings indicate that PBPK modeling significantly enhances the prediction of pharmacokinetic profiles, optimizing dosing regimens while minimizing the need for extensive clinical trials. Regulatory agencies have recognized this utility, with the FDA and EMA developing frameworks to integrate in silico methods into drug evaluations. However, challenges such as study heterogeneity and publication bias may limit the generalizability of results. Conclusions: This review highlights the critical need for standardized protocols and robust regulatory guidelines to facilitate the integration of virtual bioequivalence methodologies into pharmaceutical practices. By embracing these advancements, the industry can improve drug development efficiency and patient outcomes, paving the way for innovative therapeutic solutions. Continued research and adaptive regulatory frameworks will be essential in navigating this evolving field.
Keywords:
pharmaceutical industry
; physiologically based pharmacokinetic (PBPK) modelling
; regulatory guidelines
; virtual bioequivalence
1. Introduction
Virtual bioequivalence (VBE) is an evolving pharmaceutical concept that leverages computational modeling and simulation techniques to assess the bioequivalence of generic drugs to their reference or innovator counterparts [1,2]. Bioequivalence (BE) studies are a cornerstone of generic drug development, typically conducted to demonstrate that a generic drug exhibits pharmacokinetic (PK) and pharmacodynamic (PD) properties comparable to the original branded drug [3,4]. Traditionally, these studies require clinical trials involving human subjects, which can be costly, time-consuming, and often raise ethical concerns [5]. Such trials generally involve extensive sampling and PK testing to prove that the systemic drug exposure between the generic and branded drug does not differ significantly [6,7]. In contrast, virtual bioequivalence offers an alternative pathway by employing advanced computational techniques to predict the bioequivalence of generic drugs without the need for large-scale clinical trials [8]. This approach involves developing and applying mathematical models that simulate the drug absorption, distribution, metabolism, and excretion (ADME) processes in humans. Physiologically based pharmacokinetic (PBPK) modeling, computational fluid dynamics (CFD), and in silico dissolution modeling are key tools in this domain [9,10]. These models integrate drug properties (e.g., solubility, permeability), formulation characteristics, and physiological parameters, providing a mechanistic framework that helps predict how drugs behave in the human body [11,12]. PBPK models, in particular, are gaining prominence due to their ability to simulate complex physiological processes with high predictive power. They replace empirical approaches, offering a more nuanced understanding of drug absorption and bioavailability [13,14]. These models are increasingly used in drug discovery, development, and regulatory submissions due to their capacity to integrate a wide array of data, including genetic variability and individual patient characteristics, thus enhancing prediction accuracy [15].
Regulatory agencies such as the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and other global regulatory bodies have recognized the potential of virtual bioequivalence. These agencies have begun to introduce guidelines that support the use of modeling and simulation in bioequivalence studies [16]. The FDA, for instance, has published guidance documents encouraging applying PBPK models in regulatory submissions, particularly for complex generic drug products. Similarly, the EMA has issued draft guidelines acknowledging the role of in silico methods in demonstrating bioequivalence, especially when conventional approaches may be infeasible due to ethical or practical constraints [17,18,19]. The acceptance of virtual bioequivalence by regulatory bodies relies heavily on the validation of computational models and the availability of accurate, comprehensive data inputs. Model validation is essential to ensure that the virtual predictions align closely with in vivo results [1]. The reliability of virtual bioequivalence studies also hinges on the quality of data used to populate these models, such as precise drug physicochemical properties, human physiology parameters, and clinical trial data [2]. While virtual bioequivalence holds immense promise, several challenges must be addressed. One of the critical challenges is the validation of computational models across a wide range of drugs and formulations. Ensuring that in silico results correlate strongly with clinical outcomes is essential in gaining regulatory acceptance [20]. Additionally, the lack of standardized methodologies and limited regulatory approval in certain regions can hinder the broader adoption of virtual bioequivalence [21,22]. Furthermore, there is a need for ongoing research to establish robust correlations between virtual bioequivalence models and in vivo outcomes, particularly for complex drug formulations such as biologics and combination products [23]. Despite these challenges, the future of virtual bioequivalence looks promising. Emerging technologies such as artificial intelligence (AI) and machine learning (ML) are poised to enhance further the predictive capabilities of PBPK and other computational models. These technologies can help optimize model performance by learning from large datasets, identifying patterns, and refining simulations to improve prediction accuracy [24,25]. Integrating AI and ML with virtual bioequivalence offers the potential for personalized medicine, where models can be tailored to individual patient characteristics, enabling more precise predictions of drug behavior.
This systematic review aimed to assess the current state of virtual bioequivalence in the pharmaceutical industry by examining the computational methods and regulatory perspectives involved. The review detailed the methodologies and tools used in virtual bioequivalence, highlighted successful applications, and discussed the advantages and challenges of this approach. Additionally, it explored future perspectives on integrating virtual bioequivalence with emerging technologies such as AI and ML. The sources for this review included major databases such as PubMed, Scopus, Web of Science, and Cochrane Library, as well as regulatory websites from organizations like the FDA and EMA.
2. Materials and Methods
2.1. Eligibility Criteria
This review focused exclusively on clinical trials involving pharmaceutical drugs and bioequivalence studies, specifically those evaluating computational methods for virtual bioequivalence. The inclusion criteria were limited to studies using in silico modeling, pharmacokinetics/pharmacodynamics simulations, and other digital platforms for virtual bioequivalence assessments. Comparisons were made between these computational methods and traditional clinical bioequivalence trials where available and among different computational tools. The primary outcomes of interest were these methods’ efficacy, accuracy, and reliability, along with their compliance with regulatory standards set by agencies such as the FDA and EMA. Systematic reviews, narrative reviews, and studies lacking a focus on computational methods or regulatory perspectives were excluded.
2.2. Information Sources
To ensure comprehensive coverage of relevant literature, the review utilized several key databases: PubMed, Scopus, Web of Science, Embase, and the Cochrane Library. These databases provided access to peer-reviewed articles, conference proceedings, and systematic reviews. Additionally, Google Scholar was used to identify grey literature, including theses and reports not indexed in traditional databases. Regulatory agency websites, including those of the FDA and EMA, were also consulted for guidelines and standards related to virtual bioequivalence. The review focused on studies published from 2014 to 2024 to capture recent advancements and trends. Only studies published in English were included to ensure the accessibility and comprehensibility of the reviewed literature.
2.3. Search Strategy
A comprehensive search strategy was employed using a combination of key terms related to virtual bioequivalence and computational methods. The terms included “Virtual Bioequivalence,” “In Silico Bioequivalence,” “Computational Modelling,” “Simulation,” “Physiologically Based Pharmacokinetic (PBPK) Modelling,” “Computational Fluid Dynamics (CFD),” “In Silico Dissolution Modelling,” “Generic Drugs,” “Innovator Drugs,” “Bioequivalence Assessment,” “Pharmaceutical Industry,” “Drug Development,” and “Regulatory Guidelines.” Boolean operators (AND, OR), truncation, and MeSH (Medical Subject Headings) terms were applied to refine the search and ensure comprehensive coverage.
2.4. Study Selection
The study selection process involved an initial screening of titles and abstracts to determine relevance based on the inclusion criteria. Studies that met these criteria proceeded to a full-text review. The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram was used to document the number of studies at each stage of the review process, including identification, screening, eligibility, and final inclusion. This approach provided a clear and transparent overview of the selection process and ensured that only relevant studies were included.
2.5. Data Extraction
Data extraction was performed using a standardized form to ensure consistency across studies. Key data collected included study characteristics such as author, publication year, journal, and country. Details on the computational methods used, including PBPK modeling, CFD, and in silico dissolution modeling, were recorded. Regulatory frameworks considered in the studies were also documented, focusing on guidelines and standards from regulatory bodies like the FDA and EMA. Key outcomes related to accuracy, regulatory compliance, and computational challenges were extracted, along with assessments of study quality and potential biases.
3. Results
3.1. Systematic Literature Search and Study Selection Process
A systematic literature search was conducted to identify relevant studies exploring virtual bioequivalence through in silico tools. The identification phase began with a comprehensive search across databases (PubMed, Scopus, Web of Science, and Cochrane Library), resulting in 6,472 records (Figure 1). Additionally, 5 records were found through external sources, including a Google search and manual journal hand searches. After the removal of duplicates, 5,651 records remained for screening. During the screening phase, a title-based screening of the 5,651 records was performed. This stage resulted in the exclusion of 4,595 records that were deemed irrelevant based on their titles, leaving 1,056 records for abstract screening. Upon reviewing the abstracts, 839 records were further excluded for not meeting the inclusion criteria. The remaining 217 records proceeded to full-text assessment in the eligibility phase. At the full-text screening stage, 162 articles were excluded, each for specific reasons: 64 articles did not involve in silico tools, 54 articles lacked any connection to clinical trials, 32 articles were unrelated to health outcomes, and 12 articles failed to provide sufficient data. This rigorous assessment led to the inclusion of 55 studies in the final scoping review.
The results of this review demonstrate that despite the initial large number of records identified, only a small fraction of studies met the stringent inclusion criteria for assessing virtual bioequivalence. The high rate of exclusion highlights the specificity of the focus on in silico methodologies and the limited availability of studies directly comparing computational tools with traditional clinical bioequivalence trials. One significant finding was the exclusion of a substantial number of studies that did not utilize in silico tools (64 records), which underscores the emerging but still nascent nature of computational methods in bioequivalence assessment. Furthermore, the exclusion of studies not involving clinical trials (54 records) points to the challenge of integrating virtual bioequivalence tools into clinical research, as many studies may focus solely on theoretical modeling without validation in real-world clinical settings. The exclusion of studies due to insufficient data (12 records) also emphasizes the importance of robust data reporting and transparency in in silico research. For virtual bioequivalence to gain widespread regulatory acceptance, comprehensive data and validation will be crucial. In addition to the primary studies included in this scoping review, regulatory guidelines and frameworks regarding virtual bioequivalence were retrieved from major regulatory bodies, notably the FDA and EMA. Both organizations have established guidelines for bioequivalence assessments, but specific provisions related to the use of in silico tools in these assessments are still evolving.

Table 1.
A summary of the studies included in the scoping review outlines the pharmaceutical drugs, therapeutic indications, in silico tools used, and key findings related to bioequivalence.
Table 1.
A summary of the studies included in the scoping review outlines the pharmaceutical drugs, therapeutic indications, in silico tools used, and key findings related to bioequivalence.
| Study; Year | Database | Pharmaceutical Drug(s) | Drug(s) Indication Class |
In Silico Tool(s) | Bioequivalence Studies (Main Findings) |
|---|---|---|---|---|---|
| Yu, Y. et al. [26], 2017 | Pubmed | Palbociclib | Anticancer | SimCYP® version 14 | The study developed an in silico PBPK model of palbociclib, predicting that moderate CYP3A inhibitors can increase its AUC by ~40% and inducers can decrease it by ~40%, with most predicted vs observed discrepancies within 20%. |
| Cho, CK. et al. [27], 2021 | Pubmed | Tamsulosin | Benign prostatic hyperplasia (BPH) | SimCYP® | This study developed and validated an in silico PBPK model of tamsulosin for different CYP2D6 genotypes. The model predicts that tamsulosin exposure in CYP2D6*wt/10 and CYP2D610/10 genotypes is 1.23- and 1.76-fold higher, respectively than in CYP2D6wt/*wt, contributing to personalized pharmacotherapy by predicting pharmacokinetics based on CYP2D6 genotype. |
| Chen, G. et al. [28], 2023 | Pubmed | Maribavir | Anti-cytomegalovirus | SimCYP® version 17 | This study developed and validated an in silico PBPK model of maribavir, predicting that strong or moderate CYP3A4 inducers significantly reduce maribavir exposure (with rifampin decreasing AUC by 60%), while CYP3A4 inhibitors have no clinically significant effect, guiding dosing adjustments for personalized therapy in patients with CMV infection. |
| Kim YH, et al. [29], 2021 | Pubmed | Celecoxib | NSAIDs | PK-Sim® version 7.2 | This study developed an in silico PBPK model of celecoxib based on CYP2C9 genetic polymorphism. It successfully predicted pharmacokinetics across different genotypes, demonstrating its potential for personalized dosing and reducing adverse drug reactions in precision medicine. |
| Fendt R, et al. [30], 2021 | Pubmed | Caffeine | Neurostimulant | PK-Sim® version 8.0 | This study demonstrates that personalized PBPK models for caffeine, incorporating individual demographics, physiology, and CYP1A2 phenotype, significantly improve pharmacokinetic predictions, increasing accuracy from 45.8% to 66.15% within the 1.25-fold range of observed values, highlighting their potential for model-informed precision dosing in clinical practice. |
| Watanabe A, et al. [31], 2021 | Pubmed | Esaxerenone | Antimineralocorticoid | Simcyp®, version 17 | This study developed a PBPK model for esaxerenone that accurately predicts drug-drug interactions (DDIs) with CYP3A modulators in healthy subjects and those with hepatic impairment, demonstrating that the model’s predictions for plasma exposure changes—such as a 1.53-fold increase with itraconazole and a 0.31-fold decrease with rifampicin—align closely with observed data, highlighting the need for caution when coadministering CYP3A modulators, especially in patients with hepatic impairment. |
| Ou Y, et al. [32], 2018 | Pubmed | Oprozomib | Antitumor | SimCYP® version 13.2 | In this study on oprozomib, the clinical DDI study demonstrated no treatment-related adverse events leading to discontinuation. The PBPK model predicted that a 300 mg dose of oprozomib would not cause a clinically significant change in the exposure of CYP3A4 substrates (≤30%), a prediction that was confirmed by the clinical results. These findings suggest that oprozomib has a low potential to inhibit the metabolism of CYP3A4 substrates in humans, supporting its safe use in combination therapies. |
| Yee KL, et al. [33], 2020 | Pubmed | Doravirine | Antiretroviral | SimCYP® version 17 | The study on doravirine showed that coadministration with rifabutin, a moderate CYP3A4 inducer, significantly decreased doravirine exposure. A dose adjustment from 100 mg once daily to 100 mg twice daily was recommended. A PBPK modeling indicated that while CYP3A induction increased doravirine clearance by up to 4.4-fold, M9 exposure increased only by 1.2-fold. A 2.4-fold increase in M9 exposure was anticipated with the adjusted dosing. Subsequent clinical trials confirmed that doravirine and M9 exposures matched model predictions, supporting the new dosing recommendation when administered with rifabutin. |
| Jo H, et al. [34], 2021 | Pubmed | Dapagliflozin | Antidiabetic | SimCYP® version 18 | The results demonstrated that the PBPK model for dapagliflozin met the twofold acceptance criteria for model-predicted versus observed drug exposures and pharmacokinetic parameters (AUC and maximum drug concentration) across various scenarios, including monotherapy in healthy adults, patients with hepatic or renal impairment, and drug-drug interactions with UGT1A9 modulators like mefenamic acid and rifampin. |
| Posada M, et al. [35], 2017 | Pubmed | Baricitinib | Antirheumatic | SimCYP® version 13.2 | This study demonstrated that baricitinib, an oral selective Janus kinase 1 and 2 inhibitor, has its renal clearance effectively modeled using PBPK modeling, revealing that probenecid, a strong OAT3 inhibitor, increased baricitinib’s AUC(0–∞) by twofold and decreased renal clearance to 69% of control, while predictions indicated that clinically relevant drug-drug interactions with ibuprofen and diclofenac are unlikely, as their in vitro IC50 values suggested AUC(0–∞) ratios of 1.2 and 1.0 for baricitinib. |
| Zane NR, et al. [36], 2014 | Pubmed | Voriconazole | Antifungal | SimCYP® Paediatric | This study developed a PBPK model for voriconazole, accurately predicting pharmacokinetic parameters in adults within a 20% prediction error; however, the pediatric oral model initially overestimated oral bioavailability twofold, which improved after incorporating intestinal first-pass metabolism. This indicates that voriconazole undergoes differential first-pass metabolism in children compared to adults. |
| Lang J, et al. [37], 2020 | Pubmed | Ivabradine | Antianginal and antiischemic | SimCYP® version 18 | This study developed a joint parent-metabolite PBPK and pharmacodynamic model for ivabradine and its metabolite, successfully predicting pharmacokinetics and heart rate reductions after intravenous or oral administration, including drug-drug interactions with CYP3A4 inhibitors. The model predicted 92% and 85% of the AUC ratios for ivabradine and its metabolite within acceptable limits, with observed versus predicted heart rate reductions of -7.7/-5.9 bpm and -15.8/-14.0 bpm in control and ketoconazole groups, respectively, establishing a scalable framework for assessing DDI risks in different populations. |
| Hanke N, et al. [38], 2017 | Pubmed | Zoptarelin doxorubicin | Anticancer | PK-Sim® and MoBi® | This study established a PBPK model for zoptarelin doxorubicin, a fusion of doxorubicin and a luteinizing hormone-releasing hormone receptor agonist, to assess its DDI potential. The model, built in two steps—first for doxorubicin and then for zoptarelin doxorubicin—predicted minimal in vivo inhibition of drug transporters OATP1B3 (0.5%) and OCT2 (2.5%). Simulations indicated that co-administration with simvastatin and metformin would not significantly alter their plasma concentrations, demonstrating that zoptarelin doxorubicin has no potential for DDIs via these transporters. |
| Chen Y, et al. [39], 2017 | Pubmed | Gefitinib | Anticancer | SimCYP® version 14 | This study developed and validated a PBPK model to compare gefitinib exposure in CYP2D6 ultrarapid metabolizers (UM) and extensive metabolizers (EM). The model predicted a 39% decrease in gefitinib AUC in UM compared to EM, though this reduction remained above the IC90 for EGFR mutations in NSCLC. The model, calibrated with itraconazole-gefitinib interaction data, was validated with clinical data, showing that CYP2D6 system components were accurately modeled. Overall, the reduced exposure in UMs is unlikely to impact gefitinib’s clinical efficacy. |
| Gajewska M, et al. [40], 2020 | Pubmed | Alpelisib | Anticancer | GastroPlus™ version 9.6 | This study developed a PBPK model for alpelisib to simulate oral absorption and plasma pharmacokinetics in healthy subjects, successfully predicting bioequivalence outcomes between clinical and commercial formulations under various conditions (fasted, fed, and altered pH). The model incorporated in vitro dissolution data, and its predictive errors for plasma Cmax and AUC were ≤30%, making it a valuable tool for virtual bioequivalence assessments. |
| Donovan MD, et al. [41], 2018 | Pubmed | Bumetanide | Cardiovascular disease | SimCYP® version 14 | This study developed a PBPK model to predict bumetanide’s plasma and brain concentrations in adult and pediatric populations. While the model accurately predicted pharmacokinetic parameters for adults and older children within two-fold of observed values, it failed to fit well with neonatal data. The study highlights the need for more metabolic and transport parameter data before the model can be reliably used to predict bumetanide disposition and recommend dosing in neonates. |
| Fu Q, et al. [42], 2021 | Pubmed | Lenabasum | Anti-inflammatory | SimCYP® version 19 | A PBPK model for lenabasum, a synthetic CB2 agonist, was developed using clinical data and CYP metabolism parameters. The model accurately predicted lenabasum’s AUC and Cmax within 1.19- and 1.25-fold of observed values. Simulations indicated that rifampin (a CYP inducer) would decrease lenabasum’s AUC by 64%, while fluconazole (a CYP inhibitor) would increase AUC by 43%. The model effectively predicts lenabasum pharmacokinetics and can guide dose adjustments in drug-drug interaction scenarios. |
| Bergagnini-Kolev M, et al. [43], 2023 | Pubmed | Itraconazole | Antifungal | SimCYP® | A PBPK model was developed to assess the DDI potential of inhaled PUR1900, a dry powder itraconazole formulation, using midazolam as a “victim drug.” Simulations predicted minimal DDI risk, with midazolam’s Cmax and AUC increasing by only 14% and 26%, respectively, when co-administered with 40 mg PUR1900. The low systemic itraconazole exposure from PUR1900 suggests minimal CYP3A4 inhibition, indicating that PUR1900 poses a low DDI risk and may be safely used for pulmonary fungal infections alongside other medications contraindicated with oral itraconazole. |
| Nakamaru Y, et al. [44], 2015 | Pubmed | Teneligliptin | Antidiabetic | SimCYP® | A PBPK model for teneligliptin was developed and validated using the Simcyp simulator. The model accurately predicted plasma concentrations from clinical trials across various populations, including those with renal or liver impairment. The model effectively simulated drug-drug interactions, such as a 2.1—to 2.2-fold increase in teneligliptin exposure when co-administered with the CYP3A4 inhibitor ketoconazole. This robust PBPK model provides detailed insights into the pharmacokinetics of teneligliptin, allowing the prediction of drug-drug interactions and exposure changes in specific patient populations. |
| Thompson EJ, et al. [45], 2024 | Pubmed | Pantoprazole | GERD | PK-Sim® version 10.0 | This study developed a PBPK model for pantoprazole, extending it to children with obesity while accounting for genetic variation in CYP2C19 and physiological changes related to age and obesity. The model evaluated three dosing strategies and found that FDA-recommended weight-tiered dosing resulted in the most consistent pantoprazole exposure across children, regardless of obesity or CYP2C19 phenotype. The findings demonstrate the utility of PBPK models in optimizing dosing for pediatric populations where clinical trial data may be limited, particularly in children with obesity. |
| Wang HY, et al. [46], 2016 | Pubmed | Midazolam | Hypnotic-sedative | SimCYP® version 13 | This study developed and evaluated a PBPK model using the SimCYP simulator to predict the pharmacokinetics of midazolam in Chinese subjects across different age groups. The model accurately predicted midazolam plasma concentrations, AUC, and Cmax following oral administration, with predicted-to-observed clearance ratios ranging from 0.86 to 1.12. The findings demonstrate that the SimCYP PBPK model can effectively predict CYP3A4/5-mediated pharmacokinetics in the Chinese population, providing valuable insights for designing bridging clinical trials and optimizing drug dosing for different ethnic groups. |
| Callegari E, et al. [47], 2021 | Pubmed | Ertugliflozin | Antidiabetic | SimCYP® version 15 | This study utilized PBPK modeling to predict a 1.51-fold increase in the area under the plasma concentration-time curve (AUCR) for ertugliflozin when co-administered with the UGT inhibitor mefenamic acid (MFA). This demonstrated the model’s effectiveness in evaluating drug-drug interactions and virtual bioequivalence for UGT substrates. |
| Nakamura T, et al. [48], 2018 | Pubmed | Tamoxifen | Anticancer | MATLAB version 8.0.0.783 (R2012b) | This study used PBPK modeling and virtual clinical study (VCS) simulations to predict the outcomes of the Tamoxifen Response by CYP2D6 Genotype-based Treatment-1 (TARGET-1) trial, which investigated tamoxifen dosing guided by CYP2D6 genotypes. The simulations indicated an average probability of 0.469 for demonstrating the superior efficacy of escalated tamoxifen doses in CYP2D6 variant carriers, which increased to 0.674 with a larger sample size (n = 260). The analyses highlighted that variability in endoxifen levels negatively impacted the likelihood of achieving the study’s endpoint, emphasizing the value of PBPK modeling and VCS in optimizing clinical trial design. |
| Stader F, et al. [49], 2021 | Pubmed | Bictegravir | Antiretroviral | Matlab 2017a | This study developed a PBPK model for bictegravir to assess the impact of aging on its pharmacokinetics in people living with HIV (PLWH). The model validated with data from young (20-55 years) and elderly (55-85 years) PLWH indicated that bictegravir exposure remained unchanged with age. Simulations suggested a potential 40% increase in drug exposure across adulthood, consistent with age-related changes seen in other drugs. Thus, no dose adjustment for bictegravir is necessary in elderly PLWH without severe comorbidities. |
| Yang R, et al. [50], 2024 | Pubmed | Omeprazole | GERD | SimCYP® | This study developed PBPK and in vitro-in vivo relationship (IVIVR) models for enteric-coated omeprazole capsules, aiming to explore VBE for biological exemptions. The predicted pharmacokinetics matched observed data, establishing clinically relevant dissolution specifications (CRDS) for bioequivalence. It required 48 healthy subjects and ensured dissolution rates of 28%-54%, 52%, and 80% after two, three, and six hours, respectively. This approach can facilitate biological exemptions for other BCS class II generics and improve drug development efficiency. |
| Ojala K, et al. [51], 2020 | Pubmed | ODM-204 | Anticancer | GastroPlus version 9.7 | This study demonstrated that in vitro dissolution tests, the TIM-1 intestinal model, and PBPK simulations effectively predicted the absorption properties of the poorly soluble lipophilic weak base ODM-204, providing valuable insights for evaluating its bioavailability and informing formulation decisions. |
| Agyemang A, et al. [52], 2021 | Pubmed | Acumapimod | Anti-inflammatory | SimCYP® version 16.1 | The study found that co-administration of the CYP3A4 inhibitors azithromycin and itraconazole with acumapimod did not significantly affect its pharmacokinetics or safety profile, as confirmed by both clinical DDI studies and PBPK modeling. This supports the concomitant use of these inhibitors in patients. |
| Chen Y, et al. [53], 2014 | Pubmed | Amiodarone | Anti-arrhythmic | SimCYP® version 12 | The study demonstrates that a PBPK model effectively predicts DDIs involving amiodarone (AMIO) and its major metabolite, mono-desethyl-amiodarone (MDEA), by accurately simulating their pharmacokinetic profiles and accumulation. The model successfully captured the clinically significant increases in exposure of simvastatin (1.2- to 2-fold), dextromethorphan, and warfarin, highlighting the importance of considering inhibitory metabolites in DDI assessments. |
| Litou C, et al. [54], 2019 | Pubmed | Aprepitant | Antiemetic | SimCYP® version 16.1 | This study highlights the development of EMEND®, a nano-sized aprepitant formulation, using innovative biopharmaceutical tools. In vitro tests showed that native surfactants significantly enhanced the aprepitant’s solubility. A PBPK model in the Simcyp Simulator accurately simulated plasma concentrations of aprepitant after administering 80 mg and 125 mg capsules in both fasted and fed states. Parameter sensitivity analysis indicated that while nano-sizing improved in vivo performance, intestinal solubility remains a limiting factor for bioavailability, classifying aprepitant as DCS IIb. The findings underscore the value of combining in vitro and in silico methods for predicting absorption and supporting regulatory assessments of poorly soluble compounds. |
| Einolf HJ, et al. [55], 2017 | Pubmed | Sonidegib | Anticancer | SimCYP® version 13.2 | This study evaluated the effects of strong CYP3A inhibitors, ketoconazole (KTZ) and rifampin (RIF), on sonidegib (Odomzo) pharmacokinetics (PK) after a single 800 mg dose in healthy subjects. KTZ increased sonidegib exposure by 2.25-fold in terms of area under the curve (AUC), while RIF decreased it by 72%. A validated PBPK model accurately predicted these interactions and indicated that sonidegib would have a more significant drug-drug interaction (DDI) magnitude with CYP3A inhibitors at steady state, informing dosing recommendations in the product label. |
| Yamazaki S, et al. [56], 2015 | Pubmed | Crizotinib | Anticancer | SimCYP® version 13.1 | This study developed and refined a PBPK model for crizotinib (Xalkori), accurately predicting its exposure from clinical data. The model verified DDI results from single-dose studies with ketoconazole and rifampin, showing comparable fold-increases in crizotinib exposure during multiple-dose DDI studies. These findings suggest that dose adjustments in multiple-dose scenarios can rely on single-dose outcomes, with the PBPK model applicable for predicting crizotinib exposure in various clinical contexts. |
| Riddell K, et al. [57], 2020 | Pubmed | Molibresib | Anticancer | SimCYP® version 14 | This study evaluated the pharmacokinetics of molibresib (GSK525762) in a randomized DDI trial, utilizing PBPK modeling to determine safe dosing in healthy volunteers. Administering 5 mg of molibresib with the strong CYP3A inhibitor, itraconazole increased its AUC by 4.15-fold and Cmax by 66%, while the active metabolites’ AUC and Cmax decreased by 70% and 87%. Subsequently, the dose was increased to 20 mg with the strong CYP3A inducer rifampicin, resulting in a 91% reduction in molibresib’s AUC and an 80% reduction in Cmax, with the metabolites’ AUC decreasing by only 8% and Cmax increasing 2-fold. These findings confirmed that molibresib is a CYP3A4 substrate and demonstrated the efficiency of PBPK modeling in assessing drug-drug interactions. |
| Purohit V, et al. [58], 2024 | Pubmed | Tofacitinib | Antirheumatic | SimCYP® version 20 | This study demonstrated bioequivalence between a once-daily modified-release (MR) microsphere formulation of tofacitinib for pediatric patients and a 5 mg twice-daily immediate-release (IR) solution using PBPK virtual BE trials instead of a clinical trial. The verified PBPK model, utilizing the Simcyp ADAM module, incorporated the clinically observed intrasubject coefficient of variation (ICV) to assess BE. Results confirmed BE between the formulations after single and multiple doses, highlighting a strategy that minimizes unnecessary drug exposure for healthy volunteers while facilitating new formulation development. |
| Dennison TJ, et al. [59], 2017 | Pubmed | Aamlodipine and atorvastatin | Cardiovascular disease | SimCYP® version 14 | This study developed and characterized orally disintegrating tablets (ODTs) containing amlodipine (5 mg) and atorvastatin (10 mg), evaluating the bioequivalence of single versus fixed-dose combination (FDC) formulations. The ODTs rapidly disintegrated in under 30 seconds, exhibiting strong mechanical properties. In vitro dissolution tests were performed in fasted and fed-state simulated intestinal fluid, showing no significant differences in active pharmaceutical ingredient dissolution, except for amlodipine in fed-state conditions. Pharmacokinetic simulations using the Simcyp model indicated no difference in bioavailability between single and FDC ODTs. However, atorvastatin exhibited increased Cmax and AUC in fed subjects, likely due to altered gut transit and reduced CYP3A4 metabolism. |
| Knöchel J, et al. [60], 2024 | Pubmed | Midazolam | Hypnotic-sedative | SimCYP® version 20 | In this study, a validated PBPK model was developed to evaluate the impact of Atuliflapon on the pharmacokinetics of midazolam, a sensitive CYP3A4 substrate, revealing that Atuliflapon is a weak CYP3A4 inhibitor, with the model predicting increases of 27% in AUC and 23% in Cmax for midazolam upon co-administration, thus indicating that Atuliflapon’s minor inhibitory effect is unlikely to affect the pharmacokinetics of CYP3A4-metabolized drugs significantly. |
| Freise KJ, et al. [61], 2017 | Pubmed | Venetoclax | Anticancer | SimCYP® version 14 | This study developed and verified a venetoclax PBPK model to predict the impact of cytochrome P450 3A (CYP3A) inhibitors and inducers on venetoclax pharmacokinetics, demonstrating good agreement between predicted and observed pharmacokinetic parameters. The model simulations indicated that moderate and strong CYP3A inducers could significantly decrease venetoclax exposure, while moderate and strong CYP3A inhibitors could increase venetoclax AUC∞ by 100% to 390% and 480% to 680%, respectively. Consequently, recommended dose reductions of at least 50% and 75% for venetoclax are advised when coadministered with moderate and strong CYP3A inhibitors, respectively, to maintain therapeutic exposure levels. |
| Boetsch C, et al. [62], 2016 | Web of Science | Bitopertin | Neurodegenerative disease | GastroPlus™ | This study assessed the impact of strong and moderate cytochrome P450 (CYP) 3A4 inhibitors, ketoconazole and erythromycin, on the pharmacokinetics of bitopertin, a glycine reuptake inhibitor primarily metabolized by CYP3A4, through two open-label volunteer studies. Co-administration of ketoconazole increased the bitopertin AUC from 0 to 312 hours by 4.2-fold, while erythromycin increased the AUC from time zero to infinity by 2.1-fold. PBPK modeling predicted AUC ratios that closely matched the observed data, indicating that strong CYP3A4 inhibitors could increase bitopertin AUC0-inf by 7- to 8-fold, while moderate inhibitors could double its AUC0-inf. Consequently, strong CYP3A4 inhibitors should not be administered with bitopertin. |
| Katsube T, et al. [63], 2020 | Web of Science | Lusutrombopag | Thrombocytopenia | SimCYP® version 14 | This study evaluated the DDI potential of lusutrombopag, a thrombopoietin receptor agonist, on cytochrome P450 (CYP) 3A activity using midazolam as a probe substrate, and assessed the effect of cyclosporine on lusutrombopag pharmacokinetics through clinical studies and PBPK modeling. Clinical trials showed that lusutrombopag did not significantly affect midazolam’s pharmacokinetics, with mean ratios for maximum plasma concentration (Cmax) and AUC being 1.01 and 1.04, respectively. In contrast, cyclosporine slightly increased lusutrombopag’s Cmax and AUC by 18% and 19%, respectively. Overall, both in vitro and in vivo findings indicated that lusutrombopag has no clinically significant DDI potential with other drugs via CYP3A or P-glycoprotein pathways. |
| Andreas CJ, et al. [64], 2017 | Web of Science | Zolpidem | Hypnotic-sedative | Simcyp® and GastroPlus™ | This study explored the absorption of zolpidem, a BCS class I compound, revealing a negative food effect on its pharmacokinetics when administered as immediate or modified release formulations. Using in vitro and in silico methods, including PBPK modeling with Simcyp® and GastroPlus™, the simulations achieved average fold error (AFE) values of less than 1.5. The results indicated that absorption in the fasted state is formulation-controlled, while gastric emptying influences absorption in the fed state, with meal interactions possibly causing incomplete drug release, thereby reducing Cmax and AUC. |
| Post TM, et al. [65], 2016 | Web of Science | Nomegestrol acetate | Hormone therapy | PK-Sim® | This study compared the pharmacokinetics of nomegestrol acetate (NOMAC) in adolescent and adult women after a single dose of NOMAC/E2. No statistically significant differences in NOMAC PK parameters—Cmax, AUC, and half-life (t1/2)—were observed between the two age groups. Additionally, the WB-PBPK model accurately predicted NOMAC AUC and Cmax values for both groups. The findings suggest that NOMAC pharmacokinetics are comparable in adolescents and adults following a single dose, highlighting the model’s utility in addressing ethical challenges in adolescent PK studies. |
| Li J, et al. [66], 2020 | Web of Science | Eliglustat | Gaucher’s disease | SimCYP® version 13 | This study evaluated the pharmacokinetics of eliglustat in adults with Gaucher disease type 1 (GD1) and varying CYP2D6 metabolizer phenotypes, particularly those with hepatic and renal impairment. In two Phase 1 studies, a single 84-mg dose of eliglustat was administered. Compared to healthy extensive metabolizers (EM), Cmax and AUC were not significantly different in EMs with mild hepatic impairment, higher in EMs with moderate hepatic impairment, and similar in EMs with severe renal impairment. Based on these results, the eliglustat drug label was revised for patients with hepatic or renal impairment. |
| Samant TS, et al. [67], 2018 | Web of Science | Ribociclib | Anticancer | SimCYP® version 13 | This study evaluated the effect of proton pump inhibitors (PPIs) on the bioavailability of ribociclib (KISQALI), a cyclin-dependent kinase 4/6 inhibitor for HR+/HER2- advanced breast cancer, which can be taken with gastric pH-elevating agents and food. Through solubility tests, PBPK modeling, and clinical trial data analysis, results showed no impact of gastric pH on ribociclib pharmacokinetics. This supports labeling that allows coadministration with PPIs. Additionally, bioequivalence with or without a high-fat meal was confirmed, enabling flexible dosing to enhance patient compliance and outcomes. |
| Morcos PN, et al. [68], 2023 | Web of Science | Copanlisib | Anticancer | PK-Sim® | This study evaluated copanlisib, a PI3K inhibitor, in pediatric patients with relapsed/refractory solid tumors. A model-informed approach supported a starting dose of 28 mg/m² for patients ≥1 year old, representing 80% of the adult dose. An adult PBPK model, adapted for pediatric patients, predicted that this dose would achieve comparable exposures to the approved adult dose of 60 mg. Clinical pharmacokinetic data from a Phase I study confirmed that the 28 mg/m² dose provided consistent exposures across the pediatric age range. This approach successfully validated the pediatric dose recommendation for copanlisib. |
| Traver E, et al. [69], 2024 | Web of Science | Leriglitazone | CNS diseases | SimCYP® version 17 | This study assessed leriglitazone, a PPARγ agonist, for its pharmacokinetics and CNS efficacy in neurodegenerative diseases. A Phase 1 trial in healthy male volunteers, involving single and multiple ascending doses, showed that leriglitazone is rapidly absorbed with no food effect and has a linear dose-exposure relationship. A PBPK model was developed using Phase 1 data, incorporating CYP3A4 and CYP2C8 metabolism and biliary clearance. The model successfully predicted pediatric doses, which were preliminarily verified in five pediatric patients. |
| Tsamandouras N, et al. [70], 2015 | Web of Science | Simvastatin | Cardiovascular disease | SimCYP® version 13 | This study developed a population PBPK model for simvastatin (SV) and its active metabolite, simvastatin acid (SVA), to predict their concentrations in liver (efficacy) and muscle (toxicity). Plasma concentrations from 34 healthy volunteers were analyzed using a mechanistic model that incorporates SV/SVA inter-conversion in different tissues. The model successfully described SV/SVA plasma data and predicted the effects of OATP1B1 polymorphism and drug-drug interactions on concentrations. It also aligned with observed clinical efficacy and toxicity outcomes, supporting its use in assessing drug interactions and myopathy risk. |
| Salerno S, et al. [71], 2017 | Cochrane Library | Solithromycin | Antibiotic | PK-Sim and MoBi version 6.2 | This study developed a whole-body PBPK model for solithromycin in adults using PK-Sim and MoBi, incorporating time-dependent CYP3A4 auto-inhibition. Plasma and epithelial lining fluid (ELF) concentration data from 100 healthy subjects and 22 patients with community-acquired bacterial pneumonia (CABP) were used for model evaluation. Population simulations showed that 11% and 23% of observations fell outside the 90% prediction interval for plasma and ELF, respectively. The oral regimen (800 mg on day 1, 400 mg daily on days 2–5) was predicted to be effective, with ≥97% of simulated adults achieving the target AUC/MIC ratios for ELF. |
| Venuto C, et al. [72], 2020 | Cochrane Library | Nilotinib | Anticancer | SimCYP® | This study aims to develop a PBPK model to predict nilotinib concentrations in plasma and cerebrospinal fluid (CSF) and compare the predictions to observed data from the NILO-PD clinical trial. Nilotinib, a c-Abl inhibitor, was investigated in patients with Parkinson’s disease. Serum and CSF concentrations were measured, showing low CSF-to-serum ratios (0.002-0.003) for the 150mg and 300mg doses. Using the Simcyp Simulator, a whole-body PBPK model will simulate nilotinib pharmacokinetics in serum, CSF, and brain and compare the results to clinical trial data to validate the model’s accuracy. |
| Rhee SJ, et al. [73], 2018 | Cochrane Library | Fimasartan, Amlodipine, and Hydrochlorothiazide | Cardiovascular disease | SimCYP® version 15 | This study aimed to develop a PBPK model for fimasartan, amlodipine, and hydrochlorothiazide and assess DDI potentials. Using Simcyp software, the PBPK model was constructed with data from literature and in vitro studies and validated by comparing predicted pharmacokinetics with observed data in healthy subjects. The model predicted no significant DDI for co-administration of fimasartan with amlodipine or hydrochlorothiazide, which is consistent with clinical observations. The simulation at steady-state showed a 24.5% increase in fimasartan exposure with no changes in amlodipine and hydrochlorothiazide exposures. The model effectively predicts DDI potential. |
| Hwang S, et al. [74], 2024 | Cochrane Library | Methotrexate | Antirheumatic | SimCYP® version 21 | This study developed a robust PBPK model to quantitatively assess drug-drug interactions (DDIs) for methotrexate (MTX) mediated by transporters, demonstrating that co-administration with rifampicin and febuxostat increased MTX systemic exposure by 33% and 17%, respectively, and by 52% when combined, validating the model’s predictive capability for transporter-mediated DDIs. |
| Samant TS, et al. [75], 2020 | Cochrane Library | Ribociclib | Anticancer | SimCYP® version 18 | This study developed a PBPK model for ribociclib, used in combination with endocrine therapy for HR-positive and HER2-negative advanced breast cancer. The model integrated in vitro, preclinical, and clinical data. Key findings included ritonavir increasing ribociclib’s AUC by 3.2-fold, while rifampin decreased it by 89%. Additionally, ribociclib raised midazolam’s AUC by 3.8-fold and caffeine’s by 1.2-fold. Predictions showed that multiple ribociclib doses could increase midazolam AUC by 5.85-fold in cancer patients, with ritonavir increasing ribociclib AUC by 1.31-fold. The study recommends avoiding strong CYP3A inhibitors or inducers and exercising caution with CYP3A substrates with narrow therapeutic indices. |
| Chen B, et al. [76], 2022 | Cochrane Library | Acalabrutinib | Anticancer | SimCYP® version 19 | This study aimed to evaluate the pharmacokinetic interactions between acalabrutinib and moderate CYP3A inhibitors, fluconazole and isavuconazole, using both experimental data and a PBPK model. The effect on acalabrutinib and its active metabolite, ACP-5862, was investigated in an open-label, randomized, 2-period study. Co-administration with fluconazole and isavuconazole increased acalabrutinib’s maximum plasma concentration and area under the curve, while reducing ACP-5862 exposure. The PBPK model accurately predicted these PK profiles. There were minimal safety concerns, and no dose adjustments were deemed necessary for co-administration with moderate CYP3A inhibitors. |
| Djebli N, et al. [77], 2015 | Cochrane Library | Clopidogrel | Antiplatelet | SimCYP® version 10.2 | This study developed and validated a dynamic PBPK model in Simcyp for clopidogrel and its active metabolite clopi-H4 across four CYP2C19 phenotypic populations, accurately predicting the area under the curve (AUC) values for each group and demonstrating reliable predictions of pharmacokinetics and drug-drug interactions, making it the first model to simultaneously predict the pharmacokinetics of a prodrug and its metabolites based on genetic variability in metabolizing enzymes. |
| Xiao Q, et al. [78], 2015 | Cochrane Library | Repaglinide and pioglitazone | Antidiabetic | SimCYP® version 14.1 | This study investigated the inhibitory effect of repaglinide on pioglitazone metabolism using in vitro, in silico, and in vivo methods. In vitro studies demonstrated a strong inhibitory effect of repaglinide (Ki = 0.0757 µM, [I]/Ki > 0.1) on pioglitazone metabolism, while an IVIVE-PBPK model using Simcyp® predicted AUC and Cmax ratios of approximately 1.01 between treatment groups. However, clinical trials with 12 healthy volunteers revealed no significant difference in pioglitazone pharmacokinetics (p > 0.05) when coadministered with repaglinide. This discrepancy was attributed to extensive plasma protein binding and high clearance of repaglinide, leading to lower in vivo concentrations compared to in vitro conditions. |
| Chen J, et al. [79], 2022 | Cochrane Library | Salvianolic acid A | Cardiovascular disease | GastroPlus® Version 9.8 | This study developed a PBPK model that successfully predicted the pharmacokinetics of salvianolic acid A (SAA), particularly its plasma concentrations, in healthy subjects. The results demonstrated a lack of dose proportionality after single doses, with the area under the curve (AUC₀₋ₜ) showing higher-than-expected increases at higher doses. Specifically, the 90% confidence intervals for the slope of AUC₀₋ₜ (1.222 [1.156-1.288]) exceeded the predefined bioequivalence range, suggesting saturation of transport mechanisms such as hepatic OATP1B1 and P-glycoprotein (P-gp) at higher doses. These findings highlight potential challenges in achieving dose proportionality in SAA pharmacokinetics, which the PBPK model was able to predict and simulate accurately. |
| Kaur N, et al. [80], 2020 | Cochrane Library | Irbesartan | Cardiovascular disease | GastroPlus™ | This study mechanistically examined the oral absorption behavior of the weakly basic drug irbesartan (IRB) by investigating its pH-dependent solubility, supersaturation, and precipitation patterns. Initial simulations using equilibrium solubility were inadequate for accurately predicting oral absorption. However, the use of a multi-compartment biorelevant dissolution testing model, simulating conditions in the stomach and duodenum, allowed for sustained intestinal supersaturation (2-4-fold) across varying gastric-to-intestinal transfer rates. When combined with dissolution data, GastroPlus™ simulations predicted plasma exposure with greater accuracy (within ± 15% prediction error). The study found that amorphous precipitate formation with significant particle size reduction (about 10-fold) contributed to maintaining intestinal supersaturation, improving oral pharmacokinetics predictions for irbesartan. |
3.2. Utilization of In Silico Modeling Tools in Virtual Bioequivalence Studies
The systematic review revealed that various in silico tools had been employed to assess virtual bioequivalence, with SimCYP® emerging as the most widely used, appearing in 41 out of the 55 selected studies (74.5%), as presented in Figure 2. SimCYP® is a sophisticated PBPK modeling platform renowned for its ability to simulate the complex processes of drug ADME within diverse virtual populations. Its strength lies in its detailed modeling of drug-drug interactions, particularly those involving cytochrome P450 enzymes and key transporter proteins [81]. This capability allows researchers to explore how genetic variability, disease conditions, and demographic factors, such as age and ethnicity, influence drug pharmacokinetics and dynamics [82]. One of the main advantages of SimCYP® is its predictive power in modeling drug interactions. It can accurately simulate the impact of co-administered medications or dietary components on drug clearance and bioavailability. It has proven especially useful in predicting how CYP enzyme inhibitors or inducers modify drug exposure, providing critical insights into the risks of toxicity or therapeutic failure [83,84]. This is particularly valuable for bioequivalence studies, where the goal is to determine whether a generic drug performs similarly to an innovator drug in various patient populations without conducting large-scale clinical trials. Moreover, the ability of SimCYP® to integrate population variability makes it a favored choice for predicting bioequivalence outcomes across different subpopulations, such as those with liver or kidney impairment, the elderly, or individuals with specific genetic polymorphisms. These simulations reduce the need for multiple clinical trials in distinct patient groups, making virtual bioequivalence studies more efficient, faster, and cost-effective [85,86]. SimCYP®‘s broad application in bioequivalence studies highlights its versatility and regulatory acceptance. It has been widely used to support regulatory submissions to agencies like the FDA and EMA, as it complies with their requirements for bioequivalence assessments, particularly in cases involving complex drug-drug interactions or populations at the extremes of pharmacokinetic variability [87,88]. Thus, SimCYP® continues to play a critical role in virtual bioequivalence assessments, contributing to a more streamlined drug development process.
Following SimCYP®, PK-Sim® was the next most commonly used in silico tool, appearing in 7 studies (12.7%). PK-Sim® is another widely adopted PBPK modeling platform known for its detailed representations of whole-body physiology and customizable drug kinetic profiles. Unlike SimCYP®, which excels in virtual population simulations and drug-drug interaction predictions, PK-Sim® has a notable strength in its integration with MoBi®. This systems biology modeling tool allows for advanced mechanistic and dynamic modeling. This capability makes PK-Sim® particularly effective in studying complex drug interactions, multi-compartmental pharmacokinetics, and situations where drugs demonstrate non-linear kinetics [13,89]. One key advantage of PK-Sim® is its flexibility in simulated physiological scenarios beyond typical bioequivalence conditions. For instance, it has been employed to simulate pharmacokinetic profiles in special populations, such as pediatric, elderly, or renal-impaired patients, where clinical data may be sparse or difficult to obtain. This strength is particularly relevant in regulatory settings, where simulations can be used to predict pharmacokinetic behaviors under conditions that are difficult to test in traditional clinical trials [90]. Additionally, PK-Sim®‘s capacity to model multi-compartment systems, where drugs may exert effects at multiple sites, encounter varying enzyme activities, or exhibit complex absorption and clearance patterns, further demonstrates its utility in bioequivalence studies that involve non-linear kinetics or intricate pharmacological behaviors [91].
GastroPlus™, utilized in 5 studies (9.1%), stands out as a specialized software primarily focusing on predicting drug dissolution, absorption, and pharmacokinetics within the gastrointestinal tract. GastroPlus™ integrates PBPK and in silico dissolution models to simulate how drug formulations behave within the human digestive system, making it particularly useful for evaluating oral bioavailability and the influence of various formulation factors on drug performance. This tool has been employed to simulate bioequivalence for modified-release formulations or drugs with low solubility, where gastrointestinal transit times and dissolution play critical roles in pharmacokinetics [92,93]. Two studies (3.6%) used MATLAB for pharmacokinetic modeling. While MATLAB is not a dedicated PBPK software, it offers advanced computational capabilities and flexibility for creating custom pharmacokinetic and pharmacodynamic (PK/PD) models. MATLAB is frequently employed for exploratory analysis, custom data fitting, and simulations of drug kinetics in scenarios where commercial software might not provide the necessary specificity. For virtual bioequivalence, MATLAB can be used to build highly tailored models that simulate drug behavior under varying conditions or in populations not easily represented by standard tools [48,49].
3.3. Overview of Disease Type Distribution in Virtual Bioequivalence Studies
The systematic review conducted on the application of virtual bioequivalence from 2014 to 2024 reveals distinct trends in disease type prevalence among the studies analyzed. Cancer and tumors emerged as the most extensively researched categories, accounting for a total of 16 studies (Figure 3). This significant focus underscores the critical need for effective therapeutic interventions and the potential role of virtual bioequivalence in optimizing drug formulations for oncology. Following cancer, cardiovascular diseases were the second most studied category, with 10 studies identified. This emphasis highlights the ongoing challenges in managing cardiovascular health and the potential for virtual bioequivalence methodologies to contribute to the development of safer and more effective cardiovascular therapies. Infectious diseases and neurodegenerative diseases each comprised 6 studies. The interest in infectious diseases reflects the global health priorities and the urgent need for innovative approaches in combating various pathogens. Similarly, the focus on neurodegenerative diseases indicates a growing recognition of their complex treatment landscapes and the necessity for advanced computational strategies to enhance drug development in this area. Additionally, metabolic diseases were addressed in 5 studies, while inflammatory diseases were covered in 4 studies. The relatively lower number of studies in these categories suggests potential areas for further exploration within the context of virtual bioequivalence, as they represent significant public health concerns. Lastly, a collective of 8 studies examined various other diseases, illustrating the versatility and applicability of virtual bioequivalence across a broader range of medical conditions. This diversity in research underscores the potential for virtual bioequivalence to inform drug development and optimization across multiple therapeutic areas.
The emerging field of virtual bioequivalence studies in the context of cancer and tumor treatment has witnessed significant advancements over recent years, largely facilitated by the application of PBPK modeling. Various studies have explored the pharmacokinetics of specific anticancer drugs, enhancing our understanding of their interactions, absorption characteristics, and overall therapeutic efficacy. For instance, Yu et al. (2017) developed a PBPK model for palbociclib, an anticancer agent, predicting that moderate CYP3A inhibitors could elevate its AUC by approximately 40%, while inducers might reduce it by a similar margin. This model demonstrated a strong correlation between predicted and observed data, establishing a reliable tool for assessing drug interactions [26]. Similarly, Chen et al. (2017) utilized PBPK modeling to analyze gefitinib, revealing a 39% reduction in AUC for CYP2D6 ultrarapid metabolizers compared to extensive metabolizers. Importantly, the reduced exposure remained above the inhibitory concentration for EGFR mutations in non-small cell lung cancer, indicating that the drug’s efficacy would likely remain intact in clinical settings [39]. In another study, Gajewska et al. (2020) assessed the oral absorption of alpelisib through a PBPK model, successfully simulating bioequivalence between clinical and commercial formulations under varying conditions. The predictive capacity of the model was validated with errors in plasma Cmax and AUC below 30%, making it a critical asset for formulation development. Moreover, studies involving the prediction of DDIs have highlighted the utility of PBPK modeling [40]. Freise et al. (2017) demonstrated that the pharmacokinetics of venetoclax could be significantly altered by CYP3A inhibitors and inducers, advising necessary dose adjustments to maintain therapeutic levels [61]. Similarly, Yamazaki et al. (2015) validated a PBPK model for crizotinib, affirming that results from single-dose studies could guide multiple-dose scenarios, thereby enhancing the efficiency of drug administration strategies [56]. Recent studies have extended these methodologies to pediatric populations, as illustrated by Morcos et al. (2023), who employed model-informed approaches to support dosing recommendations for copanlisib in children with relapsed/refractory solid tumors [68].
The exploration of virtual bioequivalence studies has gained considerable traction in the context of cardiovascular diseases, emerging as the second most researched area after cancer and tumor studies. For instance, Donovan et al. (2018) developed a PBPK model to predict the plasma and brain concentrations of bumetanide in different populations, highlighting the model’s limited applicability in neonates due to insufficient metabolic and transport parameter data [41]. Similarly, Dennison et al. (2017) assessed the bioavailability of fixed-dose combination orally disintegrating tablets containing amlodipine and atorvastatin. Their findings indicated no significant differences in bioavailability between the single and fixed-dose formulations, although atorvastatin showed increased exposure in fed subjects, suggesting the influence of gut transit dynamics [59]. Furthermore, Tsamandouras et al. (2015) created a population PBPK model for simvastatin, successfully predicting plasma concentrations and the effects of genetic polymorphisms on drug interactions and toxicity [70]. Rhee et al. (2018) focused on potential drug-drug interactions involving fimasartan, amlodipine, and hydrochlorothiazide, finding no significant interactions that were consistent with clinical observations [73]. Additionally, Chen et al. (2022) investigated salvianolic acid A using a PBPK model that uncovered challenges related to dose proportionality in pharmacokinetics, specifically saturation of transport mechanisms at higher doses [79]. Lastly, Kaur et al. (2020) examined the oral absorption of irbesartan, utilizing biorelevant dissolution testing and PBPK modeling to enhance the accuracy of plasma exposure predictions [80]. These studies collectively underscore the growing importance of virtual bioequivalence methodologies in cardiovascular pharmacology. They facilitate more precise dosing recommendations and improve understanding of drug behaviors across different populations and formulations.
Research into virtual bioequivalence for infectious diseases, including bacterial, fungal, and viral infections, has positioned itself as a significant area of study, ranking third after cardiovascular and cancer-related investigations. This field has benefitted from advanced in silico tools, particularly PBPK modeling, which has been pivotal in understanding drug interactions and optimizing dosing regimens for various antimicrobial agents. For example, Yee et al. (2020) utilized PBPK modeling to analyze doravirine, revealing that coadministration with rifabutin led to a substantial decrease in doravirine exposure. Their findings necessitated a dosing adjustment from 100 mg once daily to 100 mg twice daily to achieve the desired pharmacokinetic profile, confirming the accuracy of model predictions through subsequent clinical trials [33]. In the antifungal domain, Zane et al. (2014) developed a PBPK model for voriconazole, which initially overestimated oral bioavailability in pediatric patients but improved significantly upon accounting for intestinal first-pass metabolism. This highlights the need for tailored pharmacokinetic assessments in different age groups [36]. Similarly, Bergagnini-Kolev et al. (2023) examined inhaled itraconazole and its potential for drug-drug interactions, predicting minimal risk when co-administered with midazolam due to low systemic itraconazole exposure, underscoring the safety of this formulation for treating pulmonary fungal infections [43]. Additionally, Stader et al. (2021) focused on bictegravir, a drug used in HIV treatment, and found that aging did not significantly affect its pharmacokinetics, indicating that no dose adjustment is needed for elderly individuals without severe comorbidities [49]. Salerno et al. (2017) created a PBPK model for solithromycin, effectively predicting plasma and epithelial lining fluid concentrations, with simulations showing high efficacy in achieving target exposure ratios in patients with community-acquired bacterial pneumonia [71].
3.4. Regulatory Perspectives of Virtual Bioequivalence in the Pharmaceutical Industry
The rise of virtual bioequivalence studies in drug development, particularly concerning cardiovascular diseases, has been increasingly recognized by regulatory agencies such as FDA and EMA. As the pharmaceutical landscape evolves, these agencies have begun to incorporate advanced computational modeling and simulation techniques to enhance the evaluation of bioequivalence, ensuring the safety and efficacy of generic formulations. The FDA has developed a framework for the use of in silico methods in pharmacokinetic studies, particularly under its Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products — General Considerations. This guidance acknowledges the role of PBPK modeling in predicting the pharmacokinetics of drugs in various populations, including special populations such as pediatrics and the elderly. The FDA encourages the use of PBPK models to simulate drug ADME, which can significantly reduce the need for extensive clinical trials, particularly in cases where conducting such trials would be ethically or logistically challenging. For instance, in the studies highlighted in our systematic review, researchers like Dennison et al. (2017) and Kaur et al. (2020) employed PBPK modeling to predict bioavailability and oral absorption profiles for combination therapies and individual drugs, respectively [59,80]. Their findings align with FDA recommendations, demonstrating how in silico approaches can provide reliable data that inform dosing regimens and improve therapeutic outcomes without the need for exhaustive clinical testing. The EMA similarly recognizes the potential of virtual bioequivalence studies in its Guideline on the Investigation of Bioequivalence. The EMA encourages the use of modeling and simulation as part of the drug development process, particularly for complex formulations such as fixed-dose combinations and modified-release products. The agency emphasizes that robust PBPK models can facilitate a better understanding of the impact of physiological variability and drug-drug interactions on bioavailability. In the context of the studies reviewed, Rhee et al. (2018) successfully employed PBPK modeling to assess drug-drug interaction potentials for fimasartan, amlodipine, and hydrochlorothiazide [73]. This study exemplifies the EMA’s approach, as it combined literature data, in silico tools, and in vitro studies to build a validated model that accurately predicted pharmacokinetic outcomes in healthy subjects. Such predictive capabilities not only streamline the development process but also ensure compliance with regulatory standards. Moreover, the review points out the critical need for regulatory bodies to provide clear frameworks that support the integration of these advanced methodologies into standard bioequivalence assessments. As evidenced by the ongoing studies and their implications for clinical practice, establishing a robust regulatory framework that accommodates virtual bioequivalence can lead to more efficient drug development processes, reduced costs, and improved patient outcomes.
4. Discussion
4.1. Significance of the Systematic Review Results and Correlation with Other Studies
The integration of virtual bioequivalence methodologies, particularly through PBPK modeling, has significant implications for the pharmaceutical industry. By leveraging these advanced computational techniques, companies can streamline the drug development process, leading to reduced time and costs associated with bringing new medications to market [94,95,96]. One of the most notable impacts is the enhanced efficiency in predicting pharmacokinetic profiles. PBPK modeling allows for the simulation of drug ADME in diverse populations, including vulnerable groups such as children and the elderly [97]. This capability not only optimizes dosing regimens but also minimizes the need for extensive clinical trials, which can be logistically challenging and ethically complex. For example, the use of PBPK models has been shown to effectively predict bioavailability in complex drug formulations, facilitating faster regulatory approval and more timely patient access to therapies. The findings from this systematic review underscore the growing importance of virtual bioequivalence studies in the pharmaceutical industry, particularly in optimizing the drug development process. The systematic review highlights several key insights that correlate with existing literature and reflect the shifting paradigms in regulatory perspectives on bioequivalence. The review highlights that the incorporation of PBPK modeling and simulation techniques substantially improves the efficiency of drug development processes. For example, several research studies illustrate that PBPK models can reliably predict the pharmacokinetic profiles of drugs, enabling researchers to optimize dosing regimens without the need for extensive clinical trials [98,99]. This finding reinforces our review’s assertion that virtual bioequivalence can effectively streamline drug approval processes while ensuring adherence to safety and efficacy standards. Moreover, the alignment between our review’s findings and the evolving regulatory perspectives is significant. Both the FDA and EMA are increasingly acknowledging the value of in silico methods, as evidenced by the comprehensive frameworks they are developing for the application of PBPK modeling. For instance, several studies discuss the FDA’s proactive approach to facilitating the integration of computational modeling into drug evaluations, emphasizing its importance in assessing complex formulations [100,101]. This underscores our review’s call for robust regulatory guidelines to accommodate virtual bioequivalence methodologies, ultimately fostering innovation within the pharmaceutical landscape. Additionally, the integration of virtual bioequivalence with real-world evidence (RWE) is a significant development. A study advocates for combining RWE with computational modeling to validate bioequivalence claims further, thereby addressing the variability observed in clinical settings [102]. This perspective reinforces the importance of our review’s findings, as it suggests a holistic approach to drug development that incorporates various data sources to optimize therapeutic strategies.
4.2. Limitations of the Systematic Review
While this systematic review provides valuable insights, several limitations must be acknowledged. One key limitation is the heterogeneity among the included studies. Variability in study designs, populations, and methodologies can impact the comparability of results. For instance, differences in how PBPK modeling is implemented across studies may lead to inconsistencies in findings. This heterogeneity could restrict the generalizability of our conclusions and underscores the necessity for standardized protocols in virtual bioequivalence research. Another limitation pertains to the potential for publication bias. Studies that report successful outcomes for virtual bioequivalence are often more likely to be published, while those with negative or inconclusive results may remain unpublished. This bias can distort the overall understanding of the efficacy and reliability of virtual bioequivalence methodologies, a concern also highlighted in similar systematic reviews. Addressing publication bias requires greater transparency in research reporting, including the publication of negative results. Additionally, the search strategy employed in this systematic review may present limitations. Although comprehensive, it is possible that relevant studies were overlooked due to the inclusion criteria. For instance, studies published in languages other than English or less prominent journals may not have been captured, potentially omitting valuable data and perspectives in the field. Finally, the rapid evolution of regulatory frameworks concerning virtual bioequivalence poses another challenge. As the FDA and EMA continue to refine their guidelines, the findings of this review may need to be revisited and updated regularly. This dynamic regulatory landscape implies that the implications of this systematic review could change over time, emphasizing the importance of ongoing research and monitoring in this area.
5. Conclusions
In conclusion, this systematic review underscores the significant advancements in virtual bioequivalence, particularly through the adoption of computational methods such as PBPK modeling. These innovative approaches enhance the efficiency and reliability of drug development processes, allowing for more accurate predictions of pharmacokinetic profiles and optimized dosing regimens. The growing recognition of the value of in silico methods by regulatory agencies like the FDA and EMA further solidifies their importance in evaluating complex drug formulations while ensuring safety and efficacy standards. Moreover, the review highlights the critical need for robust regulatory guidelines to support the integration of virtual bioequivalence methodologies into standard practices. This support will facilitate innovation within the pharmaceutical landscape, ultimately improving patient outcomes and streamlining drug approval processes. While acknowledging the limitations of the current body of research, including study heterogeneity and potential publication bias, the findings point to a promising future for virtual bioequivalence. Continued efforts in standardization, transparency in research reporting, and adaptive regulatory frameworks will be essential to harness the full potential of these computational techniques in drug development.
Author Contributions
Conceptualization, N.A. and D.D.; Data curation, N.A.; Formal analysis, D.D.; Funding acquisition, N.A.; Investigation, D.D.; Methodology, N.A. and D.D.; Project administration, N.A.; Resources, N.A.; Software, D.D.; Supervision, N.A.; Validation, N.A. and D.D.; Visualization, D.D.; Writing—original draft, N.A. and D.D.; Writing—review and editing, N.A. and D.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported and funded by the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University (IMSIU) (grant number IMSIU-RG23154).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Material, and further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sowmya C, Abrar H, Prakaash K. Virtual Bioequivalence in Pharmaceuticals: Current Status and Future Prospects. International Journal of Applied Pharmaceutics. 2023:1-9. [CrossRef]
- Kollipara S, Martins FS, Jereb R, Krajcar D, Ahmed T. Advancing Virtual Bioequivalence for Orally Administered Drug Products: Methodology, Real-World Applications and Future Outlook. Pharmaceuticals. 2024;17(7):876. [CrossRef]
- Chow SC. Bioavailability and Bioequivalence in Drug Development. Wiley Interdiscip Rev Comput Stat. 2014;6(4):304-12. [CrossRef] [PubMed] [PubMed Central]
- Glerum PJ, Neef C, Burger DM, Yu Y, Maliepaard M. Pharmacokinetics and Generic Drug Switching: A Regulator’s View. Clin Pharmacokinet. 2020;59(9):1065-9. [CrossRef] [PubMed] [PubMed Central]
- Hirano M, Yamada M, Tanaka T, Koue T, Saito T, Higashimori M, et al. Surveys/Research Exploring Japanese Phase I Studies in Global Drug Development: Are They Necessary Prior to Joining Global Clinical Trials? Clin Pharmacol Drug Dev. 2021;10(12):1410-8. Epub 20211126. [CrossRef] [PubMed] [PubMed Central]
- Tian Y, Reichardt B, Dunkler D, Hronsky M, Winkelmayer WC, Bucsics A, et al. Comparative effectiveness of branded vs. generic versions of antihypertensive, lipid-lowering and hypoglycemic substances: a population-wide cohort study. Sci Rep. 2020;10(1):5964. Epub 20200406. [CrossRef] [PubMed] [PubMed Central]
- Kesselheim AS, Misono AS, Lee JL, Stedman MR, Brookhart MA, Choudhry NK, Shrank WH. Clinical equivalence of generic and brand-name drugs used in cardiovascular disease: a systematic review and meta-analysis. Jama. 2008;300(21):2514-26. [CrossRef] [PubMed] [PubMed Central]
- Papadopoulos D, Karalis VD. Introducing an Artificial Neural Network for Virtually Increasing the Sample Size of Bioequivalence Studies. Applied Sciences [Internet]. 2024; 14(7).
- Chen A, Yarmush ML, Maguire T. Physiologically based pharmacokinetic models: integration of in silico approaches with micro cell culture analogues. Curr Drug Metab. 2012;13(6):863-80. [CrossRef] [PubMed] [PubMed Central]
- Zhuang X, Lu C. PBPK modeling and simulation in drug research and development. Acta Pharm Sin B. 2016;6(5):430-40. Epub 20160623. [CrossRef] [PubMed] [PubMed Central]
- Lin L, Wong H. Predicting Oral Drug Absorption: Mini Review on Physiologically-Based Pharmacokinetic Models. Pharmaceutics. 2017;9(4). Epub 20170926. [CrossRef] [PubMed] [PubMed Central]
- Jones HM, Chen Y, Gibson C, Heimbach T, Parrott N, Peters SA, et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97(3):247-62. Epub 20150109. [CrossRef] [PubMed]
- Deepika D, Kumar V. The Role of “Physiologically Based Pharmacokinetic Model (PBPK)” New Approach Methodology (NAM) in Pharmaceuticals and Environmental Chemical Risk Assessment. Int J Environ Res Public Health. 2023;20(4). Epub 20230216. [CrossRef] [PubMed] [PubMed Central]
- Rowland M, Peck C, Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45-73. [CrossRef] [PubMed]
- Siebinga H, de Wit-van der Veen BJ, Stokkel MDM, Huitema ADR, Hendrikx J. Current use and future potential of (physiologically based) pharmacokinetic modelling of radiopharmaceuticals: a review. Theranostics. 2022;12(18):7804-20. Epub 20221114. [CrossRef] [PubMed] [PubMed Central]
- Mackie C, Arora S, Seo P, Moody R, Rege B, Pepin X, et al. Physiologically Based Biopharmaceutics Modeling (PBBM): Best Practices for Drug Product Quality, Regulatory and Industry Perspectives: 2023 Workshop Summary Report. Mol Pharm. 2024;21(5):2065-80. Epub 20240410. [CrossRef] [PubMed] [PubMed Central]
- Shebley M, Sandhu P, Emami Riedmaier A, Jamei M, Narayanan R, Patel A, et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin Pharmacol Ther. 2018;104(1):88-110. Epub 20180202. [CrossRef] [PubMed] [PubMed Central]
- Sun Z, Zhao N, Zhao X, Wang Z, Liu Z, Cui Y. Application of physiologically based pharmacokinetic modeling of novel drugs approved by the U.S. food and drug administration. European Journal of Pharmaceutical Sciences. 2024;200:106838. [CrossRef]
- Manolis E, García-Arieta A, Lindahl A, Kotzagiorgis E, Limberg J, Holte Ø, et al. Using mechanistic models to support development of complex generic drug products: European Medicines Agency perspective. CPT Pharmacometrics Syst Pharmacol. 2023;12(5):556-9. Epub 20230111. [CrossRef] [PubMed] [PubMed Central]
- Tsakalozou E, Alam K, Babiskin A, Zhao L. Physiologically-Based Pharmacokinetic Modeling to Support Determination of Bioequivalence for Dermatological Drug Products: Scientific and Regulatory Considerations. Clinical Pharmacology & Therapeutics. 2021;111. [CrossRef]
- Lee M, Ly H, Möller CC, Ringel MS. Innovation in Regulatory Science Is Meeting Evolution of Clinical Evidence Generation. Clin Pharmacol Ther. 2019;105(4):886-98. [CrossRef] [PubMed] [PubMed Central]
- Marques L, Costa B, Pereira M, Silva A, Santos J, Saldanha L, et al. Advancing Precision Medicine: A Review of Innovative In Silico Approaches for Drug Development, Clinical Pharmacology and Personalized Healthcare. Pharmaceutics. 2024;16(3):332. [CrossRef]
- Alomari N, Alhussaini W. Update on the advances and challenges in bioequivalence testing methods for complex topical generic products. Front Pharmacol. 2024;15:1330712. Epub 20240208. [CrossRef] [PubMed] [PubMed Central]
- Jiang J, Ma X, Ouyang D, Williams RO, 3rd. Emerging Artificial Intelligence (AI) Technologies Used in the Development of Solid Dosage Forms. Pharmaceutics. 2022;14(11). Epub 20221022. [CrossRef] [PubMed] [PubMed Central]
- Kapustina O, Burmakina P, Gubina N, Serov N, Vinogradov V. User-friendly and industry-integrated AI for medicinal chemists and pharmaceuticals. Artificial Intelligence Chemistry. 2024;2(2):100072.
- Yu Y, Loi CM, Hoffman J, Wang D. Physiologically Based Pharmacokinetic Modeling of Palbociclib. J Clin Pharmacol. 2017;57(2):173-84. Epub 20160822. [CrossRef] [PubMed]
- Cho CK, Kang P, Park HJ, Lee YJ, Bae JW, Jang CG, Lee SY. Physiologically based pharmacokinetic (PBPK) modelling of tamsulosin related to CYP2D6*10 allele. Arch Pharm Res. 2021;44(11):1037-49. Epub 20211109. [CrossRef] [PubMed]
- Chen G, Sun K, Michon I, Barter Z, Neuhoff S, Ghosh L, et al. Physiologically Based Pharmacokinetic Modeling for Maribavir to Inform Dosing in Drug-Drug Interaction Scenarios with CYP3A4 Inducers and Inhibitors. J Clin Pharmacol. 2024;64(5):590-600. Epub 20231214. [CrossRef] [PubMed]
- Kim YH, Kang P, Cho CK, Jung EH, Park HJ, Lee YJ, et al. Physiologically based pharmacokinetic (PBPK) modeling for prediction of celecoxib pharmacokinetics according to CYP2C9 genetic polymorphism. Arch Pharm Res. 2021;44(7):713-24. Epub 20210725. [CrossRef] [PubMed]
- Fendt R, Hofmann U, Schneider ARP, Schaeffeler E, Burghaus R, Yilmaz A, et al. Data-driven personalization of a physiologically based pharmacokinetic model for caffeine: A systematic assessment. CPT Pharmacometrics Syst Pharmacol. 2021;10(7):782-93. Epub 20210626. [CrossRef] [PubMed] [PubMed Central]
- Watanabe A, Ishizuka T, Yamada M, Igawa Y, Shimizu T, Ishizuka H. Physiologically based pharmacokinetic modelling to predict the clinical effect of CYP3A inhibitors/inducers on esaxerenone pharmacokinetics in healthy subjects and subjects with hepatic impairment. Eur J Clin Pharmacol. 2022;78(1):65-73. Epub 20210820. [CrossRef] [PubMed] [PubMed Central]
- Ou Y, Xu Y, Gore L, Harvey RD, Mita A, Papadopoulos KP, et al. Physiologically-based pharmacokinetic modelling to predict oprozomib CYP3A drug-drug interaction potential in patients with advanced malignancies. Br J Clin Pharmacol. 2019;85(3):530-9. Epub 20181225. [CrossRef] [PubMed] [PubMed Central]
- Yee KL, Cabalu TD, Kuo Y, Fillgrove KL, Liu Y, Triantafyllou I, et al. Physiologically Based Pharmacokinetic Modeling of Doravirine and Its Major Metabolite to Support Dose Adjustment With Rifabutin. J Clin Pharmacol. 2021;61(3):394-405. Epub 20200928. [CrossRef] [PubMed]
- Jo H, Pilla Reddy V, Parkinson J, Boulton DW, Tang W. Model-Informed Pediatric Dose Selection for Dapagliflozin by Incorporating Developmental Changes. CPT Pharmacometrics Syst Pharmacol. 2021;10(2):108-18. Epub 20210113. [CrossRef] [PubMed] [PubMed Central]
- Posada MM, Cannady EA, Payne CD, Zhang X, Bacon JA, Pak YA, et al. Prediction of Transporter-Mediated Drug-Drug Interactions for Baricitinib. Clin Transl Sci. 2017;10(6):509-19. Epub 20170727. [CrossRef] [PubMed] [PubMed Central]
- Zane NR, Thakker DR. A physiologically based pharmacokinetic model for voriconazole disposition predicts intestinal first-pass metabolism in children. Clin Pharmacokinet. 2014;53(12):1171-82. [CrossRef] [PubMed]
- Lang J, Vincent L, Chenel M, Ogungbenro K, Galetin A. Simultaneous Ivabradine Parent-Metabolite PBPK/PD Modelling Using a Bayesian Estimation Method. Aaps j. 2020;22(6):129. Epub 20201008. [CrossRef] [PubMed]
- Hanke N, Teifel M, Moj D, Wojtyniak JG, Britz H, Aicher B, et al. A physiologically based pharmacokinetic (PBPK) parent-metabolite model of the chemotherapeutic zoptarelin doxorubicin-integration of in vitro results, Phase I and Phase II data and model application for drug-drug interaction potential analysis. Cancer Chemother Pharmacol. 2018;81(2):291-304. Epub 20171204. [CrossRef] [PubMed]
- Chen Y, Zhou D, Tang W, Zhou W, Al-Huniti N, Masson E. Physiologically Based Pharmacokinetic Modeling to Evaluate the Systemic Exposure of Gefitinib in CYP2D6 Ultrarapid Metabolizers and Extensive Metabolizers. J Clin Pharmacol. 2018;58(4):485-93. Epub 20171128. [CrossRef] [PubMed]
- Gajewska M, Blumenstein L, Kourentas A, Mueller-Zsigmondy M, Lorenzo S, Sinn A, et al. Physiologically Based Pharmacokinetic Modeling of Oral Absorption, pH, and Food Effect in Healthy Volunteers to Drive Alpelisib Formulation Selection. Aaps j. 2020;22(6):134. Epub 20201018. [CrossRef] [PubMed]
- Donovan MD, Abduljalil K, Cryan JF, Boylan GB, Griffin BT. Application of a physiologically-based pharmacokinetic model for the prediction of bumetanide plasma and brain concentrations in the neonate. Biopharm Drug Dispos. 2018;39(3):125-34. Epub 20180222. [CrossRef] [PubMed]
- Fu Q, Jones HM, Sun G, Atamas SP. A Physiologically Based Pharmacokinetic and Drug-Drug Interaction Model for the CB2 Agonist Lenabasum. Eur J Drug Metab Pharmacokinet. 2021;46(4):513-25. Epub 20210618. [CrossRef] [PubMed]
- Bergagnini-Kolev M, Kane K, Templeton IE, Curran AK. Evaluation of the Potential for Drug-Drug Interactions with Inhaled Itraconazole Using Physiologically Based Pharmacokinetic Modelling, Based on Phase 1 Clinical Data. Aaps j. 2023;25(4):62. Epub 20230621. [CrossRef] [PubMed]
- Nakamaru Y, Emoto C, Shimizu M, Yamazaki H. Human pharmacokinetic profiling of the dipeptidyl peptidase-IV inhibitor teneligliptin using physiologically based pharmacokinetic modeling. Biopharm Drug Dispos. 2015;36(3):148-62. Epub 20150128. [CrossRef] [PubMed]
- Thompson EJ, Jeong A, Helfer VE, Shakhnovich V, Edginton A, Balevic SJ, et al. Physiologically-based pharmacokinetic modeling of pantoprazole to evaluate the role of CYP2C19 genetic variation and obesity in the pediatric population. CPT Pharmacometrics Syst Pharmacol. 2024;13(8):1394-408. Epub 20240604. [CrossRef] [PubMed] [PubMed Central]
- Wang HY, Chen X, Jiang J, Shi J, Hu P. Evaluating a physiologically based pharmacokinetic model for predicting the pharmacokinetics of midazolam in Chinese after oral administration. Acta Pharmacol Sin. 2016;37(2):276-84. Epub 20151123. [CrossRef] [PubMed] [PubMed Central]
- Callegari E, Lin J, Tse S, Goosen TC, Sahasrabudhe V. Physiologically-Based Pharmacokinetic Modeling of the Drug-Drug Interaction of the UGT Substrate Ertugliflozin Following Co-Administration with the UGT Inhibitor Mefenamic Acid. CPT Pharmacometrics Syst Pharmacol. 2021;10(2):127-36. Epub 20201230. [CrossRef] [PubMed] [PubMed Central]
- Nakamura T, Toshimoto K, Lee W, Imamura CK, Tanigawara Y, Sugiyama Y. Application of PBPK Modeling and Virtual Clinical Study Approaches to Predict the Outcomes of CYP2D6 Genotype-Guided Dosing of Tamoxifen. CPT Pharmacometrics Syst Pharmacol. 2018;7(7):474-82. Epub 20180619. [CrossRef] [PubMed] [PubMed Central]
- Stader F, Courlet P, Decosterd LA, Battegay M, Marzolini C. Physiologically-Based Pharmacokinetic Modeling Combined with Swiss HIV Cohort Study Data Supports No Dose Adjustment of Bictegravir in Elderly Individuals Living With HIV. Clin Pharmacol Ther. 2021;109(4):1025-9. Epub 20210227. [CrossRef] [PubMed] [PubMed Central]
- Yang R, Lin Y, Chen K, Huang J, Yang S, Yao A, et al. Establishing Virtual Bioequivalence and Clinically Relevant Specifications for Omeprazole Enteric-Coated Capsules by Incorporating Dissolution Data in PBPK Modeling. Aaps j. 2024;26(4):82. Epub 20240712. [CrossRef] [PubMed]
- Ojala K, Schilderink R, Nykänen P, van Veen B, Malmström C, Juppo A, Korjamo T. Predicting the effect of prandial stage and particle size on absorption of ODM-204. Eur J Pharm Biopharm. 2020;156:75-83. Epub 20200819. [CrossRef] [PubMed]
- Agyemang A, Farrell C, Moore W, Parkin J. A Physiologically Based Pharmacokinetic Model to Predict Potential Drug-Drug Interactions and Inform Dosing of Acumapimod, an Oral p38 MAPK Inhibitor. CPT Pharmacometrics Syst Pharmacol. 2021;10(1):30-9. Epub 20201230. [CrossRef] [PubMed] [PubMed Central]
- Chen Y, Mao J, Hop CE. Physiologically based pharmacokinetic modeling to predict drug-drug interactions involving inhibitory metabolite: a case study of amiodarone. Drug Metab Dispos. 2015;43(2):182-9. Epub 20141016. [CrossRef] [PubMed]
- Litou C, Patel N, Turner DB, Kostewicz E, Kuentz M, Box KJ, Dressman J. Combining biorelevant in vitro and in silico tools to simulate and better understand the in vivo performance of a nano-sized formulation of aprepitant in the fasted and fed states. Eur J Pharm Sci. 2019;138:105031. Epub 20190803. [CrossRef] [PubMed]
- Einolf HJ, Zhou J, Won C, Wang L, Rebello S. A Physiologically-Based Pharmacokinetic Modeling Approach To Predict Drug-Drug Interactions of Sonidegib (LDE225) with Perpetrators of CYP3A in Cancer Patients. Drug Metab Dispos. 2017;45(4):361-74. Epub 20170125. [CrossRef] [PubMed]
- Yamazaki S, Johnson TR, Smith BJ. Prediction of Drug-Drug Interactions with Crizotinib as the CYP3A Substrate Using a Physiologically Based Pharmacokinetic Model. Drug Metab Dispos. 2015;43(10):1417-29. Epub 20150715. [CrossRef] [PubMed]
- Riddell K, Patel A, Collins G, Zhou Y, Schramek D, Kremer BE, Ferron-Brady G. An Adaptive Physiologically Based Pharmacokinetic-Driven Design to Investigate the Effect of Itraconazole and Rifampicin on the Pharmacokinetics of Molibresib (GSK525762) in Healthy Female Volunteers. J Clin Pharmacol. 2021;61(1):125-37. Epub 20200820. [CrossRef] [PubMed] [PubMed Central]
- Purohit V, Sagawa K, Hsu HJ, Kushner J, Dowty ME, Tse S, et al. Integrating Clinical Variability into PBPK Models for Virtual Bioequivalence of Single and Multiple Doses of Tofacitinib Modified-Release Dosage Form. Clin Pharmacol Ther. 2024;116(4):996-1004. Epub 20240526. [CrossRef] [PubMed]
- Dennison TJ, Smith JC, Badhan RK, Mohammed AR. Fixed-dose combination orally disintegrating tablets to treat cardiovascular disease: formulation, in vitro characterization and physiologically based pharmacokinetic modeling to assess bioavailability. Drug Des Devel Ther. 2017;11:811-26. Epub 20170316. [CrossRef] [PubMed] [PubMed Central]
- Knöchel J, Panduga V, Nelander K, Heijer M, Lindstedt EL, Ali H, et al. A drug-drug interaction study and physiologically based pharmacokinetic modelling to assess the effect of an oral 5-lipoxygenase activating protein inhibitor on the pharmacokinetics of oral midazolam. Br J Clin Pharmacol. 2024;90(9):2180-7. Epub 20240603. [CrossRef] [PubMed]
- Freise KJ, Shebley M, Salem AH. Quantitative Prediction of the Effect of CYP3A Inhibitors and Inducers on Venetoclax Pharmacokinetics Using a Physiologically Based Pharmacokinetic Model. J Clin Pharmacol. 2017;57(6):796-804. Epub 20170104. [CrossRef] [PubMed]
- Boetsch C, Parrott N, Fowler S, Poirier A, Hainzl D, Banken L, et al. Effects of Cytochrome P450 3A4 Inhibitors-Ketoconazole and Erythromycin-on Bitopertin Pharmacokinetics and Comparison with Physiologically Based Modelling Predictions. Clin Pharmacokinet. 2016;55(2):237-47. [CrossRef] [PubMed]
- Katsube T, Inoue Y, Fukuhara T, Kano T, Wajima T. Evaluation of drug-drug interaction of lusutrombopag, a thrombopoietin receptor agonist, via metabolic enzymes and transporters. Eur J Clin Pharmacol. 2020;76(12):1659-65. Epub 20200714. [CrossRef] [PubMed] [PubMed Central]
- Andreas CJ, Pepin X, Markopoulos C, Vertzoni M, Reppas C, Dressman JB. Mechanistic investigation of the negative food effect of modified release zolpidem. Eur J Pharm Sci. 2017;102:284-98. Epub 20170310. [CrossRef] [PubMed]
- Post TM, Gerrits M, Kerbusch T, de Greef R. Prediction of nomegestrol acetate pharmacokinetics in healthy female adolescents and adults by whole-body physiology-based pharmacokinetic modelling and clinical validation. Contraception. 2016;93(2):133-8. Epub 20150910. [CrossRef] [PubMed]
- Li J, Chen J, Kanamaluru V, Gaemers SJM, Peterschmitt MJ, Hou AW, et al. Impact of hepatic and renal impairment on the pharmacokinetics and tolerability of eliglustat therapy for Gaucher disease type 1. Mol Genet Metab. 2020;129(2):117-24. Epub 20191114. [CrossRef] [PubMed]
- Samant TS, Dhuria S, Lu Y, Laisney M, Yang S, Grandeury A, et al. Ribociclib Bioavailability Is Not Affected by Gastric pH Changes or Food Intake: In Silico and Clinical Evaluations. Clin Pharmacol Ther. 2018;104(2):374-83. Epub 20171208. [CrossRef] [PubMed] [PubMed Central]
- Morcos PN, Schlender J, Burghaus R, Moss J, Lloyd A, Childs BH, et al. Model-informed approach to support pediatric dosing for the pan-PI3K inhibitor copanlisib in children and adolescents with relapsed/refractory solid tumors. Clin Transl Sci. 2023;16(7):1197-209. Epub 20230418. [CrossRef] [PubMed] [PubMed Central]
- Traver E, Rodríguez-Pascau L, Meya U, Pina G, Pascual S, Poli S, et al. Clinical pharmacokinetics of leriglitazone and a translational approach using PBPK modeling to guide the selection of the starting dose in children. CPT Pharmacometrics Syst Pharmacol. 2024;13(6):982-93. Epub 20240329. [CrossRef] [PubMed] [PubMed Central]
- Tsamandouras N, Dickinson G, Guo Y, Hall S, Rostami-Hodjegan A, Galetin A, Aarons L. Development and Application of a Mechanistic Pharmacokinetic Model for Simvastatin and its Active Metabolite Simvastatin Acid Using an Integrated Population PBPK Approach. Pharm Res. 2015;32(6):1864-83. Epub 20141202. [CrossRef] [PubMed]
- Salerno SN, Edginton A, Cohen-Wolkowiez M, Hornik CP, Watt KM, Jamieson BD, Gonzalez D. Development of an Adult Physiologically Based Pharmacokinetic Model of Solithromycin in Plasma and Epithelial Lining Fluid. CPT Pharmacometrics Syst Pharmacol. 2017;6(12):814-22. Epub 20171025. [CrossRef] [PubMed] [PubMed Central]
- C. Venuto TS, B. Fiske, C. Coffey, H. Matthews, R. Wyse, P. Brundin, D. Simon, M. Schwarzschild, D. Weiner, J. Adams, L. Trusso, M. Kostrzebski, T. Ward, G. Rafaloff, K. Merchant. Physiologically-based pharmacokinetic modeling of nilotinib to determine serum, cerebrospinal fluid, and brain exposures. Mov Disord. 2020;35.
- Rhee SJ, Lee HA, Lee S, Kim E, Jeon I, Song IS, Yu KS. Physiologically Based Pharmacokinetic Modeling of Fimasartan, Amlodipine, and Hydrochlorothiazide for the Investigation of Drug-Drug Interaction Potentials. Pharm Res. 2018;35(12):236. Epub 20181015. [CrossRef] [PubMed]
- Hwang S, Lee Y, Jang Y, Cho JY, Yoon S, Chung JY. Comprehensive Evaluation of OATP- and BCRP-Mediated Drug-Drug Interactions of Methotrexate Using Physiologically-Based Pharmacokinetic Modeling. Clin Pharmacol Ther. 2024;116(4):1013-22. Epub 20240611. [CrossRef] [PubMed]
- Samant TS, Huth F, Umehara K, Schiller H, Dhuria SV, Elmeliegy M, et al. Ribociclib Drug-Drug Interactions: Clinical Evaluations and Physiologically-Based Pharmacokinetic Modeling to Guide Drug Labeling. Clin Pharmacol Ther. 2020;108(3):575-85. Epub 20200725. [CrossRef] [PubMed]
- Chen B, Zhou D, Wei H, Yotvat M, Zhou L, Cheung J, et al. Acalabrutinib CYP3A-mediated drug-drug interactions: Clinical evaluations and physiologically based pharmacokinetic modelling to inform dose adjustment strategy. Br J Clin Pharmacol. 2022;88(8):3716-29. Epub 20220328. [CrossRef] [PubMed]
- Djebli N, Fabre D, Boulenc X, Fabre G, Sultan E, Hurbin F. Physiologically based pharmacokinetic modeling for sequential metabolism: effect of CYP2C19 genetic polymorphism on clopidogrel and clopidogrel active metabolite pharmacokinetics. Drug Metab Dispos. 2015;43(4):510-22. Epub 20150121. [CrossRef] [PubMed]
- Xiao Q, Tang L, Xu R, Qian W, Yang J. Physiologically based pharmacokinetics model predicts the lack of inhibition by repaglinide on the metabolism of pioglitazone. Biopharm Drug Dispos. 2015;36(9):603-12. [CrossRef] [PubMed]
- Chen J, Ruan Z, Lou H, Yang D, Shao R, Xu Y, et al. First-in-human study to investigate the safety and pharmacokinetics of salvianolic acid A and pharmacokinetic simulation using a physiologically based pharmacokinetic model. Front Pharmacol. 2022;13:907208. Epub 20221104. [CrossRef] [PubMed] [PubMed Central]
- Kaur N, Thakur PS, Shete G, Gangwal R, Sangamwar AT, Bansal AK. Understanding the Oral Absorption of Irbesartan Using Biorelevant Dissolution Testing and PBPK Modeling. AAPS PharmSciTech. 2020;21(3):102. Epub 20200309. [CrossRef] [PubMed]
- Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami-Hodjegan A. The Simcyp population-based ADME simulator. Expert opinion on drug metabolism & toxicology. 2009;5:211-23. [CrossRef]
- Aithal P A, Aithal S, Aithal S. Case Study on Certara’s Simcyp PBPK Simulator to Eliminate Lengthy Clinical Trials. 2022;6:69-109. [CrossRef]
- Gufford BT, Barr JT, González-Pérez V, Layton ME, White JR, Jr., Oberlies NH, Paine MF. Quantitative prediction and clinical evaluation of an unexplored herb-drug interaction mechanism in healthy volunteers. CPT Pharmacometrics Syst Pharmacol. 2015;4(12):701-10. Epub 20151128. [CrossRef] [PubMed] [PubMed Central]
- Demeester C, Robins D, Edwina AE, Tournoy J, Augustijns P, Ince I, et al. Physiologically based pharmacokinetic (PBPK) modelling of oral drug absorption in older adults – an AGePOP review. European Journal of Pharmaceutical Sciences. 2023;188:106496. [CrossRef]
- Jamei M, Turner D, Yang J, Neuhoff S, Polak S, Rostami-Hodjegan A, Tucker G. Population-based mechanistic prediction of oral drug absorption. Aaps j. 2009;11(2):225-37. Epub 20090421. [CrossRef] [PubMed] [PubMed Central]
- Cristofoletti R, Dressman J. Use of Physiologically Based Pharmacokinetic Models Coupled with Pharmacodynamic Models to Assess the Clinical Relevance of Current Bioequivalence Criteria for Generic Drug Products Containing Ibuprofen. Journal of Pharmaceutical Sciences. 2014;103. [CrossRef]
- Kollipara S, Martins FS, Jereb R, Krajcar D, Ahmed T. Advancing Virtual Bioequivalence for Orally Administered Drug Products: Methodology, Real-World Applications and Future Outlook. Pharmaceuticals [Internet]. 2024; 17(7).
- Darwich AS, Ogungbenro K, Vinks AA, Powell JR, Reny JL, Marsousi N, et al. Why has model-informed precision dosing not yet become common clinical reality? lessons from the past and a roadmap for the future. Clin Pharmacol Ther. 2017;101(5):646-56. Epub 20170404. [CrossRef] [PubMed]
- Sager JE, Yu J, Ragueneau-Majlessi I, Isoherranen N. Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation Approaches: A Systematic Review of Published Models, Applications, and Model Verification. Drug Metab Dispos. 2015;43(11):1823-37. Epub 20150821. [CrossRef] [PubMed] [PubMed Central]
- Willmann S, Höhn K, Edginton A, Sevestre M, Solodenko J, Weiss W, et al. Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn. 2007;34(3):401-31. Epub 20070313. [CrossRef] [PubMed]
- Schmitt W, Willmann S. Physiology-based pharmacokinetic modeling: ready to be used. Drug Discovery Today: Technologies. 2004;1(4):449-56.
- Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50 Suppl 1:S41-67. [CrossRef] [PubMed]
- Bolger MB, Lukacova V, Woltosz WS. Simulations of the nonlinear dose dependence for substrates of influx and efflux transporters in the human intestine. Aaps j. 2009;11(2):353-63. Epub 20090512. [CrossRef] [PubMed] [PubMed Central]
- Loisios-Konstantinidis I, Cristofoletti R, Fotaki N, Turner D, Dressman J. Establishing virtual bioequivalence and clinically relevant specifications using in vitro biorelevant dissolution testing and physiologically-based population pharmacokinetic modeling. Case example: Naproxen. European Journal of Pharmaceutical Sciences. 2019;143:105170. [CrossRef]
- Dermawan D, Bahtiar R, Sofian FF. Implementation of Green Supply Chain Management (GSCM) in the pharmaceutical industry in Indonesia: feasibility analysis and case studies. J Ilm Farm. 2019;15:23-9.
- Pepin XJH, Parrott N, Dressman J, Delvadia P, Mitra A, Zhang X, et al. Current State and Future Expectations of Translational Modeling Strategies to Support Drug Product Development, Manufacturing Changes and Controls: A Workshop Summary Report. Journal of Pharmaceutical Sciences. 2021;110(2):555-66.
- Darwich AS, Ogungbenro K, Hatley OJ, Rostami-Hodjegan A. Role of pharmacokinetic modeling and simulation in precision dosing of anticancer drugs. Translational Cancer Research. 2017:S1512-S29.
- Abla N, Howgate E, Rowland-Yeo K, Dickins M, Bergagnini-Kolev MC, Chen KF, et al. Development and application of a PBPK modeling strategy to support antimalarial drug development. CPT Pharmacometrics Syst Pharmacol. 2023;12(9):1335-46. Epub 20230816. [CrossRef] [PubMed] [PubMed Central]
- Khalil F, Läer S. Physiologically Based Pharmacokinetic Modeling: Methodology, Applications, and Limitations with a Focus on Its Role in Pediatric Drug Development. BioMed Research International. 2011;2011(1):907461. [CrossRef]
- Vora LK, Gholap AD, Jetha K, Thakur RRS, Solanki HK, Chavda VP. Artificial Intelligence in Pharmaceutical Technology and Drug Delivery Design. Pharmaceutics. 2023;15(7). Epub 20230710. [CrossRef] [PubMed] [PubMed Central]
- Yadav S, Singh A, Singhal R, Yadav JP. Revolutionizing drug discovery: The impact of artificial intelligence on advancements in pharmacology and the pharmaceutical industry. Intelligent Pharmacy. 2024;2(3):367-80.
- Dagenais S, Russo L, Madsen A, Webster J, Becnel L. Use of Real-World Evidence to Drive Drug Development Strategy and Inform Clinical Trial Design. Clin Pharmacol Ther. 2022;111(1):77-89. Epub 20211128. [CrossRef] [PubMed] [PubMed Central]
Figure 1.
PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) flow diagram illustrating the study selection process, from identification through inclusion, outlining the number of records screened, excluded, and included in the final scoping review.
Figure 1.
PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) flow diagram illustrating the study selection process, from identification through inclusion, outlining the number of records screened, excluded, and included in the final scoping review.
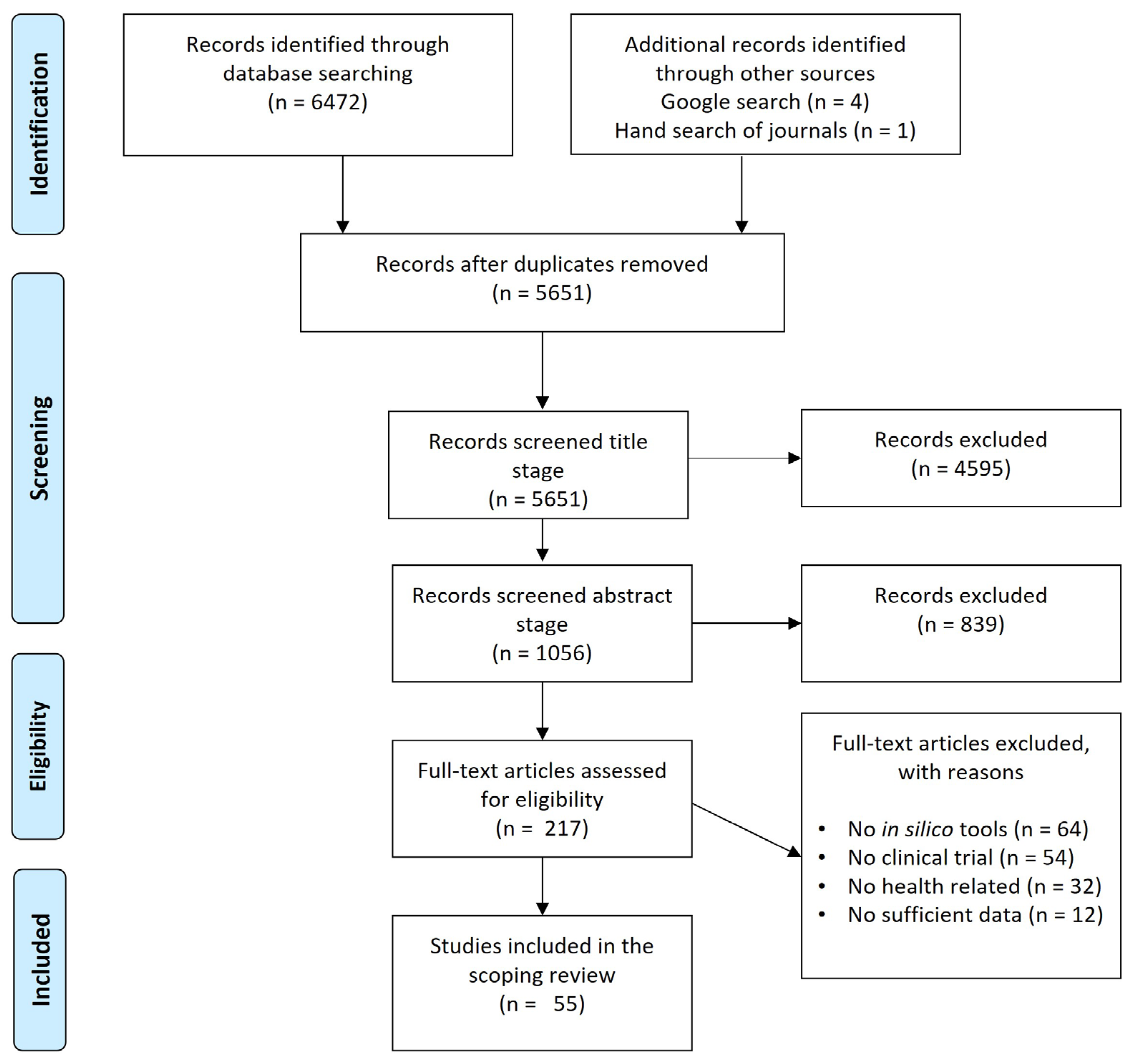
Figure 2.
Pie chart depicting the distribution of in silico modeling tools used in virtual bioequivalence studies, as identified in the systematic review. The chart highlights the percentage utilization of each tool, including SimCYP®, PK-Sim®, GastroPlus™, and MATLAB®.
Figure 2.
Pie chart depicting the distribution of in silico modeling tools used in virtual bioequivalence studies, as identified in the systematic review. The chart highlights the percentage utilization of each tool, including SimCYP®, PK-Sim®, GastroPlus™, and MATLAB®.
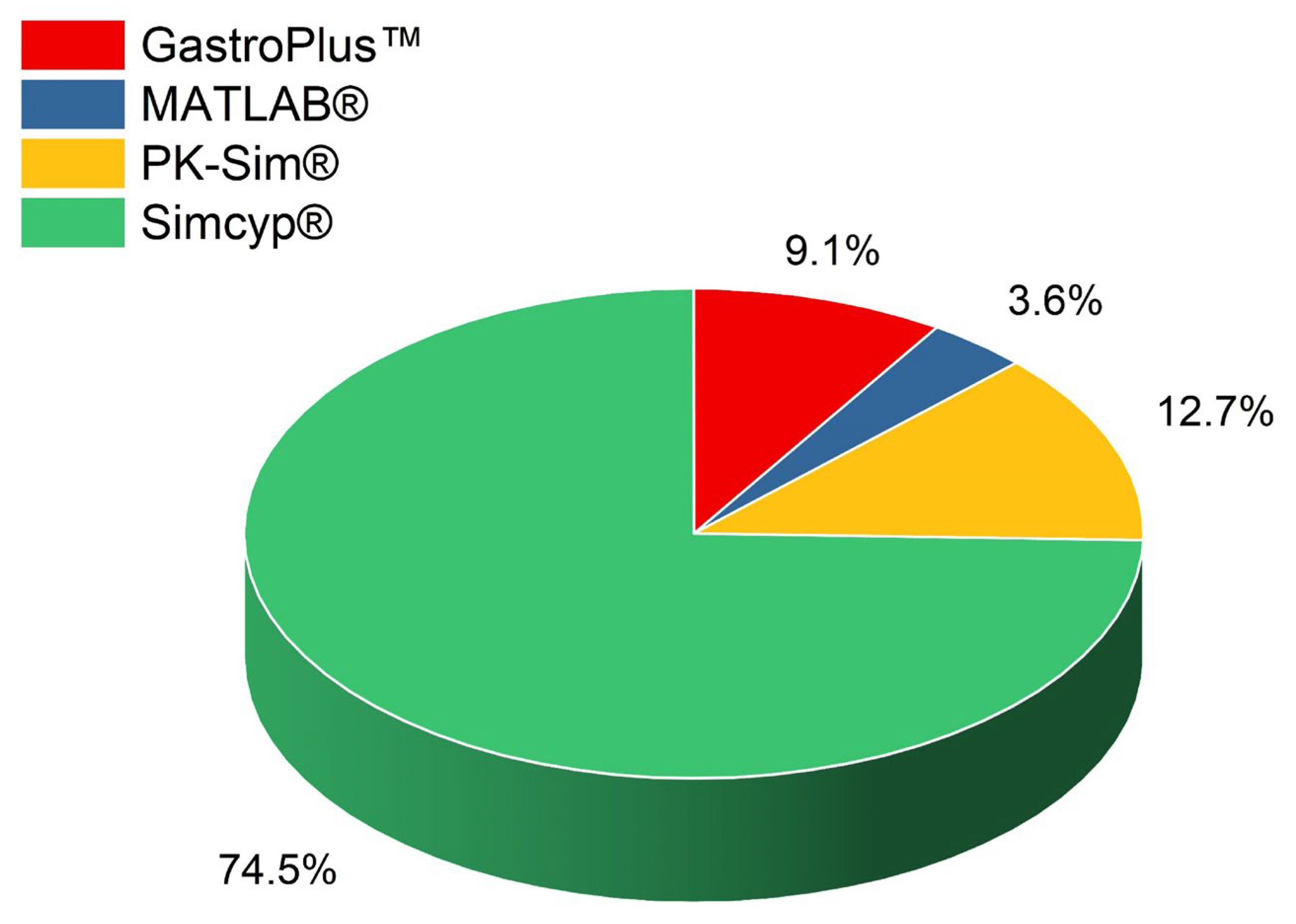
Figure 3.
Prevalence of disease types in virtual bioequivalence research (2014-2024). This figure illustrates the distribution of various disease types investigated in studies utilizing virtual bioequivalence over the past decade.
Figure 3.
Prevalence of disease types in virtual bioequivalence research (2014-2024). This figure illustrates the distribution of various disease types investigated in studies utilizing virtual bioequivalence over the past decade.
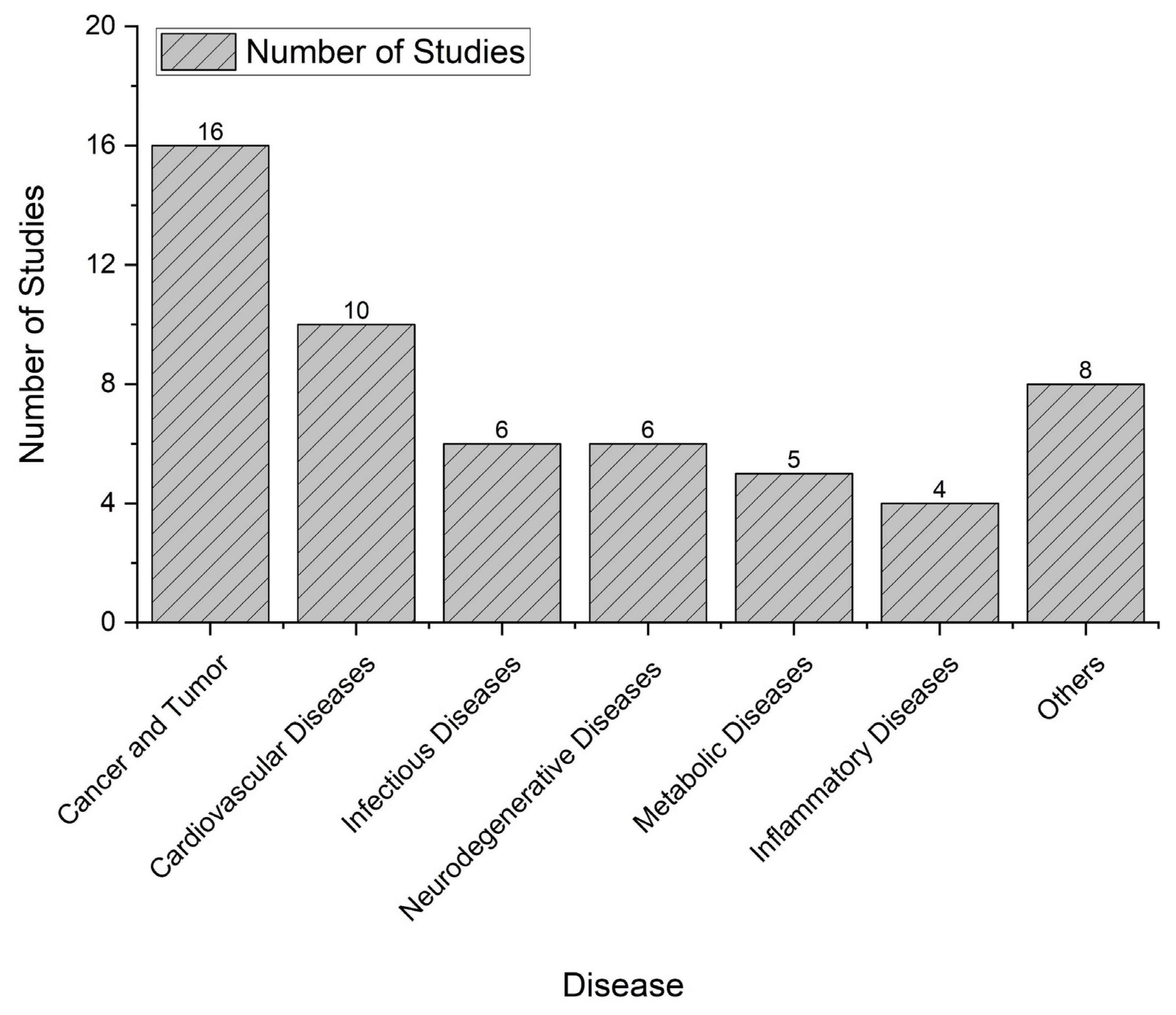
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



