
Submitted:
28 October 2024
Posted:
30 October 2024
You are already at the latest version
Abstract
Background: During normal development, epithelial cells can experience alterations at transcriptional and morphological levels, which can trigger a process named the epithelial-mesenchymal transition (EMT). The EMT underpins changes in cytoskeleton dynamics that are characteristic of neoplastic transformation. Described in many tumors, including colorectal cancer (CRC), over the past ten years, although it is an essential process in cancer development it is still poorly understood. Once initiated, the EMT regulates metastasis, the microenvironment and immune system resistance in CRC. Overexpression of the pro-inflammatory molecule inorganic polyphosphate (iPolyP) has been shown to play important role in CRC progression, alongside its binding receptor called transient receptor potential cation channel subfamily M (melastatin) member 8 (TRPM8). In this study we tested whether iPolyP/TRPM8 axis regulates EMT changes as well as NLRP3 inflammasome activation within the tumor microenvironment (TME) of CRC. Methods: To investigate these issues, western blotting, fixed and live cells immunofluorescence, 2D and 3D cell culture on CRC-patient derived biopsies, ELISA and wound healing assays were performed. Results: iPolyP triggers the expression of several EMT markers in CRC cell lines compared to control. Pharmacological inhibition of TRPM8 receptor restricts the migratory effects of cancer cells due to the presence of iPolyP. Moreover, the iPolyP/TRPM8 axis also displays an indirect pro-tumorigenic performance through shaping the immune microenvironment. By triggering NLRP3 inflammasome activation, iPolyP ensures high levels of the pro-inflammatory cytokine IL-1, which foster CRC carcinogenesis. Conclusion: This study suggests that the iPolyP/TRPM8 signaling axis is a promising target for impeding CRC progression and spread.
Keywords:
Inorganic polyphosphate
; TRPM8
; colorectal cancer
; epithelial-mesenchymal transition
; NLRP3 inflammasome
1. Introduction
Colorectal cancer (CRC) is among the major public health concerns worldwide, with an incidence of approximately 10% of all cancer cases [1]. Gradually accumulating genetic or epigenetic alterations in healthy colonic epithelial cells underpin the transition from adenomas to invasive adenocarcinomas [2]. However, while the onset and development of CRC requires genetic modifications that mostly fall into three distinct classes of aberrations, namely chromosomal instability, microsatellite instability and the CpG island methylation phenotype [3,4,5,6,7,8,9], invasive, or metastatic, these properties follow the acquisition of mesenchymal traits from epithelial cells, a process called the epithelial-mesenchymal transition (EMT) [10]. By conferring aggressiveness, the EMT triggers apical-basal polarity disruption, tight junction dissolution as well as cytoskeletal rearrangements, thus providing an infiltration and migratory ability to CRC cancer cells [10,11,12,13,14,15,16,17,18,19,20,21,22]. Extracellular stimuli from the tumor microenvironment (TME) affect the complex molecular network that initiate and support the EMT. Particularly relevant in this context are the Transforming Growth Factor beta 1/Suppressor of Mothers against Decapentaplegic (TFG-β1/SMAD), Wingless/Integrated (Wnt)/β-catenin and Neurogenic locus notch homolog protein 1 (Notch 1) pathways [23]. Not least, independent groups have shown that a key role in EMT progression and CRC invasion is mediated by the pro-inflammatory cytokines, such as interleukin-1β (IL-1β), released within the TME [24,25,26]. Notoriously, the CRC microenvironment is characterized by elevated levels of several pro-inflammatory cytokines, among which IL-1β, considered the major mediator of inflammation, stands out and fuels tumor invasion through immunosuppressive activities [27,28]. Bioactive IL-1β originates as a direct consequence of inflammasome activation in the innate immune cells [29]. Inflammasomes are supramolecular cytosolic structures found in all innate immune cells, that auto-assemble into micrometer-size complexes upon endogenous or exogenous insults, and trigger the inflammatory response. Among the several known mammalian inflammasomes, the NLRP3 inflammasome is the best studied. After sensing and binding the threat, monomers of NLRP3 organize into a wheel-shaped multimeric scaffold that signals to an adaptor protein, named apoptosis-associated speck-like protein containing a CARD (ASC) which, in turn, activates pro-Caspase-1, forming the core of the inflammasome. Active Caspase-1 cleaves pro-IL-1β, pro-IL-18 and gasdermin D (GSDMD) into their mature forms (IL-1β, IL-18 and N-terminus GSDMD, respectively). While the N-terminus GSDMD generates plasma membrane pores, allowing the release of IL-1β and IL-18 into the bloodstream, concomitantly an inflammatory form of cell death, called pyroptosis, occurs. Morphologically denominated as a “speck” or punctum, the active NLRP3 inflammasome colocalizes with the centrosome, also known as the microtubule-organizing center (MTOC), located in the perinuclear area [30]. Aberrant NLRP3 inflammasome activation has been reported to contribute, alongside other multiple risk factors, to the onset of several inflammation-driven diseases, including cancer [31]. Given the influence on CRC pathophysiology of the inflammasome-dependent and/or -independent EMT, intensive research is now underway. It has been recently reported that the pro-inflammatory molecule, named inorganic polyphosphate (iPolyP), is upregulated in neoplastic CRC tissues compared to the corresponding normal counterpart and contributes to the development and progression of CRC via its binding receptor called transient receptor potential cation channel subfamily M (melastatin) member 8 (TRPM8) [32]. Being composed by hundreds of orthophosphates linked together by ATP-like bonds, iPolyP is considered an optimal source of energy with roles in disparate pathophysiological processes, including inflammation-driven diseases [33,34,35,36,37], tumorigenesis [38], tumor metastasis [39,40] and cellular proliferation [41,42]. Both iPolyP and the receptor TRPM8 are gathering attention in CRC pathophysiology nowadays, because while the former might be described as food additive, deriving from the intestinal microbiota or released by cellular organelles within the TME and promote a pro-inflammatory niche favorable for tumor growth [43], the latter has been associated with poor prognosis in CRC subjects, where it has been found to be overexpressed [44]. Through in-vitro and ex-vivo experimental approaches, the aim of our study was firstly to investigate whether the iPolyP/TRPM8 axis is involved in the EMT program. Additionally, due to its inflammatory properties we wished to rule out the potential role of iPolyP in the activation of the NLRP3 inflammasome in the CRC context. Thus, we tested the expression of several well-characterized EMT markers, such as Vimentin (Vim) [45], phospho-Cofilin (phCFL) [46], Matrix Metalloprotease-2 (MMP2) [47], Alpha Smooth Muscle Actin (α-SMA) [48], Type 1 Collagen (COL1A1) [49], Connective Tissue Growth Factor (CTGF) [50], Fibronectin [51] and Filamentous Actin (F-actin) [52] in the presence or absence of iPolyP and TRPM8 inhibitor in a CRC cell line and CRC cell line-derived spheroids, as well as in CRC-patient derived organoids. Moreover, using the THP-1 human monocyte cell line, we investigated the possible influence of the iPolyP/TRPM8 axis on NLRP3 inflammasome activation. Together, our findings provide important insights into the involvement of the iPolyP/TRPM8 axis in CRC progression and could potentially pave the way for the development of novel anticancer agents as supplements to conventional chemotherapy.
2. Results
2.1. The iPolyP/TRPM8 Signaling Axis Promotes the Expression of the Epithelial to Mesenchymal Transition Markers in Colorectal Cancer Cells
It is believed that metastatic spread relies on genetic defects and the EMT program underpinning cellular plasticity. To determine the role of iPolyP on EMT onset we treated Caco-2 and SW620 with iPolyP for 48 hours and tested the expression level of EMT protein markers such as Vimentin, phospho-Cofilin, Metallopeptidase-2, Alpha Smooth Muscle Actin, Type 1 Collagen, Connective Tissue Growth Factor, Fibronectin and Filamentous Actin, that resulted enhanced compared to untreated (UT) cells, while no such difference was observed in the control cells HCEC-1CT (Figure 1a). In addition, immunofluorescence analysys showed an increment of Fibronectin and F-actin in iPolyP-treated cancer cells compared to HCEC-1CT (Supplementary Figure S1a,b). Pharmacological inhibition of the TRPM8 channel re-established the resting???quiescent? condition (Figure 1A and Supplementary Figure S1a,b). Similarly, patients-derived CRC organoids challenged with iPolyP underwent a significant increase in Vimentin, Metallopeptidase-2, Alpha Smooth Muscle Actin, Type 1 Collagen and Connective Tissue Growth Factor, which was dampened by blocking the TRPM8 receptor (Figure 1b). Lastly, upon iPolyP administration, HCEC-, Caco-2- and SW620-derived spheroids showed an Alpha Smooth Muscle Actin phenotype comparable to that detected in 2D culture, as mentioned above, with a complete recovery of the baseline level following treatment with the TRMP8 inhibitor (Supplementary Figure S1c,d). Overall, these results indicate the involvement of the iPolyP/TRPM8 signaling axis in the develoment of the EMT program in the CRC context.
2.2. iPolyP Stimulates Colorectal Cell Migration via TRPM8 Receptor
Cancer progression is characterized by an increased cell motility and invasiveness. To assess the migratory activity of the iPolyP/TRPM8 axis on CRC cells, we performed wound healing assays on Caco-2 cells, compared with HCEC-1CT. In brief, cells were firstly seeded in a 12-well plate until a confluent monolayer was reached. Then, using a pipette tip, an artificial gap was generated among the cells. Scratch closure was measured after 24 hours as the assay readout. The presence of iPolyP encouraged the complete sealing of both sides by Caco-2 cells, whereas no effect was observed in HCEC-1CT cells. Moreover, TRPM8 blockage prevented the migratory ability of iPolyP (Figure 2a,b). Thus, all together these results demonstrate a role of the iPolyP/TRPM8 pathway in supporting CRC cells invasiveness.
2.3. iPolyP Triggers the NLRP3 Inflammasome Activation via TRPM8 Receptor
It is well-established nowadays that patients with colorectal cancer have high levels of circulating cytokines, such as IL-1β, responsible, in turn, for fostering the invasiveness of CRC. In line with the literature, we detected high levels of IL-1β in the plasma of 10 subjects with CRC as compared to healthy individuals (Supplementary Figure S2a), suggesting, therefore, an underlying upregulation of the inflammasome complex. iPolyP is considered a pro-inflammatory molecule which contributes to shape the tumor immune microenvironment in several different types of cancers, including CRC, although the molecular mechanism has not yet been defined. Hence, we firstly explored whether iPolyP could be, at least partially, responsible for the production, maturation and/or secretion of IL-1β through the inflammasome NLRP3, often misregulated in the CRC context [53]. Similarly to Caco-2 and SW620, human monocyte THP-1 cells express the TRPM8 receptor (Supplementary Figure S3A), which makes them suitable for the study of a possible role of the iPolyP/TRPM8 axis in NLRP3 inflammasome priming or activation. Strikingly, iPolyP-primed THP-1 cells showed enhanced cytosolic pro-IL-1β, seen as protein or mRNA, whose level was analogous to those observed in LPS primed samples (Figure 3a,b), so far the best standardized first-step signal commonly used to trigger the TLR4-NLRP3 axis in monocytes and macrophages. Moreover, the presence of the TRPM8 inhibitor prevented the synthesis of pro-IL-1β (Figure 3a,b), thus assigning a role for iPolyP as a novel and unexpected player in the NLRP3 inflammasome priming pathway through the TRPM8 receptor. Consequently, iPolyP-primed THP-1 cells, followed by ATP stimulus, displayed a similar activation threshold to LPS-primed and Nigericin- or ATP-activated THP-1, used as positive controls, in terms of the canonical readouts normally explored in the inflammasome field, such as the release of mature IL-1β in the culture medium (Figure 3c), the cleavage of pro-Caspase-1 into its active form (p20 fragment) (Figure 3d), the formation of the so-called perinuclear “speck” or punctum (Figure 3e,f and Supplementary Figure S3b,c), as well as pyroptotic cell death (Figure 3g). A normal physiology of THP-1 cells, characterized by the presence of large electron-transparent autophagic vacuoles upon NLRP3 engagement, was observed under iPolyP/ATP stimulation. The presence of vacuoles was suppressed when the stimulation was combined with the TRPM8 inhibitor (Figure 3h). We next performed time-lapse video-microscopy experiments on stably expressing pro-IL-1β-mNeonGreen (mNG) (Excitation, 506 nm; Emission, 517 nm) and ASC-mScarlet-I (Excitation, 569 nm; Emission, 593 nm) THP-1, and stimulated them in the presence or absence of the TRPM8 inhibitor. Specks appearance was in line with the time-window previously reported in literature for canonical NLRP3 inflammasome activation (roughly 10-15 minutes) (Supplementary Figure S3d and Supplementary Movie 1, Supplementary Movie 2 and Supplementary Movie 3). However, no speck was detected in the presence of the TRPM8 inhibitor. All together these results delineate, for the first time, a mechanistic interpretation underpinning the onset of the iPolyP/TRPM8-mediated pro-inflammatory niche within the CRC microenvironment.
3. Discussion
Despite significant efforts to develop new therapeutic strategies, colorectal cancer (CRC) remains one of the deadliest cancers in humans. This is largely due to the complex interplay of factors such as age, environmental and genetic predisposition, which underpin each stage of tumorigenesis [53,54,55]. The EMT is a cellular process that plays a crucial role in the development and progression of CRC, thus now considered as one of the major targets for preventing neoplastic cells from acquiring invasive traits. The EMT enables the transformation of polarized epithelial cells, characterized by tight junctions and apical-basal polarity, into motile mesenchymal cells with enhanced invasive properties, thus contributing to tumor heterogeneity as well as to the onset of niches in distant organs [56]. As a consequence, cancer cells display stem-like characteristics that confer resistance to conventional therapeutic approaches. Multiple extracellular signaling pathways, synergistically or independently, converge toward the EMT. These include agents like: Transforming Growth Factor-β1 (TGF-β1), which promotes the level of several transcription factors like Snail family transcriptional repressor 1 (SNAIL), Twist Family BHLH Transcription Factor 1 (Twist1) and Zinc finger E-box-binding homeobox 1/2 (Zeb1/2); Epidermal Growth Factor (EGF); Hypoxia, which upregulates genes associated with the mesenchymal phenotype; the Wnt Signaling pathway [57], and inflammatory cytokines, such as IL-1β [58]. With its ATP-like bonds, accumulating evidence is contextualizing the inorganic polyphosphate in the cancer background as an energy supplier [59]. iPolyP has been enzymatically linked to two main sources, bacterial and human, that have different lengths. While bacterial iPolyP consists of 100 to 1,000 units, and is synthesized by polyphosphate kinase 1 (ppk1), human-derived iPolyP averages around 60 to 100 phosphate residues of unknown enzymatic origin. Niu and colleague showed in 2023 that the enzyme inorganic polyphosphatase, upregulated in CRC and known to hydrolyze long iPolyP chains into smaller molecules, is capable of activating the phosphatidylinositol 3-kinase/Protein kinase B (PI3K/AKT) pathway [60] which showed pivotal regulatory tasks in the epithelial to mesenchymal transition development [61], although the evidence remains indirect. In this study we demonstrate, through in-vitro and ex-vivo approaches, a direct involvement of the iPolyP/TRPM8 axis in the EMT process, which ultimately confers migratory properties to colorectal cancer cells, as well as a novel and unexpected involvement in the inflammatory pathway through the stimulation of the NLRP3 inflammasome. This conclusion is based on the following evidence, summarized in Figure 4A: (i) iPolyP upregulates EMT markers, such as Vimentin, phospho-Cofilin, Metallopeptidase-2, Alpha Smooth Muscle Actin, Type 1 Collagen (COL1A1), Connective Tissue Growth Factor (CTGF), Fibronectin and Filamentous Actin (F-actin) by interacting with the TRPM8 receptor in the CRC context; (ii) the iPolyP/TRPM8 axis governs CRC cell migration; (iii) iPolyP is able to shape the tumor microenvironment by priming the NLRP3 inflammasome, which is then fully activated by ATP. Canonical NLRP3 inflammasome activation, a pathway specifically found in innate immune cells, requires two consecutive steps. The priming step (signal 1) is generally mediated by the engagement of the Toll-like receptor (TLR) on the macrophage surface, which recognizes structurally conserved microbial molecules (such as lipopolysaccharide, LPS, from Gram-negative bacteria) and leads to NF-kB-mediated upregulation of intracellular levels of NLRP3, pro-IL-1β and pro-IL-18, whose initial concentrations are inadequate to initiate the assembly of NLRP3 in resting conditions [62]. Following signal 1, the activation step (signal 2), is provided by a broad range of pathogens-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), which include particulate matter, extracellular ATP and pore forming toxins. When paired, the two signals allow the oligomerization of NLRP3, Caspase-1 activation as well as the maturation of the executor proteins Gasdermin D, IL-1β and IL-18 into their bioactive, mature forms [63]. Here we identified a parallel mechanism, existing in innate immune cells, which involves the highly abundant molecule iPolyP and its matched receptor TRPM8, thus mimicking the well-known priming pathway. This novel signal 1 model, in combination with ATP, whose concentration is significantly enhanced within extracellular spaces and interstitial fractions in CRC [64], is capable, in turn, of allowing the maturation and secretion of IL-1β, inducing a similar efficiency to that seen upon LPS/ATP treatment. Hence, although our findings point out, for the first time, the involvement of the iPolyP/TRPM8 signaling axis in CRC growth and spread, more studies, including those involving in-vivo approaches, are needed. Therefore, ongoing CRC animal models are currently running in our laboratory aiming to confirm our above-described findings. In addition, we will apply these findings to other cancer types which might help to classify iPolyP/TRPM8-sensitive/insensitive neoplasms. All together, these results uncover a novel, functional axis in the CRC context, shedding light on new directions for study and paving the way for the development of new therapeutic strategies for CRC patients.
4. Materials and Methods
4.1. Patients Samples
Patients provided written informed consent to the collection of biopsy tissue specimens under Prot. No. 397/C.E. of 16/09/2020 of the Local Ethics Committee “Gabriella Serio” IRCCS Istituto Tumori “Giovanni Paolo II”, Bari, Italy. Biopsy tissue specimens were provided by the Histopathology Unit of IRCCS “S. de Bellis”. The inclusion criteria were a confirmed diagnosis by colonoscopy, biopsy, or imaging studies, for which surgery was considered beneficial. Patients with a grade of at least 2, i.e., cancer cells with more abnormal features, were also considered. The patients to undergo surgery also had health conditions sufficiently good to undergo surgery. This included a reasonable performance status and no serious comorbidities that would significantly increase the risk of developing complications post-surgery. However, the patient needed to be willing and able to consent to surgery after being informed of the risks, benefits, and potential outcomes. Samples collected in the operating room were temporarily stored in HypoThermosol FRS (for human cell and tissue preservation—BioLife Solutions Inc, Bothell, Washington, USA.; Cat. No.: 101102), sectioned, passed through liquid nitrogen, and stored dry in −80°C within 3 hours.
4.2. Cell Culture and Reagents
Human colorectal adenocarcinoma Caco-2 and SW620 cells were purchased from the American Tissue Culture Collection (ATCC, Manassas, Virginia, U.S.A.; Cat. No.: HTB-37 and CCL-227, respectively). Human Colonic Epithelial Cells 1 transduced with CDK4 and Telomerase HCEC-1CT cells were purchased from Evercyte GmbH (Vienna, Austria; Cat. No.: CkHT-039-0229). Human embryonic kidney 293T (HEK293T) were purchased from the American Tissue Culture Collection (ATCC, Manassas, Virginia, U.S.A.; Cat. No.: CRL-3216). Human monocytic THP-1 cells, isolated from peripheral blood from an acute monocytic leukemia patient, were purchased from the American Tissue Culture Collection (ATCC, Manassas, Virginia, U.S.A.; Cat. No.: TIB-202). Caco-2, SW620 and HEK293T cells were grown in Dulbecco’s Modified Eagle’s medium (DMEM), (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 11965092), supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: A5256701), 1 mM Sodium Pyruvate (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No: 11360039), 25 mM HEPES (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 15630056) and 100 U/mL Antibiotic-Antimycotic (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 15240062). Human Colonic Epithelial Cells 1 transduced with CDK4 and Telomerase (HCEC-1CT) were grown in ColoUp medium ready to use (Evercyte GmbH, Vienna, Austria; Cat. No.: MHT-039), supplemented with 100 U/mL Antibiotic-Antimycotic (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 15240062). THP-1 cells were maintained in Roswell Park Memorial Institute medium (RPMI), Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 11875093), supplemented with 10% FBS, 100 U/mL Antibiotic-Antimycotic (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 15240062) and 0.05 mM 2-mercaptoethanol (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: M6250-100 mL). All cell lines were maintained in a humidified atmosphere at 37°C with 5% CO2. Cells were passaged and medium was changed every other day.
4.3. Immunoblotting
HCEC-1CT, Caco-2 and SW-620 cell lines were seeded into 6-well plates (Corning, New York, New York, U.S.A.; Cat. No.: 3516) at a density of 0.5 x 106 cells/well in 2 mL of complete cell culture medium. Seeded cells were treated with 0.5 μM of sodium phosphate glass (iPolyP) (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: S4379-500mg), or with 10 μM of TRPM8 receptor inhibitor N-(3-Aminopropyl)-2-[(3-methylphenyl)methoxy]-N-(2-thienylmethyl)benzamide hydrochloride (AMTB), (Santa Cruz Biotechnology, Dallas, Texas, U.S.A.; Cat. No.: 926023-82-7), or in combination for 72 hours. Dimethyl sulfoxide (DMSO), (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: D8418-100mL) was added to the control cells. Pharmacological inhibition of the iPolyP/TRPM8 axis was performed by adding TRPM8 inhibitor to the HCEC-1CT, Caco-2 and SW620 cell lines. THP-1 cells were employed for NLRP3 inflammasome studies. THP-1 cells were seeded into 6-well plates at a density of 2 x 105 cells/mL in 2 mL of complete cell culture medium and treated overnight with 300 ng/mL phorbol myristate acetate (PMA), (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: P8139-5MG). The following day, the priming step was performed by adding 1 μg/mL of lipopolysaccharides (LPS), (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: L4524-5MG), considered as positive control, or 0.5 μM iPolyP, to the cells for 4 hours. Pharmacological inhibition of the priming step of NLRP3 inflammasome was performed by adding 10 μM of TRPM8 inhibitor for 4 hours. NLRP3 inflammasome activation was triggered by adding 5 mM of adenosine 5′-triphosphate (ATP) disodium salt hydrate (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: FLAAS-1VL), or 20 μM of Nigericin sodium salt (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: N7143). Pharmacological inhibition of NLRP3 inflammasome activation step was performed by adding 0.1 μM of the NLRP3 direct inhibitor MCC950 (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: 5.38120) 1 hour before the activation step. Following incubation/activation, adherent cells were detached using 0.05 % Trypsin-EDTA (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 25300-054), centrifuged at 1100 rpm for 5 minutes at 4°C and washed with 1x sterile Dulbecco’s phosphate-buffered saline 1 (1x DPBS) (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 14190-094) twice. Dried pellets were frozen at −80°C or resuspended and lysed in 200 µL of T-PERTM Tissue Protein Extraction Reagent (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 78510) supplemented with Halt™ Protease and Phosphatase Inhibitor Single-Use Cocktail, EDTA-Free (100x) (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 78443), for immunoblotting analysis. Cellular lysates were incubated on ice for 30 minutes and vortexed every 10 minutes. Samples were then centrifuged at 16,000 rpm at 4°C for 20 minutes to clarify and precipitate insoluble debris. Total extracted proteins were assayed to measure concentrations using the Bio-Rad protein assay dye reagent concentrate (Bio-Rad Laboratories, Hercules, California, U.S.A.; Cat. No.: 5000006EDU). Then, proteins were mixed with 4 x Laemmli Sample Buffer (Bio-Rad Laboratories, Hercules, California, U.S.A.; Cat. No.: 1610747) and 10% of β-mercaptoethanol (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: M6250-100mL) and denatured at 95°C for 5 minutes. 25 μg of proteins were loaded onto precast polyacrylamide 4-20% gels (Bio-Rad Laboratories, Hercules, California, U.S.A.; Cat. No.: 4568094), subsequently blotted on a polyvinylidene fluoride (PVDF) membrane (Bio-Rad Laboratories, Hercules, California, U.S.A.; Cat. No.: 1704156) using the trans-blot turbo transfer system (Bio-Rad Laboratories, Hercules, California, U.S.A.; Cat. No.: 1704150). Membranes were blocked using Pierce™ Protein-Free Blocking Buffer (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 37571) for 1 hour and stained overnight with primary antibodies. The next day membranes were washed three times with 1x Tris Buffered saline (1x TBS), (Bio-Rad Laboratories, Hercules, California, U.S.A.; Cat. No.: 1706435 diluted in ddH2O to reach 1x)/TWEEN20 (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: P9416-100mL) incubated for 1 hour with the respective horseradish peroxidase-conjugated secondary antibodies. Proteins were detected using the Clarity Max Western Enhanced Chemiluminescence (ECL) Substrate (Bio-Rad Laboratories, Hercules, California, U.S.A.; Cat. No.: 1705062) and the signals were obtained using the Chemidoc MP Imaging System (Bio-Rad Laboratories, Hercules, California, U.S.A.; Cat. No.: 1708280). The following primary antibodies were used: anti-TRPM8, 1:1000 (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: MA5-35474); anti-Vimentin, 1:1000 (Cell Signaling Technology, Danvers, Massachusetts, U.S.A.; Cat. No.: 57415); anti-phospho-Cofilin (Ser3), 1:500 (Cell Signaling Technology, Danvers, Massachusetts, U.S.A.; Cat. No.: 33115); anti-Cofilin, 1:500 (Cell Signaling Technology, Danvers, Massachusetts, U.S.A.; Cat. No.: 33185); anti-MMP2, 1:500 (Abcam, Cambridge Biomedical Campus, Cambridge, U.K.; Cat. No.: ab86607); anti-α-SMA, 1:500 (Abcam, Cambridge Biomedical Campus, Cambridge, U.K.; Cat. No.: ab5694); anti-COL1A1 1:1000 (Cell Signaling Technology, Danvers, Massachusetts, U.S.A.; Cat. No.: 911445); anti-CTGF, 1:500 (Abcam, Cambridge Biomedical Campus, Cambridge, U.K.; Cat. No.: ab6992); anti-TRPM8, 1:1000 (Thermo Fisher Scientific, Waltham, MA, USA; Cat. No.: MA5-35474); anti-pro-IL-1β, 1:1000 (Abcam, Cambridge Biomedical Campus, Cambridge, U.K.; Cat. No.: ab226918); anti-pro-Caspase-1, 1:1000 (Cell Signaling Technology, Danvers, Massachusetts, U.S.A.; Cat. No.: 3866S); anti-GAPDH, 1:1000 (Santa Cruz Biotechnology, Dallas, Texas, U.S.A.; Cat. No.: SC-47724). GAPDH was used as loading controls. The following secondary antibodies were employed: Anti-rabbit IgG, HRP-linked Antibody, 1:2000 (Cell Signaling Technology, Danvers, Massachusetts, U.S.A.; Cat. No.: 7074S); Goat anti-Mouse IgG (H+L)-HRP Conjugate (Bio-Rad Laboratories, Hercules, California, U.S.A.; Cat. No.: 1706516).
4.4. CRC Tumor Organoids
CRC specimens from surgically resected tumor tissue were washed three times with 1x sterile DPBS (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 14190-094) and digested with collagenase/hyaluronidase mixture (Stemcell Technologies, Vancouver, Canada Cat. No.: 07912), diluted in HBSS solution (with CaCl2 and MgCl2, Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 14025-050) for 5 hours under gentle rocking at 37°C. A single cell suspension was obtained and cells were embedded in matrigel matrix basement membrane (Corning, New York, New York, U.S.A.; Cat. No.: 356231), and cultured in IntestiCult-SF (ICT-SF) medium (Stemcell Technologies, Vancouver, British Columbia, Canada; Cat. No.: 100-0340) medium diluted 1:2 in Advanced DMEM/F-12 (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 12634-010), supplemented with N-2 (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 17502-048), B27 supplement (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 12587-010), 0.01% bovine serum albumin (BSA), antibiotic/antimycotic, and HEPES. Medium was renewed every two days until the organoids were fully developed (7-10 days). Mature organoids were split using TrypLE Select dissociation agent (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: A12177-01) according to [65] and the suspended cells obtained were re-cultured at a lower density in matrigel.
4.5. Immunofluorescence of Tumor Cells Derived from CRC Tumor Organoids
Cells obtained from organoids splitting were seeded at low density on microscopy slides (Nunc Lab-Tek II, Merck Life Sciences, Milan, Italy; Cat. No.: 154534) previously coated with matrigel diluted 1:10 in 1x sterile DPBS (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 14190-094), and left to attach and grow for at least 7 days in ICT-SF medium until the formation of large islets in a humidified incubator set to 37°C and 5% CO2. Cells were treated with Dimethyl sulfoxide (DMSO, indicated as untreated, UT) (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: D8418-100 mL), or 0.5 μM iPolyP, or TRPM8 inhibitor 10 μM or iPolyP + TRPM8 inhibitor for 48 hours, and fixed with 4% paraformaldehyde solution (PFA) (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: P6148-500G) in 1x sterile DPBS (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 14190-094) for 15 minutes at 4°C, permeabilized with 0.15% Triton X-100 (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: 101895731) for 7 minutes at room temperature, blocked with 2% BSA (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: A7030-100G) in 1x sterile DPBS (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 14190-094) for 1 hour at room temperature, and incubated overnight with primary antibodies (anti-Vimentin, 1:1000; anti-MMP2, 1:1000; anti-α-SMA, 1:500; anti-COL1A1, 1:1000; anti-CTGF; antibodies details are reported in the Immunoblotting section). The following day cells were washed three times with 1x sterile DPBS (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 14190-094), incubated for 1 hour at room temperature with fluorophore-conjugated secondary antibodies for the detection of the antigens of interest and subjected to microscopy analysis. The following secondary antibodies were used: Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 488, 1:500 (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: A-32731TR); Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488, 1:500 (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: A-11029). Images were collected and analyzed as described in the Cell lines immunofluorescence and confocal microscopy section.
4.6. Immunofluorescence of Cell Lines-Derived Spheroids
1 x 103 Caco-2, SW620, and HCEC-1CT cells were seeded into 3D low attachment 96-well cell culture plates (Corning, New York, New York, U.S.A.; Cat. No.: 4520) to obtain 3D cell-line spheroids with 100 µL of growth medium in each well. Cells were kept in culture in the above-described conditions. After seeding, all cell lines were treated with 0.5 μM iPolyP or with 10 μM TRPM8 inhibitor or both, while untreated cells were used as controls (untreated, UT). Spheroid cultures were observed at 24 and 96 hours and maintained in a humidified incubator set to 37°C and 5% CO2. After 96 hours, cells were treated with Dimethyl sulfoxide (DMSO, indicated as untreated, UT) (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: D8418-100 mL), or 0.5 μM iPolyP, or TRPM8 inhibitor 10 μM or iPolyP + TRPM8 inhibitor for 48 hours, and fixed with 4% paraformaldehyde solution (PFA) (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: P6148-500G) in 1x sterile DPBS (Thermo Fisher Scientific, Waltham, MA, USA; Cat. No.: 14190-094) for 15 minutes at 4°C, permeabilized with 0.15% Triton X-100 (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: 101895731) for 7 minutes at room temperature, blocked with 2% BSA (Sigma-Aldrich, St. Louis, MO, USA; Cat. No.: A7030-100G) in 1x sterile DPBS (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 14190-094) for 1 hour at room temperature, and incubated overnight with α-SMA antibody. The following day cells were washed three times with 1x sterile DPBS (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 14190-094), incubated for 1 hour at room temperature with fluorophore-conjugated secondary antibody: Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 488, 1:500 (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: A-32731TR). Nuclei were stained using PureBlu DAPI (Bio-Rad Laboratories, Hercules, California, U.S.A.; Cat. No.: 1351303). Spheroids were then subjected to microscopy analysis. Images were collected and analyzed as described in the Cell lines immunofluorescence and confocal microscopy section.
4.7. Cell Lines Immunofluorescence and Confocal Microscopy
2 x 105/mL HCEC-1CT, Caco-2, SW620 and THP-1 cells were grown in 35-mm petri dishes, no. 1.5 coverglass (MatTek, Ashland, Massachusetts, U.S.A.; Cat. No: P35G-1.5-14-C). HCEC-1CT, Caco-2, SW620 cells were treated with 0.5 μM iPolyP, or with 10 μM TRPM8 inhibitor or both for 48 hours. THP-1 cells were treated overnight with 300 ng/mL phorbol myristate acetate (PMA), (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: P8139-5MG). The following day, the priming step was performed by adding 1 μg/mL of lipopolysaccharides (LPS), (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: L4524-5MG), considered as positive control, or 0.5 μM iPolyP, to the cells for 4 hours. Pharmacological inhibition of the priming step of the NLRP3 inflammasome was performed by adding 10 μM of TRPM8 inhibitor for 4 hours. NLRP3 inflammasome activation was triggered by adding 5 mM of adenosine 5′-triphosphate (ATP) disodium salt hydrate (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: FLAAS-1VL), or 20 μM of Nigericin sodium salt (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: N7143). Pharmacological inhibition of the NLRP3 inflammasome was performed by adding 0.1 μM of the NLRP3 direct inhibitor MCC950 (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: 5.38120) 1 hour before the activation step. Fixative, permeabilization, and blocking buffers were prepared in 1x sterile DPBS (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 14190-094). Cells were fixed with 4% PFA for 30 minutes at 4°C and then washed twice using 1x sterile DPBS. Permeabilization was performed for 7 minutes at room temperature using 0.15% Triton X-100 (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: 101895731) diluted in 1x DPBS. Washing was performed to remove permeabilization buffer. Cells were then blocked for 1 hour at room temperature using blocking buffer (3% BSA), (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: A7030-100G)/1x sterile DPBS). Cells were incubated with primary (overnight) and secondary (1 hour) antibodies. Nuclei were stained using PureBlu DAPI (Bio-Rad Laboratories, Hercules, California, U.S.A.; Cat. No.: 1351303). In between, extensive washing steps were performed to remove unbound antibodies and stains. The following primary antibodies were employed: anti-Fibronectin, 1:1000 (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: MA5-11981); rhodamine phalloidin, for the F-actin detection, 1:250 (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: R415); anti-ASC (anti-ASC/TMS1 (E1E3I), 1:1000 (Cell Signaling Technology, Danvers, Massachusetts, U.S.A.; Cat. No.: 13833S). The following secondary antibodies were used: Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 488, 1:500 (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: A-32731TR); Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488, 1:500 (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: A-11029). Images were collected with a Nikon Ti2 inverted laser-scanning confocal microscope equipped with Plan Apo 20x (0.75 numerical aperture) in bright-field and analyzed with NIS-Elements software (version 5.11.01) and Fiji software (version 2.16.0). Prior to acquisition, cells were placed in the microscope CO2 chamber. All images were collected with a confocal Yokogawa spinning-disk on a Nikon Ti inverted microscope equipped with Plan Fluor 40x (0.60 numerical aperture) lens. Images were acquired with a Hamamatsu ORCA ER cooled CCD camera controlled with NIS-Elements software (version 5.11.01). For time-lapse experiments, images were collected using an exposure time of 700 milliseconds. At each time point, z-series optical sections were collected with a step size of 1 μm at the indicated time intervals. Gamma, brightness, and contrast were adjusted on displayed images (identically for comparative image sets) using NIS-Elements software (version 5.11.01). The Perfect Focus System was kept running for continuous maintenance of focus. DMEM without phenol red (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 21063-045), or RPMI without phenol red (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 11835063) were used during image acquisition.
4.8. Wound Healing Assay
HCEC-1CC, Caco-2 and SW620 cells were seeded in a 12-well plate (Corning, New York, New York, U.S.A.; Cat. No.: 3513) in 10% FBS medium and grown to reach an 80–90% confluence. Then, a scratch was made on the cell monolayer using a p200 pipette tip. The plate was washed with 1x sterile DPBS to remove the debris and markings were created on the outer bottom of the plate with a tip marker, to be used as reference points. Each cell line was incubated with medium without FBS, containing either 0.5 μM iPolyP, 10 μM TRPM8 inhibitor or both. Images were acquired at 0 and 24 hours using a confocal microscope Nikon Eclipse Ti2 in bright field microscopy equipped with Plan Fluor 10x (0.25 numerical aperture) lens and Fiji software (version 2.16.0) was used to measure the scratched area. Cell migratory ability for wound-healing was assessed by applying the following formula: [(wound area at 0 hour) – (wound area at 24 hours)] / (wound area at 0 hour) [66].
4.9. Detection of Cytokines by ELISA
The detection kit for human IL-1β (Abcam, Cambridge Biomedical Campus, Cambridge, U.K.; Cat. No.: ab214025) was used at the specified temperature and conditions according to the manufacturer’s instructions. Briefly, 50 μL plasma derived from Normal and Pathological individuals as well as supernatants derived from THP-1 cells at various conditions were placed into 96-well plates (Corning, New York, New York, U.S.A.; Cat. No. 3599), added with an additional 50 μL of antibody cocktail. The plates were then incubated for 2 hours at room temperature on a plate shaker set to 400 rpm. After extensive washing, 100 μL of TMB development solution were added to each well for 10 minutes in the dark on a plate shaker set to 400 rpm. Finally, 100 μL of stop solution were added to each well before recording the OD at 450 nm.
4.10. Propidium Iodide Fluorescence Cell Death Assay
After inflammasome priming by LPS or iPolyP for 4 hours and activation by LPS + nigericin, LPS + ATP, LPS + ATP + AMTB, iPolyP + ATP, iPolyP + ATP + AMTB, THP-1 cells were collected, alongside the untreated condition, and washed twice in 1x sterile DPBS. Pellets were then re-suspended in 2 μg/mL PI (ImmunoChemistry Technologies LLC, Davis, California, U.S.A.; Cat. No.: 638). The percentage of cells which took up propidium iodide (PI) was measured by flow cytometry (Navios), (Beckman Coulter, Brea, California, U.S.A.; Cat. No.: B83535) and the results were analyzed with FlowJo version 10 Software (FlowJo LLC, Ashland, Oregon, U.S.A.).
4.11. Cloning
Human ASC (a gift from Eicke Latz (Addgene plasmid # 41840; http://n2t.net/addgene:41840; RRID:Addgene_41840) was subcloned into pLV-mScarlet (a gift from Pantelis Tsoulfas (Addgene plasmid # 159172; http://n2t.net/addgene:159172; RRID:Addgene_159172)), plasmid between SalI-HF and EcoRV-HF (New England Biolabs Ipswich, Massachusetts, U.S.A.; Cat. No.: R0138M and R3195M respectively) restriction sites using the following primers (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.; Cat. No.: 10629186): forward primer of 5′ - ATACGTCGACGGATCTATGGGGCGC - 3′ and reverse primer of 5 ′- TTCAGATATCTAACTCGATGGTAGC - 3′.
4.12. Generation Stable Cell Lines
To generate stable cell lines, on day 0, lentivirus was produced using HEK293T cells by cotransfecting 1 mg of pLV plasmid containing the gene: 1. IL-1β: pLV-mTurquoise2-IL-1β-mNeonGreen was a gift from Hao Wu (Addgene plasmid # 166783; http://n2t.net/addgene:166783; RRID:Addgene_166783) or 2. pLV mScarlet-I-ASC with 750 ng of psPAX2 packaging plasmid (a gift from Didier Trono (Addgene plasmid # 12260; http://n2t.net/addgene:12260; RRID:Addgene_12260), and 250 ng of pMD2.G envelope plasmid (a gift from Didier Trono (Addgene plasmid # 12259; http://n2t.net/addgene:12259; RRID:Addgene_12259)). The transfected cells were incubated overnight. The following day (day 1), the medium was removed and the cells were replenished with 1 mL of fresh medium and incubated for another day. On day 2, the supernatant containing the virus was filtered using a 0.45-mm filter (Biosigma, Cona, Italy; Cat. No.: 051230) and directly used to infect THP-1 with a spinfection protocol to increase the efficacy. Spinfection was performed at 2500 x g for 90 minutes at room temperature using 8 mg/mL polybrene (EMD Millipore Corp., Burlington, Massachusetts, U.S.A.; Cat. No.: TR-1003-G). Positive clones were selected by cell sorting and colonies were expanded from single clones. Positive clones were extensively validated by immunofluorescence microscopy.
4.13. Transmission Electron Microscopy
For transmission electron microscopy (TEM), THP-1 cells, following activation with ATP or nigericin activation, were processed for plastic embedding. Cells were first incubated in fixative for 1 hour at room temperature. To prevent cellular shock and facilitate gentle fixation, a 2X fixative mixture was added to the culture medium at a 1:1 ratio in the dish containing the cells. This approach ensured optimal preservation of cellular structures for subsequent TEM analysis. Fresh fixative was prepared using 1.25% PFA, 2.5% glutaraldehyde (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: 354400), and 0.03% picric acid (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No: P6744-1GA) in a 0.1 M sodium cacodylate buffer (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: 97068), pH 7.4. After fixation, the cells were washed three times in 0.1 M sodium cacodylate buffer. Subsequently, they were incubated with 1% osmium tetroxide/1.5% potassium ferrocyanide for 1 hour at room temperature. Following this step, the cells were washed three times with water and then incubated in aqueous solution with 1% uranyl acetate (Electron Microscopy Science, Hatfield, Pennsylvania, U.S.A.; Cat. No.: 541-09-3) for 30 minutes. This was followed by another three rounds of washing with water to ensure thorough removal of excess staining agents. Dehydration steps were performed twice in different grades of alcohol (70% ethanol for 15 minutes, 90% ethanol for 15 minutes, and 100% ethanol for 15 minutes). Samples were then placed in propyleneoxide (Sigma-Aldrich, St. Louis, Missouri, U.S.A.; Cat. No.: 110205) for 1 hour and infiltration was performed with Epon mixed 1 + 1 with propyleneoxide for 3 hours at room temperature. Samples were moved to the embedding mold filled with freshly mixed Epon and allowed to polymerize for 24 to 48 hours at 60°C. Ultrathin sections (~ 60 nm) were cut on a Reichert Ultracut-S microtome, placed on copper grids, and stained with lead citrate. The grids from the above-described electron microscopy procedures were examined using a JEOL Jem-1011 transmission electron microscope (JEOL U.S.A., Inc, Peabody, Massachusetts, U.S.A.) operating at an accelerating voltage of 100 kV. Images were acquired using an Olympus Quemesa Camera (11 Mpx) (Olympus, Shinjuku-ku, Tokyo, Japan) to ensure high-quality image capture.
5. Conclusions
The finding regarding the involvement of the iPolyP/TRPM8 axis in assisting the EMT program, alongside the onset of a pro-inflammatory niche, might pave the way for the development of novel therapeutic strategies against CRC that curtail TRPM8 activity, as a complement to the conventional chemotherapy protocols nowadays approved, potentially improving their efficacy. Further investigation involving TRPM8-deficient Apc Min/+ mice model of CRC is currently underway in our laboratory to corroborate these results. Moreover, we will perform experiments to test the presence of iPolyP in body fluids or stool samples. Early diagnosis of CRC usually employs invasive methods with several systematic limitations and requires specialized skills. Steps forward have been achieved with liquid biopsy screening, which does not involve invasive procedures and allows follow-up of the disease in response to therapy. Thus, by promoting proliferation, spread and a pro-inflammatory environment in the CRC context, iPolyP can potentially be considered as a novel non-invasive biomarker used in liquid biopsy for the early detection and monitoring of the disease, which might significantly mitigate the current dependence on invasive biopsy methods, offer long-term benefits and improve healthcare, and these patients' lives, thus having a significant social impact.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Figure S1: iPolyP induces ɑ-SMA expression and fosters Caco-2- and SW620-derived spheroids growth. A. Immunofluorescence images performed on HCEC-1CT- (upper panel), Caco-2- (middle panel) and SW620-derived (lower panel) spheroids for ɑ-SMA detection upon treatment with iPolyP, TRPM8 inhibitor or both for 48 hours. Scale bar 100 µm. B. Quantification relative to panel A. Fold changes versus control, untreated (UT). Statistical analysis performed by Student’s t-test (**** p < 0.0001). C. Immunofluorescence images showing F-actin and Fibronectin level performed on HCEC-1CT (upper panel), Caco-2 (middle panel) and SW620 (lower panel) cell line upon treatment with iPolyP, TRPM8 inhibitor or both. D. Quantification relative to panel C. Fold changes versus control; untreated (UT). Statistical analysis performed by Student’s t-test (**** p < 0.0001, ** p < 0.01). Cells were treated with iPolyP and TRPM8 inhibitor for 48 hours. Supplementary Figure S2: A. CRC patients display elevated level of circulating IL-1β. Evaluation of plasma-derived IL-1β performed by ELISA on normal (n = 10) and CRC subjects (n = 10). Fold changes of Pathological subjects versus healthy individuals. Statistical analysis performed by Student’s t-test (*** p < 0.001). Supplementary Figure S3: iPolyP + ATP trigger the NLRP3 inflammasome, mimicking the canonical activation mediated by LPS and ATP. A. Cellular extracts from THP-1 monocyte, treated with LPS, iPolyP or iPolyP + TRPM8 inhibitor for 4 hours were analyzed by immunoblotting against pro-IL-1β and TRPM8, normalized on GAPDH. B. Representative confocal micrographs on THP-1 cells treated with LPS (4 hours) + ATP (30 minutes) or LPS (4 hours) + Nigericin (30 minutes), used as positive control, untreated (UT). White angle brackets indicate the presence of ASC specks. Scale bar 10 µm. Images are representative of three independent experiments. C. Quantification of the number of ASC specks, relative to panel B. D. Upper panel, confocal micrographs of time-lapse confocal microscopy performed on THP-1 cells, stably expressing IL-1β-mNG, upon treatment with iPolyP (4 hours) + ATP (30 minutes). Yellow angle brackets indicate ASC specks; Middle panel, time-lapse experiment in bright-field merged with fluorescence channel on THP-1 cell, stably expressing ASC-mRuby3, upon treatment with iPolyP (4 hours) + ATP (30 minutes). Black angle brackets indicate ASC-mScarlet-I specks; Lower panel, time-lapse confocal microscopy performed on THP-1 cells, stably expressing IL-1β-mNG, upon treatment with iPolyP (4 hours) + ATP (30 minutes) + TRPM8 inhibitor (4 hours). Scale bar 10 µm. Data are presented as mean ± SD for triplicate wells from three independent experiments. Supplementary Videos: Time-lapse confocal microscopy on THP-1 cells treated with iPolyP. Supplementary Video 1: THP-1, IL-1β-mNG, iPolyP (4 hours) + ATP (30 minutes); Scale bar = 10 µm. Supplementary Video 2: THP-1 ASC-mScarlet-I, iPolyP (4 hours) + ATP (30 minutes); Scale bar = 5 µm. Supplementary Video 3: THP-1, IL-1β-mNG, iPolyP (4 hours) + ATP (30 minutes) + TRPM8 inhibitor (4 hours); Scale bar 10 µm.
Author Contributions
V.A. and R.N. conceived the project, designed the research, and supervised the experimental work; V.A., F.D., A.A., M.D.L., F.B., L.V., F.A., G.P. and R.N. performed the experiments; V.A., G.G. and R.N. analyzed the data and interpreted the results; V.A., G.G. and R.N. wrote and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Italian Ministry of Health, Ricerca Corrente 2024 (DDG 851/23).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Prot. No. 397/C.E. of 16/09/2020. Local Ethics Committee “Gabriella Serio” IRCCS Istituto Tumori “Giovanni Paolo II”, Bari, Italy.
Data Availability Statement
The original raw data presented in the study are openly available at Supplementary Materials.
Acknowledgments
We thank the Electron Microscopy Core Facility, Harvard Medical School for help with transmission electron microscopy. Figures 4A in this manuscript was created with BioRender (https://app.biorender.com/).
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Siegel R.L., Wagle N.S., Cercek A., Smith R.A., Jemal A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [CrossRef]
- Jung G., Hernández-Illán E., Moreira L., Balaguer F., Goel A. Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 111–130. [CrossRef]
- Joung J.-G., Oh B.Y., Hong H.K., Al-Khalidi H., Al-Alem F., Lee H.-O., Bae J.S., Kim J., Cha H.-U., Alotaibi M., et al. Tumor Heterogeneity Predicts Metastatic Potential in Colorectal Cancer. Clin. Cancer Res. 2017, 23, 7209–7216.
- Ciardiello F., Ciardiello D., Martini G., Napolitano S., Tabernero J., Cervantes A. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA Cancer. J. Clin. 2022, 72, 372–401. [CrossRef]
- Moosavi S.H., Eide P.W., Eilertsen I.A., Brunsell T.H., Berg K.C.G., Røsok B.I., Brudvik K.W., Bjørnbeth B.A., Guren M.G., Nesbakken A., et al. De novo transcriptomic subtyping of colorectal cancer liver metastases in the context of tumor heterogeneity. Genome Med. 2021, 13. [CrossRef]
- Nguyen L.H., Goel A., Chung D.C. Pathways of Colorectal Carcinogenesis. Gastroenterology. 2020, 158, 291–302. [CrossRef]
- Wang W., Kandimalla R., Huang H., Zhu L., Li Y., Gao F., Goel A., Wang X. Molecular subtyping of colorectal cancer: Recent progress, new challenges and emerging opportunities. Semin. Cancer Biol. 2019, 55, 37–52.
- Schmitt M., Greten F.R. The inflammatory pathogenesis of colorectal cancer. Nat. Rev. Immunol. 2021, 21, 653–667. [CrossRef]
- Andrei P., Battuello P., Grasso G., Rovera E., Tesio N., Bardelli A. Integrated approaches for precision oncology in colorectal cancer: The more you know, the better. Semin. Cancer Biol. 2022, 84, 199–213. [CrossRef]
- Vu T., Datta P.K. Regulation of EMT in Colorectal Cancer: A Culprit in Metastasis. Cancers (Basel). 2017, 9.
- Zhang N., Ng A.S., Cai S., Li Q., Yang L., Kerr D. Novel therapeutic strategies: targeting epithelial-mesenchymal transition in colorectal cancer. Lancet Oncol. 2021, 22, 358–368.
- Singh M., Yelle N., Venugopal C., Singh S.K. EMT: Mechanisms and therapeutic implications. Pharmacol. Ther. 2018, 182, 80–94. [CrossRef]
- Stemmler M.P., Eccles R.L., Brabletz S., Brabletz T. Non-redundant functions of EMT transcription factors. Nat. Cell. Biol. 2019, 21, 102–112. [CrossRef]
- Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Dongre A., Weinberg R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [CrossRef]
- Shin A.E., Giancotti F.G., Rustgi A.K. Metastatic colorectal cancer: mechanisms and emerging therapeutics. Trends Pharmacol. Sci. 2023, 44, 222–236. [CrossRef]
- Aiello N.M., Kang Y. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 2019, 216, 1016–1026.
- Ye X., Weinberg R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686.
- Lambert A.W., Weinberg R.A. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat. Rev. Cancer. 2021, 21, 325–338. [CrossRef]
- Morin C., Moyret-Lalle C., Mertani H.C., Diaz J.-J., Marcel V. Heterogeneity and dynamic of EMT through the plasticity of ribosome and mRNA translation. Biochim. Biophys. Acta Rev. Cancer. 2022, 1877. [CrossRef]
- Shibue T., Weinberg R.A. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [CrossRef]
- Derynck R., Weinberg R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell. 2019,49, 313–316. [CrossRef]
- Khanbabaei H., Ebrahimi S., García-Rodríguez J.L., Ghasemi Z., Pourghadamyari H., Mohammadi M., Kristensen L.S. Non-coding RNAs and epithelial mesenchymal transition in cancer: molecular mechanisms and clinical implications. J. Exp. Clin. Cancer Res. 2022, 41. [CrossRef]
- Chen Y., Yang Z., Deng B., Wu D., Quan Y., Min Z. Interleukin 1β/1RA axis in colorectal cancer regulates tumor invasion, proliferation and apoptosis via autophagy. Oncol. Rep. 2020, 43, 908–918.
- Marandi Y., Hashemzade S., Tayebinia H., Karimi, J. Zamani A., I. Khodadadi. NLRP3-inflammasome activation is associated with epithelial-mesenchymal transition and progression of colorectal cancer. Iran J. Basic Med. Sci. 2021, 24, 483–492. [CrossRef]
- Li Y., Wang L., Pappan L., Galliher-Beckley A., Shi J. IL-1β promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer. 2012, 11.
- Li W., Chen F., Gao H., Xu Z., Zhou Y., Wang S., Lv Z., Zhang Y., Xu Z., Huo J., et al. Cytokine concentration in peripheral blood of patients with colorectal cancer. Front. Immunol. 2023, 14.
- Xie J., Zhang Y., Jiang L. Role of Interleukin-1 in the pathogenesis of colorectal cancer: A brief look at anakinra therapy. Int. Immunopharmacol. 2022, 105. [CrossRef]
- Martinon F., Burns K., Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002, 10, 417–426.
- Magupalli V.G., Negro R., Tian Y., Hauenstein A.V., Di Caprio G., Skillern W., Deng Q., Orning P., Alam H.B., Maliga Z. et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science. 2020, 369. [CrossRef]
- Arrè V., Scialpi R., Centonze M., Giannelli, G., Scavo M.P., Negro R. The 'speck'-tacular oversight of the NLRP3-pyroptosis pathway on gastrointestinal inflammatory diseases and tumorigenesis. J. Biomed. Sci. 2023, 30. [CrossRef]
- Arrè V., Balestra F., Scialpi R., Dituri F., Donghia R., Coletta S., Bianco A., Vincenti L., Fedele S., et al. Inorganic Polyphosphate Promotes Colorectal Cancer Growth via TRPM8 Receptor Signaling Pathway. Cancers. 2024, 16. [CrossRef]
- Bae J.-S., Lee W., Rezaie A.R. Polyphosphate elicits pro-inflammatory responses that are counteracted by activated protein C in both cellular and animal models. J. Thromb. Haemost. 2012, 10, 1145–1151.
- Müller F., Mutch N.J., Schenk W.A., Smith S.A., Esterl L., Spronk H.M., Schmidbauer S., Gahl W.A., Morrissey J.H., Renné T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009, 139, 1143–1456. [CrossRef]
- Hassanian S.M., Dinarvand P., Smith S.A., Rezaie A.R. Inorganic polyphosphate elicits pro-inflammatory responses through activation of the mammalian target of rapamycin complexes 1 and 2 in vascular endothelial cells. J. Thromb. Haemost. 2015, 13, 860–871. [CrossRef]
- Semeraro F., Ammollo C.T., Morrissey J.H., Dale G.L., Friese P., Esmon N.L., Esmon C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011, 118, 1952–1961. [CrossRef]
- Wat J.M., Foley J.H., Krisinger M.J., Ocariza L.M., Lei V., Wasney G.A., Lameignere E., Strynadka N.C., Smith S.A., Morrissey J.H. et al. Polyphosphate suppresses complement via the terminal pathway. Blood. 2014, 123, 768–776. [CrossRef]
- Kulakovskaya E.V., Zemskova M.Y., Kulakovskaya T.V. Inorganic Polyphosphate and Cancer. Biochemistry (Mosc). 2018; 83, 961–968. [CrossRef]
- Galasso A., Zollo M. The Nm23-H1-h-Prune complex in cellular physiology: a 'tip of the iceberg' protein network perspective. Mol. Cell. Biochem. 2009, 329, 149–159.
- Tammenkoski M., Koivula K., Cusanelli E., Zollo M., Steegborn C., Baykov A.A., Lahti R. Human metastasis regulator protein H-prune is a short-chain exopolyphosphatase. Biochemistry. 2008, 47, 9707–9713. [CrossRef]
- Wang L., Fraley C.D., Faridi J., Kornberg A., Roth R.A. Inorganic polyphosphate stimulates mammalian TOR, a kinase involved in the proliferation of mammary cancer cells. Proc. Natl. Acad. Sci. U S A. 2003, 100, 11249–12254. [CrossRef]
- Ito T., Yamamoto S., Yamaguchi K., Sato M., Kaneko Y., Goto S., Goto Y., Narita I. Inorganic polyphosphate potentiates lipopolysaccharide-induced macrophage inflammatory response. J. Biol. Chem. 2020, 295, 4014–4023.
- Olanbiwonnu T., Holden R.M. Inorganic phosphate as a potential risk factor for chronic disease. C.M.A.J. 2018, 190, E784–E785. [CrossRef]
- Pagano E., Romano B., Cicia D., Iannotti F.A., Venneri T., Lucariello G., Nanì M.F., Cattaneo F., De Cicco P., D'Armiento M., et al. TRPM8 indicates poor prognosis in colorectal cancer patients and its pharmacological targeting reduces tumour growth in mice by inhibiting Wnt/β-catenin signalling. Br. J. Pharmacol. 2023, 180, 235–251. [CrossRef]
- Usman S., Waseem N.H., Nguyen T.K.O., Mohsin S., Jamal A., The M.-T. Waseem A. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers. 2021, 13. [CrossRef]
- Sousa-Squiavinato A.C.M., Rocha M.R., Barcellos-de-Souza P., Barcellos-de-Souza W., Morgado-Diaz J.A. Cofilin-1 signaling mediates epithelial-mesenchymal transition by promoting actin cytoskeleton reorganization and cell-cell adhesion regulation in colorectal cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 418–429. [CrossRef]
- Pezeshkian Z., Nobili S., Peyravian N., Shojaee B., Nazari H., Soleimani H., Asadzadeh-Aghdaei H., Bonab M.A., Nazemalhosseini-Mojarad E., Mini E. Insights into the Role of Matrix Metalloproteinases in Precancerous Conditions and in Colorectal Cancer. Cancers (Basel). 2021, 13. [CrossRef]
- Nomura S. Identification, Friend or Foe: Vimentin and α-Smooth Muscle Actin in Cancer-Associated Fibroblasts. Ann. Surg. Oncol. 2019, 26, 4191–4192.
- Hosper N.A., van den Berg P.P., de Rond S., Popa E.R., Wilmer M.J., Masereeuw R., Bank R.A. Epithelial-to-mesenchymal transition in fibrosis: collagen type I expression is highly upregulated after EMT, but does not contribute to collagen deposition. Exp. Cell Res. 2013, 319, 3000-3009. [CrossRef]
- Lun W., Wu X., Deng Q., Zhi F. MiR-218 regulates epithelial-mesenchymal transition and angiogenesis in colorectal cancer via targeting CTGF. Cancer Cell Int. 2018, 18.
- Niknami Z., Eslamifar A. Emamirazavi A., Ebrahimi A., Shirkoohi R. The association of vimentin and fibronectin gene expression with epithelial-mesenchymal transition and tumor malignancy in colorectal carcinoma. EXCLI J. 2017, 16, 1009–1017. [CrossRef]
- Datta A., Deng S., Gopal V., Yap K.C.H., Halim C.E., Lye M.L., Ong M.S., Tan T.Z., Sethi G., Hooi S.C., et al. Cytoskeletal Dynamics in Epithelial-Mesenchymal Transitions: Insight into Therapeutic Targets for Cancer Metastasis. Cancers (Basel). 2021, 13.
- Hossain, M.S.; Karuniawati, H.; Jairoun, A.A.; Urbi, Z., Ooi, D.J.; John, A.; Lim, Y.C.; Kibria, K.M.K.; Mohiuddin, A.K.M.; Ming, L.C.; et al. Colorectal Cancer: A Review of Carcinogenesis, Global Epidemiology, Current Challenges, Risk Factors, Preventive and Treatment Strategies. Cancers (Basel). 2022, 14,1732. [CrossRef]
- Hibberd, A.A.; Lyra, A.; Ouwehand, A.C.; Rolny, P.; Lindegren, H.; Cedgård, L.; Wettergren, Y. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol. 2017, 4, e000145. [CrossRef]
- Zackular, J.P.; Baxter, N.T.; Iverson, K.D.; Sadler, W.D.; Petrosino, J.F.; Chen, G.Y.; Schloss, P.D. The gut microbiome modulates colon tumorigenesis. mBio. 2013, 4, e00692-13. [CrossRef]
- Lu J., Kornmann M., Traub B. Role of Epithelial to Mesenchymal Transition in Colorectal Cancer. Int. J. Mol. Sci. 2023, 24. [CrossRef]
- Ribatti D., Tamma R., Annese T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13. [CrossRef]
- Li R., Ong S.O., Tran L.M., Jing Z., Liu B., Park S.J., Huang Z.L., Walser T.C., Heinrich E.L. Lee G. et al. Chronic IL-1β-induced inflammation regulates epithelial-to-mesenchymal transition memory phenotype via epigenetic modifications in non-small cell lung cancer. Sci. Rep. 2020,10.
- Kulakovskaya E.V., Zemskova M.Y., Kulakovskaya T.V. Inorganic Polyphosphate and Cancer. Biochemistry (Mosc). 2018, 83, 961-968. [CrossRef]
- Niu T., Zhu, J., Dong L., Yuan P., Zhang L., Liu D. Inorganic pyrophosphatase 1 activates the phosphatidylinositol 3-kinase/Akt signaling to promote tumorigenicity and stemness properties in colorectal cancer. Cell. Signal. 2023, 108.
- Moghbeli M. PI3K/AKT pathway as a pivotal regulator of epithelial-mesenchymal transition in lung tumor cells. Cancer Cell Int. 2024, 24. [CrossRef]
- Bauernfeind F.G., Horvath G., Stutz A., Alnemri E.S., MacDonald K., Speert D., et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009,183,787–791.
- Kelley N., Jeltema D., Duan Y., He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019,20.
- Shatat A.-A.S., Mahgoup E.M., Rashed M.H., Saleh I.G., Akool E.-S. Molecular mechanisms of extracellular-ATP-mediated colorectal cancer progression: Implication of purinergic receptors-mediated nucleocytoplasmic shuttling of HuR. Purinergic Signal. 2024, 24. [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science, 2018, 359, 920–926. [CrossRef]
- Centonze M., Di Conza G., Lahn M., Fabregat M., Dituri F., Gigante I., Serino G., Scialpi R., Carrieri L., Negro R. Autotaxin inhibitor IOA-289 reduces gastrointestinal cancer progression in preclinical models. J. Exp. Clin. Cancer Res. 2023, 42. [CrossRef]
Figure 1.
iPolyP induces the expression of stress fibers and proteins involved in the epithelial-to-mesenchymal transition. A. Cellular extracts from HCEC-1CT, Caco-2 and SW620, treated with iPolyP, or iPolyP + TRPM8 inhibitor for 48 hours, were analyzed by immunoblotting for Vimentin, phCofilin, Cofilin, MMP2, ɑ-SMA, COL1A1 and CTGF expression levels (from top to bottom). GAPDH was used as loading control. B. Immunofluorescence on CRC patients-derived organoids, treated with iPolyP, AMTB or iPolyP + TRPM8 inhibitor to detect the expression of the above-mentioned proteins. Scale bar 100 µm. C. Quantification relative to panel B. Statistical analysis performed by Student’s t-test (*** p < 0.001). Fold changes versus control, untreated (UT).
Figure 1.
iPolyP induces the expression of stress fibers and proteins involved in the epithelial-to-mesenchymal transition. A. Cellular extracts from HCEC-1CT, Caco-2 and SW620, treated with iPolyP, or iPolyP + TRPM8 inhibitor for 48 hours, were analyzed by immunoblotting for Vimentin, phCofilin, Cofilin, MMP2, ɑ-SMA, COL1A1 and CTGF expression levels (from top to bottom). GAPDH was used as loading control. B. Immunofluorescence on CRC patients-derived organoids, treated with iPolyP, AMTB or iPolyP + TRPM8 inhibitor to detect the expression of the above-mentioned proteins. Scale bar 100 µm. C. Quantification relative to panel B. Statistical analysis performed by Student’s t-test (*** p < 0.001). Fold changes versus control, untreated (UT).
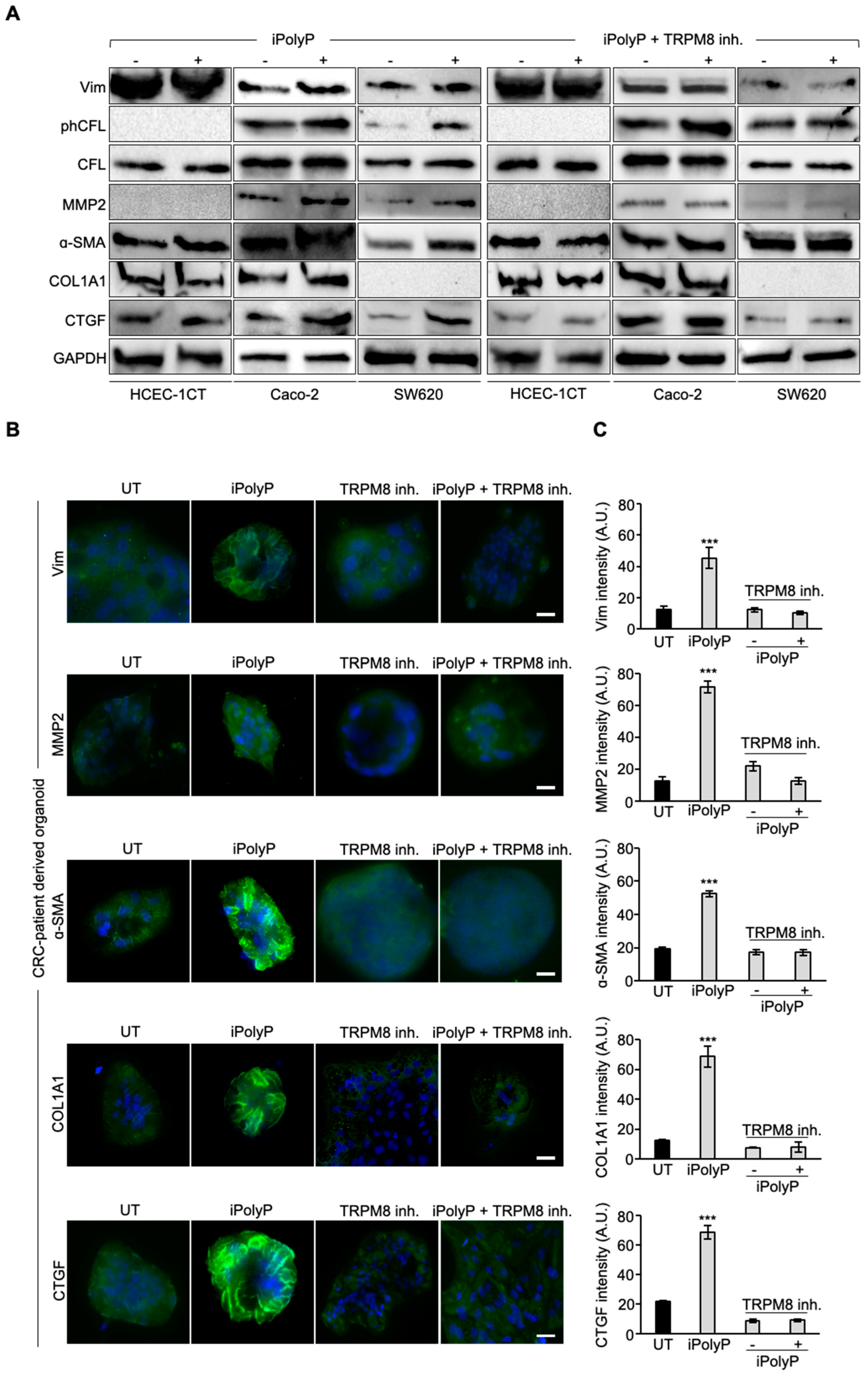
Figure 2.
Effect of the iPolyP-TRPM8 axis on cell migration. A Representative phase-contrast microscope images taken at 0 and 24 hours of a wound healing assay in cell cultures treated with iPolyP, TRPM8 inhibitor as well as the combination of both. Cell migration was promoted in Caco-2 cells, but not in HCEC-1CT cells, by the presence of iPolyP. Red line delineates the cell-free region. B. Quantification relative to panel A. Fold changes versus control, untreated (UT). Statistical analysis performed by Student’s t-test (**** p < 0.0001).
Figure 2.
Effect of the iPolyP-TRPM8 axis on cell migration. A Representative phase-contrast microscope images taken at 0 and 24 hours of a wound healing assay in cell cultures treated with iPolyP, TRPM8 inhibitor as well as the combination of both. Cell migration was promoted in Caco-2 cells, but not in HCEC-1CT cells, by the presence of iPolyP. Red line delineates the cell-free region. B. Quantification relative to panel A. Fold changes versus control, untreated (UT). Statistical analysis performed by Student’s t-test (**** p < 0.0001).
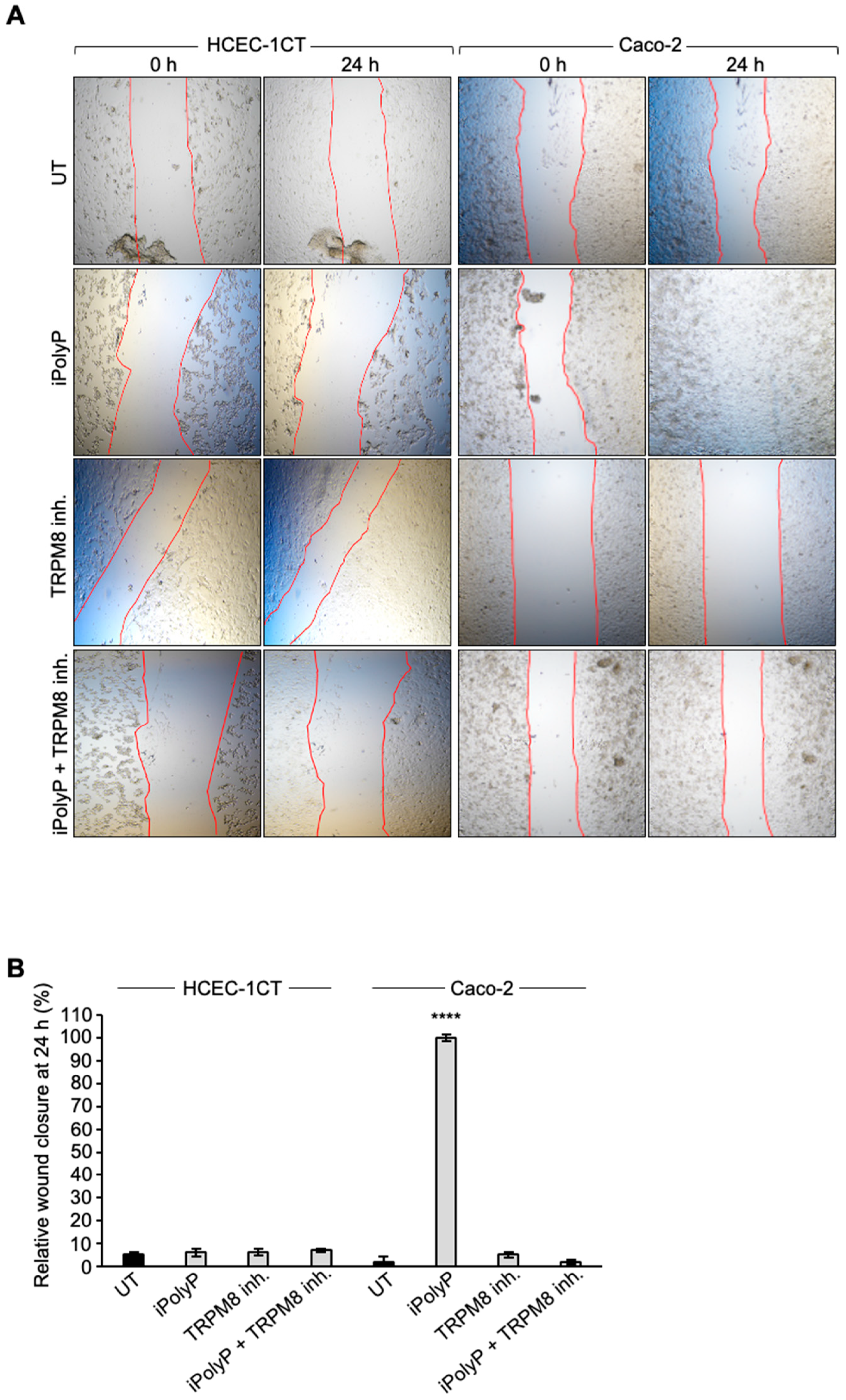
Figure 3.
iPolyP triggers NLRP3 inflammasome priming in human monocytes. A. Cellular extracts from THP-1 monocytes, treated with LPS, iPolyP or iPolyP + TRPM8 inhibitor for 4 hours, were analyzed by immunoblotting against pro-IL-1β and TRPM8, normalized to GAPDH. B. Real-Time PCR on 4 hours LPS-, iPolyP- or iPolyP + TRPM8 inhibitor-treated THP-1 cells. Statistical analysis performed by Student’s t-test (*** p < 0.001). C. Evaluation of IL-1β performed by ELISA on the supernatant of THP-1 cells treated with iPolyP (4 hours), TRPM8 inhibitor (4 hours), LPS (4 hours) + Nigericin (30 minutes), LPS (4 hours) + ATP (30 minutes), LPS (4 hours) + ATP (30 minutes) + MCC950 (4 hours), iPolyP (4 hours) + ATP (30 minutes) or iPolyP (4 hours) + ATP (30 minutes) + TRPM8 inhibitor (4 hours). Statistical analysis performed by Student’s t-test (*** p < 0.001). D. Immunoblotting on THP-1 cells treated with LPS (4 hours), iPolyP (4 hours), LPS (4 hours) + ATP (30 minutes), LPS (4 hours) + ATP (30 minutes) + MCC950 (4 hours), iPolyP (4 hours) + ATP (30 minutes), or iPolyP (4 hours) + ATP (30 minutes) + TRPM8 inhibitor (4 hours), and probed against pro-Caspase-1. Normalization was performed on GAPDH. E. Representative confocal micrographs showing the inflammasome speck (visualized as ASC punctum, white angle bracket) on THP-1 cells treated with LPS (4 hours) + ATP (30 minutes), iPolyP (4 hours) + ATP (30 minutes), iPolyP (4 hours) + ATP (30 minutes) + TRPM8 inhibitor (4 hours). Scale bar 10 µm. Images are representative of three independent experiments. F. Speck quantification relative to panel E. G. Cell death assay performed by FACS-assisted PI staining on THP-1 cells treated with iPolyP (4 hours), LPS (4 hours) + Nigericin (30 minutes), LPS (4 hours) + ATP (30 minutes), LPS (4 hours) + ATP (30 minutes) + MCC950 (4 hours), iPolyP (4 hours) + ATP (30 minutes), iPolyP (4 hours) + ATP (30 minutes) + TRPM8 inhibitor (4 hours). Statistical analysis performed by Student’s t-test (*** p < 0.001). H. Representative electron microscopy micrographs of THP-1 cells treated with LPS (4 hours) + Nigericin (30 minutes), LPS (4 hours) + ATP (30 minutes), iPolyP (4 hours) + ATP (30 minutes), iPolyP (4 hours) + ATP (30 minutes) + TRPM8 inhibitor (4 hours). Yellow asterisks denote the formation of vacuoles typical of NLRP3 inflammasome activation. Red asterisks denote the presence of the speck between centrioles, located near the nucleus. Scale bar 1 µm. Images are representative of three independent experiments. Fold changes versus control, untreated (UT). Data are presented as mean ± SD for triplicate wells from three independent experiments.
Figure 3.
iPolyP triggers NLRP3 inflammasome priming in human monocytes. A. Cellular extracts from THP-1 monocytes, treated with LPS, iPolyP or iPolyP + TRPM8 inhibitor for 4 hours, were analyzed by immunoblotting against pro-IL-1β and TRPM8, normalized to GAPDH. B. Real-Time PCR on 4 hours LPS-, iPolyP- or iPolyP + TRPM8 inhibitor-treated THP-1 cells. Statistical analysis performed by Student’s t-test (*** p < 0.001). C. Evaluation of IL-1β performed by ELISA on the supernatant of THP-1 cells treated with iPolyP (4 hours), TRPM8 inhibitor (4 hours), LPS (4 hours) + Nigericin (30 minutes), LPS (4 hours) + ATP (30 minutes), LPS (4 hours) + ATP (30 minutes) + MCC950 (4 hours), iPolyP (4 hours) + ATP (30 minutes) or iPolyP (4 hours) + ATP (30 minutes) + TRPM8 inhibitor (4 hours). Statistical analysis performed by Student’s t-test (*** p < 0.001). D. Immunoblotting on THP-1 cells treated with LPS (4 hours), iPolyP (4 hours), LPS (4 hours) + ATP (30 minutes), LPS (4 hours) + ATP (30 minutes) + MCC950 (4 hours), iPolyP (4 hours) + ATP (30 minutes), or iPolyP (4 hours) + ATP (30 minutes) + TRPM8 inhibitor (4 hours), and probed against pro-Caspase-1. Normalization was performed on GAPDH. E. Representative confocal micrographs showing the inflammasome speck (visualized as ASC punctum, white angle bracket) on THP-1 cells treated with LPS (4 hours) + ATP (30 minutes), iPolyP (4 hours) + ATP (30 minutes), iPolyP (4 hours) + ATP (30 minutes) + TRPM8 inhibitor (4 hours). Scale bar 10 µm. Images are representative of three independent experiments. F. Speck quantification relative to panel E. G. Cell death assay performed by FACS-assisted PI staining on THP-1 cells treated with iPolyP (4 hours), LPS (4 hours) + Nigericin (30 minutes), LPS (4 hours) + ATP (30 minutes), LPS (4 hours) + ATP (30 minutes) + MCC950 (4 hours), iPolyP (4 hours) + ATP (30 minutes), iPolyP (4 hours) + ATP (30 minutes) + TRPM8 inhibitor (4 hours). Statistical analysis performed by Student’s t-test (*** p < 0.001). H. Representative electron microscopy micrographs of THP-1 cells treated with LPS (4 hours) + Nigericin (30 minutes), LPS (4 hours) + ATP (30 minutes), iPolyP (4 hours) + ATP (30 minutes), iPolyP (4 hours) + ATP (30 minutes) + TRPM8 inhibitor (4 hours). Yellow asterisks denote the formation of vacuoles typical of NLRP3 inflammasome activation. Red asterisks denote the presence of the speck between centrioles, located near the nucleus. Scale bar 1 µm. Images are representative of three independent experiments. Fold changes versus control, untreated (UT). Data are presented as mean ± SD for triplicate wells from three independent experiments.
Figure 4.
iPolyP triggers the epithelial-to-mesenchymal transition. The iPolyP/TRPM8 axis leads to functional changes that culminate in the expression of EMT markers, thus promoting cancer cell migration and invasiveness.
Figure 4.
iPolyP triggers the epithelial-to-mesenchymal transition. The iPolyP/TRPM8 axis leads to functional changes that culminate in the expression of EMT markers, thus promoting cancer cell migration and invasiveness.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



