
Submitted:
29 October 2024
Posted:
30 October 2024
You are already at the latest version
Abstract
Over the past few decades, the landscape for multiple myeloma (MM) therapy has significantly advanced, largely due to the approval and introduction of new-generation proteasome inhibitors (PIs) and immunomodulatory drugs (IMiDs). Despite these advancements, MM remains incurable. In March 2021, the U.S. FDA approved the chimeric antigen receptor T-cell (CAR-T) therapy idecabtagene vicleucel (ide-cel) for relapsed/refractory multiple myeloma (R/R MM), heralding the advent of cellular therapies for R/R MM. However, due to factors such as the downregulation or loss of tumor antigen expression, T-cell exhaustion, and the influence of the tumor immune microenvironment, most R/R MM patients inevitably experience relapse following CAR-T cell therapy. Consequently, salvage therapy in the post-CAR-T setting has emerged as a critical area of research. This review discusses the potential factors leading to CAR-T therapy failure in R/R MM patients and discusses subsequent salvage therapeutic strategies, offering recommendations for addressing treatment failure in this context.
Keywords:
Multiple Myeloma
; Chimeric Antigen Receptor T-cells
; Cellular Therapy Failure
; Salvage Therapy
Visual Abstract
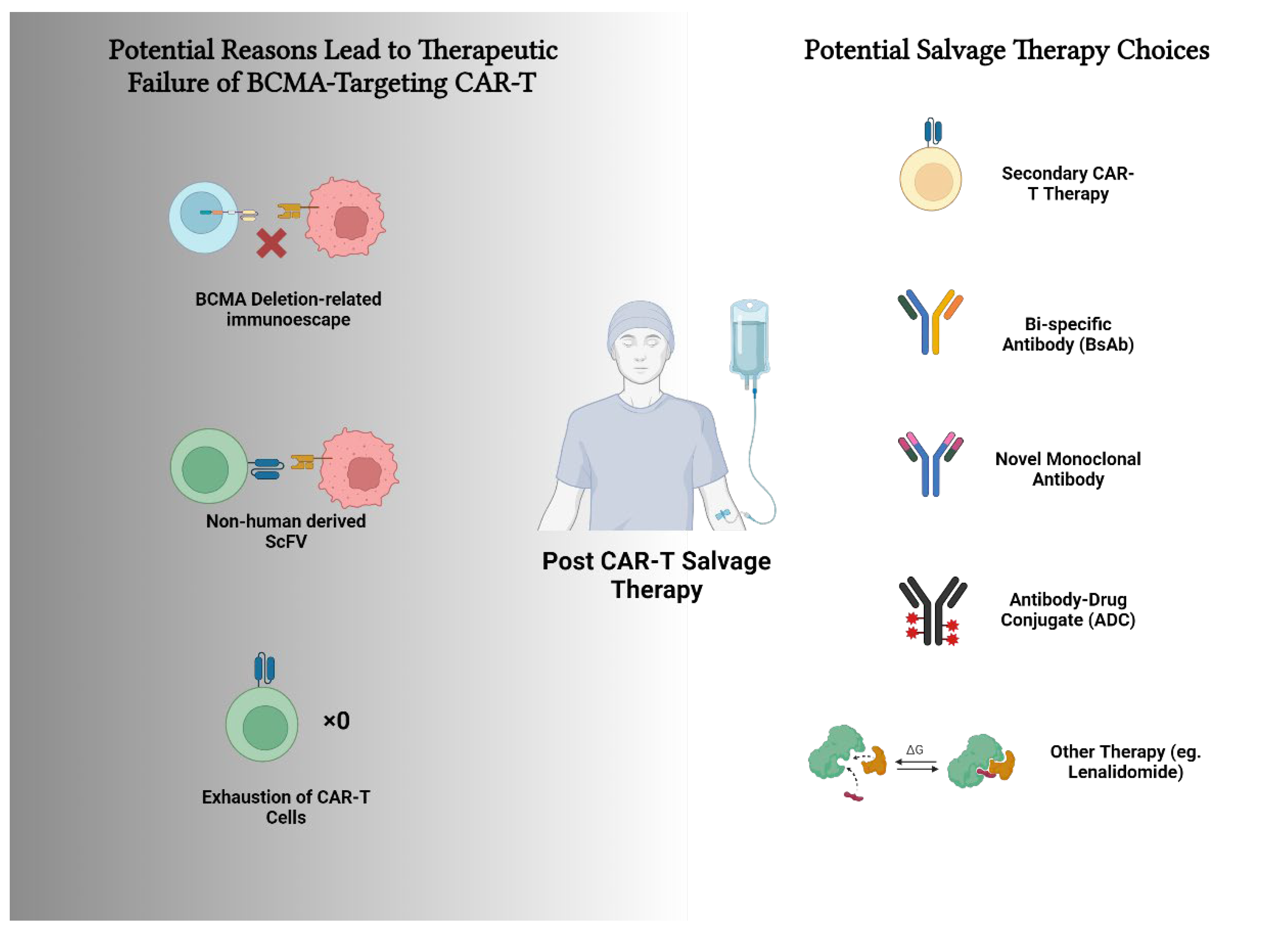
Multiple factors could resulted in the therapeutic failure of BCMA CAR-T therapy, while various choices for the salvage therapy could be applied.
1. Introduction
Multiple myeloma (MM) is a malignant proliferative disorder of plasma cells and represents the second most common hematologic malignancy. It is estimated that in 2022, 33,463 newly cases of MM in the United States were diagnosed and 22,450 in China [1], respectively. The NCCN 2024 V2 guidelines recommend combination regimens as the prior-line treatment, including proteasome inhibitors (or CD38 monoclonal antibodies), bortezomib (PI), immunomodulatory drugs (IMiDs), and dexamethasone. For patients who relapse or exhibit refractory disease after one to three lines of therapy, it is advised to switch to drug combinations with different mechanisms of action. For those who have undergone four lines of treatment, novel immunotherapy such as CAR-T cell therapy or bispecific antibody (BsAb) therapy is then recommended [2]. On April 5, 2024, after rigorous evaluation, the U.S. FDA approved ciltacabtagene autoleucel (cilta-cel) for the treatment of relapsed/refractory MM (R/R MM) in patients who have previously undergone at least one line of therapy, including a proteasome inhibitor and an immunomodulatory drug, and who are refractory to lenalidomide. Consequently, cilta-cel has become the only approved BCMA-targeted therapy (encompassing CAR-T therapy, bispecific antibodies, and antibody-drug conjugates) for second-line treatment in MM patients, therefore making it regarded as one of the most promising CAR-T therapies for R/R MM. However, a meta-analysis [3] involving 761 R/R MM patients revealed that although the objective response rate (ORR) for anti-BCMA CAR-T cell therapy was a rather desirable 87%, the median progression-free survival (PFS) was only 8.77 months. Meanwhile, unfortunately, the majority of those patients demonstrated relapse within 12 months following CAR-T therapy. At present, no consensus on subsequent salvage therapy for these patients has ever reached, despite vast kinds of clinical trials have explored various approaches, including non-BCMA CAR-T cells, bispecific antibodies (BsAb), monoclonal antibodies (mAb), antibody-drug conjugates (ADC), and combination chemotherapy, with certain kinds of success [4; 5; 6; 7; 8; 9; 10; 11; 12; 13; 14; 15; 16]. Therefore, in this review, we attempt to discuss the potential factors contributing to CAR-T therapy failure, the salvage therapeutic strategies that have been attempted, and the clinical outcomes, along with recommendations for some possible approaches to address these challenges.
2. Literature Review
The mechanism of CAR-T cell therapy involves the binding of the CAR molecule on the surface of CAR-T cells to the corresponding antigen on the surface of tumor cells, which subsequently activates the intracellular signaling pathways in CAR-T cells, leading to the cytotoxic elimination of tumor cells. In the treatment of R/R MM, several target antigens have been identified for CAR-T cell therapy. B-cell maturation antigen (BCMA) is considered as one of the most promising targets due to its expression on mature B cells, normal and malignant plasma cells, and plasmacytoid dendritic cells, while being absent on early B cells, memory B cells, and normal hematopoietic stem cells (HSCs), which makes BCMA an ideal antigen of targeting. Additionally, G protein-coupled receptor class C group 5 member D (GPRC5D) is frequently utilized as a target antigen in MM CAR-T cell therapy that is highly expressed on primary MM cells but is restricted to the immune-privileged hair follicle regions in normal tissues. Notably, GPRC5D expression is independent of BCMA, allowing for the development of single or dual-targeted cellular therapies [17]. Other antigens, such as CD138, CD38, and SLAMF7, are also being evaluated in ongoing clinical studies [18]. As to June 2024, regulatory agencies in major global pharmaceutical markets have approved four CAR-T products for the treatment of R/R MM: ide-cel, cilta-cel, equecabtagene autoleucel (equ-cel), and zevorcabtagene autoleucel (zevor-cel). Additionally, numerous other candidate CAR-T therapies are undergoing preclinical and clinical evaluation.
2.1. Current Landscape of CAR-T Cell Therapy towards Multiple Myeloma
While CAR-T cell therapy has achieved remarkable success in treating R/R MM, the challenge of overcoming therapy failure remains a significant clinical hurdle. The 5-year follow-up data from the LEGEND-2 study on cilta-cel [19] reported an overall response rate (ORR) of 87.8%, with a 5-year progression-free survival (PFS) rate of 21.0%, and the longest duration of response extending up to 6.4 years. Meanwhile, in the phase I CRB-401 trial of ide-cel, with a median follow-up of 18.1 months, ORR reached 75.8%, with a median duration of response of 10.3 months and a median PFS of 8.8 months [20]. Similarly, the FUMANBA-1 study of equecabtagene autoleucel (equ-cel) [21], with a median follow-up of 13.8 months, demonstrated an ORR of 96%, although the median duration of response and median PFS have yet to be determined; the 12-month PFS rate was 78.8% (95% CI: 68.6-85.97). Additionally, the 3-year follow-up from the LUMMICAR-1 study of zevorcabtagene autoleucel (zevor-cel) [22] reported an ORR of 100%, with a median duration of response of 24.1 months and a median PFS of 25 months . Details on these outcomes of CAR-T cell therapies for R/R MM are provided in Table 1. Although these results can be regarded as promising, it is also important to acknowledge that there is a general tendency of approximately 10-20% of the patient exhibit resistance to CAR-T therapy, and many patients who demonstrated a response might relapse. Research has identified several factors associated with less desirable PFS in the context of CAR-T therapy failure, including a history of extramedullary disease, prior BCMA-targeted therapy, elevated ferritin levels during lymphodepletion, plasma cell leukemia, and the presence of the t(4;14) translocation [23].
2.2. Potential Factors Contributing to CAR-T Therapy Failure
The mechanisms underlying the failure of CAR-T therapy in treating R/R MM are not yet fully elucidated, with various factors potentially contributing to treatment failure. These factors include the downregulation or loss of tumor cell target antigen expression, T-cell exhaustion, and the tumor immune microenvironment. Baseline heterozygous deletion of BCMA has been reported as a possible contributor to BCMA loss following immunotherapy, which could serve as a potential mechanism for immune escape [29]. In patients who received commercial anti-BCMA CAR-T cell therapy, the median BCMA expression decreased from 670 (interquartile range [IQR] 380-850) mol/cell prior to treatment to 390 (IQR 290-490) mol/cell post-treatment (n=15, p=0.011). In vitro studies have shown that as antigen levels decline, the tumor-lytic and cytokine production functions of CAR-T cells are compromised, and when antigen levels fall below 50 mol/cell, CAR-T cell functionality is lost [30]. Beyond BCMA, a reduction or loss of GPRC5D expression has also been observed in patients who experienced disease progression after achieving remission with anti-GPRC5D CAR-T therapy [31,32].
The expansion and persistence of CAR-T cells are closely linked to the efficacy of CAR-T therapy. Studies indicate that circulating CAR-T cell expansion exceeding 180/mm3 following ide-cel infusion is associated with prolonged PFS [33]. In patients with sustained best responses after cilta-cel infusion, CAR-T cell persistence was notably longer (median duration of 535.3 days to 261.6 days, p=0.01) [19]. However, the human anti-mouse antibody (HAMA) response, induced by murine-derived scFv, has been shown to diminish CAR-T cell persistence [34]. The tumor microenvironment in MM further influences T-cell functionality, with CD8+ T cells expressing several molecules associated with T-cell exhaustion (PD-1, CTLA-4, 2B4, CD160) and T-cell senescence (CD57, loss of CD28), a phenotype correlated with reduced cell proliferation and impaired function [35]. Moreover, high CAR expression levels are linked to increased signaling and an exhaustion phenotype, characterized by the expression of multiple inhibitory receptors, including PD-1, CTLA-4, LAG3, TIM3, and TIGIT, which is associated with diminished responses and poorer long-term PFS outcomes [36].
2.3. Salvage Therapies
The failure of CAR-T cell therapy in relapsed/refractory multiple myeloma (R/R MM) presents a critical challenge, particularly as these therapies are often administered in late-stage disease where patients have developed resistance to conventional treatments. The median overall survival (OS) for patients with triple-refractory R/R MM is approximately 9 months, and even shorter for those with penta-refractory disease, highlighting the urgency for effective salvage strategies [37]. Following relapse post-CAR-T therapy, the limited efficacy of subsequent treatments underscores the need for innovative approaches [7,38].
2.3.1. Secondary CAR-T Cell Therapy
Secondary CAR-T therapy, whether repeating anti-BCMA CAR-T or switching to alternative targets like GPRC5D, remains a viable option for salvage therapy. However, the results from re-administering anti-BCMA CAR-T vary [39]. While some patients respond positively [40], the benefits tend to decrease for those previously treated with BCMA-targeted therapies [41]. In contrast, GPRC5D-targeted CAR-T cells have shown promise in phase I trials [32], achieving durable responses even after anti-BCMA therapy failure [24,31,42]. Although the responses are likely reduced, compared to BCMA-naive patients, GPRC5D CAR-T cells continue to be a viable salvage option. Meanwhile, dual-target CAR-T cells, aiming to overcome single-target limitations, have also gained attention as potential choice of salvage therapy. Approaches like bispecific CS1-BCMA CAR-T have demonstrated strong responses [27], while a phase I/II trial of BCMA/CD19 CAR-T achieved an 80% ORR in relapsed patients’ post-commercial CAR-T therapy [43]. Moreover, universal CAR-T (UCAR-T) cells, derived from healthy donors, offer the advantage of immediate availability, enhanced T-cell targeting, and reduced manufacturing costs [44,45]. Despite the risk of mismatches causing GVHD, a phase I study showed a 60% ORR with no GVHD, though some patients experienced grade 2 CRS and ICANS [46].
2.3.2. Bispecific Antibodies
Teclistamab, Talquetamab, Elranatamab, and Cevostamab are FDA-approved or in clinical evaluation as bispecific antibodies for R/R MM. These BsAbs, targeting BCMA, GPRC5D, and FcRH5 through CD3, engage T cells to target malignant cells, offering a safer and effective post-CAR-T therapy option [47,48,49]. In their phase II trials, the prior ORRs for Teclistamab, Talquetamab, Elranatamab, and Cevostamab were 63% [13], 65.7% [12], 58.3% [50], and 54.5% [51], respectively. With Teclistamab and Talquetamab indicated stronger responses, their safety profiles, such as grade 3 CRS (0% for Teclistamab, 5% for Talquetamab) and ICANS, were also manageable [12,52]. Comparatively, Talquetamab exhibited stronger T-cell activation in patients post-CAR-T therapy, achieving better PFS [12] and DOR [53] than those treated with BsAbs first. Similarly, Elranatamab presented favorable outcomes, particularly in African American patients, with a median overall survival (OS) of 21.2 months post-CAR-T therapy [50]. Cevostamab, meanwhile, demonstrated notable efficacy, with ORRs ranging from 44.4% to 50%, depending on prior treatments, utilizing FcRH5 targeting to minimize severe CRS and ICANS [51].
Together, these data indicate that BsAbs are promising options in the salvage therapy landscape, especially when used sequentially after CAR-T treatment. However, optimizing their use in combination and sequencing strategies requires further research to maximize patient outcomes.
2.3.3. Novel Monoclonal Antibodies (PD-1/PD-L1)
The immunosuppressive effects of the tumor microenvironment are a key factor contributing to the failure of CAR-T cell therapy. PD-1/PD-L1 monoclonal antibodies have the potential to alleviate this immunosuppression, thereby enhancing T cell activity. Currently, researchers are investigating the clinical efficacy of Pembrolizumab (Keytruda) as a monotherapy or in combination therapies for MM patients who have not responded to anti-BCMA CAR-T therapy [11]. Studies have demonstrated that combining the PD-1 inhibitor Pembrolizumab with the anti-SLAMF7 monoclonal antibody Elotuzumab can induce re-expansion of CAR-T cells and enhance anti-myeloma effects. However, the best therapeutic outcome observed in this setting was a minimal response (MR) [10]. Similarly, the use of another PD-1 antibody, Nivolumab, in R/R MM patients who relapsed after CAR-T therapy resulted in an ORR of only 18% (2/11) [54]. Thus, the effectiveness of PD-1/PD-L1 monoclonal antibodies as a salvage therapy for R/R MM after CAR-T cell therapy failure remains an area that requires further investigation and optimization.
2.3.4. Antibody-Drug Conjugates (ADCs)
Antibody-drug conjugates (ADCs) like Blenrep (Belantamab mafodotin) are designed to target cancer cells with an antibody linked to a cytotoxic payload. Approved by the FDA in August 2020 for R/R MM patients who had undergone at least four prior therapies, Blenrep was withdrawn from the U.S. market in November 2022 due to failing to meet the primary endpoint of progression-free survival (PFS) in the phase III DREAMM-3 trial.
In clinical evaluations, Blenrep achieved an overall response rate (ORR) of 33%. However, among seven patients previously treated with anti-BCMA CAR-T therapy, none responded to Blenrep; two had stable disease and five progressed [55]. Meanwhile, in the KarMMa phase II trial, a patient resistant to a second CAR-T therapy achieved a very good partial response (VGPR) after three doses of Blenrep, with the response lasting five months [56]. Additionally, a single-center retrospective study [39], found that among 30 R/R MM patients who progressed after anti-BCMA CAR-T therapy, those receiving Blenrep had an ORR of only 25% and a median PFS of 1.8 months. These results highlight the need for further research to optimize ADC therapies in this setting.
2.3.5. Other Treatments
For R/R MM patients relapsing after CAR-T therapy, traditional therapeutic methods remain alternatives include proteasome inhibitors, immunomodulatory agents, and nuclear export inhibitors. The LEGEND-2 study [8] showed that proteasome inhibitor-based therapies were highly effective in these patients, achieving an overall response rate (ORR) of 50%, significantly higher than re-administered BCMA CAR-T (30%) or combination chemotherapy (0%). This suggests that CAR-T therapy may re-sensitize myeloma cells to previously used drugs.
Ferreri et al. [14], evaluated 30 R/R MM patients after anti-BCMA CAR-T therapy failure, exploring various salvage therapies such as repeat BCMA CAR-T therapy, stem cell transplantation, bispecific antibodies, and anti-CD38 monoclonal antibodies. The highest efficacy was observed with repeat BCMA CAR-T and stem cell transplantation [39]. Additionally, a case study by Agte et al. showed that a patient with BRAF V600E-mutated MM responded well to a BRAF inhibitor-based regimen, reducing free λ light chains by 80% [57]. Moreover, another patient with extramedullary disease (EMD) achieved a stringent complete response (sCR) following haploidentical hematopoietic stem cell transplantation (HSCT) [58]. These findings emphasize the importance of personalized treatment strategies and further research to optimize salvage therapy options for patients relapsing after CAR-T therapy.
2.4. Recommended Strategies
The approval and commercialization of Ide-cel have ushered in a new era of immune cell therapy towards R/R MM, with the subsequent approval of Cilta-cel for second-line treatment further emphasizing the pivotal role of CAR-T cell therapy in this context. However, this advancement brings forth critical research challenges, such as the need to extend progression-free survival (PFS) and overall survival (OS) associated with CAR-T therapy, as well as determining the most effective salvage treatment strategies following disease relapse or progression. Based on the findings from relevant clinical studies, we propose the following recommendations.
2.4.1. moving CAR-T therapy to earlier lines of treatment
The potential therapeutic mechanism of CAR-T cell therapy in MM might include a) At the time of disease onset or with fewer treatment lines, the patient's autologous T cells have not been significantly weakened by other therapies, hence the quality of the prepared CAR-T cells is higher, and the efficacy as an early-line treatment is also better [44,59]; b) The invasiveness of MM in the early stages of the disease is lower, with slow disease progression or low tumor burden, therefore, the use of CAR-T cell therapy can yield more benefits [60]. Studies have shown that the T cell population at the time of MM diagnosis has a higher proportion of naive T cells compared to T cells after treatment with daratumumab or after relapse, with lower proportions of exhaustion markers PD1 and LAG3, and the prepared CAR-T cells exhibit better cell proliferation and cytotoxic activity [61] Patients with fewer ( < 4) prior treatment lines before receiving anti-BCMA CAR-T cell therapy have a lower proportion of extramedullary disease (EMD), triple-drug refractory, and penta-drug refractory, and their response to subsequent salvage therapy is significantly better than that of patients with more ( ≥ 4) prior treatment lines [39]. In a phase 1, single-arm, multicenter study of ide-cel for the treatment of R/R MM, the number of prior treatment lines was 6 (range, 3-18), with an overall response rate (ORR) of 75.8%, a median progression-free survival (PFS) of 8.8 months, and a median overall survival (OS) of 34.2 months [20]. In a comparative study between ide-cel and standard therapy, the number of prior treatment lines in the ide-cel group was 3 (range, 2-4), with an ORR of 71%, a median PFS of 13.3 months, and the median OS has not been reached, while the risk of disease progression or death was reduced by 51% [62]. Further analysis indicates that patients with PFS ≥18 months had more primitive and less exhausted T cells at the time of apheresis, with a better functional T cell phenotype in the CAR-T cell preparation [20]. Therefore, the results of related studies support the advancement of CAR-T cell therapy line numbers, which can bring greater benefits to patients [63].
2.4.2. Exploring CAR-T Cells with Different Targets and Origins
For R/R MM patients after CAR-T therapy, it is recommended to initially assess the expression of antigens such as BCMA and GPRC5D on the surface of MM cells and select CAR-T cell therapy targeting the corresponding antigens. However, some studies have indicated that the response rate is not related to baseline BCMA expression [4,64]. Currently, CAR-T cells targeting BCMA have been approved for marketing, while those targeting other antigens such as GPRC5D are undergoing clinical trials. Treatments based on anti-GPRC5D CAR-T cells [31,32,42] , anti-BCMA CAR-T [39] or dual-target CAR-T cells [43] have been shown to be effective for patients who have failed previous anti-BCMA CAR-T cell therapy, and we recommend using CAR-T cells with different targets from those previously used as thefirst line of therapy. Additionally, considering that different sources and constructs of scFv can affect the efficacy of CAR-T cell therapy [64,65],it is also possible to choose humanized or fully human CAR-T as salvage therapy following the progression of CAR-T treatment.
2.4.3. Rapid-Manufactured CAR-T
During the conventional autologous CAR-T cell manufacturing process, patients with a high tumor burden often require bridging therapy to manage disease progression. However, the rapid advancement of the disease in some patients can preclude the administration of CAR-T cell therapy, potentially leading to mortality before the therapy can be administered [66]. Moreover, bridging therapy can also affect subsequent efficacy, such as longer median hospital stay, shorter median progression-free survival (PFS) (8.1 months to 11.5 months, p = 0.03), and shorter median overall survival (OS) (13.8 months to not reached, p = 0.002) [67]. Rapid CAR-T cell manufacturing processes, by shortening production time, yield T cells with lower differentiation, requiring fewer cell doses, reducing production costs, and enhancing in vivo expansion of CAR-T cells, thereby potentially offering significant advantages in the treatment of R/R MM. In addition, the advantages of rapid manufacturing also include reducing bridging therapy and mitigating adverse events following bridging therapy. Therefore, rapidly manufactured CAR-T cells are a very promising type of CAR-T cell. The results of a phase I clinical study conducted by CARsgen Therapeutics, targeting GPRC5D with rapidly manufactured CAR-T cells (CT071) for the treatment of R/R MM, were presented at the EHA Annual Meeting (Poster P941). Ten R/R MM patients were treated with CT071, achieving an ORR of 90%. Among them, two patients who had previously received BCMA/CD19 dual-target CAR-T cell therapy achieved sCR and PR, respectively, with MRD testing negative at a 10-6 threshold. NovartisTM has developed an anti-BCMA CAR-T cell product, PHE885, using its T-charge platform for rapid manufacturing (less than 2 days), which achieved an ORR of 98% in R/R MM patients who had undergone a median of four prior lines of therapy [68]. The incidence of CRS was 96%, with 11% experiencing grade 3 CRS, while ICANS occurred in 22%, with 7% experiencing grade 3 ICANS. Additionally, other companies such as Gracell Biotechnologies [28], BMS [69], and IASO Bio [70] are also developing rapidly manufactured CAR-T cells for the treatment of R/R MM, featuring a desirable landscape of the application of rapid-manufactured CAR-T towards the failure of post CAR-T therapies.
2.4.4. Sequential Selection of T-Cell Redirecting Therapies
T-cell redirecting therapies currently encompass CAR-T cells, ADCs, and BsAbs, with CAR-T cells and BsAbs being the most frequently utilized salvage treatments after CAR-T therapy failure in R/R MM [16]. Both CAR-T cells and BsAbs are forms of immunotherapy that mediate non-MHC-restricted cytotoxicity; however, BsAbs depend on the patient’s own T cells, which raises concerns about potential T-cell dysfunction [71]. Due to differences in toxicity profiles, expected response rates, and administration methods, CAR-T cell therapy is generally more suitable for younger patients, those with aggressive or relapsed disease, and those with a high tumor burden [72].
At present, there is no established consensus on the optimal sequencing of CAR-T cells, ADCs, and BsAbs. Research indicates that the effectiveness of CAR-T therapy is somewhat diminished when administered following ADC or BsAb treatment. In a real-world study of Ide-cel treatment in R/R MM patients who were ineligible for the KarMMa-1 trial, those with prior BCMA-targeted therapy (16 treated with ADCs, 1 with a trispecific T-cell activator, and 1 with an experimental CAR-T therapy) exhibited a lower median PFS compared to patients who had not received BCMA-targeted therapy (6.2 months to 9.0 months, p = 0.11) [73]. Similarly, in the phase II CARTITUDE-2 study evaluating Cilta-cel in R/R MM, cohort C focused on patients previously treated with BCMA-targeted therapies (13 with belantamab mafodotin, 7 with BsAbs, including 1 who had received both in the belantamab mafodotin group) [74]. The results showed an ORR of 60.0% and a median PFS of only 9.1 months, significantly lower than the outcomes in BCMA-targeted therapy-naive patients in the CARTITUDE-1 study. In contrast, BsAb therapy following CAR-T cell treatment has demonstrated promising efficacy. Jakubowiak et al. [12] examined the safety and effectiveness of talquetamab in 70 R/R MM patients who had undergone prior T-cell redirecting therapies, including 50 treated with CAR-T, 25 with BsAbs, and 5 with both CAR-T and BsAbs. The ORR was 72.9% in patients who had received prior BCMA CAR-T therapy, compared to 56.5% in those with prior BsAb therapy. Further analysis indicated that whether CAR-T was used as the last or a previous line of therapy had no significant impact on BsAb efficacy (ORR, 71.4% to 75.9%), whereas BsAb efficacy was significantly reduced when BsAbs were used as the last line of therapy comparing to the previous line (ORR, 28.6% to 66.7%). Given that CAR-T cell therapy can induce profound and durable responses with extended treatment-free intervals, thereby reducing the risk of T-cell exhaustion and the selective pressure from continuous therapy [16],it is recommended that CAR-T cell therapy be prioritized in the treatment course of MM, with BsAb therapy considered upon disease progression after CAR-T treatment.
2.4.5. Combination Therapy
Combining CAR-T therapy with other drugs can enhance the persistence of CAR-T cells in vivo, improve BCMA expression, and reduce T-cell exhaustion. Such drugs include immunomodulatory agents, γ-secretase inhibitors, and CRBN E3 ligase modulators.
Lenalidomide is a commonly used immunomodulatory drug (IMiD) that was approved by the FDA for the treatment of MM in June 2006. Preclinical studies have shown that lenalidomide can provide a co-stimulatory effect on CS1 CAR-T cells, enhancing their antitumor activity and persistence by improving CS1 CAR-T cell cytotoxicity, memory maintenance, Th1 cytokine production, and immunological synapse formation [75]. Lenalidomide can regulate the ICOSL/ICOS pathway, Th1/Th2 pathway, increase CAR-T cell secretion of cytokines such as IFN-γ, IL-2, and TNF-α, and enhance CAR-T cell cytotoxicity [76]. Researchers reported a case of an R/R MM patient who relapsed after multiple chemotherapies, autologous hematopoietic stem cell transplantation, murine CAR-T cell therapy, and human CAR-T cell therapy. After lymphodepletion with fludarabine and cyclophosphamide, the patient was given lenalidomide on day -1, followed by human CAR-T cell infusion on day 0, achieving VGPR within 14 days and maintaining it for over 8 months [77].
BCMA is cleaved from the surface of MM cells by γ-secretase. Preclinical studies have indicated that inhibiting γ-secretase can increase BCMA expression on MM cells, thereby enhancing the efficacy of CAR-T cell therapy [78]. A phase I clinical study evaluated the safety of crenigacestat, a γ-secretase inhibitor, in combination with anti-BCMA CAR-T cells for the treatment of R/R MM patients. The results showed that the γ-secretase inhibitor could block BCMA shedding, overcoming antigen downregulation and thereby improving CAR-T cell efficacy [79].
In vitro studies on the CRBN E3 ligase modulator iberdomide have demonstrated that iberdomide can significantly reduce the transcription factors Ikaros and Aiolos in CAR-T cells, promote the persistence and proliferation of IL-2-starved cells, significantly increase the expression of Ki67 in CAR-T cells, enhance cell viability, upregulate activation markers such as HLADR and CD69, downregulate TIGIT expression, and increase the expression of IL-2, IL-17a, and TNF-α. Therefore, iberdomide shows promise in assisting CAR-T cell therapy by overcoming CAR-T cell resistance and enhancing its efficacy [80].
Based on these findings, combining CAR-T cell therapy with related drugs may improve the efficacy of CAR-T treatment from multiple aspects, potentially extending PFS and OS in patients.
2.4.6. Maintenance Therapy
A survey conducted by the International Myeloma Society revealed that 30% of the clinicians surveyed administer maintenance therapy after CAR-T cell treatment, primarily to sustain the level of remission achieved or to deepen the response further [81]. However, a survey by the American Society for Transplantation and Cellular Therapy (ASTCT) Practice Guidelines Committee found that 86% of clinicians would not consider maintenance therapy in the absence of biochemical or clinical evidence of myeloma, with only 18% considering it for patients who achieved a partial response (PR) after CAR-T cell therapy but did not convert to a complete response (CR) after several months of follow-up [82]. Therefore, it is recommended to identify risk factors before initiating CAR-T cell therapy, assess which patients might benefit from maintenance therapy, and determine the appropriate regimens. Continuous monitoring of disease status post-CAR-T cell therapy is crucial, with maintenance therapy being initiated at the optimal time to prolong progression-free survival (PFS) and overall survival (OS).
In a study by Li et al.[27], the use of bispecific CS1-BCMA CAR-T cells in treating R/R MM patients resulted in an overall response rate (ORR) of 81% (13/16). Within the study, three patients (Patients 10, 11, and 15) received lenalidomide as maintenance therapy (10 mg/day, 21 days of a 28-day cycle) following CAR-T cell treatment. Patients 10 and 11 achieved deeper responses; however, Patients 10 and 15 eventually experienced relapse or progression, while Patient 11 continued to maintain a stringent complete response (sCR). Another case involved an R/R MM patient who developed secondary plasma cell leukemia and was treated with anti-BCMA CAR-T cells, achieving a sustained sCR for 9 months. This patient then received intermittent venetoclax treatment at 10 mg/day, maintaining the response for an additional 7 months [83]. In the phase II CARTITUDE-2 clinical study [84] evaluating Cilta-cel for newly diagnosed MM patients with suboptimal response to first-line autologous stem cell transplantation, 17 patients were assessed. Among all the patients, five received Cilta-cel alone, while 12 began continuous lenalidomide maintenance therapy (for up to 2 years) 21 days after Cilta-cel infusion. The results showed a general ORR of 94%, with both the 18-month PFS rate (as assessed by investigators) and OS rate at 94%, suggesting that a single infusion of Cilta-cel combined with lenalidomide maintenance therapy can produce a durable, deep response. These findings indicate that incorporating lenalidomide as maintenance therapy after CAR-T cell treatment may be an effective strategy to address CAR-T cell therapy failure.
3. Conclusions
Although CAR-T cell therapy has significantly improved the efficacy of treatment for R/R MM, factors such as downregulation or loss of tumor antigen expression, T-cell exhaustion, and the tumor immune microenvironment can lead to CAR-T therapy failure. Various salvage therapies, including repeat CAR-T cell treatments, have been employed in clinical practice with positive outcomes. However, there is currently no consensus on the optimal salvage treatment strategies or their sequencing for these patients. It is recommended to develop individualized treatment plans based on the patient's specific condition, selecting the most appropriate therapies to help R/R MM patients who have experienced CAR-T failure achieve prolonged remission. Most of the current literature consists of small retrospective case series, highlighting the need for larger studies in the future to establish standardized salvage treatment protocols for R/R MM patients following CAR-T therapy failure.
Conflict of Interests
The authors declare no competing interests.
Acknowledgments
This study is funded by Technical Innovation Guidance Program of Jiangxi Province, China, (20212BDH80014) and the Science and Technology Innovation Action Plan of Shanghai, China, (23S11905500).
References
- Xia, C.; Dong, X.; Li, H.; Cao, M.; Sun, D.; He, S.; Yang, F.; Yan, X.; Zhang, S.; Li, N.; et al. Cancer statistics in China and United States, 2022: profiles, trends, and determinants. Chin Med J (Engl) 2022, 135, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Callander, N.S.; Adekola, K.; Anderson, L.D., Jr.; Baljevic, M.; Baz, R.; Campagnaro, E.; Castillo, J.J.; Costello, C.; D'Angelo, C.; et al. Multiple Myeloma, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2023, 21, 1281–1301. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Chen, L.; Yan, D.; Dong, W.; Chen, M.; Niu, S.; Wang, S.; Zhang, J.; Nie, X.; Fang, Y. Effectiveness and safety of anti-BCMA chimeric antigen receptor T-cell treatment in relapsed/refractory multiple myeloma: a comprehensive review and meta-analysis of prospective clinical trials. Front Pharmacol 2023, 14, 1149138. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wei, R.; Guo, S.; Min, C.; Zhong, X.; Huang, H.; Cheng, Z. An alternative fully human anti-BCMA CAR-T shows response for relapsed or refractory multiple myeloma with anti-BCMA CAR-T exposures previously. Cancer Gene Ther 2024, 31, 420–426. [Google Scholar] [CrossRef]
- Keller, A.L.; Parzych, S.E.; Reiman, L.T.; Walker, Z.J.; Forsberg, P.A.; Sherbenou, D.W. BCMAxCD3 Bispecific Antibody Elranatamab Is Effective in Patient Myeloma Relapsed after BCMA CAR-T. Blood 2023, 142, 4684. [Google Scholar] [CrossRef]
- Reyes, K.R.; Liu, Y.-C.; Huang, C.-Y.; Banerjee, R.; Martin, T.; Shah, N.; Wong, S.W.; Wolf, J.L.; Arora, S.; Chung, A. Clinical Outcomes and Salvage Therapies in Patients with Relapsed/Refractory Multiple Myeloma Following Progression on BCMA-Targeted CAR-T Therapy. Blood 2022, 140, 617–619. [Google Scholar] [CrossRef]
- Van Oekelen, O.; Nath, K.; Mouhieddine, T.H.; Farzana, T.; Aleman, A.; Melnekoff, D.T.; Ghodke-Puranik, Y.; Shah, G.L.; Lesokhin, A.; Giralt, S.; et al. Interventions and outcomes of patients with multiple myeloma receiving salvage therapy after BCMA-directed CAR T therapy. Blood 2023, 141, 756–765. [Google Scholar] [CrossRef]
- Liu, R.; Yang, R.; Xu, X.; Zhao, W.; Wang, F.; Zhang, W.; Lei, B.; Yang, R.; Wang, Y.; He, A.; et al. Outcomes in patients with multiple myeloma receiving salvage treatment after BCMA-specific CAR-T therapy: A retrospective analysis of LEGEND-2. Br J Haematol 2024. [Google Scholar] [CrossRef]
- Snyder, J.; Bellman, P.; Alsaddi, Z.; Alkharabsheh, O.; Paul, B.; Hashmi, H.; Lutfi, F.; Ahmed, N.; Mahmoudjafari, Z.; Abdallah, A.-O.; et al. Patient Outcomes Following First and Second Exposure to BCMA-Directed Therapies Including CAR-T Cell Therapy in Relapsed/Refractory Multiple Myeloma. Transplantation and Cellular Therapy 2024, 30, S394–S395. [Google Scholar] [CrossRef]
- Bernabei, L.; Garfall, A.L.; Melenhorst, J.J.; Lacey, S.F.; Stadtmauer, E.A.; Vogl, D.T.; Gonzalez, V.; Plesa, G.; Young, R.M.; Waxman, A.; et al. PD-1 Inhibitor Combinations As Salvage Therapy for Relapsed/Refractory Multiple Myeloma (MM) Patients Progressing after Bcma-Directed CAR T Cells. Blood 2018, 132, 1973–1973. [Google Scholar] [CrossRef]
- Chung, A.; Huang, C.-Y.; Martin, T.; Wolf, J.L.; Wong, S.W.; Shah, N. Phase II Study of Pembrolizumab in Multiple Myeloma Patients Relapsing after or Refractory to Anti-BCMA CAR-T Therapies. Blood 2022, 140, 12651–12652. [Google Scholar] [CrossRef]
- Jakubowiak, A.J.; Anguille, S.; Karlin, L.; Chari, A.; Schinke, C.; Rasche, L.; San-Miguel, J.; Campagna, M.; Hilder, B.W.; Masterson, T.J.; et al. Updated Results of Talquetamab, a GPRC5D×CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma with Prior Exposure to T-Cell Redirecting Therapies: Results of the Phase 1/2 MonumenTAL-1 Study. Blood 2023, 142, 3377. [Google Scholar] [CrossRef]
- Grajales-Cruz, A.F.; Castaneda, O.; Hansen, D.K.; Vazquez-Martinez, M.A.; Blue, B.; Khadka, S.; Liu, H.; Ochoa-Bayona, J.L.; Freeman, C.L.L.; Locke, F.L.; et al. Teclistamab Induces Favorable Responses in Patients with Relapsed and Refractory Multiple Myeloma after Prior BCMA-Directed Therapy. Blood 2023, 142, 3351. [Google Scholar] [CrossRef]
- Ferreri, C.J.; Hildebrandt, M.A.T.; Hashmi, H.; Shune, L.O.; McGuirk, J.P.; Sborov, D.W.; Wagner, C.B.; Kocoglu, M.H.; Rapoport, A.; Atrash, S.; et al. Real-world experience of patients with multiple myeloma receiving ide-cel after a prior BCMA-targeted therapy. Blood Cancer J 2023, 13, 117. [Google Scholar] [CrossRef]
- Popat, R.; Zweegman, S.; Cavet, J.; Yong, K.; Lee, L.; Faulkner, J.; Kotsopoulou, E.; Al-Hajj, M.; Thomas, S.; Cordoba, S.P.; et al. Phase 1 First-in-Human Study of AUTO2, the First Chimeric Antigen Receptor (CAR) T Cell Targeting APRIL for Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2019, 134, 3112–3112. [Google Scholar] [CrossRef]
- Reyes, K.R.; Liu, Y.C.; Huang, C.Y.; Banerjee, R.; Martin, T.; Wong, S.W.; Wolf, J.L.; Arora, S.; Shah, N.; Chari, A.; et al. Salvage therapies including retreatment with BCMA-directed approaches after BCMA CAR-T relapses for multiple myeloma. Blood Adv 2024, 8, 2207–2216. [Google Scholar] [CrossRef]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med 2019, 11. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Lan, H.; Wu, J.; Xiao, Y. CAR-T cell therapy in multiple myeloma: Current limitations and potential strategies. Front Immunol 2023, 14, 1101495. [Google Scholar] [CrossRef]
- Xu, J.; Wang, B.Y.; Yu, S.H.; Chen, S.J.; Yang, S.S.; Liu, R.; Chen, L.J.; Hou, J.; Chen, Z.; Zhao, W.H.; et al. Long-term remission and survival in patients with relapsed or refractory multiple myeloma after treatment with LCAR-B38M CAR T cells: 5-year follow-up of the LEGEND-2 trial. J Hematol Oncol 2024, 17, 23. [Google Scholar] [CrossRef]
- Lin, Y.; Raje, N.S.; Berdeja, J.G.; Siegel, D.S.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Massaro, M.; et al. Idecabtagene vicleucel for relapsed and refractory multiple myeloma: post hoc 18-month follow-up of a phase 1 trial. Nat Med 2023, 29, 2286–2294. [Google Scholar] [CrossRef]
- Li, C.; Wang, D.; Song, Y.; Huang, H.; Li, J.; Chen, B.; Liu, J.; Dong, Y.; Hu, K.; Liu, P.; et al. CT103A, a novel fully human BCMA-targeting CAR-T cells, in patients with relapsed/refractory multiple myeloma: Updated results of phase 1b/2 study (FUMANBA-1). Journal of Clinical Oncology 2023, 41, 8025–8025. [Google Scholar] [CrossRef]
- Fu, C.; Chen, W.; Cai, Z.; Yan, L.; Wang, H.; Shang, J.; Wu, Y.; Yan, S.; Gao, W.; Shi, X.; et al. Three-Year Follow-up on Efficacy and Safety Results from Phase 1 Lummicar Study 1 of Zevorcabtagene Autoleucel in Chinese Patients with Relapsed or Refractory Multiple Myeloma. Blood 2023, 142, 4845–4845. [Google Scholar] [CrossRef]
- Hashmi, H.; Hansen, D.K.; Peres, L.C.; Puglianini, O.C.; Freeman, C.; De Avila, G.; Sidana, S.; Shune, L.; Sborov, D.W.; Davis, J.; et al. Factors associated with refractoriness or early progression after idecabtagene vicleucel in patients with relapsed/ refractory multiple myeloma: US Myeloma Immunotherapy Consortium real world experience. Haematologica 2024, 109, 1514–1524. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Hu, Y.; Zhang, M.; Wei, G.; Zhou, L.; Fu, S.; Feng, J.; Hong, R.; Cui, J.; Huang, S.; et al. OriCAR-017, a novel GPRC5D-targeting CAR-T, in patients with relapsed/refractory multiple myeloma: Long term follow-up results of phase 1 study (POLARIS). 2024, 42, 7511–7511. [CrossRef]
- Xia, J.; Li, H.; Yan, Z.; Zhou, D.; Wang, Y.; Qi, Y.; Cao, J.; Li, D.; Cheng, H.; Sang, W.; et al. Anti-G Protein-Coupled Receptor, Class C Group 5 Member D Chimeric Antigen Receptor T Cells in Patients With Relapsed or Refractory Multiple Myeloma: A Single-Arm, Phase Ⅱ Trial. J Clin Oncol 2023, 41, 2583–2593. [Google Scholar] [CrossRef]
- Mei, H.; Li, C.; Jiang, H.; Zhao, X.; Huang, Z.; Jin, D.; Guo, T.; Kou, H.; Liu, L.; Tang, L.; et al. A bispecific CAR-T cell therapy targeting BCMA and CD38 in relapsed or refractory multiple myeloma. J Hematol Oncol 2021, 14, 161. [Google Scholar] [CrossRef]
- Li, C.; Xu, J.; Luo, W.; Liao, D.; Xie, W.; Wei, Q.; Zhang, Y.; Wang, X.; Wu, Z.; Kang, Y.; et al. Bispecific CS1-BCMA CAR-T cells are clinically active in relapsed or refractory multiple myeloma. Leukemia 2024, 38, 149–159. [Google Scholar] [CrossRef]
- Du, J.; Fu, W.-J.; Jiang, H.; Dong, B.; Gao, L.; Liu, L.; Ge, J.; He, A.; Li, L.; Lu, J.J.J.C.O. Updated results of a phase I, open-label study of BCMA/CD19 dual-targeting fast CAR-T GC012F for patients with relapsed/refractory multiple myeloma (RRMM). 2023, 41, 8005.
- Da Via, M.C.; Dietrich, O.; Truger, M.; Arampatzi, P.; Duell, J.; Heidemeier, A.; Zhou, X.; Danhof, S.; Kraus, S.; Chatterjee, M.; et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat Med 2021, 27, 616–619. [Google Scholar] [CrossRef]
- Perica, K.; Nataraj, S.; Sjojstrand, M.L.; Nagel, K.; Pavkovic, E.; Payson, E.; Cuenca, A.; Patel, A.; Chung, D.; Mailankody, S.; et al. Low Target Antigen Expression Mediates Resistance to BCMA CAR T Cell Therapy. Blood 2023, 142, 2124. [Google Scholar] [CrossRef]
- Mailankody, S.; Devlin, S.M.; Landa, J.; Nath, K.; Diamonte, C.; Carstens, E.J.; Russo, D.; Auclair, R.; Fitzgerald, L.; Cadzin, B.; et al. GPRC5D-Targeted CAR T Cells for Myeloma. N Engl J Med 2022, 387, 1196–1206. [Google Scholar] [CrossRef]
- Zhang, M.; Wei, G.; Zhou, L.; Zhou, J.; Chen, S.; Zhang, W.; Wang, D.; Luo, X.; Cui, J.; Huang, S.; et al. GPRC5D CAR T cells (OriCAR-017) in patients with relapsed or refractory multiple myeloma (POLARIS): a first-in-human, single-centre, single-arm, phase 1 trial. Lancet Haematol 2023, 10, e107–e116. [Google Scholar] [CrossRef]
- Caillot, L.; Sleiman, E.; Lafon, I.; Chretien, M.-L.; Gueneau, P.; Payssot, A.; Pedri, R.; Lakomy, D.; Bailly, F.; Guy, J.; et al. Early Chimeric Antigen Receptor T Cell Expansion Is Associated with Prolonged Progression-Free Survival for Patients with Relapsed/Refractory Multiple Myeloma Treated with Ide-Cel: A Retrospective Monocentric Study. Transplantation and Cellular Therapy 2024. [Google Scholar] [CrossRef] [PubMed]
- Lam, N.; Trinklein, N.D.; Buelow, B.; Patterson, G.H.; Ojha, N.; Kochenderfer, J.N. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat Commun 2020, 11, 283. [Google Scholar] [CrossRef] [PubMed]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Johrer, K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol 2016, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Marquez, P.; Calleja-Cervantes, M.E.; Serrano, G.; Oliver-Caldes, A.; Palacios-Berraquero, M.L.; Martin-Mallo, A.; Calvino, C.; Espanol-Rego, M.; Ceballos, C.; Lozano, T.; et al. CAR density influences antitumoral efficacy of BCMA CAR T cells and correlates with clinical outcome. Sci Adv 2022, 8, eabo0514. [Google Scholar] [CrossRef]
- Gandhi, U.H.; Cornell, R.F.; Lakshman, A.; Gahvari, Z.J.; McGehee, E.; Jagosky, M.H.; Gupta, R.; Varnado, W.; Fiala, M.A.; Chhabra, S.; et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia 2019, 33, 2266–2275. [Google Scholar] [CrossRef]
- Chinese Medical Doctor Association, H.B.; Chinese Society of Hematology, C.M.A. [The Chinese consensus for the CAR-T cell therapy in multiple myeloma (2022 version)]. Zhonghua Xue Ye Xue Za Zhi 2022, 43, 265–271. [Google Scholar] [CrossRef]
- Subramanian, N.G.; Garcia Pleitez, H.; Pasvolsky, O.; Kalariya, N.; Patel, K.K.; Ferreri, C.J. Clinical outcomes of patients with multiple myeloma following disease progression after BCMA-targeted CAR T-cell therapy. Journal of Clinical Oncology 2023, 41, e20002–e20002. [Google Scholar] [CrossRef]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J Clin Oncol 2018, 36, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Guoxing, Z.; CHENG, Z.; Runhong, W.; Yi, W.; Lei, F.; Qiuling, M.; Xianhui, L. Efficacy and safety of anti-B cell maturation antigen chimeric antigen receptor T-cell for retreatment of relapsed/refractory multiple myeloma. Journal of Leukemia Lymphoma 2022, 229–234. [Google Scholar] [CrossRef]
- Bal, S.; Htut, M.; Nadeem, O.; Anderson, L.D.; Koçoğlu, H.; Gregory, T.; Rossi, A.C.; Martin, T.; Egan, D.N.; Costa, L.; et al. BMS-986393 (CC-95266), a G Protein-Coupled Receptor Class C Group 5 Member D (GPRC5D)-Targeted Chimeric Antigen Receptor (CAR) T-Cell Therapy for Relapsed/Refractory Multiple Myeloma (RRMM): Updated Results from a Phase 1 Study. Blood 2023, 142, 219. [Google Scholar] [CrossRef]
- Shi, M.; Wang, J.; Huang, H.; Liu, D.; Cheng, H.; Wang, X.; Chen, W.; Yan, Z.; Sang, W.; Qi, K.; et al. Bispecific CAR T cell therapy targeting BCMA and CD19 in relapsed/refractory multiple myeloma: a phase I/II trial. Nat Commun 2024, 15, 3371. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, X.; Xu, J.; Liu, J.; Mei, H. Treatment of multiple myeloma: What is the impact on T-cell function? Ther Adv Hematol 2024, 15, 20406207241245194. [Google Scholar] [CrossRef] [PubMed]
- Metelo, A.M.; Jozwik, A.; Luong, L.A.; Dominey-Foy, D.; Graham, C.; Attwood, C.; Inam, S.; Dunlop, A.; Sanchez, K.; Cuthill, K.; et al. Allogeneic Anti-BCMA CAR T Cells Are Superior to Multiple Myeloma-derived CAR T Cells in Preclinical Studies and May Be Combined with Gamma Secretase Inhibitors. Cancer Res Commun 2022, 2, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Dholaria, B.; Shune, L.; Kin, A.; McArthur, K.; Eskew, J.D.; Martin, C.E.; Haag, S.; McCaigue, J.; Namini, H.; DePrimo, S.; et al. Abstract CT071: Clinical activity of P-BCMA-ALLO1, a B-cell maturation antigen (BCMA) targeted allogeneic chimeric antigen receptor T-cell (CAR-T) therapy, in relapsed refractory multiple myeloma (RRMM) patients (pts) following progression on prior BCMA targeting therapy. Cancer Research %J Cancer Research 2024, 84, CT071–CT071. [Google Scholar] [CrossRef]
- P. Moreau, A.L. P. Moreau, A.L. Garfall, N. van de Donk, H. Nahi, J.F. San-Miguel, A. Oriol, A.K. Nooka, T. Martin, L. Rosinol, A. Chari, L. Karlin, L. Benboubker, M.V. Mateos, N. Bahlis, R. Popat, B. Besemer, J. Martinez-Lopez, S. Sidana, M. Delforge, L. Pei, D. Trancucci, R. Verona, S. Girgis, S.X.W. Lin, Y. Olyslager, M. Jaffe, C. Uhlar, T. Stephenson, R. Van Rampelbergh, A. Banerjee, J.D. Goldberg, R. Kobos, A. Krishnan, and S.Z. Usmani, Teclistamab in Relapsed or Refractory Multiple Myeloma. N Engl J Med 387 (2022) 495-505.
- Chari, M.C. Minnema, J.G. Berdeja, A. Oriol, N. van de Donk, P. Rodriguez-Otero, E. Askari, M.V. Mateos, L.J. Costa, J. Caers, R. Verona, S. Girgis, S. Yang, R.B. Goldsmith, X. Yao, K. Pillarisetti, B.W. Hilder, J. Russell, J.D. Goldberg, and A. Krishnan, Talquetamab, a T-Cell-Redirecting GPRC5D Bispecific Antibody for Multiple Myeloma. N Engl J Med 387 (2022) 2232-2244.
- A.M. Lesokhin, M.H. A.M. Lesokhin, M.H. Tomasson, B. Arnulf, N.J. Bahlis, H. Miles Prince, R. Niesvizky, P. Rodriotaguez-Otero, J. Martinez-Lopez, G. Koehne, C. Touzeau, Y. Jethava, H. Quach, J. Depaus, H. Yokoyama, A.E. Gabayan, D.A. Stevens, A.K. Nooka, S. Manier, N. Raje, S. Iida, M.S. Raab, E. Searle, E. Leip, S.T. Sullivan, U. Conte, M. Elmeliegy, A. Czibere, A. Viqueira, and M. Mohty, Elranatamab in relapsed or refractory multiple myeloma: phase 2 MagnetisMM-3 trial results. Nat Med 29 (2023) 2259-2267.
- Varshavsky-Yanovsky, A.; Jethava, Y.; Stevens, D.A.; Nooka, A.K.; Stiff, P.J.; Perez-Cruz, I.; Leip, E.; Lesokhin, A. Efficacy and Safety of Elranatamab in Black Patients with Relapsed/Refractory Multiple Myeloma (RRMM): A Subgroup Analysis of the Magnetismm Studies. Blood 2023, 142, 3333. [Google Scholar] [CrossRef]
- Trudel, S.; Cohen, A.D.; Krishnan, A.Y.; Fonseca, R.; Spencer, A.; Berdeja, J.G.; Lesokhin, A.; Forsberg, P.A.; Laubach, J.P.; Costa, L.J.; et al. Cevostamab Monotherapy Continues to Show Clinically Meaningful Activity and Manageable Safety in Patients with Heavily Pre-Treated Relapsed/Refractory Multiple Myeloma (RRMM): Updated Results from an Ongoing Phase I Study. Blood 2021, 138, 157–157. [Google Scholar] [CrossRef]
- Schinke, C.D.; Touzeau, C.; Minnema, M.C.; van de Donk, N.W.; Rodríguez-Otero, P.; Mateos, M.-V.; Rasche, L.; Ye, J.C.; Vishwamitra, D.; Ma, X. Pivotal phase 2 MonumenTAL-1 results of talquetamab (tal), a GPRC5DxCD3 bispecific antibody (BsAb), for relapsed/refractory multiple myeloma (RRMM). 2023.
- Mouhieddine, T.H.; Van Oekelen, O.; Melnekoff, D.T.; Li, J.; Ghodke-Puranik, Y.; Lancman, G.; Thibaud, S.; Pan, D.; Rajeeve, S.; Agte, S.; et al. Sequencing T-cell redirection therapies leads to deep and durable responses in patients with relapsed/refractory myeloma. Blood Adv 2023, 7, 1056–1064. [Google Scholar] [CrossRef]
- Banerjee, R.; Lynch, R.C.; Wu, Q.V.; Simon, S.; Ujjani, C.S.; Till, B.G.; Wuliji, N.; Gausman, D.; Dizon, J.; Kwok, M.L.; et al. Post-CAR-T Checkpoint Inhibition Can Result in Durable Responses in a Minority of Patients with Multiple Myeloma (MM) or Non-Hodgkin's Lymphoma (NHL): Results of a Phase 2 Study of Nivolumab after CAR-T Failure. Transplantation and Cellular Therapy 2024, 30, S399–S400. [Google Scholar] [CrossRef]
- Vaxman, I.; Abeykoon, J.; Dispenzieri, A.; Kumar, S.K.; Buadi, F.; Lacy, M.Q.; Dingli, D.; Hwa, Y.; Fonder, A.; Hobbs, M.; et al. "Real-life" data of the efficacy and safety of belantamab mafodotin in relapsed multiple myeloma-the Mayo Clinic experience. Blood Cancer J 2021, 11, 196. [Google Scholar] [CrossRef]
- Gazeau, N.; Beauvais, D.; Yakoub-Agha, I.; Mitra, S.; Campbell, T.B.; Facon, T.; Manier, S. Effective anti-BCMA retreatment in multiple myeloma. Blood Adv 2021, 5, 3016–3020. [Google Scholar] [CrossRef]
- Agte, S.; Elnaggar, M.; Adamopolous, C.; Melnekoff, D.; Adleman, A.; Kappes, K.; Restrepo, P.; Oekelen, O.V.; Leshchenko, V.; Poulikakos, P.I.; et al. P-090: BRAF V600E multiple myeloma patient salvaged with triple MAPK inhibition after CAR T relapse. Clinical Lymphoma Myeloma and Leukemia 2021, 21, S88. [Google Scholar] [CrossRef]
- Qian, Y.; Qian, Z.; Zhao, X.; Pan, W.; Wei, X.; Meng, H.; Yang, L.; Xiao, H. Successful Treatment of Relapsed/Refractory Extramedullary Multiple Myeloma With Anti-BCMA CAR-T Cell Therapy Followed by Haploidentical Hematopoietic Stem Cell Transplantation: A Case Report and a Review of the Contemporary Literature. Front Med (Lausanne) 2021, 8, 649824. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Dancy, E.K.; Cohen, A.D.; Hwang, W.T.; Fraietta, J.A.; Davis, M.M.; Levine, B.L.; Siegel, D.L.; Stadtmauer, E.A.; Vogl, D.T.; et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv 2019, 3, 2812–2815. [Google Scholar] [CrossRef]
- Dima, D.; Ullah, F.; Mazzoni, S.; Williams, L.; Faiman, B.; Kurkowski, A.; Chaulagain, C.; Raza, S.; Samaras, C.; Valent, J.; et al. Management of Relapsed-Refractory Multiple Myeloma in the Era of Advanced Therapies: Evidence-Based Recommendations for Routine Clinical Practice. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Abecassis, A.; Roders, N.; Fayon, M.; Choisy, C.; Nelson, E.; Harel, S.; Royer, B.; Villesuzanne, C.; Talbot, A.; Garrick, D.; et al. CAR-T cells derived from multiple myeloma patients at diagnosis have improved cytotoxic functions compared to those produced at relapse or following daratumumab treatment. EJHaem 2022, 3, 970–974. [Google Scholar] [CrossRef]
- Rodriguez-Otero, P.; Ailawadhi, S.; Arnulf, B.; Patel, K.; Cavo, M.; Nooka, A.K.; Manier, S.; Callander, N.; Costa, L.J.; Vij, R.; et al. Ide-cel or Standard Regimens in Relapsed and Refractory Multiple Myeloma. N Engl J Med 2023, 388, 1002–1014. [Google Scholar] [CrossRef]
- Wu, J.F.; Dhakal, B. BCMA-targeted CAR-T cell therapies in relapsed and/or refractory multiple myeloma: latest updates from 2023 ASCO Annual Meeting. J Hematol Oncol 2023, 16, 86. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Lin, Q.; Zhao, J.; Song, Y.; Liu, D. Recent updates on CAR T clinical trials for multiple myeloma. Mol Cancer 2019, 18, 154. [Google Scholar] [CrossRef]
- Ahmed, N.; Wesson, W.; Mushtaq, M.U.; Bansal, R.; AbdelHakim, H.; Bromert, S.; Appenfeller, A.; Ghazal, B.A.; Singh, A.; Abhyankar, S.; et al. "Waitlist mortality" is high for myeloma patients with limited access to BCMA therapy. Front Oncol 2023, 13, 1206715. [Google Scholar] [CrossRef]
- Afrough, A.; Hashmi, H.; Hansen, D.K.; Sidana, S.; Ahn, C.; Dima, D.; Freeman, C.L.; Castaneda Puglianini, O.A.; Kocoglu, M.H.; Atrash, S.; et al. Impact of bridging therapy (BT) on outcome of relapsed refractory multiple myeloma (RRMM) with Ide-cel CAR T-cell therapy: Real-world experience from the US myeloma CAR T consortium. Journal of Clinical Oncology 2023, 41, 8013–8013. [Google Scholar] [CrossRef]
- Sperling, A.S.; Derman, B.A.; Nikiforow, S.; Im, S.-Y.; Ikegawa, S.; Prabhala, R.H.; Rodriguez, D.H.; Li, Y.; Quinn, D.S.; Pearson, D.; et al. Updated phase I study results of PHE885, a T-Charge manufactured BCMA-directed CAR-T cell therapy, for patients (pts) with r/r multiple myeloma (RRMM). 2023, 41, 8004–8004. [CrossRef]
- Costa, L.J.; Kumar, S.K.; Atrash, S.; Liedtke, M.; Kaur, G.; Derman, B.A.; Bergsagel, P.L.; Mailankody, S.; McCarthy, P.L.; Limones, J.; et al. Results from the First Phase 1 Clinical Study of the B-Cell Maturation Antigen (BCMA) Nex T Chimeric Antigen Receptor (CAR) T Cell Therapy CC-98633/BMS-986354 in Patients (pts) with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2022, 140, 1360–1362. [Google Scholar] [CrossRef]
- Liu, H.; Wang, T.; Yang, Y.; Feng, R.; Li, J.; Zhang, C.; Bai, J.; Ding, Y.; Liu, G.; Wu, F.; et al. Phase I Study of a BCMA-Directed CAR-T Cell Therapy for Relapsed/Refractory Multiple Myeloma Manufactured in <3 Days Using the High-Performance Platform. Blood 2022, 140, 10307–10308. [Google Scholar] [CrossRef]
- Choi, T.; Kang, Y. Chimeric antigen receptor (CAR) T-cell therapy for multiple myeloma. Pharmacol Ther 2022, 232, 108007. [Google Scholar] [CrossRef] [PubMed]
- Kegyes, D.; Constantinescu, C.; Vrancken, L.; Rasche, L.; Gregoire, C.; Tigu, B.; Gulei, D.; Dima, D.; Tanase, A.; Einsele, H.; et al. Patient selection for CAR T or BiTE therapy in multiple myeloma: Which treatment for each patient? J Hematol Oncol 2022, 15, 78. [Google Scholar] [CrossRef]
- Dima, D.; Rashid, A.; Davis, J.A.; Shune, L.; Abdallah, A.O.; Li, H.; DeJarnette, S.; Khouri, J.; Williams, L.; Hashmi, H.; et al. Efficacy and safety of idecabtagene vicleucel in patients with relapsed-refractory multiple myeloma not meeting the KarMMa-1 trial eligibility criteria: A real-world multicentre study. Br J Haematol 2024, 204, 1293–1299. [Google Scholar] [CrossRef]
- Cohen, A.D.; Mateos, M.V.; Cohen, Y.C.; Rodriguez-Otero, P.; Paiva, B.; van de Donk, N.; Martin, T.; Suvannasankha, A.; De Braganca, K.C.; Corsale, C.; et al. Efficacy and safety of cilta-cel in patients with progressive multiple myeloma after exposure to other BCMA-targeting agents. Blood 2023, 141, 219–230. [Google Scholar] [CrossRef]
- Wang, X.; Walter, M.; Urak, R.; Weng, L.; Huynh, C.; Lim, L.; Wong, C.W.; Chang, W.C.; Thomas, S.H.; Sanchez, J.F.; et al. Lenalidomide Enhances the Function of CS1 Chimeric Antigen Receptor-Redirected T Cells Against Multiple Myeloma. Clin Cancer Res 2018, 24, 106–119. [Google Scholar] [CrossRef]
- Works, M.; Soni, N.; Hauskins, C.; Sierra, C.; Baturevych, A.; Jones, J.C.; Curtis, W.; Carlson, P.; Johnstone, T.G.; Kugler, D.; et al. Anti-B-cell Maturation Antigen Chimeric Antigen Receptor T cell Function against Multiple Myeloma Is Enhanced in the Presence of Lenalidomide. Mol Cancer Ther 2019, 18, 2246–2257. [Google Scholar] [CrossRef]
- Zhao, G.; Wei, R.; Feng, L.; Wu, Y.; He, F.; Xiao, M.; Cheng, Z. Lenalidomide enhances the efficacy of anti-BCMA CAR-T treatment in relapsed/refractory multiple myeloma: a case report and revies of the literature. Cancer Immunol Immunother 2022, 71, 39–44. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. gamma-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.J.; Pont, M.J.; Sather, B.D.; Turtle, C.J.; Till, B.G.; Libby, E.N., 3rd; Coffey, D.G.; Tuazon, S.A.; Wood, B.; Gooley, T.; et al. gamma-Secretase inhibitor in combination with BCMA chimeric antigen receptor T-cell immunotherapy for individuals with relapsed or refractory multiple myeloma: a phase 1, first-in-human trial. Lancet Oncol 2023, 24, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Aleman, A.; Kogan-Zajdman, A.; Upadhyaya, B.; Van Oekelen, O.; Chen, L.; Leshchenko, V.; Jagannath, S.; Parekh, S. P-175 Improving Anti-BCMA CAR-T functionality with novel immunomodulatory agent Iberdomide (CC220) in Multiple Myeloma. Clinical Lymphoma Myeloma and Leukemia 2023, 23, S131–S132. [Google Scholar] [CrossRef]
- Hassan, H.; Samur, M.K.; Cohen, A.D.; Moreau, P.; Rodríguez Otero, P.; Kumar, S.K.; Mateos, M.V.; Avigan, D.; Sperling, A.S.; Nadeem, O.; et al. Heterogeneity in Access and Toxicity Management of Commercially Available BCMA-Directed CAR-T and Bispecific T-Cell Engager Therapy Among the International Myeloma Community. Blood 2023, 142, 7248–7248. [Google Scholar] [CrossRef]
- Hashmi, H.; Kumar, A.; Kharfan-Dabaja, M.A.; Munshi, P.N.; Inamoto, Y.; DeFilipp, Z.M.; Dholaria, B.; Jain, T.; Perales, M.A.; Carpenter, P.A.; et al. ASTCT Committee on Practice Guidelines Survey on Evaluation & Management of Relapsed Refractory Multiple Myeloma after Failure of Chimeric Antigen Receptor T-Cell Therapy. Transplant Cell Ther 2024. [Google Scholar] [CrossRef]
- Deng, J.; Lin, Y.; Zhao, D.; Tong, C.; Chang, A.H.; Chen, W.; Gao, W. Case report: Plasma cell leukemia secondary to multiple myeloma successfully treated with anti-BCMA CAR-T cell therapy. Front Oncol 2022, 12, 901266. [Google Scholar] [CrossRef]
- Arnulf, B.; Kerre, T.; Agha, M.E.; Delforge, M.; Braunschweig, I.; Shah, N.; Richard, S.; Alsina, M.; Einsele, H.; Mistry, P.; et al. Efficacy and safety of ciltacabtagene autoleucel ± lenalidomide maintenance in newly diagnosed multiple myeloma with suboptimal response to frontline autologous stem cell transplant: CARTITUDE-2 cohort D. 2024, 42, 7505–7505. [CrossRef]

Table 1.
Some Clinical Trial Result of CAR-T Therapy.
| Trial Code/Number | CAR-T Product | CAR-T Target | Median Follow-up (months) | Efficacy | Safety | |||
|---|---|---|---|---|---|---|---|---|
| ORR | Median PFS (months) | Median OS (months) | CRS | ICANS | ||||
| LEGEND-2 (NCT03090659) |
LCAR-B38M [19] | BCMA | 65.4 | 87.8% | 18 | 55.8 | - | - |
| CRB401 (NCT02658929) |
bb2121 [20] | BCMA | 18.1 (1.5-44.5) | 75.8% | 8.8 (95% CI, 5.9-11.9) | 34.2 (95% CI, 19.2-NE) | 75.8% | - |
| FUMANBA-1 (NCT05066646) |
CT103A [21] | BCMA | 13.8 (0.4-27.2) | 96% | NR | - | - | - |
| LUMMICAR-1 (NCT03975907) |
CT053 [22] | BCMA | 37.7 (14.8-44.2) | 100% | 25 | NR | 92.9% | 0 |
| POLARIS (NCT05016778) |
OriCAR-017 [24] | GPRC5D | >24 (all patients) | 100% | 11.37 (95% CI, 5.93-18.00) | NR | 100% | 0 |
| ChiCTR2100048888 | anti-GPRC5D CAR-T [25] | GPRC5D | 5.2 (3.2-8.9) | 91% | NR | NR | 76% | 6% |
| ChiCTR1800018143 | BM38 CAR-T [26] | BCMA & CD38 | 9.0 (0.5-18.5) | 87% | 17.2 (95% CI, 7.5-26.8) | NR | 87% | - |
| NCT04662099 | CS1-BCMA CAR-T [27] | CS1 & BCMA | 246 days (55-547) | 81% | 9.0 (95% CI, 2.1-NR) | NR | 38% | 0 |
| NCT04236011 & NCT04182581 | GC012F [28] | BCMA & CD19 | 30.7 (14.6-43.6) | 93.1% | 38.0 (95% CI, 11.8-NE) | - | 86.2% | 0 |
IQR, interquartile range; NE, not evaluable.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



