
Submitted:
28 October 2024
Posted:
29 October 2024
You are already at the latest version
Abstract
Senescence-associated secretory phenotype (SASP) is a particular expression profile portrayed by senescent cells, which includes the secretion of cytokines, proteases, and chemokines, among other signalling molecules, exerting great influence on local as well as systemic environment which participates in cell senescence, immunoregulation, angiogenesis, cell proliferation, tumor invasion, etc. We performed a clinic-pathological, immunohistochemical and ultrastructural analysis with the aim of studying the association between the different histological characteristics of craniopharyngioma, the effects of OMF exposure and its potential role in the induction of SASP. We observed that cells of the outer epithelium can secrete lipoproteins and glycoproteins into the external tumor environment, inducing an intense inflammatory response. Neoplastic cells protrude out of the tumor and are able to grow rapidly and form wet keratin, which calcifies afterward. This limited growth ability is acquired by interacting with the aforementioned growth factors, proinflammatory molecules, angioblast and fibroblast activation, angiogenesis, cell proliferation, and brain invasion, associated with glycoproteins secretion of tumor cells associated with SASP.
Keywords:
senescence-associated secretory phenotype
; craniopharyngioma
; oil machinery fluid
; immunohistochemistry
; electron microscopy.
1. Introduction
Craniopharyngioma (CP) is a slow-growing tumor that arises in the sellar or para-sellar regions of the brain. It represents 1 % to 5% of all intracranial neoplasms, with an incidence of 0.16 cases per 100,000 people [1,2,3]. According to the WHO classification of brain tumors, are considered benign, classified as grade I [4,5,6]. CPs does not have a specific age of presentation. In children, they account for 1.2- 4% of intracranial tumors, typically occurring between the ages of 5-14. In adults, they are most commonly seen between 50-74 years. [3,7,8,9,10]. Histologically, are categorized into two types: adamantinomatous (AdaCP) and papillary (PaCP). However, molecular studies have suggested that these tumors have different origins [1].
The AdaCP type is characterized by squamous epithelium arranged in irregular cords, lobes, and trabeculae surrounded by palisade columnar epithelium. These densely packed cells clusters blend with loosely organized aggregates of squamous cells known as stellate reticulum. Nodules of "wet keratin," which are remnants of pale nuclei embedded in an eosinophilic keratinous mass, are present in both compact and loose regions. This tumor is most commonly cystic, and inside the cysts, an eosinophilic proteinaceous substance called oil machinery fluid (OMF), can be observed, noted for its high density under microscopic examination [11]. The cystic cavities, filled with squamous debris, are lined by flattened epithelium [2,3,7].
Cellular senescence (CS) is a state of permanent cell cycle arrest triggered by cellular stress. It serves as a crucial defense mechanism against tumorigenesis by regulating the proliferation of damage or neoplastic cells. Senescent cells exhibit a distinct secretion profile knowns as the Senescence-Associated Secretory Phenotype (SASP), which consists of a complex mixture of soluble factors, including cytokines, growth factors, chemokines, and matrix metalloproteinases. [12,13]. These cells are continually subjected to various stimuli, including oxidative stress (OS), stress-induced premature senescence (SIPS) and proteasome inhibition-induced premature senescence (PIIPS), which collectively contribute to cellular quiescence [14]. Cellular senescence is a multifaceted process involving growth arrest and altered cytokine signalling. The presence of other components within the tissue microenvironment is also essential for this effect to manifest. Recent evidence has shown that NF-κB signalling is the primary pathway responsible for driving the development of the SASP [14].
The aim of this study was to analyze the different histopathological findings, immunohistochemistry, and electron microscopy in AdaCP to investigate the secondary effects of exposure to OMF and its potential role in inducing the SASP.
2. Materials and Methods
2.1. Clinical Data
This is a descriptive, retrospective, and observational study in which we included patients admitted at the National Institute of Neurology and Neurosurgery, in Mexico City (INNN) diagnosed with craniopharyngioma following surgical resection during the period between 2014 and 2016. This study was approved by the Ethics Committee of National Institute of Neurology and Neurosurgery, in Mexico City (PINNN20-88). The study was conducted in accordance with the Declaration of Helsinki. We analyzed the following clinical variables: age, gender, time of symptom onset, relapse, time of follow-up since the last surgery, and death. The cases were divided into recurrence and non-recurrence tumors.
2.2. Histopathology
Tissue samples were formalin-fixed, alcohol dehydrated and paraffin-embedded. Four μm histology sections were H&E stained, and histopathological features were analyzed: dystrophic calcifications (DC), wet keratin (WK), word-like arrays (WLA), external epithelium (EE) in the palisading cell layer, cords, trabeculae and lobules of well-differentiated epithelium, and stellate reticulum (SR), blood vessels (V), adjacent brain tissue (ABT), Rosenthal Fibers (RFs) nest of epithelial cells (NEC), finger-like tumor protrusions (FLTP) and inflammatory areas (IA). In AdaCP and PaCP were the epithelium's superficial, middle, and basal layers. Both types were evaluated by immunohistochemical analysis.
2.3. Immunohistochemistry
Four μm histology sections were stained by Immunohistochemistry using as primary antibodies: cytokeratin 5/6 (M7237; DAKO), Cytokera AE1/AE3 (PDM072, Diagnostc BioSystems), cytokeratin 8 (CK8, Mob050, Diagnostc BioSystems), EMA (Epithelial Membrane Antigen Dako M0613), claudin 5 (ARG52881, Arigo Biolaboratories), N-cadherin (Human N Cadherin protein; ab112268; abcam), E cadherin (GTX100443; Genetex), ß-catenin (MUB01-UC, Biogenex), EGFR (PU335-UP Biogenex), PDGFR-beta (phospho Tyr751; GeneTex GTX133457), βFGF (AM359-5M Biogenex), GFAP (AM387-5M, BioGenex), Nestin (GeneTex GTX30671), Osteonectin (Biogenex AM387-5M); Osteopontin (AKm2A1, Santa Cruz Biotechnology sc-21742), GLUT1 (MU505-UC, BioGenex), and GLUT3 (GeneTex GTX15311), CD34 (QR093, QUARTETT). The immune reaction was detected by streptavidin-biotin system with the Peroxidase Mouse & Rabbit kit (Diagnostic BioSystems, Pleasanton, CA) and revealed with diaminobenzidine using a 2 Component DAB Pack kit (BioGenex Carpinteria, CA) according to manufacturer’s instructions; by last sections were hematoxylin counterstained.
Periodic Acid-Schiff (PAS) stains were also performed and contrasted with immunohistochemistry stains. We considered the protein expression in the tumor tissue and in the tumor-adjacent brain parenchyma.
2.4. Electron Microscopy
Ultrastructural observations of AdaCp were done by transmission electron microscopy (TEM). AdaCp samples were cut into 3mm3 cubes; tissues were formalin fixed, post-fixed in 2.5% glutaraldehyde-cacodylic acid buffer 0.1M pH 7.4, for 1h; later were post-fixed in 1% Osmium tetroxide- 0.1M pH 7.2 cacodylic acid buffer for 1 h. Finally, the tissues were washed in cacodylic acid buffer, alcohol-dehydrated, and embedded in EPON resin. Semi-thin sections of one µm thickness were stained with toluidine blue and observed under the optic microscope; fine sections of 60 nm thick were contrasted with uranyl acetate - lead citrate and examined at 60 KV on a JEOL Jem-1400 Plus transmission electron microscope. Ultrastructure of external epithelium, stellate reticulum, wet keratins, ghost cells, swirl-like arrays, and blood vessels was assessed.
2.5. Statistical Analysis
Data were analyzed using SPSS version 21.0.0 for Windows (SPSS Inc., Chicago, IL). Scale variables were presented as the mean ± standard deviation (mean ± SD) or percentages as appropriate. Fisher’s exact test was used to test the difference in immunohistochemical expression of each marker between ACP and PCP and recurrence and non-recurrence tumors. The P values were reported, and the significance level was 0.05.
3. Results
3.1. Clinical Characteristics
Twenty eight patients with CP were included in the study. Twenty six cases were diagnosed as AdaCP and 2 cases were PaCP. Fifty-four percent were recurrent tumors which are larger and had a shorter progression-free survival time (12 months) than non-recurrence tumors (24 months). (The clinical characteristics of the cases are presented in Table 1).
3.2. Histopathological Findings
We observed that AdaCPs, unlike PaCPs, are made up of various components and histopathological structures that are summarized in Table 2. We found that different proteins are expressed in these structures, which were detected by immunohistochemistry (Table 3).
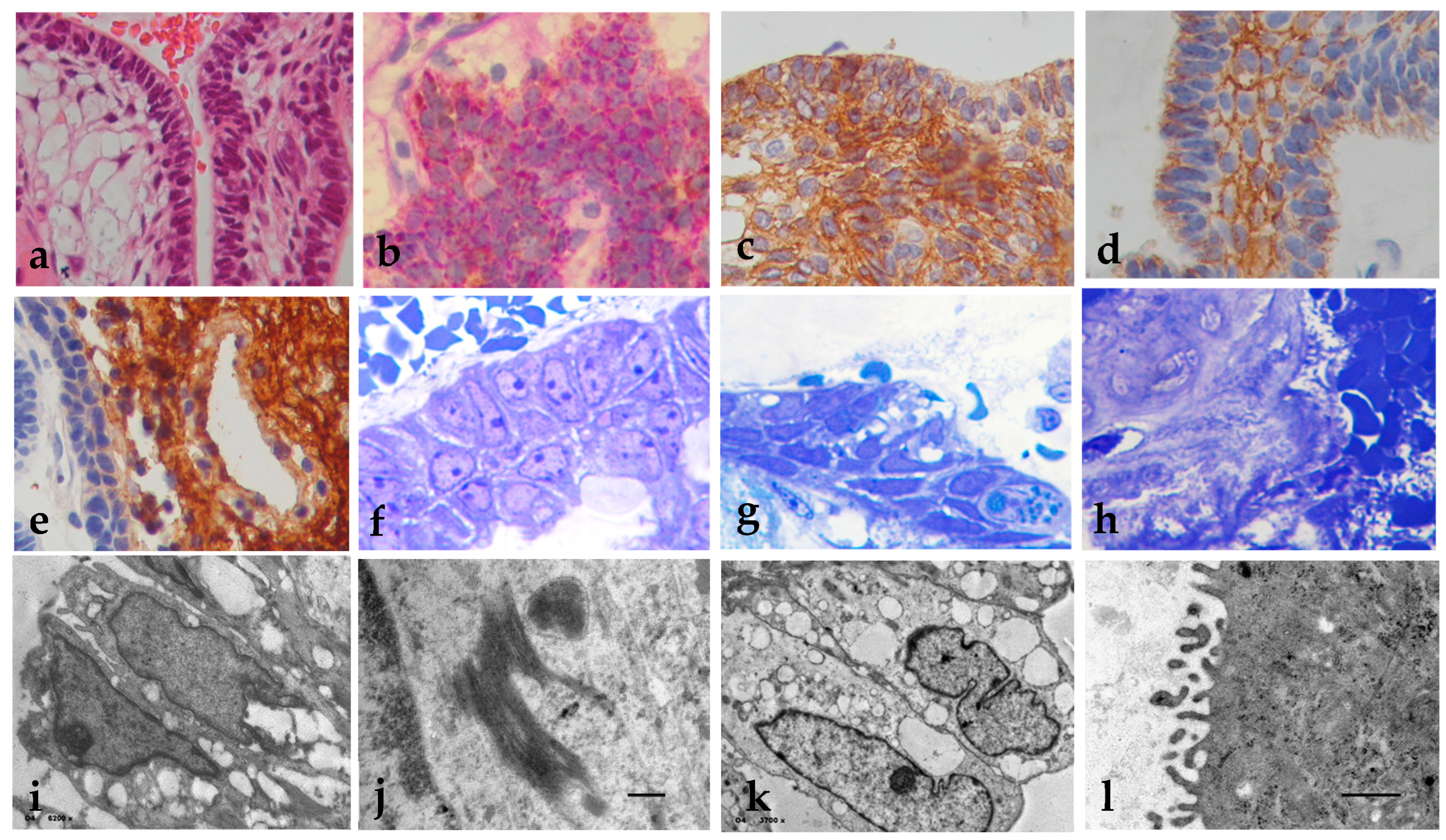
AdaCP show an external epithelium which is covered by stratified epithelium (Figure 1a) that is disposed in cords, lobules, and irregular trabeculae bordered by palisaded columnar epithelium, which forms cystic and solid structures. The external epithelium may present deposits of mucinous material in the form of fine PAS+ red drops (Figure 1b), which is usually positive for cytokeratin 5/6 (Figure 1c), claudin 5, N-cadherin and membranal E-cadherin, (Figure 1d). EE and SR are slightly positive for EGFR, was negative for PDGFR-beta, BFGF, GLUT1, GLUT3, osteonectin, osteopontin, and nestin. GFAP was strong positive immunoreaction in the external zone of the external epithelium (Figure 1e). In the semi-thin sections, we observe that the cells of the external epithelium become distant from one to another and fill up with intracytoplasmic vacuole of variable sizes, and they lose intercellular junctions (Figure1f); the external basement membrane is filled with a proteinaceous material (Figure1g) that diffusely spreads out of the epithelium (Figure1h). In electron microscopy (EM) sections, the epithelial cells fill intracytoplasmic vacuoles (Figure1i) and tonofibrils (Figure 1j), the intercellular junctions open (1k) and the cilia become short and thick with tortuous protrusions (l). In this stage, epithelial cells start folding, breaking and filling up with the OMF, making cells stick together and forming the sword-like arrays, at the same time, peripheral cells die and form wet keratin. Arranged cells show hyperchromatic nuclei with prominent nucleoli, intranuclear inclusions, loss of tonofilaments, and abnormal basal membranes.
In the stellate reticulum, we can observe ghost cells, wet keratin, and dystrophic calcifications (Figure2a). It was positive for CK5/6 (Figure2b, 2c). With CK8 cytokeratin+PAS stain, we observed that basal cells of wet keratin were positive and also produce a secretion of PAS+ material (Figure 2d). The basal cells of the wet keratins were positive for E-cadherin (Figure 2e), N-cadherin (Figure 2f), and β-Caterin (Figure 2g), osteonectin and thinly in membrane form for claudin 5 (Figure 2h), and they were harmful to GFAP, PDGFR-beta, BFGF, and nestin. The semi-thin sections show intracytoplasmic vacuoles like the external epithelium. By electron microscopy, we observe that basal cells show thick chromatin nuclei and lose basal membranes and desmosomes (2i). They also have a higher lipid concentration and intracytoplasmic vacuoles (Fig 2j); other organelles disappear (2k) and fill up with amorphous crystals and glycogen granules (Fig 2l).
The WLA findings, which are variable-sized concentric formations, were present in tumor and epithelial cell nests (Figure 3a). Beta-catenin show nuclear immunoexpression (Figure 3b), positive detection was observed with Osteopontin (Figure 3c), E-cadherin (Figure 3d), claudin 5, and GLUT-3 (Figure 3e). With PAS+ staining, intensely positive is also observed in membrane form as in small clusters (Figure 3f). The staining expressed anomalous E-Cadherin in correlation with PAS+ (Figure 3g), and nuclear E-cadherin cells also expressed mucinous PAS+ material (Figure 3h). Semi-thin sections showed the arrangement of the epithelial cell with mucinous production (Figure 3i). The nucleus showed varied degrees of chromatin changes (Figure 3j). Degraded basal membrane was observed with EM, and cytoplasmic vacuoles (Figure 3k) form concentric structures. And loss of adherent structures (Figure 3l). There is a wide variety of nuclear morphology, from no nuclei at all to cells with prominent nucleoli with glycogen/lipid inside the nucleus (3ll), which may appear as inclusion bodies (Figure 3m), chromatin becomes thicker, and nuclei show peripheral pores that seem to exert its content into the cytoplasm. Note that the cells show changes in the nucleus, showing different density of the chromatin (Figure 3n).
The blood vessels are usually small inside of the tumors, but when there is basal membrane rupture with the release of PAS+ material, these become thicker with endothelial cell atypia, showing a PAS+ membrane, and we can observe rupture of cell junction intracellular pore (Fig 4a). With PAS staining, we observed the OMF material and PAS+ particles (Fig 4b). The vessel wall was positive for nestin (4c), CD34 (4d), VEGF (4e), PDGFRB (4f), EGFR, GLUT 1(Figure 4g) and GLUT-3. With EM, we can observe nuclear alteration, with intracytoplasmic vesicles, pore rupture, and exit of the OMF into the vessel, with a wide loss of endothelial cells (Figure 4h) and OMF in adjacent brain tissue (Figure 4i). By electron microscopy, observe the endothelial cells with chromatin alterations and vacuoles (Figure 4j), the presence of OMF in the cells (Figure 4k), and the precipitation of OMF forming microcalcifications (Figure 4l).
In the fingers, like protrusions or nests in the adjacent brain tissue, we observe that they are surrounded by fibrils and Rosenthal fibers (Figure 5a); they also contain drops of PAS positive material (Figure 5b) and were positive with PAS + cytokeratin (Figure 5c), they are positive for E-cadherin (Figure 5d), PDGFR-beta was positive in the loose peripheral cells in the neuropil (Figure 5e), BFGF also shows fibroblasts and RF positive (Figure 5f), they are B-catenin positive in the stroma or very occasional expression nuclei (Figure 5g), and were negative immunoreaction to GFAP, with the adjacent brain tissue being positive (Figure 5h). In the semi-thin sections, the epithelial cells show nucleoli and globes of amorphous material that suggest OMF and open desmosomes and vacuoles (Figure 5i). We also observed that these cells show intermediate filaments in differentiation, the formation of vacuoles and nucleus with electron-dense chromatin clumps (Figure 5j).
4. Discussion
CPs are tumors considered benign. They originate from remnants of Rathke's pouch or the craniopharyngeal duct. Two subtypes are known: adamantinomatous CP, characterized by a mutation in the β-catenin gene, and papillary CP, with a mutation in the BRAF V600E gene [15,16]
CPs comprises neoplastic epithelial cells with keratinized cell nests that are often in contact with fibrous connective tissue, and stromal cells denominated reticule stellate cells [17]. They show epithelial cysts with motor-oil-like content (oil machinery fluid OMF), that are lined with elongated cells with numerous microvilli and blebs, and some cysts are lined with polyhedral cells. Basal cells reveal tonofilaments and desmosomes, forming aggregates and demonstrating smooth surface topography [15,16,19]. The epithelial cells of the cystic structures show microvilli, surface lining material and a well-developed basement membrane that is continuously observed beneath the epithelial cells [18,20].
In our cases, we observed that the cells of the outer epithelium, as well as the stellate cells of the reticulum, can produce secretory granules of PAS+ material, which protrudes from the cysts possibly through mechanisms such as the growth of adjacent structures. Overproduction of OMF was also found with disarrangement of tonofilaments, loss of basement membrane and desmosomes, exerting a structural modification on the stellate cells of the reticulum, forcing them to arrange themselves in a swirl, forming the WLA. This array induces cell death by forming wet keratin, which calcifies to form large basophilic acellular structures. Immunohistochemistry results suggest that basal cells can produce osteopontin, osteonectin, GLUT-1, GLUT-3, growth factors and nestin, meaning that these cells grow rapidly but die slowly. Zhu [21] observed that these tumor cells can die in clusters displaying altered morphology with nuclei of disrupted membranes with densely clumped chromatin.
We also observed that the epithelial cells are arranged in groups and may present short microvilli with deformed morphology. These cells have a pleomorphic nucleus with a preserved membrane with uniformly distributed chromatin; however some of them have chromatin arranged towards the nuclear membrane which may be related to an apoptotic process (Zhu). The presence of mitochondria, ribosomes and endoplasmic reticulum was distinguished in their cytoplasm, although they were not abundant. These features may be suggesting that the cells still retain their synthetic activity. These observations are in agreement with those described by Zhu [21] who describe cells with abundant mitochondria, as well as endoplasmic reticulum and ribosomes, indicating an active synthetic activity. However, in our observations we find a less differentiated cytoplasm with structural changes that may indicate that they are in the beginning of a malignant transformation. We also observed that the basal membrane gets thinner, at the same time that OMF is released, which we suggest that disrupts the basal membrane. Furthermore, we observed that at this moment, the aberrant expression of beta-catenin due to the entry of OMF to the nucleus.
It has been reported that although CPs show different histological characteristics, as observed in this work, ultrastructurally they have been shown to be similar, showing cells with abundant mitochondria and well-developed endoplasmic reticulum, demonstrating that these cells are metabolically active and secretory [22,23]. We observed that this secretion starts at the level of external endothelial cells, which lose polarity and start producing blisters, loss of desmosomes and intercellular junctions, secretion of a dense proteinaceous material which is expelled from the cells and hyalinized, gets thicker and induces a secondary inflammatory response.
Eosinophilic cells called ghost cells (GC) are characterized by the absence of nuclei and abundant eosinophilic cytoplasm, forming adjacent to cuboidal basal cells or transitional cells surrounding the whorl-like arrays adjacent to clusters [24]. Viable cells still present nuclei surrounding some GC, those cells expressing membranous solid expression of GLUT-1 and β-catenin nuclear, cytoplasmic and membranous expression, AE1/AE3, and enamel-related proteins, but they are harmful to CK8, and by electron microscopy aspects the transitional cells showing central small structures correspondent to the pyknotic nuclei an empty central area, and a network of multiple irregular fibrils filling the cytoplasm, ultrastructural studies of shown bundles of fibrils within the cytoplasm of GC, interpreted as tonofilaments, probably related to keratins and most authors consider it an unusual or aberrant form of cellular keratinization. The GC showed loss of the nucleus in the cytoplasm, usually interpreted as keratin, and absence of organelles; GC generates an empty round central space identified. Higher magnification shows GC separated by artefacts from the adjacent transitional cells. Rumayor et al. [24] described those changes as artifacts.
In our analysis we found histological and ultrastructural alterations of blood vessels. Rupture of cell junctions of a thicker basal membrane with release of PAS+ and OMF material was observed. Vessels with increased vesicles were also found, which are transported from edema. An alteration of endothelial junctions has been reported in cases of brain tumors and trauma, in which mechanisms have been proposed by which micropinocytic vesicles are transported through cell junctions. They propose that endothelial junctions open to establish a route for the transfer of edema fluid [25]. The rupture of the basal membrane and the release of its contents causing the release of inflammatory factors such as cytokines, immunomodulators such as the enzyme IDO-I [26] and expression of angiogenic factors, such as those found here by immunohistochemistry. The vessel damage is also related to PAS+ material, which promotes the release of immature fibroblast proliferation, and endothelial cell death.
We observed widely variable changes in blood vessels. VEGF-positive fenestrated endothelium with hydropic changes, pyknotic endothelial cells with nuclei loss and empty cytoplasm, with ripped-off aspect, intracytoplasmic vesicles, cell membrane loss, and content protrusion into the vessel lumen, open or ripped-off pores, some vessels show a haemosiderin deposit, as well as monocytes, which suggests microangiopathic hemolysis. It is noteworthy that high vascularization and ultrastructural vessel changes have been suggested as a factor of aggressiveness in recurrent tumors [20].
In endothelial cells, OMF precipitation was found forming micro calcifications. Several diseases have been associated with the presence of calcium crystals, with the formation of apoptotic bodies, as well as with intracellular and matrix external vesicles (EV) that may contain DNA fragments, mRNAs, lipids, miRNAs, small peptides and minerals that can be released into the extracellular space. MiRNAs that are involved in the regulation of cellular senescence and aging; they have been found packaged in EVs from where they intervene in osteogenic differentiation that is associated with age [27,28] CPs are among the most frequently calcified tumors of the CNS, with a rate of 60-96% [27,29,30]. It has been reported that whorl-like arrays cells and wet keratin are considered as important structure for calcification [15].
Calcium is deposited forming different morphological patterns such as lumps, popcorn-type or in an eggshell pattern, but until now the molecular processes that form them are not known [31]. Calcium, phosphorus, and carbon element content influence the calcification in CPs [32]. The calcified plaques are constituted of hydroxyapatite crystals and some amorphous materials. These plaques correspond to linear keratinization, which corresponds to wet keratin, which fills with eosinophilic calcic or mineralized particles and forms crystals. It is widely known that ectopic calcification is a typical response to injury and systemic mineral imbalance [33]. We observed that ectopic calcifications are associated with an intense inflammatory response and the OMF's protrusion to the tumor's external environment.
Ectopic calcification is inhibited by osteopontin, which is a phosphorylated glycoprotein. In macrophages and giant cells, osteopontin stimulates the resorption of ectopic calcification. In mice, lack of osteopontin causes susceptibility to ectopic calcification. It has been reported that the level of osteopontin expression correlates with the degree of calcification. [31,33,34] In CPs calcification take place in membrane-bound vesicles, which are considered to be derived from osteoblastic mesenchymal cells that are surrounded by matrix and collagen fibrils. The ossification process begins with the accumulation of hydroxyapatite inside the vesicles. Osteoblastic mesenchymal cells originate from multipotent mesenchymal cells that are located in the border area of the keratinized cell nest and fibrous connective tissue [31]
The CP invasion begins with the epithelial-mesenchymal transition (EMT) process. EMT was first described for embryological processes and was later recognized in cancer and metastasis. In this process, epithelial cells lose their morphological characteristics of cell adhesion capacity by detachment of desmosomes, and develop those of mesenchymal cells, acquiring invasive capacity by changes in the actin cytoskeleton, formation of pseudopodia and alteration of cell-matrix by proteolytic enzymes. These were some of the characteristics observed in our cases [35]. The epithelial-mesenchymal transition is a transcriptional process where the epithelial markers E-cadherin and β-catenin and the mesenchymal cell marker vimentin are regulated. It has been reported that vimentin, E-cadherin and β-catenin expression is associated with tumor recurrence; vimentin expression correlate with poor prognosis and vimentin expression with low/absent E-cadherin expression and high cytoplasmic and/or nuclear expression of β-catenin is marker of mesenchymal origin [36] The participation of other molecules in the EMT process has been found, such as transforming growth factor beta (TGF-B), microRNA and transcription factors. In the establishment of the EMT process, the tumor microenvironment is an important factor in the spread of cancer. This microenvironment includes elements such as the extracellular matrix, immune cells, fibroblasts and other elements. In this environment, tumor cells interact and, through autocrine and/or paracrine mechanisms, induce EMT. These mechanisms involve the secretion of growth factors, cytokines and extracellular matrix proteins. [37,38]. This secretory apparatus is called senescence-associated secretory phenotype (SASP) which can promote EMT, leading to tumor invasion, vascular proliferation and suppression of immune activity, developing metastasis and even inducing cells to develop resistance to therapies. [39]. This secretory phenotype is characteristic of senescent cells, which has been seen to be the product of external stimuli such as stress from therapies (radiotherapy or chemotherapy), exposure to reactive oxygen species, alteration in mitochondrial function or oncogenic stress and DNA damage which causes a DNA damage response (DDR) in which the NF-κB and C/EBPβ11 pathways are involved [40]. In the case of CP, it may be the product of exposure to EVs and the cystic content since it has been reported that it has high inflammatory activity with lipids, proteins and peptides elements. [41,42].
Senescence is a mechanism that cells adopt to protect tissues by stopping uncontrolled cell proliferation and preventing the dissemination of tumor cells. In some senescent cells, the SASP-associated cytokines IL-6 and IL-8 have been shown to participate in this process. It has been observed that the phenotype of senescence varies molecularly depending on the cell type ( senescence-associated phenotypes, SA), the origin of the damage that can be a function of time, the microenvironment in which the cells are located and the interaction with the immune system [40,43]. It has been reported that the p53/p21WAF1 and p16INK4a/Rb suppressor pathways are involved in SA-phenotype (Malaquin). Cellular senescence can also participate in aging and tissue repair, in addition to tumor suppression and promotion [44]. Therefore, SASP may be the target of therapeutic treatments targeting SA phenotypes [45,46] SASP exhibits increased expression of stem cell markers and regenerative capacity. It has been reported that transient exposure to SASP stimulates cellular plasticity promoting tissue regeneration; therefore it has been proposed that it may have a therapeutic application [47]. It has been proposed that pro- and anti-senescence treatments may have beneficial effects, such as those directed against SASP to eliminate senescent cells in order to reduce their harmful effects. The therapies seek to reduce damage, particularly in brain invasion, as well as tumor progression through escape or reversal of senescence [48]. Creating a pro-tumorigenic microenvironment leads to cell transformation of a non-tumor cell and tumor formation, modulating the immune response to produce an immunosuppressive environment. Conversely, anti-senescent therapies may help eliminate accumulated senescent cells and recover tissue function; therapies against these brain pathologies or in some brain tumors [49].
5. Conclusions
In this study, we describe how epithelial cells and cells from the RE secrete OMF, which through specific mechanisms, form cystic structures. The OMF plays a crucial role in tumor dynamics, promoting the extrusion of OMF along with neoplastic cells that can grow, invade, regenerate, and ultimately undergo cell death. This process results in the formation of wet keratin, dystrophic calcifications, and a marked inflammatory response. Additionally, we explain how the different histological features develop about the mechanisms associated with the SASP.
Author Contributions
Conceptualization, Martha Tena-Suck and Alma Ortiz Plata; Data curation, Martha Tena-Suck and Alma Ortiz Plata; Formal analysis, Martha Tena-Suck and Alma Ortiz Plata; Investigation, Martha Tena-Suck; Methodology, Martha Tena-Suck, Francisca Fernández-Valverde, Aurora Sánchez-García and Alma Ortiz Plata; Validation, Martha Tena-Suck; Visualization, Martha Tena-Suck and Alma Ortiz Plata; Writing – original draft, Martha Tena-Suck and Alma Ortiz Plata; Writing – review & editing, Eliezer Villanueva-Castro, Marco Munuzuri-Camacho, Erick Gomez-APO, Citaltepetl Salinas-Lara, Carlos Sánchez-Garibay and Alma Ortiz Plata.
Funding
This research received no external funding. The authors contributed to the development of the publication. Transmission electron microscope was acquired by support of National Council of Science and Technology of Mexico from the call for support for the strengthening and development of scientific and technological infrastructure 2014 (CONACyT, Grant number 226201)
Institutional Review Board Statement
This study was approved by the Ethics Committee of National Institute of Neurology and Neurosurgery, in Mexico City (PINNN20-88). The study was conducted in accordance with the Declaration of Helsinki.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available upon request from the corresponding authors.
Conflicts of Interest
The authors declared no conflicts of interest.
References
- Campanini, M.L.; Almeida, J.P.; Martins, C.S.; de Castro, M. The molecular pathogenesis of craniopharyngiomas. Arch Endocrinol Metab. 2023, 67, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Cuny, T.; Buchfelder, M.; Dufour, H.; Grossman, A.; Gatta-Cherifi, B.; Jouanneau, E.; Raverot, G.; Vasiljevic, A.; Castinetti, F. The Challenging Management of Craniopharyngiomas in Adults: Time for a Reappraisal? Cancers 2022, 14, 3831–3846. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.L.; Merchant, T.E.; Warmuth-Metz, M.; Martinez-Barbera, J.P.; Puget, S. Craniopharyngioma Nat. Rev. Dis. Primers. 2019, 5, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Rushing, E.J.; Giangiaspero, F.; Paulus, W.; Burger, P.C. Craniopharyngioma. In World Health Organization classification of tumours: pathology and genetics of tumours of the Nervous System; Louis, D.N., Ohgaki, H., Wiestler, O.D., Cavenee, W.K., Eds.; IARC Press: Lyon, France, 2007; pp. 238–240. [Google Scholar]
- Luo, S.P.; Zhang, H.W.; Yu, J.; Jiao, J.; Yang, J.H.; Leim, Y.; Lin, F. A rare case of giant cystic adamantinomatous craniopharyngioma in an adult. Radiol. Case Rep. 2020, 15, 846–849. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Bratm, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; Soffietti, R.; von Deimling, A.; Ellison, D.W. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Larkin, S.J.; Ansorge, O. Pathology and pathogenesis of Craniopharyngiomas. Pituitary 2013, 16, 9–17. [Google Scholar] [CrossRef]
- Fiorito, C.M.; Giglione, E.; Bellone, S.; Peretta, P.; Bertin, D.; Basso, M.E.; Bona, G. Craniopharyngioma in children: importance of a multidisciplinary approach and therapeutic strategies in the treatment of relapsing. Minerva Pediatr. 2013, 65, 673–676. [Google Scholar] [PubMed]
- Van Effenterre, R.; Boch, A.L. Craniopharyngioma in adults and children: a study of 122 surgical cases. J. Neurosurg. 2002, 97, 3–11. [Google Scholar] [CrossRef]
- Müller, H.L. Craniopharyngioma. Endocrine Reviews 2014, 35, 513–543. [Google Scholar] [CrossRef]
- Desiderio, C.; Martelli, C.; Rossetti, D.V.; Di Rocco, C.; D'Angelo.; L, Caldarelli, M.; Tamburrini, G.; Iavarone, F.; Castagnola, M.; Messana, I.; Cabras, T.; Faa, G. Identification of thymosins β4 and β 10 in paediatric craniopharyngioma cystic fluid. Childs. Nerv. Syst. 2013, 29, 951-60. [CrossRef]
- Barajas-Gómez, B.A.; Rosas-Carrasco, O.; Morales-Rosales, S.L.; Pedraza-Vázquez, G.; González-Puertos, V.Y; Juárez-Cedillo, T.; García-Álvarez, J.A.; López-Diazguerrero, N.E.; Damián-Matsumura, P.; Königsberg, M.; Luna-López, A. Relationship of inflammatory profile of elderly patients serum and senescence-associated secretory phenotype with human breast cancer cells proliferation: Role of IL6/IL8 ratio. Cytokine 2017, 91, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Konja, D.; Singh, S.; Zhang, B.; Wang, Y. Endothelial Senescence: From Macro- to Micro-Vasculature and Its Implications on Cardiovascular Health. Int. J. Mol. Sci. 2024, 25, 1978–1999. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal 2012, 24, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Sato, K,; Kubota, T. Fine structure of ossification in Craniopharyngiomas. Ultrastruct Pathol. 1999, 23, 395–9. [CrossRef]
- Harada, H,; Takeuchi, K,; Nagata, Y,; Inoshita, N,; Ito, E,; Okumura, E,; Kondo, T,; Sato, Y,; Saito, R. A Case of Papillary Craniopharyngioma Mimicking Rathke's Cleft Cyst. NMC Case Rep J. 2024, 27:191-194. [CrossRef]
- Xu, C,; Wu, J,; Ye, J,; Si, Y,; Zhang, J,; Wu, B,; Pan, L,; Fu, J,; Ren, Q,; Xie, S,; Tang, B,; Xiao, Y,; Hong, T. Multiomics integration-based immunological characterizations of adamantinomatous craniopharyngioma in relation to keratinization. Cell Death Dis. 2024,15:439. [CrossRef]
- Oka, H.; Kawano, N.; Yagishita, S.; Kobayashi, I.; Saegusa, H.; Fujii, K. Ciliated craniopharyngioma indicates histogenetic relationship to Rathke cleft epithelium. Clin. Neuropathol. 1997, 16, 103–106 PMID: 9101113. [Google Scholar] [PubMed]
- Vilches, J.; Lopez, A.; Martinez, M.C.; Gomez, J.; Barbera, J. Scanning and transmission electron microscopy of a craniopharyngioma: x-ray microanalytical study of the intratumoural mineralized deposits. Ultrastruct. Pathol. 1981, 2, 343–356. [Google Scholar] [CrossRef]
- Agozzino, L.; Ferraraccio, F.; Accardo, M.; Esposito, S.; Agozzino, M.; Cuccurullo, L. Morphological and ultrastructural findings of prognostic impact in craniopharyngiomas. Ultrastruct. Pathol. 2006, 30, 143–150. [Google Scholar] [CrossRef]
- Zhu, J,; You, C. Craniopharyngioma: Survivin expression and ultrastructure. Oncol Lett. 2015, 9:75-80. [CrossRef]
- Szeifert, G.T.; Julow, J.; Szabolcs, M.; Slowik, F.; Bálint, K.; Pásztor, E. Secretory component of cystic craniopharyngiomas: a mucino-histochemical and electron-microscopic study. Surg. Neurol. 1991, 36, 286–293. [Google Scholar] [CrossRef]
- Kawano, N.; Oka, H.; Suwa, T.; Ito, H.; Yada, K.; Kameya, T.; Yagishita, S. Origin of craniopharyngioma: an electron microscopic study. Noshuyo Byori 1993, 10, 117–123 PMID: 8220791. [Google Scholar] [PubMed]
- Rumayor, A.; Carlos, R.; Kirsch, H.M.; de Andrade, B.A.; Romañach, M.J.; de Almeida, O.P. Ghost cells in pilomatrixoma, craniopharyngioma, and calcifying cystic odontogenic tumor: histological, immunohistochemical, and ultrastructural study. J. Oral Pathol. Med. 2015, 44, 284–290. [Google Scholar] [CrossRef]
- Castejón, O.J. Electron microscopic observations of endothelial junctions in perifocal human brain edema. An interpretative study. J. Submicrosc. Cytol. 1985, 17, 105–114 PMID: 3973952. [Google Scholar] [PubMed]
- Donson, A.M.; Apps, J.; Griesinger, A.M.; Amani, V.; Witt, D.A.; Anderson, R.C.; Niazi, T.N.; Grant, G.; Souweidane, M.; Johnston, J.M.; Jackson, E.M.; Kleinschmidt-DeMasters, B.K.; et al. Molecular Analyses Reveal Inflammatory Mediators in the Solid Component and Cyst Fluid of Human Adamantinomatous Craniopharyngioma. J. Neuropathol. Exp. Neurol. 2017, 76, 779–788. [Google Scholar]
- Sun, Y.; Zeng, X.R.; Wenger, L.; Cheung, H.S. Basic calcium phosphate crystals stimulate the endocytotic activity of cells--inhibition by anti-calcification agents. Biochem. Biophys. Res. Commun. 2003, 312, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Terlecki-Zaniewicz, L,; Lämmermann, I,; Latreille, J,; Bobbili, MR,; Pils, V,; Schosserer, M,; Weinmüllner, R,; Dellago, H,; Skalicky, S,; Pum, D,; Almaraz, JCH,; Scheideler, M,; Morizot, F;, Hackl, M,; Gruber, F,; Grillari, J.; Small extracellular vesicles and their miRNA cargo are anti-apoptotic members of the senescence-associated secretory phenotype. Aging (Albany NY). 2018, 10:1103-1132. [CrossRef]
- Azari, F.; Vali, H.; Guerquin-Kern, J.L; Wu, T.D.; Croisym, A.; Sears, S.K.; Tabrizian, M.; McKee, M.D. Intracellular precipitation of hydroxyapatite mineral and implications for pathologic calcification. J. Struct. Biol. 2008, 162, 468–479. [Google Scholar] [CrossRef]
- Song-Tao, Q,; Xiao-Rong, Y,; Jun, P,; Yong-Jian, D,; Jin, L,; Guang-Long, H,; Yun-Tao, L,; Jian, R,; Xiang-Zhao, L,; Jia-Ming, X. Does the calcification of adamantinomatous craniopharyngioma resemble the calcium deposition of osteogenesis/odontogenesis? Histopathology. 2014, 64:336-47. [CrossRef]
- Qi, S.; Huang, G.; Pan, J.; Li, J.; Zhang, X.; Fang, L.; Liu, B.; Meng, W.; Zhang, Y.; Liu, X. Involvement of osteopontin as a core protein in craniopharyngioma calcification formation. J. Neurooncol. 2010, 98, 21–30. [Google Scholar] [CrossRef]
- Peng, J,; Qi, S,; Pan, J,; Zhang, X,; Huang, G,; Li, D. Preliminary Study on Composition and Microstructure of Calcification in Craniopharyngiomas. J Craniofac Surg. 2016, 27:e409-13. [CrossRef]
- iachelli, CM. Inducers and inhibitors of biomineralization: lessons from pathological calcification. Orthod Craniofac Res. 2005, 8:229-31. [CrossRef]
- Giachelli, CM. Ectopic calcification: new concepts in cellular regulation. Z Kardiol. 2001;90 Suppl 3:31-7. [CrossRef]
- Huang, Z,; Zhang Z,, Zhou, C,; Liu, L,; Huang, C. Epithelial-mesenchymal transition: The history, regulatory mechanism, and cancer therapeutic opportunities. MedComm. 2022 3:e144. [CrossRef]
- Qi, ST,; Zhou, J,; Pan, J,; Zhang, C,; Silky, C,; Yan, XR. Epithelial-mesenchymal transition and clinicopathological correlation in craniopharyngioma. Histopathology. 2012, 61:711-25. [CrossRef]
- Iwatsuki, M,; Mimori, K,; Yokobori, T,; Ishi, H,; Beppu, T,; Nakamori, S,; Baba, H,; Mori, M. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010, 101:293-9. [CrossRef]
- Gonzalez-Meljem, J.M.; Martinez-Barbera, J.P. Adamantinomatous craniopharyngioma as a model to understand paracrine and senescence-induced tumourigenesis. Cell Mol. Life Sci. 2021, 78, 4521–4544. [Google Scholar] [CrossRef]
- Chambers, C.R.; Ritchie, S.; Pereira, B.A.; Timpson, P. Overcoming the senescence-associated secretory phenotype (SASP): a complex mechanism of resistance in the treatment of cancer. Mol Oncol. 2021, 15, 3242–3255. [Google Scholar] [CrossRef]
- Gonzalez-Meljem, JM,; Haston, S,; Carreno, G,; Apps, JR,; Pozzi, S,; Stache, C,; Kaushal, G,; Virasami, A,; Panousopoulos, L,; Mousavy-Gharavy, SN,; Guerrero, A,; Rashid, M,; Jani, N,; Goding, CR,; Jacques, TS,; Adams, DJ,; Gil, J,; Andoniadou, CL,; Martinez-Barbera, JP. Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun. 2017, 8:1819. [CrossRef]
- Donson, AM,; Apps, J,; Griesinger, AM,; Amani, V,; Witt, DA,; Anderson, RCE,; Niazi, TN,; Grant, G,; Souweidane, M,; Johnston, JM,; Jackson, EM,; Kleinschmidt-DeMasters, BK,; Handler, MH,; Tan, AC,; Gore, L,; Virasami, A,; Gonzalez-Meljem, JM,; Jacques, TS,; Martinez-Barbera, JP,; Foreman, NK,; Hankinson, TC. Advancing Treatment for Pediatric Craniopharyngioma Consortium. Molecular Analyses Reveal Inflammatory Mediators in the Solid Component and Cyst Fluid of Human Adamantinomatous Craniopharyngioma. J Neuropathol Exp Neurol. 2017, 76:779-788.
- Desiderio, C,; Rossetti, DV,; Castagnola, M,; Massimi, L,; Tamburrini, G. Adamantinomatous craniopharyngioma: advances in proteomic research. Childs Nerv Syst. 2021, 37:789-797. [CrossRef]
- Gonzalez-Meljem, JM,; Martinez-Barbera, JP. Implications of cellular senescence in paediatric pituitary tumours. EBioMedicine. 2024, 99:104905. [CrossRef]
- Rodier, F,; Campisi, J. Four faces of cellular senescence. J Cell Biol. 2011, 192(4):547-56. [CrossRef]
- Malaquin, N,; Martinez, A,; Rodier, F. Keeping the senescence secretome under control: Molecular reins on the senescence-associated secretory phenotype. Exp Gerontol. 2016, 82:39-49. [CrossRef]
- Watanabe, S,; Kawamoto, S,; Ohtani, N,; Hara E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017, 108:563-569. [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef]
- Song, S,; Lam, EW,; Tchkonia, T,; Kirkland, JL,; Sun, Y. Senescent Cells: Emerging Targets for Human Aging and Age-Related Diseases. Trends Biochem Sci. 2020, 45:578-592. [CrossRef]
- Carreno, G.; Guiho, R.; Martinez-Barbera, J.P. Cell senescence in neuropathology: A focus on neurodegeneration and tumours. Neuropathol. Appl. Neurobiol. 2021, 47, 359–378. [Google Scholar] [CrossRef]
Figure 1.
Histopathology, immunohistochemistry and EM of the external epithelium. (a) The external epithelium is covered by a stratified epithelium (H&E, x200); (b) EE may show deposits of mucinous material in the form of fine red droplets PAS+ (PAS Stain x400); (c) Positive immunoreaction for cytokeratin 5/6; (d) E-cadherin membrane expression; (e) GFAP positive in the external zone of the external epithelium (x400); (f) Semi-thin sections of the external epithelium cells open and fill with vacuoles between the intracellular junctions (x1000); (g) External basement membrane filled with a proteinaceous material that diffusely spreads out of the epithelium (h) (Toluidin blue x400). (i) EM sections of epithelial cells fill with intracytoplasmic vacuoles (i: x6200); (j) Tonofibrils were observed in the cytoplasm (x30k); (k) Open intercellular junctions (x3700); (l) Cilia become tortuous with short protrusions in (X10k). Uranyl acetate-lead citrate stain.
Figure 1.
Histopathology, immunohistochemistry and EM of the external epithelium. (a) The external epithelium is covered by a stratified epithelium (H&E, x200); (b) EE may show deposits of mucinous material in the form of fine red droplets PAS+ (PAS Stain x400); (c) Positive immunoreaction for cytokeratin 5/6; (d) E-cadherin membrane expression; (e) GFAP positive in the external zone of the external epithelium (x400); (f) Semi-thin sections of the external epithelium cells open and fill with vacuoles between the intracellular junctions (x1000); (g) External basement membrane filled with a proteinaceous material that diffusely spreads out of the epithelium (h) (Toluidin blue x400). (i) EM sections of epithelial cells fill with intracytoplasmic vacuoles (i: x6200); (j) Tonofibrils were observed in the cytoplasm (x30k); (k) Open intercellular junctions (x3700); (l) Cilia become tortuous with short protrusions in (X10k). Uranyl acetate-lead citrate stain.
| EE | SR | WK | DC | WLA | ABT | V | RFs | |
|---|---|---|---|---|---|---|---|---|
| CK5/6 | +++ | ++ | + | + | ++ | --- | --- | --- |
| CK8 | ++ | ++ | + | -- | + | --- | --- | --- |
| AE1/3 | + | -- | --- | --- | --- | --- | --- | --- |
| EMA | ++ | ++ | + | -- | + | --- | --- | --- |
| Claudin 5 | + | + | + | - | + | --- | --- | --- |
| N-cadherin | ++ | + | + | + | + | --- | --- | --- |
| E-Cadherin | +++ | ++ | + | + | ++ | + | --- | --- |
| B-cat | ++ | + | + | + | *** | + | --- | --- |
| EGFR, | + | + | + | + | + | + | + | -- |
| PDGFR-beta | --- | --- | + | + | + | + | + | -- |
| BFGF | --- | --- | + | + | + | + | --- | --- |
| GFAP | +++ | --- | --- | --- | --- | +++ | --- | + |
| Nestin | --- | --- | --- | --- | --- | --- | + | --- |
| Osteonectin | --- | --- | + | + | + | --- | --- | --- |
| Osteopontin | --- | --- | + | + | + | --- | --- | --- |
| CD34 | + | + | + | + | + | +++ | + | --- |
| AdaCps N=26 (%) |
PaCPs N=2(%) |
Recurrence N=15(%) |
Non-recurrence N=13(%) |
|
|---|---|---|---|---|
| Cyts formation | 8(31%) | 0 | 8(53%) | 5(38%) |
| Solid stellate reticulum | 9(35%) | 2(100) | 7(47%) | 3(23%) |
| Atypia in EE | 7(27%) | 0 | 5(33%) | 2(15%) |
| Atypia in SR | 7(27%) | 0 | 5(33%) | 2(15%) |
| Mitosis figures | 7(27%) | 0 | 5(33%) | 2(15%) |
| Metaplastic changes | 7(27%) | 0 | 5(33%) | 2(15%) |
| Wet keratin | 17(65%) | 0 | 15(100%) | 2(15%) |
| Dystrophic calcification | 24(92%) | 0 | 15(100%) | 9(69%) |
| Sword like arrays | 26(100%) | 0 | 13(87%) | 5(35%) |
| Inflammation | 26(100%) | 0 | 15(100%) | 8(62%) |
| Macrophages | 20(77%) | 0 | 15(100%) | 5(38%) |
| Granuloma formation | 8(31%) | 0 | 8(53%) | 0 |
| Cleft cholesterol | 9(35%) | 0 | 7(47%) | 2(15%) |
| Reactive gliosis | 22(85%) | 1(50%) | 15(100%) | 2(15%) |
| Rosenthal Fibers | 20(77%) | 1(50%) | 15(100%) | 0 |
| Nest form of invasión | 26(100%) | 0 | 15(100%) | 5(38%) |
| DC form of invasión | 20(77%) | 0 | 13(87%) | 5(38%) |
| Necrosis | 7(27%) | 0 | 3(20%) | 0 |
| Clinical data | Recurrence Tumors 15 (54%) |
Non- recurrence Tumors 13 (46%) |
P value |
|---|---|---|---|
| Age (years) | 31 | 33 | .051 |
| Female Male |
9 6 |
3 10 |
.757 |
| Patient weight | 80 | 75 | .186 |
| Diabetes mellitus | 7 | 3 | .184 |
| Time of onset of symptoms (months) | 7.7 | 9 | .443 |
| Tumor’s size (mm) | 42 | 24 | .004 |
| Progression-free survival (months) | 5 | 12 | .000 |
Figure 2.
Histopathology, immunohistochemistry and EM of (a) wet keratin and dystrophic calcifications (H&E x200); (b) Positive Immunoexpression of cytokeratin 5/6 in wet keratins and (c) dystrophic calcifications (x400); (d) Immunoexpression of Cytokeratin 8+PAS in basal cells of wet keratins formations (x400); (e) Positive Immunoexpression of E-cadherin in the basal cells of the wet keratins, (f) N-cadherin, (g), and B-Caterin (x400); (h) Membrane immunoexpression of claudin 5 (x400); (i) Electron microscopy of basal cells showing nucleus with thick chromatin and lose basal membranes and desmosomes (x1850); (j) Lipid content and intracytoplasmic vacuoles (x400); (k) Other organelles disappear (x600); (l) Amorphous crystals and glycogen granule (x600). Uranyl acetate-lead citrate stain.
Figure 2.
Histopathology, immunohistochemistry and EM of (a) wet keratin and dystrophic calcifications (H&E x200); (b) Positive Immunoexpression of cytokeratin 5/6 in wet keratins and (c) dystrophic calcifications (x400); (d) Immunoexpression of Cytokeratin 8+PAS in basal cells of wet keratins formations (x400); (e) Positive Immunoexpression of E-cadherin in the basal cells of the wet keratins, (f) N-cadherin, (g), and B-Caterin (x400); (h) Membrane immunoexpression of claudin 5 (x400); (i) Electron microscopy of basal cells showing nucleus with thick chromatin and lose basal membranes and desmosomes (x1850); (j) Lipid content and intracytoplasmic vacuoles (x400); (k) Other organelles disappear (x600); (l) Amorphous crystals and glycogen granule (x600). Uranyl acetate-lead citrate stain.
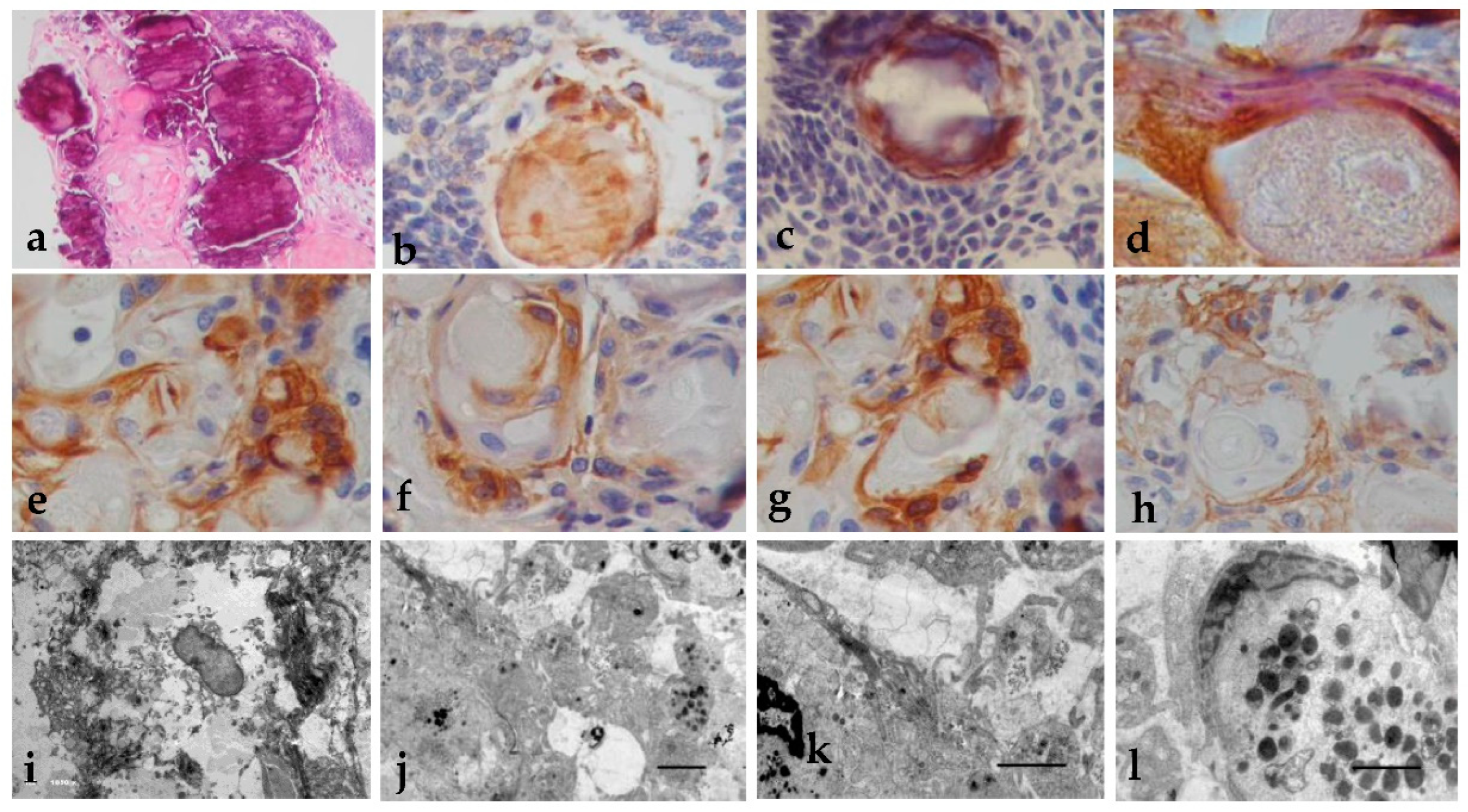
Figure 3.
Histopathology, immunohistochemistry, and EM of the WLA findings: (a) WLA epithelial cell nests (H&E x200); (b), beta-catenin with nuclear Immunoexpression (arrow, x200); (c) osteopontin positive reaction (x100); (d) E-cadherin immunoexpression (x400); (e) GLUT-3 immunoexpression (x400); (f) Positive PAS staining in membrane as in small clusters (x400); (g) Positive Immunoexpression of E-cadherin + PAS stain (x400); (h) Nuclear E-cadherin cells also expressed mucinous PAS+ (x400); (i) Semi-thin sections showing the arrangement of epithelial cell with mucinous production (x400); (j) Nucleus showed varied degrees of chromatin changes (x400); (k) Degraded basal membrane and cytoplasmic vacuoles (x8000); (l) Cells in formation of concentric structures (x2500); (ll) Glycogen/lipid inside the nucleus (arrow, x20k); (m) Glycogen/lipid may appear as inclusion bodies (arrow, x6200); (n) Nucleus with different density of the chromatin (arrow, x6200). Uranyl acetate-lead citrate stain. .
Figure 3.
Histopathology, immunohistochemistry, and EM of the WLA findings: (a) WLA epithelial cell nests (H&E x200); (b), beta-catenin with nuclear Immunoexpression (arrow, x200); (c) osteopontin positive reaction (x100); (d) E-cadherin immunoexpression (x400); (e) GLUT-3 immunoexpression (x400); (f) Positive PAS staining in membrane as in small clusters (x400); (g) Positive Immunoexpression of E-cadherin + PAS stain (x400); (h) Nuclear E-cadherin cells also expressed mucinous PAS+ (x400); (i) Semi-thin sections showing the arrangement of epithelial cell with mucinous production (x400); (j) Nucleus showed varied degrees of chromatin changes (x400); (k) Degraded basal membrane and cytoplasmic vacuoles (x8000); (l) Cells in formation of concentric structures (x2500); (ll) Glycogen/lipid inside the nucleus (arrow, x20k); (m) Glycogen/lipid may appear as inclusion bodies (arrow, x6200); (n) Nucleus with different density of the chromatin (arrow, x6200). Uranyl acetate-lead citrate stain. .
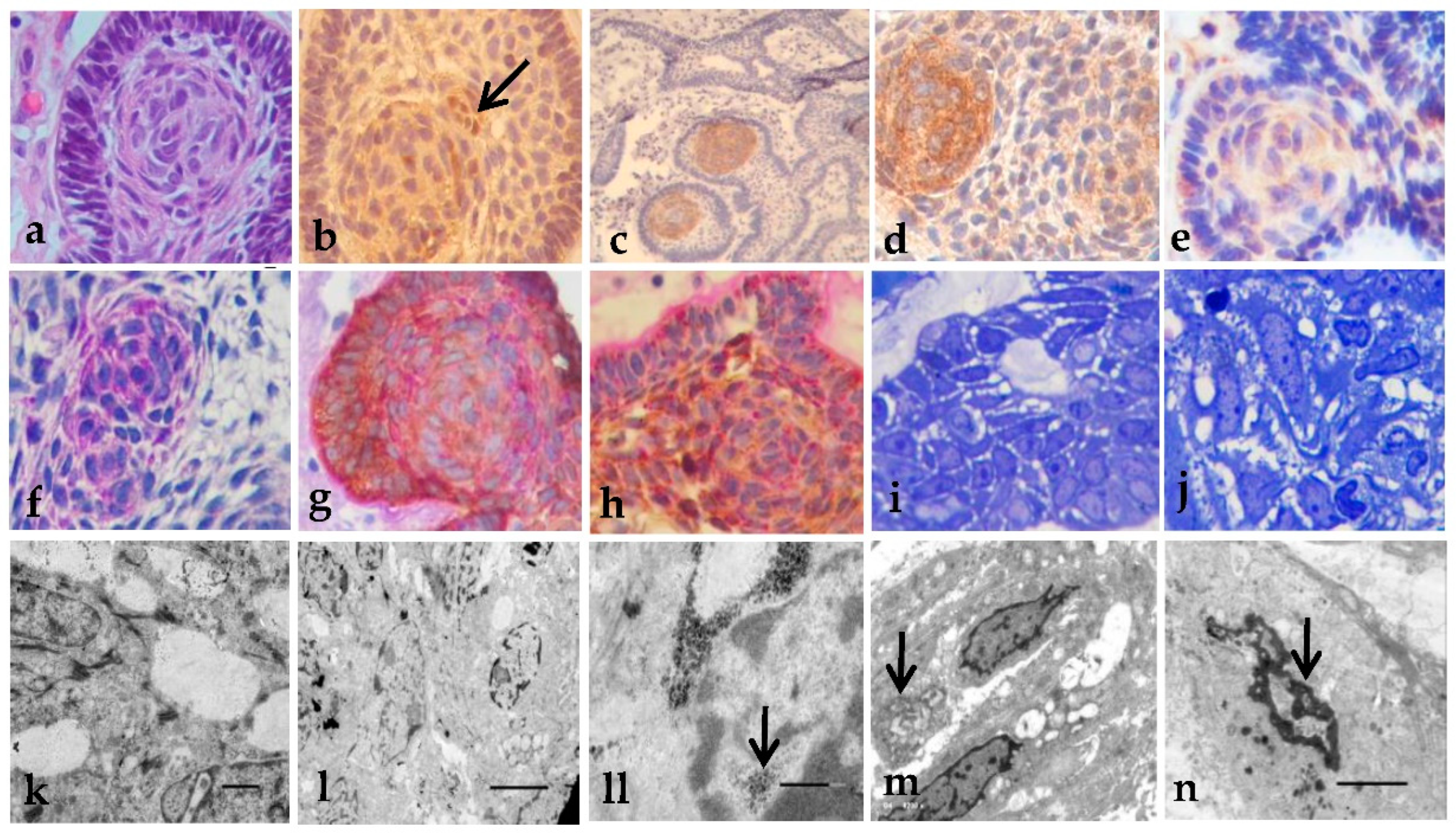
Figure 4.
Histology of the blood vessels. (a) Basal membrane rupture with release of PAS+ material (x400); (b) Cytokeratin staining show the OMF material and PAS+ particles (PAS stain x400). (c) Wall of the vessels was nestin-positive (x400); (d) CD34 positive Immunoexpression (x400); (e) positive VEGF (x400); (f) positive PDGFR Immunoexpression (x400); (g) GLUT-1 positive immunoreaction (x400); (h) Semi-thin sections show vast loss of endothelial cells, with nuclear alteration, intracytoplasmic vesicles, pore rupture, and exit of the OMF into the vessel (toluidine blue x400); (i) OMF in adjacent brain tissue (x400); (j) Electron microscopy of endothelial cells with chromatin alterations and vacuoles (x500); (k) Presence of the OMF in the cells (x500); (l) Precipitation of the OMF forming microcalcifications (x3700).
Figure 4.
Histology of the blood vessels. (a) Basal membrane rupture with release of PAS+ material (x400); (b) Cytokeratin staining show the OMF material and PAS+ particles (PAS stain x400). (c) Wall of the vessels was nestin-positive (x400); (d) CD34 positive Immunoexpression (x400); (e) positive VEGF (x400); (f) positive PDGFR Immunoexpression (x400); (g) GLUT-1 positive immunoreaction (x400); (h) Semi-thin sections show vast loss of endothelial cells, with nuclear alteration, intracytoplasmic vesicles, pore rupture, and exit of the OMF into the vessel (toluidine blue x400); (i) OMF in adjacent brain tissue (x400); (j) Electron microscopy of endothelial cells with chromatin alterations and vacuoles (x500); (k) Presence of the OMF in the cells (x500); (l) Precipitation of the OMF forming microcalcifications (x3700).
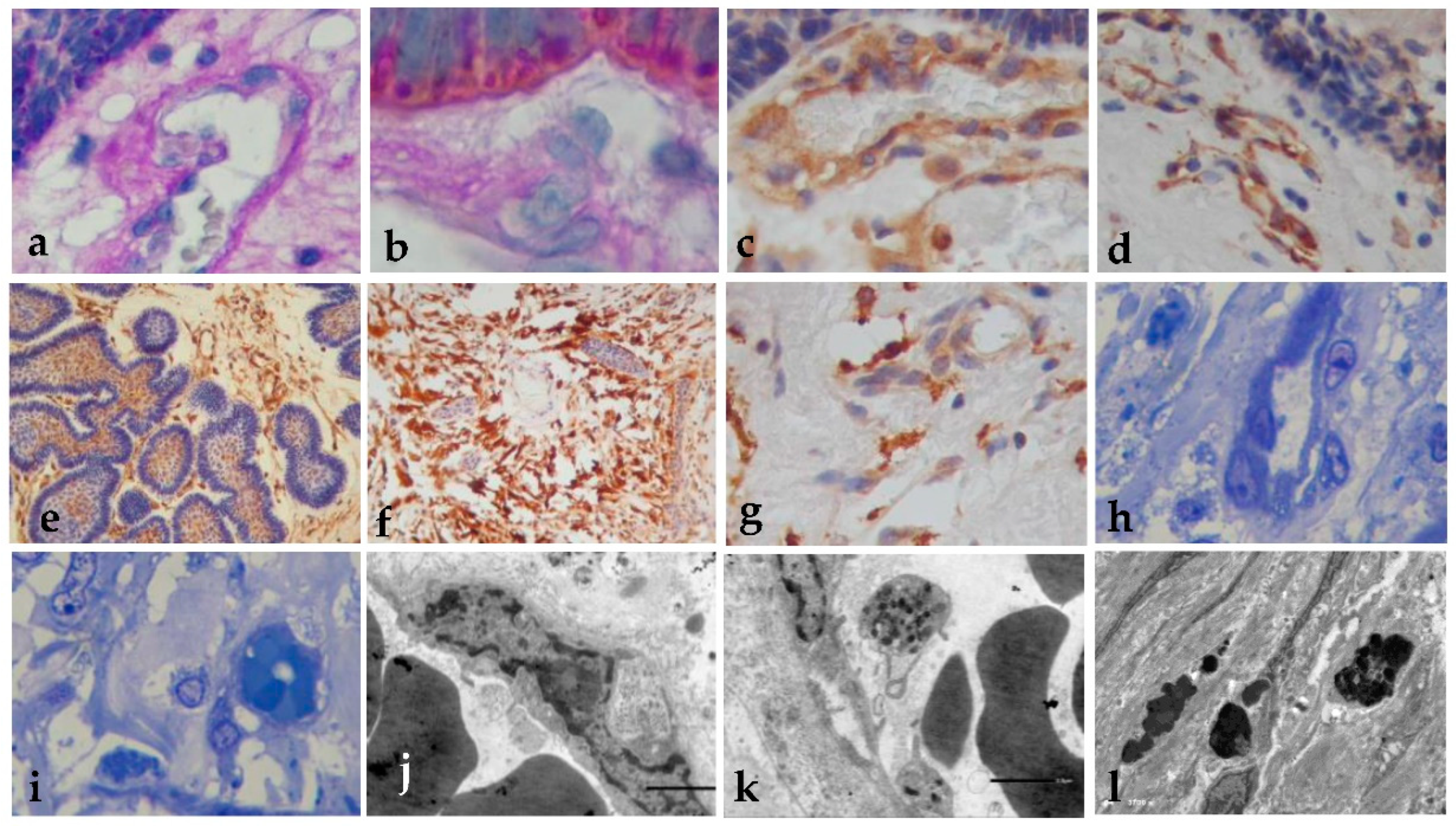
Figure 5.
Histology of the fingers, adjacent brain tissue. (a) Nest was surrounded by fibrils and Rosenthal fibers (H&E x200); (b) drops of PAS+ material (x400); (c) PAS+ with Immunoexpression of cytokeratin (x400). (d) E-cadherin Immunoexpression (x400); (e) PDGFR-beta positive cells in loose peripheral cells and the neuropil (x400); (f) BFGF positive reaction in fibroblasts and RF (x400); (g) positive beta-catenin in the stroma and occasional nuclei expression (x400); (h) GFAP positive at the adjacent brain tissue (x400); (i) Semi-thin sections, of epithelial cells with nucleoli, open desmosomes, vacuoles and globes of amorphous material that suggest OMF (toluidine blue x400); (j) EM shows cells with vacuoles and nuclei with electron-dense chromatin clumps (x4000).
Figure 5.
Histology of the fingers, adjacent brain tissue. (a) Nest was surrounded by fibrils and Rosenthal fibers (H&E x200); (b) drops of PAS+ material (x400); (c) PAS+ with Immunoexpression of cytokeratin (x400). (d) E-cadherin Immunoexpression (x400); (e) PDGFR-beta positive cells in loose peripheral cells and the neuropil (x400); (f) BFGF positive reaction in fibroblasts and RF (x400); (g) positive beta-catenin in the stroma and occasional nuclei expression (x400); (h) GFAP positive at the adjacent brain tissue (x400); (i) Semi-thin sections, of epithelial cells with nucleoli, open desmosomes, vacuoles and globes of amorphous material that suggest OMF (toluidine blue x400); (j) EM shows cells with vacuoles and nuclei with electron-dense chromatin clumps (x4000).
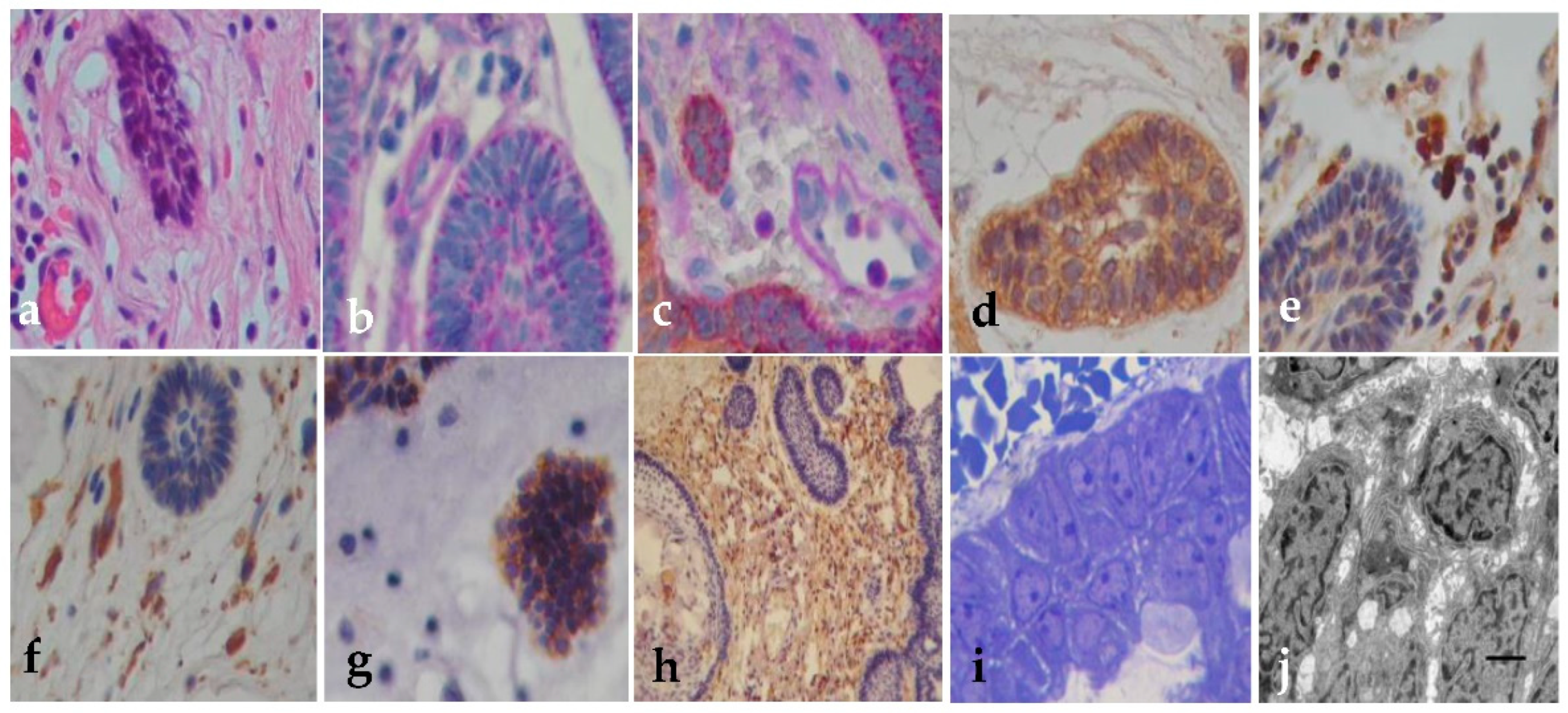

Table 1.
Clinical data of patients with craniopharyngioma are included in this study.
| Clinical data | Recurrence Tumors 15 (54%) |
Non- recurrence Tumors 13 (46%) |
P value |
|---|---|---|---|
| Age (years) | 31 | 33 | .051 |
| Female Male |
9 6 |
3 10 |
.757 |
| Patient weight | 80 | 75 | .186 |
| Diabetes mellitus | 7 | 3 | .184 |
| Time of onset of symptoms (months) | 7.7 | 9 | .443 |
| Tumor’s size (mm) | 42 | 24 | .004 |
| Progression-free survival (months) | 5 | 12 | .000 |
Table 2.
Histopathological features of craniopharyngiomas.
| AdaCps N=26 (%) |
PaCPs N=2(%) |
Recurrence N=15(%) |
Non-recurrence N=13(%) |
|
|---|---|---|---|---|
| Cyts formation | 8(31%) | 0 | 8(53%) | 5(38%) |
| Solid stellate reticulum | 9(35%) | 2(100) | 7(47%) | 3(23%) |
| Atypia in EE | 7(27%) | 0 | 5(33%) | 2(15%) |
| Atypia in SR | 7(27%) | 0 | 5(33%) | 2(15%) |
| Mitosis figures | 7(27%) | 0 | 5(33%) | 2(15%) |
| Metaplastic changes | 7(27%) | 0 | 5(33%) | 2(15%) |
| Wet keratin | 17(65%) | 0 | 15(100%) | 2(15%) |
| Dystrophic calcification | 24(92%) | 0 | 15(100%) | 9(69%) |
| Sword like arrays | 26(100%) | 0 | 13(87%) | 5(35%) |
| Inflammation | 26(100%) | 0 | 15(100%) | 8(62%) |
| Macrophages | 20(77%) | 0 | 15(100%) | 5(38%) |
| Granuloma formation | 8(31%) | 0 | 8(53%) | 0 |
| Cleft cholesterol | 9(35%) | 0 | 7(47%) | 2(15%) |
| Reactive gliosis | 22(85%) | 1(50%) | 15(100%) | 2(15%) |
| Rosenthal Fibers | 20(77%) | 1(50%) | 15(100%) | 0 |
| Nest form of invasión | 26(100%) | 0 | 15(100%) | 5(38%) |
| DC form of invasión | 20(77%) | 0 | 13(87%) | 5(38%) |
| Necrosis | 7(27%) | 0 | 3(20%) | 0 |
External epithelium (EE); Stellate Reticulum (SR); Distrophic Calcification (DC).
Table 3.
Immunohistochemical detection of the different proteins in AdaCPs.
| EE | SR | WK | DC | WLA | ABT | V | RFs | |
|---|---|---|---|---|---|---|---|---|
| CK5/6 | +++ | ++ | + | + | ++ | --- | --- | --- |
| CK8 | ++ | ++ | + | -- | + | --- | --- | --- |
| AE1/3 | + | -- | --- | --- | --- | --- | --- | --- |
| EMA | ++ | ++ | + | -- | + | --- | --- | --- |
| Claudin 5 | + | + | + | - | + | --- | --- | --- |
| N-cadherin | ++ | + | + | + | + | --- | --- | --- |
| E-Cadherin | +++ | ++ | + | + | ++ | + | --- | --- |
| B-cat | ++ | + | + | + | *** | + | --- | --- |
| EGFR, | + | + | + | + | + | + | + | -- |
| PDGFR-beta | --- | --- | + | + | + | + | + | -- |
| BFGF | --- | --- | + | + | + | + | --- | --- |
| GFAP | +++ | --- | --- | --- | --- | +++ | --- | + |
| Nestin | --- | --- | --- | --- | --- | --- | + | --- |
| Osteonectin | --- | --- | + | + | + | --- | --- | --- |
| Osteopontin | --- | --- | + | + | + | --- | --- | --- |
| CD34 | + | + | + | + | + | +++ | + | --- |
(---) negative, (+) weak), (++) moderate and (+++) strong) cytoplasmic and membranal immunoreaction. *** nuclear positive reaction. External epithelium (EE), stellate reticulum (SR), wet keratins (WK), dystrophic calcification (DC), world likes arrays (WLA), adjacent brain tissue (ABT), blood vessels (V), Rosenthal Fibers (RFs). Cytokeratin (CK), cytokeratin High molecular weight (CK5/6), epithelial membrane antigen (EMA), Beta catenin (B-cat), Epithermal growth factor receptor (EGFR), plaqueless derivate growth factor receptor beta (PDGFR-b), Beta fibroblast growth factor (BFGF).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



