
Submitted:
23 October 2024
Posted:
24 October 2024
You are already at the latest version
Abstract
The distortions and instability of high-symmetry configurations of polyatomic systems in nondegenerate states are usually ascribed to the pseudo-Jahn-Teller effect (PJTE). The geometries of hypericin, isohypericin, and fringelite D were optimized within various symmetry groups. Group-theoretical treatment and (TD-)DFT calculations were used to identify the corresponding electronic states during the symmetry descent. The symmetry descent paths (up to the stable structures without any imaginary vibrations) were determined using the corresponding imaginary vibrations as their kernel subgroups starting from the highest possible symmetry group. The vibronic interaction between the ground and excited electronic states relates to an increasing energy difference of both states during the symmetry decrease. This criterion was used to identify possible PJTE. We have shown that the PJTE in these naturally occurring compounds explains only the symmetry descent paths C2v → C2 and C2v → Cs in hypericin, and the D2h → C2v, D2h → C2v → Cs, and D2h → C2h ones in fringelite D. The electric dipole moments of hypericin and its analogs are determined prevailingly by the mutual orientations of the hydroxyl groups. The same holds for the energies of frontier orbitals in these systems, but their changes during the symmetry descent are less significant.
Keywords:
M06-2X hybrid functional
; natural compounds
; molecular symmetry
; non-degenerate electronic states
; excitation energy
1. Introduction
Straintronics represents a progressive field of modern condensed matter physics which investigates how physical effects in solids related to mechanical strain under the influence of external controlling fields in a layer of two-dimensional materials can change their electric, magnetic, optical, and other physical properties [1]. The aim of this investigation is to create prerequisites for the implementation of the new construction of sensors and electronics applicable in information technologies. Historically, the first materials, where these effects were studied, were of inorganic origin. These were mainly crystalline forms of germanium and silicon. Significant advances in this direction have been associated with two-dimensional materials such as graphene, hexagonal boron nitride, ZnO, ultrathin films of transition metal chalcogenides, semiconductor monolayers, and planar heterostructures on them [2]. The development of molecular electronics and instrumentation now allows for operation at distances less than 100 nanometers [3]. One of the biggest technical problems with working on single molecules is establishing reproducible electrical contact with only one molecule. Despite these difficulties, it is plausible to assume that spintronics will start to develop to the molecular level in the future. At this level, physical properties can be controlled by van der Waals interactions, acid-base processes, charge-transfer processes, or a combination of these. The use of this research can be expected, for example, in the design and control of molecular motors or robots [4]. The identification and investigation of organic molecules with a chemical structure suitable for potential applications in straintronics represents a significant challenge for theoretical chemistry. The desired structural motif may be based not only on synthetic molecules, but compounds of natural origin are also proving to be potentially interesting. The hypericin molecule, its derivatives and corresponding tautomers turn out to be very interesting in this respect. This natural molecule is a derivative of anthraquinone and has antibacterial and antiviral effects [5]. It is a highly powerful biologically active compound. The existence of a large chromophore system in the molecule implies its photosensitivity. Hypericin was previously under research as an agent in photodynamic therapy.
According to single-crystal X-ray measurements the structure of hypericin 1,3,4,6,8,13-hexahydroxy-10,11-dimethylphenanthro[1,10,9,8-opqra]perylene-7,14-dione (I) [6] (see Figure 1) is non-planar. Ab initio Møller-Plesset method up to the second order perturbation theory (MP2) [7] and numerous density functional theory (DFT) studies [8,9,10,11,12,13,14,15,16,17,18,19,20,21] found stable ’propeller’ and ‘double butterfly’ conformations, the latter slightly lower in energy, which is consistent with X-ray structure data [6]. The nonplanar conformations are explained by steric repulsion between the hydroxyl groups in 3, 4 positions and the methyl groups in 10, 11 positions. The interconversion between them may occur at room temperature; the theoretically estimated barrier height is 6.7 kcal/mol [12]. Concerning possible hydrogen bonds, the most stable structure among 10 tautomers has quinone oxygens at 7 and 14 positions. Many of these studies are focused on the hypericin spectral properties [10,11,12,15,17,18] with the aim of explaining its photodynamic activity.
To our best knowledge, no theoretical study dealing with the structure of isohypericin 1,3,4,6,8,10,13-hexahydroxy-4,11-dimethylphenanthro[1,10,9,8-opqra]perylene-7,14-dione (II) (see Figure 1) is available in literature.
Their fully hydroxylated symmetric analogue fringelite D, 1,3,4,6,8,10,11,13-octahydroxyphenanthro[1,10,9,8-opqra]perylene-7,14-dione (III), (see Figure 1) has been investigated only in a single DFT study [12].
Although the distance between oxygen atoms O3-O4 as a measure of their mutual repulsion is evidently comparable with the remaining O-O distances in hypericins, their non-planarity is ascribed to the steric repulsion of hydroxyl groups in 3 and 4 positions. An alternative explanation of this symmetry descent might be based on the pseudo-Jahn-Teller (PJT) effect (see later). According to Bersuker [22,23], ‘the pseudo-Jahn−Teller effect is the only source of instability and distortions of high-symmetry configurations of polyatomic systems in non-degenerate states’. The aim of our recent study is to verify this hypothesis in the case of hypericin, isohypericin, and fringelite D. We will use a group-theoretical treatment and quantum-chemical calculations at the DFT level for this purpose. Our methodology is of more general character to distinguish which distortions can be explained by the PJT effect and which not. Because these two-dimensional molecules represent suitable model compounds with potential future applications in spintronics, it is of interest to understand the extent to which the PJT effect or steric repulsion modulate molecular properties.
2. Theoretical Background
A potential energy surface (PES) of a molecule, an ion, a crystal, etc. describes its energy as a function of the coordinates of its atoms [24]. Geometry optimization is the process of finding PES stationary points (their gradients, i.e. the first partial derivatives of the energy with respect to all geometry parameters Qi are equal to zero), such as its minima or saddle points. PES minima represent stable or quasistable species with energies E lower than those of its surrounding species (local minimum) or the lowest on the whole PES (absolute minimum). An n-th order saddle point has n negative eigenvalues of the Hessian matrix (∂2E/∂Qi∂Qj). Saddle points represent PES maxima along the directions of the reaction coordinates, and minima along all other directions. This means that saddle points represent transition states along the reaction coordinates.
A symmetry operation will be conserved during a nuclear displacement if and only if it leaves the displacement coordinate invariant [25]. A nondegenerate distortion coordinate Qi described by a non-totally symmetric representation Γi within the symmetry group G0 causes the symmetry decrease from G0 to its kernel subgroup K(G0, Γi) which consists of all symmetry elements with characters equal to +1. A degenerate representation describes a set of distortion coordinates and consists of several components that span a distortion space. An epikernel subgroup E(G0, Γi) is conserved only in a part of the distortion space. Thus, epikernels are intermediate subgroups between the parent group G0 and the kernel subgroup, which is conserved in all distorted structures.
According to the Jahn-Teller (JT) theorem [26], any nonlinear configuration of atomic nuclei in a degenerate electronic state is unstable. Consequently, at least one stable nuclear configuration of lower symmetry must be obtained during a symmetry decrease where the degenerate electronic state is split. The PES of such systems can be described analytically using a perturbation theory treatment and its stationary points can be obtained [27]. The energy difference between the high-symmetry unstable and low-symmetry stable structures of the same compound is denoted as the Jahn-Teller stabilization energy EJT.
The classical JT effect is restricted to high-symmetry structures with degenerate electronic states, but a similar instability, known as the pseudo-Jahn-Teller (PJT) effect [23], may be observed in the case of sufficiently strong vibronic coupling between pseudodegenerate electronic states (usually ground and excited). JT active coordinate Q gives non-vanishing values of integrals between interacting electronic states Ψ1 and Ψ2, where is Hamiltonian of the system under study. The PES of two interacting electronic states Ψ1 and Ψ2 of different space symmetries can be described in the simplest case using perturbation theory as
The method of the epikernel principle [28] was originally elaborated to predict symmetry groups of stable JT structures and has been extended to PJT systems as well [29]. It is based on the representation ΓJT of the JT active coordinate Q that gives non-vanishing values of integrals between interacting electronic states Ψ1 and Ψ2 described by representations Γ1 and Γ2, respectively. For the totally symmetric Hamiltonian it implies that ΓJT is contained in the direct product of representations of both interacting electronic states
ΓJT є Γ1 ⊗ Γ2 (3)
Analogously for the classical JT effect with a degenerate electronic state Ψ1 = Ψ2 and Γ1 = Γ2 = Γ an even more strict condition is valid and the ΓJT must be contained in its symmetric direct product.
ΓJT є [Γ ⊗ Γ]+
Classical JT vibronic interactions can be identified according to a single criterion – the degenerate electronic state. On the other hand, the identification of PJT vibronic interactions is a more complicated task that consists of several steps. For simplicity, we will restrict our analysis to interactions between the ground state Ψ1 and the excited state(s) Ψ2. At first, the geometry of the studied system is optimized within its highest symmetry group, the symmetries of imaginary vibrations are identified by vibrational analysis, and subsequently the symmetries and energies of its excited states are evaluated. For known symmetries of an imaginary vibration (tentatively identified with a JT active coordinate) and of the ground state, we can determine the excited-state symmetry according to Eq. (3). In the next step(s) we repeat the geometry optimization (the energy of the system in the parent group must be higher than in its subgroups), determine the imaginary vibration symmetries as well as energies and symmetries of excited states of kernel and epikernel subgroups of the parent high-symmetric group. Subsequently, we identify the corresponding excited states in the parent group and its subgroups according to the group – subgroup relations (see Tables S1-S3 Supplementary Information) [30], similar oscillator strengths, and similar molecular orbital compositions. This step might be problematic because – based on the perturbation principle - a high similarity between the corresponding excited states is supposed. The PJT vibronic interaction causes the excitation energy of the corresponding excited state in the PJT subgroup to be higher than in the parent group (compare Eq. (1)). Otherwise, the investigated symmetry descent cannot be a consequence of the PJT vibronic interaction, and other explanations must be searched for.
3. Results and Discussion
All the molecules under study are neutral closed-shell systems in the ground singlet spin state. Thus, their ground spin states are described by total symmetric representations independent of the symmetry group. It implies that the representation of the PJT interacting excited state and of the JT active coordinate are equal (see Eq. (3)).
We started our study with geometries of the highest possible symmetry with quinone oxygens at 7 and 14 positions (see Figure 1). According to [17], this tautomer corresponds to the most stable structure of hypericin.
Although we performed time dependent (TD)-DFT calculations for 50 excited states, only the lowest ten excited states are presented due to space reasons. Our analysis is restricted to the lowest excited state of every symmetry, as the vibronic interaction with this state is supposed to be stronger than with the higher ones. The notation nmΓ(G) is used for the n-th state of Γ representation with spin multiplicity m within the G symmetry group.
3.1. Hypericin (I)
The highest possible symmetry group of hypericin (I) is C2v. Here both its hydroxyl groups at positions 3 and 4 are either in anti- or syn- mutual orientations and denoted as models Ia and Ib, respectively (Figure 2 and Figure 3). The third possible mutual orientation (one in syn- and the other in mutual anti-orientations) is denoted as Ic with the highest possible symmetry group Cs (Figure 4). The non-symmetric structure (C1) cannot be found for Ia and Ib model systems, but it corresponds to the Ic model system only. This assignment is implied by the highest gradients at the H atoms of the hydroxyl groups at the 3 and 4 positions of unstable Ia and Ib structures in the Cs group.
Table 1 illustrates the energy decrease of hypericin model systems Ia, Ib, and Ic with their decreasing symmetry as implied by imaginary vibrations (corresponding kernel subgroups). The increasing distances between hydroxyl O atoms in 3 and 4 positions dOO illustrate their decreasing mutual repulsion with symmetry decrease, which coincide with possible JT stabilization energies. The most stable Ic model systems (especially the C1 ‘propeller’) are stabilized by an additional hydrogen bond (compare Figure 4). The mutual orientations of hydroxyl groups in 3 and 4 positions affect the electric dipole moments and the frontier orbital (HOMO = the Highest Occupied Molecular Orbital, LUMO = the Lowest Unoccupied Molecular Orbital) energies (Table S4 in Supplementary Information). The Cs symmetry structures have the highest dipole moments. The orbital energies decrease in the order Ia > Ic > Ib, and their dependence on symmetry is less significant.
Individual symmetry descent paths are summarized in Table 2. The possible PJT origin of the observed symmetry descent paths can be checked with the help of Table S5 in Supplementary Information as follows:
The symmetry descent no. 1 can be ascribed to the vibronic interaction of X1A1(C2v) ground state with the 11A2(C2v) excited state which corresponds to the 41A(C2) excited state with increased excitation energy in both the Ia and Ib systems.
The two-step symmetry decrease no. 2 meets the problem with its excited state identification. Nevertheless, using an elimination method we can conclude that the HOMO-7 → LUMO electron transition generating the 11B1(C2v) excited state corresponds to the HOMO-8 → LUMO electron transition generating the 41A’(Cs) excited state with the higher excitation energy and so the PJT effect is possible. The structure obtained in this way is unstable, but the supposed vibronic interaction of the X1A’(Cs) ground state with the 11A”(Cs) leads to 21A(C1-‘propeller’) or 31A(C1-‘double butterfly’) excited state with lower excitation energies. Therefore, this second step cannot be explained by the PJT effect.
In the first step of the symmetry descent no. 3 the X1A1(C2v) ground state interacts with the 11B2(C2v) excited state which corresponds to the 11A”(Cs) excited state with higher excitation energy and the PJT vibronic interaction is possible. However, this structure is unstable and this state, corresponds to 21A(C1) state which has lower excitation energy in both ‘propeller’ and ‘double butterfly’ structures. Thus, this step cannot be explained by the PJT effect.
Within the symmetry descent no. 4 the X1A’(Cs) ground state could interact with 11A”(Cs) excited state. It is problematic to find its corresponding excited state in the C1 kernel subgroup, but it is surely lower than the 81A(C1) state and therefore its excitation energy is lower. Consequently, this symmetry descent cannot be explained by the PJT effect.
3.2. Isohypericin (II)
The highest possible symmetry group of isohypericin (II) is C2h if both of its hydroxyl groups at 3 and 10 positions are related to methyl groups at 4 and 11 positions either in anti- or syn- orientations and are denoted as models IIa and IIb, respectively (Figure 5 and Figure 6). The third possible orientation (one in syn- and the other in anti-orientations to methyl groups) is denoted as IIc with the highest possible symmetry group Cs (Figure 7).
Table 3 illustrates the energy decrease of isohypericin model systems IIa, IIb, and IIc with their decreasing symmetry as implied by imaginary vibrations (corresponding kernel subgroups). The increasing distances between hydroxyl O atoms in the 3 (11) and methyl C atoms in the 4 (10) positions dOC illustrate their decreasing mutual repulsion with symmetry decrease, which coincide with possible JT stabilization energies. The most stable are ‘propeller’ structures, especially in IIb model systems.
The centrosymmetric C2h and Ci structures have no dipole moments (Table S6 in Supplementary Information). The mutual orientation of hydroxyl groups in 3 and 10 positions affects the dipole moments (the highest dipole has the IIc structure of the symmetry Cs) and the frontier orbital energies (HOMO, LUMO) (Table S6 in Supplementary Information). The orbital energies decrease in the order IIa > IIc > IIb, and their dependence on symmetry is less significant.
The possible symmetry descent paths of isohypericin are presented in Table 3 and Table 4. The possible PJT origin of the observed symmetry descent paths can be checked with the help of Table S7 in Supplementary Information as follows:
The symmetry descent path no. 5 might be explained by vibronic interaction of the ground electronic state X1Ag(C2h) with the excited state 11Bg(C2h) (the HOMO-8 → LUMO electron transition in IIa and the HOMO-6 → LUMO one in IIb) which corresponds to the 41Ag(Ci) excited state with lower excitation energy. This excludes the PJT effect as the reason for this symmetry descent. Using an elimination method for IIb we can show that the HOMO-6 → LUMO electron transition (11Bg(C2h) excited electronic state) corresponds to the HOMO-8 → LUMO electron transition (the 41Ag(Ci) excited state) with lower excitation energy and so this symmetry descent cannot be of PJT origin.
The symmetry descent path no. 6 for both IIa and IIb systems might originate in vibronic interaction of the ground state X1Ag(C2h) with the excited state 11Bu(C2h) which corresponds to the 41A(C2) excited state with lower excitation energy. Thus, the PJT origin of this symmetry descent is excluded.
The symmetry descent no. 7 (model system IIc) meets the problem of excited state identification. The most probable assignment of the 11A’(Cs) excited state generated by the HOMO-7 → LUMO electron transition is the 71A(C1) electronic state generated mainly by the HOMO-8 → LUMO electron transition in both ‘propeller’ and ‘double butterfly’ forms of the C1 kernel subgroup, which has lower excitation energy. Thus, the PJT origin of this symmetry descent can be rejected.
3.3. Fringelite D (III)
The highest possible symmetry group of fringelite D (III) is D2h if both its hydroxyl groups at 3 and 4 as well as in 10 and 11 positions are either in anti- or syn- mutual orientations. They are denoted as models IIIa and IIIb, respectively (Figure 8 and Figure 9). Another possible mutual orientations are denoted as IIIc (syn-orientations at 3 and 10 positions, anti-orientations at 4 and 11 positions) with the highest possible symmetry group C2h and IIId (syn-orientations at 3 and 11 positions, anti-orientations at 4 and 10 positions) with the highest possible symmetry group C2v (Figure 10 and Figure 11). The structures of Cs and C2 symmetry groups cannot be found for IIIa and IIIb model systems, and so they correspond to those of IIIc and IIId systems. This assignment is supported by the highest gradients at H atoms of hydroxyl groups at 3, 4, 10 and 11 positions of unstable IIIa and IIIb structures in the C2v group.
Table 5 illustrates the energy decrease of fringelite D model systems IIIa, IIIb, IIIc, and IIId with their decreasing symmetry as implied by imaginary vibrations (corresponding kernel subgroups). The increasing distances between hydroxyl O atoms in 3 and 4 (or 10 and 11) positions dOO illustrate their decreasing mutual repulsion with symmetry decrease which coincide with possible JT stabilization energies. The increased stability of IIIc and IIId systems is caused by additional hydrogen bonds of hydroxyl groups in 3 and 4 as well as in 10 and 11 positions. The most stable are ‘propeller’ structures, especially in IIIc model systems.
The D2h, D2, C2h and Ci structures have no dipole moments (Table S8 in Supplementary Information). The mutual orientations of hydroxyl groups in 3, 4, 10 and 11 positions affect the dipole moments (the highest dipole has the IIId structure of C2v symmetry) and the frontier orbital energies (Table S8 in Supplementary Information). The orbital energies decrease in the order IIIa > IIIc ~ IIId > IIIb and their dependence on symmetry is less significant.
Individual symmetry descent paths are summarized in Table 6. The possible PJT origin of the observed symmetry descent paths can be checked with the help of Table S9 in Supplementary Information as follows:
The lowest excited 11B2g(D2h) state (no. 37) of IIIa and IIIb compounds which is able of vibronic interaction with the X1Ag(D2h) ground electronic state within the descent path no. 8 is too high (the HOMO-22 → LUMO+8 electron transition is over 6.1 eV) to be PJT active. It is confirmed by the lower excitation energy of its 81Ag(C2h) counterpart for the IIIa structure. We have not found its counterpart for the IIIb one.
The symmetry descent path no. 8 can be explained by the vibronic interaction of the X1A1g(D2h) ground state with the 11Au(D2h) excited state in the IIIb system only (the electron transition HOMO-9 → LUMO) because its corresponding 21A(D2) state in the IIIb system has lower excitation energy. It implies that there is no PJT effect.
The symmetry descent path no. 10a consists of two steps. In IIIa, the X1Ag(D2h) ground state might at first interact with the 11B3u(D2h) excited state which corresponds to the 101A1(C2v) state with higher excitation energy (the HOMO-9 → LUMO+2 electron transition with the excitation energy over 6.5 eV). In the next step, the 11B2(C2v) excited state (the HOMO-2 → LUMO electron transition) corresponds to 11A’(Cs) state for IIIc with lower excitation energy, which means no PJT interaction. In IIIb, the excited state 11B3u(D2h) (the HOMO-1 → LUMO electron transition) corresponds to the 11A1(C2v) state with higher excitation energy which allows the PJT interaction. In the 2nd step, the 11B2(C2v) excited state (the HOMO → LUMO electron transition) corresponds to the 11A”(Cs) excited state with lower excitation energy, that excludes the PJT interaction.
The first step of the 10b symmetry descent path for the IIIa and IIIb systems is the same as in 10a. In the second step, the 11A2(C2v) excited state (the HOMO → LUMO + 1 electron transition) corresponds to the 31B(C2) one with higher excitation energies of both IIIc and IIId systems, allowing the PJT interaction.
The IIIb system undergoes the symmetry descent no. 11 but it meets the problem with the excited states identification. The most probable assignment of the 11B3g(D2h) excited state generated by the HOMO-7 → LUMO electron transition corresponds to the 31Bg(C2h) electronic state generated by the HOMO-8 → LUMO electron transition with higher excitation energy which allows the PJT interaction.
The symmetry descent paths 12a and 12b of IIIb consist of two steps. In the first step the 11Bu(D2h) excited state (the HOMO- 9 → LUMO+2 electron transition, excitation state no. 46 with more than 6.3 eV excitation energy) corresponds to the 101A1(C2v) state. This higher excitation energy allows the PJT interaction. In the second step of the 12a path, the 11B2(C2v) excited state (the HOMO → LUMO excitation) corresponds to the 11A”(Cs) state of the IIId system. This lower excitation energy excludes the PJT interaction. In the second step of the 12b path the 11A2(C2v) state of IIIb (the HOMO → LUMO+1 electron excitation) corresponds to 31B(C2) state of IIId or to 31A(C2) state of IIIc with higher excitation energies. Here the PJT effect is possible. The 31B(C2) state of IIId does not satisfy the group – subgroup relations (see Table S3 in Supplementary Information ).
In the symmetry descent path no. 13 the 11B1g(D2h) state of the IIIb system (the HOMO-4 → LUMO and HOMO → LUMO+1 electron transitions) cannot correspond to the 21Au(C2h) state because of incorrect group-subgroup relations. However, the 21B1g(D2h) state obtained by similar electron transitions corresponding to the 21Bg(C2h) state of the same IIIb system meets all conditions for PJT interactions.
The symmetry descent paths 14a and 14b consist of two steps. At first, the 11B3u(D2h) excited state (the HOMO-1 → LUMO electron transition) coincides with the 11B1(C2v) state with higher excitation energy in agreement with the PJT interactions. The path 14a continues with the 11B2(C2v) electronic state of IIIb (the HOMO →LUMO electron transition) to the 11A”(Cs) state in the IIId system with lower excitation energy that excludes the PJT interaction. The path 14b continues with the 11A2(C2v) excited state of the IIIb system (the HOMO →LUMO+1 electron transition) to the corresponding 31A(C2) state of the IIIc system with higher excitation energy in agreement with the PJT interaction. The 31B(C2) state of the IIId system does not fulfill the group-subgroup relations (see Table S3 in Supplementary Information).
In the symmetry descent path no. 15 the interaction of the X1Ag(C2h) ground state with the 11Bg(C2h) excited state of the IIIc system (the HOMO-8 → LUMO electron transition) would lead to its 31Ag(Ci) state with lower excitation energy, which excludes the PJT interaction.
The symmetry descent path no. 16 might be due to 11Au(C2h) excited state. This transition comes from HOMO-9→LUMO electron excitation. It corresponds to the 41A(C2) state of the IIIc system with lower excitation energy, which is in contradiction to the PJT effect.
The symmetry descent no. 17 cannot be ascribed to the PJT effect. It corresponds to the hypothetical interaction of the X1A1(C2v) ground state with the very high excited state 11B1(C2v) (state no. 37) corresponding to the 161A’(Cs) state of the IIId system with a lower excitation energy excludes the vibronic interaction.
The same holds for the symmetry descent no. 18 where the 11A2(C2v) state (the HOMO-7 →LUMO electron transition) corresponds to the 31A(C2) state of the IIId system with lower excitation energy and thus without PJT effect.
4. Method
The geometries of the neutral hypericin (I), isohypericin (II), and fringelite D (III) in the ground spin state were optimized within various symmetry groups using the M06-2X hybrid functional [31] and standard cc-pVDZ basis sets for all atoms taken from the Gaussian library [32]. The optimized geometries were checked for imaginary vibrations by vibrational analysis. Time-dependent DFT (TD-DFT) treatment [33,34] for up to 50 vertical electronic states was used to obtain excitation energies and excited states. Gaussian16 software (Revision B.01) [32] was used for all quantum-chemical calculations. The MOLDRAW software (https://www.moldraw.software.informer.com, accessed on 9 September 2019) [35] was used for geometry manipulation and visualization purposes.
5. Conclusions
The group-theoretical treatment and quantum-chemical calculations were used to identify the corresponding electronic states of natural compounds hypericin (I), isohypericin (II), and fringelite D (III) in the structures of various symmetry groups. The symmetry descent paths (up to the stable structures without any imaginary vibrations) were determined using the corresponding imaginary vibrations as their kernel subgroups starting from the highest possible symmetry group for the given orientations of the hydroxyl groups at 3, 4, 10 and/or 11 positions. Because the vibronic interaction between the ground and excited electronic states is connected with increasing excitation energy (the energy difference of both states) during the symmetry decrease (see above), this criterion was used for the identification of the possible PJT effect (this criterion is not mandatory if the symmetry descent is due to the mutual repulsion of hydroxyl and/or methyl groups). Our results indicate that the PJT effect explains only the symmetry descent paths C2v→C2 and C2v → Cs in hypericin, the D2h → C2v, D2h → C2v → Cs, and D2h → C2h ones in fringelite D. Moreover, this criterion might be satisfied accidentally in some cases. No symmetry descent path can be ascribed to the PJT effect in isohypericin, in the Ic model systems of hypericin and in IIIc and IIId model systems of fringelite D. This finding is in contradiction with the statement of Bersuker [22,23], that ‘the pseudo-Jahn−Teller effect is the only source of instability and distortions of high-symmetry configurations of polyatomic systems in non-degenerate states’.
Our results showed that the dipole moments of hypericin and its analogs are determined prevailingly by the mutual orientations of the hydroxyl groups. The same holds for the energies of frontier orbitals in these systems, and their changes during the symmetry descent are less significant, independently on the origin of the corresponding symmetry descent paths. These findings might be important for the future development of straintronics materials.
In this study we proposed the methodology to distinguish the structural consequences of the PJT effect from those caused by mutual repulsion of relevant functional groups. We plan its applications to less symmetric naturally occurring molecules that might be interesting from the point of view of the PJT effect. Further experimental and theoretical studies in this field are desirable.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Tables S1 – S3: Correlation tables for the symmetry descent of the D2h, C2h and C2v groups to their immediate subgroups; Tables S4, S6 and S8: Dipole moments, frontier orbital energies EHOMO, ELUMO and their difference in hypericin, isohypericin and fringelite D model systems of various symmetry groups; Tables S5, S7 and S9: Symmetry groups G, the corresponding ground state symmetries Γgr, the low excited state symmetries Γexc and excitation energies Eexc, the corresponding oscillator strengths f and electron transition descriptions of hypericin, isohypericin and fringelite D model systems.
Author Contributions
Investigation, D.Š., V.L. and M.B.; writing—original draft preparation, D.Š., V.L. and M.B.; writing—review and editing, V.L. and M.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been supported by the Slovak Research and Development Agency (no. APVV-20-0213), by the Scientific Grant Agency of the Ministry of Education, Science, Research, and Sport of the Slovak Republic VEGA (nos. 1/0461/21 and 1/0324/24), and by the Operational Program Integrated Infrastructure for the project: “ Support of research activities of Excellence laboratories STU in Bratislava”, Project no. 313021BXZ1, co-financed by the European Regional Development Fund.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or supplementary materials.
Acknowledgments
The authors thank the HPC center at the Slovak University of Technology in Bratislava, which is a part of the Slovak Infrastructure of High Performance Computing (SIVVP project ITMS 26230120002 funded by the European Region Development Funds) for the computational time and resources made available.
Conflicts of Interest
The authors declare no conflicts of interest.
Sample Availability
Not applicable.
References
- Pandey, M.; Pandey, Ch.; Ahuja, R.; Kumar, R. Straining techniques for strain engineering of 2D materials towards flexible straintronic applications. Nano Energy 2023, 109, 108278. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Atulasimha, J.; Barman, A. Magnetic straintronics: Manipulating the magnetization of magnetostrictive nanomagnets with strain for energy-efficient applications. Appl. Phys. Rev. 2021, 8, 041323. [Google Scholar] [CrossRef]
- Li, T.; Bandari, V. K.; Schmidt, O. G. Molecular Electronics: Creating and Bridging Molecular Junctions and Promoting Its Commercialization. Adv. Mater. 2023, 35, 2209088. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.; Saka, S. K.; Xuan, F.; Yin, P. Molecular robotic agents that survey molecular landscapes for information retrieval. Nature Commun. 2024, 15, 3293. [Google Scholar] [CrossRef]
- Murali, M.; Gowtham, H.G.; Shilpa, N.; Krishnappa, H.K.N.; Ledesma, A.E.; Jain, A.S.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I.; Achar, R.R.; Silina, E.; Stupin, V.; Ortega-Castro, J.; Frau, J.; Flores-Holguín, N.; Amruthesh, K. N.; Shivamallu, Ch.; Kollur, Sh. P.; Glossman-Mitnik, D. Exploration of Anti-HIV Phytocompounds against SARS-CoV-2 Main Protease: Structure-Based Screening, Molecular Simulation, ADME Analysis and Conceptual DFT Studies. Molecules 2022, 27, 8288. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.; Frolow, F.; Kapinus, E.; Lavie, D.; Lavie, G.; Merueloc, D.; Mazur, Y. Acidic Properties of Hypericin and its Octahydroxy Analogue in the Ground and Excited States. J. Chem. Soc. Chem. Commun. 1994, 891–892. [Google Scholar] [CrossRef]
- Petrich,J. W.; Gordon, M. S.; Cagle, M. Structure and Energetics of Ground-State Hypericin: Comparison of Experiment and Theory. J. Phys. Chem. A 1998, 102, 1647–1651. [Google Scholar] [CrossRef]
- Uličný, J.; Laaksonen, A. Hypericin, an intriguing internally heterogenous molecule, forms a covalent intramolecular hydrogen bond. Chem. Phys. Let. 2000, 319, 396–402. [Google Scholar] [CrossRef]
- Guedes, R. C.; Eriksson, L. A. Theoretical study of hypericin. J. Photochem. Photobiol. A 2005, 172, 293–299. [Google Scholar] [CrossRef]
- Shen, L.; Ji, H-F. ; Zhang, H-Y. Anion of hypericin is crucial to understanding the photosensitive features of the pigment. Bioorg. Med. Chem. Let. 2006, 16, 1414–1417. [Google Scholar] [CrossRef]
- Guedes, R. C.; Eriksson, L. A. Photophysics, photochemistry, and reactivity: Molecular aspects of perylenequinone reactions. Photochem. Photobiol. Sci. 2007, 6, 1089–1096. [Google Scholar] [CrossRef]
- Shoaf, A. L.; Bayse, C. A. TD-DFT and structural investigation of natural photosensitive phenanthroperylene quinone derivatives. New J. Chem. 2016, 40, 413–422. [Google Scholar] [CrossRef]
- Szymanski, S.; Majerz, I. Aromaticity and Electron Density of Hypericin. J. Nat. Prod. 2019, 82, 2106–2115. [Google Scholar] [CrossRef] [PubMed]
- Siskos, M. G.; Choudhary, M. I.; Tzakos, A. G.; Gerothanassis, I. P. 1H NМR chemical shift assignment, structure and conformational elucidation of hypericin with the use of DFT calculations. The challenge of accurate positions of labile hydrogens. Tetrahedron 2016, 72, 8287–8293. [Google Scholar] [CrossRef]
- Cvetanovic Zobenica, K.; Lacnjevac, U.; Etinski, M.; Vasiljevic-Radovica, D.; Stanisavljev, D. Influence of the electron donor properties of hypericin on its sensitizing ability in DSSCs. Photochem. Photobiol. Sci. 2019, 18, 2023–2030. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wackenhut, F.; Hauler, O.; Scholz, M.; zur Oven-Krockhaus, S.; Ritz, R. ; Adam, P-M.; Brecht, M., Ed.; Meixner, A. J. Hypericin: Single Molecule Spectroscopy of an Active Natural Drug. J. Phys. Chem. A 2020, 124, 2497−2504. [Google Scholar] [CrossRef]
- Liu, Q.; Wackenhut, F.; Wang, L.; Hauler, O.; Roldao, J. C. ; Adam, P-M.; Brecht, M.; Gierschner, J.; Meixner, A. J. Direct Observation of Structural Heterogeneity and Tautomerization of Single Hypericin Molecules. J. Phys. Chem. Lett. 2021, 12, 1025–1031. [Google Scholar] [CrossRef]
- De Simone, B. C.; Mazzone, G.; Toscano, M.; Russo, N. On the origin of photodynamic activity of hypericin and its iodine-containing derivatives. J. Comput. Chem. 2022, 43, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Murali, M.; Gowtham, H.G.; Shilpa, N.; Krishnappa, H.K.N.; Ledesma, A.E.; Jain, A.S.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I.; Achar, R.R.; Silina, E.; Stupin, V.; Ortega-Castro, J.; Frau, J.; Flores-Holguín, N.; Amruthesh, K. N.; Shivamallu, Ch.; Kollur, Sh. P.; Glossman-Mitnik, D. Exploration of Anti-HIV Phytocompounds against SARS-CoV-2 Main Protease: Structure-Based Screening, Molecular Simulation, ADME Analysis and Conceptual DFT Studies. Molecules 2022, 27, 8288. [Google Scholar] [CrossRef] [PubMed]
- Peeters, S.; Losi, G.; Loehlé, S.; Righi, M.C. Aromatic molecules as sustainable lubricants explored by ab initio simulations. Carbon 2023, 203, 717–726. [Google Scholar] [CrossRef]
- Chen, W-P. ; Wang, R-Q.; Zhang, Y-R.; Song, K.; Tian, Y.; Li, J-X.; Wang, G-Y.; Shi, G-F. HPLC, fluorescence spectroscopy, UV spectroscopy and DFT calculations on the mechanism of scavenging •OH radicals by Hypericin. J. Mol. Struct. 2023, 1274, 134472. [Google Scholar] [CrossRef]
- Bersuker, I. B. The Jahn-Teller Effect; Cambridge University Press: Cambridge, U.K, 2006. [Google Scholar]
- Bersuker, I. B. Pseudo-Jahn−Teller Effect-A Two-State Paradigm in Formation, Deformation, and Transformation of Molecular Systems and Solids. Chem. Rev. 2013, 113, 1351–1390. [Google Scholar] [CrossRef]
- Lewars, E. G. Computational Chemistry. Introduction to the Theory and Applications of Molecular and Quantum Mechanics. Third Edition 2016. Springer International Publishing: Switzerland, 2016.
- Ceulemans, A.; Beyens, D.; Vanquickenborne, L. G. Symmetry aspects of Jahn-Teller activity: structure and reactivity. J. Am. Chem. Soc. 1984, 106, 5824–5837. [Google Scholar] [CrossRef]
- Jahn, H. A.; Teller, E. Stability of polyatomic molecules in degenerate electronic states. I. Orbital degeneracy. Proc. R. Soc. London A 1937, 161, 220–235. [Google Scholar] [CrossRef]
- Opik, U.; Pryce, M. H. L. Studies of the Jahn-Teller effect I. A survey of the static problem. Proc. R. Soc. London A 1957, 238, 425–447. [Google Scholar] [CrossRef]
- Ceulemans, A.; Vanquickenborne, L. G. The Epikernel Principle. Struct. Bonding 1989, 71, 125–159. [Google Scholar] [CrossRef]
- Breza, M. Group-Theoretical Treatment of Pseudo-Jahn-Teller Systems. In Vibronic Interactions and the Jahn-Teller Effect: Theory and Application. (Prog. Theor. Chem. Phys. B 23); Atanasov, M., Daul, C., Tregenna-Piggott, P.L.W., Eds.; Springer: Dordrecht, The Netherlands; Berlin/Heidelberg, Germany; London, UK; New York, NY, USA, 2012; pp. 59–82. ISBN 1567-7354. [Google Scholar]
- Salthouse, J. A.; Ware, M. J. Point group character tables and related data. Cambridge University Press, Cambridge 1972.
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Frisch, G.W.; Trucks, M.J.; Schlegel, B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J.; Mennucci, B.; Tomasi, J.; Cammi, R.; Barone, V. Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model. J. Chem. Phys. 2006, 124, 94107. [Google Scholar] [CrossRef]
- Ugliengo, P. MOLDRAW: A Program to Display and Manipulate Molecular and Crystal Structures, University Torino, Torino, 2012. Available online: https://www.moldraw.software.informer.com (accessed on 9 September 2019).
Figure 1.
Atom notation of hypericin (R1 = R2 = OH, R3 = R4 = CH3), isohypericin (R1 = R3 = OH, R2 = R4 = OH) and fringelite D (R1 = R2 = R3 = R4 = OH).
Figure 1.
Atom notation of hypericin (R1 = R2 = OH, R3 = R4 = CH3), isohypericin (R1 = R3 = OH, R2 = R4 = OH) and fringelite D (R1 = R2 = R3 = R4 = OH).
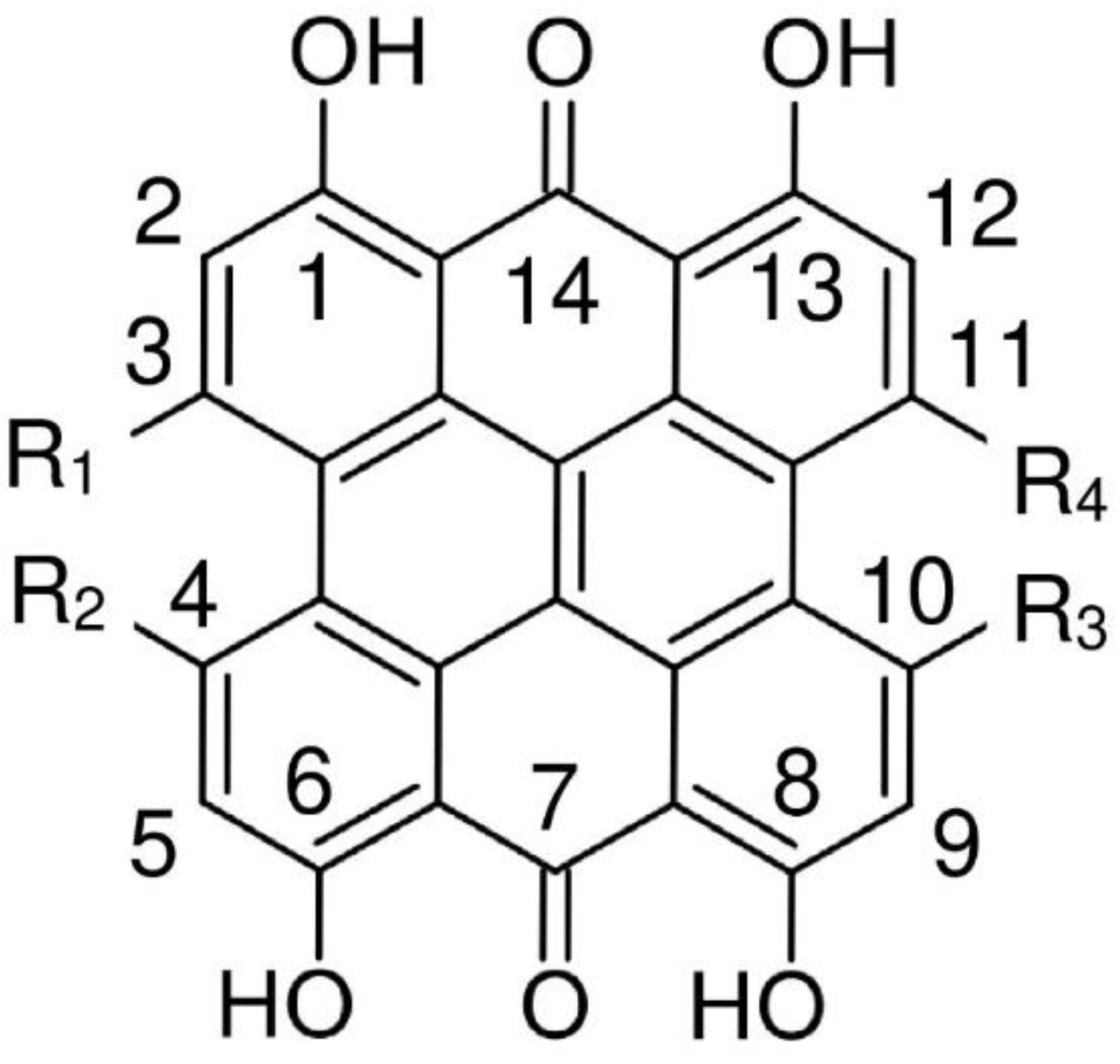
Figure 2.
Optimized structures of hypericin Ia model systems (C – black, O – red, H – white).
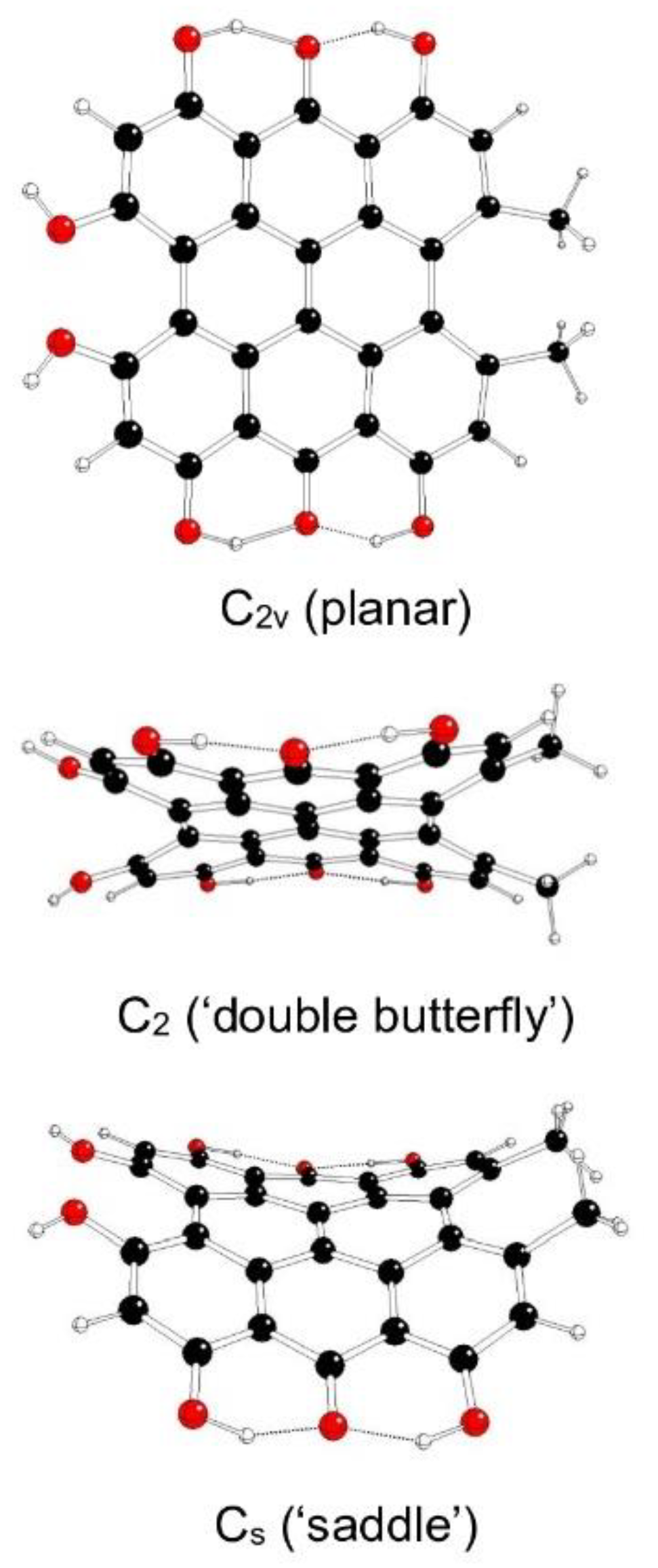
Figure 3.
Optimized structures of hypericin Ib model systems (C – black, O – red, H – white).
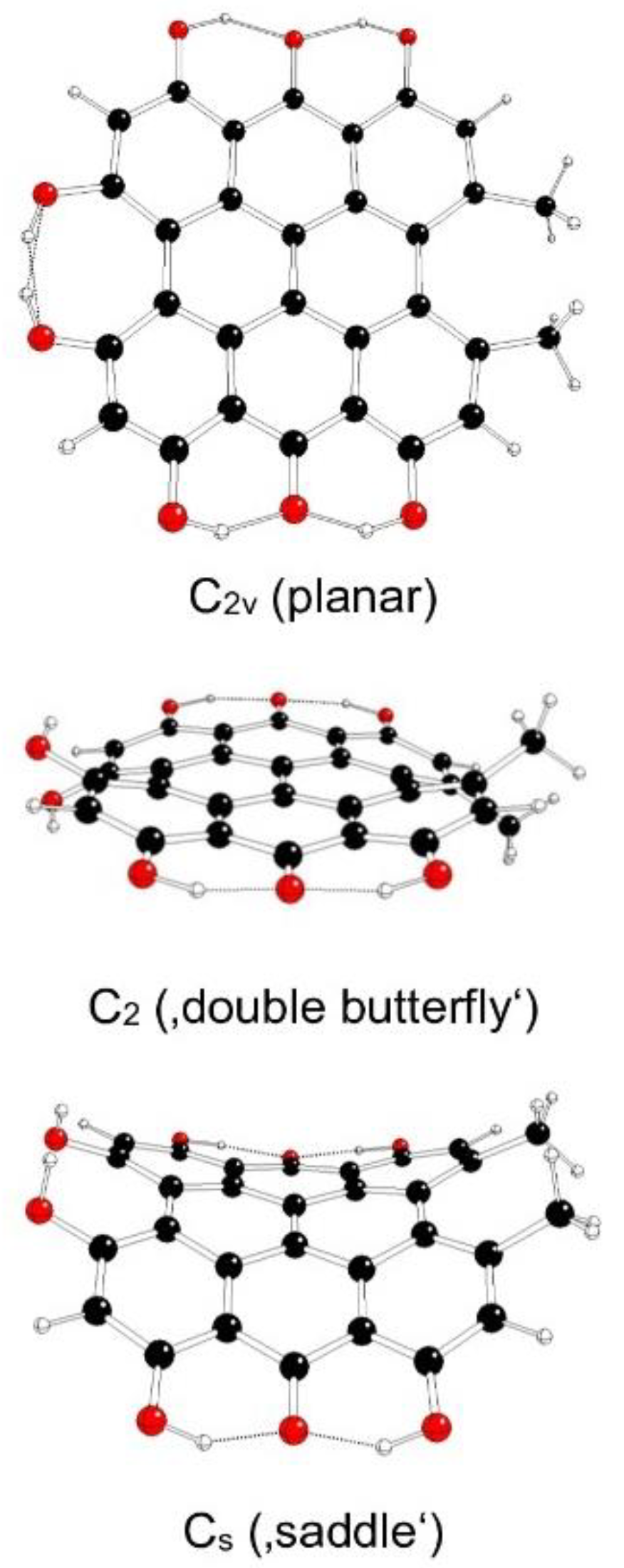
Figure 4.
Optimized structures of hypericin Ic model systems (C – black, O – red, H – white).

Figure 5.
Optimized structures of isohypericin IIa model systems (C – black, O – red, H – white).

Figure 6.
Figure 6. Optimized structures of isohypericin IIb model systems (C – black, O – red, H – white).
Figure 6.
Figure 6. Optimized structures of isohypericin IIb model systems (C – black, O – red, H – white).
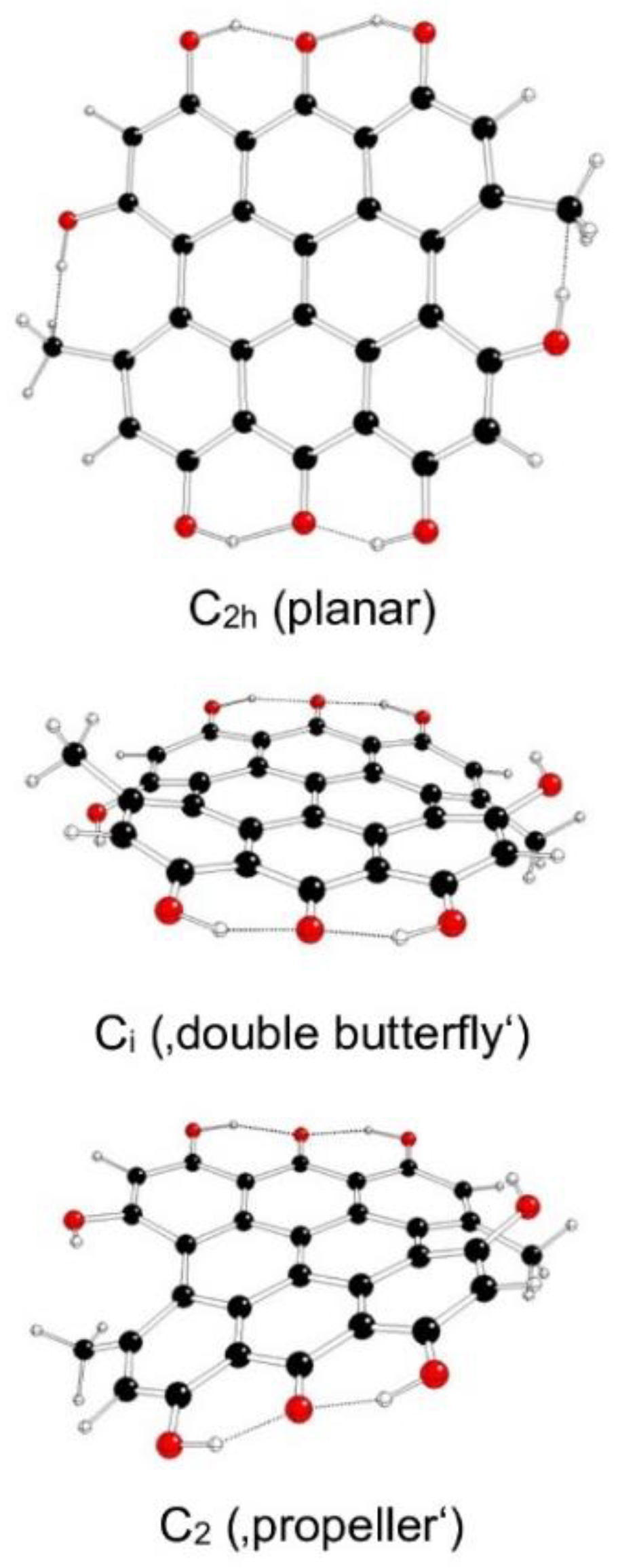
Figure 7.
Optimized structures of hypericin IIc model systems (C – black, O – red, H – white).
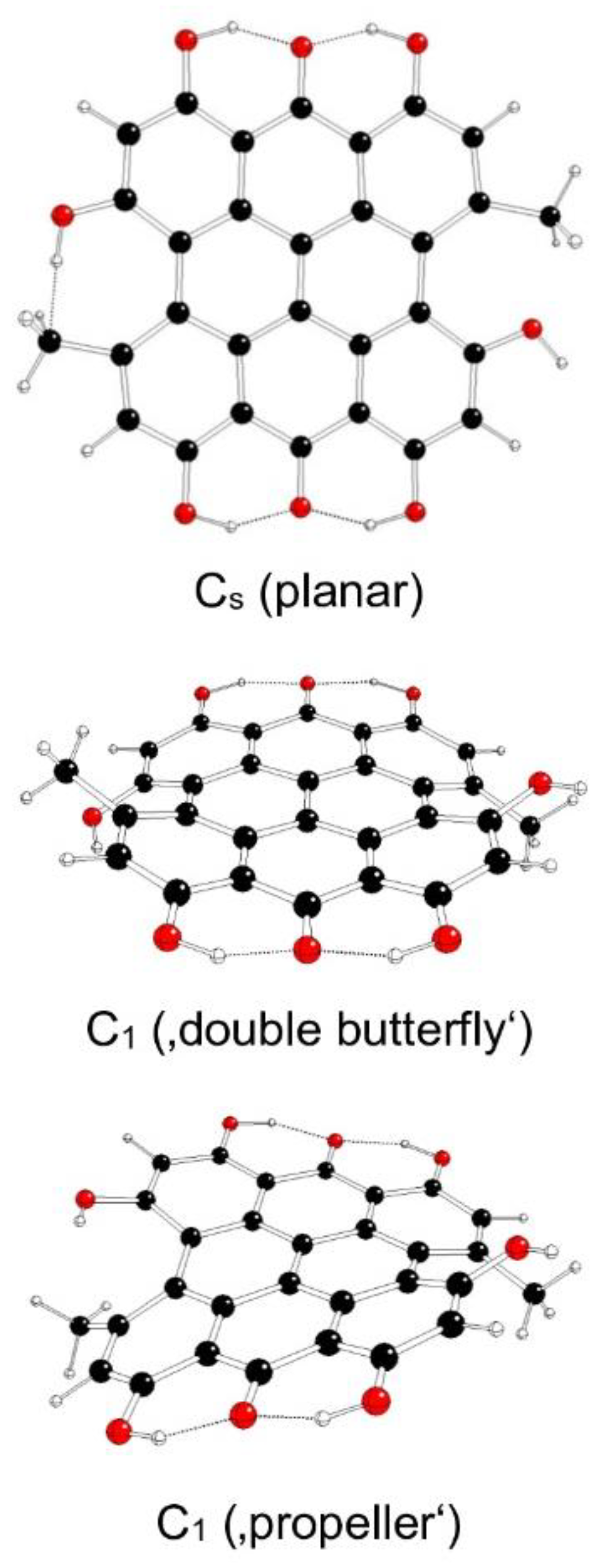
Figure 8.
Optimized structures of fringelite D IIIa model systems (C – black, O – red, H – white).
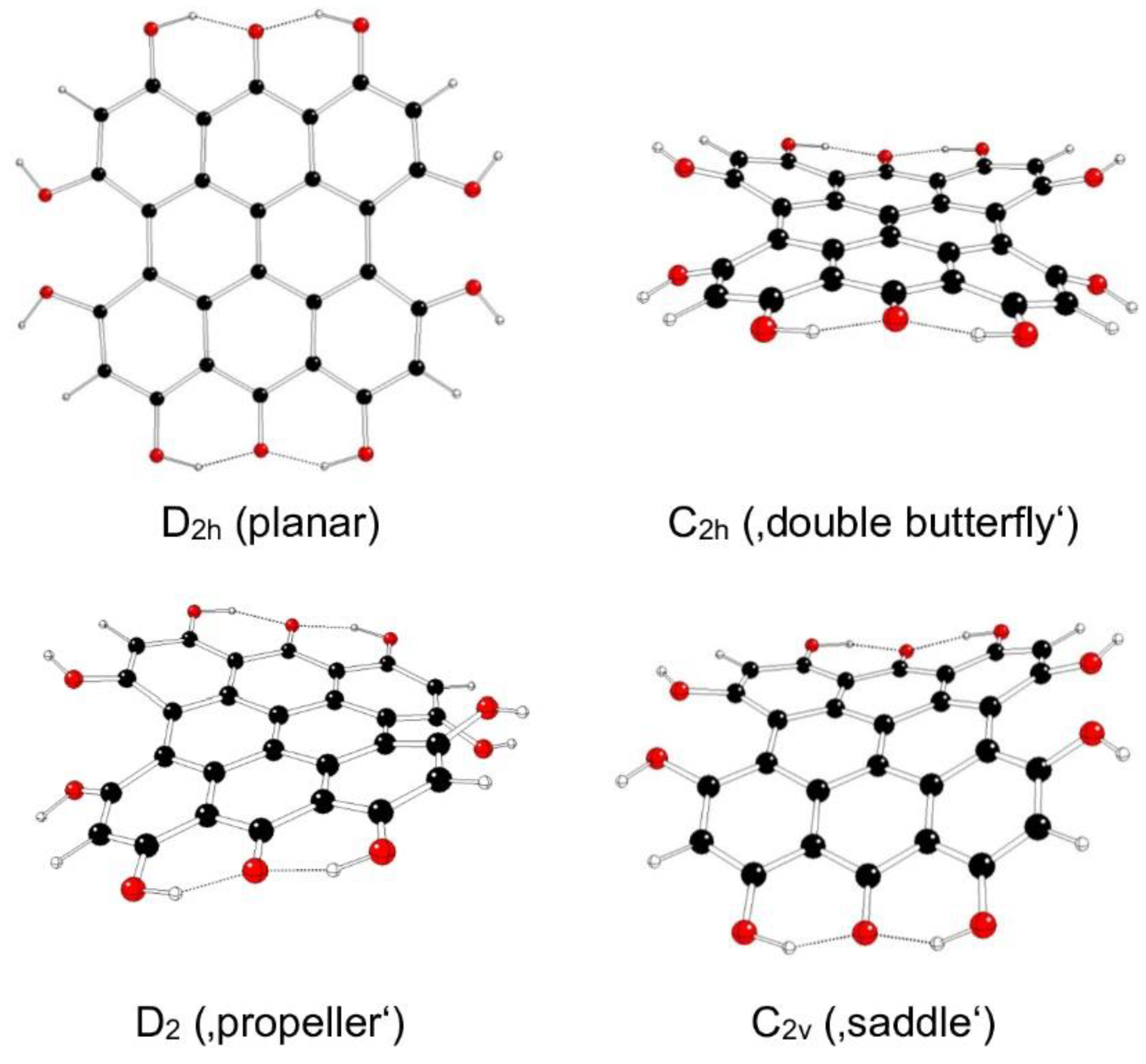
Figure 9.
Optimized structures of fringelite D IIIb model systems (C – black, O – red, H – white).
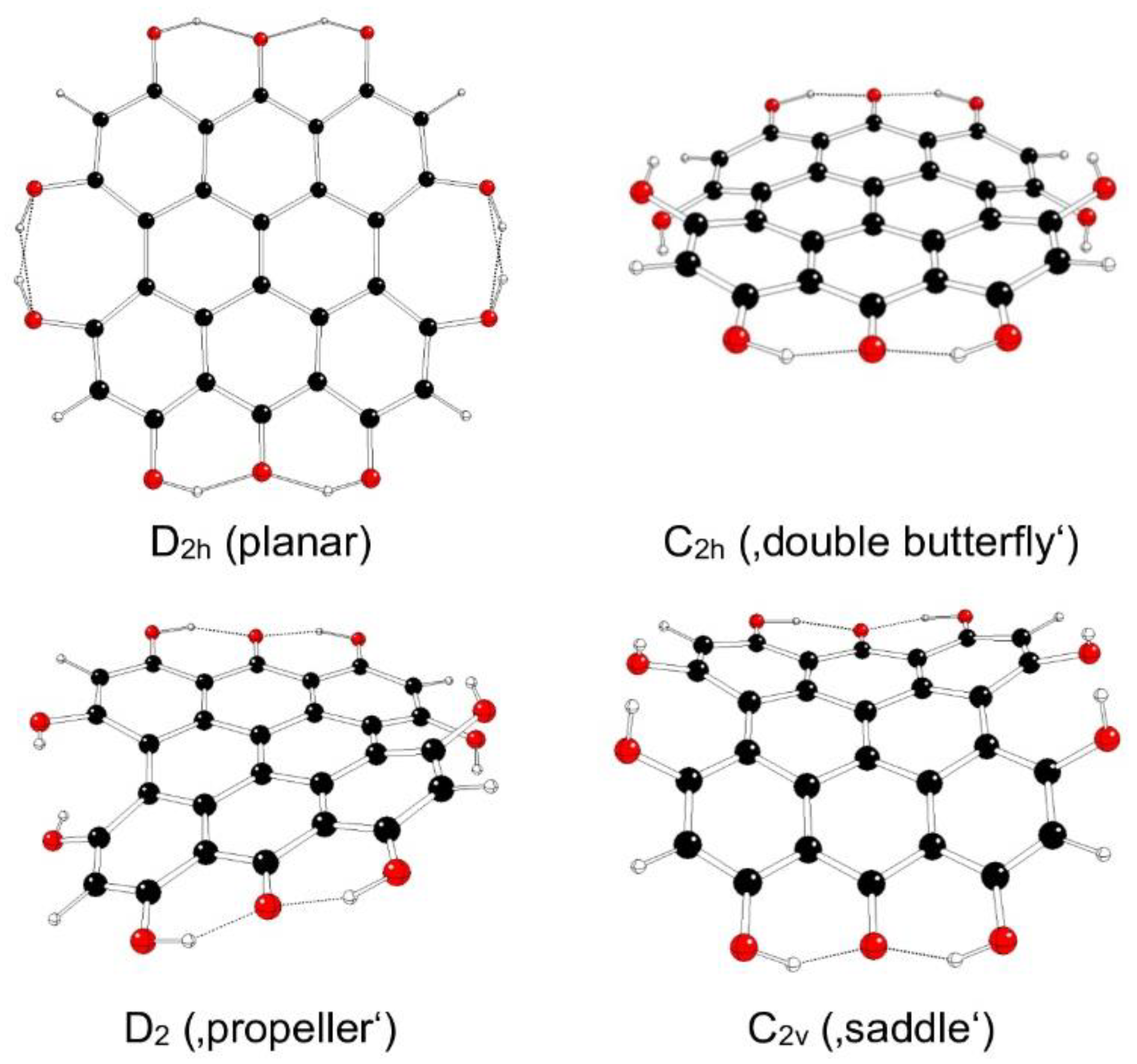
Figure 10.
Optimized structures of fringelite D IIIc model systems (C – black, O – red, H – white).
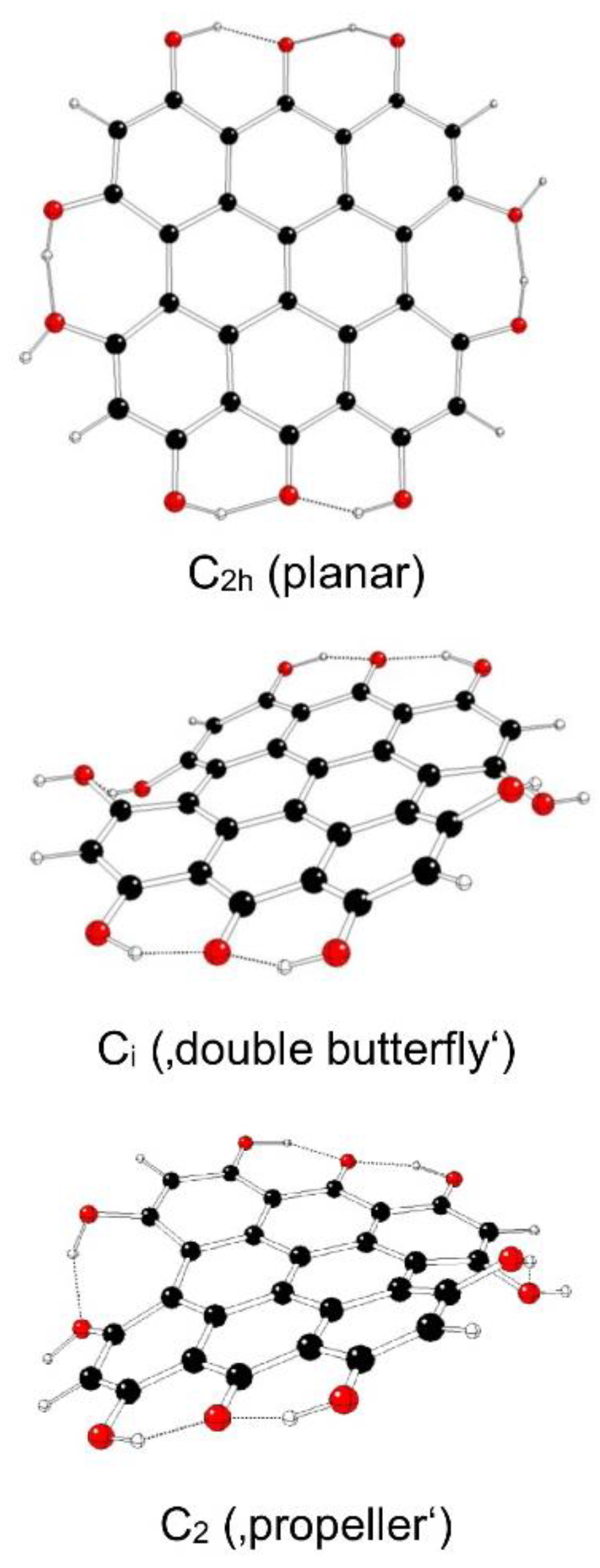
Figure 11.
Optimized structures of fringelite D IIId model systems (C – black, O – red, H – white).
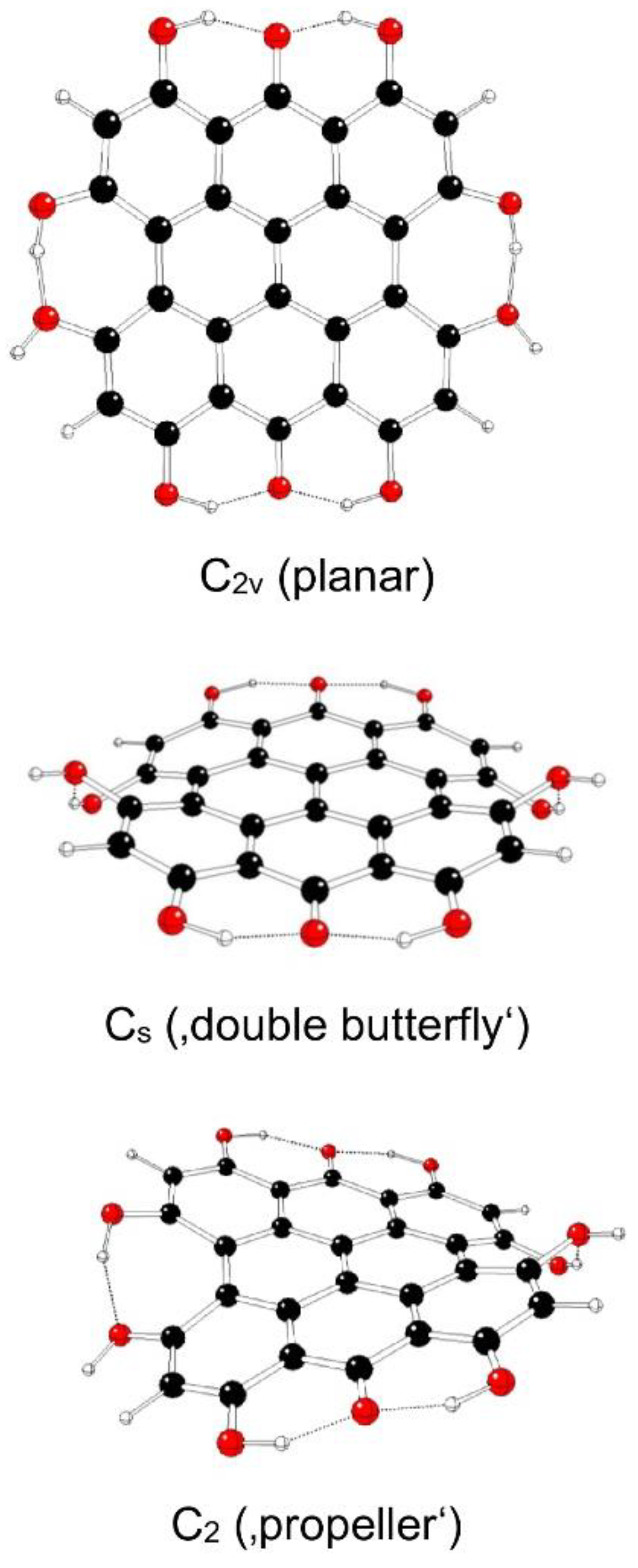

Table 1.
Hypericin model systems of the symmetry groups G and the corresponding electronic ground states Γgr, with DFT energies EDFT, JT energies EJT related to the parent group G0, distances between hydroxyl O atoms at 3 and 4 positions dOO, symmetries Γim and wavenumbers νim of the corresponding imaginary frequencies and resulting kernel subgroups K(G0, Γim).
Table 1.
Hypericin model systems of the symmetry groups G and the corresponding electronic ground states Γgr, with DFT energies EDFT, JT energies EJT related to the parent group G0, distances between hydroxyl O atoms at 3 and 4 positions dOO, symmetries Γim and wavenumbers νim of the corresponding imaginary frequencies and resulting kernel subgroups K(G0, Γim).
| Model | G | Γgr | EDFT [Hartree] | EJT [eV] | dOO [Å] | Γim | νim [cm-1] | K(G0, Γim) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| G0 = C2v (Ia) | |||||||||||
| Ia | C2v a) | X1A1 | -1754.24175 | 0.000 | 2.253 | a2 | -169, -132, -26 | C2 | |||
| b1 | -51, -35 | Cs | |||||||||
| Ia | C2 b) | X1A | -1754.31051 | 1.871 | 2.515 | - | - | - | |||
| Ia | Cs c) | X1A’ | -1754.26014 | 0.500 | 2.363 | a” | -114, -86 | C1 | |||
| Ic | C1 d) | X1A | -1754.31780 | 2.069 | 2.547 | - | - | - | |||
| Ic | C1 b) | X1A | -1754.31525 | 2.000 | 2.534 | - | - | - | |||
| G0 = C2v(Ib) | |||||||||||
| Ib | C2v a) | X1A1 | -1754.19146 | 0.00 | 2.931 | a2 | -1469, -174, -147, -35 | C2 | |||
| b2 | -660 | Cs | |||||||||
| b1 | -653, -57, -53 | Cs | |||||||||
| Ib | C2 b) | X1A | -1754.31497 | 3.361 | 2.609 | - | - | - | |||
| Ib | Cs c) | X1A’ | -1754.24669 | 1.503 | 2.566 | a” | -530, -112, -106 | C1 | |||
| Ic | C1 b) | X1A | -1754.31525 | 3.368 | 2.534 | - | - | - | |||
| Ic | C1 d) | X1A | -1754.31780 | 3.438 | 2.547 | - | - | - | |||
| G0 = Cs(Ic) | |||||||||||
| Ic | Cs a) | X1A’ | -1754.25393 | 0.000 | 2.396 | a” | -169, -123, -52, -36, -26 | C1 | |||
| Ic | C1 d) | X1A | -1754.31780 | 1.738 | 2.547 | - | - | - | |||
| Ic | C1 b) | X1A | -1754.31525 | 1.669 | 2.534 | - | - | - | |||
Remarks: a)planar conformation b)’double butterfly’ conformation c)’saddle’ conformationd)’propeller’ conformation
Table 2.
Symmetry descent paths of hypericin model systems. Possible PJT descent paths are in bold.
| No. | Systems | Descent path |
|---|---|---|
| 1 | Ia, Ib | C2v C2 |
| 2 | Ia, Ib | C2v Cs C1 |
| 3 | Ib | C2v Cs C1 |
| 4 | Ic | Cs C1 |
Table 3.
Isohypericin model systems of symmetry groups G and the corresponding ground electronic states Γgr, with DFT energies EDFT, JT energies EJT related to the parent group G0, distances between hydroxyl O atoms and methyl C atoms in 3 and 4 or 10 and 11 positions dOC, symmetries Γim and wavenumbers νim of the corresponding imaginary frequencies and resulting kernel subgroups K(G0, Γim).
Table 3.
Isohypericin model systems of symmetry groups G and the corresponding ground electronic states Γgr, with DFT energies EDFT, JT energies EJT related to the parent group G0, distances between hydroxyl O atoms and methyl C atoms in 3 and 4 or 10 and 11 positions dOC, symmetries Γim and wavenumbers νim of the corresponding imaginary frequencies and resulting kernel subgroups K(G0, Γim).
| Model | G | Γgr | EDFT [Hartree] | EJT [eV] | dOC [Å] | Γim | νim [cm-1] | K(G0, Γim) | |
|---|---|---|---|---|---|---|---|---|---|
| G0 = C2h(IIa) | |||||||||
| IIa | C2h a) | X1Ag | -1754.26464 | 0.000 | 2.423 | bg | -130, -19 | Ci | |
| au | -129, -39, -12 | C2 | |||||||
| IIa | Ci b) | X1Ag | -1754.31444 | 1.355 | 2.668 | - | - | - | |
| IIa | C2 c) | X1A | -1754.31670 | 1.417 | 2.677 | - | - | - | |
| G0 = C2h(IIb) | |||||||||
| IIb | C2h a) | X1Ag | -1754.24601 | 0.000 | 2.636 | bg | -380, -105, -15 | Ci | |
| au | -379, -103, -40, -20 | C2 | |||||||
| IIb | Ci b) | X1Ag | -1754.31792 | 1.957 | 2.745 | - | - | - | |
| IIb | C2 c) | X1A | -1754.31991 | 2.011 | 2.757 | - | - | - | |
| G0 = C2h(IIc) | |||||||||
| IIc | Cs a) | X1A’ | -1754.25521 | 0.000 | 2.419 2.641 |
a” | -393, -133, -107, -41, -20, -15 | C1 | |
| IIc | C1 b) | X1A | -1754.31619 | 1.659 | 2.744 2.666 |
- | - | - | |
| IIc | C1 c) | X1A | -1754.31834 | 1.718 | 2.753 2.677 |
- | - | - | |
Remarks: a)planar conformation b)’double butterfly’ conformation c)’propeller’ conformation
Table 4.
Symmetry descent paths of isohypericin model systems. .
| No. | Systems | Descent path |
|---|---|---|
| 5 | IIa, IIb | C2h Ci |
| 6 | IIa, IIb | C2h C2 |
| 7 | IIc | Cs C1 |
Table 5.
Fringelite D model systems of symmetry groups G and the corresponding ground electronic states Γgr, with DFT energies EDFT, JT energies EJT related to the parent group G0, distances between hydroxyl O atoms in 3 and 4 (or 10 and 11) positions dOO, symmetries Γim and wavenumbers νim of the corresponding imaginary frequencies and resulting kernel subgroups K(G0, Γim).
Table 5.
Fringelite D model systems of symmetry groups G and the corresponding ground electronic states Γgr, with DFT energies EDFT, JT energies EJT related to the parent group G0, distances between hydroxyl O atoms in 3 and 4 (or 10 and 11) positions dOO, symmetries Γim and wavenumbers νim of the corresponding imaginary frequencies and resulting kernel subgroups K(G0, Γim).
| Model | G | Γgr | EDFT [Hartree] | EJT [eV] | dOO [Å] | Γim | νim [cm-1] | K(G0, Γim) | ||
| G0 = D2h(IIIa) | ||||||||||
| IIIa | D2h a) | X1Ag | -1826.09765 | 0.000 | 2.262 | b2g | -127 | C2h | ||
| au | -126 | D2 | ||||||||
| b3u | -37 | C2v | ||||||||
| IIIa | C2h b) | X1Ag | -1826.13780 | 1.093 | 2.516 | - | - | - | ||
| IIIa | D2 d) | X1A | -1826.13963 | 1.142 | 2.526 | - | - | - | ||
| IIIa | C2v c) | X1A1 | -1826.10503 | 0.201 | 2.344 | b2 | -94 | Cs | ||
| a2 | -92 | C2 | ||||||||
| IIIc | C2 d) | X1A | -1826.15004 | 1.426 | 2.544 | - | - | - | ||
| IIId | C2 d) | X1A | -1826.14935 | 1.407 | 2.551 | - | - | - | ||
| IIIc | Cs b) | X1A’ | -1826.14682 | 1.338 | 2.539 | - | - | - | ||
| G0 = D2h(IIIb) | ||||||||||
| IIIb | D2h a) | X1Ag | -1825.99795 | 0.00 | 2.930 | b2g | -1466, -149 | C2h | ||
| au | -1466, -148, -35 | D2 | ||||||||
| b3g | -659 | C2h | ||||||||
| b1u | -657 | C2v | ||||||||
| b1g | -654, -54 | C2h | ||||||||
| b3u | -653, -53 | C2v | ||||||||
| IIIb | C2h b) | X1Ag | -1826.14666 | 4.047 | 2.610 | - | - | - | ||
| IIIb | D2 d) | X1A | -1826.14870 | 4.102 | 2.621 | - | - | - | ||
| IIIb | C2v c) | X1A1 | -1826.07720 | 2.157 | 2.552 | a2 | -531, -109 | C2 | ||
| b2 | -518, -109 | Cs | ||||||||
| IIIc | C2 d) | X1A | -1826.15004 | 4.139 | 2.544 | - | - | - | ||
| IIId | C2 d) | X1A | -1826.14935 | 4.120 | 2.551 | - | - | - | ||
| IIId | Cs b) | X1A’ | -1826.14682 | 4.051 | 2.539 | - | - | - | ||
| G0 = C2h(IIIc) | ||||||||||
| IIIc | C2h a) | X1Ag | -1826.12302 | 0.000 | 2.401 | bg | -118, | Ci | ||
| au | -117, -38 | C2 | ||||||||
| IIIc | Ci b) | X1Ag | -1826.14762 | 0.669 | 2.533 | - | - | - | ||
| IIIc | C2 d) | X1A | -1826.15004 | 0.735 | 2.544 | - | - | - | ||
| G0 = C2h(IIId) | ||||||||||
| IIId | C2v a) | X1A1 | -1826.12138 | 0.000 | 2.406 | b1 | -119, -38 | Cs | ||
| a2 | -118, -11 | C2 | ||||||||
| IIId | Cs b) | X1A’ | -1826.14682 | 0.692 | 2.539 | - | - | - | ||
| IIId | C2 d) | X1A | -1826.14935 | 0.761 | 2.551 | - | - | - | ||
Remarks: a)planar conformation b)’double butterfly’ conformation c)’saddle’ conformation d)’propeller’ conformation
Table 6.
Symmetry descent paths of fringelite D (III) model systems. Possible PJT descent paths are in bold.
Table 6.
Symmetry descent paths of fringelite D (III) model systems. Possible PJT descent paths are in bold.
| No. | Systems | Descent path |
| 8 | IIIa, IIIb | D2h C2h |
| 9 | IIIa, IIIb | D2h D2 |
| 10a | IIIa, IIIb | D2h C2v Cs |
| 10b | IIIa, IIIb | D2h C2v C2 |
| 11 | IIIb | D2h C2h |
| 12a | IIIb | D2h C2v Cs |
| 12b | IIIb | D2h C2v C2 |
| 13 | IIIb | D2h C2h |
| 14a | IIIb | D2h C2v Cs |
| 14b | IIIb | D2h C2v C2 |
| 15 | IIIc | C2h Ci |
| 16 | IIIc | C2h C2 |
| 17 | IIId | C2v Cs |
| 18 | IIId | C2v C2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



