
Submitted:
28 October 2024
Posted:
28 October 2024
You are already at the latest version
Abstract
In the information society, challenges posed by climate change and population growth have rendered the development of crop varieties with enhanced abiotic stress tolerance and improved nutritional value a prime objective in agricultural advancement. Chinese cabbage, recognized for its significant economic benefits, not only supplements the human body with vitamin C and vitamin E but also plays a crucial role in preventing cardiovascular diseases. Nonetheless, its growth and development are adversely affected by various abiotic stresses, including waterlogging and temperature fluctuations. Consequently, the identification of abiotic stress-responsive genes (SRGs) in Chinese cabbage is of paramount importance for enhancing its resilience. While transcriptome analysis is a reliable approach for identifying stress-related genes, it is inherently species-specific and can be time-consuming. In this study, we proposed a computational model utilizing machine learning techniques to predict genes in Chinese cabbage that respond to four specific abiotic stresses: cold, heat, drought, and salt. To construct this model, we compiled data from relevant studies regarding the response to these abiotic stresses, and the protein sequences encoded by abiotic SRGs were converted into numerical representations for subsequent analysis. For the selected feature set, we employed six distinct machine learning binary classification algorithms. The results demonstrate that various models can effectively predict SRGs associated with the four types of abiotic stresses, with the area under the receiver operating characteristic curve (auROC) for the models being 81.42%, 87.92%, 80.85%, and 88.87%, respectively. Furthermore, we have established an online prediction server, named MLAS (http://47.122.66.48:4433), designed to predict the response genes of Chinese cabbage under the aforementioned abiotic stresses. The computational model and the prediction tool developed in this study can serve as valuable resources for the identification of abiotic SRGs in Chinese cabbage.
Keywords:
Chinese cabbage
; stress-responsive genes
; machine learning
; binary classification
1. Introduction
Chinese cabbage (Brassica rape L. ssp. Pekinensis) is a significant vegetable in Asia, with its consumption steadily increasing in Western countries [1]. As the origin of Chinese cabbage, China possesses a rich array of germplasm resources and local varieties. Therefore, the effective utilization of these germplasm resources to enhance the quality and yield of Chinese cabbage holds considerable importance for practical agricultural production [2]. However, the growth of Chinese cabbage is negatively impacted by abiotic stress, which leads to reductions in both yield and quality, thereby significantly affecting agricultural productivity. Abiotic stress refers to the detrimental effects on the normal growth and development of plants caused by non-biological factors. Common abiotic stressors that plants frequently encounter include drought, radiation, nutrient deficiency, extreme temperatures (both high and low), metal ion toxicity, salinity, and organic pollution, all of which severely compromise the distribution and growth conditions of plants worldwide [3]. Research indicates that abiotic stresses such as drought, extreme temperatures, and high salinity influence nearly every stage of the Chinese cabbage life cycle, affecting not only the expression of relevant genes but also cellular metabolism and developmental processes [4]. Under these stress conditions, Chinese cabbage can develop a certain degree of resistance; however, as the intensity of the stress escalates, this self-generated resistance becomes insufficient to cope with more severe abiotic challenges. The consequences of such stress can result in abnormal growth or even mortality of the Chinese cabbage, leading to diminished yields and subsequent market shortages. The capacity of plants to respond to environmental changes is critical for their adaptation and survival. Hence, it is essential to investigate the mechanisms by which plants adapt to varying climatic conditions. Consequently, researching the molecular mechanisms underlying abiotic stress in Chinese cabbage is of great significance for enhancing its yield under adverse conditions. In particular, the identification of abiotic stress-responsive genes (SRGs) [5] and proteins is vital for fostering the resilience of Chinese cabbage [6].
In recent years, the advancement of high-throughput technologies has led to the rapid generation of extensive biological data. The availability of complete genome sequences of various plant species has enabled comprehensive research on biomacromolecules, thereby promoting the investigation of abiotic SRGs at the whole-genome level. In addition to the identification of SRGs through transcriptome analysis, gene expression studies serve as an additional method for recognizing these genes [7,8,9,10,11,12,13]. Specifically, 35 BrHsf genes have been identified in Chinese cabbage, and a thorough analysis has revealed their potential to enhance plant heat tolerance [10]. Furthermore, comparative genomic analyses have enhanced our comprehension of the evolutionary dynamics of Hsf genes in cabbage. A co-expression network of genes responsive to cold, drought, and salt stress in cabbage has been constructed and analyzed from multiple perspectives, leading to the identification of previously unknown genes associated with abiotic stress tolerance [4]. Additionally, through the evolutionary analysis of gene families, abiotic SRGs have also been identified within the B-Box family in cabbage. The application of advanced analytical methods to high-throughput genomic data—including genes, transcripts, proteins, and metabolites—will undoubtedly optimize data utilization and enhance the accuracy of abiotic SRGs identification [14].
In the domain of bioinformatics, various machine learning algorithms have been employed to address significant biological challenges. The integration of machine learning into biological research has introduced a novel perspective that contrasts sharply with traditional experimental and simulation methodologies. This approach has demonstrated considerable potential due to its flexibility, accuracy, and robust generalization capabilities when analyzing complex biological systems. Artificial intelligence-driven machine learning techniques have emerged as pivotal tools for data interpretation, particularly in the context of predictive modeling and plant stress responses [15]. Previous research has successfully predicted the types of stresses to which plants respond by analyzing the expression patterns of plant microRNAs (miRNAs). In this context, intricate nonlinear relationships between input variables (miRNA expression) and output variables (plant stress responses) are discerned from training datasets housed in various databases. This enables the identification of whether previously uncharacterized plant miRNAs are responsive to stress conditions [16]. Additionally, computational models utilizing machine learning have been developed to predict proteins associated with abiotic stress in plants [17], specifically focusing on the classification of abiotic stress response proteins in crops of the family Poaceae through the application of deep convolutional neural networks [14]. These investigations have illustrated the efficacy of machine learning methodologies in the identification, classification, and prediction of diverse stress response molecules in plants. However, despite the labor-intensive and time-consuming nature of identifying genes related to abiotic stress through conventional genetic techniques, there remains a lack of dedicated computational models for the identification of abiotic SRGs in Chinese cabbage. Given these considerations, the development of a computational method to predict abiotic SRGs in Chinese cabbage is warranted.
The objective of this study is to develop a machine learning-based computational model to identify the genes associated with cold, heat, drought, and salt stresses in Chinese cabbage. This endeavor aims to uncover novel abiotic SRGs within this species. To accomplish this goal, we systematically collected and organized genes responsive to the aforementioned stressors from pertinent literature, subsequently obtaining the protein sequences generated through the transcription and translation of each gene. We employed these protein sequences as a robust form of high-throughput data to analyze specific targets related to abiotic stress, consequently establishing complex linear relationships between the protein-coding gene sequences of Chinese cabbage and the four types of stress.
2. Materials and Methods
2.1. Data Collection and Pre-Processing
To date, substantial advancements have been achieved in the investigation of stress resistance in Chinese cabbage, employing various experimental methodologies to identify genes associated with cold, heat, drought, and salt stress. A comprehensive collection of 2,598 Chinese cabbage gene identifiers responsive to these four distinct types of abiotic stress has been compiled from relevant studies. This includes 600 genes related to drought, 761 to heat, 427 to salt, and 801 to cold stress. Table 1 presents a list of the most significant reference articles in this field. Given that certain genes exhibit responses to multiple stressors, Boolean operations were employed to ensure that genes respond to only a single type of stress. While differential expression and downstream analyses have resulted in a considerable number of experimentally validated abiotic stress-responsive genes (SRGs), various platforms provide differing gene sequences. To enhance specificity, we commence our approach by utilizing the protein sequences encoded by these genes and querying the gene identifiers through the BRAD database (http://brassicadb.org/brad/) to obtain the corresponding protein sequences. Additionally, to reduce potential bias in the model that may arise from similar protein sequences, the CD-HIT tool was employed, applying a threshold of 0.8 to eliminate redundant sequences, thereby constructing a non-redundant dataset for subsequent experimental analysis. The final counts of sequences obtained for cold, heat, drought, and salt stress were 527, 515, 409, and 239, respectively.
One category of abiotic stress data is utilized as the positive sample set, whereas the remaining three categories comprise the negative sample set. The quantities of positive samples are 527, 515, 409, and 239, while the quantities of negative samples are 1163, 1175, 1281, and 1451. Furthermore, the entire dataset is partitioned into a training set and a testing set in a ratio of 7:3.

Table 1.
Summary of the abiotic SRGs retrieved from related reference.
| Category | Number of Sequences | Ref. |
|---|---|---|
| Cold | 801 | [12,18,19,20] |
| Heat | 761 | [4,21,22,23,24,25] |
| Drought | 600 | [26,27,28,29,30] |
| Salt | 427 | [8,31,32] |
Table 2.
Summary of the positive and negative datasets.
| Category | Cold | Heat | Drought | Salt |
|---|---|---|---|---|
| Single stress | 728 | 663 | 449 | 328 |
| Positive set | 527 | 515 | 409 | 239 |
| Negative set | 1163 | 1175 | 1281 | 1451 |
2.2. Feature Construction and Selection
iFeatureOmega serves as a comprehensive computational tool designed for the characterization of diverse biomolecules. This platform is freely accessible and user-friendly, enabling users to generate, analyze, and visualize numerical vector representations of 189 biological sequences, structures, and ligands [33]. In our study, we employed the 6 protein feature extraction methods available through the graphical user interface (GUI) version of this tool to extract features from the collected protein sequence data.
In the context of multi-dimensional space classification and datasets characterized by feature interaction variables, there is a pressing need to enhance model classification accuracy. To address these issues, feature selection techniques are commonly utilized to reduce the complexity of the data structure. This is accomplished by identifying and selecting meaningful features that contribute positively to the model, thus obviating the need to incorporate all available features during the training process. Such an approach not only reduces the computational time required for classification but also enhances overall classification accuracy. Commonly utilized feature selection methods include backward feature selection, forward feature selection, and bidirectional feature selection [34]. Additionally, SVM-recursive feature elimination (SVM-RFE) is recognized as an effective feature selection technique. SVM-RFE facilitates the identification of relevant features while simultaneously eliminating relatively insignificant feature variables, ultimately leading to improved classification performance [35]. Empirical research demonstrates that datasets curated using SVM-RFE result in more straightforward computations and substantially improve classification accuracy [36].
2.3. Prediction Using Machine-Learning Methods
Machine learning techniques have been effectively utilized across various domains of bioinformatics [37,38], including gene discovery [39], genome annotation [40], protein classification prediction [41], and gene expression analysis [42]. Among the methodologies employed, supervised learning, unsupervised learning, reinforcement learning, and sparse dictionary learning have been extensively explored in prior research, with supervised learning emerging as a reliable and efficient approach for addressing challenges in the life sciences [43]. In this study, we evaluated several classification algorithms within the framework of supervised learning, specifically support vector machine (SVM) [44], extreme gradient boosting (XGB) [45], random forest (RF) [46], bagging (BAG) [47], adaptive boosting (ADB) [48], and gradient boosting decision trees (GBDT) [48]. The implementation of machine learning models was conducted using Python, leveraging Scikit-learn, an open-source Python library that offers straightforward and effective tools for data processing and modeling pertinent to data mining and analysis. Scikit-learn encompasses a comprehensive array of algorithms, including those for classification, regression, clustering, and dimensionality reduction, in addition to providing resources for model selection and evaluation [49]. This functionality enables the construction and optimization of machine learning models, making it a valuable asset for the models developed in this study.
2.4. Cross Validation and Performance Metrics
During the model development process, the training dataset is utilized to train the model, while the validation dataset is employed to assess the quality of the model and to select the optimal hyper-parameters. The final model derived from this process is subsequently applied to the test dataset to evaluate its real performance. However, relying solely on a training set and a validation set when the dataset is limited may lead to significant bias in the true evaluation of model performance, and allocating a separate validation set from a small dataset may further result in poor model performance. To achieve a reliable and stable model, a five-fold cross-validation method is implemented [50]. This approach eliminates the need to partition a separate validation set, as the test set is consistently reserved for the final evaluation of the model. Specifically, the entire training dataset is randomly divided into five equal-sized, mutually exclusive subsets. Four of these subsets are utilized for model training, while the remaining subset is employed to validate the established model. This procedure is repeated until each subset has served as a validation set. The accuracy across all five validation sets is averaged to provide a performance measure for the model, thereby assessing the quality of the model and facilitating the selection of the model and its corresponding parameters. Furthermore, the area under the receiver operating characteristic curve (auROC) and the area under the precision-recall curve (auPRC) are computed to evaluate predictive capability of the model [51]. The ROC curve is a graphical representation of the true positive rate (recall) plotted against the false positive rate. The area under the ROC curve (AUC-ROC) is a commonly used metric for assessing the model's performance across various thresholds. The specific formulation is as follows:
In this context, TP, FP, TN, and FN denote the quantities of positive samples accurately predicted as positive, the quantities of negative samples inaccurately predicted as positive, the quantities of negative samples accurately predicted as negative, and the quantities of positive samples inaccurately predicted as negative, respectively. All steps involved in the proposed methodology are illustrated in Figure 1.
3. Results
3.1. Preliminary Analysis of the Sequence Data
Motifs refer to conserved regions within DNA or protein sequences, as well as common sequence patterns that are postulated to have biological functions or to play a role in essential biological processes. Motif analysis is a methodological approach employed to identify and analyze recurring patterns or sequence fragments present in DNA, RNA, or protein sequences [52]. For each type of abiotic stress, we conducted motif analysis utilizing the MEME Suite [53], which facilitates the identification of recurring sequence fragments in both positive and negative sample sets. When focusing exclusively on significant motifs (p=0.05), the search was constrained to a length of 10 amino acids. Through a comparative analysis of motifs derived from both positive and negative sample sets, we identified three motifs that are associated with cold stress: NTKFCYYNNY, FCKSCRRYWT, and KALKCPRCNS. These motifs were observed to be significantly more prevalent in the positive sample set compared to the negative sample set. Employing the same analytical method for the remaining three stress datasets, we identified the following significant motifs for heat stress: FYRQLNTYGF, LPKYFKHNNF, and YRKLALKYHP. In the context of drought stress, the motifs QLTIFYGGKV, IQTAFRGYLA, and ERNRREKINE were recognized as significant. Lastly, for salt stress, the motifs FGNKWATIAR, TDNAVKNHWN, and SCRLRWCNQL were identified as significant motifs.
In conclusion, motif analysis is demonstrated to be an effective approach for identifying significant sequence characteristics. The results indicate that distinct motifs can be discerned from both positive and negative sample sets across various forms of abiotic stress. As a result, classification can be performed based on the attributes of protein sequences.
Figure 2.
Discriminative motif discovery in protein sequences under various stress conditions.

3.2. Feature Construction and Selection Analysis
In this section, six distinct feature extraction methods were employed to derive features from protein sequences, and the corresponding feature sets from the training dataset were utilized to assess the predictive accuracy of various machine learning algorithms. As illustrated in Figure 3, the feature sets produced by the CKSAAP, DDE, DPC, and TPC methods demonstrated superior performance compared to the other two methods. Specifically, when the CKSAAP method was utilized to generate the feature set, the support vector machine SVM-based model achieved the highest area under the receiver operating characteristic curve (auROC) for cold, heat, and drought stresses, with respective values of 76.60%, 80.32%, and 78.09%. In the case of salt stress, the SVM model also attained the highest auROC of 85.14%, although this feature set was derived using the DDE method; the CKSAAP method followed closely with an auROC of 82.91%. Across the four types of stress evaluated, the CKSAAP method consistently outperformed the DDE and TPC methods based on the assessment results of various machine learning algorithms. Moreover, while the DPC and TPC methods produced a greater number of features, their performance was either slightly inferior to or comparable with that of the DDE method. Consequently, to mitigate computational demands, it may be advantageous to select feature extraction methods that yield fewer features. Overall, the feature sets generated by the CKSAAP and DDE methods exhibited higher predictive accuracy than those produced by the other methods employed in this study.
The inclusion of an excessive number of features can increase the complexity of a model, potentially leading to overfitting, whereas an insufficient number of features may result in underfitting. The primary objective of feature selection is to optimize the model such that it is sufficiently complex to ensure generalizable performance, while remaining simple enough to facilitate training, maintenance, and interpretation. In this study, multiple numerical features were generated for each protein sequence. However, the presence of numerous sparse features resulted in high correlation among them, which can adversely affect classification accuracy due to the presence of correlated or redundant features. To address this issue, the SVM-recursive feature elimination (SVM-RFE) method was employed to rank the features and systematically eliminate those deemed unimportant. Additionally, combinations of features were considered, focusing on three specific feature sets: CKSAAP, DDE, and the combined CKSAAP + DDE. The findings indicated that, following the screening process, the optimal feature set for various types of abiotic stress comprised a different number of features, with the optimal set achieving the highest area under the receiver operating characteristic curve (auROC) score. For the 1,600 features generated by the CKSAAP method, the optimal features selected for cold, heat, drought, and salt stress were 1,045, 1,224, 1,016, and 1,018, respectively, with auROC scores reaching 70% (see Figure 4). The DDE method, which has a lower feature dimensionality, extracted 400 features per sequence. After the feature selection process, the number of features selected for the four types of stress were 112, 84, 175, and 167, respectively. However, the performance for drought and salt stress was suboptimal, with auROC scores only reaching 60%. By integrating the two feature extraction methods, the combined total of (1,600 + 400) features resulted in the selection of 1,237, 1,062, 375, and 589 features for the optimal feature set, respectively.
3.3. Prediction Analysis with Selected Features
In the context of the four types of abiotic stresses, the optimal parameters for each predictive model were established utilizing a selected feature set, which was evaluated through five-fold cross-validation on the training dataset. Subsequently, receiver operating characteristic (ROC) analysis was conducted on the selected models using the test dataset to ascertain the most effective model for predicting genes associated with each type of stress. Taking salt stress as a case study, ROC analysis was performed on three feature sets (CKSAAP, DDE, and CKSAAP + DDE) in conjunction with six classification algorithms (SVM, XGB, RF, GBDT, ADB and BAG) as illustrated in Figure 5. The results indicated that the GBDT classification algorithm exhibited a superior area under the ROC curve (auROC) when utilizing features derived from the CKSAAP method compared to the other algorithms. In contrast, when employing the DDE feature set and the two combined feature sets, the BAG and RF algorithms demonstrated enhanced performance, respectively. Table 3 presents the most effective machine learning classification methods for various abiotic stresses across the three feature selection approaches. A comparative analysis revealed that the CKSAAP-derived features yielded a higher overall accuracy for each category of abiotic stress than the DDE and CKSAAP + DDE feature sets. Under the feature extraction method employed, the performance metrics of the six classification algorithm models demonstrate a minimum accuracy of 70% (refer to Figure 6). This observation suggests that the sequences corresponding to the four categories are all suitable for feature extraction utilizing the CKSAAP approach. Consequently, following the analysis presented above, the most effective model for predicting genes associated with each type of stress has been determined (as shown in Table 4).
3.4. Discovery of New SRGs in Chinese Cabbage
The traditional methodology for identifying genes associated with plant stress employs microarray analysis technology, which enables the statistical evaluation of gene expression data across different treatment conditions in comparison to a control group. This methodology facilitates the screening of a subset of genes exhibiting significant expression alterations. The differentially expressed genes are likely implicated in the relevant stress processes of organisms. Consequently, this study aims to investigate the intrinsic mechanisms underlying environmentally induced changes in organisms from a molecular perspective. In the Gene Expression Omnibus (GEO) database, the dataset GSE75145 was filtered utilizing specific keywords such as cold stress and Chinese cabbage, along with a and group analysis of the samples based on the experimental content. The online analysis tool, Analyze with GEO2R, was employed for conducting the analysis, and gene data were filtered according to the criteria of and , leading to the identification of 6,212 differentially expressed genes. Based on the current experimental validation results, a total of 728 genes that respond to cold stress have been identified (as shown in Table 2). Moreover, additional 5,484 differentially expressed genes were predicted using the cold stress model developed in this study, from which the top 15 genes with the highest scores were selected (as shown in Table 5).
Among these genes, Bra010873 is noted for its significant role in the development of pollen and anthers in Chinese cabbage, as well as its involvement in carbohydrate metabolism, which is influenced by cold treatment [54]. Bra008311 (BrPDI1-4) [55] and Bra005803 [56] belong to the PDI and C2H2 ZFP gene families, respectively. The former is regarded as a candidate gene associated with low temperature and cold damage stress, while the latter is crucial for regulating cabbage growth and development in response to cold stress. Cold treatment has been observed to reduce the expression level of the Bra017871 gene, thereby decelerating the synthesis rate of indole-3-acetic acid (IAA) and resulting in a decrease in its content, which subsequently promotes flower bud differentiation [57]. Both methylation and demethylation processes occur during cold acclimation, with studies indicating that Bra031266 plays a pivotal role in downregulating methylation during this phase [58]. Bra005785 (BrC3H28) [59], a member of the CCCH-type zinc finger family, is essential for plant growth and development, as well as for the response to cold stress. Both Bra033675 and Bra005785 are implicated in biological processes associated with stress resistance mechanisms. Transport proteins are posited to have potential applications in crop breeding, and the study of plant transport proteins is critical for understanding plant stress resistance [60]. Functional analysis of Bra033675 suggests that it can influence the activity of transport proteins, thereby playing a significant role in responses to abiotic stress. Additionally, Bra005785 plays a role in facilitating calcium ion binding. Calcium ions function as ubiquitous signaling molecules that are essential to numerous biological processes, particularly in relation to plant growth and responses to stress [61]..
3.5. Online Prediction Tool
To facilitate the implementation of the proposed method, we developed a web-based tool named MLAS, designed to predict the response genes of Chinese cabbage under four distinct types of abiotic stress: cold, heat, drought, and salt. The architecture of the web-based tool comprises three layers: a presentation layer, a web API layer, and an application layer [62]. The presentation layer is constructed using HTML, CSS, and JavaScript. The application layer is primarily implemented in the Python programming language and interacts with the data layer through API calls. Furthermore, the application layer incorporates models that enhance usability for end users, thereby simplifying access and interaction with the tool. The interface of the web tool is developed using the Python Flask framework, while the backend is constructed utilizing machine learning modules within the Python framework. This online prediction server enables the rapid upload and analysis of corresponding protein sequences based on cabbage gene IDs, with results presented in a tabular format that correlates with the specified Chinese cabbage genes under the four types of abiotic stress.
Figure 6.
Interface for use of MLAS.
Users have the option to upload either a single gene ID or multiple gene IDs for analysis. The former can be directly inputted on the "Analysis" page, whereas the latter necessitates the uploading of a file in a specified format. Upon uploading the corresponding original protein sequence of the cabbage gene in FASTA format, the output will categorize the gene into the predicted classification. The accurate classification and prediction of non-biological stress genes are instrumental for biologists engaged in research aimed at crop improvement. Machine learning and deep learning models are capable of effectively capturing complex patterns and dependencies within protein sequences, thereby facilitating the rapid identification of genes associated with non-biological stress response protein sequences. Figure 6 illustrates the interface of this web implementation server.
4. Discussion
Chinese cabbage (Brassica rape L. ssp. Pekinensis) is a vegetable that is rich in cellulose, vitamins, and trace elements, rendering it highly nutritious. It plays a crucial role in vegetable cultivation due to its significant economic benefits within our country. However, the yield and quality of Chinese cabbage are negatively impacted by abiotic stressors. Depending on the developmental stage of the plant and the duration of the stress, yield losses attributed to abiotic stress can reach approximately 70%. Consequently, investigating the molecular mechanisms underlying abiotic stress responses in Chinese cabbage is essential for enhancing resistance breeding and improving yield and profitability. Given the necessity of identifying various genes that respond to abiotic stress, this study introduces a novel computational tool designed to identify proteins encoded by genes associated with cold, heat, drought, and salt stress in Chinese cabbage. This tool offers a more efficient and rapid alternative to the traditional BLAST program.
The discovery that a single gene may be associated with multiple types of stress necessitates the development of classifiers designed for multi-class prediction. However, such classifiers may lead to the erroneous conclusion that a single gene corresponds exclusively to one type of stress. Consequently, this study establishes four distinct optimal prediction models, each tailored to predict the probability of individual genes being associated with one of four different types of abiotic stress. In the comparative analysis of these models, a consistent feature extraction method is employed, thereby demonstrating the efficacy of the CKSAAP method for extracting features from proteins encoded by stress-related genes in Chinese cabbage. Notably, the most effective model does not rely on traditional feature extraction methods, further underscoring the specificity of the employed feature extraction technique. Taking cold stress as a case study, this research has identified novel cold stress-related genes within the Chinese cabbage genome, several of which have been extensively studied across various plant species due to their significant functional roles. Based on existing literature, it can be inferred that these genes may play a role in the response to cold stress. Future experiments will be conducted to further validate the functions of these predicted genes in cold environments. Additionally, in the realm of modeling, there is potential for the exploration of more advanced machine learning and deep learning techniques, such as convolutional neural networks, recurrent neural networks, and multilayer perceptron, to enhance the predictive capabilities of our models.
This research method, along with the associated online prediction server, is anticipated to enhance experimental approaches in the identification of abiotic SRGs in plants. The findings of this study may facilitate the development of additional crop varieties that exhibit resistance to abiotic stresses in the future, thereby aiding agricultural production in effectively addressing the challenges posed by climate change and environmental pressures.
Funding
This research was funded by National Natural Science Foundation of China, 11171155, the Natural Science Foundation of Jiangsu Province, China, BK20171370, and the Primary Research & Development Plan (Modern Agriculture) of Jiangsu Province, China, BE2023350.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Seong, G.-U.; Hwang, I.-W.; Chung, S.-K. Antioxidant Capacities and Polyphenolics of Chinese Cabbage (Brassica Rapa L. Ssp. Pekinensis) Leaves. Food Chemistry 2016, 199, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, D.; Cai, Z.; Wang, L.; Wang, J.; Sun, L.; Fan, X.; Shen, S.; Zhao, J. Spectral Technology and Multispectral Imaging for Estimating the Photosynthetic Pigments and SPAD of the Chinese Cabbage Based on Machine Learning. Computers and Electronics in Agriculture 2022, 195, 106814. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J.; Gong, Z.; Zhu, J.-K. Abiotic Stress Responses in Plants. Nat Rev Genet 2022, 23, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.-H.; Lee, G.-S.; Yu, J.-G.; Kim, Y.-H.; Park, Y.-D. Correlation Network Analysis of Abiotic StressrelatedGenes Reveals the Coordinated Regulationof Transcription in Chinese Cabbage. HST 2018, 36, 266–279. [Google Scholar] [CrossRef]
- Shaik, R.; Ramakrishna, W. Genes and Co-Expression Modules Common to Drought and Bacterial Stress Responses in Arabidopsis and Rice. PLoS ONE 2013, 8, e77261. [Google Scholar] [CrossRef]
- Ma, Y.; Qin, F.; Tran, L.-S.P. Contribution of Genomics to Gene Discovery in Plant Abiotic Stress Responses. Molecular Plant 2012, 5, 1176–1178. [Google Scholar] [CrossRef]
- Chen, L.; Wu, X.; Zhang, M.; Yang, L.; Ji, Z.; Chen, R.; Cao, Y.; Huang, J.; Duan, Q. Genome-Wide Identification of BrCMF Genes in Brassica Rapa and Their Expression Analysis under Abiotic Stresses. Plants 2024, 13, 1118. [Google Scholar] [CrossRef]
- Hui, J.; Zhang, M.; Chen, L.; Wang, Y.; He, J.; Zhang, J.; Wang, R.; Jiang, Q.; Lv, B.; Cao, Y. Identification, Classification, and Expression Analysis of Leucine-Rich Repeat Extensin Genes from Brassica Rapa Reveals Salt and Osmosis Stress Response Genes. Horticulturae 2024, 10, 571. [Google Scholar] [CrossRef]
- Singh, S.; Chhapekar, S.S.; Ma, Y.; Rameneni, J.J.; Oh, S.H.; Kim, J.; Lim, Y.P.; Choi, S.R. Genome-Wide Identification, Evolution, and Comparative Analysis of B-Box Genes in Brassica Rapa, B. Oleracea, and B. Napus and Their Expression Profiling in B. Rapa in Response to Multiple Hormones and Abiotic Stresses. IJMS 2021, 22, 10367. [Google Scholar] [CrossRef]
- Song, X.; Liu, G.; Duan, W.; Liu, T.; Huang, Z.; Ren, J.; Li, Y.; Hou, X. Genome-Wide Identification, Classification and Expression Analysis of the Heat Shock Transcription Factor Family in Chinese Cabbage. Mol Genet Genomics 2014, 289, 541–551. [Google Scholar] [CrossRef]
- Wang, C.; Duan, W.; Riquicho, A.R.; Jing, Z.; Liu, T.; Hou, X.; Li, Y. Genome-Wide Survey and Expression Analysis of the PUB Family in Chinese Cabbage (Brassica Rapa Ssp. Pekinesis). Mol Genet Genomics 2015, 290, 2241–2260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, F.; You, X.; Hou, X. Identification and Functional Characterization of a Cold-Related Protein, BcHHP5, in Pak-Choi (Brassica Rapa Ssp. Chinensis). IJMS 2018, 20, 93. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhao, Y.; Wu, X.; Zhang, Y.; Fu, Y.; Duan, Q.; Ma, W.; Huang, J. Genome-Wide Identification and Expression Analysis of BraGLRs Reveal Their Potential Roles in Abiotic Stress Tolerance and Sexual Reproduction. Cells 2022, 11, 3729. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, B.; Haque, M.A.; Iquebal, M.A.; Jaiswal, S.; Angadi, U.B.; Kumar, D.; Rai, A. DeepAProt: Deep Learning Based Abiotic Stress Protein Sequence Classification and Identification Tool in Cereals. Front. Plant Sci. 2023, 13, 1008756. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.; Anderson, R.; Hu, H.; Bennamoun, M.; Petereit, J.; Valliyodan, B.; Nguyen, H.T.; Batley, J.; Bayer, P.E.; Edwards, D. Machine Learning Models Outperform Deep Learning Models, Provide Interpretation and Facilitate Feature Selection for Soybean Trait Prediction. BMC Plant Biol 2022, 22, 180. [Google Scholar] [CrossRef]
- Vakilian, K.A. Machine Learning Improves Our Knowledge about miRNA Functions towards Plant Abiotic Stresses. Sci Rep 2020, 10, 3041. [Google Scholar] [CrossRef]
- Meher, P.K.; Sahu, T.K.; Gupta, A.; Kumar, A.; Rustgi, S. ASRpro: A Machine-learning Computational Model for Identifying Proteins Associated with Multiple Abiotic Stress in Plants. The Plant Genome 2022, e20259. [Google Scholar] [CrossRef]
- Wu, J.; Xu, X.-D.; Liu, L.; Ma, L.; Pu, Y.; Wang, W.; Hua, X.-Y.; Song, J.-M.; Liu, K.; Lu, G.; et al. A Chromosome Level Genome Assembly of a Winter Turnip Rape (Brassica Rapa L. ) to Explore the Genetic Basis of Cold Tolerance. Front. Plant Sci. 2022, 13, 936958. [Google Scholar] [CrossRef]
- Ma, L.; Coulter, J.; Liu, L.; Zhao, Y.; Chang, Y.; Pu, Y.; Zeng, X.; Xu, Y.; Wu, J.; Fang, Y.; et al. Transcriptome Analysis Reveals Key Cold-Stress-Responsive Genes in Winter Rapeseed (Brassica Rapa L. ). IJMS 2019, 20, 1071. [Google Scholar] [CrossRef]
- Huang, F.; Wang, J.; Tang, J.; Hou, X. Identification, Evolution and Functional Inference on the Cold-Shock Domain Protein Family in Pak-Ch. JOURNAL OF PLANT INTERACTIONS.
- Li, M.-Y.; Wang, F.; Jiang, Q.; Li, R.; Ma, J.; Xiong, A.-S. Genome-Wide Analysis of the Distribution of AP2/ERF Transcription Factors Reveals Duplication and Elucidates Their Potential Function in Chinese Cabbage (Brassica Rapa Ssp. Pekinensis). Plant Mol Biol Rep 2013, 31, 1002–1011. [Google Scholar] [CrossRef]
- Song, H.; Dong, X.; Yi, H.; Ahn, J.Y.; Yun, K.; Song, M.; Han, C.-T.; Hur, Y. Genome-Wide Identification and Characterization of Warming-Related Genes in Brassica Rapa Ssp. Pekinensis. IJMS 2018, 19, 1727. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Hu, J.; Huang, X.; Li, X.; Zhou, G.; Yan, Z. Comparative Transcriptome Analysis Reveals Heat-Responsive Genes in Chinese Cabbage (Brassica Rapa Ssp. Chinensis). Front. Plant Sci. 2016, 7, 939. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Hu, J.; Gao, C.; Chen, G.; Wang, B.; Lin, C.; Song, L.; Ding, Y.; Zhou, G. Genome-Wide Analysis of Long Non-Coding RNAs Unveils the Regulatory Roles in the Heat Tolerance of Chinese Cabbage (Brassica Rapa Ssp. Chinensis). Sci Rep 2019, 9, 5002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, P.; Su, T.; Li, P.; Xin, X.; Wang, W.; Zhao, X.; Yu, Y.; Zhang, D.; Yu, S.; et al. Comprehensive Analysis of Wall-Associated Kinase Genes and Their Expression Under Abiotic and Biotic Stress in Chinese Cabbage (Brassica Rapa Ssp. Pekinensis). J Plant Growth Regul 2020, 39, 72–86. [Google Scholar] [CrossRef]
- Saha, G.; Park, J.-I.; Ahmed, N.U.; Kayum, M.A.; Kang, K.-K.; Nou, I.-S. Characterization and Expression Profiling of MYB Transcription Factors against Stresses and during Male Organ Development in Chinese Cabbage (Brassica Rapa Ssp. Pekinensis). Plant Physiology and Biochemistry 2016, 104, 200–215. [Google Scholar] [CrossRef]
- Song, X.; Li, Y.; Hou, X. Genome-Wide Analysis of the AP2/ERF Transcription Factor Superfamily in Chinese Cabbage (Brassica Rapa Ssp. Pekinensis). BMC Genomics 2013, 14, 573. [Google Scholar] [CrossRef]
- Lu, X.; Cheng, Y.; Gao, M.; Li, M.; Xu, X. Molecular Characterization, Expression Pattern and Function Analysis of Glycine-Rich Protein Genes Under Stresses in Chinese Cabbage (Brassica Rapa L. Ssp. Pekinensis). Front. Genet. 2020, 11, 774. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, M.; Yin, F.; Cao, X.; Fan, S.; Wu, C.; Xiao, X. Genome-Wide Identification and Expression Analysis of BrATGs and Their Different Roles in Response to Abiotic Stresses in Chinese Cabbage. Agronomy 2022, 12, 2976. [Google Scholar] [CrossRef]
- Guo, Y.M.; Samans, B.; Chen, S.; Kibret, K.B.; Hatzig, S.; Turner, N.C.; Nelson, M.N.; Cowling, W.A.; Snowdon, R.J. Drought-Tolerant Brassica Rapa Shows Rapid Expression of Gene Networks for General Stress Responses and Programmed Cell Death Under Simulated Drought Stress. Plant Mol Biol Rep 2017, 35, 416–430. [Google Scholar] [CrossRef]
- Ouyang, B.; Yang, T.; Li, H.; Zhang, L.; Zhang, Y.; Zhang, J.; Fei, Z.; Ye, Z. Identification of Early Salt Stress Response Genes in Tomato Root by Suppression Subtractive Hybridization and Microarray Analysis. Journal of Experimental Botany 2007, 58, 507–520. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, X.; Chen, L.; Yang, L.; Cui, X.; Cao, Y. The RopGEF Gene Family and Their Potential Roles in Responses to Abiotic Stress in Brassica Rapa. IJMS 2024, 25, 3541. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, X.; Zhao, P.; Li, C.; Wang, Y.; Li, F.; Akutsu, T.; Bain, C.; Gasser, R.B.; Li, J.; et al. iFeatureOmega: An Integrative Platform for Engineering, Visualization and Analysis of Features from Molecular Sequences, Structural and Ligand Data Sets. Nucleic Acids Research 2022, 50, W434–W447. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Li, L.; Huang, Z.; Situ, H. Quantum-Enhanced Feature Selection with Forward Selection and Backward Elimination. Quantum Inf Process 2018, 17, 154. [Google Scholar] [CrossRef]
- Huang, M.-L.; Hung, Y.-H.; Lee, W.M.; Li, R.K.; Jiang, B.-R. SVM-RFE Based Feature Selection and Taguchi Parameters Optimization for Multiclass SVM Classifier. The Scientific World Journal 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Chen, D.; Liu, J.; Zang, L.; Xiao, T.; Zhang, X.; Li, Z.; Zhu, H.; Gao, W.; Yu, X. Integrated Machine Learning and Bioinformatic Analyses Constructed a Novel Stemness-Related Classifier to Predict Prognosis and Immunotherapy Responses for Hepatocellular Carcinoma Patients. Int. J. Biol. Sci. 2022, 18, 360–373. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, H.H.; Wang, X. Machine Learning for Big Data Analytics in Plants. Trends in Plant Science 2014, 19, 798–808. [Google Scholar] [CrossRef]
- Sun, S.; Wang, C.; Ding, H.; Zou, Q. Machine Learning and Its Applications in Plant Molecular Studies. Briefings in Functional Genomics 2020, 19, 40–48. [Google Scholar] [CrossRef]
- Cui, P.; Zhong, T.; Wang, Z.; Wang, T.; Zhao, H.; Liu, C.; Lu, H. Identification of Human Circadian Genes Based on Time Course Gene Expression Profiles by Using a Deep Learning Method. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2018, 1864, 2274–2283. [Google Scholar] [CrossRef]
- Polanski, K.; Rhodes, J.; Hill, C.; Zhang, P.; Jenkins, D.J.; Kiddle, S.J.; Jironkin, A.; Beynon, J.; Buchanan-Wollaston, V.; Ott, S.; et al. Wigwams: Identifying Gene Modules Co-Regulated across Multiple Biological Conditions. Bioinformatics 2014, 30, 962–970. [Google Scholar] [CrossRef]
- Li, X.; Liu, T.; Tao, P.; Wang, C.; Chen, L. A Highly Accurate Protein Structural Class Prediction Approach Using Auto Cross Covariance Transformation and Recursive Feature Elimination. Computational Biology and Chemistry 2015, 59, 95–100. [Google Scholar] [CrossRef]
- Kang, D.; Ahn, H.; Lee, S.; Lee, C.-J.; Hur, J.; Jung, W.; Kim, S. StressGenePred: A Twin Prediction Model Architecture for Classifying the Stress Types of Samples and Discovering Stress-Related Genes in Arabidopsis. BMC Genomics 2019, 20, 949. [Google Scholar] [CrossRef] [PubMed]
- Sohail, A.; Arif, F. Supervised and Unsupervised Algorithms for Bioinformatics and Data Science. Progress in Biophysics and Molecular Biology 2020, 151, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Chakraborty, S. Support Vector Machine in Structural Reliability Analysis: A Review. Reliability Engineering & System Safety 2023, 233, 109126. [Google Scholar] [CrossRef]
- Li, Z. Extracting Spatial Effects from Machine Learning Model Using Local Interpretation Method: An Example of SHAP and XGBoost. Computers, Environment and Urban Systems 2022, 96, 101845. [Google Scholar] [CrossRef]
- Wang, H.; Wang, G. Improving Random Forest Algorithm by Lasso Method. Journal of Statistical Computation and Simulation 2021, 91, 353–367. [Google Scholar] [CrossRef]
- Ngo, G.; Beard, R.; Chandra, R. Evolutionary Bagging for Ensemble Learning. Neurocomputing 2022, 510, 1–14. [Google Scholar] [CrossRef]
- Mienye, I.D.; Sun, Y. A Survey of Ensemble Learning: Concepts, Algorithms, Applications, and Prospects. IEEE Access 2022, 10, 99129–99149. [Google Scholar] [CrossRef]
- Abraham, A.; Pedregosa, F.; Eickenberg, M.; Gervais, P.; Mueller, A.; Kossaifi, J.; Gramfort, A.; Thirion, B.; Varoquaux, G. Machine Learning for Neuroimaging with Scikit-Learn. Front. Neuroinform. 2014, 8. [Google Scholar] [CrossRef]
- Jiang, G.; Wang, W. Error Estimation Based on Variance Analysis of k -Fold Cross-Validation. Pattern Recognition 2017, 69, 94–106. [Google Scholar] [CrossRef]
- Canbek, G.; Taskaya Temizel, T.; Sagiroglu, S. BenchMetrics: A Systematic Benchmarking Method for Binary Classification Performance Metrics. Neural Comput & Applic 2021, 33, 14623–14650. [Google Scholar] [CrossRef]
- Xiong, H.; Capurso, D.; Sen, Ś.; Segal, M.R. Sequence-Based Classification Using Discriminatory Motif Feature Selection. PLoS ONE 2011, 6, e27382. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, Z.; Li, C.; Zhang, Y.; Feng, H. Comparative Transcriptome Analysis of Fertile and Sterile Buds from a Genetically Male Sterile Line of Chinese Cabbage. In Vitro Cell.Dev.Biol.-Plant 2016, 52, 130–139. [Google Scholar] [CrossRef]
- Kayum, A. Genome-Wide Characterization and Expression Profiling of PDI Family Gene Reveals Function as Abiotic and Biotic Stress Tolerance in Chinese Cabbage (Brassica Rapa Ssp. Pekinensis). 2017. [CrossRef]
- Ma, L.; Xu, J.; Tao, X.; Wu, J.; Wang, W.; Pu, Y.; Yang, G.; Fang, Y.; Liu, L.; Li, X.; et al. Genome-Wide Identification of C2H2 ZFPs and Functional Analysis of BRZAT12 under Low-Temperature Stress in Winter Rapeseed (Brassica Rapa). IJMS 2022, 23, 12218. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, X.; Zhu, Z.; Sun, M.; Li, M.; Hou, L. Expression Analysis of Genes Related to Auxin Metabolism at Different Growth Stages of Pak Choi. Horticultural Plant Journal 2020, 6, 25–33. [Google Scholar] [CrossRef]
- Liu, T.; Li, Y.; Duan, W.; Huang, F.; Hou, X. Cold Acclimation Alters DNA Methylation Patterns and Confers Tolerance to Heat and Increases Growth Rate in Brassica Rapa. Journal of Experimental Botany 2017, 68, 1213–1224. [Google Scholar] [CrossRef]
- Rameneni, J.J.; Dhandapani, V.; Paul, P.; Devaraj, S.P.; Choi, S.R.; Yi, S.Y.; Kim, M.-S.; Hong, S.; Oh, S.H.; Oh, M.-H.; et al. Comprehensive Analysis of CCCH Zinc-Finger-Type Transcription Factors in the Brassica Rapa Genome. Hortic. Environ. Biotechnol. 2018, 59, 729–747. [Google Scholar] [CrossRef]
- Li, P.; Luo, T.; Pu, X.; Zhou, Y.; Yu, J.; Liu, L. Plant Transporters: Roles in Stress Responses and Effects on Growth and Development. Plant Growth Regul 2021, 93, 253–266. [Google Scholar] [CrossRef]
- Naz, M.; Afzal, M.R.; Raza, M.A.; Pandey, S.; Qi, S.; Dai, Z.; Du, D. Calcium (Ca2+) Signaling in Plants: A Plant Stress Perspective. South African Journal of Botany 2024, 169, 464–485. [Google Scholar] [CrossRef]
- Orovwode, H.; Ibukun, O.; Abubakar, J.A. A Machine Learning-Driven Web Application for Sign Language Learning. Front. Artif. Intell. 2024, 7, 1297347. [Google Scholar] [CrossRef]
Figure 1.
Schematic workflow for model development.
Figure 3.
Scatterplot heatmaps of auROC for different machine learning algorithms and feature extraction methods.
Figure 3.
Scatterplot heatmaps of auROC for different machine learning algorithms and feature extraction methods.

Figure 4.
Plots of the area under the receiver operating characteristic curve (auROC) were created using CKSAAP methods with SVM-RFE for feature selection.
Figure 4.
Plots of the area under the receiver operating characteristic curve (auROC) were created using CKSAAP methods with SVM-RFE for feature selection.
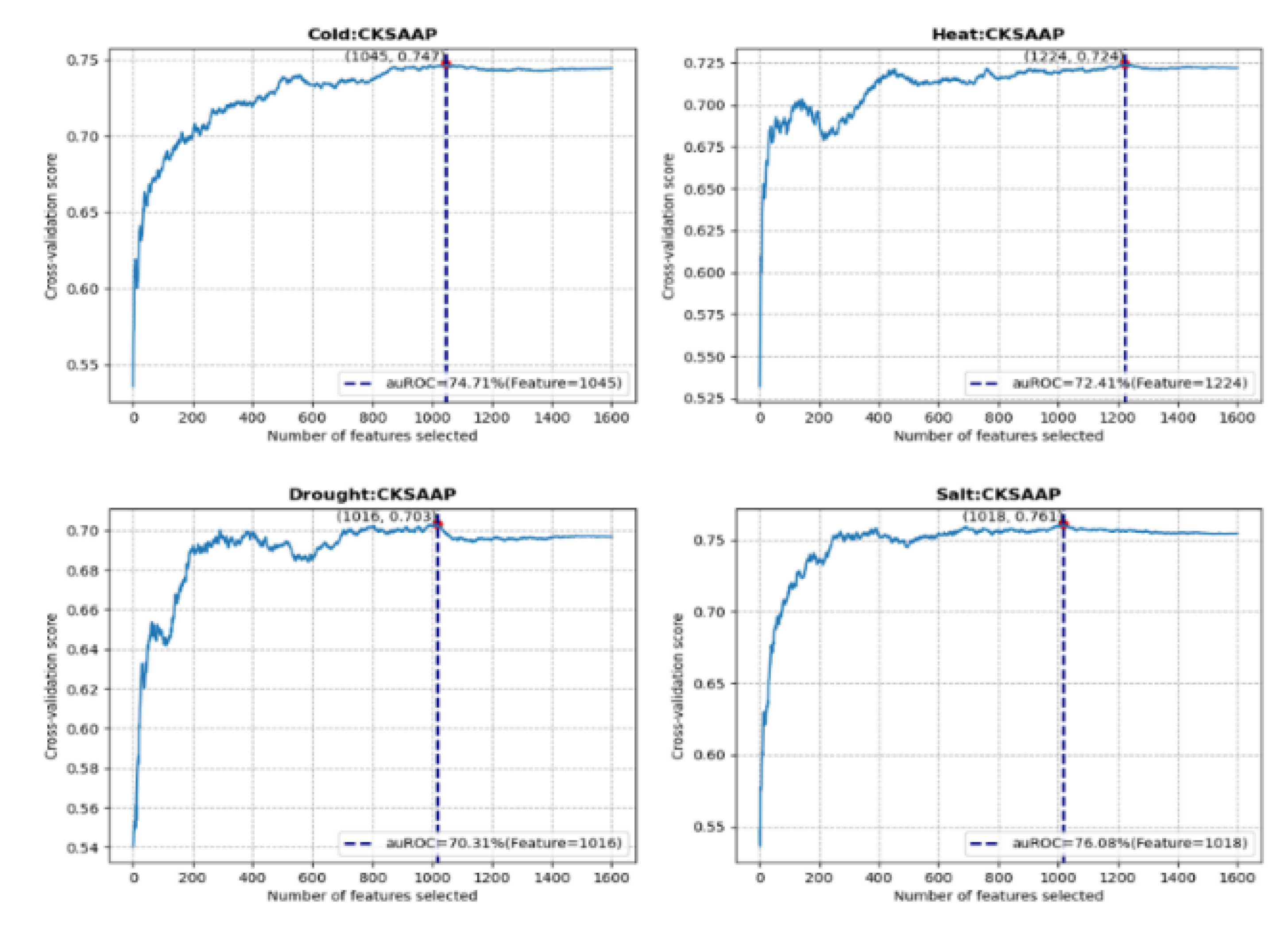
Figure 5.
ROC curves for salt stress show evaluation results from different feature extraction methods and classification algorithms.
Figure 5.
ROC curves for salt stress show evaluation results from different feature extraction methods and classification algorithms.
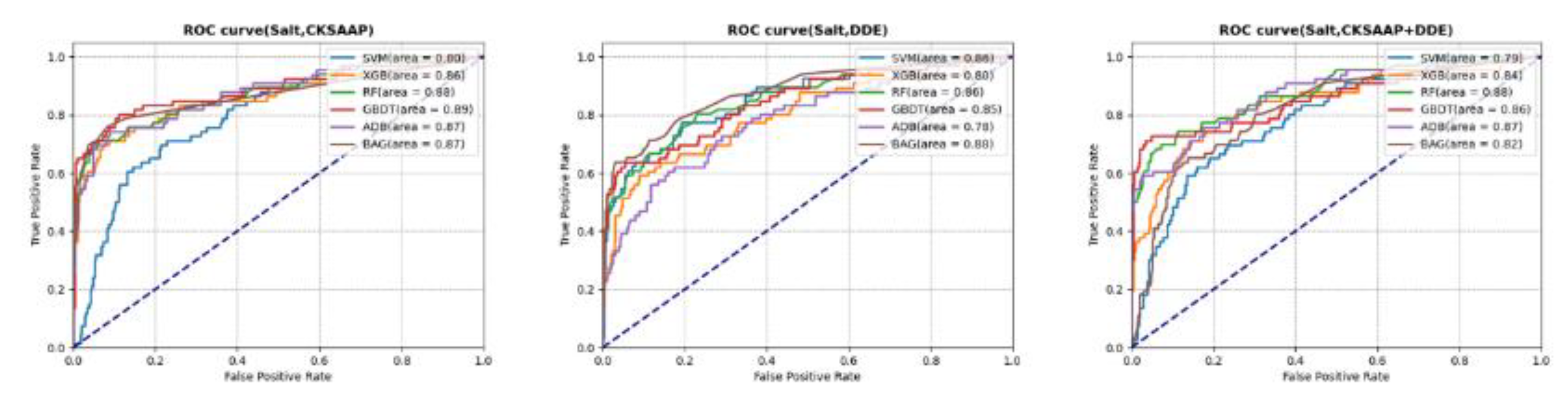
Table 3.
The best machine learning method for classifying abiotic stress with three feature selection techniques.
Table 3.
The best machine learning method for classifying abiotic stress with three feature selection techniques.
| Stress | Feature | Model | Accuracy (%) | auROC (%) | auPRC (%) |
|---|---|---|---|---|---|
| Cold | CKSAAP | RF | 74.26 | 81.42 | 70.92 |
| DDE | BAG | 76.92 | 77.50 | 63.92 | |
| CKSAAP+DDE | RF | 73.67 | 80.86 | 71.22 | |
| Heat | CKSAAP | GBDT | 82.84 | 87.92 | 81.76 |
| DDE | RF | 73.08 | 79.59 | 65.73 | |
| CKSAAP+DDE | GBDT | 77.51 | 85.72 | 76.66 | |
| Drought | CKSAAP | XGB | 81.36 | 80.85 | 63.11 |
| DDE | SVM | 79.88 | 78.78 | 56.21 | |
| CKSAAP+DDE | RF | 75.74 | 80.62 | 62.54 | |
| Salt | CKSAAP | GBDT | 88.48 | 88.87 | 79.63 |
| DDE | BAG | 89.04 | 87.97 | 74.29 | |
| CKSAAP+DDE | RF | 83.15 | 88.15 | 76.79 |
Table 4.
Predicting the best model for gene responses to abiotic stress.
| Stress | Feature | Model | Number of Features |
|---|---|---|---|
| Cold | CKSAAP | RF | 1045 |
| Heat | CKSAAP | GBDT | 1224 |
| Drought | CKSAAP | XGB | 1016 |
| Salt | CKSAAP | GBDT | 1018 |
Table 5.
The 15 cold stress-related genes with the highest predicted scores.
| Rank | BRAD gene ID |
|---|---|
| 1 | Bra031865 |
| 2 | Bra010873 |
| 3 | Bra022362 |
| 4 | Bra031763 |
| 5 | Bra008311 |
| 6 | Bra005803 |
| 7 | Bra017871 |
| 8 | Bra031266 |
| 9 | Bra011277 |
| 10 | Bra035084 |
| 11 | Bra033675 |
| 12 | Bra010302 |
| 13 | Bra023302 |
| 14 | Bra005785 |
| 15 | Bra005543 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



