
Submitted:
29 October 2024
Posted:
30 October 2024
You are already at the latest version
Abstract
Iron-sulfur (Fe-S) clusters are essential cofactors found in many proteins in the mitochondria, cytosol, and nucleus of the cell. These versatile cofactors may undergo reversible oxidation-reduction reactions to enable electron transfers; they may be structural and confer stability to a folded protein; they may be regulatory and transduce an iron signal that alters the function or stability of a recipient protein. Of the nearly 70 proteins described in mammalian cells that bind Fe-S clusters, about half localize exclusively or partially to the nucleus, where they are required for DNA replication and repair, telomere maintenance, transcription, mitosis, and cell cycle control. Most nuclear Fe-S cluster proteins interact with DNA, including DNA polymerases, primase, helicases, and glycosylases. However, the specific roles of the clusters in the enzymatic activities of these proteins and their interplay with DNA remain a matter of debate. Defects in the metallation of nuclear Fe-S proteins cause genome instability and alter the regulation of cell division and proliferation, which are hallmarks of various genetic diseases and cancers. Here, we provide an inventory of the nuclear Fe-S cluster binding proteins and discuss cluster types, binding sites, the process of cluster acquisition, and potential roles of the cluster in the function of the proteins. However, many questions remain unresolved. We highlight critical gaps in our understanding of cluster delivery to nuclear client proteins, the potential for cluster repair, and the mechanistic roles that clusters play in these enzymes. Taken together, this review brings the focus to the nucleus of the human cell as a hotspot for Fe-S cluster proteins and aims to inspire new research in the roles of iron in DNA metabolism and the maintenance of genome integrity.
Keywords:
Iron-sulfur cluster
; nucleus
; iron trafficking
; DNA replication
; DNA repair
1. Introduction
Iron-sulfur (Fe-S) clusters are essential cofactors found in a wide variety of proteins across all domains of life. These clusters consist of iron and sulfur ions with different compositions, structures, redox states, and coordination ligands in proteins. The most frequently found clusters are the rhombic [2Fe-2S] and the cubic or cubane [4Fe-4S], but [3Fe-4S] and [4Fe-3S] clusters have also been described. The chemistry of these clusters makes them ideal for sensing redox signals, as they can accommodate different electronic configurations by accepting or donating unpaired electrons. Consequently, these clusters serve a wide spectrum of functions within proteins, including electron transfer, enzymatic catalysis, protein structural stability, cofactor transfer, oxygen and iron sensing, as well as redox stress [1]. These functions render Fe-S proteins crucial roles in multiple cellular processes, including energy production, metabolite transformation, DNA maintenance, gene expression regulation, protein synthesis, mitosis, and antiviral defense [2].
In the early stages of identifying and characterizing Fe-S clusters in the human proteome, the proteins discovered were primarily associated with mitochondrial and cytosolic functions, such as ferredoxins, aconitases, and respiratory complexes [3]. When the first Fe-S cluster bound to a helicase was reported in 2006, the authors stated “Fe-S clusters are rare in nuclear proteins” [4]. Nearly two decades later, we see that this perspective has completely changed, with the nucleus now known to contain about 35 proteins that bind Fe-S clusters, turning the nuclear compartment into a hot spot for Fe-S-related processes. The inventory of nuclear Fe-S cluster binding proteins described to date is listed in Table 1.
The link between iron metabolism and nucleic acid metabolism is becoming increasingly clear. This is evident in processes related to genome integrity, with Fe-S clusters found in many proteins involved in DNA replication and repair, as well as in transcription, with Fe-S clusters present in RNA polymerase, transcription factors, and RNA modification proteins. Although the precise role of these Fe-S clusters often remains elusive, their prevalence in proteins associated to nucleic acid transactions, and the fact that they have not been replaced by less redox-active metals throughout evolution, suggest that they play a crucial role in nucleic acid metabolism.
In this review, we discuss the current knowledge of nuclear Fe-S cluster proteins, examine accumulating evidence suggesting that Fe-S clusters may have additional and unexpected roles in cells, and highlight areas of research that may have practical significance but for which no definitive evidence has yet emerged.
2. Metallation of Nuclear Fe-S Cluster Proteins
The assembly of Fe-S clusters in mammalian cells involves a complex interplay between two machineries: the mitochondrial Iron-Sulfur Cluster (ISC) pathway and the Cytosolic Iron-Sulfur Assembly (CIA) pathway. This organized, multistep process relies on the functions of multiple proteins located inside and outside the mitochondria and consists of common underlying mechanisms of cluster synthesis, trafficking, and insertion into the final recipient proteins. Cofactor maturation of the nuclear [4Fe-4S] proteins depends on the function of both the ISC and the CIA pathways, as evidenced by the defects in the metallation of nuclear Fe-S proteins when depleting either ISC or CIA components [11,65,73,74,75,76,77,78,79]. Excellent recent reviews have focused on the known aspects of the de novo assembly of clusters inside and outside the mitochondria [2,80,81]. Although numerous studies in yeast and mammalian cells indicate that mitochondria provide the raw materials for the de novo assembly of clusters, studies in mammalian cells suggest that de novo assembly of Fe-S clusters also occurs in the cytosol via a dual localization of some ISC components in the mitochondria and the cytosol [66,68,82,83,84]. However, the relative contribution of each pathway to the assembly of clusters in mammalian cells remains unclear.
Similarly, the source of the iron used for the assembly of clusters has not been determined. In the cytosol, the iron chaperone poly C-binding protein 1 (PCBP1) binds iron from the kinetically labile, exchangeable iron pool (called the labile iron pool) and delivers it to ferritin and non-heme iron enzymes [85,86,87,88]. The co-chaperone complex comprised of PCBP1 and BOLA2, formed via a bridging Fe ligand, serves as an intermediate for the assembly of [2Fe-2S] clusters on the chaperone complex formed by BOLA2 and the monothiol glutaredoxin GLRX3 [89]. In mitochondria, however, the source of iron for cluster assembly is still an open question due to the lack of known iron chaperones residing in the mitochondrial compartment. Although some early studies suggested that frataxin could act as the mitochondrial iron donor for the ISC [90,91,92,93], mounting evidence does not support an iron chaperone function for frataxin, but instead indicates a role as an allosteric activator of the persulfide transfer process during the initial steps of Fe-S cluster assembly [81,94,95]. Consequently, the source of iron for mitochondrial ISC-mediated cluster synthesis remains unknown.
A fully assembled Fe-S cluster may be transferred to a recipient apoprotein that requires the cluster for function. This associative transfer occurs via protein-protein interactions, which protect the labile cofactor from chemical attack during delivery and prevent the cofactor from engaging in harmful redox chemistry. More than one system may be involved in the transfer of clusters to recipient apoproteins. The insertion of Fe-S clusters into apoproteins is facilitated by chaperone proteins such as HSC20 [82,84] or co-chaperone systems of the CIA-targeting complex (CTC), composed of three proteins: CIAO1, MMS19, and FAM96B/CIAO2B [74,77,78,96,97]. To assist the incorporation of Fe-S clusters into target proteins, HSC20 recognizes target proteins containing a short peptide (LYR) motif [82]. In contrast, the CTC guides the Fe-S clusters from the CIA to client apoproteins containing a conserved [LIM]-[DES]-[WF] C-terminal tripeptide motif, called the CTC recognition motif, which facilitates the identification of about a quarter of the known CTC clients or their adaptors [98]. However, this C-terminal signature is only present in a handful of the known Fe-S cluster-binding proteins (Table 1). It is still unclear how the apo-client recognition occurs for the remaining 75% of the Fe-S cluster proteins that do not contain the described CTC recognition motif. Alternative recognition motifs and new adaptor proteins mediating cluster transfer from CTC to apoproteins are likely yet to be discovered.
It is broadly assumed that Fe-S clusters are inserted into client apoproteins during protein synthesis and folding in the cytosol and remain bound during the lifetime of the Fe-S cluster protein. This seems necessary for proteins in which the cluster is buried deeply within the tertiary structure of the protein. For example, xanthine oxidase (XDH) contains 2 Fe-S clusters buried in the interior of the protein. The free exchange of these clusters seems unlikely due to steric hindrance inherent in the structure of the folded protein (Figure 1A). However, evidence is lacking that shows Fe-S metallation steps must occur exclusively during folding in the cytosol and before the nuclear protein is translocated to the nucleus. An additional hypothesis points to the possibility of cluster transfer after full translation and folding of the apoproteins. Two interesting observations may shed light in this regard: the evidence of Fe-S cluster transfer via adaptor-mediated protein-protein interactions [98], and the presence of multiple CIA components in the nucleus of the cell.
First, emerging evidence suggests that a subset of Fe-S proteins require an adaptor protein for their recognition by the CTC [98,99]. This is the case for the DNA primase subunit 2 (PRIM2) with its adaptor PRIM1, the elongator subunit ELP3 with its adaptor ELP4, and the ribosome recycling factor ABCE1 with its adaptor LTO1 [98,99]. These three [4Fe-4S] cluster proteins receive clusters from the CTC but do not contain the C-terminal [LIM]-[DES]-[WF] CTC recognition motif in their sequences. Instead, the CTC recognition motif is present in the C-terminus of the corresponding adaptor proteins [98]. This suggests that the transfer of the metalloclusters through adaptor-mediated protein-protein interactions should occur once the proteins have been synthesized and folded, and the client-adaptor complexes have been formed. Supporting this idea, the PRIM2/PRIM1 complex appears to form prior to Fe-S cluster incorporation into PRIM2 [100]. Both PRIM2 and PRIM1, while in complex, directly interact with the CIA targeting complex for cluster transfer [101]. This may only be possible for proteins harboring readily accessible, solvent-exposed cluster binding sites. Figure 1B-D shows that the three proteins, ABCE1, PRIM2, and ELP3, that use an adaptor protein for CTC recruitment have, in fact, solvent-exposed clusters. Thus, it is probable that the CTC donates cofactors to fully- or partially folded proteins with accessible cluster binding sites, while proteins with buried clusters may utilize a co-translational system that delivers clusters during protein folding. Further studies are needed to clarify how this parallel process occurs.
Although solvent-exposed clusters are inherently more susceptible to oxidative damage [104,105,106,107] and may require replacement or repair to maintain their function, proteins with exposed clusters may also exchange cofactors more readily. This lability could confer to these proteins the capacity to sense and respond to environmental cues. For example, the variable metallation status of the iron-responsive protein 1 (IRP1/ACO1) [108], the nuclear receptor coactivator 4 (NCOA4) [109], or the F-box and leucine-rich repeat protein 5 (FBXL5) [50], determine the biological activity of these three proteins. They are central to the regulation of intracellular iron metabolism and Fe-S cluster binding allows for coordination of the intracellular iron supply with their cellular activities. These three proteins also harbor solvent-exposed clusters. However, it remains to be demonstrated whether other Fe-S proteins, including nuclear Fe-S enzymes with solvent-exposed cluster binding sites, may exist as a pool of fully folded proteins that incorporate the cluster only when required to respond to cellular needs. Interestingly, although most of the proteins involved in the assembly and delivery of clusters are present in the cytosol, the nucleus of the cell also houses many of them including, MMS19 [21,110], CIAPIN1 [56], CIAO1 [110], CIAO2B [21], GLRX3 [62], NFU1 [66], NFS1 [68], NUBP2 [63], GLRX2 [71], and PCBP1 [5]. Whether these proteins assist in the in-situ transfer of iron or Fe-S clusters to client proteins in the nucleus remains elusive.
3. Fe-S Clusters Proteins Involved in Nuclear DNA Transactions
Defects in Fe-S cluster biogenesis result in DNA damage and genome instability. This is due to the requirement of Fe-S clusters for the function of multiple enzymes involved in DNA transactions [76,77,78,111,112,113,114]. In fact, the fastest-growing category of Fe-S binding proteins consists of enzymes that participate in nucleic acid metabolism, including DNA polymerases, primases, glycosylases, helicases, nucleases, transcription factors, RNA polymerases, and RNA-modifying enzymes.
3.1. Fe-S Clusters and DNA Replication
DNA replication in eukaryotic cells relies entirely on several Fe-S cluster-binding proteins, whose function and cluster binding properties are summarized below. Fe-S clusters may link the initiation of DNA replication to intracellular iron availability through licensing factors that ensure replication occurs only when the cell’s iron levels are sufficient to meet the high demand for this essential cofactor during DNA synthesis.
3.1.1. DNA Polymerases
The human genome encodes at least 14 DNA-dependent DNA polymerases [115]. Among these, DNA polymerases Pol α, δ, and ε, all located in the nucleus, are considered the replicative DNA polymerases. These multisubunit complexes are classified as class B DNA polymerases and play crucial roles in faithful nuclear DNA duplication [115]. Pol α initiates DNA replication at origins by synthesizing an RNA-DNA hybrid primer of 20-30 nucleotides. This primer is further extended by the highly processive Pol δ and ε on the lagging and leading strands of DNA, respectively [115] (Figure 2A). The catalytic subunits of all eukaryotic replicative DNA polymerases (POLA1, POLD1, and POLE) harbor a [4Fe-4S] cluster that is essential for replisome stability [11,116,117]. The fourth and final member of the class B DNA polymerase family, the error-prone DNA polymerase Pol ζ (zeta) [115], also binds a [4Fe-4S] cluster using a Cys-rich motif at the C-terminal domain of its catalytic subunit, REV3L [11]. Mutations that prevent cluster binding to Pol ζ abrogate its activity in UV mutagenesis repair [118]. Fe-S cluster binding has not been described for other classes of DNA polymerases [11]. Interestingly, although all class B DNA polymerases bind the cubane [4Fe-4S] cluster via a classical 4 Cys arrangement, the site of cluster binding varies. The cysteine motifs CysA and CysB, located at the C-terminus, are conserved among the four members of the family. CysB serves as the cluster binding site for Pol δ and Pol ζ [118]. In contrast, the catalytic domain of Pol ε binds the [4Fe-4S] cluster at an additional Cys-rich motif in the N-terminus, known as CysX, which is unique to Pol ε (C651, C654, C663 and C747) [13,116]. This positions the cluster in an exposed area, away from the DNA template, at the base of the processivity domain (P-domain), near the junction of the P and Palm domains [11,13,116,117]. Mutation of the cluster binding site in Pol ε abolishes DNA binding and proofreading exonuclease activities [119].
The metalation of all class B DNA polymerases depends on the function of the CIA pathway. Mutations in the CIA machinery lead to the loss of clusters in DNA polymerases, resulting in reduced protein stability and activity that causes genome instability [11,74,77,78]. Although the precise roles of Fe-S clusters in these proteins remain enigmatic, increasing evidence suggests that they may function as redox switches controlling the speed of DNA synthesis. Faster synthesis is associated with the reduced form of the [4Fe-4S] cluster present in Pol δ. This may provide a rapid and reversible mechanism to respond to replication stress by slowing down the DNA polymerase through cluster oxidation, allowing the accumulation and activation of DNA repair factors at lesion sites. Once DNA is repaired, reduction of the polymerase cluster may enable replication to resume [120]. This model needs further validation.
The presence of the cluster in Pol α is somewhat controversial because structural data shows Zn [2]+ bound at the position of the Fe-S cluster, which suggests that the presence of a cluster in Pol α might prevent its interaction with the corresponding B subunits [118,121,122,123,124,125]. Although no structures of Pol α showing a bound [4Fe-4S] cluster are currently available in the Protein Data Bank (PDB), it is not uncommon to find zinc replacing bona fide Fe-S clusters in protein structures due to the intrinsic sensitivity of these clusters to oxidative damage under aerobic conditions [116,126,127,128,129,130,131]. Resolving these discrepancies is essential to bridging a critical gap in our understanding of the role of Fe-S cluster in DNA polymerases.
3.1.2. DNA Primase
DNA replication begins with the synthesis of a short RNA primer of 8–12 nucleotides catalyzed by DNA primase. The primer is then extended by the catalytic subunit of Pol α [115] (Figure 2A). The primosome consists of four subunits: the catalytic subunit POLA1, the accessory subunit POLA2, and two primase subunits, the small catalytic subunit PRIM1 and the large regulatory subunit PRIM2. Each is located in the nucleoplasm, [5] where DNA replication occurs. In addition to the cluster in POLA1, the primase complex contains an additional [4Fe-4S] cluster bound to the C-terminal domain of PRIM2, which is essential for primer synthesis [8,9,132]. None of the smaller non-catalytic subunits of Pol δ or Pol ε harbor Fe-S clusters [11]. PRIM2, which is present only in archaeal and eukaryotic primases [132,133,134], bridges the two catalytic domains of the primosome–POLA1 and PRIM1, thereby facilitating the transition from RNA primer synthesis to DNA elongation [132].
The metalation of PRIM2 relies on the CIA machinery [16,77,97,132]. It has been proposed that PRIM1 may act as an adaptor protein between PRIM2 and the CIA targeting complex (Figure 1C), aiding in the client protein recognition necessary for cluster transfer [98,101]. The [4Fe-4S] cluster in PRIM2 is bound via a canonical four-Cys motif (C287, C367, C384, C424), located at the junction of the N-terminal and C-terminal domains, with each domain contributing two binding Cys residues [8,9,121,133,134,135] (Figure 1C). This arrangement suggests that the Fe-S cluster serves a structural rather than a redox-active role in the protein. However, one might question why such a complex cofactor would be used solely to preserve the structural integrity of a small domain. Recent evidence indicates that the [4Fe-4S] cluster in DNA primase functions as a redox switch, utilizing DNA charge transport to regulate the protein's DNA binding activity [136,137]. Briefly, oxidized [4Fe-4S] [3]+ PRIM2 binds more tightly to DNA than its reduced counterpart [136,137]. Electrostatic analyses revealed that the oxidation/reduction of the cluster, along with the accumulation of negative charges on the newly synthesized primers, induces structural changes at the DNA-protein interface [138]. The redox switch model proposes that the redox state of the [4Fe-4S] cluster in DNA primase acts as a reversible switch for DNA binding. The oxidized cluster activates the primase, enhancing its binding to DNA to initiate primer synthesis. As the primase extends the RNA primer, Pol α interacts with the RNA/DNA primer-template, and its [4Fe-4S] facilitates DNA-mediated redox signaling for primer truncation. This promotes PRIM2 dissociation by reducing its cluster, which allows the handoff of the primed template to Pol α [136,137,139]. This is a model and further research is needed to determine whether a DNA-mediated electron transfer relay coordinates the association, transfer, and dissociation steps of replication.
3.1.3. Fe-S Clusters and DNA Replication Origins
Nuclear receptor coactivator 4 (NCOA4), is the protein responsible for targeting ferritin for degradation during iron starvation, reintroducing iron into the labile iron pool. Under iron-replete conditions, iron-bound NCOA4 is targeted for proteasomal degradation by the ubiquitin ligase HERC2, which maintains ferritinophagy at basal levels [140,141]. It was recently shown that NCOA4 binds a 3Fe-4S cluster [48]. The insertion of the Fe-S cluster in NCOA4 promotes its recognition by HERC2, triggering its degradation and inhibiting ferritinophagy [47]. How NCOA4 acquires the Fe-S cluster is unknown. In addition to its role in ferritinophagy, NCOA4 is a multifunctional protein also implicated in coordination between cellular iron availability and the DNA replication machinery [109]. Federico et al. demonstrated that under low-iron conditions, apo-NCOA4 binds to DNA replication origins, inhibiting their activation and preventing replication stress [109]. NCOA4 specifically interacts with MCM7, a subunit of the MCM2-7 helicase complex that unwinds DNA during replication. Binding of NCOA4 to the MCM complex hinders its helicase activity [46] (Figure 2B). This mechanism may allow mammalian cells to respond to low iron by delaying initiation of DNA replication, an adaptive response when the pool of metallated Fe-S enzymes is limited. By finely tuning DNA replication to fluctuations in intracellular iron levels, this mechanism may help to safeguard genome stability during periods of diminished bioavailable iron and replicative stress [109].
3.1.4. Fe-S Clusters in Helicase Activity
Helicases are motor proteins that move along linear nucleic acid tracks using the energy from the hydrolysis of a nucleoside triphosphate. They function by destabilizing the hydrogen bonds between the complementary base pairs of unwinding DNA or RNA strands to generate the single-stranded intermediates required for processes like replication, repair, recombination, transcription, and splicing. Helicases are classified into six superfamilies, SF1-6 [142]. From the about 24 DNA helicases present in human cells, only five are known to bind Fe-S clusters. Those belong to the two largest superfamilies of DNA helicases (SF1 and SF2) [143]. Yet not all members of those families bind a cluster. Four nuclear DNA helicases from the SF2 family (Rad3/XPD group) contain Fe–S clusters in mammalian cells: the Fanconi Anemia (FA) protein J FANCJ, the Warsaw Breakage Syndrome protein ChlR1/DDX11, the telomere maintenance helicase RTEL1, and the component of the TFIIH complex involved in transcription and nucleotide excision repair XPD/ERCC2 [4,19,20,77,78,143]. XPD, the founding member of the Fe-S helicase family, is a central component of the general transcription factor TFIIH which plays major roles in transcription and nucleotide excision repair (NER), discussed below. FANCJ participates in the homologous recombination pathway of DNA double strand break repair, and it is part of the FA pathway proposed to function downstream of FANCD2. FANCJ also interacts with the breast cancer type 1 susceptibility protein BRCA1 and contributes to its DNA repair function. DDX11 has roles in replication of DNA, establishment of sister chromatid cohesion, and maintenance of chromosome architecture. RTEL1 is essential for preserving telomere length as discussed below. The SF1 helicase/nuclease DNA2 is the fifth helicase known to bind a Fe-S cluster in mammalian cells [23,143,144]. Although initially reported to be exclusively in the mitochondria [145], DNA2 also localizes to the nucleus [22]. DNA2 participates in the resolution of stalled replication forks during DNA synthesis [24] (Figure 2A). All these Fe-S helicases have been linked to multiple cancers and hereditary disorders characterized by genome instability [4,21,146,147,148,149,150] and mutations resulting in loss of the cluster are associated with the onset of disease. For example, mutations adjacent to the Fe-S cluster-binding Cys residues (M299I and A349P) in FANCJ are linked to breast cancer and Fanconi anemia, respectively [15,151,152,153].
The four SF2 helicases bind a single cubane [4Fe-4S] cluster using a canonical 4Cys arrangement (Table 1) located within the helicase core at the N-terminus of the proteins [4,17]. The cluster binding to DNA2 is unusual, as one of the four Cys residues coordinating the cubane [4Fe-4S] is located a few hundred amino acids upstream of the other three (C136, C393, C396, C402) [24]. Structurally speaking, there are two different types of helicases: those that form ring-like hexameric structures and those that do not. None of the known Fe-S cluster binding helicases form hexameric ring-like structures.
All known eukaryotic Fe-S cluster-containing helicases have a 5'-3' polarity and unwind DNA substrates. Although the clusters are essential for the DNA unwinding activity of the SF2 helicases [17,146,153], they do not affect the DNA binding and ATP hydrolysis capacities [153]. In contrast, loss of the cluster in DNA2 not only impairs DNA binding, but also impairs the nuclease and ATPase activities [24]. The Fe-S helicases exhibit an apparent specialization for the resolution of non-canonical DNA structures, including G4-quadruplexes (G4s), D-loops, and R-loops [154]. However, not all G4-resolving helicases have been reported to bind an Fe-S cluster, such as the helicases BLM, and WRN [146]. Non-canonical DNA structures play roles in genome organization, gene regulation, replication initiation and progression, recombination, and telomere metabolism [155,156]. However, unscheduled accumulation of these secondary DNA structures stall replication forks, causing genome instability. FANCJ efficiently resolves G4 DNA structures to enable smooth DNA synthesis during replication [157,158,159] (Figure 2A). RTEL1 resolves R-loops and G4s [160,161,162] and binds to the rG4 formed by the Telomeric Repeat-Containing RNA (TERRA) for to the maintenance of TERRA-containing R-loops, enhancing telomere stability [163]. DDX11 also resolves G4 DNA structures [164] to support sister chromatid cohesion [165]. And finally, G4 unwinding activity is also sustained by the archaeal XPD helicase [166]. Although it is clear, at least for FANCJ, that the presence of the Fe-S cluster is essential for this G4 unwinding activity [153], the mechanism by which the Fe-S cluster facilitates the unwinding of non-B DNA structures catalyzed by these helicases is still poorly understood.
The metallation of the helicases likely depends on the CIA machinery, as suggested by the interaction of MMS19 with XPD, FANCJ, DDX11, RTEL1, and DNA2 [16,21,77,78,100,110,167,168] and the decreased protein levels of these helicases when MMS19 is depleted [16,77,78]. The interaction of HSC20 with XPD, and DDX11 has been also confirmed [84]. However, several questions remain regarding the cluster insertion on those enzymes. For example, when the interaction of these helicases with other components of the CIA targeting complex was examined, XPD, DNA2, and RTEL1 were found to interact with CIAO1 and CIAO2B [97], however FANCJ did not interact with CIAO1 [16]. CIAO1 is the CTC component that offers the docking site for CIA client proteins to interact with the CIA delivery system. In general, it is not known how these Fe-S proteins are recognized for cluster delivery as they all lack the C-terminal CTC recognition motif proposed to facilitate the recognition of CIA clients. Further investigation is required to identify the factors that mediate the delivery of cluster to all the nuclear DNA helicases.
Surprisingly, Fe-S cluster binding in RNA helicases has not been described in mammalian cells. The only example of an RNA helicase binding an Fe-S cluster is the viral 5′-3′ SF1 helicase nsp13, the metalation of which depends on the host Fe-S cluster assembly machinery [127]. However, helicases that are currently annotated as Zn-binding proteins may in fact bind Fe-S clusters.
3.1.5. Fe-S Clusters and Telomere Maintenance
Telomeres, the protective caps at the ends of chromosomes that prevent degradation and fusion of chromosomes, also rely on functional Fe-S cluster enzymes for their maintenance. Replication of the telomeres is mechanistically challenging due to their repetitive nature and the presence of non-canonical DNA structures, such as G4s. Ultimately this results in the shortening of the telomeres during cell division. Key among the proteins involved in telomere maintenance is the ATP-dependent DNA helicase RTEL1, that unwinds G-quadruplex structures at telomeric DNA to facilitate telomere replication, preventing telomere loss [169] (Figure 2C). RTEL1 is located in the nucleoplasm and in nuclear speckles [5] and binds a redox-active cubane [4Fe-4S] cluster that is essential for its helicase activity. The cluster is bound at the N-terminus via a four-Cys arrangement (C145, C163, C172, C207) [20]. Although the role of the cluster in RTEL1 activity has not been determined, the physical interaction of RTEL1 with MMS19 [77,78] and the altered telomere length observed in MMS19 mutants [170] confirm that cluster insertion in RTEL1 depends on the CIA machinery.
3.2. Fe-S Clusters and DNA Repair
An estimate of 50,000 DNA-damaging events occur in our cells per day [171]. These lesions block replication-fork progression, cause cell cycle arrest, and constitute a major source for genome instability. At least six major DNA repair systems are required to remove the distinct types of DNA damage: base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR), non-homologous end joining (NHEJ), and translesion DNA synthesis (TLS) [172]. Fe-S clusters are integral components of the molecular machinery that repair DNA to ensure the faithful replication and distribution of genetic material during cell division (Figure 3). In addition to the Fe-S DNA polymerases and helicases discussed above, which are central to all DNA repair mechanisms [172,173], two Fe-S cluster DNA glycosylases, MUTYH and NTHL1, play central roles in the repair of oxidative DNA damage [174,175,176]. These enzymes cleave the glycosidic bond between bases and sugars to remove damaged or mispaired bases from the DNA duplex and act as initiators of BER [177]. MUTYH is specific for removing mismatched adenines and 8-dihydro-8-oxodeoxyguanine (8-oxoG) [178] and NTHL1 removes oxidatively-damaged pyrimidines in DNA [179]. They bind a redox-active cubane [4Fe-4S] cluster in the catalytic domain using a four-cysteinyl motif located in a solvent-exposed loop, referred to as the Fe-S cluster loop (FCL) motif (Table 1) [28,180,181,182,183]. Although originally assumed to play purely structural roles, the Fe-S clusters in DNA glycosylases are required for DNA binding and affect the detection and removal of the damaged bases [183,184,185]. Electrochemical studies using purified proteins indicate that cluster oxidation in the bacterial DNA glycosylases EndoIII and MutY promotes an increase in binding affinity [186,187]. The hypothesis is that the cluster binding motif is positioned for interaction with the DNA backbone [27,182,188] and the redox potential of the Fe-S clusters may aid glycosylases in scanning the DNA duplex for damaged bases [184]. Finally, although the metallation of these nuclear DNA glycosylases relies on the CIA machinery [110], they lack the C-terminal motif believed to guide the CIA targeting complex to client apo-proteins.
3.2.1. Fe-S Clusters, Transcription, and Nuclear RNA Transactions
Fe-S proteins are also involved in RNA processing pathways by modulating the activity of transcription factors, RNA polymerases, and RNA modifying enzymes. These proteins can be found as subunits of the RNA polymerase complex III, the transcription factor IIH, the elongator complex, and the pre-mRNA processing complex in mammalian cells. Although Fe-S binding transcriptional regulators that sense intracellular iron levels have not been described in mammalian cells, examples from yeast will be reviewed below to illustrate the transfer of Fe-S clusters between regulators in the nucleus of eukaryotic cells.
3.2.2. Fe-S Cluster Binding Fe-Dependent Transcriptional Regulators
Fe-dependent transcriptional regulators that bind Fe-S clusters and thereby sense intracellular iron levels have been described in yeast species but are not yet found in mammalian cells. In this discussion, we focus on fungal transcriptional regulators that sense intracellular iron levels, as their metalation appears to occur in the nucleus, suggesting the presence of dynamic Fe-S trafficking within the nuclear space. Although the iron regulatory mechanisms in the nonpathogenic fungi Saccharomyces cerevisiae and Schizosaccharomyces pombe are fundamentally distinct, both rely on Fe-S clusters as signals to regulate iron acquisition, storage, and utilization within the cell. In S. cerevisiae, the Fe-S cluster-binding transcriptional activators Aft1 and Aft2 regulate the expression of genes involved in iron uptake and use. Under iron-sufficient conditions, a heterodimeric [2Fe-2S] chaperone complex comprised of a monothiol glutaredoxin (Grx3/4) and a BolA-like protein (Bol2) binds and delivers the cluster to the DNA-bound transcriptional activators, altering their oligomeric state and leading to their dissociation from DNA, thereby suppressing the transcription of iron uptake genes [189,190,191]. In S. pombe, the transcriptional repressors Fep1 and Php4 regulate the expression of iron uptake and iron utilization genes, respectively. Under iron-depleted conditions, Php4 accumulates in the nucleus and associates with the DNA-binding complex Php2/3/5 to suppress the expression of iron utilization genes. As iron levels increase, a [2Fe-2S] cluster-dependent interaction between Php4 and Grx4 promotes the dissociation of Php4 from the Php2/3/5 complex, leading to the de-repression of iron-usage gene expression [192,193,194]. In contrast, the transcriptional repressor Fep1 binds a [2Fe-2S] cluster under iron-sufficient conditions, which facilitates its binding to DNA and halts the expression of iron uptake genes [195,196]. Under iron-depleted conditions, the cluster is unidirectionally transferred from Fep1 to Grx4-Fra2, causing the apo form of Fep1 to detach from DNA, allowing the expression of iron uptake genes [195]. The mechanism by which Fep1 acquires the cluster remains unclear. Notably, Fep1 localizes to the nucleus under both iron-replete and iron-deficient conditions [197], and remains bound to DNA when iron is sufficient. This suggests that the transfer of the cluster between Fep1 and the Grx4-Fra2 chaperone system occurs within the nucleus.
The [2Fe-2S]-coordinating complex formed by the glutaredoxin protein Glrx3 and the BolA-like protein BolA2 is also present in the cytosol of mammalian cells, where it participates in the maturation of cytosolic Fe-S cluster proteins [61]. Although mammals, unlike fungi, do not have iron-responsive transcriptional regulators such as Aft1/2, Fep1, or Php4, it is tempting to infer from the fungal evidence that the Glrx3-[2Fe-2S]-BolA2 system may constitute a pool of bioavailable, exchangeable clusters that are transferred to client partners for signaling purposes. However, these proposed roles remain to be experimentally tested.
3.2.3. RNA Polymerase III
Pol III is a DNA-dependent RNA polymerase specialized for the transcription of tRNAs and other short, essential RNAs. It is responsible for transcribing approximately 15% of total cellular RNA, including tRNA, 5S ribosomal RNA, U6 spliceosomal RNA, and other small ubiquitous RNAs. Misregulation of human Pol III is associated with tumor transformation, neurodegenerative and developmental disorders, and increased susceptibility to viral infections [198,199]. Pol III is a multi-subunit complex composed of 17 subunits, with a ten-subunit catalytic core and a peripheral heterodimeric stalk [35,200]. RPC6 (POLR3F), the subunit that binds a cubane [4Fe-4S] cluster, is part of the peripheral heterotrimeric complex RPC3/RPC6/RPC7 that modulates Pol III dynamics during transcription initiation [36,37,201]. The Fe-S cluster binds to the C-terminus of RPC6 via the thiol groups of four Cys residues (C287, C290, C296, C307). The Fe-S cluster is sandwiched between RPC3, RPC7, and the RPC1 clamp, tethering the interactions between the catalytic core and the RPC3-RPC6-RPC7 heterotrimer, thereby stabilizing Pol III [37]. Interestingly, although the Fe-S binding residues of RPC6 are widely conserved in many organisms, they are missing in Saccharomyces cerevisiae [199]. Distinct from other RNA polymerases, Pol III exhibits functional activities in both the nucleus and the cytoplasm. The canonical function of Pol III involves transcribing tRNAs, 5S rRNAs, and other small RNAs within the nucleus. Interestingly, cytoplasmic Pol III plays a vital role as a viral DNA sensor, synthesizing RNA from AT-rich viral DNA templates and triggering innate immune responses [202,203]. The current model suggests that Pol III is assembled in the cytosol, with some of it translocated into the nucleus and some remaining in the cytosol to participate in viral DNA sensing and response [203]. However, the actual factors facilitating Fe-S cluster insertion in RPC6 remain to be elucidated.
3.2.4. Transcription Factor IIH
The general transcription factor TFIIH participates in transcription initiation and nucleotide excision repair [204]. It is required for promoter opening and the transition from the pre-initiation complex to active transcription. TFIIH is a protein complex composed of ten subunits, divided into two subcomplexes: the core (XPB, p62, p52, p44, p34, p8) and the CDK-activating kinase (CDK7, cyclin H, MAT1) [205]. These subcomplexes are bridged by the helicase XPD, which connects p44 and MAT1. XPD (ERCC2) is a mostly nuclear helicase [5] that binds a [4Fe-4S] cluster via 4 Cys (C116, C134, C155, C190) in its N-terminal domain [4]. Mutations in XPD affect the interaction between XPD and p44, resulting in dissociation of XPD from the TFIIH complex. This disruption results in defects in the phosphorylation of various transcriptional activators, causing aberrant transcriptional activation [206,207,208].
3.2.5. RNA Modifying Proteins
In addition to capping and splicing, pre-mRNA undergoes 3' end maturation before protein translation. The cleavage and polyadenylation specific factor complex (CPSF) plays a crucial role in this pre-mRNA processing step by adding a poly-A tail to the mRNA. CPSF consists of six different proteins that recognize the highly conserved polyadenylation signal in the pre-mRNA, cleave the 3’ end, and recruit additional polyadenylation factors. CPSF4, one of the subunits of the complex, binds one [2Fe-2S] cluster and four Zn [2]+ ions as cofactors. In vitro analyses have shown that both metal cofactors are necessary to recognizing the polyadenylation signal in the pre-mRNA [129]. CPSF4 contains 5 CCCH domains, which are known to bind Zn and Fe-S clusters. The binding site for the [2Fe-2S] cluster in CPSF4 exhibits flexibility due to redundant iron-binding sites, allowing the cluster to be bound to an alternative site when the preferred site is mutated [129,209]. It is currently unknown which CCCH domain of CPSF4 is the preferred binding site for the [2Fe-2S] cluster in vivo. It is of interest to determine whether such flexibility in cluster binding sites have a meaningful functional effect on the biological role of the protein. Notably, CPSF4 is the only protein, exclusively localized in the nucleus known to bind a [2Fe-2S] cluster. Although a physical interaction between CPSF4 and the Fe-S chaperone HSC20 has been described [84], the mechanism by which CPSF4 acquires its cluster is not well established.
The multi-functional Elongator complex facilitates transcription by RNA Pol II [32], but is also responsible for the post-transcriptional modification of tRNAs at the wobble uridine (U34), a crucial modification that enhances translation efficiency [34]. It is composed of two copies of each of its six individual protein subunits, ELP1-ELP6, that form two discrete subcomplexes known as ELP123 and ELP456 [210]. ELP3 is the catalytic subunit of the complex with tRNA acetyltransferase activity, which is necessary for multiple tRNA modifications, including formation of 5-methoxycarbonylmethyl uridine (mcm5U) and its derivates [34]. ELP3 harbors a cubane [4Fe-4S] cluster bound to its N-terminus via 3 cysteines (C99, C109, C112) and an exchangeable S-adenosyl-L-methionine (SAM) (Figure 1D) [33,210,211,212]. Interestingly, ELP3 is the only Elongator subunit that has orthologs present in eukaryotes, archaea, bacteria, and viruses [211]. ELP3 interacts with MMS19 [78,110], and deficiency in ELP3, and other Fe-S proteins, was observed in cell lines derived from patients with biallelic loss of function in CIAO1 [213], confirming that ELP3 acquires its cluster from the CIA complex. As discussed above, ELP4 may serve as an adaptor protein between CTC and ELP3 (Figure 1D), exposing the C-terminal tryptophan motif recognized by the CIA targeting complex for the delivery of [4Fe-4S] clusters to client proteins [98]. However, demonstration of the involvement of other CIA factors and ELP4 in facilitating the metallation of ELP3 is still lacking.
Humans have three other tRNA-modifying enzymes in the nucleus that bind Fe-S clusters. The SAM-dependent tRNA 4-demethylwyosine synthases, TYW1 and TYW1B, are involved in the wybutosine biosynthesis pathway and are responsible for forming imidazoline rings. This RNA modification is important for stabilizing the codon-anticodon interaction, maintaining the reading frame and preventing errors during translation [34,214]. Information reported in databases suggest that TYW1 localizes to the nucleus (reason for the inclusion in Table 1). However, a recent publication shows that TYW1 mainly localizes in the cytosol in human cells [215], which is the compartment where the type of tRNA modification catalyzed by this enzyme is assumed to occur [216]. The third protein, CDK5RAP1, is a methylthiotransferase responsible for modifying four mitochondrial DNA-encoded tRNAs to optimize mitochondrial translation [217]. CDK5RAP1 is primarily located in the mitochondria but can also be found in the nucleoplasm and cytosol. Its RNA modification activity is not limited to mitochondrial tRNA, as there is evidence that it also introduces modification into nuclear RNA species [42]. These three proteins bind cubane [4Fe-4S] clusters via 3 cysteines and an exchangeable SAM (Table 1).
4. Fe-S Cluster Proteins in Mitosis
Once DNA is fully replicated, cells enter mitosis following an intricate and highly regulated process that ensures each new cell receives an exact copy of the genetic blueprint. It has long been established that MMS19 and CIA2B localize to the mitotic spindle during mitosis and that their knockdown disrupts the mitotic pathway, leading to poor alignment of chromosomes, improper segregation, accumulation of nuclei with abnormal shapes, and mislocalization of key mitotic factors [21]. However, it was only a few years ago that a mitotic factor was identified as binding an Fe-S cluster. The chromokinesins KIF4A, which is involved in faithful chromosome segregation during mitosis, binds an Fe-S cluster [45] (Figure 2D). Molecular motor kinesins are the primary source of cellular forces and drive many types of intracellular movement along microtubule networks. KIF4A and KIF4B are the two variants of the kinesin family 4 present in humans. They share 98% homology in their DNA sequence with different chromosomal location [218]. This ATP-dependent microtubule-based motor kinesin resides in the nucleoplasm during interphase and on the arms of condensed chromosomes during mitosis. KIF4A accumulates at the central spindle that connects the two daughter cells during the late stages of mitosis, from late anaphase through cytokinesis [44,219,220]. This stabilizes the bipolar mitotic spindle, which is essential for successful midzone formation and cytokinesis. In KIF4-deficient cells, the central spindle becomes disorganized, leading to cytokinesis failure [221,222], chromosome misalignment, spindle defects, and chromosome missegregation. This results in shorter and wider chromosomes, lagging chromosomes, aneuploidy, and ultimately tumor formation [220,223].
Unlike other motor kinesins, KIF4A resides in the nucleus during interphase where it performs additional non-mitotic functions. KIF4A has been proposed to play a role in the DNA damage response by modulating the BRCA2/Rad51 pathway. Its C-terminal cargo-binding domain interacts with the conserved C-terminal region of BRCA2, facilitating the translocation of BRCA2 to damaged DNA regions following injury [224]. Depletion of KIF4A results in a significant slowdown of DNA replication and decreased homologous recombination [224]. KIF4 also acts as a modulator of large-scale chromatin architecture. It binds globally to chromatin and its depletion leads to chromatin decondensation and loss of heterochromatin domains [223]. Overall, KIF4 deficiency has been associated with developmental delays and intellectual disabilities [225] and it is abnormally expressed in multiple cancers [218]. KIF4A has been also implicated in the infection process of Hepatitis B and D viruses by regulating the transport of the NTCP receptor from the cytoplasm to the cell surface, where it functions as a receptor for viral entry [218,226].
Structurally speaking, KIF4A features a highly conserved ATPase/motor domain at the N-terminus that binds to microtubules and provides the mechanochemical force for movement. This is followed by a central α-helical stalk domain and a C-terminal cargo-docking domain responsible for capturing cargoes [227,228]. The [4Fe-4S] cluster in KIF4A is bound to a conserved cysteine-rich domain located at the C-terminus [45]. However, it remains unclear which of the nine conserved Cys residues located in that region coordinate the Fe-S cluster. Whether the cluster is redox-active is also unknown.
In mitotic cells, KIF4A colocalizes with the CIA components CIAO2B and MMS19 at the spindle midzone and midbody between separating chromosomes [45] (Figure 2D). KIF4A also co-immunoprecipitates with CIAO2B and MMS19, confirming their physical interaction [45]. This interaction is enhanced after iron chelation, suggesting a more stable association between the CIA complex and the apo client protein [45]. Similar enhancements in interactions between CIA and client proteins under iron deprived conditions has been observed for other Fe-S proteins, including XPD, FANCJ, and Pol δ [78]. The mislocalization of the KIF4A variant lacking the conserved C-terminal cysteine domain that binds the cluster, along with the reduced association of endogenous KIF4A with the midzone/midbody following CIAO3 depletion, suggests that the Fe-S cluster in KIF4A modulates the proper subcellular localization of the protein at the mitotic spindle. This likely influences the protein-protein interactions essential for the mitotic process [45].
Although the cluster in KIF4A is quite labile in [55]Fe radiolabeling experiments, its absence does not appear to affect the stability of the protein [45], as seen for other Fe-S cluster binding proteins. This dynamic nature may be biologically relevant and allow for the assembly and disassembly of the Fe-S cluster in KIF4A to regulate the progression of mitosis [45], which could explain why the late-acting CIA factors colocalize at the midbody with components of the mitotic machinery. This suggests that the cluster is transferred to a folded client protein at the site of function, rather than co-translationally transferred. Further mechanistic studies are needed to elucidate the role of the Fe-S cluster in modulating KIF4A activity and whether the metallation of KIF4A occurs only after the breakdown of the nuclear membrane when cells enter mitosis, or if KIF4A residing in the nucleus during interphase also binds an Fe-S cluster.
KIF4A and KIF4B are currently the only known components of the mitotic machinery that bind an Fe-S cluster. However, the significant mitotic defects observed when silencing the CIA, along with the colocalization of late-acting CIA factors with components of the mitotic machinery, suggest the potential existence of other, yet-to-be-discovered Fe-S cluster proteins that are essential for successful mitosis.
5. Other Fe-S Cluster Proteins in the Nucleus
The inventory of nuclear Fe-S cluster proteins presented in Table 1 includes some unexpected proteins whose functions are typically associated with other cellular compartments. These proteins are involved in post-translational modifications (ATE1, DPH1, and DPH2), nucleotide metabolism (XDH), maturation of cytosolic Fe-S proteins (CIAPIN1, GLRX3, and BOLA2), immune modulation and restriction of viral replication (RSAD2), and mitochondrial function (SDHB, GLRX2, NFS1, and NFU1). It remains unclear why these proteins are also localized in the nucleus and whether they retain the function already characterized in other subcellular localizations or have alternative roles while in the nucleus.
6. Conclusions and Open Questions
About half of all known Fe-S binding proteins in human cells localize to the nucleus, with many identified only recently. A few decades ago, the presence of iron cofactors in the nucleus was considered unlikely due to the threat that the reactive iron poses to the genetic material. However, chaperoning and trafficking systems that safely mobilize these cofactors facilitate their incorporation in DNA metabolic enzymes, leveraging their redox capabilities while minimizing risks. The connection between iron cofactors and genome integrity is now well established, with an increasing number of nuclear Fe-S proteins, particularly those involved in nucleic acid metabolism such as DNA replication and repair. The high metabolic expense associated with incorporating clusters into proteins suggest that they play essential roles. However, the mechanistic benefit of incorporating Fe-S clusters into nucleic acid-binding proteins remains unclear. Mismetallation of these nuclear Fe-S cluster enzymes often leads to genome instability, which is linked to various diseases, including breast and ovarian cancer, Fanconi anemia, Xeroderma pigmentosum, and the Cockayne, Warsaw breakage and Hoyeraal-Hreidarsson syndromes.
Most nuclear Fe-S cluster proteins bind cubane [4Fe-4S] clusters using a canonical four-cysteinyl ligand structure. A few exceptions exhibit alternative architectures for cluster binding sites and stoichiometries, including the [2Fe-2S] proteins CPSF4 and FBXL5, the [3Fe-4S] protein NCOA4, and the radical SAM enzymes that bind the [4Fe-4S] cluster using three cysteines and an exchangeable S-adenosyl-L-methionine. In some cases, such as CPSF4 and KIF4A/B, the exact residues responsible for cluster binding have yet to be determined.
There is clear evidence that most nuclear Fe-S proteins depend on the CIA machinery for their metallation. However, a systematic survey of the specific CIA subcomplexes and adaptors required for the metallation of each nuclear Fe-S protein is necessary to fully understand how, when, and where Fe-S proteins acquire their clusters, as well as how this process is regulated. Notably, nuclear Fe-S proteins appear to bind the clusters at solvent-exposed loops. New evidence suggests that cluster transfer can occur to fully folded proteins forming complexes. Investigation of newly obtained structures of Fe-S proteins would help us understand whether solvent accessible clusters are a general characteristic of [4Fe-4S] proteins and if this has functional implications. One can speculate that these accessible positions may allow for sensory roles of the clusters, enabling dynamic cluster disassembly and reassembly in response to intracellular cues like redox status or iron levels, thus allowing cells to modulate Fe-S enzyme activity according to their needs. Do solvent-accessible, redox-active clusters play a role in sensing structural changes in DNA, e. g. DNA damage? [229] The possibility of re-metallation or repair of exchangeable clusters for reactivating apo proteins raises important questions about the dynamic trafficking of bioavailable Fe-S clusters and whether an equilibrium exists between the apo and holo forms of these proteins. Trafficking systems, such as the Glrx3-[2Fe-2S]-BolA2 system that exchanges Fe-S clusters with nuclear iron-responsive transcriptional regulators in fungi, may also traffic a pool of bioavailable Fe-S clusters in the nucleus of mammalian cells. Alternatively, CIA targeting complexes may deliver clusters in-situ when and where needed, as observed for the mitotic factor KIF4A, which colocalizes with MMS19 at the mitotic spindle. All these hypotheses remain to be experimentally tested.
The discovery of Fe-S clusters in viral proteins, including the SARS-CoV-2 RNA-dependent RNA polymerase [128] and the helicase nsp13 [127], as well as the hepatitis B virus protein HBx, which localizes to the nucleus of mammalian cells during infection [230,231], indicates a broad role for Fe-S clusters in viral infection. It seems likely that additional nuclear Fe-S binding proteins will be identified, particularly among those automatically annotated in the proteome as Zn-binding proteins. We hope this review encourage further research on the unresolved questions regarding Fe-S clusters in human cells, potentially leading to promising avenues for the development of novel therapeutic strategies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, L.N.A., A.L.T. and C.C.P.; original draft preparation, L.N.A. and A.L.T.; L.N.A. created in BioRender.com; review and editing, L.N.A., A.L.T. and C.C.P. All authors have made a substantial, direct, and intellectual contribution to the work and agreed to the published version of the manuscript. Figures Created in BioRender. Novoa-Aponte, L. (2024) https://BioRender.com/m09w088. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Intramural Research Program of the National Institutes of Health (NIH), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK).
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lill, R., Broderick, J. B. & Dean, D. R. Special issue on iron-sulfur proteins: Structure, function, biogenesis and diseases. Biochim Biophys Acta 1853, 1251-1252 (2015). [CrossRef]
- Braymer, J. J., Freibert, S. A., Rakwalska-Bange, M. & Lill, R. Mechanistic concepts of iron-sulfur protein biogenesis in Biology. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1868, 118863 (2021). [CrossRef]
- Bak, D. W. & Weerapana, E. Proteomic strategies to interrogate the Fe-S proteome. Biochim Biophys Acta Mol Cell Res 1871, 119791 (2024). [CrossRef]
- Rudolf, J., Makrantoni, V., Ingledew, W. J., Stark, M. J. & White, M. F. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Molecular cell 23, 801-808 (2006). [CrossRef]
- Thul, P. J. et al. A subcellular map of the human proteome. Science 356, eaal3321 (2017). [CrossRef]
- Consortium, T. U. UniProt: the universal protein knowledgebase in 2023. Nucleic acids research 51, D523-D531 (2023).
- Binder, J. X. et al. COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database 2014 (2014). [CrossRef]
- Klinge, S., Hirst, J., Maman, J. D., Krude, T. & Pellegrini, L. An iron-sulfur domain of the eukaryotic primase is essential for RNA primer synthesis. Nature structural & molecular biology 14, 875-877 (2007). [CrossRef]
- Weiner, B. E. et al. An iron-sulfur cluster in the C-terminal domain of the p58 subunit of human DNA primase. Journal of biological chemistry 282, 33444-33451 (2007). [CrossRef]
- Starokadomskyy, P. et al. DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA: DNA synthesis. Nature immunology 17, 495-504 (2016). [CrossRef]
- Netz, D. J. et al. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nature chemical biology 8, 125-132 (2012).
- Chea, J. et al. Spatiotemporal recruitment of human DNA polymerase delta to sites of UV damage. Cell cycle 11, 2885-2895 (2012). [CrossRef]
- Roske, J. J. & Yeeles, J. T. Structural basis for processive daughter-strand synthesis and proofreading by the human leading-strand DNA polymerase Pol ε. Nature Structural & Molecular Biology, 1-11 (2024). [CrossRef]
- Ræder, S. B. et al. APIM-mediated REV3L–PCNA interaction important for error free TLS Over UV-induced DNA lesions in human cells. International Journal of Molecular Sciences 20, 100 (2018). [CrossRef]
- Cantor, S. B. et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105, 149-160 (2001). [CrossRef]
- Seki, M., Takeda, Y., Iwai, K. & Tanaka, K. IOP1 protein is an external component of the human cytosolic iron-sulfur cluster assembly (CIA) machinery and functions in the MMS19 protein-dependent CIA pathway. Journal of Biological Chemistry 288, 16680-16689 (2013). [CrossRef]
- Wu, Y. et al. Fanconi anemia group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood, The Journal of the American Society of Hematology 116, 3780-3791 (2010). [CrossRef]
- Parish, J. L. et al. The DNA helicase ChlR1 is required for sister chromatid cohesion in mammalian cells. Journal of cell science 119, 4857-4865 (2006). [CrossRef]
- Simon, A. K. et al. The iron–sulfur helicase DDX11 promotes the generation of single-stranded DNA for CHK1 activation. Life science alliance 3 (2020). [CrossRef]
- Landry, A. P. & Ding, H. The N-Terminal Domain of Human DNA Helicase Rtel1 Contains a Redox Active Iron-Sulfur Cluster. BioMed research international 2014, 285791 (2014). [CrossRef]
- Ito, S. et al. MMXD, a TFIIH-independent XPD-MMS19 protein complex involved in chromosome segregation. Molecular cell 39, 632-640 (2010). [CrossRef]
- Duxin, J. P. et al. Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Molecular and cellular biology 29, 4274-4282 (2009). [CrossRef]
- Pokharel, S. & Campbell, J. L. Cross talk between the nuclease and helicase activities of Dna2: role of an essential iron–sulfur cluster domain. Nucleic acids research 40, 7821-7830 (2012). [CrossRef]
- Mariotti, L. et al. The iron–sulphur cluster in human DNA2 is required for all biochemical activities of DNA2. Communications biology 3, 322 (2020). [CrossRef]
- Sparks, J. L. et al. Human exonuclease 5 is a novel sliding exonuclease required for genome stability. Journal of Biological Chemistry 287, 42773-42783 (2012). [CrossRef]
- Komine, K. et al. Functional complementation assay for 47 MUTYH variants in a MutY-disrupted Escherichia coli strain. Human mutation 36, 704-711 (2015).
- Chepanoske, C. L., Golinelli, M.-P., Williams, S. D. & David, S. S. Positively Charged Residues within the Iron–Sulfur Cluster Loop of E. coli MutY Participate in Damage Recognition and Removal. Archives of biochemistry and biophysics 380, 11-19 (2000).
- Nuñez, N. N., Majumdar, C., Lay, K. T. & David, S. S. in Methods in enzymology Vol. 599 21-68 (Elsevier, 2018).
- Ikeda, S., Kohmoto, T., Tabata, R. & Seki, Y. Differential intracellular localization of the human and mouse endonuclease III homologs and analysis of the sorting signals. DNA repair 1, 847-854 (2002). [CrossRef]
- Kuo, C.-F. et al. Atomic structure of the DNA repair [4Fe-4S] enzyme endonuclease III. Science 258, 434-440 (1992). [CrossRef]
- Carroll, B. L. et al. Caught in motion: human NTHL1 undergoes interdomain rearrangement necessary for catalysis. Nucleic Acids Research 49, 13165-13178 (2021). [CrossRef]
- Kim, J.-H., Lane, W. S. & Reinberg, D. Human Elongator facilitates RNA polymerase II transcription through chromatin. Proceedings of the National Academy of Sciences 99, 1241-1246 (2002). [CrossRef]
- Paraskevopoulou, C., Fairhurst, S. A., Lowe, D. J., Brick, P. & Onesti, S. The elongator subunit Elp3 contains a Fe4S4 cluster and binds S-adenosylmethionine. Molecular microbiology 59, 795-806 (2006).
- Kimura, S. & Suzuki, T. Iron-sulfur proteins responsible for RNA modifications. Biochim Biophys Acta 1853, 1272-1283 (2015). [CrossRef]
- Ramsay, E. P. et al. Structure of human RNA polymerase III. Nature communications 11, 6409 (2020). [CrossRef]
- Blombach, F. et al. Archaeal TFEα/β is a hybrid of TFIIE and the RNA polymerase III subcomplex hRPC62/39. Elife 4, e08378 (2015).
- Li, L. et al. Structure of human RNA polymerase III elongation complex. Cell research 31, 791-800 (2021). [CrossRef]
- Chen, W. et al. CPSF4 activates telomerase reverse transcriptase and predicts poor prognosis in human lung adenocarcinomas. Molecular oncology 8, 704-716 (2014). [CrossRef]
- Shimberg, G. D. et al. Cleavage and polyadenylation specificity factor 30: An RNA-binding zinc-finger protein with an unexpected 2Fe-2S cluster. Proc Natl Acad Sci U S A 113, 4700-4705 (2016). [CrossRef]
- Pritts, J. D. et al. Unraveling the RNA Binding Properties of the Iron–Sulfur Zinc Finger Protein CPSF30. Biochemistry 59, 970-982 (2020). [CrossRef]
- Young, A. P. & Bandarian, V. in Methods in enzymology Vol. 606 119-153 (Elsevier, 2018).
- Reiter, V. et al. The CDK5 repressor CDK5RAP1 is a methylthiotransferase acting on nuclear and mitochondrial RNA. Nucleic Acids Res 40, 6235-6240 (2012). [CrossRef]
- Pierrel, F., Douki, T., Fontecave, M. & Atta, M. MiaB protein is a bifunctional radical-S-adenosylmethionine enzyme involved in thiolation and methylation of tRNA. Journal of Biological Chemistry 279, 47555-47563 (2004). [CrossRef]
- LEE, Y. M. et al. Human kinesin superfamily member 4 is dominantly localized in the nuclear matrix and is associated with chromosomes during mitosis. Biochemical Journal 360, 549-556 (2001).
- Ben-Shimon, L. et al. Fe-S cluster coordination of the chromokinesin KIF4A alters its subcellular localization during mitosis. Journal of Cell Science 131, jcs211433 (2018). [CrossRef]
- Bellelli, R. et al. NCOA4 transcriptional coactivator inhibits activation of DNA replication origins. Molecular cell 55, 123-137 (2014). [CrossRef]
- Kuno, S. & Iwai, K. Oxygen modulates iron homeostasis by switching iron sensing of NCOA4. Journal of Biological Chemistry 299 (2023). [CrossRef]
- Zhao, H. et al. NCOA4 requires a [3Fe-4S] to sense and maintain the iron homeostasis. Journal of Biological Chemistry 300 (2024). [CrossRef]
- Vinas-Castells, R. et al. Nuclear ubiquitination by FBXL5 modulates Snail1 DNA binding and stability. Nucleic acids research 42, 1079-1094 (2013). [CrossRef]
- Wang, H. et al. FBXL5 regulates IRP2 stability in iron homeostasis via an oxygen-responsive [2Fe2S] cluster. Molecular cell 78, 31-41. e35 (2020). [CrossRef]
- Bruening, W. et al. Expression of OVCA1, a candidate tumor suppressor, is reduced in tumors and inhibits growth of ovarian cancer cells. Cancer research 59, 4973-4983 (1999).
- Zhang, Y. et al. Diphthamide biosynthesis requires an organic radical generated by an iron–sulphur enzyme. Nature 465, 891-896 (2010). [CrossRef]
- Kwon, Y. T., Kashina, A. S. & Varshavsky, A. Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Molecular and cellular biology 19, 182-193 (1999).
- Van, V. et al. Iron-sulfur clusters are involved in post-translational arginylation. Nature Communications 14, 458 (2023). [CrossRef]
- Du, Z. et al. Structure of the human respiratory complex II. Proceedings of the National Academy of Sciences 120, e2216713120 (2023).
- Hao, Z. et al. Subcellular localization of CIAPIN1. Journal of Histochemistry & Cytochemistry 54, 1437-1444 (2006).
- Banci, L. et al. Anamorsin is a [2Fe-2S] cluster-containing substrate of the Mia40-dependent mitochondrial protein trapping machinery. Chemistry & biology 18, 794-804 (2011). [CrossRef]
- Zhang, Y., Yang, C., Dancis, A. & Nakamaru-Ogiso, E. EPR studies of wild type and mutant Dre2 identify essential [2Fe–-2S] and [4Fe–-4S] clusters and their cysteine ligands. The journal of biochemistry, mvw054 (2016). [CrossRef]
- Willems, P. et al. BOLA1 is an aerobic protein that prevents mitochondrial morphology changes induced by glutathione depletion. Antioxidants & redox signaling 18, 129-138 (2013). [CrossRef]
- Li, H., Mapolelo, D. T., Randeniya, S., Johnson, M. K. & Outten, C. E. Human glutaredoxin 3 forms [2Fe-2S]-bridged complexes with human BolA2. Biochemistry 51, 1687-1696 (2012). [CrossRef]
- Frey, A. G., Palenchar, D. J., Wildemann, J. D. & Philpott, C. C. A Glutaredoxin· BolA Complex Serves as an Iron-Sulfur Cluster Chaperone for the Cytosolic Cluster Assembly Machinery*♦. Journal of Biological Chemistry 291, 22344-22356 (2016).
- Pandya, P., Braiman, A. & Isakov, N. PICOT (GLRX3) is a positive regulator of stress-induced DNA-damage response. Cellular signalling 62, 109340 (2019). [CrossRef]
- Okuno, T., Yamabayashi, H. & Kogure, K. Comparison of intracellular localization of Nubp1 and Nubp2 using GFP fusion proteins. Molecular biology reports 37, 1165-1168 (2010). [CrossRef]
- Netz, D. J., Pierik, A. J., Stümpfig, M., Mühlenhoff, U. & Lill, R. The Cfd1–Nbp35 complex acts as a scaffold for iron-sulfur protein assembly in the yeast cytosol. Nature chemical biology 3, 278-286 (2007). [CrossRef]
- Stehling, O. et al. Human Nbp35 is essential for both cytosolic iron-sulfur protein assembly and iron homeostasis. Molecular and cellular biology 28, 5517-5528 (2008). [CrossRef]
- Tong, W.-H., Jameson, G. N., Huynh, B. H. & Rouault, T. A. Subcellular compartmentalization of human Nfu, an iron–sulfur cluster scaffold protein, and its ability to assemble a [4Fe–4S] cluster. Proceedings of the National Academy of Sciences 100, 9762-9767 (2003).
- Cai, K. et al. Structural/functional properties of human NFU1, an intermediate [4Fe-4S] carrier in human mitochondrial iron-sulfur cluster biogenesis. Structure 24, 2080-2091 (2016). [CrossRef]
- Land, T. & Rouault, T. A. Targeting of a human iron–sulfur cluster assembly enzyme, nifs, to different subcellular compartments is regulated through alternative AUG utilization. Molecular cell 2, 807-815 (1998). [CrossRef]
- Fox, N. G. et al. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nature communications 10, 2210 (2019). [CrossRef]
- Marelja, Z., Stöcklein, W., Nimtz, M. & Leimkühler, S. A novel role for human Nfs1 in the cytoplasm: Nfs1 acts as a sulfur donor for MOCS3, a protein involved in molybdenum cofactor biosynthesis. Journal of Biological Chemistry 283, 25178-25185 (2008).
- Lundberg, M. et al. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. Journal of Biological Chemistry 276, 26269-26275 (2001). [CrossRef]
- Lillig, C. H. et al. Characterization of human glutaredoxin 2 as iron–sulfur protein: a possible role as redox sensor. Proceedings of the National Academy of Sciences 102, 8168-8173 (2005). [CrossRef]
- Kispal, G., Csere, P., Prohl, C. & Lill, R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. The EMBO journal 18, 3981-3989 (1999). [CrossRef]
- Netz, D. J., Mascarenhas, J., Stehling, O., Pierik, A. J. & Lill, R. Maturation of cytosolic and nuclear iron–sulfur proteins. Trends in cell biology 24, 303-312 (2014). [CrossRef]
- Song, D. & Lee, F. S. A role for IOP1 in mammalian cytosolic iron-sulfur protein biogenesis. Journal of Biological Chemistry 283, 9231-9238 (2008). [CrossRef]
- Netz, D. J. et al. A bridging [4Fe-4S] cluster and nucleotide binding are essential for function of the Cfd1-Nbp35 complex as a scaffold in iron-sulfur protein maturation. Journal of Biological Chemistry 287, 12365-12378 (2012). [CrossRef]
- Stehling, O. et al. MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science 337, 195-199 (2012). [CrossRef]
- Gari, K. et al. MMS19 links cytoplasmic iron-sulfur cluster assembly to DNA metabolism. Science 337, 243-245 (2012). [CrossRef]
- Balk, J., Pierik, A. J., Netz, D. J. A., Mühlenhoff, U. & Lill, R. The hydrogenase-like Nar1p is essential for maturation of cytosolic and nuclear iron–sulphur proteins. The EMBO journal 23, 2105-2115 (2004). [CrossRef]
- SantaMaria, A. M. & Rouault, T. A. Regulatory and Sensing Iron–Sulfur Clusters: New Insights and Unanswered Questions. Inorganics 12, 101 (2024). [CrossRef]
- Querci, L., Piccioli, M., Ciofi-Baffoni, S. & Banci, L. Structural aspects of iron-sulfur protein biogenesis: An NMR view. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 119786 (2024). [CrossRef]
- Maio, N. et al. Cochaperone binding to LYR motifs confers specificity of iron sulfur cluster delivery. Cell metabolism 19, 445-457 (2014). [CrossRef]
- Li, K., Tong, W.-H., Hughes, R. M. & Rouault, T. A. Roles of the mammalian cytosolic cysteine desulfurase, ISCS, and scaffold protein, ISCU, in iron-sulfur cluster assembly. Journal of Biological Chemistry 281, 12344-12351 (2006). [CrossRef]
- Kim, K. S., Maio, N., Singh, A. & Rouault, T. A. Cytosolic HSC20 integrates de novo iron–sulfur cluster biogenesis with the CIAO1-mediated transfer to recipients. Human molecular genetics 27, 837-852 (2018). [CrossRef]
- Shi, H., Bencze, K. Z., Stemmler, T. L. & Philpott, C. C. A cytosolic iron chaperone that delivers iron to ferritin. Science 320, 1207-1210 (2008). [CrossRef]
- Philpott, C. C. Coming into view: eukaryotic iron chaperones and intracellular iron delivery. Journal of Biological Chemistry 287, 13518-13523 (2012). [CrossRef]
- Philpott, C. C., Ryu, M.-S., Frey, A. & Patel, S. Cytosolic iron chaperones: proteins delivering iron cofactors in the cytosol of mammalian cells. Journal of Biological Chemistry 292, 12764-12771 (2017). [CrossRef]
- Philpott, C. C., Patel, S. J. & Protchenko, O. Management versus miscues in the cytosolic labile iron pool: The varied functions of iron chaperones. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1867, 118830 (2020). [CrossRef]
- Patel, S. J. et al. A PCBP1–BolA2 chaperone complex delivers iron for cytosolic [2Fe–2S] cluster assembly. Nature chemical biology 15, 872-881 (2019).
- Adamec, J. et al. Iron-dependent self-assembly of recombinant yeast frataxin: implications for Friedreich ataxia. The American Journal of Human Genetics 67, 549-562 (2000). [CrossRef]
- Bulteau, A.-L. et al. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 305, 242-245 (2004). [CrossRef]
- Cavadini, P., O’Neill, H. A., Benada, O. & Isaya, G. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Human molecular genetics 11, 217-227 (2002). [CrossRef]
- Bencze, K. Z. et al. The structure and function of frataxin. Critical reviews in biochemistry and molecular biology 41, 269-291 (2006).
- Parent, A. et al. Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nature communications 6, 5686 (2015). [CrossRef]
- Bridwell-Rabb, J., Fox, N. G., Tsai, C.-L., Winn, A. M. & Barondeau, D. P. Human frataxin activates Fe–S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry 53, 4904-4913 (2014). [CrossRef]
- Balk, J., Aguilar Netz, D. J., Tepper, K., Pierik, A. J. & Lill, R. The essential WD40 protein Cia1 is involved in a late step of cytosolic and nuclear iron-sulfur protein assembly. Molecular and cellular biology 25, 10833-10841 (2005). [CrossRef]
- Stehling, O. et al. Human CIA2A-FAM96A and CIA2B-FAM96B integrate iron homeostasis and maturation of different subsets of cytosolic-nuclear iron-sulfur proteins. Cell metabolism 18, 187-198 (2013). [CrossRef]
- Marquez, M. D. et al. Cytosolic iron–sulfur protein assembly system identifies clients by a C-terminal tripeptide. Proceedings of the National Academy of Sciences 120, e2311057120 (2023). [CrossRef]
- Paul, V. D. et al. The deca-GX3 proteins Yae1-Lto1 function as adaptors recruiting the ABC protein Rli1 for iron-sulfur cluster insertion. Elife 4, e08231 (2015). [CrossRef]
- Odermatt, D. & Gari, K. (2017).
- Kassube, S. A. & Thomä, N. H. Structural insights into Fe–S protein biogenesis by the CIA targeting complex. Nature structural & molecular biology 27, 735-742 (2020). [CrossRef]
- Alhebshi, A., Sideri, T. C., Holland, S. L. & Avery, S. V. The essential iron-sulfur protein Rli1 is an important target accounting for inhibition of cell growth by reactive oxygen species. Molecular biology of the cell 23, 3582-3590 (2012). [CrossRef]
- Honarmand Ebrahimi, K. et al. Iron–sulfur clusters as inhibitors and catalysts of viral replication. Nature Chemistry 14, 253-266 (2022). [CrossRef]
- Jang, S. & Imlay, J. A. Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. Journal of Biological Chemistry 282, 929-937 (2007). [CrossRef]
- Mettert, E. L. & Kiley, P. J. Fe–S proteins that regulate gene expression. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1853, 1284-1293 (2015).
- Imlay, J. A. Iron-sulphur clusters and the problem with oxygen. Molecular microbiology 59, 1073-1082 (2006). [CrossRef]
- Mettert, E. L. & Kiley, P. J. How is Fe-S cluster formation regulated? Annual review of microbiology 69, 505-526 (2015).
- Lauble, H., Kennedy, M., Beinert, H. & Stout, C. Crystal structures of aconitase with isocitrate and nitroisocitrate bound. Biochemistry 31, 2735-2748 (1992). [CrossRef]
- Federico, G. et al. NCOA4 links iron bioavailability to DNA metabolism. Cell Reports 40 (2022). [CrossRef]
- van Wietmarschen, N., Moradian, A., Morin, G. B., Lansdorp, P. M. & Uringa, E.-J. The mammalian proteins MMS19, MIP18, and ANT2 are involved in cytoplasmic iron-sulfur cluster protein assembly. Journal of Biological Chemistry 287, 43351-43358 (2012). [CrossRef]
- Puig, S., Ramos-Alonso, L., Romero, A. & Martínez-Pastor, M. (2017).
- Troadec, M.-B., Loréal, O. & Brissot, P. The interaction of iron and the genome: For better and for worse. Mutation Research/Reviews in Mutation Research 774, 25-32 (2017). [CrossRef]
- Veatch, J. R., McMurray, M. A., Nelson, Z. W. & Gottschling, D. E. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell 137, 1247-1258 (2009). [CrossRef]
- Shi, R., Hou, W., Wang, Z.-Q. & Xu, X. Biogenesis of iron–sulfur clusters and their role in DNA metabolism. Frontiers in Cell and Developmental Biology 9, 735678 (2021). [CrossRef]
- Loeb, L. A. & Monnat Jr, R. J. DNA polymerases and human disease. Nature Reviews Genetics 9, 594-604 (2008).
- Ter Beek, J. et al. Structural evidence for an essential Fe–S cluster in the catalytic core domain of DNA polymerase ϵ. Nucleic acids research 47, 5712-5722 (2019). [CrossRef]
- Jain, R. et al. An iron–sulfur cluster in the polymerase domain of yeast DNA polymerase ε. Journal of molecular biology 426, 301-308 (2014).
- Baranovskiy, A. G. et al. DNA polymerase δ and ζ switch by sharing accessory subunits of DNA polymerase δ. Journal of Biological Chemistry 287, 17281-17287 (2012).
- Lisova, A. E. et al. The iron-sulfur cluster is essential for DNA binding by human DNA polymerase ε. Scientific reports 12, 17436 (2022). [CrossRef]
- Bartels, P. L., Stodola, J. L., Burgers, P. M. & Barton, J. K. A redox role for the [4Fe4S] cluster of yeast DNA polymerase δ. Journal of the American Chemical Society 139, 18339-18348 (2017). [CrossRef]
- Baranovskiy, A. G. et al. Mechanism of concerted RNA-DNA primer synthesis by the human primosome. Journal of Biological Chemistry 291, 10006-10020 (2016). [CrossRef]
- Suwa, Y. et al. Crystal structure of the human Pol α B subunit in complex with the C-terminal domain of the catalytic subunit. Journal of Biological Chemistry 290, 14328-14337 (2015). [CrossRef]
- Klinge, S., Núñez-Ramírez, R., Llorca, O. & Pellegrini, L. 3D architecture of DNA Pol α reveals the functional core of multi-subunit replicative polymerases. The EMBO journal 28, 1978-1987 (2009). [CrossRef]
- Zhang, Y., Baranovskiy, A. G., Tahirov, T. H. & Pavlov, Y. I. The C-terminal domain of the DNA polymerase catalytic subunit regulates the primase and polymerase activities of the human DNA polymerase α-primase complex. Journal of Biological Chemistry 289, 22021-22034 (2014). [CrossRef]
- Baranovskiy, A. G. & Tahirov, T. H. Elaborated action of the human primosome. Genes 8, 62 (2017). [CrossRef]
- Pritts, J. D. & Michel, S. L. Fe-S clusters masquerading as zinc finger proteins. Journal of Inorganic Biochemistry 230, 111756 (2022). [CrossRef]
- Maio, N. et al. An iron–sulfur cluster in the zinc-binding domain of the SARS-CoV-2 helicase modulates its RNA-binding and-unwinding activities. Proceedings of the National Academy of Sciences 120, e2303860120 (2023).
- Maio, N. et al. Fe-S cofactors in the SARS-CoV-2 RNA-dependent RNA polymerase are potential antiviral targets. Science 373, 236-241 (2021).
- Shimberg, G. D. et al. Cleavage and polyadenylation specificity factor 30: An RNA-binding zinc-finger protein with an unexpected 2Fe–2S cluster. Proceedings of the National Academy of Sciences 113, 4700-4705 (2016). [CrossRef]
- Conlan, A. R. et al. Crystal structure of Miner1: The redox-active 2Fe-2S protein causative in Wolfram Syndrome 2. Journal of molecular biology 392, 143-153 (2009). [CrossRef]
- Cutone, A. et al. Pichia pastoris Fep1 is a [2Fe-2S] protein with a Zn finger that displays an unusual oxygen-dependent role in cluster binding. Scientific Reports 6, 31872 (2016). [CrossRef]
- Liu, L. & Huang, M. Essential role of the iron-sulfur cluster binding domain of the primase regulatory subunit Pri2 in DNA replication initiation. Protein & Cell 6, 194-210 (2015). [CrossRef]
- Baranovskiy, A. G. et al. Crystal structure of the human primase. Journal of Biological Chemistry 290, 5635-5646 (2015). [CrossRef]
- Sauguet, L., Klinge, S., Perera, R. L., Maman, J. D. & Pellegrini, L. Shared active site architecture between the large subunit of eukaryotic primase and DNA photolyase. PLoS One 5, e10083 (2010). [CrossRef]
- Agarkar, V. B., Babayeva, N. D., Pavlov, Y. I. & Tahirov, T. H. Crystal structure of the C-terminal domain of human DNA primase large subunit: Implications for the mechanism of the primase-polymerase α switch. Cell Cycle 10, 926-931 (2011).
- O’Brien, E. et al. The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport. Science 355, eaag1789 (2017). [CrossRef]
- O’Brien, E. et al. Yeast require redox switching in DNA primase. Proceedings of the National Academy of Sciences 115, 13186-13191 (2018).
- Amin, M. & Brooks, B. R. The oxidation of the [4Fe-4S] cluster of DNA primase alters the binding energies with DNA and RNA primers. Biophysical Journal (2024). [CrossRef]
- O’Brien, E., Holt, M. E., Salay, L. E., Chazin, W. J. & Barton, J. K. Substrate binding regulates redox signaling in human DNA primase. Journal of the American Chemical Society 140, 17153-17162 (2018). [CrossRef]
- Mancias, J. D., Wang, X., Gygi, S. P., Harper, J. W. & Kimmelman, A. C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105-109 (2014). [CrossRef]
- Dowdle, W. E. et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nature cell biology 16, 1069-1079 (2014). [CrossRef]
- Singleton, M. R., Dillingham, M. S. & Wigley, D. B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76, 23-50 (2007). [CrossRef]
- Wu, Y. & Brosh Jr, R. M. DNA helicase and helicase–nuclease enzymes with a conserved iron–sulfur cluster. Nucleic acids research 40, 4247-4260 (2012).
- Yeeles, J. T., Cammack, R. & Dillingham, M. S. An iron-sulfur cluster is essential for the binding of broken DNA by AddAB-type helicase-nucleases. Journal of Biological Chemistry 284, 7746-7755 (2009).
- Zheng, L. et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Molecular cell 32, 325-336 (2008). [CrossRef]
- Estep, K. N. & Brosh Jr, R. M. RecQ and Fe–S helicases have unique roles in DNA metabolism dictated by their unwinding directionality, substrate specificity, and protein interactions. Biochemical Society Transactions 46, 77-95 (2018).
- Matsuzaki, K., Borel, V., Adelman, C. A., Schindler, D. & Boulton, S. J. FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes & Development 29, 2532-2546 (2015). [CrossRef]
- Capo-Chichi, J. M. et al. Identification and biochemical characterization of a novel mutation in DDX11 causing W arsaw breakage syndrome. Human mutation 34, 103-107 (2013).
- Deng, Z. et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal–Hreidarsson syndrome. Proceedings of the National Academy of Sciences 110, E3408-E3416 (2013). [CrossRef]
- Falquet, B. et al. Disease-associated DNA2 nuclease–helicase protects cells from lethal chromosome under-replication. Nucleic acids research 48, 7265-7278 (2020). [CrossRef]
- Sommers, J. A. et al. FANCJ uses its motor ATPase to destabilize protein-DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. Journal of Biological Chemistry 284, 7505-7517 (2009). [CrossRef]
- Levran, O. et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nature genetics 37, 931-933 (2005). [CrossRef]
- Odermatt, D. C. et al. Cancer-associated mutations in the iron-sulfur domain of FANCJ affect G-quadruplex metabolism. PLoS genetics 16, e1008740 (2020). [CrossRef]
- Bharti, S. K. et al. Specialization among iron-sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. Journal of Biological Chemistry 288, 28217-28229 (2013). [CrossRef]
- Wulfridge, P. & Sarma, K. Intertwining roles of R-loops and G-quadruplexes in DNA repair, transcription and genome organization. Nature Cell Biology, 1-12 (2024). [CrossRef]
- Hänsel-Hertsch, R., Di Antonio, M. & Balasubramanian, S. DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nature reviews Molecular cell biology 18, 279-284 (2017). [CrossRef]
- Wu, Y., Shin-ya, K. & Brosh Jr, R. M. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Molecular and cellular biology 28, 4116-4128 (2008).
- London, T. B. et al. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. Journal of Biological Chemistry 283, 36132-36139 (2008). [CrossRef]
- Wu, C. G. & Spies, M. G-quadruplex recognition and remodeling by the FANCJ helicase. Nucleic acids research 44, 8742-8753 (2016). [CrossRef]
- Vannier, J.-B. et al. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 342, 239-242 (2013). [CrossRef]
- Uringa, E.-J., Youds, J. L., Lisaingo, K., Lansdorp, P. M. & Boulton, S. J. RTEL1: an essential helicase for telomere maintenance and the regulation of homologous recombination. Nucleic acids research 39, 1647-1655 (2011). [CrossRef]
- Wu, W. et al. RTEL1 suppresses G-quadruplex-associated R-loops at difficult-to-replicate loci in the human genome. Nature structural & molecular biology 27, 424-437 (2020). [CrossRef]
- Ghisays, F. et al. RTEL1 influences the abundance and localization of TERRA RNA. Nature communications 12, 3016 (2021). [CrossRef]
- Wu, Y., Sommers, J. A., Khan, I., de Winter, J. P. & Brosh, R. M. Biochemical characterization of Warsaw breakage syndrome helicase. Journal of Biological Chemistry 287, 1007-1021 (2012). [CrossRef]
- van Schie, J. J. et al. Warsaw Breakage Syndrome associated DDX11 helicase resolves G-quadruplex structures to support sister chromatid cohesion. Nature communications 11, 4287 (2020).
- Gray, L. T., Vallur, A. C., Eddy, J. & Maizels, N. G quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nature chemical biology 10, 313-318 (2014). [CrossRef]
- Kou, H., Zhou, Y., Gorospe, R. C. & Wang, Z. Mms19 protein functions in nucleotide excision repair by sustaining an adequate cellular concentration of the TFIIH component Rad3. Proceedings of the National Academy of Sciences 105, 15714-15719 (2008). [CrossRef]
- Seroz, T. et al. Cloning of a human homolog of the yeast nucleotide excision repair gene MMS19 and interaction with transcription repair factor TFIIH via the XPB and XPD helicases. Nucleic acids research 28, 4506-4513 (2000). [CrossRef]
- Ding, H. et al. Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell 117, 873-886 (2004).
- Askree, S. H. et al. A genome-wide screen for Saccharomyces cerevisiae deletion mutants that affect telomere length. Proceedings of the National Academy of Sciences 101, 8658-8663 (2004). [CrossRef]
- Friedberg, E. C., Walker, G. C., Siede, W. & Wood, R. D. DNA repair and mutagenesis. (American Society for Microbiology Press, 2005).
- Knijnenburg, T. A. et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell reports 23, 239-254. e236 (2018). [CrossRef]
- Fuss, J. O., Tsai, C.-L., Ishida, J. P. & Tainer, J. A. Emerging critical roles of Fe–S clusters in DNA replication and repair. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1853, 1253-1271 (2015). [CrossRef]
- Cunningham, R. P. et al. Endonuclease III is an iron-sulfur protein. Biochemistry 28, 4450-4455 (1989). [CrossRef]
- Hinks, J. A. et al. An iron-sulfur cluster in the family 4 uracil-DNA glycosylases. Journal of Biological Chemistry 277, 16936-16940 (2002). [CrossRef]
- Parker, A., Gu, Y. & Lu, A.-L. Purification and characterization of a mammalian homolog of Escherichia coli MutY mismatch repair protein from calf liver mitochondria. Nucleic acids research 28, 3206-3215 (2000). [CrossRef]
- Jacobs, A. L. & Schär, P. DNA glycosylases: in DNA repair and beyond. Chromosoma 121, 1-20 (2012). [CrossRef]
- McGoldrick, J. P., Yeh, Y.-C., Solomon, M., Essigmann, J. M. & Lu, A.-L. Characterization of a mammalian homolog of the Escherichia coli MutY mismatch repair protein. Molecular and cellular biology 15, 989-996 (1995). [CrossRef]
- Aspinwall, R. et al. Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proceedings of the National Academy of Sciences 94, 109-114 (1997). [CrossRef]
- Lukianova, O. A. & David, S. S. A role for iron–sulfur clusters in DNA repair. Current opinion in chemical biology 9, 145-151 (2005).
- Trasviña-Arenas, C. H., Lopez-Castillo, L. M., Sanchez-Sandoval, E. & Brieba, L. G. Dispensability of the [4Fe-4S] cluster in novel homologues of adenine glycosylase MutY. The FEBS journal 283, 521-540 (2016). [CrossRef]
- Guan, Y. et al. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nature structural biology 5, 1058-1064 (1998). [CrossRef]
- Boal, A. K. et al. DNA-bound redox activity of DNA repair glycosylases containing [4Fe-4S] clusters. Biochemistry 44, 8397-8407 (2005). [CrossRef]
- Romano, C. A., Sontz, P. A. & Barton, J. K. Mutants of the base excision repair glycosylase, endonuclease III: DNA charge transport as a first step in lesion detection. Biochemistry 50, 6133-6145 (2011). [CrossRef]
- Porello, S. L., Cannon, M. J. & David, S. S. A substrate recognition role for the [4Fe-4S] 2+ cluster of the DNA repair glycosylase MutY. Biochemistry 37, 6465-6475 (1998). [CrossRef]
- Bartels, P. L. et al. Electrochemistry of the [4Fe4S] cluster in base excision repair proteins: tuning the redox potential with DNA. Langmuir 33, 2523-2530 (2017). [CrossRef]
- Tse, E. C., Zwang, T. J. & Barton, J. K. The oxidation state of [4Fe4S] clusters modulates the DNA-binding affinity of DNA repair proteins. Journal of the American Chemical Society 139, 12784-12792 (2017). [CrossRef]
- Bruner, S. D., Norman, D. P. & Verdine, G. L. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. nature 403, 859-866 (2000). [CrossRef]
- Poor, C. B. et al. Molecular mechanism and structure of the Saccharomyces cerevisiae iron regulator Aft2. Proc Natl Acad Sci U S A 111, 4043-4048 (2014). [CrossRef]
- Ueta, R., Fujiwara, N., Iwai, K. & Yamaguchi-Iwai, Y. Iron-induced dissociation of the Aft1p transcriptional regulator from target gene promoters is an initial event in iron-dependent gene suppression. Mol Cell Biol 32, 4998-5008 (2012). [CrossRef]
- Li, H. et al. The yeast iron regulatory proteins Grx3/4 and Fra2 form heterodimeric complexes containing a [2Fe-2S] cluster with cysteinyl and histidyl ligation. Biochemistry 48, 9569-9581 (2009).
- Mercier, A., Watt, S., Bähler, J. r. & Labbé, S. Key function for the CCAAT-binding factor Php4 to regulate gene expression in response to iron deficiency in fission yeast. Eukaryotic cell 7, 493-508 (2008). [CrossRef]
- Vachon, P., Mercier, A., Jbel, M. & Labbé, S. The monothiol glutaredoxin Grx4 exerts an iron-dependent inhibitory effect on Php4 function. Eukaryotic Cell 11, 806-819 (2012). [CrossRef]
- Dlouhy, A. C., Beaudoin, J., Labbé, S. & Outten, C. E. Schizosaccharomyces pombe Grx4 regulates the transcriptional repressor Php4 via [2Fe–2S] cluster binding. Metallomics 9, 1096-1105 (2017). [CrossRef]
- Hati, D. et al. Iron homeostasis proteins Grx4 and Fra2 control activity of the Schizosaccharomyces pombe iron repressor Fep1 by facilitating [2Fe-2S] cluster removal. J Biol Chem 299, 105419 (2023). [CrossRef]
- Jacques, J. F., Mercier, A., Brault, A., Mourer, T. & Labbe, S. Fra2 is a co-regulator of Fep1 inhibition in response to iron starvation. PLoS One 9, e98959 (2014). [CrossRef]
- Pelletier, B., Trott, A., Morano, K. A. & Labbé, S. Functional characterization of the iron-regulatory transcription factor Fep1 from Schizosaccharomyces pombe. Journal of Biological Chemistry 280, 25146-25161 (2005). [CrossRef]
- Ogunjimi, B. et al. Inborn errors in RNA polymerase III underlie severe varicella zoster virus infections. The Journal of clinical investigation 127, 3543-3556 (2017). [CrossRef]
- Girbig, M. et al. Cryo-EM structures of human RNA polymerase III in its unbound and transcribing states. Nature structural & molecular biology 28, 210-219 (2021). [CrossRef]
- Wang, Q. et al. Structural insights into transcriptional regulation of human RNA polymerase III. Nature structural & molecular biology 28, 220-227 (2021). [CrossRef]
- Wang, Z. & Roeder, R. G. Three human RNA polymerase III-specific subunits form a subcomplex with a selective function in specific transcription initiation. Genes & development 11, 1315-1326 (1997). [CrossRef]
- Chiu, Y.-H., MacMillan, J. B. & Chen, Z. J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576-591 (2009).
- Tian, K., Wang, R., Huang, J., Wang, H. & Ji, X. Subcellular localization shapes the fate of RNA polymerase III. Cell Reports 42 (2023). [CrossRef]
- Zurita, M. & Merino, C. The transcriptional complexity of the TFIIH complex. TRENDS in Genetics 19, 578-584 (2003). [CrossRef]
- Giglia-Mari, G. et al. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nature genetics 36, 714-719 (2004). [CrossRef]
- Keriel, A., Stary, A., Sarasin, A., Rochette-Egly, C. & Egly, J.-M. XPD mutations prevent TFIIH-dependent transactivation by nuclear receptors and phosphorylation of RARα. Cell 109, 125-135 (2002). [CrossRef]
- Dubaele, S. et al. Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Molecular cell 11, 1635-1646 (2003). [CrossRef]
- Compe, E. et al. Dysregulation of the peroxisome proliferator-activated receptor target genes by XPD mutations. Molecular and cellular biology 25, 6065-6076 (2005). [CrossRef]
- Pritts, J. D. & Michel, S. L. J. Fe-S clusters masquerading as zinc finger proteins. J Inorg Biochem 230, 111756 (2022). [CrossRef]
- Abbassi, N.-e.-H. et al. Cryo-EM structures of the human Elongator complex at work. Nature Communications 15, 4094 (2024). [CrossRef]
- Selvadurai, K., Wang, P., Seimetz, J. & Huang, R. H. Archaeal Elp3 catalyzes tRNA wobble uridine modification at C5 via a radical mechanism. Nature chemical biology 10, 810-812 (2014). [CrossRef]
- Dalwadi, U. & Yip, C. K. Structural insights into the function of Elongator. Cellular and Molecular Life Sciences 75, 1613-1622 (2018). [CrossRef]
- Maio, N. et al. CIAO1 loss of function causes a neuromuscular disorder with compromise of nucleocytoplasmic Fe-S enzymes. The Journal of Clinical Investigation 134 (2024). [CrossRef]
- Romero, A. M., Martinez-Pastor, M. T. & Puig, S. Iron in Translation: From the Beginning to the End. Microorganisms 9 (2021). [CrossRef]
- Sun, C. et al. Wybutosine hypomodification of tRNAphe activates HERVK and impairs neuronal differentiation. Iscience 27 (2024). [CrossRef]
- Kessler, A. C., Silveira d'Almeida, G. & Alfonzo, J. D. The role of intracellular compartmentalization on tRNA processing and modification. RNA biology 15, 554-566 (2018).
- Wei, F. Y. et al. Cdk5rap1-mediated 2-methylthio modification of mitochondrial tRNAs governs protein translation and contributes to myopathy in mice and humans. Cell Metab 21, 428-442 (2015). [CrossRef]
- Sheng, L., Hao, S.-L., Yang, W.-X. & Sun, Y. The multiple functions of kinesin-4 family motor protein KIF4 and its clinical potential. Gene 678, 90-99 (2018). [CrossRef]
- Lee, Y. M. & Kim, W. Kinesin superfamily protein member 4 (KIF4) is localized to midzone and midbody in dividing cells. Experimental & Molecular Medicine 36, 93-97 (2004). [CrossRef]
- Mazumdar, M., Sundareshan, S. & Misteli, T. Human chromokinesin KIF4A functions in chromosome condensation and segregation. The Journal of cell biology 166, 613-620 (2004). [CrossRef]
- Zhu, C. & Jiang, W. Cell cycle-dependent translocation of PRC1 on the spindle by Kif4 is essential for midzone formation and cytokinesis. Proceedings of the National Academy of Sciences 102, 343-348 (2005). [CrossRef]
- Kurasawa, Y., Earnshaw, W. C., Mochizuki, Y., Dohmae, N. & Todokoro, K. Essential roles of KIF4 and its binding partner PRC1 in organized central spindle midzone formation. The EMBO journal 23, 3237-3248 (2004). [CrossRef]
- Mazumdar, M., Sung, M.-H. & Misteli, T. Chromatin maintenance by a molecular motor protein. Nucleus 2, 591-600 (2011). [CrossRef]
- Wu, G. et al. A novel role of the chromokinesin Kif4A in DNA damage response. Cell Cycle 7, 2013-2020 (2008). [CrossRef]
- Kalantari, S. et al. Expanding the KIF4A-associated phenotype. American Journal of Medical Genetics Part A 185, 3728-3739 (2021).
- Gad, S. A. et al. The kinesin KIF4 mediates HBV/HDV entry through the regulation of surface NTCP localization and can be targeted by RXR agonists in vitro. PLoS Pathogens 18, e1009983 (2022). [CrossRef]
- Mazumdar, M. & Misteli, T. Chromokinesins: multitalented players in mitosis. Trends in cell biology 15, 349-355 (2005). [CrossRef]
- Wu, G. & Chen, P.-L. Structural requirements of chromokinesin Kif4A for its proper function in mitosis. Biochemical and biophysical research communications 372, 454-458 (2008). [CrossRef]
- Barton, J. K., Silva, R. M. & O'Brien, E. Redox chemistry in the genome: emergence of the [4Fe4S] cofactor in repair and replication. Annual review of biochemistry 88, 163-190 (2019). [CrossRef]
- Ueda, C., Langton, M., Chen, J. & Pandelia, M.-E. The HBx protein from hepatitis B virus coordinates a redox-active Fe-S cluster. Journal of Biological Chemistry 298 (2022). [CrossRef]
- Henkler, F. et al. Intracellular localization of the hepatitis B virus HBx protein. Journal of General Virology 82, 871-882 (2001). [CrossRef]
Figure 1.
Comparison of solvent-exposed and buried Fe-S clusters in proteins. Structures (left) and close-ups of the Fe-S binding domains (square, right). A. X-ray structure of human xanthine oxidase (XDH) showing the two 2Fe-2S clusters deeply buried within the protein structure (PDB 2CKJ). B. Cryo-EM structure of the human ribosome recycling factor ABCE1 showing the two 4Fe-4S clusters situated in solvent-exposed loops at the N-terminal domain (PDB 7A09). The clusters in ABCE1 are prone to degradation [102]. Each cluster in ABCE1 is coordinated by three nearby Cys residues and one distant Cys residue in the primary sequence, suggesting a structural role for these clusters [103]. C. Cryo-EM structure of human DNA primase subunits, PRIM1 (green) and PRIM2 (blue) (PDB 8QJ7). The 4Fe-4S cluster bound to PRIM2 locates at the junction of the N-terminal and C-terminal domains close to the protein surface. PRIM1 is proposed to serve as an adaptor for the recognition of PRIM2 by the CTC to facilitate its metallation. D. Cryo-EM structure of the yeast elongator complex proteins ELP3 (green) and ELP4 (yellow) (PDB 8ASV). The radical-SAM enzyme ELP3 has a [4Fe-4S](Cys)3 cluster, which is exposed to solvent via two channels [103]. ELP4 is proposed to serve as an adaptor for the recognition of ELP3 by the CTC to facilitate its metallation.
Figure 1.
Comparison of solvent-exposed and buried Fe-S clusters in proteins. Structures (left) and close-ups of the Fe-S binding domains (square, right). A. X-ray structure of human xanthine oxidase (XDH) showing the two 2Fe-2S clusters deeply buried within the protein structure (PDB 2CKJ). B. Cryo-EM structure of the human ribosome recycling factor ABCE1 showing the two 4Fe-4S clusters situated in solvent-exposed loops at the N-terminal domain (PDB 7A09). The clusters in ABCE1 are prone to degradation [102]. Each cluster in ABCE1 is coordinated by three nearby Cys residues and one distant Cys residue in the primary sequence, suggesting a structural role for these clusters [103]. C. Cryo-EM structure of human DNA primase subunits, PRIM1 (green) and PRIM2 (blue) (PDB 8QJ7). The 4Fe-4S cluster bound to PRIM2 locates at the junction of the N-terminal and C-terminal domains close to the protein surface. PRIM1 is proposed to serve as an adaptor for the recognition of PRIM2 by the CTC to facilitate its metallation. D. Cryo-EM structure of the yeast elongator complex proteins ELP3 (green) and ELP4 (yellow) (PDB 8ASV). The radical-SAM enzyme ELP3 has a [4Fe-4S](Cys)3 cluster, which is exposed to solvent via two channels [103]. ELP4 is proposed to serve as an adaptor for the recognition of ELP3 by the CTC to facilitate its metallation.
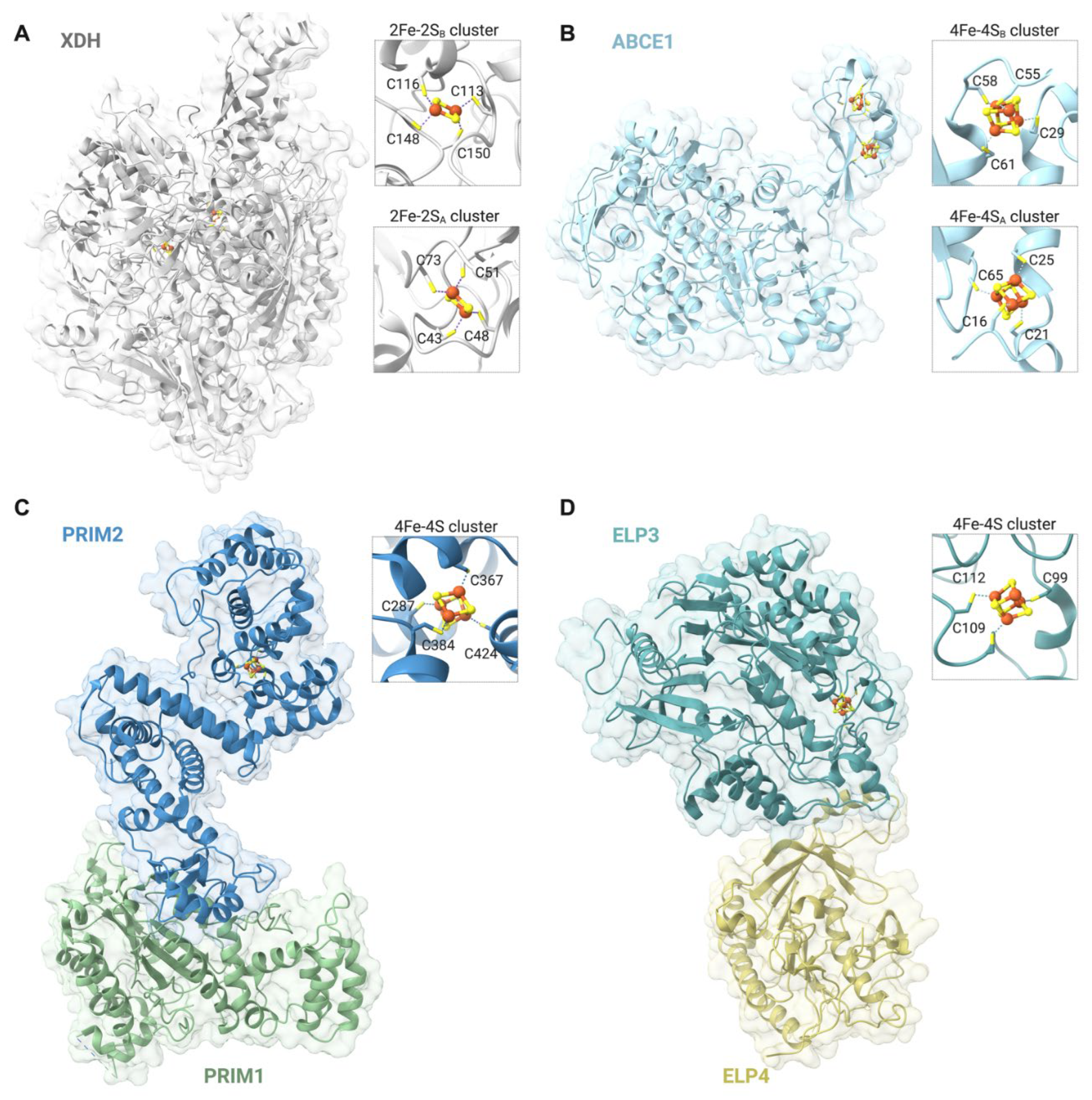
Figure 2.
Essential roles of nuclear Fe-S cluster-binding proteins in DNA replication, telomere maintenance, and mitosis. A. Representation of a eukaryotic DNA replication fork. The mini-chromosome maintenance (MCM2-7) helicase complex unwinds DNA at replication origins, separating the double helix to generate the leading and lagging strands. All replicative DNA polymerases, Pol α, δ, and ε, bind a 4Fe-4S cluster. DNA replication initiates with an RNA primer (orange) synthesized by DNA primase, with the regulatory subunit PRIM2 also binding a 4Fe-4S cluster. Pol ε further extends the primer on the leading strand, while Pol δ does so on the lagging strand. The proliferating cell nuclear antigen sliding clamp PCNA enhances the processivity of DNA polymerases and facilitates DNA repair. The 4Fe-4S cluster DNA helicases FANCJ and DDX11 unwind G4s to prevent stalling of the replisome. DNA lesions obstruct replication, triggering monoubiquitylation of PCNA, which recruits the DNA damage bypass DNA polymerase Pol ζ. The catalytic subunit of Pol ζ also binds a 4Fe-4S cluster. Finally, the 4Fe-4S cluster nuclease-helicase DNA2 cleaves exposed single stranded DNA ends from Okazaki fragments and stalled replication forks. B. Under Fe-sufficient conditions, the MCM helicase moves in opposite directions from the activated origin of replication. This results in the formation of two functional replication forks, enabling DNA synthesis. Under iron-depleted conditions, apo NCOA4 accumulates in the nucleus and binds to the helicase subunit MCM7 hindering MCM helicase activity, and inactivating replication origins. This ensures that DNA synthesis only occurs when there is a sufficient pool of metallated Fe-S enzymes, thereby maintaining genomic integrity and preventing replication under suboptimal conditions. C. The 4Fe-4S cluster helicase RTEL1 unwinds G4s and R-loops at the telomeric repeats, facilitating telomere replication by telomerase and preventing telomere shortening. D. During the late stages of mitosis (metaphase, anaphase, and telophase), the nuclear 4Fe-4S cluster mitotic factor KIF4A binds the arms of condensed chromosomes. As a kinesin, KIF4A moves along microtubules to mobilize cargoes. KIF4A localizes at the central spindle, accumulating at the spindle midzone and midbody. The close-up image on the bottom illustrates the co-localization of the CIA targeting factors MMS19 and CIAO2B with KIF4A during mitosis, suggesting an in situ metallation of KIF4A during function.
Figure 2.
Essential roles of nuclear Fe-S cluster-binding proteins in DNA replication, telomere maintenance, and mitosis. A. Representation of a eukaryotic DNA replication fork. The mini-chromosome maintenance (MCM2-7) helicase complex unwinds DNA at replication origins, separating the double helix to generate the leading and lagging strands. All replicative DNA polymerases, Pol α, δ, and ε, bind a 4Fe-4S cluster. DNA replication initiates with an RNA primer (orange) synthesized by DNA primase, with the regulatory subunit PRIM2 also binding a 4Fe-4S cluster. Pol ε further extends the primer on the leading strand, while Pol δ does so on the lagging strand. The proliferating cell nuclear antigen sliding clamp PCNA enhances the processivity of DNA polymerases and facilitates DNA repair. The 4Fe-4S cluster DNA helicases FANCJ and DDX11 unwind G4s to prevent stalling of the replisome. DNA lesions obstruct replication, triggering monoubiquitylation of PCNA, which recruits the DNA damage bypass DNA polymerase Pol ζ. The catalytic subunit of Pol ζ also binds a 4Fe-4S cluster. Finally, the 4Fe-4S cluster nuclease-helicase DNA2 cleaves exposed single stranded DNA ends from Okazaki fragments and stalled replication forks. B. Under Fe-sufficient conditions, the MCM helicase moves in opposite directions from the activated origin of replication. This results in the formation of two functional replication forks, enabling DNA synthesis. Under iron-depleted conditions, apo NCOA4 accumulates in the nucleus and binds to the helicase subunit MCM7 hindering MCM helicase activity, and inactivating replication origins. This ensures that DNA synthesis only occurs when there is a sufficient pool of metallated Fe-S enzymes, thereby maintaining genomic integrity and preventing replication under suboptimal conditions. C. The 4Fe-4S cluster helicase RTEL1 unwinds G4s and R-loops at the telomeric repeats, facilitating telomere replication by telomerase and preventing telomere shortening. D. During the late stages of mitosis (metaphase, anaphase, and telophase), the nuclear 4Fe-4S cluster mitotic factor KIF4A binds the arms of condensed chromosomes. As a kinesin, KIF4A moves along microtubules to mobilize cargoes. KIF4A localizes at the central spindle, accumulating at the spindle midzone and midbody. The close-up image on the bottom illustrates the co-localization of the CIA targeting factors MMS19 and CIAO2B with KIF4A during mitosis, suggesting an in situ metallation of KIF4A during function.
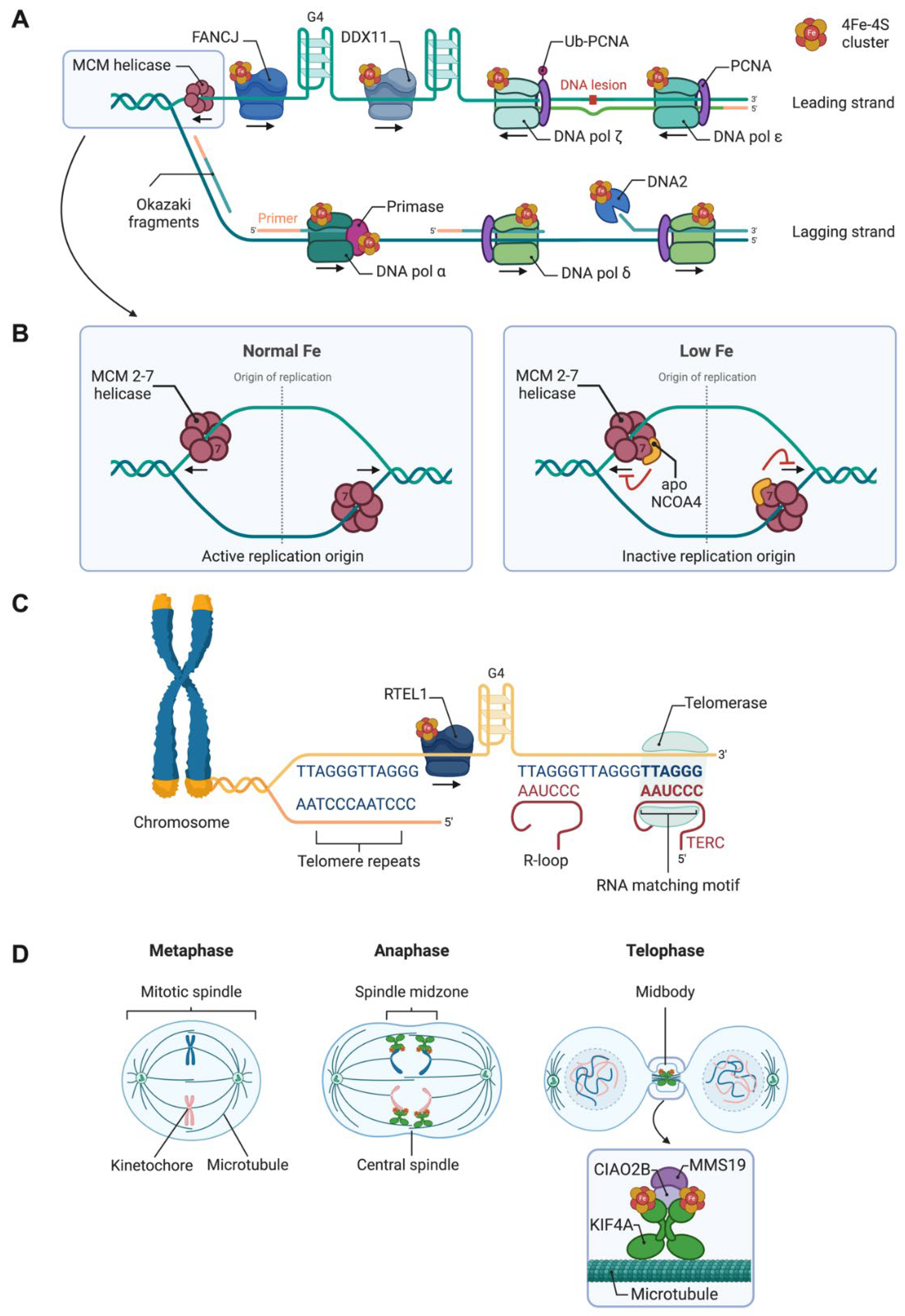
Figure 3.
Fe-S cluster proteins within the six major DNA repair pathways: base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR), non-homologous end joining (NHEJ), and translesion DNA synthesis (TLS). Schematic representation of DNA lesions and the corresponding repair pathways. Key proteins involved in each pathways are organized according Knijnenburg et al. [172]. DNA repair proteins that bind 4Fe-4S clusters are shown in a blue square at the bottom. * While DDX11 and EXO5 are not formally categorized under HR, their functions align with this repair mechanism.
Figure 3.
Fe-S cluster proteins within the six major DNA repair pathways: base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR), non-homologous end joining (NHEJ), and translesion DNA synthesis (TLS). Schematic representation of DNA lesions and the corresponding repair pathways. Key proteins involved in each pathways are organized according Knijnenburg et al. [172]. DNA repair proteins that bind 4Fe-4S clusters are shown in a blue square at the bottom. * While DDX11 and EXO5 are not formally categorized under HR, their functions align with this repair mechanism.
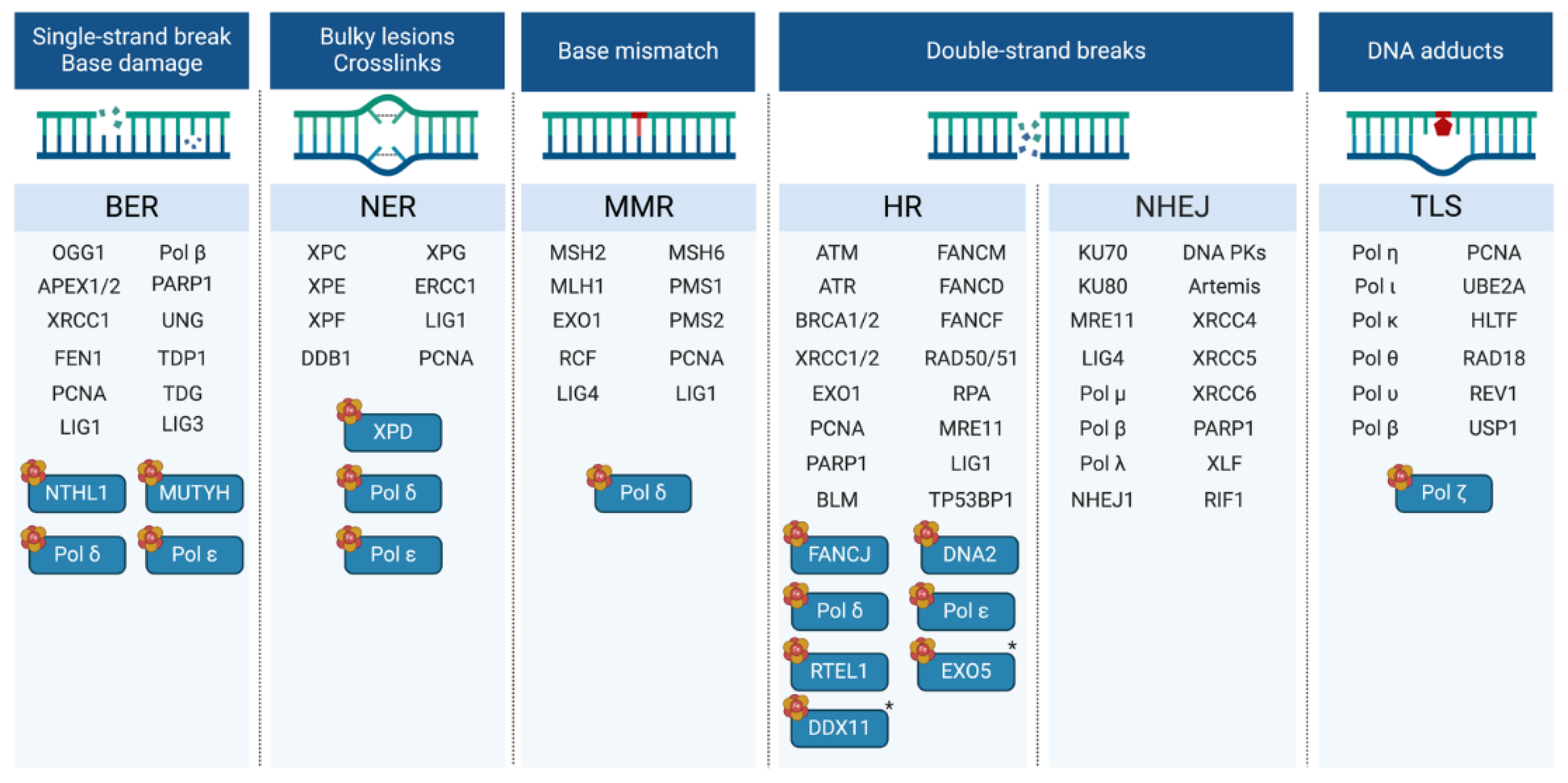

Table 1.
Inventory of Fe-S cluster binding proteins that localize to the nucleus of mammalian cells. The localization of the proteins reported here is based on consolidating information from references (when available) and from three different databases: The Human Protein Atlas [5] (https://www.proteinatlas.org), The UniProt Knowledgebase [6] (https://www.uniprot.org), and COMPARTMENTS [7] (https://compartments.jensenlab.org). An extended version of the table, including the residues that coordinate the clusters, is available as Supplementary Information. The asterisks (*) denote that Fe-S cluster ligands are shared between oligomeric partners from the CIA or ISC pathways. Question marks indicate unknown or debatable cluster binding residues, cluster types, or stoichiometries. SAM: S-adenosyl-L-methionine.
Table 1.
Inventory of Fe-S cluster binding proteins that localize to the nucleus of mammalian cells. The localization of the proteins reported here is based on consolidating information from references (when available) and from three different databases: The Human Protein Atlas [5] (https://www.proteinatlas.org), The UniProt Knowledgebase [6] (https://www.uniprot.org), and COMPARTMENTS [7] (https://compartments.jensenlab.org). An extended version of the table, including the residues that coordinate the clusters, is available as Supplementary Information. The asterisks (*) denote that Fe-S cluster ligands are shared between oligomeric partners from the CIA or ISC pathways. Question marks indicate unknown or debatable cluster binding residues, cluster types, or stoichiometries. SAM: S-adenosyl-L-methionine.
| Participates in | Name | Consolidated Localization | Fe-S cluster type |
|---|---|---|---|
| DNA replication DNA repair | PRIM2 | Nucleus | [4Fe-4S] (Cys)4 [8,9] |
| POLA1 | Nucleus, Cytosol [10] | [4Fe-4S] (Cys)4 [11] | |
| POLD1 | Nucleus, Cytosol [12] | [4Fe-4S] (Cys)4 [11] | |
| POLE | Nucleus | [4Fe-4S] (Cys)4 [11,13] | |
| REV3L | Nucleus, Cytosol [14] | [4Fe-4S] (Cys)4 [11] | |
| FANCJ | Nucleus, Cytosol [15,16] | [4Fe-4S] (Cys)4 [4,17] | |
| DDX11 | Nucleus [18] | [4Fe-4S] (Cys)4 [19] | |
| RTEL1 | Nucleus [16] | [4Fe-4S] (Cys)4 [20] | |
| XPD | Nucleus, Cytosol [16,21] | [4Fe-4S] (Cys)4 [4] | |
| DNA2 | Nucleus, Mitochondria [22] | [4Fe-4S] (Cys)4 [23,24] | |
| EXO5 | Nucleus, Cytosol [25] | [4Fe-4S] (Cys)4 [25] | |
| MUTYH | Nucleus, Mitochondria [26] | [4Fe-4S] (Cys)4 [27,28] | |
| NTHL1 | Nucleus, Mitochondria [29] | [4Fe-4S] (Cys)4 [30,31] | |
| RNA transactions | ELP3 | Nucleus, Cytosol [32] | [4Fe-4S] (Cys)3 SAM [33,34] |
| RPC6 | Nucleus [35] | [4Fe-4S] (Cys)4 [36,37] | |
| CPSF4 | Nucleus [38] | [2Fe-2S] (Cys)3 (His)? [39,40] | |
| TYW1 | Nucleus, Cytosol | [4Fe-4S] (Cys)3 SAM [34,41] | |
| TYW1B | Nucleus | [4Fe-4S] (Cys)3 SAM [34,41] | |
| CDK5RAP1 | Nucleus, Mitochondria, Cytosol [42] | [4Fe-4S] (Cys)4, [4Fe-4S] (Cys)3 SAM [34,43] | |
| Mitosis | KIF4A | Nucleus [44] | [4Fe-4S] (Cys)4? [45] |
| KIF4B | Nucleus [44] | [4Fe-4S] (Cys)4? [45] | |
| Iron metabolism | NCOA4 | Nucleus, Cytosol [46] | [3Fe-4S] (Cys)4? [47,48] |
| FBXL5 | Nucleus, Perinuclear region [49] | [2Fe-2S] (Cys)4 [50] | |
| Post-translational modifications | DPH1 | Nucleus, Cytosol [51] | [4Fe-4S] (Cys)3 SAM [52] |
| DPH2 | Nucleus, Cytosol | [4Fe-4S] (Cys)3 SAM [52] | |
| ATE1 | Nucleus, Cytosol [53] | [4Fe-4S] (Cys)4 [54] | |
| Respiration | SDHB | Mitochondria, Nucleus [55] | [2Fe-2S] (Cys)4, [3Fe-4S] (Cys)3, [4Fe-4S] (Cys)4 [55] |
| Unknown | RFESD | Nucleus | [2Fe-2S] (Cys)2 (His)2 |
| CIA | CIAPIN1 | Nucleus, Mitochondria, Cytosol [56] | [2Fe-2S] (Cys)4, [4Fe-4S] (Cys)4? [57,58] |
| BOLA2 | Nucleus, Cytosol [59] | [2Fe-2S]* Ligands shared with GLRX3 [60,61] | |
| GLRX3 | Nucleus, Cytosol [62] | [2Fe-2S]* Ligands shared with BOLA2 [60,61] | |
| NUBP2 | Nucleus, Cytosol [63] | [4Fe-4S]* Ligands shared with NUBP1 [64,65] | |
| ISC | NFU1 | Nucleus, Mitochondria, Cytosol [66] | [4Fe-4S]* Ligands shared between dimers [66,67] |
| NFS1 | Nucleus, Mitochondria, Cytosol [68] | [2Fe-2S]* Ligands shared with ISCU [69,70] | |
| GLRX2 | Nucleus, Mitochondria [71] | [2Fe-2S]* Ligands shared between dimers [72] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



