
Submitted:
21 October 2024
Posted:
22 October 2024
You are already at the latest version
Abstract
Background/Objectives: Emerging data support an essential role of glucagon for lipid metabolism. However, data on the role of dietary fat intake for glucagon secretion is limited. This analysis investigated whether altering nutritional fat intake affects glucagon levels in healthy subjects. Methods: 92 twins (age: 31±14 years, BMI: 23±3 kg/m2) consumed two 6-week diets: first a low fat, high carbohydrate diet (LFD) followed by an isocaloric high fat, low carbohydrate diet (HFD). 24 twins (age: 39±15 years, BMI: 24±2 kg/m2) continued with a high protein diet (HPD). Clinical investigation days were performed after 6 weeks of LFD, after 1 and 6 weeks of HFD and after 6 weeks of HPD. Results: The LFD caused a significant decrease of fasting glucagon (-27 %, p<0.001) compared to baseline. After 6 weeks of HFD glucagon increased (117 %, p<0.001 vs. LFD) while free fatty acids decreased. 6 weeks of HPD further increased glucagon levels (72 %, p=0.502 vs. HFD) although fasting amino acid levels remained constant. Fasting insulin and HOMA-IR moderately increased after one week of HFD, while 6 weeks of HPD significantly decreased both. The fasting glucagon-to-insulin ratio decreased through LFD (p<0.001) but increased after HFD (p<0.001) and even further after HPD (p=0.018). Liver fat, triglycerides and blood glucose did not increase during the HFD. The heritability of glucagon levels was 45% with the LFD. Conclusions: A HFD increases glucagon levels and the glucagon-to-insulin ratio under isocaloric conditions compared to a LFD in healthy lean subjects. This rise in glucagon may represent a favorable metabolic response as neither glucose levels nor insulin resistance or liver fat showed clinically relevant increases.
Keywords:
Glucagon
; Insulin
; High fat diet
; Low fat diet
; High Protein Diet
; Heritability
; Healthy twins
1. Introduction
Glucagon (GCGN) levels are closely related to insulin levels in healthy subjects, which together control fasting and postprandial plasma glucose levels in an intra-islet interplay of alpha- and beta-cells [1,2,3]. GCGN increases hepatic glucose production by glycogenolysis or gluconeogenesis in the fasting state to prevent hypoglycemia [4]. Insulin was shown to suppress GCGN postprandially as reflected by an inverse relationship of insulin to GCGN levels in humans [1,2] and experimental systems [3,4]. Nevertheless, alpha-cell GCGN was recently shown to stimulate insulin release from beta-cells through actions on beta-cell GCGN- and GLP1-receptors which was required for intact beta-cell function in mice [4,5].
Glucose acutely suppresses GCGN levels in healthy subjects after oral intake or intravenous application [6], whereas amino acids (AA) are strong secretagogues for GCGN and were identified as powerful stimulators of alpha-cell proliferation [7,8]. In reverse, GCGN stimulates AA degradation by the urea cycle in the liver which plays a primary role to maintain physiological AA levels [9,10,11]. Inhibition of GCGN signaling by antagonists or GCGN-receptor deletion leads to hyperaminoacidemia which further induces the proliferation of alpha-cells [7] while GCGN excess due to glucagonomas reduces circulating AA levels.
Emerging data indicates that GCGN is moreover involved in lipid metabolism since GCGN receptor antagonists were found to increase liver fat accumulation and LDL levels in early phase clinical trials [12,13]. GCGN was shown to increment hepatic lipolysis and hepatic ß-oxidation in mice, rats and humans via stimulation of the inositol trisphosphate receptor (INSP3R), thereby reversing hepatic steatosis in rodent models [14,15]. The question whether and how lipids conversely regulate the secretion of GCGN is still debated. Free fatty acids (FFA) were shown to stimulate GCGN secretion in isolated alpha-cells which may function directly via activation of FFA receptors such as the G protein-coupled receptor 40 (GPR40) and the GPR119, or indirectly via increased fatty acid oxidation in pancreatic islets [13,16,17]. Mice fed a high-fat diet show elevated GCGN levels and decreased secretion of somatostatin, a potent inhibitor of insulin and GCGN release, revealing that intra-islet somatostatin signaling may also play a role for FFA mediated GCGN increases [18,19]. Others observed that mice fed a high fat diet demonstrate increased alpha cell mass [20]. In rats, on the contrary, a high fat diet reduced GCGN levels or had no effect [21]. In healthy lean men neither oral nor intravenous administration of a lipid emulsion led to significant GCGN level alterations, although GCGN tended to increase during the first 30 min after oral intake [22]. In contrast, ingestion of oleic acid caused a modest acute stimulation of GCGN secretion [23]. A study comparing fatty acids of different chain length in meal challenge tests observed the greatest increases of GCGN in response to olive oil (containing long-chain fatty acids) followed by C-8 dietary oil (digested to medium-chain fatty acids), but no change in response to tributyrin (containing short-chain fatty acids) [24], thus confirming the acute glucagonotropic effect of lipids may depend on fat composition. Heparin-induced elevation of FFA decreased GCGN secretion while nicotinic acid-induced suppression of FFA elevated GCGN secretion in humans [25]. Despite some contrary findings, it appears that alongside the predominant roles of glucose and amino acids, lipids may also influence the physiological secretion of glucagon. To our knowledge, no long-term effects of high fat diets on GCGN levels have been reported in healthy subjects.
Given the underexplored role of dietary fat intake on GCGN levels in humans, we investigated the response of GCGN in 92 lean, healthy adult twins to low and high fat intake for 6 weeks each. We moreover tested the further response to 6 weeks of high protein, low fat intake in 24 participants who agreed to continue the study. We assessed the interaction with insulin secretion, circulating free fatty acids and amino acids as well as hepatic fat content.
2. Materials and Methods
2.1. Study Protocol and Participants
The NutriGenomic Analysis in Twins (NUGAT) study protocol was approved by the ethics committee of the Charité-Universitätsmedizin Berlin and conducted in accordance with the principles of the Helsinki Declaration of 1975, as revised in 2000. Prior to the study all participants gave written informed consent. The NUGAT study was registered at ClinicalTrials.gov: NCT01631123. Details of recruitment and enrollment of study participants, the initial screening visit and exclusion criteria have been published elsewhere [26]. Since genetic variance analyses were the primary endpoint of this study, a randomized controlled design was deliberately avoided. 92 subjects (46 pairs of twins – 34 monozygotic and 12 dizygotic), 58 females and 34 males, age 18 to 70 years and body mass index (BMI) 18 to 29 kg/m2 with BMI difference <3 kg/m2 between twins completed the study between September 2009 and September 2012. To standardize food intakes, the study participants were first asked to consume a healthy diet for 6 weeks containing 30 %Energy (%E) fat, one third each saturated fatty acids (SFA), mono-unsaturated fatty acids (MUFA), and poly-unsaturated fatty acids (PUFA). The protein content was 15 %E and carbohydrates were 55 %E, corresponding to the low fat recommendations of the German Society of Nutrition at that time. To increase compliance and ensure a standardized dietary pattern, approximately 70 % of food was supplied together with detailed meal plans for the last week before the first clinical investigation day (CID1). The participants were then switched to a high fat diet (HFD) for 6 weeks containing 45 %E fat (18% SFA, 17% MUFA and 10% PUFA), 40 %E carbohydrates and 15 %E protein. Clinical investigation days were performed after 1 week of the HFD (CID2), in which food was mainly supplied, and after another 5 weeks of HFD (CID3), again with supply of most foods eaten for the week before the CID3. Thereafter, 24 twins continued the study with the intake of a low fat (30 %E) but high protein (30 %E) and moderate carbohydrate (40%E) diet for 6 weeks until CID4 (Figure 1). At each CID, anthropometric measurements were performed. Additionally, magnetic resonance spectroscopy was performed on a Magnetom Avanto 1.5T whole-body scanner (Siemens Healthcare, Erlangen, Germany) for quantification of liver fat content (intrahepatic lipids, IHL) at CID1, CID2 and CID3 [27]. All three diets were isocaloric to avoid significant changes of body weight which may affect GCGN levels independently from food composition. To obtain homogenous food intakes, all participants received detailed dietary advice and meal plans by an experienced dietician. The subjects completed 6 dietary records of 5-6 days at specified times during the study as published previously [26] and the HPD subgroup completed an additional food record during the high protein intervention period.
2.2. Blood Parameters
At each CID, blood samples were drawn in the fasted state (>10 h since last food intake) in the morning from the forearm vein, centrifuged at 1.800 x g for 10 min at 4 °C and serum was stored at -80 °C until analysis. Determination of routine serum parameters (e.g., total cholesterol, triglycerides, free fatty acids) was performed using an automated analyzer (ABX Pentra 4000; ABX, Montpellier, France). LDL cholesterol concentrations were calculated using the Friedewald equation. Fasting levels of the following amino acid were measured in EDTA plasma samples of CID1-4 via liquid chromatography-mass spectrometry (LC–MS): Alanine, Arginine, Asparagine, Aspartic acid, Citrulline, Cystine, Glutamine, Glutamic acid, Glycine, Histidine, Leucine, Lysine, Methionine, Ornithine, Phenylalanine, Proline, Serine, Threonine, Tryptophan, Tyrosine, Valine. Isoleucine was not determined and is in consequence not reported here. GCGN and insulin were measured in the fasting serum of all participants at each CID with specific human ELISA kits (Mercodia, Uppsala, Sweden). In addition, at CID 1,2 and 3 a subgroup of 14 participants randomly taken from the entire study group consumed a test meal (Fresubin® Energy drink) at noon and we measured GCGN serum levels before and 240 min after the test meal consumption.
The homeostasis model assessment-estimated insulin resistance (HOMA-IR) was calculated as fasting insulin [mU/L] multiplied with fasting glucose [mmol/L] divided by 22.5 [28].
2.3. Heritability
Heritability was estimated by applying the “ACE structural equation model” for every CID. This model analyzes covariance based on comparing the degree of concordance in form of the correlation coefficient within and between monozygous versus dizygous twin pairs. The proportion of variance is partitioned into (A) additive genetic influences, (C) common environmental, and (E) individual environmental influences.
2.4. Statistical Analysis
Prior to data analysis a test of plausibility was performed and unusual values that were outside of 3-fold interquartile range were declared as extreme outliers and were not considered in further analysis. The Shapiro-Wilk test was used to assess variables for normal distribution. Mean values for continuous data were compared using repeated measures ANOVA followed by Bonferroni adjusted post hoc test. The requirements for the ANOVA were tested by the Shapiro-Wilk-test and Mauchly’s sphericity-test with ln- and/or Greenhouse-Geisser transformation if necessary. The Friedman test as nonparametric equivalent of the ANOVA was used to verify significant results for non-normally distributed data.
To analyze whether the age of the subjects had an impact on high fat diet induced GCGN changes we split the cohort in 3 age subgroups (tertiles: 18-23 years, 24-30 years, 31-70 years) and conducted Friedman tests followed by Bonferroni adjusted post hoc test for each age group again. A nonparametric one-way ANOVA (Kruskal-Wallis-test) was applied for the comparison between the age tertiles. To test for sex differences, we split the cohort by sex and the Friedman test with Bonferroni adjusted post hoc test was applied for males and females separately. Mann-Whitney-U test was used to check for significant differences between GCGN levels in females and males.
Pearson’s and Spearman’s rank correlation coefficients were used for correlation analysis of variables with normal and skewed distribution, respectively. In cases of missing data pairwise deletion was applied. Statistical analyses were processed using SPSS 28.0 (SPSS Inc., Chicago, IL, USA) and p<0.05 were considered significant. Values are expressed as mean ± SEM, unless otherwise stated. The graphs were generated with GraphPad Prism 9 (GraphPad, California, USA).
3. Results
3.1. Participants’ Characteristics and Compliance
We analyzed 92 healthy participants whose baseline anthropometric and clinical characteristics are shown in Table 1. The macronutrient intake, calculated from dietary records with the PRODI®4.5 software (Nutri-Science GmbH, Freiburg, Germany), was 35 E% fat, 50 E% carbohydrates and 15 E% protein prior to the study, 29 E% fat, 55 E% carbohydrates and 16 E% protein during the LFD, 44 E% fat, 42 E% carbohydrates and 14 E% protein during the HFD and 29 E% fat, 40 E% carbohydrates and 31 E% protein for the HPD, reflecting a high dietary compliance of the participants during all study phases (Table S1). Although the study followed an isocaloric approach, the bodyweight of our subjects slightly decreased in response to the LFD (-0.98 kg, p<0.001), and increased again after 6 weeks of HFD (+0.40 kg, p=0.124 vs. LFD) and minutely again with the HPD (+0.12 kg, p=1.00 vs. HFD6). However, these weight changes where within a range of natural fluctuation and thus the effects of weight alterations were successfully reduced to a minimum in the present study.
As expected, we observed a rise in total cholesterol as well as its LDL- and HDL-fraction in response to the HFD (Table S2), which again represents the good compliance of the participants to the dietary intervention.
3.2. Changes of Serum GCGN and Metabolic Parameters in Response to High Dietary Fat Intake
Fasting GCGN levels increased within 1 week after onset of the HFD and increased further over the 6 weeks of HFD to almost double of the values after the LFD (Figure 2). To identify factors involved in the increases of GCGN we first determined correlations with anthropometric parameters and insulin resistance measured as HOMA-IR. The HFD had remarkably modest effects on insulin resistance in this lean and healthy population. Insulin levels increased slightly after 1 week of HFD and diminished again to baseline levels after 6 weeks of HFD, while glucose levels remained unchanged, resulting in a small but significant increase in HOMA-IR (Figure 2). However, the marginal increase in HOMA-IR did not correlate with the changes in GCGN levels (Table S4).
We did not observe a significant correlation between IHL and GCGN neither before nor after the HFD (Table S3). It should be noted that the lean and healthy twins had low IHL at baseline which did not change in response to the dietary interventions (Table S2).
The GCGN-to-insulin ratio decreased in response to the LFD and increased in response to the HFD without changes of glucose (Figure 2). Thus, the higher fasting GCGN after the HFD did not elevate fasting blood sugar levels as might be expected, particularly since changes of insulin and insulin resistance were rather small.
The high fat diet significantly reduced circulating FFA in the fasted state over the 6 weeks of the study (Figure 2). Notably, the previous LFD significantly increased FFA as compared to screening values, which might be due to the slightly higher SFA intake (13 E%) of the twins on their habitual diet before the study. The HFD-induced decrease in FFA might be related to the increase in GCGN. However, we did not find a correlation between the absolute values or the changes of FFA and GCGN (Tables S3 and S4).
The sum of all measured circulating AA did not change in response to the HFD. Only Proline showed a significant increase after one week of high dietary fat intake but decreased again after 6 weeks. None of the other plasma AA showed significant changes after 1 week of HFD. After 6 weeks of HFD asparagine slightly decreased, whereas tryptophan and valine increased compared to LFD levels (Table S5). Interestingly, the total level of measured circulating AA correlated with the GCGN levels after 6 weeks of HFD, however the changes of AA and GCGN in response to the HFD did not correlate (Tables S6 and S7).
3.3. Impact of Age and Sex on GCGN Levels
To investigate whether age and sex play a role for GCGN responses to food intake, we conducted an age-stratified and sex-stratified analysis. A comparison of age tertiles showed a GCGN decline after 6 weeks of LFD and an increase in response to the HFD in all 3 groups independent of age and no significant differences between the age tertiles were found (Figure S1). Thus, in the present lean population, age appears to have no influence on diet-mediated GCGN changes. In contrast, sex seems to play a role for GCGN levels, as we observed significantly higher GCGN levels in males compared to females at baseline, after 6 weeks of LFD and after 6 weeks of HFD. Nevertheless, both sexes showed a significant increase of GCGN levels in response to 6 weeks of high dietary fat intake (Figure S2).
3.4. Changes of Postprandial GCGN in Response to the HFD
In a subgroup of 14 participants randomly taken from the entire study group, we further assessed whether GCGN levels changed in response to a test meal (Fresubin® Energy drink) that was administered at noon after 6 weeks of LFD, after 1 week of HFD and after 6 weeks of HFD. We measured serum GCGN before the test meal consumption (0 min) and 240 min after the meal. In this subgroup, GCGN showed a slight, but not significant increase before intake of the test meal at noon after 6 weeks of HFD. However, the levels after the meal increased significantly, suggesting that both, fasted and postprandial levels of GCGN, increase upon prolonged intake of a HFD in healthy people (Figure S3).
3.5. Serum GCGN and Metabolic Parameter Changes in Response to High Dietary Protein Intake
The further switch to a low fat, high protein diet was studied in a subgroup of 24 participants who agreed to another 6 weeks of controlled dietary intake. This subgroup was, on average, 8 years older and had a slightly higher BMI of 24 ± 2 kg/m², which is still in the range of normal weight (Table 1). The responses of this subgroup to LFD and HFD were similar to those of the 92 twins (Figure 3) with a pronounced decrease of GCGN under the LFD compared to screening and an increase with the HFD. The HPD further increased GCGN values compared to the HFD despite of the lower fat content. The FFA profile (Figure 3) and HDL-cholesterol levels were similar to those under the low fat diet, confirming the adherence to lower fat intake (Table S8). This suggests that the HPD further increased fasting GCGN values independently from the effects of fat.
Insulin levels were significantly reduced under the HPD, resulting in a pronounced increase (more than 2-fold compared to after LFD) of the GCGN-to-insulin ratio (Figure 3). Nevertheless, glucose levels remained unchanged, which resulted in an improvement of insulin sensitivity as calculated by HOMA-IR values. The high GCGN levels thus did not impair glucose metabolism during the 6 weeks of high protein intervention.
The total concentration of AA that was measured in this study did not alter with the HPD. Nevertheless, significant changes in response to the HPD diet occurred in several AA, but as some decreased (alanine, glutamine, glycine, proline) and others increased (glutamic acid, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine) no change in the overall level was observed. Except for glutamic acid, all AA that increased in response to the HPD were essential AA (Table S9). Surprisingly, we found less correlations between AA and GCGN with the HPD compared to the LFD and HFD, as only serine correlated with GCGN after the HPD (Table S6). Instead, the change of total AA in response to the HPD was negatively correlated with the change of GCGN (Table S7).
3.6. Heritability of GCGN Levels
The twin design allowed a calculation of the heritability of the GCGN levels at screening and in the presence of the different diets (Figure S4). The GCGN serum levels at screening were not heritable, despite a slight but significant correlation among monozygotic twin pairs (r=0.353, p=0.041). After 6 weeks of low fat diet the significant correlation of GCGN levels (r=0.534, p=0.001) among the monozygotic twins was more pronounced, but no correlation was observed among the dizygotic twins (r=0.308, ns), resulting in an estimated heritability of 45%. Remarkably, this heritability and the significant correlation among monozygotic twin pairs was lost after 6 weeks of HFD suggesting that non-heritable factors play a pronounced role in this condition. In addition, the change of GCGN levels (delta GCGN) in response to altered nutrition was not heritable at any time of the study. The high protein diet included 10 monozygotic and 2 dizygotic pairs of twins and thus did not allow a calculation of heritability. Nevertheless, the GCGN levels showed a high correlation in the monozygotic pairs (r=0.733, p=0.025) after 6 weeks of high protein intake.
4. Discussion
We report extensive diet-dependent changes of serum GCGN in lean healthy young subjects. GCGN showed a pronounced decrease in response to the LFD. In contrast, GCGN progressively increased after 1 and 6 weeks of HFD, which has not been reported before. The increased fat intake was in exchange for carbohydrates which were reduced from 55 to 40 %E. This corresponds to a moderate restriction of carbohydrates which is unlikely to increase GCGN levels extensively. Previous studies showed low carbohydrate intake to significantly increase GCGN levels [29,30,31], however these trials used considerably lower carbohydrate portions than our present study. We hypothesize the here observed rise in GCGN in response to the HFD diet is not solely due to the moderate carbohydrate restriction, but to its combination with higher nutritional fat intake. The ingestion of fat alone has previously been shown to modestly increase prandial GCGN release in healthy subjects and subjects with type 2 diabetes [32]. However, the ingestion of solely protein was found to increase GCGN to a higher extent than fat [23,32]. These results are in line with our data which show the highest fasting GCGN levels in response to 6 weeks of HPD, reflecting the potent stimulation of GCGN secretion through AA that has been reported since the 1960s [7,33]. Taken together, this supports the hypothesis that fat intake plays a role for GCGN secretion, even though it appears to be not as potent as protein/AA intake to stimulate GCGN release.
The total fasting blood AA concentration did not alter with the high protein intake in our study, and we found a negative correlation between the change of GCGN and the change of the total AA in response to 6 weeks of HPD. This may appear contradictory at first, but appears reasonable under consideration of the liver-alpha-cell axis: postprandial AA function as glucagon secretagogues causing a rise of GCGN which in reverse leads to elevated AA degradation by the urea cycle in the liver, thereby normalizing the circulating AA concentrations [7]. A previous study in healthy men showed lower fasting blood AA in a high protein intake group compared to a normal protein intake group, except for leucine, methionine and tyrosine [34]. In line with these findings, leucine and methionine levels, among others, were significantly elevated after the HPD in our cohort, whereas alanine, glutamine, glycine, and proline concentrations diminished after the HPD. Fernstrom et al. have also reported lower fasting glycine and alanine plasma concentrations in subjects that consumed 150 g egg protein/day compared to those consuming 75 or 0 g protein daily for 5 days [35]. In contrast to our findings Forslund et al. reported fasting lysine, phenylalanine, threonine, tryptophan, and valine to be lower in the high protein group [34], whereas the fasting levels of these essential AA increased significantly after the HPD in our study. Notably, the increase of branched chain amino acids may be explained by the lowered insulin levels which regulate branched chain amino acid degradation[36,37]. Several studies did not show an upregulation of branched chain amino acids by higher intakes [38,39]. These differences in the AA responses may be due to varying nutritional intake of the specific AA and different study duration. However, besides the amount and composition of AA supplied, several tissues and metabolic pathways impact the concentrations of circulating plasma AA, making it very difficult to identify the complete metabolic background of the observed AA alterations. A drop of fasting glucogenic AA in response to high nutritional protein consumption has been described in rats and related to their sustained catabolism [40]. In addition to increased amino acid catabolism and renal clearance, decreased de novo synthesis and restrained body protein breakdown have been postulated as responsible for the inverse response of glutamine and alanine concentrations to high protein intake in humans [41,42]. Interestingly, glutamine, alanine, glycine and proline have been described as potent glucagon secretagogues [7,43,44]. Moreover, alanine and proline were found to be involved in the acute regulation of the liver-alpha-cell axis in female mice [44], which may play a role in their significant decrease after 6 weeks of HPD.
Studies comparing high fat vs. low fat meals confirmed a greater acute stimulation of GCGN by the high fat meal in healthy [45,46], and overweight subjects [47]. Raben and coworkers [48] submitted 10 healthy lean and 8 normal-weight postobese women to high fat, high starch or high sucrose diets for 2 weeks in a crossover design and did not find significant changes of fasting or postprandial GCGN levels, although fasting levels of GCGN increased by 33%. The participants consumed low fat diets for 2 weeks containing 28 %E of fat prior to the HFD with 46 %E fat. These results are compatible with our data which show a time-dependent further increase of GCGN levels over the 6 weeks. This suggests a delayed metabolic adaptation similar to the alterations of LDL- and HDL-cholesterol. The mechanisms involved are unclear.
The role of GCGN in lipid metabolism has been established by the increases of liver fat induced by GCGN antagonists in human trials [12,13] which most likely relates to the induction of hepatic lipolysis and lipid oxidation by GCGN [14,15]. Thus, the elevation of GCGN in the present study might be interpreted as a metabolic response to prevent hepatic fat accumulation upon high dietary fat intake. This may explain the absence of increases of liver fat by the HFD in our healthy subjects which contrasts previous reports in obese subjects [49]. The diet-induced changes of GCGN found in the present study are also contrary to observations in diabetes patients who did not show decreases of GCGN upon intake of a low fat diet containing 30 %E as fat compared to 42 %E before start of the intervention, or increases in response to a high protein diet for 6 weeks. Notably, plasma AA levels did not change in this study [39,50]. However, chronic hyperglycemia is associated with profound alterations of GCGN secretion which may account for the differences [51].
GCGN levels were shown to be correlated with hepatic fat content in obese individuals with [50] and without type 2 diabetes [52]. This correlation was not observed in our healthy population who had low levels of liver fat content and included only 7 individuals with IHL above the threshold of 5.56%, defining non-alcoholic fatty liver disease [53]. Moreover, the lean and healthy twins were resistant to high fat diet induced increases in liver fat over the 6 weeks of the isocaloric study.
The GCGN-to-Insulin ratio increased in response to 6 weeks of HFD and even further after 6 weeks of HPD. Remarkably these distinct increases occurred without changes in blood glucose levels. This indicates that healthy people may desensitize to the glucose producing effects of GCGN or counteract the hepatic glucose production via unclear mechanisms upon permanently elevated GCGN levels. Capozzi and Coworkers [54] have already reported that under prandial conditions GCGN functions more as an insulinotropic agent ensuring euglycemia rather than further elevating blood glucose levels via hepatic glucose production.
Major strengths of the current study are the isocaloric approach, enabling results independent from presumably confounding body weight changes, and the high dietary compliance of the subjects. Limitations of the study include the moderate number of subjects analyzed and the restriction to Caucasian ethnicity, therefore its conclusions may not apply to other populations because of ethnic or genetic differences. Additionally, the study was not randomized and targeted similar dietary conditions for all twins using the differences in pair-wise responses to address the inheritance to dietary changes. Moreover, we only measured total serum FFA without further differentiation, though the ability of FFAs to stimulate GCGN secretion may depend on fatty acid length and degree of saturation [13,24].
5. Conclusions
In conclusion, a high fat, low carbohydrate diet strongly raises GCGN levels and the GCGN-to-insulin ratio in healthy lean subjects in comparison to an isocaloric low fat, high carbohydrate diet. Interestingly, blood glucose levels, liver fat and insulin resistance showed no clinically relevant increases. Thus, the observed rise in GCGN might be interpreted as metabolic response to high dietary fat intake to prevent hepatic fat accumulation, as increases in GCGN have been shown previously to induce hepatic lipolysis and fat oxidation [14,15]. This allows an alternative perspective to position elevated GCGN secretion observed in type 2 diabetes as a mechanism to compensate, rather than induce, dysregulated homeostasis.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Glucagon changes in age subgroups.; Figure S2: Glucagon changes grouped by sex.; Figure S3: Glucagon levels before and after a test meal.; Figure S4: Heritability of Glucagon.; Table S1: Target and actual macronutrient composition for each intervention phase.; Table S2: Characteristics of all participants (n=92) in response to the high fat diet.; Table S3: Correlation analysis of clinical parameters with fasting glucagon.; Table S4: Correlation analysis of the change (delta %) of fasting glucagon with the change of clinical parameters.; Table S5: Changes of amino acids of all participants in response to the high fat diet.; Table S6: Correlation analysis of fasting circulating amino acids with fasting glucagon.; Table S7: Correlation analysis of the change (delta) of fasting glucagon with the change of fasting circulating amino acids.; Table S8: Characteristics of the subgroup of participants that continued with the high protein intervention (n=24) at each clinical investigation day.; Table S9: Changes of fasting amino acids in the subgroup of participants that continued with the high protein intervention (n=24).
Author Contributions
Conceptualization, Andreas Busjahn and Andreas Pfeiffer; Data curation, Anne-Cathrin Ost; Formal analysis, Bettina Schuppelius, Rita Schüler, Olga Pivovarova-Ramich and Jürgen Machann; Funding acquisition, Andreas Pfeiffer; Investigation, Silke Hornemann, Michael Kruse, Soyoung Park and Anne-Cathrin Ost; Methodology, Silke Hornemann, Jürgen Machann and Anne-Cathrin Ost; Project administration, Andreas Pfeiffer; Supervision, Andreas Pfeiffer; Visualization, Bettina Schuppelius; Writing – original draft, Bettina Schuppelius and Andreas Pfeiffer; Writing – review & editing, Olga Pivovarova-Ramich, Stefan Kabisch and Marta Csanalosi. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by a grant of the German Federal Ministry of Education and Research (BMBF, grant no. 0 315 424). The funding organization had no influence in study design; in the collection, analysis, interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the ethics committee of the Charité-Universitätsmedizin Berlin (EA4/021/09, date of approval 26 September 2009).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study prior to their participation.
Data Availability Statement
Data available on request due to privacy restrictions.
Acknowledgments
The authors thank all study participants for their cooperation. We would also like to thank the staff of our research ambulance, Melanie Hannemann, Alexandra Ullrich, Dominique Zschau, Anja Henkel as well as Katrin Ritter, Andrea Borchert and Tanja Ahrens for their excellent technical assistance and Daniela Hoffmann for providing participants with excellent nutritional guidance.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Rohrer, S.; Menge, B.A.; Gruber, L.; Deacon, C.F.; Schmidt, W.E.; Veldhuis, J.D.; Holst, J.J.; Meier, J.J. Impaired crosstalk between pulsatile insulin and glucagon secretion in prediabetic individuals. J Clin Endocrinol Metab 2012, 97, E791–795. [Google Scholar] [CrossRef] [PubMed]
- Menge, B.A.; Gruber, L.; Jorgensen, S.M.; Deacon, C.F.; Schmidt, W.E.; Veldhuis, J.D.; Holst, J.J.; Meier, J.J. Loss of inverse relationship between pulsatile insulin and glucagon secretion in patients with type 2 diabetes. Diabetes 2011, 60, 2160–2168. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Kjems, L.L.; Veldhuis, J.D.; Lefebvre, P.; Butler, P.C. Postprandial suppression of glucagon secretion depends on intact pulsatile insulin secretion: further evidence for the intraislet insulin hypothesis. Diabetes 2006, 55, 1051–1056. [Google Scholar] [CrossRef]
- Moede, T.; Leibiger, I.B.; Berggren, P.O. Alpha cell regulation of beta cell function. Diabetologia 2020, 63, 2064–2075. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Dattaroy, D.; Pham, J.; Wang, L.; Barella, L.F.; Cui, Y.; Wilkins, K.J.; Roth, B.L.; Hochgeschwender, U.; Matschinsky, F.M.; et al. Intra-islet glucagon signaling is critical for maintaining glucose homeostasis. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Meier, J.J.; Deacon, C.F.; Schmidt, W.E.; Holst, J.J.; Nauck, M.A. Suppression of glucagon secretion is lower after oral glucose administration than during intravenous glucose administration in human subjects. Diabetologia 2007, 50, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Dean, E.D. A Primary Role for alpha-Cells as Amino Acid Sensors. Diabetes 2020, 69, 542–549. [Google Scholar] [CrossRef]
- Dean, E.D.; Li, M.; Prasad, N.; Wisniewski, S.N.; Von Deylen, A.; Spaeth, J.; Maddison, L.; Botros, A.; Sedgeman, L.R.; Bozadjieva, N.; et al. Interrupted Glucagon Signaling Reveals Hepatic alpha Cell Axis and Role for L-Glutamine in alpha Cell Proliferation. Cell Metab 2017, 25, 1362–1373.e1365. [Google Scholar] [CrossRef]
- Okun, J.G.; Rusu, P.M.; Chan, A.Y.; Wu, Y.; Yap, Y.W.; Sharkie, T.; Schumacher, J.; Schmidt, K.V.; Roberts-Thomson, K.M.; Russell, R.D.; et al. Liver alanine catabolism promotes skeletal muscle atrophy and hyperglycaemia in type 2 diabetes. Nat Metab 2021, 3, 394–409. [Google Scholar] [CrossRef]
- Gerich, J.E.; Charles, M.A.; Grodsky, G.M. Characterization of the effects of arginine and glucose on glucagon and insulin release from the perfused rat pancreas. J Clin Invest 1974, 54, 833–841. [Google Scholar] [CrossRef]
- Wewer Albrechtsen, N.J.; Pedersen, J.; Galsgaard, K.D.; Winther-Sorensen, M.; Suppli, M.P.; Janah, L.; Gromada, J.; Vilstrup, H.; Knop, F.K.; Holst, J.J. The Liver-alpha-Cell Axis and Type 2 Diabetes. Endocr Rev 2019, 40, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J.; Paquot, N.; Lefebvre, P.J. Investigational glucagon receptor antagonists in Phase I and II clinical trials for diabetes. Expert Opin Investig Drugs 2017, 26, 1373–1389. [Google Scholar] [CrossRef] [PubMed]
- Galsgaard, K.D.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Wewer Albrechtsen, N.J. Glucagon Receptor Signaling and Lipid Metabolism. Front Physiol 2019, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Zhang, D.; Guerra, M.T.; Brill, A.L.; Goedeke, L.; Nasiri, A.R.; Rabin-Court, A.; Wang, Y.; Peng, L.; Dufour, S.; et al. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature 2020, 579, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Mehal, W.Z.; Shulman, G.I. Glucagon promotes increased hepatic mitochondrial oxidation and pyruvate carboxylase flux in humans with fatty liver disease. Cell Metabolism 2024. [Google Scholar] [CrossRef]
- Briant, L.J.B.; Dodd, M.S.; Chibalina, M.V.; Rorsman, N.J.G.; Johnson, P.R.V.; Carmeliet, P.; Rorsman, P.; Knudsen, J.G. CPT1a-Dependent Long-Chain Fatty Acid Oxidation Contributes to Maintaining Glucagon Secretion from Pancreatic Islets. Cell Reports 2018, 23, 3300–3311. [Google Scholar] [CrossRef]
- Armour, S.L.; Frueh, A.; Chibalina, M.V.; Dou, H.; Argemi-Muntadas, L.; Hamilton, A.; Katzilieris-Petras, G.; Carmeliet, P.; Davies, B.; Moritz, T.; et al. Glucose Controls Glucagon Secretion by Regulating Fatty Acid Oxidation in Pancreatic α-Cells. Diabetes 2023, 72, 1446–1459. [Google Scholar] [CrossRef]
- Gromada, J.; Franklin, I.; Wollheim, C.B. α-Cells of the Endocrine Pancreas: 35 Years of Research but the Enigma Remains. Endocrine Reviews 2007, 28, 84–116. [Google Scholar] [CrossRef]
- Kellard, J.A.; Rorsman, N.J.G.; Hill, T.G.; Armour, S.L.; van de Bunt, M.; Rorsman, P.; Knudsen, J.G.; Briant, L.J.B. Reduced somatostatin signalling leads to hypersecretion of glucagon in mice fed a high-fat diet. Mol Metab 2020, 40, 101021. [Google Scholar] [CrossRef]
- Ellingsgaard, H.; Ehses, J.A.; Hammar, E.B.; Van Lommel, L.; Quintens, R.; Martens, G.; Kerr-Conte, J.; Pattou, F.; Berney, T.; Pipeleers, D.; et al. Interleukin-6 regulates pancreatic α-cell mass expansion. Proceedings of the National Academy of Sciences 2008, 105, 13163–13168. [Google Scholar] [CrossRef]
- Eisenstein, A.B.; Strack, I.; Steiner, A. Increased hepatic gluconeogenesis without a rise of glucagon secretion in rats fed a high fat diet. Diabetes 1974, 23, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, O.; Carr, R.D.; Deacon, C.F.; Holst, J.J.; Pacini, G.; Mari, A.; Ahrén, B. Incretin Hormone and Insulin Responses to Oral Versus Intravenous Lipid Administration in Humans. The Journal of Clinical Endocrinology & Metabolism 2011, 96, 2519–2524. [Google Scholar] [CrossRef]
- Carr, R.D.; Larsen, M.O.; Winzell, M.S.; Jelic, K.; Lindgren, O.; Deacon, C.F.; Ahren, B. Incretin and islet hormonal responses to fat and protein ingestion in healthy men. Am J Physiol Endocrinol Metab 2008, 295, E779–784. [Google Scholar] [CrossRef] [PubMed]
- Mandøe, M.J.; Hansen, K.B.; Hartmann, B.; Rehfeld, J.F.; Holst, J.J.; Hansen, H.S. The 2-monoacylglycerol moiety of dietary fat appears to be responsible for the fat-induced release of GLP-1 in humans. Am J Clin Nutr 2015, 102, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E.; Langlois, M.; Schneider, V.; Karam, J.H.; Noacco, C. Effects of alternations of plasma free fatty acid levels on pancreatic glucagon secretion in man. J Clin Invest 1974, 53, 1284–1289. [Google Scholar] [CrossRef]
- Schuler, R.; Osterhoff, M.A.; Frahnow, T.; Seltmann, A.C.; Busjahn, A.; Kabisch, S.; Xu, L.; Mosig, A.S.; Spranger, J.; Mohlig, M.; et al. High-Saturated-Fat Diet Increases Circulating Angiotensin-Converting Enzyme, Which Is Enhanced by the rs4343 Polymorphism Defining Persons at Risk of Nutrient-Dependent Increases of Blood Pressure. J Am Heart Assoc 2017, 6. [Google Scholar] [CrossRef]
- Machann, J.; Thamer, C.; Schnoedt, B.; Stefan, N.; Haring, H.U.; Claussen, C.D.; Fritsche, A.; Schick, F. Hepatic lipid accumulation in healthy subjects: a comparative study using spectral fat-selective MRI and volume-localized 1H-MR spectroscopy. Magn Reson Med 2006, 55, 913–917. [Google Scholar] [CrossRef]
- Wallace, T.M.; Levy, J.C.; Matthews, D.R. Use and Abuse of HOMA Modeling. Diabetes Care 2004, 27, 1487–1495. [Google Scholar] [CrossRef]
- Gannon, M.C.; Nuttall, F.Q. Effect of a high-protein, low-carbohydrate diet on blood glucose control in people with type 2 diabetes. Diabetes 2004, 53, 2375–2382. [Google Scholar] [CrossRef]
- Fujita, Y.; Gotto, A.M.; Unger, R.M. Basal and Postprotein Insulin and Glucagon Levels During a High and Low Carbohydrate Intake and Their Relationships to Plasma Triglycerides. Diabetes 1975, 24, 552–558. [Google Scholar] [CrossRef]
- Shimy, K.J.; Feldman, H.A.; Klein, G.L.; Bielak, L.; Ebbeling, C.B.; Ludwig, D.S. Effects of Dietary Carbohydrate Content on Circulating Metabolic Fuel Availability in the Postprandial State. J Endocr Soc 2020, 4, bvaa062. [Google Scholar] [CrossRef] [PubMed]
- Alsalim, W.; Tura, A.; Pacini, G.; Omar, B.; Bizzotto, R.; Mari, A.; Ahrén, B. Mixed meal ingestion diminishes glucose excursion in comparison with glucose ingestion via several adaptive mechanisms in people with and without type 2 diabetes. Diabetes Obes Metab 2016, 18, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Ohneda, A.; Parada, E.; Eisentraut, A.M.; Unger, R.H. Characterization of response of circulating glucagon to intraduodenal and intravenous administration of amino acids. The Journal of Clinical Investigation 1968, 47, 2305–2322. [Google Scholar] [CrossRef] [PubMed]
- Forslund, A.H.; Hambræus, L.; van Beurden, H.; Holmbäck, U.; El-Khoury, A.E.; Hjorth, G.; Olsson, R.; Stridsberg, M.; Wide, L.; Åkerfeldt, T.; et al. Inverse relationship between protein intake and plasma free amino acids in healthy men at physical exercise. American Journal of Physiology-Endocrinology and Metabolism 2000, 278, E857–E867. [Google Scholar] [CrossRef] [PubMed]
- Fernstrom, J.D.; Wurtman, R.J.; Hammarstrom-Wiklund, B.; Rand, W.M.; Munro, H.N.; Davidson, C.S. Diurnal variations in plasma concentrations of tryptophan, tryosine, and other neutral amino acids: effect of dietary protein intake. Am J Clin Nutr 1979, 32, 1912–1922. [Google Scholar] [CrossRef]
- Nie, C.; He, T.; Zhang, W.; Zhang, G.; Ma, X. Branched Chain Amino Acids: Beyond Nutrition Metabolism. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef]
- Vanweert, F.; Schrauwen, P.; Phielix, E. Role of branched-chain amino acid metabolism in the pathogenesis of obesity and type 2 diabetes-related metabolic disturbances BCAA metabolism in type 2 diabetes. Nutr Diabetes 2022, 12, 35. [Google Scholar] [CrossRef]
- Prodhan, U.K.; Milan, A.M.; Thorstensen, E.B.; Barnett, M.P.G.; Stewart, R.A.H.; Benatar, J.R.; Cameron-Smith, D. Altered Dairy Protein Intake Does Not Alter Circulatory Branched Chain Amino Acids in Healthy Adults: A Randomized Controlled Trial. Nutrients 2018, 10. [Google Scholar] [CrossRef]
- Markova, M.; Hornemann, S.; Sucher, S.; Wegner, K.; Pivovarova, O.; Rudovich, N.; Thomann, R.; Schneeweiss, R.; Rohn, S.; Pfeiffer, A.F.H. Rate of appearance of amino acids after a meal regulates insulin and glucagon secretion in patients with type 2 diabetes: a randomized clinical trial. Am J Clin Nutr 2018, 108, 279–291. [Google Scholar] [CrossRef]
- Moundras, C.; Remesy, C.; Demigne, C. Dietary protein paradox: decrease of amino acid availability induced by high-protein diets. American Journal of Physiology-Gastrointestinal and Liver Physiology 1993, 264, G1057–G1065. [Google Scholar] [CrossRef]
- Matthews, D.E.; Campbell, R.G. The effect of dietary protein intake on glutamine and glutamate nitrogen metabolism in humans. Am J Clin Nutr 1992, 55, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.D.; Matthews, D.E.; Bier, D.M.; Wen, Z.M.; Young, V.R. Response of alanine metabolism in humans to manipulation of dietary protein and energy intakes. Am J Physiol 1986, 250, E39–46. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.A.; Faloona, G.R.; Unger, R.H. The effect of alanine on glucagon secretion. J Clin Invest 1971, 50, 2215–2218. [Google Scholar] [CrossRef] [PubMed]
- Galsgaard, K.D.; Jepsen, S.L.; Kjeldsen, S.A.S.; Pedersen, J.; Wewer Albrechtsen, N.J.; Holst, J.J. Alanine, arginine, cysteine, and proline, but not glutamine, are substrates for, and acute mediators of, the liver-α-cell axis in female mice. American Journal of Physiology-Endocrinology and Metabolism 2020, 318, E920–E929. [Google Scholar] [CrossRef]
- Dandona, P.; Ghanim, H.; Abuaysheh, S.; Green, K.; Batra, M.; Dhindsa, S.; Makdissi, A.; Patel, R.; Chaudhuri, A. Decreased insulin secretion and incretin concentrations and increased glucagon concentrations after a high-fat meal when compared with a high-fruit and -fiber meal. Am J Physiol Endocrinol Metab 2015, 308, E185–191. [Google Scholar] [CrossRef]
- Radulescu, A.; Gannon, M.C.; Nuttall, F.Q. The Effect on Glucagon, Glucagon-Like Peptide-1, Total and Acyl-Ghrelin of Dietary Fats Ingested with and without Potato. The Journal of Clinical Endocrinology & Metabolism 2010, 95, 3385–3391. [Google Scholar] [CrossRef]
- Sloth, B.; Due, A.; Larsen, T.M.; Holst, J.J.; Heding, A.; Astrup, A. The effect of a high-MUFA, low-glycaemic index diet and a low-fat diet on appetite and glucose metabolism during a 6-month weight maintenance period. Br J Nutr 2009, 101, 1846–1858. [Google Scholar] [CrossRef]
- Raben, A.; Holst, J.J.; Madsen, J.; Astrup, A. Diurnal metabolic profiles after 14 d of an ad libitum high-starch, high-sucrose, or high-fat diet in normal-weight never-obese and postobese women. Am J Clin Nutr 2001, 73, 177–189. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Sädevirta, S.; Zhou, Y.; Kayser, B.; Ali, A.; Ahonen, L.; Lallukka, S.; Pelloux, V.; Gaggini, M.; Jian, C.; et al. Saturated Fat Is More Metabolically Harmful for the Human Liver Than Unsaturated Fat or Simple Sugars. Diabetes Care 2018, 41, 1732–1739. [Google Scholar] [CrossRef]
- Zhang, J.; Pivovarova-Ramich, O.; Kabisch, S.; Markova, M.; Hornemann, S.; Sucher, S.; Rohn, S.; Machann, J.; Pfeiffer, A.F.H. High Protein Diets Improve Liver Fat and Insulin Sensitivity by Prandial but Not Fasting Glucagon Secretion in Type 2 Diabetes. Front Nutr 2022, 9, 808346. [Google Scholar] [CrossRef]
- Knudsen, J.G.; Hamilton, A.; Ramracheya, R.; Tarasov, A.I.; Brereton, M.; Haythorne, E.; Chibalina, M.V.; Spegel, P.; Mulder, H.; Zhang, Q.; et al. Dysregulation of Glucagon Secretion by Hyperglycemia-Induced Sodium-Dependent Reduction of ATP Production. Cell Metab 2019, 29, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Gar, C.; Haschka, S.J.; Kern-Matschilles, S.; Rauch, B.; Sacco, V.; Prehn, C.; Adamski, J.; Seissler, J.; Wewer Albrechtsen, N.J.; Holst, J.J.; et al. The liver-alpha cell axis associates with liver fat and insulin resistance: a validation study in women with non-steatotic liver fat levels. Diabetologia 2021, 64, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Szczepaniak, L.S.; Nurenberg, P.; Leonard, D.; Browning, J.D.; Reingold, J.S.; Grundy, S.; Hobbs, H.H.; Dobbins, R.L. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab 2005, 288, E462–468. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, M.E.; Wait, J.B.; Koech, J.; Gordon, A.N.; Coch, R.W.; Svendsen, B.; Finan, B.; D’Alessio, D.A.; Campbell, J.E. Glucagon lowers glycemia when β cells are active. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
Figure 1.
Design of the study. Following an initial screening, the 92 subjects underwent two 6-week interventions: first, they consumed a low fat, high carbohydrate diet (LFD), which was carried out to standardize the diet of the subjects and second the participants consumed an isocaloric high fat diet (HFD). 24 of the subjects additionally participated in a third 6-week intervention, a high protein diet (HPD). Clinical investigation days (CID) took place at the end of each intervention and after one week of HFD, to examine possible short-term effects. A standardized diet was provided one week prior to each CID to increase participant’s compliance.
Figure 1.
Design of the study. Following an initial screening, the 92 subjects underwent two 6-week interventions: first, they consumed a low fat, high carbohydrate diet (LFD), which was carried out to standardize the diet of the subjects and second the participants consumed an isocaloric high fat diet (HFD). 24 of the subjects additionally participated in a third 6-week intervention, a high protein diet (HPD). Clinical investigation days (CID) took place at the end of each intervention and after one week of HFD, to examine possible short-term effects. A standardized diet was provided one week prior to each CID to increase participant’s compliance.
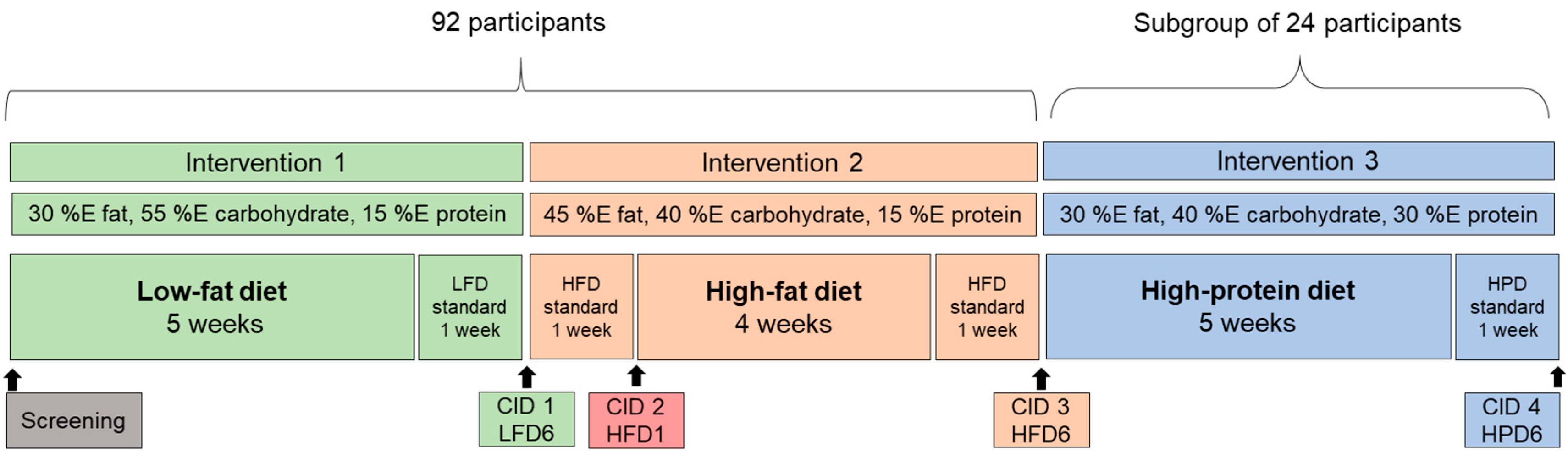
Figure 2.
Effects of varying dietary fat intake on glucose metabolism. All parameters are shown for screening (Scr, grey), after low fat diet for 6 weeks (LFD6, green), after high fat diet for 1 week (HFD1, red) and after high fat diet for 6 weeks (HFD6, orange). Values are shown as mean ± SEM. A: Fasting serum glucagon (GCGN) values. GCGN showed a pronounced decrease in response to 6 weeks of low fat intake and increased significantly after 1 week of high fat intake and even further after 6 weeks. B: Fasting serum insulin. Fasting insulin levels increased slightly but significant after 1 week of high fat intake, however returned to baseline levels after 6 weeks. C: Fasting serum free fatty acids (FFA). FFAs changed opposite to GCGN levels and increased after the LFD but decreased again after HFD. D: Homeostasis model assessment-estimated insulin resistance. The HOMA-IR increased slightly due to high dietary fat intake, nevertheless the subjects remained very insulin sensitive at all time points. E: GCGN-to-insulin ratio. As insulin levels did not change extensively the GCGN-to-insulin ratio acts similar to the fasting GCGN levels, thus decreased in response to the LFD and increased in response to the HFD.
Figure 2.
Effects of varying dietary fat intake on glucose metabolism. All parameters are shown for screening (Scr, grey), after low fat diet for 6 weeks (LFD6, green), after high fat diet for 1 week (HFD1, red) and after high fat diet for 6 weeks (HFD6, orange). Values are shown as mean ± SEM. A: Fasting serum glucagon (GCGN) values. GCGN showed a pronounced decrease in response to 6 weeks of low fat intake and increased significantly after 1 week of high fat intake and even further after 6 weeks. B: Fasting serum insulin. Fasting insulin levels increased slightly but significant after 1 week of high fat intake, however returned to baseline levels after 6 weeks. C: Fasting serum free fatty acids (FFA). FFAs changed opposite to GCGN levels and increased after the LFD but decreased again after HFD. D: Homeostasis model assessment-estimated insulin resistance. The HOMA-IR increased slightly due to high dietary fat intake, nevertheless the subjects remained very insulin sensitive at all time points. E: GCGN-to-insulin ratio. As insulin levels did not change extensively the GCGN-to-insulin ratio acts similar to the fasting GCGN levels, thus decreased in response to the LFD and increased in response to the HFD.

Figure 3.
Effects of high protein intake on glucose metabolism. All parameters are shown for screening (Scr, grey), after low fat diet for 6 weeks (LFD6, green), after high fat diet for 1 week (HFD1, red), after high fat diet for 6 weeks (HFD6, orange) for the subgroup of 24 participants that additionally conducted 6 weeks of high protein diet (HPD6, blue). In general, the responses of this subgroup to LFD and HFD were similar to those of the total cohort (see Figure 2). Values are shown as mean ± SEM. A: Fasting serum Glucagon (GCGN) values. GCGN showed a pronounced increase after 6 weeks of HPD. B: Fasting serum insulin. Fasting insulin levels decreased significantly after 6 weeks of high protein intake. C: Fasting serum free fatty acids (FFA). FFAs were highest in response to 6 weeks of HPD. D: Homeostasis model assessment-estimated insulin resistance. The HOMA-IR decreased slightly but significantly through the HPD. E: GCGN-to-insulin ratio. The GCGN-to-insulin ratio increased most intensively with the HPD.
Figure 3.
Effects of high protein intake on glucose metabolism. All parameters are shown for screening (Scr, grey), after low fat diet for 6 weeks (LFD6, green), after high fat diet for 1 week (HFD1, red), after high fat diet for 6 weeks (HFD6, orange) for the subgroup of 24 participants that additionally conducted 6 weeks of high protein diet (HPD6, blue). In general, the responses of this subgroup to LFD and HFD were similar to those of the total cohort (see Figure 2). Values are shown as mean ± SEM. A: Fasting serum Glucagon (GCGN) values. GCGN showed a pronounced increase after 6 weeks of HPD. B: Fasting serum insulin. Fasting insulin levels decreased significantly after 6 weeks of high protein intake. C: Fasting serum free fatty acids (FFA). FFAs were highest in response to 6 weeks of HPD. D: Homeostasis model assessment-estimated insulin resistance. The HOMA-IR decreased slightly but significantly through the HPD. E: GCGN-to-insulin ratio. The GCGN-to-insulin ratio increased most intensively with the HPD.
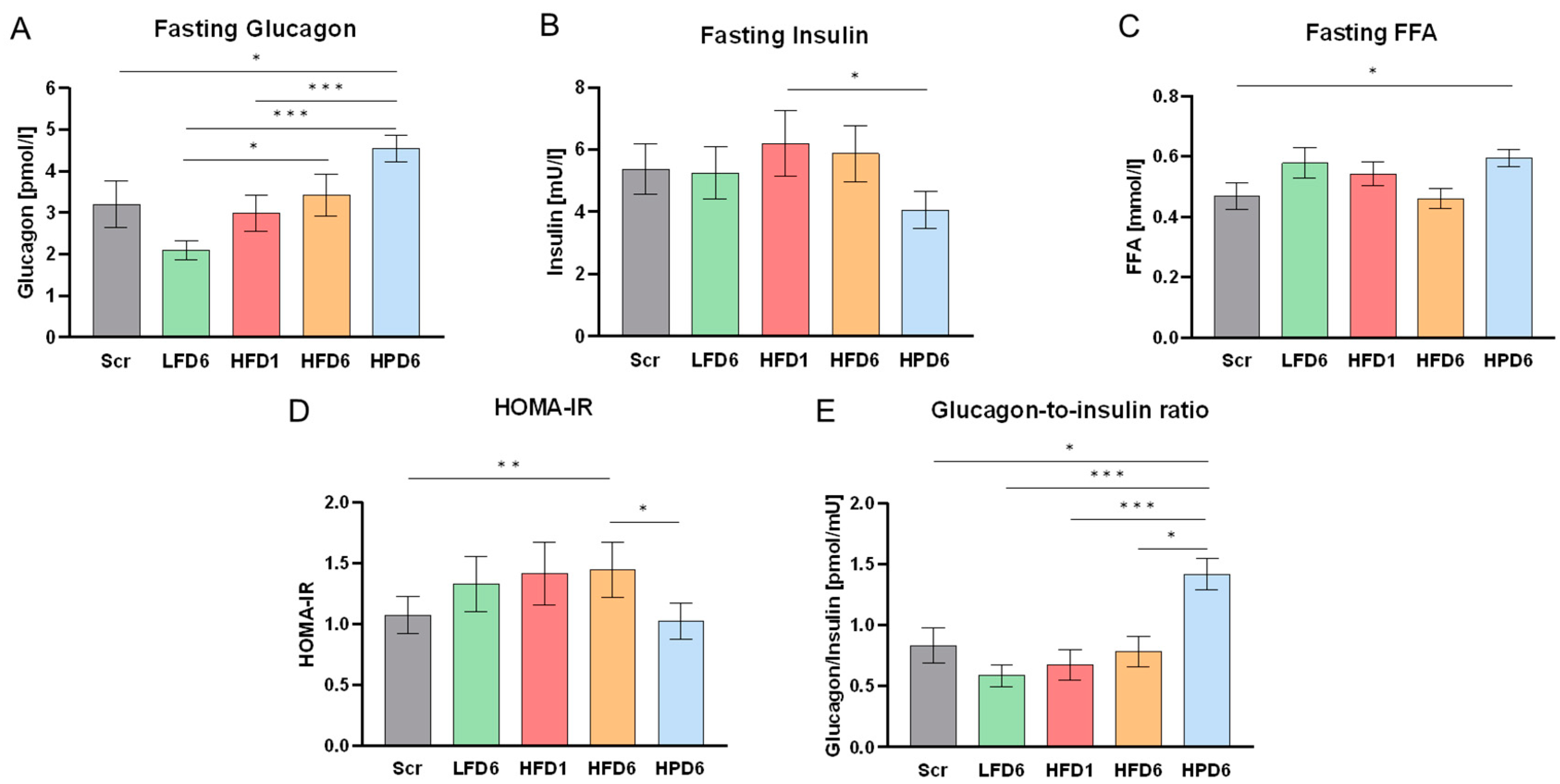

Table 1.
Baseline clinical parameters of all subjects and the subgroup that additionally conducted. the high protein intervention.
Table 1.
Baseline clinical parameters of all subjects and the subgroup that additionally conducted. the high protein intervention.
| Baseline characteristics (mean ± SD) |
All participants n=92 |
HPD subgroup n=24 |
|---|---|---|
| Sex [female/male] | 58/34 | 10/14 |
| Zygosity [mono/di] | 68/24 | 20/4 |
| Age [y] | 31 ± 14 | 39 ± 15 |
| BMI [kg/m²] | 23 ± 3 | 24 ± 2 |
| WHR | 0.81 ± 0.07 | 0.84 ± 0.06 |
| Systolic BP [mm Hg] | 118 ± 13 | 117 ± 11 |
| Diastolic BP [mm Hg] | 74 ± 9 | 76 ± 6 |
| Fasting insulin [mU/L] | 5.21 ± 3.68 | 5.38 ± 4.00 |
| Fasting glucose [mmol/L] | 4.78 ± 0.48 | 5.05 ± 0.46 |
| HbA1c [%] | 5.0 ± 0.4 | 5.0 ± 0.4 |
| HbA1c [mmol/mol] | 31 ± 5 | 31 ± 5 |
| Fasting glucagon [pmol/L] | 3.97 ± 2.71 | 3.21 ± 2.75 |
| Fasting total cholesterol [mmol/L] | 4.58 ± 0.93 | 4.69 ± 0.98 |
| Fasting HDL cholesterol [mmol/L] | 1.38 ± 0.35 | 1.32 ± 0.34 |
| Fasting LDL cholesterol [mmol/L] | 2.73 ± 0.77 | 2.89 ± 0.83 |
| Fasting triglycerides [mmol/L] | 0.99 ± 0.44 | 0.94 ± 0.38 |
| Fasting FFA [mmol/L] | 0.52 ± 0.26 | 0.47 ± 0.22 |
Values are shown as mean ± SD. BP, Blood pressure; FFA, Free fatty acids; HPD, High protein diet; WHR, Waist-to-hip-ratio.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



