
Submitted:
22 October 2024
Posted:
24 October 2024
You are already at the latest version
Abstract
This paper examined the chemoselectivity and diastereoslectivity of silyl nitronate alkenyn-nitroethers in Intramolecular Silyl Nitronate Cycloadditions (ISNC) to produce isoxazole derivatives with interesting medicinal properties. These reactions resulted in the formation of either dihydrofuro[3,4-c]isoxazolines/isoxazolidines and/or alkynyl moieties attached to 2,5-dihydrofuryl carbonyls. It also discerned the diastereoselectivity of the resulting cyclic adducts and compared those to previous findings. The reactions were also investigated with Spartan molecular modeling computations to aide in the understanding of any displayed chemo- and/or stereoselectivity. These [3+2]-cycloaddition reactions demonstrated excellent to complete chemospecificity. The cycloadditions also demonstrated remarkable diastereospecificity in that each diastereomer of the nitroethers resulted in the formation of only one of four possible diastereomeric outcomes. The stereochemistry of the major diastereomers did not agree with previously published findings.
Keywords:
PM6
; dihydrofuro-2-isoxazolines
; ISNC
; alkenynyl nitroethers
; 3-(2
; 5-dihydrofuryl)carbonyls
; chemoselectivity
; diastereoselectivity
1. Introduction
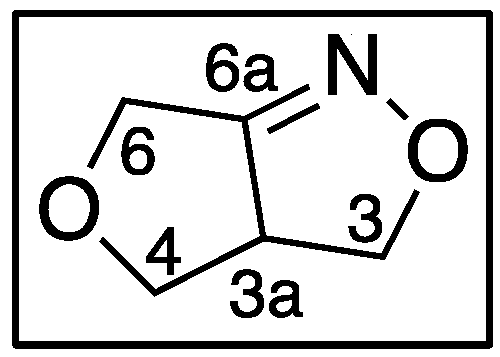
Nitrile Olefin Cycloadditions and Silyl Nitronate Olefin Cycloaddition reactions have been studied extensively by many groups as viable routes for stereoselective preparation of isoxazoles, isoxazolines, and isoxazolidines [1,2,3,4,5,6,7]. Isoxazoles, isoxazolines, and isoxazolidines are well known for their medicinal value as antibacterial, anticancer, antifungal, antiviral, anti-inflammatory, ectoparasiticide and anti-tuberculosis agents [8,9,10,11,12]. Unlike the Intramolecular Nitrile Oxide Olefin Cycloaddition (INOC), the Intramolecular Silyl Nitronate Olefin Cycloaddition (ISOC) reaction differs in the reaction of alkenyl- and alkynyl- nitroethers as viewed in Scheme 1. Alkenyl-nitroethers (1) when treated under INOC conditions yield the expected 3a,4-dihydro-3H,6H-furo[3,4-c]isoxazoles (2). Alkynyl-nitroethers (3) when treated under INOC conditions produce the expected dihydrofuroisoxazoles (4) [8,13]. However, alkynyl-nitroethers (3) surprisingly yield 3-dihydrofuranylcarbonyls (5) under ISOC conditions [13,14]. Thus the label ISNC (Intramolecular Silyl Nitronate Cycloaddition) is used by the principal author to describe these reactions with either alkene or alkyne moieties. The common name for an H,H-isoxazole is isoxazoline and isoxazolidine is commonly used for H,H,H,H-isoxazoles. The numbering for the furoisoxazoles is shown in Figure 1.
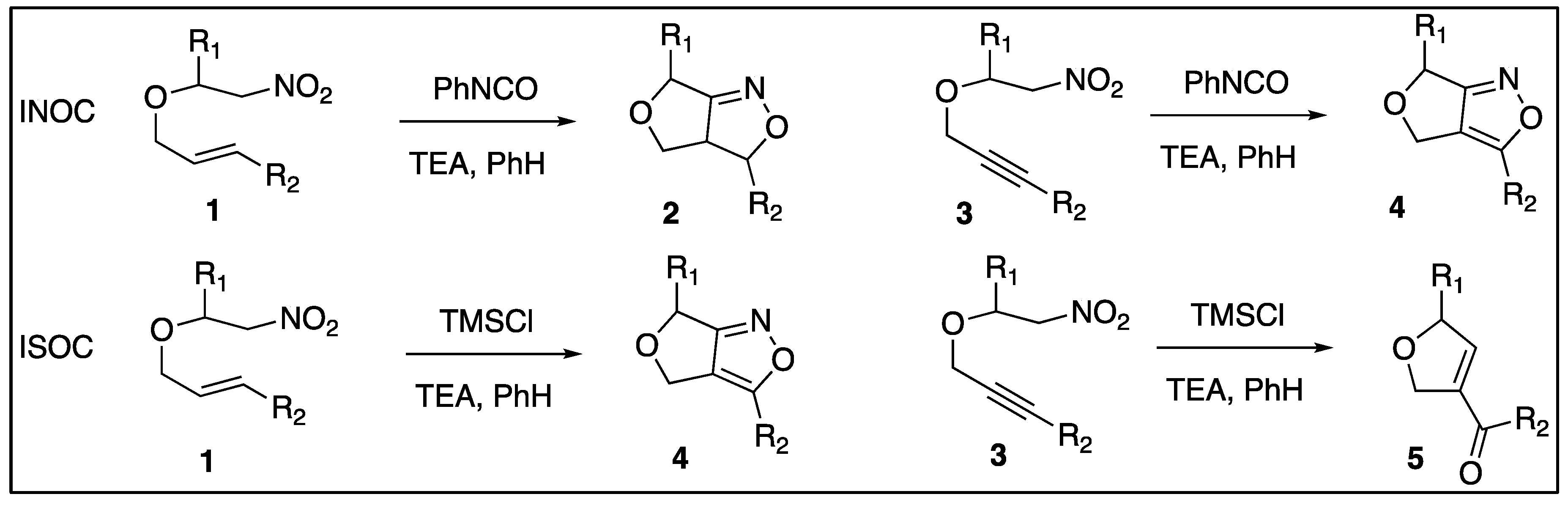
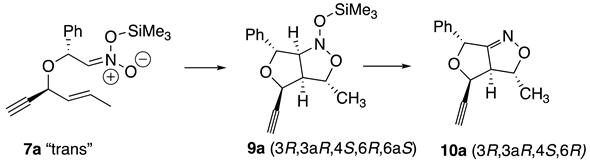
This paper examined the diastereoselectivity and chemoselectivity of nitroethers in INSC reactions with both alkenyl and alkynyl side chains. The chemoselectivity for the INSC reactions is examined more closely in Scheme 2. The trimethylsiloxy-nitronates (7a,b) are in the correct conformation to undergo ring closing with the alkenyls to make N-trimethylsiloxy-dihydrofuro-3H,6H-isoxazoles (9a,b) and consequently the propenyl-furo-4H,6H-isoxazoles (10a,b) after acidic workup. A similar series of reactions were expected for the trimethylsiloxy-nitronates (8a,b) to form the trimethylsiloxy intermediates (11a,b) that corresponded from reactions with the triple bonds. After acidic workup these intermediates produced the dihydrofuro-carbonyl products (12a,12b) from the ISNC reactions.
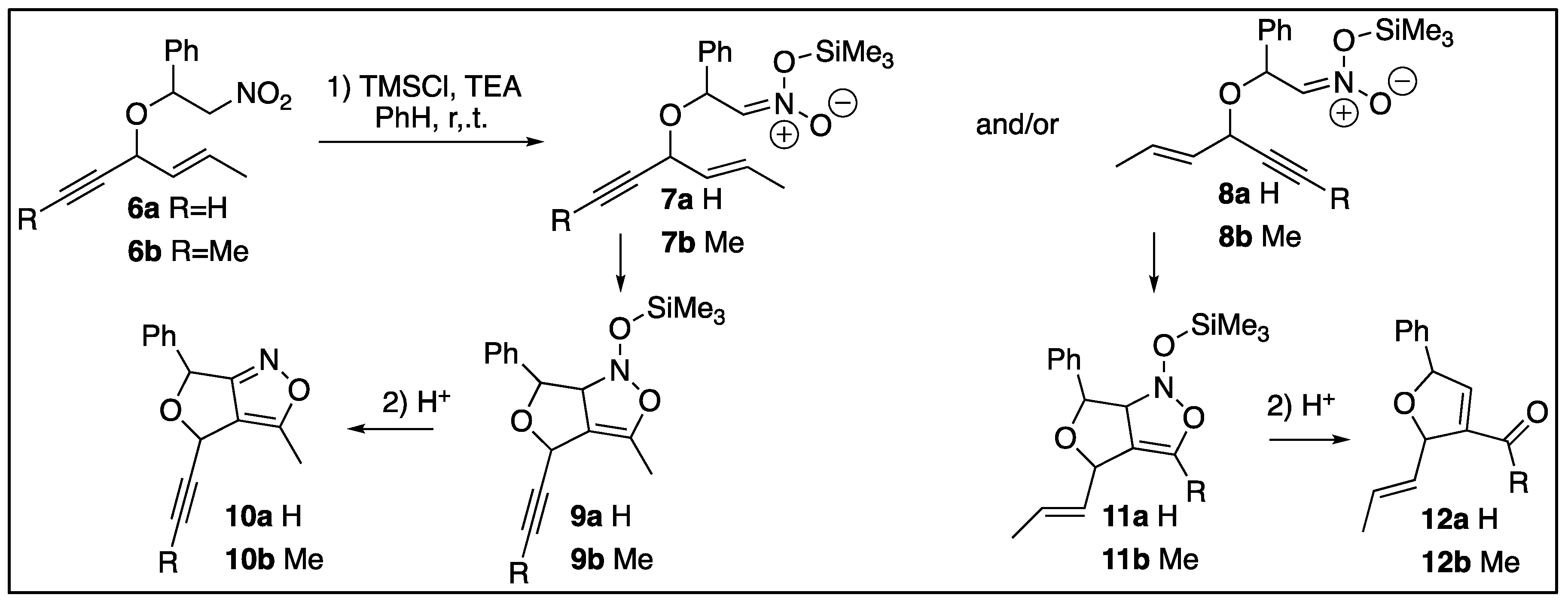
The synthetic scheme is demonstrated in Scheme 3. The alkenynols (14) were prepared from Grignard reactions with either ethynylmagnesium or propynylmagnesium bromides and crotonaldehyde (13). Nitrostyrene (15) was prepared in a Henry reaction between nitromethane and benzaldehye.[8] Disappointingly, these allyllic/propargrylic systems do not react cleanly in the tandem one pot reaction of olefinic silylnitronates reported by Hassner [15] and Cheng [16]. Complex reaction mixtures were obtained instead. The nitroethers (5) were instead prepared via Micheal Additions between sodium alkoxides and unsaturated nitrocompounds [17]. These compounds were then treated to ISNC reactions to yield furo-2-isoxazolines (10) and/or carbonyl dihdyrofurans (12).
This work examined if the alkenynl-nitroethers demonstrated selectivity for either the double or triple bonds during ISNC conditions. The chemoselectivity of the alkenynl-silylnitronates were determined based on the ratio of the isoxazole:carbonyl products. The disasteroselectivities of the cycloadditions were also of interest. Cycloadditions to form the dihydrofuro-2-isoxazolines produces 3 new stereocenters in the trimethylsiloxy-furoisoxazole intermediates (9,11) and retains 2 of the new stereocenters in the products (10). The stereochemistry for each enantiomeric pair will be labeled with the configurations/name of one of the enantiomers, with a (±) sign in front to indicate the racemic mixture. The identification and diastereoselectivity of the isoxazoline/isoxazolidine and/or furo-carbonyl adducts were obtained primarily via NMR experiments (1H, 13C, COSY, HMQC, NOE). Computational modeling was utilized to probe any displayed chemoselectivity and/or diastereoselectivity in the products.
2. Results
The synthetic scheme was repeated over the years by several different undergraduate students. Sometimes the 1H NMR analysis of the crude reaction mixture of 10a would show very small singlet peaks between 9-10 ppm. This would seem to indicate that a small amount of the dihydrofuro-carbonyl adducts (3%) were formed. However, these crude NMRs did not have the expected doublet (or doublet of doublets) between 6-7 ppm that would have indicated the presence of the expected vinylic proton on the tetrahydrofuran ring as based on previous work by the authors.[14] The crude NMR for 10a displayed evidence of small amounts of the nitroether, nitrostyrene, and the corresponding alcohol but not any of the expected peaks for the carbonyl compounds other than these singlets. Column chromatography was used to try to isolate compounds for the 9-10 ppm peaks. Analysis of these fractions showed a complex mixture that had many peaks that could not be seen in the beginning crude NMR. Therefore, while some evidence of small amounts of aldehydes were present, the authors are not convinced that the dihydrofuro-carbaldehydes for 10a were formed. If the authors are incorrect, than this cycloaddition showed excellent chemoselectivity for the double bond (97%). The crude 1H NMR for 10b did not display any evidence of carbonyl products as no singlets near 2.1 ppm were evident. Based on the crude 1H NMR data, both products (10a,b) resulted from chemospecific reactions of the double bonds over the triple bonds. NOE experiments were used to confirm the major and minor diastereomers that were found for the cycloadditions as shown in Table 1. These finding show that the “cis” and “trans”- diastereomers of the nitroether only produce one diastereomer each in the cycloadditions. Unexpectedly the assignments of the major and minor diastereomers were switched based on previous findings of the principal author and others.
3. Discussion
The stereochemistry of the ISOC (ISNC) cycloaddition reaction has been extensively studied. The diastereoselectivity of the resulting dihydrofuro-isoxazolidines at C3 and C3a are controlled by the formation of the ring system and the stereochemistry of the alkene is retained [18]. The relationship of substituents on carbon 4 to the hydrogen on carbon 3a and were shown to have a selectively cis relationship (trans hydrogens) if there is no substituent on C6 [8,13,17,18]. Kurth/Duffy showed that the ISOC has complete cis isomer preferences for the hydrogen on C3a and C6 substituents in systems without C3 substituents [19]. Kim’s work [20] with allylic and homoallylic nitroethers gave complete regioselectivity as predicted by the findings of Duffy [19] but gave a mixture of stereoisomers that under ISOC conditions slightly favored the trans relationship between substituents on C4 and C6 (no substituent on C3). Duffy-Matzner’s investigation into nonterminal propargylic systems also displayed a stronger preference for trans isomers between C4 and C6 [14]. Kurth/Duffy also gave evidence that the major stereoisomer had a trans relationship between C4/C6 substituents in systems with substituents in C3, C4 and C6 positions as shown in Scheme 2. The assignment of the major diastereomer in the 4th row in Figure 2 was also proven with crystal X-ray evidence for the diphenyl product [18].
It was expected that the “trans”-nitroether would control the major stereoisomer in this work as well. Trans refers to the stereochemical relationship of the two methine hydrogens alpha to the ether’s oxygen. Unexpectedly the results of the alkenynl-nitroethers (6a,6b) showed that the “cis”-nitroethers were responsible for the stereochemical outcomes of the major diastereomers. As mentioned previously, cycloadditions to form the dihydrofuro-isoxazolidines produces 3 new stereocenters (C3,C3a,C6a) in the silyl-nitronate intermediates (8) and retains 2 of the new stereocenters (C3,C3a) in the products (10). The numbering of the ring systems is displayed in Figure 3.
Figure 3 demonstrates that the cycloaddition for the “cis’-nitroether (R,R) would produce two new stereocenters at C3 and C3a to give an end product with four stereocenters. 32 possible diastereomers could be present in the formation of the ring systems from the two nitroethers, based on the four stereocenters present in each ring. Since the configurations of C4 and C6 are determined by the formation of the nitroethers, only 8 diastereomers are of interest for the newly defined stereocenters. Furthermore, the two hydrogens on C3 and C3a must be trans to each other for the furo-2-isoxazoline due to the configuration of the double bond. This narrows the investigation down to 4 possible diastereomers for each diastereomer of the nitroether.
Computational studies were performed on the silyl-nitronates (7a,7b,8a,8b) to form the trimethylsiloxy-isoxazolidines (9a,b) and trimethylsiloxy-2-isoxazolines (11a,b) intermediates. Analyses of these compounds were examined since they have same numbers of atoms which makes it easier to compare the results. Multiple Spartan calculations were employed, with the focus of the research centered on semi-empirical (PM6) and density functional (wB97X-D 6-31G*) calculations. For the purposes of conserving time, the density functional calculations were performed with the molecule in the gas phase. Future work will carry out the DFT calculations in suitable nonpolar solvents.
The semi-empirical (PM6) and density functional theory (wB97X-D 6-31G*) for the ISNC silyl nitronate intermediates provided useful information as can be seen in Figure 4. The calculations originated with the lowest energy conformer of the trimethylsilyloxy-nitroether (7a). This was then rearranged to reflect the correct orientations for the corresponding transition states (19a&20a) which were calculated. Finally, the final intermediate trimethylsilyloxy-furoisoxazoline (11a) and furoisoxazolidine (9a) were examined. The results showed that the triple bond intermediates (20a) had the higher energy transition state with the lowest enthalpy of formation as compared to the transition state involving the double bond (19a). This suggests the formation of the isoxazoline intermediate (11a) is favored by thermodynamic conditions. The reactions with the double bond to form the isoxazolidine (9a) demonstrated that these products may be formed under kinetic conditions. The DFT and PM6 calculations suggest that under the experimental conditions for this report, the kinetic product was selectively formed.
It was noted that the trends present for the reactions were clearer to see in the PM6 calculations. Some would argue that the semi-empirical quantum chemistry method of James J.P. Stewart calculates heat of formation of a wide variety of molecules with an accuracy arguably better than Hartree-Fock and better overall than Density Functional Theory [21,22]. This accuracy is accomplished by using experimental parameters along with quantum chemistry calculations. The Hartree-Fock and Density Functional methods rely solely on complex calculations derived from the Born-Oppenheimer approximation. It was decided to examine all the possible intermediates for the terminal and non-terminal alkyne systems via PM6 experiments for the remaining calculations. The computations were run on species generated from the “cis” and “trans”-siloxynitroethers to reflect the stereochemical outcome of the major cyclic diastereomers. These calculations also displayed that the reactions with the double bond produced a product with a lower transition state energy but higher enthalpy of formation. This was true independent of the cis or trans orientation and the substituent placed on the triple bond. It is also interesting to note that the transition states of the terminal alkyne intermediates had higher transition state energies and that the trans isomers were slightly higher in energy than the cis isomers.
These calculations show that the reaction with the double bond is favored over the triple bond. This explains the chemoselectivity of the reactions and slightly supports the surprising stereoselectivity of the major diastereomerst. It was unexpected because cycloadditions of nitroethers have been reported to prefer the (±)-(3R,3aR,4S,6S)-furoisoxazolines adducts for silyl nitronate cycloadditons instead of our reulsts reported in Table 1 [9,13,14,15]. The investigation then examined all the possible diastereomers to try to find an explanation.
Four possible diastereomers for the trimethylsiloxy-dihydrofuroisoxazolidine (9a) can be expected for one “cis”-nitroether. These arise from the different faces that are available for the double bond to react with and the orientation of the C=C bond and the imino moiety (C=N) of the trimethysiloxy-nitronate.
Table 2 demonstrates that four possible diastereomers are generated from different orientations of the alkenyl or imino silyl nitronate groups in the four possible transition states from the “cis”-nitroether. I & II show the same face selectivity for all the prochiral centers, this demonstrates that these processes are suprafacial. III & IV show the prochiral centers reacting from different faces of the system, thus these cycloadditions are antarafacial. The suprafacial processes demonstrate a less strained transition state for the formation of the new isoxazole and would be favored over the antarafacial ones. Selectivity was also seen in that the si facial attack was favored over the re for the C3a and C6a prochiral centers. The si facial strike led to a chair-like transition state, while the re led to a boat-like transition state. Thus, the major stereoisomer for the “cis”-nitroether cyloaddition would be the (±)-(3S,3aS,4R,6R,6aR) silyl intermediate and the (±)-(3S,3aS,4R,6R)-adduct. These results agree with the experimental NOE data for the major diastereomer of 10a. This reasoning is also supported by the PM6 calculations which clearly show that intermediate I is the lowest energy intermediate. It results from a “chair” transition state with all substituents in “equatorial” positions as shown in Table 2.
The diastereomer of the “trans”-nitroether was also considered. Table 3 shows the outcomes of the analysis of its facial selectivity to yield a suprafacial, si transition state. The “trans”-nitroether (7a) would result in the (3S,3aS,4S,6R,6aS)-silyl intermediate (9a) and the (3S,3aS,4S,6R)-dihydrofuro-2-isoxazolidine (10a). These results agree with the experimental NMR NOE data for the minor diastereomer of 10a. Similar results were obtained for both the major and minor diastereoisomers (10b) of the cycloadditions from the propenyl nitroethers (7b). It is unlikely that the stereochemistry at the C4 and C6 positions change during the ISNC process, since anionic intermediates are unlikely to experience epimerization rearrangements. This leads the authors to conclude that the formation of the alkenynyl-nitroethers must favor the cis orientation for substitutents on the carbons alpha to the ether’s oxygen unlike alkenyl [13] or alkynyl-nitroethers [9,10].
4. Materials and Methods
4.1. Physical and Spectral Data
- Low resolution mass spectra were determined on a Hewlett Packard G1800A GCD system.
- Proton and Carbon NMR spectrra were obtained either on a JEOL (400MHz) spectrometer or a Varian (300 MHz) system. Listed proton NMR data are given in the following order: ppm (multiplicity, coupling constants, integrated number of protons and assignment). Listed carbon NMR data are given by chemical shift and assignment. All the spectra were run in deuterated chloroform with TMS unless stated otherwise.
- Infared spectra were recorded on a Nicolet Avatar 361 FT-IR with Gateway 2000 data system and were either neat NaCl plates (liquids) or KBr pellet (solids).
4.2. Chromatography
- Flash column chromatography refers to the resolution technique of W. Clark Still [J. Organic Chem. 1978, 43, 2923]. A glass column is filled with a slurry of dry 40-62 μm silica gel and solvent. The same solvent is used as an eluent to push a sample through the column, with pressure from a nitrogen inlet to speed the elution to a rate of 2 in./min.
- TLC refers to thin layer chromatography, which was done on Sigma Chemical Co. plates made of 250 μm silica gel on polyester with a 254 nm fluorescent indicator added. Visualization was performed via iodine chambers or UV lamp.
- Capillary gas chromatography (GC) was performed on a HP G1800A GCMS, equipped with a HP-5 Crosslinked 5% PH ME Siloxane column, 30m x 0.25mm x 0.25 μm.
Solvents
Tetrahydrofuran was distilled under a nitrogen atmosphere from sodium and stored over 4 Å molecular sieves. Hexane was distilled under a nitrogen atmosphere from sodium and stored over 4 Å molecular sieves. Benzene was distilled under a nitrogen atmosphere from sodium and stored over 4 Å molecular sieves.
4.3. Reactions
- Concentration under reduced pressure refers to solvent removal on a Büchi RE 111 rotary evaporator connected to a water aspirator and an ethylene glycol cooling system.
- All reactions were run under a nitrogen atmosphere unless otherwise stated.
- Unless otherwise stated, all other solvents and reagents were reagent grade and used without further purification.
General Procedures
General Procedure A: Alkenynols
The Grignard reagent (0.1M, 1.2 mol) was placed in a round-bottomed flask with a stir bar under nitrogen. Then dry THF was added to dilute the solution (0.01M) while the flask was in an ice bath, crotonaldehyde (neat, 1 mol) was added dropwise through a syringe. After the addition is complete the solution was slowly allowed to reach room temperature. At this point TLC showed no starting material. The reaction was quenched with HCl (3M, 2 mol). Diethyl ether is added. The organic and aqueous layers were separated. The aqueous layer was extracted three times with diethyl ether. The combined organic layers were washed with 5% sodium bicarbonate, brine and then dried over magnesium sulfate. After gravity filtration the product was reduced under vacuum to yield an oil.
(4E)-hex-4-en-1-yn-3-ol (14a):
General procedure A after vacuum distillation (0.1 Torr) gave 5.465 g (54.65%) of 14a as a clear pale yellow liquid from 6.48 g (0.09247 mol) of crotonaldehyde (11). [FTIR (neat): 3300cm-1 (OH v), 3032 (=C-H v), 2975, 2941 (C-H asym v), 2879 (C-H sym v), 2159 (C≡C v), 1447, 1378 (C-H δ), 1016 (COC v), 964 (oop δ trans CH=CH)]; [1H NMR (400 MHz, CDCl3): δ= [1.75 ppm (m, 3H, CH3), 1.96 (s, 1H, OH), 2.57 (d, J=1.6Hz, 1H, C≡C-H), 4.83 (m, 1H, CH-O), 5.64 (ddq, J=15.3,6.4,1.6Hz,1H, CH=CH-CH3), 5.94 (dqd, J=15.3, 6.4, 1.6Hz, 1H, =CH-CH3)]. [13C NMR (100 MHz, CDCl3): δ= [17.4 ppm (CH3), 62.3 (CH-O), 73.8 (C≡C-H), 83.7 (C≡C-H), 128.6 (CH3-CH=CH), 130.1 (CH3-CH=CH)]; GCMS (M+ C6H8O 96.15 m/z).
(E)-5-heptyn-2-en-4-ol (14b):
General procedure A gave 10.9 grams (82% yield) of 14b as dark brown oil from 8.468 g (0.1207 mol) of crotonaldehyde (16). [Rf = 0.50, (1:4 EtOAc: Pentane), [FTIR (neat): 3363cm-1 (OH ν), 2919 (sp3 C-Hν)), 2237 (C≡Cν)), 1673 (CH=CHδ); [1H NMR (300 MHz, CDCl3): δ 1.73ppm (m, 3H, CH3-CH=), 1.86 (d, J = 1.8Hz, 3H, CH3- C≡), 3.26 (s, 1H. OH), 4.77 (m, 1H, CH-OH), 5.60 (m, 1H, =CH-CHOH), 5.84 (dq, J = 15, 6.6Hz, 1H, CH3-CH= )]; [13C NMR (75.4ppm MHz, CDCl3) δ = 3.6 (CH3-C≡C), 17.4 (CH3-C=), 62.9 (CH-OH), 79.3 (CH3-C≡C), 81.9 (C≡C-CHOH), 127.9 (=CH-CH3), 131.2 (=CH-CHOH)]; GCMS (M+ C7H10O 110.05 m/z).
General Procedure B: Nitroethers (6)
Sodium hydride (0.21 mol) was washed with distilled hexanes (15 mL, 5 times) in a dry round-bottomed flask with a stir bar and dried under nitrogen. The alkenynol (.20 mol) was added slowly at room temperature. The flask was placed in a dry ice and isopropanol bath at -30 °C bath. A solution of nitrostyrene in THF was prepared (0.1 M). Using a syringe-pump set at 20 mL/hour, nitrostyrene was added to the alkoxide solution (0.10 mol). When all of the nitrostyrene was added, the reaction was quenched with HCl (3M, 0.3mol). At this point TLC showed no starting material. The aqueous layer was extracted three times with diethyl ether (30 mL). The organic layer was then dried over magnesium sulfate. After gravity filtration, the solution was reduced under vacuum to yield an oil. The nitroether was purified through column chromatography in a solvent of 1:9 ethyl
acetate:hexanes that slowly transitioned to 1:6. Unfortunately clean nitroethers tend to under retro-Micheal Additions and were not suitable for elemental or HRMS analysis.
(±)-(3R,4E)-3-[(1R)-2-nitro-1-phenyleth-1-oxy]hex-4-en-1-yne (6a):
General Procedure B gave 6a as a yellow oil 1.970 g in a crude 100% yield. After column chromatography 6a was isolated as a yellow oil, 1.611 g (81.8%) from 1.198 g (0.008032 mol) of nitrostyrene (18) as two inseparable diastereomers (1.5:1 ratio): I (±)-(3R,4E)-3-[(1R)-2-nitro-1-phenyleth-1-oxy]hex-4-en-1-yne, II. (±)-(3S,4E)-3-[(1R)-2-nitro-1-phenyleth-1-oxy]hex-4-en-1-yne. [Rf = 0.40, (1:6 EtOAc: hexanes)]; FTIR (KBr): [3290cm-1 (≡CHν), 3050 (sp2 C-Hν), 2940, 2919 (sp3 C-Hν), 2100 (C≡Cν), 1556, 1380 (NO2ν), 1061 (C-O-Cν)]; 1H NMR (400 MHz, CDCl3): δ= [1.70 ppm (d, J=6.5Hz, 0.40H, =C-CH3 II), 1.75 (d, J=6.5Hz, 0.60H =C-CH3 I), 2.45 (d, J=2.4Hz, 0.60H, H-C≡C I), 2.59 (d, J=2.4Hz, .40H, H-C≡C II), 4.42 (m, 1.2H, O-CH-C=C I, CHNO2 I), 4.42 (m, 0.80H, O-CH-C=C II, CH2NO2 II), 4.69 (m, 1H, CHNO2 I&II), 5.26 (dd, J=10.4, 3.2Hz, 0.60H, CH-Ph I), 5.48 (m, 1.40H, CH=CH-CH3 I&II, CH-Ph II), 5.74 (dq, J=15.2, 6.4Hz, 0.60H, =CH-CH3 I), 5.86 (dq, J=15.2, 6.4Hz, 0.40H, =CH-CH3 II), 7.39 (m, 5H, Ph-H, I&II).] [13C NMR (100 MHz, CDCl3): δ= [17.6ppm (CH3 I), 68.9 (CH-O I), 74.7 (H-C≡C I), 75.3 (H-C≡C I), 76.7 (CH-Ph I), 126.9, 127.0, 127.1, 129.0 129.1, 129.2, 129.3 (Ar CH I&II, CH2-NO2 I), 132.0 (=CH-CH3 I), 136.2 (Ar C I).] δ= [17.6 (CH3 II), 67.5 (CH-O II), 75.6 (H-C≡C II), 75.8 H-C≡C II), 77.3 (CH-Ph II), 80.1 (CH2-NO2 II), 81.1 (CH2-NO2 II), 125.9 (CH=CH-CH3 II), 126.9, 127.0, 127.1, 129.0 129.1, 129.2, 129.3 (Ar CH I&II, CH2-NO2 I), 130.6 (=CH-CH3 II), 135.7 (Ar C II).]
(±)-(2E,4R)-4-[(1R)-2-nitro-1-phenyleth-1-oxy]hept-2-en-4-yne (6b).
General Procedure B yielded a crude yellow oil with a large amount of starting alcohol present. After column chromatography 6b was isolated as a white powder, 2.2054 g (82%) from 1.6355 g (0.01096 mol) of nitrostyrene (15) as two inseparable diastereomers (1.2:1): I. (±)-(2E,4R)-3-[(1R)-2-nitro-1-phenyleth-1-oxy]hept-2-en-4-yne, II. (±)-(2E,4S)-3-[(1R)-2-nitro-1-phenyleth-1-oxy]hept-2-en-4-yne. [Rf = 0.23 (1:10 EtOAc:Hexane)]; FTIR (KBr) [3032cm-1 (sp2 C-H ν), 2979, 2919, 2875 (sp3 C-H ν), 2251 (C≡C ν), 1557, 1381 (NO2 ν), 1086 (C-O-C ν)]. 1H NMR (500 MHz, CDCl3): δ= [1.69 ppm (d, 6.5Hz, 0.34H, CH3-C= II), 1.74 (d, J = 6.5Hz, .64H, CH3-C= I),1.76 (d, J=2.0Hz, 0.64H, CH3-C≡C I), 1.90 (d, J=1.5Hz, 0.36H, CH3-C≡C II), 4.40 (m, 1.64H, =C-CH-O I, CH-NO2 I& II,II), 4.68 (m, 1.64H, CH-NO2 I, CH-CPh I&II), 5.24 (dd, J = 9.5Hz, 3.5Hz, 0.64H, CH-C≡C I), 5.44 (m, 0.72H, =CH-CHO II, CH-C≡C II), 5.50 (m, 0.64H, =CH-CHO I ), 5.68 (dq, J = 15 .0Hz, 6.5Hz, 0.64H, CH3-CH=CH I), 5.81 (dq, J=15.0, 6.5 Hz, 0.34H, CH3-CH=CH II ), 7.38 (m, 5H, ArH I&II]. [13C NMR (75.4 MHz, CDCl3) δ= [ 3.6ppm (CH3-C≡ I), 17.4 (CH3-C= I), 69.4 (CH-Ph I), 74.9 (=C-CH-O I), 76.7 (C≡CH I), 80.1 (CH2-NO2 I), 83.2 (C≡CH I), 126.9, 127.0 (CH Ar), 128.0 (=CH-CH-O I), 128.8, 128.9, 129.0, 129.1 (Ar CH I), 131.0 (=C-CH3 I), 136.6 (Ar C I)]. δ= [ 3.6 (CH3-C≡ II), 17.4 (CH3-C= II), 68.1 (CH-Ph II), 75.3 ((=C-CH-O II), 75.4 (C≡CH II), 80.3 (CH2-NO2 II), 84.1(C≡CH II), 126.9, 127.0 (CH Ar), 128.1 (=CH-CH-O II), 128.8, 128.9, 129.0, 129.1 (Ar CH II), 129.7 (=C-CH3 II), 136.3 (Ar C II)].
General Procedure C, Intramolecular Silyl Nitronate Cycloaddition (ISNC)
The nitroether (0.1 mol) was dissolved in enough dried benzene to make a 0.2M solution under nitrogen and with a stirbar in a dry round bottom flask. Distilled triethylamine (0.2 mol) and then tetramethylsilylchloride (TMSCl) (0.2 mol) was added to the solution and the nitrogen lead was removed. The solution was stirred for 2 days in room temperature and TLC showed no evidence of the nitroether. Hydrochloric acid (1M, 0.4 mol) was added as well as some diethyl ether. The organic and aqueous layers were separated. The aqueous layer was extracted with diethyl ether 3 times (30mL per extraction). The combined organic layer was washed with 5% sodium bicarbonate and brine. The solution was dried with anhydrous magnesium sulfate, filtered and concentrated first with a rotatory evaporator and then a vacuum pump.
(±)-(3,3a-dihydro-3-methyl-6-phenyl-4-[eth-1-ynyl]-4H,6H-furo[3,4-c]isoxazole (10a).
General procedure C gave 0.930 grams of the crude 10a (2.1:1 ratio of diastereomers via 1H NMR) with THF still visible. It was then treated with column chromatography to yield an inseparable mixture of the two diastereomers 8a (I&II) (0.8147 g, 89.3%) from (0.985 g, .004016 mol) 6a. Solvent washing and large plate TLC (1:6 EtOAc:hexanes) allowed a small portion of each diastereomer to be isolated for NMR analysis.
10a I. (±)-(3S, 3aS, 4R, 6R)-3,3a-dihydro-3-methyl-6-phenyl-4-[eth-1-ynyl]-4H,6H-furo[3,4-c]isoxazole.
Rf = 0.27 (1:6 EtOAc:hexanes); GCMS (7.51 min, ramp 30, 60-280˚C) 51, 77, 103, 130, 227 M+; FTIR (KBr pellet) 3236 cm-1 (≡C-H v cm-1), 3050 (=C-H v), 2983, 2972 (C-H asym v), 2894 cm-1, 2857 (C-H sym v), 2126 (C≡C v), 1457 (C=N-O v), 1006 (C-OC v), 968 (oop δ trans C=C), 757,704 (oop δ monosub Ph); 1H NMR [400MHz, CDCl3]: I δ= 1.53ppm (d, J=6.0Hz, 3H, CH3), 2.67 (d, 2.0 Hz, 1H, H-C≡C), 3.95 ( ddd, J=11,8.0 1 Hz, 1H, CH-CHCH3), 4.98 (dd, J=9.6, 1.6Hz, 1H, CH- C≡C), 4.70 (dq, 12.4, 6.0 Hz, 1H, CH-CH3), 5.62 (s, 1H, CH-Ph), 7.40 (m, 5H, Ar CH). [13C NMR (100 MHz, CDCl3) δ= 18.3ppm (CH3), 65.2 (CH-CHCH3), 69.6 (CH-C≡C), 74.0 (CH-Ph), 75.9 (C≡C-H), 79.6 (C≡C-H), 83.0 (CH-CH3), 125.8, 128.6, 128.7 (Ar CH), 136.9 (Ar C), 169.4 (C=N).] HRMS C14H13NO2 [M+] calculated 227.09464, found 227.09464.
10a II. (±),(±),(±),(±)-(3S, 3aS, 4S, 6R)-3,3a-dihydro-3-methyl-6-phenyl-4-[eth-1-ynyl]-4H,6H-furo[3,4-c]isoxazole.
Rf = 0.26 (1:6 EtOAc:hexanes); GCMS (7.80 min, ramp 30, 60-280˚C), 51, 77, 103, 130, 227 M+; FTIR (KBr pellet) 3236 cm-1 (≡C-H v cm-1), 3066 (=C-H v), 2983, 2920 (C-H asym v), 2855 (C-H sym v), 2109 (C≡C v), 1446 (C=N-O v), 1008 (C-OC v), 967 (oop δ trans CH=CH), 757,701 (oop δ monosub Ph); 1H NMR [400MHz, CDCl3]: minor trans-diastereomer II δ= 1.54ppm (d, J=6.0Hz, 3H, CH3), 2.82 (d, 1.2 Hz, 1H, H-C≡C), 3.85 ( ddd, J=12.4,9.6,1.6 Hz, 1H, CH-CHCH3), 4.98 (dq, J=11,6.4 Hz, 1H, CH-CH3), 5.07(dd, J=8,2.2 Hz, 1H, CH-C≡C), 5.68 (apparent s, 1H, CH-Ph), 7.36 (m, 5H, Ar CH). [13C NMR (100 MHz, CDCl3) δ= 18.4ppm (CH3), 62.8 (CH-CHCH3), 67.7 (CH-CH3), 72.6 (CH-Ph), 78.0 (C≡C-H), 79.6 (C≡C-H), 82.3 (CH-C≡C), 125.9,128.6,128.8 (Ar CH), 136.4 (Ar C), 169.3 (C=N)].
(±)-3,3a-dihydro-3-methyl-6-phenyl-4-[prop-1-ynyl]-4H,6H-furo[3,4-c]isoxazole (10b).
General Procedure C gave the crude isoxazole (1.8:1 ratio of diastereomers via 1H NMR) as a yellow oil, 2.056 g, 110%). Column chromatography yielded 10b clean but inseparable mixture (1.86:1) of the two diastereomers as a yellow oil, 1.869 grams (81% yield) from 1.8843g (0.007266 mol) of 5b.
10b I. (±)-(3S, 3aS, 4R, 6R)-3,3a-dihydro-3-methyl-6-phenyl-4-[prop-1-ynyl]-4H,6H-furo[3,4-c]isoxazole. 10b II. (±)-(3S, 3aS, 4S, 6R)-3,3a-dihydro-3-methyl-6-phenyl-4-[prop-1-ynyl]-4H,6H-furo[3,4-c]isoxazole.
Rf = 0.628, 0.545 (1:4 EtOAc : Pentane); FTIR (NaCl) 3032cm-1, 3063 (sp2 C-H ν), 2960, 2922, 2873 (sp3 C-H ν), 2254 (C≡C ν), 1602 (Ar C=C ν), 1051 (C-O-C ν); [1H NMR (400MHz, CDCl3): δ= [1.54 (d, J=6.4Hz, 1.05H, CH3-CH II), 1.55ppm (d, J=6.4Hz, 1.95H, CH3-CH I), 1.91 (d, J=6.4 Hz, 1.95H, CH3- C≡C I), 1.97 (d, 2.0 Hz, 1.05H, CH3-C≡C II), 3.79 ( ddd, 11, 9.2, J=1.0 Hz, 0.65H, CH-CHC-C≡C I), 3.92 ( ddd, J=10, 8, 1.6 Hz, 0.35H, CH-CH-C≡C II), 4.60 (dq, J=9.2, 2.0Hz, 0.65H, CH-OCHPh I), 4.69 (dq, J=11, 6.4 Hz, 0.65H, CH-CH3 I), 4.95 (dq, J=10, 6.4Hz, 1H, CH-CH3 II), 5.06 (m, 0.35H, CH-OCHPh II), 5.59 (s (apparent d), J=1 Hz, 0.65H, CH-Ph I), 5.68 (m, 0.35H, CH-Ph II), 7.32 (dt, J=7.2, 1.3 Hz, 0.35H, Ar CH para II), 7.34 (dt, J=7.2, 1.3 Hz, 0.65H, Ar CH para I), 7.39 (tt, 7.2, 1.3 Hz, 2H, Ar CH meta I&II), 7.46 (m, 2H, Ar CH ortho I&II)]; [13C NMR(100 MHz, CDCl3) δ= [ 3.7ppm (CH3-C≡C I), 18.4 (CH3-CH I), 65.6 (CH-CHCH3 I), 70.5 (CH-C≡C I), 73.9 (CH-Ph I), 82.9 (CH-CH3 I), 84.5 (C≡C-CH3 I), 88.3 (C≡C-CH3 I), 126.0, 126.0, 128.5, 128.7 (Ar CH 1 & II), 137.2 (Ar C I), 170.3 (C=N I)]; δ= [ 3.7 (CH3-C≡C II), 18.3 (CH3-CH II), 63.2 (CH-CHCH3 II), 68.4 (CH-C≡C II), 72.4 (CH-Ph II), 75.1 (C≡C-CH3 II), 79.8 (C≡C-CH3 II), 82.3 (CH-CH3 II), 126.0, 126.0, 128.5, 128.7 (Ar CH 1 & II), 136.8 (Ar C II), 170.0 (C=N II)];GCMS M+ 241.10; HRMS for C15H15NO2 [M+H] calculated 242.1181, found 242.1181.
5. Conclusions
This work examined the chemoselectivity and diastereoslectivity of alkenynyl-nitroethers in Intramolecular Silyl Nitronate Cycloadditions (ISNC). Two different systems were tested in the alkenyl-nitroethers: terminal and non-terminal alkynes. The non-terminal alkyne nitroether was expected to be more chemoselective since reactions of alkenyl nitroethers are much faster than reactions of alkenyl nitroethers under INSC conditions, as shown in previous works by the authors. It was found that the non-terminal system was shown to be chemospecific in that only the C=C bond reacted. The terminal system at worst shows excellent chemoselectivity (97%). While crude 1H NMR did indicate that some aldehydes were present with very small peaks between 9-10.1 ppm, it did not show any evidence of the vinylic dihydrofuran hydrogens. Thus, the authors conclude that the two systems were chemospecific for the reaction of the double bonds. Spartan semi-empirical (PM6) and DFT (wB97X-D 6-31G*) analyses indicated that the chemoselective reactions could be controlled under kinetic and/or thermodynamic conditions. It was found that these cycloadditions demonstrated chemospecific interactions with the double over the triple bonds of the alkenylalkynyl-nitroethers. This indicates that the kinetic products were formed under the room temperature experimental conditions. The authors assumed that the major diastereomers for the two systems would have (±)-(4S,6R)-configurations. Instead, the results indicate that the major diastereomers for the two systems instead have (±)-(4R,6R)-configurations. Therefore, the alkenynyl-nitroethers formation reactions must choose the cis orientation for the methines alpha to the oxygen atoms over the trans for the major diastereomer. This is hard to prove since the nitroethers (oils) will readily undergo retro-Michael Additions and the non-cyclic structures are not favorable for NOE analysis. However, since epimerization is extremely unlikely with the anionic intermediates, the stereochemistry of the C4 and
C6 carbons of the rings systems must be set by the nitroethers. Analyses of the possible transition states for the four possible diastereomers that could be formed by each nitroether diastereomer agrees with the stereochemical outcomes found by NOE analysis of the cycloaddition adducts. This leads the authors to conclude that reaction of each diastereomer was diastereospecific, in that only one of the four diastereomers were formed. Thus, the ISNC reactions of these alkenynyl-nitroethers was found to be highly chemoselective and diastereoselective. Since the stereochemical outcomes were unexpected, the authors intend to further investigate the outcomes of these reactions with a variety of electron donating and electron withdrawing groups present at C4 and C6 for the furoisoxazolines and furoisoxazolidines.
Supplementary Materials
The following supporting information can be downloaded at the websitr of this paper posted on Preprints.org. 1H NMR, 13C{1H} NMR information are provided for compounds 6a, 6b, 10a, & 10b. 14a, 14b COSY, DEPT and HMQC information are provides for compounds 6a, 6b, 10a, & 10b. NOE information is provided for 10a,b. Also a crude 1H NMR is provided for 10a. PM6 computational methods and information are provided for cis/trans compounds: 6ab, 7ab, 7ab*, 8ab*, 9ab, 11ab, 19ab, 20ab; and for cis studies of 10a, I, II, III, IV. DFT computational methods and information are provided for cis: 7a, 9a, 11a, 19a, 20a.
Author Contributions
Synthesis and characterization of the terminal alkyne reactions: Katelyn Stevens, Katie Hassebroek, Matthew Grandbois, Synthesis and characterization of the methyl alkyne reactions: Shik Ki Li, Annika Schull, Katie Hassebroek, Joseph Stevens. Computational chemistry: Emily Kaufman, Katie Hassebroek, Matthew Grandbois. Supervision of computational chemistry: Dr. Arlen Viste. Conceptualization, synthesis and characterization supervision, writing, editing, checking computational work, funds acquisition: Dr. Jetty Duffy-Matzner.
Funding
Research reported in this publication was supported in part by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health [P20GM103443]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The Augustana Research and Artists Fund at Augustana University also supported this work.
Data Availability Statement
All data provided in supplementary materials.
Acknowledgments
The principal author would like to thank Dr. Mark Kurth for his continuing friendship and guidance. The authors would also like to thank Dr. Charlie Weiss for editing draft documents; and Cameron Jensen and Brandon Gustafson for their assistance in the preparation of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Young, D.G.J.; Gomez-Bengoa, E.; Hoveyda, A.H. Diastereoselective Intramolecular Cycloaddition of Vinylsilanes and Silyl Nitronates. Effective Control of Remote Acyclic Asymmetry. J. Org. Chem. 1999, 64, 692–693. [CrossRef]
- Marrugo, H.; Dogbéavou, R.; Breau, L. Diastereoselective synthesis of 2-isoxazolines via silaketal tethered 1,3-dipolar cycloadditions. Tetrahedron Lett. 1999, 40, 8979–8983. [CrossRef]
- Hassner, A.; Friedman, O.; Dehaen, W. Cycloadditions, 55. – Substituent Effects in Tandem Intramolecular Silyl Nitronate Olefin Cycloadditions (ISOC) Leading to Functionalized Tetrahydrofurans. Eur. J. Org. Chem. 1997, 1997, 587–594. [CrossRef]
- Galley, G.; Jones, P.G.; Pätzel, M. Enantiomerically pure isoxazolines by stereoselective 1,3-dipolar cycloaddition of silyl nitronates. Tetrahedron: Asymmetry 1996, 7, 2073–2082. [CrossRef]
- Kim, B.H.; Lee, J.Y.; Kim, K.; Whang, D. Asymmetric induction in silyl nitronate cycloadditions to Oppolzer's chiral sultam derivatives. Tetrahedron: Asymmetry 1991, 2, 27–30. [CrossRef]
- Torssell, K.; Zeuthen, O. ChemInform Abstract: REACTIONS OF TERT-BUTYL NITRONES AND TRIMETHYLSILYL NITRONATES. SYNTHESIS AND REACTIONS OF ISOXAZOLIDINES AND 2-ISOXAZOLINES. Chem. Informationsdienst 1978, 9. [CrossRef]
- Gottlieb, L.; Hassner, A. Cycloadditions. 53. Stereoselective Synthesis of Functionalized Pyrrolidines via Intramolecular 1,3-Dipolar Silyl Nitronate Cycloaddition. J. Org. Chem. 1995, 60, 3759–3763. [CrossRef]
- Dehaen, W.; Hassner, A. Stereoselectivity in intramolecular 1,3-dipolar cycloadditions. Nitrile oxides versus silyl nitronates. Tetrahedron Lett. 1990, 31, 743–746. [CrossRef]
- Kumar, A.; Fernandes, J.; Kumar, P. Synthesis and Biological Evaluation of Some Novel Isoxazoline Derivatives of Carbostyril. World J. Pharm. Pharmacuetical Sci. 2014, 3 (2), 1267–1277.
- Kaur, K.; Kumar, V.; Sharma, A.K.; Gupta, G.K. Isoxazoline containing natural products as anticancer agents: A review. Eur. J. Med. Chem. 2014, 77, 121–133. [CrossRef]
- Zhou, X.; Hohman, A.E.; Hsu, W.H. Current review of isoxazoline ectoparasiticides used in veterinary medicine. J. Veter- Pharmacol. Ther. 2021, 45, 1–15. [CrossRef]
- Indorkar, D.; Chourasia, O. P.; Limaye, S. N. Synthesis, Characterization, Antimicrobial, Antifungal Activity of Some s-Triazine Derivatives of Isoxazoline, Pyrazoline and PC Model Computational Studies. Res. J. Pharm. Sci. 2012, 1 (4), 10–16.
- Duffy, J.L.; Kurth, M.J. A Novel Intramolecular Silyl Nitronate Cycloaddition Route to Dihydrofuraldehydes and Dihydropyranaldehydes. J. Org. Chem. 1994, 59, 3783–3785. [CrossRef]
- Grandbois, M.L.; Betsch, K.J.; Buchanan, W.D.; Duffy-Matzner, J.L. Synthesis of novel 2H,5H-dihydrofuran-3-yl ketones via ISNC reactions. Tetrahedron Lett. 2009, 50, 6446–6449. [CrossRef]
- Namboothiri, I.N.N.; Hassner, A.; Gottlieb, H.E. A Highly Stereoselective One-Pot Tandem Consecutive 1,4-Addition−Intramolecular 1,3-Dipolar Cycloaddition Strategy for the Construction of Functionalized Five- and Six-Membered Carbocycles,1. J. Org. Chem. 1997, 62, 485–492. [CrossRef]
- Cheng, Y.; Wang, J.; Hu, Z.; Zhong, S.; Huang, N.; Zhao, Y.; Tao, Y.; Liang, Y. Preparation of norfloxacin-grafted chitosan antimicrobial sponge and its application in wound repair. Int. J. Biol. Macromol. 2022, 210, 243–251. [CrossRef]
- Duffy, J.L.; Kurth, J.A.; Kurth, M.J. Lithium, potassium and sodium alkoxides: Donors in the Michael addition reaction of α-nitroolefins.. Tetrahedron Lett. 1993, 34, 1259–1260. [CrossRef]
- Kim, H.R.; Kim, H.J.; Duffy, J.L.; Olmstead, M.M.; Ruhlandt-Senge, K.; Kurth, M.J. Double diastereoselectivity in the intramolecular nitrile oxide-olefin cycloaddition (INOC) reaction.. Tetrahedron Lett. 1991, 32, 4259–4262. [CrossRef]
- Duffy, Jetty L. An Investigation into the Diastereoselectivity of the Intramolecular Nitrile Oxide Olefin Cycloaddition vs. the Intramolecular Silyl Nitronate Olefin Cycloaddition. Ph.D. Thesis, University of California, Davis, Davis, CA, 1993.
- Kim, H.R.; Kim, K.M.; Kim, J.N.; Ryu, E.K. Regioselectivity and Stereoselectivity in the Intramolecular Nitrile Oxide-Olefin Cycloaddition (INOC) Reaction and the Intramolecular Silyl Nitronate-Olefin Cycloaddition (ISOC) Reaction. Synth. Commun. 1994, 24, 1107–1116. [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2012, 19, 1–32. [CrossRef]
Figure 1.
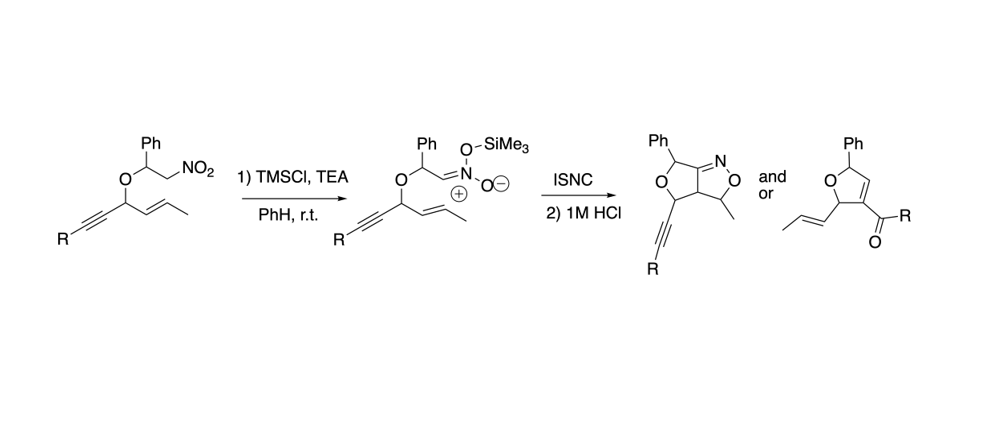
The numbers for carbons in the dihydrofuroisoxazole system following IUPAC rules are displayed. This ring can also be commonly described as dihydrofuro-2-isoxazoline.
Figure 1.
The numbers for carbons in the dihydrofuroisoxazole system following IUPAC rules are displayed. This ring can also be commonly described as dihydrofuro-2-isoxazoline.
Scheme 1.
INOC reactions yield the furoisoxazoles/furoisoxazolines with alkenyl- or alkynyl-nitroethers. ISOC reactions will yield 3-dihydrofuro-carbonyls with the alkynyl-nitroethers and isoxazolines when reacting with alkenyl-nitroethers.
Scheme 1.
INOC reactions yield the furoisoxazoles/furoisoxazolines with alkenyl- or alkynyl-nitroethers. ISOC reactions will yield 3-dihydrofuro-carbonyls with the alkynyl-nitroethers and isoxazolines when reacting with alkenyl-nitroethers.
Scheme 2.
Chemoselectivity options for proposed alkenynl-nitroethers.
Scheme 3.
Overall synthetic scheme for the production of the nitroethers and ISNC adducts.
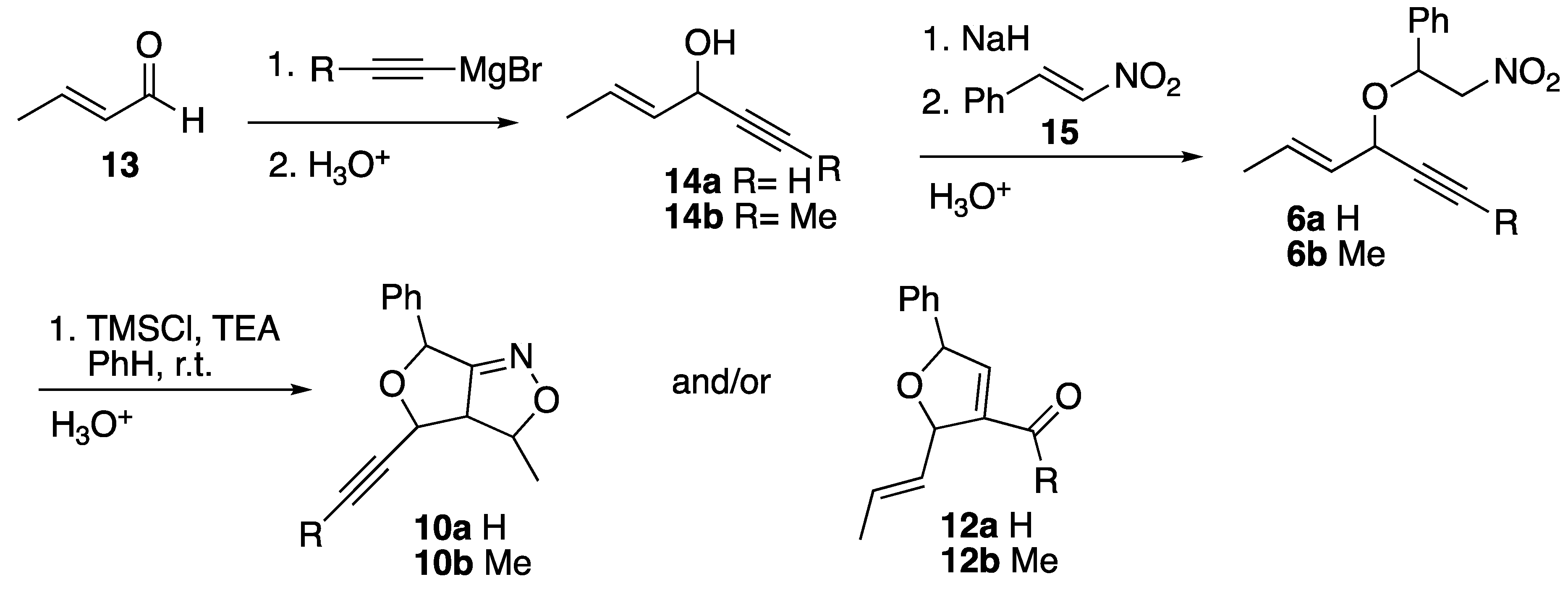
Figure 2.
Past evidence of major/minor diastereoisomers’ assignments in ISOC/INOC reactions.
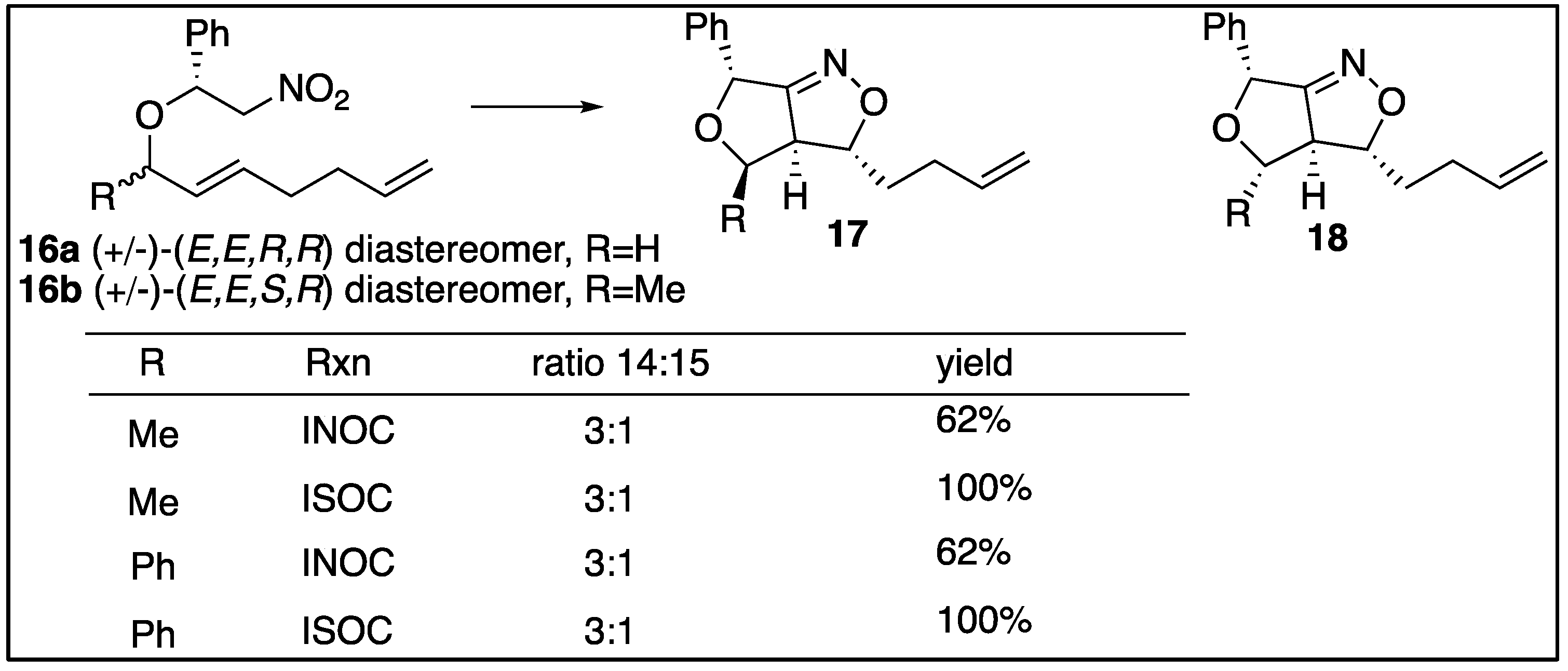
Figure 3.
New stereocenters due to ring closure.
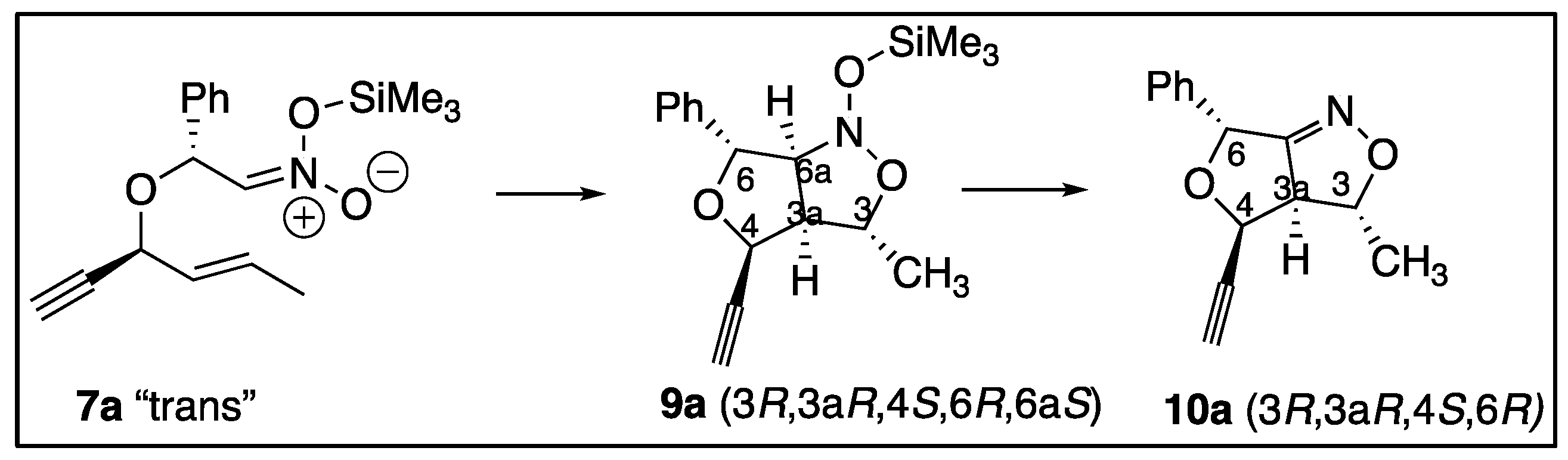
Figure 4.
Comparison of the HF/DFT vs PM6 calculations for the terminal alkyne system.
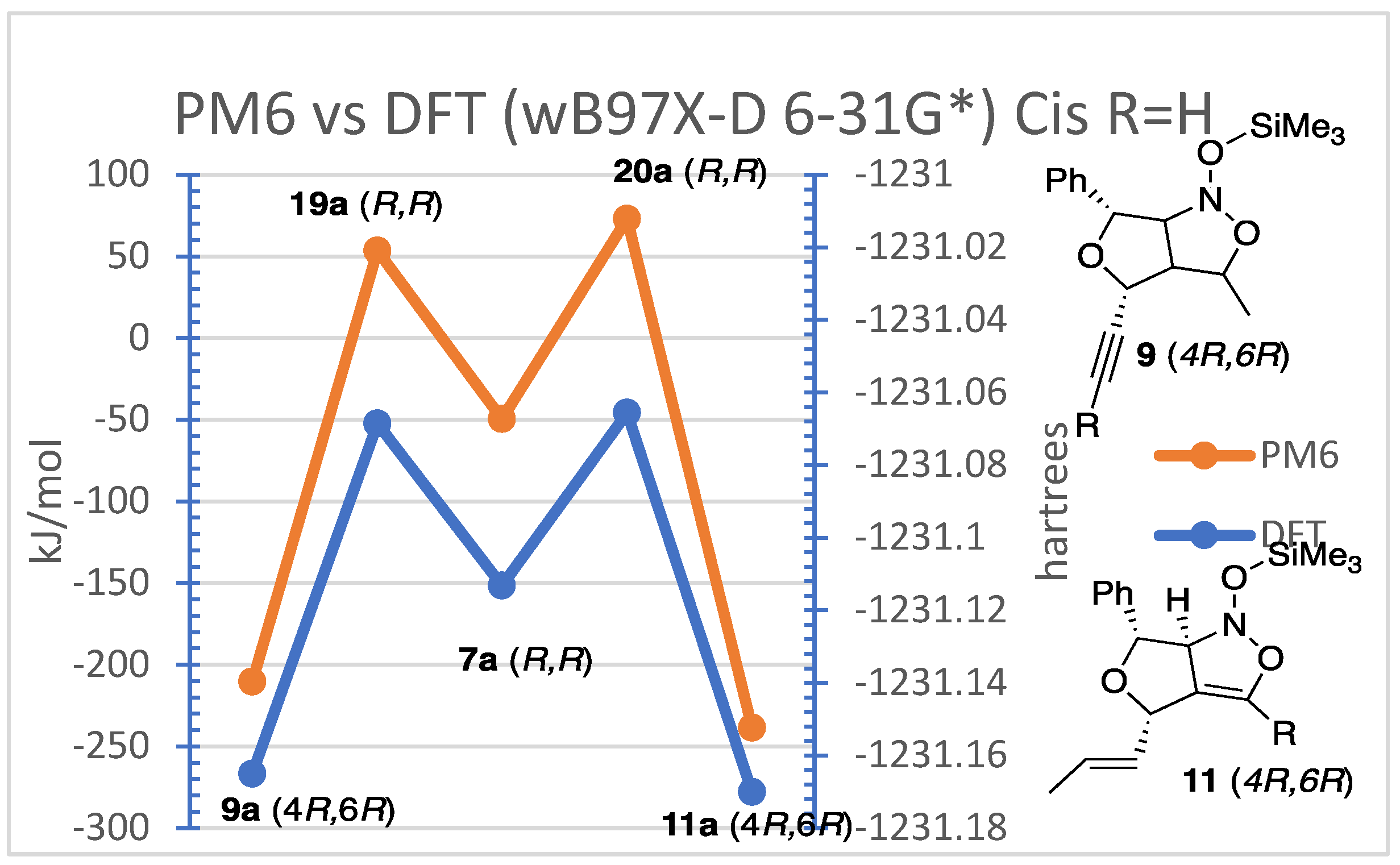
Figure 5.
Semi-empirical calculations (PM6) for the trimethylsilyl intermediates.
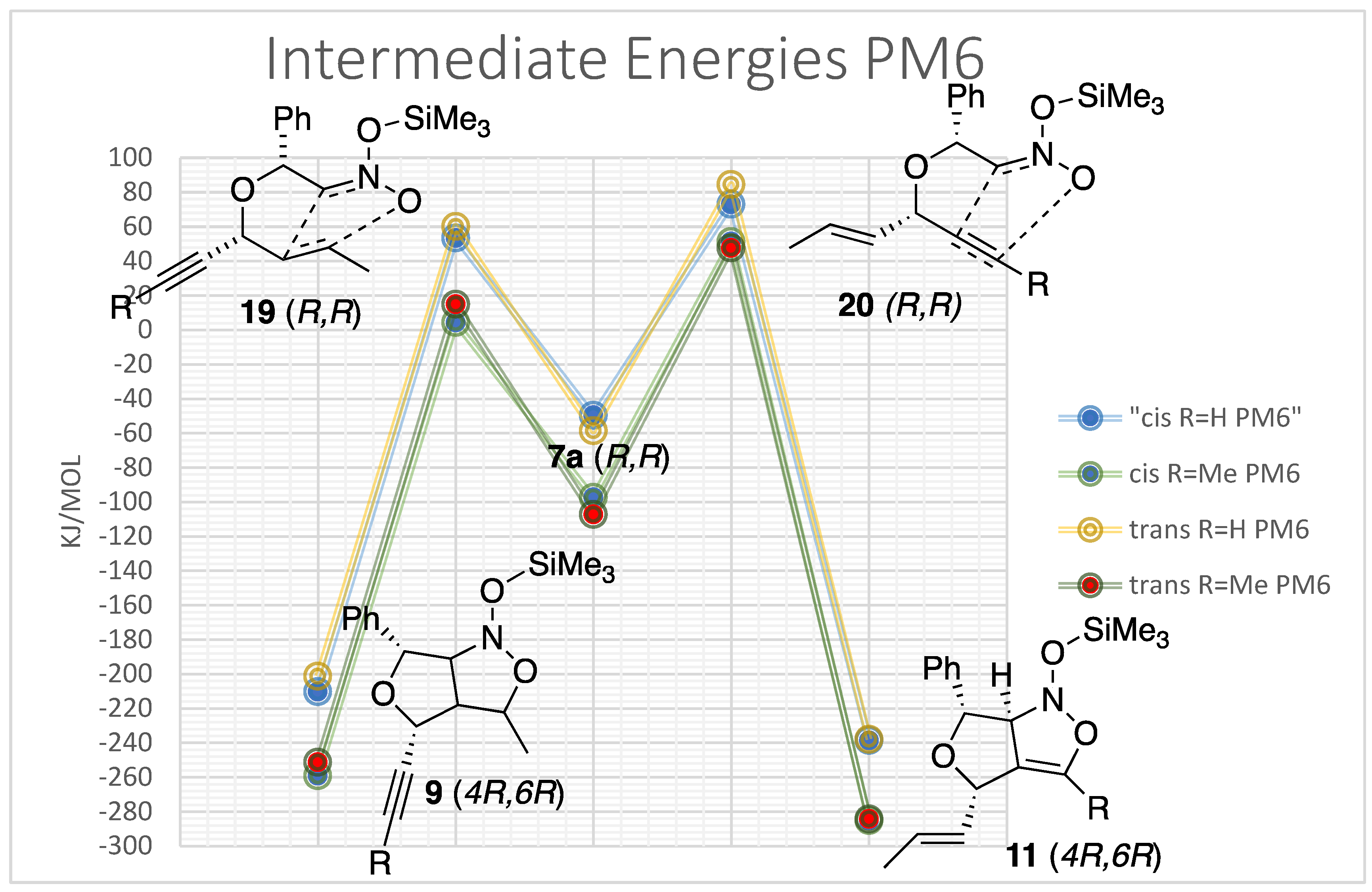

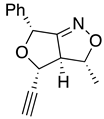
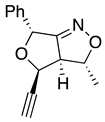
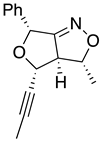
Table 1.
stereochemical outcomes for the major/minor diastereomers of the ISNC reactions.
| 10a major | 10a minor | 10b major | 10b minor | |
 |
 |
 |
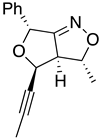 |
|
| (±)-(3R,3aR,4R,6R) | (±)-(3R,3aR,4S,6R) | (±)-(3R,3aR,4R,6R) | (±)-(3R,3aR,4S,6R) |
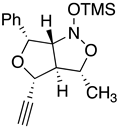
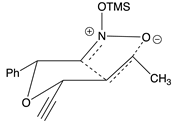
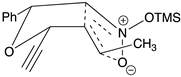
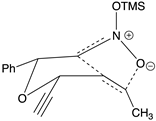
Table 2.
Images to explain the formation of four possible diastereomers of the silyl nitronate from the “cis”-nitroether (7a) 3rd Row lists enthalpies of formation for the cyclic intermediates based on PM6 calculations.
Table 2.
Images to explain the formation of four possible diastereomers of the silyl nitronate from the “cis”-nitroether (7a) 3rd Row lists enthalpies of formation for the cyclic intermediates based on PM6 calculations.
| I 3R 3aR 4R 6R 6aS | II 3S 3aS4R 6R 6aR | III 3S 3aS 4R 6R 6aS | IV 3R 3aR 4R 6R6aR |
| Intermediates | |||
| ∆Hf˙= -210.26 kJ/mol | -189.11 kJ/mol | -132.63 kJ/mol | -117.22 kJ/mol |
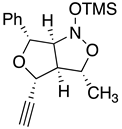 |
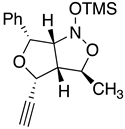 |
 |
 |
| Transition states | |||
| ‡alkene up/imino up | ‡alkene down/imino down | ‡alkene down/imino up | ‡alkene up/imino down |
 |
 |
 |
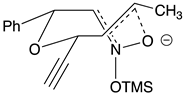 |
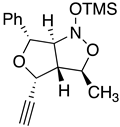
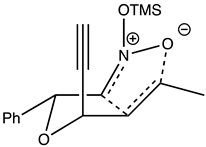
Table 3.
Justification for the major furoisoxazoline diastereomers of the “trans”-nitroether. (±)-(3S,4E)-3-[(1R)-1-phenyl-2-O-trimethylsilylnitro)-2-ethoxy]hex-4-en-1-yne, 7a.
Table 3.
Justification for the major furoisoxazoline diastereomers of the “trans”-nitroether. (±)-(3S,4E)-3-[(1R)-1-phenyl-2-O-trimethylsilylnitro)-2-ethoxy]hex-4-en-1-yne, 7a.
| Rxn scheme | Transition state |
 |
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



