
Submitted:
27 October 2024
Posted:
28 October 2024
You are already at the latest version
Abstract
Amid the current multi-country mpox outbreak, analyzing monkeypox virus (MPXV) and vaccinia virus (VACV) genomes is vital for understanding evolutionary processes that may impact vaccine efficacy and design. This study aimed to elucidate the phylogenetic relationships and structural features of viral antigens, which are crucial for developing effective vaccines. By aligning 1,903 MPXV genomes from the NCBI Virus repository (released between 2022 and 2024), an increase in phylogenetic diversity was observed compared to previous studies. These genomes grouped into Clade I (25 genomes) and Clade IIB (1,898 genomes), with a new Clade I sub-lineage emerging from samples collected in Sud-Kivu province, Democratic Republic of the Congo. Homology-based modeling of six key neutralization determinants encoded by the highly conserved Clade I MPXV genes revealed several amino acid variants near potential antibody binding sites compared to those of vaccinia and variola virus templates. Despite these structural differences, the long-standing vaccinia vaccines, initially created for smallpox, still play a crucial role in safeguarding humanity against mpox today. The findings have deepened our understanding of disparate evolutionary mechanisms for MPXV and VACV, influencing vaccine efficacy. The workflows for viral genomic analysis and structural modeling in CLC Genomics Workbench will facilitate MPXV research and vaccinology. Finally, this study highlights the critical role of MPXV metagenomic surveillance in identifying viral evolution and potential vaccine resistance.
Keywords:
bioinformatics
; disease outbreaks
; monkeypox
; monkeypox virus
; phylogeny
; poxvirus
; taxonomic classification
; vaccinia virus
; virus database
1. Introduction
On August 14, 2024, the World Health Organization (WHO) declared mpox a public health emergency of international concern (PHEIC) due to a surge in cases and fatalities, as well as its multi-country spread across the African continent [1]. Mpox, an infectious disease caused by the Monkeypox virus (MPXV) of the Orthopoxvirus genus, has been endemic to Central and West Africa for over five decades [2,3,4,5,6]. The unprecedented global outbreak of mpox, declared a PHEIC on July 23, 2022, and its resurgence in 2024, marked a significant shift in transmission dynamics and the affected population, expanding from primarily gay/bisexual men to include women, children, and infants [2,7]. Consequently, concerns about viral mutagenesis and evolution have been raised. MPXV mutations have been linked to the activity of the APOBEC3 (apolipoprotein B mRNA-editing catalytic polypeptide-like 3) enzymes, which induce cytidine deamination during viral replication, leading to mutations in the viral genome [8,9,10,11]. These mutations may inadvertently provide a fitness advantage during infection by altering viral proteins to facilitate transmission and immune evasion [9]. Therefore, the interaction between APOBEC3 and MPXV may be contributing to viral diversity and evolutionary fitness. This is indicated by the emergence of a new sub-lineage within Clade I of MPXV in 2023 and the upsurge in cases, particularly in the Democratic Republic of the Congo (DRC) (formerly Zaire) [12,13,14]. The latest WHO Situation Report from October 2024 disclosed alarming year-to-date figures for the DRC, with 31,350 suspected cases and 992 deaths among them [15].
Today’s mpox prophylactic vaccines are sourced from the live, attenuated vaccinia virus (VACV) originally developed for smallpox [2]. Currently, there are only three licensed vaccines available globally, each subject to country-specific regulations and restrictions [2]. The most widely available vaccine is the Modified Vaccinia Ankara-Bavarian Nordic (MVA-BN), developed in Denmark and patented in 2007 for use in adults (18 years and older) [2,16,17]. In 2019, the United States Food and Drug Administration (FDA) approved MVA-BN for mpox prophylaxis in adults and granted emergency use authorization (EUA) in 2022 for individuals under 18 years of age [18]. On October 8, 2024, the WHO granted prequalification to the MVA-BN vaccine for individuals aged 12 to 17 [15]. In Japan, the LC16m8 vaccine was initially licensed in 1975 for smallpox prophylaxis and was expanded in 2022 to include mpox prophylaxis without age restrictions [2,19,20,21]. The third vaccine, ACAM2000, was developed by the United States for biological defense and approved by the FDA for smallpox prophylaxis [22]. It is available for use against mpox only under an Expanded Access Investigational New Drug (EA IND) protocol [23]. These vaccines originated from distinct lineages of VACV that have been propagated over the last century, resulting in differences in their genomes, antigens, and safety profiles [24]. The milestones in the development of smallpox inoculation and their relevance in modern vaccinology are briefly summarized below [25].
The practice of inoculation against smallpox dates to ancient China and the Ottoman Empire [25,26,27,28,29]. The earliest known reference in China is from a book published in 1549, which describes using powdered pox scabs for nasal insufflation [25,26,27]. In the Ottoman Empire, the method involved pricking the skin with a needle that had been dipped in the pox blister of an infectee [25,29]. This practice likely dispersed along trade routes through Africa, the Middle East, and India [25]. In 1721, Lady Mary Wortley Montagu introduced variolation to Britain after witnessing Turkish women in Constantinople using the technique [25,29]. In 1796, Edward Jenner made a significant advancement by developing the smallpox vaccine using the cowpox virus, which is antigenically similar but less virulent (a concept known as homology-based vaccine selection) [25,30]. Louis Pasteur further advanced the field of vaccinology by attenuating (weakening) live vaccines through serial passage in different host species and conditions. This method, now recognized as a form of genomic reductive evolution, played a crucial role in developing effective and safe vaccines [25,31,32]. Over the last century, three generations of live attenuated VACV vaccines have been developed in several countries [24]. Various laboratory and propagation techniques, such as using different host species, cell cultures, and passage numbers, have resulted in variations in VACV genomes, genes, and antigens [24]. In recent years, genome sequencing has been instrumental in decoding several ancestral poxviruses and VACV genomes [21,33,34,35,36]. These advancements have now enabled lineage tracing and comparative analysis of VACV genomes and antigens.
The emergence of a new sub-lineage within Clade I of MPXV is concerning due to potential structural changes in virion epitopes caused by nonsynonymous mutations. Epitope alterations may result in ineffective binding by host cross-neutralizing antibodies induced by prophylactic vaccines [25,30]. Consequently, this study aimed to explore the evolution of MPXV and VACV genomes and to compare their present antigenic determinants. Focusing on the highly virulent MPXV Clade I genome, we analyzed the encoded protein sequences of six key neutralization determinants shared between MPXV and VACV virions and performed 3D structural modeling for variant profiling and visualization (Figure 1) [37].
2. Materials and Methods
2.1. MPXV Customized Reference Database and Genome Dataset (2022-2024)
Our initial MPXV genome reference database included the MPXV RefSeq and 217 additional genomes obtained from NCBI Virus (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/) [6,40]. These sequence files were derived from human (n = 203) and animal (n = 15) hosts, with collection dates spanning from 1958 to July 2022. For this study, we conducted another search in the NCBI Virus repository for complete MPXV genomes under Taxid: 10244, restricting the release dates from July 2022 to June 2024. After identifying the genomes and their accession numbers (AN), we downloaded the raw sequencing files and metadata directly into CLC Genomics Workbench Premium 23.0.4 (Redwood City, CA, USA) using the “Search for Sequences at NCBI” tool with the search term “monkeypox virus complete genome.” The genome files (n = 1,903) underwent whole genome alignment (WGA), annotation, and clade/sub-lineage determination as described in section 2.3. The annotations adhered to the Clade I MPXV RefSeq (NC_003310) convention. The clade/sub-lineage nomenclature is based on two publications, as the International Committee on Taxonomy of Viruses has not classified beyond the species level [5,6,41]. Figure 2 displays the MPXV RefSeq along with six genes encoding major neutralization determinants that are homologous to those of VACV [6,37].
2.2. Vaccinia Virus (VACV) Genome Dataset and Major Neutralization Determinants
The Vaccinia Virus genome dataset was compiled by searching the NCBI Virus repository (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/) for complete Vaccinia Virus genomes using the term “vaccinia virus”[40]. After identifying both historical and currently relevant genomes (n = 9), we downloaded the raw sequencing files and metadata directly into CLC Genomics Workbench using the “Search for Sequences at NCBI” tool with NCBI GenBank AN. The genomes downloaded were: VACV RefSeq (NC_006998), VACV-WR (Western Reserve) (AY243312), VACV-Tian Tan (AF095689), VACV-Copenhagen (M35027), VACV-Acam2000 (AY313847), VACV-Lister (OR837118), VACV-LC16m8 (Lister Clone 16, m8) (AY678275), VACV-MVA (Modified Vaccinia Ankara) (U94848), and VACV-MVA-BN (Modified Vaccinia Ankara-Bavarian Nordic) (DQ983238). Additionally, the cowpox virus (CPXV) and horsepox virus (HPXV) genomes—(NC_003663.2), CPXV-GRI-90 (X94355), and HPXV RefSeq (NC_066642)—were downloaded to serve as ancestral pox genomes for lineage tracing in the phylogenomic analyses. The Vaccinia Virus genome dataset then underwent WGA, pairwise comparison (PWC), and phylogenomic analysis as described in section 2.3. Notably, the NCBI RefSeq for VACV and CPXV originated from the VACV-WR and CPXV Brighton Red strains, respectively [40]. As for the naming convention of VACV ORF or genes, it is based on the Copenhagen strain using three characters: letters A to O for HindIII restriction fragments, numbers 1 to 56 for ORFs within these fragments, and R or L for transcription direction (omitted for corresponding proteins) [38].
Functionally, the six antigenic VACV membrane proteins are crucial for cell attachment and entry [38]. MV proteins A27, D8, and H3 bind to heparan sulfate or chondroitin sulfate for cell attachment. The MV L1 protein, part of the entry-fusion complex (EFC), is essential for cell entry through fusion with the plasma or endocytic membrane. A33 and B5 proteins are present on both EV and MV. EV, less abundant than MV, is involved in cell-to-cell spread within the host. EV adhere to cell surfaces to prevent superinfection through a “repulsion mechanism” induced by A33. Finally, B5 disrupts the plasma membrane to aid EV entry. The corresponding proteins for VACV are A27, A33, B5, D8, H3, and L1, and for MPXV, they are A29, A35, B6, E8, H3, and M1 [37].
2.3. Whole Genome Alignment, Pairwise Comparison and Phylogenomic Analysis
CLC Genomics Workbench Premium 23.0.4 (Redwood City, CA, USA) was installed on an HP notebook with Windows 10, an Intel i7-7500U processor, and 8 GB RAM. The “Whole Genome Alignment” plugin tools were used for automated data analysis, involving three steps: 1) importing sequences, 2) selecting alignment parameters, and 3) optionally copying annotations from a reference genome (Figure 3A, B). The WGA output shows aligned, conserved regions as synteny blocks across the genomes, connected by a phylogenetic tree.
The WGA output file was inputted into the “Create Average Nucleotide Identity Comparison” tool to quantify genome similarity (Figure 3A, C). For each genome pair, the aligned regions were identified to calculate two metrics: 1) Alignment Percentage (AP), which is the average percentage of alignment between two genomes, and 2) Average Nucleotide Identity (ANI), which is the average percentage of matching nucleotides in the aligned regions. The AP or ANI pairwise comparison table generated was utilized in the “Create Tree from Comparison” tool (Figure 3A) to build a Neighbor Joining (NJ) tree. The geographical map of sample collection sites was generated using Wolfram Mathematica 13.0 (Champaign, IL, USA).
2.4. Antigenic Protein Structure Modeling
The “Find and Model Structure” tool in the CLC Protein Analysis tool kit (Figure 3A) was used for protein structural modeling. A homology-based model of an amino acid (AA) sequence of interest was generated using a BLAST-identified template. The workflow included four steps: 1) importing the AA sequence, 2) performing a BLAST search against the protein structure sequence database, 3) filtering out low-quality hits (identity < 20%) and poor resolution (< 4 Å) PDB structures, and 4) ranking the structures. The “Download 3D protein structure database” tool was used to install the necessary database within CLC before running the “Find and Model Structure” tool. The output lists the available Protein Data Bank (PDB) (https://www.rcsb.org/) [43] structures for creation of the structural model, BLAST statistics, and model rankings. The PDB was also searched for crystallographic structures of VACV MV (A27, D8, H3, L1) and EV (A33, B5) proteins. Structures preferably complexed with targeted antibodies were used for reconstructing and visualizing epitope/paratope binding sites in CLC Genomics Workbench.
3. Results
3.1. MPXV Genomes from 2022-2024 Clustered with Clades I and IIB, with a New Clade I Sub-Lineage Emerging from Sud-Kivu, DRC
The alignment of 1,903 MPXV genomes was performed in batches of 200, alongside reference genomes from Clades I (NC_003310), IIA (AY741551), and IIB (NC_063383). Phylogenomic analysis showed 25 genomes aligned with Clade I and 1,898 with Clade IIB. The 25 Clade I genomes were added to our initial MPXV database (218 genomes) for further analysis and displayed as a phylogram with 243 MPXV genomes in Figure 4A. MPXV Clades I and II, originating from Central and West Africa respectively, have distinct branches: groups I-V and A-B [5,6,41]. The outermost ring of the phylogram shows three attributes for each genome: sequence AN, 3-letter country code, and year, enhancing the visualization of geotemporal data. Of the 25 new Clade I genomes, all hosts were human except for AN OR943698, which came from a captive chimpanzee during a 2016 outbreak at Mefou Primate Sanctuary in Cameroon and clustered with Group I [44]. Another sample, AN OP498046, collected from a 9-month-old girl in Gabon in 1988 but sequenced in 2022 also clustered with Group I [45]. Twelve samples (AN OQ621553, OQ729808, and PP601183-PP601206), unclassified by the authors, closely aligned with Group II [12]. Eleven additional unclassified genomes (AN PP601207-PP601228) by the same authors collected in Sud-Kivu formed a new Clade I sub-lineage (Figure 4A) [12].
The MPXV Clade I genomes from the complete set of 243 genomes were extracted and displayed as a radial phylogram by group, country, and city/province (if available) in Figure 4B. The map correlates the phylogenetic and geospatial relationship. The metadata for the 243 genomes is provided in Supplementary Table S1. The new sub-lineage emerging from the Kamituga Health Zone of Sud-Kivu province can be easily identified on the eastern border of the DRC near Tanzania. In contrast, genomes from Sud-Ubangi, Tshopo, and Equateur, DRC, as well as South Sudan, aligned with the pre-existing Group II genomes. Furthermore, the limited number of genomes (n = 11) from Sud-Kivu in the NCBI Virus does not reflect the disease’s extent and virulence, with 31,350 suspected cases reported in the DRC between January and October 2024 [15]. The distinct, novel sub-lineage has raised concerns about APOBEC3-mediated, intra-host mutagenesis and viral microevolution [12]. Consequently, we selected the most ancestral genome from Sud-Kivu (PP601216) as the representative genome, along with the Clade I RefSeq (NC_003310), for downstream comparative analysis and antigen modeling. Phylograms from Figure 4, displayed at page width, are shown in Supplementary Figure S1. The 1,898 genomes released between 2022-2024 that grouped with Clade IIB are displayed as phylograms in Supplementary Figure S2, with metadata provided in Supplementary Table S2.
3.2. VACV Genomes Show a Close Alignment with HPXV, Featuring a Conserved Central Region, Variable Terminal Regions, and Shortened Lengths
Genome alignment and comparative analysis of 9 representative VACV, 2 CPXV, and 1 HPXV genomes revealed a conserved central region, variable terminal synteny blocks, and shortened genome lengths (Supplementary Figure S3). The longest VACV genome was VACV-ACAM2000 (199,234 bp), while the shortest was VACV MVA-BN (165,041 bp). The VACV RefSeq genome length (194,711 bp) was 17,922-29,788 bp shorter than the RefSeqs for HPXV (212,633 bp) and CPXV (224,499 bp). The distal synteny blocks beyond the 200,000 bp position, which are homologous to those in the CPXV, HPXV, and VACV RefSeqs, were truncated, rearranged, or deleted in later generations of VACV. Remarkably, the genes for the 6 neutralization determinants were conserved in the genomes of two current vaccines, VACV MVA-BN and LC16m8.
The unrooted API NJ phylogenetic tree illustrates genetic distances, showing VACV is closer to HPXV than CPXV (Figure 5A). Pairwise genome comparisons revealed alignment similarities of 82-87% between VACV and HPXV, and 80-85% between VACV and CPXV (Figure 5B). Within the VACV clade, the distinct divergent branches indicated genetic differences. PWC showed alignment similarity exceeding 87% between VACV genomes, with nucleotide identity surpassing 98% within aligned regions (Figure 5B).
3.3. MPXV Clade I Genomes Are Longer Than Current VACV Vaccines, with over 85% Alignment Similarity and More Than 97% Average Nucleotide Identity
Genome alignment revealed a highly conserved central region in MPXV Clades I and II, spanning approximately from 6,500 to 175,000 bp (Figure 6A). Significant variability was observed at the genome’s distal ends. In contrast, the VACV genomes (MVA-BN and LC16m8) are shorter, with genes at the distal ends curtailed, rearranged, or deleted, as indicated by the pink, blue, and brown synteny blocks in Figure 6A. These deletions align with the characteristics of attenuated, replication-proficient (LC16m8) and replication-deficient (MVA-BN) vaccines, which have undergone extensive serial passages. A phylogenetic tree was constructed from the pairwise comparison of representative MPXV and VACV (MVA-BA and LC16m8) genomes. Figure 6B shows the two distinct clades formed by MPXV and VACV genomes with genetic differences evident between individual genomes (divergent branches). The representative genome from Sid Kivu (PP601216), closely aligned with the Clade I RefSeq, showing 95% alignment similarity and 99.88% ANI (Figure 6C). Pairwise comparisons between MPXV Clade I and the genomes of VACV MVA-BN and VACV-LC16m8 showed alignment similarities exceeding 85-86% and ANI over 97% (Figure 6C).
3.4. Variations in Sequence and Structure between Homologous MPXV and VACV Antigens May Impact Epitope/Paratope Binding
Table 1 presents a summary of six crystallographic structures identified for the surface antigenic proteins of MPXV Clade I (NC 003310) by the “Find and Model Structure” tool. The MV proteins (A29L, E8L, H3L, and M1R) and EV proteins (A35R and B6R) showed highly significant matches (E-value range, 3.1E-20 to 1.7E-158) and match identities (range, 84 to 99%) with homologous proteins on VACV, except for B6R. The MPXV B6R had a satisfactory match (E-value, 1.0E-11; match identity, 30%) with the structure of the Smallpox inhibitor of complement (SPICE). The percentage coverage, indicating the portion of the full-length AA sequence covered by the ectodomain sequence encoding the crystallographic structures, ranged from 38.7% to 75.4%. Remarkably, the structural modeling results for the representative MPXV genome from Sud-Kivu (PP601216) matched those of the 1996 Clade I RefSeq (NC_003310) (Supplementary Tables S3 and S4), highlighting well-preserved neutralization determinants.
For MPXV antigen modeling, top homologous structures, preferably complexed with monoclonal antibodies, were selected. This approach visualized AA differences between the constructed model and the template, along with epitope-paratope binding sites. The vaccinia virus surface antigens, shown as a collage of ectodomains in Figure 7, served as templates for MPXV 3D modeling. The VACV proteins homologous to MPXV included MV proteins (A27, D8, H3, L1) and the EV protein (A33). For MPXV B6, the top homologous structure was the Variola virus protein (VARV D15). Each ectodomain is shown as a surface and an AA-labeled backbone model in Figure 7. The Fab regions of monoclonal antibodies are shown in complex with the ectodomains. The epitope/paratope binding sites serve as a point of reference for homologous MPXV antigens. The VARV D15 is shown as an ectodomain-complement C3b complex.
In Figure 8A, the VACV ectodomain templates are juxtaposed with the homologous MPXV 3D models. The VACV templates are depicted as stick and backbone models in monochrome gold. The superimposed MPXV stick and backbone models highlight AA variants (labeled) in non-gold colors. The MPXV surface models superimposed over the gold templates reveal AA variants in orange. For example, the MPXV A29 models display four labeled AA variants (ILE, CYS, CYS, HIS) on the backbone model and as orange molecules on the surface model.
Protein analysis of the six MPXV antigenic proteins was also conducted. The complete AA sequence of each protein was entered into the “Predict Secondary Structure” tool, and the resulting predicted structure (strand or helix) was displayed above the sequence. Additional “protein information” tracks were selected to view the predicted antigenic potential of an AA or segment (Supplementary Figure S4). A representation of protein analysis using MPXV A29 as an example is shown in Figure 8B. The Kolaskar-Tongaonkar, Surface Probability and Chain Flexibility tracks are predictions of antigenic regions based on 1) hydrophilicity, surface accessibility and flexibility, 2) surface probability, and 3) backbone chain flexibility, respectively [47,48,49,50]. Generally, increased surface accessibility and chain flexibility correlate with antigenicity. A surface residue is usually one having more than 20 Å (2.0 nm) of water-accessible surface area [49]. The surface probability (S) at a specific sequence position (n) for a random hexapeptide sequence is set at 1.0. Probabilities > 1.0 suggest a higher likelihood of the residue being on the surface [49]. For protein chain flexibility, B-Factors (Temperature Factors) are indicative of atomic displacement or flexibility within the protein structure. Higher B-factors (> 1.0) suggest greater flexibility [50]. The Kolaskar-Tongaonkar composite measure defines potential antigenic residues as having an average antigenic propensity (Ap) value ≥ 1.0 for 7 consecutive residues [48]. In Figure 8B, the MPXV A29 full length AA sequence is shown. The truncated A29 segment with the predicted antigenic potential of four variant AAs (ILE, CYS, CYS, HIS) corresponds to the surface model and variants in Figure 8A.
4. Discussion
In this study, we investigated the evolution of MPXV and VACV genomes and compared homologous antigens relevant to current vaccine efficacy and design. The alignment of 1,903 newly deposited MPXV genomes from 30 countries revealed greater phylogenetic diversity compared to our previous study [6]. We identified 25 genomes clustering with Clade I and 1,898 with Clade IIB. Eleven genomes from Sud-Kivu, DRC formed a new sub-lineage within Clade I presumably due to human APOBEC3 [8,9,10,11,51]. Remarkably, the six neutralization determinants encoded by the 2024 novel sub-lineage genome (PP601216) remained conserved compared to the Clade I RefSeq (NC_003310) from 1996. These viral surface proteins, essential for cell attachment and entry, are typically conserved due to their critical function, structural necessity, and strong evolutionary pressures to maintain viral infectivity and transmissibility. Thus, the six conserved neutralization determinants are ideal candidates as antigens in future vaccine design. Nevertheless, continuous MPXV metagenomic surveillance is essential for detecting changes in viral evolution and dynamics within individuals or populations. Mutations that provide a functional selective advantage could eventually dominate, potentially leading to resistance against current vaccines.
Comparison of the structural models of the six neutralization determinants from MPXV RefSeq (NC_003310) with five VACV and one VARV crystallographic templates revealed a few but significant amino acid differences near potential epitope/paratope binding sites. Quantitatively, four of the MPXV models exhibited approximately 90% identity to the VACV template. MPXV M1 had an almost perfect match at 99%, whereas the top homologous B6 template (variola SPICE) was less than ideal, with only 29.6% identity. Considering the essential functions of these viral surface proteins in cell attachment and entry, it is probable that they have evolved to adapt to host-specific cell membrane proteins and structures, leading to the observed differences. Although imperfect, the long-standing vaccinia vaccines, initially created for smallpox, still play a crucial role in safeguarding humanity against mpox today. Computationally, we have showcased the simplicity and effectiveness of in-silico modeling for investigating structural differences. These findings can guide experimental testing and validate their significance through antigen-antibody (Ag-Ab) binding assays, which are essential for vaccine design.
A comparative analysis of three generations of VACV genomes with HPXV and CPXV reference genomes has provided evolutionary insights. It was found that VACV is distinct but phylogenetically closer to HPXV than to CPXV, consistent with prior studies [52,53]. However, the limited availability of HPXV and CPXV genomes from primary hosts, along with the age of the samples, may introduce bias into the findings. In particular, the RefSeq for HPXV is the only field isolate (MNR-76) available in NCBI Virus, collected during a horsepox outbreak in 1976 in Khentii province of northeastern Mongolia [33]. Another HPXV genome (VK05) was sequenced from historical vaccination kits stored at the Mütter Museum in Philadelphia, dating back to the American Civil War era (circa 1859–1873) [34]. The CPXV RefSeq (Brighton Red strain) was isolated in 1937 from a milker in Brighton, England, while the CPXV GRI-90 strain was isolated in 1990 from a four-year-old Russian child infected by a mole (mammal) [54,55]. Regarding the VACV genomes, we noted variations in sequence length, as well as deletions and rearrangements of synteny blocks at the terminal regions. VACV MVA-BN, a third-generation vaccine, exhibited the shortest genome with a truncated distal synteny block, but still retained the six neutralization determinants. These genomic changes underscore the profound effect of serial passage as a process for reductive evolution.
The strength of this study lies in the extensive number of MPXV genomes analyzed, enabling the identification and analysis of novel Clade I genomes. Additionally, the complete coding sequences were utilized for structural modeling and comparative analysis of antigenic proteins. Another key strength is the streamlined CLC workflows for whole genome analysis and structural modeling. The user-friendly, step-by-step process for investigating newly sequenced or publicly deposited samples will facilitate scientific discovery and enhance surveillance and monitoring efforts.
We acknowledge that our study has limitations, particularly the scarcity of fully sequenced, novel Clade I genomes available in the NCBI repository. Small sample sizes may not adequately capture the scope of viral diversity within the population, potentially leading to skewed results. Additionally, experimental crystal structures of the six MPXV surface proteins were not found in the PDB. Crystal structures are crucial for validating our homology-based models for downstream applications such as antigen selection or vaccine design. Despite these limitations, the results of this study have enriched our understanding of MPXV and VACV genome evolution and highlighted structural differences between key neutralization determinants relevant and valuable to vaccine design.
5. Conclusions
This study has advanced our understanding of MPXV and VACV genome evolution and their implications for vaccine design and efficacy. The workflows for viral whole genomic analysis and homology-based structural modeling will facilitate discoveries and advancements in MPXV research and vaccinology.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: A) MPXV phylogram by clades and AN (enlarged) and, B) MPXV phylogram by clades and geo-locations (enlarged), Figure S2: MPXV phylograms of 1,898 genomes grouping with Clade IIB, Figure S3: WGA of VACV, HPXV and, CPXV genomes, Figure S4: MPXV Clade I antigenic protein analysis tracks, Table S1: MPXV database (243 genomes), Table S2: MPXV dataset (1,878 genomes), Table S3: Summary of BLAST hits and structures for MPXV (NC_003310) proteins, Table S4: Summary of BLAST hits and structures for MPXV (PP601216) proteins
Author Contributions
Conceptualization, J.SG.; methodology, J.SG. and Y.W.; database, J.SG.; validation, J.SG. and Y.W.; formal analysis, J.SG., H.C., and Y.W.; investigation, J.SG.; resources, J.SG. and Y.W.; data curation, J.SG., H.C., and Y.W.; writing – original draft preparation, J.SG.; writing – review & editing, J.SG., H.C. and Y.W.; visualization, J.SG. and Y.W.; supervision, J.SG. and Y.W.; project administration, J.SG. and Y.W.; funding acquisition, J.SG. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The MPXV and VACV genomes are publicly available from NCBI Virus (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/) under respective Taxid numbers: 10244 and 10245 (accessed on 26 June 2024). The crystallographic structures used for structural modeling are publicly available from PDB (https://www.rcsb.org/) under respective structure IDs: VACV A27 (3VOP); VACV 27-Fab 1G6 (5EOQ); VACV D8 (5USH); VACV H3 (5EJ0); VACV L1 (2I9L); VACV A33 (4LU5); VARV (5FOB) (accessed on 23 August 2024).
Acknowledgments
We sincerely thank the CDC PHIL for providing images in the public domain as an educational resource. CDC PHIL Intracellular mpox virions [Photograph] is publicly available online: https://phil.cdc.gov/Details.aspx?pid=26503 (accessed on 1 August 2024). The graphical abstract and figure icons (depicting viruses, a horse, and a cow) were created using BioRender.com.
Conflicts of Interest
The Defense Health Agency (DHA) of the U.S. Department of Defense has licensed the customized MPXV database described herein to QIAGEN Digital Insights. The inventor of the customized taxonomy is J.SG. No potential conflicts of interest were disclosed by the other authors. DHA and QIAGEN had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
AA, amino acid; AP, alignment percentage; APOBEC3, apolipoprotein B mRNA-editing catalytic polypeptide-like 3; ANI, average nucleotide identity; CLC MGM, CLC Microbial Genomics Module; CPXV, Cowpox virus; DRC, Democratic Republic of the Congo; dsDNA, double-stranded DNA; DB, database; EA IND, Expanded Access Investigational New Drug protocol; ICTV, International Committee on Taxonomy of Viruses; MPXV, Monkeypox virus; NCBI, National Center for Biotechnology Information; NJ, Neighbor Joining; ORF, open reading frame; PHEIC, Public Health Emergency of International Concern; PDB, Protein Data Bank; PWC, pairwise comparison; RefSeq, reference sequence; RepSeq, representative sequence; VACV, Vaccinia virus; VARV, Variola virus; WGA, whole genome alignment; WHO, World Health Organization
References
- World Health Organization (WHO). Available online: https://www.who.int/news-room/speeches/item/who-director-general-s-opening-remarks-at-the-ihr-emergency-committee-meeting-regarding-the-upsurge-of-mpox-2024---14-august-2024 (accessed on 14 August 2024).
- World Health Organization (WHO). Smallpox and mpox (orthopoxviruses) vaccine position paper. Weekly Epidemiological Record, 2024, 99 (34), 429-456.
- Shchelkunov, S.N.; Totmenin, A.V.; Babkin, I.V.; Safronov, P.F.; Ryazankina, O.I.; Petrov, N.A.; Gutorov, V.V.; Uvarova, E.A.; Mikheev, M.V.; Sisler, J.R.; et al. Human monkeypox and smallpox viruses: genomic comparison. FEBS Lett. 2001, 509, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Li, G.; Liszewski, M.K.; Atkinson, J.P.; Jahrling, P.B.; Feng, Z.; Schriewer, J.; Buck, C.; Wang, C.; Lefkowitz, E.J.; et al. Virulence differences between monkeypox virus isolates from West Africa and the Congo basin. Virology 2005, 340, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Berthet, N.; Descorps-Declère, S.; Besombes, C.; Curaudeau, M.; Meyong, A.A.N.; Selekon, B.; Labouba, I.; Gonofio, E.C.; Ouilibona, R.S.; Tchetgna, H.D.S.; et al. Genomic history of human monkey pox infections in the Central African Republic between 2001 and 2018. Sci. Rep. 2021, 11, 13085. [Google Scholar] [CrossRef] [PubMed]
- Shen-Gunther, J.; Cai, H.; Wang, Y. A Customized Monkeypox Virus Genomic Database (MPXV DB v1.0) for Rapid Sequence Analysis and Phylogenomic Discoveries in CLC Microbial Genomics. Viruses 2022, 15, 40. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-statement-on-the-press-conference-following-IHR-emergency-committee-regarding-the-multi--country-outbreak-of-monkeypox--23-july-2022 (accessed on day month year).
- Isidro, J.; Borges, V.; Pinto, M.; Sobral, D.; Santos, J.D.; Nunes, A.; Mixão, V.; Ferreira, R.; Santos, D.; Duarte, S.; et al. Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus. Nat. Med. 2022, 28, 1569–1572. [Google Scholar] [CrossRef]
- Chen, Y.; Li, M.; Fan, H. The monkeypox outbreak in 2022: adaptive evolution associated with APOBEC3 may account for. Signal Transduct. Target. Ther. 2022, 7, 323. [Google Scholar] [CrossRef] [PubMed]
- Forni, D.; Cagliani, R.; Pozzoli, U.; Sironi, M. An APOBEC3 Mutational Signature in the Genomes of Human-Infecting Orthopoxviruses. mSphere 2023, 8, e0006223. [Google Scholar] [CrossRef] [PubMed]
- O’toole. ; Neher, R.A.; Ndodo, N.; Borges, V.; Gannon, B.; Gomes, J.P.; Groves, N.; King, D.J.; Maloney, D.; Lemey, P.; et al. APOBEC3 deaminase editing in mpox virus as evidence for sustained human transmission since at least 2016. Science 2023, 382, 595–600. [Google Scholar] [CrossRef]
- Vakaniaki, E.H.; Kacita, C.; Kinganda-Lusamaki, E.; O’toole. ; Wawina-Bokalanga, T.; Mukadi-Bamuleka, D.; Amuri-Aziza, A.; Malyamungu-Bubala, N.; Mweshi-Kumbana, F.; Mutimbwa-Mambo, L.; et al. Sustained human outbreak of a new MPXV clade I lineage in eastern Democratic Republic of the Congo. Nat. Med. 2024, 30, 2791–2795. [Google Scholar] [CrossRef] [PubMed]
- Masirika, L.M.; Kumar, A.; Dutt, M.; Ostadgavahi, A.T.; Hewins, B.; Nadine, M.B.; Steeven, B.K.; Mweshi, F.K.; Mambo, L.M.; Mbiribindi, J.B.; et al. Complete Genome Sequencing, Annotation, and Mutational Profiling of the Novel Clade I Human Mpox Virus, Kamituga Strain. J. Infect. Dev. Ctries. 2024, 18, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Masirika, L.M.; Udahemuka, J.C.; Schuele, L.; Ndishimye, P.; Otani, S.; Mbiribindi, J.B.; Marekani, J.M.; Mambo, L.M.; Bubala, N.M.; Boter, M.; et al. Ongoing mpox outbreak in Kamituga, South Kivu province, associated with monkeypox virus of a novel Clade I sub-lineage, Democratic Republic of the Congo, 2024. Eurosurveillance 2024, 29, 2400106. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Available online: https://www.who.int/publications/m/item/multi-country-outbreak-of-mpox--external-situation-report-40--13-october-2024 (accessed on day month year).
- Chaplin P, Howley P, Meisinger C. (2007). Modified Vaccinia Ankara virus variant (US 7,189,536 B2) USPTO. Available online: https://patentcenter.uspto.gov/applications/10440073/ifw/docs?application= (accessed on day month year).
- Grabenstein, J.D.; Hacker, A. Vaccines against Mpox: MVA-BN and LC16m8. Expert Rev. Vaccines 2024, 23, 796–811. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration (FDA). Available online: https://www.fda.gov/vaccines-blood-biologics/jynneos (accessed on day month year).
- Eto, A.; Saito, T.; Yokote, H.; Kurane, I.; Kanatani, Y. Recent advances in the study of live attenuated cell-cultured smallpox vaccine LC16m8. Vaccine 2015, 33, 6106–6111. [Google Scholar] [CrossRef] [PubMed]
- Kenner, J.; Cameron, F.; Empig, C.; Jobes, D.V.; Gurwith, M. LC16m8: An attenuated smallpox vaccine. Vaccine 2006, 24, 7009–7022. [Google Scholar] [CrossRef]
- Morikawa, S.; Sakiyama, T.; Hasegawa, H.; Saijo, M.; Maeda, A.; Kurane, I.; Maeno, G.; Kimura, J.; Hirama, C.; Yoshida, T.; et al. An Attenuated LC16m8 Smallpox Vaccine: Analysis of Full-Genome Sequence and Induction of Immune Protection. J. Virol. 2005, 79, 11873–11891. [Google Scholar] [CrossRef]
- Monath TP, Caldwell JR, Mundt W, Fusco J, Johnson CS, Buller M, Liu J, Gardner B, Downing G, Blum PS, Kemp T, Nichols R, Weltzin R. ACAM2000 clonal Vero cell culture vaccinia virus (New York City Board of Health strain)--a second-generation smallpox vaccine for biological defense. Int J Infect Dis. 2004 Oct;8 Suppl 2:S31-44. [CrossRef] [PubMed]
- Center for Disease Control and Prevention (CDC). Available online: https://www.cdc.gov/mpox/hcp/vaccine-considerations/vaccination-overview.html (accessed on day month year).
- Jacobs, B.L.; Langland, J.O.; Kibler, K.V.; Denzler, K.L.; White, S.D.; Holechek, S.A.; Wong, S.; Huynh, T.; Baskin, C.R. Vaccinia virus vaccines: Past, present and future. Antivir. Res. 2009, 84, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Davies DH, Schmidt CS, Sheikh NA. Concept and Scope of Modern Vaccines. In Vaccinology Principles and Practice; Morrow WJW, Sheikh NA, Schmidt CS, Ed.; Wiley-Blackwell: West Sussex, UK, 2012; pp. 3–11. [Google Scholar]
- Boylston, A. The origins of inoculation. J. R. Soc. Med. 2012, 105, 309–313. [Google Scholar] [CrossRef]
- Fang, Q.; Yang, L.; Zhu, W.; Liu, L.; Wang, H.; Yu, W.; Xiao, G.; Tien, P.; Zhang, L.; Chen, Z. Host range, growth property, and virulence of the smallpox vaccine: Vaccinia virus Tian Tan strain. Virology 2005, 335, 242–251. [Google Scholar] [CrossRef]
- Indiana University Libraries. Available online: https://collections.libraries.indiana.edu/iulibraries/s/smallpox-vaccine-exhibit/page/great-qing (accessed on day month year).
- Evered, E. .; Evered, K.T. Mandating immunity in the Ottoman Empire: A history of public health education and compulsory vaccination. Heliyon 2020, 6, e05488. [Google Scholar] [CrossRef]
- Carter, D. Sequence-based computational approaches to vaccine discovery and design. In Vaccinology Principles and Practice; Morrow WJW, Sheikh NA, Schmidt CS, Ed.; Wiley-Blackwell: West Sussex, UK, 2012; pp. 133–149. [Google Scholar]
- Barranco, C. The first live attenuated vaccines. Nature Portfolio. 2020 Nov S7. doi: https://www.nature.com/articles/d42859-020-00008-5.
- Wolf, Y.I.; Koonin, E.V. Genome reduction as the dominant mode of evolution. BioEssays 2013, 35, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Tulman, E.R.; Delhon, G.; Afonso, C.L.; Lu, Z.; Zsak, L.; Sandybaev, N.T.; Kerembekova, U.Z.; Zaitsev, V.L.; Kutish, G.F.; Rock, D.L. Genome of Horsepox Virus. J. Virol. 2006, 80, 9244–9258. [Google Scholar] [CrossRef] [PubMed]
- Duggan, A.T.; Klunk, J.; Porter, A.F.; Dhody, A.N.; Hicks, R.; Smith, G.L.; Humphreys, M.; McCollum, A.M.; Davidson, W.B.; Wilkins, K.; et al. The origins and genomic diversity of American Civil War Era smallpox vaccine strains. Genome Biol. 2020, 21, 175. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liang, M.; Evans, D.H. Genomic analysis of vaccinia virus strain TianTan provides new insights into the evolution and evolutionary relationships between Orthopoxviruses. Virology 2013, 442, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Landsberger, M.; Quick, J.; Mercer, J. Coding-Complete Genome Sequences of Copenhagen and Copenhagen-Derived vP811 Strains of Vaccinia Virus Isolated from Cell Culture. Genome Announc. 2023, 12, e0009023. [Google Scholar] [CrossRef] [PubMed]
- Riccardo, V.; Pablo, G.-C. Neutralization Determinants on Poxviruses. Viruses 2023, 15, 2396. [Google Scholar] [CrossRef] [PubMed]
- Moss B, Smith GL. Poxviridae: The Viruses and Their Replication. In Fields Virology. DNA Viruses., 7th ed.; Howley PM, Knipe DM., Ed.; Wolters Kluwer: Philadelphia, USA, 2022; pp. 573–613. [Google Scholar]
- Condit, RC. Poxviruses. In Fundamental of Molecular Virology., 2nd ed.; Acheson NH. Ed.; John Wiley & Son: Hoboken, USA, 2011; pp. 312–324. [Google Scholar]
- National Center for Biotechnology Information (NCBI). Virus. Available online: https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/ (accessed on day month year).
- Happi, C.; Adetifa, I.; Mbala, P.; Njouom, R.; Nakoune, E.; Happi, A.; Ndodo, N.; Ayansola, O.; Mboowa, G.; Bedford, T.; et al. Urgent need for a non-discriminatory and non-stigmatizing nomenclature for monkeypox virus. PLOS Biol. 2022, 20, e3001769. [Google Scholar] [CrossRef] [PubMed]
- CDC Public Health Image Library (PHIL). Colorized transmission electron microscopic image of mpox virus particles. 2022. Available online: https://phil.cdc.gov/Details.aspx?pid=26503 (accessed on day month year).
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et. al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. Available online: http://www.rcsb.org/ (accessed on 23 August 2024). [CrossRef]
- Brien, S.C.; LeBreton, M.; Doty, J.B.; Mauldin, M.R.; Morgan, C.N.; Pieracci, E.G.; Ritter, J.M.; Matheny, A.; Tafon, B.G.; Tamoufe, U.; et al. Clinical Manifestations of an Outbreak of Monkeypox Virus in Captive Chimpanzees in Cameroon, 2016. J. Infect. Dis. 2024, 229, S275–S284. [Google Scholar] [CrossRef]
- NCBI GenBank. Available online: https://www.ncbi.nlm.nih.gov/nuccore/OP498046.1 (accessed on day month year).
- Qiagen Digital Insights: CLC Genomics Workbench 24.0.2 Find and Model Structure. Available online: https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Find_Model_Structure.html (accessed on day month year).
- Qiagen Digital Insights: CLC Genomics Workbench 24.0.2 Antigenicity. Available online: https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Antigenicity.html (accessed on day month year).
- Kolaskar, A.S.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef]
- A Emini, E.; Hughes, J.V.; Perlow, D.S.; Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839. [Google Scholar] [CrossRef] [PubMed]
- Karplus, P.A.; Schulz, G.E. Prediction of chain flexibility in proteins. Sci. Nat. 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Sadeghpour, S.; Khodaee, S.; Rahnama, M.; Rahimi, H.; Ebrahimi, D. Human APOBEC3 Variations and Viral Infection. Viruses 2021, 13, 1366. [Google Scholar] [CrossRef] [PubMed]
- Schrick, L.; Tausch, S.H.; Dabrowski, P.W.; Damaso, C.R.; Esparza, J.; Nitsche, A. An Early American Smallpox Vaccine Based on Horsepox. New Engl. J. Med. 2017, 377, 1491–1492. [Google Scholar] [CrossRef] [PubMed]
- Molteni, C.; Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Genetic ancestry and population structure of vaccinia virus. npj Vaccines 2022, 7, 1–9. [Google Scholar] [CrossRef]
- Downie, AW. The Immunological Relationship of the Virus of Spontaneous Cowpox to Vaccinia Virus. Br J Exp Pathol. 1939 Apr;20(2):158–76.
- Marennikova, S.S.; Gashnikov, P.V.; A Zhukova, O.; I Riabchikova, E.; Strel'Tsov, V.V.; I Riazankina, O.; Chekunova, E.V.; Ianova, N.N.; Shchelkunov, S.N. [The biotype and genetic characteristics of an isolate of the cowpox virus causing infection in a child]. . 1996, 6–10. [Google Scholar]
Figure 1.
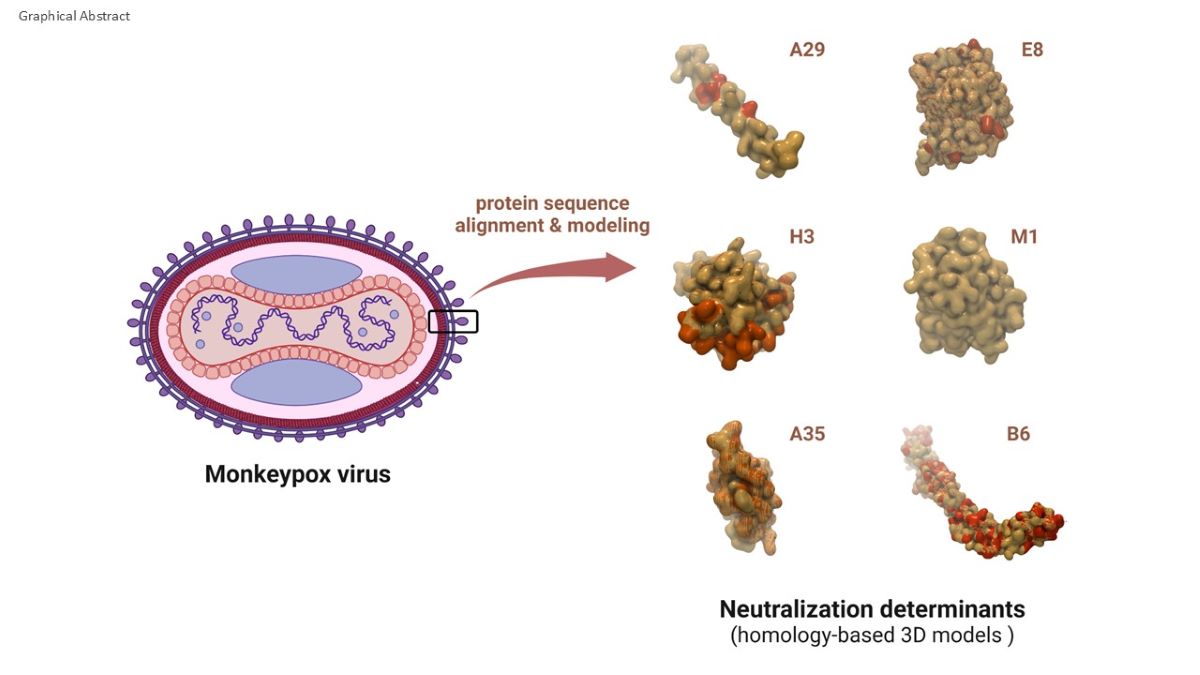
Monkeypox viral lifecycle and antigenic surface proteins. A) The viral lifecycle comprises several distinct stages: attachment to the host cell, entry, uncoating, protein expression, DNA replication, assembly, maturation, and release. Pox virions exist as either infectious Mature Virions (MV) or the less abundant Enveloped Virions (EV) [38,39]. B) The MV features an outer membrane composed of lipids and viral proteins, while the EV is encased in an additional lipid bilayer with unique virus-encoded proteins. The proteins in the EV envelope (A35, B6) and MV membrane (A29, E8, H3, M1) are immunogenic and can be neutralized by cross-reactive antibodies derived from vaccination with attenuated vaccinia virus [37,38]. EC, ectodomain; IV, intraviral domain; TM, transmembrane domain (figure created with BioRender.com).
Figure 1.
Monkeypox viral lifecycle and antigenic surface proteins. A) The viral lifecycle comprises several distinct stages: attachment to the host cell, entry, uncoating, protein expression, DNA replication, assembly, maturation, and release. Pox virions exist as either infectious Mature Virions (MV) or the less abundant Enveloped Virions (EV) [38,39]. B) The MV features an outer membrane composed of lipids and viral proteins, while the EV is encased in an additional lipid bilayer with unique virus-encoded proteins. The proteins in the EV envelope (A35, B6) and MV membrane (A29, E8, H3, M1) are immunogenic and can be neutralized by cross-reactive antibodies derived from vaccination with attenuated vaccinia virus [37,38]. EC, ectodomain; IV, intraviral domain; TM, transmembrane domain (figure created with BioRender.com).
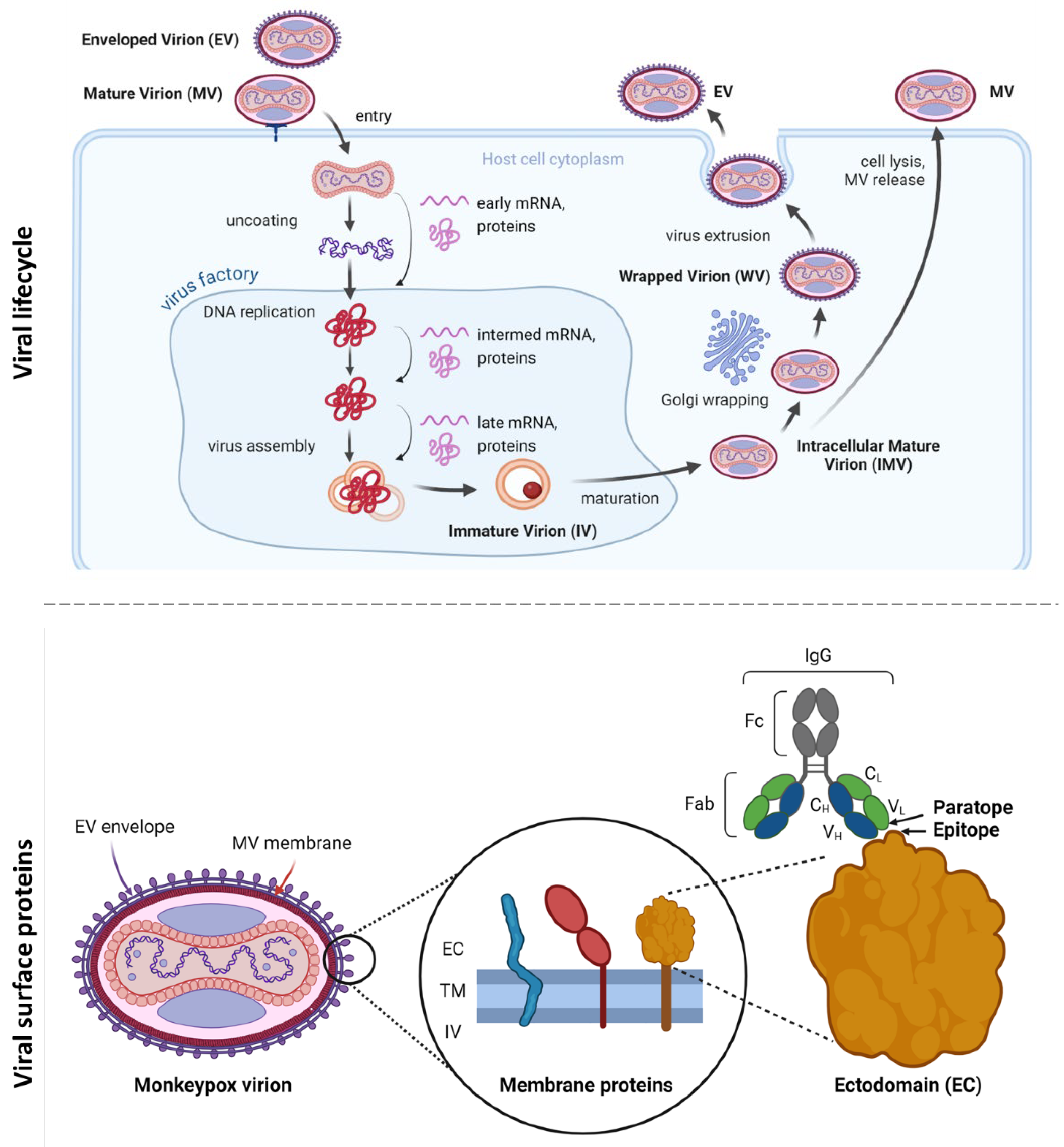
Figure 2.
Representative Monkeypox virus genome. The linear, reference genome (NC_003310) from the ZAI-96-I-16 (MPV-ZAI) isolate was collected from a patient during the 1996 outbreak in Zaire (now DRC) [3,4]. This 196,858-bp double-stranded DNA sequence encodes 190 open reading frames (ORFs), featuring a highly conserved central region and bilateral terminal variable regions enclosed in hairpin loops. ORFs (A29L, A35R, B6R, E8L, H3L, M1R) encoding the antigenic proteins, homologous to those of the Vaccinia Virus, are indicated by red arrows [37]. Three genes that are either deleted (D14L) or mutated (B10R and B14R) in the less virulent, prototypic Clade II MPXV isolate (SL-V70) are marked by yellow triangles [4]. Electron micrograph of two MPXV virions reveals the genome-containing dumbbell-shaped inner core [42].
Figure 2.
Representative Monkeypox virus genome. The linear, reference genome (NC_003310) from the ZAI-96-I-16 (MPV-ZAI) isolate was collected from a patient during the 1996 outbreak in Zaire (now DRC) [3,4]. This 196,858-bp double-stranded DNA sequence encodes 190 open reading frames (ORFs), featuring a highly conserved central region and bilateral terminal variable regions enclosed in hairpin loops. ORFs (A29L, A35R, B6R, E8L, H3L, M1R) encoding the antigenic proteins, homologous to those of the Vaccinia Virus, are indicated by red arrows [37]. Three genes that are either deleted (D14L) or mutated (B10R and B14R) in the less virulent, prototypic Clade II MPXV isolate (SL-V70) are marked by yellow triangles [4]. Electron micrograph of two MPXV virions reveals the genome-containing dumbbell-shaped inner core [42].
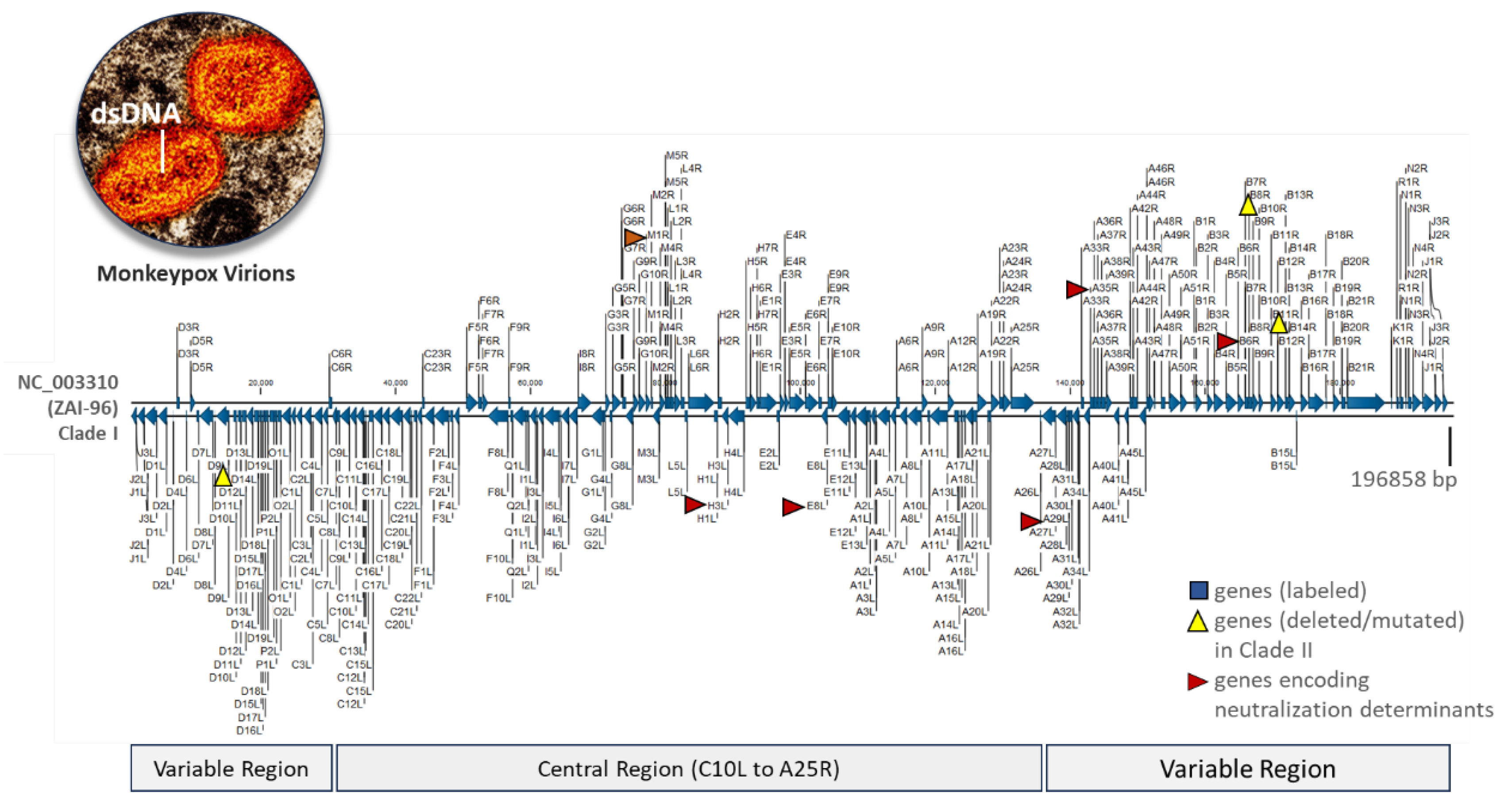
Figure 3.
Bioinformatics methods (A) CLC Microbial Genomics Module, database and datasets used for Monkeypox Virus (MPXV), Vaccinia Virus (VACV) Whole Genome Alignment (WGA) and protein analysis. Primary workflows and tools used for this study are designated by the blue virus icon. A MPXV dataset of 25 new Clade I genomes is shown; (B) WGA workflow steps (1-4) with parameter settings for WGA (bold) and annotation, e.g., MPXV REF genome; (C) The pairwise comparison workflow uses the WGA file as input to measure genome similarity and produces a matrix as output.
Figure 3.
Bioinformatics methods (A) CLC Microbial Genomics Module, database and datasets used for Monkeypox Virus (MPXV), Vaccinia Virus (VACV) Whole Genome Alignment (WGA) and protein analysis. Primary workflows and tools used for this study are designated by the blue virus icon. A MPXV dataset of 25 new Clade I genomes is shown; (B) WGA workflow steps (1-4) with parameter settings for WGA (bold) and annotation, e.g., MPXV REF genome; (C) The pairwise comparison workflow uses the WGA file as input to measure genome similarity and produces a matrix as output.
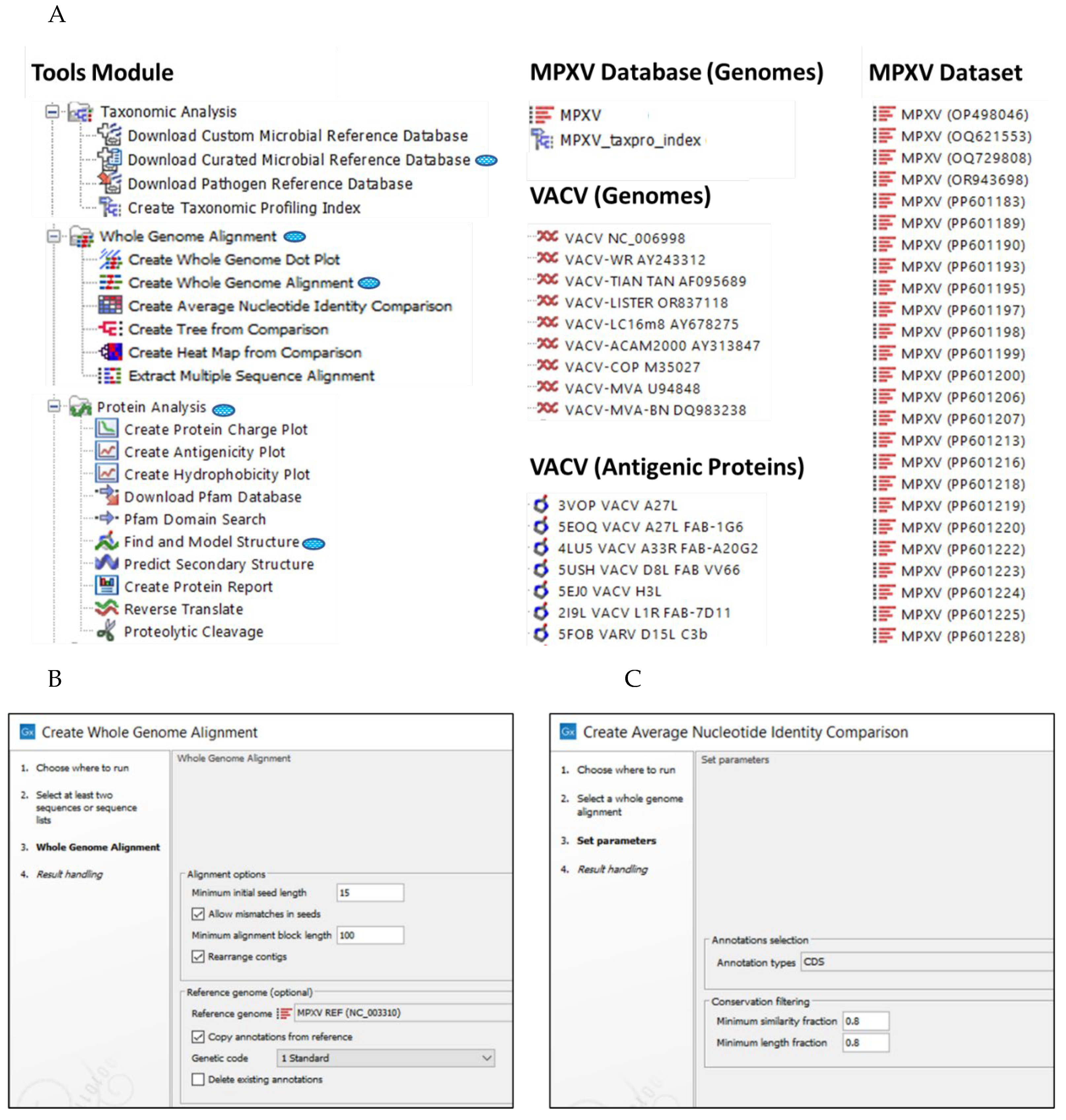
Figure 4.
Phylograms of MPXV genomes for visualization of evolutionary and geo-temporal relationships (A) Phylogram of complete MPXV genomes (n = 243) by clades. Clades I and II originated from Central and West Africa, respectively, with divergent branches. The sub-lineages (groups) reveal the relatedness of its member samples. Three attributes of each genome (sequence AN, 3-letter country code, and collection year) are displayed as the outermost ring. Arrowheads indicate the MPXV reference sequences (RefSeq) for Clades I and II, as well as the representative sequence (RepSeq) (PP601216) from the novel sub-lineage; (B) The radial phylogram of Clade I MPXV genomes, annotated with country and city/province attributes, reveals phylogeography. The map links the colors of the clades and groups to respective countries and cities/provinces (drop pins). The ANI NJ unrooted trees were constructed from the pairwise comparison table.
Figure 4.
Phylograms of MPXV genomes for visualization of evolutionary and geo-temporal relationships (A) Phylogram of complete MPXV genomes (n = 243) by clades. Clades I and II originated from Central and West Africa, respectively, with divergent branches. The sub-lineages (groups) reveal the relatedness of its member samples. Three attributes of each genome (sequence AN, 3-letter country code, and collection year) are displayed as the outermost ring. Arrowheads indicate the MPXV reference sequences (RefSeq) for Clades I and II, as well as the representative sequence (RepSeq) (PP601216) from the novel sub-lineage; (B) The radial phylogram of Clade I MPXV genomes, annotated with country and city/province attributes, reveals phylogeography. The map links the colors of the clades and groups to respective countries and cities/provinces (drop pins). The ANI NJ unrooted trees were constructed from the pairwise comparison table.
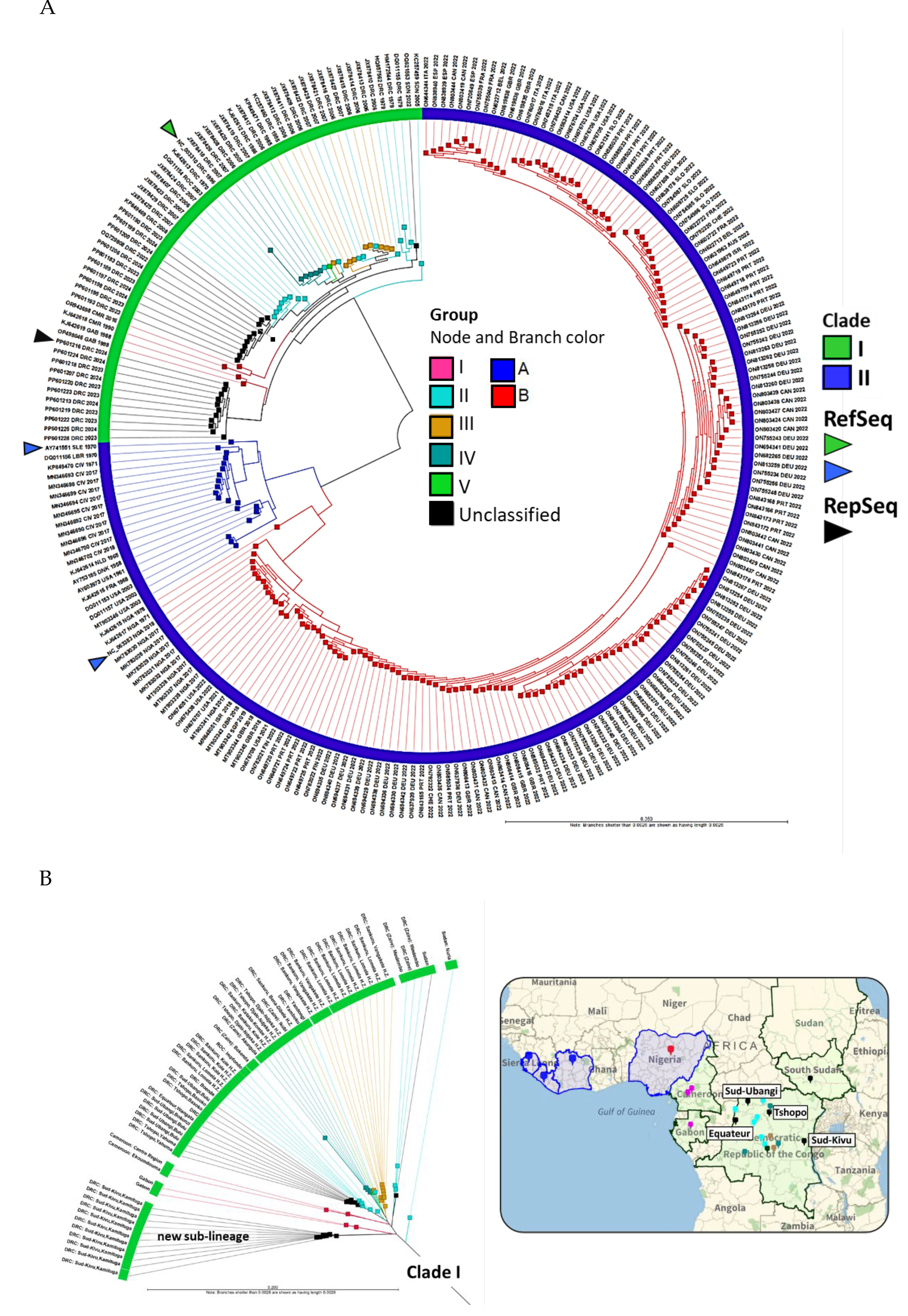
Figure 5.
Comparative analysis of representative Vaccinia, Cowpox, and Horsepox viral genomes (A) The phylogenetic tree depicts the genetic distances between VACV and ancestral zoonotic pox genomes, indicating a closer relationship to HPXV compared to CPXV (unrooted AP NJ tree). Within the VACV clade, genetic variations are apparent, with divergent branches. The labels display four attributes of each genome (virus-strain, sequence AN, 3-letter country code, and approximate collection year); (B) The pairwise comparison table reveals alignment statistics. The VACV RefSeq (NC_006998) exhibited greater similarity to HPXV compared to CPXV, with alignment percentages of 87% vs 85%, respectively. The alignment similarity between VACV genomes exceeded 87%, and within these aligned regions, the nucleotide identity surpassed 98%. The current Bavarian Nordic vaccine (VACV-MVA-BN) compared to its ancestral Modified Vaccinia Ankara genome was highly conserved, with over 99% AP and ANI (red rectangle). AP, alignment percentage; ANI, average nucleotide identity; CPXV, Cowpox virus; HPXV, Horsepox virus; NJ, neighbor-joining; VACV, Vaccinia virus.
Figure 5.
Comparative analysis of representative Vaccinia, Cowpox, and Horsepox viral genomes (A) The phylogenetic tree depicts the genetic distances between VACV and ancestral zoonotic pox genomes, indicating a closer relationship to HPXV compared to CPXV (unrooted AP NJ tree). Within the VACV clade, genetic variations are apparent, with divergent branches. The labels display four attributes of each genome (virus-strain, sequence AN, 3-letter country code, and approximate collection year); (B) The pairwise comparison table reveals alignment statistics. The VACV RefSeq (NC_006998) exhibited greater similarity to HPXV compared to CPXV, with alignment percentages of 87% vs 85%, respectively. The alignment similarity between VACV genomes exceeded 87%, and within these aligned regions, the nucleotide identity surpassed 98%. The current Bavarian Nordic vaccine (VACV-MVA-BN) compared to its ancestral Modified Vaccinia Ankara genome was highly conserved, with over 99% AP and ANI (red rectangle). AP, alignment percentage; ANI, average nucleotide identity; CPXV, Cowpox virus; HPXV, Horsepox virus; NJ, neighbor-joining; VACV, Vaccinia virus.
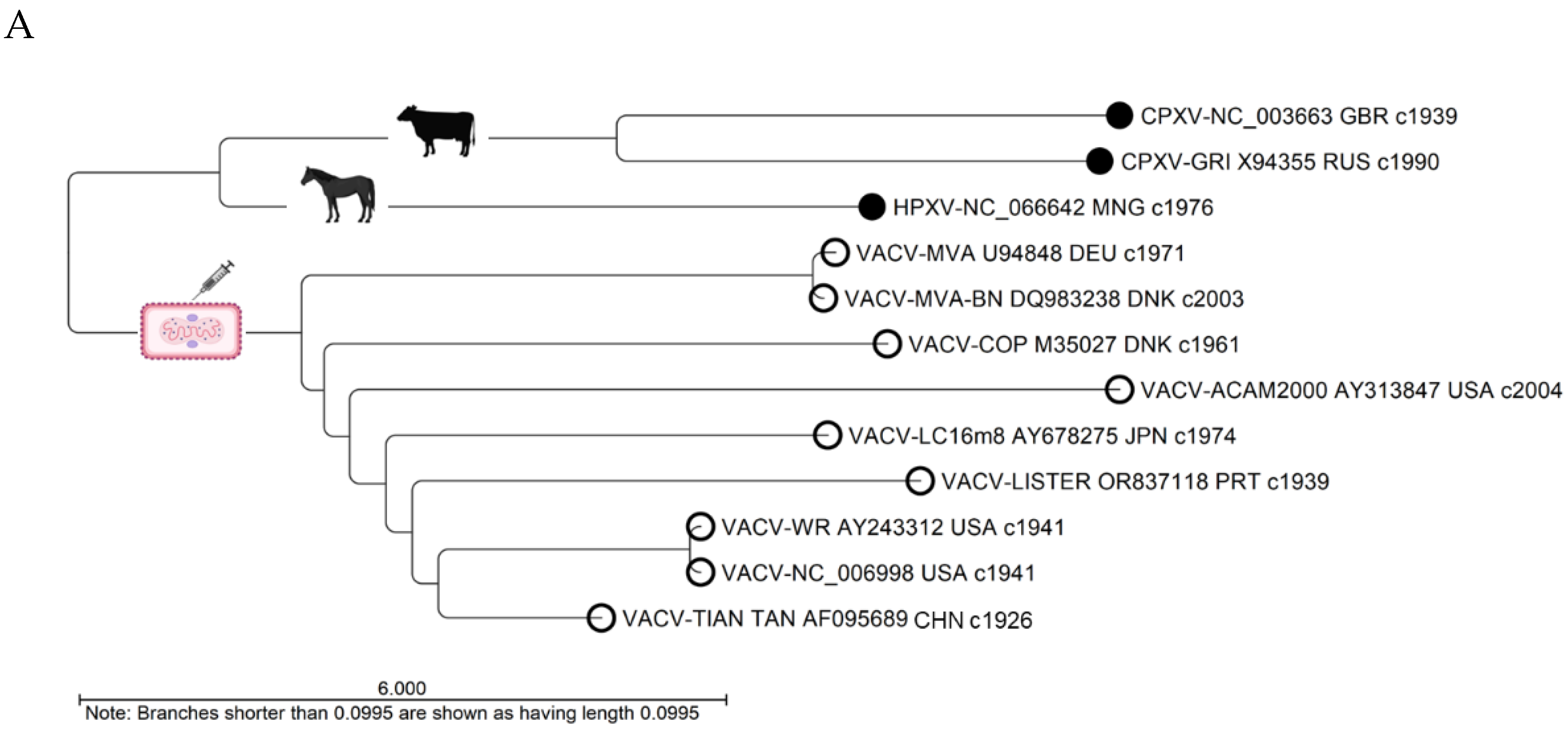
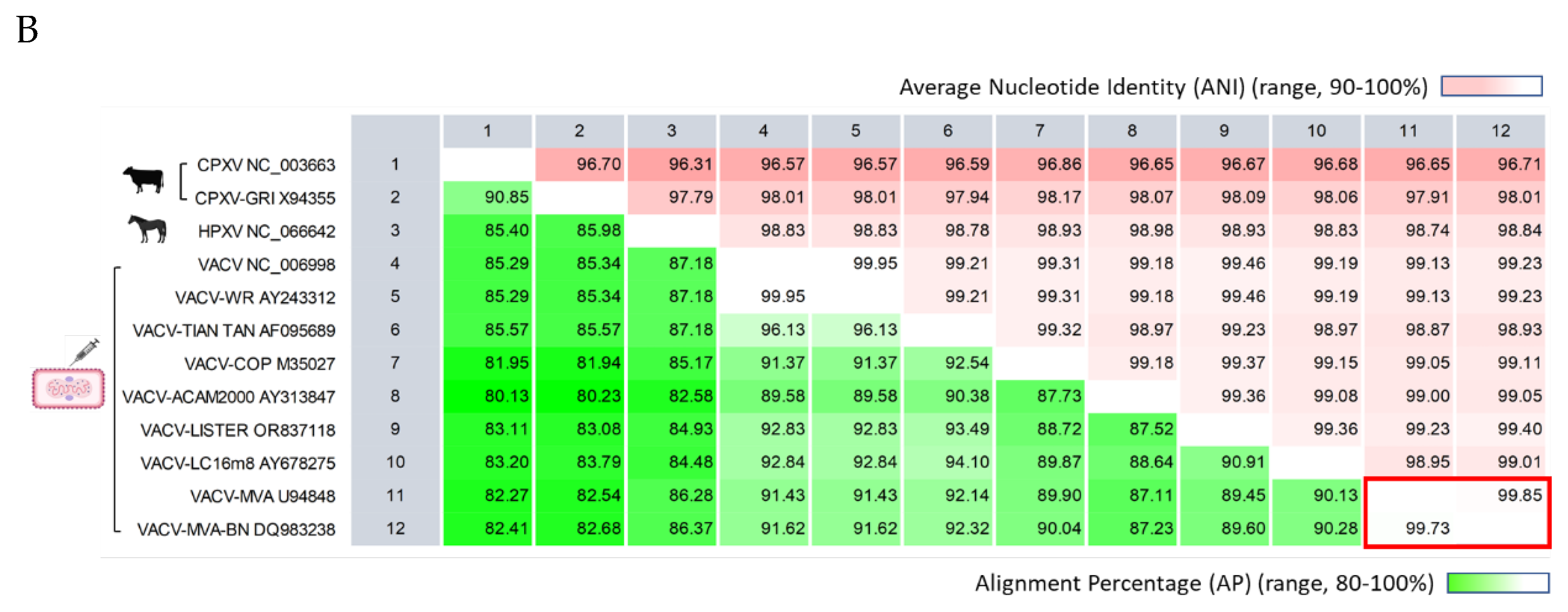
Figure 6.
Figure 6. Comparative analysis of representative Monkeypox and Vaccinia viral genomes (A) WGA revealed a highly conserved central region for representative MPXV Clades I and II genomes (green synteny blocks). Significant variability was observed at the distal ends, before 6,500 bp and beyond 190,000 bp (black lines). In comparison, the VACV-MVA-BN and VACV-LC16m8 are shortened, with synteny blocks at the distal ends being truncated, deleted or rearranged (pink, blue and brown synteny blocks); (B) The unrooted AP NJ tree illustrates the genetic distances between representative MPXV and VACV genomes, forming two distinct clades. Genetic differences are evident between the MPXV Clades and current vaccines (VACV-MVA-BN and VACV-LC16m8); (C) Pairwise comparison between the MPXV Clade I and VACV MVA-BN genomes quantified alignment similarity at over 85-86% and ANI at over 97% (red rectangles). The statistics for VACV-LC16m8 were nearly identical to those for VACV MVA-BN. AP, alignment percentage; ANI, average nucleotide identity; NJ, neighbor-joining; VACV, Vaccinia virus.
Figure 6.
Figure 6. Comparative analysis of representative Monkeypox and Vaccinia viral genomes (A) WGA revealed a highly conserved central region for representative MPXV Clades I and II genomes (green synteny blocks). Significant variability was observed at the distal ends, before 6,500 bp and beyond 190,000 bp (black lines). In comparison, the VACV-MVA-BN and VACV-LC16m8 are shortened, with synteny blocks at the distal ends being truncated, deleted or rearranged (pink, blue and brown synteny blocks); (B) The unrooted AP NJ tree illustrates the genetic distances between representative MPXV and VACV genomes, forming two distinct clades. Genetic differences are evident between the MPXV Clades and current vaccines (VACV-MVA-BN and VACV-LC16m8); (C) Pairwise comparison between the MPXV Clade I and VACV MVA-BN genomes quantified alignment similarity at over 85-86% and ANI at over 97% (red rectangles). The statistics for VACV-LC16m8 were nearly identical to those for VACV MVA-BN. AP, alignment percentage; ANI, average nucleotide identity; NJ, neighbor-joining; VACV, Vaccinia virus.
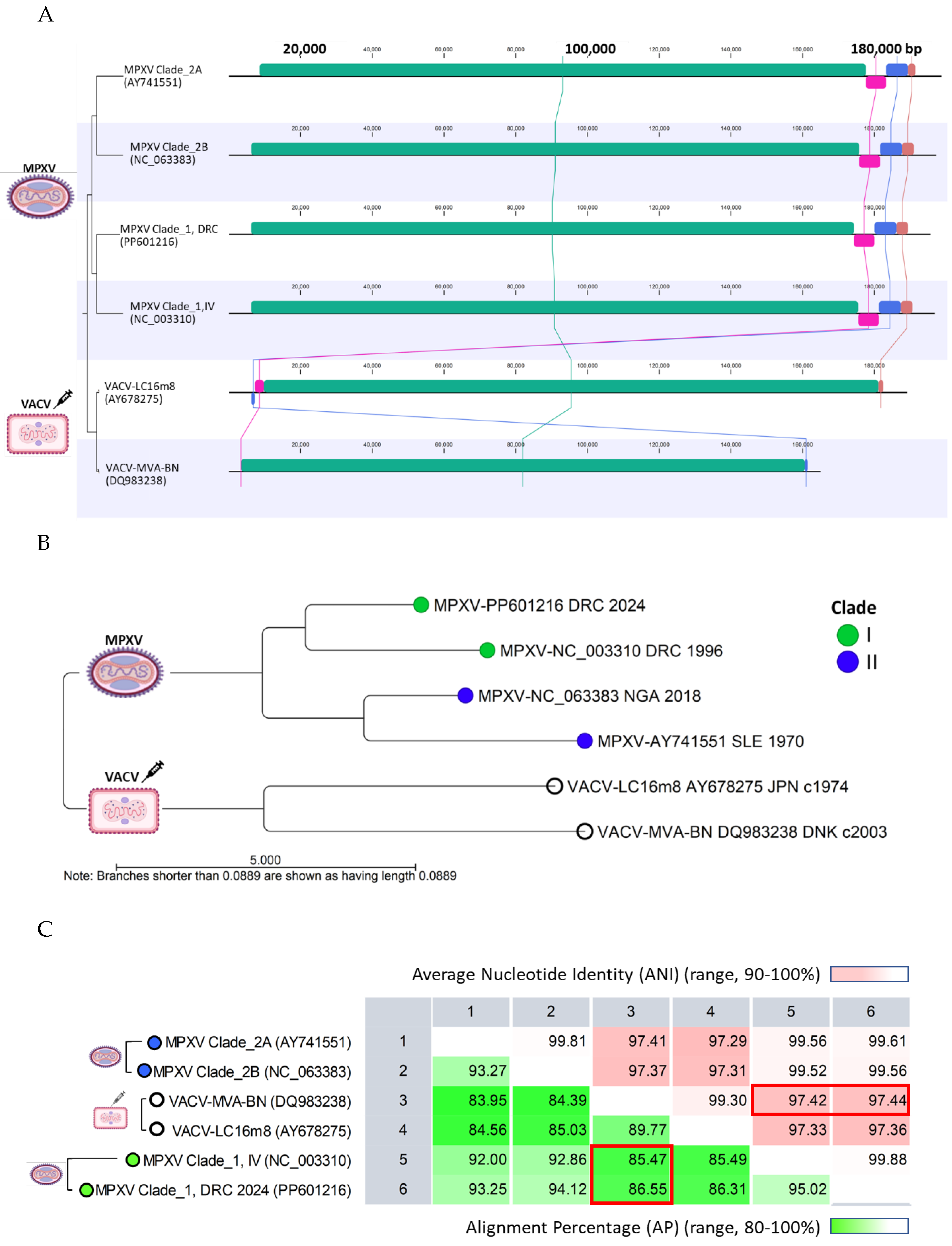
Figure 7.
Vaccinia and variola virus template structures. A collage of five vaccinia and one variola virus antigenic ectodomains used as templates for MPXV 3D modeling. Each ectodomain is shown as a surface model (left) and an AA-labeled backbone model (right). The Fab regions of monoclonal antibodies are depicted as backbone models, with VH (blue) or VL (green) binding to the surface ectodomains. VACV A33 is shown as a homo-dimer. For VACV H3 and VARV D15, only an ectodomain and an ectodomain-C3b complex, respectively, were available in the PDB. (PDB Entry IDs listed in Data Availability Statement).
Figure 7.
Vaccinia and variola virus template structures. A collage of five vaccinia and one variola virus antigenic ectodomains used as templates for MPXV 3D modeling. Each ectodomain is shown as a surface model (left) and an AA-labeled backbone model (right). The Fab regions of monoclonal antibodies are depicted as backbone models, with VH (blue) or VL (green) binding to the surface ectodomains. VACV A33 is shown as a homo-dimer. For VACV H3 and VARV D15, only an ectodomain and an ectodomain-C3b complex, respectively, were available in the PDB. (PDB Entry IDs listed in Data Availability Statement).
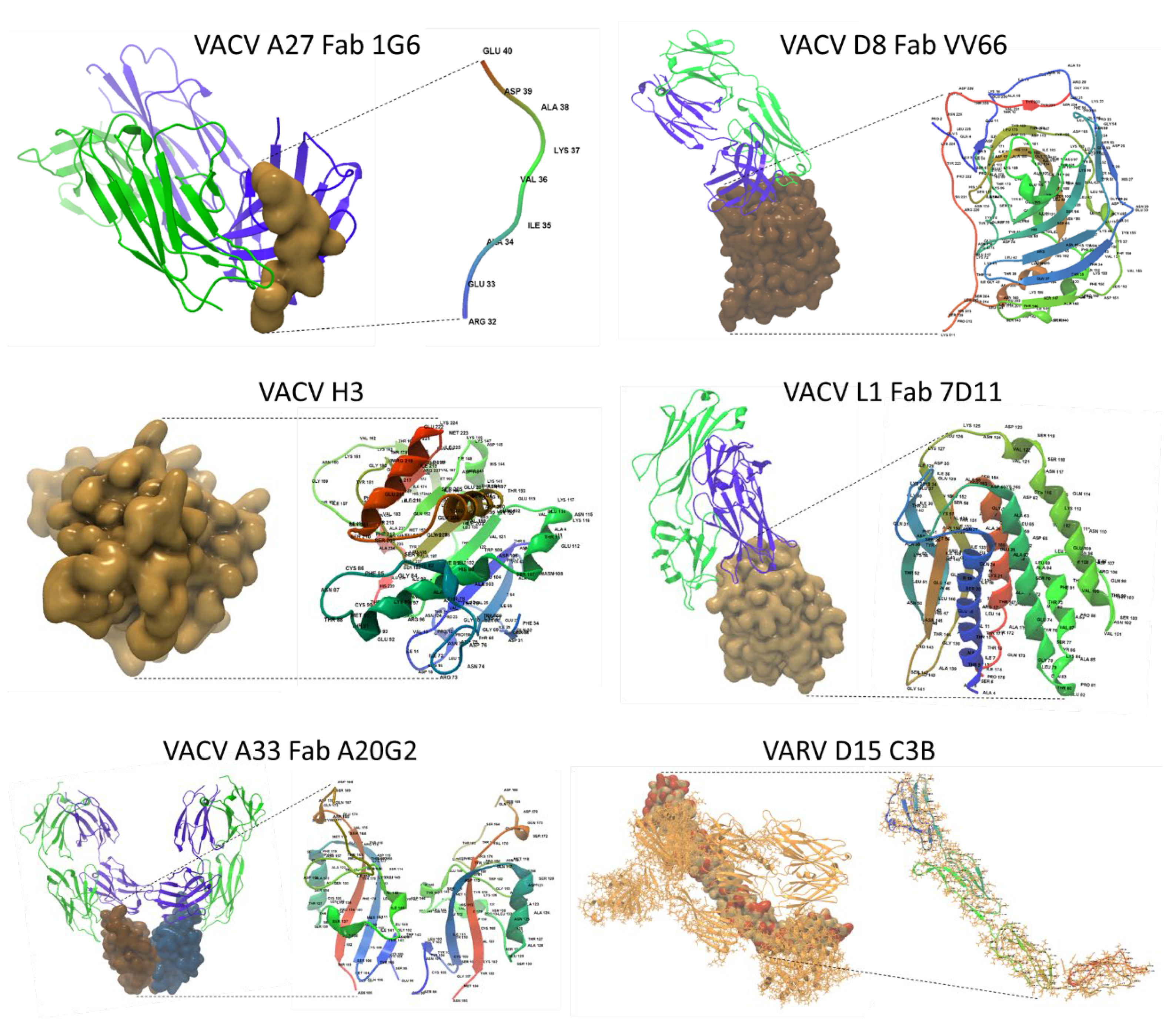
Figure 8.
Monkeypox virus surface antigen models with protein analysis. (A) Collage of six MPXV antigenic ectodomains generated by homology-based 3D modeling. The top ranking VACV and VARV ectodomain templates are shown as stick and backbone models in monochrome gold (left column). The superimposed MPXV backbone models highlight non-synonymous AA in non-gold colors (middle column). The MPXV surface models reveal variant AA (orange) superimposed over the gold template (right column). For MPXV B6R, the highest ranking template was VARV D15, a structure of smallpox inhibitor of complement (SPICE). All models are shown as monomers; (B) Protein analyses are displayed as tracks, showcasing the AA sequence, structural features, antigenicity, and physicochemical properties [47,48,49,50]. The representative, complete sequence of MPXV A29, along with its predicted secondary structure (strand or helix), is displayed over the sequence. The Kolaskar-Tongaonkar, Surface Probability, and Chain Flexibility tracks (colored) indicate predicted antigenic regions based on: 1) hydrophilicity, surface accessibility, and flexibility; 2) surface probability; and 3) backbone chain flexibility, respectively [47,48,49,50]. The zoomed-in truncated segment with the predicted antigenic potential of four variant AA (*) (ILE, CYS, CYS, HIS) corresponds to the surface model and variants depicted in panel (A).
Figure 8.
Monkeypox virus surface antigen models with protein analysis. (A) Collage of six MPXV antigenic ectodomains generated by homology-based 3D modeling. The top ranking VACV and VARV ectodomain templates are shown as stick and backbone models in monochrome gold (left column). The superimposed MPXV backbone models highlight non-synonymous AA in non-gold colors (middle column). The MPXV surface models reveal variant AA (orange) superimposed over the gold template (right column). For MPXV B6R, the highest ranking template was VARV D15, a structure of smallpox inhibitor of complement (SPICE). All models are shown as monomers; (B) Protein analyses are displayed as tracks, showcasing the AA sequence, structural features, antigenicity, and physicochemical properties [47,48,49,50]. The representative, complete sequence of MPXV A29, along with its predicted secondary structure (strand or helix), is displayed over the sequence. The Kolaskar-Tongaonkar, Surface Probability, and Chain Flexibility tracks (colored) indicate predicted antigenic regions based on: 1) hydrophilicity, surface accessibility, and flexibility; 2) surface probability; and 3) backbone chain flexibility, respectively [47,48,49,50]. The zoomed-in truncated segment with the predicted antigenic potential of four variant AA (*) (ILE, CYS, CYS, HIS) corresponds to the surface model and variants depicted in panel (A).
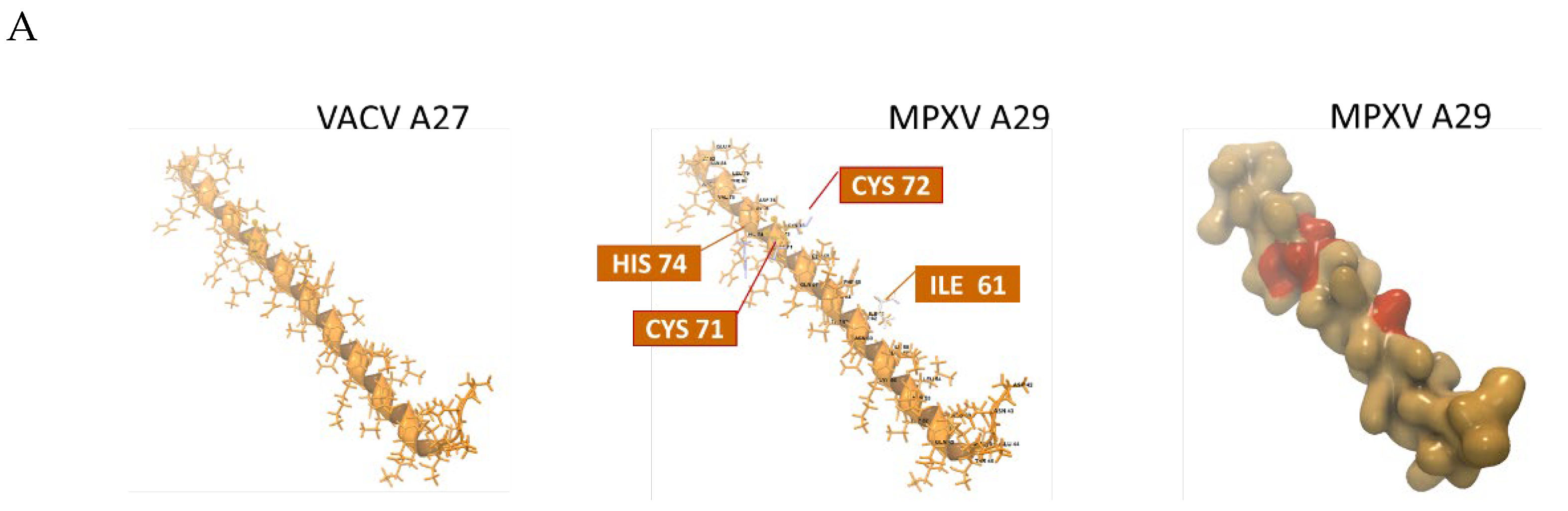
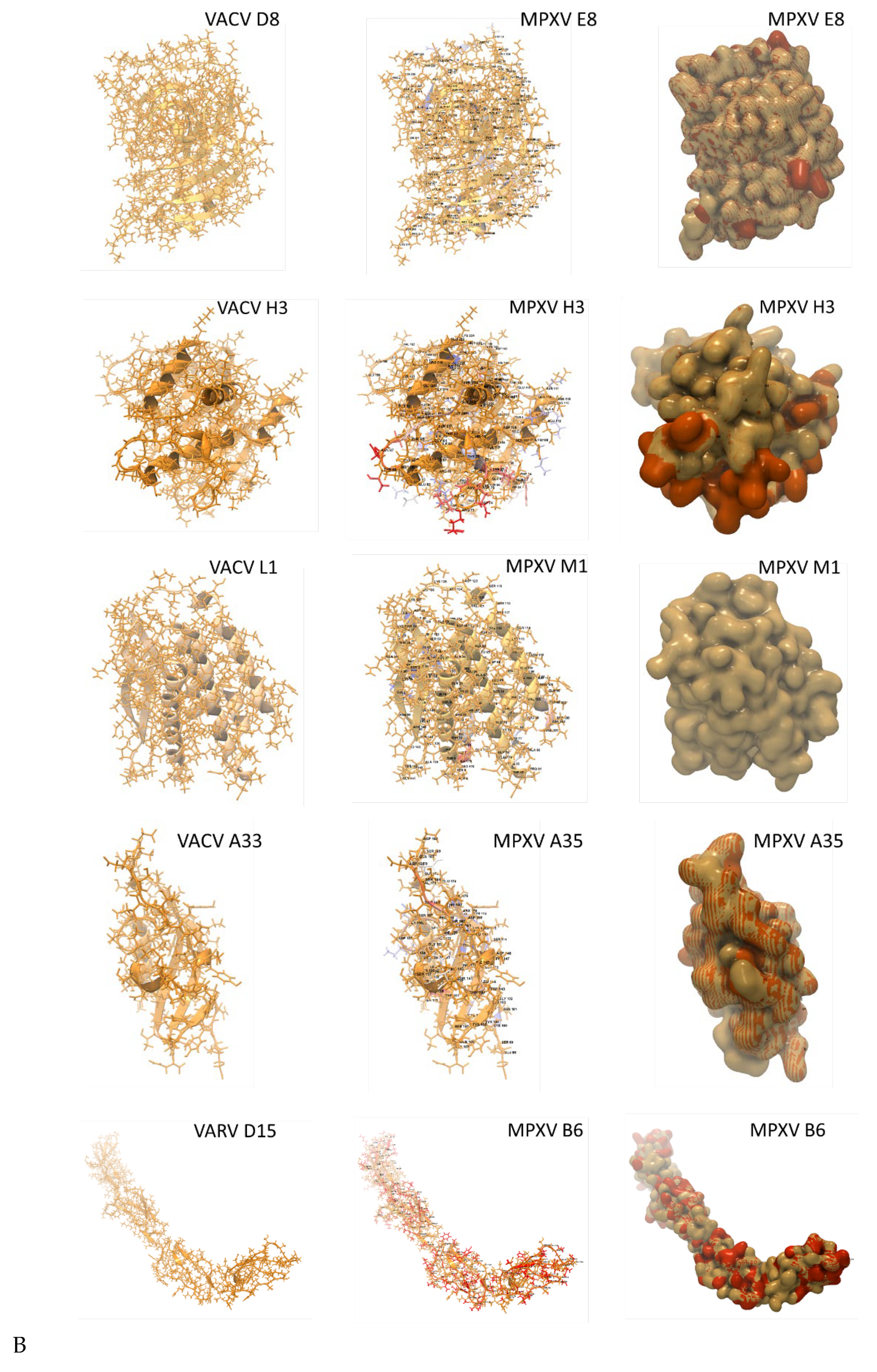
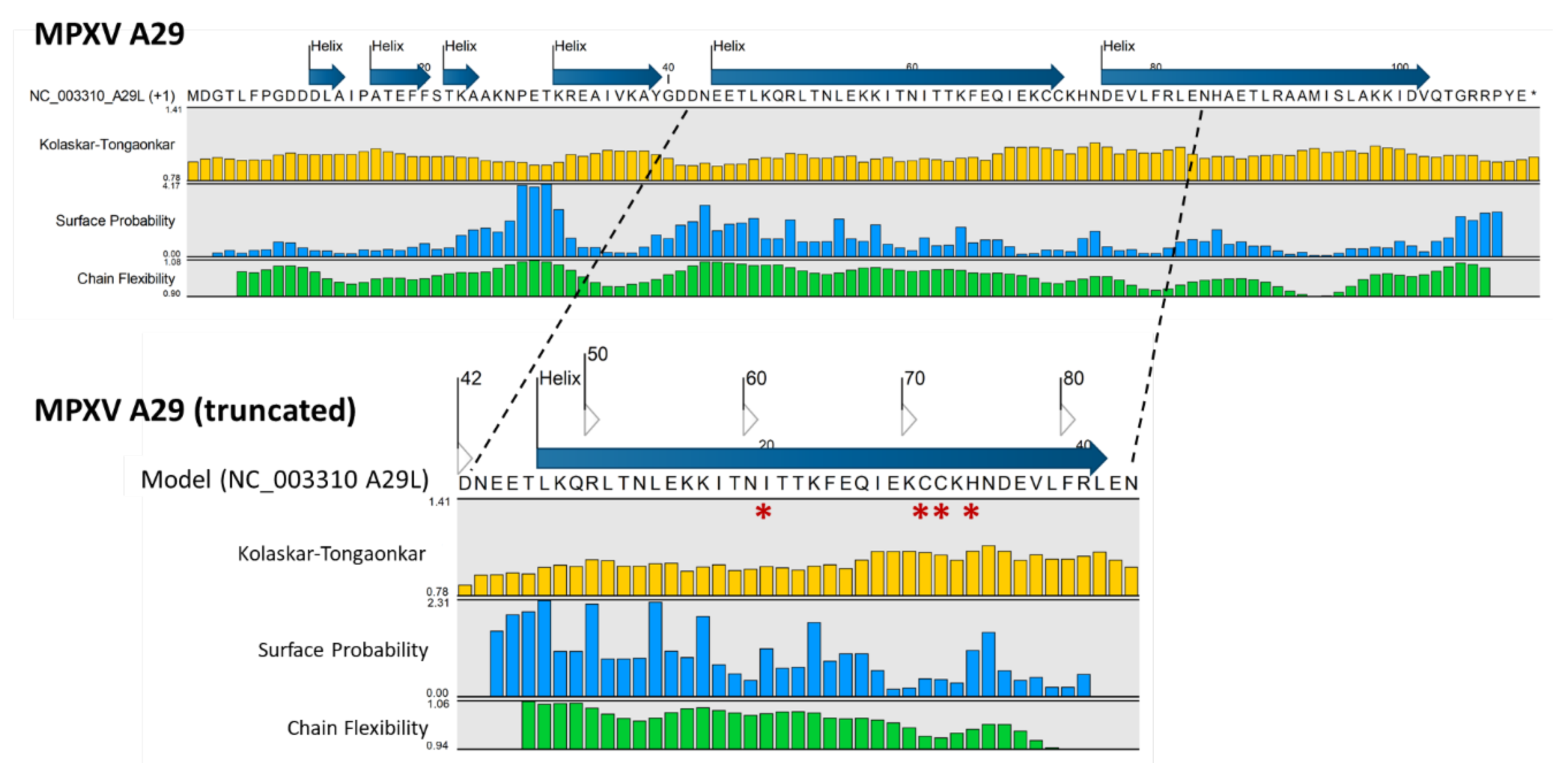

Table 1.
Summary of structures found for MPXV Clade I (NC_003310) antigenic proteins.
| MPXV virion |
MPXV gene (protein) |
Structures found (n) |
Available structures1 |
Rank2 | E-value | Match ID (%) |
Coverage (%) |
|---|---|---|---|---|---|---|---|
| MV | A29L (A29) | 1 | 3VOP3 | 1 | 3.1E-20 | 90.7 | 38.7 |
| MV | E8L (E8) | 1,309 | 5USH4 | 3 | 1.7E-158 | 92.7 | 75.4 |
| MV | H3L (H3) | 1 | 5EJ05 | 1 | 1.6E-142 | 84.4 | 65.2 |
| MV | M1R (M1) | 6 | 2I9L6 | 3 | 4.5E-125 | 99.4 | 68.9 |
| EV | A35R (A35) | 7 | 4LU57 | 5 | 1.1E-50 | 90.4 | 45.1 |
| EV | B6R (B6) | 36 | 5FOB8 | 1 | 1.0E-11 | 29.6 | 67.9 |
EV, enveloped virion; ID, identity; MPXV, Monkeypox virus; MV, mature virion; VACV, vaccinia virus. 1 Available crystallographic structure(s) found in RCSB Protein Data Bank (PDB) [43]. All structures are limited to the ectodomains of the antigenic proteins. The complete table is provided in Supplementary Table S3. 2 The rank of structure(s) found is scored based on homology to the query sequence and the structural quality. Specifically, the variables include three BLAST statistics (E-value, % Match identity, % Coverage), resolution (of crystal structure), and free R-value (R-free of crystal structure) [46]. 3 3VOP: PDB Entry ID for structure of VACV A27 antigenic protein. 4 5USH: PDB Entry ID for structure of VACV D8 antigenic protein bound to human Fab vv66. 5 5EJ0: PDB Entry ID for structure of VACV H3 antigenic protein. 6 2I9L: PDB Entry ID for structure of VACV L1 antigenic protein bound to Fab 7D11. 7 4LU5: PDB Entry ID for structure of VACV A33 antigenic protein bound to murine IgG2a Fab A20G2. 8 5FOB: PDB Entry ID for structure of Smallpox inhibitor of complement (SPICE) bound to complement C3b.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



