
Submitted:
21 October 2024
Posted:
22 October 2024
You are already at the latest version
Abstract
Background: There is paucity of evidence on the association of genetic propensity for hippocampal atrophy with cognitive outcomes. Therefore, we examined the relationship of a Polygenic Risk Score for hippocampal atrophy (PRShp) with the incidence of amnestic mild cognitive impairment (aMCI) and Alzheimer’s disease (AD) as well as the rates of cognitive decline. Methods: Participants were drawn from the population based HELIAD cohort. Comprehensive neuropsychological assessments were performed at baseline and follow-up. PRShp was derived from the summary statistics of a large GWAS for hippocampal volume. Cox proportional hazards models as well as generalized estimating equations (GEEs) were used to evaluate the association of PRShp with the combined incidence of aMCI/AD and cognitive changes over time, respectively. All models were adjusted for age, sex, education, and Apolipoprotein E (APOE) genotype. Results: Our analysis included 618 older adults, among whom 73 developed aMCI/AD after an average follow-up of 2.96±0.8 years. Each additional SD of PRShp elevated the relative hazard for incident aMCI/AD by 46%. Participants at the top quartile of PRShp had almost three times higher risk of converting to aMCI/AD compared to the lowest quartile group. Higher PRShp scores were also linked to steeper global cognitive and memory decline. The impact of PRShp was greater among women and younger adults. Conclusions: Our findings support the association of PRShp with aMCI/AD incidence and with global cognitive and memory decline over time. PRS association was sex and age dependent, suggesting that these factors should be considered in genetic modelling for AD.
Keywords:
hippocampal atrophy
; Alzheimer’s disease
; cognitive decline
; polygenic risk score
1. Introduction
The Amyloid Tau Neurodegeneration (ATN) framework reflects the pathological hallmarks of Alzheimer’s disease (AD): Aβ amyloid deposition (A), tau aggregation (T) and neurodegeneration (N). These markers differentiate AD pathology from other neurocognitive disorders[1]. Unfortunately, the evaluation of ATN markers is limited by the costs or invasive nature of associated procedures.
Of note, novel findings from genetic association studies suggest that AD-related pathology is strongly linked to genetics, leading to a deeper understanding of how genetics contribute to AD-related neurodegeneration [2,3]. Although genetic measures (such as polygenic risk scores -PRS) fail to encapsulate the full and changing landscape of neurodegeneration, they present important advantages in that they capture the time-invariant risk-conferring constituent of AD-related pathology (regardless of the age and time of assessment) and by providing better large-scale applicability. There is, however, paucity of published evidence on the direct association between genetics and clinical outcomes, as the majority of studies have focused on endophenotypes rather than clinical manifestations [4,5].
Hippocampal volume is a well-established structural marker of AD-related neurodegeneration [6]. Numerous studies have already related hippocampal atrophy to more precipitous trajectories of cognitive decline and greater risk of incident AD [7,8]. Of note, hippocampal volumes are highly heritable with genetic factors accounting for 50-70% of the variability in hippocampal size- [4,9]. In a genome-wide association study (GWAS), genes linked to neuronal differentiation, locomotive and exploratory behavior, AD and schizophrenia were associated with hippocampal subfields volumes and the reported SNP-based heritability estimates ranged between 0.1 to 0.3 [10]. However, to our knowledge, only one study has found an association between genetic propensity for higher hippocampal volumes with better executive function and slower verbal fluency decline [4].
Therefore, the aim of the current study was to explore the longitudinal association of genetic predisposition towards smaller hippocampal volumes (hippocampal atrophy) with a) the incidence of Alzheimer’s disease dementia (AD) or amnestic mild cognitive impairment (aMCI) and b) rates of cognitive decline. For this purpose, we capitalized on data from the population-based HELIAD (Hellenic Longitudinal Investigation of Aging and Diet) cohort, a randomly selected sample of community-dwelling older adults. PRS for hippocampal volume (PRShp) derived from the summary statistics of a large GWAS [10]. Potential dependence of genetic risk for hippocampal atrophy on age and sex was examined, as well.
2. Materials and Methods
2.1. Settings and Participants
Our sample was drawn from the prospective HELIAD study, a multidisciplinary population-based cohort that primarily explores the epidemiology of dementia and other neuropsychiatric disorders in the older Greek population [11]. Study procedures were approved by the Institutional Ethics Review Boards of the National and Kapodistrian University of Athens and the University of Thessaly and conducted, in accordance with principles of Good Clinical Practice and the Helsinki Declaration or later amendments. All participants or surrogates provided informed consent, prior to participation.
Older participants (>64 years) were randomly selected from the rosters of two Greek municipalities, one in the rural area of Larissa (province of Thessaly) and one in the urban area of Marousi (suburb of Athens). Collaborative assessments were conducted by certified neurologists, trained neuropsychologists and dieticians. Relevant information was collected either from participants themselves or from participants’ carers (first-degree relatives, spouses, etc.), whenever deemed necessary. Participants underwent baseline and follow-up evaluations after approximately 3-year intervals. Extensive details about the design and key features of the HELIAD study have been described previously [12].
2.2. Neuropsychological Evaluation
Cognition was evaluated by trained neuropsychologists according to a designated approach involving the comprehensive assessment of all major cognitive domains [13]. Episodic memory was assessed through the Greek verbal learning test (GVLT; verbal memory) and the Medical College of Georgia Complex Figure Test (MCG; non-verbal memory); language on the semantic and phonological fluency tasks and subtests of the Greek version of the Boston Diagnostic Aphasia Examination short form (BDAE; the Boston Naming Test-short form, and selected items from the Complex Ideational Material Subtest, to assess verbal comprehension and repetition of words and phrases); visuospatial ability on the Judgment of Line Orientation abbreviated form, the MCG copy condition and the clock drawing test; attention and processing speed on the Trail Making Test - Part A (TMT-A); executive functioning on the Trail Making Test - Part B (TMT-B), Anomalous Sentence Repetition (created for the present investigation), Graphical Sequence Test and Motor Programming. Individual test raw scores were converted into z-scores using mean and standard deviation (SD) values of the participants who were cognitively normal (without MCI or dementia) at baseline [14]. Higher scores were consistent with better cognitive performance.
2.3. Diagnostic Approach
All participants underwent a standard physical and neurological examination. Particular focus was placed on identifying potential comorbidities that could interfere with cognition, through screening the participants for depression, anxiety, essential tremor, behavioural symptoms, Parkinson disease, dementia with Lewy bodies (DLB), as well as personal history of cerebrovascular disease accounting for the onset or deterioration of cognitive decline. Information was also gathered on comorbidities, regular medication intake, sleep and dietary habits, mental and physical activity, as well as laboratory tests (imaging studies and blood examinations) when available.
The diagnostic classification of the participants according to their cognitive status was established during expert consensus meetings involving senior neurologists and neuropsychologists. Dementia and AD were diagnosed according to the Diagnostic and Statistical Manual of Mental Disorders -IV-text revision criteria [15] and the National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer Disease and Related Disorders Association criteria [16], respectively. The diagnosis of vascular dementia was based on a history or clinical evidence of stroke, the presence of a clear temporal relation between stroke and the onset of dementia and the Hachinski Ischemia Scale score [17]. Lewy body and frontotemporal dementias were diagnosed based on the respective criteria [18,19]. MCI and its subtypes were diagnosed according to the Petersen criteria [20]. MCI was categorized as amnestic in cases of isolated memory impairment or multi-domain impairment involving memory, and as non-amnestic in cases of isolated or combined language, attention, executive, or visuo-perceptual impairment (not involving memory).
2.4. Genotyping and Imputation
Genome-wide genotyping was performed at Life & Brain facilities (Germany) using the Illumina Infinium Global Screening Array and calling was generated by the “centre national de recherche en génétique humaine” (France) using the data generated by the centres involved in genotyping (Life & Brain, CNRGH and Amsterdam). Details has been described elsewhere (Supplementary Material S1.1) [21].
2.5. Polygenic Risk Score Calculation
PRS for hippocampal volume was derived from the summary statistics of a large GWAS on the genetic architecture of hippocampal volumes. The data for this study was derived from 21,297 participants across 16 different cohorts who had available T1-weighted brain scans [10]. In the GWAS summary statistics, each SNP is associated with hippocampal volume levels at a certain p-value threshold.
Details on the polygenic risk calculation are in Supplementary material S1.2. For each subject, we computed 10 different genome-wide PRS for hippocampal volumes based on a priori set of 10 P-value thresholds (PT). To calculate each PRS, we consecutively used reduced sets of genetic variants, summing over all SNPs meeting each of the following significance thresholds: p<0.0001, p<0.001, p<0.01, p<0.05, p<0.1, p<0.2, p<0.3, p<0.4, p<0.5 and p<1.0. Given that hippocampal volumes are inversely related with the risk of incident AD, PRS for hippocampal volumes were multiplied by -1 to align a higher risk score with smaller hippocampal volumes. To facilitate interpretation, PRS were standardized to have a mean zero and SD of 1.
Since each PRS threshold consists of a different set of SNPs, we constructed a set of logistic regression models with the presence of AD-aMCI as the outcome, and the different PRS thresholds as the main predictors. Then, Receiver Operator Characteristic (ROC) curves were constructed for each model. Area under the curve (AUC) and p-values were estimated. The PRS threshold of the model with the best classification accuracy, estimated by calculating the area under the ROC curve, was considered the one that can predict better hippocampal volumes and as such it was used as the measure of genetic predisposition for smaller hippocampal volumes (Table S1).
2.6. Statistical Analyses
Analyses were performed using SPSS 26 (SPSS, Chicago, Ill., USA). Baseline characteristics between groups were compared via independent sample t-test for continuous variables and Pearson’s chi-squared test for categorical variables. The significance level was set at α ≤ 0.05.
PRS were normally distributed according to the Kolmogorov Smirnoff and Shapiro Wilk tests for normality, so we initially treated standardized PRS as scale variable. In turn, to evaluate potential threshold effects we trichotomized our cohort according to PRS quartiles into low (1st quartile) vs. intermediate (2nd and 3rd quartiles) vs. high (4th quartile) PRS. Adjusted models included age and education (in years of formal schooling) as covariates in a continuous scale together with sex (female vs male as reference) and ApoE genotype (ApoE4 carriers vs ApoE4 non carriers as reference) treated as categorical variables. To correct for cryptic relatedness between subjects or unexpected genotyping batch-related errors [22], we included in the models as covariates the first two principal components (PCs) of the whole sample, similarly to previous studies [5].
2.6.1. Cox Proportional Hazard Models
In Cox models, the combined incidence of clinical aMCI (among all MCI types) and AD (among all dementia types) was used as dichotomous outcome. We used this composite event due to the substantially small number of incident AD cases that would considerably undermine the power our analyses. We selected aMCI and no other MCI types because aMCI has been defined as a prodromal stage of AD, since a considerably higher rate of aMCI cases will be converted to AD compared to other types of MCI. Participants with baseline diagnosis of dementia or aMCI were removed from the analysis. Participants that converted to other dementia entities at follow-up were excluded as well, due to the competing nature of different dementia diagnoses. Those who did not develop aMCI or dementia were censored at follow-up. The interval between the baseline evaluation and follow-up was inserted as the time-to-event variable.
First, we examined the association of PRS for hippocampal atrophy with risk of aMCI or AD over time through Cox models adjusted for age, sex, educational attainment, ApoE genotype, PC1 and PC2. The proportionality of hazards assumption was confirmed using Cox regression analyses with time-dependent covariates. In particular, for each PRS an extended Cox model including the term PRS*time along with the PRS was analysed. To verify that the proportionality of hazards assumption was not violated, the coefficient of the time interaction product had to be statistically insignificant. Afterwards, the proportionality of hazards for different PRS strata (quartile approach) over time was confirmed via the same approach.
Next, we performed subgroup analyses, to examine for potential sex and age cohort effects [5]: 1) men vs. women, 2) according to age, participants were divided into older vs. younger, using the median of our sample as cut-off (72.67 years).
2.6.2. Generalized Estimating Equations
The effect of PRS for hippocampal atrophy on rates of cognitive decline was explored using generalised estimating equations (GEE) analyses. GEE accounts for the potential correlation of repeated measurements in the same individual. We treated each participants’ baseline and follow-up evaluations as a cluster. Both autoregressive and exchangeable covariance matrices were used as working correlation structures with comparable results. Six consecutive GEE models were explored using the composite and individual domain cognitive measurements (memory, language, executive function, visuospatial perception, attention) as the dependent scale variables. GEE analyses featured the main effects of PRS (first as scale and followingly as trichotomous variable) and time from baseline as well as PRS by time interaction terms. Models were again controlled for age, educational attainment, sex, ApoE genotype, PC1 and PC2.
Finally, to explore for potential disparities regarding the impact of PRS on different sex and age, we performed subgroup analyses based on the same approach described above.
3. Results
3.1. Participant Characteristics and Missing Data Analysis
There were 1017 unrelated, older adults, with available genetic data and without aMCI or dementia at baseline. Follow-up information were not available for 378 participants, 52 converted to aMCI, 21 progressed to AD, 4 developed other dementias and 16 had missing follow-up values on MCI-dementia. Thus, the present analysis involved a total of 619 participants at baseline, 73 of whom developed aMCI or AD at follow-up. Compared to those included (n=619), those without available follow-up information (n=378) were older (74.7 ±5.3 vs. 73.4 ±5.0 years, p<0.001) and less educated (6.5 ±4.1 vs. 7.3 ±4.7 years, p=0.002). Sex (p= 0.612) and ApoE4 (p=0.659) distributions were similar between the two groups. No differences were found with respect to PRS.
The average follow-up of included participants was 2.96 ±0.80 years (between 1.28 and 6.93 years). The demographic and genetic characteristics of the total sample, based on the incidence of aMCI or AD, are presented in Table 1. Those who developed aMCI or AD at follow-up were older, less educated and had higher PRS for hippocampal atrophy.
3.2. Polygenic Risk Score for Hippocampal Atrophy and Incident Amnestic MCI or AD
The proportionality of hazards assumption was confirmed for both scale and trichotomous PRS. First, we tested the unadjusted relationship between the ten PRS constructed at different PT and the risk of aMCI or AD (Supplementary Table 1). Results showed a significant unadjusted association between the PRS calculated at PT=0.01 (8475 SNPs included) and aMCI-AD status at follow-up. Higher PRS values were associated with a significantly higher hazard for progressing to aMCI or AD; one SD increased the relative risk by 39% (p=0.005). When participants were trichotomized according to PRS quartiles, higher PRS strata were again related to significantly higher aMCI-AD risk at the 0.01 PT (p for trend=0.036). Results showed that those who belong to the highest quartile of PRS for hippocampal atrophy had ~2.57 higher risk of developing aMCI or AD (p=0.011) compared to individuals belonging to the bottom quartile of PRS.
Subsequently, adjusted Cox models were tested using PRS calculated at PT=0.01 (Table 2). Adjusted associations were even more prominent, with one SD elevating the relative hazard for incident aMCI or AD by 46% (p=0.002). At the same time, the top quartile of PRS exhibited almost three (~2.81, p=0.006) times higher risk of progressing to aMCI or AD compared to the lowest quartile (Figure 1).
In subgroup analyses (Table 2), it was revealed that the association of PRS for hippocampal atrophy with incident aMCI-AD was mostly driven by women, with one SD of PRS enhancing the relative hazard by 60% in women. In parallel, the upper PRS quartile had almost thrice (~2.89, p=0.033) the risk of developing aMCI or AD at follow-up compared to the lowest quartile. By contrast, results were not significant in men. Regarding age, genetic propensity for hippocampal atrophy was found to affect younger individuals more than older ones. In specific, one additional SD of PRS increased the risk of future aMCI-AD by almost two (~1.87) times in those younger than 72.67 years while associations did not achieve statistical significance in the older age group.
3.3. Polygenic Risk Score for Hippocampal Atrophy and Rates of Cognitive Decline
First, we tested adjusted GEE models using PRS as scale variable (Table 3). Results suggested that for each extra SD of PRS the rates of yearly global cognitive decline increased by 1.3% of a SD. Although comparable associations were captured for memory (1.5%) executive function (1.2%) and language (1.2%), these estimates were less precise and did not reach statistical significance. Subsequently, we tested adjusted GEE models using PRS as trichotomous variable (Table 3). It was found that the top PRS quartile experienced more precipitous memory (by 5.1% per year) and global cognitive decline (by 3.8% annualy) compared to the low quartile.
In subgroup analyses, it was revealed once again that the relationship between genetic predisposition to hippocampal atrophy and cognitive decline was mostly driven by women (Supplementary Table 1). In specific, higher PRS was related to steeper global cognitive, executive, memory, language, as well as visuoperceptual decline in older female but not male adults. The association between PRS and cognitive decline was differentiated only with respect to executive function between younger and older adults: executive function diminished in a steeper fashion among those younger than 72.67 years (Supplementary Table 2).
4. Discussion
In the present study, we found that PRS for hippocampal atrophy was related to greater risk of incident aMCI or AD, as well as to steeper global cognitive and memory decline. It was revealed that these associations were more prominent in women compared to men and in younger versus older adults. Of note, in the female subgroup, apart from memory, higher PRS was associated with steeper global cognitive, executive function, language, and visuoperceptual skills decline.
Hippocampus is the central hub of a complex brain network supporting episodic memory [23]. Episodic memory impairment is the neuropsychological correlate of typical AD, while aMCI (involving episodic memory impairment) is the MCI precursor of AD [24,25]. Initial pathological alterations in AD take place in the medial temporal lobes including the hippocampal formations [26]. Subsequently, hippocampal volume loss is an early structural change in CU individuals at high risk of developing AD and is considered a specific marker of AD-related neurodegeneration [27]. As AD-related pathology accumulates, brain reserve -a term that encompasses the structural characteristics of the brain which enable individuals to better cope with neuropathological alterations- assumes a crucial role in the individual susceptibility to disease [28]. In this general framework, genetic predisposition towards hippocampal atrophy could not only reflect increased propensity for AD-related neurodegeneration but also decreased resilience leading to unsuccessful adaptation to these changes.
Amyloid-β deposition has been longitudinally related to hippocampal volume loss spreading from selective subfields across the hippocampus and concurring memory decline [29]. Recent evidence suggests that large hippocampal volumes may represent a measure of brain reserve that allows older adults to maintain normal cognition in spite of amyloid accumulation [30]. According to this scenario, in the presence of comparable pathological changes, individuals with larger hippocampal volumes have greater brain resilience, will remain asymptomatic for longer periods of time and progress towards aMCI/AD in a slower manner. However, until additional evidence is acquired, the concept that hippocampal volumes constitute a measure of brain reserve will remain purely theoretical.
Irrespective of the neurobiological underpinnings, hippocampal atrophy is an early structural change in CU individuals at high risk of developing AD and is considered a specific marker of AD-related neurodegeneration [27]. Structural magnetic resonance imaging (MRI) is the standard modality to quantify hippocampal volumes. The routine utilization of structural MRI, especially in cases of CU older adults (for research purposes) is hindered by a number of limitations. PRS for hippocampal atrophy admittedly outweigh the standard modality in terms of procedure-related limitations (e.g., tolerability or applicability in cases of metal implants), re-evaluation requirements (PRS values are interchangeable regardless of the age and timing of assessment) and large-scale applicability. Hippocampal volumes constitute a highly heritable trait [4,9] and our findings suggest that a PRS for hippocampal atrophy could be useful in a research setting as a potential alternative to structural MRI in studies conducted in large cohorts with available genetic data such as UKBiobank [31].
Of note, the impact of PRS was found to be sex-dependent and more pertinent to women. The neurobiology of cognition differs between men and women, with sex differences likely being influenced by a great number of factors including psychosocial, cultural, behavioural and most notably biological parameters (e.g., sex hormones). Previous research examining sex interactions with AD-related pathology has demonstrated that the long-standing female advantage in episodic memory and verbal fluency (which is heavily based on executive skills) is moderated or even vanished in the presence of biomarkers of AD-related pathology in non-demented adults (e.g., positive amyloid positron emission tomography, high amyloid level in the cerebrospinal fluid, lower temporal lobe glucose metabolic rates or smaller hippocampal volumes) [32,33,34]. Notably, a published survey has specifically found that the female supremacy in episodic memory is diminished with smaller hippocampal volumes among individuals with aMCI [35]. -In line with these findings and using PRS for hippocampal atrophy as proxy for smaller hippocampal volumes we -found that greater hippocampal atrophy is related to steeper cognitive changes in female compared to male older adults. Further studies should examine if this association could in time moderate or eliminate the long-standing female advantage in cognition.
As for age, although previous research has shown that hippocampal volumes are longitudinally related to cognitive -especially episodic memory- changes over time in older adults above 65 years of age, potential differences between younger versus older individuals in this age group have not been explored [36]. Therefore, the reproducibility of our findings cannot be tested.
Strengths and Limitations
The present article has several strengths. To our knowledge this is the first study examining the association between PRS for hippocampal atrophy and incidence of aMCI and AD. Subgroup analyses were performed to identify the potential exaggerated (or deflated) influence of genetic predisposition to hippocampal atrophy in different demographic subgroups. Another strength is the longitudinal design of our study that enabled us to eliminate any life-long genetic effects on our subgroup of older individuals and focus on the contemporary impact of genetics, during the follow-up period of the study. To address the potential impact of advanced undergoing neurodegenerative processes on the rates of cognitive decline we analyzed only participants with normal cognition throughout the follow-up. An expert-consensus clinically established diagnosis of dementia based on standard criteria and supported by a comprehensive neuropsychological evaluation augmented the accuracy of the diagnostic categorization of the participants.
However, the current survey has several limitations, as well. First, volumetric measurements from MRI scans were not available and thus information on the intermediate phenotype was lacking. Second, we did not have access to ATN biomarkers to corroborate the clinically-established neuropsychiatric diagnoses; such biomarkers are considered to increase the precision in the identification and differential diagnosis of dementia. Therefore, misclassification bias may yet be present. Third, non-response and attrition biases may have influenced our findings since follow-up assessments were not conducted for a non-trivial proportion of the original sample. Additionally, the moderate follow-up period of the present study led to the documentation of a small number of events and may have underpowered our analyses. Finally, HELIAD participants are of Greek ancestry and results may not be applied to other ethnic groups. Further studies should be performed in other ethnic groups to confirm these findings.
Supplementary Materials
Author Contributions
Conceptualization, I.L.; methodology, I.L. and V.S.; investigation, I.L., formal analysis, I.L. and S.N.S.; writing—original draft preparation, I.L., writing—review and editing, N.M., S.C., S.N.S., I.F., A.H., A.R., J.C.L., M.Y., M.H.K., E.D., G.M.H., P.S., K.R. and N.S.; supervision, N.S..; project administration, N.S..; funding acquisition, N.S. and E.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the following grants: IIRG-09-133014 from the Alzheimer’s Association; 189 10276/8/9/2011 from the ESPA-EU program Excellence Grant (ARISTEIA), which is co-funded by the European Social Fund and Greek National resources, and DY2b/oik.51657/14.4.2009 from the Ministry for Health and Social Solidarity (Greece). The funders had no role in the design, analysis or writing of this article.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Boards of the National and Kapodistrian University of Athens (Approval Code: 256) and the University of Thessaly (Approval Code: 138).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author [N.S.], upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Koychev, I.; Jansen, K.; Dette, A.; Shi, L.; Holling, H. Blood-Based ATN Biomarkers of Alzheimer’s Disease: A Meta-Analysis. J Alzheimers Dis. 2021, 79, 177–195. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Jung, S.H.; Kim, J.; Jang, H.; Kang, S.H.; Hwangbo, S.; et al. Identifying novel genetic variants for brain amyloid deposition: A genome-wide association study in the Korean population. Alzheimer’s Research & Therapy. 2021, 13, 117. [Google Scholar]
- Seo, J.; Byun, M.S.; Yi, D.; Lee, J.H.; Jeon, S.Y.; Shin, S.A.; et al. Genetic associations of in vivo pathology influence Alzheimer’s disease susceptibility. Alzheimer’s Research & Therapy. 2020, 12, 156. [Google Scholar]
- Armstrong, N.M.; Dumitrescu, L.; Huang, C.W.; An, Y.; Tanaka, T.; Hernandez, D.; et al. Association of Hippocampal Volume Polygenic Predictor Score with Baseline and Change in Brain Volumes and Cognition Among Cognitively Healthy Older Adults. Neurobiol Aging. 2020, 94, 81–88. [Google Scholar] [CrossRef]
- Mourtzi, N.; Charisis, S.; Tsapanou, A.; Ntanasi, E.; Hatzimanolis, A.; Ramirez, A.; et al. Genetic propensity for cerebral amyloidosis and risk of mild cognitive impairment and Alzheimer’s disease within a cognitive reserve framework. Alzheimers Dement. 2023. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Gorbach, T.; Pudas, S.; Bartrés-Faz, D.; Brandmaier, A.M.; Düzel, S.; Henson, R.N.; et al. Longitudinal association between hippocampus atrophy and episodic-memory decline in non-demented APOE ε4 carriers. Alzheimers Dement (Amst). 2020, 12, e12110. [Google Scholar] [CrossRef]
- Li, J.Q.; Tan, L.; Wang, H.F.; Tan, M.S.; Tan, L.; Xu, W.; et al. Risk factors for predicting progression from mild cognitive impairment to Alzheimer’s disease: A systematic review and meta-analysis of cohort studies. J Neurol Neurosurg Psychiatry. 2016, 87, 476–484. [Google Scholar] [CrossRef]
- den Braber, A.; Bohlken, M.M.; Brouwer, R.M.; van’t Ent, D.; Kanai, R.; Kahn, R.S.; et al. Heritability of subcortical brain measures: A perspective for future genome-wide association studies. Neuroimage. 2013, 83, 98–102. [Google Scholar] [CrossRef]
- van der Meer, D.; Rokicki, J.; Kaufmann, T.; Córdova-Palomera, A.; Moberget, T.; Alnæs, D.; et al. Brain scans from 21,297 individuals reveal the genetic architecture of hippocampal subfield volumes. Mol Psychiatry. 2020, 25, 3053–3065. [Google Scholar] [CrossRef]
- Liampas, I.; Siokas, V.; Kyrozis, A.; Sakoutis, G.; Yannakoulia, M.; Kosmidis, M.H.; et al. Prevalence and Determinants of Restless Legs Syndrome (Willis-Ekbom Disease) in an Older Greek Population. Behav Sleep Med. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liampas, I.; Hatzimanolis, A.; Siokas, V.; Yannakoulia, M.; Kosmidis, M.H.; Sakka, P.; et al. Antihypertensive Medication Class and the Risk of Dementia and Cognitive Decline in Older Adults: A Secondary Analysis of the Prospective HELIAD Cohort. J Alzheimers Dis. 2022, 89, 709–719. [Google Scholar] [CrossRef]
- Bougea, A.; Maraki, M.I.; Yannakoulia, M.; Stamelou, M.; Xiromerisiou, G.; Kosmidis, MH.; et al. Higher probability of prodromal Parkinson disease is related to lower cognitive performance. Neurology. 2019, 92, e2261–72. [Google Scholar] [CrossRef]
- Liampas, I.; Siokas, V.; Ntanasi, E.; Kosmidis, M.H.; Yannakoulia, M.; Sakka, P.; et al. Cognitive trajectories preluding the imminent onset of Alzheimer’s disease dementia in individuals with normal cognition: Results from the HELIAD cohort. Aging Clin Exp Res. 2023, 35, 41–51. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition. Washington, DC; 2000.
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, EM. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984, 34, 939–939. [Google Scholar] [CrossRef]
- Hachinski, V.C.; Iliff, L.D.; Zilhka, E.; Du Boulay, G.H.; McAllister, V.L.; Marshall, J.; et al. Cerebral Blood Flow in Dementia. Archives of Neurology. 1975, 32, 632–637. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Galasko, D.; Kosaka, K.; Perry, E.K.; Dickson, D.W.; Hansen, LA.; et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology. 1996, 47, 1113–1124. [Google Scholar] [CrossRef]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; et al. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology. 1998, 51, 1546–1554. [Google Scholar] [CrossRef]
- Petersen, RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004, 256, 183–194. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.; Andrade, V.; Moreno-Grau, S.; Amin, N.; et al. New insights on the genetic etiology of Alzheimer’s and related dementia. medRxiv. 2020, 20200659. [Google Scholar]
- Collister, J.A.; Liu, X.; Clifton, L. Calculating Polygenic Risk Scores (PRS) in UK Biobank: A Practical Guide for Epidemiologists. Front Genet. 2022, 13, 818574. [Google Scholar] [CrossRef] [PubMed]
- Tulving, E.; Markowitsch, HJ. Episodic and declarative memory: Role of the hippocampus. Hippocampus. 1998, 8, 198–204. [Google Scholar] [CrossRef]
- Economou, A.; Routsis, C.; Papageorgiou, S.G. Episodic Memory in Alzheimer Disease, Frontotemporal Dementia, and Dementia With Lewy Bodies/Parkinson Disease Dementia: Disentangling Retrieval From Consolidation. Alzheimer Disease & Associated Disorders 2016, 30, 47–52. [Google Scholar]
- Petersen, R.C.; Stevens, J.C.; Ganguli, M.; Tangalos, E.G.; Cummings, J.L.; DeKosky, ST. Practice parameter: Early detection of dementia: Mild cognitive impairment (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001, 56, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Schröder, J.; Pantel, J. Neuroimaging of hippocampal atrophy in early recognition of Alzheimer´s disease – a critical appraisal after two decades of research. Psychiatry Research: Neuroimaging 2016, 247, 71–78. [Google Scholar] [CrossRef]
- Whitepaper: Defining and investigating cognitive reserve, brain reserve and brain maintenance. Alzheimers Dement. 2020, 16, 1305–1311. [CrossRef]
- Zhang, L.; Mak, E.; Reilhac, A.; Shim, H.Y.; Ng, K.K.; Ong, M.Q.W.; et al. Longitudinal trajectory of Amyloid-related hippocampal subfield atrophy in nondemented elderly. Human Brain Mapping 2020, 41, 2037–2047. [Google Scholar] [CrossRef]
- Yildirim, Z.; Delen, F.; Berron, D.; Baumeister, H.; Ziegler, G.; Schütze, H.; et al. Brain reserve contributes to distinguishing preclinical Alzheimer’s stages 1 and 2. Alzheimer’s Research & Therapy 2023, 15, 43. [Google Scholar]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018, 562, 203–209. [Google Scholar] [CrossRef]
- Caldwell, J.Z.K.; Berg, J.L.; Cummings, J.L.; Banks, S.J.; Alzheimer’s Disease Neuroimaging Initiative. Moderating effects of sex on the impact of diagnosis and amyloid positivity on verbal memory and hippocampal volume. Alzheimers Res Ther. 2017, 9, 72. [Google Scholar] [CrossRef]
- Koran, M.E.I.; Wagener, M.; Hohman, TJ. Sex Differences in the Association between AD Biomarkers and Cognitive Decline. Brain Imaging Behav. 2017, 11, 205–213. [Google Scholar] [CrossRef]
- Sundermann, E.E.; Maki, P.M.; Rubin, L.H.; Lipton, R.B.; Landau, S.; Biegon, A. Female advantage in verbal memory. Neurology. 2016, 87, 1916–1924. [Google Scholar] [CrossRef]
- Sundermann, E.; Biegon, A.; Rubin, L.; Lipton, R.; Landau, S.; Maki, P. Sex Differences in the Relationship between Hippocampal Volume and Verbal Memory Performance (P6.177). Neurology [Internet]. 2015 Apr 6 [cited 2023 Mar 26];84(14 Supplement). Available from: https://n.neurology.org/content/84/14_Supplement/P6.177.
- Gorbach, T.; Pudas, S.; Lundquist, A.; Orädd, G.; Josefsson, M.; Salami, A.; et al. Longitudinal association between hippocampus atrophy and episodic-memory decline. Neurobiology of Aging. 2017, 51, 167–176. [Google Scholar] [CrossRef]
- Grove, M.L.; Yu, B.; Cochran, B.J.; Haritunians, T.; Bis, J.C.; Taylor, KD.; et al. Best Practices and Joint Calling of the HumanExome BeadChip: The CHARGE Consortium. PLoS ONE. 2013, 8, e68095. [Google Scholar] [CrossRef]
- Abraham, G.; Qiu, Y.; Inouye, M. FlashPCA2: Principal component analysis of Biobank-scale genotype datasets. Bioinformatics. 2017, 33, 2776–2778. [Google Scholar] [CrossRef]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016, 48, 1279–1283. [Google Scholar] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; et al. Next-generation genotype imputation service and methods. Nat Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef]
- Loh, P.R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; A Reshef, Y.; K Finucane, H.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; O’Reilly, PF. PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience. 2019, 8, giz082. [Google Scholar] [CrossRef] [PubMed]
- International Schizophrenia Consortium, Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan MC.; et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009, 460, 748–752. [CrossRef] [PubMed]
Figure 1.
Cumulative survival curves depicting the combined aMCI/AD risk of participants with different levels of PRShp (p for trend= 0.021). The figure was derived from a model adjusted for age, sex, education, PC1, PC2 and APOE ε4 genotype.
Figure 1.
Cumulative survival curves depicting the combined aMCI/AD risk of participants with different levels of PRShp (p for trend= 0.021). The figure was derived from a model adjusted for age, sex, education, PC1, PC2 and APOE ε4 genotype.
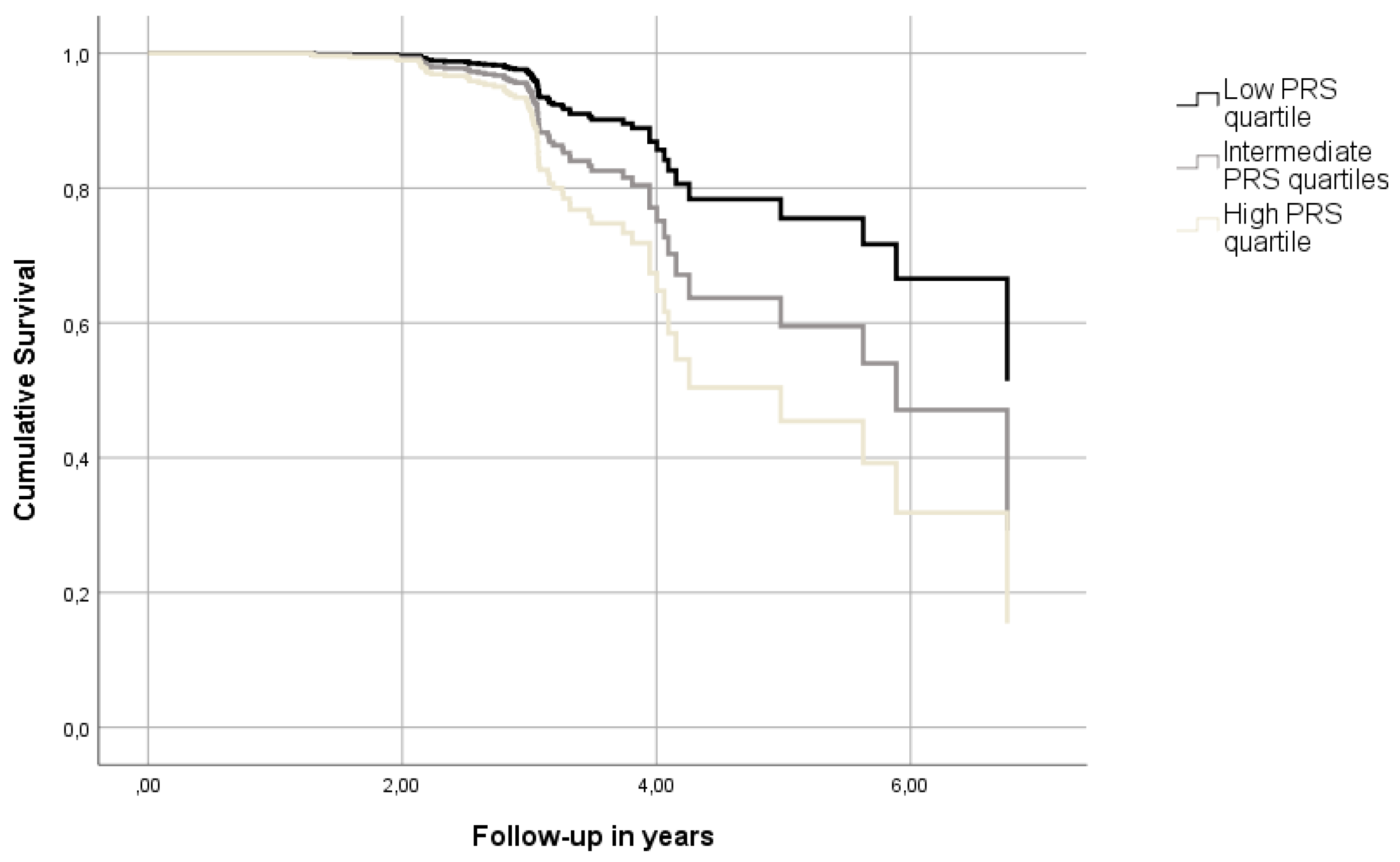

Table 1.
Participants’ baseline clinical and genetic characteristics by incidence of aMCI/AD.
| Parameters | Total sample | Non aMCI1/AD2 at follow-up |
aMCI1/AD2 at follow-up |
|
|---|---|---|---|---|
| N = 619 | N = 546 | N = 73 | p-value | |
| Age (years), mean ± SD | 73.4 ± 5.0 | 73.1 ± 4.9 | 75.1 ± 5.4 | 0.001 |
| Sex, female (%) | 361 (58.3%) | 321 (58.8%) | 40 (54.8%) | 0.515 |
| Education (years), mean ± SD | 7.3 ± 4.5 | 7.5 ± 4.5 | 5.8 ± 4.3 | 0.002 |
| ApoE ε4 carrier, n (%) | 98 (15.8%) | 86 (15.8%) | 12 (16.4%) | 0.880 |
| PRShp3, mean ± SD | -0.01 ± 0.98 | -0.05 ± 0.98 | 0.33 ± 0.96 | 0.002 |
| PRS strata using quartiles | ||||
| High | 154 (24.9%) | 128 (23.4%) | 26 (35.6%) | 0.018 |
| Intermediate | 311 (50.2%) | 274 (50.2%) | 37 (50.7%) | |
| Low | 154 (24.9%) | 144 (26.4%) | 10 (13.7%) |
1 Amnestic Mild Cognitive Impairment, 2 Alzheimer’s Disease, 3 Polygenic Risk Score Scale variables are presented in mean ±SD; categorical variables are presented as absolute numbers (percentages); p-values refer to differences between the non-aMCI-AD and aMCI-AD groups. Bold denotes statistical significance.
Table 2.
Cox regression models for aMCI-AD incidence using PRS as scale and trichotomous variable. Models were adjusted for age, sex, and years of education, PC1, PC2 and ApoE ε4 genotype. Subgroup analyses abide by the same approach.
Table 2.
Cox regression models for aMCI-AD incidence using PRS as scale and trichotomous variable. Models were adjusted for age, sex, and years of education, PC1, PC2 and ApoE ε4 genotype. Subgroup analyses abide by the same approach.
| Total Sample | |||
|---|---|---|---|
| PRShp1 | HR2 (95% CI3) | P-value | |
| PRS (scale) | 1.46 (1.14 – 1.86) | 0.002 | |
| PRS strata using quartiles | 0.021 | ||
| High | 2.81 (1.34 – 5.90) | 0.006 | |
| Intermediate | 1.85 (0.91 – 3.77) | 0.091 | |
| Low | Reference | ||
| Men (N = 258) | Women (N = 361) | ||
| PRShp1 | HR2 (95% CI3), P-value | HR2 (95% CI3), P-value | |
| PRS (scale) | 1.18 (0.78 – 1.78), .442 | 1.60 (1.17 – 2.19), 0.003 | |
| PRS strata using quartiles | 0.216 | 0.103 | |
| High | 2.38 (0.72 – 7.84), 0.154 | 2.89 (1.09 – 7.64), 0.033 | |
| Intermediate | 1.28 (0.42 – 3.91), 0.664 | 2.13 (0.82 – 5.50), 0.119 | |
| Low | Reference | Reference | |
|
Younger than 72.67 years (N = 310) |
Older than 72.67 years (N = 309) |
||
| PRShp1 | HR2 (95% CI 3), P-value | HR2 (95% CI 3), P-value | |
| PRS (scale) | 1.87 (1.21 – 2.90), 0.005 | 1.31 (0.96 – 1.78), 0.089 | |
| PRS strata using quartiles | 0.087 | 0.162 | |
| High | 3.71 (1.09 – 12.68), 0.037 | 2.52 (0.97 – 6.54), 0.058 | |
| Intermediate | 1.88 (0.59 – 6.02), 0.289 | 1.86 (0.74 – 4.70), 0.187 | |
| Low | Reference | Reference | |
1 Polygenic Risk Score for hippocampal atrophy 2 Hazard Ratio, 3 Confidence Interval. Bold denotes statistical significance. For each subgroup the number of (events/subgroup size) is provided.
Table 3.
GEE (generalized estimating equations) -predicted rates of cognitive decline in cognitively unimpaired older adults. For high and intermediate PRS by time interactions the reference group was the low PRS by time product. Models were adjusted for age, sex and years of education, PC1, PC2, ApoE4 genotype and incidence of aMCI at follow-up.
Table 3.
GEE (generalized estimating equations) -predicted rates of cognitive decline in cognitively unimpaired older adults. For high and intermediate PRS by time interactions the reference group was the low PRS by time product. Models were adjusted for age, sex and years of education, PC1, PC2, ApoE4 genotype and incidence of aMCI at follow-up.
| Parameter | PRShp1 (scale) by time interaction (β2, 95% CI3, p-value) |
High PRShp1 by time interaction (β2, 95% CI3, p-value) |
Intermediate PRShp1 by time interaction (β2, 95% CI3, p-value) |
|---|---|---|---|
| Global cognition | -0.013 (-0.025, -0.000), 0.043 | -0.038 (-0.075, -0.002), 0.038 | -0.017 (-0.045, 0.011), 0.236 |
| Memory | -0.015 (-0.032, 0.001), 0.069 | -0.051 (-0.096, 0.006), 0.025 | -0.011 (-0.052, 0.030), 0.606 |
| Visuospatial | -0.003 (-0.028, 0.021), 0.791 | -0.024 (-0.091, 0.043), 0.486 | -0.015 (-0.070, 0.040), 0.586 |
| Executive | -0.012 (-0.025, 0.001), 0.079 | -0.033 (-0.069, 0.002), 0.067 | -0.031 (-0.063, 0.002), 0.066 |
| Language | -0.012 (-0.025, 0.001), 0.076 | -0.022 (-0.062, 0.017), 0.269 | -0.010 (-0.046, 0.026), 0.588 |
| Attention | -0.007 (-0.032, 0.017), 0.556 | -0.010 (-0.082, 0.061), 0.780 | -0.026 (-0.088, 0.036), 0.414 |
1 Polygenic Risk Score for hippocampal atrophy, 2 Regression Coefficient, 3 Confidence Interval. Bold denotes statistical significance.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



