
Submitted:
30 October 2024
Posted:
30 October 2024
You are already at the latest version
Abstract
This article provides a comprehensive overview of lung cancer from a genetic perspective, emphasizing the molecular pathways involved in its pathogenesis. This review presents the genetic syndromes and key genomic variations, including alterations and instabilities, that contribute to tumor development and progression. Furthermore, advancements in genomic testing, such as liquid biopsy, are explored, highlighting their utility in early diagnosis, prognostic evaluation, and personalized treatment strategies. The role of biomarkers in informing therapeutic decisions and optimizing patient outcomes is also examined, offering insights into their potential to refine clinical interventions. This review aims to enrich the understanding of the genetic landscape of lung cancer, providing insights that may lead to improved diagnostic and therapeutic approaches.
Keywords:
lung cancer
; NSCLC
; SCLC
; genetics
; pathways
; alterations
; biomarkers
; diagnosis
; therapies
; personalized medicine
1. Introduction
Lung cancer, a malignant neoplasm originating from the epithelial cells of the lung, is characterized by its aggressive growth and poor prognosis, making it a leading cause of cancer-related mortality globally [1,2]. In the European Union (EU), lung cancer represents a significant public health challenge, accounting for approximately one-fifth of all cancer-related deaths. In 2020, lung cancer was responsible for the deaths of nearly a quarter of a million individuals within the EU, constituting 4.5% of the overall mortality rate and 19.8% of all cancer-related deaths. The epidemiology of lung cancer exhibits a gender disparity, with males demonstrating a higher mortality rate at 5.9%, nearly double that of females at 3.0% [3,4].
2. Classification of Lung Cancers
From a histological perspective, lung cancer is primarily divided into two main subtypes: small cell lung cancer (SCLC) and non-small cell lung carcinoma (NSCLC). Additionally, there are less common forms of lung cancer, such as carcinoid tumors, bronchial gland carcinomas and sarcomatoid carcinomas (Figure 1). NSCLC constitutes approximately 85% of all lung cancer cases, while SCLC represents 10-15% of cases [5].
In 2021, the World Health Organization (WHO) revised the classification of lung tumors based on morphological characteristics, molecular abnormalities, and immunohistochemical profiles. The main categories of lung cancer classification now include epithelial tumors, lung neuroendocrine neoplasms, tumors of ectopic tissues, mesenchymal tumors specific to the lungs, and hematolymphoid tumors [6].
3. Genetic Predisposition and Associated Genes
Smoking is the principal cause of lung cancer, accounting for the majority of cases, though other factors such as air pollution, ionizing radiation, chronic pulmonary conditions, and occupational exposures also contribute, either independently or synergistically [7,8]. Additionally, a significant number of lung tumors occur in non-smokers, often without identifiable external risk factors. Individuals with a family history of lung cancer, especially those with early-onset disease, exhibit an elevated risk, indicating that genetic factors play a crucial role in the development of lung cancer.
Lung tumors cases associated with inherited genetic variations typically exhibit an autosomal dominant inheritance pattern. Most lung cancer cases are not linked to inherited genetic mutations, but rather arise from somatic mutations specifically within lung cells. However, there are several genetic syndromes known to present a risk for developing lung tumors (Figure 2). These genetic conditions vary in their level of association with lung cancer, reflecting a range of potential risks [8,9,10,11].
Cancer cells emerge from non-malignant tissues through the sequential accumulation of molecular alterations that drive proliferation, bypass growth suppression and apoptosis, and enable processes such as angiogenesis, invasion, and metastasis. Typically, cancer-related genes are categorized as either oncogenes, which promote tumorigenesis through gain-of-function mutations, or tumor suppressor genes (TSGs), which require loss-of-function mutations to lose their protective roles against cancer [12].
In lung adenocarcinoma, key oncogenes such as KRAS, EGFR, ALK, ERBB2, and BRAF are frequently mutated and are found in over 50% of cases [13,14]. Alongside these alterations, tumor suppressor genes like TP53, STK11, and CDKN2A are significantly impacted, with other suppressors such as NF1, ATM, APC, and RB1 also playing crucial roles in the development and progression of lung adenocarcinoma [14,15].
However, some genes can exhibit both oncogenic and tumor-suppressive functions depending on the cellular context, due to the expression of multiple isoforms and post-translational modifications [15]. This dual functionality underscores the complexity of cancer biology, where single genetic events can shift the balance from tumor suppression to oncogenesis.
However, some genes can exhibit both oncogenic and tumor-suppressive functions depending on the cellular context, due to the expression of multiple isoforms and post-translational modifications [15]. This dual functionality underscores the complexity of cancer biology, where single genetic events can shift the balance from tumor suppression to oncogenesis.
In addition to the roles of oncogenes and TSGs in cancer development, another pivotal mechanism is the emergence of gene fusions, known as oncofusions (Figure 3). These hybrid genes result from genomic rearrangements such as deletions, inversions, translocations, or duplications, where two previously independent genes are juxtaposed [16,17].
Oncofusions arise primarily from these rearrangements and contribute to tumorigenesis by either deregulating existing cellular pathways or creating novel, oncogenic activities. With over 10,000 gene fusions identified in various human cancers, they represent a significant category of driver alterations in cancer biology [15,16,17]. The most common targetable fusions involve the anaplastic lymphoma kinase (ALK) gene, followed by those involving ROS1 and RET proto-oncogenes. These fusions provide critical targets for precision therapies in NSCLC patients [18].
4. Molecular Pathways and Therapeutic Strategies
Lung cancer is a complex and multifactorial disease, driven by intricate molecular mechanisms that regulate cell proliferation, survival, and metastasis. This complexity arises from numerous interconnected signaling pathways, each playing a distinct role in driving cancer progression and shaping the tumor microenvironment. Among the most critical pathways implicated in its pathogenesis are those governing cellular responses to growth factors, energy stress, and immune evasion [19,20]. These signaling cascades, including MAPK, JAK-STAT, and EGFR/PI3K/Akt/mTOR, orchestrate key oncogenic processes, while dysregulation in pathways like Wnt/β-catenin, VEGF/VEGFR, and NF-κB promotes tumor progression and angiogenesis.
Moreover, aberrations in hypoxia signaling, apoptosis regulation, and immune checkpoints further enable cancer cells to adapt and evade therapeutic interventions, making these pathways essential targets in lung cancer treatment and research. By thoroughly examining how these disrupted pathways interact with one another, we can gain a deeper understanding of the complex mechanisms driving tumor biology, which not only enhances our knowledge of cancer progression but also uncovers new and potentially more effective therapeutic targets that could be exploited for treatment interventions.
4.1. The MAPK pathway, also known as the RAS-RAF-MEK-ERK pathway, is pivotal in transmitting external signals into cellular actions, regulating processes such as proliferation, survival, and metastasis. The pathway's four primary cascades—ERK1/2, JNK, p38, and ERK5—each play specialized roles in response to stimuli like growth factors or environmental stresses (Figure 4). Active at all stages of tumor development, this pathway contributes significantly to cancer progression, especially through mutations like KRAS, which are found in 30% of lung adenocarcinomas [19,20,21]. Such mutations lead to uncontrolled growth and resistance to apoptosis.
The dysregulation of the RAS-RAF-MEK-ERK pathway contributes to the aggressive nature of these cancers, making it a key target for therapeutic strategies. The interaction between the RAS and TGF-β signaling pathways drives epithelial-mesenchymal transition (EMT) and extracellular matrix (ECM) remodeling, which are key processes in cancer invasion and metastasis. The RREB1 gene, activated by RAS, primes EMT and fibrogenic gene enhancers, which are then activated by the TGF-β/SMAD pathway. Blocking RREB1 disrupts this process, preventing metastasis and presenting RREB1 as a potential target for cancer therapy [22,23].
4.2. The JAK-STAT pathway is a highly conserved signaling mechanism that is crucial for transmitting extracellular signals into the nucleus, ultimately leading to changes in gene expression (Figure 5). This pathway involves four JAK proteins (JAK1, JAK2, JAK3, and TYK2) and seven STAT proteins (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6). The pathway is primarily activated by cytokines binding to their respective cell-surface receptors. This binding event triggers the activation of receptor-associated Janus kinases (JAKs). Once activated, JAKs phosphorylate specific tyrosine residues on the receptor, facilitating the recruitment of Signal Transducers and Activators of Transcription (STATs). In addition to cytokines, growth factors such as EGF can also activate STATs, expanding the pathway’s role in cellular signaling. Phosphorylated STATs then dimerize, typically through interactions between their SH2 domains and phosphorylated tyrosine residues [24]. These dimers translocate to the nucleus, where they bind to DNA and modulate the transcription of target genes. In the context of NSCLC, the JAK-STAT pathway, particularly STAT3 and STAT5, is frequently aberrantly activated. Persistent activation of STAT3 has been linked to enhanced tumor proliferation, survival, and metastasis. This constitutive activation supports oncogenic processes such as resistance to apoptosis, angiogenesis, and increased invasive potential, contributing to the aggressive nature of NSCLC [25].
4.3. The LKB1/AMPK/mTOR pathway is a critical signaling cascade that regulates energy homeostasis, cell growth, and survival. Liver Kinase B1 (LKB1) is a key regulator of this pathway, activating AMPK in response to cellular energy changes, acting as a metabolic checkpoint. This pathway also helps maintain redox balance, particularly through NADPH regulation. AMPK inhibits the mTOR pathway, which controls cell growth and metabolism, conserving energy during metabolic stress (Figure 6). Additionally, the pathway influences cell polarity, the cytoskeleton, and the cell cycle, and functions as a tumor suppressor [26]. In NSCLC, the LKB1/AMPK/mTOR pathway plays a dual role. While LKB1 acts as a tumor suppressor by regulating cell polarity, energy metabolism, and inhibiting cell proliferation, its dysregulation is associated with tumor progression and metastasis. Loss of LKB1 function disrupts these regulatory mechanisms, leading to increased invasive and migratory capabilities of cancer cells.
Notably, while LKB1/AMPK signaling typically acts as a barrier to tumorigenesis by maintaining energy balance and inhibiting mTOR activity, its dysregulation can also confer survival advantages to cancer cells, particularly in the nutrient-deprived tumor microenvironment. Furthermore, genetic variations within this pathway, such as mutations in AKT and TSC1, have been associated with differential responses to chemotherapy in NSCLC patients, suggesting that the LKB1/AMPK/mTOR axis may also impact treatment outcomes [26,27].
4.4. The EGFR/PI3K/Akt/mTOR pathway is integral to the progression of various cancers, including NSCLC. This pathway controls vital cellular processes such as metabolism, migration, cell survival, and proliferation and its impact extends to modifying the tumor microenvironment [28]. Specifically, it facilitates the recruitment of inflammatory cells and stimulates angiogenesis, which is essential for tumor growth and metastasis. (Figure 7). In lung cancer, mutations and amplifications in the PIK3CA gene are frequently observed, particularly in squamous cell carcinoma, and significantly contribute to tumorigenesis and disease progression.
Moreover, the activation of Akt and mTOR, driven by mutations in PIK3CA, EGFR, or KRAS, is frequently associated with resistance to conventional therapies, further complicating treatment strategies. The EGFR/PI3K/Akt/mTOR pathway governs several processes in lung cancer, including genomic stability, cell survival, senescence, angiogenesis, proliferation, metastasis, and cellular metabolism. Therefore, any dysregulation or mutation within this pathway can substantially contribute to lung cancer progression. In NSCLC, one of the primary mutations occurs in receptor tyrosine kinases (RTKs), which act as the initial trigger for the pathway. Similarly, KRAS mutations can activate this pathway through alternate mechanisms. The pathway’s importance is further highlighted by its role in tumors with activating mutations, particularly those involving EGFR. Research shows that the Akt/mTOR signaling cascade remains constitutively active in approximately 67% of patients with EGFR mutations [28,29].
For patients with EGFR mutations, several targeted therapies have been developed, offering a range of benefits based on the specific mutation and patterns of resistance observed. One notable example is Osimertinib, a third-generation EGFR tyrosine kinase inhibitor (TKI), which has shown significant efficacy, especially in patients with T790M-positive mutations. Due to its favorable progression-free survival (PFS) and overall survival (OS) outcomes, Osimertinib is often considered a preferred first-line treatment for such patients [30,31,32].
4.5. The Keap1-Nrf2 pathway plays a important role in cellular defense by regulating the expression of detoxifying and antioxidant enzymes. Nrf2 (Nuclear factor erythroid 2-related factor 2) is a transcription factor that drives the expression of these protective genes, while Keap1 (Kelch-like ECH-associated protein 1) serves as its primary regulator. Under normal conditions, Keap1 binds to Nrf2, promoting its ubiquitination and degradation, thereby maintaining low Nrf2 activity [33]. However, under oxidative stress, modifications in Keap1 prevent this degradation, allowing Nrf2 to accumulate and activate the transcription of target genes involved in cellular protection (Figure 8).
In lung cancer, alterations in the Keap1-Nrf2 pathway, such as mutations or loss of Keap1 function, are common and significantly contribute to tumor development. These mutations often disrupt the Keap1-Nrf2 interaction, leading to increased Nrf2 activity and the upregulation of genes that promote tumor survival and progression [34]. Studies have shown that Keap1 mutations are frequent in lung squamous cell carcinoma, leading to persistent Nrf2 activation, which promotes drug resistance and tumor growth [33,34]. Furthermore, Nrf2 mutations, particularly in regions interacting with Keap1, further enhance Nrf2's protective role in cancer cells, contributing to their survival under stress conditions.
4.6. The Wnt/β-catenin pathway is triggered when Wnt ligands bind to cell receptors, stabilizing β-catenin and allowing it to accumulate in the nucleus, where it promotes gene expression related to cell growth and survival. Without Wnt signaling, β-catenin is degraded (Figure 9).
In lung adenocarcinoma cells, WIF1 overexpression has been shown to inhibit migration and invasion by downregulating Wnt/β-catenin signaling. Similarly, the secreted frizzled-related protein (sFRP) family antagonizes Wnt signaling by competing with Fzd receptors for Wnt ligands, while Dickkopf (DKK) family members like DKK-1 are often overexpressed in lung cancer tissues and associated with tumor progression and metastasis [35]. Additionally, the DKK-3 gene, when reactivated, can induce cell cycle arrest and apoptosis, and improve cisplatin sensitivity in resistant cell lines. Oncogenes like TIPE and high mobility group protein 3 (HMGB3) also influence this pathway; TIPE promotes NSCLC cell proliferation by downregulating Wnt/β-catenin signaling, while HMGB3 is linked to poor prognosis due to its role in enhancing Wnt signaling activity [36,37].
4.7. VEGF/VEGFR Pathway - Vascular endothelial growth factor (VEGF), predominantly secreted by tumor cells, stromal cells, and endothelial cells within the tumor microenvironment (TME), consists of several family members, including VEGF-A, VEGF-B, and VEGF-C. These factors exert their effects by binding to specific receptors, mainly tyrosine kinase receptors (VEGFRs) and neuropilin receptors (NRPs).
VEGF-A, the most important for angiogenesis, primarily binds to VEGFR-2, leading to receptor dimerization and activation of downstream signaling pathways essential for endothelial cell proliferation and migration (Figure 10). When VEGF-A binds to VEGFR-2, it triggers several intracellular signaling cascades, including phospholipase C γ (PLC-γ) and phosphoinositide 3-kinase (PI3K). PLC-γ hydrolyzes phosphatidylinositol-4,5-bisphosphate to produce inositol triphosphate (IP3) and diacylglycerol (DAG), leading to an increase in intracellular calcium and the activation of protein kinase C (PKC). PKC, in turn, stimulates the Raf1-MEK1/2-ERK1/2 pathway, promoting endothelial cell proliferation. Concurrently, PI3K activation results in AKT phosphorylation, which enhances endothelial nitric oxide synthase (eNOS) activity, supporting cell proliferation and migration. These processes are critical for the angiogenic response and tumor growth [38,39].
In lung cancer, VEGF promotes the formation of new blood vessels, which are necessary for tumors to grow and metastasize. VEGF achieves this by binding to its primary receptor, VEGFR-2, on endothelial cells, activating pathways that promote cell proliferation and survival. In NSCLC, high VEGF levels are associated with poor prognosis, increased tumor recurrence, and metastasis. VEGF also directly impacts tumor cells by promoting their proliferation and survival. Bevacizumab, a treatment for lung cancer, inhibits VEGF-A activity, which is often upregulated by hypoxia-inducible factor 1 (HIF-1). This inhibition prevents the abnormal angiogenesis driven by onco-metabolic stress, making Bevacizumab a cytostatic rather than a cytotoxic agent [40,41].
4.8. NF-κB Pathway - Nuclear factor-kappa B is a critical transcription factor that regulates inflammation, cell survival, and proliferation. Its dysregulation is implicated in various cancers, including lung cancer. The NF-κB family consists of five proteins—p65 (RelA), RelB, c-Rel, p50/p105 (NF-κB1), and p52 (NF-κB2)—that form dimers and bind DNA to regulate gene expression. p65, RelB, and c-Rel act as transcriptional activators, while p50 and p52 primarily function as repressors. NF-κB controls numerous genes related to key cellular processes, making it a central player in lung cancer by modulating both inflammation and cell survival [42].
In lung cancer, particularly K-ras-driven tumors, NF-κB activation plays a crucial role in tumor progression. K-ras mutations, which are common in lung adenocarcinomas, correlate with aggressive disease. NF-κB is activated through the canonical pathway involving IKK-β and promotes tumor growth by enhancing cell proliferation and survival (Figure 11). It regulates key targets like Cyclin D1, driving the cell cycle, and PTEN, a tumor suppressor that NF-κB suppresses. The suppression of PTEN activates the PI3K/AKT pathway, further promoting cell survival and therapy resistance, both critical factors in lung cancer development and maintenance [42,43].
4.9. PD-1/PD-L1 Pathway is a crucial immune checkpoint mechanism that regulates immune tolerance within the tumor microenvironment. PD-1 (Programmed Cell Death Protein 1) is an inhibitory receptor expressed on T cells, while PD-L1 (Programmed Death-Ligand 1) is its ligand, found on various tumor cells and in the TME. When PD-1 interacts with PD-L1, it inhibits T cell activation and function, leading to a diminished anti-tumor immune response, allowing cancer cells to evade immune surveillance. This pathway plays a significant role in cancer progression by facilitating immune evasion and tumor persistence through suppression of effective immune responses [44]. Several signaling pathways, such as PI3K/AKT, MAPK, and JAK-STAT, influence the PD-1/PD-L1 axis, impacting its role in tumorigenesis. For instance, the PI3K/AKT pathway can upregulate PD-L1 expression, enhancing immune evasion by tumors. Similarly, the MAPK pathway regulates PD-L1 levels through both transcriptional and post-translational mechanisms. The JAK-STAT pathway also plays a role in regulating PD-L1 expression, and JAK2 inhibitors have shown potential in suppressing PD-L1 levels to improve anti-tumor immunity. Additionally, the Wnt and NF-κB pathways also modulate PD-L1 expression, influencing the efficacy of immunotherapies [45,46].
The introduction of anti-PD-1/PD-L1 inhibitors has transformed the treatment of advanced NSCLC, offering patients significantly extended survival and prolonged disease control. A subset of patients even achieves long-term progression-free survival or no detectable disease. However, the response to PD-1/PD-L1 blockade remains limited to a portion of patients, leaving many unresponsive to monotherapy. For NSCLC patients with high PD-L1 expression, Atezolizumab has been shown to significantly extend overall survival compared to platinum-based chemotherapy [47].
4.10. Apoptosis Pathway involves two primary pathways in lung cancer: the extrinsic death receptor pathway and the intrinsic mitochondrial pathway. The extrinsic pathway is initiated when death ligands, such as Fas ligand (Fas-L), bind to their receptors, such as Fas receptor (Fas-R), leading to the recruitment and activation of caspase-8. Caspase-8, in turn, activates caspase-3, which cleaves proteins and fragments DNA, driving apoptosis.
Additionally, caspase-8 can activate Bid, increasing mitochondrial membrane permeability and releasing cytochrome c, thereby promoting the intrinsic pathway of apoptosis [48].
In non-small cell lung cancer , disruptions in these apoptotic pathways are common. Loss of key apoptotic factors like FasL and Apaf-1, as well as mutations in death receptors such as TRAIL receptor 2, can impair apoptosis and contribute to therapy resistance. Elevated levels of anti-apoptotic proteins, including Bcl-2 and pro-caspase-3, are frequently observed in NSCLC and are associated with poor prognosis and reduced treatment effectiveness [48,49]. Growth factor signaling pathways, such as those involving IGF-1R, EGFR, and K-Ras, exacerbate anti-apoptotic signaling in lung cancer, promoting cell survival and resistance to therapy. Additionally, deficiencies in MAPK pathways, such as JNK and p38, impair radiation-induced apoptosis. Elevated levels of MKP1/CL100 phosphatase may also impair JNK activity, further contributing to therapy resistance. Understanding these disruptions is crucial for developing targeted therapies to overcome resistance in NSCLC [49].
4.11. HIF (Hypoxia-Induced) Pathway plays a key role in cellular metabolism, especially in tumor biology. HIF is a transcription factor composed of α- and β-subunits, with HIF-1α being crucial for oxygen-sensitive responses (Figure 12). Under normal oxygen levels, HIF-1α is rapidly degraded through hydroxylation and ubiquitination, mediated by the von Hippel-Lindau (pVHL) protein, resulting in a short half-life. This degradation prevents the activation of downstream genes involved in glycolysis and hypoxic adaptation. However, in low-oxygen (hypoxic) environments, HIF-1α stabilizes and translocates to the nucleus, where it activates genes that promote glycolysis and other metabolic changes that support tumor growth [50,51].
The regulation of HIF-1α is influenced by both oxygen-dependent mechanisms, such as prolyl hydroxylase domain proteins (PHDs) and factor inhibiting HIF (FIH), and oxygen-independent mechanisms, including NF-κB, SP1, and STAT3. Together, these pathways contribute to the metabolic shift observed in tumors. In NSCLC, HIF-1α is overexpressed, driving glycolysis even in the presence of oxygen (the Warburg effect) and facilitating tumor adaptation to hypoxia. HIF-1α also enhances the expression of glycolytic enzymes and pro-angiogenic factors, promoting tumor progression and therapy resistance. Elevated HIF-1α levels are linked to poor prognosis in NSCLC, making it an important therapeutic target [50,51,52,53,54].
4.12. The Notch signaling pathway plays a complex role in NSCLC, affecting both cell growth and apoptosis. In NSCLC, Notch1 is often overexpressed, especially under low-oxygen (hypoxic) conditions, which support tumor growth and survival (Figure 13). However, its effects vary by cell type. For instance, Notch1 inhibition can increase cell death and decrease growth in certain NSCLC cells, like A549, while in others, such as squamous cell carcinoma (SCC), it may have the opposite effect. This shows that the effects of Notch1 are cell-type dependent [55].
Notch1 also interacts with other pathways, such as PI3K/AKT and hypoxia signaling, to further promote cancer growth. Blocking Notch1, for example, with γ-secretase inhibitors, can reduce NSCLC viability by inducing apoptosis. Notch3 also plays a role, particularly in its interaction with the EGFR-MAPK and Wnt signaling pathways. Targeting Notch3 has been shown to reduce tumor growth by promoting apoptosis and may work well in combination therapies aimed at multiple pathways [56,57]. In summary, Notch1 and Notch3 are crucial in NSCLC development, and targeting them offers promising therapeutic potential, particularly when combined with inhibitors for other pathways.
5. Genomic Alterations and Instabilities in Lung Cancer
Lung cancer is driven by various genomic alterations including point mutations, insertions-deletions, copy number variations, gene fusions, and rearrangements, all of which contribute to tumor initiation and progression. These mutations frequently affect key oncogenes and tumor suppressor genes, leading to dysregulated processes like uncontrolled proliferation, evasion of apoptosis, and increased metastatic potential [58].
KRAS mutations, especially at codon 12 (e.g., G12C, G12D, G12V), result in the continuous activation of the RAS signaling pathway, driving uncontrolled proliferation. Mutations like KRAS G12D are associated with poorer clinical outcomes, including shorter progression-free survival (PFS) and overall survival (OS). Certain variants, like KRAS G>T, have been linked to more favorable outcomes [58,59].
EGFR mutations, mainly in exons 18–21, affect the tyrosine kinase domain, with around 90% involving microdeletions in exon 19 and exon 21. These mutations make tumors highly responsive to EGFR tyrosine kinase inhibitors (TKIs), although their role in surgically resectable lung cancer is still debated [59].
ALK gene mutations, such as His694Arg (H694R) and Glu1384Lys (E1384K), activate downstream pathways like STAT3 and AKT, promoting oncogenesis. This has made ALK inhibitors like Lorlatinib, Ceritinib, and Brigatinib essential for treating ALK-positive tumors [60,61].
In addition to specific mutations, genomic instability—particularly chromosomal instability (CIN) and microsatellite instability (MSI)—plays a key role in lung cancer progression:
- CIN contributes to chromosomal aberrations such as gain of chromosome 5p (enhancing TERT expression for tumor growth) or loss of chromosome 9p21 (inactivating CDKN2A, leading to unchecked proliferation). CIN promotes karyotype heterogeneity, fostering genetic diversity within tumors, complicating treatment, and enabling adaptation to evolving conditions. CIN also supports the creation of an inflammatory tumor microenvironment that promotes pro-tumor signaling and metabolic changes that favor cancer cell survival and immune evasion [62].
- MSI is less common in lung cancer, but when present, it often arises due to defects in DNA mismatch repair (MMR) genes like MSH2 and MLH1, leading to a hypermutated state and increased neoantigen formation, making MSI-high tumors more responsive to immunotherapy. MSI mutations, such as those in TGFBR2, further promote genomic instability and cancer progression [63].
6. Genetic Testing and Biomarkers in Lung Cancer
One of the most significant advancements in lung cancer diagnosis and treatment over the past decade has been the transition towards personalized medicine. In this approach, therapeutic decisions are guided by the unique histologic and genetic characteristics of the patient’s tumor, allowing for more targeted treatment strategies [64]. Genetic testing is now a standard practice for patients with NSCLC.
In lung cancer, the detection of specific molecular changes helps classify tumors more accurately, guiding clinicians in determining the most appropriate course of treatment. By identifying these genetic alterations, oncologists can prescribe therapies that target the molecular mechanisms driving cancer growth [65]. This approach not only improves treatment efficacy but also reduces unnecessary interventions, thereby minimizing the potential side effects of broader therapies. Drawing on the Centers of Disease Control Genetics Working Group framework, the applicability of genetic testing for lung cancer prevention can be evaluated based on several critical criteria. Notably, a high proportion of the population carries genetic variants linked to an increased risk of developing lung cancer, suggesting a broad potential for genetic testing.
However, there are significant challenges, including the need to maintain high-quality standards in genetic testing laboratories and the costs associated with widespread genetic testing. Furthermore, the strength of the association between the identified genotype and lung cancer risk has not been prospectively validated, raising concerns about the clinical utility of these tests. Therefore, while genetic testing holds promise, further research and refinement are needed to meet the rigorous standards required for its effective use in lung cancer prevention [66].
Lung cancer screening, as recommended by the National Comprehensive Cancer Network (NCCN), provides several significant benefits (Figure 14). Early detection of lung cancer can reduce mortality by enabling timely interventions that improve survival rates. Patients diagnosed at earlier stages often require less aggressive treatment, which helps to reduce the side effects of both the disease and its treatments [67]. Additionally, lung cancer screening can motivate healthier lifestyle changes, such as smoking cessation, and reduce the psychosocial burden by relieving anxiety related to lung cancer risk. Screening also helps to identify tumors when they are smaller and less invasive, contributing to a better overall quality of life for patients.
Despite its advantages, genetic testing in the context of lung cancer carries certain risks. One concern is the potential to identify indolent tumors—those that may not cause harm during the patient’s lifetime—which could lead to unnecessary treatment, potentially reducing quality of life. The psychological impact of test results, including anxiety and stress, is also a key consideration. There are physical risks associated with follow-up diagnostic procedures, such as biopsies, and the possibility of false-positive or false-negative results. False positives may lead to unnecessary treatments, while false negatives could result in missed opportunities for early intervention. Furthermore, the cost of genetic testing and subsequent follow-up procedures, including radiation exposure during diagnostic imaging, adds an additional layer of complexity.
Next-generation sequencing (NGS) has revolutionized lung cancer diagnostics and treatment strategies. In the era of precision medicine, this technology enables comprehensive tumor molecular profiling, providing critical insights into a wide range of cancer-related mutations. This increases the possibility of identifying germline mutations that may have implications for both treatment and prevention [68]. NGS facilitates a comprehensive examination of the genome or exome in any type of cancer by enabling the parallel sequencing of millions of short reads from various nucleic acids, such as micro-RNA and other non-protein-coding DNA species [69]. NGS applications include whole-genome sequencing (WGS), whole-exome sequencing (WES), RNA expression profiling, and targeted oncology panels that may cover anywhere from a few to hundreds of genes.
While NGS offers significant advantages that have made it indispensable in both research and clinical practice, it also presents certain limitations, notably the requirement for sophisticated bioinformatics tools and highly trained personnel to manage the experimental procedures and data analysis. Some advantages of NGS include lower costs, quick processing times, a wide range of applications, and utility in both research and clinical environments [70]. Additionally, commercial NGS platforms and specialized kits are widely available. However, NGS also has disadvantages, such as the need for specialized software and computing resources for data analysis, a lack of standardized protocols or materials for clinical applications, and remaining cost barriers in certain developing countries. Beyond NGS, several other genetic testing techniques are crucial in lung cancer diagnostics. Fluorescence in situ hybridization (FISH) can identify chromosomal abnormalities when traditional sequencing methods fail, making it a valuable complementary technique, especially in cases where limited tumor tissue is available. Additionally, polymerase chain reaction (PCR) remains a gold standard for detecting specific mutations in genes like EGFR, KRAS, and BRAF [70,71].
Emerging technologies like liquid biopsy have gained considerable attention in recent years due to its minimal invasiveness and ability to provide continuous, real-time insights into tumor biology [72]. This technique allows for repeated sampling of biological material such as circulating tumor cells (CTCs), extracellular vesicles (EVs), cell-free DNA (cfDNA), circulating tumor DNA (ctDNA), non-coding RNAs (ncRNAs), and proteins, among others [73].
These biomarkers, detectable in bodily fluids such as blood, cerebrospinal fluid, saliva, pleural fluid, and urine, offer essential information for the early diagnosis of lung cancer, therapeutic monitoring, and prognostic assessment. The concentrations of these tumor markers can vary depending on factors like tumor stage, metastasis, and genomic instability, making them valuable for tracking tumor progression and therapy response [74,75].
Circulating free DNA (cfDNA) consists of fragmented DNA released into the bloodstream through natural processes like apoptosis, necrosis, or active secretion from both healthy and damaged cells, including those from tumors. This fragmented DNA, often derived from multiple tissues, plays a pivotal role as a non-invasive biomarker in cancer research. In NSCLC, elevated cfDNA levels have been associated with poorer prognosis, especially when linked to additional markers like the neutrophil-to-lymphocyte ratio (NLR) [76,77].
Circulating tumor DNA (ctDNA), a subset of circulating free DNA (cfDNA), offers significant promise as a tumor biomarker due to its ability to provide real-time insights into the tumor's genetic makeup. ctDNA is released into the bloodstream primarily through apoptosis and necrosis of tumor cells and represents only a small fraction of total cfDNA, typically ranging from 0.01% to 90% of cfDNA in peripheral blood [59]. Its unique properties, such as a short half-life of approximately 15 minutes to 2.5 hours, make ctDNA a dynamic indicator of tumor activity, as it can reflect changes in tumor status much earlier than traditional protein markers, which may take weeks to appear [78].
The genetic content of ctDNA includes vital information on tumor mutations, methylation patterns, and microsatellite instability, allowing for detailed genomic analyses that can support early diagnosis, monitor disease progression, and track the emergence of drug resistance in cancer patients. For instance, ctDNA has proven to be particularly valuable in non-small cell lung cancer, where it helps detect EGFR mutations, enabling clinicians to tailor targeted therapies more effectively and monitor patients' responses to treatment [79,80].
The clinical utility of ctDNA is further enhanced by advanced detection techniques such as digital PCR, quantitative PCR (qPCR), and next-generation sequencing, which offer high sensitivity and specificity. These methods allow for both qualitative and quantitative analysis of ctDNA, even when the circulating levels are low, which is a common challenge in early-stage lung cancer and minimal residual disease settings. Moreover, ctDNA testing is less influenced by tumor heterogeneity than tissue biopsies, making it a more reliable tool for comprehensive genomic profiling. In lung cancer, ctDNA-based liquid biopsies enable the detection of emerging genetic mutations related to drug resistance, offering critical insights for modifying treatment plans.
Despite its low abundance in blood, ctDNA's association with tumor burden has been well established, with studies showing elevated levels correlating with advanced cancer stages. The ability of ctDNA to predict recurrence and metastasis, alongside its application in non-invasive liquid biopsy technologies, positions it as a next-generation biomarker for early diagnosis, prognosis, and real-time monitoring of tumor dynamics [81,82].
Circulating tumor cells (CTCs) have emerged as a significant biomarker in the context of liquid biopsy for non-small cell lung cancer (NSCLC), providing valuable insights into disease progression and treatment response. CTCs, alongside other liquid biopsy analytes like circulating tumor DNA and extracellular vesicles , offer a non-invasive method for monitoring cancer and detecting therapeutic resistance mechanisms. Notably, CTCs have been shown to predict patient outcomes, with specific markers like PD-L1+ CTCs being linked to poorer progression-free and overall survival. This biomarker also aids in assessing resistance to immunotherapy. While ctDNA is useful for identifying mutations and guiding targeted therapies, CTCs offer a more comprehensive analysis by representing whole cells, making them a more reliable indicator of tumor behavior, especially when combined with PD-L1+ EV analysis. Further research is necessary to standardize liquid biopsy procedures and confirm these findings, but the integration of multiple liquid biopsy markers, including CTCs, holds promise for improving personalized cancer treatment strategies [83,84].
The research on CTCs and angiogenesis highlights their critical roles in cancer progression and metastasis. Angiogenesis, regulated by factors like VEGF, leads to the formation of leaky, immature blood vessels, facilitating CTC intravasation and aiding metastasis. Studies show that higher microvessel density in tumors correlates with increased metastasis risk, particularly in cancers like breast cancer. CTCs, which can form clusters with enhanced metastatic potential, are abundant in aggressive cancers such as small cell lung cancer (SCLC), where they contribute to the rapid spread of the disease. Despite advancements in detecting CTCs, their clinical utility as biomarkers is limited due to detection challenges, lack of standardization, and their heterogeneity. In cancers such as non-small cell lung cancer, where CTC counts are typically lower, the prognostic value of these cells remains unclear. Moreover, while CTCs offer valuable insights into tumor biology, their use in guiding therapy lags behind other biomarkers like cell-free DNA. Overall, the study of CTCs continues to evolve, but more standardized approaches are needed to fully harness their potential in clinical practice [84,85].
Extracellular vesicles (EVs) are small, lipid bilayer-bound vesicles secreted by cells, involved in intercellular communication and the regulation of tumor processes. EVs, including exosomes and microvesicles, carry genetic materials such as miRNAs, lncRNAs, and proteins, making them vital for tumor progression and immune modulation. In lung cancer, EVs are emerging as promising liquid biopsy tools due to their stability and capacity to reflect real-time tumor dynamics [86,87]. Specific miRNAs, like miR-934, miR-186-5p, miR-29a-3p and miR-497-5p, carried by EVs have been linked to lung cancer progression. Additionally, EVs-derived circRNAs such as circUSP7 and circSATB2 and lncRNAs like ZEB2-AS1, UFC1 have shown significant potential for early lung cancer detection. These biomarkers, by regulating various signaling pathways, offer promising avenues for disease diagnosis and monitoring. Despite their diagnostic promise, the low abundance of EVs in biological samples presents a challenge, necessitating the refinement of current isolation and characterization techniques to improve clinical applications [88].
The versatility of liquid biopsy lies in its ability to detect a wide range of molecular markers, including epigenetically modified DNA, long non-coding RNAs, microRNAs, tumor-associated antigens, and metabolites [89]. These components not only provide critical insights into the genetic and molecular characteristics of the tumor but also help identify mutations in tumor-associated genes, enabling personalized treatment strategies in precision medicine. In clinical practice, liquid biopsies are proving highly effective in monitoring disease recurrence and guiding treatment decisions, positioning them as a promising alternative to traditional tissue biopsies. As research progresses, the use of liquid biopsy is expected to expand, further enhancing lung cancer management through improved early detection and personalized therapeutic interventions.
7. Conclusions
Lung cancer continues to be a leading cause of cancer mortality globally, driven by a combination of environmental exposures and complex genetic alterations. Recent advancements in understanding its molecular underpinnings have shed light on the pivotal role of oncogenes and tumor suppressor genes. These discoveries have transformed the diagnostic landscape, enabling the use of precision medicine through targeted therapies that inhibit specific molecular pathways involved in tumor progression. Techniques such as next-generation sequencing and liquid biopsies now allow for real-time monitoring of genetic mutations, providing valuable insights into the disease's behavior and guiding more personalized treatment approaches.
These innovations have contributed significantly to improving early diagnosis and prognostic evaluations, particularly in cases of non-small cell lung cancer. The expanding molecular understanding not only helps refine prognosis but also paves the way for more effective interventions. However, despite these significant strides, the treatment of lung cancer faces ongoing challenges, particularly in overcoming drug resistance, tumor heterogeneity and the identification of actionable mutations in all patients. The integration of biomarkers in clinical practice has improved early diagnosis and treatment outcomes. While targeted therapies and immunotherapies have shown promise, their efficacy is often hampered by the development of resistance mechanisms, necessitating further research into combinatorial treatment strategies.
Future directions in lung cancer management will likely prioritize refining personalized therapies by exploring novel molecular targets and enhancing the effectiveness of existing treatments. Expanding the use of real-time monitoring through genetic and liquid biopsy technologies will enable dynamic adjustments to treatment plans, improving both efficacy and timing. This approach is expected to allow for more precise tailoring of therapies to individual patient profiles, potentially reducing side effects and increasing survival rates. At the same time, efforts should focus on developing combinatory therapeutic strategies that target multiple signaling pathways and molecular mechanisms simultaneously. These multifaceted approaches may provide a more durable and sustained therapeutic effect. Continued innovation in genetic testing and molecular diagnostics will be crucial for guiding precision medicine, ensuring that therapies remain adaptable to the evolving nature of cancer. Ultimately, integrating molecular advancements with therapeutic innovation holds great promise for significantly improving long-term outcomes and enhancing the quality of life for lung cancer patients.
Abbreviation.
SCLC - small cell lung cancer
NSCLC - non-small cell lung carcinoma
WHO - World Health Organization
TSG – tumor suppressor gene
EMT - epithelial-mesenchymal transition
ECM - extracellular matrix
STAT - signal transducers and activators of transcription
LKB1 - liver kinase B1
RTK - receptor tyrosine kinase
TKI - tyrosine kinase inhibitor
PFS - progression-free survival
EU - European Union
OS - overall survival
DKK – Dickkopf
VEGF - vascular endothelial growth factor
TME - tumor microenvironment
PKC - protein kinase C
DAG – diacylglycerol
eNOS - endothelial nitric oxide synthase
JNK - c-Jun N-terminal kinases
HIF - hypoxia-induced
pVHL - von Hippel-Lindau protein
MSI - microsatellite instability
CIN - chromosomal instability
NGS - next-generation sequencing
WES - whole-exome sequencing
WGS - whole-genome sequencing
FISH - fluorescence in situ hybridization
CTC - circulating tumor cell
cfDNA - cell-free DNA
ctDNA - circulating tumor DNA
EV - extracellular vesicle
ncRNA - non-coding RNA
NLR - neutrophil-to-lymphocyte ratio
Author Contributions
Conceptualization, A.D. and V.-E.R. ; methodology, V.-E.R.; software, A.D.; validation, V.-E.R., A.D. and L.-M.B.; formal analysis, L.-M.B.; investigation, A.D. and M.-C.C.; resources, M.-C.C. and S.D.-I.; data curation, I.-O.F. and R.C.; writing—original draft preparation, A.D and L.-M.B.; writing—review and editing, V.-E.R., A.D. and L.-M.B.; visualization, P.-C.C., A.-G.Z. and I.-A.H.; supervision,M.-I.S and L.-C.B. All authors have read and agreed to the published version of the manuscript.
Funding
Publication of this paper was supported by the University of Medicine and Pharmacy Carol Davila through the institutional program Publish not Perish.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
If needed, we will find a way to provide further information.
Acknowledgments
Publication of this paper was supported by the University of Medicine and Pharmacy Carol Davila through the institutional program Publish not Perish.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kanwal, M.; Ding, X.J.; Cao, Y. Familial risk for lung cancer. Oncol. Lett. 2017, 13, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://ec.europa.eu/eurostat/statistics-explained/index.php?title=Cancer_statistics_-_specific_cancers#Lung_cancer (accessed on 28 August 2024).
- Powell, H.A.; Iyen-Omofoman, B.; Hubbard, R.B.; Baldwin, D.R.; Tata, L.J. The association between smoking quantity and lung cancer in men and women. Chest 2013, 143, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Majumdar, S.; Bhattacharyya, P. Lung Cancer. In Pulmonomics: Omics Approaches for Understanding Pulmonary Diseases; Springer: Singapore, 2023. [Google Scholar] [CrossRef]
- Available online: https://tumourclassification.iarc.who.int/ (accessed on 28 August 2024).
- Lubin, J.H.; Boice, J.D.; Edling, C.; Hornung, R.W.; Howe, G.R.; Kunz, E.; Kusiak, R.A.; Morrison, H.I.; Radford, E.P.; Samet, J.M.; et al. Lung cancer in radon-exposed miners and estimation of risk from indoor exposure. JNCI J. Natl. Cancer Inst. 1995, 87, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Spyratos, D.; Zarogoulidis, P.; Porpodis, K.; Tsakiridis, K.; Machairiotis, N.; Katsikogiannis, N.; Kougioumtzi, I.; Dryllis, G.; Kallianos, A.; Rapti, A.; et al. Occupational exposure and lung cancer. J. Thorac. Dis. 2013, 5 (Suppl. 4), S440–S445. [Google Scholar] [CrossRef]
- Panagiotou, E.; Vathiotis, I.A.; Makrythanasis, P.; Hirsch, F.; Sen, T.; Syrigos, K. Biological and therapeutic implications of the cancer-related germline mutation landscape in lung cancer. Lancet Respir. Med. Published online June 14, 2024. [CrossRef]
- Alencar, V.T.L.; Formiga, M.N.; de Lima, V.C.C. Inherited Lung Cancer: A Review. Ecancermedicalscience 2020, 14, 1008. [Google Scholar] [CrossRef]
- Serio, V.B.; Rosati, D.; Maffeo, D.; Rina, A.; Ghisalberti, M.; Bellan, C.; Spiga, O.; Mari, F.; Palmieri, M.; Frullanti, E. The Personalized Inherited Signature Predisposing to Non-Small-Cell Lung Cancer in Non-Smokers. Cancers 2024, 16, 2887. [Google Scholar] [CrossRef]
- Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 30 August 2024).
- Kosaka, T.; Yatabe, Y.; Onozato, R.; Kuwano, H.; Mitsudomi, T. Prognostic implication of EGFR, KRAS, and TP53 gene mutations in a large cohort of Japanese patients with surgically treated lung adenocarcinoma. J. Thorac. Oncol. 2009, 4, 22–29. [Google Scholar] [CrossRef]
- Greulich, H. The genomics of lung adenocarcinoma: opportunities for targeted therapies. Genes Cancer 2010, 1(12), 1200–1210. [Google Scholar] [CrossRef]
- Barros-Filho, M.C.; Guisier, F.; Rock, L.D.; Becker-Santos, D.D.; Sage, A.P.; Marshall, E.A.; Lam, W.L. Tumour Suppressor Genes with Oncogenic Roles in Lung Cancer. In Genes and Cancer; Guy-Joseph, L., Ed.; IntechOpen: Rijeka, Croatia, 2019. [Google Scholar] [CrossRef]
- Bruno, R.; Fontanini, G. Next Generation Sequencing for Gene Fusion Analysis in Lung Cancer: A Literature Review. Diagnostics 2020, 10, 521. [Google Scholar] [CrossRef] [PubMed]
- Dashi, G.; Varjosalo, M. Oncofusions – shaping cancer care. Oncologist 2024, oyae126. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P. Targeting Gene Fusions in Non-Small Cell Lung Cancer—A Ceaseless Success Story? Transl. Lung Cancer Res. 2023, 12, 1358–1360. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, N.; Bawari, S.; Burcher, J.T.; Sinha, D.; Tewari, D.; Bishayee, A. Targeting Cell Signaling Pathways in Lung Cancer by Bioactive Phytocompounds. Cancers 2023, 15, 3980. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Sánchez-Rivera, F.J.; He, L.; et al. TGF-β and RAS Jointly Unmask Primed Enhancers To Drive Metastasis. Cell published online. 2024. [Google Scholar] [CrossRef]
- Drosten, M.; Barbacid, M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef]
- Chiosea, S.I.; Sherer, C.K.; Jelic, T.; Dacic, S. KRAS Mutant Allele-Specific Imbalance in Lung Adenocarcinoma. Mod. Pathol. 2011, 24, 1571–1577. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Xue, C.; Yao, Q.; Gu, X.; et al. Evolving Cognition of the JAK-STAT Signaling Pathway: Autoimmune Disorders and Cancer. Sig Transduct Target Ther 2023, 8, 204. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; et al. The JAK/STAT Signaling Pathway: From Bench to Clinic. Sig Transduct Target Ther 2021, 6, 402. [Google Scholar] [CrossRef]
- Choi, S.H.; Do, S.K.; Lee, S.Y.; et al. Genetic Variants in LKB1/AMPK/mTOR Pathway Are Associated with Clinical Outcomes of Chemotherapy in Non-Small Cell Lung Cancer. Thorac. Cancer 2022, 13, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Ciccarese, F.; Zulato, E.; Indraccolo, S. LKB1/AMPK Pathway and Drug Response in Cancer: A Therapeutic Perspective. Oxid. Med. Cell. Longev. 2019, 2019, 8730816. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.J.; Razi, S.; Pourbagheri-Sigaroodi, A.; Bashash, D. The PI3K/Akt/mTOR Pathway in Lung Cancer: Oncogenic Alterations, Therapeutic Opportunities, Challenges, and a Glance at the Application of Nanoparticles. Transl. Oncol. 2022, 18, 101364. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C. Targeting the PI3K/Akt/mTOR Pathway in Non-Small Cell Lung Cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Fernandez-Bruno, M.; Bracht, J.W.P.; Rosell, R. EGFR First- and Second-Generation TKIs—There Is Still Place for Them in EGFR-Mutant NSCLC Patients. Transl. Cancer Res. 2019, 8 (Suppl 1), S23–S47. [Google Scholar] [CrossRef]
- Kishikawa, T.; Kasai, T.; Okada, M.; et al. Osimertinib, a Third-Generation EGFR Tyrosine Kinase Inhibitor: A Retrospective Multicenter Study of Its Real-World Efficacy and Safety in Advanced/Recurrent Non-Small Cell Lung Carcinoma. Thorac. Cancer 2020, 11, 935–942. [Google Scholar] [CrossRef]
- Ayoup, M.S.; Shawki, I.; Abdel-Hamid, H.; et al. Targeting EGFR/PI3K/AKT/mTOR Signaling in Lung and Colon Cancers: Synthesis, Antitumor Evaluation of New 1,2,4-Oxdiazoles Tethered 1,2,3-Triazoles. RSC Adv. 2024, 14, 16713–16726. [Google Scholar] [CrossRef]
- Tong, Y.H.; Zhang, B.; Fan, Y.; Lin, N.M. Keap1-Nrf2 Pathway: A Promising Target Towards Lung Cancer Prevention and Therapeutics. Chronic Dis. Transl. Med. 2015, 1, 175–186. [Google Scholar] [CrossRef]
- Oh, Y.S.; Jun, H.S. Effects of Glucagon-Like Peptide-1 on Oxidative Stress and Nrf2 Signaling. Int. J. Mol. Sci. 2017, 19, 26. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, H.; Zhu, D. Wnt/β-Catenin Signaling Pathway in Lung Cancer. Med. Drug Discov. 2022, 13, 100113. [Google Scholar] [CrossRef]
- Zhang, Z.; Westover, D.; Tang, Z.; et al. Wnt/β-catenin signaling in the development and therapeutic resistance of non-small cell lung cancer. J Transl Med 2024, 22, 565. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Enta, A.; Maruya, Y.; et al. Wnt/β-Catenin Signaling and Resistance to Immune Checkpoint Inhibitors: From Non-Small-Cell Lung Cancer to Other Cancers. Biomedicines 2023, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Barr, M.P.; Gray, S.G.; Gately, K.; et al. Vascular Endothelial Growth Factor Is an Autocrine Growth Factor, Signaling through Neuropilin-1 in Non-Small Cell Lung Cancer. Mol. Cancer 2015, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Nilsson, M.B.; Le, X.; Cascone, T.; Jain, R.K.; Heymach, J.V. Molecular Mechanisms and Future Implications of VEGF/VEGFR in Cancer Therapy. Clin. Cancer Res. 2023, 29, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Ascha, M.S.; Wang, J.F.; Kumthekar, P.; et al. Bevacizumab for the Treatment of Non-Small Cell Lung Cancer Patients with Synchronous Brain Metastases. Sci. Rep. 2019, 9, 17792. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, S.; Deng, J.; et al. VEGF/VEGFR-Targeted Therapy and Immunotherapy in Non-Small Cell Lung Cancer: Targeting the Tumor Microenvironment. Int. J. Biol. Sci. 2022, 18, 3845–3858. [Google Scholar] [CrossRef]
- Cai, Z.; Tchou-Wong, K.-M.; Rom, W.N. NF-KappaB in Lung Tumorigenesis. Cancers 2011, 3, 4258–4268. [Google Scholar] [CrossRef]
- Chen, W.; Li, Z.; Bai, L.; Lin, Y. NF-KappaB in Lung Cancer, a Carcinogenesis Mediator and a Prevention and Therapy Target. Front. Biosci. 2011, 16, 1172–1185. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; et al. Atezolizumab for First-Line Treatment of PD-L1-Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef]
- Shimoji, M.; Shimizu, S.; Sato, K.; Suda, K.; Kobayashi, Y.; Tomizawa, K.; Takemoto, T.; Mitsudomi, T. Clinical and pathologic features of lung cancer expressing programmed cell death ligand 1 (PD-L1). Lung Cancer 2016, 98, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Santini, F.C.; Hellmann, M.D. PD-1/PD-L1 Axis in Lung Cancer. Cancer J. 2018, 24, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The PD-1: PD-L1/2 Pathway from Discovery to Clinical Implementation. Front. Immunol. 2016, 7, 550. [Google Scholar] [CrossRef] [PubMed]
- Viktorsson, K.; Lewensohn, R. Apoptotic Signaling Pathways in Lung Cancer. J. Thorac. Oncol. 2007, 2, 175–179. [Google Scholar] [CrossRef]
- Shimoda, L.A.; Semenza, G.L. HIF and the Lung: Role of Hypoxia-Inducible Factors in Pulmonary Development and Disease. Am. J. Respir. Crit. Care Med. 2011, 183, 152–156. [Google Scholar] [CrossRef]
- García-Del Río, A.; Prieto-Fernández, E.; Egia-Mendikute, L.; et al. Factor-Inhibiting HIF (FIH) Promotes Lung Cancer Progression. JCI Insight 2023, 8, e167394. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, Y.; Zhang, J.; et al. Apoptotic signaling pathways in bone metastatic lung cancer: a comprehensive analysis. Discov Onc 2024, 15, 310. [Google Scholar] [CrossRef]
- Shimoji, K.; Nakashima, T.; Masuda, T.; et al. Hypoxia-inducible factor 1α modulates interstitial pneumonia-mediated lung cancer progression. J Transl Med 2023, 21, 857. [Google Scholar] [CrossRef]
- Shi, Y.; Lin, X.; Wang, J.; Zhou, Z.; Chen, S.; Chen, G. Advances of HIF 1α/Glycolysis Axis in Non-Small Cell Lung Cancer (Review). Oncol. Rep. 2024, 51, 55. [Google Scholar] [CrossRef]
- Westhoff, B.; Colaluca, I.N.; D’Ario, G.; Donzelli, M.; Tosoni, D.; Volorio, S.; Pelosi, G.; Spaggiari, L.; Mazzarol, G.; Viale, G.; et al. Alterations of the Notch Pathway in Lung Cancer. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 22293–22298. [Google Scholar] [CrossRef]
- Zou, B.; Zhou, X.L.; Lai, S.Q.; Liu, J.C. Notch Signaling and Non-Small Cell Lung Cancer. Oncol. Lett. 2018, 15, 3415–3421. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wu, H.; Han, N.; et al. Notch Signaling and EMT in Non-Small Cell Lung Cancer: Biological Significance and Therapeutic Application. J. Hematol. Oncol. 2014, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Bayramova, J.; Tarakci, E.; Huseynova, G. Genetic Alterations in Lung Cancer. Turk. J. Oncol. 2024, 39, 234–243. [Google Scholar] [CrossRef]
- Tang, Y.; Pu, X.; Yuan, X.; et al. Targeting KRASG12D Mutation in Non-Small Cell Lung Cancer: Molecular Mechanisms and Therapeutic Potential. Cancer Gene Ther. 2024, 31, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Ostrem, J.; Pellini, B.; et al. Overcoming KRAS-Mutant Lung Cancer. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 1–11. [Google Scholar] [CrossRef]
- He, Q.; Xin, P.; Zhang, M.; et al. The Impact of Epidermal Growth Factor Receptor Mutations on the Prognosis of Resected Non-Small Cell Lung Cancer: A Meta-Analysis of Literatures. Transl. Lung Cancer Res. 2019, 8, 124–134. [Google Scholar] [CrossRef]
- Zheng, S.; Guerrero-Haughton, E.; Foijer, F. Chromosomal Instability-Driven Cancer Progression: Interplay with the Tumor Microenvironment and Therapeutic Strategies. Cells 2023, 12, 2712. [Google Scholar] [CrossRef]
- Hu, Z.; Liu, Z.; Zheng, J.; et al. Microsatellite Instability-Related Prognostic Risk Score (MSI-pRS) Defines a Subset of Lung Squamous Cell Carcinoma (LUSC) Patients with Genomic Instability and Poor Clinical Outcome. Front. Genet. 2023, 14, 1061002. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef]
- Jordan, E.J.; Kim, H.R.; Arcila, M.E.; et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017, 7, 596–609. [Google Scholar] [CrossRef]
- Marcy, T.W.; Stefanek, M.; Thompson, K.M. Genetic Testing for Lung Cancer Risk: If Physicians Can Do It, Should They? J. Gen. Intern. Med. 2002, 17, 946–951. [Google Scholar] [CrossRef] [PubMed]
- NCCN Guidelines. Available online: https://www.nccn.org/guidelines/ (accessed on Sept 2, 2024).
- Farinea, G.; Crespi, V.; Listì, A.; et al. The Role of Germline Mutations in Thoracic Malignancies: Between Myth and Reality. J. Thorac. Oncol. 2023, 18, 1146–1164. [Google Scholar] [CrossRef] [PubMed]
- Cainap, C.; Balacescu, O.; Cainap, S.S.; Pop, L.-A. Next Generation Sequencing Technology in Lung Cancer Diagnosis. Biology 2021, 10, 864. [Google Scholar] [CrossRef] [PubMed]
- Nooreldeen, R.; Bach, H. Current and Future Development in Lung Cancer Diagnosis. Int. J. Mol. Sci. 2021, 22, 8661. [Google Scholar] [CrossRef]
- Bhattarai, A.; Shah, S.; Abu Serhan, H.; Sah, R.; Sah, S. Genomic Profiling for Non-Small Cell Lung Cancer: Clinical Relevance in Staging and Prognosis. Medicine (Baltimore) 2023, 102, e36003. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.B.; Hou, L.K.; et al. Liquid Biopsy in Lung Cancer: Significance in Diagnostics, Prediction, and Treatment Monitoring. Mol. Cancer 2022, 21, 25. [Google Scholar] [CrossRef]
- Li, L.; Jiang, H.; Zeng, B.; et al. Liquid Biopsy in Lung Cancer. Clin. Chim. Acta 2024, 554, 117757. [Google Scholar] [CrossRef]
- Souza, V.G.P.; Forder, A.; Brockley, L.J.; et al. Liquid Biopsy in Lung Cancer: Biomarkers for the Management of Recurrence and Metastasis. Int. J. Mol. Sci. 2023, 24, 8894. [Google Scholar] [CrossRef]
- Corvigno, S.; Johnson, A.M.; Wong, K.K.; et al. Novel Markers for Liquid Biopsies in Cancer Management: Circulating Platelets and Extracellular Vesicles. Mol. Cancer Ther. 2022, 21, 1067–1075. [Google Scholar] [CrossRef]
- Peng, W.W.; Liu, Y.; Sha, H.H.; et al. Relationship between Plasma Circulating Cell-Free DNA Concentration and Treatment Outcomes Including Prognosis in Patients with Advanced Non-Small Cell Lung Cancer. BMC Pulm. Med. 2023, 23, 348. [Google Scholar] [CrossRef]
- Shu, Y.; Wu, X.; Tong, X.; et al. Circulating Tumor DNA Mutation Profiling by Targeted Next Generation Sequencing Provides Guidance for Personalized Treatments in Multiple Cancer Types. Sci. Rep. 2017, 7, 583. [Google Scholar] [CrossRef] [PubMed]
- Capuozzo, M.; Ferrara, F.; Santorsola, M.; Zovi, A.; Ottaiano, A. Circulating Tumor Cells as Predictive and Prognostic Biomarkers in Solid Tumors. Cells 2023, 12, 2590. [Google Scholar] [CrossRef] [PubMed]
- Pécuchet, N.; Zonta, E.; Didelot, A.; et al. Base-Position Error Rate Analysis of Next-Generation Sequencing Applied to Circulating Tumor DNA in Non-Small Cell Lung Cancer: A Prospective Study. PLoS Med. 2016, 13, e1002199. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Fei, Q.; Qiu, K.; et al. Liquid Biopsy Techniques and Lung Cancer: Diagnosis, Monitoring, and Evaluation. J. Exp. Clin. Cancer Res. 2024, 43, 96. [Google Scholar] [CrossRef]
- Eslami-S, Z.; Cortés-Hernández, L.E.; Sinoquet, L.; et al. Circulating Tumor Cells and PD-L1-Positive Small Extracellular Vesicles: The Liquid Biopsy Combination for Prognostic Information in Patients with Metastatic Non-Small Cell Lung Cancer. Br. J. Cancer 2024, 130, 63–72. [Google Scholar] [CrossRef]
- Deng, Z.; Wu, S.; Wang, Y.; Shi, D. Circulating Tumor Cell Isolation for Cancer Diagnosis and Prognosis. EBioMedicine 2022, 83, 104237. [Google Scholar] [CrossRef]
- Hamilton, G.; Rath, B.; Stickler, S. Significance of Circulating Tumor Cells in Lung Cancer: A Narrative Review. Transl. Lung Cancer Res. 2023, 12, 877–894. [Google Scholar] [CrossRef]
- Hsu, X.R.; Wu, J.E.; Wu, Y.Y.; et al. Exosomal Long Noncoding RNA MLETA1 Promotes Tumor Progression and Metastasis by Regulating the miR-186-5p/EGFR and miR-497-5p/IGF1R Axes in Non-Small Cell Lung Cancer. J. Exp. Clin. Cancer Res. 2023, 42, 283. [Google Scholar] [CrossRef]
- Hsu, C.C.; Yang, Y.; Kannisto, E.; et al. Simultaneous Detection of Tumor-Derived Exosomal Protein-MicroRNA Pairs with an Exo-PROS Biosensor for Cancer Diagnosis. ACS Nano 2023, 17, 8108–8122. [Google Scholar] [CrossRef]
- Zang, X.; Gu, J.; Zhang, J.; et al. Exosome-Transmitted lncRNA UFC1 Promotes Non-Small-Cell Lung Cancer Progression by EZH2-Mediated Epigenetic Silencing of PTEN Expression. Cell Death Dis. 2020, 11, 215. [Google Scholar] [CrossRef]
- Bibikova, M.; Fan, J. Liquid Biopsy for Early Detection of Lung Cancer. Chin. Med. J. Pulm. Crit. Care Med. 2023, 1, 200–206. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Classification of lung cancers.
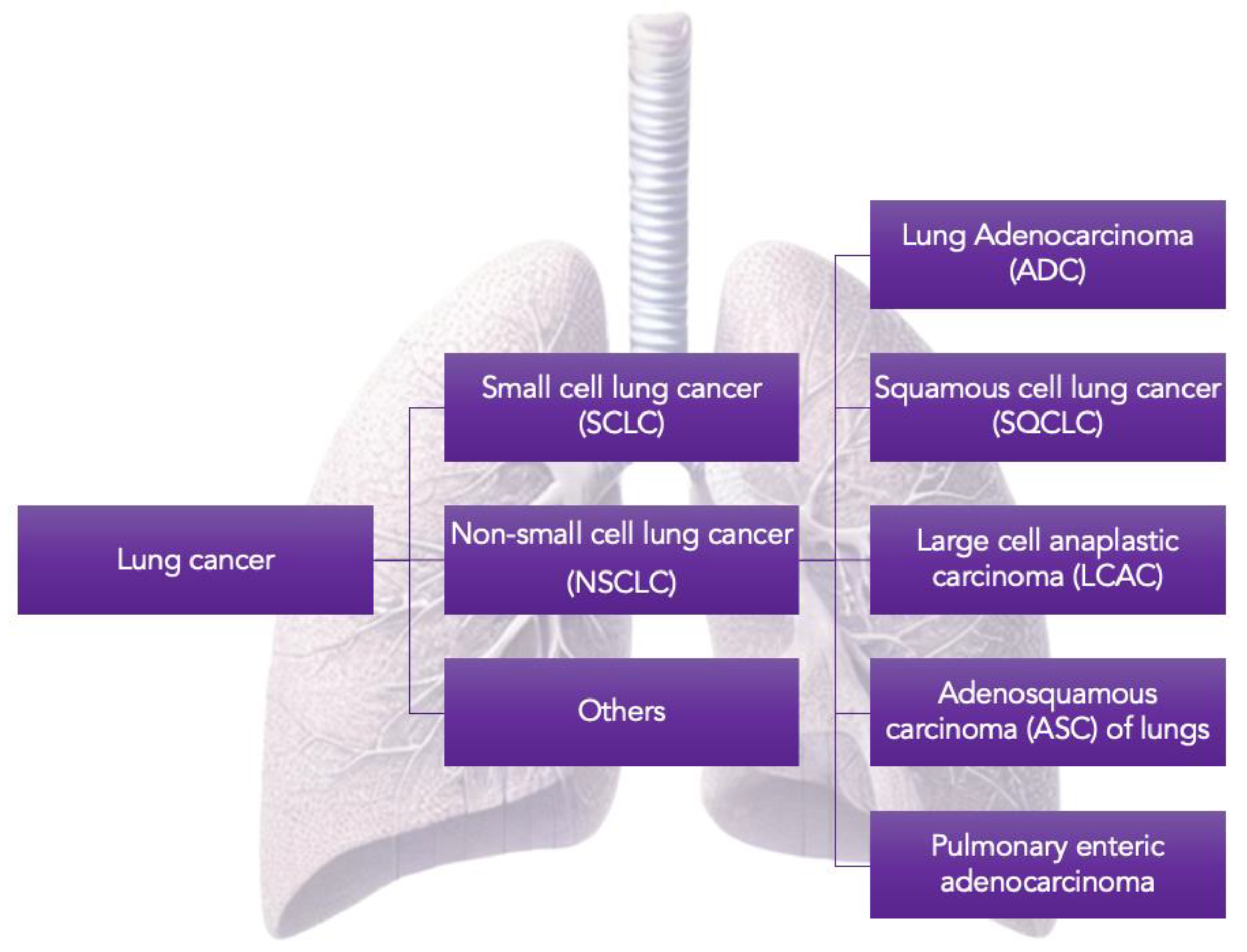
Figure 2.
Lung cancer predisposition syndromes.

Figure 3.
Genes involved in lung carcinogenesis.
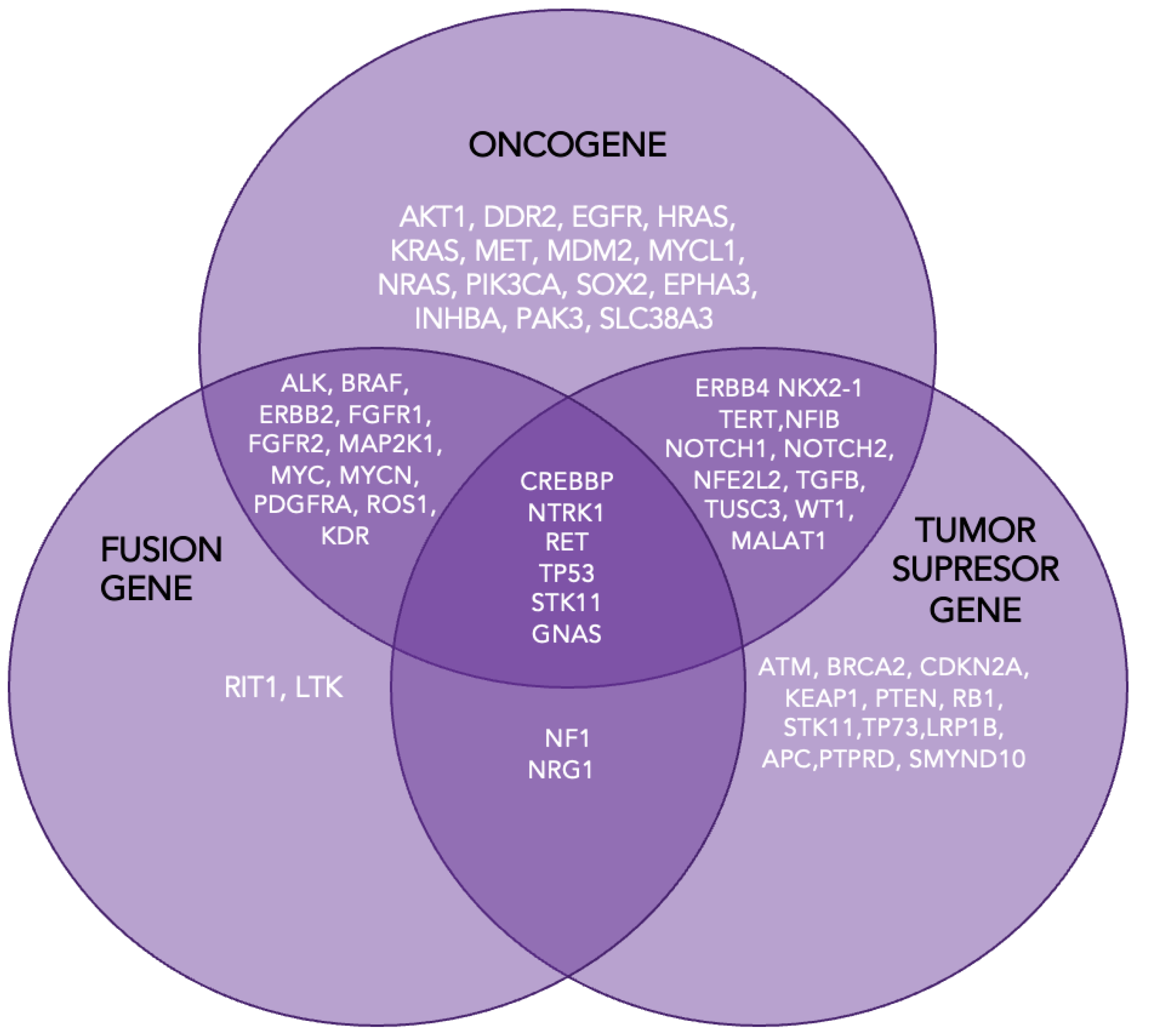
Figure 4.
The MAPK pathway.
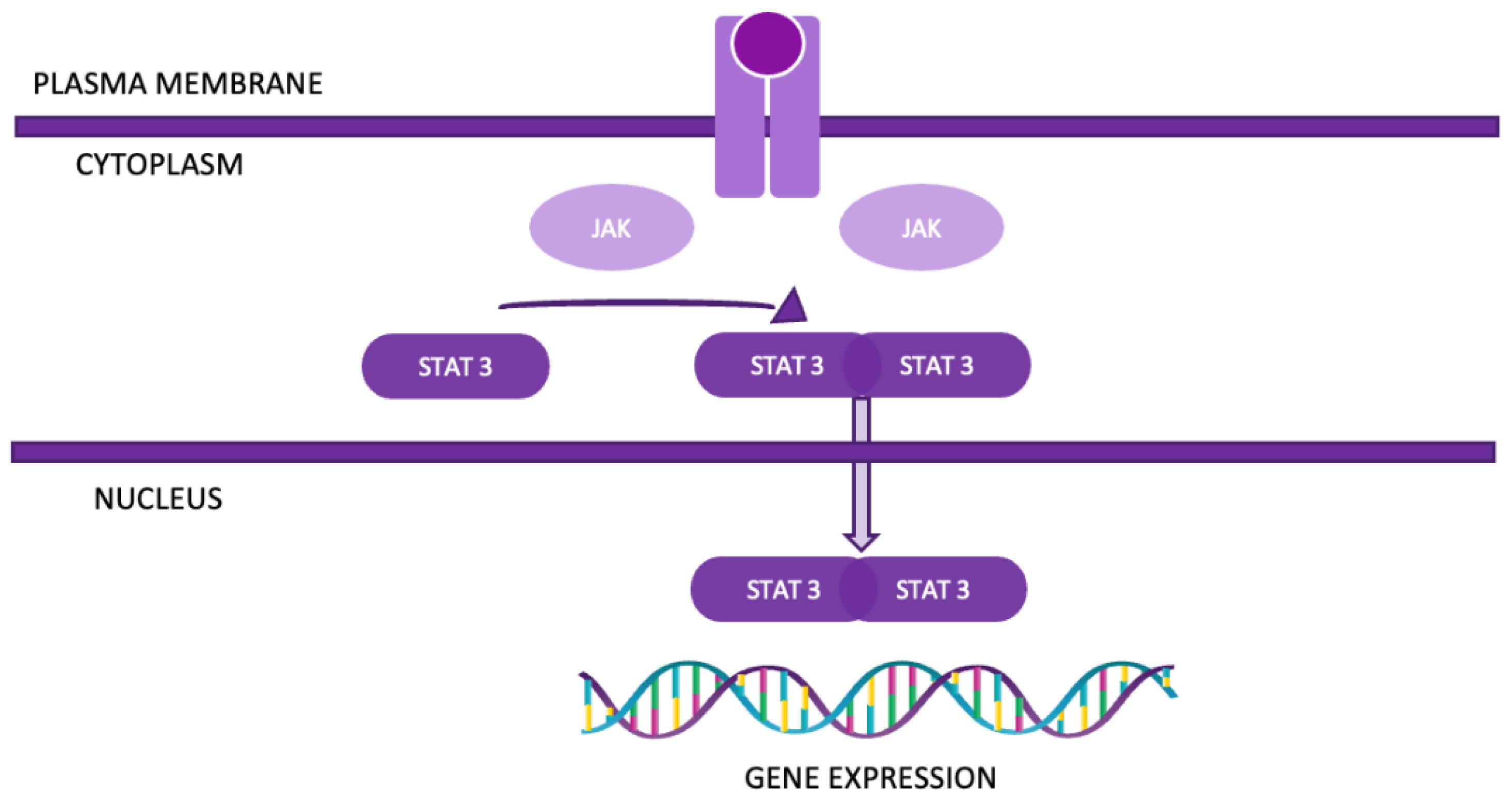
Figure 5.
The JAK-STAT pathway.

Figure 6.
The LKB1/AMPK/mTOR pathway.
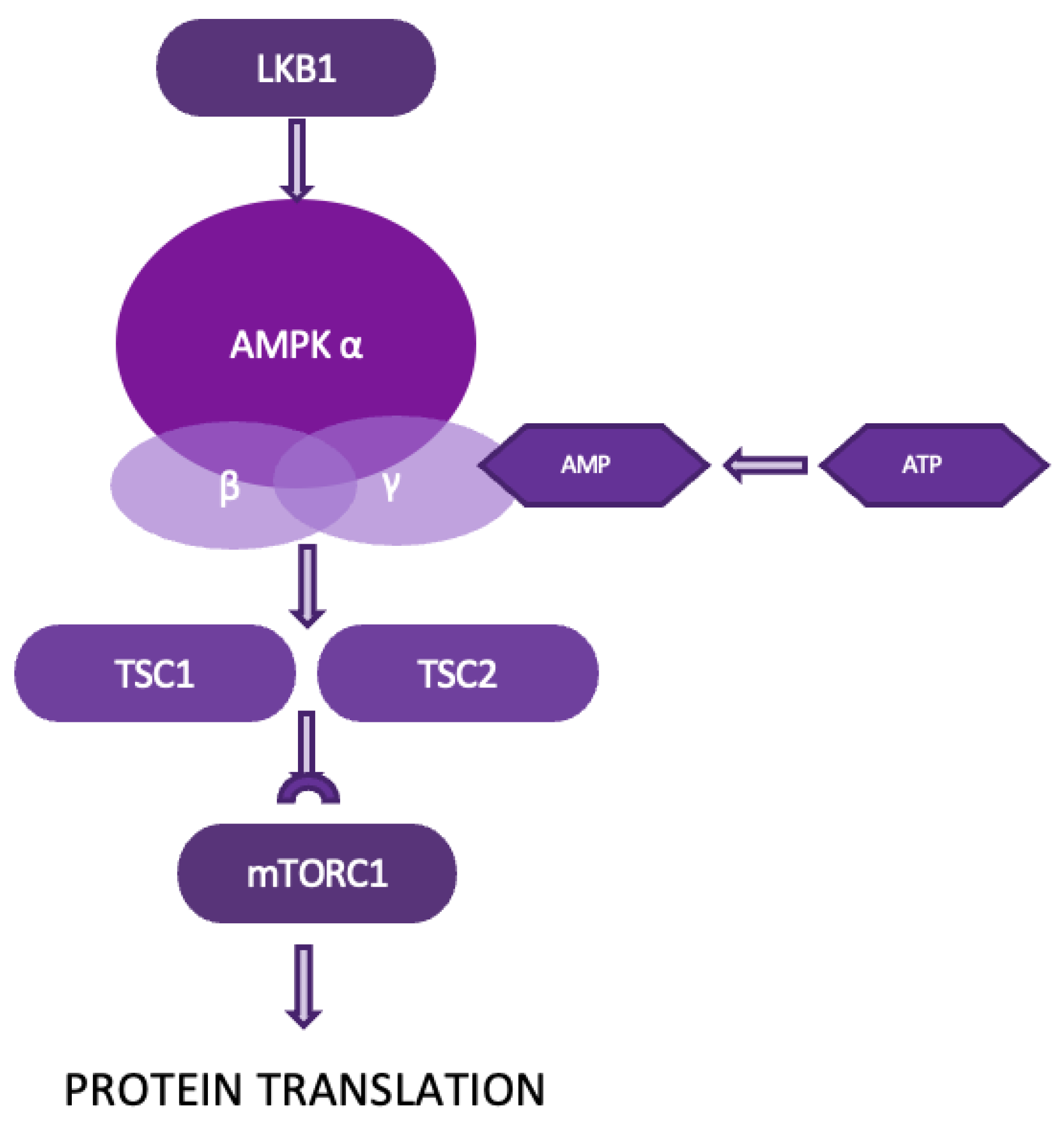
Figure 7.
The EGFR/PI3K/Akt/mTOR pathway.
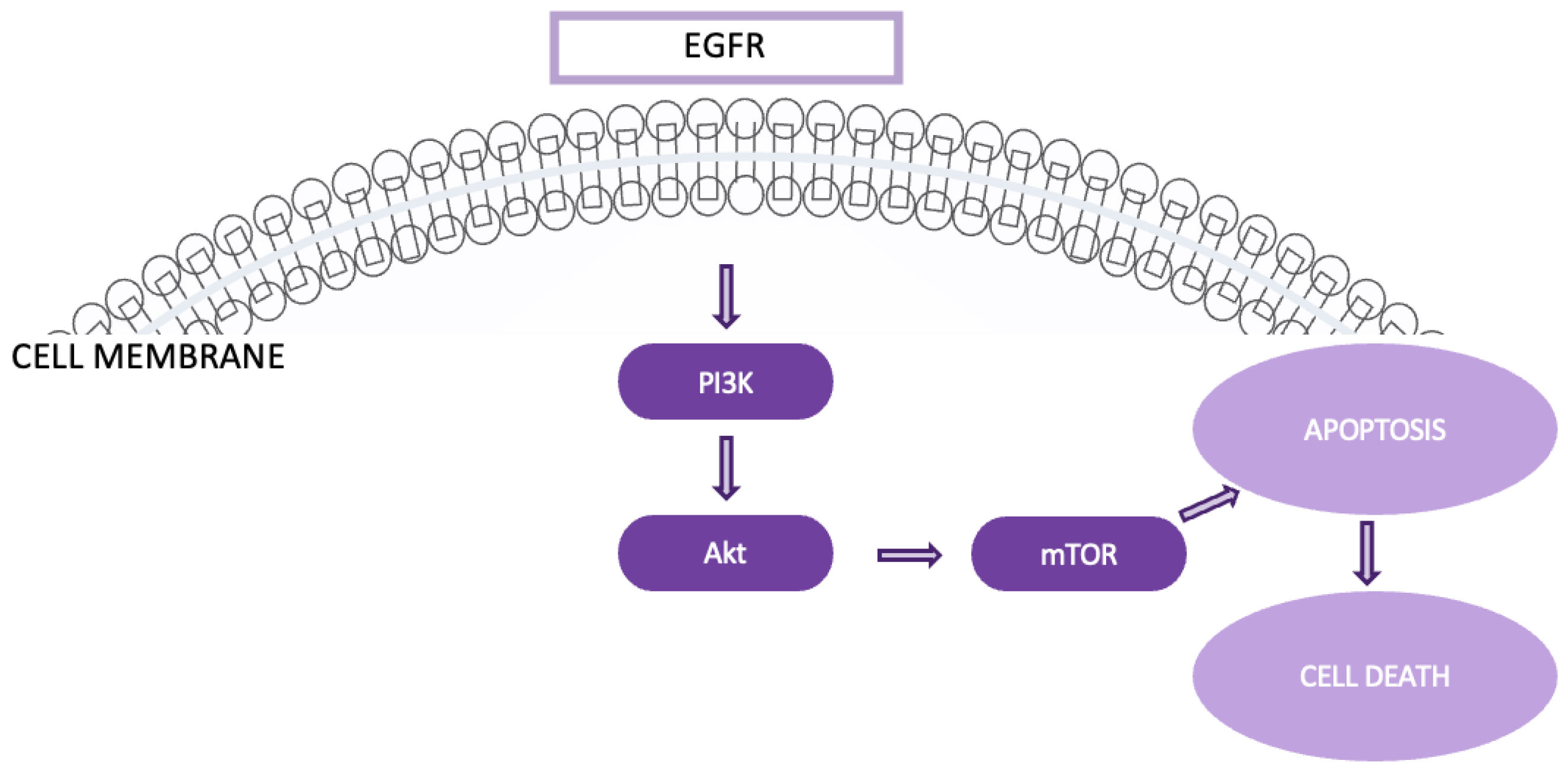
Figure 8.
The Keap1-Nrf2 pathway.
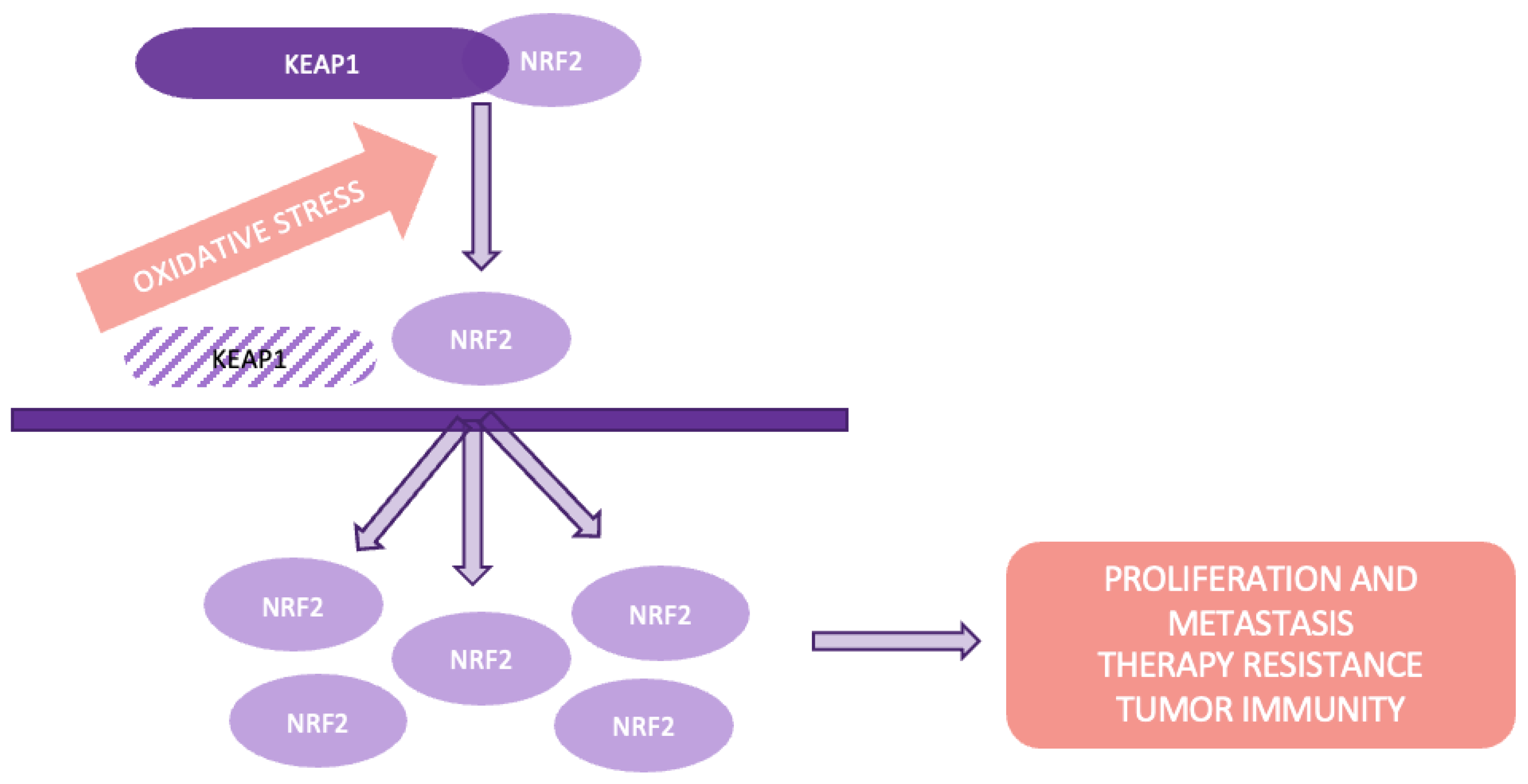
Figure 9.
The Wnt/β-catenin pathway.
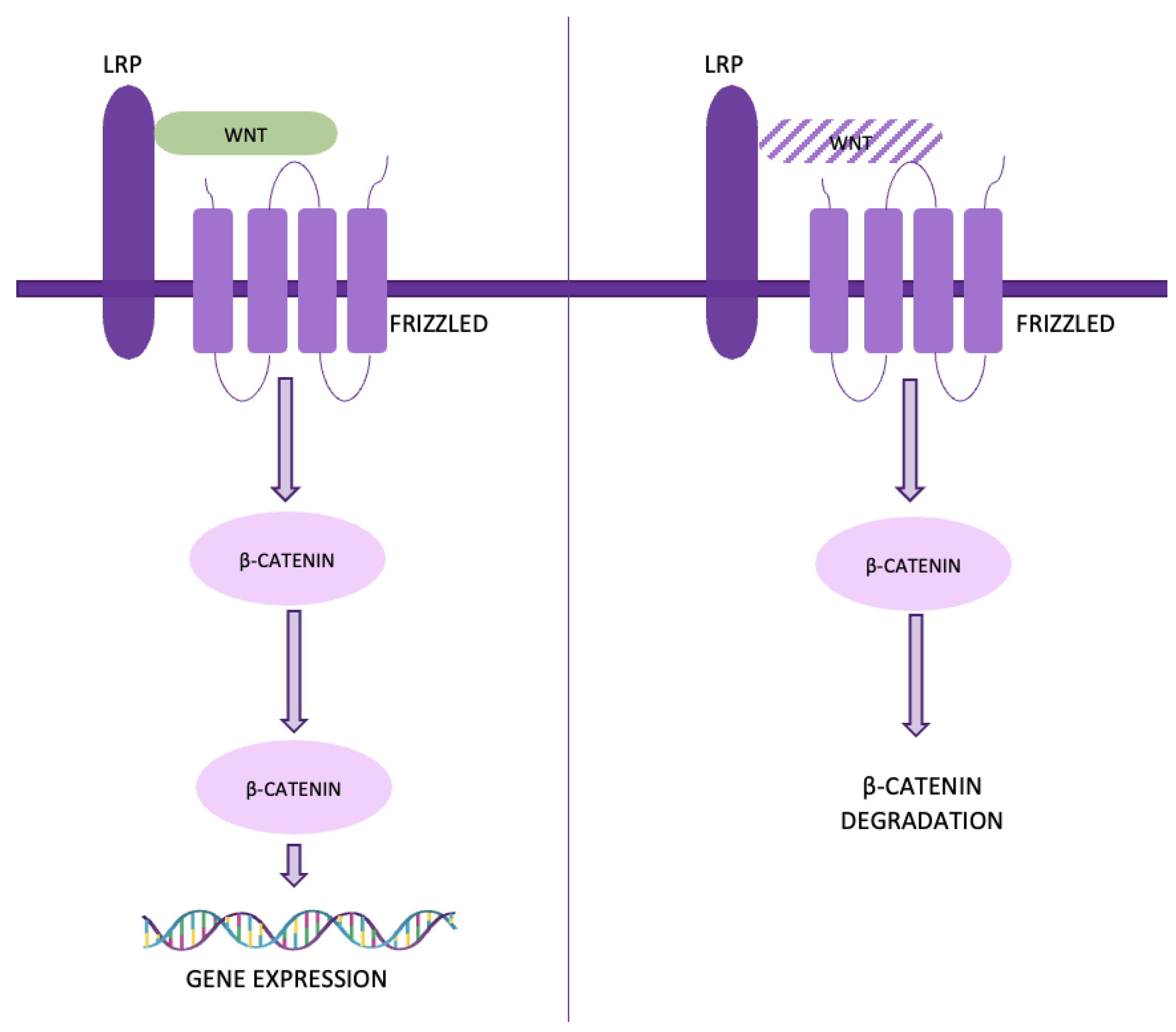
Figure 10.
The VEGF/VEGFR pathway.
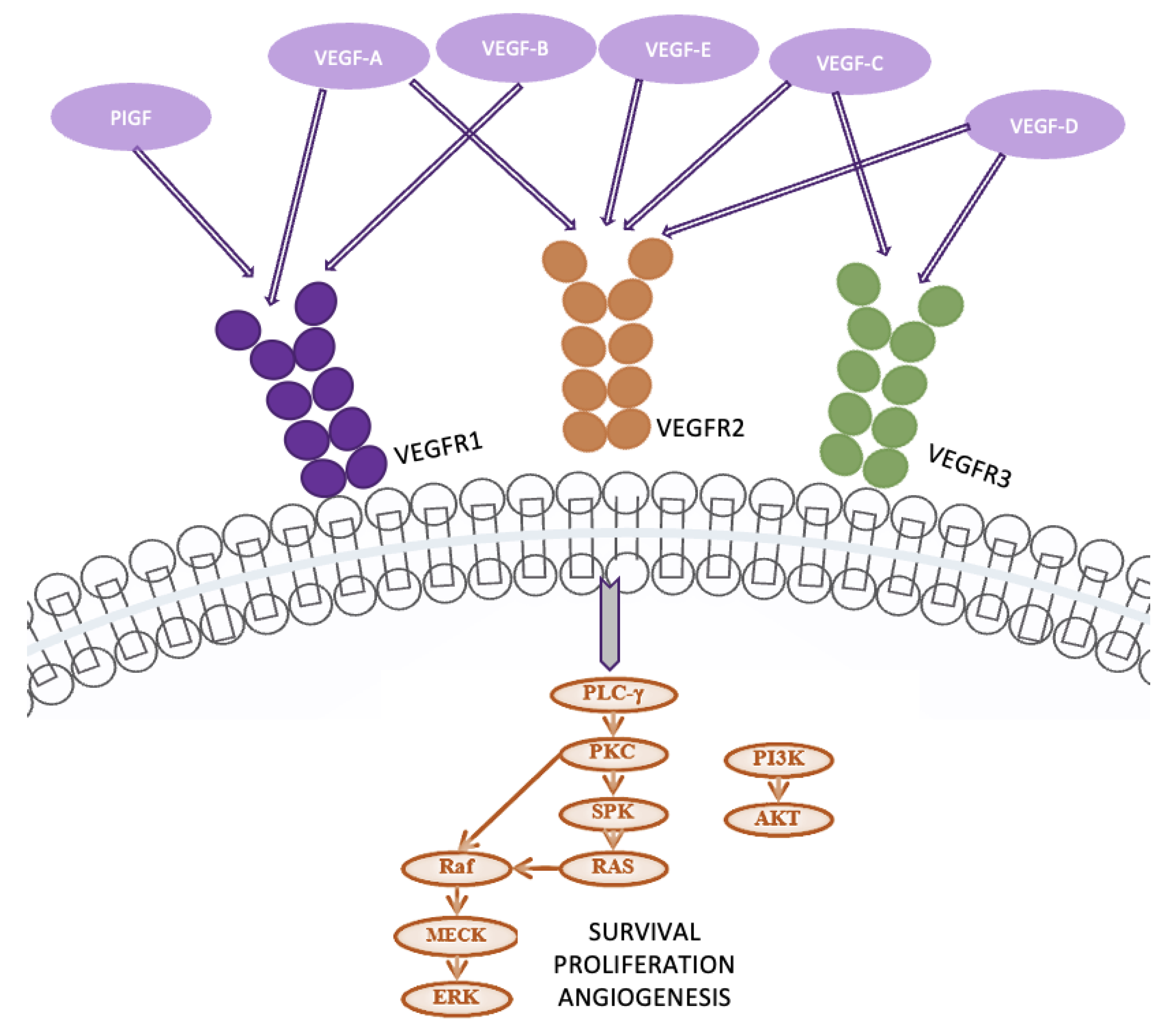
Figure 11.
The NF-κB pathway.
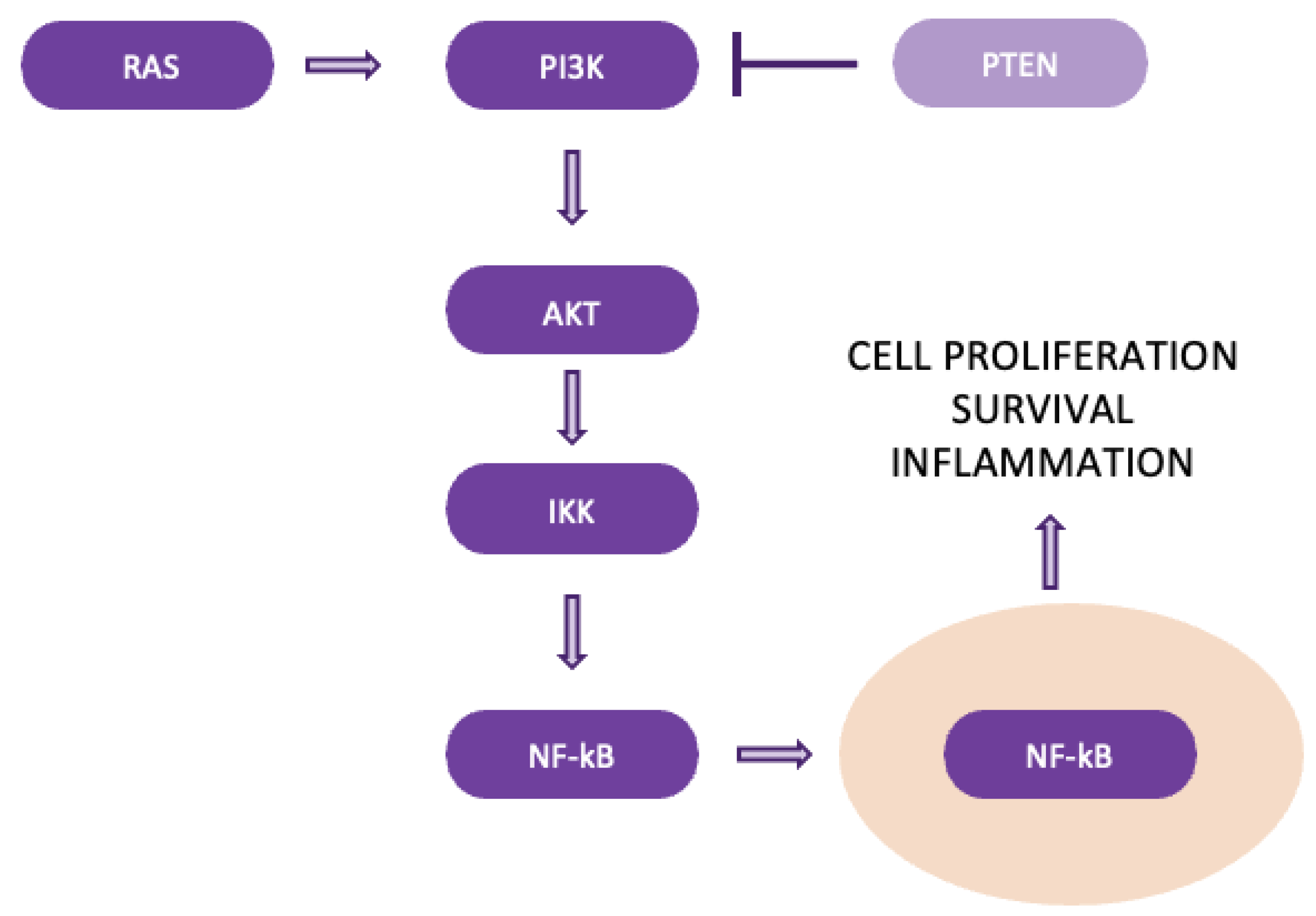
Figure 12.
The Hypoxia-Induced pathway.
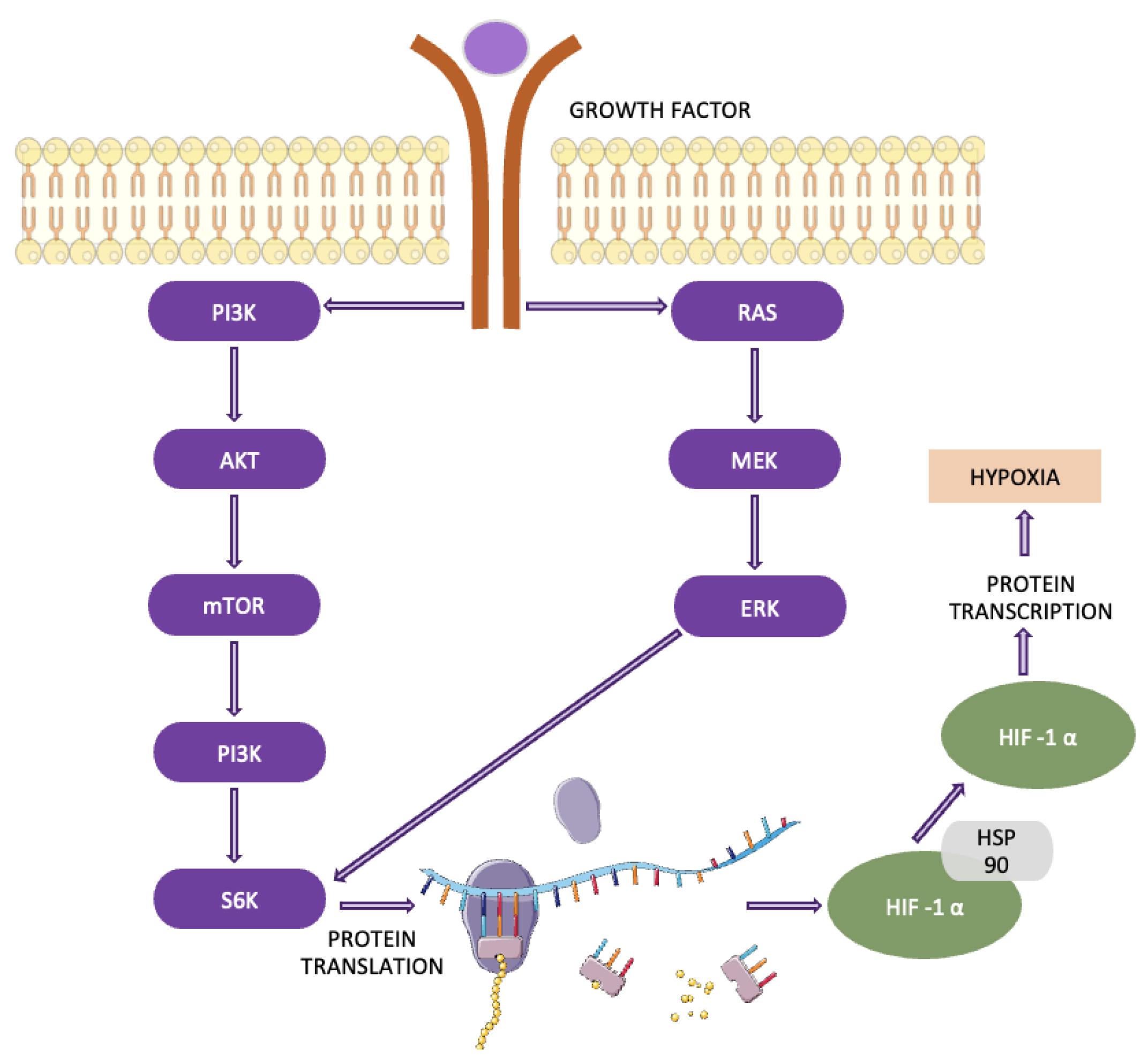
Figure 13.
NOTCH pathway.
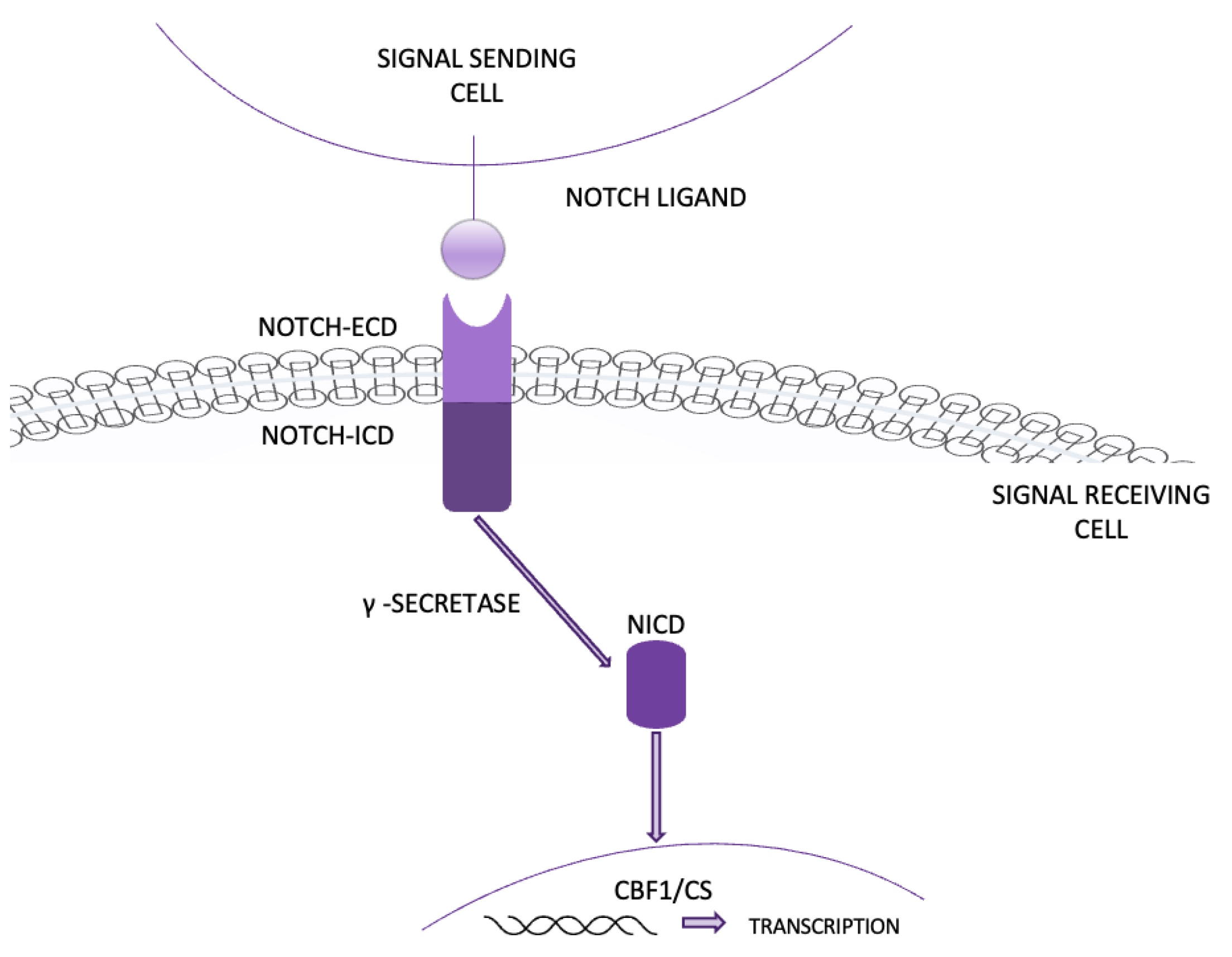
Figure 14.
Benefits and risks of genetic testing.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



