
Submitted:
29 October 2024
Posted:
30 October 2024
You are already at the latest version
Abstract
African swine fever (ASF) has widely spread around the world in the last 100 years since its discovery. The African swine fever virus (ASFV) particles are composed of more than 150 proteins, with the p17 protein encoded by the D117L gene serving as one of the major capsid proteins and playing a crucial role in the virus's morphogenesis and immune evasion. Therefore, monoclonal antibody (mAb) targeting p17 is useful for the research and detection of ASFV infection. Here, we generated two specific mAbs against p17, named 1G2 and 6G3, respectively, both of which have been successfully applied in ELISA, Western blotting and immunofluorescence assay. Moreover, we found that both 1G2 and 6G3 mAbs recognize a new epitope of 72–78 amino acids of p17 protein, which is highly conserved across different ASFV strains, including all genotype I and II strains. Based on the recognized epitope, an indirect ELISA has been established to effectively detect antibodies during ASFV infection, and it exhibits high consistency with commercial ASFV ELISA kit. In summary, the production of the specific p17 mAbs and the identification of the corresponding epitope will significantly promote the serological diagnosis of ASFV and vaccine research.
Keywords:
African swine fever virus
; p17
; monoclonal antibody
; epitope
; diagnosis
1. Introduction
African swine fever (ASF) is a highly contagious and fatal hemorrhagic disease in pigs caused by the African swine fever virus (ASFV). Since its first report in China in 2018, ASF has spread rapidly and has posed a devastating impact on the pig industry in the following years [1,2]. Despite the domestic and international efforts to develop a vaccine, there is currently no safe and effective commercial vaccine available [3,4,5,6]. Therefore, the prevention and control of ASF mainly rely on rapid detection and early diagnosis.
ASFV is a double-stranded DNA virus, with a large genome of approximately 170-190 kb, encoding over 150 viral proteins [7]. The structure of ASFV is complex, which can be divided from the outside to the inside into the outer membrane, capsid, inner membrane, core shell, and nucleoid [8]. The p17 protein encoded by ORF D117L is an important component of the ASFV capsid [9]. ASFV p17 plays a critical role in viral morphogenesis, the absence of p17 hinders the proteolysis of the polyproteins pp220 and pp62, thereby affecting the assembly of the viral particles [10].
ASFV p17 not only affects the morphogenesis and assembly of viral particles but also participates in regulation of host cell function and immune evasion. In eukaryotic cells, p17 is located on the endoplasmic reticulum (ER) membrane, and its expression can cause ER stress, leading to the release of reactive oxygen species, which inhibits cell proliferation by arresting the cell cycle [11]. The binding of p17 to TOMM70 promotes the binding of the autophagy receptor SQSTM1 to TOMM70, resulting in mitophagy and decreased expression of MAVS, thereby suppressing the innate immune response [12]. The p17 is involved in regulation of the innate immune response mediated by cGAS-STING signaling pathway. On the one hand, p17 binds to STING, inhibiting the interactions between STING and TBK1/IKKɛ, and blocking the production of type I IFN [13]. On the other hand, p17 downregulates the phosphorylation of TBK1 and IRF3 by recruiting the host scaffold protein PR65A and inducing partial degradation of STING via apoptosis [14].
As one of the main capsid proteins, p17 has a high abundance in ASFV particles and possesses a good immunogenicity [7], making it a potential target for detecting ASFV infection. There has been a report of p17 expression in CHO cells and establishment of a specific indirect ELISA based on the purified p17 from CHO cells [15]. The p17 monoclonal antibodies (mAbs) were generated from eukaryotic p17 protein immunized mice and the mAb recognized linear epitopes are located at the N-terminus of p17 [15,16]. In this study, we expressed and purified the prokaryotic p17 to produce p17 mAbs from immunized mice. Two p17 mAbs generated could recognize a conserved linear epitope at 72-78 amino acids in the middle of p17. In addition, the epitope-based indirect ELISA was established, and it was able to specifically detect ASFV antibodies in the serum samples of pigs with ASFV infection. Our results not only provide useful tools for ASFV research, but also establish a new method for the detection and prevention of ASF.
2. Materials and Methods
2.1. Mice, Cells, Viruses, Sera and Reagents
The BALB/c mice of 6- to 8-weeks old were from the Laboratory Animal Center of Yangzhou University. The animal experiment was in strict accordance with the Guidance for the Care and Use of Laboratory Animals of Yangzhou University (SYXK(JS)-2021-0026). HEK-293T cells (ATCC Cat # CRL-3216), Marc-145 cells (Cellosaurus Cat # CVCL_4540), Vero cells (ATCC Cat # CCL-81), PK-15 cells (ATCC Cat # CCL-33), MDCK cells (ATCC Cat # CCL-34) and myeloma cell line SP2/0 (ATCC Cat # CRL-1581) were cultured in Dulbecco modified Eagle medium (DMEM, Hyclone Laboratories, USA) containing 10% fetal bovine serum (FBS) and 100 IU/ml of penicillin plus 100 μg/ml streptomycin. Primary porcine alveolar macrophages (PAMs) and 3D4/21 cells (ATCC cat# CRL-2843) were cultured in RPMI 1640 medium (Hyclone Laboratories) which contains 100 IU/mL of penicillin plus 100 μg/mL streptomycin and 10% FBS. All cells were grown at 37°C in a 5% CO2 humidified incubator. The ASFV strain (genotype II, GenBank accession ON456300) was stored in the animal biosafety level 3 (ABSL-3) of Yangzhou University approved by the Ministry of Agriculture and Rural Affairs (07140020201109-1), whereas the porcine reproductive and respiratory syndrome virus (PRRSV), recombinant PRRSV expressing p17 (PRRSV-p17), porcine epidemic diarrhea virus (PEDV), porcine delta coronavirus (PDCoV), swine influenza virus (SIV) and porcine serum samples were kepted in our lab. The African swine fever virus ELISA antibody detection kit was bought from Putai Biology Technology Co., Ltd. The RFP mAb (ab185921) was acquired from Abcam (Cambridge, UK). The anti-β-actin mAb (5057) were acquired from Cell Signaling Technology (Boston, MA, USA). The anti-PRRSV N mAb, anti-PEDV N mAb and anti-SIV NP mAb were produced and stored in our lab. The anti-PDCoV NS7 mAb was a gift from Prof. Zhenhai Chen of Yangzhou University.
2.2. Expression and Purification of p17 Protein
The ASFV p17 C-terminal intramembrane region (61-171 aa) gene coding sequence D117L was amplified by PCR from pCAGGS-p17-HA stored in our laboratory and cloned into Xho I and EcoR I sites of pCold-TF vector or Sal I and Xba I sites of pCold-MBP vector by Seemless/In-Fusion Cloning (2×MultiF Seamless Assembly Mix, Abclonal, Wuhan, China). The plasmids were transformed into BL21/DE3 E. coli competent cells and treated with 1 mM isopropyl-β-d-1-thiogalactoside (IPTG) at 16 °C or 37 °C for 24 h to induce p17 expressions. The expressed p17 fusion proteins were purified by gel recovery method. Specifically, the bacteria pellet was collected after centrifugation, resuspended in PBS and sonicated on ice. Following centrifugation, the supernatant was retained and run in SDS-PAGE as much as possible. After staining with Coomassie brilliant blue and subsequent complete de-staining, the target gel piece was cut out and the ground gel was placed in a dialysis bag for electrophoresis at 120 V for 2 h. Finally, the liquid in the dialysis bag was transferred and dialyzed in PBS overnight at 4°C, and concentrated with PEG 20000.
2.3. Production of Anti-p17 Monoclonal Antibodies (mAbs)
BALB/c mice were first immunized subcutaneously at multiple points on the back with 100 μg purified TF-p17 protein mixed with Montanide gel (SEPPIC SA Cedex, France) at a volume ratio of 10:1. The second and third immunizations were performed at 14 d and 21 d after the first immunization, with each 50 µg protein mixed with same adjuvant. Five days later, tail vein blood was drawn and serum p17 antibody was measured by p17 protein indirect ELISA. Mouse exhibiting the highest antibody titer was chosen for the final boost (50 µg p17 protein injection intraperitoneally without adjuvant). Subsequently, the mouse was sacrificed three days later for cell fusion with SP2/0 cells. The hybridomas secreting p17 specific antibodies were screened out by MBP-p17 based indirect ELISA, followed by Western blotting confirmation. Positive hybridomas were subjected for limited dilution subcloning three times and tested further for antibody secretion.
2.4. Mapping of the Precise Linear B Cell Epitope of p17 Protein
The pCAGGS-p17-HA and pDsRed-C1-p17 plasmids were previously constructed and used in this study [11,13].First, the C-terminal intramembrane region of p17 (aa 61-117) was separated into three fragments which were tested for the reactivity with p17 mAbs. Second, based on the reaction information of the fragment, progressive truncations at both N and C terminal ends were performed from full length p17. The reactivity of different fragments with p17 mAbs was tested, the critical amino acids of both N and C ends for reactivity with p17 mAbs were determined, and thus, the minimal epitope was deduced. Totally, 14 p17 fragments (P1-P14) were designed, with all cloning PCR primers listed in Table S1. All p17 fragments were amplified by PCR using the template pCAGGS-p17-HA, and then cloned into the Bgl II and EcoR I sites of pDsRed-C1-express vector by Seemless/In-Fusion Cloning. All the constructed plasmids were transfected into 293T cells, and the reactivity of different truncated p17 proteins expressed in transfected 293 T cells with p17 mAbs was tested using Western blotting.
2.5. Western Blotting
The reactivity of anti-p17 mAbs with p17 expressed in transfected 293T cells, PRRSV-p17 infected Marc-145 cells and ASFV infected PAMs, as well as the p17 fragments in transfected 293T cells was evaluated using Western blotting. The cell protein samples were separated by 6-10 % SDS-PAGE, and transferred to PVDF membranes. The membranes were then blocked using 5% non-fat dry milk TBS solution for 1 h, with 0.1% Tween-20 (TBST). Next, the membrane was incubated with the primary mAbs (1 ∶ 1000 anti-RFP and p17 hybridoma ascites) at 4 ℃ overnight. Next, the membrane was incubated with HRP-conjugated Goat Anti-Mouse IgG (1: 10000, BBI, Shanghai, China) for 1 h. Protein signals was visualized and captured by Western blot imaging system.
2.6. Immunofluorescence Assay
3D4/21 cells were transfected with pCAGGS-p17-HA or pdsRed-C1-p17/truncated mutants for 24 h, while Marc-145 cells were infected with PRRSV-p17 with a multiplicity of infection (MOI) of 0.1 for 72 h. The cells were fixed in 4 % paraformaldehyde for 30 min, permeabilized with 0.5% Triton X-100, and blocked with 5% BSA. The treated cells were incubated with p17 mAbs (1: 200 ascites) overnight, and Donkey anti-Mouse IgG (H+L) Alexa Fluor 488 (1: 500, Invitrogen, Shanghai, China) for 1 h, followed by DAPI staining (Beyotime, Shanghai, China) for 15 min. Cells were visualized by fluorescence microscope at the excitation wavelengths 405 nm, 488 nm and 535 nm, respectively.
2.7. Enzyme-Linked Immunosorbent Assay
For p17 protein indirect ELISA, the ELISA wells were coated with p17 purified protein (MBP-p17) diluted in PBS at a concentration of 0.3125 μg/mL at 4°C overnight, followed by washing and blocking with 5% skim milk at 37°C for 2 h. Hybridoma supernatant (100 μL/well) was added to each well and incubated for 2 h at 37 °C. After washing with PBST, secondary antibody Goat anti-Mouse IgG-HRP (1:10000 dilution, 100 μL/well, TransGen Biotech, Beijing, China) was added and incubated at 37°C for 1 h. Next, TMB (50 μL/well) was added and incubated for 15 min at 37 °C in the dark. The reaction was stopped with 0.5 M H2SO4 and the optical density at 450 nm (OD450) was measured. The ratios of hybridoma supernatants to SP2/0 supernatant (P/N) were calculated, with P/N ≥ 2.0 considered as positive.
For p17 epitope indirect ELISA, ELISA wells were coated with antigenic epitope peptide diluted with PBS at the concentration of 0.3125-10 μg/mL at 4°C overnight, followed by washing and blocking with 5% BSA at 37 °C for 2 h. Diluted porcine serum (1:5-1:800, 100 μL/well) was added and incubated at 37 °C for 2 h. After washing with PBST, secondary antibody Goat anti-Swine IgG-HRP (1:10000, 100 μL/well, Proteintech, Wuhan, China) was added and incubated at 37 °C for 1 h, followed by addition of substrate TMB, stopping with 0.5 M H2SO4 and OD450 measurement. The ratios of positive sera to negative normal serum (P/N) were calculated.
2.8. Bioinformatics Analysis
The ASFV p17 protein transmembrane (TM) was analyzed and predicted by online software TMHMM-2.0 (https://services.healthtech.dtu.dk/services/TMHMM-2.0/). The analysis of hydrophobicity or hydrophilicity scales of p17 was performed by the online software ProtScale (https://web.expasy.org/protscale/). The B cell epitopes in p17 protein were predicted via the online tool (http://tools.iedb.org/main/bcell/). To verify the consensus of identified p17 epitope, the p17 amino acid sequences of 207 ASFV strains from GenBank were downloaded. The alignment of amino acid sequences and conservation analysis was performed by DNAMAN software, version 9.0 (San Ramon, CA, USA). The spatial distribution and structure of epitope in p17 protein was predicted by Alphafold2 (https://colab.research.google.com/github/sokrypton/colabFold/blob/main/Alphafold2.ipynb) and visualized by the PyMOL Molecular Graphics System (Version 2.4.0, Schrödinger, LLC).
2.9. Quantification and Statistical Analysis
Statistical analyses was performed by GraphPad Prism 8 software (GraphPad Software, Inc.), with the data presented as means ± SEM. The p values were calculated by using unpaired t test. Statistical significance was represented as: not significant (NS): p > 0.05, *: p ≤ 0.05, and **: p ≤ 0.01. The p ≤ 0.05 was considered as statistically significant.
3. Results
3.1. Characterization and Production of Recombinant p17 Protein
Bioinformatic analysis showed that the ASFV p17 protein is a transmembrane protein, with the transmembrane (TM) region located at 38-60 amino acids (aa) (Figure 1A), which is corresponding to the highly hydrophobic region (Figure 1B). Both extramembrane region (1-37 aa) and intramembrane region (61-117 aa) contain the B cells antigenic epitopes (Figure 1C). Given that the C terminal intramembrane region contains more antigenicity (Figure 1C), this hydrophilic region (61-117 aa) was selected for cloning and fusion expression with soluble tags, E. coli chaperone proteins trigger factor (TF) and maltose binding protein (MBP), respectively. The constructed recombinant prokaryotic expression plasmids, pCold-TF-p17 jd and pCold-MBP-p17 jd, were transformed into DE3/BL21 competent E. coli and cultured at 37 ℃ or 16 ℃ with the induction of 1mM IPTG for 24 h. The soluble expressions of both TF-p17 jd (Figure 1D) and MBP-p17 jd (Figure 1E) were significantly induced by IPTG at 16 ℃ rather than 37 ℃. SDS-PAGE analysis showed that the purified TF-p17 jd and MBP-p17 jd proteins had high purity, and exhibited the molecular weights of approximately 68 kD and 53 kD, respectively, as expected (Figure 1F).
3.2. Generation of Specific Monoclonal Antibodies (mAbs) Against p17 Protein
The hybridomas were obtained by regular procedure of cell fusion between spleen cells of immunized mice and myeloma SP2/0 cells, and positive hybridoma clones were selected by MBP-p17 protein indirect ELISA. We determined the optimal coating concentration of p17 protein using ASFV positive pig serum as 0.3125 μg/mL (Figure 2A). After screening by indirect ELISA and three rounds of subcloning, two hybridoma cell clones named 1G2 and 6G3 were generated (Figure 2B). The monoclonal antibodies (mAbs) produced by both 1G2 and 6G3 clones are both the IgG2b subclass (Figure 2C). The titers of ascites 1G2 and 6G3 mAbs were measured by ELISA to be 400000-800000 (Figure 2D). The specific reactivity of these two mAbs with the eukaryotic expressed p17 proteins from 293T cells transfected with pCAGGS-p17-HA (Figure 3A) and pDsRed-p17 (Figure 3B), from Marc-145 cells infected with PRRSV-p17 (Figure 3C) and from primary PAMs infected with ASFV (Figure 3D) was determined by using Western blotting. These results illustrated that both 1G2 and 6G3 mAbs specifically react with not only exogenous p17 (Figure 3A-3C), but also endogenous p17 (Figure 3D).
Further tests were conducted to assess the reactivity of the two mAbs with various swine viruses. The results showed that both 1G2 and 6G3 only reacted with AFSV infected samples, but not other porcine viruses including PRRSV, PEDV, PDCoV and SIV (Figure 3E). Similarly, immunofluorescence analysis indicated that both mAbs could react specifically with the p17 expressed in transfected 3D4/21 cells (Figure 4A and Figure S1) and in PRRSV-p17-infected Marc-145 cells (Figure 4B), with the detected p17 primarily located in the cytoplasm. These results collectively demonstrated that the produced mAbs are specific to ASFV p17, successfully detecting p17 in various immune assays.
3.3. Identification and Bioinformatics Analysis of the Antigenic Epitope Recognized by Two p17 mAbs
The precise mapping of antigenic epitopes for two mAbs was conducted by segmental and gradient truncation of the p17, each of which was cloned into the pDsRed-C1 expression vector (Figure 5A). Western blotting results showed that the deletion of N-terminal K72 and C-terminal Y78 would disrupt the reactivity of two mAbs with expressed p17, indicating that the epitope recognized by two p17 mAbs is 72KPPPSYY78 (Figure 5B). Similarly, in immunofluorescence assay, the deletion of K72 or Y78 in p17 completely abolished the reactivity with the mAbs (Figure S2). As such, the results clearly showed that both 1G2 and 6G3 mAbs recognize the linear epitope 72KPPPSYY78.
In order to evaluate conservation of the antigenic epitope cross different ASFV strains, the p17 protein sequences of 207 ASFV strains were downloaded from GenBank and aligned with the epitope sequence we identified. The results showed that the amino acids 72-78 of all genotype I and II ASFV p17 are identical, some of which are presented as representatives (Figure 6A). However, the amino acid sequences in other genotypes of ASFV p17 proteins have differences, hence exhibiting only relative conservation (Table S2). Due to the lack of precise crystal information for ASFV p17 protein in the Protein Data Bank (PDB), we utilized the Alphafold2 web tools to predict the tertiary structure of the p17 protein, followed by visualization with PyMOL. The results indicated that the epitope recognized by the two mAbs is located at the junction of an alpha-helix and a random coil (Figure 6B).
3.4. Establishment of Epitope Indirect ELISA for Detection of ASFV Antibodies
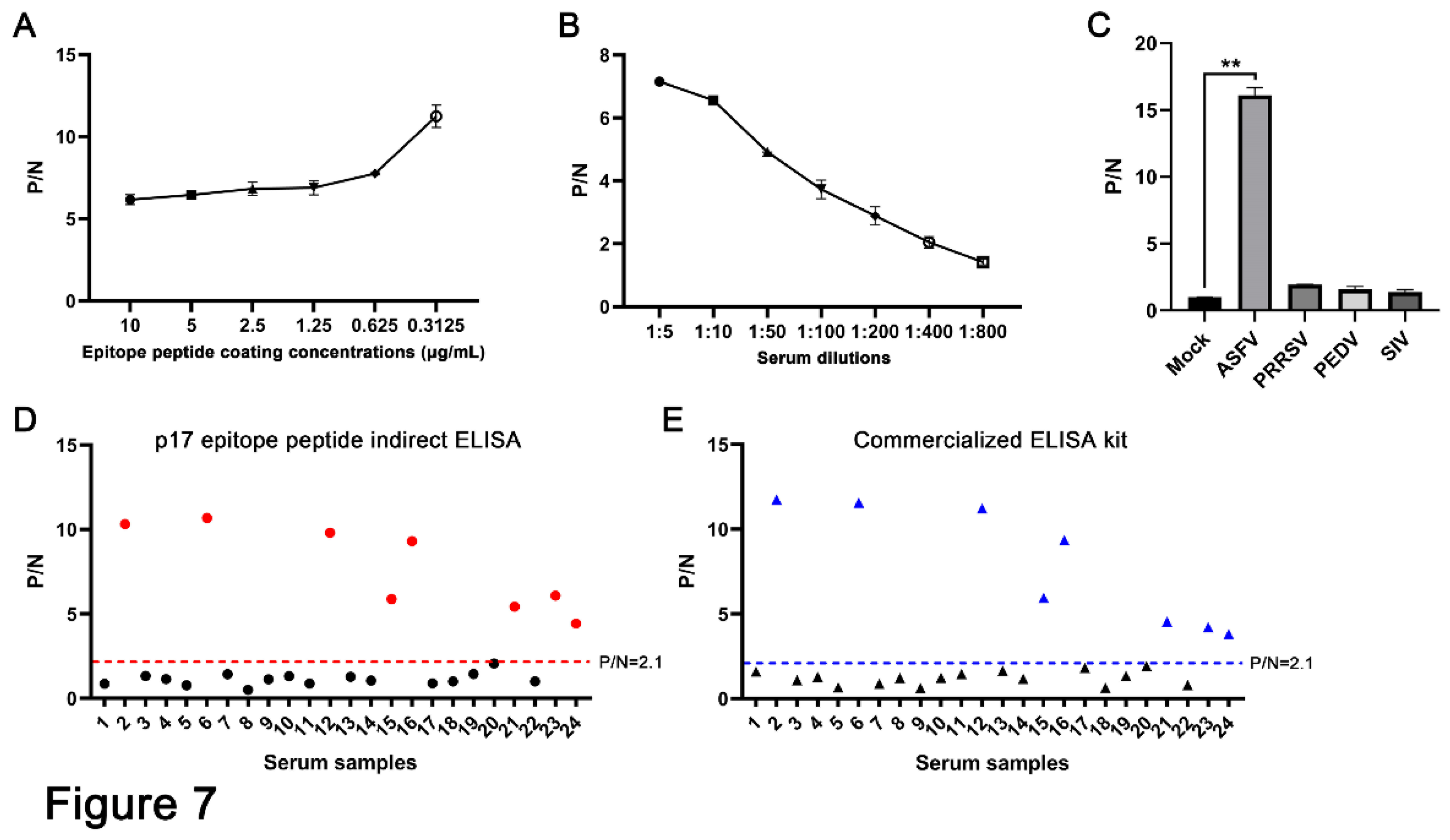
To verify the diagnostic ability of the epitope recognized by the p17 mAbs, we developed an epitope-based indirect ELISA to detect the ASFV antibodies. First optimization of the indirect ELISA showed that the highest positive-to-negative (P/N) ratio was achieved when the p17 epitope peptide was coated at a concentration of 0.3125 μg/mL for the detection of ASFV-positive serum (Figure 7A). Second, the dilution of ASFV-positive serum at 1:5 yielded the highest P/N ratio (Figure 7B). The specificity of the epitope-based indirect ELISA was tested with different positive serum samples of PRRSV, PEDV, SIV and ASFV. The ELISA results showed that only ASFV positive serum, rather than others, gives a positive P/N ratio (Figure 7C). Our developed epitope ELISA was applied for detection of 24 pig clinical serum samples, and 9 sera were detected as positive (Figure 7D). In comparison, a commercial ELISA kit was also used for parallel detection, and both detection methods achieved a 100% consistency of positive sample detection rate (Figure 7D and 7E). Therefore, our preliminary results suggested that the epitope recognized by the p17 mAbs can be used in ELISA for the detection of ASFV infection.
4. Discussion
African Swine Fever (ASF) has a history of over 100 years since its first discovery in Kenya in 1921 [1]. Over the following decades, ASF made the leap from Africa to Europe, spreading widely until that it entered China in 2018 [2]. It is well known that China is a major country for pig farming and product consumption, with pig farming and inventory accounting for more than half of the global total [17]. The prevalence of ASF in China has caused the mutation and recombination of the ASFV [18,19]. At present, the recombinant ASFV of genotype I and II have been found in China, some of which, although classified as genotype I, have inserted multiple virulence genes of genotype II and exhibit high virulence and transmissibility in pigs [18]. These pose a huge challenge to the prevention and control of ASF.
The inactivated vaccine for ASF has shown poor protective efficacy [20], and the gene-deleted vaccine has been observed to undergo gene recombination in the field, carrying the risk of virulence reversion [19,21]. For the prevention and control of ASF, detection has become a crucial procedure [22]. Currently, the detection of ASF primarily focuses on the detection of the viral genome or particles, as well as the detection of antibodies related to ASFV [23]. In subacute and chronic infections, serological detection of ASFV antibodies is a reliable means [23]. The production of mAbs against ASFV structural proteins, the identification of antigenic epitopes, and the subsequent establishment of detection methods mainly concentrated on structural proteins such as p72 [24,25,26,27], p54 [28,29,30,31,32], p30 [33,34,35], and CD2v [36,37,38], with only few reports on p17. ASFV p17 closely surrounds p72 in a trimeric form and is crucial for the assembly of the ASFV capsid and the formation of the icosahedral morphology [9]. Besides mediating viral morphogenesis and immune evasion, the expression of p17 also affects various host cell events, including host cell endocytosis, ubiquitin-mediated proteolysis, N-glycan biosynthesis, and apoptosis [39].
Due to the poor prokaryotic expression of full length p17 (not shown), we discarded the hydrophobic transmembrane region and selected the hydrophilic intramembrane region for fusion with two molecular chaperone proteins, TF and MBP, respectively (Figure 1). By immunizing with TF-p17 protein and screening of the hybridomas by MBP-p17 based indirect ELISA to reduce the non-specificity, we obtained two p17 specific mAbs, which work successfully in multiple immune assays including ELISA, Western blotting and Immunofluorescence (Figure 2-4 and Figure S1), demonstrating the value of broad application. Notably, the mAb recognized p17 in transfected cells and PRRSV-17 infected cells are about 23 kD, whereas the mAb recognized p17 in ASFV infected cells is about 17 kD (Figure 3A). The size difference of p17 in different expression systems indicated the disparity of post translational modification of p17 protein under different conditions, which is interesting and deserves for further investigation.
We identified the minimal antigenic epitope recognized by these two p17 mAbs that is 72KPPPSYY78 (Figure 5 and Figure S2). Previous studies identified two antigenic epitopes in p17 that are 3TETSPLLSH11 and 8LLSHNLSTREGIK20, by using p17 mAbs [15,16]. Consistent with our study, a recent study predicted and confirmed that 63TIDCKSSIPKPPPSYYVQQPEPHH86 is one of most immunogenic and immunoreactive epitopes [40], further confirmed the validity of this antigenic epitope. The identification of precise antigenic epitope justified the utilization of the epitope in the serological detection of ASFV infection. Based on the highly conserved antigenic epitope recognized by p17 mAbs (Figure 6), we established an indirect ELISA to detect the antibodies against ASFV (Figure 7). In detection of 24 clinical serum samples from pigs, 9 of 24 serum samples were detected as positive (Figure 7D). The results were totally consistent with that of commercial ELISA kit (Figure 7E), suggesting the effectiveness and validity of our peptide ELISA assay and the potential for practical application in clinic. Recently, our team has developed both RPA-LbCas12a and RPA-LwCas13a detection methods based on the ASFV structural protein p17 gene (D117L), reaching a sensitivity as high as two gene copies, showing no cross-reactivity with other common pig viruses [41,42]. Thus, the combined detections of nucleic acid and antibody based on the ASFV structural D117L gene / p17 protein can greatly improve the detection of ASFV infection, achieving convenience and precision.
Collectively, we generated two specific mAbs of ASFV p17, identified a highly conserved B-cell epitope of p17, and established an epitope based ELISA detecting ASFV antibodies. These tools not only provide effective means for in-depth research on the structural p17 protein of ASFV, but also contribute to the prevention and control of ASF.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
J.Z. (Jianzhong Zhu) conceived and designed the experiments; N.X., Q.C., A.L., J.Z. (Jiajia Zhang), H.H., J.J., P.H., Z.S., and Z.X. performed the experiments; W.Z. and S.J. analyzed the data; N.C. and J.B. provided the resources; N.X. and J.Z. (Jianzhong Zhu) drafted the paper. All authors have read and agreed the manuscript.
Funding
This work was partly supported by the National Key Research and Development Program of China (2021YFD1800105), the 111 Project under Grant D18007, and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). N.X is supported by Research Innovation Project of Jiangsu Province Graduate Students (KYCX23_3596). Z.X is supported by Innovation and Entrepreneurship Training Program for College Students in Jiangsu Province (XCX20240816).
Data Availability Statement
All data are available in the article.
Disclosure Statement
The authors declare no conflict of interest.
References
- Li, Z.; Chen, W.; Qiu, Z.; Li, Y.; Fan, J.; Wu, K.; Li, X.; Zhao, M.; Ding, H.; Fan, S.; Chen, J. , African Swine Fever Virus: A Review. Life (Basel), 2022, 12, (8).
- Wu, K.; Liu, J.; Wang, L.; Fan, S.; Li, Z.; Li, Y.; Yi, L.; Ding, H.; Zhao, M.; Chen, J. , Current State of Global African Swine Fever Vaccine Development under the Prevalence and Transmission of ASF in China. Vaccines (Basel), 2020, 8, (3).
- Urbano, A. C.; Ferreira, F. , African swine fever control and prevention: an update on vaccine development. Emerg Microbes Infect 2022, 11 (1), 2021–2033. [Google Scholar] [CrossRef] [PubMed]
- Tran, X. H.; Le, T. T. P.; Nguyen, Q. H.; Do, T. T.; Nguyen, V. D.; Gay, C. G.; Borca, M. V.; Gladue, D. P. , African swine fever virus vaccine candidate ASFV-G-ΔI177L efficiently protects European and native pig breeds against circulating Vietnamese field strain. Transbound Emerg Dis 2022, 69 (4), e497–e504. [Google Scholar]
- Liu, W.; Li, H.; Liu, B.; Lv, T.; Yang, C.; Chen, S.; Feng, L.; Lai, L.; Duan, Z.; Chen, X.; Li, P.; Guan, S.; Chen, L. , A new vaccination regimen using adenovirus-vectored vaccine confers effective protection against African swine fever virus in swine. Emerg Microbes Infect 2023, 12 (2), 2233643. [Google Scholar] [CrossRef] [PubMed]
- Vu, H. L. X.; McVey, D. S. , Recent progress on gene-deleted live-attenuated African swine fever virus vaccines. NPJ Vaccines 2024, 9 (1), 60. [Google Scholar] [CrossRef]
- Alejo, A.; Matamoros, T.; Guerra, M.; Andrés, G. , A Proteomic Atlas of the African Swine Fever Virus Particle. Journal of virology, 2018, 92, (23).
- Xian, Y.; Xiao, C. , The Structure of ASFV Advances the Fight against the Disease. Trends Biochem Sci 2020, (4), 276–278. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhao, D.; Wang, J.; Zhang, Y.; Wang, M.; Gao, Y.; Li, F.; Wang, J.; Bu, Z.; Rao, Z.; Wang, X. , Architecture of African swine fever virus and implications for viral assembly. Science 2019, 366 (6465), 640–644. [Google Scholar] [CrossRef]
- Suárez, C.; Gutiérrez-Berzal, J.; Andrés, G.; Salas, M. L.; Rodríguez, J. M. , African swine fever virus protein p17 is essential for the progression of viral membrane precursors toward icosahedral intermediates. Journal of virology 2010, 84 (15), 7484–99. [Google Scholar] [CrossRef]
- Xia, N.; Wang, H.; Liu, X.; Shao, Q.; Ao, D.; Xu, Y.; Jiang, S.; Luo, J.; Zhang, J.; Chen, N.; Meurens, F.; Zheng, W.; Zhu, J. , African Swine Fever Virus Structural Protein p17 Inhibits Cell Proliferation through ER Stress-ROS Mediated Cell Cycle Arrest. Viruses, 2020, 13, (1).
- Hu, B.; Zhong, G.; Ding, S.; Xu, K.; Peng, X.; Dong, W.; Zhou, J. , African swine fever virus protein p17 promotes mitophagy by facilitating the interaction of SQSTM1 with TOMM70. Virulence 2023, 14 (1), 2232707. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Xia, N.; Zhang, J.; Cao, Q.; Jiang, S.; Luo, J.; Wang, H.; Chen, N.; Zhang, Q.; Meurens, F.; Zhu, J. , African Swine Fever Virus Structural Protein p17 Inhibits cGAS-STING Signaling Pathway Through Interacting With STING. Front Immunol 2022, 13, 941579. [Google Scholar] [CrossRef]
- Wang, S.; Xiang, Z.; Gao, P.; Zhang, Y.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Yang, H. , African swine fever virus structural protein p17 inhibits IRF3 activation by recruiting host protein PR65A and inducing apoptotic degradation of STING. Front Microbiol 2024, 15, 1428233. [Google Scholar] [CrossRef]
- Li, L.; Qiao, S.; Li, G.; Tong, W.; Dong, S.; Liu, J.; Guo, Z.; Zheng, H.; Zhao, R.; Tong, G.; Zhou, Y.; Gao, F. , The Indirect ELISA and Monoclonal Antibody against African Swine Fever Virus p17 Revealed Efficient Detection and Application Prospects. Viruses, 2022, 15, (1).
- Li, L.; Qiao, S.; Wang, S.; Liu, J.; Zhao, K.; Zhou, Y.; Li, G.; Jiang, Y.; Liu, C.; Tong, G.; Tong, W.; Gao, F. , Expression of ASFV p17 in CHO cells and identification of one novel epitope using a monoclonal antibody. Virus Res 2023, 336, 199194. [Google Scholar] [CrossRef] [PubMed]
- Blome, S.; Franzke, K.; Beer, M. , African swine fever - A review of current knowledge. Virus Res 2020, 287, 198099. [Google Scholar] [CrossRef]
- Zhao, D.; Sun, E.; Huang, L.; Ding, L.; Zhu, Y.; Zhang, J.; Shen, D.; Zhang, X.; Zhang, Z.; Ren, T.; Wang, W.; Li, F.; He, X.; Bu, Z. , Highly lethal genotype I and II recombinant African swine fever viruses detected in pigs. Nat Commun 2023, 14 (1), 3096. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.; Zhang, Z.; Wang, Z.; He, X.; Zhang, X.; Wang, L.; Wang, W.; Huang, L.; Xi, F.; Huangfu, H.; Tsegay, G.; Huo, H.; Sun, J.; Tian, Z.; Xia, W.; Yu, X.; Li, F.; Liu, R.; Guan, Y.; Zhao, D.; Bu, Z. , Emergence and prevalence of naturally occurring lower virulent African swine fever viruses in domestic pigs in China in 2020. Sci China Life Sci 2021, 64 (5), 752–765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, S.; Zhang, H.; Qin, Z.; Shan, H.; Cai, X. , Vaccines for African swine fever: an update. Front Microbiol 2023, 14, 1139494. [Google Scholar] [CrossRef] [PubMed]
- Deutschmann, P.; Forth, J. H.; Sehl-Ewert, J.; Carrau, T.; Viaplana, E.; Mancera, J. C.; Urniza, A.; Beer, M.; Blome, S. , Assessment of African swine fever vaccine candidate ASFV-G-∆MGF in a reversion to virulence study. NPJ Vaccines 2023, 8 (1), 78. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Fernández-Pinero, J.; Arias, M. , African swine fever (ASF) diagnosis, an essential tool in the epidemiological investigation. Virus Res 2019, 271, 197676. [Google Scholar] [CrossRef]
- Hu, Z.; Tian, X.; Lai, R.; Wang, X.; Li, X. , Current detection methods of African swine fever virus. Front Vet Sci 2023, 10, 1289676. [Google Scholar] [CrossRef] [PubMed]
- Caixia, W.; Songyin, Q.; Ying, X.; Haoyang, Y.; Haoxuan, L.; Shaoqiang, W.; Chunyan, F.; Xiangmei, L. , Development of a Blocking ELISA Kit for Detection of ASFV Antibody Based on a Monoclonal Antibody Against Full-Length p72. J AOAC Int 2022, 105 (5), 1428–1436. [Google Scholar] [CrossRef]
- Chang, Z.; Du, Y.; Li, R.; Sun, X.; Chen, Y.; Li, M.; Fan, L.; Liu, S.; Wang, S.; Ding, P.; Zhang, G. , Development and characterization of monoclonal antibody against the critical loop structure of african swine fever virus P72 protein. Vet Microbiol 2023, 283, 109776. [Google Scholar] [CrossRef]
- Duan, X.; Liu, Y.; Chen, Z.; Xie, Z.; Tian, C.; Li, Y.; Lv, L.; Wang, R.; Liu, J.; Chen, H. , Identification of monoclonal antibody targeting epitope on p72 trimeric spike of African swine fever virus. Virus Genes 2023, (4), 582–590. [Google Scholar] [CrossRef] [PubMed]
- Tesfagaber, W.; Wang, W.; Wang, L.; Zhao, R.; Zhu, Y.; Li, F.; Sun, E.; Liu, R.; Bu, Z.; Meng, G.; Zhao, D. , A highly efficient blocking ELISA based on p72 monoclonal antibody for the detection of African swine fever virus antibodies and identification of its linear B cell epitope. Int J Biol Macromol 2024, 268 (Pt 1), 131695. [Google Scholar] [CrossRef]
- Wang, A.; Jiang, M.; Liu, H.; Liu, Y.; Zhou, J.; Chen, Y.; Ding, P.; Wang, Y.; Pang, W.; Qi, Y.; Zhang, G. , Development and characterization of monoclonal antibodies against the N-terminal domain of African swine fever virus structural protein, p54. Int J Biol Macromol 2021, 180, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, G.; Dong, H.; Wu, S.; Du, Y.; Wan, B.; Ji, P.; Wu, Y.; Jiang, D.; Zhuang, G.; Duan, H.; Zhang, G.; Zhang, A. , Identification of a Linear B Cell Epitope on p54 of African Swine Fever Virus Using Nanobodies as a Novel Tool. Microbiol Spectr 2023, 11 (3), e0336222. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Han, D.; Zhang, Y.; Zhang, K.; Du, N.; Tong, W.; Li, G.; Zheng, H.; Liu, C.; Gao, F.; Tong, G. , Identification of one novel epitope targeting p54 protein of African swine fever virus using monoclonal antibody and development of a capable ELISA. Res Vet Sci 2021, 141, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Li, C.; Hou, H.; Chen, Y.; Zhang, A.; Han, S.; Wan, B.; Wu, Y.; He, H.; Wang, N.; Du, Y. , A Novel Linear B-Cell Epitope on the P54 Protein of African Swine Fever Virus Identified Using Monoclonal Antibodies. Viruses, 2023, 15, (4).
- Gao, Y.; Xia, T.; Bai, J.; Zhang, L.; Zheng, H.; Jiang, P. , Preparation of Monoclonal Antibodies against the Viral p54 Protein and a Blocking ELISA for Detection of the Antibody against African Swine Fever Virus. Viruses, 2022, 14, (11).
- Wu, P.; Lowe, A. D.; Rodríguez, Y. Y.; Murgia, M. V.; Dodd, K. A.; Rowland, R. R.; Jia, W. , Antigenic regions of African swine fever virus phosphoprotein P30. Transbound Emerg Dis 2020, 67 (5), 1942–1953. [Google Scholar] [CrossRef]
- Zhou, J.; Ni, Y.; Wang, D.; Fan, B.; Zhu, X.; Zhou, J.; Hu, Y.; Li, L.; Li, B. , Development of a Competitive Enzyme-Linked Immunosorbent Assay Targeting the-p30 Protein for Detection of Antibodies against African Swine Fever Virus. Viruses, 2023, 15, (1).
- Tian, P.; Sun, Z.; Wang, M.; Song, J.; Sun, J.; Zhou, L.; Jiang, D.; Zhang, A.; Wu, Y.; Zhang, G. , Identification of a novel linear B-cell epitope on the p30 protein of African swine fever virus using monoclonal antibodies. Virus Res 2024, 341, 199328. [Google Scholar] [CrossRef]
- Ren, D.; Ding, P.; Liu, S.; Zhang, N.; Chen, Y.; Li, Q.; Fan, L.; Chang, Z.; Zhang, G. , Development and characterization of recombinant ASFV CD2v protein nanoparticle-induced monoclonal antibody. Int J Biol Macromol 2022, 209 (Pt A), 533–541. [Google Scholar] [CrossRef]
- Jia, R.; Zhang, G.; Bai, Y.; Liu, H.; Chen, Y.; Ding, P.; Zhou, J.; Feng, H.; Li, M.; Tian, Y.; Wang, A. , Identification of Linear B Cell Epitopes on CD2V Protein of African Swine Fever Virus by Monoclonal Antibodies. Microbiol Spectr 2022, 10 (2), e0105221. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, Q.; Li, L.; Zhang, R.; Chang, Y.; Zhao, J.; Liu, S.; Zhao, X.; Chen, X.; Sun, Y.; Zhao, Q. , Nanobodies against African swine fever virus p72 and CD2v proteins as reagents for developing two cELISAs to detect viral antibodies. Virol Sin 2024, 39 (3), 478–489. [Google Scholar] [CrossRef]
- Chen, Y.; Ni, J.; Wang, C.; Zhai, X.; Luo, T.; Li, Y. P.; Wei, Y.; Liu, Y. , The proteomic analysis uncovers the cellular responses to the African swine fever virus membrane proteins p54, p17, and pB117L. Microbes Infect, 2024, 26, (5-6), 105348.
- Lu, H.; Shao, J.; Liu, W.; Gao, S.; Zhou, G.; Ning, X.; Huang, H.; Liu, Y.; Chang, H. , Screening and identification of linear B-cell epitopes on structural proteins of African Swine Fever Virus. Virus Res 2024, 350, 199465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jiang, S.; Xia, N.; Zhang, Y.; Zhang, J.; Liu, A.; Zhang, C.; Chen, N.; Meurens, F.; Zheng, W.; Zhu, J. , Rapid Visual Detection of African Swine Fever Virus with a CRISPR/Cas12a Lateral Flow Strip Based on Structural Protein Gene D117L. Animals (Basel), 2023, 13, (23).
- Zhang, D.; Jiang, S.; Xia, N.; Zhang, J.; Liu, A.; Deng, D.; Zhang, C.; Sun, Y.; Chen, N.; Kang, X.; Pan, Z.; Zheng, W.; Zhu, J. , Development of visual detection of African swine fever virus using CRISPR/LwCas13a lateral flow strip based on structural protein gene D117L. Vet Microbiol 2024, 293, 110073. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Production and identification of the p17 truncated fusion proteins. (A-C) Analysis of the transmembrane region (A), hydrophilicity (B), and B cell epitopes (C) of p17 protein using online tools, as described in Methods. (D and E) The TF-p17 jd (D) and MBP-p17 jd (E) were induced by IPTG at a final concentration of 1mM at 37 ℃ and 16 ℃. Whole cell lysate, supernatant, and precipitate were examined for p17 protein expressions by SDS-PAGE and Coomassie blue staining, with the major band about 68 kD (D) and 53 kD (E) which are red boxed. (F) The purified p17 fusion proteins were verified by SDS-PAGE and Coomassie blue staining against standard bovine serum albumin (BSA). The concentration of TF-p17 jd protein is greater than 1 μg/μL, while the concentration of MBP-p17 jd protein is approximately 500 ng/μL.
Figure 1.
Production and identification of the p17 truncated fusion proteins. (A-C) Analysis of the transmembrane region (A), hydrophilicity (B), and B cell epitopes (C) of p17 protein using online tools, as described in Methods. (D and E) The TF-p17 jd (D) and MBP-p17 jd (E) were induced by IPTG at a final concentration of 1mM at 37 ℃ and 16 ℃. Whole cell lysate, supernatant, and precipitate were examined for p17 protein expressions by SDS-PAGE and Coomassie blue staining, with the major band about 68 kD (D) and 53 kD (E) which are red boxed. (F) The purified p17 fusion proteins were verified by SDS-PAGE and Coomassie blue staining against standard bovine serum albumin (BSA). The concentration of TF-p17 jd protein is greater than 1 μg/μL, while the concentration of MBP-p17 jd protein is approximately 500 ng/μL.

Figure 2.
Characterization of anti-p17 monoclonal antibodies. (A) Optimization of the amount of coating MBP-p17 jd protein in indirect ELISA for detection of ASFV positive serum. (B) The mAbs were tested in MBP-p17 protein based indirect ELISA. The cell supernatants of hybridoma clones 1G2 and 6G3 were used as the primary antibodies, the SP2/0 cell supernatant was used as the negative control, and the serum of immunized mice was used as positive control. **p < 0.01. (C) Subclass of p17 mAbs 1G2 and 6G3 was determined by the monoclonal antibody subclass identification kit (C060101) from CELLWAY-LAB (Luoyang, China). (D) Measurement of the titers of ascite mAbs 1G2 and 6G3 by MBP-p17 protein indirect ELISA. The dotted line denotes the P/N value of 2.1.
Figure 2.
Characterization of anti-p17 monoclonal antibodies. (A) Optimization of the amount of coating MBP-p17 jd protein in indirect ELISA for detection of ASFV positive serum. (B) The mAbs were tested in MBP-p17 protein based indirect ELISA. The cell supernatants of hybridoma clones 1G2 and 6G3 were used as the primary antibodies, the SP2/0 cell supernatant was used as the negative control, and the serum of immunized mice was used as positive control. **p < 0.01. (C) Subclass of p17 mAbs 1G2 and 6G3 was determined by the monoclonal antibody subclass identification kit (C060101) from CELLWAY-LAB (Luoyang, China). (D) Measurement of the titers of ascite mAbs 1G2 and 6G3 by MBP-p17 protein indirect ELISA. The dotted line denotes the P/N value of 2.1.
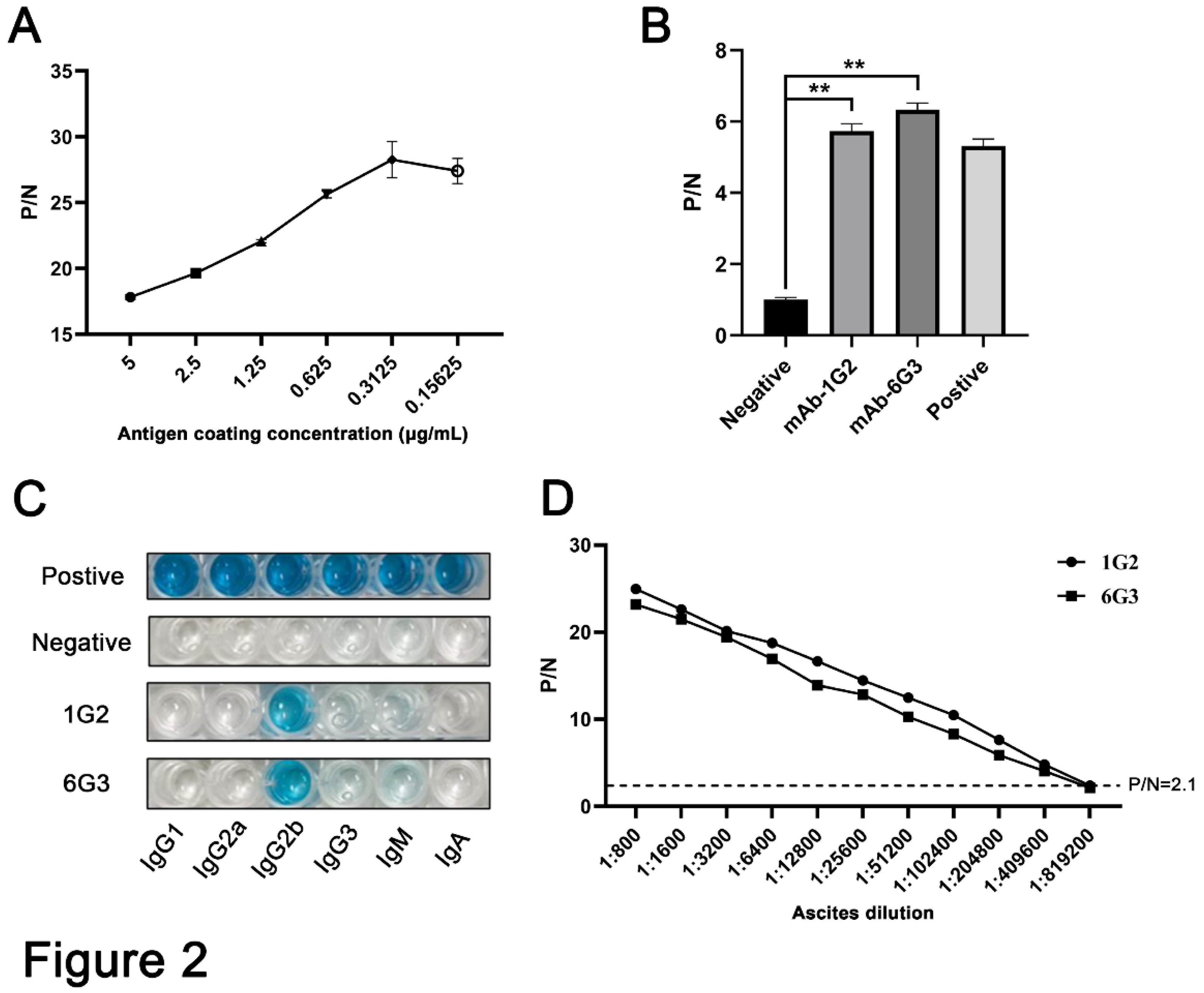
Figure 3.
Analysis of specific reactivity of p17 mAbs by Western blotting. (A-C) 293T cells were transfected with pCAGGS-p17-HA (1μg/mL) (A) and pDsRed-p17 (1μg/mL) (B) with pCAGGS vector and pDsRed vector as the controls. Marc-145 cells were infected with PRRSV-p17 at a multiplicity of infection (MOI) of 0.1 for 72 h (C). Cell were harvested and cell lysates were detected for exogenous p17 by Western blotting with 1G2 and 6G3 mAbs as primary antibodies. (D) Primary PAMs were infected with ASFV (0.1 MOI) or mock infected for 96 h, and cell lysates were detected for endogenous p17 by Western blotting with the mAbs 1G2 and 6G3 as primary antibodies. (E) Primary PAMs, Marc-145 cells, Vero cells, PK15 cells and MDCK cells were infected with ASFV (MOI 0.1), PRRSV (MOI 0.1), PEDV (MOI 0.1), PDCoV (MOI 0.1) and SIV (MOI 0.1), respectively. Cell were harvested and cell lysates were detected the specificity of p17 mAbs by Western blotting with 1G2 and 6G3 mAbs as primary antibodies. The replications of PRRSV, PEDV, PDCoV and SIV were detected by Western blotting with the indicated antibodies.
Figure 3.
Analysis of specific reactivity of p17 mAbs by Western blotting. (A-C) 293T cells were transfected with pCAGGS-p17-HA (1μg/mL) (A) and pDsRed-p17 (1μg/mL) (B) with pCAGGS vector and pDsRed vector as the controls. Marc-145 cells were infected with PRRSV-p17 at a multiplicity of infection (MOI) of 0.1 for 72 h (C). Cell were harvested and cell lysates were detected for exogenous p17 by Western blotting with 1G2 and 6G3 mAbs as primary antibodies. (D) Primary PAMs were infected with ASFV (0.1 MOI) or mock infected for 96 h, and cell lysates were detected for endogenous p17 by Western blotting with the mAbs 1G2 and 6G3 as primary antibodies. (E) Primary PAMs, Marc-145 cells, Vero cells, PK15 cells and MDCK cells were infected with ASFV (MOI 0.1), PRRSV (MOI 0.1), PEDV (MOI 0.1), PDCoV (MOI 0.1) and SIV (MOI 0.1), respectively. Cell were harvested and cell lysates were detected the specificity of p17 mAbs by Western blotting with 1G2 and 6G3 mAbs as primary antibodies. The replications of PRRSV, PEDV, PDCoV and SIV were detected by Western blotting with the indicated antibodies.
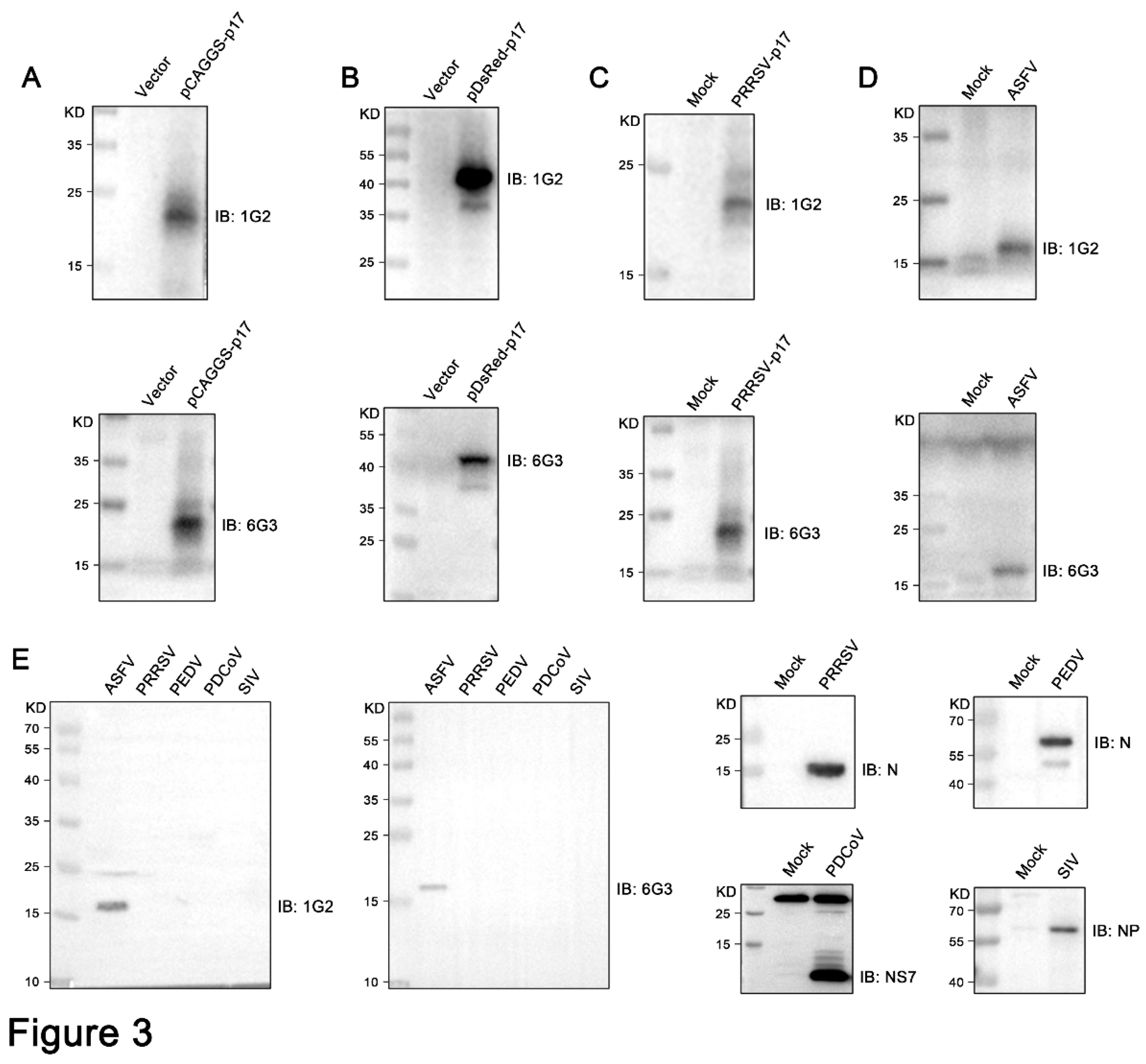
Figure 4.
Analysis of specific reactivity of p17 mAbs by Immunofluorescence. (A) 3D4/21 cells were transfected pCAGGS-p17-HA and pCAGGS vector, respectively. Cells were fixed at 24 h post-transfection and stained with 1G2 or 6G3 mAbs, together with Goat anti-mouse IgG H&L Alexa Fluor 488. Cellular nuclei were counterstained with 4’,6’-diamidino-2-phenylindole (DAPI). (B) Marc-145 cells were infected with PRRSV-p17 (MOI 0.1). Cells were fixed at 72 h post-infection and stained with 1G2 or 6G3, together with secondary antibody and DAPI.
Figure 4.
Analysis of specific reactivity of p17 mAbs by Immunofluorescence. (A) 3D4/21 cells were transfected pCAGGS-p17-HA and pCAGGS vector, respectively. Cells were fixed at 24 h post-transfection and stained with 1G2 or 6G3 mAbs, together with Goat anti-mouse IgG H&L Alexa Fluor 488. Cellular nuclei were counterstained with 4’,6’-diamidino-2-phenylindole (DAPI). (B) Marc-145 cells were infected with PRRSV-p17 (MOI 0.1). Cells were fixed at 72 h post-infection and stained with 1G2 or 6G3, together with secondary antibody and DAPI.
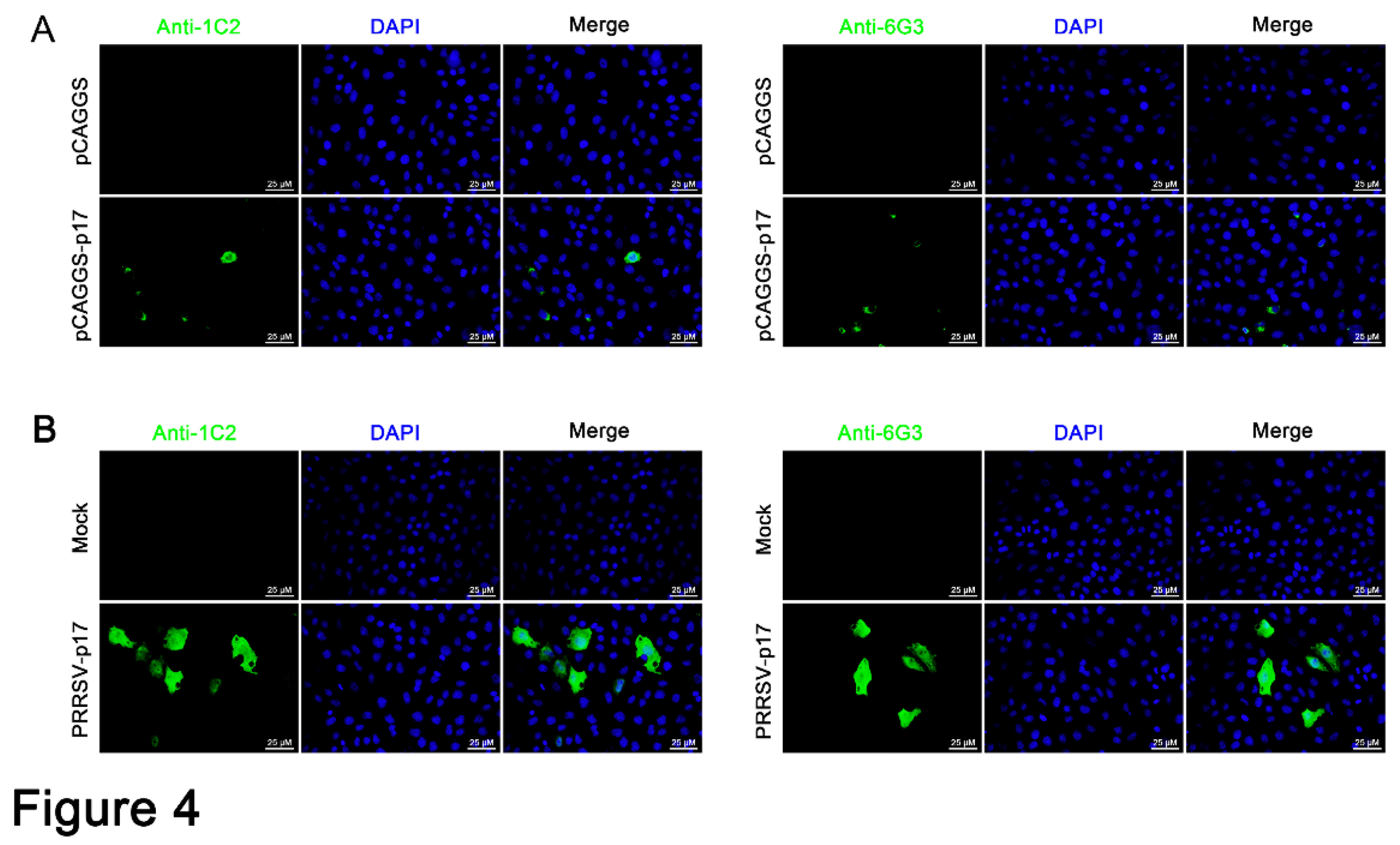
Figure 5.
Precise mapping of the antigenic epitopes recognized by p17 monoclonal antibodies. (A) Schematic strategy for precise mapping the epitope. The fragments with reactivity with p17 mAbs are blue marked, whereas those without reactivity with mAbs are black marked. (B) Western blotting analysis of the critical C terminal amino acid (left) and N terminal amino acid (right) for reactivity of p17 proteins with the two mAbs 1G2 and 6G3. The aa is an abbreviation for amino acid.
Figure 5.
Precise mapping of the antigenic epitopes recognized by p17 monoclonal antibodies. (A) Schematic strategy for precise mapping the epitope. The fragments with reactivity with p17 mAbs are blue marked, whereas those without reactivity with mAbs are black marked. (B) Western blotting analysis of the critical C terminal amino acid (left) and N terminal amino acid (right) for reactivity of p17 proteins with the two mAbs 1G2 and 6G3. The aa is an abbreviation for amino acid.
Figure 6.
Conservation analysis of the identified novel linear epitope of the p17 protein. (A) Alignment of the epitope (72KPPPSYY78) in 30 representative genotypes I and II ASFV strains. The red box indicated the identified conserved epitope. (B) Prediction of the p17 structure by using Alphafold2 online web tools and visualization using PyMOL. The epitope recognized by two mAbs is displayed in pink color, and shown in the front view (a) and in flipping 180° along the X-axis (b), respectively.
Figure 6.
Conservation analysis of the identified novel linear epitope of the p17 protein. (A) Alignment of the epitope (72KPPPSYY78) in 30 representative genotypes I and II ASFV strains. The red box indicated the identified conserved epitope. (B) Prediction of the p17 structure by using Alphafold2 online web tools and visualization using PyMOL. The epitope recognized by two mAbs is displayed in pink color, and shown in the front view (a) and in flipping 180° along the X-axis (b), respectively.

Figure 7.
Establishment of epitope indirect ELISA for detecting of ASFV antibodies. (A) The p17 epitope peptide (synthesized by GeneCreate Wuhan China) was used for coating at the concentrations from 0.3125-10 μg/mL. The epitope ELISA was tested for detection of ASFV positive serum (1:5 dilution) and negative normal pig serum to determine the optimized concentration of coating epitope peptide. (B) The optimization of serum dilution in indirect ELISA with peptide coating concentration at 0.3125 μg/mL. (C) The specificity of the established epitope based indirect ELISA. PRRSV, PEDV, SIV and ASFV positive pig sera and negative pig serum were used. **p < 0.01. (D and E) 24 clinical pig serum samples were tested using our established indirect epitope peptide ELISA (D) and the commercial ASFV ELISA detection kit (E), with pig negative serum as the control. The samples marked in red (D) and blue (E) are those with positive detection results.
Figure 7.
Establishment of epitope indirect ELISA for detecting of ASFV antibodies. (A) The p17 epitope peptide (synthesized by GeneCreate Wuhan China) was used for coating at the concentrations from 0.3125-10 μg/mL. The epitope ELISA was tested for detection of ASFV positive serum (1:5 dilution) and negative normal pig serum to determine the optimized concentration of coating epitope peptide. (B) The optimization of serum dilution in indirect ELISA with peptide coating concentration at 0.3125 μg/mL. (C) The specificity of the established epitope based indirect ELISA. PRRSV, PEDV, SIV and ASFV positive pig sera and negative pig serum were used. **p < 0.01. (D and E) 24 clinical pig serum samples were tested using our established indirect epitope peptide ELISA (D) and the commercial ASFV ELISA detection kit (E), with pig negative serum as the control. The samples marked in red (D) and blue (E) are those with positive detection results.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



