
Submitted:
28 October 2024
Posted:
30 October 2024
You are already at the latest version
Abstract
Background/Objectives: Camptothecin (CPT) is a well-known chemical compound recognized for its significant anticancer properties. However, its clinical application remains limited due to challenges related to CPT’s high hydrophobicity and the instability of its active form. To address these difficulties, our research focused on the development of four novel nanoparticulate systems intended for either oral or intravenous administration; Methods: These nanosystems were based on a poly(amidoamine) (PAMAM) dendrimer/CPT complex, which had been coated with biodegradable homo- and copolymers, designed with appropriate physicochemical properties and chain microstructures; Results: The resulting nanomaterials, with diameters ranging from 110 to 406 nm and dispersity values between 0.10 and 0.67, exhibited a positive surface charge and were synthesized using biodegradable poly(L-lactide) (PLLA), poly(L-lactide-co-ε-caprolactone) (PLACL), and poly(glycolide-co-ε-caprolactone) (PGACL). Biological assessments, including cell viability and hemolysis tests, indicated that all polymers demonstrated less than 5% hemolysis, confirming their hemocompatibility for potential intravenous use. Furthermore, fibroblasts exposed to these matrices showed concentration-dependent viability. The entrapment efficiency (EE) of CPT reached up to 27%, with drug loading (DL) values as high as 17%. The in vitro drug release studies lasted over 400 h with the use of phosphate buffer solutions at two different pH levels, demonstrating that time-dependent processes allowed for a gradual and controlled release of CPT from the developed nanosystems. The release kinetics of the active compound at pH 7.4 ± 0.05 and 6.5 ± 0.05 followed near-first-order or first-order models, with diffusion and Fickian/non-Fickian transport mechanisms. Importantly, the nanoparticulate systems enabled the stabilization of the pharmacologically active form of CPT, while providing protection against hydrolysis, even in physiological environments; Conclusions: In our opinion, these results underscore the promising future of biodegradable nanosystems as effective drug delivery systems (DDSs) for targeted cancer treatment, offering stability and efficacy over short, medium, and long-term applications.
Keywords:
nanopharmacy
; camptothecin
; PAMAM dendrimer
; biodegradable polyesters
; controlled drug delivery systems
; targeted therapy
1. Introduction
The field of nanotechnology focuses on the molecular creation of a wide range of functional systems. These systems exhibit unique physical, electrical, and optical properties that make them appealing in a wide range of applications [1]. Nanopharmacy is one of the most well-known nanotechnology study areas. It employs nanotechnology to provide precisely tailored pharmaceutical treatments for illness diagnosis, prevention, and therapy. Over the last several decades, there has been an increase in nanopharmacy research, which is now being translated into commercialization efforts around the world, culminating in the selling of a variety of products [2,3]. Drug delivery systems (DDSs) currently dominated nanopharmacy, accounting for more than 75% of total sales [4].
The term DDSs refers to a wide variety of vehicles developed to increase the selectivity of action and improve the drug's pharmacological and pharmacokinetic characteristics. Most of the drugs in use today are classified as low molecular weight compounds. These substances are characterized by their quick metabolism and elimination from the human body, along with a lack of selectivity in their pharmacodynamic effects. This highlights the necessity for the development of formulations that can address these challenges [5]. The idea of DDSs originated as early as 1891, when German scientist Paul Erlich proposed a paradigm for selectively delivering drugs to their site of action, in the right concentration and at the appropriate time [6].
Traditional ways of drug administration are frequently inefficient and pose a significant risk of side effects. DDSs allow for the alteration of drug characteristics, such as increasing solubility, preventing the drug from being transformed into an inactive form, and improving pharmacokinetic parameters and biodistribution [7]. DDSs are most commonly employed to develop systems that provide controlled or prolonged release of the active substance, known as therapeutic systems. Therapeutic systems are designed to maintain a constant drug concentration throughout time and to make the release rate independent of the amount of drug left in the system (achieving zero-order kinetics). The time required for the therapeutic system to release the drug is highly associated with the type of carrier used, which is the factor that controls the release [2]. The polymeric, inorganic, or composite material that acts as the drug's carrier must fulfill certain requirements, including biocompatibility, non-toxicity, non-immunogenicity, and no accumulation in the body. Furthermore, its size, surface character, method of binding the active substance and presence of functional groups on the surface all play an important role in DDSs functioning [8].
Recent focus has been directed towards the chemistry of biocompatible and biodegradable polymers, often referred to as "biomedical polymers." These materials are advantageous due to their ability to be easily hydrolyzed into non-toxic, removable products that can be eliminated through metabolic processes [9,10]. Furthermore, it is essential that biomedical polymers are synthesized with the use of natural initiators, co-initiators, and/or catalysts that are both effective and safe for the environment and human health [11]. Most significantly, these materials have great potential for various medical applications, including tissue engineering, gene therapy, temporary implanted devices, regenerative medicine, nanopharmacy, and coatings on implants [12]. Biodegradable and/or bioresorbable polymers such as polyglycolide (PGA), polylactide (PLA), poly(ε-caprolactone) (PCL), poly(trimethylene carbonate) (PTMC) and copolymers of glycolide (GL), L-lactide (LA), rac-lactide (rac-LA), ε-caprolactone (CL), trimethylene carbonate (TMC) or others cyclic esters and carbonates are most commonly used in nanopharmacy to develop DDSs and other therapeutic systems [13].
Dendrimers, especially poly(amidoamine) (PAMAM) dendrimers, also known as tree-shaped, stellar, hyperbranched, or cascading polymers, are unique carriers that require particular attention. These are spherical structures with greatly branching arms that terminate in functional groups [14]. The presence of peripheral functional groups (e.g. amine, hydroxyl) as well as internal cavities in PAMAM dendrimers can be adopted to encapsulate drugs or other active compounds. The encapsulation of the active component within the PAMAM dendrimer cavity is a typical idea for drugs with a low molecular weight and poor water solubility. A host-guest interaction occurs inside dendrimer cavities due to the hydrogen bonds between dendrimer and the drug molecules, resulting in a highly soluble complex [15]. Furthermore, the empty spaces in the dendrimer structure are frequently hydrophobic, allowing for interactions with poorly soluble molecules via hydrophobic interactions [16]. The active substance might be additionally conjugated to the peripheral functional groups via covalent bonding. This strategy is preferred, since it provides more precise control over the drug release, enabling its delivery to the target site [17]. Last but not least, the main benefits of using PAMAM dendrimers as drug carriers include improved solubility of hydrophobic drugs, drug protection during transport in the body, a longer period of the drug activity, improved pharmacodynamics, reduced side effects, and the possibility of prolonged and controlled drug release [18].
Cancer is a prevalent cause of mortality worldwide. The American Cancer Society predicts that in 2024, 2,001,140 new cancer cases and 611,720 cancer deaths will occur in the United States [19]. According to the National Cancer Registry data, the overall number of malignant tumor cases in Poland in 2020 was over 146,000, with about 100,000 deaths. Unfortunately, the disease structure in Poland is remarkably similar to that of other countries [20].
The main objective of oncological therapy is to bring the tumor under control to ensure better life expectancy and quality. In addition to surgical therapy and radiotherapy, chemotherapy includes a wide range of drugs from various groups, each with a different mechanism of action. All three treatment procedures carry a significant risk of major side effects, as well as limited specificity and selectivity [21].
Chemotherapeutic drug treatment is associated with high toxicity, which is a severe clinical issue that has a considerable impact on the patient's quality of life [22]. Furthermore, the anticancer drugs utilized exhibit rapid renal clearance, making it difficult to achieve the appropriate therapeutic concentration, thus necessitating the use of higher doses. Also, the majority of drugs taken systemically are lipophilic, tend to accumulate in the liver and, as a result, may lead to hepatic steatosis [23]. In order to improve the efficacy and safety of therapy, scientists are searching for carriers that would allow targeted drug delivery to cancer cells as well as controlled drug release.
One of the most promising anticancer drugs is camptothecin (CPT), a pentacyclic alkaloid isolated from the Camptotheca acuminata tree [24]. The α-hydroxylactone ring in the CPT's structure plays a crucial role in sustaining the anticancer efficiency, since its opening leads to deactivation [25]. CPT provides high cytotoxic effect, and its mechanism of action relies on the suppression of topoisomerase I activity, which results in DNA damage and, consequently, cell death [26]. Nonetheless, CPT's therapeutic usage, however, is challenging due to its low solubility in water, high cellular toxicity and hydrolytic cleavage of the lactone ring to the inactive carboxyl form in vivo [27]. In recent years, extensive research has been conducted to generate novel formulations of CPT, which could address the aforementioned concerns [24]. It involves the development of polymer nanoparticles, micelles, liposomes, and lipid nanoparticles [28,29,30,31,32]. Another alternative strategy is to encapsulate CPT inside the cavity of a PAMAM (polyamidoamine) dendrimer, which has minimal cytotoxicity and is able to penetrate biological membranes [33].
In our previous investigation, we examined PAMAM dendrimer/CPT complex for non-small-cell lung cancer cell targeting [34]. Considering that the findings we obtained were highly promising and revealed an opportunity for further study, we decided to develop innovative PAMAM dendrimer/biodegradable polymer nanosystems as a CPT carrier in the present research. To date, there have been no reports in the literature of nanosystems that encapsulate both CPT and its derivatives inside a PAMAM dendrimer, with a biodegradable polymer serving as the carrier. We suspected that encapsulating CPT inside a PAMAM dendrimer would improve its solubility and the therapeutic bioavailability, while utilizing a biodegradable material with a predicted microstructure and topology would allow for controlled and sustained drug release. Furthermore, the acidic microenvironment formed by the hydrolytic degradation of the biodegradable matrix will inhibit the active lactone form of CPT from converting to its inactive carboxyl form. As a result, the developed novel nanosystems may allow for oral or intravenous administration, while maintaining the therapy's effectiveness and biosafety.
2. Materials and Methods
2.1. Materials
(S)-(+)-Camptothecin ((S)-4-Ethyl-4-hydroxy-1H-pyrano-[3′,4′:6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione) (CPT) (purity: >97.0% (HPLC)) was purchased from TCI Europe N.V. (Zwijndrecht, Belgium). PAMAM dendrimer, ethylenediamine core, generation 4.0, 10 wt.% solution in methanol was obtained from Merck, Poznan, Poland. L-lactide ((3S)-cis-3,6-dimethyl-1,4-dioxane-2,5-dione) (LA) (purity: 100%), -caprolactone (2-oxepanone) (CL) (purity: 100%), glycolide (1,4-dioxane-2,5-dione) (GL) (purity: 100%), diethylzinc solution 1.0 M in hexanes (Et2Zn), poly(ethylene glycol) average Mn 400 (PEG-400), Tween® 80 (polyethylene glycol sorbitan monooleate, polysorbate 80) were also obtained from Merck, Poznan, Poland. N,N-dimethylformamide (DMF, anhydrous, 99%, Avantor Performance Materials S.A., Gliwice, Poland), dimethyl sulphoxide (DMSO, anhydrous, 99%, Avantor Performance Materials S.A., Gliwice, Poland), methanol (anhydrous, 99.9%, Avantor Performance Materials S.A., Gliwice, Poland), dichloromethane (DCM, anhydrous, 99.8%, Chempur, Piekary Śląskie), chloroform (pure, stabilized with amylen, 98.5%, Chempur, Piekary Śląskie, Poland), petroleum ether boiling range 6090°C (Chempur, Piekary Śląskie, Poland), liquid paraffin (Chempur, Piekary Śląskie, Poland), hydrochloric acid (37%, Merck, Poznan, Poland) and acetonitrile ((ACN), anhydrous, 99.8%, Avantor Performance Materials S.A., Gliwice, Poland) were used as received. Dimethyl-d6-sulfoxide (DMSO-d6) in ampoules, for NMR measurements (99.9 atom% D), was purchased from ARMAR Chemicals (Döttingen, Switzerland), whereas chloroform-d (deuterochloroform, CDCl3 99.8 atom% D) was purchased from Merck, Poznan, Poland. Phosphate buffer solutions (PBS, pH 7.40 ± 0.05 and pH 6.50 ± 0.05, 20°C, Avantor Performance Materials S.A., Gliwice, Poland) were also used as received.
2.2. Synthesis of PAMAM Dendrimer/CPT Complex
The synthesis was carried out according to a previously established method by Oledzka et al. [34]. Briefly, 9.1 mg of CPT was placed in a 5.0 mL round-bottom flask and dissolved in 2 mL of DMF. Then, 1 mL of PAMAM dendrimer (G4, 10 wt.% in methanol) was added, and the prepared reaction mixture was incubated in the dark for 24 h at 37°C, at rotational speed of 130 rpm. Following that, the DMF was evaporated, and 2.0 mL of distilled water was added to the solid residue and stirred for 1 h in order to remove the unreacted CPT. The obtained suspension was filtered using a 0.45 μm syringe filter, and the supernatant was evaporated using a rotary evaporator. The resulting solid compound was dried in a vacuum oven at 40°C for 72 h, until the constant weight was achieved. The PAMAM dendrimer/CPT complex synthesis yielded 84.85%. The synthesised complex was stored in the dark in an inert gas atmosphere until it was used. Subsequent analyses were conducted within 24 h of synthesis.
The 1H NMR spectrum of the synthesized PAMAM dendrimer/CPT complex (DMSO-d6, 400 MHz; δH): 8.69 ppm (G, B-ring of CPT), 7.93 ppm (H and K, A-ring of CPT), 7.70 ppm (I and J, A-ring of CPT), 7.34 ppm (A, D-ring of CPT), 5.42 ppm (E, E-ring of CPT), 5.29 ppm (F, C-ring of CPT), 4.87 ppm (6 (-CH2-CH2-C(O)-N(H)-) of PAMAM dendrimer), 4.67 ppm (7 (-C(O)-N(H)-CH2-CH2-NH2) of PAMAM dendrimer), 2.00 – 3.62 ppm (2 (-CH2-CH2-C(O)-N(H)-), 3 (-CH2-CH2-C(O)-N(H)-), 4 (-C(O)-N(H)-CH2-CH2-) and 5 (-C(O)-N(H)-CH2-CH2-NH2) of PAMAM dendrimer), 1.86 ppm (B, E-ring of CPT, methylene group), 1.22 ppm (1 (-N-CH2-CH2-N-) and 1’ (-C(O)-N(H)-CH2-CH2-) of PAMAM dendrimer), 0.87 ppm (C, E-ring of CPT, methylene group) (Figure S1, Supplementary Materials).
2.3. Synthesis of Biodegradable Polymers
The Ring-Opening Polymerisation (ROP) process was applied to produce four polymers, with ZnEt2 acting as a catalyst and PEG-400 serving as a co-initiator. The reactions were carried out in a bulk. Briefly, carefully weighted amounts of monomers - LA (M1), LA and CL (M2), CL and GL (M3, M4) (1 g total) - were placed in glass ampoules and vacuum dried to remove trace amounts of moisture. After that, calculated amounts of ZnEt2 and PEG-400 were added in an argon atmosphere via the Schlenk line, and the ampoules were safely sealed. Reactions were carried out in an oil bath at 130°C for 24 h. The post-reaction mixtures of M1 and M2 were then dissolved in 6.0 mL of DCM, while M3 and M4 were dissolved in 6.0 mL of a 1:1 mixture consisting of DCM and chloroform. Following that, the dissolved polymers were transferred to beakers and precipitated using an ice-cold 5% HCl solution in methanol. The precipitates were subsequently washed twice more with pure, ice-cold methanol. The collected products were vacuum-dried for 24 h at 30°C.
The 1H NMR spectrum of the synthesized PLLA (M1, CDCl3, 400 MHz; δH): 5.15 ppm (2, (-O(O)C-(H)C(CH3)-)), 4.31 ppm (5, (-O(O)C-(H)C(CH3)-OH)), 3.63 ppm (PEG, (-CH2-CH2-)), 2.17 ppm (1, (-O(O)C-(H)C(CH3)-OH)), 1.42 ppm (3, (-O(O)C-(H)C(CH3)-)), 1.25 ppm (4, (-O(O)C-(H)C(CH3)-OH)) (Figure S2, Supplementary Materials).
The 13C NMR spectrum of the synthesized PLLA (M1, CDCl3, 400 MHz; δH): 169.7 ppm (-O(O)C-(H)C(CH3)-), 69.0 ppm (-O(O)C-(H)C(CH3)-), 16.7 ppm (2, (-O(O)C-(H)C(CH3)-)) (Figure S3, Supplementary Materials).
The 1H NMR spectrum of the synthesized PLACL (M2, CDCl3, 400 MHz; δH): 5.15 ppm (2, (-O(O)C-(H)C(CH3)-)), 5.06 ppm (3, (-O-(H)C(CH3)-C(O)O-(CH2)5-)), 4.12 ppm (5, (-(H)C(CH3)-C(O)O-CH2-CH2-CH2-CH2-CH2-C(O)O-)), 4.05 ppm (10, (-(CH2)5-C(O)O-CH2-CH2-CH2-CH2-CH2-)), 3.64 ppm (PEG, (-CH2-CH2-)), 2.38 ppm (9, (-(H)C(CH3)-C(O)O-CH2-CH2-CH2-CH2-CH2-C(O)O-)), 2.29 ppm (14, (-(CH2)5-C(O)O-CH2-CH2-CH2-CH2-CH2-)), 1.45 - 1.71 ppm (1, (-O(O)C-(H)C(CH3)-), 4, (-O-(H)C(CH3)-C(O)O-(CH2)5-), 6, (-(H)C(CH3)-C(O)O-CH2-CH2-CH2-CH2-CH2-C(O)O-), 8, (-(H)C(CH3)-C(O)O-CH2-CH2-CH2-CH2-CH2-C(O)O-), 11, (-(CH2)5-C(O)O-CH2-CH2-CH2-CH2-CH2-), 13 (-(CH2)5-C(O)O-CH2-CH2-CH2-CH2-CH2-)), 1.39 ppm (7, (-(H)C(CH3)-C(O)O-CH2-CH2-CH2-CH2-CH2-C(O)O-), 12, (-(CH2)5-C(O)O-CH2-CH2-CH2-CH2-CH2-)) (Figure S4, Supplementary Materials).
The 13C NMR spectrum of the synthesized PLACL (M2, CDCl3, 400 MHz; δH): 172.6 – 173.9 ppm (1, (-(CH2)5-C(O)O-(CH2)5-), carbonyl carbon atoms of oxycaproyl unit), 169.3 – 171.2 ppm (2, (-O(O)C-(H)C(CH3)-), carbonyl carbon atoms of lactidyl unit), 67.8 – 69.9 ppm (4, (-O(O)C-(H)C(CH3)-), methine carbon atoms of lactidyl unit), 63.4 – 65.9 ppm (9, (-C(O)O-CH2-CH2-CH2-CH2-CH2-), -carbon atoms of oxycaproyl unit), 33.1 – 33.4 ppm (5, (-C(O)O-CH2-CH2-CH2-CH2-CH2-), -carbon atoms of oxycaproyl unit)), 28.2 ppm (8, (-C(O)O-CH2-CH2-CH2-CH2-CH2-), -carbon atoms of oxycaproyl unit), 23.9 – 25.9 ppm (6, (-C(O)O-CH2-CH2-CH2-CH2-CH2-), -carbon atoms of oxycaproyl unit, 7, (-C(O)O-CH2-CH2-CH2-CH2-CH2-), -carbon atoms of oxycaproyl unit), 16.6 ppm (3, (-O(O)C-(H)C(CH3)-), methyl carbon atoms of lactidyl unit) (Figure S5, Supplementary Materials).
The 1H NMR spectrum of the synthesized PGACL (M3 and M4, CDCl3, 400 MHz; δH): 4.60 ppm (1, (-O-CH2-C(O)-), glycolidyl unit), 4.05 ppm (2, (-C(O)O-CH2-CH2-CH2-CH2-CH2-), caproyl unit), 3.65 ppm (PEG, (-CH2-CH2-)), 2.30 ppm (6, (-C(O)O-CH2-CH2-CH2-CH2-CH2-), caproyl unit), 1.65 ppm (6, (-C(O)O-CH2-CH2-CH2-CH2-CH2-), caproyl unit, 5, (6, (-C(O)O-CH2-CH2-CH2-CH2-CH2-), caproyl unit), 1.38 ppm (4, (-C(O)O-CH2-CH2-CH2-CH2-CH2-), caproyl unit) (Figure S6, Supplementary Materials).
The 13C NMR spectrum of the synthesized PGACL (M3 and M4, CDCl3, 400 MHz; δH): 172.8 – 173.5 ppm ((-O-CH2-C(O)-), glycolidyl unit), 167.9 ppm ((-C(O)O-CH2-CH2-CH2-CH2-CH2-), caproyl unit), 64.2 – 65.2 ppm ((-C(O)O-CH2-CH2-CH2-CH2-CH2-), -carbon atoms of oxycaproyl unit), 60.2 – 60.5 ppm ((-O-CH2-C(O)-), glycolidyl unit), 33.6 – 34.1 ppm ((-C(O)O-CH2-CH2-CH2-CH2-CH2-), -carbon atoms of oxycaproyl unit)), 28.2 ppm ((-C(O)O-CH2-CH2-CH2-CH2-CH2-), -carbon atoms of oxycaproyl unit), 25.4 ppm ((-C(O)O-CH2-CH2-CH2-CH2-CH2-), -carbon atoms of oxycaproyl unit), 24.4 ppm ((-C(O)O-CH2-CH2-CH2-CH2-CH2-), -carbon atoms of oxycaproyl unit) (Figure S7, Supplementary Materials).
The conversion of monomers was determined from 1H NMR spectra of post-reaction, by comparing the integrated signals of equivalent protons from the monomer and polymer, according to the following formula (Equation 1):
where Ii and II represent the integral intensities of signals from equivalent protons in the monomer and polymer, respectively.
The microstructural analysis of the obtained polymers was carried out with 13C NMR spectra in the methine and carbonyl regions for PLLA and the carbonyl region for PLACL. The microstructure of PGACL matrices was examined using 1H NMR spectra in the region of methylene protons of glycolidyl units (GG) and -methylene proton region of caproyl units (Cap). By comparison with the available literature, relevant spectral lines were assigned to the respective sequences [35,36,37].
With the use of 1H and 13C spectra, the experimental lengths of glycolidyl units (LeGG) and lactydyl units (LeLL) were calculated according to Equation (2), whereas experimental lengths of caproyl units were calculated with the use of Equation (3) for 1H NMR spectrum and Equation (4) for 13C NMR spectrum, respectively:
where X represents lactydyl unit -OCH(CH3)CO- (L) or glycolidyl unit -OCH2CO- (G), Cap represents caproyl unit -O(CH2)5CO-, and CapXX, CapXCap, XCap, CapCapX, and so on represent particular sequences in the polymeric chain.
The transesterification of the second mode (TII) may cause break in the L or G units within the copolymeric chain, resulting in the formation of distinct CapLCap or CapGCap sequences. To quantitatively determine the yield of TII in the copolymeric chains following Equation (5) was applied:
where [CapXCap] represents the experimental concentration of CapXCap sequences, whereas [CapXCap]R is the concentration of CapXCap sequences in a completely random copolymeric chain.
The [CapXCap]R can be described using following formula (Equation 6), where the ratio of [Cap]/[X] is denoted as k’:
The degree of the randomness (R) of the copolymeric chain was calculated according to the Equation 7:
where LRXX represents the average lengths of lactydyl or glycolidyl blocks (Equation 8) and caproyl blocks (Equation 9) respectively, in a completely randomized copolymeric chain.
2.4. In Vitro Hydrolytic Degradation of Polymerics
In brief, 10 mg of the produced polymers were weighed into prepared and labelled glass vials. The experiment was conducted simultaneously in two series: 10.0 mL of buffer pH 7.4 ± 0.05 was added to the first series, and 10.0 mL of buffer pH 6.5 ± 0.05 was used in the second series. The prepared samples were placed in an incubator set to 37°C with a rotational speed of 100 rpm. The measurement points for each sample were set at 7, 14, 21, 28, 56, and 77 days. The pH of the sample was determined at each time point using the Volcraft pH-100ATC pH-meter.
2.5. Synthesis of the Nanosystems Composed of PAMAM Dendrimer/CPT Complex and Biodegradable Polymer
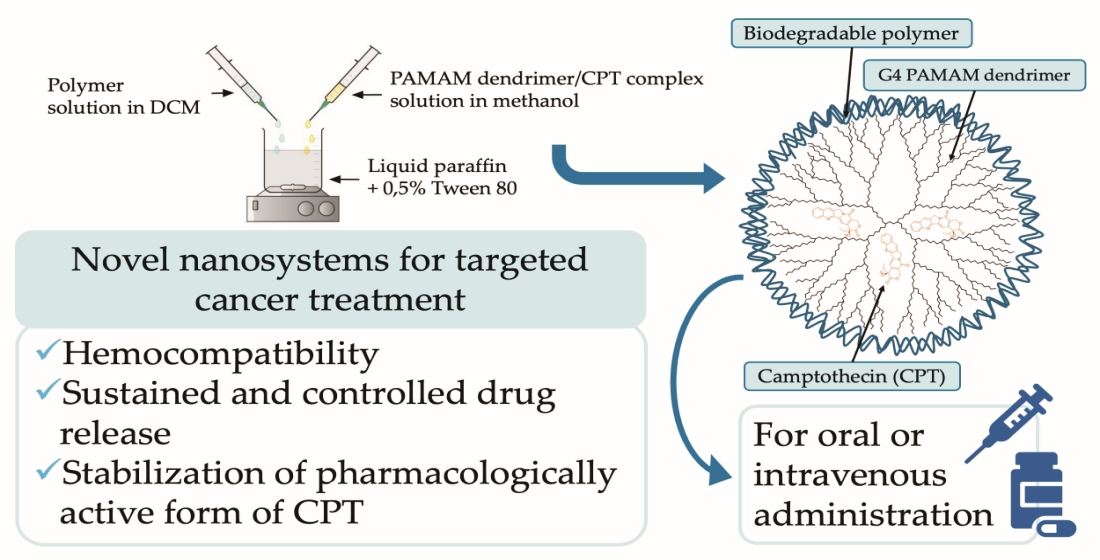
The beaker was filled with 49.5 mL of liquid paraffin and 0.5 mL of Tween 80 to produce a 0.5% solution, which was then placed on a magnetic stirrer. Two glass vials were weighed with 200 mg of the proper biodegradable polymer (M1, M2, M3 or M4) and 20 mg of the PAMAM/CPT complex. The appropriate amount of DCM was poured to the vials with the matrices, followed by methanol to the vial with complex. All vials spent 30 min in an ultrasonic cleaner. The contents of the vials were then mixed and withdrawn using a syringe. The contents of the syringe were added in a dropwise manner to the Tween 80 solution in liquid paraffin at a constant mixing speed of 1000 rpm. Subsequently, the beaker was placed in an ultrasonic homogeniser for 5 min, while cooling in an ice bath. The beaker was then put back on the magnetic stirrer at 400 rpm, covered with parafilm, and left for about 3 h. Following this, n-hexane was poured to the beaker and kept at room temperature for 24 h. The precipitate solution was then decanted, and the resulting products (denoted as NP1, NP2, NP3, NP4, Table 3) were washed seven times with petroleum ether before drying.
To determine the drug loading (DL) and the entrapment efficiency (EE) of CPT in the obtained nanosystems, the accurately weighted amounts of the developed products ( 10 mg) were dissolved in 5 mL of ACN and sonicated for 30 min in an ice bath. The resulting solutions were filtered through 0.45 µm syringe filters to the HPLC vials.
The DL was calculated using following Equation 10:
whereas the EE was determined using the Equation 11:
2.6. In Vitro Release Study of CPT from the Developed Nanosystems
The obtained nanosystems (~50 mg) were suspended in PBS at pH 6.50 ± 0.05 and 7.40 ± 0.05. The resulting suspensions were transferred to dialysis membranes with pore diameters of 3.50 kDa and immersed in appropriate buffer solution. The release experiment lasted 23 days and involved constant rotating at 100 cycles per min at 37°C. At each time point, 1.5 mL of samples were collected and supplemented with 1.5 mL of fresh pH-appropriate PBS. The collected samples were then subjected to HPLC analysis to assess the amount of CPT released as well as the percentage amount of lactone and carboxylic form of CPT. The analysis was performed using gradient elution and UV detector at a wavelength of 363 nm. The calibration curve was designed between 0.46 and 46.55 g/mL (R2 = 1.000, y = 2.3926x – 0.2040). Each of the collected samples was tested three times, and the outcome of the analysis was expressed as an amount of CPT released (in g).
2.7. Mathematical Models
The release data points were subjected to zero-order, first-order kinetics, Higuchi and Korsmeyer–Peppas models, respectively. The calculations were performed according to the formulas provided below:
Zero-order:
First-order:
Higuchi model:
Korsmeyer-Peppas model:
where:
F is the fraction of drug released from the matrix after time t;
F0 is the initial amount of the drug;
2.8. Hemolysis
Defibrinated sheep blood was purchased from Graso, Poland. Blood was centrifuged at 2000 rpm for 10 min (RT). The supernatant was carefully removed without disturbing the RBC pellet. Samples were resuspended in 5 mL of PBS (pH 7.40 ± 0.05) and centrifuged at 2000 rpm for 10 min. The washing step was repeated three times until the supernatant was clear. The remaining RBC pellet was diluted in PBS (pH 7.4, RT) to obtain a 2% suspension. Next, 2% RBC suspension was incubated with serial dilution of the proper matrix (M1, M2, M3 or M4, 0.05-0.5 mg/mL) in a 1:1 ratio for 1 h at 37°C. Positive control (100% hemolysis) was prepared with distilled water and negative control (0% hemolysis) with PBS (pH 7.40 ± 0.05). Next, samples were centrifuged at 2000 rpm for 5 min (RT) and 100 µL of supernatant from each sample was transferred to a 96-well F-bottom plate. Optical density (OD) was measured at 540 nm in a microplate reader (BioTek Synergy HTX Multimode Reader, Agilent, USA). The value of compound-induced hemolysis was calculated according to the Equation 16 [42]:
where A is the absorbance of the sample, A100% is the absorbance of the positive control (100% hemolysis), and A0% is the absorbance of the negative control (0% hemolysis).
Hemolysis [%] = (A - A0%)/(A100% - A0%)·100%
2.9. Cell Culture and Viability Assay
Normal human dermal fibroblasts (Promocell) were maintained in Dulbecco's Modified Eagle Medium (DMEM) (Biowest, Nuaillé, France) containing 10% fetal bovine serum (FBS) (Biowest, Nuaillé, France) and 1% penicillin-streptomycin solution (Biowest, Nuaillé, France). Cells were incubated at 37°C in humidifying conditions with 5% CO2. At confluency of 80% cells were harvested and seeded in 96-well plates at a density of 3·103 per well and incubated overnight. The growth media was then changed to DMEM at pH 7.40 ± 0.05 or DMEM at pH 6.50 ± 0.05 with serial dilutions of M1, M2, M3, or M4 (0.05-50 µg/mL). Plates were incubated at 37°C for 72 h. After the incubation period, 20 µL of CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) (Promega, Walldorf, Germany)was added to each well, and the plates were incubated for additional 2 h at 37°C. The absorbance was measured at ƛ = 490 nm. The viability of untreated cells (negative control) was defined as 100%.
2.10. Statistical Analysis
The in vitro experiment findings were provided as a mean and standard deviation of mean (SEM). The normality of distribution was tested first, followed by one-way ANOVA or the non-parametric Kruskal-Walli’s test to determine the statistical significance of mean differences. To compare the different circumstances, two-way ANOVA was employed, followed by Bonferroni's multiple comparisons. P<0.05 was deemed statistically significant. The data was analysed with GraphPad Prism 5.0 (GraphPad Software, San Diego, California, USA).
2.11. Molecular Modelling of the Nanosystems Composed of PAMAM Dendrimer/CPT Complex and Biodegradable Polymer
The structure of PAMAM dendrimer generation 4.0 was built using the Build Polymers module within the Biovia Materials Studio 2020 software suite (https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-materials-studio/, accessed on 20 October 2024). The terminal, primary amino groups of dendrimers are demonstrated to be protonated and thereby positively charged at pH ranges between 6.5 ± 0.05 and 9.0 ± 0.05 [43]. Since the dissolution analyses conducted for this study were done at pH 6.5 ± 0.05 and 7.4 ± 0.05, all of the terminal amino groups of the modeled PAMAM structure have been protonated to improve comparability with those studies.
The Forcite Plus module has been used to optimize the structures, applying ultra-fine quality settings of geometry optimization and smart algorithm. The convergence tolerance values were set to 2 · 10-5 kcal/mol for energy, 1 · 10-3 kcal/(Å · mol) for force and 1 · 10-5 Å for displacement, with 5 · 104 maximum iterations. All calculations have been performed using the COMPASS II forcefield [44], which has been shown to provide accurate results when modeling PAMAM dendrimers [45]. As charge assignments are one of the characteristics provided in this forcefield specification, the charges have been allocated using the COMPASS II forcefield. The electrostatic and van der Waals summation methods were both atoms based with cubic spline truncation of non-bond energy terms, 18.5 Å cutoff distance, 1 Å spline width and 0.5 Å buffer width.
Molecular docking calculations were performed using the Adsorption Locator module within the Biovia Materials Studio 2020 software suite. The Adsorption Locator module identifies possible adsorption sites by carrying out Monte Carlo searches of the configurational space of the substrate (PAMAM) – adsorbate (CPT) system as the temperature is slowly decreased within the simulated annealing process of a molecular dynamics run. The benefit of such an approach is that it enables simultaneous docking of the specified numbers of CPT, in this work 3 molecules of API. The Adsorption Locator makes use of a simulation of annealing with geometry adjustments in between repeating heat-cool cycles. For repeatable results, 10 cycles with 100 000 steps each and annealing temperatures ranging from 100 to 5000 K were utilized. For "conformer," "rotate," and "translate," the Monte Carlo parameters were set to a probability of 0.32 (ratio = 1), whereas "regrow" was set to 0.1 (ratio = 0.03). The computations employed the identical geometry optimization parameters from the preceding paragraph's detailed description of geometry optimization and the COMPASS II force field.
To calculate the stoichiometry of the PAMAM dendrimer/CPT complex, the formula below has been used (Equation 17):
where L is the loading (average number of CPT molecules per one PAMAM molecule), mCPT is the mass of added CPT (9.1 · 10-3 g), MPAMAM is the molar mass of PAMAM (14214.17 g/mol), EE is the encapsulation efficiency (84.85%), MCPT is the molar mass of CPT (348.35 g/mol), VPAMAM is the volume of added PAMAM methanol solution (1 · 10-3 dm3), dPAMAM is the density of the PAMAM methanol solution (0.813 g/cm3) and CPAMAM is the percentage concentration of PAMAM methanol solution (10%).
The optimized systems containing three molecules of CPT were then subjected to molecular dynamics (MD) simulations under periodic boundary conditions. First, the MODELS 1-4 were created using the Amorphous Cell module of Materials Studio. This application provides a comprehensive set of tools to construct three-dimensional periodic structures of polymeric systems. The module builds molecules in a cell in a Monte Carlo fashion, by minimizing close contacts between atoms, whilst ensuring a realistic distribution of torsion angles for any given forcefield. The output of such simulations is a single periodic structure, which in the case of present study served as the basic input to MD simulations. The composition of the cubic unit cells of MODELS 1-4 included PAMAM G4.0/3 molecules of CPT complex, surrounded by the polymer as defined in Table 5. After construction, the systems MODELS 1-4 were subjected to geometry optimization, including unit cell dimensions optimization, using the parameters from the preceding paragraph's detailed description of geometry optimization setup and the COMPASS II force field as well.
Subsequently, a simulated annealing process was undertaken in 5 cycles of 10 000 steps, within a temperature range of 300-500 K, using the NVT ensemble mode, 5 heating ramps per cycle, 100 dynamics steps per run and the Berendsen thermostat with 0.1 ps decay constant. A longer (100 ns) production run, in NVT mode, with the Nosé-Hoover thermostat for temperature control with a Q-ratio of 2 was performed with a 1 fs time step. For the MD calculations, the COMPASS II force field was used with the parameters the same as those used for geometry optimization.
2.12. Measurements
All 1H and 13C NMR measurements were performed using a Varian 300 MHz (Palo Alto, Santa Clara, CA, USA) and Agilent Technologies 400 MHz (Santa Clara, CA, USA) spectrometer.
The quantitative analysis of CPT content in the samples was determined using previously developed and described method by Oledzka et al. [34]. The HPLC apparatus (Beckman Coulter, Miami, Florida USA) was equipped with an autosampler (Triathlon 900, Spark Holland B.V., Emmen, Netherlands), pump (Beckman Coulter System Gold® 125NM Solvent Module, Fullerton, CA, USA), and UV/Vis detector (Beckman Coulter System Gold® 166, Fullerton, CA, USA). The analysis was performed at 363 nm using C18 column (Luna 25 cm, 5 μm, 100 A, Phenomenex, Basel, Switzerland) using a gradient mobile phase of acetonitrile (ACN) : phosphate buffer solution (pH 6.50 ± 0.05 or 7.40 ± 0.05) (v/v), delivered at a flow rate of 1.0 mL/min (ACN concentration varied with time: 5% ACN after 0 min, 15% ACN after 5 min, 35% ACN after 15 min, 50% ACN after 20 min, and 5% ACN after 22 and 25 min). The column was placed at 30◦C, and the injection volume was 20 μL. The retention time of CPT was 13.90 ± 0.1 min for carboxylic form of CPT and 20.69 ± 0.1 min for the lactone form.
The number-average molecular weight (Mn) and dispersity index (Ð) of the obtained polymers were measured by SEC-MALLS instrument (Wyatt Technology Corporation, Santa Barbara, CA, USA), composed of an 1100 Agilent isocratic pump, autosampler, degasser, thermostatic box for columns, a photometer MALLS DAWN EOS (Wyatt Technology Corporation, Santa Barbara, CA, USA) and differential refractometer Optilab Rex (Wyatt Technology Corporation, Santa Barbara, CA, USA). ASTRA 4.90.07 software (Wyatt Technology Corporation, Santa Barbara, CA, USA) was used for data collecting and processing. Two 2 x PLGel 5 microns MIXED-C columns were used for separation. The samples were injected as a solution in methylene chloride. The volume of the injection loop was 100 mL. Methylene chloride was used as a mobile phase at a flow rate of 0.8 mL/min.
Zetasizer Nano ZS (Malvern, UK) was applied for dynamic light scattering mea- surements of hydrodynamic diameters and zeta potential. Excitation wavelength of DLS instrument was 633 nm (He-Ne laser, power = 5 W) and measurement angle was 173. Polystyrene disposable cuvettes were used during hydrodynamic diameter determination. Measurements were conducted at 25◦C in aqueous solutions. Samples were briefly sonicated before measurement. Zeta potential was measured using standard dip cell for zeta potential measurements equipped with palladium electrodes. All experiments were conducted in three replications.
The transmission electron microscopy (TEM) has been used to assess the shape morphology and size of the developed nanosystems. The developed products that had been synthesized were collected on TEM grids. TEM studies were carried out using equipment installed in the Laborarory of Electron Microscopy, Nencki Institute of Experimental Biology, Polish Academy of Sciences, Warsaw, Poland. Electron micrographs were taken using a CCD MORADA G2 (EMSIS GmBH, Germany) camera on a JEM 1400 (JEOL Co., Japan) transmission electron microscope sponsored by the EU Structural Funds: Centre of Advanced Technology BIM – Equipment purchase for the Laboratory of Biological and Medical Imaging.
Concerning confocal fluorescence microscopy, the samples were mounted (Chemland, Stargard, Poland) and sealed with #1.5 coverslips (Epredia, Braunschweig, Germany). The fluorescent signal was acquired with an Olympus Fluoview FV1000 confocal microscope (EVIDENT Europe GmbH, Hamburg, Germany) and with 10x and 60x objectives. Fluorescence was excited with 488 nm line of an Argon laser and the emission was detected at 461 nm.
3. Results and Discussion
3.1. Synthesis and Characterization of Biodegradable Polymers
As a first approach, four biodegradable homo- and copolymers were synthesized via ROP of LA, CL and GL in the presence of ZnEt2 as a catalyst and PEG-400 as a biosafe co-initiator. The polymerization process was carried out at 130°C for 24 h. The molar ratio of the monomers to the catalyst and co-initiator was constant and equal to 100:1:1. The reactions were carried out in bulk. The polymers were synthesized with a moderate high monomer conversion, satisfactory yield, reasonably narrow Ɖ and Mn ranged from 8700 to 21300 g/mol (Table 1). It is well established in the scientific literature that polymeric matrices characterized by a higher Mn tend to demonstrate slower drug release profiles compared to those with lower Mn values [46]. Therefore, our primary intention in this study was to develop PAMAM dendrimer/biodegradable polymer nanosystems that are proficient in releasing CPT in a controlled manner over varying durations, including short, medium, and long periods.
The 1H and 13C NMR spectra of the synthesized materials were utilized to determine the structural properties of the polymeric matrices (Figure S2, 1H NMR spectra of PLLA; Figure S3, 13C NMR spectra of PLLA; Figure S4, 1H NMR spectra of PLACL; Figure S5, 13C NMR spectra of PLACL; Figure S6, 1H NMR spectra of PGACL; Figure S7, 13C NMR spectra of PGACL, Supplementary Materials). The characteristic signals observed in the spectra were assigned according to the literature, which confirmed the structures of the produced PLLA, PLACL, and PGACL [35,36,37,47].
As seen in Figure S8 (Supplementary Materials), which displays the PLLA carbonyl and methine regions, the only spectral lines that could be assigned represent iiiii hexad and iii tetrad. Accordingly, our data indicates that transesterification has no effect on the stereoregularity of the produced PLLA. Consequently, the synthesized PLLA exhibited high isotacticity.
The examination of the copolymer spectra, particularly the 13C NMR spectrum of PLACL (M2) (Figure 1) and the 1H NMR spectra of PGACL (M3 and M4) (Figure 2), supported in the identification of spectral lines corresponding to distinct comonomeric sequences within the PGACL and PLACL copolymeric chains. The 1H NMR spectra of M3 and M4 revealed the presence of a signal generated from the -GCapG- triad, with G representing the glycolidyl unit -OCH2CO- and Cap (C) representing the caproyl unit -O(CH2)5CO-. The presence of this signal indicated that type II intermolecular transesterification occurred during the synthesis. Furthermore, the presence of the -CapLCap- triad in the 13C NMR spectrum of PLACL, where L represents the lactydyl unit -OCH(CH3)CO-, suggests that in this case type II intermolecular transesterification also took place. The distribution of comonomeric units in the polymer chains was calculated according to the literature and using the relevant equations (2-8, Experimental section) [36,37]. The average block lengths, transesterification of the second mode, and degree of randomness are provided in Table 2. The estimated findings indicate that the resultant copolymers have an atactic chain microstructure.
Given that it is commonly known that materials used in the medical and pharmaceutical fields must fulfil certain criteria, the resulting polymer matrices were subjected to biological tests, namely cell viability and hemolysis.
The level of RBC hemolysis was matrix-dependent (P = 0.0982), but concentration-independent (Figure 3). Among tested polymeric matrices, the lowest hemolysis was induced by M3 (max. 0.3%) followed by M1 (max. 1%) and M4 (max. 1.2%). The highest hemolytic activity was observed for M2 (max. 2.85%). No statistically significant differences in the level of hemolysis were observed between all matrices except the highest concentration. At a concentration of 0.5 mg/mL, M2-induced hemolysis was significantly higher compared to M1, M3, and M4.
A hemolysis assay is an indispensable initial step in evaluating the blood compatibility of polymers to identify severe acute toxic reactions in RBCs in vivo [48]. Furthermore, many studies have reported that in vitro hemolysis assays have good correlations with in vivo toxicity considering the hemolytic effect [49,50]. Importantly, if the hemolysis rate is less than 5%, medicinal products are termed non-hemolysis under national biological safety guidelines. In our study, all tested matrices revealed results of less than 5%, meeting the national regulatory requirements for biological materials and establishing their hemocompatibility for possible intravenous application (Figure 3 and Figure S9, Supplementary Materials).
As seen in Figure 4, the fibroblasts treated with polymers exhibited comparable concentration-dependent viability. The pH of the cultures had no significant influence on the activity of the polymer samples, except for M4, which caused a greater decrease in viability at pH 6.5±0.05 (see Figure S10, Supplementary Materials). The viability of polymers-treated cells reached approximately 90% viability level of control cells (the concentration was ranged from 0.01 to 5 µg/mL). However, at higher concentrations (10-50 µg/mL), the fibroblast viability was reduced to around 80% after 72 h of treatment. A statistically significant difference in cell viability was noted for M1 and M2 matrices at a concentration of 0.5 µg/mL and between M3 and M4 at 10 µg/mL.
3.2. Synthesis and Characterization of the Nanosystems Composed of PAMAM Dendrimer/CPT Complex and Biodegradable Polymers
In the second step of the investigation, the synthesized and characterized polymers, as well as the PAMAM dendrimer/CPT complex, were employed to develop nanoparticulate systems for targeted therapy. Based on the previous optimization findings, four nanosystems were produced, each employing a different biodegradable polymer (Table 3).

Table 3.
Characterization of the developed nanosystems composed of PAMAM dendrimer/CPT complex and biodegradable polymer.
Table 3.
Characterization of the developed nanosystems composed of PAMAM dendrimer/CPT complex and biodegradable polymer.
| Code | Polymer used |
Entrapment Efficiency (EE)a [%] | Drug Loading (DL)a [%] | Sizeb [nm] | Zeta Potentialb [mV] | PDIb |
|---|---|---|---|---|---|---|
| NP1 | PLLA (M1) | 25 | 16 | 190 | 5.24 ± 2.34 | 0.51 |
| NP2 | PLACL/40:60 (M2) | 27 | 17 | 110 | 34.40 ± 2.14 | 0.52 |
| NP3 | PGACL/15:85 (M3) | 22 | 14 | 284 | 23.43 ± 3.11 | 0.67 |
| NP4 | PGACL/10:90 (M4) | 11 | 7 | 406 | 26.83 ± 1.79 | 0.10 |
a – determined by HPLC; b – determined by DLS.
As shown in Table 3, the EE value ranged from 11 to 27%. The highest value was observed for the NP2 sample (27%), and the lowest for the NP4 (11%). We suspect that these variations are most likely caused by the varied types of polymeric materials used to obtain nanosystems. Furthermore, the DLS analysis indicated that the developed nanosystems had an average size of 190 nm (NP1), 110 nm (NP2), 284 nm (NP3), and 406 nm (NP4). The dispersity values ranged from 0.10 to 0.67, showing a rather broad particle size distribution. Whereas zeta potential experiments indicated that all produced nanoparticulate systems have a positive surface charge (ranging from 5.24 to 24.4 mV), which is most likely explained by the presence of protonated primary amino groups in PAMAM dendrimer macromolecules [51]. It is also important to note that the TEM images of the produced nanomaterials supported the DLS results. Figure 5 shows that the size range of the nanosystems was below 500 nm.
To confirm the presence of CPT more clearly in the prepared nanosystems and roughly determine its location, the confocal microscopy has been applied (Figure 6). Given that CPT is a naturally fluorescent substance (Figure 6a) [52], this technique appears to be suitable for this specific application. The confocal microscopic images showed an effective entrapment of this drug into PAMAM dendrimer cavity as fluorescence was detected in all nanoparticulate materials.
3.3. CPT Release Study from the Nanosystems Composed of PAMAM Dendrimer/CPT Complex and Biodegradable Polymmer
The in vitro release study of CPT was conducted for more than 400 h, utilizing phosphate buffer solutions at two distinct pH levels: 7.40 ± 0.05, which simulates physiological conditions found in the bloodstream, and 6.50 ± 0.05, representing the acidic environment characteristic of endosomes in cancer cells. This study aimed to assess stability and elucidate the CPT release characteristics from the developed nanosystems. The drug release profiles for the samples NP1, NP2, NP3, NP4 are illustrated as a correlation between cumulative drug release and time (Figure 7 and Figure 8). As it can be clearly seen in Figure 7, the drug release rate at a pH of 6.50 ± 0.05 reached a plateau after 120 h for NP4, 216 h for NP1 and NP2, and after 360 h for NP3. The highest drug release rate was observed for the samples NP3 and NP4 (78%), while the NP1 material released only 60% of the active substance. Figure 8 depicts the release rates at a pH of 7.40 ± 0.05. It can be observed that a plateau was achieved after 48 h for the samples NP1, NP2 and NP4, whereas sample NP2 reached its plateau after 168 h. The highest release rate was observed for the NP4 sample (91%), while the NP2 nanosystem demonstrated the lowest drug release rate (45%).
The findings from the two distinct release media are notably consistent, with the highest release rates recorded for the nanosystems obtained using PGACL (NP3 and NP4). This observation may be attributed to the presence of the GL units within the copolymeric chain. The degradation rates of GL copolymers are closely related to the GL composition, as these copolymers exhibit significant hydrophilicity, rendering them more susceptible to hydrolytic degradation [53].
To summarize these findings, it is important to highlight that coating the PAMAM dendrimer/CPT complex with a biodegradable polymer significantly influenced the drug release profile. The time-dependent mechanisms facilitated a gradual and controlled release of the drug from the formulated nanosystems. This phenomenon may be attributed to several factors, including the dissociation during dilution, the competitive displacement of the drug from the PAMAM dendrimer cavity by components of the release medium, the adsorption of the free drug onto the nanoparticle surface, and the diffusion of the free drug from the polymer chain structure [54].
The release rates illustrated in Figure 7 and Figure 8 include both the carboxylic form of CPT, which is pharmacologically inactive, and the lactone form, which is pharmacologically active. According to existing literature, these two forms exist in a state of equilibrium [55]. However, the percentage content of the lactone form of CPT is a critical factor to consider, especially in predicting tumor response. Given that physiological conditions facilitate the hydrolysis of the lactone ring, utilizing biodegradable polymers as a vehicle for CPT presents a viable solution to the issue of CPT instability. As can be seen in Figure S11 (Supplementary Materials), both forms of CPT were detected during the release studies conducted at pH 7.4 ± 0.05 (Figure S11a) and pH 6.5 ± 0.05 (Figure S11b). A comparative analysis of the lactone conformation percentage at pH 7.4 ± 0.05 reveals a notable increase in its content over the duration of incubation (Figure 9). This increase is probably attributable to the acidic degradation products of the polymer carriers present in the incubation environment, which effectively lower the pH and enhance the stability of the pharmacologically active form of CPT. A similar trend was observed in the release study conducted at pH 6.5 ± 0.05 (Figure 10). In all samples, the percentage of the lactone form of CPT was significantly higher than what has been reported in the existing literature [55]. Additionally, it is important to note that, in contrast to the findings of Liu and coworkers, who reported no observable conversion from lactone to carboxylate structures in CPT-loaded poly(ω-pentadecalactone-co-butylene-co-succinate) (PPBS) nanoparticles over a 24 h period [56], our study demonstrates a significant increase in the lactone form of CPT over a duration of 450 h. This increase is particularly pronounced at 6.50 ± 0.05, where all analyzed nanosystems exhibited a lactone form content exceeding 70%.
Based on these findings it can be concluded that the developed nanoparticulate systems enabled the stabilization of the pharmacologically active form of CPT and provided its protection against hydrolysis, even under physiological conditions.
In the next step, the data collected from the CPT release studies were analyzed using zero-order, first-order, Higuchi, and Korsmeyer-Peppas models to assess the kinetics and mechanism of CPT release from the developed nanosystems (Table 4).
The release kinetics of CPT at a pH of 7.4 ± 0.05 from the nanosystem NP3 conformed to a near-first-order model, as indicated by an R² value of 0.740. Additionally, drug release at a pH of 6.5 ± 0.05 from the samples NP1, NP2, and NP3 exhibited near-first-order kinetics, with R² values of 0.717, 0.779, and 0.877, respectively. It is also likely that CPT was released from the nanosystems NP1, NP2, and NP4 at pH 7.4 ± 0.05, as well as from NP4 at pH 6.5 ± 0.05, following first-order kinetics; however, this conclusion is made with caution due to the relatively low R² values (R² < 0.7). Analysis of the CPT release data using the Higuchi model indicated that CPT was released from the sample NP3 at pH 7.4 ± 0.05 and from all nanosystems at pH 6.5 ± 0.05 via a diffusion mechanism, as evidenced by R² values exceeding 0.7. Furthermore, the analysis of CPT release data from all nanosystems using the Korsmeyer-Peppas model suggested that the transport mechanism was either Fickian (n = 0.23 - 0.32, R² > 0.7) or non-Fickian (n = 0.45 - 0.62, R² > 0.9).
3.4. Molecular Modeling of the Nanosystems Composed of PAMAM Dendrimer/CPT Complex and Biodegradable Polymer
3.4.1. MODELS Preparation
Molecular simulations of dendrimer-drug complexes, while inherently challenging, provide valuable insights into the conformational flexibility of dendrimers and the atomic-level interactions that occur during the association of dendrimers with drugs. In our prior research, which detailed the molecular modeling of the PAMAM dendrimer/CPT complex [34], we established that the most stable configuration for the PAMAM dendrimer of generation 4.0 corresponds to a molar ratio of 1:3 for PAMAM dendrimer to CPT. This finding aligns with experimental data.
In our current study, we have enhanced our model by incorporating the specific environments for each of the four types of prepared nanosystems. This development led to the creation of MODELS 1-4, which are detailed in the Materials and Methods section. Each of these models consists of a PAMAM dendrimer/CPT complex, featuring three CPT molecules for every PAMAM dendrimer of generation 4.0, all set within a polymer matrix that aligns with the experimental composition. It's important to highlight that, given the size of the experimentally obtained systems, which are several hundred nanometers, we conducted our calculations under periodic boundary conditions. This approach allows for a more accurate representation of the intramolecular forces that influence the structure and behavior of the system. The structures of MODELS 1-4 can be found in Figure 11, Figure 12, Figure 13 and Figure 14.
The structures presented exhibit similarities in the shape and size of the PAMAM dendrimer/CPT complex within the nanosystems, with the exception of MODEL 3. In this particular one, the PAMAM dendrimer structure is notably more branched and less compact, allowing the PGACL chains to easily interweave with the PAMAM dendrimer chains. This suggests that the interactions between the matrix components, PGACL and PAMAM dendrimer, are stronger in this nanosystem, resulting in reduced surface tension and enhanced miscibility. Furthermore, these interactions likely contribute to the aggregation observed in some nanosystems, as confirmed by the DLS graph analysis.
The structures of the MODELS align with the findings from the in vitro CPT release evaluation. MODELS 1 and 2, which feature a more compact arrangement of the PAMAM dendrimer/CPT components, exhibit a notably slower release of CPT in vitro. In contrast, MODELS 3 and 4 have a less dense structure, with copolymer molecules integrated even within the core of the PAMAM dendrimer. This structural difference accounts for the more efficient drug release observed in these two models, particularly at a pH of 7.4 ± 0.05. These insights suggest that the design of the optimized system may play a crucial role in determining drug release characteristics, especially when comparing multiple systems.
3.4.2. Molecular Dynamics (MD) Simulations
Based on the MD simulation results for the MODELS 1-4, chosen geometric features of the complexes were determined, including the mean gyration radius (Rg), solvent-accessible surface area (SASA), the radial distribution functions G(r), and root-mean-square deviation of atomic positions (RMSD).
Gyration radius (Rg) represents a measure of how much the polymer chains curl and their average dimensions. Opitz and Wagner used molecular dynamics simulations to study the molecular structure of PAMAM dendrimers and discovered that the gyration radius determined by the experiment was in good agreement with the simulation outcomes [57]. Also, in our previous work, we have successfully determined the Rg values using this computational approach [34]. The experimentally determined Rg of PAMAM dendrimer generation 4.0 at neutral pH was found to be 22.5 Å [58], while in this study the Rg for MODELS 1-4 PAMAM dendrimer/CPT complex were 20.4 Å, 21.0 Å; 24.5 Å, and 23.6 Å, respectively. The decrease of the Rg upon complexation, when compared with the free PAMAM dendrimer molecule, was observed in MODELS 1 and 2. This can be explained as resulting from the intermolecular forces (van der Waals, hydrogen bonds) existing between the host and guest (CPT) molecules that are located in the cavity of PAMAM dendrimer. The expansion of PAMAM dendrimer dimensions observed in MODELS 4 and especially MODEL 3, results from the higher affinity of PAMAM dendrimer molecules towards the polymer matrix particles simulated in that model. Therefore, instead of existing in their spherical shape, the molecules of PAMAM dendrimer tend to increase their specific surface. It also explains the different behavior of nanosystems, observed in the dissolution studies. To confirm this observation, we have calculated the SASA for the nanosystems in each model.
To obtain the SASA, a probe radius of 1.4 Å was used as the van der Waals radius of a water molecule of 2.75 Å. The obtained results were 13960.65, 14969.37, 17326.98 Å2, and 16414.81 Å2 for the MODELS 1-4, respectively. These results are comparable to those obtained for PAMAM at pH=7.0, namely 14862.4 Å2 [59]. Again, similar as for the Rg, the increase of SASA has been observed for MODELS 3 and 4, which results from the more branched shape that the PAMAM dendrimer forms in these systems.
The radial distribution function (RDF) represents the features of the microstructure. This function is frequently employed to explore the structure and unique interactions of the condensed matter [60]. Its physical meaning relates to the ratio of the probability density of another atom at a distance from the center atom to the random distribution density. In the g(r)-r diagram (Figure 15), the chemical bonds and hydrogen bond interactions distances are mostly within 3.1 Å, and the non-bonding interactions such as Coulomb forces and van der Waals forces are primarily within 3.1-10.0 Å. The interactions greater than 10.0 Å are very weak. The radial distribution function, g(r), was calculated by averaging the distribution of interatomic vector lengths on a sphere of radius (r) and thickness (Δr). We used Δr=0.1 Å with the radius, r, of the sphere varying in the range 1–20 Å. The interactions between PAMAM dendrimer and CPT molecules can be analyzed by means of RDFs that were created for each model (Figure 15). The highest peak exists slightly after 1.0 Å, indicating the presence of covalent bonds. Other peaks are visible in the region of 2.0 to 3.5 Å, which means that the H-bonds are formed between PAMAM dendrimer and CPT molecules. As there aren’t any peaks in the region above 5.0 Å, this indicates no major electrostatic interactions. Also, the RDFs were found to be similar, regardless of the studied model.
Figure 16 shows the root-mean-square deviation of atomic positions (RMSD) plots obtained for CPT in the studied models during the 100 ns MD simulations. RMSD analysis can indicate the dynamic stability of a molecule in a studied system, larger values of RMDS indicate more intense molecular motions and more efficient diffusion. All the models studied showed an increase in RMSD throughout the simulation time. However, the values reached after 100 ns depend on the model studied. The highest final RMSD was observed in MODEL 3, and the lowest in MODEL 1, 2.5 Å and 0.2 Å, respectively. This indicates that the polymer matrix has a major influence on the behavior of CPT in the nanosystem, which was also observed experimentally.
4. Conclusions
The development of DDSs has been influential in transforming innovative therapies into effective medical interventions. As the therapeutic environment has progressed, these systems and their underlying technologies have quickly evolved to accommodate the changing requirements of drug delivery. The physicochemical attributes of small molecules largely determine their delivery mechanisms, which in turn influence their bioavailability. Therefore, drug delivery strategies prioritize not only the improvement of solubility, but also the management of release profiles, the augmentation of therapeutic effects, and the customization of pharmacokinetic behaviors.
In line with this focus, our research concentrated on the development of forward-looking PAMAM dendrimer/biodegradable polymer nanosystems intended for the delivery of CPT. Despite the fact that this drug has been recognized as one of the most promising anticancer agents in recent years, challenges related to its physiological stability and solubility remain unresolved. Initially, four biodegradable homo- and copolymeric matrices were synthesized and extensively characterized, focusing on the microstructural aspects of their chains. The biological assessment conducted on these biodegradable materials, specifically hemolysis tests, revealed that all matrices exhibited hemolysis rates below 5%, thereby conforming to national safety standards for biological materials. Furthermore, fibroblasts exposed to the synthesized polymeric matrices demonstrated viability that was comparable and dependent on concentration. In the subsequent phase of our research, four nanosystems with diameters ranging from 110 to 406 nm and an entrapment efficiency of up to 27% were developed. These nanoparticulate systems were formulated from a PAMAM dendrimer/CPT complex, which was subsequently coated with an appropriate biodegradable matrix. Notably, confocal microscopy confirmed the successful entrapment of CPT within the PAMAM dendrimer cavity, as evidenced by the detection of fluorescence from the drug in all nanoparticulate systems. The research demonstrated that CPT was released from the engineered nanosystems in a sustained and controlled manner, exhibiting near-first-order and first-order kinetics without any burst release effect. Additionally, the biodegradable polymer matrices played a significant role in shaping the drug release profile. Notably, the nanoparticulate systems we have developed facilitated the stabilization of the pharmacologically active form of CPT, offering protection against hydrolysis even under physiological conditions. Through molecular modeling techniques, we constructed four MODELS, each comprising a PAMAM dendrimer/CPT complex with three CPT molecules associated with each generation 4.0 PAMAM dendrimer, all integrated within a polymer matrix that corresponds to the experimental setup. Our analysis revealed that the structures of the MODELS displayed similarities in the shape and size of the PAMAM dendrimer/CPT complex within the nanosystems, with MODEL 3 being an exception. In this particular model, the PAMAM dendrimer structure was significantly more branched and less compact, which facilitates the interweaving of PGACL matrix chains with the PAMAM dendrimer chains. This indicates that the interactions between the matrix components, PGACL and PAMAM dendrimer, were more robust in this nanosystem, leading to decreased surface tension and improved miscibility. The structural characteristics of the MODELS were consistent with the results obtained from the in vitro evaluation of CPT release.
In summary, we can assert that the nanoparticulate systems developed may serve as a novel platform for the delivery of significant hydrophobic anticancer drugs. Consequently, these nanomaterials hold great promise as delivery systems for cancer therapy, and further investigation of the in vivo behavior of these nanoparticulate materials is warranted in animal models.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: 1H NMR spectrum of the synthesized PAMAM dendrimer/CPT complex, Figure S2: 1H NMR spectrum of the PLLA (matrix M1), Figure S3: 13C NMR spectrum of the PLLA (matrix M1), Figure S4: 1H NMR spectrum of the PLACL 40:60 (matrix M2), Figure S5: 13C NMR spectrum of the PLACL 40:60 (matrix M2), Figure S6: 1H NMR spectrum of the PGACL (matrices M3 and M4), Figure S7: 13C NMR spectrum of the PGACL (matrices M3 and M4), Figure S8: Expansion of the domain of interest in 13C NMR spectrum of PLLA (matrix M1), Figure S9: Hemolytic activity of individual matrices. The graph depicts the level of hemolysis of RBC treated with increasing concentrations of the complex after 1 h of treatment. Statistical analysis using two-way ANOVA followed by the Bonferroni post-test revealed no significant differences (P > 0.05) between the obtained results, Figure S10: The viability of normal fibroblasts treated for 72 h with individual matrices at pH 7.4±0.05 and 6.5±0.05. The graphs depict differences in the susceptibility of cells to the complex. The MTS assay was used to determine the relative cell number. The results are given as mean ± SEM. Two-way ANOVA was used for statistical analysis, followed by Bonferroni post-tests. When the following conditions were met, the results were considered statistically significant: *P<0.05; **P<0.01; ***P<0.005, Figure S11: HPLC chromatograms of the CPT released in the lactone and carboxylic forms (a) pH = 7.4 ± 0.05; (b) pH = 6.5 ± 0.05.
Author Contributions
K.S.: methodology investigation, chemical research, formal analysis, and writing—original draft and editing; A.K.: analytical research - HPLC method; T.B.: analytical research - GPC method; A.B-J.: analytical research - DLS method; A.L.: investigation, methodology, formal analysis - cell viability assay and hemolysis, and writing—original draft and editing; Ł.S.: investigation, formal analysis—molecular modeling, visualization, and writing—original draft and editing; M.G.: analytical research – confocal microscopy; M.S.: conceptualization, mathematical models investigation, formal analysis, and writing—original draft and editing; E.O.: conceptualization, supervision, methodology investigation, chemical research, formal analysis, and writing—original draft and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors would like to thank Dr. Urszula Piotrowska for her suggestions on the preparation of the nanosystems.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mazayen, Z.M.; Ghoneim, A.M.; Elbatanony, R.S.; Basalious, E.B.; Bendas, E.R. Pharmaceutical Nanotechnology: From the Bench to the Market. Future J. Pharm. Sci. 2022, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- The Emergence of Nanopharmacy: From Biology to Nanotechnology and Drug Molecules to Nanodrugs. In Pharmaceutical Nanotechnology: Innovation and Production; John Wiley & Sons, Ltd, 2017; pp. 43–62 ISBN 978-3-527-80068-1.
- Weissig, V.; Pettinger, T.; Murdock, N. Nanopharmaceuticals (Part 1): Products on the Market. Int. J. Nanomedicine 2014, 4357. [Google Scholar] [CrossRef] [PubMed]
- Afzal, O.; Altamimi, A.S.A.; Nadeem, M.S.; Alzarea, S.I.; Almalki, W.H.; Tariq, A.; Mubeen, B.; Murtaza, B.N.; Iftikhar, S.; Riaz, N.; et al. Nanoparticles in Drug Delivery: From History to Therapeutic Applications. Nanomaterials 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Haag, R.; Kratz, F. Polymer Therapeutics: Concepts and Applications. Angew. Chem. Int. Ed. 2006, 45, 1198–1215. [Google Scholar] [CrossRef]
- Silverstein, A.M. Paul Ehrlich’s Passion: The Origins of His Receptor Immunology. Cell. Immunol. 1999, 194, 213–221. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Drug Delivery Systems: Entering the Mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef]
- Niemirowicz, K.; Car, H. Nanonośniki jako nowoczesne transportery w kontrolowanym dostarczaniu leków. Chemik 2012, 66, 868–881. [Google Scholar]
- Mukherjee, C.; Varghese, D.; Krishna, J.S.; Boominathan, T.; Rakeshkumar, R.; Dineshkumar, S.; Rao, C.V.S.B.; Sivaramakrishna, A. Recent Advances in Biodegradable Polymers – Properties, Applications and Future Prospects. Eur. Polym. J. 2023, 192, 112068. [Google Scholar] [CrossRef]
- Satchanska, G.; Davidova, S.; Petrov, P.D. Natural and Synthetic Polymers for Biomedical and Environmental Applications. Polymers 2024, 16. [Google Scholar] [CrossRef]
- Chen, J. Design and Synthesis of Biomedical Polymer Materials. Int. J. Mol. Sci. 2024, 25. [Google Scholar] [CrossRef]
- Deb, P.K.; Kokaz, S.F.; Abed, S.N.; Paradkar, A.; Tekade, R.K. Chapter 6 - Pharmaceutical and Biomedical Applications of Polymers. In Basic Fundamentals of Drug Delivery; Tekade, R.K., Ed.; Advances in Pharmaceutical Product Development and Research; Academic Press, 2019; pp. 203–267 ISBN 978-0-12-817909-3.
- Morsada, Z.; Hossain, M.M.; Islam, M.T.; Mobin, M.A.; Saha, S. Recent Progress in Biodegradable and Bioresorbable Materials: From Passive Implants to Active Electronics. Appl. Mater. Today 2021, 25, 101257. [Google Scholar] [CrossRef]
- Nikzamir, M.; Hanifehpour, Y.; Akbarzadeh, A.; Panahi, Y. Applications of Dendrimers in Nanomedicine and Drug Delivery: A Review. J. Inorg. Organomet. Polym. Mater. 2021, 31, 2246–2261. [Google Scholar] [CrossRef]
- Abdou, E.M. PAMAM DENDRIMERS AS DRUG DELIVERY CARRIERS , BENEFITS AGAINST RISKS.; 2017.
- Cheng, Y.; Li, M.; Xu, T. Potential of Poly(Amidoamine) Dendrimers as Drug Carriers of Camptothecin Based on Encapsulation Studies. Eur. J. Med. Chem. 2008, 43, 1791–1795. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, S.; Das, M.K. Dendrimers and Their Applications as Novel Drug Delivery Carriers.; 2013.
- Huang, D.; Wu, D. Biodegradable Dendrimers for Drug Delivery. Mater. Sci. Eng. C 2018, 90, 713–727. [Google Scholar] [CrossRef]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA. Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Wojciechowska, U.; Barańska, K.; Miklewska, M.; Didkowska, J.A. Cancer Incidence and Mortality in Poland in 2020. Nowotw. J. Oncol. 2023, 73, 129–145. [Google Scholar] [CrossRef]
- Cegieła, U.; Janiec, W.; Folwarczna, J.; Janas, A.; Janiec, R.; Kaczmarczyk-Sedlak, I.; Londzin, P.; Nowińska, B.; Podwińska, E.; Pytlik, M.; et al. Kompendium Farmakologii; PZWL, 2021; ISBN 978-83-200-6052-2.
- Strąg-Lemanowicz, A.; Leppert, W. Rola Onkologicznego Leczenia Systemowego u Pacjentów z Zaawansowaną Chorobą Nowotworową. Palliat. Med. Pract. 2014, 8, 11–22. [Google Scholar]
- Ramadori, G.; Cameron, S. Effects of Systemic Chemotherapy on the Liver. Ann. Hepatol. 2010, 9, 133–143. [Google Scholar] [CrossRef]
- Strzelecka, K.; Piotrowska, U.; Sobczak, M.; Oledzka, E. The Advancement of Biodegradable Polyesters as Delivery Systems for Camptothecin and Its Analogues—A Status Report. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Ziomkowska, B.; Kruszewski, S.; Siuda, R.; Cyrankiewicz, M. Deactivation Rate of Camptothecin Determined by Factor Analysis of Steady-State Fluorescence and Absorption Spectra. Opt. Appl. 2006, 36, 137–146. [Google Scholar]
- Pommier, Y. Topoisomerase I Inhibitors: Camptothecins and Beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Botella, P.; Rivero-Buceta, E.M. Safe Approaches for Camptothecin Delivery: Structural Analogues and Nanomedicines. J. Controlled Release 2017, 247, 28–54. [Google Scholar] [CrossRef] [PubMed]
- Alshammari, M.K.; Alshehri, M.M.; Alshehri, A.M.; Alshlali, O.M.; Mahzari, A.M.; Almalki, H.H.; Kulaybi, O.Y.; Alghazwni, M.K.; Kamal, M.; Imran, M. Camptothecin Loaded Nano-Delivery Systems in the Cancer Therapeutic Domains: A Critical Examination of the Literature. J. Drug Deliv. Sci. Technol. 2023, 79, 104034. [Google Scholar] [CrossRef]
- Min, K.H.; Park, K.; Kim, Y.-S.; Bae, S.M.; Lee, S.; Jo, H.G.; Park, R.-W.; Kim, I.-S.; Jeong, S.Y.; Kim, K.; et al. Hydrophobically Modified Glycol Chitosan Nanoparticles-Encapsulated Camptothecin Enhance the Drug Stability and Tumor Targeting in Cancer Therapy. J. Controlled Release 2008, 127, 208–218. [Google Scholar] [CrossRef]
- Watanabe, M.; Kawano, K.; Toma, K.; Hattori, Y.; Maitani, Y. In Vivo Antitumor Activity of Camptothecin Incorporated in Liposomes Formulated with an Artificial Lipid and Human Serum Albumin. J. Controlled Release 2008, 127, 231–238. [Google Scholar] [CrossRef]
- Kawano, K.; Watanabe, M.; Yamamoto, T.; Yokoyama, M.; Opanasopit, P.; Okano, T.; Maitani, Y. Enhanced Antitumor Effect of Camptothecin Loaded in Long-Circulating Polymeric Micelles. J. Controlled Release 2006, 112, 329–332. [Google Scholar] [CrossRef]
- Martins, S.M.; Sarmento, B.; Nunes, C.; Lúcio, M.; Reis, S.; Ferreira, D.C. Brain Targeting Effect of Camptothecin-Loaded Solid Lipid Nanoparticles in Rat after Intravenous Administration. Eur. J. Pharm. Biopharm. 2013, 85, 488–502. [Google Scholar] [CrossRef]
- Kubiak, M. Dendrymery – fascynujące nanocząsteczki w zastosowaniu w medycynie. Chemik 2014, 68, 141–150. [Google Scholar]
- Oledzka, E.; Paśnik, K.; Domańska, I.; Zielińska-Pisklak, M.; Piotrowska, U.; Sobczak, M.; Szeleszczuk, Ł.; Laskowska, A. Poly(Amidoamine) Dendrimer/Camptothecin Complex: From Synthesis to In Vitro Cancer Cell Line Studies. Molecules 2023, 28, 2696–2717. [Google Scholar] [CrossRef]
- Bero, M.; Kasperczyk, J.; Jedlinski, Z.J. Coordination Polymerization of Lactides, 1. Structure Determination of Obtained Polymers. Makromol. Chem. 1990, 191, 2287–2296. [Google Scholar] [CrossRef]
- Kasperczyk, J.; Bero, M. Coordination Polymerization of Lactides, 2.Microstructure Determination of Poly[(L,L-Lactide)-Co-(ε-Caprolactone)] with 13C Nuclear Magnetic Resonance Spectroscopy. Makromol. Chem. 1991, 192, 1777–1787. [Google Scholar] [CrossRef]
- Bero, M.; Czapla, B.; Dobrzyński, P.; Janeczek, H.; Kasperczyk, J. Copolymerization of Glycolide Andε-Caprolactone, 2. Random Copolymerization in the Presence of Tin Octoate. Macromol. Chem. Phys. 1999, 200, 911–916. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.; Nath, L.; Chowdhury, P. Kinetic Modeling on Drug Release from Controlled Drug Delivery Systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar] [PubMed]
- Singhvi, G.; Singh, M. Review: In Vitro Drug Release Characterization Models. Int. J. Pharm. Stud. Res. 2011, 2, 77–84. [Google Scholar]
- Bialik, M.; Proc, J.; Zgadzaj, A.; Mulas, K.; Kuras, M.; Sobczak, M.; Oledzka, E. Development and Comprehensive Characteristics of Thermosensitive Liquid Suppositories of Metoprolol Based on Poly(Lactide-Co-Glycolide) Nanoparticles. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Bialik, M.; Kurkowski, P.; Strzelecka, K.; Kuras, M.; Sobczak, M.; Mulas, K.; Zgadzaj, A.; Czerwińska, M.E.; Gniadek, M.; Oledzka, E. Dual-Controlled Delivery of Furosemide and Atenolol Using a Biodegradable Nanosystem for Antihypertensive Therapy. J. Drug Deliv. Sci. Technol. 2023, 89, 105006. [Google Scholar] [CrossRef]
- Sæbø, I.P.; Bjørås, M.; Franzyk, H.; Helgesen, E.; Booth, J.A. Optimization of the Hemolysis Assay for the Assessment of Cytotoxicity. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Otto, D.P.; Villiers, M.M. de All-atomistic Molecular Dynamics (AA-MD) Studies and Pharmacokinetic Performance of PAMAM-dendrimer-furosemide Delivery Systems. Int. J. Pharm. 2018, 547, 545–555. [Google Scholar] [CrossRef]
- Sun, H.; Jin, Z.; Yang, C.; Akkermans, R.L.C.; Robertson, S.H.; Spenley, N.A.; Miller, S.; Todd, S.M. COMPASS II: Extended Coverage for Polymer and Drug-like Molecule Databases. J. Mol. Model. 2016, 22, 1–10. [Google Scholar] [CrossRef]
- Wang, L.; Lv, C.; Liu, Z.; Zhang, N.; Zhou, W.; Wang, J. Study on the Mechanism of PAMAM(DETA as the Core) against Silica Scale. J. Mol. Model. 2021, 27, 304. [Google Scholar] [CrossRef]
- Al Ameri, J.; Alsuraifi, A.; Curtis, A.; Hoskins, C. Effect of Poly(Allylamine) Molecular Weight on Drug Loading and Release Abilities of Nano-Aggregates for Potential in Cancer Nanomedicine. J. Pharm. Sci. 2020, 109, 3125–3133. [Google Scholar] [CrossRef] [PubMed]
- Domańska, I.M.; Zgadzaj, A.; Kowalczyk, S.; Zalewska, A.; Oledzka, E.; Cieśla, K.; Plichta, A.; Sobczak, M. A Comprehensive Investigation of the Structural, Thermal, and Biological Properties of Fully Randomized Biomedical Polyesters Synthesized with a Nontoxic Bismuth(III) Catalyst. Molecules 2022, 27, 1139. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; McNeil, S.E. Understanding the Correlation between in Vitro and in Vivo Immunotoxicity Tests for Nanomedicines. J. Controlled Release 2013, 172, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Lu Senlin; Duffin Rodger; Poland Craig; Daly Paul; Murphy Fiona; Drost Ellen; MacNee William; Stone Vicki; Donaldson Ken Efficacy of Simple Short-Term in Vitro Assays for Predicting the Potential of Metal Oxide Nanoparticles to Cause Pulmonary Inflammation. Environ. Health Perspect. 2009, 117, 241–247. [CrossRef]
- Jeong, H.; Hwang, J.; Lee, H.; Hammond, P.T.; Choi, J.; Hong, J. In Vitro Blood Cell Viability Profiling of Polymers Used in Molecular Assembly. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.; Fan, J.; Claudel, M.; Sonntag, T.; Didier, P.; Ronzani, C.; Lebeau, L.; Pons, F. Density of Surface Charge Is a More Predictive Factor of the Toxicity of Cationic Carbon Nanoparticles than Zeta Potential. J. Nanobiotechnology 2021, 19, 5. [Google Scholar] [CrossRef]
- Li, F.; Jiang, T.; Li, Q.; Ling, X. Camptothecin (CPT) and Its Derivatives Are Known to Target Topoisomerase I (Top1) as Their Mechanism of Action: Did We Miss Something in CPT Analogue Molecular Targets for Treating Human Disease Such as Cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar]
- Díaz-Celorio, E.; Franco, L.; Rodríguez-Galán, A.; Puiggalí, J. Study on the Hydrolytic Degradation of Glycolide/Trimethylene Carbonate Copolymers Having Different Microstructure and Composition. Polym. Degrad. Stab. 2013, 98, 133–143. [Google Scholar] [CrossRef]
- Memiṣoǧlu, E.; Bochot, A.; Özalp, M.; Şen, M.; Duchěne, D.; Hincal, A.A. Direct Formation of Nanospheres from Amphiphilic β-Cyclodextrin Inclusion Complexes. Pharm. Res. 2004, 20, 117–125. [Google Scholar] [CrossRef]
- Fassberg, J.; Stella, V.J. A Kinetic and Mechanistic Study of the Hydrolysis of Camptothecin and Some Analogues. J. Pharm. Sci. 1992, 81, 676–684. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, Z.; Zhang, S.; Saltzman, W.M. Poly(ω-Pentadecalactone-Co-Butylene-Co-Succinate) Nanoparticles as Biodegradable Carriers for Camptothecin Delivery. Biomaterials 2009, 30, 5707–5719. [Google Scholar] [CrossRef] [PubMed]
- Opitz, A.W.; Wagner, N.J. Structural Investigations of Poly(Amido Amine) Dendrimers in Methanol Using Molecular Dynamics. J. Polym. Sci. Part B 2006, 44, 3062–3077. [Google Scholar] [CrossRef]
- Pourianazar, N.T.; Mutlu, P.; Gunduz, U. Bioapplications of Poly(Amidoamine) (PAMAM) Dendrimers in Nanomedicine. J. Nanoparticle Res. 2014, 16, 1–38. [Google Scholar] [CrossRef]
- Liu, Y.; Bryantsev, V.S.; Diallo, M.S.; Goddard III, W.A. PAMAM Dendrimers Undergo pH Responsive Conformational Changes without Swelling. J. Am. Chem. Soc. 2009, 131, 2798–2799. [Google Scholar] [CrossRef]
- Canetta, E.; Maino, G. Molecular Dynamic Analysis of the Structure of Dendrimers. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2004, 213, 71–74. [Google Scholar] [CrossRef]
Figure 1.
Expansion of the domain of interest in 13C NMR spectrum of carbonyl region for PLACL copolymer (M2).
Figure 1.
Expansion of the domain of interest in 13C NMR spectrum of carbonyl region for PLACL copolymer (M2).
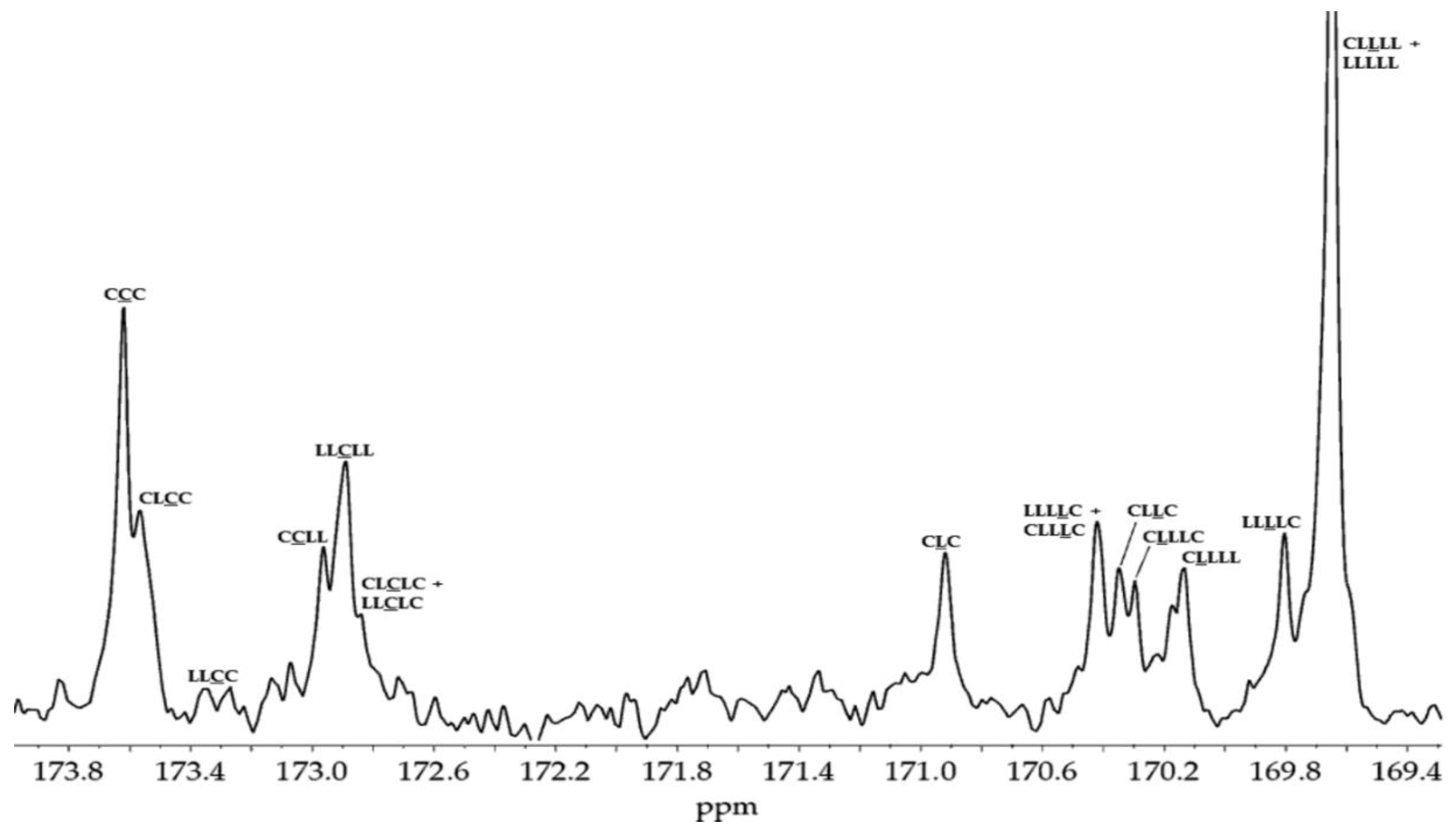
Figure 2.
Expansion of the domain of interest in 1H NMR spectrum of PGACL copolymer (M3 and M4).
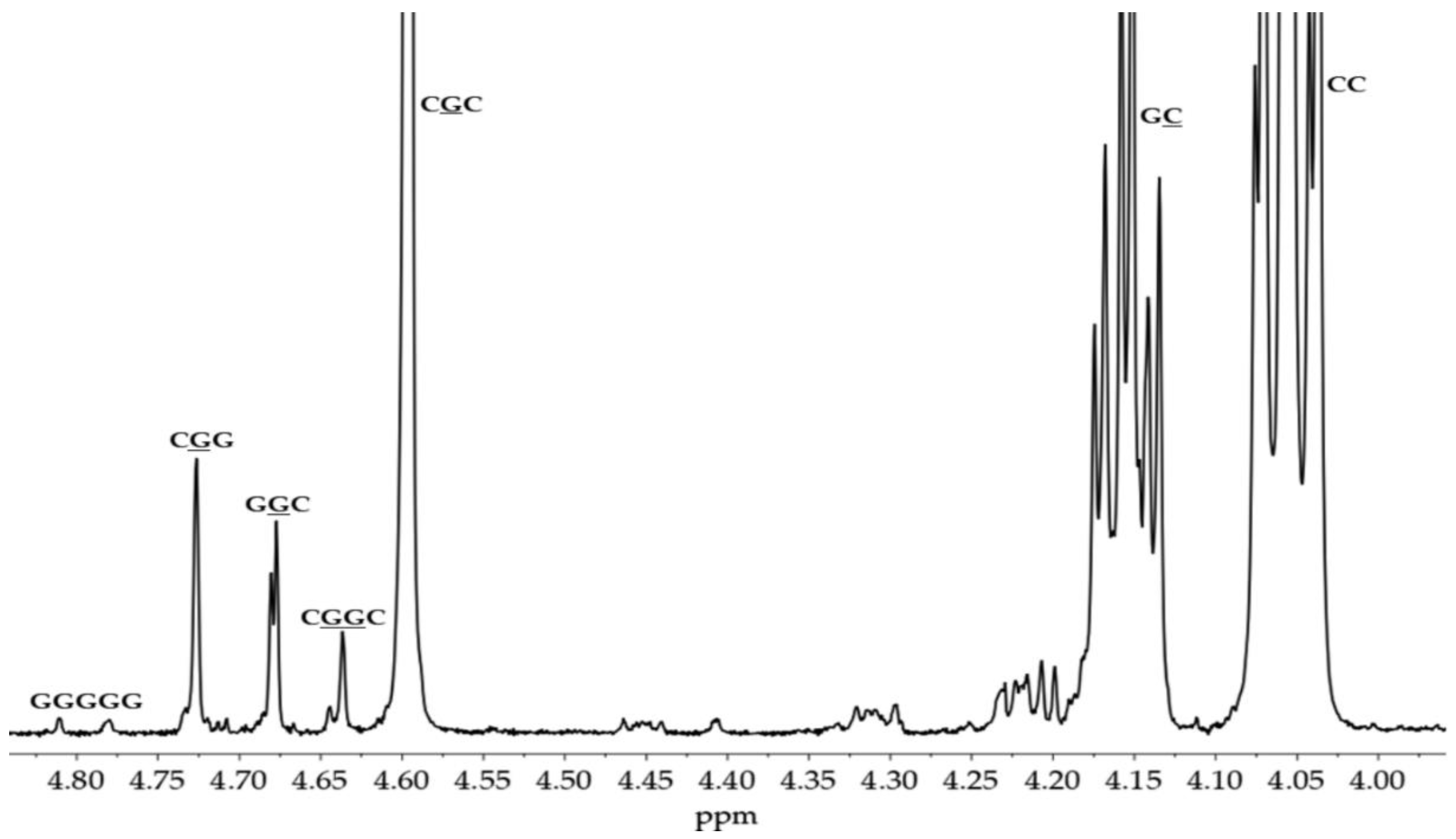
Figure 3.
Hemolytic activity of the synthesized biodegradable polymers: M1, M2, M3 and M4 after 1 h of incubation. The graph depicts the level of hemolysis of RBC treated with increasing concentrations of the complex after 1 of treatment. Statistical analysis using two-way ANOVA followed by the Bonferroni post-test revealed no significant differences (p > 0.05) between the obtained results.
Figure 3.
Hemolytic activity of the synthesized biodegradable polymers: M1, M2, M3 and M4 after 1 h of incubation. The graph depicts the level of hemolysis of RBC treated with increasing concentrations of the complex after 1 of treatment. Statistical analysis using two-way ANOVA followed by the Bonferroni post-test revealed no significant differences (p > 0.05) between the obtained results.
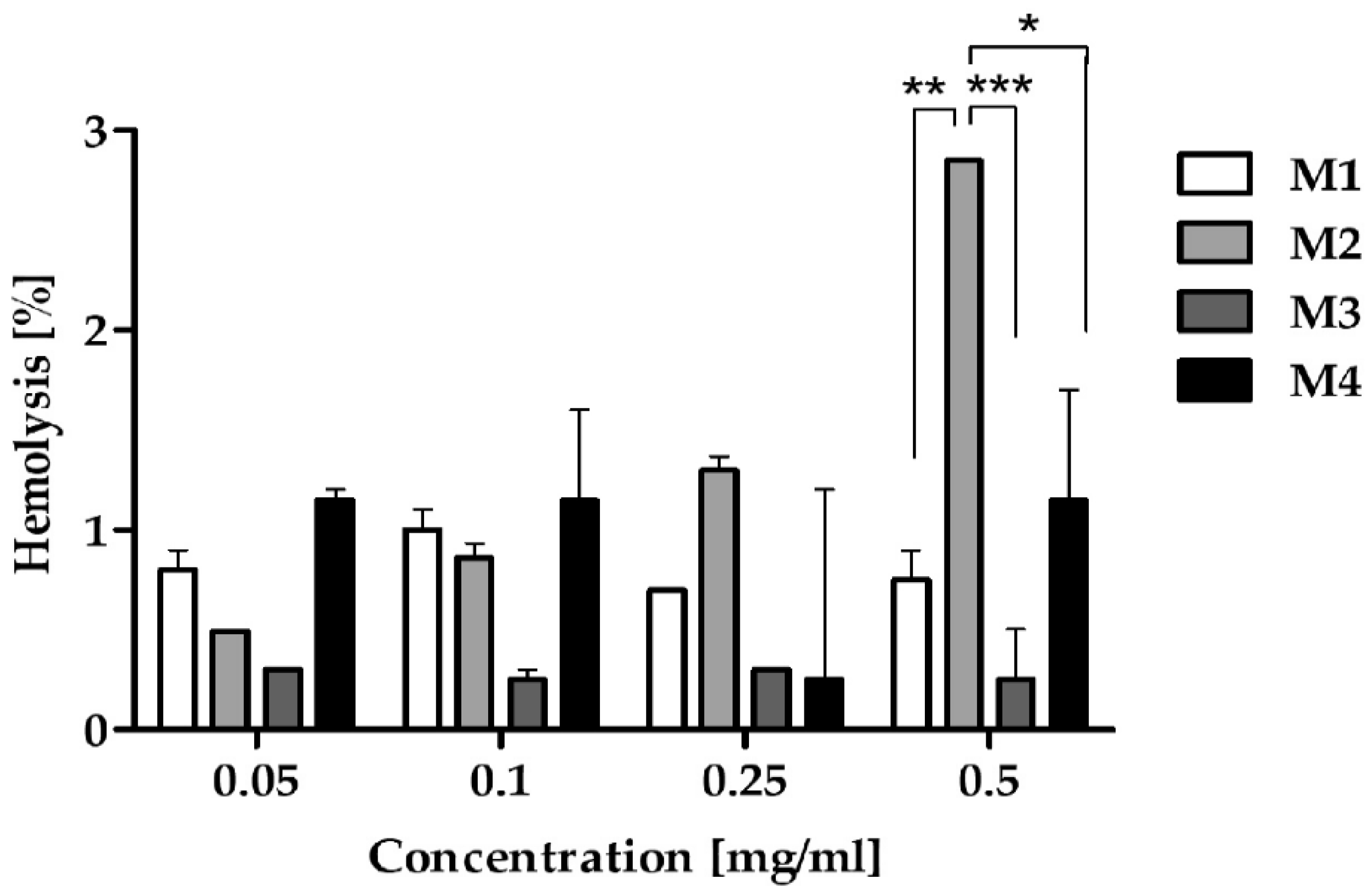
Figure 4.
The viability of normal fibroblasts treated for 72 h with the synthesized biodegradable polymers at pH 7.4 ± 0.05 (a) and pH 6.5 ± 0.05 (b). The graphs depict differences in the susceptibility of cells to the complex. The MTS assay was used to determine the relative cell number. The results are given as mean ± SEM. Two-way ANOVA was used for statistical analysis, followed by Bonferroni post-tests. When the following conditions were met, the results were considered statistically significant: * p < 0.05; ** p < 0.01; *** p < 0.005.
Figure 4.
The viability of normal fibroblasts treated for 72 h with the synthesized biodegradable polymers at pH 7.4 ± 0.05 (a) and pH 6.5 ± 0.05 (b). The graphs depict differences in the susceptibility of cells to the complex. The MTS assay was used to determine the relative cell number. The results are given as mean ± SEM. Two-way ANOVA was used for statistical analysis, followed by Bonferroni post-tests. When the following conditions were met, the results were considered statistically significant: * p < 0.05; ** p < 0.01; *** p < 0.005.
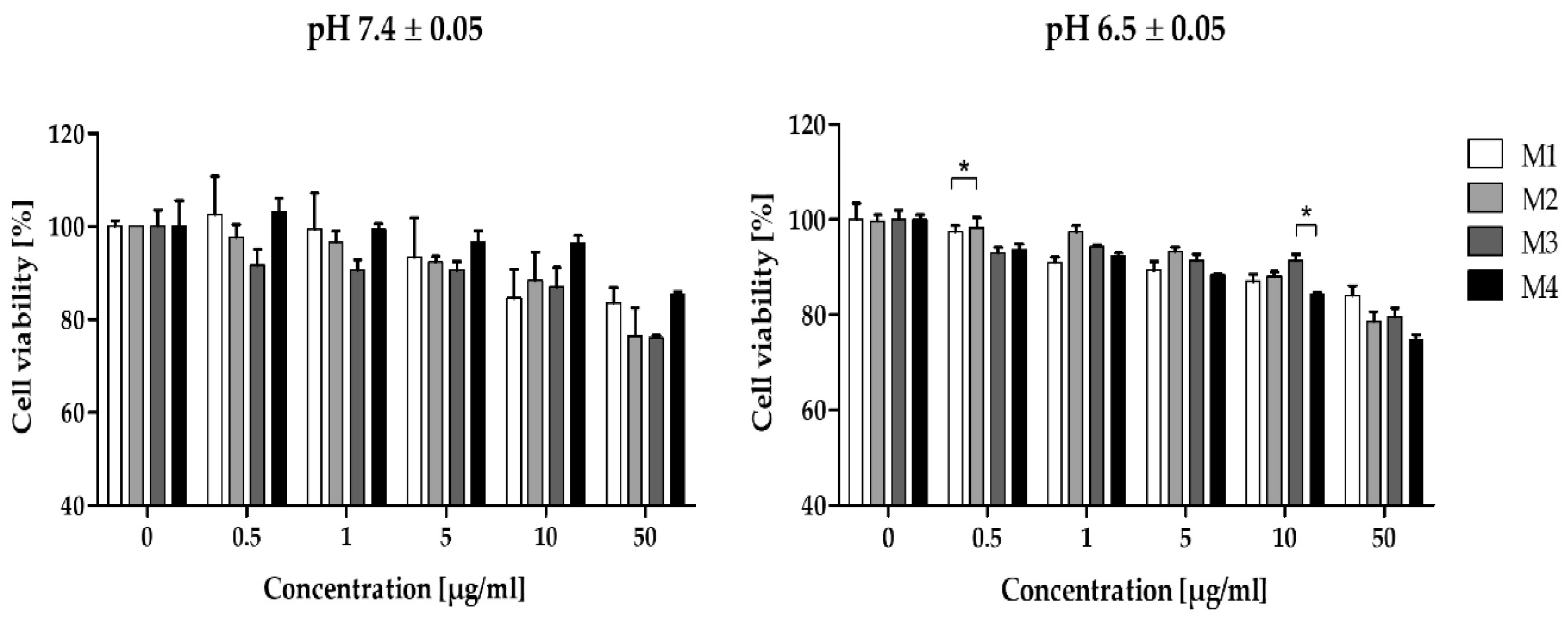
Figure 5.
TEM image of: (a) NP1 sample, (b) NP2 sample, (c) NP3 sample, (d) NP4 sample.
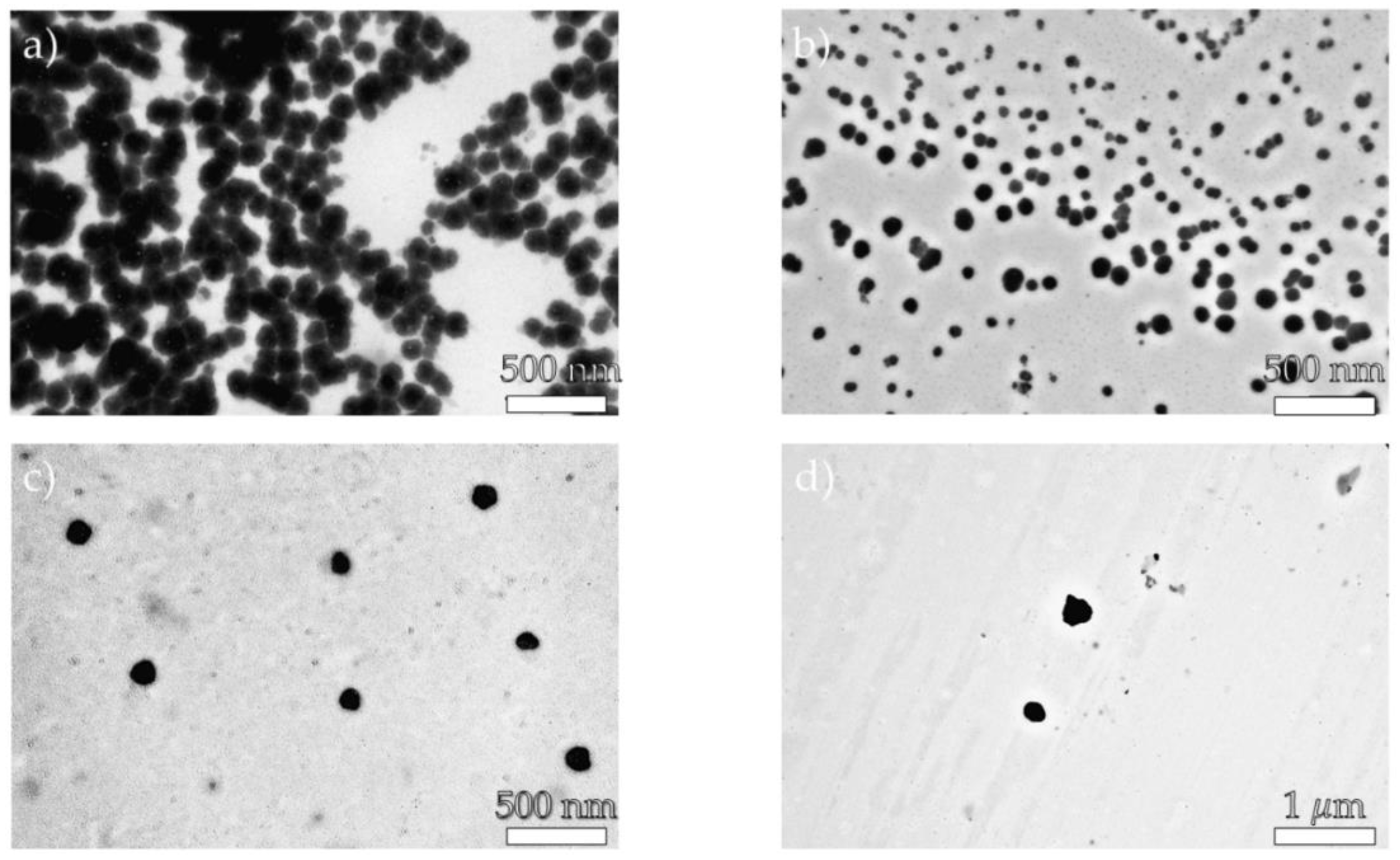
Figure 6.
Confocal fluorescence imaging of: (a) CPT, (b) NP1 sample, (c) NP2 sample, (d) NP3 sample, (e) NP4 sample.
Figure 6.
Confocal fluorescence imaging of: (a) CPT, (b) NP1 sample, (c) NP2 sample, (d) NP3 sample, (e) NP4 sample.

Figure 7.
CPT release profiles from the developed nanosystems at pH 6.50 ± 0.05.
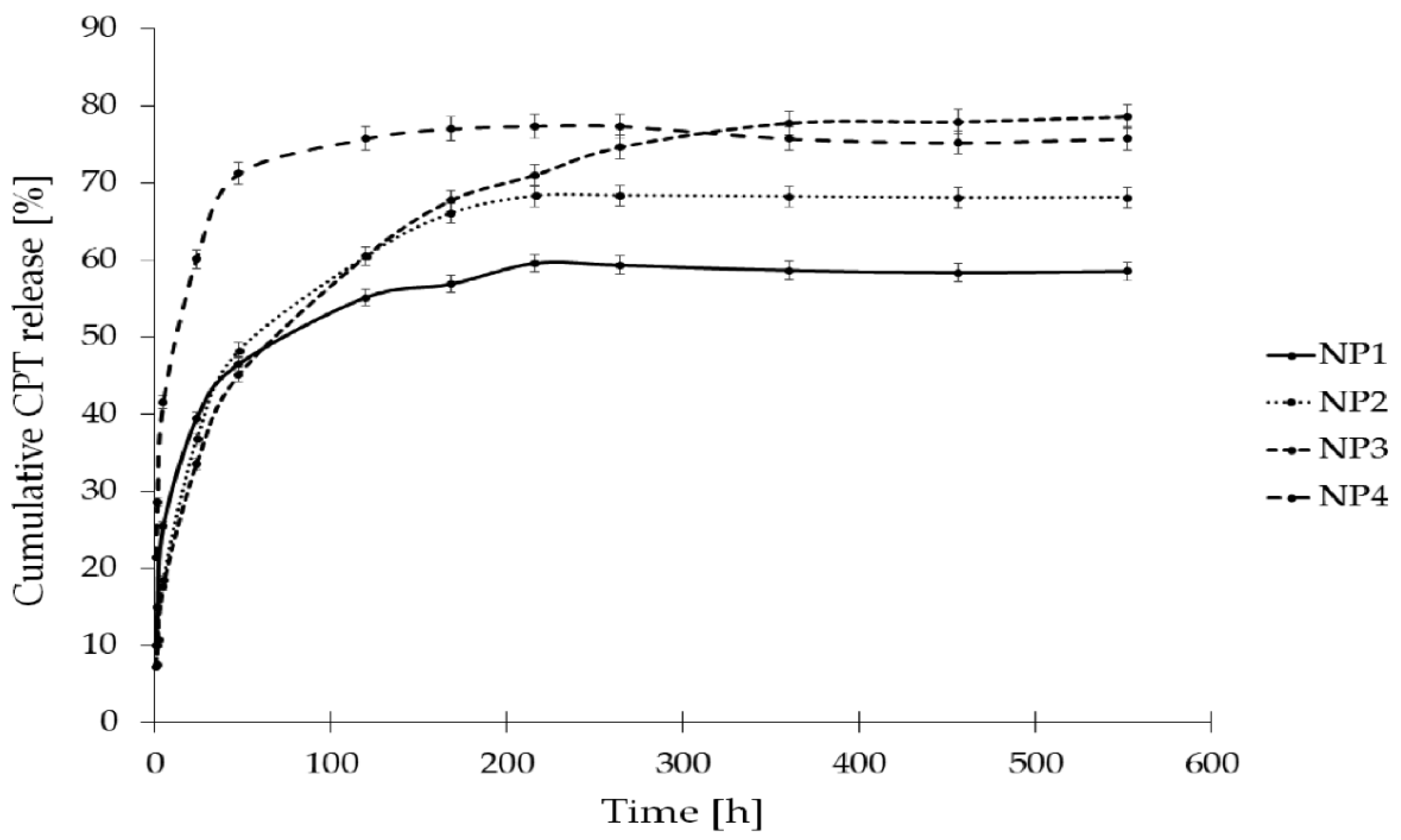
Figure 8.
CPT release profiles from the developed nanosystems at pH 7.40 ± 0.05.
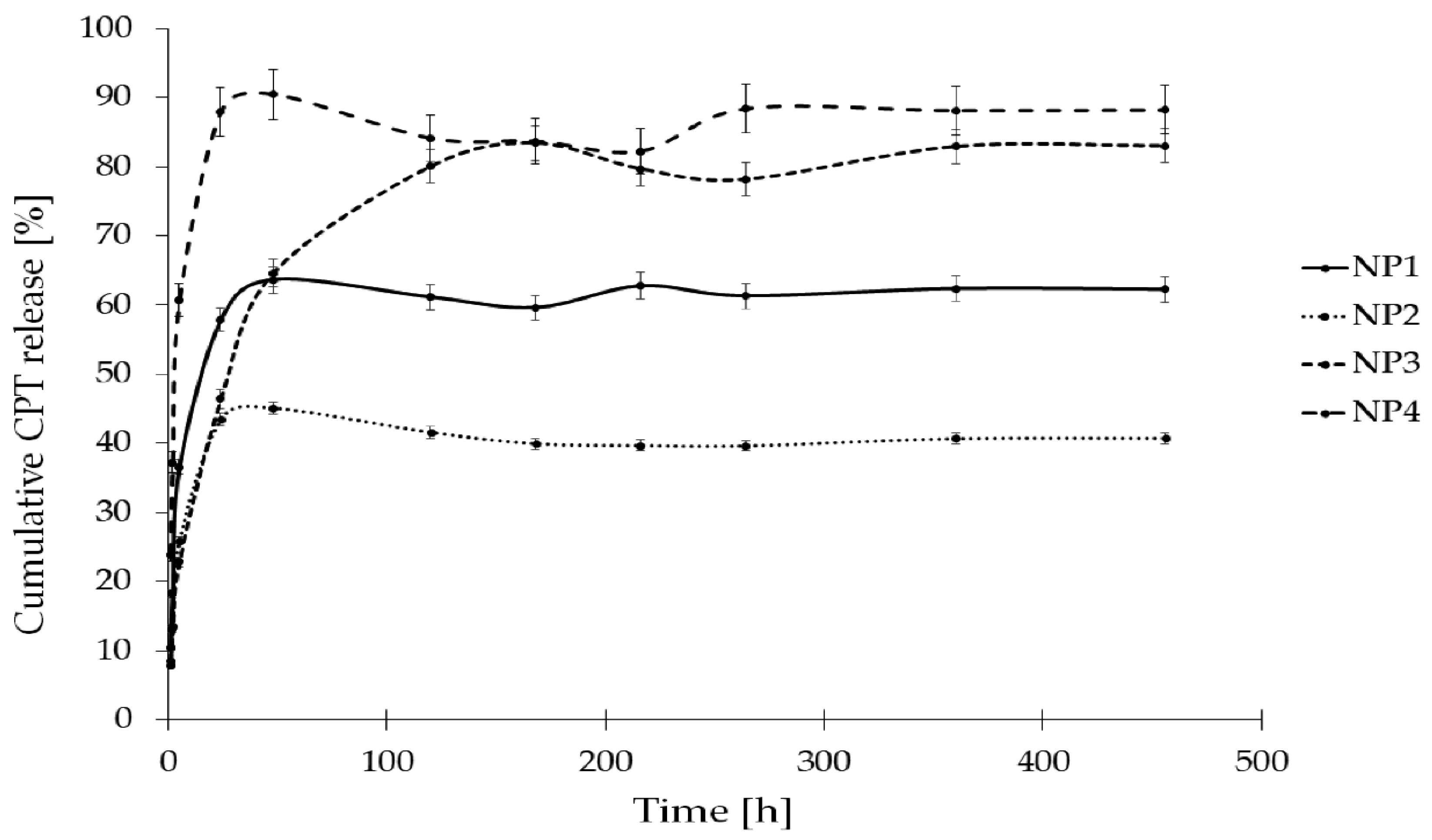
Figure 9.
Correlation between cumulative CPT release and percentage of the lactone form of CPT at 7.40 ± 0.05.
Figure 9.
Correlation between cumulative CPT release and percentage of the lactone form of CPT at 7.40 ± 0.05.
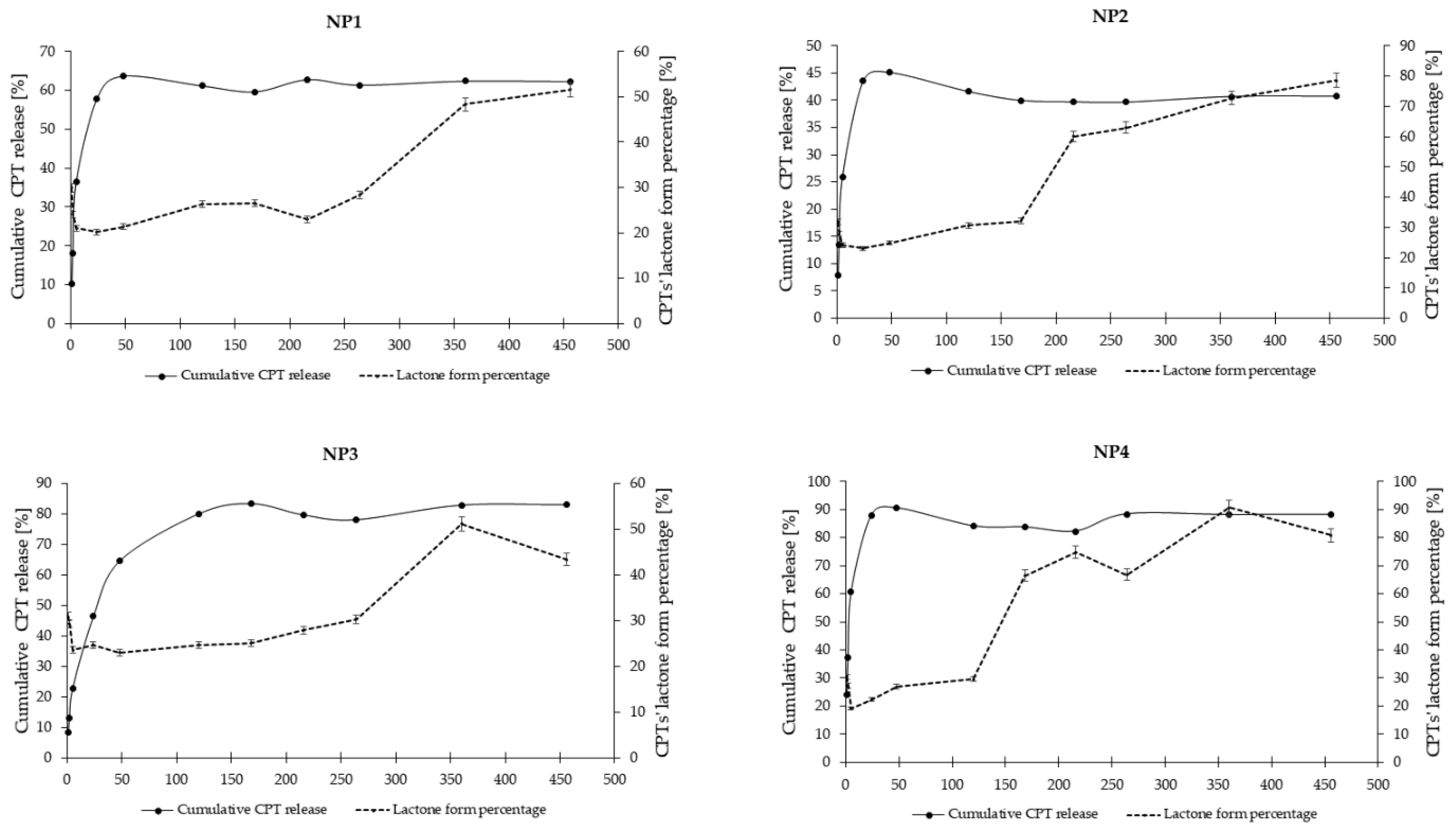
Figure 10.
Correlation between cumulative CPT release and percentage of the lactone form of CPT at 6.50 ± 0.05.
Figure 10.
Correlation between cumulative CPT release and percentage of the lactone form of CPT at 6.50 ± 0.05.
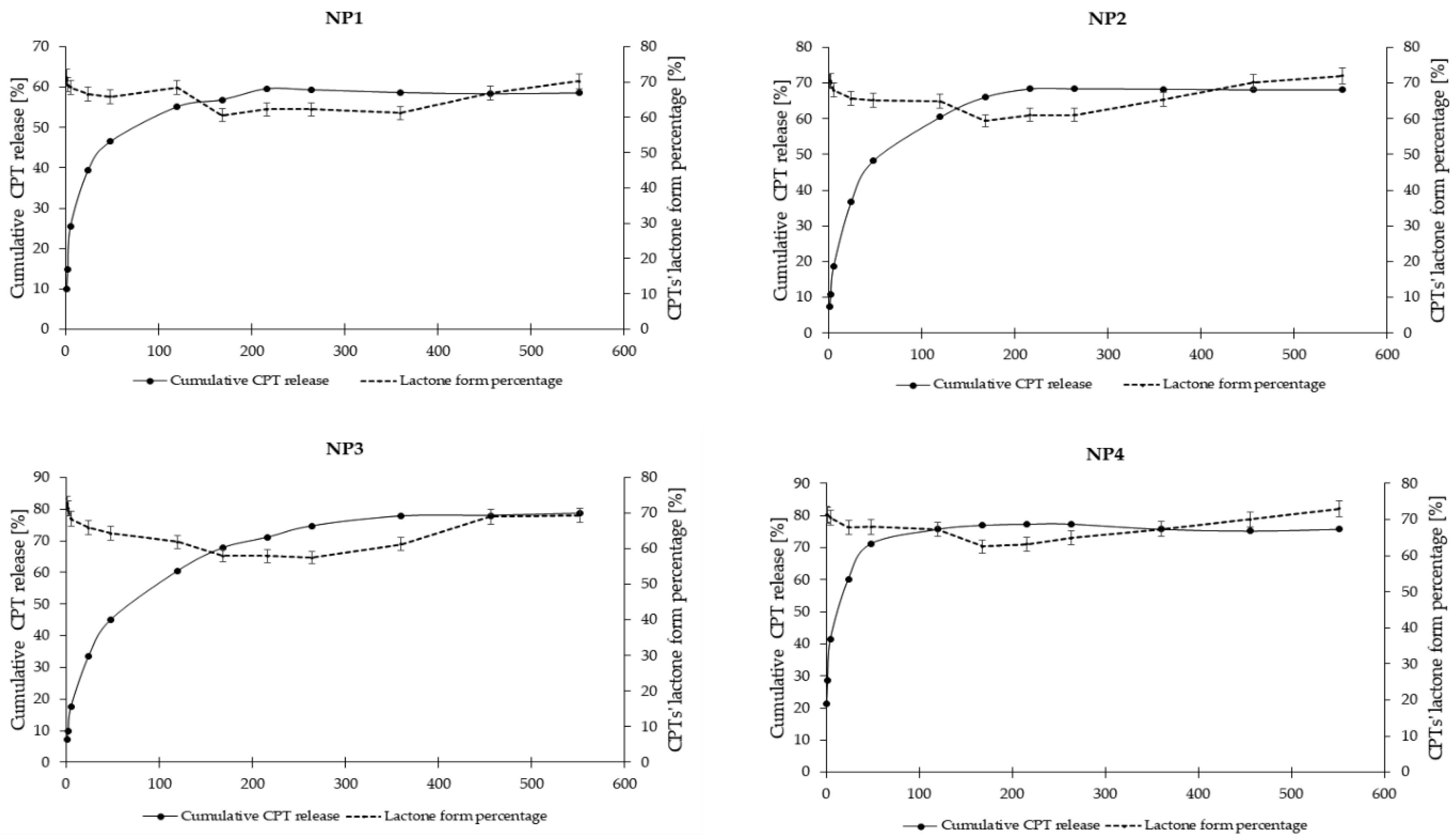
Figure 11.
The structure of MODEL 1 - PAMAM dendrimer generation 4.0 loaded with 3 molecules of CPT and surrounded by PLLA matrix. Atom coloring: Hydrogen-light blue, Nitrogen-dark blue, Carbon-grey, Oxygen-red. Rendering: the atoms and bonds of PLLA are rendered as sticks, the atoms of PAMAM dendrimer generation 4.0 are rendered as solid cylinders, CPT atoms are rendered as spheres with radii that are related to the van der Waals radii of its atoms.
Figure 11.
The structure of MODEL 1 - PAMAM dendrimer generation 4.0 loaded with 3 molecules of CPT and surrounded by PLLA matrix. Atom coloring: Hydrogen-light blue, Nitrogen-dark blue, Carbon-grey, Oxygen-red. Rendering: the atoms and bonds of PLLA are rendered as sticks, the atoms of PAMAM dendrimer generation 4.0 are rendered as solid cylinders, CPT atoms are rendered as spheres with radii that are related to the van der Waals radii of its atoms.
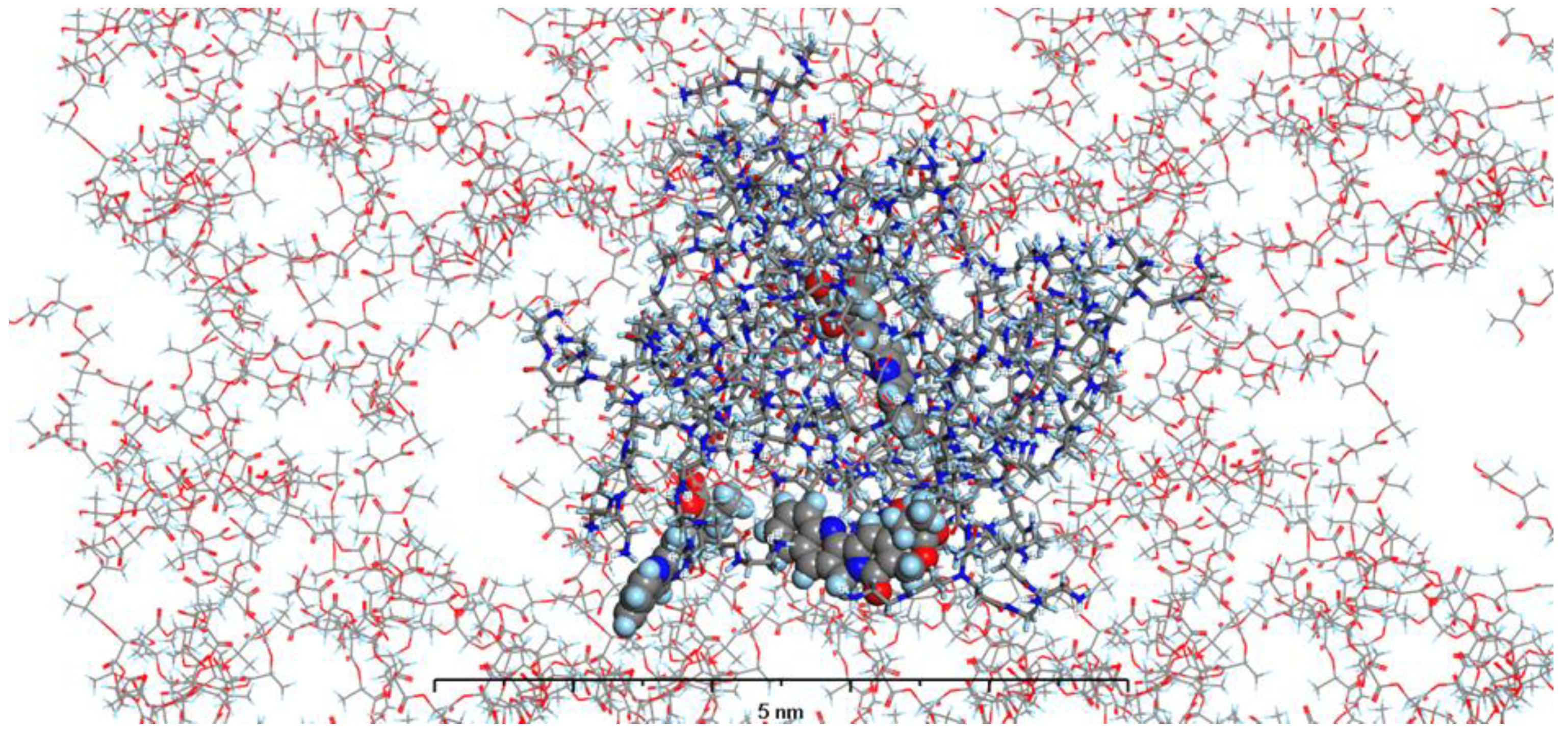
Figure 12.
The structure of MODEL 2 - PAMAM dendrimer generation 4.0 loaded with 3 molecules of CPT and surrounded by PLACL matrix. Atom coloring: Hydrogen-light blue, Nitrogen-dark blue, Carbon-grey, Oxygen-red. Rendering: the atoms and bonds of PLLA are rendered as sticks, the atoms of PAMAM dendrimer generation 4.0 are rendered as solid cylinders, CPT atoms are rendered as spheres with radii that are related to the van der Waals radii of its atoms.
Figure 12.
The structure of MODEL 2 - PAMAM dendrimer generation 4.0 loaded with 3 molecules of CPT and surrounded by PLACL matrix. Atom coloring: Hydrogen-light blue, Nitrogen-dark blue, Carbon-grey, Oxygen-red. Rendering: the atoms and bonds of PLLA are rendered as sticks, the atoms of PAMAM dendrimer generation 4.0 are rendered as solid cylinders, CPT atoms are rendered as spheres with radii that are related to the van der Waals radii of its atoms.
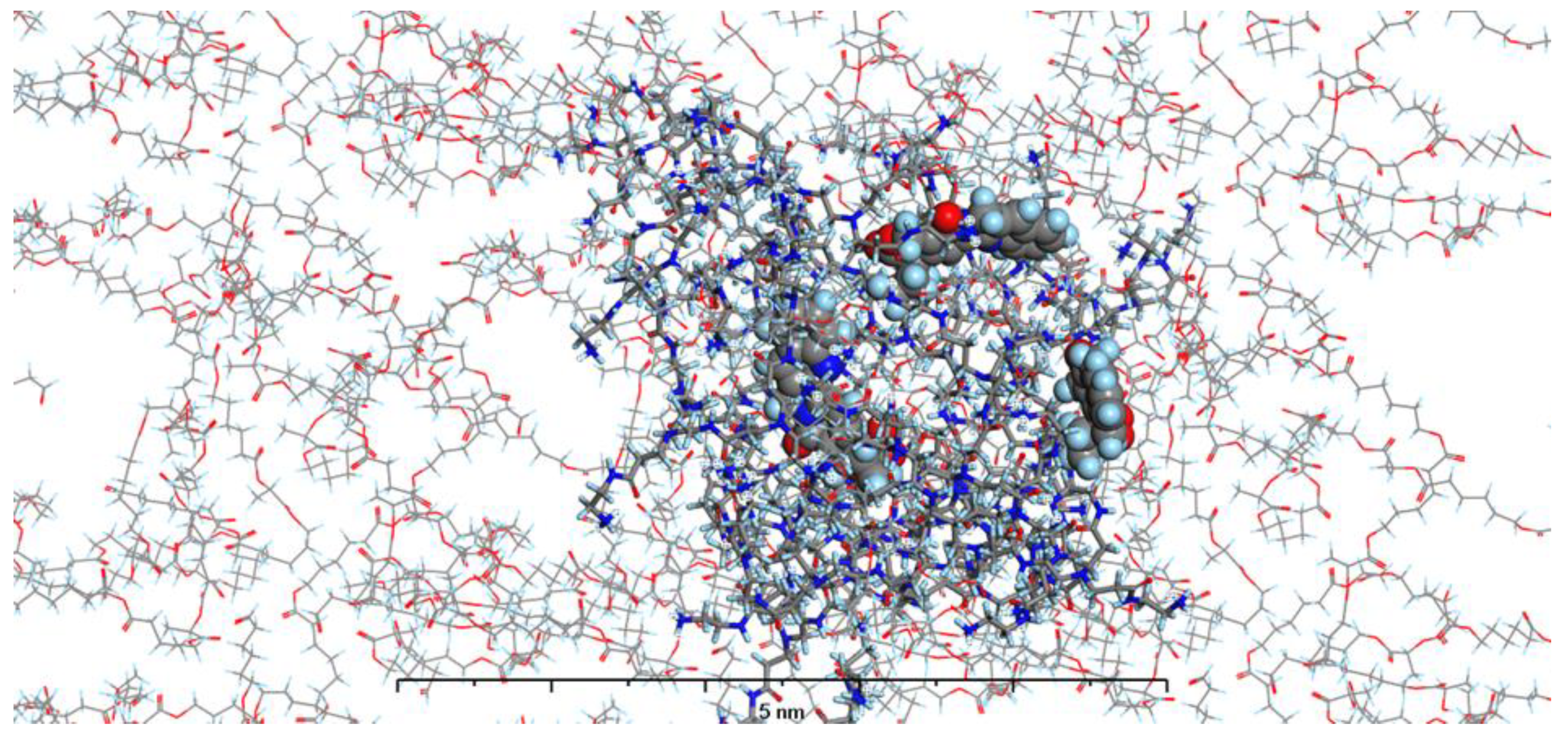
Figure 13.
The structure of MODEL 3 - PAMAM dendrimer generation 4.0 loaded with 3 molecules of CPT and surrounded by PGACL 85:15 (ε-CL:GL) matrix. Atom coloring: Hydrogen-light blue, Nitrogen-dark blue, Carbon-grey, Oxygen-red. Rendering: the atoms and bonds of PLLA are rendered as sticks, the atoms of PAMAM dendrimer generation 4.0 are rendered as solid cylinders, CPT atoms are rendered as spheres with radii that are related to the van der Waals radii of its atoms.
Figure 13.
The structure of MODEL 3 - PAMAM dendrimer generation 4.0 loaded with 3 molecules of CPT and surrounded by PGACL 85:15 (ε-CL:GL) matrix. Atom coloring: Hydrogen-light blue, Nitrogen-dark blue, Carbon-grey, Oxygen-red. Rendering: the atoms and bonds of PLLA are rendered as sticks, the atoms of PAMAM dendrimer generation 4.0 are rendered as solid cylinders, CPT atoms are rendered as spheres with radii that are related to the van der Waals radii of its atoms.
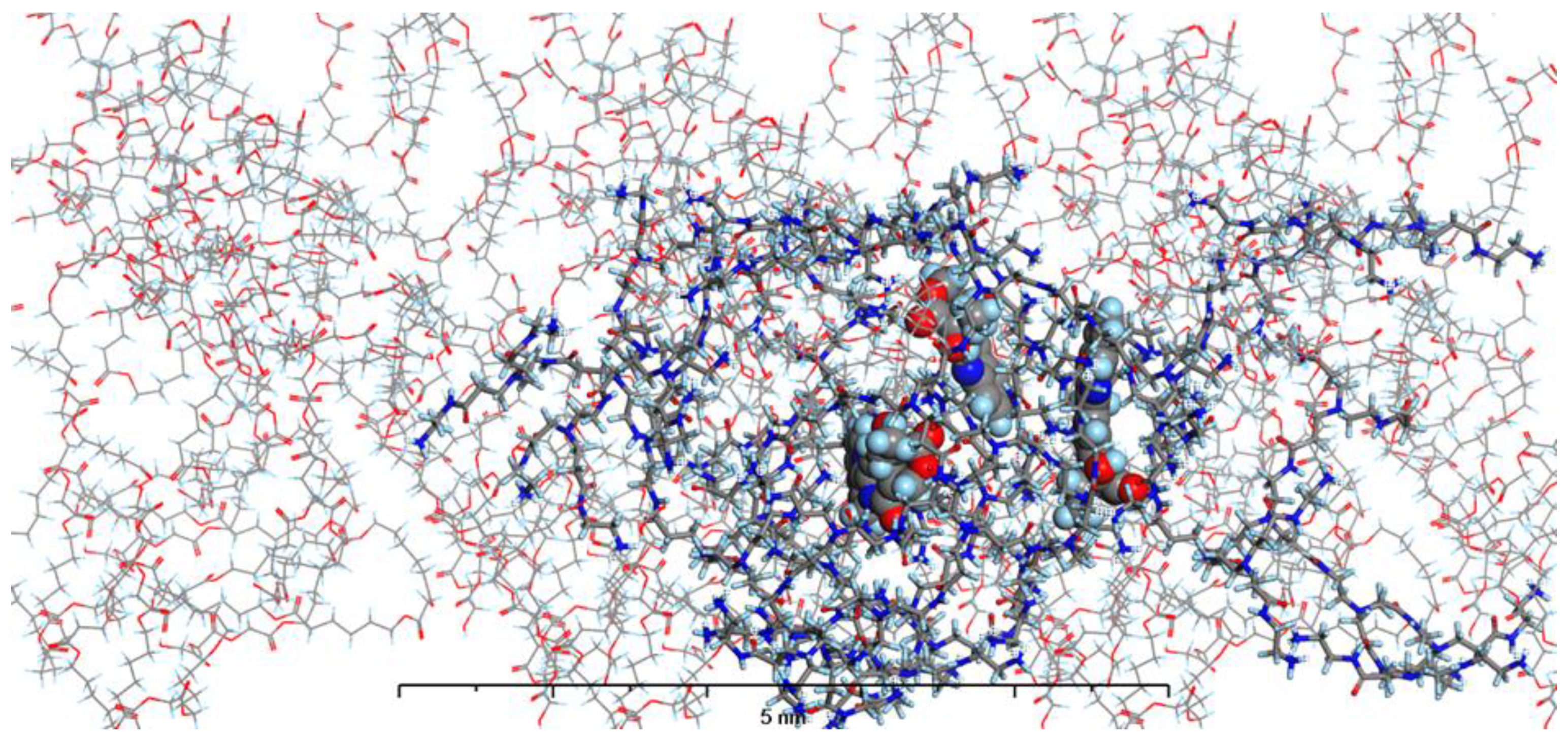
Figure 14.
The structure of MODEL 4 - PAMAM dendrimer generation 4.0 loaded with 3 molecules of CPT and surrounded by PGACL 90:10 (ε-CL:GL) matrix. Atom coloring: Hydrogen-light blue, Nitrogen-dark blue, Carbon-grey, Oxygen-red. Rendering: the atoms and bonds of PLLA are rendered as sticks, the atoms of PAMAM dendrimer generation 4.0 are rendered as solid cylinders, CPT atoms are rendered as spheres with radii that are related to the van der Waals radii of its atoms.
Figure 14.
The structure of MODEL 4 - PAMAM dendrimer generation 4.0 loaded with 3 molecules of CPT and surrounded by PGACL 90:10 (ε-CL:GL) matrix. Atom coloring: Hydrogen-light blue, Nitrogen-dark blue, Carbon-grey, Oxygen-red. Rendering: the atoms and bonds of PLLA are rendered as sticks, the atoms of PAMAM dendrimer generation 4.0 are rendered as solid cylinders, CPT atoms are rendered as spheres with radii that are related to the van der Waals radii of its atoms.
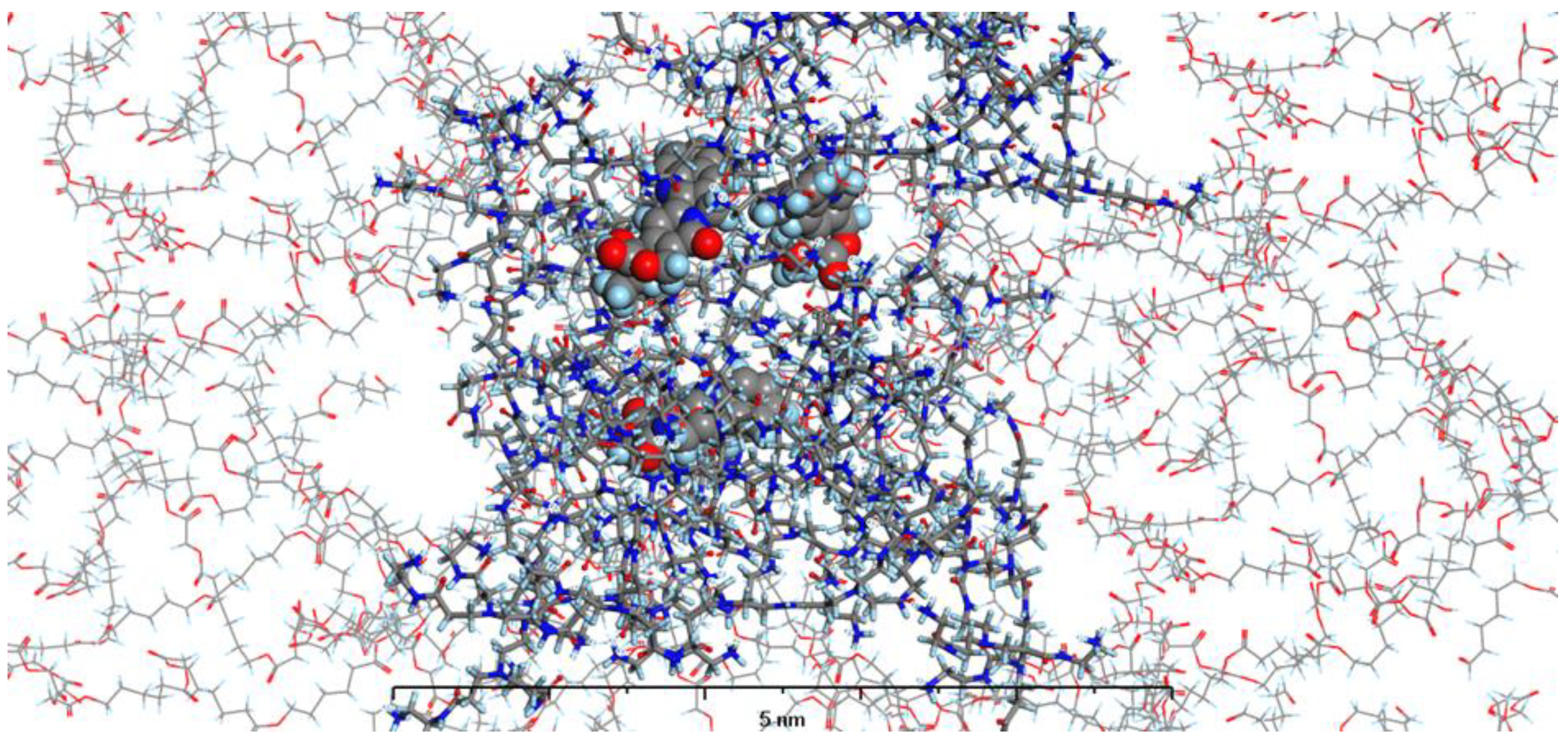
Figure 15.
The RDFs between PAMAM and CPT molecules.
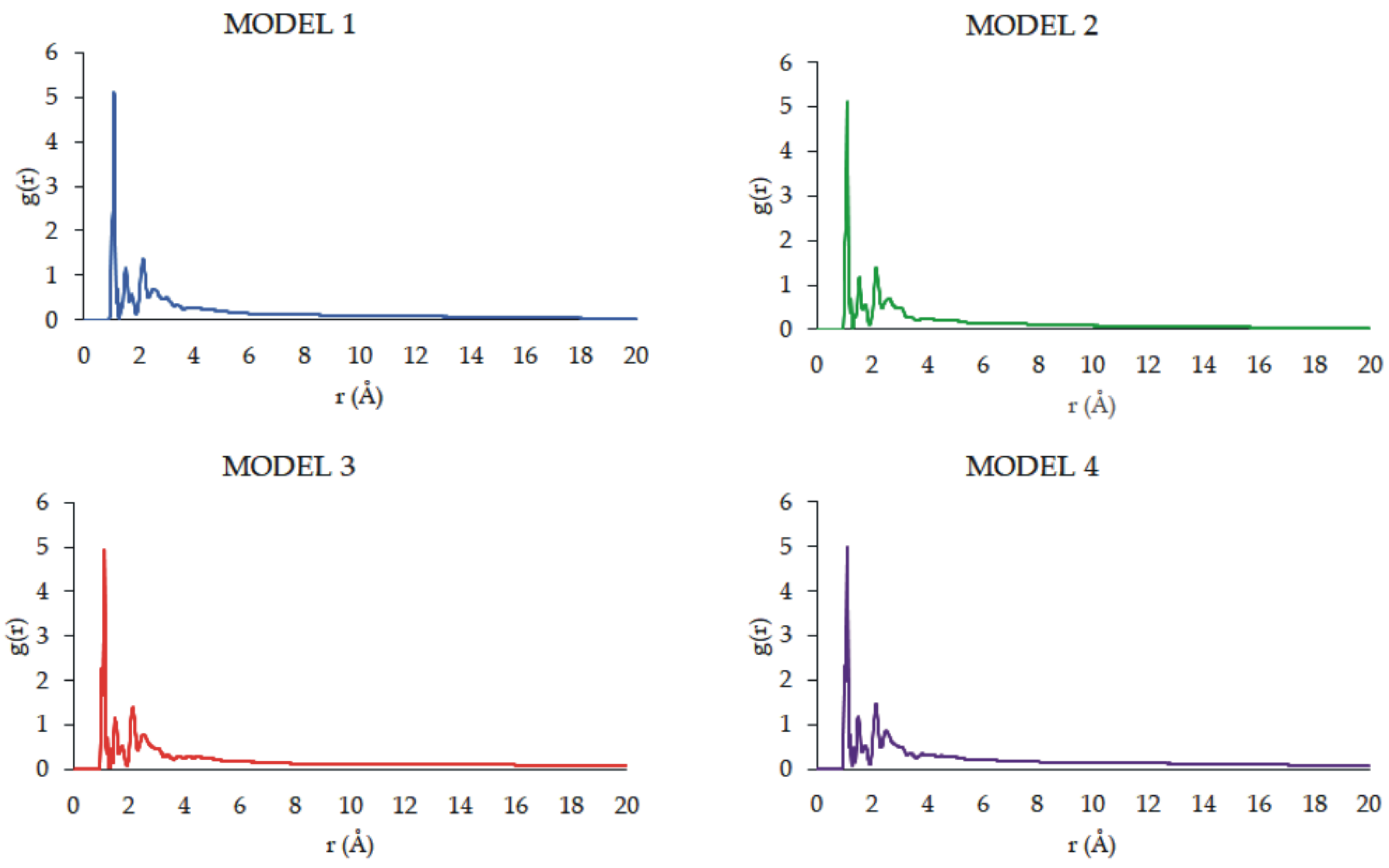
Figure 16.
RMSD plots obtained for CPT in the MODELS 1-4 during 100 ns MD simulation.
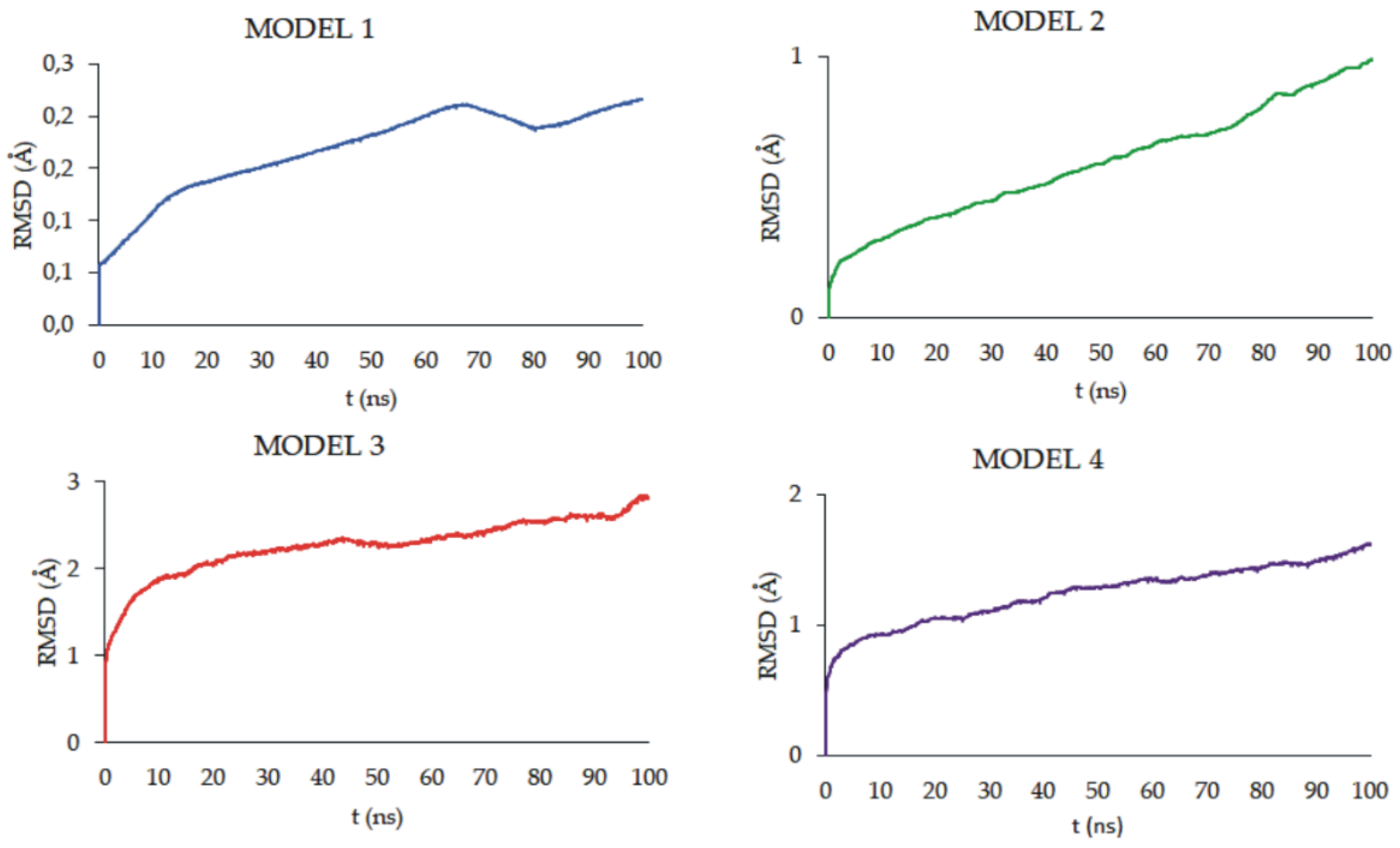
Table 1.
Homo- and copolymerization of CL, GL and LA. The characteristic of the obtained biodegradable polymers.
Table 1.
Homo- and copolymerization of CL, GL and LA. The characteristic of the obtained biodegradable polymers.
| Code | Matrix | Monomer molar ratio | Yield [%] | Convia [%] | Mnb [g/mol] | Ɖb |
|---|---|---|---|---|---|---|
| M1 | PLLA poly(L-lactide) |
LA = 1.0 | 78 | 97 | 13600 | 1.23 |
| M2 | PLACL poly(L-lactide-co --caprolactone) |
LA = 0.40 CL = 0.60 |
78 | 83 (LA) 82 (CL) |
8700 | 1.77 |
| M3 | PGACL poly(glycolide-co --caprolactone) |
CL = 0.85 GL = 0.15 |
83 | 86 (CL) 74 (GL) |
16100 | 1.57 |
| M4 | CL = 0.90 GL = 0.10 |
90 | 85 (CL) 63 (GL) |
21300 | 1.52 |
a – calculated from 1H NMR; b – determined by SEC-MALLS.
Table 2.
Analysis of the chain microstructure of the synthesized copolymers.
| Code | Polymer | Average block length | R | |
|---|---|---|---|---|
| M2 | poly(L-lactide-co --caprolactone) PLACL/40:60 |
= 1.27 = 0.99 |
0.49 | 1.00 |
| M3 | poly(glycolide-co --caprolactone) PGACL/15:85 |
= 1.67 = 4.43 |
0.73 | 0.60 |
| M4 | poly(glycolide-co --caprolactone) PGACL/10:90 |
= 1.46 = 8.81 |
0.94 | 0.68 |
– experimental average length of lactydyl blocks; – experimental average length of caproyl blocks; – experimental average length of glycolidyl blocks; – yield of the second mode of transesterification; R – the degree of randomness.
Table 4.
Analysis data of CPT release from obtained NPs.
| Sample | pH | Zero-order model |
First- order model |
Higuchi model |
Kosmeyer-Peppas model |
Drug transport mechanism | |
|---|---|---|---|---|---|---|---|
| R2 | R2 | R2 | R2 | n | |||
| NP1 NP2 NP3 NP4 |
7.4±0.05 7.4±0.05 7.4±0.05 7.4±0.05 |
0.465 0.400 0.620 0.401 |
0.542 0.445 0.740 0.575 |
0.688 0.593 0.845 0.616 |
0.915 0.707 0.993 0.999 |
0.45 0.23 0.53 0.62 |
non-Fickian transport Fickian diffusion non-Fickian transport non-Fickian transport |
| NP1 NP2 NP3 NP4 |
6.5±0.05 6.5±0.05 6.5±0.05 6.5±0.05 |
0.628 0.684 0.758 0.517 |
0.717 0.779 0.877 0.653 |
0.844 0.893 0.939 0.744 |
0.952 0.987 0.992 0.978 |
0.32 0.45 0.45 0.32 |
Fickian diffusion non-Fickian transport non-Fickian transport Fickian diffusion |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



