Submitted:
22 October 2024
Posted:
24 October 2024
You are already at the latest version
Abstract
Evolving or "re-emerging" viral deviation remains to pretense substantial worldwide public health threats. Influenza A virus (IAV), the main contributor to respiratory infections and related death in humans, is a prime example. Universally well-known as ‘flu’, it has disturbed our daily breathing, leaving a breakthrough till now. This virus can transmit from animals to humans and familiarize human hosts, resulting in ongoing transmission and the rise of novel viral strains. The processes through which viruses evolve within one host, cause animal-to-human transmission, and acclimatize to different host species are not yet fully understood. This review scans influenza antiviral resistance, highlighting the fundamental concepts behind its development and clinical impact. Influenza A represents a significant burden on healthcare systems and communities. While antiviral drugs are crucial for managing influenza effectively, healthcare providers and policymakers may need to be cautious due to the potential for antiviral-resistant strains to develop. Adamantanes and neuraminidase inhibitors targeting the viral components of the M2 ion channel and NA have been approved to treat IAV. However, the ongoing dissemination of drug-resistant virus variants due to mutation of these viral components poses a challenge and could undermine the effectiveness of primary antiviral therapies. Therefore, this review provides the susceptibility status of IAV subtypes to currently available antiviral drugs, factors to anti-IAV resistance, and soon-to-be-available control measures to emerging drug resistance IAV.
Keywords:
Anti-influenza agents
; Anti-influenza resistance
; Influenza A virus
; M2 ion channel inhibitor
; Neuraminidase inhibitors
1. Introduction
Influenza A viruses (IAV) of avian and swine origin have resulted in 5 pandemics in the aforementioned 2 centuries. An age-old doctrine of IAV biology identified swine as a mixing vessel and vivacious to the emergence of human pandemic IAV by auxiliary reassortment that could lead to antigenic shift[1]. Because influenza viruses are efficient at evading the host's immunity against infections, they continuously alter their antigenicity. In addition to causing the formation of drug-resistant strains, genetic alterations in the influenza virus genome produce antigenic drift[2] but also evade antibody-mediated viral neutralization[3]. An illness that has currently been identified and is largely caused by influenza viruses A or B is referred to as "flu" informally. People may come into touch with an infectious infection from a patient's cough or sneeze. Influenza signs comprise nasal discharge, non-productive cough, sore throat, elevated temperature, chills, and myalgia [4]. IAV is a cause of common respiratory infections in humans, spreading as seasonal epidemics and sporadic pandemics. IAV belongs to the family Orthomyxoviridae. Its viral genome is organized in eight, negative-sense RNA segments [1]. They are enveloped viruses with a single-stranded negative-sense RNA genome, composed of seven or eight segments that code for nine to 11 viral proteins. Based on differences in antigenicity among HA and NA proteins, IAV can be further classified into 16 HA and 9 NA subtypes. Among these subtypes, only three HA (H1, H2, H3) and two NA (N1, N2) subtypes have efficiently infected humans in the last century, with the seasonal H1N1 and H3N2 viruses as well as the recently described 2009 pandemic H1N1 (pH1N1) strain as the most relevant at present. Nevertheless, intermittent cases of transmission of avian IAV of the H5, H7, or H9 subtype to humans have been reported, with inconstant clinical outcomes. Influenza viruses are major human pathogens with a worldwide dissemination, causing yearly outbreaks with a seasonal pattern in cold areas [5].
Antiviral medications are the most important component of orders for mitigating pandemic strains of IAV for which vaccines are not immediately available[6]. The genomic foundation for adamantine resistance has been characterized well and substitutions of amino acids at residues L26, V27, A30, S31, and G34 within the transmembrane domain of the M2 protein are associated with adamantane resistance[7]. Up to now, L26F, G34E, V27A, A30V, S31N, A30T, and L38F in the transmembrane region of M2 are the identified mutations that confer adamantine resistance[8]. Some studies reported that out of 7000 influenza A isolates collected from countries in Africa, Asia, Europe, the Americas, and Oceania between 1994 and 2005, the occurrence of adamantane resistance fluctuations rose from 0.4% in 1994–1995 to 12.3% in 2003–2004[9]. The swift rise and blowout of variants of adamantane-resistant influenza H1N1, H3N2, recent H7N9 viruses from China, and H5N1 viruses have headed to changes in recommendations of CDC for the use of adamantanes in the regulator of IAV infections[10].
The whole genome of IAV size is approximately 13.5 kb, whereas the viral RNA varies in size from around 0.9 to 2.3 kb. Every vRNA displays the same structure: Two brief untranslated regions (UTRs) surround the core open reading frame, which encodes one or more proteins (in the antisense orientation). PB2, PB1, PA, HA, NP, NA, M, and NS are the names given to segments 1 through 8 based on the protein that is encoded in them. Based on the glycoproteins on their surface, IAVs can be separated into antigenic subtypes: Classifications for HA proteins are H1 through H18, while classifications for A proteins are N1 through N11. Only a limited number of these HAs and NAs have been isolated from viruses known to infect humans[11].
In addition to having membrane fusion activity and receptor binding, HA facilitates viral entrance into cells. In the advanced stages of invasion, NA promotes cleavage of the viral receptor enzymatically, facilitating progeny virion release. M2 is a multipurpose, proton-selective ion channel involved in assembly, budding, and virus entry. The M1 protein gives the virion structure and mediates connections between the lipid membrane and the RNP core of the virus. The NP facilitates the binding and packaging of its genome, and PB1, PB2, and PA make up the RNP core. Non-structural protein 1 is a multi-functional protein with a significant role in avoiding the host's immune system. NS2 plays a vital role in facilitating the export of viral RNPs from the cell nucleus during replication. Furthermore, many strains of the influenza virus, though not all, produce a protein called PB1-F2 which is transcribed from an alternative reading frame in PB1. The PB1-F2 protein is implicated in triggering host-cell apoptosis[12].
IAV attacks several backbone-animals. IAV from various host species can 'reassort' their segmented genomes, generating antigenically unique pandemic strains that are otherwise well-suited to humans. The Great Influenza pandemic of 1918 is still the most severe infectious disease outbreak in the past. There is apprehension that highly pathogenic avian influenza viruses of the H5 and H7 subtypes might evolve to trigger similar pandemics. In humans, influenza viruses infect the respiratory epithelium. The HA proteins of IAV and IBV, or the haemagglutinin-esterase-fusion proteins of ICV, bind SA, causing endocytosis. Strangely among RNA viruses, the viral genome replicates in the nucleus. New viruses assemble at the cell surface and are released by the receptor-cleaving neuraminidase (NA) proteins of IAV and IBV or the ICV hemagglutinin-esterase-fusions protein[13].
IAV is the etiology of major ill health and death in the bargained immune systems of patients. IAV infection in organ transplant recipients is associated with a higher rate of respiratory complications, extra pulmonic signs, an augmented risk of graft dysfunction and rejection, and high attributable death[14].
M2 ion channel blockers and inhibitors of neuraminidase have been sanctioned for treating IAV infections. However, similar to antibiotic resistance, the emergence of antiviral drug resistance in the influenza virus is a significant concern. As a result, NAIs are the only class of anti-influenza medications currently in use since most circulating influenza viruses have developed resistance to M2 ion channel blockers. Nonetheless, many circulating influenza viruses have also developed resistance to NAIs. This review emphasizes the emergence of drug-resistance mutations in M2 and NA, with a focus on influenza A virus[15].
Antiviral resistance development in IAV is driven by the virus's high mutation rate and genetic diversity, which enable it to rapidly adapt to selective pressures imposed by antiviral therapies. Mutations in key viral proteins, such as neuraminidase and polymerase complex subunits, can reduce the effectiveness of current antiviral drugs, rendering them less effective or even obsolete. This pretenses a substantial challenge to public health, as resistant strains can lead to treatment failures, prolonged illness, and increased transmission. Despite the critical importance of this issue, there remain significant cracks in our understanding of the molecular mechanisms underlying antiviral resistance in Influenza A. Specifically; there is a need for comprehensive research to identify resistance-associated mutations, understand their impact on viral fitness and drug susceptibility, and develop strategies to counteract resistance.
With viral RNA polymerase's low proofreading activity and inherently high error rate, IAVs exist as populations of quasispecies. Chance mutations can be rapidly selected for or against, depending upon the evolutionary pressures applied, including a novel host environment, response to pre-existing immunity leading to antigenic drift, or antiviral drug pressure leading to resistance.
Therefore, the key objective of this review is
- ⮚
- To assess the current and future alternative antiviral drugs used to treat IAV
- ⮚
- To describe the various mechanisms by which IAV develops resistance to commonly used anti-influenza drugs
2. Literature Review
2.1. Genomic Organization of the Influenza a Virus RNA
IAV encompasses a segmented and negative-sense-single-stranded RNA genome, with each RNA segment packaged into a viral vRNP complex[16] Within a vRNP, a single RNA-dependent RNA polymerase molecule captures the 3′ and 5′ termini of the viral RNA, forming a pseudo-circularised piece of RNA[11]. The rest of the vRNA is associated with nucleoprotein, with an estimated ratio of 24 nucleotides per NP. NP oligomerizes, forming a helical framework that packages and protects the viral genome[17]. In the course of infection, the virus binds to a cell-surface receptor and enters the host cell via endocytosis. Afterward, vRNPs are released into the cytoplasm and are trafficked to the nucleus. Once inside the nucleus, the viral polymerase transcribes and replicates the viral genome[18].
Associations between the function and structure of RNA have been the subject of rigorous research for centuries[19]. Several studies have demonstrated the biological significance of the viral RNA secondary structure in critical phases of the pathogen's replication cycle. Influenza RNA secondary structure motifs have been meticulously examined in vitro[20,21].
Based on complex sample studies and bioinformatics studies, structural models have been proposed for full-length naked vRNA5, 7, and 8, so far[20]. They have unveiled the complicated nature of vRNA secondary structures, which are organized into domains. Identified secondary structure motifs may form locally or involve long-range interactions. In addition to the panhandle motif, formed by partial base-pairing of the 5′ and 3′ vRNA terminal nucleotides, whose functional role and structural dynamics in influenza transcription and replication have been well-defined, other motifs have also been described [21,22,23]. vRNAs helical regions are separated with bulges and single-stranded loops, which are potential sites for interactions of functional importance. RNA secondary structure significance of the influenza virus has been supported by sequence-structure bioinformatics studies showing the conservation of structural motifs, despite the high genetic variability of the virus. Therefore, compensatory mutations consistent with the predicted structure can be observed among strains. Mutations that occur in conservative regions regularly permit the maintenance of canonical base pairs[24].
IAV every segment is enclosed by NP. The RNA of IAV is enveloped around NP monomers and assembled into viral RNPs along with PB2, PB1, and PA in virions. A heterotrimer is formed from polymerase proteins and attached to a short hairpin structure formed by the partially complementary terminal 5' and 3' untranslated regions of each RNA segment. PB1 functions as the RNA-dependent RNA polymerase. PB2 is involved in mRNA synthesis by binding to host mRNA caps. It may have additional proteolytic activity and might also function as an elongation factor during RNA synthesis. NP primarily acts as a single-strand RNA-binding protein and serves as the structural component in the RNPs. Moreover; it theatres a critical role in transcription and the trafficking of RNPs between the nucleus and cytoplasm. IAV RNA transcription and replication occur in the host nucleus, as IAV relies on the RNA processing machinery of the host cell. The processes of import into the nucleus, export back to the cytoplasm, and prevention of re-entry into the nucleus all appear to depend on the interaction of NP with host proteins[25].
Figure 1.
Graphic depiction of the influenza A virion architecture.
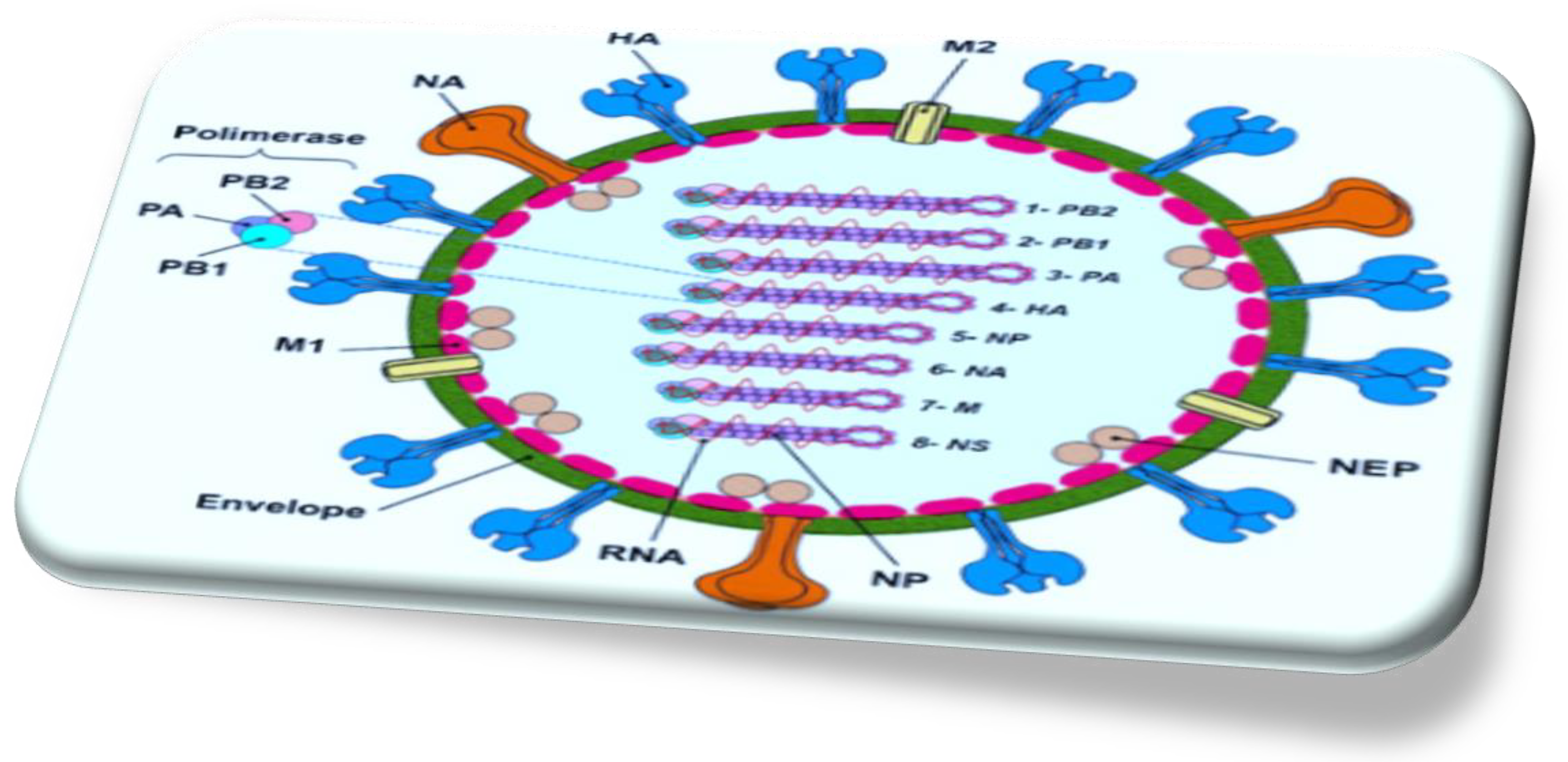
HA and NA, alongside small numbers of M2 ion channel protein, are surrounded by a lipid bilayer. The M1 protein underlies the envelope and interacts with the surface proteins and also with the RNPs. RNPs comprise the eight negative-stranded RNA segments and nucleoprotein (NP) and the polymerase complex heterotrimer (PB2, PB1, and PA) [25].
The structure of NA is composed of four indistinguishable polypeptides, resembling a mushroom. The four domains of NA: are an N-terminal cytoplasmic sequence, followed by a hydrophobic trans-membrane domain that anchors it to the membrane. This is connected to a slim branch of variable length, which culminates in a globular head domain where the SA binding site is located[26].
HA is a glycosylated type I integral membrane protein that functions both as the fusion protein and viral receptor-binding protein. The M1 protein is located beneath the membrane and interacts with the cytoplasmic domains of the surface glycoproteins as well as with the viral ribonucleoprotein (RNP) complexes. HA recognizes SA (N-acetylneuraminic acid) attached to core sugars on the tips of host cell glycoproteins. IAVs possess HAs with different specificities for the disaccharide composed of SAs and the penultimate sugar (galactose or N-acetylgalactosamine [GalNAc]) with varying glycosidic bond isomerization[27]. IAVs adapted to birds have an HA receptor-binding preference for α2-3 SA, whereas HAs from IAVs adapted to humans exhibit higher specificity for α2-6 SA. Upon binding its receptors, the virus is internalized, and the acidic pH in the endosomal compartment triggers a conformational change in HA, facilitating the fusion of the viral and endosomal membranes and allowing the release of viral RNPs into the cytoplasm. Mature HA is a trimer, and each monomer undergoes proteolytic cleavage to generate disulfide-bonded HA1 and HA2 polypeptide chains before activation. Influenza A virus requires exogenous serine proteases (trypsin-like enzymes) for activation, recognizing a conserved Q/E-X-R motif found at the HA cleavage site. In other mammals and humans, this is possibly the tryptase Clara produced by cells of the bronchiolar epithelium. In avian intestinal and respiratory cells, cleavage activation of HA likely requires similar proteases. IAV of H5 and H7 subtypes can obtain insertional mutations at the HA cleavage site, changing their protease recognition site to a furin-like recognition sequence, R-X-R/K-R. This polybasic cleavage site broadens protease specificity, allowing intracellular cleavage activation and systemic replication, resulting in a highly pathogenic avian influenza[28].
2.2. Mechanism of Action of IAV Replication
Attachment and Entrance: IAV binds to SA residues on the host's glycoprotein or glycolipid receptors to enter the host cell via the hemagglutinin glycoprotein present in the viral envelope. Once the virus is internalized by the cell, the hemagglutinin protein undertakes a conformational change and merges with the endosomal membrane in the acidic environment of the endosome[29]. Complexes of vRNP are subsequently discharged into the host cell's cytoplasm and transported to the nucleus; they photocopy and produce viral mRNA transcripts. This viral entry stage presents a promising antiviral target strategies aimed at inhibiting ion channels, thereby preventing vRNP discharge into the cytoplasm[30].
Synthesis of RNA and Protein: complexes of viral vRNP conduct primary transcription in the host's nucleus, producing the proteins required for replication. The primary transcription mechanism is known as “cap snatching,” where the 5′ methylguanosine cap of the host’s mRNA is bound by PB2, followed by the removal of the cap and 10–13 nucleotides by the viral endonuclease, polymerase acidic[31]. The resulting oligonucleotide serves as a primer for transcription by viral polymerase protein 1. This phase of processing of the virus is also a potential target for antiviral therapy. During the inhibition of the cap-snatching process, viral mRNA transcription can be halted, thereby preventing the production of the proteins necessary for virion assembly[32]. Eight genome segments translation in influenza A produces ten primary proteins, including a component of the vRNP complex, two M proteins, two NS proteins, NA, NP, and HA. Another key protein, RNA-dependent RNA polymerase, can be selectively inhibited by antiviral agents, making it a potential target for viral inhibition strategies[33].
Packaging and export: After initial protein production, the eight negative-sense viral RNA segments in influenza A are used to generate eight positive-sense complementary RNA strands. These strands lack the 5′ capped primer and the 3′ poly (A) tail found in mRNA. Negative-sense vRNA is then synthesized from the cRNA. Several viral proteins produced afterward assemble and assist in the export of this vRNA from the nucleus into the host cytoplasm [12].
At the packaging and export stage, HA and NA undergo glycosylation, polymerization, and acylation in the cytoplasm. Along with M2, they are transported to the plasma membrane, where they become anchored. The proteins then interact with the matrix protein M1, initiating the budding process. The virus buds once at least eight RNA segments reach the site through an unidentified means. Lastly, the SA of the HA protein of the progenitor is cleaved by neuraminidase, allowing the virus to exit the cell. This step of NA-mediated is a promising target for inhibition, as inhibiting the cleavage of SA residues would reduce viral infectivity by limiting the dispersal of progeny virus within mucosal secretions[34].
2.3. Natural History and Epidemiology of IAV
IAV from seals[35], whales[36], horses[37], and pigs have been found[38]. SA is the receptor for IAV; the viral HA binds to the receptor. Avian and mammalian influenza viruses preferentially bind SA molecules with specific oligosaccharide side chains with a2-3 and a2-6 linkages, respectively. Receptor specificity is thought to be a vital determinant of host range. Reports of natural human infections with avian influenza viruses have been sporadic and experimental infections have been difficult to achieve[39], leading to the vision that avian influenza viruses are limited in their ability to replicate in human hosts. It was thought that avian IAV would have to gain one or more genes from a human IAV to cross the species barrier. Pigs tracheal epithelium bears SA molecules with both a2,3 and a2,6 linkages to galactose that can be infected with avian in addition to mammalian IAV[40]. Simultaneous infection of pigs with avian and mammalian influenza A viruses were consequently proposed to be the intermediate hosts in which genetic reassortment would take place, giving rise to novel IAV pandemic potential[41].
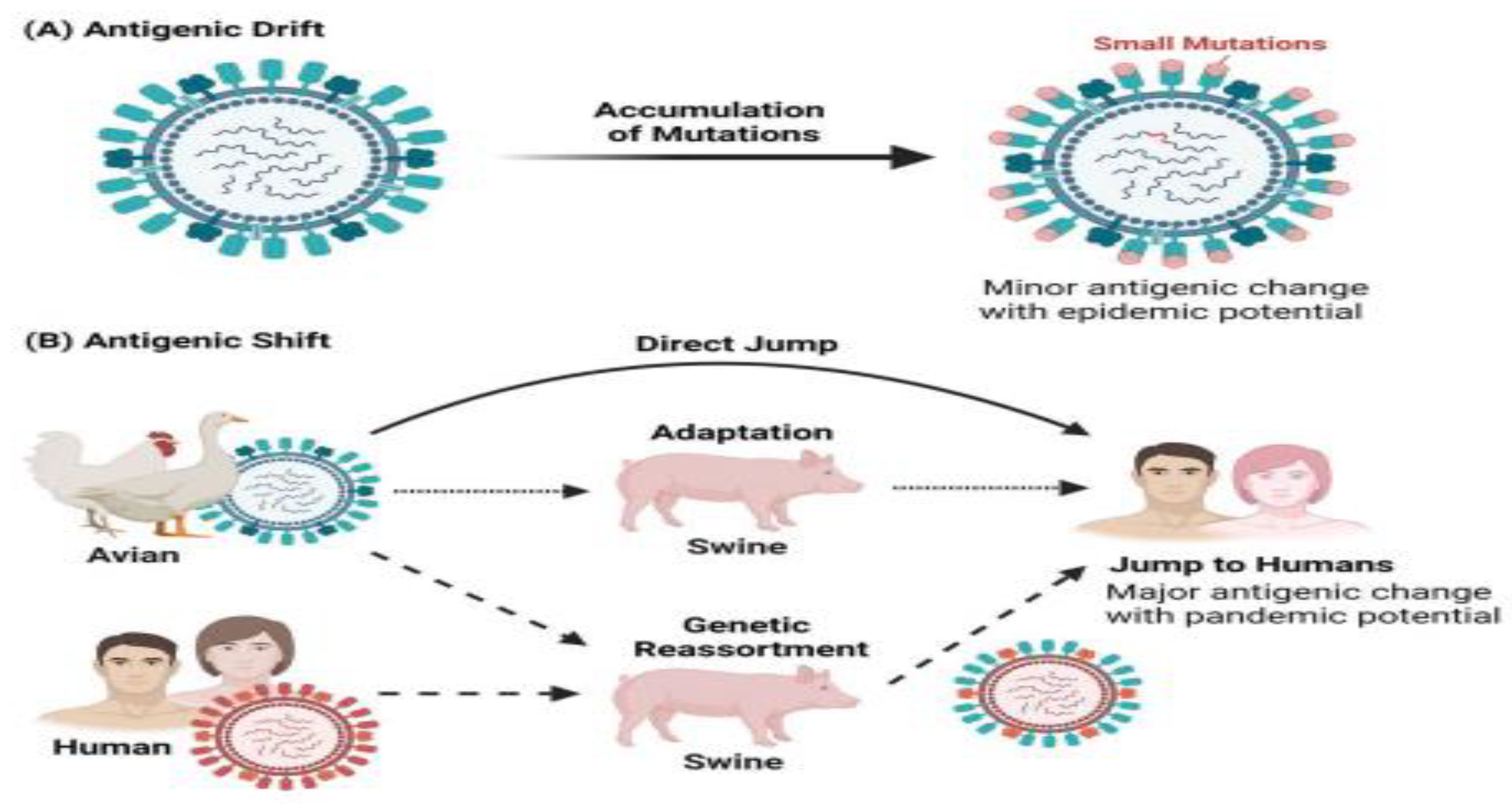
Figure 2.
Influenza A Virus Mutation and Reassortment. (A) Antigenic Drift results in minor antigenic changes from an accumulation of mutations. (B) Antigenic Shift results in major antigenic change via direct jump, adaptation, and genetic reassortment. Figure credited to BioRender.com.
Figure 2.
Influenza A Virus Mutation and Reassortment. (A) Antigenic Drift results in minor antigenic changes from an accumulation of mutations. (B) Antigenic Shift results in major antigenic change via direct jump, adaptation, and genetic reassortment. Figure credited to BioRender.com.
Influenza A virus virion particles exhibit both spherical and filamentous shapes and contain a negative-sense, segmented, single-stranded RNA genome. Each of the eight IAV gene segments encodes at least one primary viral protein. However, some IAV segments produce more than one viral protein through mechanisms such as leaky ribosomal scanning, alternative splicing, ribosomal frameshifting, and the use of alternative start codons[42]. To date, IAV has been reported to encode at least 17 viral proteins, though not all IAV subtypes encode each protein. IAV is an enveloped virus, and each virion displays approximately about 40 NA and 300 HA glycoprotein spikes on its surface. HA serves as the receptor-binding protein and aids in IAV entry into the host cell, while NA helps in the release of newly produced virions from the host cell.
M2 forms an ion channel and is critically involved in virus entrance, is also embedded in the viral envelope, which is derivative from the host cell plasma membrane. Underneath the envelope is a rigid layer comprised of M1, which maintains the shape and integrity of the IAV virion. M1 also interacts with the cytoplasmic domains of IAV envelope proteins and viral ribonucleoprotein (vRNP) core[43]. The vRNP core primarily comprises the viral genome, nucleoprotein (NP), and polymerase complex, which consists of PA, PB1, and PB2 proteins.
IAV has a worldwide occurrence and broad host range, including humans, seals, horses, pigs, dogs, cats, and birds. The aquatic birds, such as waterfowl and shorebirds, are the reservoir hosts of IAV[44]. IAV is subtyped based on the type and antigenicity of its surface glycoproteins, HA, and NA. So far, 18 HA and 11 NA subtypes have been defined, of which 16 HA and 9 NA have been found to circulate in avian species, however 2 HA and 2 NA subtypes have been detected in bats[45]. Nevertheless, the bat IAV subtypes, H17N10 and H18N11, are remarkably different from other IAV subtypes prompting the proposal that these bat viruses should be labeled as influenza-like viruses[46].
Between-species interaction of IAV occurs frequently and significantly between humans and pigs, and between poultry and pigs, while it is infrequent in other cases. The capacity of IAV to spread among species depends on its ability to alter its specificity to infect different species. IAV is also adept at fostering antigenic diversity through two distinct mechanisms called antigenic drift and antigenic shift.[47]. Antigenic drift causes mutations in HA and NA resulting in antigenic variants, which can reinfect a host and avoid the pre-existing immunity.
The error-prone nature of viral RNA polymerase is the major sponsor of antigenic drift, which along with frequent reassortment and natural selection is the main cause of recurring seasonal influenza epidemics[48]. These epidemics are capable of lasting at least 6 to 12 weeks, with observed infection rates of 10–30% in adults and 20–50% in children[49]. Alternatively, antigenic shift is the reassortment of gene segments between two different parental viruses within the same host, giving rise to a novel pandemic IAV. The H1N1 subtype, which caused the first recorded IAV pandemic in 1918, originated from the reassortment between a human H1 subtype and an avian N1 subtype[50]. The IAV pandemic of 1957 was caused by the H2N2 subtype, which originated from the reassortment of the mixing 1918 H1N1 subtype with an avian H2N2 subtype. The subsequent IAV pandemic in 1968 was caused by the H3N2 subtype. The Influenza A virus H3N2 subtype caused a pandemic in 1968, which rose when the circulating H3 subtype was reassorted with the 1957 H2N2 subtype. The latest IAV pandemic, in 2009, was caused by the H1N1 subtype which originated from swine. This subtype originated from sequential reassortment events between swine H1N1, human H3N2, and avian H1N2 subtypes from both North American and Eurasian lineages[51].
Subsequently, all four pandemics together led to the mortality of lots of persons globally. Lately, several exclusively avian-origin influenza A virus subtypes have been identified as a cause of deadly outbreaks in humans. Many of these IAV subtypes lead to a disease that is clinically different and more severe compared to that caused by human IAV subtypes, resulting in a significantly high mortality rate[51]. IAV uses SA, carbohydrate moieties present on the host cell surface, as its receptor. The HA of human IAV subtypes specifically binds to SA with α-2,6-linkages, while the HA of avian IAV subtypes binds to SA with α-2,3-linkages. In humans, the upper respiratory tract predominantly contains SA with α-2-6linkages, whereas SA with α-2,3-linkages is found in the lower respiratory tract. This enables avian IAV subtypes to infect humans upon exposure. Interestingly, both α-2-6 and α-2-3 linkages are present in the upper respiratory tract of pigs, making them “mixing vessels” for the generation of reassortant IAV subtypes between human and avian strains. Fortunately, none of these avian IAV subtypes have acquired the capability of direct human-to-human transmission by aerosol; they mainly spread through direct contact. The transmission of avian IAV to humans is limited by differing receptor specificities[52].
2.4. Spread and Epidemiology of Drug-Resistant Human Influenza Viruses
Pandemics, epidemics, outbreaks, and sporadic, isolated instances are all possible outcomes of influenza. In the coldest parts of the northern and southern hemispheres, seasonal epidemics frequently occur in the winter[53]. In hot zones, influenza outbreaks can occur in all seasons[54]. The WHO has estimated that annual influenza epidemics result in about 4 million severe infection cases and about half of a million deaths each year[55]. The infection's epidemiologic pattern reflects the virus's changing antigenic properties, resulting in new viral strains that change, the virus's capability to transmit, and the population's susceptibility. An antigenic shift occurs when quick and significant changes in surface glycoproteins (particularly HA of Influenza A virus) occur due to a genetic transfer from some animal strains to human strains[56]. Over the previous period, substantial changes in the circulation of antiviral-resistant influenza viruses have occurred. The emergence and continued circulation of A (H1N1)pdm09 and adamantine-resistant A (H3N2) viruses mean that the adamantanes are no longer recommended for use. Resistance to the newer class of drugs, the neuraminidase inhibitors, is typically associated with poorer viral replication and transmission. But ‘permissive’ mutations, which compensated for the impairment of viral function in A (H1N1) viruses during 2007/2008, enabled them to acquire the H275Y NA resistance mutation without fitness loss, resulting in their rapid global spread. Permissive mutations now appear to be present in A(H1N1)pdm09 viruses thereby increasing the risk that oseltamivir-resistant A(H1N1)pdm09 viruses may also spread globally, a concerning scenario given that oseltamivir is the most widely used influenza antiviral[57].
2.5. Pathogenesis of IAV
IAV is renowned for initiating complications, ranging from bronchitis and otitis media to death[58]. Possibly most are afraid is when replication of the virus spreads to the lower airways and (severe) inflammation of the lung unfolds[59]. This can potentially lead to significant lung injury, low blood oxygen level, circulatory collapse, kidney failure, and acute respiratory disease syndrome.
From a viewpoint of its pathology, the hallmark of alveolar alterations in IAV-induced pneumonia triggered by cytokine rainstorms includes capillary clotting, alveolar wall congestion, hyaline membrane formation, fluid accumulation in the lung, and bleeding around the bronchi[60]. The deviations distinctive of severe IAV pneumonia include interstitial edema and inflammatory infiltrate, capillary and small vessel coagulation, the formation of hyaline membranes in alveoli and alveolar ducts, diffuse alveolar damage and varying degrees of acute interalveolar edema and hemorrhage in addition to necrotizing bronchitis and bronchiolitis. In advanced stages: diffuse alveolar damage, fibrosis, epithelial regeneration, and squamous metaplasia may encountered[60].
Production of Cytokines in IAV
IAV infection is linked with a storm of cytokines and a blown-up intrinsic immune response[61]. IAV-infected cells of epithelium and leukocytes respond to the infection by producing chemokines, proinflammatory, and other immunoregulatory cytokines[62].
Figure 3.
Production of cytokine in influenza A virus-infected epithelial cells and macrophages.
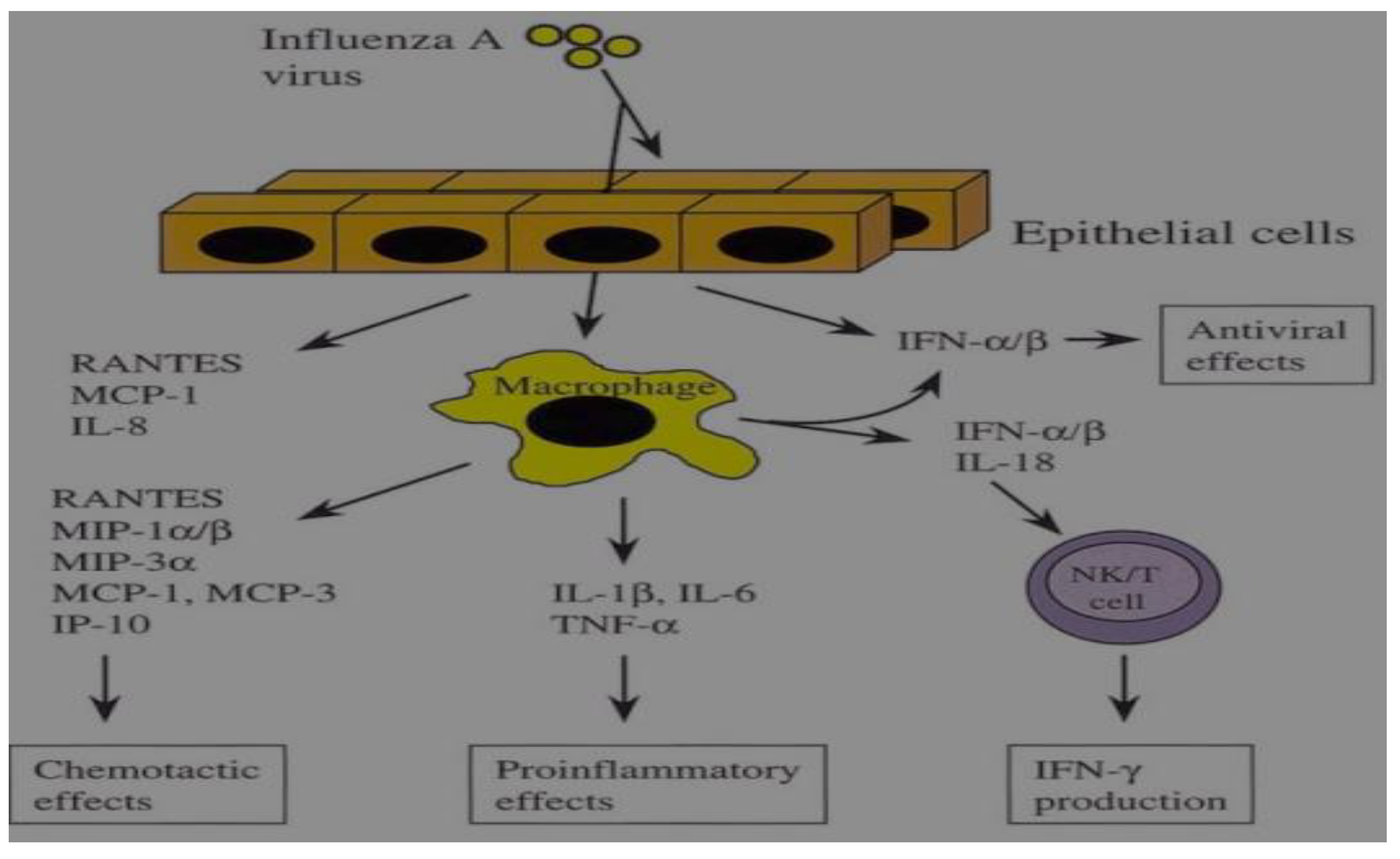
Respiratory tract epithelial cells and macrophages are the principal aims of IAV infection. In answer to the infection, epithelial cells generate a limited range of cytokines, including antiviral IFN-1 and chemokines. Conversely, a wide variety of proinflammatory cytokines and chemokines is produced by macrophages. Macrophages infected with IAV secrete IFN and IL-18, which together boost NK and T cell IFN production and promote the development of immune response of Th1-type [63,64].
Although activated NF-κB signaling promotes the production of proinflammatory cytokines, it also induces the expression of IκB proteins and anti-inflammatory cytokines to dampen NF-κB signaling and inflammation. Therefore, the outcome of an IAV infection is influenced by the balance between proinflammatory and anti-inflammatory responses in the lungs. Proinflammatory cytokines, such as IFNs, ILs, TNFα, and chemokines play crucial roles in antiviral defense. They facilitate communication between immune and non-immune cells, recruit leukocytes to stimulate epithelial cells by enhancing major histocompatibility complex expression, promote cell growth and differentiation, and mediate antigen-specific immune responses to drive several critical reactions against IAV infection. On the other hand, these proinflammatory cytokines can also damage the airway epithelium. Thus, virus-related inflammation is associated with not only viral clearance but also pathological injury in the lungs. During the process of an IAV infection, there are different cytokine waves from the initial early responses of IL-8, IFN-α, IFN-β, and IFN-κ[65] to the advanced responses of IL-6, IL-1α/β, IL-18, TNF-α, and IFN type I which are mainly secreted by infected macrophages in the lower respiratory tract[66]. Unrestrained inflammatory responses, together with viral virulence, may lead to severe lung injury[67]. Consequently, the levels of cytokine storms will determine the severity of the disease during an influenza infection[68].
IAV targets endothelial cells, alveolar macrophages, and epithelial cells, to generate the initial cytokine waves, particularly type I interferon which enhance the expression of various IFN-induced genes (ISGs). Succeeding the type I IFNs released, higher expression of ISGs initiates downstream antiviral responses and consequent inflammatory cytokine production by innate immune cells. Cells of the adaptive immune system are activated and regulated to secrete a subsequent wave of cytokines that promote tissue homeostasis, clearance of viruses, and lung restoration[69].
PB2 and HA proteins are the primary factors determining pathogenicity in influenza. Highly virulent strains possess several basic amino acid residues at the cleavage site where the precursor protein, HA0, is cleaved into HA1 and HA2. In human and highly pathogenic avian influenza viruses, variations in the amino acid residue at position 627 affect replication efficiencies in the upper and lower respiratory tracts of various animals. Other pathogenicity factors include the NS1 protein, which disrupts cell responses triggered by interferon and inhibits the activation of innate immune response pathways.
2.6. Assortment of Coding Strategies in Influenza a Viruses
Influenza viruses have abused a diversity of tactics to upsurge their genome coding capacities. These include unspliced, spliced, alternatively spliced, and bicistronic mRNAs, translation from overlapping reading frames, and a coupled stop-start translation of tandem cistrons[70].
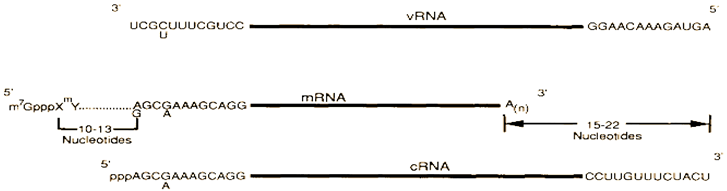
The figure illustrates the distinctions among influenza virus vRNA segments, mRNAs, and full-length cRNA or template RNA. The conserved 12 nucleotides at the 3'-end and 13 nucleotides at the 5'-end of each influenza A virus vRNA segment are shown. Similar conserved sequences are present at the 3'- and 5'-ends of influenza B and C virus RNA segments.
mRNAs Splicing and Reading Frames Overlapping
Evidence of genetics and biochemistry indicated that vRNA segment 8 (890 nucleotides), the smallest of the influenza virus RNA segments, encodes two distinct proteins, NS1 (26 kDa) and NS2 (14 kDa), with NS2 being translated from a short mRNA (around 350 nucleotides). Nucleotide sequencing revealed that the NS1 mRNA is not spliced, directly encoding the NS1 protein (237 amino acids), while the NS2 pre-mRNA includes a 473 nucleotide intron. NS1 and NS2 utilize the same AUG codon for the initiation of protein synthesis and share nine amino acids before the intron; thereafter, translation of the NS2 mRNA continues in the +1 reading frame, which overlaps the NS frame by approximately 70 amino acids[71].
Figure 4.
Prototypical for the organization of (A) NS 1 and NS 2 mRNAs and their coding regions and (B) M1, M 2 mRNAs and mRNA and their coding regions [72].
Figure 4.
Prototypical for the organization of (A) NS 1 and NS 2 mRNAs and their coding regions and (B) M1, M 2 mRNAs and mRNA and their coding regions [72].
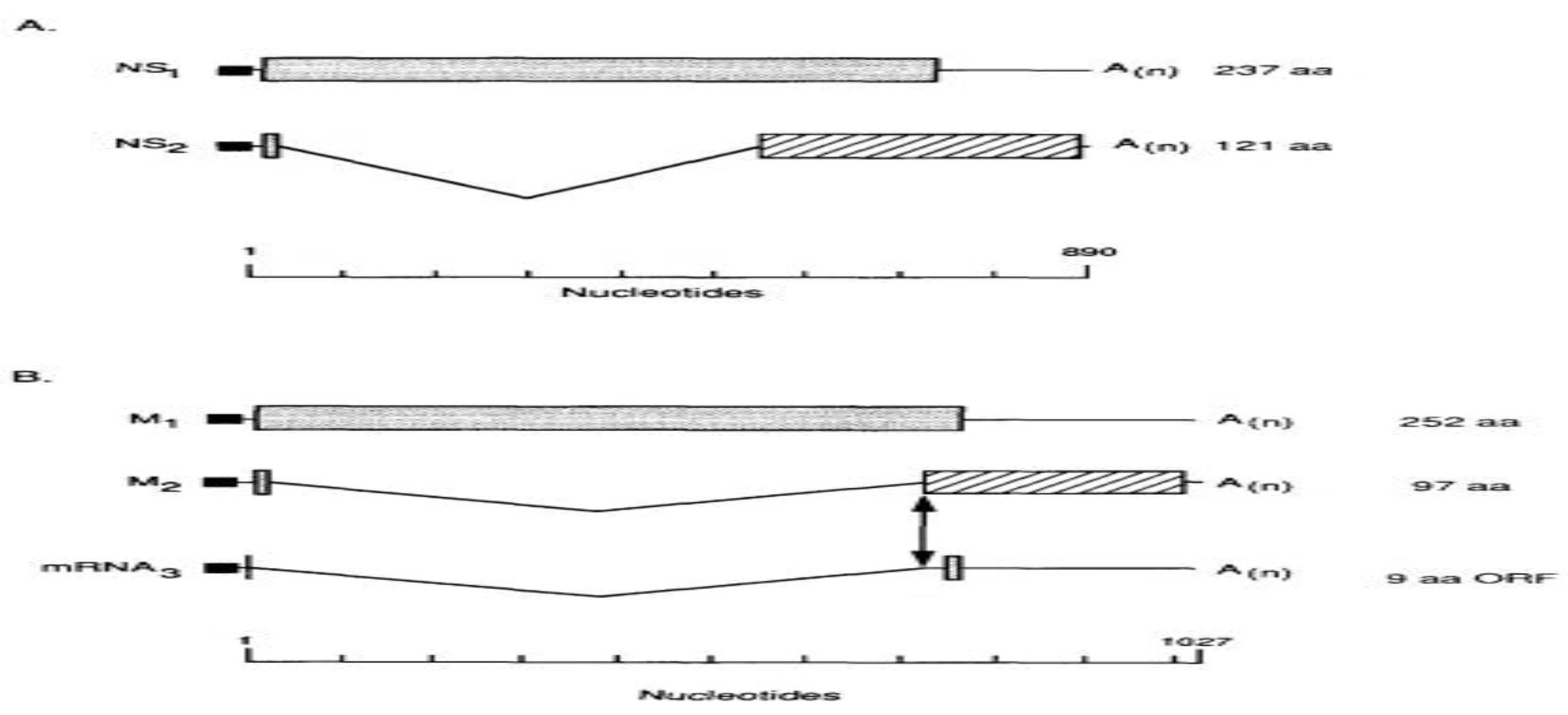
Thin lines at the 5' and 3' ends of the mRNAs denote untranslated regions. Shaded or striped areas indicate coding regions in 0 or +1 reading frames, respectively. Introns in the mRNAs are depicted by V-shaped lines, and filled rectangles at the 5'-ends of mRNAs represent diverse nucleotides from cellular RNAs that are covalently attached to viral sequences. The viral factors, including polymerase proteins and host factor SF2, regulate splicing. M1, M2, and M3 are coded by segment 7 of influenza A.
2.7. Clinical Presentation of IAV Infection
IAV typically takes 18 to 72 hours to incubate, though this might vary from case to case. The commonly encountered symptoms of IAV include chills and high fever, weakness, headache, throughout the body, agonizing pain in the joints and muscles, red eyes, and respirational signs including sore throat, dry cough, and rhinitis. Influenza symptoms frequently manifest abruptly[73]. The systemic signs usually last up to 7 days, but cough and weakness may last for weeks[74]. Fever (38–41°C) typically lasts about 3 days in adults[75]. A severe generalized, usually a frontal lobe headache, is common with influenza, worsening with sudden head movement[76]. IAV cases commonly convalesce from simple infection after a few days, but prolonged signs may indicate complications. For case in point, a headache with persistent fever may be due to sinusitis[77]. Pain in eye motion and photophobia may develop in many patients; the eyes can become red, watery, and congested[78]. A sore throat due to influenza usually lasts about 5 days[73].
Inflammation in the lung is renowned as the most critical influenza complication, especially in the elderly[79]. Complicated influenza infection frequently manifests as primary viral pneumonia, secondary bacterial pneumonia, and/or combined viral and bacterial pneumonia. Primary influenza pneumonia symptoms include high fever, dry cough, headache, sore throat, fatigue, dyspnea, and cyanosis[80]. Adults and children at high risk can develop some extrapulmonary complications such as heart problems, sinusitis, muscle inflammation, ear infections, and myoglobinuria may develop in addition to pneumonia[81]. Neurologic complications of influenza include Reye's syndrome, aseptic meningitis, encephalomyelitis, and Guillain-Barré syndrome (GBS). Research shows an association of influenza infections with myocarditis and pericarditis[82]. Myoglobinuria infrequently occurs following acute infection with symptoms suggesting an upper respiratory infection (URI) and has been associated with influenza, adenovirus, and parainfluenza[83]. Reye's syndrome is an uncommon and potentially life-threatening disease, distinguished by hepatic encephalopathy[84]. Long-term use of aspirin in children is another well-recognized risk factor for Reye's syndrome[85].
2.8. Diagnosis of IAV
IAV diagnosis is regularly made on clinical grounds, laboratory testing, epidemiology information, and influenza infection symptoms[86]. Cough and fever are the most critical symptomatic predictors of influenza infection, that is, before laboratory confirmation[87]. The widely held influenza patients are diagnosed based on clinical criteria during an epidemic because diagnosis based on clinical symptoms accuracy during outbreaks is high. Rapid diagnostic testing for influenza, PCR, virus culture, and nucleic acid amplification tests are examples of laboratory techniques[88]. Loop-mediated isothermal amplification has high accuracy and is very rapid (about 30 min) to deliver results but is costly[89]. A multiplex real-time PCR assay has been developed in recent years to detect Types A and B influenza strains[90].
IAV antigen detection in respiratory samplings and immunoassays such as rapid antigen tests have been developed. These tests are simple to use, take less than 30 minutes to complete, have a high specificity, and are attributed to lower infection activity in the event of false-positive test results.[91]. Nonetheless, they have unfortunate sensitivity in contrast to molecular assays and viral culture methods. The sensitivity of rapid antigen tests depends on the disease's course, delivering high sensitivity to 2 days from the signs' onset. Nasopharyngeal samples also bring high sensitivity to the test. Because of the poor sensitivity, rapid antigen tests may not be suitable for making diagnostic and treatment decisions[91].
Figure 5.
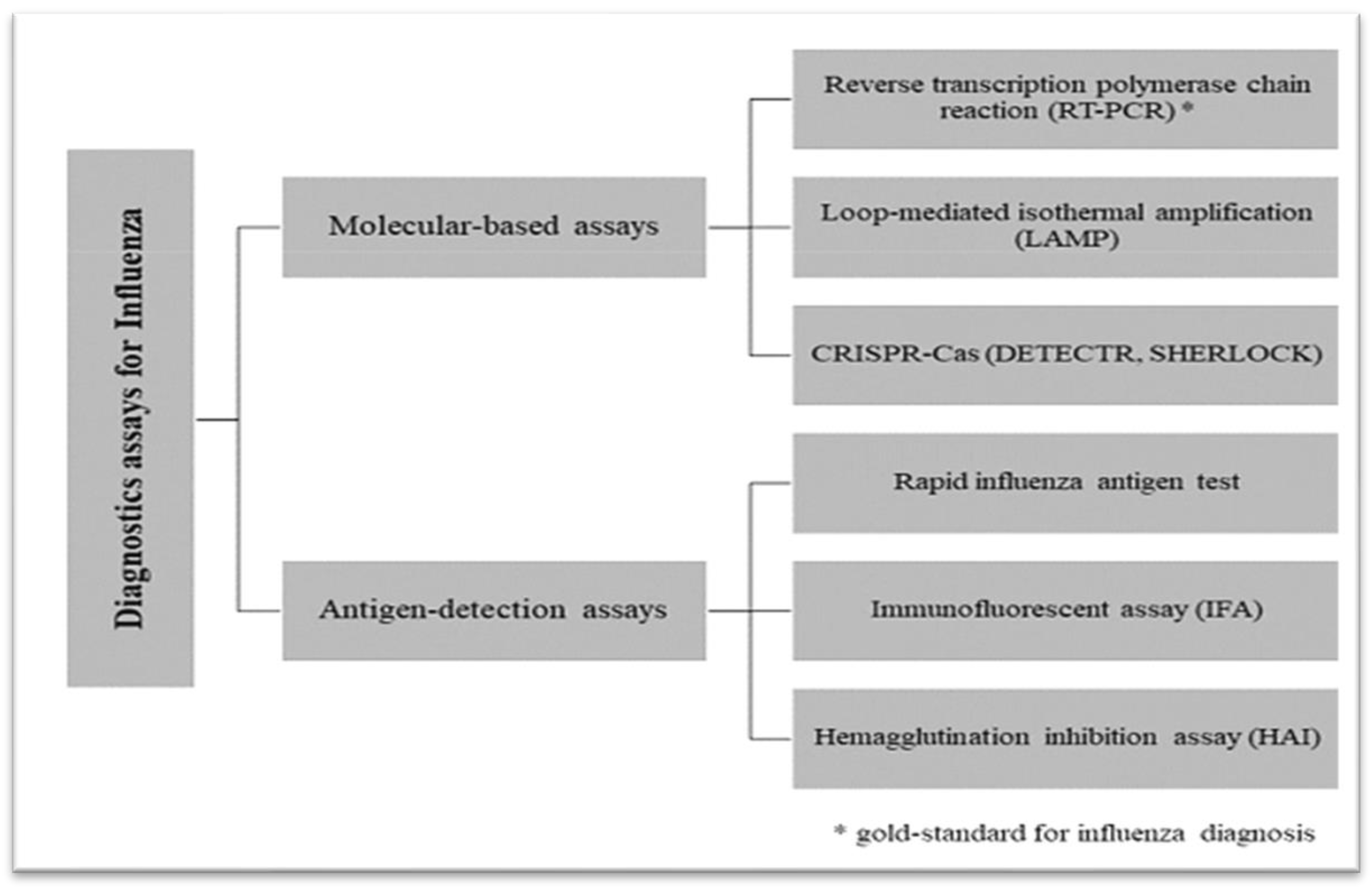
An outline of current diagnostics assays for influenza[92].
Figure 5.
An outline of current diagnostics assays for influenza[92].
2.9. Antiviral Drugs for IAV
Management for uncomplicated influenza and low-risk individuals should involve symptomatic and supportive care. To counteract the fluid loss that usually occurs with a fever, maintaining adequate hydration is crucial. Instances of NSAIDs that can be used to improve symptoms such as fever, headaches, and muscle pain include aspirin, naproxen, diclofenac sodium, and ibuprofen[93].
Among the antivirals, currently, only four have been approved and recommended for use in chemoprophylaxis and treatment of influenza: zanamivir, oseltamivir, baloxavir marboxil, and peramivir[94]. Oseltamivir, zanamivir, and peramivir are neuraminidase inhibitors and act by inhibiting the NA enzyme's function and inhibiting the virus from leaving the infected cells[95].
Orally directed oseltamivir is recommended to treat simple acute influenza up to 48 hours after the onset of symptoms in adults and children over 2 weeks old. For chemoprophylaxis in adults and children over 1 year old oseltamivir is recommended[96]. Research confirmed that treatment with oseltamivir initiated 36 hours after the start of signs resulted in a 40% reduction in the illness's severity[96].
Zanamivir inhalation is suggested for handling uncomplicated acute influenza within 2 days after the beginning of signs in adults and children aged over 7 years old and for chemoprophylaxis of influenza in adults and children aged over 5 years old[97]. Zanamivir in severe milk protein allergy is contraindicated. Early therapy of unsophisticated influenza with zanamivir resulted in a reduction in duration (by up to 2 days) and severity of symptoms.
M2 inhibitors act as molecular plugs by using a positively charged amino group to seal the ion channel of M2, creating electrostatic resistance and preventing the flow of H+ ions into the virion[98].
The pro-drug baloxavir marboxil derivative, baloxavir, is a cap-dependent endonuclease (PA) inhibitor that blocks the cycle of viral replication by hindering the mRNA synthesis initiation. It is effective against influenza A and B viruses, including oseltamivir-resistant strains as well as avian strains H7N9 and H5N1[99].
Food and Drug Administration (FDA) agreed neuraminidase inhibitors (oseltamivir, peramivir, and zanamivir) are the results of N-acetyl-neuraminic acid (2, 3-dehydro-2-deoxy-N-acetylneuraminic acid, “DANA”). These inhibitors attach to the active site of neuraminidase (NA), competitively blocking its interaction with N-acetyl-neuraminic acid. This inhibition prevents NA from cleaving N-acetyl-neuraminic acid from host proteins, thereby halting the release of virion [100].
Figure 6.
A diagram depicting the crucial steps of influenza virus infection and antiviral treatment [101].
Figure 6.
A diagram depicting the crucial steps of influenza virus infection and antiviral treatment [101].
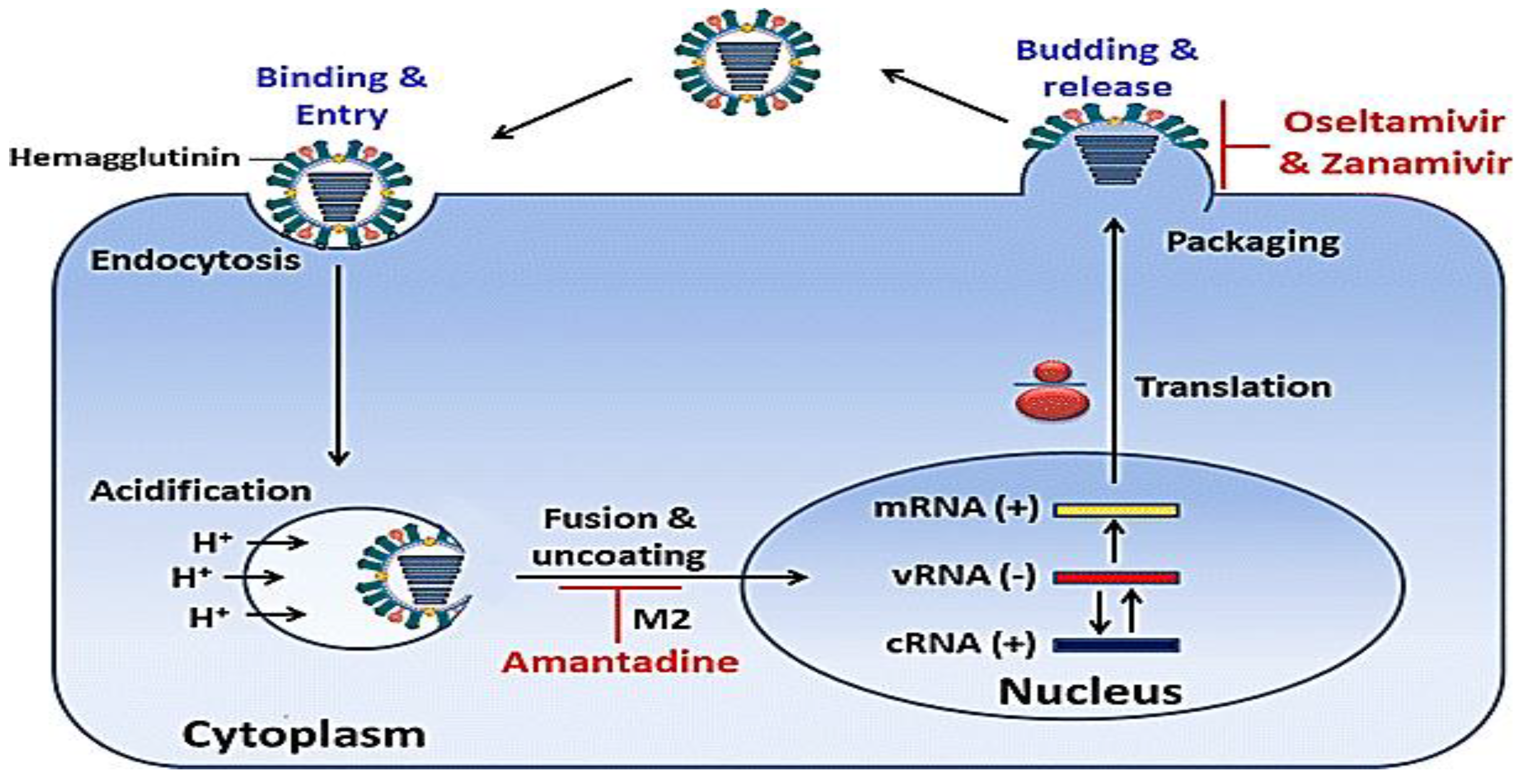
IAV particles adhere to SA on the cell surface and are internalized via endocytosis. Acidification within the endosome facilitates virion breakdown. DAS181 eliminates sialic acids from the cell surface, hindering viral infection. Blockage of M2 ion channel by adamantanes, inhibiting H+ transport into the virion and its disassembly. Fusion between the viral and endosomal membranes is mediated by HA, while agents like Amantadine inhibit this fusion. Following fusion, RNP complexes are transported into the nucleus, where viral transcription and replication occur. Genome-associated RdRP complex carried out initial transcription. PB2 cap-binding function is blocked by VX-787, while endonuclease activity is inhibited by S-033188 and L-742,001, both impeding viral mRNA transcription. Export of Viral mRNA is done from the nucleus, leading to the production of viral proteins. Nucleozin disrupts NP functions by promoting NP aggregation. Newly formed viral proteins are transported to the plasma membrane, delivering HA, NA, and M2. Nitazoxanide affects HA processing and maturation. A newly produced RdRP protein derives replication of the viral genome, with nucleoside analogs like Favipiravir and Ribavirin disrupting RdRP function. The conserved RNA binding site in NP is bound by Curcumin and Naproxen, obstructing RNA binding.
2.10. Alternative Antiviral Approach That Targets the Genome
Despite the discovery of new class drugs such as Baloxavir; and Marboxil, the rapid emergence of resistant IAV poses a serious concern to the already limited therapeutic options. This calls for an alternative approach to mitigate the rapid evolution of IAV, especially on the recurrent annual influenza infection (cold flu). The use of RNA-targeting technologies such as RNA interference (RNAi) and antisense oligonucleotides (ASO) are the most widely used nucleic acids that cause RNA cleavage/degradation or block mRNA synthesis and translation[102].
2.10.1. Interference RNA (RNAi)
RNAi is a small RNA engaged for sequence-specific mRNA degradation elicited by long double-stranded RNA ([103]. Abundant virus-derived siRNAs, PIWI-interacting RNAs (piRNAs), and transfer RNAs (tRNAs are produced by mammalian host cells upon RNA virus infection in reply to antiviral immunity; influenza infection is not excluded [104].
The use of small interfering RNA molecules (siRNAs) by characterizing three siRNAs (M747, M776, and M832) targeting the influenza matrix 2 genes and three (NS570, NS595, and NS615) targeting the nonstructural protein 1 and 2 genes have been investigated. siRNAs, M331 and M950, which target the matrix 1 and 2 genes have also been examined. Treatment with M331-, M776-, M832-, and M950-siRNAs attenuated influenza titer[105].
Figure 7.
The mechanism of RNA interference (RNAi) in contradiction of IAV.
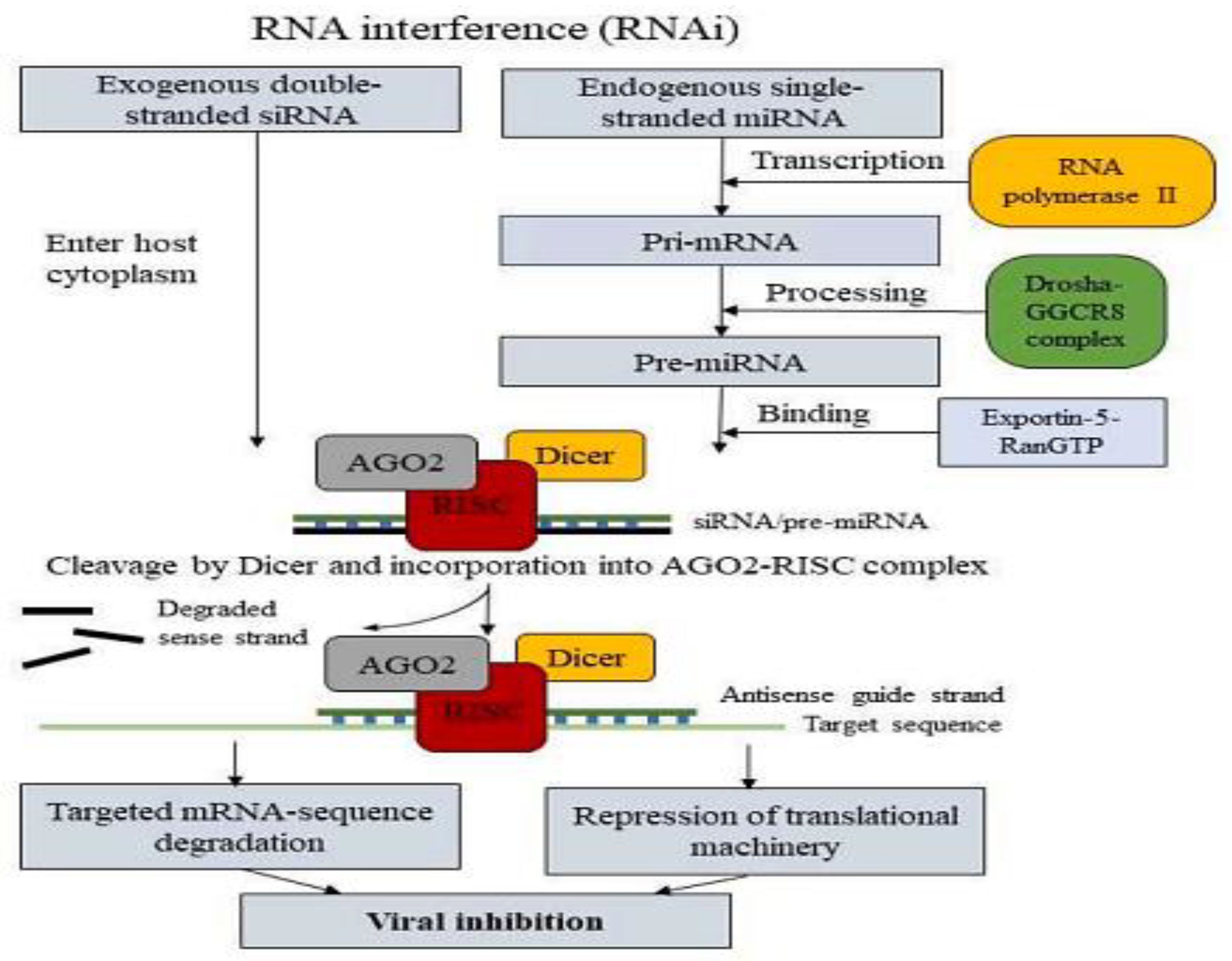
The Argonaute2-RISC complex activation facilitates sequence-specific gene silencing by attaching to and cleaving the target mRNA sequence according to Watson-Crick base pairing, aided by an antisense guide strand. miRNAs function similarly but involve additional transcriptional steps. RNA polymerase II synthesizes miRNA, this Pri-miRNA contains a 5′-cap structure and a polyA tail. The interaction of Pri-miRNA with the Drosha–DGCR8 complex produces a 70-nucleotide long pre-miRNA structure with 2-nucleotide overhangs at the 3′ end and a 5′ phosphate group. The pre-miRNA associates with exportin-5–RanGTP, enters the cytoplasm, and is processed by Dicer into a 22-nucleotide long microRNA capable of binding to the AGO-RISC complex, similar to siRNA. Unlike siRNA, however, the miRNA RISC complex (miRISC) binds to the target mRNA, creates a bulge sequence, and accumulates in P-bodies, thereby inhibiting the machinery of translation.
2.10.2. ASO
Antisense oligonucleotides (ASO), as the name suggests, a short single-stranded DNA oligonucleotides (12–25 nucleotides) designed to complement the mRNA targets by the Watson-Crick base pairing. The use of ASO for gene silencing or suppression is no stranger since the FDA approval of the Fomivirsen drug for cytomegalovirus retinitis in August 1998[106]. DNA-RNA heteroduplex which in turn recruits RNASEH1 for mRNA degradation is formed by the binding of ASOs to the flanking region of target mRNA. ASO blockage sterically, alternatively, can bind to the AUG region and disrupt the translational activity of the primary open reading frame of mRNA by introducing upstream open reading frames (uORFs) or blocking the attachment of ribosomes for translation[107].
Currently, using of ASOs for IAV studies encompasses targeting vRNA segments 5, 7, and 8. For instance, research done by Michalak and coworkers targeting vRNA segment 5 has revealed a surprising 88% viral titer reduction and 55% vRNA reduction with ASO 883–11L (targeting region 878–888 nucleotide)[20]. Additionally, attachment of steric ASO to the pre-mRNA can influence alternative splicing to encourage exon inclusion/splicing, leading to mRNA that reduces abnormal protein production (see figure below). Simply put, the small compound ASO is used to stop or modify protein production, thereby decreasing harmful protein synthesis in various diseases mentioned earlier.
Figure 8.
Antisense oligonucleotides (ASO) as an antiviral approach for IAV.
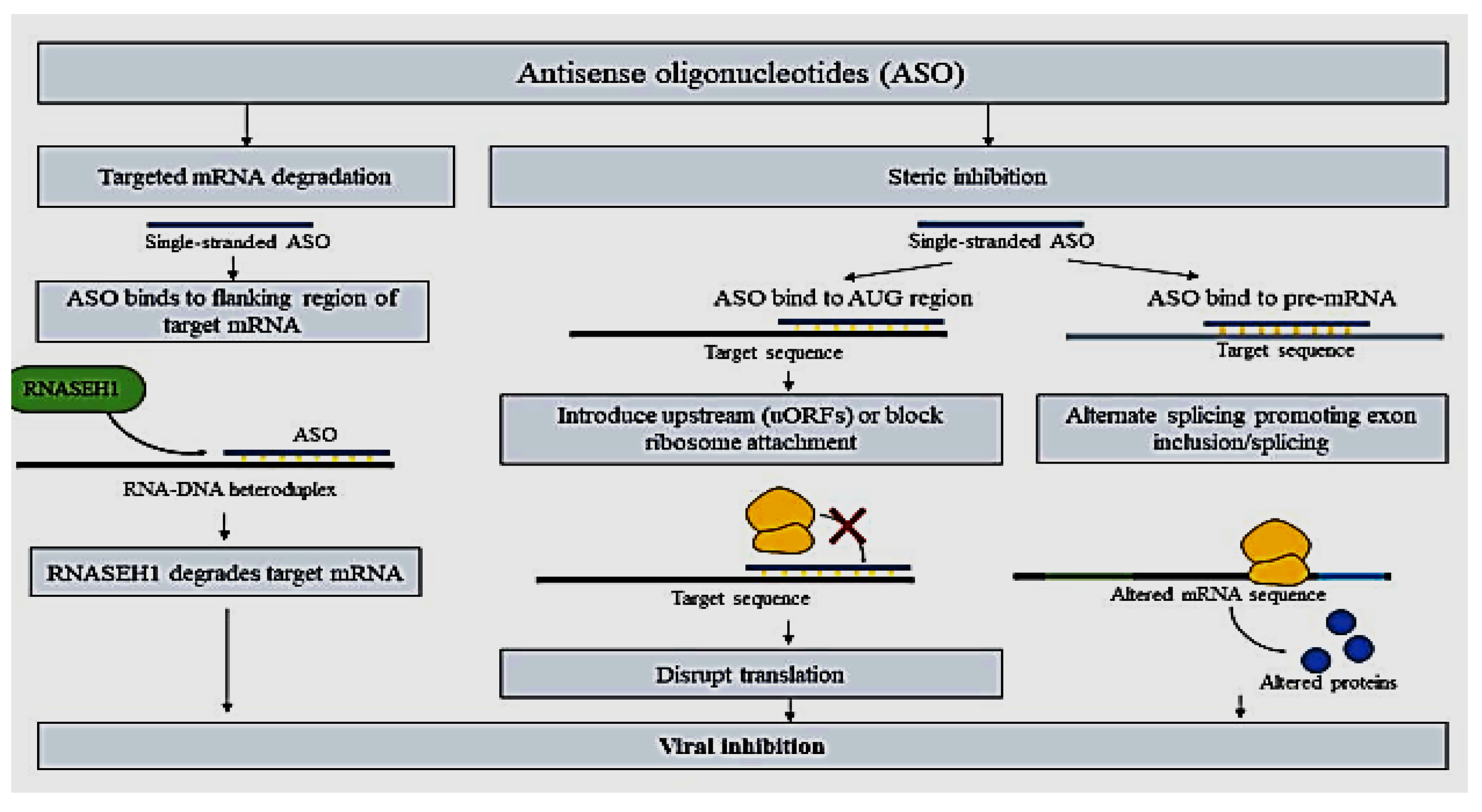
2.11. Evolution of Influenza a Virus
Influenza viruses often change their antigens, resulting in their rapid evolution. The mechanisms by which the virus changes and produces novel strains are referred to as antigenic drift and shift. Major deviations in the HA protein antigenicity in mixing IAV strains are referred to as antigenic shift, a process through which human influenza strains can reassort to incorporate animals of the IAV variants[108]. The change in the genetic makeup of the IAV may be introduced by at least three evolutionary mechanisms[109] such as alterations due to reassortment, recombination, and RNA polymerase mistakes.
Similar to other viral RNA polymerases, influenza viruses RNA polymerase lacks proofreading ability. Consequently, the error-prone nature of RNA polymerases leads to a substantially higher rate of mutation for influenza and other RNA viruses compared to viruses of DNA, where polymerases have proofreading mechanisms. Consequently, the rate of mutation for RNA genomes can be up to a hundred thousand times greater than that for DNA genomes. Each replicated genome may acquire several point mutations per replication cycle; these changes can have neutral, detrimental, or beneficial effects on the virus's viability[110].
The most noticeable consequence of these gradual changes driven by positive selection is the phenomenon known as antigenic drift in the glycoproteins neuraminidase and hemagglutinin, which are located on the virion surface. Production of antibodies against the original strain may fail to recognize and neutralize the viral variants resulting from these incremental changes in antigenic drift, affecting the antigenicity of these proteins. These new antigenic variants can potentially trigger outbreaks. For quite a few years, these strains usually dominate human or animal populations before being replaced by new antigenic variations. Although hemagglutinin and neuraminidase display these changes most prominently, other genes are also likely to undergo similar gradual alterations due to the lack of proofreading activity of RNA-dependant RNA polymerase.
As the genome of an IAV comprises of eight discrete RNA segments, the coinfection of one host cell with two different IAVs can result in progeny viruses containing gene segments of both parental viruses. There are theoretically 256 (28) possible combinations of the 8 gene segments from reassortment between 2 parental viruses. Mutually shared and significant in IAV evolution are reassortment[111] and events of host switch[112].
2.12. Significance of Anti-IAV Resistance
IAV is the primary cause of irregular influenza pandemics and severe zoonotic outbreaks and accounts for at least half of the annual influenza epidemics in humans. Two classes of anti-IAV medications, adamantanes, and NAIs, targeting the viral components M2 ion channel and NA, respectively, have been approved for treating IAV infections. However, IAV quickly developed resistance to both drug classes through mutations in these viral components. The adamantane-resistant IAV has become established in the environment, and most IAV subtypes, particularly the prevalent H3N2 and H1N1 strains, circulating worldwide are resistant to adamantanes[113]. Blockers of M2 ion channels and NAIs have been approved for treating influenza virus infections. However, akin to antibiotic resistance, the rise of antiviral drug resistance in influenza viruses is a significant issue. As a result, NAIs are the only class of anti-influenza drugs presently in use Since most circulating influenza viruses have developed resistance to M2 ion channel blockers[15].
2.13. Factors Driving Antiviral Resistance
Among all influenza viruses, IAV is the most clinically relevant, as it causes annual epidemics in humans. Various avian and mammalian species both wild and domesticated are vulnerable to this zoonotic virus. IAV's evolutionary success is partly due to its ability to switch between hosts, occasionally leading to pandemic outbreaks[114]. Several factors contribute to the development of antiviral resistance in influenza viruses, including intrinsic characteristics of the virus itself. Due to its RNA-dependent RNA polymerase and RNA genome, which lacks proofreading capability, the influenza virus exhibits high mutagenicity, leading to ‘quasispecies’ creation which is a collection of genetically diverse viral variants centered on a common genomic sequence. This diversity enables the AIV to withstand selection pressures, such as antiviral treatments. Antigenic drift occurs when mutations enable the AIV to evade immune exposure and regulators. Additionally, to avoid immune recognition the surface glycoproteins continually change, resulting in antigenically distinct influenza strains. The segmented genome also allows for genome mixing when different strains infect the same host, a process known as genetic reassortment. This can lead to the emergence of new strains with ant-influenza virus resistance if a delicate strain acquires a segment with unaffected traits. These ongoing mechanisms of genetic variation enable AIV to acclimatize to selective pressures, comprising antiviral treatments [100].

Table 1.
Anti-viral resistance deriving factors.
| Factors driven by virus | Factors driven by a host | ||
| Issue | Potential Explication | Problem | Solution |
| Error-prone polymerase, quasispecies, antigenic drift | Target host pathways | Chemoprophylaxis, use of sub-therapeutic doses | Confined and checked the use of chemoprophylaxis with full dosage |
| Antigenic shift | Watchful surveillance | Persistent shedding owing to virulent strain or contagion of a high-risk group. | Hospitalized isolation, treatment, and maintenance to prevent nosocomial communication |
2.14. Resistance to Anti-Influenza Therapy
As of now, the U.S. Food and Drug Administration has approved only four antiviral drugs for therapy or prevention of influenza. These include two adamantanes—amantadine and rimantadine—which inhibit the M2 protein channel in the viral envelope, and two neuraminidase (NA) inhibitors—oseltamivir and zanamivir. Currently, influenza viruses that are mostly circulating are either naturally resistant to adamantanes or do not respond to this drug class [100]. In addition, most circulating influenza viruses are either inherently adamantane resistant (influenza A) or not receptive to this drug class (influenza B)[115]. In a recent paper by[116], the incidence of current NAI resistance reports has been conducted in a worldwide multicenter study between 2008 and 2012. For IAV, oseltamivir resistance was found to be rare (2.9%) and detected only relative to treatment. The emergence of oseltamivir resistance was uppermost in children between the ages of 1 and 5 years (14/138 (10.1%)). The study did not detect NAI-resistant influenza B viruses. No zanamivir resistance was detected either in this study. Nevertheless, others have reduced zanamivir susceptibility to influenza A viruses[117] and B viruses [118].
Table 2.
Resistance to Anti-influenza Therapy.
| Subtypes of Influenza | M2 Ion Channel Inhibitors (Adamantanes) | Neuraminidase Inhibitors | ||||||
| Oseltamivir | Zanamivir | |||||||
| %Resistant | Major M2 Mutation | Refs | %Resistant to NAIs | Major NA Mutations | Compensatory Mutations | Major NA Mutation* | Refs. | |
| H3N2 | >96% | S31N | [119] | 0.2% | R292K E119V N294S | Unknown | Q136K | [120] |
|
Pre-2009 seasonal H1N1 |
16% | S31N | [121] | >99% | H274Y | R354G K329E V234M, R222Q, D344N, |
Q136K | [122] |
| 2009 Pandemic H1N1 | 100% (USA) | S31N | [119] | 0.5% | H274Y I223R | V240I, N368K | I223R | [123] |
| H5N1 | Differs by clade | S31N V27A | [124] | Rare | H274Y N294S | Unknown | Unknown | [125] |
2.15. Mode of Resistance to NAIs and M2
In the course of the development of resistance neuraminidase inhibitors, most amino acids in or around the active site of NA virus have changed in IAV [126], including R292K, E119V, I222V, N294S, and H274Y. The NA active site shape of these mutations directly or indirectly changes, drastically reducing NAI binding. Among different influenza viruses, there is the highly conserved active site of the NA [127]. Nonetheless, minor variances in the active sites of B, N1, and N2 neuraminidases, along with variations between the NAIs, create different possibilities for the progress of resistance changes of most amino acid changes responsible for NAI resistance situated in or near the active site[128]. Nearly 95 percent of resistant adamantane IAV subtypes have been revealed to port an S31N mutation[129]. Cause resistance to baloxavir in influenza A viruses has been shown at isoleucine 38 mutation in the PA catalytic site[130].
2.16. Alternate Options Contrary to the Increasing Resistance of Influenza a Antivirals
The wide-ranging host of IAV and within-species transmission are critical factors for its continuous circulation and evolution in nature[131]. Pigs, birds, and horses serve as crucial intermediate hosts that sustain the IAV in the wild and facilitate its transmission to humans. Effective management of these hosts is essential to prevent the emergence and spread of new, more virulent IAV strains. Alongside continuous surveillance, universal expansion of vaccines, and the creation of potent antiviral treatments, it is challenging to eradicate IAV from zoonotic hosts with current methods and technologies. There is a need to understand at the molecular and genetic levels why certain animals (e.g., sheep and rabbits) resist IAV infection, while others (e.g., ducks) are resilient to IAV disease. Identifying the genes responsible for such resistance could enable the development of gene-editing strategies to make intermediate hosts resistant to IAV. This approach could potentially reduce or even eliminate IAV from these hosts, decreasing its presence in nature and preventing its transmission to humans. Initial proof-of-concept studies have begun to explore this strategy by attempting to restrict IAV transmission or replication in genetically modified animals [132,133,134].
2.16.1. CRISPR
Clustered regularly interspaced short palindromic repeats (CRISPR) application increased reputation during the SARS-COV-2 pandemic. The CRISPR-Cas9-induced genome-wide gene knockout is a great instrument for digging out viral essential host factors[135]. CRISPR-Cas 12a/Cas13a systems have been adapted to detect the M gene and the H1 and H2 genes in influenza A/B. The system of CRISPR-Cas 13d employs 22-nucleotide spacer CRISPR-associated RNAs (crRNAs), also known as guide RNAs (gRNAs), to guide Cas13d to specific RNA targets, facilitating their cleavage and consequent degradation, directed by a protospacer flanking sequence(PFS)[136].
Six of the eight IAV viral genome segments were highly conserved. Subsequently, the crRNA-transfected cells were challenged with a fluorescent reporter strain of influenza. This result implies a robust 72% and 52% reduction in viral titer at MOI 2.5 and MOI 5 for the crRNA pool targeting segment 6 (encodes for NA), respectively[136].
2.16.2. Natural Occurring Compounds
Compounds that occur naturally can contain a hidden trove of anti-viral agents. Active ingredient identification and purification from natural sources like plants present an attractive and logical strategy for novel drug innovation. Compounds of plant origin have long been scrutinized for their antioxidant, anti-inflammatory, and antitumor activity[137]. For example, by applying the benchmark to the ZINC Natural Product database, monitored by ligand-based virtual screening and molecular docking, research suggested the most hopeful candidate as a potential NS1 inhibitor. Consequently, the selected natural compound was experimentally evaluated, enlightening measurable virus replication inhibition activity in cell culture. The approach of this offers a hopeful avenue for emerging new anti-influenza agents targeting the NS1 protein[138].
2.16.3. Directing Towards Host Molecules
To achieve effective viral inhibition, inhibitors targeting host mechanisms should be prioritized due to the rapid mutation of viral proteins and the dependence on the virus-host life cycle. For example, research has demonstrated promising results with Diltiazem, a calcium channel blocker used for hypertension, in combination with Oseltamivir, a neuraminidase inhibitor against IAV. This combination was identified from an initial screening of 35 potential candidates and transcriptomic analysis of 1,309 FDA-approved bioactive compounds[137]. The study demonstrated that the aforementioned combination achieved a greater than 3-log reduction in viral replication in Human Airway Epithelia (HAE) at an MOI of 0.1, 48 hours after infection, compared to the untreated control. Notably, Diltiazem alone considerably saved 40% (4/10) of the mice in an in vivo experiment, a more impressive result than the 20% rescue rate observed with Oseltamivir[139].
3. Conclusion and Recommendation
Humans and animals are severely threatened by influenza A virus infection globally. The IAV suffers continuous change, segment reassortment, and genome recombination. Its segmented genome and the high mutation rate during replication across multiple hosts contribute to frequent epidemics and occasional pandemics. Consequently, new outbreaks are likely to occur, making the eradication of the disease difficult. While antiviral medications are crucial for managing influenza, their effectiveness is often compromised by human misuse and the virus's ability to evade antiviral mechanisms. This difficulty of resistance to both M2 and neuraminidase inhibitors warns of the need to promote research on the prevalence of resistance and the development of new antivirals. Community well-being impacts the mitigation of influenza, and emerging treatments should be chosen for their ability to address not only the disease itself but also the issue of anti-influenza resistance. Thus, the most effective strategy for treating influenza infection involves combination therapy or immunomodulators that employ distinct mechanisms of action. In addition, alternative interventions such as RNAi, ASO, naturally occurring compounds, CRISPR, and targeted host signaling mechanisms have to be implemented to overcome resistance to IAV.
References
- Long, J.S., et al., Host and viral determinants of influenza A virus species specificity. 2019. 17(2): p. 67-81. [CrossRef]
- Taubenberger, J.K., J.C.J.C.h. Kash, and microbe, Influenza virus evolution, host adaptation, and pandemic formation. 2010. 7(6): p. 440-451. [CrossRef]
- Jang, Y.H. and B.L.J.Y.m.j. Seong, Cross-protective immune responses elicited by live attenuated influenza vaccines. 2013. 54(2): p. 271-282.
- Tong, S., et al., New world bats harbor diverse influenza A viruses. 2013. 9(10): p. e1003657. [CrossRef]
- Pizzorno, A., Y. Abed, and G. Boivin. Influenza drug resistance. In Seminars in respiratory and critical care medicine. 2011. © Thieme Medical Publishers.
- Ferguson, N.M., et al., Strategies for containing an emerging influenza pandemic in Southeast Asia. 2005. 437(7056): p. 209-214. [CrossRef]
- Abed, Y., et al., Generation and characterization of recombinant influenza A (H1N1) viruses harboring amantadine resistance mutations. 2005. 49(2): p. 556-559. [CrossRef]
- Cheung, C.-L., et al., Distribution of amantadine-resistant H5N1 avian influenza variants in Asia. 2006. 193(12): p. 1626-1629. [CrossRef]
- Bright, R.A., et al., Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. 2005. 366(9492): p. 1175-1181. [CrossRef]
- Bright, R., et al., High levels of adamantane resistance among influenza A (H3N2) viruses and interim guidelines for use of antiviral agents--United States, 2005-06 influenza season. 2006. 55(2).
- Hutchinson, E.C., et al., Conserved and host-specific features of influenza virion architecture. 2014. 5(1): p. 1-11. [CrossRef]
- Rossman, J.S. and R.A.J.V. Lamb, Influenza virus assembly and budding. 2011. 411(2): p. 229-236. [CrossRef]
- Van Reeth, K., I.H. Brown, and C.W.J.D.o.s. Olsen, Influenza virus. 2012. 10: p. 557-571.
- Ison, M.G. and F.G.J.C.o.i.i.d. Hayden, Viral infections in immunocompromised patients: what's new with respiratory viruses? 2002. 15(4): p. 355-367.
- Webster, R.G. and E.A.J.A.o.t.N.Y.A.o.S. Govorkova, Continuing challenges in influenza. 2014. 1323(1): p. 115-139.
- Eisfeld, A.J., G. Neumann, and Y.J.N.R.M. Kawaoka, At the center: influenza A virus ribonucleoproteins. 2015. 13(1): p. 28-41.
- Moeller, A., et al., Organization of the influenza virus replication machinery. 2012. 338(6114): p. 1631-1634. [CrossRef]
- Wandzik, J.M., T. Kouba, and S.J.C.S.H.p.i.m. Cusack, Structure and function of influenza polymerase. 2021. 11(9): p. a038372. [CrossRef]
- Ferhadian, D., et al., Structural and functional motifs in influenza virus RNAs. 2018. 9: p. 354580. [CrossRef]
- Michalak, P., et al., Secondary structure of the segment 5 genomic RNA of influenza A virus and its application for designing antisense oligonucleotides. 2019. 9(1): p. 3801. [CrossRef]
- Wang, J., et al., Dual roles of the hemagglutinin segment-specific noncoding nucleotides in the extended duplex region of the influenza A virus RNA promoter. 2017. 91(1): p. 10.1128/jvi. 01931-16. [CrossRef]
- Pflug, A., et al., Structural insights into RNA synthesis by the influenza virus transcription-replication machine. 2017. 234: p. 103-117. [CrossRef]
- Anchisi, S., et al., Mismatches in the influenza A virus RNA panhandle prevent retinoic acid-inducible gene I (RIG-I) sensing by impairing RNA/RIG-I complex formation. 2016. 90(1): p. 586-590. [CrossRef]
- Soszynska-Jozwiak, M., et al., Influenza virus segment 5 (+) RNA-secondary structure and new targets for antiviral strategies. 2017. 7(1): p. 15041. [CrossRef]
- Chauhan, R.P. and M.L.J.V.G. Gordon, An overview of influenza A virus genes, protein functions, and replication cycle highlighting important updates. 2022. 58(4): p. 255-269. [CrossRef]
- Ellis, D., et al., Structure-based design of stabilized recombinant influenza neuraminidase tetramers. 2022. 13(1): p. 1825. [CrossRef]
- Sun, Y., et al., Hemagglutinin Glycosylation Pattern-Specific Effects: Implications for The Fitness of H9. 4.2. 5-branched H9N2 Avian Influenza Viruses: Effect of HA glycosylation on the fitness of H9N2 AIV. 2024(just-accepted): p. 2364736.
- Gaymard, A., et al., Functional balance between neuraminidase and haemagglutinin in influenza viruses. 2016. 22(12): p. 975-983. [CrossRef]
- Richard, M., et al., Rescue of a H3N2 influenza virus containing a deficient neuraminidase protein by a hemagglutinin with a low receptor-binding affinity. 2012. 7(5): p. e33880. [CrossRef]
- Lo, C.-Y., et al., Structure and function of influenza virus ribonucleoprotein. 2018: p. 95-128.
- Domínguez, L.M., et al., Antiviral resistance in influenza viruses. 2023. 69(13): p. 16-23. [CrossRef]
- Noda, T. and Y.J.R.i.m.v. Kawaoka, Structure of influenza virus ribonucleoprotein complexes and their packaging into virions. 2010. 20(6): p. 380-391.
- Dawson, A.R., et al., Post-translation regulation of influenza virus replication. 2020. 7(1): p. 167-187. [CrossRef]
- McNicholl, I.R. and J.J.J.A.o.P. McNicholl, Neuraminidase inhibitors: zanamivir and oseltamivir. 2001. 35(1): p. 57-70.
- Geraci, J., et al., Mass mortality of harbor seals: pneumonia associated with influenza A virus. 1982. 215(4536): p. 1129-1131. [CrossRef]
- Hinshaw, V., et al., Characterization of two influenza A viruses from a pilot whale. 1986. 58(2): p. 655-656. [CrossRef]
- Yuanji Guo, Y.G., et al., Characterization of a new avian-like influenza A virus from horses in China. 1992. [CrossRef]
- Guan, Y., et al., Emergence of avian H1N1 influenza viruses in pigs in China. 1996. 70(11): p. 8041-8046. [CrossRef]
- Beare, A., and R.J.A.o.v. Webster, Replication of avian influenza viruses in humans. 1991. 119: p. 37-42. [CrossRef]
- Cox, N. and Y. Kawaoka, Thomyxo Viruses: Influenza in Topely and Will Sons Microbiology and Microbial Infections. 1998.
- Ito, T., et al., Molecular basis for the generation in pigs of influenza A viruses with pandemic potential. 1998. 72(9): p. 7367-7373. [CrossRef]
- Nayak, D.P., et al., Influenza virus morphogenesis and budding. 2009. 143(2): p. 147-161. [CrossRef]
- Shtykova, E.V., et al., Structural analysis of influenza A virus matrix protein M1 and its self-assemblies at low pH. 2013. 8(12): p. e82431. [CrossRef]
- Webster, R.G., et al., Evolution and ecology of influenza A viruses. 1992. 56(1): p. 152-179. [CrossRef]
- Tong, S., et al., A distinct lineage of influenza A virus from bats. 2012. 109(11): p. 4269-4274. [CrossRef]
- Wu, Y., et al., Bat-derived influenza-like viruses H17N10 and H18N11. 2014. 22(4): p. 183-191.
- Hope-Simpson, R., D.J.E. Golubev, and Infection, A new concept of the epidemic process of influenza A virus. 1987. 99(1): p. 5-54. [CrossRef]
- Rambaut, A., et al., The genomic and epidemiological dynamics of human influenza A virus. 2008. 453(7195): p. 615-619. [CrossRef]
- Neuzil, K.M., et al., Illness among schoolchildren during influenza season: effect on school absenteeism, parental absenteeism from work, and secondary illness in families. 2002. 156(10): p. 986-991.
- Worobey, M., G.-Z. Han, and A.J.P.o.t.N.A.o.S. Rambaut, Genesis and pathogenesis of the 1918 pandemic H1N1 influenza A virus. 2014. 111(22): p. 8107-8112. [CrossRef]
- Husain, M.J.I., Genetics and Evolution, Avian influenza A (H7N9) virus infection in humans: epidemiology, evolution, and pathogenesis. 2014. 28: p. 304-312. [CrossRef]
- Bouvier, N.M. and P.J.V. Palese, The biology of influenza viruses. 2008. 26: p. D49-D53. [CrossRef]
- Tamerius, J., et al., Global influenza seasonality: reconciling patterns across temperate and tropical regions. 2011. 119(4): p. 439-445. [CrossRef]
- Paules, C.I. and A.S.J.T.J.o.i.d. Fauci, Influenza vaccines: good, but we can do better. 2019. 219(Supplement_1): p. S1-S4.
- Clayville, L.R.J.P. and Therapeutics, Influenza update: a review of currently available vaccines. 2011. 36(10): p. 659.
- Shao, W., et al., Evolution of influenza a virus by mutation and re-assortment. 2017. 18(8): p. 1650. [CrossRef]
- Hurt, A.C.J.C.o.i.v., The epidemiology and spread of drug-resistant human influenza viruses. 2014. 8: p. 22-29.
- Short, K.R., et al., Influenza-induced inflammation drives pneumococcal otitis media. 2013. 81(3): p. 645-652. [CrossRef]
- Kaiser, L., et al., Impact of oseltamivir treatment on influenza-related lower respiratory tract complications and hospitalizations. 2003. 163(14): p. 1667-1672. [CrossRef]
- Lê, V.B., et al., Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. 2015. 191(7): p. 804-819. [CrossRef]
- Koutsakos, M., K. Kedzierska, and K.J.t.J.o.I. Subbarao, Immune responses to avian influenza viruses. 2019. 202(2): p. 382-391. [CrossRef]
- Zlotnik, A. and O.J.I. Yoshie, Chemokines: a new classification system and their role in immunity. 2000. 12(2): p. 121-127.
- Pirhonen, J., et al., Virus infection activates IL-1β and IL-18 production in human macrophages by a caspase-1-dependent pathway. 1999. 162(12): p. 7322-7329. [CrossRef]
- Gong, J.-H., et al., Influenza A virus infection of macrophages. Enhanced tumor necrosis factor-alpha (TNF-alpha) gene expression and lipopolysaccharide-triggered TNF-alpha release. 1991. 147(10): p. 3507-3513. [CrossRef]
- Le Goffic, R., et al., Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. 2007. 178(6): p. 3368-3372. [CrossRef]
- Jewell, N.A., et al., Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. 2010. 84(21): p. 11515-11522. [CrossRef]
- Kuiken, T., et al., Pathogenesis of influenza virus infections: the good, the bad and the ugly. 2012. 2(3): p. 276-286. [CrossRef]
- Marion, T., et al., Respiratory mucosal proteome quantification in human influenza infections. 2016. 11(4): p. e0153674. [CrossRef]
- Guo, X.-z.J. and P.G. Thomas. New fronts emerge in the influenza cytokine storm. In Seminars in immunopathology. 2017. Springer. [CrossRef]
- Lamb, R.A. and C.M.J.T.i.G. Horvath, Diversity of coding strategies in influenza viruses. 1991. 7(8): p. 261-266.
- Hale, B.G., et al., The multifunctional NS1 protein of influenza A viruses. 2008. 89(10): p. 2359-2376. [CrossRef]
- Lamb, R.A. and C.-J.J.V. Lai, Expression of unspliced NS1 mRNA, spliced NS2 mRNA, and a spliced chimera mRNA from cloned influenza virus NS DNA in an SV40 vector. 1984. 135(1): p. 139-147. [CrossRef]
- Gibson, S., et al., Three cases of acute myositis in adults following influenza-like illness during the H1N1 pandemic. 2013. 4(1): p. 51. [CrossRef]
- Taubenberger, J.K. and D.M.J.A.R.P.M.D. Morens, The pathology of influenza virus infections. 2008. 3: p. 499-522.
- Chughtai, A., et al., The presence of fever in adults with influenza and other viral respiratory infections. 2017. 145(1): p. 148-155. [CrossRef]
- Popescu, C.P., et al., Neurologic complications of influenza B virus infection in adults, Romania. 2017. 23(4): p. 574. [CrossRef]
- Milne, M., et al., Influenza, hay fever, and sinusitis: Know the differences. 2018. 85(6): p. 19-26.
- Kong, W.J.T.L.I.D., Influenza virus associated with ocular complications. 2018. 18(6): p. 602-603. [CrossRef]
- Wilhelm, M.J.T.A.j.o.m.c., Influenza in older patients: a call to action and recent updates for vaccinations. 2018. 24(2 Suppl): p. S15-S24.
- Rello, J. and A.J.C.C. Pop-Vicas, Clinical review: primary influenza viral pneumonia. 2009. 13: p. 1-6.
- Odio, C.D., et al., Severe influenza A (H1N1) virus infection complicated by myositis, refractory rhabdomyolysis, and compartment syndrome. 2019. 2019(1): p. 1540761.
- Pandey, Y., et al., Acute influenza infection presenting with cardiac tamponade: a case report and review of the literature. 2019. 23. [CrossRef]
- Henrickson, K.J.J.C.m.r., Parainfluenza viruses. 2003. 16(2): p. 242-264.
- Noor, A. and E.J.O.J. Gradidge, A case of Reye syndrome caused by influenza A virus. 2018. 18(4): p. 425-427. [CrossRef]
- Schrör, K.J.P.D., Aspirin and Reye syndrome: a review of the evidence. 2007. 9: p. 195-204.
- Binnicker, M.J., et al., Direct detection of influenza A and B viruses in less than 20 minutes using a commercially available rapid PCR assay. 2015. 53(7): p. 2353-2354. [CrossRef]
- Anderson, K.B., et al., Clinical and laboratory predictors of influenza infection among individuals with influenza-like illness presenting to an urban Thai hospital over five years. 2018. 13(3): p. e0193050. [CrossRef]
- Ghebrehewet, S., P. MacPherson, and A.J.T.B. Ho, Clinical Updates: Influenza. 2016. 355.
- Mahony, J., et al., Multiplex loop-mediated isothermal amplification (M-LAMP) assay for the detection of influenza A/H1, A/H3, and influenza B can provide a specimen-to-result diagnosis in 40 min with single genome copy sensitivity. 2013. 58(1): p. 127-131.
- Zhang, H., et al., Development of a multiplex real-time RT-PCR assay for simultaneous detection and differentiation of influenza A, B, C, and D viruses. 2019. 95(1): p. 59-66.
- Trombetta, V.K., et al., Are rapid influenza antigen tests still clinically useful in today's molecular diagnostics world? 2018. 77(9): p. 226.
- Low, Z.Y., et al., The convergent evolution of influenza A virus: Implications, therapeutic strategies and what we need to know. 2023: p. 100202. [CrossRef]
- Eyers, S., et al., The effect on mortality of antipyretics in the treatment of influenza infection: a systematic review and meta-analysis. 2010. 103(10): p. 403-411.
- Ng, K.E.J.P. and Therapeutics, Xofluza (Baloxavir marboxil) for the treatment of acute uncomplicated influenza. 2019. 44(1): p. 9.
- Kosik, I. and J.W.J.V. Yewdell, Influenza hemagglutinin, and neuraminidase: Yin–Yang proteins coevolving to thwart immunity. 2019. 11(4): p. 346. [CrossRef]
- McLean, H.Q., et al. Impact of late oseltamivir treatment on influenza symptoms in the outpatient setting: results of a randomized trial. In Open forum infectious diseases. 2015. Oxford University Press. [CrossRef]
- Eiland, L.S., E.H.J.T. Eiland, and C.R. Management, Zanamivir for the prevention of influenza in adults and children age 5 years and older. 2007. 3(3): p. 461-465.
- Pielak, R.M., K. Oxenoid, and J.J.J.S. Chou, Structural investigation of rimantadine inhibition of the AM2-BM2 chimera channel of influenza viruses. 2011. 19(11): p. 1655-1663. [CrossRef]
- Baker, D.E.J.H.P., Baloxavir marboxil. 2019. 54(3): p. 165-169.
- Han, J., et al., Influenza virus: small molecule therapeutics and mechanisms of antiviral resistance. 2018. 25(38): p. 5115-5127. [CrossRef]
- Kumar, B., et al., Potent intracellular knock-down of influenza A virus M2 gene transcript by DNAzymes considerably reduces viral replication in host cells. 2015. 57: p. 836-845. [CrossRef]
- Tarn, W.-Y., et al., Antisense oligonucleotide-based therapy of viral infections. 2021. 13(12): p. 2015. [CrossRef]
- Svoboda, P.J.F.i.p.s., Key mechanistic principles and considerations concerning RNA interference. 2020. 11: p. 1237.
- Takahashi, T., S.M. Heaton, and N.F.J.P.p. Parrish, Mammalian antiviral systems directed by small RNA. 2021. 17(12): p. e1010091. [CrossRef]
- McMillen, C.M., et al., Inhibition of influenza A virus matrix and nonstructural gene expression using RNA interference. 2016. 497: p. 171-184. [CrossRef]
- Henahan, S.J.I.W., Fomivirsen focuses on the future of CMV retinitis. 1998. 1138(1): p. 11-12. [CrossRef]
- Roberts, T.C., R. Langer, and M.J.J.N.r. D.d. Wood, Advances in oligonucleotide drug delivery. 2020. 19(10): p. 673-694. [CrossRef]
- Chen, Y.-Q., et al., Influenza infection in humans induces broadly cross-reactive and protective neuraminidase-reactive antibodies. 2018. 173(2): p. 417-429. e10.
- Palese, P.J.V., Orthomyxoviridae. 2007: p. 1647-1740.
- Szewczyk, B., K. Bieńkowska-Szewczyk, and E.J.A.B.P. Król, Introduction to molecular biology of influenza A viruses. 2014. 61(3): p. 397-401.
- Dugan, V.G., et al., The evolutionary genetics and emergence of avian influenza viruses in wild birds. 2008. 4(5): p. e1000076.
- Garten, R.J., et al., Antigenic and genetic characteristics of swine-origin 2009 A (H1N1) influenza viruses circulating in humans. 2009. 325(5937): p. 197-201.
- Hussain, M., et al., Drug resistance in influenza A virus: the epidemiology and management. 2017: p. 121-134.
- Kessler, S., et al., Influenza A viruses and zoonotic events—are we creating our reservoirs? 2021. 13(11): p. 2250.
- Van der Vries, E., Influenza Resistance to Antiviral Drugs: Virus characterization, mechanism and clinical impact. 2014.
- Whittle, J.R., et al., Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. 2011. 108(34): p. 14216-14221.
- LeGoff, J., et al., I223R mutation in influenza A (H1N1) pdm09 neuraminidase confers reduced susceptibility to oseltamivir and zanamivir and enhanced resistance with H275Y. 2012.
- Wang, D., et al., Neuraminidase inhibitor susceptibility testing of influenza type B viruses in China during 2010 and 2011 identifies viruses with reduced susceptibility to oseltamivir and zanamivir. 2013. 97(3): p. 240-244.
- Deyde, V.M., et al., Surveillance of Resistance to Adamantanes among Influenza A(H3N2) and A(H1N1) Viruses Isolated Worldwide. The Journal of Infectious Diseases, 2007. 196(2): p. 249-257.
- Hurt, A.C., et al., Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2014–2015. 2016. 132: p. 178-185.
- Deyde, V.M., et al., Surveillance of resistance to adamantanes among influenza A (H3N2) and A (H1N1) viruses isolated worldwide. 2007. 196(2): p. 249-257.
- Dharan, N.J., et al., Infections with oseltamivir-resistant influenza A (H1N1) virus in the United States. 2009. 301(10): p. 1034-1041.
- Butler, J., et al., Estimating the fitness advantage conferred by permissive neuraminidase mutations in recent oseltamivir-resistant A (H1N1) pdm09 influenza viruses. 2014. 10(4): p. e1004065.
- Baranovich, T., R.G. Webster, and E.A.J.C.O.i.V. Govorkova, Fitness of neuraminidase inhibitor-resistant influenza A viruses. 2011. 1(6): p. 574-581.
- Schünemann, H.J., et al., WHO Rapid Advice Guidelines for pharmacological management of sporadic human infection with avian influenza A (H5N1) virus. 2007. 7(1): p. 21-31.
- Duan, S., et al., Novel genotyping and quantitative analysis of neuraminidase inhibitor resistance-associated mutations in influenza viruses by single-nucleotide polymorphism analysis. 2011. 55(10): p. 4718-4727.
- Russell, R.J., et al., The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. 2006. 443(7107): p. 45-49.
- van der Vries, E., M. Schutten, and C.A.J.C.o.i.i.d. Boucher, The potential for multidrug-resistant influenza. 2011. 24(6): p. 599-604.
- Dong, G., et al., Adamantane-resistant influenza viruses in the world (1902–2013): frequency and distribution of M2 gene mutations. 2015. 10(3): p. e0119115.
- Omoto, S., et al., Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. 2018. 8(1): p. 9633.
- Donatelli, I., et al., Human-animal interface: the case for influenza interspecies transmission. 2017: p. 17-33.
- Lyall, J., et al., Suppression of avian influenza transmission in genetically modified chickens. 2011. 331(6014): p. 223-226.
- Lee, H.J., et al., Germline modification and engineering in avian species. 2015. 38(9): p. 743-749.
- Furuse, Y., et al., Large-scale sequence analysis of M gene of influenza A viruses from different species: mechanisms for the emergence and spread of amantadine resistance. 2009. 53(10): p. 4457-4463.
- Yi, C., et al., Genome-wide CRISPR-Cas9 screening identifies the CYTH2 host gene as a potential therapeutic target of influenza viral infection. 2022. 38(13).
- Abbott, T.R., et al., Development of CRISPR as an antiviral strategy to combat SARS-CoV-2 and influenza. 2020. 181(4): p. 865-876. e12.
- Owen, L., K. Laird, and M.J.L.i.A.M. Shivkumar, Antiviral plant-derived natural products to combat RNA viruses: Targets throughout the viral life cycle. 2022. 75(3): p. 476-499.
- Perovic, V., et al., Exploring the Antiviral Potential of Natural Compounds against Influenza: A Combined Computational and Experimental Approach. 2024. 25(9): p. 4911.
- Pizzorno, A., et al., Repurposing of drugs as novel influenza inhibitors from clinical gene expression infection signatures. 2019. 10: p. 60.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



