
Submitted:
25 October 2024
Posted:
28 October 2024
You are already at the latest version
Abstract
Solar driven hydrogen generation is one of the promising technologies to a address the world’s growing energy demand in an sustainable way. While, for hydrogen generation (otherwise water splitting), photocatalytic, photoelectrochemical and PV-integrated water splitting systems employing conventional semiconductor oxides materials and their electrodes are under investigation over a decade, lead (Pb)- halide perovskites (HPs) made their debut in 2016. Since then, the exceptional characteristics of these materials, such as their tunable optoelectronic properties, ease of processing, high absorption coefficients, and long diffusion lengths, have positioned them as a highly promising material for solar-driven water splitting. Like in solar photovoltaics, in solar driven water splitting field is also dominated by Pb-HPs with on-going efforts to improve the material stability and improving the hydrogen evolution/generation rate (HER). Despite this, with the unveiling potential of various Pb-free HP compositions in the photovoltaics and optoelectronics inspired researchers to explore the potential of these materials in water splitting. In this current review, we outlined the fundamentals of the water splitting, summary of Pb HPs in this field and the associated issues are presented. Subsequently, Pb-free HPs compositions and strategies employed for improving the photocatalytic and/or electrochemical activity of the material are discussed in detail. Finally, this review presents existing issues and future potential of lead-free HPs, which show potential for enhancing productivity of solar-to-hydrogen conversion technologies.
Keywords:
halide perovskite
; solar to hydrogen
; efficiency
; photocatalysis
; photo electrochemical
1. Introduction
Photocatalytic water splitting is one of the efficient and green energy technologies to produce hydrogen fuel with less to no toxic emissions. Ever since the first demonstration of the photoelectrochemical water splitting by Honda and Fujishima, solar energy driven hydrogen generation by employing semiconductors as an active material created a buzz in the research community. Several materials including TiO2, ZrO2, hetero-phase-mixed oxides, other white titanates and zirconates and oxide perovskite semiconductors have been under constant investigation for hydrogen generation; however, most of these semiconductors are active in UV region and there had been an ongoing demand for investigates materials with absorption in the visible region or competent with a broader absorption spectrum to utilize a substantial amount of solar radiation. While doping of the existing active materials to tune the band gap and achieve absorption in the visible region is one approach, alternatively the materials which are already proven to be satisfactory for other photovoltaic applications (like LEDs and solar cells) intrigued the researchers.
Materials with AMX3 (A being the large cation M being the small metal cation and X being halide anion) are often referred to as Halide-perovskites (HPs), while the term perovskite pertains to the materials crystal structure. These materials are composed by two cations: A the larger cation, B the smaller cations, usually metal ion and X the halide anion. The investigation of Pb-HP CsPbX3 is dated back to 1958 by Moller.[1] The following decade Pb- HPs fascinated the researchers with their intriguing optoelectronic and structural properties. Pb-HPs are known for their high light absorption coefficient and high carrier mobility with low excitonic binding energy, which make them potential candidates for photon-induced activities. In 2009, with Miyasaka’s report of replacing dye with organic-inorganic halide perovskite CH3NH3PbI3 as a sensitizer in solar cell, HPs made a dynamic entry to the photovoltaic applications.[2] These materials have been extensively studied and employed for solar cells, LEDs, photodetectors, gas sensors, switching devices, FETs, metal-ion batteries etc., In 2016, the most promising Pb-HP perovskite made its debut into solar driven hydrogen generation.[3] However, realizing the inevitable stability and toxicity issues associated with lead HP, there have been constant efforts to develop sustainable and stable HP perovskites for renewable hydrogen generation.
In the current review, we summarize the Pb-free HPs for solar driven hydrogen generation (/evolution) from the aqueous solutions. As a part of this discussion, a general introduction to the solar driven hydrogen generation systems, status and issues associated with Pb-HPs in hydrogen generation are elaborated. A detailed discussion is provided on the novel Pb-free HP compositions developed for hydrogen generation, strategies to improve the performance well as stability of these materials.
2. Solar-Driven Hydrogen Generation Systems
Figure 1 shows the HP-based water splitting process and systems. Conventionally solar-driven hydrogen generation systems are classified into three systems based on the operating mechanisms- photocatalytic, photoelectrochemical (PEC) and photovoltaic-powered electrolysis (PV-E) hydrogen generation
Figure 1a summarizes the basic principle of photocatalytic water splitting. To split water into its constituent H2 and O2 molecules via photocatalytic process requires a visible-light-responsive photocatalyst with a sufficient potential to overcome the positive Gibbs free energy of splitting reaction of the water. Therefore, the photocatalyst should have a suitable a sufficiently narrow band gap to harvest visible photons and its band gap must encompass both the water reduction and oxidation potentials. These potentials stand at +0 V and +1.23 V respectively, referenced to the normal hydrogen electrode (NHE). Generally, the photocatalysis mechanism involves three major steps: (1) photogeneration of charge carriers (electron-hole pairs) which can be enhanced by bandgap tunability and high absorption coefficient (2) Diffusion of photogenerated charge carriers towards the redox-active sites on the surface. Charge separation and collection efficiency, can be enhanced with high carrier lifetime, defect and surface passivation hetero/homo-junctions ensuring that more electrons and holes successfully participate in the desired redox reactions. These systems are characterized by two-step excitation and/or charge separation process and mimic the natural photosynthesis process, harnessing wide range of visible light for splitting because a change in Gibbs free energy (∆G0= 237.18 kJ·mol−1) required to drive each photocatalyst can be reduced as compared to the one-step water splitting system and that the separation of evolved H2 and O2 is possible. (3) The final step is the photocatalytic oxidation and reduction of the water molecules for evolution of hydrogen and oxygen. As the photocatalyst is dispersed in aqueous solutions, the stability of these materials plays a crucial role with limited choices.
Figure 1c shows the schematic of the PEC water splitting system consisting of a photocathode and photoanode immersed in an aqueous solution, where the half-cell reactions constitute oxidation at anode and reduction at cathode – resulting in water splitting into H2 and O2. The performance of the PEC water splitting system is quantified by Solar-to-hydrogen (STH) efficiency (ηSTH), usually measured under standard solar irradiance of AM 1.5G with no applied bias. ηSTH is calculated from the photocurrent density generated Jsc (in mA.cm-2) and Faradaic efficiency (ηF) contribution to the hydrogen evolution reaction (HER) and 1.23 V represents the standard potential required for the electrolysis of water. Alternatively, H2 production/evolution can be measured by gas chromatography or mass spectrometry.
Figure 1d shows the schematic of a PV-E based water splitting system wherein a PV device provides the required potential to drive oxidation-reduction reactions for H2 evolution.
3. Lead-Halide Perovskites for Hydrogen Generation
In 2016, Park et al.[3] pioneered the use of HPs for hydrogen generation through hydroiodic (HI) acid splitting, by leveraging the dynamic equilibrium of ions in methylammonium lead iodide (MAPI) perovskite in aqueous HI and HBr reaction mediums as shown in Figure 2a. The study demonstrated continuous photocatalytic activity of MAPI for approximately 6.5 days, showcasing robust phase stability and potential for hydrogen generation. Dissolution of the PbI3- ions in aqueous solution requires controlled accessibility of I- ions and with an excess of I- ions in the reaction medium leads to the formation of PbI3- and PbI4-2 ions. The availability of H+ ions is to be monitored to restrict the formation of hydrated phases and induce forced precipitation of MAPI crystals. Therefore, MAPI, in aqueous solution, forms hydrated phases or disassociates into it constitute metallic salt and requires a specific range of concentration of H+ and I- ions for precipitation and stabilization. This understanding led Park et al. to employ aqueous HI and HBr reaction mediums to produce hydrogen gas by photocatalytic activity of MAPI. The study highlights a continuous photocatalytic activity of MAPI under illumination for about 6 and a half days. MAPI exhibited a robust phase stability due to the dynamic equilibrium of the material and the saturated reaction system. A series of experiments conducted under variable conditions, including variable intensities and wavelengths of visible light, and in presence of the Pt co-catalyst and H3PO2 reducing agent, indicated the potential of employing MAPI in aqueous environment for hydrogen generation by its photocatalytic activity.
Following this, a series of mixed halide perovskites, hydrogen generation from aqueous hydrohalic acids. Wu et.al significantly enhanced the photocatalytic hydrogen evolution activity by incorporating Br ion into MAPI.[4] They prepared powdered samples of MAPbBr3–xIx with a gradient of iodide concentration, gradually increasing from the interior to the surface of each particle to a facile light-assisted halide exchange, resulting in a band gap funnel structure as shown in Figure 2b(i). The distribution of Cl and I ions within the perovskite crystal is as shown in Figure 2b(ii). In this unique architecture, photoluminescence (PL) experiments revealed swift charge carrier transport from the bromide-rich region to the iodide-rich region. The iodide-rich sites acted as the primary locations for charge recombination, while the bromide-rich sites served as the absorbing and carrier generation sites. This funnel structure effectively minimized carrier recombination, leading to improved H2 photogeneration. To demonstrate this, the experiments were conducted under aqueous mixed HI/HBr acid-saturated solutions, along with H3PO2 selectively reducing I– ions that could interfere with light absorption. Under visible light irradiation (λ ≥ 420 nm, 100 mW cm–2), MAPbBr3–xIx exhibited H2 evolution of 1021.20 μmol h–1g–1 (compared to 11.20 μmol h–1g–1 for pristine MAPbBr3). This efficiency was further enhanced to 2604.80 μmol h–1g–1 by incorporating a 3 wt % Pt loading to separate electron–hole pairs. The solar-to-light chemical efficiency at the start of the reaction without H3PO2 was found to be 1.05%, and the apparent quantum efficiency of the MAPbBr3–xIx/Pt system under 450 nm irradiation reached 8.10%. Remarkably, this system demonstrated excellent stability over 30 hours of continuous experiments, repeated in 6 cycles of 5 hours each.[4] The authors extended their band gap funnel approach to explore all-inorganic mixed perovskite, specifically the CsPbBr3–xIx system, using the same preparation method as before.[5] The band gap alignment and photographic images of the synthesized perovskites are shown in Figure 2c(i,ii). In this case, CsPbBr3–xIx was also loaded with Pt co-catalyst. Under visible light irradiation (λ ≥ 420 nm, 120 mW cm–2) in an aqueous HBr solution saturated with CsPbBr3, the CsPbBr3–xIx system demonstrated remarkable H2 photogeneration, reaching 896.0 μmol h–1 g–1, while maintaining high stability for up to 50 hours. The apparent quantum efficiency for this system was found to be 2.15% under 450 nm irradiation.[5]
Employing composites, a traditional strategy to improve photocatalytic activity of semiconductors, is adapted by research in case halide perovskites as well. Wu and his team developed a composite of MAPI with reduced graphene oxide (rGO) by dispersing GO powders in an aqueous HI acid solution saturated with MAPbI3.[6] Upon exposure to visible light (λ≥420 nm) during the photoreaction, GO was reduced to rGO, as confirmed by Raman spectroscopy. Further analyses using Transmission Electron Microscopy (TEM) and Fourier transform IR (FTIR) validated the attachment of r-GO to MAPbI3 and the formation of new chemical bonds between them. Figure 2d shows the SEM image of the MAPI/rGO; inset shows the schematic of photocatalytic activity involved. Photocatalytic H2 evolution reactions were carried out on powder samples of the MAPbI3/r-GO composite in an aqueous HI solution saturated with MAPbI3 (along with H3PO2, in order to reduce I3−). Compared to pristine MAPbI3, the composite exhibited a remarkable 67-fold improvement in photoactivity, reaching a rate of 939.90 μmol h−1g−1. Interestingly, the authors found that rGO performed even better than Pt as cocatalysts, as measured by hydrogen photogeneration in Pt-loaded MAPI. Moreover, the composite displayed excellent stability over 20 cycles of repeated experiments, with each cycle lasting 10 hours. Photoluminescence (PL) measurements confirmed that the composite facilitated more rapid charge transfer and effective electron−hole separation, leading to an increased H2 evolution rate.[6]
Wang and colleagues employed a similar strategy to enhance HI splitting by MAPbI3, using Pt/TiO2 nanoparticles as nanoscale electron-transporting channels to efficiently extract electrons when in contact with MAPI. To improve the interaction between the metal halide perovskite (MHP) and Pt/TiO2 nanoparticles, they heated the solution containing both semiconductors to promote MAPI dissolution and subsequent reprecipitation during cooling. This process strengthened the dynamic attachment of MAPI to Pt/TiO2, facilitating their interaction. Remarkable enhancement in hydrogen evolution was achieved with a 50% loading of Pt (0.75 wt %)/TiO2, resulting in an 89-fold improvement compared to pristine Pt/MAPI and leading to the evolution of 436.6 μmol of hydrogen in 6 hours. The composite’s stability was confirmed over four successive photocatalytic cycles. Based on photoluminescence (PL) measurements, the authors proposed a mechanism in which MAPI particles dispersed in a saturated solution acted as light absorbers, generating electrons and holes upon light irradiation. The proper band alignment between Pt/TiO2 and MAPbI3 facilitated the transfer of electrons generated on MAPI to Pt/TiO2, where they reduced protons to form H2. Simultaneously, the holes on MAPI oxidized I− to I3−, enhancing HI splitting efficiency. The authors also experimented with a composite using commercial Ta2O5 and Nb2O5 nanoparticles loaded with Pt. While Ta2O5 showed some effectiveness in promoting H2 evolution, its potentially unfavourable band alignment resulted in negligible catalytic activity compared to TiO2. However, it is important to note that these findings were preliminary, and further investigations could be pursued to explore the potential of such composites.[7]
While the traditional PV devices suffer from low open circuit voltages and does not overcome the thermodynamic driving forces and overpotentials of the water splitting reactions, HP solar cells possess sufficiently high Vocs are advantages for water splitting. [3,8,9,10] However, integration of PV systems with the electrochemical cell is a challenging task. Designing multijunction/tandem junctions is an effective strategy to improve power conversion efficiency of the solar cells. Combining wide band gap HP with a narrow-band gap crystalline Si for a tandem solar cells recently surpassed Shockley Queisser limit, researchers from KAUST and Helmholtz Zentrum Berlin’s (HZB) demonstrated perovskite-Si tandem solar cells with efficiencies beyond 32%. With the PV-assisted water splitting which require high operating potential, these devices with high open circuit voltage (Voc > 1.23 V) are of special interest. For the first time Dutta et.al integrated electrochemical cell and a monolithic lead HP- Si tandem solar cell to achieve solar-to-hydrogen (STH) efficiency > 21%.[9] With the operation of electrochemical cell operating at high photocurrent of 18 mA and operating potential over 1.4 V which is close to the maximum power point of the employed solar cell, the designed water splitting system is conveniently without light concentrating techniques.[9] K. Fehr et.al developed a durable HP photochemical cell fabricated with the conductive adhesive-barrier (CAB). [10] The CAB protected the HP from the corrosion and degradation of the material from the water splitting reaction and the stability and the Solar-to-hydrogen (STH) efficiencies are demonstrated for two electrochemical systems- one with a co-planar photoelectrodes and the other with a HP-Si photoanode. For a Co-planar photoelectrodes system, the researchers achieved a solar-to-hydrogen (STH) efficiency of 13.4% and a lifetime of 16.3 hours to reach t60 (60% of initial photocurrent). Monolithic silicon-perovskite tandem photoanode outperformed its counterpart with a remarkable STH efficiency of 20.8% and a lifetime of 102 hours to reach t60. [10]
4. Issues with Lead HPs for Water Splitting
4.1. Toxicity
Halide perovskite energy conversion systems, solar cells as well as water splitting systems are dominated exhibit superior power conversion efficiency when conventional efficiency and figure of merit. However, it is worth noting that the most efficient PSCs are constructed using lead halide salts (PbI2 and PbBr2), which renders them potentially hazardous due to their toxic nature. Figure 3 shows the possible ways of lead into ground water, followed by entering the food chain and eventually into human intake.[11] Based on energy market projections for 2050, we can estimate the required amount of lead to produce these solar cells. Ram et al. conducted calculations and predicted that the electricity energy demand from PVs in 2050 would be 12,210 TWh/year, accounting for 60% of the total electricity demand. Assuming that PSCs will contribute approximately 30% of the PV energy share, the demand for lead for PSCs would amount to 1000 tons per year for the European Union (EU) alone. [12]
Despite the amount of lead used in PSCs is comparably low (as compared to other applications), there is a concern arises from the relatively high-water solubility of lead halide salts, higher than other heavy metal compounds commonly employed in the PV industry, such as CdS, PbS, and CdTe. For instance, the water solubility of PbI2 is approximately one billion times higher than that of PbS and PbSe.[13] This solubility poses a problem as water/ moisture (as well as oxygen) can cause damage to solar cell, leading to the decomposition of perovskite into PbI2, hydroiodic acid, and methylamine.[14] The interaction between MA+ and H2O forms a strong hydrogen bond, weakening the original bond between MA+ and the Pb-I octahedral. This accelerates the deprotonation of the organic cation, and the proton can transfer to I− via H2O, resulting in the production of volatile species like CH3NH2 and HI leaving behind PbI2 on the device.[15] Particularly failure of encapsulation of photoanode leads to the leaching out of lead into ground water ingress and is associated with elevated bioavailability, means a mere 10% increase in lead contamination from perovskite in soil could result in a staggering over 100% increase in lead contamination within plants. Consequently, plants grown in soils contaminated with lead, even at concentrations within stringent international limits for agricultural soils, exhibit evident lead intoxication.[13,16]
4.2. Stability
Photo-energy conversion systems used in practical applications must be designed to withstand diverse weather conditions, with temperature and humidity being critical factors. The International Electrotechnical Commission (IEC) standard includes three stability tests: thermal cycling, damp heat, and humidity freeze, which closely relate to these two factors. Ensuring stability against temperature and humidity can be assessed by considering various aspects, including the perovskite material, functional layers, interfaces, and back electrode. These elements collectively contribute to the overall performance and durability of solar cells under challenging environmental conditions. However, popular lead perovskite halides, include MA-based and FA-based ones face severe thermal instability.[17] Thermogravimetric analysis reveals that organic lead halide perovskites exhibit a high decomposition temperature beyond 85 °C, questioning their application for terrestrial photovoltaics. Reports indicate that decomposition can occur at 85 °C due to the volatilization and loss of various organic species in an open space.[18] Using less volatile alternatives like FA+ and inorganic Cs+ instead of MA+ is a common approach. However, the non-photoactive yellow δ-phase of FAPbI3 and CsPbI3, rather than the black phase, is thermodynamically stable at room temperature.[19]
4.2.1. Moisture induced Degradation
Despite their popularity, phase transitions, instability to the thermal stress and exposure to the ambient air and moisture have been constantly viewed as potential barriers for the organic-inorganic lead halide perovskite to be adopted of for technology. Earlier in 2014, researchers identified the degradation of CH3NH3PbI2 into aqueous HI and Methyl amine and followed by the interference of O2, the HI decomposes into I2 and H2O.[20,21,22,23,24] Figure 4a shows the proposed decomposition mechanism of MAPI in presence of water molecules. Further Snaith et.al established the roots of degradation of the perovskite to the dangling hydrogen bond and escaping of the MAI from the lattice structure, leaving behind PbI2. [25] While Yang et.al investigated the degradation of MAPI using in-Situ GIXRD and absorption and found out the first step to degradation is formation of perovskite dihydrate containing isolated PbI64– octahedra,[26] Frost et al. suggested reversible degradation mechanism that involves the phase transition of the perovskite molecule into its mono hydrate and successive breaking down into organic salt and the and PbI2.[20]
4.2.2. Oxygen and Photo- Induced Degradation
In 2015, Aristidou et.al accounted for the degradation of CH3NH3I3 in moisture-free condition to the oxygen present in the atmosphere.[27] Despite careful encapsulation, the material tends to decompose in the presence of oxygen and light; the photo-exited perovskite results in generation of the superoxide by transferring an electron to oxygen molecule, in return this superoxide attacks perovskite and decomposes into the organic and inorganic salts with water as by-product. The decomposition can be summarized as follows:
Degradation mechanism of CH3NH3PbI3 in presence of oxygen
* photo-excited spices of MAPI perovskite
The following year, the same group identified that contrary to reports suggesting adverse effects of the moisture induced degradation, the rate of light and oxygen induced degradation is higher. Figure 4b shows the degradation of MAPI under various conditions and clearly the degradation of the material in dry air and open air under light condition doesn’t exhibit much difference, indicating moisture not being the major culprit alone.[21]
Tang et.al studied the photo degradation of the perovskite thin film under various conditions (in nitrogen, vacuum, and air) and unveiled the photoinduced degradation of the CH3NH3PbI3 under various experimental conditions (N2, vacuum and atmospheric conditions under light and dark environments). Figure 4(c) shows the XRD patterns of MAPI films exposed to various conditions. The light-exposed sample under vacuum shows no sign of absorption, ascertaining the detrimental effect of photons on the perovskite. This investigation identified the similarity of the photo-degradation of CH3NH3PbI3 and photolysis of PbI2. While earlier discussed reported suggests that the degradation of MAPI under vacuum conditions primarily involves the formation of non-ionic molecular (MAI) defects, rather than ionic defects, implies that the loss of the CH3NH3+ cation is closely linked to the sequestration of iodide from the anion octahedron lattice (PbI6)4-, thereby creating iodine vacancies within the MAPI films. This study highlights that the initiation of photoinduced degradation in MAPI within a vacuum arises from the generation of iodine vacancies due to MAI loss.[22]
Sun et.al studied the photo-induced degradation of perovskite processed using two different precursors- Pb.Ac2 (Ac being acetate) and solution-engineered (sol-eng) processed perovskite films and co-related the diffusion of O2 and variation in the microstructure of the material. Interestingly sol-eng perovskite showed faster degradation and as compared to the Pb.Ac2 perovskite and this attributed to the small gains of the sol-eng perovskite with high defect density at the boundaries and thereby making the material more susceptible to the oxygen diffusion (Figure 4d). On the other hand Pb.Ac2 perovskite are characterized by large gains and O2 attacks the grain boundaries followed the propagation of the defects throughout the material. Figure 4d shows the SEM images of the Pb.Ac2 perovskite and diffusion for O2 across the grains and the relative degradation mechanism.[23]
4.3. Band Gap of Pb-HPs and Need of Pb-Free HPs in PECs
Conventional lead perovskite with iodide halide has a narrow band gap of 1.54 eV and is known to deliver high saturation current. However, the material is not an ideal choice for water splitting due to the photoelectric voltage which barely supports the water oxidation and charge injection potentials. On the other hand, chlorine-based perovskites are limited by the low absorption spectrum. Alternatively, bromide perovskites exhibit suitable energy band gaps and are apt for designing of low-onset potential photoanodes. Particularly, cesium lead bromide (CsPbBr3) has the larger band gap at 2.4 eV, yet the lowest theoretical saturation current, which required combating strategies to compensate the saturation current. Moreover, while all inorganic perovskites require high temperature synthesis, methylammonium lead bromide (MAPbBr3) have poor thermal and moisture stability. This opened doors for formamidinium lead bromide (FAPbBr3) as a candidate material for photoanodes for water splitting. Recently, Yang et.al investigated monolithic FAPbBr3 photoanode for water splitting. The photoanode delivered high photon-to-current efficiency of 8.5% with an open circuit voltage of 1.4 V. Interestingly, the energy band off-set allowed the coupling of the photoanode with carbon/graphite protective layer, improving the stability of the of the system to operational 100 hours.[28] On the other hand, 2D perovskites inherently have higher band gaps, which ensure sufficiently high potential to facilitate water oxidation/ splitting. For example (HDA)2PbI4 perovskite, a lead compound with hexadecylammonium (HDA) organic cations incorporated between the metal halide nanosheets, exhibit a bandgap of 2.4 eV and offer high stability in water environment because of the hydrophobicity of the long chain organic molecule.[29] Similarly, long chain alkyl amine perovskites (CnH2n+1NH3)2PbI4 (n = 14, 16 and 18) have absorption edge extended to ~ 540 nm and demonstrated excellent stability up to 96 hours when dispersed in water.[30] However, lead-based 2D perovskites for water splitting are not significantly unexplored, probably due to the limited charrier mobility governed by structural low dimensionality.
While instability of conventional (organic) lead halide perovskite is a significant problem that needs to be addressed, some progress has been made in moving towards low –dimensional perovskites as well as all-inorganic perovskites. Proposing the development of 2D/3D heterojunction perovskites that could provide a trade-off between band gap, stability, and charge mobility emerges as a potential solution.[31] However, the toxicity concern primarily centers around lead, and efforts have been made to identify alternative materials that can substitute it.
5. Lead-Free Perovskites in Water Splitting/Hydrogen Generation
5.1. Classification of HP Perovskites (Based on Their Structure)
Based on the ionic radius of the cations and anions, perovskite materials adapt either of the following structural dimensionalities- 0-D or 2-D or 1-D or 3-D structures. Figure 5 shows the structure of different dimensional perovskites. Considering a 3-D perovskite with a general formula of AMX3, a material has continuous corner-sharing metal halide [MX6]4− octahedra and a 3-D perovskite can be transformed into 2-D perovskite by incorporating a spacer cation which separate the metal-halide octahedra. Whereas 1-D perovskites consist of either face-sharing or corner sharing octahedra and in case of the 0-D perovskites, the metal-halide octahedral clusters exist individually and are surrounded by the A type inorganic or organic cations. Considering the strictly periodical spatial arrangement the metal-halide octahedra and wrapping/ surrounding of the A-type cation species of the octahedra, the lower dimensional (2-D, 1-D, and 0-D) perovskites can therefore be treated as assemblies of 2D quantum wells or 1D quantum wires or 0D molecules/cluster. Thus, lower dimensional perovskites are structurally unique and usually differ from morphological forms of 3-D perovskites like nanosheets/nanoplatelets, nanowires/nanorods, and nanoparticles/quantum dots based on 3D AMX3.[32] For example, the 1-D perovskites morphologically resemble nanorods or wires with a strong interaction between the metal-halide species. These interactions lead to the formation of the electronic band formation, yet with the material being limited in length, favoring the quantum confinement effect.[33] Contrary to this, in a molecular level 1-D perovskites anionic metal halide species are surrounded by organic cations and are isolated from one another. This framework results in bulk assemblies of quantum wire-like structures, that is macroscopic crystals that exhibit properties of nanomaterials. However, lead-based 1-D perovskites did not progress greatly and therewith Pb-free 1-D perovskites have not been investigated extensively.[34,35] For instance Chenkun et.al attempted to synthesize 1D (C4N2H14)SnBr4 alongside 0D (C4N2H14Br)4SnBr6 and observed the photoinduced degradation into 0-D perovskite. [36]
Furthermore, the dimensionality of the perovskite plays a crucial role in the stability, electrical and optoelectronic properties of the material. Typically, 3-D perovskites with small (alkyl chain organic) cations are susceptible to moisture, temperature and/or photo–induced degradation. Incorporating a bulky/long chain organic cation into 3-D perovskite matrix not only transforms it into a lower dimensional 2-D perovskite, but also improves the air/ environmental stability of the materials. Higher formation energy and hydrophobicity of the A site cations of the lower dimensional perovskites, particularly 2-D perovskites offer greater stability over its 3-D counter parts. On the other has 0-D perovskites, owing to their isolated and undisturbed metal-halide cores, offer highest stability; in fact, unstable tin-based perovskites on subjected to photodegradation or oxidation of Sn end up stabilizing into stable 0-D perovskites.[36,37] To understand the electronic and optoelectronic properties, it is essential to acknowledge the origin of the electronic band formation. Generally, with reduction in the dimensionality the bandgap and excitonic binding energy increases, which hinders their application in solar cells. However, with the quantum confinement(-like) effect due to electronic bands generated by the alternative A-type cation spices and metal-halide species, lower dimensional (particularly 2-D) perovskites are prominent in LED and photodetector applications. For a perovskite, the conduction band and valency band are formed from the hybridized orbitals of the metal cation and halide anion, with a little to no contribution from the A-type (organic and/or smaller) cation; with the reduction in the dimensionality the connection between the metal-halide framework is weakened, which is the basic reason for increase in the bandgap as well as reduction in the electronic dimensionality, otherwise low charge mobility (higher effect mass of the charge) of material. Despite this, the electronic dimensionality and effective mass of the charges of the perovskite or even the band gap can be improved by wisely choosing/replacing the inactive A-type cation otherwise orienting/tilting the metal-halide octahedra.[38,39] This viability 2-D as well as 0-D perovskites is greatly exploited in tuning these materials for specific applications.[40,41]
Apart from classification of perovskite in terms of the structural dimensionality perspective, replacement of the Pb in by different metal cations leads to the following perovskite/ perovskite-inspired materials. When Pb+2 is replaced with either trivalent metal cations (like Bi+3 or Sb+3) leading to the formation of perovskite- inspired materials, which are often structurally 0-D and have a general formula of A3B2X9, these 0-D perovskites be structurally tuned to from a layered 2-D perovskite as well; Lee et.al introduced polyethylene glycols (PEGs) ligands to a 0-D Cs3-nBi2X9 to synthesize a 2-D [(PEG6-NH3+)nCs3-nBi2X9, perovskite.[42] On the other hand, when Pb+2 is replaced with a pair of metal cations, M+1 and M+3, forms the vacancy order double perovskites with a general formula of A2M(I)M(III)X6. Also, other class of the vacancy order perovskites consist of metal cation with tetravalent metal cation and have a general formula of A2MX6; Tin- based (Sn-) HPs, Cs2SnX6, are one such popular vacancy order perovskite known for their air-stability and explored for solar cells as well as photocatalysis.
5.2. Structure and Bandgap in (Lead-Free) Perovskite
Consider a 3-D perovskite AMX3 such as CsSnX3 (X = I, Br, Cl), Huang et.al studied the electronic band structure of the material using the quasiparticle self-consistent GW method (QSGW). For a cubic phased perovskite, the VB maxima is comprised of the mostly the hybridized X’s ns states and a band which is antibonding between Sn 5s and X’s np orbitals and the CB minima is mainly comprised of the Sn’s 5p states. Furthermore, conduction band minima (CBM) exhibits threefold degeneration without SO coupling and splits into a doublet and a quadruplet (including spin) when SO is included.[43] Similarly for a 3-D MASnX3 and 2-D perovskite the contribution from the larger (/organic) cation is insignificant. In fact, the hybridized orbitals of the Sn-s to X−p form strong band dispersion at the valence band maxima (VBM) and lead to the higher hole mobility, thus the material was first reported as solid hole transport layer for the dye sensitized solar cells.[44] However, absence of the spin-orbital and band splitting at the CBM, the electron mobility of is not in par with the conventional Pb-perovskite. Moreover, the stabilization of tetravalent ions of the metal ions increases from Pb to Sn to Ge to Si over their divalent forms, which argues the synthesis and applications of the 3-D Ge and Si perovskites.
Conventional method to tune the band gap by partial to full replacement of halide anion and the A-type cation in the traditional lead-based perovskite is equally effective for the 3-D lead-free perovskites. Typically, for a 3-D tin-based perovskite MASnCl3 the bandgap is reported to be 1.1 eV and replacement of MA with FA or Cs led to increasing band gap to 1.4 and 1.36 eV respectively.[45] Although literature suggest the non-involvement of the A-type cation in the CB and VB formation, the A-type cation is responsible for the titling of the metal-halide octahedra, thereby affecting the band gap.[46] Other arguments validated the signature of the C- and N- orbitals of the organic cation in the formation of the VB.[47] Replacement of MA with a larger cation like dimethylammonium not only increased the band gap (2.05eV to 2.9) but also transformed the crystal structure from pure cubic to orthorhombic.[48]
The exploration of group-VA cations (M3+=Bi3+ and Sb3+) as potential replacements for Pb2+ aims to maintain the chemistry of the lone-pair ns2 state, which is known for its benefits in achieving high photovoltaic performance. However, due to their higher +3 oxidation state, the normal AMX3 perovskite structure, consisting of corner-sharing [MX6] octahedra, cannot form. Instead, A3M2X9 emerges as the stable stoichiometry in this context. Generally, researchers have experimentally reported two main phases of A3M2X9: the hexagonal phase, featuring zero-dimensional (0D) bi-octahedral face-sharing clusters [M2I9]3− (also known as the dimer phase, and the phase composed of two-dimensional (2D) corrugated layers with partially corner-sharing MX6 octahedra (referred to as the layered phase,). The 0D dimer phase can be easily synthesized through low-temperature synthesis methods.
Perovskites with two metal cations with a general formula A2M(I)M(III)X6 or A2M(IV)X6, (otherwise called as vacancy order perovskites) and A cation typically being a large monovalent ion, often Cs⁺ cation, M(I) and M(III) cation often being transition metal ions, such as Ag+ (K+ or Tl+ ) combined with Bi3+ (Sb3+) or it would be a simple tetravalent metal ion like Sn4+ (Te4+) in case vacancy order perovskite. The crystal structure of double perovskites can be visualized as a framework of corner-sharing octahedra. The larger A-site cations occupy the voids between the octahedra. The smaller metal cations are located at the center of the octahedra. The octahedra share their corners, forming a three-dimensional network. This arrangement provides structural stability to the material. The double perovskite structure is highly flexible, allowing for the incorporation of different combinations of cations at the M(I) and M(III) sites. This flexibility gives rise to a wide range of properties and functionalities in double perovskite materials. Furthermore, in A2M(IV)X6, the octahedron is no longer connected by the halide anion resulting from the alternative arrangement of vacancies and M(IV) cations. Generally, considering their valence atomic orbitals, the possible substituting for metal cations can be metals- 1. with the semi-core d states, 2. with the lone pair s state, and 3. without the d or s states. And the presence of lone pair due to the ns2 electrons from the metal cation determines the nature of the bandgap. For suppose, the predominant Ag based double perovskite systems Cs2AgBiX6 exhibit indirect bandgaps with mismatch of the CBM and VBM at L point and X point respectively and angular momentum mismatch of the orbitals that comprise the CBM and VBM. Moreover, With M(I) being monovalent and M(III) being a trivalent metal cation, the band edges are determined by the [M(III)X6] octahedra rather than M(I). The CB and VB arise from the np orbital of the M(III) and antibonding orbitals of the M(III)’s ns and X’s np orbitals. This leads to the isolation of the [B(I)X6] octahedra and is responsible for the electronically 0-D structure. Thus, the indirect and large bandgap alongside the reduced electronic dimensionality leading to the relatively large carrier effective masses and reduced mobility makes the Cs2AgBiX6 not an ideal choice for solar cells. However, the material has significant importance in LEDs and in recent times explored for photocatalysis.
In case of double perovskite A2M(I)M(III)X6, VBM of formed is by bonding orbitals of MIII (ns), MI (nd), and X (np), whereas the conduction band minimum (CBM) is formed by antibonding orbitals MIII (np) and X (np). This electronic band structure is similar to that of a 3-D perovskite as well as GaAs or CdSe. [49] On the other hand, absence of the antibonding orbitals at either of the band edges, leads to poorer defect-tolerant materials. To understand the electronic band of a 3-D double perovskite, let us consider novel Bi-Ag perovskite system, say Cs2AgBiX6. The outer shell electronic configuration of Bi3+ is 6s26p0 and is same as Pb+2. The VB is predominated by the X[np] and the Ag [4d] while the CB is formed by X[np], Ag [5s] and Bi [6p] states. Further, the mismatch in the localization of the CBM and VBM gives rise to the indirect bandgap in these materials. An ordered Cs2AgBiBr6 exhibits a indirect bandgap of 2.04 eV and easily be switched to direct bandgap disordered Cs2AgBiBr6. It is known the direct bandgap materials have a higher mobility (lesser effective mass of electrons) as compared to that of the than their counter part with of the same bandgap. Thus, disordered Cs2AgBiBr6 has suitable direct band gap of 1.59 eV with higher mobility and is apt for photovoltaic and photocatalytic applications. [50]
Stability of the A2M(I)M(III)X6 is mostly dependent on the cations A and M(I) and the halide X. The theoretical calculations suggest a perovskite system with larger A+ cations (e.g., Cs+ is preferred over Li+) and smaller halides (e.g., F– is preferred over I–) has higher stability. Most investigated double perovskites and contain Cs cation and either Cl or Br anions, and to the best of our knowledge, the bandgap of these systems is suitable for PV and PEC applications. For example, Cs2AgFeCl6 exhibited a bandgap of ∼1.55 eV and replacing the Fe with Ga lead to reduction in the bandgap to ∼1.37 eV. [51] Also, stability is influenced by the size of the metal cation M(I) and often perovskite systems with Ag+ have higher stability compared to Cu+. Introducing larger cation like iodine into the perovskite system for tuning the bandgap could be an unfavorable approach and can lead to collapsing of perovskite structure due to crystal instability. Interestingly, alloying the trivalent metal cations can be adopted for finely tuned by bandgap and switching between direct and indirect bandgap material.
Furthermore, in case of double perovskites, a rational substitution of the metal cations allow tuning from indirect to direct bandgap materials. The key to choose M(I) /M(III) cations in A2M(I) M(III)X6 with lone-pair in ns orbital, resulting in strong s–p coupling between the ns orbital and X p orbitals, or induce a disordered metal alloy at the respective metal ion sites. Alternatively, double perovskites with metal cations fully occupied pseudo-closed s2 shells have similar band structure as lead perovskite and pose direct bandgap, high mobility and high absorption coefficient. However, unlike Bi/Sb- Ag based double perovskite, not many of these perovskites are realized. Zhao et.al reported design of the double perovskites for photovoltaic application, which reveals that Cs2InBiCl6 and Cs2InSbCl6 exhibits a direct bandgap, with the CBM formed from antibonding states of metal cations’ np orbitals and Cl 3p orbitals, and VBM from the antibonding states of metal cations’ ns orbitals and Cl 3p orbitals.[52] Theoretical studies identified that (MA)2TlBiBr6 is shows the similar electronic structure as conventional lead perovskite, and experimental results confirmed the direct band of the material to be 2.16 eV. However, the presence of Tl, which is again a toxic element, is the major setback for this material.[53]
Perovskites crystallizing in 2-dimensional units separated by interlayer species are termed layered perovskites. Depending on the spacer cations the layered perovskites can fall in on the two categories – Ruddlesden-Popper (RP) perovskite (with a chemical composition An−1MnX3n+1) and Dion-Jacobson (DJ) perovskite (with a chemical composition F′An−1MnX3n+1 where F and F′ are monovalent (+1) and divalent (+2) organic spacer cations, respectively. To simplify the understanding, RP perovskites have spacer molecules (generally significantly larger organic cations) interlayered between the edge-sharing inorganic Octahedra to create a 3-D network. whereas DJ perovskites still have organic spacer layers but connected in a zig-zag fashion and corner-sharing inorganic Octahedra resulting in 2-D framework. Interestingly, the structural stability of the RP perovskite is vulnerable due to variable thickness, on the other hand DJ perovskites are limited by tolerance factor due to restricted movement of corner sharing species. Apart from these structural differences, in both the layered perovskites share the similar traits- Metal-halide framework is mainly responsible for the optoelectronic properties, the spacer cations play role in physical properties like exciton binding energy and charge mobility and exhibit wider bandgap. better stability and moisture resistance compared to their 3-D counter parts. It’s worth noting that in 1994, D.B. Mitzi in his Communications about synthesis of the Sn layered perovskite, commented about the tuning of electronic and magnetic properties in layered perovskites without disturbing the core active layers.[54] Thanks to the interlayer arrangement of the organic and inorganic layers in the layered/ 2-D perovskites, the material exhibits better overall stability because of the following factors- 1. Restriction of the movement of the ions 2. Structural confinement and maintained integrity of the material 3. Restriction of the moisture ingress by the organic spacer layers with tunable length 4. Tolerance to mechanical strain and thermal cycles. In fact, incorporating/ introducing a small amount (0.08 M) of layered (Sn) perovskite in the 3-D FASnI3 perovskite improved the crystallinity of material and reduced the trap states, which are otherwise caused by the oxidation of the Sn+2 to Sn+4 in 3-D tin perovskites. This 2D/3D hybrid tin perovskite devices exhibited 9 % PCE (Vs. 6% PCE for the 3-D Sn perovskite device) with enhanced light and environmental stability. [55]
For a (2-D Ruddlesden–Popper) layered perovskites, the inorganic metal-halide [MX6]4− layers are separated by insulating organic (/separator layers), and these separator layers are connected via van der Waals force. Provided the thickness of the inorganic sheet is comparable to the de Broglie wavelength of carriers, 2-D materials experience a strong quantum confinement and quantum size effects.[56] This results in unique quantum well like electronic band structures in with a formation of strongly bound exciton and increased photon absorption.[57] Thus, 2D perovskites exhibit different optical properties compared to 3D ones. Evidently, the size and shape of the organic and the spacer cations determine the tilting and relative positioning of the [MX6]4−, interfering with the optoelectronic properties of the material. Madal et.al studied the influence of the positioning of the fluorine substituted PEA in the phenyl ring of (PEA)2SnI4. However, the study revealed that the bandgap variation is insignificant.[58]
By understanding of 3-D, 2-D and 0-D perovskite structures and structure-bandgap correlation and stability, researchers identified various potential halide-perovskite compositions for hydrogen generation. Based on the conduction and valence band potential levels with respect to the NHE, these lead-free perovskites are employed either in photocatalytic or photo-electro chemical systems. The following discussion provides a summary of Pb-free compositions used for hydrogen generation.
5.3. Photocatalytic Water Splitting Using Pb-Free Halide Perovskites
Figure 6 shows the band gap alignment of various Pb-free HP with rest to NHE that exhibited promising results in the solar-driven hydrogen generation from the water and/or aqueous solution.
5.3.1. Bismuth and Antimony HPs
Stable oxidation state of bismuth and antimony (Bi+3 and Sb+3) is isoelectronic with Pb2+ with comparable ionic radius and similar valence band configuration (6s2), making bismuth a potential replacement of Pb+2 in conventional perovskite. However, owing to the hetero-valent metal replacement, the perovskite adapts a 0-D structure with A3Bi(/Sb)2X9 formula. Thus 0-D perovskites are often difficult blend into a continuous uniform film and possess a indirect band gap. These factors restrict their application in photovoltaic applications. However, they are promising lead-free halide perovskites for photocatalytic activities. Table. 1 lists out the 0-D Bi and Sb HPs employed for the photocatalytic water splitting.

Table 1.
0-D bismuth and antimony HP photocatalytic systems discussed in Section 5.3.
Table 1.
0-D bismuth and antimony HP photocatalytic systems discussed in Section 5.3.
| No | Material | Reaction Medium | Illumination | HER/ Photocurrent | Stability | Ref |
|---|---|---|---|---|---|---|
| 1 | MA3Bi2I9/ Pt | Aqueous HI/H3PO2 | 300 W Xe-lamp with a 400 nm cutoff filter | 169.21 µmol g−1 h−1 | 10 hrs of 7 cycles | 59 |
| 2 | Cs3Bi2I9 | Aqueous HI/H3PO2 | Visible Light | 22.5 μmol h-1 11.7 H2 molecules per second |
5 hrs of 3 cycles | 60 |
| 3 | Cs3Bi2I9 | HI in n ethyl acetate | 100 mW cm-2 | 1504.5 μmol h-1 g-1 | 2 hrs of 4 cycles | 61 |
| 4 | Cs3Bi2I9/Pt | Aqueous HI/H3PO2 Aqueous Me.OH |
100 mW cm-2 (λ > 420 nm) | 2304 μmol g-1 35.5 μmol g-1 |
4 hrs | 62 |
| 5 | MA3Sb2I9/ Pt | Aqueous HI/H3PO2 | 100 mW cm-2 (λ > 400 nm) | 883 | 3 hrs of 4 cycles | 63 |
| 6 | Cs3Sb2I9/ Pt | Aqueous HI/H3PO2 | 100 mW cm-2 (λ ≥ 400 nm) | 804.54 μmol g−1 | 50 h | 64 |
| 7 | 2-AMPSbI5-1 | Sodium sulfate: H2O | 150 W xenon lamp | 106.7 | 4 cycles | 65 |
| 8 | 2-AMPSbI5-2 | Sodium sulfate: H2O | 150 W xenon lamp | 96.3 | 4 cycles | 65 |
| 9 | PtSA/Cs2SnI6 | Aqueous HI | 100 mW cm−2 (λ ≥ 420 nm, | 430 μmol h−1 g−1 | 180 h | 66 |
Guo et al. (2019) successfully synthesized bright-red colored bismuth perovskite MA3Bi2I9 using a simple solvothermal method for the photocatalytic hydrogen generation from aqueous HI solution (hypophosphorous acid as a selective reducing agent). The perovskite powder has a VBM of −5.76 eV and a CBM of −3.78 eV, which are suitable for photocatalytic HI splitting. The researchers demonstrated the exceptional phase stability of MA3Bi2I9 in hydriodic acid (HI) aqueous solutions with varying concentrations under visible light irradiation. The photocatalytic activity of the material with the addition of Pt as a cocatalyst is significantly enhanced the photocatalytic rate for H2 evolution, reaching approximately 169.21 µmol g−1 h−1, a 14-fold improvement compared to the bare perovskite. The solar chemical conversion efficiency was measured at 0.48%. The synthesized perovskite exhibited excellent phase and photocatalytic stability, even after 70 hours of repeated H2 evolution the material remained robust in the reaction solution. The enhanced stability of MA3Bi2I9 was attributed to the oxidation state of bismuth (III).[59]
As photocatalytic activity takes place on the surface of the photocatalyst, nanostructure and morphology- which influence the surface area of the catalyst, and anisotropic properties-which vary based on the orientation of the catalyst, play a crucial role in the photocatalytic activity. Choudary et.al., (2022) study the Cs3Bi2I9 nanodiscs (NDs) for photocatalytic aqueous HI splitting as well the photoelectrochemical conversion. The authors were successful in stabilizing the NDs at various HI by maintaining a dynamic equilibrium between the saturated perovskite solution and the material precipitated. The perovskite NDs exhibited exceptional stability at a diluted concentration of HI for about 8 hours. The synthesized nanostructures exhibited a remarkable H2 evolution rate of 22.5 μmol h-1 under visible light in an aqueous HI with apparent quantum efficiency of 1.3%. Furthermore, these NDs demonstrate a low overpotential of 533 mV at -100 mA per square centimeter for electrocatalytic H2 evolution in the same 6.34 M HI solution, with a turnover frequency of 11.7 H2 molecules per second. Notably, the NDs display exceptional recyclability and durability for both photocatalytic and electrocatalytic HI splitting processes.[60] Li et.al., (2022) studied the anisotropic charge transfer of in the bismuth perovskite and indicated the favorable facets for the effective charge separation and enhanced photocatalytic hydrogen generation. While (100) and (006) exhibited higher photocatalytic activity, the researchers developed a simple solvothermal process to achieve defined morphologies and facet-specific Cs3Bi2I9 perovskites to capture this advantageous anisotropic properties. Perovskite hexagonal prisms showed superior photocatalytic H2 evolution performance, splitting of HI in n ethyl acetate (EA) reached 1504.5 μmol h-1g-1 of H2 evolution rate, which is 22.1 times that of a disordered Cs3Bi2I9 photocatalyst compared to their counterpart samples with disordered structures and low (100)/(006) ratios. The stability and morphology of the perovskite were maintained after 8 h of photocatalytic reaction, with trace iodine particles formed on the (100) facets because of iodide photo-oxidation.[61] Miodynska et.al., (2023) attempted to understand the of effect of the larger cation in Bi- based 0-D perovskite in terms of morphology, stability and band edge variation. As series of Bi-perovskite, A3Bi2I9 (A = Cs, Rb, MA, FA) are investigated for this purpose and identified that although the crystal structure and band gap are not affected, morphology and greatly influenced by the A type cation. While Cs forming hexagonal prisms, Rb forming irregular aggregates, and MA and FA forming irregular structures of aggregated particles with a band gap varying between 1.82-1.88. Owing to the higher degree of crystallinity and defined morphology, Cs3Bi2I9 exhibited superior photocatalytic performance in both methanolic and acidic electrolytes: The Cs3Bi2I9 photocatalyst yielded approximately 35.5 μmol g-1 and 2304 μmol g-1. in the reaction when employed with 10% MeOH and HI/H3PO2 electrolytes, respectively under 4 hours of illumination.[62]
Recently, Sb-based 0-D perovskite turned out to be an attractive option for photovoltaic and photocatalytic applications owing to its stability and non-toxic nature. Particularly, DFT studies of MA3Sb2I9 perovskite reveal high charge mobility, direct band gap and good defect tolerance. This motivated researchers to exploit the material properties for hydrogen generation. Ahmad et.al (2023) synthesized stable MA3Sb2I9 via solvothermal reaction for the hydrogen generation from aqueous HI using H3PO2 as a scavenger. The material produces 300 μmol g-1 h-1 of H2 under visible light irradiation. The addition of Pt as a co-catalyst enhances the H2 production rate up to 883 μmol g-1 h-1. The material exhibits a high stability and maintains its activity after five cycles.[63] The same group (2023), reported Cs3Sb2I9 with a comparable performance as compared to the MA-Sb based perovskite. Cs3Sb2I9 showed hydrogen generation of 95.54 μ mol g−1. Platinum was used as bi-catalyst and obtained results for Cs3Sb2I9/Pt (2 mg) showed excellent hydrogen generation of 804.54 μ mol g−1. As anticipated the material exhibited higher stability as compared to MA-based counterpart owing to its all-inorganic composition.[64] Recently Rokesh et.al., (2023) reported 2-(aminomethyl pyridine)SbI5 perovskite inspired material for photocatalytic and PEC hydrogen generation. Two photocatalysts, AMPS-1 and AMPS-2 are essentially synthesized from two Sb- precursors, iodide and oxide and their hydrogen evolution rates are compared. The band of AMPS-2 is lightly larger than AMPS-1, which could possibly due to the presence of oxide impurities. However, these impurities have not significantly affected the photocatalytic performance of the material: while AMPS-1 produced about 106.7 μmol h−1 g−1 hydrogen, AMPS-2 yielded 96.3 μmol h−1 g−1.[65]
5.3.2. Tin and Germanium HPs
While tin-halide perovskites established their prominence in replacing the lead perovskites in solar cell devices, they phase instability and low band gaps remain detrimental for their potential utilization in photocatalytic applications. On the other hand, Ge, being from the same group as Pb, Ge is looked up as prospective replacement for Pb in conventional perovskites. However, as far as we know very little literature is available for conventional 3-D tin and Ge perovskites used in hydrogen generation. Fortunately, like that of lead perovskite, the electronic structure of tin perovskites is strongly influenced by the organic cation and halogen, which can be exploited to tune their electronic energy levels and photo reactivity. Based on this strategy, Ricciarelli et.al., (2022) suggested modifying conversional 3-D tin perovskite by replacing MA cation with DMA, resulting in a stable perovskite with a suitable band gap and energy level alignment for hydrogen generation.[67] Furthermore, PEA2SnBr4, DMASnBr3 and PhBz2GeBr4 showed remarkable stability in water and are employed in conjunction with g-C3N4 as heterojunction photocatalysts for water splitting.[68,69,70] These are discussed in heterojunction photocatalysts with g-C3N4 section.
5.3.2. Vacancy-Order HPs
Another class of perovskites, vacancy-order (double) perovskites have recently been investigated in various optoelectronic, electronic as well as energy conversion systems owing to their robust crystal structure and optoelectronic properties in par with conventional lead perovskites. Particularly, Sn- based perovskite Cs2SnX6, Bi-Ag based perovskites Cs2AgBiX6 have proven to be suitable substitutes for the Pb-HPs for hydrogen generation.
Zhou et al. (2021) synthesized all-inorganic Cs2SnI6 perovskite material that can anchor single-atom platinum (Pt) sites with a unique Pt-I3 configuration on its surface (referred to as PtSA/Cs2SnI6) enhancing the photocatalytic activity and stability of hydrogen evolution reaction. The Pt single-atom sites on the perovskite surface decorated by using hydrothermal treatment, impregnation, and activation techniques. The PtSA/Cs2SnI6 catalyst has exceptional photocatalytic performance in hydrogen production from aqueous HI solution, achieving a remarkable turnover frequency of 70.6 h−1 per Pt. This value is approximately 176.5 times higher than that seen for Pt nanoparticles supported on Cs2SnI6 perovskite. Here is a summary of the photocatalytic test results. This superior performance of PtSA/Cs2SnI6 is attributed to the unique Pt–I3 configuration and the strong metal-support interaction effect, which enhanced the charge separation and transfer, and reduced the energy barrier for hydrogen production.[66]
5.3. Pb-Free HPs for Water Splitting via PEC Systems
Table 2 summaries the Pb-free HP based photoelectrode systems used in PEC for hydrogen generation. Often 3-D perovskites (ABX3) suffer with instability issues in aqueous and are usually employed with multiple protection layers to avoid direct contact with electrolytic medium, particularly in PCE water splitting systems. Usually employed protection layers are InBiSn alloy, metal foils including titanium and nickel, carbon materials and atomic layer deposited oxides. On the other hand, vacancy-order Pt, Ag-Bi, Re based-perovskites exhibited resistance to degradation in harsh aqueous mediums which enabled the use of perovskite electrodes without protection layers, opening new doors to PEC water splitting in variable pH mediums.
Cs2PtX6 vacancy order perovskite is known for its exceptional stability over a wide range of pH, variable thermal and prolonged ambient exposure conditions. Hamdan et.al., (2020), studied the electrochemistry of Cs2PtI6 in electrolytic solution of pH varying from 1 to 14. Cyclic voltammetry (CV) is conducted to investigate the redox processes and catalytic activity of the materials in aqueous solutions of different pH values. The key finding was that Cs2PtI6 showed reversible oxidation/reduction peaks corresponding to I-/I3- and Pt4+/Pt2+, and that it was stable in acidic, neutral, and basic media. Interestingly, Cs2PtI6 had mixed valency of Pt4+ and Pt2+, and that the redox processes involved the formation and reduction of triiodide (I3-). Furthermore, Cs2PtI6 exhibited a photocurrent of 0.8 mA cm-2 at 1.23 V(vs. RHE) and over 12 hours of PEC stability without loss of performance. However, this value is much lower than the reported values for lead halide perovskites which is possibly due to low charge carrier mobility and lifetime of Cs2PtI6. The Cs2PtI6 material faces multiple challenges: its high production cost due to platinum usage limits its commercial viability; its 1.4 eV band gap isn’t optimal for single-junction photovoltaics, but this could be improved through halide/chalcogen substitution in the X anion; and its poor contact with conducting glass leads to high series resistance and a low fill factor, future work on optimizing deposition methods and charge transport layers.[71] Sikarwar et.al (2023) et.al explored stable Ag-M (M= In, Bi, Sb) vacancy-order perovskite photoelectrodes for PEC water splitting. The synthesized materials exhibit remarkable resistance to oxidation retaining their structural stability over 100 cycles of electrochemical cycling. This allows for the utilization of these substances in the process of photoelectrochemical (PEC) water oxidation in both CH3CN and H2O, with and without the presence of an IrOx cocatalyst.[74]
Nandigana et.al., (2023) studied two dimensional variants of bismuth perovskite vacancy-order 2-D Cs2AgBiCl6 and 0-D Cs3Bi2Cl9. The hydrothermally synthesized double perovskite Cs2AgBiCl6 exhibits multifaceted crystal structures, featuring uniform di-pyramidal units, while Cs3Bi2Cl9 particles lack the defined structural formation. Interestingly both the perovskites exhibit high thermal stability, the TGA studies reveal that both the materials are stable upto 470 oC. The perovskite photoanodes are tested for the photoelectrochemical performance using three electrode electrochemical workstation in the 1 M KOH (pH ∼13.7) alkaline electrolyte with Pt counter electrode and Ag/AgCl (Sat. KCl) as the counter electrode. The dark and light current densities of the Cs2AgBiCl6 are 3.30 mA cm-2 and 3.85mA cm-2 and that of Cs3Bi2Cl9 are 1.78mA cm-2 and 2.18mA cm-2. The photo-response of both the photoanodes is instantaneous and persisted for about 300 sec. The PCE for the photoelectrochemical system is determined by the following equation and PCE of Cs3Bi2Cl9 and Cs2AgBiCl6 is achieved to be 0.09% and 0.13% at the 0.93 V vs Ag/AgCl. The materials exhibited steady performance over 10 hours. The low charge transfer resistance and high charge transfer density of Cs2AgBiCl6 as compared to the Cs3Bi2Cl9 resulted in its superior performance, which is understandably from the high crystallinity and defined morphology of the nanocrystals.[73]
Chandra et.al., (2023) the first time investigated for Re-based vacancy-ordered perovskite, Cs2ReX6 (X= Cl and Br) for PEC generation hydrogen. The hydrothermally synthesized Re-perovskites have similar structure to Cs2PtX6 compounds and exhibited great thermal stability 600 °C. Furthermore, the Cs2ReX6 materials show panchromatic light absorption covering the entire visible region hinting high photon harnessing ability and with their valence band positions are below the water oxidation potential, they are suitable for photoanodes for solar water oxidation. Cs2ReX6 materials displayed their impressive electrochemical stability in a pH 11 solution, with no oxidation currents even at high potentials (up to 1 V vs. Ag/AgCl). The materials consistently yield photocurrent densities of 0.15–0.20 mA cm−2 at 0.4 V vs. Ag/AgCl under AM1.5G illumination, remaining stable under both interrupted and continuous illumination. Also, Re-Br perovskite, due to its higher and suable flat-band potential and lower charge transfer resistance generated better photocurrent than Re-I perovskite.[75]
Recently, Liu et.al., (2023) studies the Cu-Ag based perovskite inspired material photoelectrodes for PEC water splitting. The synthesized Cu1.4Ag0.6BiI5 nanocrystals exhibited a direct band gap of 2.19 eV and band structures and well aligned with the redox potentials of water splitting reaction. To demonstrate the PEC from the material the photoanode is fabricated without an protective layer, and impressively the photoanode showed –0.05 VRHE (RHE = reversible hydrogen electrode) onset potential and a photocurrent density of 4.62 mA cm–2 at 1.23 VRHE, as well as an applied bias photon-to-current efficiency (ABPE) of 2.94%. The charge recombination dynamics and transient absorption studies reveal the n-type nature of the material. Furthermore, the material displayed good stability in both solvent and water, which is attributed to the hydrophobic capping ligands involved during the synthesis process. The NCs-based photoanode for photoelectrochemical (PEC) water splitting exhibits a T50% lifetime of ~310 min, which is one of the longest reported lifetimes for lead-free perovskite-inspired materials.[72]
6. Enhancing Photocatalytic Performance of Pb-Free HPs
Improving the photocatalytic performance and/or photocurrent of the electrode is crucial for ensuring sustainability of the lead-perovskite materials for hydrogen generation. The photocatalytic activity is highly dependent on 1. Ability to generate electron-hole pairs by harnessing maximum solar energy 2. Effective separation of the generated electron-hole pairs. 3. Supporting redox reactions (on the surface in case of photocatalysis or photoelectrode immersed in the reaction solution. Approaches including the 1. band gap engineering by adapting suitable composition, 2. Forming heterojunctions are essentially employed for improving the HER from the system.
Compositional engineering, on the other hand, is a promising tool to design stable perovskites. Careful choice of larger cation has a significant effect on the moisture induced degradation of the perovskite. Molecules like DMA and PEA exhibit strong hydrophobicity, which protects the refrain the penetration of the water molecules into the perovskite lattice/structure. Unlike the usual Sn-perovskites, DMASnI3 exhibited exceptional stability. The material remained unaffected even after continuous exposure to the water for several hours.
6.1. Band Gap Tuning by Compositional Engineering
While the optical band gap, absorption co-efficient and the nature of the band gap are responsible for absorbing the photons and generating the charge carriers, tuning these inherent properties in the (lead-) halide perovskites is widely explored by developing mixed halide perovskite compositions and doping metal ions. In general, adjusting the band gaps by varying the halide composition of the perovskite is the most explored method and interestingly mixed halide perovskites have superior optoelectronic properties- while bromide and chloride-based perovskites have long charge-carrier lifetimes, exceeding milisecond timescale, while iodide-based HPs have short charge-carrier lifetimes, not exceeding ns timescale however the absorption range is in the inverse order. Therefore, designing a mixed halide perovskites to balance out the properties based on the application is essential. For example, in lead HPs, creating funneling band gap by varying the halide composition, creates a favorable energy band gradient for the effective charge transfer across the perovskite material.[76] Similarly, attempts are made to adjust the band gap energy of CsPbX3 by anion exchange method resulting in the interfacial charge transfer and lifetime between the perovskite-TiO2 heterojunction. In addition to the band gap tuning the studies revealed that chloride substitution reduces the trapping states, while iodide substitution leads to slower charge-carrier relaxation and indirect excitons with longer lifetime at the HPs/TiO2 interface.[77] This approach of tuning the band gap and/or introducing the funneling band gap to lead-free perovskites to improve the photocatalytic activity has been studied by few researchers.
Tang et.al (2022) created a bandgap funneling effect for charge carriers within the bismuth-based double halide perovskites MA3Bi2Cl9–xIx by synthesizing the perovskite material via facile solvothermal anion-exchange technique. The synthesized double halide perovskite exhibited cubic crystal structure with lattice expansion, attributed to the I- substitution. Furthermore, the EDS mapping showed the gradient distribution of I- from the surface to the interior of the perovskites, indicating a bandgap funnel structure. Figure 7a shows the schematic representation of band gap funneling effect in the MA3Bi2Cl9–xIx. The charge carried dynamics suggest a lower recombination rate and longer carrier lifetime. These eventually lead to higher photocurrent in mixed halide perovskites as compared to that of MA3Bi2Cl9. The photocatalytic performance of the perovskite samples was assessed for H2 production in a saturated HCl/HI solution (with H3PO2 as a reducing agent) under visible light irradiation (λ ≥ 420 nm, 100 mW cm−2), MA3Bi2Cl8.8I0.2 perovskite demonstrated highly efficient H2 evolution, yielding a rate of ≈214 µmol h−1g−1. To further enhance H2 generation and inhibit the reverse reaction, 3.0 wt.% Pt cocatalyst was incorporated and the system exhibited enhanced photocatalytic activity, achieving a hydrogen evolution rate of ≈341 ± 61.7 µmol h−1g−1.[78]
Chen et al. (2020) developed a novel lead-free perovskite material, Cs3Bi2xSb2–2xI9 (CBSI-x), with potential applications as a photocatalyst for generating hydrogen from aqueous HI solutions. The researchers investigated the effects of Sb doping on the crystal structure, electronic structure, optical absorption, charge transfer, and defect properties of Cs3Bi2xSb2–2xI9 and the photocatalytic activity of the synthesized perovskite is compared to the conventional MAPI’s performance. The series of perovskites with variable Sb- dopant concentrations were synthesized through a co-precipitation method. The perovskite exhibited a hexagonal structure with space group P63/mmc with the peaks shifting to higher 2θ values with the increase of Sb in the perovskite. Figure 7b shows the absorption spectra of the Cs3Bi2xSb2–2xI9 doped with variable concentration of Sb. While the doping of Sb in Cs3Bi2xSb2–2xI9 effectively reduces the contribution of Bi3+ on the conduction band, this resulted in smaller bandgaps of the Bi-SB perovskites with stronger optical absorption than individual metal perovskites, particularly CBSI-0.3 posed smallest bandgap (≈1.63 eV) and the smallest absorption tail. Moreover, the incorporation of Sb effectively reduced mid gap states, thereby leading to reduced non-radiative defects and improving charge transfer and separation mechanisms. Consequently, this material exhibited superior photocatalytic activity with enhanced photocurrent (when compared to the conventional lead-based perovskite).[79]
Yin et.al. (2021), synthesized a series of stable and multifunctional Pt doped Sn-based perovskites Cs2PtxSn1-xCl6 (0<x<1) via a simple hydrothermal method. The perovskites demonstrated the switchable function for optoelectronics and photocatalysis applications. Figure 7c(i) and (ii) shows the Crystal structure and transformation relationship of Cs2PtxSn1−xCl6 and its charge-carrier dynamics model for x= 0.25 and 0.75. The perovskites have a face-centered cubic crystal structure with Fm3¯m space group and form a series of continuous Cs2PtxSn1-xCl6 solid solutions with isolated [SnCl6]2- and [PtCl6]2- octahedra. The study reveals that the Pt substitution not only enhances the thermodynamic stability and the thermal stability of Cs2PtxSn1-xCl6 but also positively impacts the charge-carrier dynamics and the radiative recombination process of Cs2PtxSn1-xCl6. With the Pt incorporation, the electron density surrounding chlorine improved the Pt-Cl bonding improving the stability of the crystal lattice. Additionally, the sub-band gap states control the photoluminescence and photocatalytic functions of Cs2PtxSn1-xCl6. The samples with low Pt content have higher sub-band gap state-density and longer charge-carrier lifetime than those with high Pt content. In other words, as the Pt content increases, efficient self-trapping occurs, resulting in an enhanced radiative transition process. The substitution of Pt proves to be an effective method for adjusting the radiative recombination process and extended lifetime of photogenerated charge carriers. This has a direct correlation with the ultimate improvement of photocatalytic activity. The photocatalytic hydrogen evolution activity of Cs2PtxSn1-xCl6 with triethanolamine (TEOA) as a sacrificial reagent under simulated solar light illumination. The hydrogen production rate followed an opposite trend of the photoluminescence intensity and quantum yield, indicating that different types of photogenerated charge carriers were involved in the two processes. This concludes that the hydrogen evolution activity to the sub-band gap states in Cs2PtxSn1-xCl6, which could capture or respond to the photoexcited carriers and facilitate the electron transfer process. The highest rate was 16.11 mmol g-1h-1 for Cs2Pt0.05Sn0.95Cl6.[80]
6.2. Pb-Free HP Heterojunctions
Effective charge separation by suppressing the recombination losses is one of the key factors to improve the photocatalytic (photoelectrochemical) activity of the hydrogen generation/production system. The electron-hole recombination is inevitable phenomenon due to the Coulombic force between oppositely polar charges, moreover this swift process, with time limited to nanoseconds, is challenging to regulate. However, this can be minimized by engineering the photocatalyst and the optimizing the system. Introducing heterojunctions is a classic and promising strategy to address the (surface) recombination in photocatalysts and is well explored in the traditional solar-driven hydrogen-generation systems. Band alignment plays a crucial role in developing an appropriate heterojunction that ensures rapid charge separation, thereby improving the overall photocatalytic performance of the system. Lead-perovskite- heterojunctions with TiO2, rGO, g-C3N4. etc.., have been investigated and employed for photocatalytic activities including hydrogen generation, CO reduction and organic contaminants degradation.[81,82,83,84,85,86,87] These studies have inspired researchers to adapt lead-free perovskite-based heterojunction architectures for hydrogen generation.
6.2.1. Semiconductor/Pb-Free Heterojunctions
Table 3 summarizes the semiconductor/Pb-free HP heterojunctions employed for the H2 generation. Jayaraman et.al (2021) investigated heterojunction of hydrothermally synthesized vacancy-order Pt-perovskites A2PtI6 (A= Cs+, Rb+, or K+) with BiVO4 for photoelectrochemical oxidation of water. While synthesized Pt-perovskites exhibited an impressive stability for about 2 weeks under ambient conditions, Cs2PtI6 showed higher thermal stability with decomposition temperature at ~364 oC. Furthermore, Cs2PtI6 exhibited remarkable stability in both acidic and alkaline environments, while Rb2PtI6 and K2PtI6 demonstrated instability. The heterojunction between BiVO4 and Cs2PtI6 led to enhanced photocurrent in comparison to BiVO4 alone, primarily attributed to improved charge separation and enhanced light absorption. Figure 8(a) shows the photocurrent response of BiVO4 and BiVO4/Cs2PtI6 heterojunction (Inset- Band alignment of the photoanode as a function of the wavelength). Furthermore, an IrOx cocatalyst further boosted the photocurrent, achieving a level of 2 mA cm−2 at 1.23 V (vs RHE). However, the study is confined to the oxidation of water and can be potentially exploited for hydrogen generation for either acidic or basic reaction solutions.[88]
Tang et.al (2020) demonstrated a novel perovskite/perovskite heterojunction catalyst by establishing interface between methylammonium bismuth iodide (MA3Bi2I9) and tri(dimethylammonium) hexa-iodobismuthate (DMA3BiI6) via a facile solvent engineering technique; a series of heterojunction perovskites (BBP-0, BBP-1, BBO-5 and BBP10) by reacting MAI, HI and Bi(NO3) in IPA and variable amounts of DMF. The synthesis process and the formation of the MA3Bi2I9 and DMA3BiI6 is schematically represented as shown in Figure 8(b) The VBM and CBM positions of MA3Bi2I9 and DMA3BiI6 showed a well-matched type-II heterostructure, which can facilitate the interfacial charge transfer between the two phases. It is suggested that the photoexcitation results in electron transportation from the conduction band of DMA3BiI6 to that of MA3Bi2I9, while the holes on the valence bands migrate in the opposite direction. Thus, this perovskite-perovskite heterojunction enhances the charge separation and thereby improves the photocatalytic active. The synthesized photocatalysts are employed for the H2 evolution from aqueous HI solution and identified that BBP-5 exhibited superior performance- BBP-5’s H2 production rate is 198.4 µmol h−1g−1, which is 15- and 8-fold increases relative to BBP-0 and BBP-10, respectively.[89]
Jiang et.al (2021) attempted to synthesize Cs2AgBiBr6 supported on nitrogen-doped carbon (N-C) for efficient photocatalytic hydrogen evolution from aqueous HBr solution under visible light irradiation. Conventional one-pot synthesis is employed for the synthesis of the Cs2AgBiBr6/N-C) composited, schematic representation of the synthesis process is shown in Figure 8c(i). The crystal structure of the perovskite is determined to be cubic and is not effected by the N-C interaction. The band alignment of the various heterojunction photocatalysts are shown in Figure 8c(ii)- the CBM of Cs2AgBiBr6 is higher than the Fermi level of N-C, which enables photoexcited electron transfer from Cs2AgBiBr6 to N-C for subsequent H2 generation. The energy level difference is the largest for Cs2AgBiBr6/N-C-140, resulting in the strongest driving force of charge transfer. Furthermore, BET analysis reveals that N-C-140 has the highest surface area and nitrogen content, which facilitate electron diffusion and interfacial charge separation implicating that Cs2AgBiBr6/N-C-140 is expected to outperform. The photocatalytic activity of the photocatalyst is measured by the HER reaction from aqueous HBr solution. Cs2AgBiBr6/N-C-140 achieves a high hydrogen evolution rate of 380 μmol g−1 h−1, which is about 19 times faster than that of pure Cs2AgBiBr6 and 4 times faster than that of a physical mixture of Cs2AgBiBr6 and N-C-140. And the material exhibited a decent stability for 6 cycles with 3 hours of hydrogen reaction per cycle.[90]
Lin et.al (2023) studied a new 0-D perovskite halide, Cs3Rh2I9 (with dimer unit [Rh2I9]3− separated by Cs ions) functionalized by nitrogen-doped carbon (NC) for hydrogen evolution for the chlorine-alkali electrolyte. The Rb perovskite is prepared via solid state reaction using CsI as the flux followed by dissolution-precipitation method to form Cs3Rh2I9 nanoclusters on a polar nitrogen-doped carbon (NC) as shown in Figure 8d(i). Cs3Rh2I9, due to their 0-D, experience downsizing to smaller clusters creating active sites of the NC due to repeated dissolution and precipitation process in aqueous DMF solution. The perovskite material undergoes self-electrochemical self-reduction and a bottom-up transformation, resulting in the formation of distinctive Rh nanoparticles. The fully reconstructed Cs3Rh2I9/NC-R catalyst significantly lowers the barrier for water dissociation in alkaline HER. The process is schematically represented in Figure 8d ii and iii. Consequently, Cs3Rh2I9/NC-R exhibits an impressive mass activity of 772.1 mA mg−1Rh in a chlor-alkali electrolyte. This figure is approximately 2.5 times greater than that of liquid-reduced Rh/NC catalysts with similar particle sizes and 35.5 times higher than electrochemically reduced Cs3Rh2I9-R catalysts with larger particle sizes.[91]
6.2.2. g-C3N4/Pb-Free HP Heterojunction
Carbon based semiconductors like reduced graphene or g-C3N4 have played a promising role in photocatalytic activities. Developing g-C3N4 based composite/ heterojunction photocatalysts by anchoring the halide perovskite onto the g-C3N4 have an advantage of energy band alignment at the interfaces for enhancing the charge separation. Also, with surface functionalization of the g-C3N4 with amino or carboxyl groups, stronger chemical bond forms with the perovskite material rather than a mere physical adsorption, which further improves the charge injection at the interface. Moreover, g-C3N4 being a synthetic polymer, enables surface passivation of the perovskites and partially addresses the stability issues. In recent years, several researchers have reported several g-C3N4/Pb-free HP heterojunctions for hydrogen generation as listed in Table 4.
Researchers have exploited the stability of 2-D perovskites to develop superior photocatalyst for hydrogen generation. However, 2-D perovskites are known for high band gap and are to couple with the suitable co-catalysts to facilitate the HER and in this case g-C3N4 based heterojunction is used to broaden the absorption spectra as well as support the HER. Romani et.al (2020) demonstrated PEA2SnBr4, a water-resistant 2D Pb-free HP, coupled with g-C3N4 co-catalyst system is employed for hydrogen photogeneration and organic dye degradation under visible light. The synthesized material retains its crystal structure and 2.67 eV band gap even in contact with water for 4 hours. The band alignment of the PES2SnBr4/ g-C3N4 heterojunction is shown in Figure 10a. The hydrogen evolution rates (HERs) showed that the PEA2SnBr4/g-C3N4 composites had a remarkable enhancement of photocatalytic hydrogen production compared to pure g-C3N4 or PEA2SnBr4, with an optimal loading of 5 wt% perovskite. The composites also showed high HERs with glucose as a sacrificial agent. PEA2SnBr4/g-C3N4 composites exhibit synergistic effects, achieving high hydrogen evolution rates (1600 mmol g-1 h-1). This novel, water-resistant perovskite opens new possibilities for catalytic applications.[69] The group (2023) also introduced 2-D Ge- perovskites PhBz2GeBr4 (with band gap 3.64 eV) and PhBz2GeI4 (with band gap 3.32 eV) for the H2 production from aqueous triethanolamine (TEOA). The HER showed synergistic enhancement with heterojunction and Pt co-catalyst when compared to the pure perovskites. For both PhBz2GeBr4 and PhBz2GeI4, the optimal perovskite loading was found to be 2.5 wt%, resulting in impressive hydrogen evolution rates of 81 mmol g-1 h-1 and 1,200 mmol g−1 h−1, respectively. The iodine-based perovskite exhibited superior performance owing to its lower band and better band alignment with g-C3N4 which lead to effective charge transfer and exhibited stability upto 4 cycles.[70]
Bresolina et.al (2020) synthesized Cs3Bi2I9/ g-C3N4 binary photocatalyst for hydrogen generation as well as degradation of organic compounds. A simple processing by ultrasonication of Cs3Bi2I9 and g- C3N4 nanosheets resulted in nitrogen-iodine chemical interactions between the two semiconductors The estimated band gaps of g- C3N4 and Cs3Bi2I9 were about 2.68 eV and 1.88 eV, respectively. The band alignment of Cs3Bi2I9/ g- C3N4 is shown in Figure 9b. The pristine g-C3N4 sample exhibits solar-light photocatalytic activity through the hydrogen evolution reaction (HER) at a rate of 496.85 µmol g-1 h-1, attributed to the favorable bandgap and distinctive electronic structure of g-C3N4. Upon integration with metal halide perovskite, the photocatalytic performance of the heterojunction demonstrates an enhancement of approximately 46%, yielding a HER rate of approximately 920.76 µmol g-1 h-1.[92]
Wang et.al (2020) combined Cs2AgBiBr6 (CABB) with rGO by synthesizing the perovskite solid-state reaction followed by photo-reduction. The SEM image of the synthesized photocatalyst is shown in Figure 9c(i)-inset. The schematic representation of the mechanism involved in the photocatalytic H2 evolution from aqueous HBr via CABB/2.5%RGO is shown in Figure 9c(i). In the saturated solution, CABB perovskite on the surface of RGO, undergoes dynamic precipitation-dissolution equilibrium process and RGO could act as an attachment for CABB particles. Under illumination, CABB generates charge carries and the generated electrons are transferred to conductive RGO through the M–O–C bonds and reduce H+ to produce H2 at the active sites of rGO. Meanwhile Br− was oxidized to Br3− by the holes on CABB particles. For CABB/2.5% rGO, the H2 evolution was observed to be 80 times greater than that achieved with pure CABB when both were immersed in a saturated HBr:H3PO2 solution uninterruptedly for 120-hour.[93]
Romani et.al (2020) introduced water stable DMASnBr3 as a co-catalyst for g-C3N4 for hydrogen evolution from 10% v/v aqueous triethanolamine (TEOA) reaction medium with the glucose sacrificial agent. Figure 9d shows the band alignment of the synthesized photocatalyst and the crystal structures of the DMASnBr3 and g-C3N4. Owing to the hydrophobic nature of the dimethylammonium cation, heterojunction showed an impressive stability of 4 hour when dispersed in water. The composite showed an impressive HER of >1700 μmol g-1 h-1 in 10% (v/v) aqueous triethanolamine (TEOA) solution without any metal co-catalyst, under simulated solar light irradiation. This is a nearly 10-fold improvement compared to pure g-C3N4 and a 100-fold improvement compared to pure DMASnBr3. The composite also showed a high HER of 300 μmol g-1 h-1 in 0.1 M aqueous glucose solution, with 3 wt% Pt as co-catalyst.[68] The same group adapted a similar scheme for the synthesis of stable Bi- based perovskite and fabrication of its heterojunctions with g-C3H4 and demonstrated their application in photocatalytic hydrogen generation. [70,93,94] Medina-Llamas et.al (2023) employed simple and scalable methods such as wet ball milling and thermal exfoliation to produce nanocrystalline Cs3Bi2Br9 and g-C3N4 nanosheets, respectively. The composites showed significant improvement of H2 production compared to the pure components. The highest HER is achieved at low perovskite loading (0.02 wt.%), while higher loadings result in a detrimental effect due to self-trapping phenomena in Cs3Bi2Br9. Also, this work highlights the importance of the use of photocatalysts in the nanocrystals and nanosheets, which enhanced the photocatalytic by improving the surface area.[94]
Song et al., (2022) employed an in situ self-assembly method to yield CABB/g-C3N4 composite, which resulted in a type II heterojunction structure. The band alignment of the synthesized composited is shown in Figure 9e. The optimal CABB/g-C3N4 composite exhibited an impressive hydrogen evolution rate of 60 μmol g−1 h−1, a notable 2.5-fold increase compared to pristine Cs2AgBiBr6.[95]
Figure 9.
g-C3N4/Pb-free HP Heterojunctions: (a) Band alignment of the PEA2SnBr4 and g-C3H4 relative to NHE potential. Reproduced with permission: Copyright 2020, Royal Society of Chemistry.[69] (b) Band alignment of the Cs3Bi2I9 and g-C3H4 relative to NHE potential. Reproduced with permission: Copyright 2020, Elsevier B.V.[92] (c) i. Schematic representation of H2 evolution mechanism by Cs2AgBiBr6-rGO under visible light irradiation (inset: SEM image of the photocatalyst). Reproduced with permission: Copyright 2019, Elsevier B.V.[93] (d) Band alignment of g-C3N4 and DMASnBr3 aligned with respect to water, H+/H2 and TEOA/TEOA+ redox level. Reproduced with permission: Copyright 2020, Wiley-VCH.[68] (e) Band alignment of the Cs2AgBiBr6 and g-C3H4 relative to NHE potential. Reproduced with permission: Copyright 2020, Springer Nature.[95].
Figure 9.
g-C3N4/Pb-free HP Heterojunctions: (a) Band alignment of the PEA2SnBr4 and g-C3H4 relative to NHE potential. Reproduced with permission: Copyright 2020, Royal Society of Chemistry.[69] (b) Band alignment of the Cs3Bi2I9 and g-C3H4 relative to NHE potential. Reproduced with permission: Copyright 2020, Elsevier B.V.[92] (c) i. Schematic representation of H2 evolution mechanism by Cs2AgBiBr6-rGO under visible light irradiation (inset: SEM image of the photocatalyst). Reproduced with permission: Copyright 2019, Elsevier B.V.[93] (d) Band alignment of g-C3N4 and DMASnBr3 aligned with respect to water, H+/H2 and TEOA/TEOA+ redox level. Reproduced with permission: Copyright 2020, Wiley-VCH.[68] (e) Band alignment of the Cs2AgBiBr6 and g-C3H4 relative to NHE potential. Reproduced with permission: Copyright 2020, Springer Nature.[95].
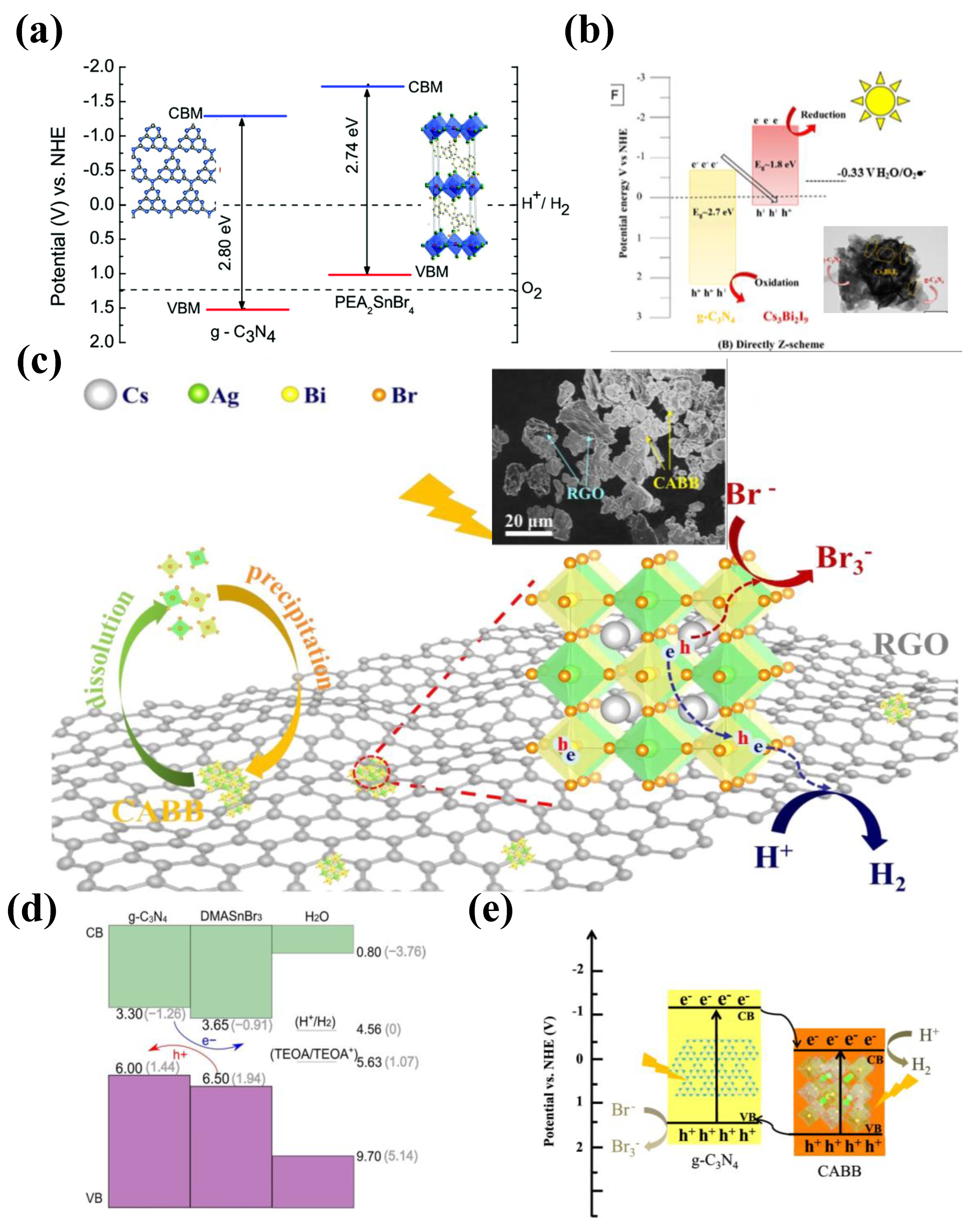
7. Prospectives and Conclusions
Lead-free perovskites turned out to be attractive active materials for solar-driven hydrogen generation owing to their excellent and tunable optoelectrical properties, ease of synthesis and thin-film fabrication. In this review, the status of the lead-free perovskite for hydrogen generation using photocatalytic and photoelectrochemical systems and state-of-art strategies to improve the performance of the active material in-terms of increasing the HER are discussed. Based on the literature available we take the liberty to conclude and comment on the following observations:
- (1).
- Dimensionality and bandgap: In general, most of the non-lead metals (excluding Sn and Ge) tend to crystallize in lower-dimensional perovskite structures. These 0-D and 2-D perovskites inherently exhibit higher band gaps, making them suitable for water splitting applications.
- (2).
- Stability: Unlike conventional lead perovskite with MA cation, lead-free perovskites with Cs cation are structurally stable and allow crystallization of materials into its low dimensional perovskite phases. All inorganic 0-D Bi (/Sb) perovskite and vacancy order Sn, Ag-Bi etc.., exhibited excellent stability in water medium for several hours, proving their potential application in photocatalytic systems. Alternatively, polymer encapsulation, hydrophobic ligand assisted nanoparticle stabilization and core-shell perovskites also enhance stability and can be exploited for photocatalytic water splitting.
- (3).
- Co-catalysts: Loading Pt co-catalyst to improve the HER has become trivial, however this modification in the system improved hydrogen evolution drastically. Considering the total system cost, it is essential to explore alternatives to Pt. While halide-perovskite/Pt based photocatalytic systems are extensively studied, serval other metal (Ni, Cu, Mn) and oxide (-perovskite) co-catalysts in conjunction with lead-free perovskites can be explored.
- (4).
- Lead-free perovskites are usually employed in photocatalytic systems rather than in photoelectrochemical water splitting. Most possible reason would be the challenges in formation of the uniform thin films for the fabrication of photoanode due to their low dimensionality.
- (5).
- Owing to their wide gap, several lead-free perovskite compositions can be excellent choices for coupling with Si cells to develop tandem photoanodes for photoelectrochemical water splitting.
Overall, lead-free halide perovskites water splitting systems need further developments to be considered effectively as sustainable technology for hydrogen generation. With the ongoing dynamic and persistent research on the lead-free perovskite material study, innovative approaches to address the stability and improve the performance, these materials have a great scope in water splitting or solar driven energy conversion applications altogether.
Acknowledgments
NSF CAREER Award 2046944References.
Conflicts of interest
There is no conflict to declare.
References
- Møller, C.K. Crystal structure and photoconductivity of caesium plumbohalides. Nature 1958, 182, 1436. [Google Scholar] [CrossRef]
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc 2009, 131, 6050–6051. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Chang, W.J.; Lee, C.W.; Park, S.; Ahn, H.-Y.; Nam, K.T. Photocatalytic hydrogen generation from hydriodic acid using methylammonium lead iodide in dynamic equilibrium with aqueous solution. Nat Energy 2016, 2, 1–8. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, P.; Guan, Z.; Liu, J.; Wang, Z.; Zheng, Z.; Jin, S.; Dai, Y.; Whangbo, M.-H.; Huang, B. Enhancing the Photocatalytic Hydrogen Evolution Activity of Mixed-Halide Perovskite CH3NH3PbBr3–xIx Achieved by Bandgap Funneling of Charge Carriers. ACS Catal 2018, 8, 10349–10357. [Google Scholar] [CrossRef]
- Guan, Z.; Wu, Y.; Wang, P.; Zhang, Q.; Wang, Z.; Zheng, Z.; Liu, Y.; Dai, Y.; Whangbo, M.-H.; Huang, B. Perovskite photocatalyst CsPbBr3-xIx with a bandgap funnel structure for H2 evolution under visible light. Appl Catal B 2019, 245, 522–527. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, P.; Zhu, X.; Zhang, Q.; Wang, Z.; Liu, Y.; Zou, G.; Dai, Y.; Whangbo, M.; Huang, B. Composite of CH3NH3PbI3 with Reduced Graphene Oxide as a Highly Efficient and Stable Visible-Light Photocatalyst for Hydrogen Evolution in Aqueous HI Solution. Advanced Materials 2018, 30, 1704342. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Zhang, H.; Yu, W.; Wang, X.; Zhao, Y.; Zong, X.; Li, C. Dynamic Interaction between Methylammonium Lead Iodide and TiO2 Nanocrystals Leads to Enhanced Photocatalytic H2 Evolution from HI Splitting. ACS Energy Lett 2018, 3, 1159–1164. [Google Scholar] [CrossRef]
- Wang, Y.; Sharma, A.; Duong, T.; Arandiyan, H.; Zhao, T.; Zhang, D.; Su, Z.; Garbrecht, M.; Beck, F.J.; Karuturi, S. Direct solar hydrogen generation at 20% efficiency using low-cost materials. Adv Energy Mater 2021, 11, 2101053. [Google Scholar] [CrossRef]
- Datta, K.; Branco, B.; Zhao, Y.; Zardetto, V.; Phung, N.; Bracesco, A.; Mazzarella, L.; Wienk, M.M.; Creatore, M.; Isabella, O. Efficient Continuous Light-Driven Electrochemical Water Splitting Enabled by Monolithic Perovskite-Silicon Tandem Photovoltaics. Adv Mater Technol 2023, 8, 2201131. [Google Scholar] [CrossRef]
- Fehr, A.M.K.; Agrawal, A.; Mandani, F.; Conrad, C.L.; Jiang, Q.; Park, S.Y.; Alley, O.; Li, B.; Sidhik, S.; Metcalf, I. Integrated halide perovskite photoelectrochemical cells with solar-driven water-splitting efficiency of 20.8%. Nat Commun 2023, 14, 3797. [Google Scholar] [CrossRef]
- Li, J.; Cao, H.-L.; Jiao, W.-B.; Wang, Q.; Wei, M.; Cantone, I.; Lü, J.; Abate, A. Biological impact of lead from halide perovskites reveals the risk of introducing a safe threshold. Nat Commun 2020, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Ponti, C.; Nasti, G.; Di Girolamo, D.; Cantone, I.; Alharthi, F.A.; Abate, A. Environmental lead exposure from halide perovskites in solar cells. Trends Ecol Evol 2022, 37, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Nain, P.; Kumar, A. Theoretical evaluation of metal release potential of emerging third generation solar photovoltaics. Solar Energy Materials and Solar Cells 2021, 227, 111120. [Google Scholar] [CrossRef]
- Hailegnaw, B.; Kirmayer, S.; Edri, E.; Hodes, G.; Cahen, D. Rain on methylammonium lead iodide based perovskites: possible environmental effects of perovskite solar cells. J Phys Chem Lett 2015, 6, 1543–1547. [Google Scholar] [CrossRef]
- Leguy, A.M.A.; Hu, Y.; Campoy-Quiles, M.; Alonso, M.I.; Weber, O.J.; Azarhoosh, P.; van Schilfgaarde, M.; Weller, M.T.; Bein, T.; Nelson, J.; Docampo, P.; Barnes, P.R.F. Reversible Hydration of CH3NH3PbI3 in Films, Single Crystals, and Solar Cells. Chemistry of Materials 2015, 27, 3397–3407. [Google Scholar] [CrossRef]
- Li, J.; Cao, H.-L.; Jiao, W.-B.; Wang, Q.; Wei, M.; Cantone, I.; Lü, J.; Abate, A. Biological impact of lead from halide perovskites reveals the risk of introducing a safe threshold. Nat Commun 2020, 11, 310. [Google Scholar] [CrossRef]
- Conings, B.; Drijkoningen, J.; Gauquelin, N.; Babayigit, A.; D’Haen, J.; D’Olieslaeger, L.; Ethirajan, A.; Verbeeck, J.; Manca, J.; Mosconi, E. Intrinsic thermal instability of methylammonium lead trihalide perovskite. Adv Energy Mater 2015, 5, 1500477. [Google Scholar] [CrossRef]
- Ma, L.; Guo, D.; Li, M.; Wang, C.; Zhou, Z.; Zhao, X.; Zhang, F.; Ao, Z.; Nie, Z. Temperature-dependent thermal decomposition pathway of organic–inorganic halide perovskite materials. Chemistry of Materials 2019, 31, 8515–8522. [Google Scholar] [CrossRef]
- Masi, S.; Gualdrón-Reyes, A.F.; Mora-Sero, I. Stabilization of Black Perovskite Phase in FAPbI3 and CsPbI3. ACS Energy Lett 2020, 5, 1974–1985. [Google Scholar] [CrossRef]
- Frost, J.M.; Butler, K.T.; Brivio, F.; Hendon, C.H.; van Schilfgaarde, M.; Walsh, A. Atomistic origins of high-performance in hybrid halide perovskite solar cells. Nano Lett 2014, 14, 2584–2590. [Google Scholar] [CrossRef]
- Bryant, D.; Aristidou, N.; Pont, S.; Sanchez-Molina, I.; Chotchunangatchaval, T.; Wheeler, S.; Durrant, J.R.; Haque, S.A. Light and oxygen induced degradation limits the operational stability of methylammonium lead triiodide perovskite solar cells. Energy Environ Sci 2016, 9, 1655–1660. [Google Scholar] [CrossRef]
- Tang, X.; Brandl, M.; May, B.; Levchuk, I.; Hou, Y.; Richter, M.; Chen, H.; Chen, S.; Kahmann, S.; Osvet, A. Photoinduced degradation of methylammonium lead triiodide perovskite semiconductors. J Mater Chem A Mater 2016, 4, 15896–15903. [Google Scholar] [CrossRef]
- Sun, Q.; Fassl, P.; Becker-Koch, D.; Bausch, A.; Rivkin, B.; Bai, S.; Hopkinson, P.E.; Snaith, H.J.; Vaynzof, Y. Role of microstructure in oxygen induced photodegradation of methylammonium lead triiodide perovskite films. Adv Energy Mater 2017, 7, 1700977. [Google Scholar] [CrossRef]
- Niu, G.; Li, W.; Meng, F.; Wang, L.; Dong, H.; Qiu, Y. Study on the stability of CH3NH3PbI3 films and the effect of post-modification by aluminum oxide in all-solid-state hybrid solar cells. J Mater Chem A Mater 2014, 2, 705–710. [Google Scholar] [CrossRef]
- Habisreutinger, S.N.; Leijtens, T.; Eperon, G.E.; Stranks, S.D.; Nicholas, R.J.; Snaith, H.J. Carbon nanotube/polymer composites as a highly stable hole collection layer in perovskite solar cells. Nano Lett 2014, 14, 5561–5568. [Google Scholar] [CrossRef]
- Yang, J.; Siempelkamp, B.D.; Liu, D.; Kelly, T.L. Investigation of CH3NH3PbI3 Degradation Rates and Mechanisms in Controlled Humidity Environments Using in Situ Techniques. ACS Nano 2015, 9, 1955–1963. [Google Scholar] [CrossRef]
- Aristidou, N.; Sanchez-Molina, I.; Chotchuangchutchaval, T.; Brown, M.; Martinez, L.; Rath, T.; Haque, S.A. The role of oxygen in the degradation of methylammonium lead trihalide perovskite photoactive layers. Angewandte Chemie 2015, 127, 8326–8330. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Y.; Ding, Y.; Li, F.; Wang, L.; Cai, B.; Zhang, F.; Liu, T.; Boschloo, G.; Johansson, E.M.J. Monolithic FAPbBr3 photoanode for photoelectrochemical water oxidation with low onset-potential and enhanced stability. Nat Commun 2023, 14, 5486. [Google Scholar] [CrossRef]
- Hong, Z.; Chong, W.K.; Ng, A.Y.R.; Li, M.; Ganguly, R.; Sum, T.C.; Soo, H.S. Hydrophobic Metal Halide Perovskites for Visible-Light Photoredox C−C Bond Cleavage and Dehydrogenation Catalysis. Angewandte Chemie International Edition 2019, 58, 3456–3460. [Google Scholar] [CrossRef]
- Kore, B.P.; Gardner, J.M. Water-resistant 2D lead (ii) iodide perovskites: correlation between optical properties and phase transitions. Mater Adv 2020, 1, 2395–2400. [Google Scholar] [CrossRef]
- Metcalf, I.; Sidhik, S.; Zhang, H.; Agrawal, A.; Persaud, J.; Hou, J.; Even, J.; Mohite, A.D. Synergy of 3D and 2D perovskites for durable, efficient solar cells and beyond. Chem Rev 2023, 123, 9565–9652. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhou, C.; Tian, Y.; Siegrist, T.; Ma, B. Low-dimensional organometal halide perovskites. ACS Energy Lett 2018, 3, 54–62. [Google Scholar] [CrossRef]
- Zhang, D.; Eaton, S.W.; Yu, Y.; Dou, L.; Yang, P. Solution-phase synthesis of cesium lead halide perovskite nanowires. J Am Chem Soc 2015, 137, 9230–9233. [Google Scholar] [CrossRef] [PubMed]
- Hintermayr, V.A.; Richter, A.F.; Ehrat, F.; Döblinger, M.; Vanderlinden, W.; Sichert, J.A.; Tong, Y.; Polavarapu, L.; Feldmann, J.; Urban, A.S. Tuning the optical properties of perovskite nanoplatelets through composition and thickness by ligand-assisted exfoliation. Advanced Materials 2016, 28, 9478–9485. [Google Scholar] [CrossRef]
- Sadhanala, A.; Ahmad, S.; Zhao, B.; Giesbrecht, N.; Pearce, P.M.; Deschler, F.; Hoye, R.L.Z.; Gödel, K.C.; Bein, T.; Docampo, P.; Dutton, S.E.; De Volder, M.F.L.; Friend, R.H. Blue-green color tunable solution processable organolead chloride–bromide mixed halide perovskites for optoelectronic applications. Nano Lett 2015, 15, 6095–6101. [Google Scholar] [CrossRef]
- Zhou, C.; Tian, Y.; Wang, M.; Rose, A.; Besara, T.; Doyle, N.K.; Yuan, Z.; Wang, J.C.; Clark, R.; Hu, Y.; Siegrist, T.; Lin, S.; Ma, B. Low-dimensional organic tin bromide perovskites and their photoinduced structural transformation. Angewandte Chemie 2017, 129, 9146–9150. [Google Scholar] [CrossRef]
- Qiu, X.; Cao, B.; Yuan, S.; Chen, X.; Qiu, Z.; Jiang, Y.; Ye, Q.; Wang, H.; Zeng, H.; Liu, J.; Kanatzidis, M.G. From unstable CsSnI3 to air-stable Cs2SnI6: A lead-free perovskite solar cell light absorber with bandgap of 1.48 eV and high absorption coefficient. Solar Energy Materials and Solar Cells 2017, 159, 227–234. [Google Scholar] [CrossRef]
- Prasanna, R.; Gold-Parker, A.; Leijtens, T.; Conings, B.; Babayigit, A.; Boyen, H.G.; Toney, M.F.; McGehee, M.D. Band gap tuning via lattice contraction and octahedral tilting in perovskite materials for photovoltaics. J Am Chem Soc 2017, 139, 11117–11124. [Google Scholar] [CrossRef]
- Marchenko, E.I.; Korolev, V.V.; Fateev, S.A.; Mitrofanov, A.; Eremin, N.N.; Goodilin, E.A.; Tarasov, A.B. Relationships between distortions of inorganic framework and band gap of layered hybrid halide perovskites. Chemistry of Materials 2021, 33, 7518–7526. [Google Scholar] [CrossRef]
- Pious, J.K.; Muthu, C.; Dani, S.; Saeki, A.; Nair, V.C. Bismuth-based zero-dimensional perovskite-like materials: effect of benzylammonium on dielectric confinement and photoconductivity. Chemistry of Materials 2020, 32, 2647–2652. [Google Scholar] [CrossRef]
- Zhou, C.; Lin, H.; He, Q.; Xu, L.; Worku, M.; Chaaban, M.; Lee, S.; Shi, X.; Du, M.-H.; Ma, B. Low dimensional metal halide perovskites and hybrids. Mater. Sci. Eng. R Rep. 2018, 137, 38–65. [Google Scholar] [CrossRef]
- Lee, J.T.; Seifert, S.; Sardar, R. Colloidal Synthesis of Single-Layer Quasi-Ruddlesden–Popper Phase Bismuth-Based Two-Dimensional Perovskite Nanosheets with Controllable Optoelectronic Properties. Chemistry of Materials 2021, 33, 5917–5925. [Google Scholar] [CrossRef]
- Huang, L.; Lambrecht, W.R.L. in APS March Meeting Abstracts 2013, vol. 2013, pp. U23–008.
- Chung, I.; Lee, B.; He, J.; Chang, R.P.H.; Kanatzidis, M.G. All-solid-state dye-sensitized solar cells with high efficiency. Nature 2012, 485, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Di Liberto, G.; Pacchioni, G. Density functional theory estimate of halide perovskite band gap: When spin orbit coupling helps. Journal of Physical Chemistry C 2022, 126, 2184–2198. [Google Scholar] [CrossRef]
- Prasanna, R.; Gold-Parker, A.; Leijtens, T.; Conings, B.; Babayigit, A.; Boyen, H.-G.; Toney, M.F.; McGehee, M.D. Band gap tuning via lattice contraction and octahedral tilting in perovskite materials for photovoltaics. J Am Chem Soc 2017, 139, 11117–11124. [Google Scholar] [CrossRef]
- Ferrara, C.; Patrini, M.; Pisanu, A.; Quadrelli, P.; Milanese, C.; Tealdi, C.; Malavasi, L. Wide band-gap tuning in Sn-based hybrid perovskites through cation replacement: the FA 1− x MA x SnBr 3 mixed system. J Mater Chem A Mater 2017, 5, 9391–9395. [Google Scholar] [CrossRef]
- Pisanu, A.; Speltini, A.; Quadrelli, P.; Drera, G.; Sangaletti, L.; Malavasi, L. Enhanced air-stability of Sn-based hybrid perovskites induced by dimethylammonium (DMA): synthesis, characterization, aging and hydrogen photogeneration of the MA1−xDMAxSnBr3 system. J Mater Chem C Mater 2019, 7, 7020–7026. [Google Scholar] [CrossRef]
- Li, Z.; Kavanagh, S.R.; Napari, M.; Palgrave, R.G.; Abdi-Jalebi, M.; Andaji-Garmaroudi, Z.; Davies, D.W.; Laitinen, M.; Julin, J.; Isaacs, M.A. Bandgap lowering in mixed alloys of Cs 2 Ag (Sb x Bi 1− x) Br 6 double perovskite thin films. J Mater Chem A Mater 2020, 8, 21780–21788. [Google Scholar] [CrossRef]
- Gebhardt, J.; Elsässer, C. The Electronic Structure of Cs2AgBiBr6 at Room Temperature. Phys. Status Solidi (B) 2022, 259, 2200124. [Google Scholar] [CrossRef]
- Yin, H.; Xian, Y.; Zhang, Y.; Chen, W.; Wen, X.; Rahman, N.U.; Long, Y.; Jia, B.; Fan, J.; Li, W. An Emerging Lead-Free Double-Perovskite Cs2AgFeCl6:In Single Crystal. Adv Funct Mater 2020, 30, 2002225. [Google Scholar] [CrossRef]
- Zhao, X.-G.; Yang, J.-H.; Fu, Y.; Yang, D.; Xu, Q.; Yu, L.; Wei, S.-H.; Zhang, L. Design of lead-free inorganic halide perovskites for solar cells via cation-transmutation. J Am Chem Soc 2017, 139, 2630–2638. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wei, F.; Sun, S.; Kieslich, G.; Cheetham, A.K.; Bristowe, P.D. Exploring the properties of lead-free hybrid double perovskites using a combined computational-experimental approach. J Mater Chem A Mater 2016, 4, 12025–12029. [Google Scholar] [CrossRef]
- Mitzi, D.B.; Feild, C.A.; Harrison, W.T.A.; Guloy, A.M. Conducting tin halides with a layered organic-based perovskite structure. Nature 1994, 369, 467–469. [Google Scholar] [CrossRef]
- Shao, S.; Liu, J.; Portale, G.; Fang, H.; Blake, G.R.; Brink, G.H.T.; Koster, L.J.A.; Loi, M.A. Highly reproducible Sn-based hybrid perovskite solar cells with 9% efficiency. Adv Energy Mater 2018, 8, 1702019. [Google Scholar] [CrossRef]
- Zhu, X.; Xu, Z.; Zuo, S.; Feng, J.; Wang, Z.; Zhang, X.; Zhao, K.; Zhang, J.; Liu, H.; Priya, S. Vapor-fumigation for record efficiency two-dimensional perovskite solar cells with superior stability. Energy Environ Sci 2018, 11, 3349–3357. [Google Scholar] [CrossRef]
- Hong, X.; Ishihara, T.; Nurmikko, A.V. Dielectric confinement effect on excitons in PbI4-based layered semiconductors. Phys Rev B 1992, 45, 6961. [Google Scholar] [CrossRef]
- Mandal, S.; Khan, B.A.; Sarkar, P. 2D lead free Ruddlesden-Popper phase perovskites as efficient photovoltaic materials: A first-principles investigation. Comput Mater Sci 2022, 211, 111545. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, G.; Li, Z.; Lou, Y.; Chen, J.; Zhao, Y. Stable Lead-Free (CH3NH3)3Bi2I9 Perovskite for Photocatalytic Hydrogen Generation. ACS Sustain Chem Eng 2019, 7, 15080–15085. [Google Scholar] [CrossRef]
- Chaudhary, S.P.; Bhattacharjee, S.; Hazra, V.; Shyamal, S.; Pradhan, N.; Bhattacharyya, S. Cs3Bi2I9 nanodiscs with phase and Bi (iii) state stability under reductive potential or illumination for H 2 generation from diluted aqueous HI. Nanoscale 2022, 14, 4281–4291. [Google Scholar] [CrossRef]
- Li, M.; Xu, S.; Wu, L.; Tang, H.; Zhou, B.; Xu, J.; Yang, Q.; Zhou, T.; Qiu, Y.; Chen, G.; Waterhouse, G.I.N.; Yan, K. Perovskite Cs3Bi2I9 Hexagonal Prisms with Ordered Geometry for Enhanced Photocatalytic Hydrogen Evolution. ACS Energy Lett 2022, 7, 3370–3377. [Google Scholar] [CrossRef]
- Miodyńska, M.; Klimczuk, T.; Lisowski, W.; Zaleska-Medynska, A. Bi-based halide perovskites: Stability and opportunities in the photocatalytic approach for hydrogen evolution. Catal Commun 2023, 177, 106656. [Google Scholar] [CrossRef]
- Ahmad, K.; Niyitanga, T.; Chaudhary, A.; Raza, W.; Khan, R.A.; Kim, H. Hydrothermal Synthesis of MA3Sb2I9 for Hydrogen Production Applications. ChemPhotoChem 2023, 7, e202300104. [Google Scholar] [CrossRef]
- Ahmad, K.; Raza, W.; Alsulmi, A.; Kim, H. Improved hydrogen production using lead-free and air stable perovskite-like Cs3Sb2I9. Mater Chem Phys 2023, 307, 128159. [Google Scholar] [CrossRef]
- Rokesh, K.; Sakar, M.; Do, T.O. 2-(Aminomethyl pyridine) SbI5: An emerging visible-light driven organic–inorganic hybrid perovskite for photoelectrochemical and photocatalytic applications. Mater Lett 2019, 242, 99–102. [Google Scholar] [CrossRef]
- Zhou, P.; Chen, H.; Chao, Y.; Zhang, Q.; Zhang, W.; Lv, F.; Gu, L.; Zhao, Q.; Wang, N.; Wang, J.; Guo, S. Single-atom Pt-I3 sites on all-inorganic Cs2SnI6 perovskite for efficient photocatalytic hydrogen production. Nat Commun 2021, 12, 4412. [Google Scholar] [CrossRef]
- Ricciarelli, D.; Kaiser, W.; Mosconi, E.; Wiktor, J.; Ashraf, M.W.; Malavasi, L.; Ambrosio, F.; De Angelis, F. Reaction mechanism of photocatalytic hydrogen production at water/tin halide perovskite interfaces. ACS Energy Lett 2022, 7, 1308–1315. [Google Scholar] [CrossRef]
- Romani, L.; Speltini, A.; Ambrosio, F.; Mosconi, E.; Profumo, A.; Marelli, M.; Margadonna, S.; Milella, A.; Fracassi, F.; Listorti, A.; De Angelis, F.; Malavasi, L. Water-Stable DMASnBr3 Lead-Free Perovskite for Effective Solar-Driven Photocatalysis. Angewandte Chemie - International Edition 2021, 60, 3611–3618. [Google Scholar] [CrossRef]
- Romani, L.; Bala, A.; Kumar, V.; Speltini, A.; Milella, A.; Fracassi, F.; Listorti, A.; Profumo, A.; Malavasi, L. PEA2SnBr4: a water-stable lead-free two-dimensional perovskite and demonstration of its use as a co-catalyst in hydrogen photogeneration and organic-dye degradation. J Mater Chem C Mater 2020, 8, 9189–9194. [Google Scholar] [CrossRef]
- Romani, L.; Speltini, A.; Chiara, R.; Morana, M.; Coccia, C.; Tedesco, C.; Armenise, V.; Colella, S.; Milella, A.; Listorti, A. Air-and water-stable and photocatalytically active germanium-based 2D perovskites by organic spacer engineering. Cell Rep Phys Sci 2023, 4, 101214. [Google Scholar] [CrossRef]
- Hamdan, M.; Chandiran, A.K. Cs2PtI6 Halide Perovskite is Stable to Air, Moisture, and Extreme pH: Application to Photoelectrochemical Solar Water Oxidation. Angewandte Chemie - International Edition 2020, 59, 16033–16038. [Google Scholar] [CrossRef]
- Liu, M.; Grandhi, G.K.; Al-Anesi, B.; Ali-Löytty, H.; Lahtonen, K.; Grisorio, R.; Vivo, P. Water-resistant perovskite-inspired copper/silver pnictohalide nanocrystals for photoelectrochemical water splitting. Electrochim Acta 2023, 462, 142734. [Google Scholar] [CrossRef]
- Nandigana, P.; Pari, S.; Sujatha, D.; Shidhin, M.; Jeyabharathi, C.; Panda, S.K. Lead-Free Bismuth-Based Halide Perovskites with Excellent Stability for Visible-light-Driven Photoelectrochemical Water Splitting. ChemistrySelect 2023, 8, e202204731. [Google Scholar] [CrossRef]
- Sikarwar, P.; Koneri, I.T.; Appadurai, T.; Chandiran, A.K. Highly Efficient Photoelectrochemical Water Oxidation Using Cs2AgMCl6(M = In,Bi,Sb) Halide Double Perovskites. Phys Rev Appl 2023, 19, 044083. [Google Scholar] [CrossRef]
- Chandra, N.V.P.; Hamdan, M.; Chandiran, A.K. Stable Cs 2 ReX 6 (X–Cl, Br) vacancy-ordered perovskites for solar water splitting. Sustain Energy Fuels 2023, 7, 949–955. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, P.; Guan, Z.; Liu, J.; Wang, Z.; Zheng, Z.; Jin, S.; Dai, Y.; Whangbo, M.H.; Huang, B. Enhancing the Photocatalytic Hydrogen Evolution Activity of Mixed-Halide Perovskite CH3NH3PbBr3–xIx Achieved by Bandgap Funneling of Charge Carriers. ACS Catal 2018, 8, 10349–10357. [Google Scholar] [CrossRef]
- Knezevic, M.; Quach, V.D.; Lampre, I.; Erard, M.; Pernot, P.; Berardan, D.; Colbeau-Justin, C.; Ghazzal, M.N. Adjusting the band gap of CsPbBr3−yXy (X= Cl, I) for optimal interfacial charge transfer and enhanced photocatalytic hydrogen generation. J Mater Chem A Mater 2023, 11, 6226–6236. [Google Scholar] [CrossRef]
- Tang, Y.; Mak, C.H.; Wang, C.; Fu, Y.; Li, F.; Jia, G.; Hsieh, C.; Shen, H.; Colmenares, J.C.; Song, H. Bandgap Funneling in Bismuth-Based Hybrid Perovskite Photocatalyst with Efficient Visible-Light-Driven Hydrogen Evolution. Small Methods 2022, 6, 2200326. [Google Scholar] [CrossRef]
- Chen, G.; Wang, P.; Wu, Y.; Zhang, Q.; Wu, Q.; Wang, Z.; Zheng, Z.; Liu, Y.; Dai, Y.; Huang, B. Lead-free halide perovskite Cs3Bi2xSb2–2xI9 (x≈ 0.3) possessing the photocatalytic activity for hydrogen evolution comparable to that of (CH3NH3) PbI3. Advanced Materials 2020, 32, 2001344. [Google Scholar] [CrossRef]
- Yin, H.; Chen, J.; Guan, P.; Zheng, D.; Kong, Q.; Yang, S.; Zhou, P.; Yang, B.; Pullerits, T.; Han, K. Controlling Photoluminescence and Photocatalysis Activities in Lead-Free Cs2PtxSn1−xCl6 Perovskites via Ion Substitution. Angewandte Chemie International Edition 2021, 60, 22693–22699. [Google Scholar] [CrossRef]
- Dong, Z.; Zhang, Z.; Jiang, Y.; Chu, Y.; Xu, J. Embedding CsPbBr3 perovskite quantum dots into mesoporous TiO2 beads as an S-scheme heterojunction for CO2 photoreduction. Chemical Engineering Journal 2022, 433, 133762. [Google Scholar] [CrossRef]
- Li, N.; Chen, X.; Wang, J.; Liang, X.; Ma, L.; Jing, X.; Chen, D.-L.; Li, Z. ZnSe Nanorods–CsSnCl3 Perovskite Heterojunction Composite for Photocatalytic CO2 Reduction. ACS Nano 2022, 16, 3332–3340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, M.; Chi, Z.; Li, W.; Yu, H.; Yang, N.; Yu, H. Internal electric field engineering step-scheme–based heterojunction using lead-free Cs3Bi2Br9 perovskite–modified In4SnS8 for selective photocatalytic CO2 reduction to CO. Appl Catal B 2022, 313, 121426. [Google Scholar] [CrossRef]
- Liu, Z.-L.; Liu, R.-R.; Mu, Y.-F.; Feng, Y.-X.; Dong, G.-X.; Zhang, M.; Lu, T.-B. In Situ Construction of Lead-Free Perovskite Direct Z-Scheme Heterojunction Cs3Bi2I9/Bi2WO6 for Efficient Photocatalysis of CO2 Reduction. Solar RRL 2021, 5, 2000691. [Google Scholar] [CrossRef]
- Bai, Z.-J.; Mao, Y.; Wang, B.-H.; Chen, L.; Tian, S.; Hu, B.; Li, Y.-J.; Au, C.-T.; Yin, S.-F. Tuning photocatalytic performance of Cs3Bi2Br9 perovskite by gC3N4 for C(sp3)—H bond activation. Nano Res 2023, 16, 6104–6112. [Google Scholar] [CrossRef]
- Paul, T.; Das, D.; Das, B.K.; Sarkar, S.; Maiti, S.; Chattopadhyay, K.K. CsPbBrCl2/g-C3N4 type II heterojunction as efficient visible range photocatalyst. J Hazard Mater 2019, 380, 120855. [Google Scholar] [CrossRef] [PubMed]
- Low, J.; Yu, J.; Jaroniec, M.; Wageh, S.; Al-Ghamdi, A.A. Heterojunction photocatalysts. Advanced Materials 2017, 29. [Google Scholar] [CrossRef]
- Jayaraman, J.P.; Hamdan, M.; Velpula, M.; Kaisare, N.S.; Chandiran, A.K. BiVO4/Cs2PtI6 Vacancy-Ordered Halide Perovskite Heterojunction for Panchromatic Light Harvesting and Enhanced Charge Separation in Photoelectrochemical Water Oxidation. ACS Appl Mater Interfaces 2021, 13, 16267–16278. [Google Scholar] [CrossRef]
- Tang, Y.; Mak, C.H.; Liu, R.; Wang, Z.; Ji, L.; Song, H.; Tan, C.; Barrière, F.; Hsu, H. In situ formation of bismuth-based perovskite heterostructures for high-performance cocatalyst-free photocatalytic hydrogen evolution. Adv Funct Mater 2020, 30, 2006919. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, K.; Wu, X.; Zhu, M.; Zhang, H.; Zhang, K.; Wang, Y.; Loh, K.P.; Shi, Y.; Xu, Q.H. In Situ Synthesis of Lead-Free Halide Perovskite Cs2AgBiBr6 Supported on Nitrogen-Doped Carbon for Efficient Hydrogen Evolution in Aqueous HBr Solution. ACS Appl Mater Interfaces 2021, 13, 10037–10046. [Google Scholar] [CrossRef]
- Lin, G.; Zhang, Z.; Ju, Q.; Wu, T.; Segre, C.U.; Chen, W.; Peng, H.; Zhang, H.; Liu, Q.; Liu, Z. Bottom-up evolution of perovskite clusters into high-activity rhodium nanoparticles toward alkaline hydrogen evolution. Nat Commun 2023, 14, 280. [Google Scholar] [CrossRef]
- Bresolin, B.-M.; Sgarbossa, P.; Bahnemann, D.W.; Sillanpää, M. Cs3Bi2I9/g-C3N4 as a new binary photocatalyst for efficient visible-light photocatalytic processes. Sep Purif Technol 2020, 251, 117320. [Google Scholar] [CrossRef]
- Wang, T.; Yue, D.; Li, X.; Zhao, Y. Lead-free double perovskite Cs2AgBiBr6/RGO composite for efficient visible light photocatalytic H2 evolution. Appl Catal B 2020, 268, 118399. [Google Scholar] [CrossRef]
- Medina-Llamas, M.; Speltini, A.; Profumo, A.; Panzarea, F.; Milella, A.; Fracassi, F.; Listorti, A.; Malavasi, L. Preparation of Heterojunctions Based on Cs3Bi2Br9 Nanocrystals and gC3N4 Nanosheets for Photocatalytic Hydrogen Evolution. Nanomaterials 2023, 13, 263. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Gou, J.; Yang, L.; Zeng, C. Environmentally Stable Mesoporous gC3N4 Modified Lead-Free Double Perovskite Cs2AgBiBr6 for Highly Efficient Photocatalytic Hydrogen Evolution. Catal Letters 2023, 153, 534–543. [Google Scholar] [CrossRef]
Figure 1.
HP-based water splitting process and systems (a) The process of the photocatalytic water splitting employing two cocatalysts A and B- cocatalyst A is responsible for the H2O oxidation into O2 and cocatalyst B is responsible for the hydrogen evolution from the reduction of the H+ in the oxidation process. (b) Photocatalysis system, showing the dispersed the photocatalyst in an aqueous medium. (c) Photo-electrocatalytic system, showing the perovskite- based photoelectrode, counter and reference electrodes immersed in the aqueous medium. (d) Photovoltaic-electrocatalytic system, showing the (perovskite-based) solar cell electrically connected to aqueous medium through the electrodes immersed in the medium.
Figure 1.
HP-based water splitting process and systems (a) The process of the photocatalytic water splitting employing two cocatalysts A and B- cocatalyst A is responsible for the H2O oxidation into O2 and cocatalyst B is responsible for the hydrogen evolution from the reduction of the H+ in the oxidation process. (b) Photocatalysis system, showing the dispersed the photocatalyst in an aqueous medium. (c) Photo-electrocatalytic system, showing the perovskite- based photoelectrode, counter and reference electrodes immersed in the aqueous medium. (d) Photovoltaic-electrocatalytic system, showing the (perovskite-based) solar cell electrically connected to aqueous medium through the electrodes immersed in the medium.
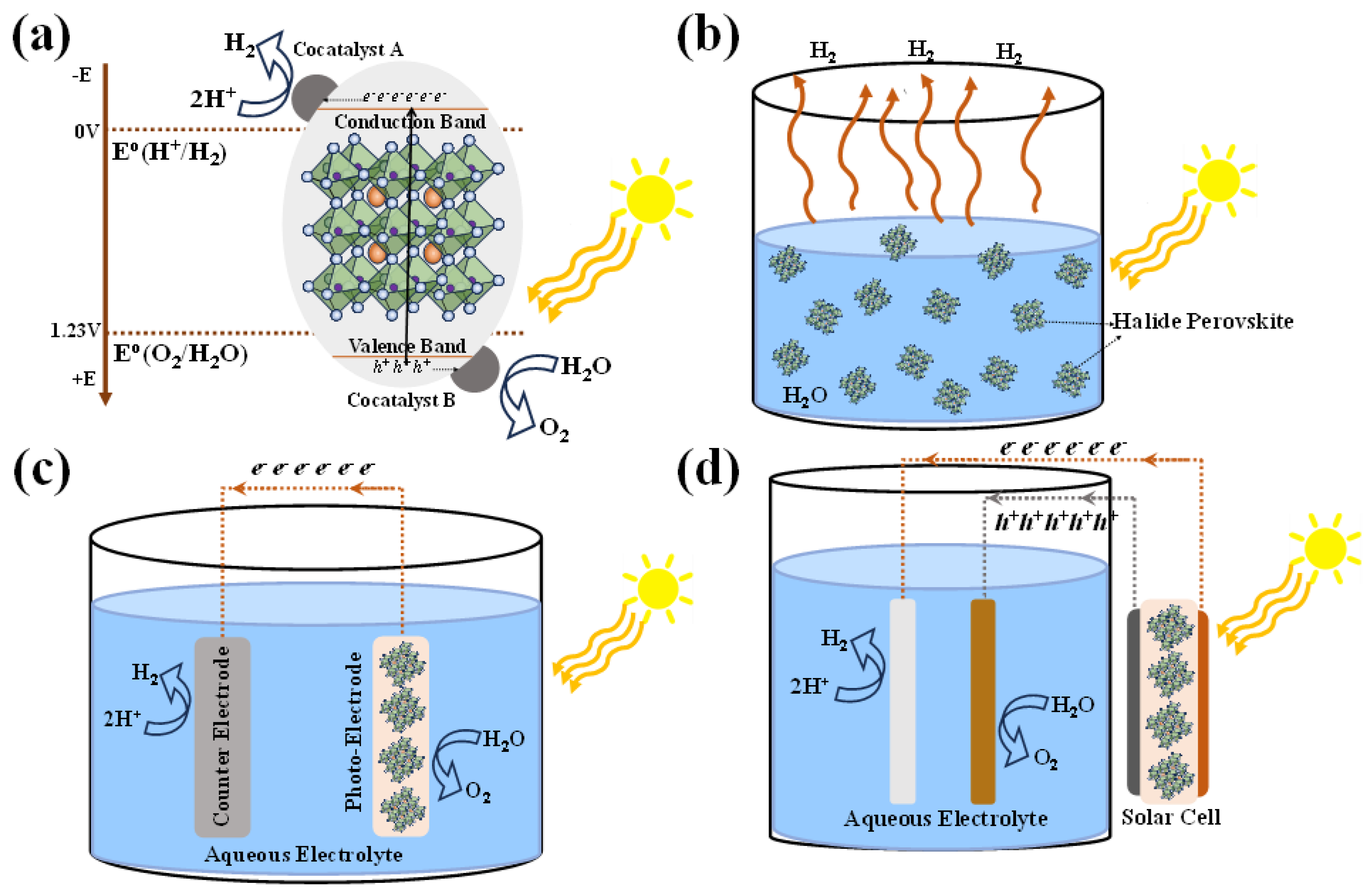
Figure 2.
Photocatalytic Water Splitting using Pb-HPs: (a) Band alignment of MAPbI3 with respect to NHE. Reproduced with permission: Copyright 2016, Springer Nature Limited.[3] (b) i. Bandgap funnel structure of MAPbBr3–xIx, with the bandgap narrowing to the surface, with Pt co-catalyst loaded on the surface of the perovskite. ii. Cross sectional SEM image MAPbBr3–xIx particle with the corresponding EDX mapping along the yellow line profile with I-element distribution profile. Reproduced with permission: Copyright 2018, American Chemical Society.[4] (c) i. Band diagram of the CsPbBr3, CsPbBr3−xIx and CsPbI3. ii. Photographic images of CsPbBr3-xIx powders with different halide exchange reaction times. Reproduced with permission: Copyright 2019, Elsevier.[5] (d) i. SEM image of MAPbI3/rGO (inset- Schematic illustration MAPbI3/rGO composite photocatalyst. Reproduced with permission: 2018 WILEY-VCH.[6].
Figure 2.
Photocatalytic Water Splitting using Pb-HPs: (a) Band alignment of MAPbI3 with respect to NHE. Reproduced with permission: Copyright 2016, Springer Nature Limited.[3] (b) i. Bandgap funnel structure of MAPbBr3–xIx, with the bandgap narrowing to the surface, with Pt co-catalyst loaded on the surface of the perovskite. ii. Cross sectional SEM image MAPbBr3–xIx particle with the corresponding EDX mapping along the yellow line profile with I-element distribution profile. Reproduced with permission: Copyright 2018, American Chemical Society.[4] (c) i. Band diagram of the CsPbBr3, CsPbBr3−xIx and CsPbI3. ii. Photographic images of CsPbBr3-xIx powders with different halide exchange reaction times. Reproduced with permission: Copyright 2019, Elsevier.[5] (d) i. SEM image of MAPbI3/rGO (inset- Schematic illustration MAPbI3/rGO composite photocatalyst. Reproduced with permission: 2018 WILEY-VCH.[6].
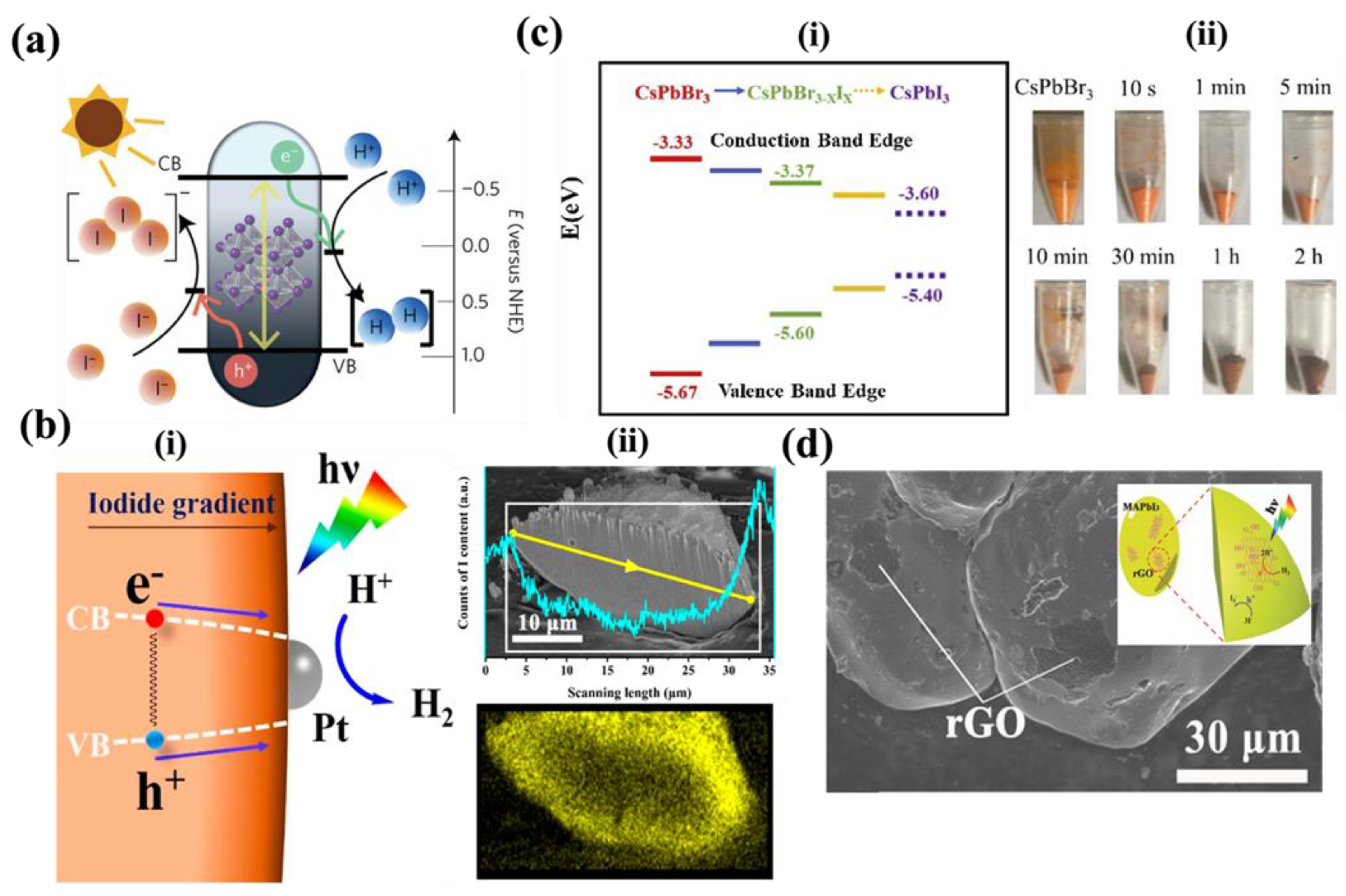
Figure 3.
Toxicity of Pb-HP devices: (a) Illustration of possible leaching of lead into ground water (b) The photographs of mint plants grown on control soil (left) and 250 mg kg−1 Pb2+ perovskite-contaminated soil (right). Reproduced with permission: Copyright 2020, Springer Nature.[11] (c) Assessment of the lead contamination on the human Lead Weekly intake level, considering the data for the world population and the total PSC lead necessary for electricity generation in 2050 Vs the adult Lead Weekly intake limit in 2010 and 3,000 to 5,000 years ago. Reproduced with permission: Copyright 2022, Elsevier Ltd.[12].
Figure 3.
Toxicity of Pb-HP devices: (a) Illustration of possible leaching of lead into ground water (b) The photographs of mint plants grown on control soil (left) and 250 mg kg−1 Pb2+ perovskite-contaminated soil (right). Reproduced with permission: Copyright 2020, Springer Nature.[11] (c) Assessment of the lead contamination on the human Lead Weekly intake level, considering the data for the world population and the total PSC lead necessary for electricity generation in 2050 Vs the adult Lead Weekly intake limit in 2010 and 3,000 to 5,000 years ago. Reproduced with permission: Copyright 2022, Elsevier Ltd.[12].
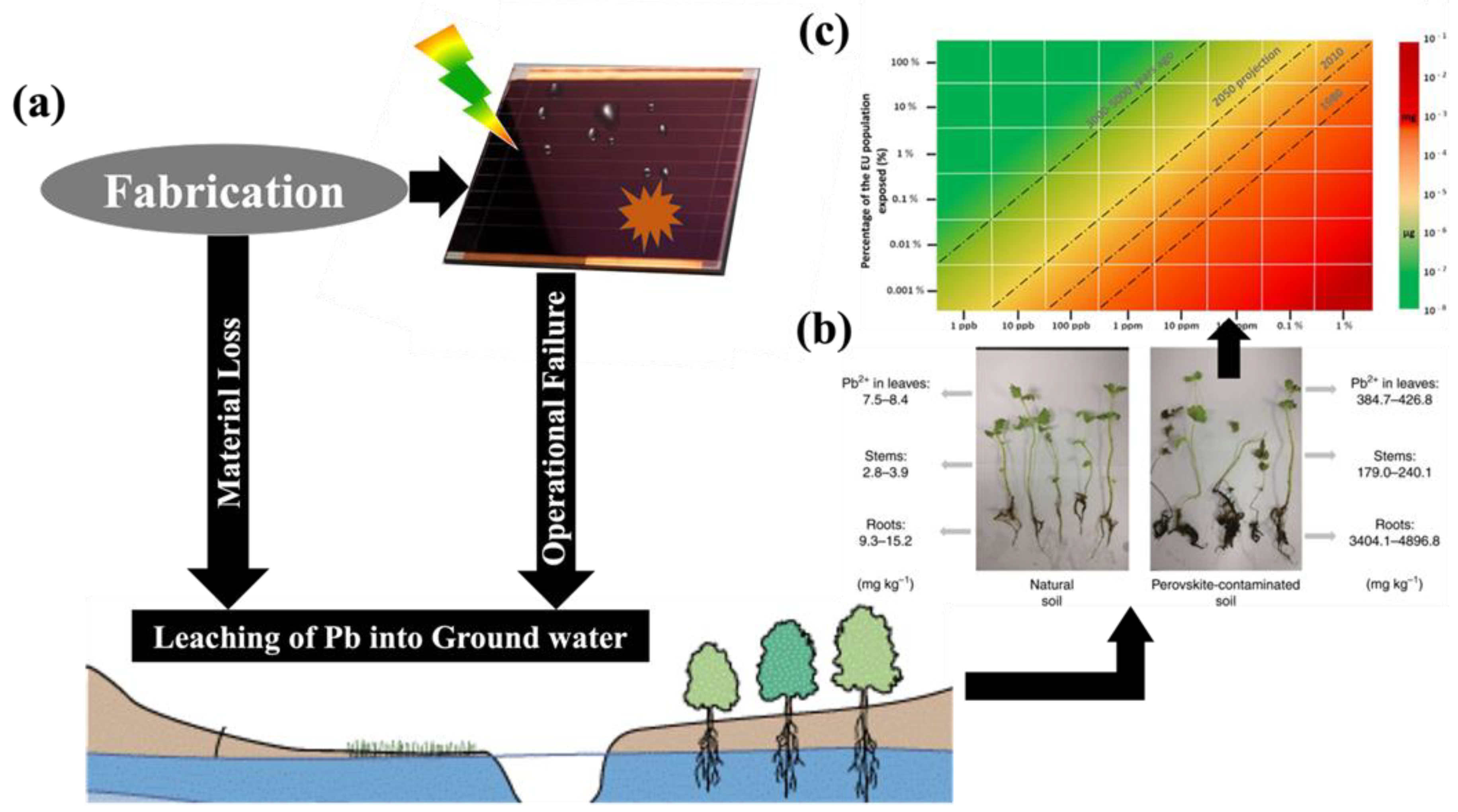
Figure 4.
Stability of CH3NH3PbI3 Perovskite: (a) Decomposition of path of the perovskite in presence of water molecules (A water molecule H2O is required to initiate the process with the decomposition, leading to the formation of hydrated MAPI and eventually to Pb and PbI2 with the gases HI and CH3NH2 leaving the substrate. Reproduced with permission: Copyright 2014, American Chemical Society.[20] (b) absorption spectra for the perovskite films on glass before (black) and after (red) ageing in different conditions. The inset shows the photographs of films before (left) and after (right) ageing. Reproduced with permission: Copyright 2016, The Royal Society of Chemistry.[21] (c) X-ray diffractograms of the perovskite films degraded in different atmospheres for 24 h. (The inset shows the photographs of films). Reproduced with permission: Copyright 2016, The Royal Society of Chemistry.[22] (d) SEM pictures of perovskite on ITO: pristine layers, initial stages of degradation, and later stages of degradation. (The scale bars represent 200 nm) followed by the illustration of the progression of oxygen induced degradation from the grain boundaries. Reproduced with permission: Copyright 2017, WILEY-VCH.[23].
Figure 4.
Stability of CH3NH3PbI3 Perovskite: (a) Decomposition of path of the perovskite in presence of water molecules (A water molecule H2O is required to initiate the process with the decomposition, leading to the formation of hydrated MAPI and eventually to Pb and PbI2 with the gases HI and CH3NH2 leaving the substrate. Reproduced with permission: Copyright 2014, American Chemical Society.[20] (b) absorption spectra for the perovskite films on glass before (black) and after (red) ageing in different conditions. The inset shows the photographs of films before (left) and after (right) ageing. Reproduced with permission: Copyright 2016, The Royal Society of Chemistry.[21] (c) X-ray diffractograms of the perovskite films degraded in different atmospheres for 24 h. (The inset shows the photographs of films). Reproduced with permission: Copyright 2016, The Royal Society of Chemistry.[22] (d) SEM pictures of perovskite on ITO: pristine layers, initial stages of degradation, and later stages of degradation. (The scale bars represent 200 nm) followed by the illustration of the progression of oxygen induced degradation from the grain boundaries. Reproduced with permission: Copyright 2017, WILEY-VCH.[23].
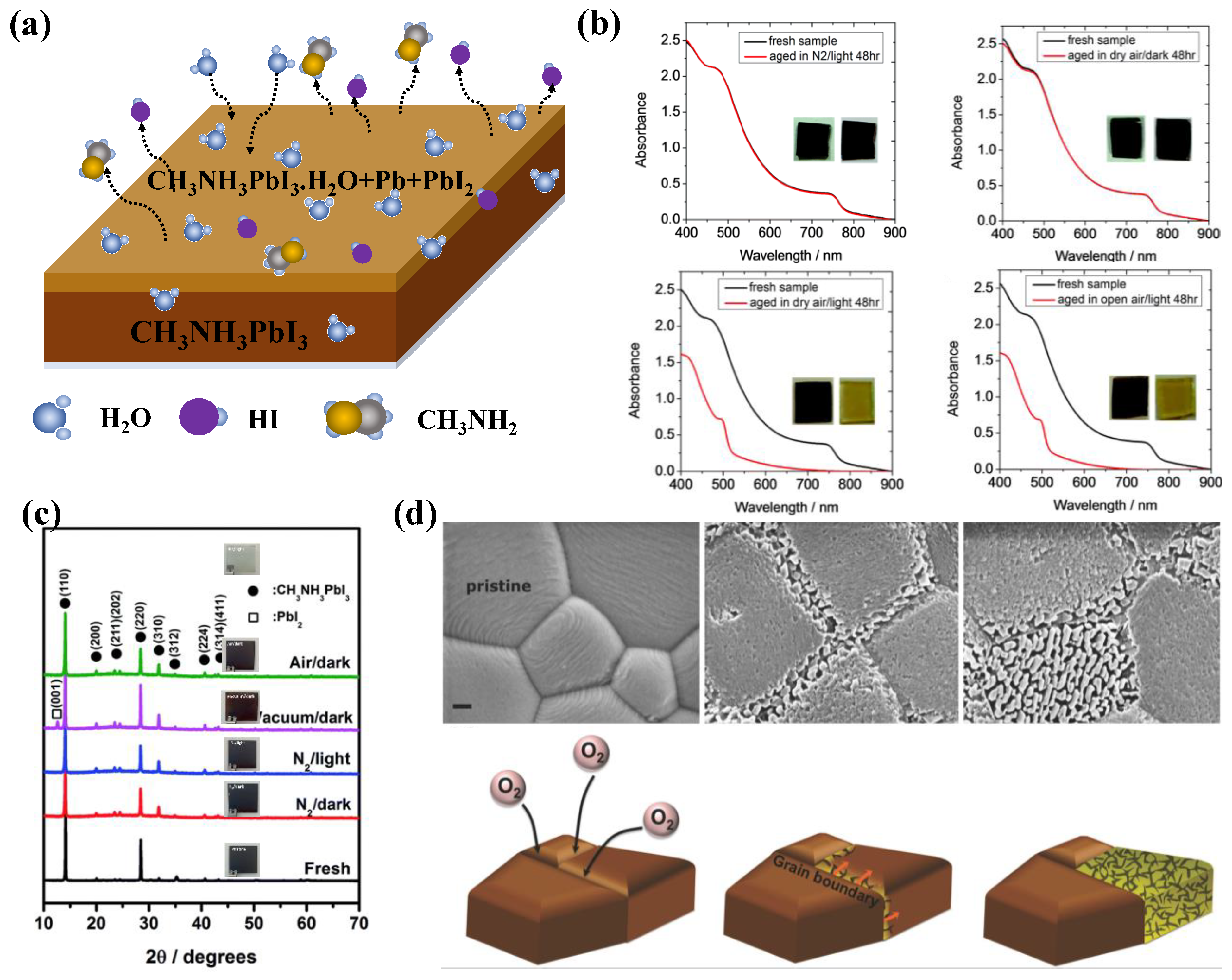
Figure 5.
Structure of different dimensional perovskites (a) 3-D (b) 2-D (c) 1-D and (d) 0-D.
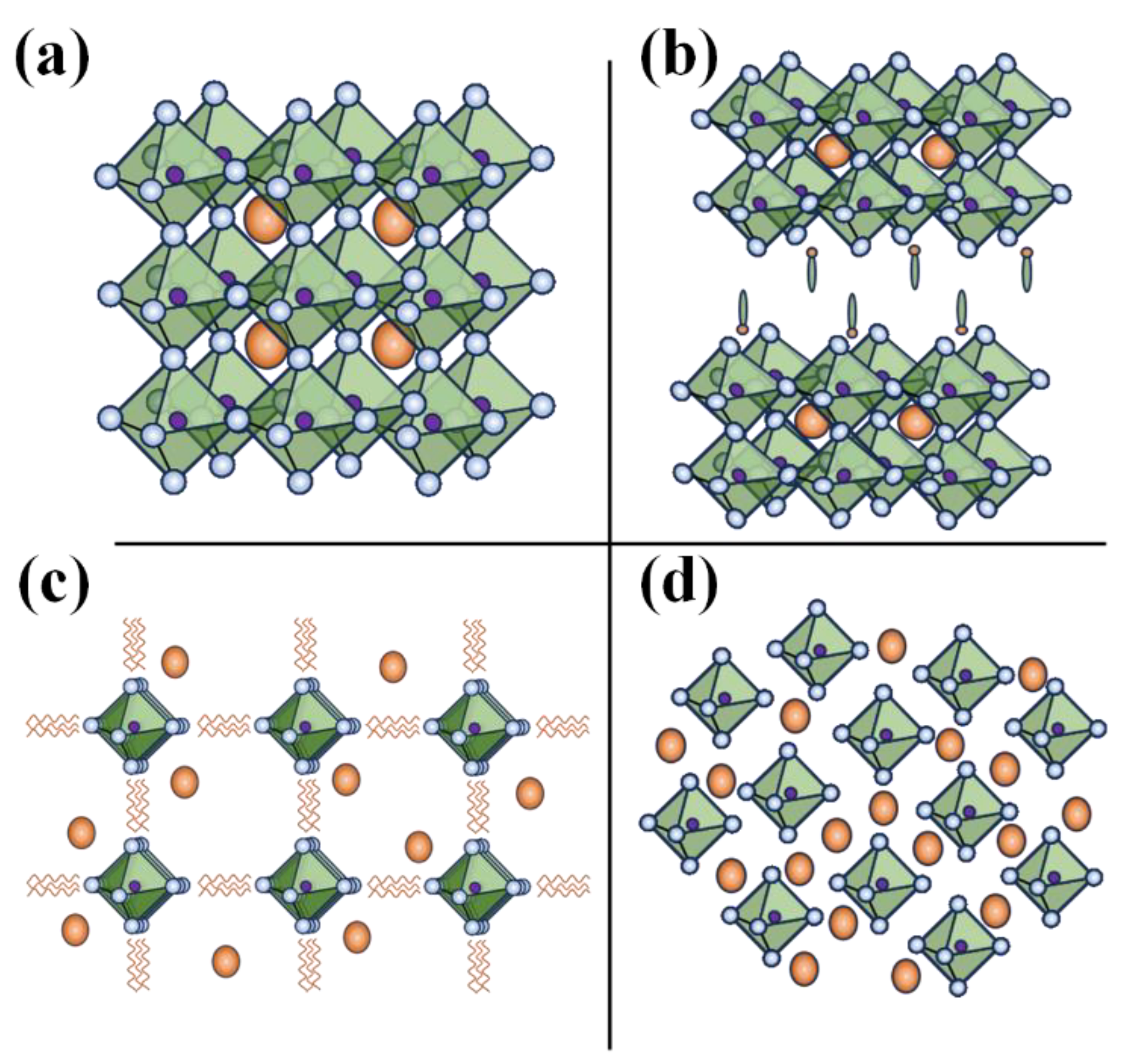
Figure 6.
Band gaps of various Lead-free HP compositions discussed in this section.

Figure 7.
Compositional Engineering: (a) Schematic of Interfacial interaction of MA3Bi2Cl9–xIx with the bandgap funnel structure. Reproduced with permission: Copyright 2022, Wiley-VCH.[78] (b) Absorption spectra of of the as-prepared Cs3Bi2xSb2–2xI9 (x = 0.9, 0.7, 0.5, 0.3, 0.1). Reproduced with permission: Copyright 2020, Wiley-VCH.[79] (c)i. Crystal structure and transformation relationship of Cs2PtxSn1−xCl6. ii. Charge-carrier dynamics model of Cs2Pt0.05Sn0.95Cl6 and Cs2Pt0.75Sn0.25Cl6. Reproduced with permission: Copyright 2021, Wiley-VCH.[80].
Figure 7.
Compositional Engineering: (a) Schematic of Interfacial interaction of MA3Bi2Cl9–xIx with the bandgap funnel structure. Reproduced with permission: Copyright 2022, Wiley-VCH.[78] (b) Absorption spectra of of the as-prepared Cs3Bi2xSb2–2xI9 (x = 0.9, 0.7, 0.5, 0.3, 0.1). Reproduced with permission: Copyright 2020, Wiley-VCH.[79] (c)i. Crystal structure and transformation relationship of Cs2PtxSn1−xCl6. ii. Charge-carrier dynamics model of Cs2Pt0.05Sn0.95Cl6 and Cs2Pt0.75Sn0.25Cl6. Reproduced with permission: Copyright 2021, Wiley-VCH.[80].
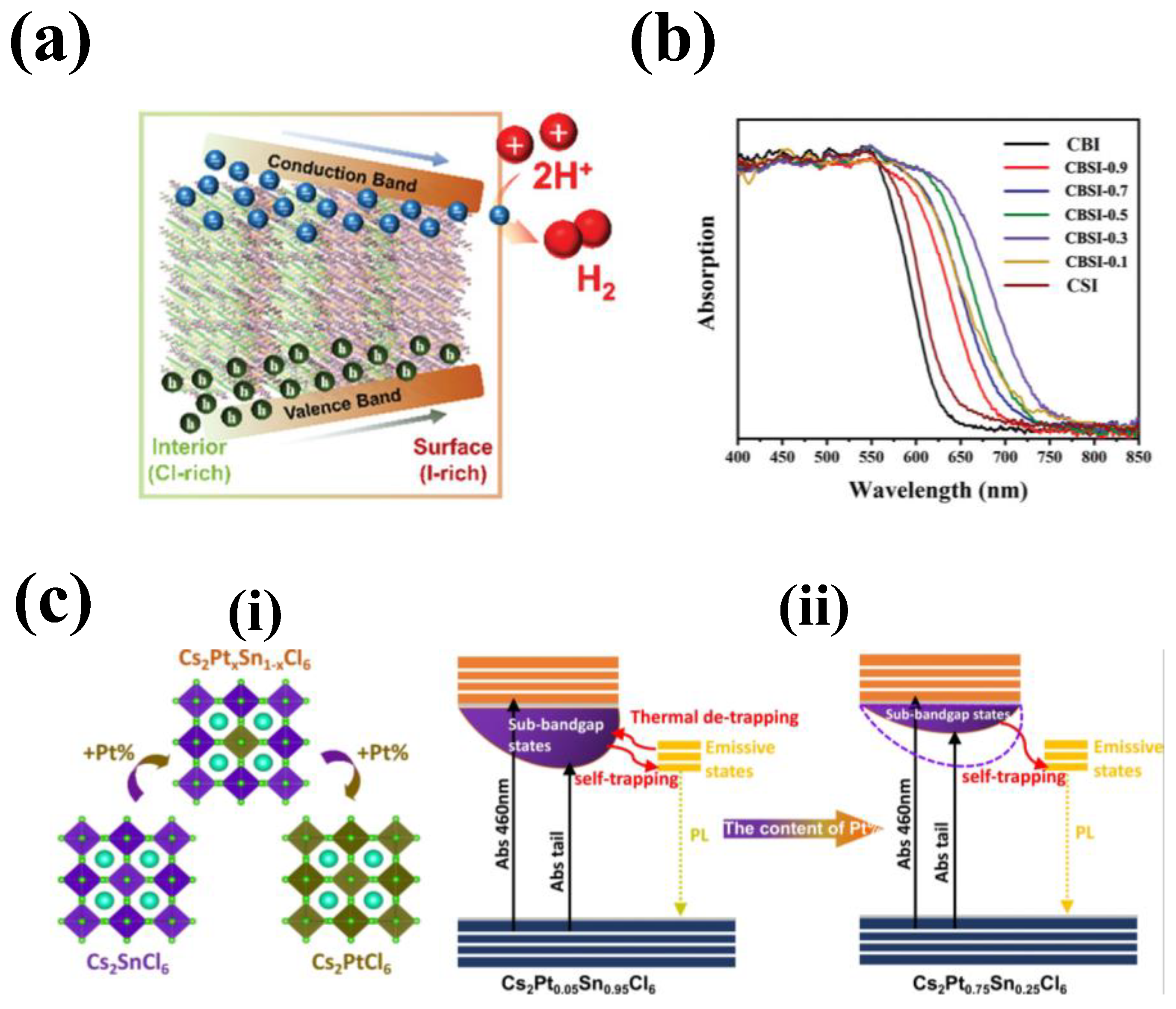
Figure 8.
Semiconductor/Pb-free HP heterojunctions: (a) Photocurrent response of BiVO4 and BiVO4/Cs2PtI6 heterojunction (Inset- Band alignment of the photoanode as a function of the wavelength). Reproduced with permission: Copyright 2021, American Chemical Society.[88] (b) Schematic representation of the formation of MA3Bi2I9/DMA3BiI6 heterojunctions. Reproduced with permission: Copyright 2020, Wiley-VCH.[89] (c) Schematic representation of synthesis of Cs2AgBiBr6/N-C Photocatalyst. Reproduced with permission: Copyright 2021, American Chemical Society.[90] (d) i. Schematic representation of Cs3Rh2I9 bulk crystal. ii. Electrochemical reduction of Cs3Rh2I9 clusters on NC (Cs3Rh2I9/NC) to form Cs3Rh2I9/NC-R iii. Electrochemical reduction of bulk Cs3Rh2I9 to form Cs3Rh2I9-R with large particle size. Reproduced with permission: Copyright 2023, Springer Nature.[91].
Figure 8.
Semiconductor/Pb-free HP heterojunctions: (a) Photocurrent response of BiVO4 and BiVO4/Cs2PtI6 heterojunction (Inset- Band alignment of the photoanode as a function of the wavelength). Reproduced with permission: Copyright 2021, American Chemical Society.[88] (b) Schematic representation of the formation of MA3Bi2I9/DMA3BiI6 heterojunctions. Reproduced with permission: Copyright 2020, Wiley-VCH.[89] (c) Schematic representation of synthesis of Cs2AgBiBr6/N-C Photocatalyst. Reproduced with permission: Copyright 2021, American Chemical Society.[90] (d) i. Schematic representation of Cs3Rh2I9 bulk crystal. ii. Electrochemical reduction of Cs3Rh2I9 clusters on NC (Cs3Rh2I9/NC) to form Cs3Rh2I9/NC-R iii. Electrochemical reduction of bulk Cs3Rh2I9 to form Cs3Rh2I9-R with large particle size. Reproduced with permission: Copyright 2023, Springer Nature.[91].
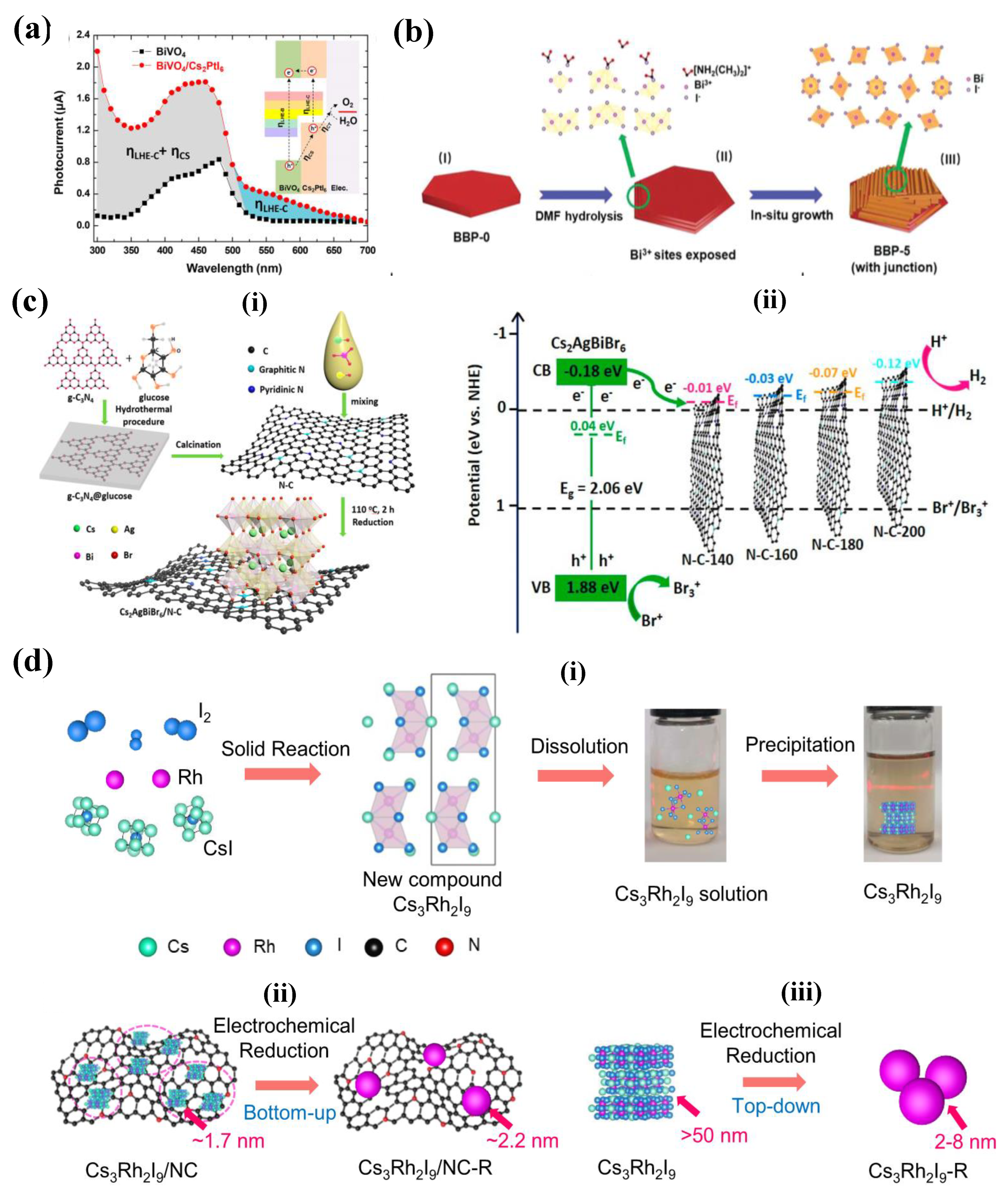
Table 2.
List of Pb-free HPs for PEC systems for hydrogen generation discussed in Section 5.4.
| No. | Material | Photoanode Area | Electrolyte/ Illumination | Photocurrent | Stability | Ref |
|---|---|---|---|---|---|---|
| 1 | Cs2PtI6 | pH -11 1 sun (AM 1.5 G, 100 mW cm–2) |
0.8 mAcm@2 at 1.23 V | 12 h | 71 | |
| 2 | Cu1.4Ag0.6BiI5 | 0.785 cm2 | 1 sun (AM 1.5 G, 100 mW cm–2) | 4.62 mA cm–2 at 1.23 VRHE | ~5 h | 72 |
| 3 | Cs2AgBiCl6 | 1 cm2 | 1 M KOH 1 Sun |
3.85 mA @ 1.0 V (Vs Ag/AgCl) | 10 h | 73 |
| 4 | Cs3Bi2Cl9 | 1 cm2 | 1 M KOH 1 Sun |
3.85 mA @ 1.0 V (Vs Ag/AgCl) | 10 h | 73 |
| 5 | Cs2AgInCl6 | - | water and acetonitrile |
0.75 mA cm−2 @ 600 mV (Vs RHE) | 2 h | 74 |
| 6 | Cs2ReBr6 | 25 mm2 | 1.5 mM KOH solution 1 Sun |
0.20 mA cm−2 0.4 V vs. Ag/AgCl | 75 | |
| 7 | Cs2ReI6 | 25 mm2 | 1.5 mM KOH solution 1 Sun |
0.14 mA cm−2 0.4 V vs. Ag/AgCl | 75 |
Table 3.
List of Semiconductor/Pb-free HP heterojunctions discussed in section 6.2.1.
| Heterojunction | Reaction Solution | Light Source | HER (µmol g-1 h-1) | Stability | Photocurrent | Ref |
|---|---|---|---|---|---|---|
| BiVO4/Cs2PtI6 | H2O:KOH | 500 Wm-2 , AM 1.5G filter | - | 2 mA cm−2 at 1.23 V (vs RHE) | 88 | |
| Cs2AgInCl6/ IrOx | CH3CN:H2O | 1 Sun | 2 h | 155.8 mA @ 600mV (vs RHE) | 74 | |
| MA3Bi2I9/DMA3BiI6 | H2O:HBr | 100 mW cm−2 (λ ≥ 420 nm) | 198.2 | 10 h / 10 cycles | 89 | |
| 2-AMPSbI5/ GO | sodium sulfate:H2O | 150 W xenon lamp | 185.8 | 4 cycles | 65 | |
| Cs2AgBiBr6/N-C | H2O:HBr | λ ≥ 420 nm | 380 | 3 h / 6 cycles | 90 | |
| Cs3Rh2I9/NC-R | H2O:KOH | 50h | mass activity of 772.1 mA mg−1 (10mA cm−2 at 1.23 V (vs RHE) |
91 |
Table 4.
List of g-C3N4/Pb-free HP heterojunctions discussed in section 6.2.3.
| Material | Reaction Solution | Light Source | Hydrogen Evolution Rate (µmol g-1 h-1) | Stability | Ref |
|---|---|---|---|---|---|
| PEA2SnBr4 | H2O/ 10 % TEOA | 500 Wm-2 , AM 1.5G filter | 1613 | - | 69 |
| PhBz2GeI4 | H2O/ 10 % TEOA | 500 Wm-2 , AM 1.5G filter | 1200 | 6h / 4 cycles | 70 |
| Cs3Bi2I9 | H2O/ 10 % Me.OH | 450 W Xe lamp | 920.76 | 6 h | 92 |
| Cs2AgBiBr6 -rGO |
H2O/HBr | >420 nm | 48.9 | 10h / 12 cycles | 93 |
| DMASnX3 | H2O/10 % TEOA | 500 Wm-2 , 300-800 nm | 1730 | 4 h | 68 |
| Cs3Bi2Br9 | H2O/10 % TEOA | 500 Wm-2 , 300-800 nm | 4 593 | - | 94 |
| Cs2AgBiBr6 | HBr/ 20% H3PO2 | 300 W (λ ≥ 420 nm) | 60 | 3h/ 14 cycles | 95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



