
Submitted:
30 October 2024
Posted:
31 October 2024
You are already at the latest version
Abstract
Acute pancreatitis (AP) is a potentially fatal organ disease that is widespread globally. Although significant progress has been made in the previous decade, the study of mechanisms and therapeutic strategies is still far from being completed. Xanthine oxidase (XO) is an enzyme that generating urate and also responsible to produce reactive oxygen species (ROS). Considerable clinical and mechanical research has been conducted over many decades to explore the function of XO in AP and its promising therapeutic applications. Unquestionably, XO is a key part of starting and progressing AP by ROS production. Not fully known, though, are XO's many other functions besides ROS resource in both local and global processes, such as its role in cellular signaling or metabolism regulation. This review seeks to summarize the function of XO in AP pathophysiology by analyzing the inherent biological and chemical properties of XO, and lastly provide fresh concepts for future research.
Keywords:
Xanthine oxidase
; acute pancreatitis
; drug target
; cellular mechanism
; multiple organ failure
1. The Major Challenge: Mechanism, Target, and Drug Discovery of Severe AP
Acute pancreatitis (AP) is a prevalent gastrointestinal disorder marked by intense pain1. The estimation of AP incidence ranges from 110 to 140 per 100,000 population, which has been increasing2. Gallstones, alcohol and triglyceridemia rank as the top etiologies3. The clinical guidance highlighted the need of supportive therapy including goal-directed fluid resuscitation, early oral feeding, enteral nutrition fluid control, and nutrition as aspects of the current therapeutic approach4. It is well acknowledged that 80% of AP patients are mild cases, which are mostly self-limited. But there still remains 20% AP patients developed severe AP (SAP), with local or systematic complications5. The high mortality of SAP, ranging from 20% to 40%, has been regarded as a critical problem5. Nevertheless, the most recent Cochrane review6 indicates that medication development is generally impeded resulting in restricted clinical intervention for AP, particularly in severe cases7. Therefore, a key component in achieving therapeutic success is to hinder the transition from mild to SAP.
By now, pathological research based on laboratory studies of AP has become the foundation of efforts. In summary, the mechanisms can be categorized into two primary processes: disturbance of acinar cell homeostasis and the amplification of the inflammatory cascade7. The major pathological events in acinar cells including calcium overload, trypsinogen activation, mitochondrial dysfunction and autophagy failure8. When the local inflammatory damage progresses to systemic alterations, it will lead to the activation of multiple organ failure, primarily respiratory and circulatory failure, which in turn contributes to the high fatality rate in SAP9. Therefore, identifying the pathogenic processes and associated targets that contribute to and worsen the development of AP is the crucial step in achieving a substantial decrease in clinical mortality.
2. What Makes XO a Drug Target
2.1. General Profile of XO and Target Characteristics
XO is a molybdoflavo, dimeric protein with a net molecular weight of approximately 290 kDa10. Each monomeric unit is composed of 3 major domains in (Figure 1), a small Fe/S center (amino acid: 3-165, molecular weight: 20 kDa), an intemediate Flavin Adenine Dinucleotide (FAD) domain (amino acid: 226-531, molecular weight: 40 kDa), and the Molybdopterin (Mo-Pt) center (amino acid: 590-1332, molecular weight: 85 kDa), which is the most prominent domain, the domain where hydro-oxidation takes place11.
XO is the oxidase form of xanthine oxidoreductase (XOR), and xanthine dehydrogenase (XDH) represents the dehydrogenase form of XOR. These two proteins are both encoded by the XDH gene and belong to the molybdenum hydroxylase flavoprotein family. Structurally, the main difference between XO and XDH is the FAD-binding domain. They can convert to each other during different conditions. Generally, XDH may be converted to XO via sulfhydryl side group oxidation reversibly, and via proteolysis by serine proteases irreversibly. The former situation, usually induced by hypoxia conditions, and the other transformation path is usually induced by prolonged ischemic conditions. XO transfers electrons to oxygen molecules and produces hydrogen peroxide (H2O2) and a superoxide anion, meanwhile, XDH transfers 2 electrons generated during the oxidation of the substrate in each reaction step to one molecule of NAD+(Figure 2). In this case, XO uses molecular oxygen as electron acceptor and consequently generates the superoxide anion, a molecule that participates in the generation of other ROS including hydrogen peroxide, hydroxyl radical and peroxynitrite12. Under physiologic conditions, the enzyme exists mainly as XDH, which utilizes NAD+ as an electron acceptor and primarily functions as a dehydrogenase. Conversely, in pathological circumstances, cellular processes such as alterations in intracellular pH or calcium entry lead to the conversion of XOR to its oxidase form, XO, due to restricted proteolytic cleavage.
As a superoxide-producing enzyme, XO was found in serum and various organs. XOR is present in all cell types, acting mainly as dehydrogenase, and in most cases, with a low level of activity. The gene expression of XOR was found highest in the intestine and liver, with the highest enzyme activities correspondingly13 in other tissues including the brain, heart, lung, kidney, skeletal muscle14, and pancreas as well15. Additionally, it has been reported that plasma XOR activity is much lower in humans than in animals. XOR enzyme catalyzes the final 2 steps of purine degradation, namely the oxidation of hypoxanthine and xanthine to uric acid. Circulative XOR activity is associated with obesity, smoking, liver dysfunction, hyperuricemia, dyslipidemia, insulin resistance, and adipokines16. Two of the most significant contributions of XO to pathophysiology are the production of ROS and the buildup of uric acid. A recent prospective cohort study involving 124,316 people found a linear correlation between uric acid levels and the risk of AP17, suggesting an essential role of XO in AP progression.
2.2. Inhibitors of XO
Due to the crucial role of XO in the development of acute pancreatitis, the focus has been on identifying potential therapeutic agents that can inhibit this enzyme. A multitude of different compounds have been discovered that can inhibit XO with varying degrees of selectivity and potency. These compounds can be classified into purine analogs, which closely resemble the structure of xanthine, and nonpurine analogs, which differ structurally18. Moreover, several synthetic and natural compounds with high selectivity are summarized and are presented (Table 1).
- (1)
- Purine analogs
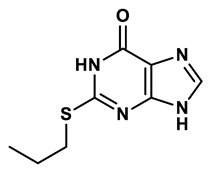
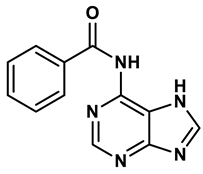
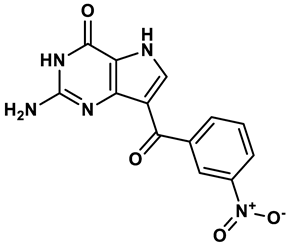
Purine analogs, such as allopurinol (1), have been one focus of development for XO inhibitors. Allopurinol was the first US’ Food and Drug Administration (FDA) approved purine based XO inhibitor. At low concentrations, allopurinol competes with the enzyme as a substrate and inhibitor, while at higher concentrations, it acts as a noncompetitive inhibitor, with an IC50 ranging from 0.2-50 μmol·L19. Its active metabolite, oxypurinol, binds to XOR and leads to enzyme inhibition. However, the use of allopurinol has been associated with Drug Reactions with Eosinophilia and Systemic Symptoms (DRESS syndrome) and inhibits other enzymes such as purine nucleoside phosphorylase (PNP) and orotidine-5′-monophosphate decarboxylase (OMPDC)20. Other compounds with purine structures, such as, monosubstituted 2-(thioalkyl)purines21 (2), 6-(N-benzoylamino)purine22 (3), 9-Benzoyl 9-deazaguanines23 (4) also been reported; However, these compounds have also been associated with severe side effects, including hypersensitivity with fever, skin rash and eosinophilia, renal toxicity, and gastrointestinal distress24. Therefore, the broad therapeutic use of these compounds has been limited, highlighting the need for novel, potent non-purine XO inhibitors with fewer adverse effects.
- (2)
- Nonpurine analogs
Synthetic Nonpurine Analogs
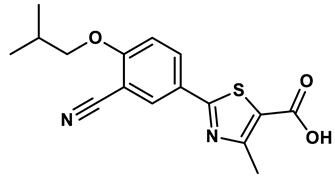
Nonpurine analogs, such as febuxostat (5) and topiroxostat (6), have been widely studied as XO inhibitors25,26. Febuxostat, a thiazole derivative, was one of the first non-purine XO inhibitors approved by the FDA. It acts as a highly potent non-competitive inhibitor of human XO25. However, febuxostat has been found to have adverse side effects, such as acute liver dysfunction and an increased risk of heart-related deaths27. Investigation into the inhibition mechanism of febuxostat founded that it exhibits a mixed-type inhibition, probably because of its tight binding to the molibdopterin cofactor of XO, which impedes the entry of the substrate into the active site of the enzyme24. Likewise, topiroxostat also demonstrated a hybrid-type inhibition mechanism by forming a covalent linkage with the active site of the enzyme28.
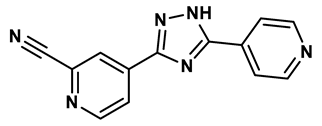
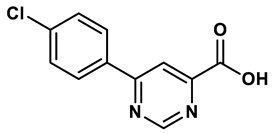
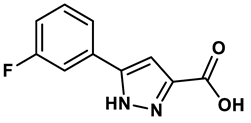
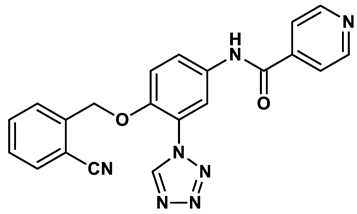
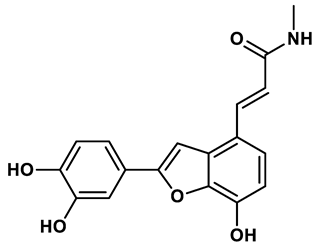
In response to the limitations of existing nonpurine XO inhibitors, researchers have explored the development of new molecules that are effective and specific in inhibiting XO. During a virtual screening study aimed at the discovery of novel inhibitors targeting another oxidoreductase, superoxide dismutase, four compounds with non-purine-like structures (compound 7, compound 8, compound 9, compound 10), were identified being capable of causing direct inhibition of XO28. Following investigation of the structure–activity relationship around the thiazole core, compound 11, which was structurally related to febuxostat, was identified as a potent XO inhibitor with a novel 2-phenylthiazole-4-carboxylic acid scaffold, with an IC50 value of 0.0486 μM29. 1,2,3-triazoles derivatives based on the structural features of topiroxostat are found effective inhibitors of XO resulted in reduced production of UA in the body. Zhang et. al. also investigated a series of 4-(phenoxymethyl)-1H-1,2,3-triazole derivatives, with compound 12 emerging as the most effective XO inhibitor with an IC50 value of 0.70 μM, approximately 14 times more potent than allopurinol. In vitro results, compound 12 showed potent hypouricemic effects at an oral dose of 20 mg/kg in a rat hyperuricemia model induced by potassium oxonate30. They also conducted a study focusing on the potential of N-(1-alkyl-3-cyano-1H-indol-5-yl) aromatic heterocyclic carboxamide derivatives as XO inhibitors. They identified an optimized compound 13 with an IC50 value of 0.018 μM, demonstrating comparable potency to the known XO inhibitor topiroxostat31. Building on this work, they proceeded to design and synthesize a series of N-(4-alkoxy-3-(1H-tetrazol-1-yl) phenyl) heterocyclic aromatic amide derivatives based on the framework of a previously reported compound, identifying compound 14 with an IC50 value of 0.022 μM as the most promising inhibitor. This compound possesses a unique 1H-imidazole-5-carboxamide scaffold and comparable potency to topiroxostat32. Moreover, recent studies have reported the development of XO inhibitors based on benzo[b]furan scaffolds (15), 3-Phenylcoumarins (16), pyrimidine (17) and coumarin (18), further expanding the potential pool of efficacious XO inhibitors18,33–35.
Natural Nonpurine Analogs
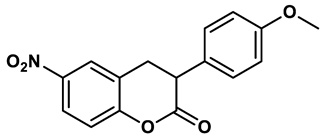
Natural nonpurine analogs such as flavonoids, chalcones, terpenoids, alkaloids, coumarins, phenolic acids, and glycosides have been found to effectively inhibit XO. Flavonoids, a type of natural phenolic compound, have received the most attention, have been found to strongly inhibit XO in several studies36. An integrated method combining enzyme kinetics, multi-spectroscopic spectra, and molecular simulations was used to investigate the structure-activity relationship and interaction mechanism between different flavonoids and XO. The findings suggest that active flavonoids can bind to XO's active center, block the entrance of substrates, and induce significant structural changes, leading to inhibition37. Among them, flavanols including luteolin, quercetin38, myricetin39 and galangin40 exhibited the remarkable inhibitory effect due to the hydroxylation pattern (hydroxylated number and position) on the core scaffold of flavonoids played a key role in their biological activity and affinity on XO, while the hydrogenation of the C2═C3 double bond were unfavorable for its inhibition potency. An ethanolic extract obtained from A. microcarpus flowers containing luteolin 7-O-glucoside (19) displayed a higher XO inhibitory activity compared to the standard inhibitor allopurinol, with an IC50 value of 4.8 μg/mL as opposed to 11.5 μg/mL, respectively in vitro studies41. A computational approach using molecular docking and free energy calculations led to the discovery of several natural compounds with XO inhibitory properties, including previously known inhibitors as well as newly identified substances like flavonol (20, IC50 = 8.45 ± 0.68 μM) and 5,7-dihydroxycoumarin (21, IC50 = 10.91 ± 0.71 μM) displayed the great potency as mixed-type inhibitors. Further analysis through docking studies and molecular dynamics simulations revealed that these hits interacted with key active site residues of XO, including ARG880, MOS1328, and ASN76842. Notably, some novel natural products with different chemotypes have been observed with XO inhibition, which provide novel scaffolds for developing new XO inhibitors. An extract from the endophytic fungus Alternaria alternata GDZZ-J6 showed the most potent inhibition of XO, with one of its secondary metabolites, alternariol (AOH, 22), a dibenzo-α-pyrone derivative, exhibiting strong inhibitory activity on XO. The potency of XO inhibition by AOH was more than 12 times higher compared to the standard inhibitor allopurinol43. An integrated analytical model was developed for screening XO inhibitors, leading to the identification of polydatin, a stilbene compound, as a potential active ingredient for XO44. These findings present promising potential for the development of natural XO inhibitors with improved efficacy.
3. Role of XO in AP Onset and Deterioration
3.1. Changes of XO Specifically Occur in Necrotic SAP Model Rather Than Edematous Model
To investigate the alterations in XO activity in AP, Devenyi45 initially quantified the XO activity of dehydrogenase and oxidase in pancreatic homogenates of mice. The present study documented that following 3 injections of cerulein at a dosage of 50 μg/kg, the activity of pancreatic XO remained in its dehydrogenase form rather than its oxidative form. The study proposed that the oxidative form of XO could be transformed by intrapancreatic protease and concluded that, for the first time, this conversion may take place in other models of AP rather than cerulein-induced AP mice. It is reasonable to conclude that XO does not contribute to the development of mild edematous AP model46.
The subsequent investigations demonstrated the obvious enhancement of XO in SAP models. The study conducted by D.Closa47 revealed a notable increase in XO activity in rats with sodium taurocholate (NaT)-induced SAP in contrast to rats with cerulein-induced AP. Moreover, in NaT-induced cell death, the XDH was fully transformed into XO within the initial 5 minutes, thereby generating oxygen free radicals (OFRs). This observation implies that XO is the primary origin of OFRs that contribute to further damage of the pancreas48. Other studied also reported that in NaT-SAP models, XO appears to undergo substantial alterations and is favorably correlated with pancreatic MPO levels49. These dysregulations can directly lead to organ failure associated with AP50. Therefore, it looks like XO has a distinct correlation with SAP pathogenesis. Moreover, it has been determined that XO-generated toxicity plays a significant role in AP models that are clinically relevant, such as fatty acid infusion (which corresponds to the cause of hyperlipemia in clinical settings), ischemia (shock), pancreatic duct blockage (gallstone), and acetaldehyde after ischemia (alcohol), rather than the cerulein AP model, which lacks a clear clinical equivalent51. However, the role of XO in ethanol-induced AP is still a subject of debate. Studies have shown that while ethanol played a significant role in the production of OFRs, the activity of XO in ethanol-induced AP was moderate52.
Our latest work53 revealed that in a model of cerulein-induced edematous AP, the expression of the pancreas Xdh gene showed a substantial rise 8 hours after the initial injection. However, there was no significant increase observed in the XO protein level. The pancreatic mRNA level and serum enzyme activity of XO increased dramatically in an L-Arginine-induced severe necrotic AP model within 8 hours of modeling, while XO protein levels significantly increased at 72 hours. Furthermore, the severe AP model caused by NAT exhibited a notable rise in serum XO activity within 3 hours after modeling. Taken together, these data unequivocally indicated that XO was particularly upregulated in the SAP model rather than in mild examples.
3.2. Targeting XO in Experimental AP
The classic XO inhibitors, allopurinol and its metabolite oxypurine, have been widely used in treatment and prevention of AP (Table 2). According to the current literatures, the effect of XO inhibitors on AP models seems controversial. However, this result is due to following reasons including the time and dose of drug administration, and most importantly, type of AP model.
In severe necrotic AP models, both therapeutic and prophylactic administration of XO inhibition showed obvious beneficial effect on pancreatic injury and systematic inflammation. In cases of therapeutical administration, 10 mg/kg of allopurinol intravenous infusion successfully inhibited pancreatic SOD increasing in 15% ethyl alcohol-induced dog AP model54. Similarly, in L-Arginine-induced SAP mice model, 200 mg/kg of allopurinol treatment could significantly ameliorated decease severity55,56 and cytokines levels including TNF-α and IL-657. The protective effect of allopurinol has been also confirmed in pancreatic duct obligation and ischemia with 20 mg/kg administration58, and in choline-deficient ethionine-supplemented diet-induce-induced AP with 24 mg/kg59, and in FFA, POSS and ischemia60 and also ischemia with acetaldehyde61-induced AP with 100 mg administration. Meanwhile, prophylactic administration also showed beneficial effects on SAP models. Pretreatment with allopurinol could prevent pancreatic edema and the serum amylase in oleic acid induced-AP mice62, and could decrease histologic changes of the pancreatic parenchyma, serum amylase and lipase of pancreatography-induced pancreatitis in dogs63. Inhibiting XO could significantly decrease 3.5% sodium taurocholate-induced pancreatic and lung injury64, and systematic oxidative stress in AP rats50,65–67. In addition, Lankisch68 investigated the effect of allopurinol on 5% sodium taurocholate-induced AP rats, and choline-deficient ethionine-supplemented diet-induced AP mice, with administration of allopurinol at 100 mg/kg bodyweight 1h before, and 12, 24, 36 h after surgery for rat, and 50 mg/kg bodyweight 1h before starting diet and every 24 h during diet up to 5 days for mice. This study did not achieve positive results, which may be attributed by the time and dose of drug administration, lower dose of therapeutic usage of inhibitors may be effective according to the previous study59.
However, the effect of XO inhibitors in mild AP models did not achieve consistence. Pre-treatment of 5 mg/kg oxypurinol significantly decreased ERCP-induced pancreatitis in canine model63. In cerulein-induced edematous AP, a continuous intravenous infusion of allopurinol (20 mg/kg/h) for 6 hours significantly improved cerulein-induced AP rats model69. And in vitro cerulein model, pre-treatment of allopurinol (1μM) also showed beneficial effect70. On the contrary, Devenyi45 reported that 0.7-7mg/kg allopurinol did not improve pancreas edema. Similarly, Takafumi71 reported that 50 mg/kg allopurinol did not improve serum amylase, lipase, trypsin or pancreatic histological damage.
Collectively, in SAP models including L-Arginine and NaT models, prophylactic or therapeutic usage of XO inhibitors with a dose range from 20 to 200 mg/kg may largely be effective. For choline-deficient ethionine-supplemented diet-induced necrotic AP, therapeutic administration of lower dose around 24 mg/kg is recommended. This result is closely related to the XO increase during different models. Our study53 revealed that XO significantly increased in L-Arginine-induced SAP as early as 8 h after modeling, and also increased in NaT-induced SAP at 3 h after modeling, with no significant changes was observed in cerulein-induced edematous AP model.
3.3. Role of XO in Pancreatic Injury During AP
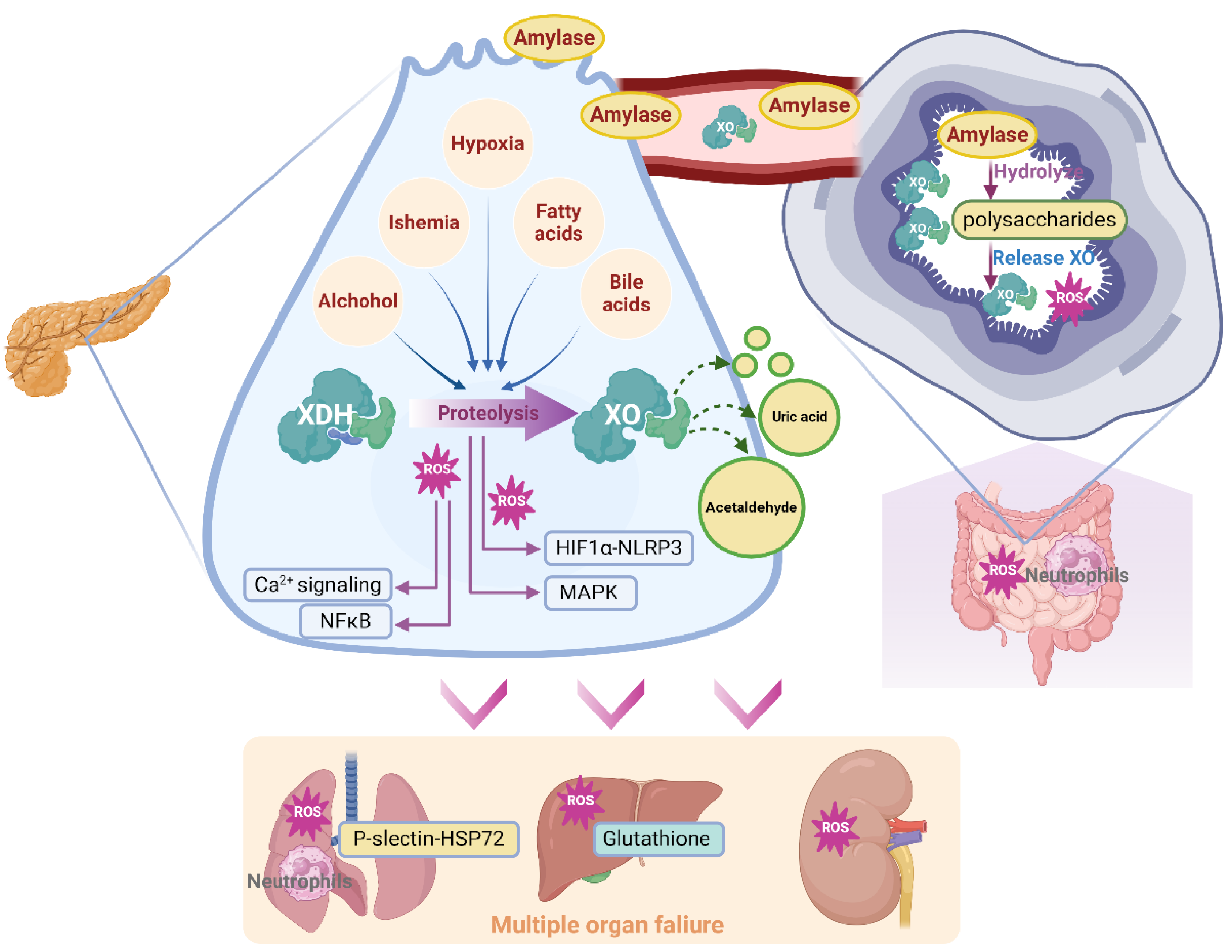
As a longstanding target in AP, XO was recognized as a ROS resource in pancreatic acinar cells that activating inflammatory signals and oxidative stress-related cell death, and effectively increasing pancreatic enzymes72. However, the advance evidence revealed that, beside ROS, the process during oxygen consumption, like hypoxia conditions for pancreatic acinar cells, also triggers multiple signaling such as NLRP3 and composes the cellular mechanism of XO activation53.
It has been well established that the XO molecule, functioning as a crucial redox response component, has the potential to cause harm to tissues and cells through oxidative stress. ROS are byproducts of aerobic metabolism. Under normal conditions, ROS are safely neutralized by the antioxidant defense system. However, when ROS production exceeds the capacity of the antioxidant defense system, the uncontained ROS cause cellular injury and dysfunction by attacking biomolecules, modulating redox-sensitive signal transduction pathways and transcription factors73,74. The generation of oxygen radicals may occur through the action of XO that exists in the epithelial cells of pancreatic tissues. Claus Niederau75 observed that an in vitro model exposed to exogenous XO (20 mU/mL) induces iron-catalyzed (50 μM FeCl3) reactions on hypoxanthine (500 μM), leading to time-dependent damage in freshly isolated rat pancreatic acinar cells. The main cytotoxic oxidant species produced in this process is H2O2. Hence, the process by which XDH is converted to XO produces massive OFRs, which serve as a significant damaging resource. Neutralization of OFRs significantly ameliorated pancreatic injury caused by free fatty acid, pancreatic duct obligation, and ischemia76. During AP, multiple pathologies could trigger XDH convert to XO like ischemia51. It is worth to note that, OFRs produced by exogenous hypoxanthine/SOD alone does not induce the typical enzymatic and morphologic changes of AP, this is more like an aggregating factor rather than initial pathology77. Oxygen radicals could induce tissue damage via lipid peroxidation during AP78. Mechanisms of oxidative stress and redox status participating systematically in AP including glutathione depletion, xanthine oxidase activation, and thiol oxidation in proteins79. Additionally, these alterations were partially influenced by the calcium excess established by oxygen radicals80.
SAP patients generally suffered from deterioration of oxygen supply and consumption81. Hyperbaric oxygen could improve pancreatic micro circulation82 and inflammation83, and could ameliorate pancreatic duct ligation-induced AP rats by inducing caspase-dependent apoptosis84. In a dose-dependent way, the hypoxanthine-related oxidative system has been shown to activate trypsin in pancreatic acinar cells of rats85. During oxidative stress, the excessive ROS lead to hypoxia condition, which directly up-regulates HIF-1α, and activated HIF-1α subsequently induces multiple inflammatory signaling including NLRP3 inflammasome86, and triggers glycolytic reprogramming by increasing LDHA53. Thus, XDH activation-induced hypoxia and newly generated ROS could accelerate pancreatic injury by these inflammatory response and metabolic disorder during AP.
Apart from the oxidative stress or hypoxia that mediated by the conversion of XO, the metabolism disorder that triggered via XO activation also participates in AP. Another important role of XO in pancreatic function responding to purine metabolism. XOR is known as the rate-limiting enzyme in purine catabolism. It can also metabolize xenobiotics, including some anticancer compounds, which make XOR an important enzyme in inflammatory diseases. Nordback51,61 reported that, XO may participate in acute alcoholic pancreatitis by disturbing ethanol metabolism that triggers oxidizing acetaldehyde, the first product of and a substrate of XO.
Figure 3.
Schematic diagram of XO-mediated cellular mechanism during AP.
3.4. Effect of XO in AP-Associated Multiple Organ Failure
XO could be generated extracellularly as emission of damaged cells and into circulation87. Thus, XO has been reported to participate in systemic pathology by ROS generating during AP progression79,88. Due to the significant higher levels of antioxidants in mild AP patients than SAP patients, oxidative stress plays an important role in SAP rather than mild AP89. Notably, since XO is the major source of circulating ROS, it may leave significant effect in the systematic influences of AP90. ROS is an crucial pathology related to the onset of multiple organ failure in SAP91. ROS play an important role in ischemia-reperfusion injury, sepsis, acute respiratory distress, and multiple organ dysfunction syndrom92, by multiple mechanisms including affecting the rate of apoptosis in tissue and endothelial cells of varies organs93. Additionally, oxidative stress immediately triggers ferroptosis via multiple mechanism like deubiquitinating and stabilizing Sirt394. And it has been widely accepted that ferroptosis participates in AP95 through exacerbating lipid peroxidation96.
The XO-mediated oxidative stress contributes to the multiple organ failure associated with SAP, and lung injury is the most common organ that be attacked during AP progression. A system conducted by XO infusion with a dosage of 4 U/h and rate of 0.5 mL/h/100g body weight over a catheter inserted in the abdominal aorta, along with 10 mM hypoxanthine intravenous infusion, was previously reported to significantly recruit polymorphonuclear leukocytes in pancreas after 3 h, which may be attributed to the effect of oxygen radicals in releasing platelet activating factor and leukocyte B497. Correspondingly, inhibition of XO with 10 mM oxypurinol infusion significantly decreased SAP associated lung injury and P-selectin expression of lung, suggested that free radicals released by XO actions as mediators inducing lung P-selectin, which mediate neutrophil infiltration and organ inflammatory injury98. Further evidence suggested that circulating XO might induce, in part, the synthesis of platelet activating factor, and subsequently mediate lung neutrophil infiltration by combination with P-selectin, subsequently triggered inflammatory injury and resulted in AP associated lung injury99. Among polymorphonuclear leukocytes, neutrophil infiltration is a main group mediating lung injury during AP. XO-meditated free oxygen radicals participated in neutrophil recruitment in AP98. Inhibition of XO by 10 mM oxypurinol decreased NaT-induced pancreatitis in SAP rats, as well as lung HSP72 protein expression, which was mediated by neutrophil infiltration in early stage of SAP100. During physiological conditions, XDH/XO is bound to the polysaccharide chains of heparin-like proteoglycans, which are located on the cell membrane of endothelial cells of gastrointestinal system. Heparin could aggravate AP-induced lung injury by inhibiting the binding between XO and endothelia cells, which produces a free radical-generating system that induces an inflammatory response in the lungs49. Susana Granell90 reported that, during AP, massive amylase has been released in to circulative system, due to its ability to hydrolyze the internal α-1,4 linkages of polysaccharides, which may disrupt the binding-site of XDH/XOD and release XDH/XOD from endothelia cell surface in gastrointestinal system, amylase absorbed from ascitic fluid was involved in XO mobilization during AP. On the other hand, AP associated lung inflammation occurred due to the activation of proteolytic enzymes, which mainly contributed to the XDH-XO conversion and following free radical-dependent oxidative damage. During the early stage of AP, pulmonary dysfunction had generally occurred during to the high sensitivity of lung endothelium, and this process was at least partly mediated by XO-dependent free radicals. Notably, the oxygen in alveolar space is involved in XO-mediated lung inflammation, which use XO as substrate101.
Other organs including liver and kidney have also been associated with XO. Recent findings indicate that in L-Arginine-induced SAP model, the MDA activity significantly increased in pancreas and kidney. This improvement was notably achieved by administering 200 mg/kg of allopurinol. Furthermore, the oxidative stress in the kidney showed nearly the same level of evidence as in the pancreas, without any pathological changes. The pancreas and kidney exhibited a clear reduction in endogenous scavengers, while the liver showed a marginal decrease55. Under the NaT-induced SAP paradigm, pancreatitis occurs simultaneously with the rise in XO activity in plasma and the reduction in glutathione levels in both plasma and liver50. Collectively, the lung is the most likely affected organ during AP by XO-mediated injury, mechanisms involving ROS and circulative immune response. In case of liver and kidney, XO seems to injury these organs by accelerating local and systematic ROS. The effect of XO on other organs during SAP remains further research.
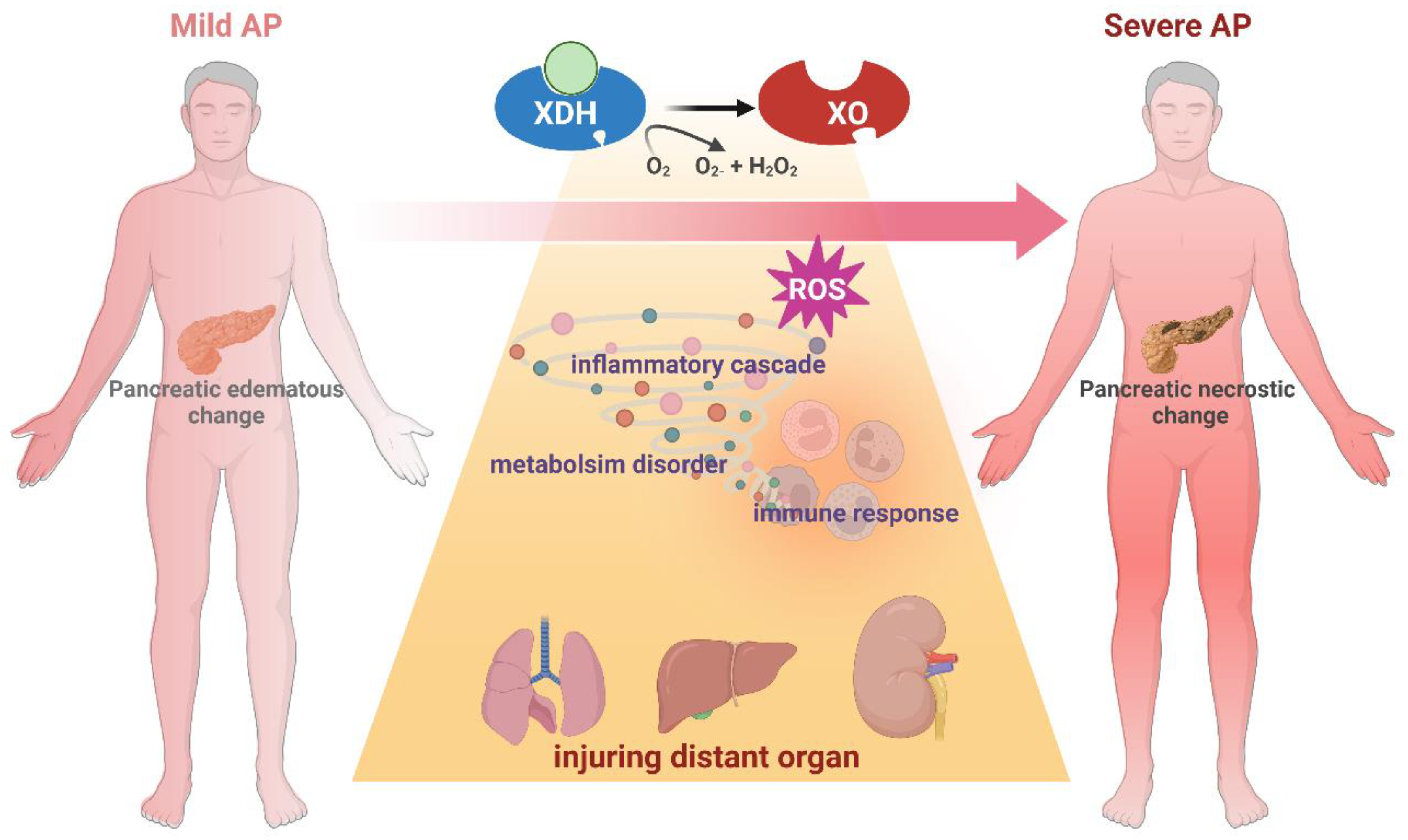
Figure 4.
Schematic diagram of XO-mediated acceleration from AP to SAP.
4. Clinical Trials of XO Inhibitors in AP Patients
The clinical administration of XO inhibitors focused on ERCP-induced AP (Table 3). According to meta-analysis, prophylactic use of allopurinol failed to prevent ERCP-induced acute pancreatitis102,103. Oral administration of 40, 200, or 300 mg of allopurinol 1-15 h prior to ERCP exhibited non beneficial effect on the incidence or severity of post-ERCP pancreatitis104–108. On the contrary, high-dose of 600 mg orally administration of allopurinol could protect ERCP-induced clinical AP, including promoting pancreatic injury and inhibiting incidences of complications109. And another RCT using 300 mg showed protective effect on the incidences of hyperamylasemia and pancreatitis in patients submitted to high-risk procedures110. Thus, the diverse severity and etiology of AP, the dose and time window of drug administration, all contribute to the clinical outcomes of RCT. It cannot be concluded that if XO inhibition emerges beneficial effects on ERCP or other etiology-induced AP patients, more RCTs are needed in this field.
5. Limitations and Perspectives
Elevated evidence has revealed that XO functioned as an enhancer during the progression of SAP. Multiple risk factors during AP can activate XO including fatty acids, ischemia, hypoxia, alcohol and bile acids. These factors trigger XO conversion to the oxidative state, companied by massive ROS generation, which accelerate local injury. It has been concluded that, therapeutically administration of relative lower dose of XO inhibitors like allopurinol shows beneficial effect on severe AP models instead of mild cases. However, the regulatory variables in the diverse etiology and time course during AP progression, and the upstream of XO remain still unclear in terms of processes. Due to the lack of genetic tools, parallel comparison between diverse models regarding role of XO in pancreatic acinar cells and systematic circulation, the application of natural products that inhibiting XO, as well as the inconsistence in drug administration methods, the current research remains to resolve the question that how XO functions as in inflammatory response, immune activation, metabolism disturbance or other mechanisms. In this case, the future experimental should explore the cellular mechanisms of XO and the therapeutic effects of inhibitors with suitable dose using severe AP models. Regarding to clinical research, the current studies merely focused on the ERCP-induced AP, which probably failed to integrate the significant enhancement of XO in severe cases. Future studies with various etiology and large sample size are needed to testify the effectiveness of XO as a promising target in AP treatment.
Author Contributions
Conceptualization, D.D. and Q.X..; methodology, J.R.; writing—original draft preparation, C.H. and Y.W.; writing—review and editing, D.D.; funding acquisition, D.D. and C.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of Sichuan Province (No.2023NSFSC1755 to D.D.) and Key Research and Development Project of Sichuan Province (No. 2024YFFK0153 to C.H.).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S.; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012, Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Garg SK, Sarvepalli S, Campbell JP, Obaitan I, Singh D, Bazerbachi F, et al. Incidence, Admission Rates, and Predictors, and Economic Burden of Adult Emergency Visits for Acute Pancreatitis: Data From the National Emergency Department Sample, 2006 to 2012. J Clin Gastroenterol 2019, 53, 220–5.
- Mederos MA, Reber HA, Girgis MD. Acute Pancreatitis: A Review. JAMA 2021, 325, 382–90. [Google Scholar] [CrossRef] [PubMed]
- Crockett, S.D.; Wani, S.; Gardner, T.B.; Falck-Ytter, Y.; Barkun, A.N.; Feuerstein, J.; Flamm, S.; Gellad, Z.; Gerson, L.; Gupta, S.; et al. American Gastroenterological Association Institute Clinical Guidelines Committee, American Gastroenterological Association Institute Guideline on Initial Management of Acute Pancreatitis. Gastroenterology 2018, 154, 1096–1101. [Google Scholar] [CrossRef]
- Boxhoorn L, Voermans RP, Bouwense SA, Bruno MJ, Verdonk RC, Boermeester MA, et al. Acute pancreatitis. Lancet 2020, 396, 726–34. [Google Scholar] [CrossRef]
- Moggia, E.; Koti, R.; Belgaumkar, A.P.; Fazio, F.; Pereira, S.P.; Davidson, B.R.; Gurusamy, K.S. Pharmacological interventions for acute pancreatitis. Cochrane Database Syst. Rev. 2017, 4, CD011384. [Google Scholar] [CrossRef]
- Barreto, S.G.; Habtezion, A.; Gukovskaya, A.; Lugea, A.; Jeon, C.; Yadav, D.; Hegyi, P.; Venglovecz, V.; Sutton, R.; Pandol, S.J. Critical thresholds: key to unlocking the door to the prevention and specific treatments for acute pancreatitis. Gut 2020, 70, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Lee PJ, Papachristou GI. New insights into acute pancreatitis. Nat Rev Gastroenterol Hepatol 2019, 16, 479–96. [Google Scholar] [CrossRef]
- Shi, N.; Liu, T.; de la Iglesia-Garcia, D.; Deng, L.; Jin, T.; Lan, L.; Zhu, P.; Hu, W.; Zhou, Z.; Singh, V.; et al. Duration of organ failure impacts mortality in acute pancreatitis. Gut 2019, 69, 604–605. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Matsumoto, K.; Hille, R.; Eger, B.T.; Pai, E.F.; Nishino, T. The crystal structure of xanthine oxidoreductase during catalysis: Implications for reaction mechanism and enzyme inhibition. Proc. Natl. Acad. Sci. USA 2004, 101, 7931–7936. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.V.; Bedi, P.M.S.; Singh, H.; Sharma, S. Xanthine oxidase inhibitors: patent landscape and clinical development (2015–2020). Expert Opin. Ther. Patents 2020, 30, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Parks DA, Granger DN. Xanthine oxidase: biochemistry, distribution and physiology. Acta Physiol Scand Suppl 1986, 548, 87–99. [Google Scholar]
- Saksela, M.; Lapatto, R.; Raivio, K.O. Xanthine Oxidoreductase Gene Expression and Enzyme Activity in Developing Human Tissues. Neonatology 1998, 74, 274–280. [Google Scholar] [CrossRef] [PubMed]
- A Pritsos, C. Cellular distribution, metabolism and regulation of the xanthine oxidoreductase enzyme system. Chem. Interactions 2000, 129, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.M.; Vaitaitis, G.M.; Wilson, C.M.; Repine, T.B.; Terada, L.S.; E Repine, J. cDNA cloning, characterization, and tissue-specific expression of human xanthine dehydrogenase/xanthine oxidase. Proc. Natl. Acad. Sci. USA 1993, 90, 10690–10694. [Google Scholar] [CrossRef]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: role of xanthine oxidoreductase activity. Am. J. Physiol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef]
- Su A, Yuan X, Zhu G, Jiang X, Shu R, Yang W, et al. Association Between Baseline Uric Acid and the Risk of Acute Pancreatitis: A Prospective Cohort Study. Pancreas 2022, 51, 966–71. [Google Scholar] [CrossRef]
- Era, B.; Delogu, G.L.; Pintus, F.; Fais, A.; Gatto, G.; Uriarte, E.; Borges, F.; Kumar, A.; Matos, M.J. Looking for new xanthine oxidase inhibitors: 3-Phenylcoumarins versus 2-phenylbenzofurans. Int. J. Biol. Macromol. 2020, 162, 774–780. [Google Scholar] [CrossRef]
- Pacher, P.; Nivorozhkin, A.; Szabó, C. Therapeutic Effects of Xanthine Oxidase Inhibitors: Renaissance Half a Century after the Discovery of Allopurinol. Pharmacol. Rev. 2006, 58, 87–114. [Google Scholar] [CrossRef]
- Thankachen J, Agarwal V. Challenges in Diagnosis, Management, and Treatment of Allopurinol-Induced DRESS Syndrome: Case Report and Literature Review. Am J Ther 2015, 22, e77–e83. [Google Scholar] [CrossRef]
- Biagi, G.; Costantini, A.; Costantino, L.; Giorgi, I.; Livi, O.; Pecorari, P.; Rinaldi, M.; Scartoni, V. Synthesis and Biological Evaluation of New Imidazole, Pyrimidine, and Purine Derivatives and Analogs as Inhibitors of Xanthine Oxidase. J. Med. Chem. 1996, 39, 2529–2535. [Google Scholar] [CrossRef] [PubMed]
- Tamta, H.; Thilagavathi, R.; Chakraborti, A.K.; Mukhopadhyay, A.K. 6-(N-benzoylamino)purine as a novel and potent inhibitor of xanthine oxidase: Inhibition mechanism and molecular modeling studies. J. Enzym. Inhib. Med. Chem. 2005, 20, 317–324. [Google Scholar] [CrossRef]
- Rodrigues MV, Barbosa AF, da Silva JF, dos Santos DA, Vanzolini KL, de Moraes MC, et al. 9-Benzoyl 9-deazaguanines as potent xanthine oxidase inhibitors. Bioorg Med Chem 2016, 24, 226–31. [Google Scholar] [CrossRef] [PubMed]
- 37. Šmelcerović, A.; Tomović, K.; Šmelcerović, Ž.; Petronijević, Ž.; Kocić, G.; Tomašič, T.; Jakopin, Ž.; Anderluh, M. Xanthine oxidase inhibitors beyond allopurinol and febuxostat; an overview and selection of potential leads based on in silico calculated physico-chemical properties, predicted pharmacokinetics and toxicity. Eur. J. Med. Chem. 2017, 135, 491–516. [Google Scholar] [CrossRef]
- Malik, U.Z.; Hundley, N.J.; Romero, G.; Radi, R.; Freeman, B.A.; Tarpey, M.M.; Kelley, E.E. Febuxostat inhibition of endothelial-bound XO: Implications for targeting vascular ROS production. Free. Radic. Biol. Med. 2011, 51, 179–184. [Google Scholar] [CrossRef]
- Sato T, Ashizawa N, Matsumoto K, Iwanaga T, Nakamura H, Inoue T, et al. Discovery of 3-(2-cyano-4-pyridyl)-5-(4-pyridyl)-1,2,4-triazole, FYX-051 - a xanthine oxidoreductase inhibitor for the treatment of hyperuricemia [corrected]. Bioorg Med Chem Lett 2009, 19, 6225–9.
- Jordan, A.; Gresser, U. Side Effects and Interactions of the Xanthine Oxidase Inhibitor Febuxostat. Pharmaceuticals 2018, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Rullo, R.; Cerchia, C.; Nasso, R.; Romanelli, V.; De Vendittis, E.; Masullo, M.; Lavecchia, A. Novel Reversible Inhibitors of Xanthine Oxidase Targeting the Active Site of the Enzyme. Antioxidants 2023, 12, 825. [Google Scholar] [CrossRef]
- Xu, X.; Deng, L.; Nie, L.; Chen, Y.; Liu, Y.; Xie, R.; Li, Z. Discovery of 2-phenylthiazole-4-carboxylic acid, a novel and potent scaffold as xanthine oxidase inhibitors. Bioorganic Med. Chem. Lett. 2019, 29, 525–528. [Google Scholar] [CrossRef]
- Zhang, T.-J.; Zhang, Y.; Zhang, Z.-H.; Wang, Z.-R.; Zhang, X.; Hu, S.-S.; Lu, P.-F.; Guo, S.; Meng, F.-H. Discovery of 4-(phenoxymethyl)-1H-1,2,3-triazole derivatives as novel xanthine oxidase inhibitors. Bioorganic Med. Chem. Lett. 2022, 60, 128582. [Google Scholar] [CrossRef]
- Zhang, T.-J.; Tu, S.; Zhang, X.; Wang, Q.-Y.; Hu, S.-S.; Zhang, Y.; Zhang, Z.-H.; Wang, Z.-R.; Meng, F.-H. Amide-based xanthine oxidase inhibitors bearing an N-(1-alkyl-3-cyano-1H-indol-5-yl) moiety: Design, synthesis and structure-activity relationship investigation. Bioorganic Chem. 2021, 117, 105417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-J.; Zhang, Z.-H.; Zhang, X.; Wang, Z.-R.; Xu, E.-Y.; Tu, S.; Zhang, Y.; Meng, F.-H. Design, synthesis and biological evaluation of N-(4-alkoxy-3-(1H-tetrazol-1-yl)phenyl) heterocyclic aromatic amide derivatives as xanthine oxidase inhibitors. Bioorganic Chem. 2022, 127, 105938. [Google Scholar] [CrossRef]
- Tang H-J, Li W, Zhou M, Peng L-Y, Wang J-X, Li J-H, et al. Design, synthesis and biological evaluation of novel xanthine oxidase inhibitors bearing a 2-arylbenzo b furan scaffold. European Journal of Medicinal Chemistry 2018, 151, 849–60. [Google Scholar] [CrossRef]
- Zhao J, Mao Q, Lin F, Zhang B, Sun M, Zhang T, et al. Intramolecular hydrogen bond interruption and scaffold hopping of TMC-5 led to 2-(4-alkoxy-3-cyanophenyl)pyrimidine-4/5-carboxylic acids and 6-(4-alkoxy-3-cyanophenyl)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-ones as potent pyrimidine-based xanthine oxidase inhibitors. Eur J Med Chem 2022, 229, 114086. [Google Scholar]
- Fais A, Era B, Asthana S, Sogos V, Medda R, Santana L, et al. Coumarin derivatives as promising xanthine oxidase inhibitors. International Journal of Biological Macromolecules 2018, 120, 1286–93. [Google Scholar] [CrossRef] [PubMed]
- Mohos, V.; Fliszár-Nyúl, E.; Poór, M. Inhibition of Xanthine Oxidase-Catalyzed Xanthine and 6-Mercaptopurine Oxidation by Flavonoid Aglycones and Some of Their Conjugates. Int. J. Mol. Sci. 2020, 21, 3256. [Google Scholar] [CrossRef]
- Zhao, J.; Huang, L.; Sun, C.; Zhao, D.; Tang, H. Studies on the structure-activity relationship and interaction mechanism of flavonoids and xanthine oxidase through enzyme kinetics, spectroscopy methods and molecular simulations. Food Chem. 2020, 323, 126807. [Google Scholar] [CrossRef]
- Li J, Gong Y, Li J, Fan L. In vitro inhibitory effects of polyphenols from Tartary buckwheat on xanthine oxidase: Identification, inhibitory activity, and action mechanism. Food Chem 2022, 379, 132100. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Wu, S.; Xie, F.; Yang, C.S.; Shao, P. Natural compounds lower uric acid levels and hyperuricemia: Molecular mechanisms and prospective. Trends Food Sci. Technol. 2022, 123, 87–102. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, D. Investigation of the interaction between salvianolic acid C and xanthine oxidase: Insights from experimental studies merging with molecular docking methods. Bioorganic Chem. 2019, 88, 102981. [Google Scholar] [CrossRef]
- Di Petrillo, A.; Siguri, C.; Delogu, G.L.; Fais, A.; Era, B.; Floris, S.; Pintus, F.; Kumar, A.; Fantini, M.C.; Olla, S. Exploring Asphodelus microcarpus as a source of xanthine oxidase inhibitors: Insights from in silico and in vitro studies. Chem. Interactions 2024, 397, 111087. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Du, J.; He, P.; Wang, N.; Li, L.; Liu, Y.; Yang, C.; Xu, H.; Li, Y. Identification of natural xanthine oxidase inhibitors: Virtual screening, anti-xanthine oxidase activity, and interaction mechanism. Int. J. Biol. Macromol. 2024, 259, 129286. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Sun, S.; Lv, C.; Li, Z.; Guo, M.; Yin, Y.; Wang, H.; Wang, W. Discovery of mycotoxin alternariol as a potential lead compound targeting xanthine oxidase. Chem. Interactions 2022, 360, 109948. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, S.; Wang, X.; Fan, X.; Wilson, G.; Sa, Y.; Ma, X. A strategy for inhibitors screening of xanthine oxidase based on colorimetric sensor combined with affinity chromatography technology. Biosens. Bioelectron. 2024, 261, 116510. [Google Scholar] [CrossRef]
- Devenyi, Z.J.; Orchard, J.L.; Powers, R.E. Xanthine oxidase activity in mouse pancreas: Effects of caerulein-induced acute pancreatitis. Biochem. Biophys. Res. Commun. 1987, 149, 841–845. [Google Scholar] [CrossRef]
- Nordback, I.H.; Cameron, J.L. The mechanism of conversion of xanthine dehydrogenase to xanthine oxidase in acute pancreatitis in the canine isolated pancreas preparation. . 1993, 113, 90–7. [Google Scholar]
- Closa, D.; Bulbena, O.; Hotter, G.; Roseixó-Catafau, J.; Fernández-Cruz, L.; Gelpf, E. Xanthine oxidase activation in cerulein-and taurocholate-induced acute pancreatitis in rats. Arch. Int. de Physiol. de Biochim. et de Biophys. 1994, 102, 167–170. [Google Scholar] [CrossRef]
- Closa, D.; Bulbena, O.; Rosello-Catafau, J.; Fernandez-Cruz, L.; Gelpi, E. Effect of prostaglandins and superoxide dismutase administration on oxygen free radical production in experimental acute pancreatitis. Inflammation 1993, 17, 563–571. [Google Scholar] [CrossRef]
- Granell, S.; Gironella, M.; Bulbena, O.; Panés, J.; Mauri, M.; Sabater, L.; Aparisi, L.; Gelpí, E.; Closa, D. Heparin mobilizes xanthine oxidase and induces lung inflammation in acute pancreatitis. Crit. Care Med. 2003, 31, 525–530. [Google Scholar] [CrossRef]
- Folch, E.; Gelpi, E.; Rosello-Catafau, J.; Closa, D. Free Radicals Generated by Xanthine Oxidase Mediate Pancreatitis-Associated Organ Failure. Dig. Dis. Sci. 1998, 43, 2405–2410. [Google Scholar] [CrossRef]
- Nordback, I.H.; Olson, J.L.; Chacko, V.P.; Cameron, J.L. Detailed characterization of experimental acute alcoholic pancreatitis. Surgery 1995, 117, 41–49. [Google Scholar] [CrossRef]
- Weber H, Merkord J, Jonas L, Wagner A, Schroder H, Kading U, et al. Oxygen radical generation and acute pancreatitis: effects of dibutyltin dichloride/ethanol and ethanol on rat pancreas. Pancreas 1995, 11, 382–8. [Google Scholar] [CrossRef]
- Rong, J.; Han, C.; Huang, Y.; Wang, Y.; Qiu, Q.; Wang, M.; Wang, S.; Wang, R.; Yang, J.; Li, X.; et al. Inhibition of xanthine oxidase alleviated pancreatic necrosis via HIF-1α-regulated LDHA and NLRP3 signaling pathway in acute pancreatitis. Acta Pharm. Sin. B 2024, 14, 3591–3604. [Google Scholar] [CrossRef]
- Shabanov VV, Sarbaeva NN, Milyakova MN. Generation of free oxygen radicals in the pathogenesis of experimental acute reflux pancreatitis. Bull Exp Biol Med 2002, 134, 26–7. [Google Scholar] [CrossRef] [PubMed]
- Czakó, L.; Takács, T.; Varga, I.S.; Tiszlavicz, L.; Hai, D.Q.; Hegyi, P.; Matkovics, B.; Lonovics, J. Oxidative Stress in Distant Organs and the Effects of Allopurinol During Experimental Acute Pancreatitis. J. Gastrointest. Cancer 2000, 27, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Czako, L.; Takacs, T.; Varga, I.S.; Tiszlavicz, L.; Hai, D.Q.; Hegyi, P.; Matkovics, B.; Lonovics, J. Involvement of Oxygen-Derived Free Radicals in L-Arginine-Induced Acute Pancreatitis. Dig. Dis. Sci. 1998, 43, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Czakó, L.; Takács, T.; Varga, I.S.; Hai, D.Q.; Tiszlavicz, L.; Hegyi, P.; Mándi, Y.; Matkovics, B.; Lonovics, J. The pathogenesis of L-arginine-induced acute necrotizing pancreatitis: Inflammatory mediators and endogenous cholecystokinin. J. Physiol. 2000, 94, 43–50. [Google Scholar] [CrossRef]
- Hirano, T.; Manabe, T.; Ohshio, G.; Nio, Y. Protective effects of combined therapy with a protease inhibitor, ONO 3307, and a xanthine oxidase inhibitor, allopurinol on temporary ischaemic model of pancreatitis in rats. . 1992, 61, 224–33. [Google Scholar]
- Degertekin H, Ertan A, Yater RD, Van Meter K, Akdamar K. Hyperbaric oxygen, allopurinol, and diet-induced acute pancreatitis. Ann Intern Med 1985, 103, 474–5. [Google Scholar] [CrossRef]
- Sanfey, H.; Bulkley, G.B.; Cameron, J.L. The Source and Role of Oxygen-derived Free Radicals in Three Different Experimental Models. Ann. Surg. 1985, 201, 633–639. [Google Scholar] [CrossRef]
- Nordback, I.H.; Macgowan, S.F.; Potter, J.J.B.; Cameron, J.L.M. The Role of Acetaldehyde in the Pathogenesis of Acute Alcoholic Pancreatitis. Ann. Surg. 1991, 214, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Sarr MG, Bulkley GB, Cameron JL. Temporal efficacy of allopurinol during the induction of pancreatitis in the ex vivo perfused canine pancreas. Surgery 1987, 101, 342–6. [Google Scholar]
- Marks, J.M.; Dunkin, B.J.; Shillingstad, B.L.; Youngelman, D.F.; Schweitzer, M.A.; Lash, R.H.; Singh, J.; Ponsky, L.; Ponsky, J.L. Pretreatment with allopurinol diminishes pancreatography-induced pancreatitis in a canine model. Gastrointest. Endosc. 1998, 48, 180–183. [Google Scholar] [CrossRef]
- Pereda J, Sabater L, Cassinello N, Gómez-Cambronero L, Closa D, Folch-Puy E, et al. Effect of simultaneous inhibition of TNF-alpha production and xanthine oxidase in experimental acute pancreatitis: the role of mitogen activated protein kinases. Ann Surg 2004, 240, 108–16. [Google Scholar] [CrossRef]
- Comert B, Isik AT, Aydin S, Bozoglu E, Unal B, Deveci S, et al. Combination of allopurinol and hyperbaric oxygen therapy: a new treatment in experimental acute necrotizing pancreatitis? World J Gastroenterol 2007, 13, 6203–7.
- Escobar, J.; Pereda, J.; Arduini, A.; Sandoval, J.; Moreno, M.L.; Pérez, S.; Sabater, L.; Aparisi, L.; Cassinello, N.; Hidalgo, J.; et al. Oxidative and nitrosative stress in acute pancreatitis. Modulation by pentoxifylline and oxypurinol. Biochem. Pharmacol. 2012, 83, 122–130. [Google Scholar] [CrossRef]
- Isik, A.T.; Mas, M.R.; Yamanel, L.; Aydin, S.; Comert, B.; Akay, C.; Erdem, G.; Mas, N. The role of allopurinol in experimental acute necrotizing pancreatitis. Indian J Med Res 2006, 124, 709–14. [Google Scholar]
- Lankisch, P.G.; Pohl, U.; Otto, J.; Wereszczynska-Siemiatkowska, U.; Gröne, H.-J. *Xanthine Oxidase Inhibitor in Acute Experimental Pancreatitis in Rats and Mice. Pancreas 1989, 4, 436–440. [Google Scholar] [CrossRef]
- Wisner JR, Renner IG. Allopurinol attenuates caerulein induced acute pancreatitis in the rat. Gut 1988, 29, 926–9. [Google Scholar] [CrossRef]
- Suzuki H, Suematsu M, Miura S, Asako H, Kurose I, Ishii H, et al. Xanthine oxidase-mediated intracellular oxidative stress in response to cerulein in rat pancreatic acinar cells. Pancreas 1993, 8, 465–70. [Google Scholar] [CrossRef]
- Ito, T.; Nakao, A.; Kishimoto, W.; Nakano, M.; Takagi, H. The Involvement and Sources of Active Oxygen in Experimentally Induced Acute Pancreatitis. Pancreas 1996, 12, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Mantke, R.; Rocken, C.; Schubert, D.; Pross, M.; Sokolowski, A.; Halangk, W.; Lippert, H.; Schulz, H.-U. Enzymatic and histological alterations in the isolated perfused rat pancreas under conditions of oxidative stress. Langenbeck's Arch. Surg. 2002, 387, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Robles L, Vaziri ND, Ichii H. Role of Oxidative Stress in the Pathogenesis of Pancreatitis: Effect of Antioxidant Therapy. Pancreat Disord Ther 2013, 3, 112. [Google Scholar]
- Armstrong JA, Cash N, Soares PM, Souza MH, Sutton R, Criddle DN. Oxidative stress in acute pancreatitis: lost in translation? Free Radic Res 2013, 47, 917–33.
- Niederau, C.; Schulz, H.-U.; Klonowski, H. Lazaroids Protect Isolated Rat Pancreatic Acinar Cells Against Damage Induced by Free Radicals. Pancreas 1995, 11, 107–121. [Google Scholar] [CrossRef]
- Sanfey, H.; Bulkley, G.B.; Cameron, J.L. The Role of Oxygen-derived Free Radicals in the Pathogenesis of Acute Pancreatitis. Ann. Surg. 1984, 200, 405–413. [Google Scholar] [CrossRef]
- Rau B, Poch B, Gansauge F, Bauer A, Nussler AK, Nevalainen T, et al. Pathophysiologic role of oxygen free radicals in acute pancreatitis: initiating event or mediator of tissue damage? Ann Surg 2000, 231, 352–60.
- Schoenberg, M.; Büchler, M.; Beger, H. The role of oxygen radicals in experimental acute pancreatitis. Free. Radic. Biol. Med. 1992, 12, 515–522. [Google Scholar] [CrossRef]
- Pérez, S.; Pereda, J.; Sabater, L.; Sastre, J. Redox signaling in acute pancreatitis. Redox Biol. 2015, 5, 1–14. [Google Scholar] [CrossRef]
- Weber, H.; Roesner, J.; Nebe, B.; Rychly, J.; Werner, A.; Schröder, H.; Jonas, L.; Leitzmann, P.; Schneider, K.-P.; Dummler, W. Increased Cytosolic Ca2+ Amplifies Oxygen Radical-Induced Alterations of the Ultrastructure and the Energy Metabolism of Isolated Rat Pancreatic Acinar Cells. Digestion 1998, 59, 175–185. [Google Scholar] [CrossRef]
- Raghu, M.G.; Wig, J.D.; Kochhar, R.; Gupta, D.; Gupta, R.; Yadav, T.D.; Agarwal, R.; Kudari, A.K.; Doley, R.P.; Javed, A. Lung complications in acute pancreatitis. JOP 2007, 8, 177–85. [Google Scholar] [PubMed]
- Cuthbertson, C.M.M.; Su, K.H.B.; Muralidharan, V.M.; Millar, I.M.; Malcontenti-Wilson, C.B.; Christophi, C.M. Hyperbaric Oxygen Improves Capillary Morphology in Severe Acute Pancreatitis. Pancreas 2008, 36, 70–75. [Google Scholar] [CrossRef]
- Yu X, Li YG, He XW, Li XR, Din BN, Gan Y, et al. Hyperbaric oxygen reduces inflammatory response in acute pancreatitis by inhibiting NF-kappaB activation. Eur Surg Res 2009, 42, 130–5. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ge, B.; Yuan, Y.; Wang, G. Hyperbaric Oxygen Ameliorated Acute Pancreatitis in Rats via the Mitochondrial Pathway. Dig. Dis. Sci. 2020, 65, 3558–3569. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Klonowski, H.; Schulz, H.-U.; Sarbia, M.; Lüthen, R.; Häussinger, D. Oxidative injury to isolated rat pancreatic acinar cells vs. isolated zymogen granules. Free. Radic. Biol. Med. 1996, 20, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Jiang Q, Ding Y, Li F, Fayyaz AI, Duan H, Geng X. Modulation of NLRP3 inflammasome-related-inflammation via RIPK1/RIPK3-DRP1 or HIF-1alpha signaling by phenothiazine in hypothermic and normothermic neuroprotection after acute ischemic stroke. Redox Biol 2024, 73, 103169. [Google Scholar]
- de Groot H, Littauer A. Hypoxia, reactive oxygen, and cell injury. Free Radic Biol Med 1989, 6, 541–51. [Google Scholar] [CrossRef]
- Nonaka, A.; Manabe, T.; Tamura, K.; Asano, N.; Imanishi, K.; Tobe, T. Changes of Xanthine Oxidase, Lipid Peroxide and Superoxide Dismutase in Mouse Acute Pancreatitis. Digestion 1989, 43, 41–46. [Google Scholar] [CrossRef]
- Lopez Martin A, Carrillo Alcaraz A. Oxidative stress and acute pancreatitis. Rev Esp Enferm Dig 2011, 103, 559–62. [Google Scholar] [CrossRef]
- Granell, S.; Bulbena, O.; Genesca, M.; Sabater, L.; Sastre, J.; Gelpi, E.; Closa, D. Mobilization of xanthine oxidase from the gastrointestinal tract in acute pancreatitis. BMC Gastroenterol. 2004, 4, 1–1. [Google Scholar] [CrossRef]
- Shi, C.; Andersson, R.; Zhao, X.; Wang, X. Potential role of reactive oxygen species in pancreatitis-associated multiple organ dysfunction. Pancreatology 2005, 5, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Fink, MP. Reactive oxygen species as mediators of organ dysfunction caused by sepsis, acute respiratory distress syndrome, or hemorrhagic shock: potential benefits of resuscitation with Ringer's ethyl pyruvate solution. Curr Opin Clin Nutr Metab Care 2002, 5, 167–74. [Google Scholar] [CrossRef] [PubMed]
- Papathanassoglou, E.D.E.; Moynihan, J.A.; Ackerman, M.H. Does programmed cell death (apoptosis) play a role in the development of multiple organ dysfunction in critically ill patients? A review and a theoretical framework. Crit. Care Med. 2000, 28, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sun, R.; Sun, K.; Yan, C.; Jiang, J.; Kong, F.; Shi, J. The deubiquitinase USP11 ameliorates intervertebral disc degeneration by regulating oxidative stress-induced ferroptosis via deubiquitinating and stabilizing Sirt3. Redox Biol. 2023, 62, 102707. [Google Scholar] [CrossRef]
- Li, H.; Lin, Y.; Zhang, L.; Zhao, J.; Li, P. Ferroptosis and its emerging roles in acute pancreatitis. Chin. Med J. 2022, 135, 2026–2034. [Google Scholar] [CrossRef]
- Gu, X.; Huang, Z.; Ying, X.; Liu, X.; Ruan, K.; Hua, S.; Zhang, X.; Jin, H.; Liu, Q.; Yang, J. Ferroptosis exacerbates hyperlipidemic acute pancreatitis by enhancing lipid peroxidation and modulating the immune microenvironment. Cell Death Discov. 2024, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Poch, B.; Gansauge, F.; Rau, B.; Wittel, U.; Gansauge, S.; Nüssler, A.K.; Schoenberg, M.; Beger, H.G. The role of polymorphonuclear leukocytes and oxygen-derived free radicals in experimental acute pancreatitis: mediators of local destruction and activators of inflammation. FEBS Lett. 1999, 461, 268–272. [Google Scholar] [CrossRef]
- Folch E, Salas A, Panes J, Gelpi E, Rosello-Catafau J, Anderson DC, et al. Role of P-selectin and ICAM-1 in pancreatitis-induced lung inflammation in rats: significance of oxidative stress. Ann Surg 1999, 230, 792–8. [Google Scholar] [CrossRef]
- Folch, E.; Salas, A.; Prats, N.; Panés, J.; Piqué, J.M.; Gelpı́, E.; Roselló-Catafau, J.; Closa, D. H2O2 and PARS mediate lung P-selectin upregulation in acute pancreatitis. Free. Radic. Biol. Med. 2000, 28, 1286–1294. [Google Scholar] [CrossRef]
- Folch, E.; Closa, D.; Ñeco, P.; Solé, S.; Planas, A.; Gelpí, E.; Roselló-Catafau, J. Pancreatitis Induces HSP72 in the Lung: Role of Neutrophils and Xanthine Oxidase. Biochem. Biophys. Res. Commun. 2000, 273, 1078–1083. [Google Scholar] [CrossRef]
- Granell, S.; Serrano-Mollar, A.; Folch-Puy, E.; Navajas, D.; Farre, R.; Bulbena, O.; Closa, D. Oxygen in the alveolar air space mediates lung inflammation in acute pancreatitis. Free. Radic. Biol. Med. 2004, 37, 1640–1647. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.-L.; Yan, W.-S.; Xiang, X.-H.; Chen, K.; Xia, S.-H. Prevention Effect of Allopurinol on Post-Endoscopic Retrograde Cholangiopancreatography Pancreatitis: A Meta-Analysis of Prospective Randomized Controlled Trials. PLOS ONE 2014, 9, e107350. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Chen, Y.; Bai, J.; Xin, Y.; Pan, X.; Zhao, L. Meta-Analysis of Prophylactic Allopurinol Use in Post-Endoscopic Retrograde Cholangiopancreatography Pancreatitis. Pancreas 2008, 37, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Budzyńska, A.; Marek, T.; Nowak, A.; Kaczor, R.; Nowakowska-Dulawa, E. A Prospective, Randomized, Placebo-Controlled Trial of Prednisone and Allopurinol in the Prevention of ERCP-Induced Pancreatitis. Endoscopy 2001, 33, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Mosler, P.; Sherman, S.; Marks, J.; Watkins, J.L.; Geenen, J.E.; Jamidar, P.; Fogel, E.L.; Lazzell-Pannell, L.; Temkit, M.; Tarnasky, P.; et al. Oral allopurinol does not prevent the frequency or the severity of post-ERCP pancreatitis. Gastrointest. Endosc. 2005, 62, 245–250. [Google Scholar] [CrossRef]
- Romagnuolo, J.; Hilsden, R.; Sandha, G.S.; Cole, M.; Bass, S.; May, G.; Love, J.; Bain, V.G.; McKaigney, J.; Fedorak, R.N. Allopurinol to Prevent Pancreatitis After Endoscopic Retrograde Cholangiopancreatography: A Randomized Placebo-Controlled Trial. Clin. Gastroenterol. Hepatol. 2008, 6, 465–471. [Google Scholar] [CrossRef]
- Abbasinazari, M.; Alizadeh, A.H.M.; Moshiri, K.; Pourhoseingholi, M.A.; Zali, M.R. Does allopurinol prevent post endoscopic retrograde cholangio- pancreatography pancreatitis? A randomized double blind trial.. 2011, 49, 579–83. [Google Scholar]
- Romagnuolo, J.; Hilsden, R.; Sandha, G.S.; Cole, M.; Bass, S.; May, G.; Love, J.; Bain, V.G.; McKaigney, J.; Fedorak, R.N. Allopurinol to Prevent Pancreatitis After Endoscopic Retrograde Cholangiopancreatography: A Randomized Placebo-Controlled Trial. Clin. Gastroenterol. Hepatol. 2008, 6, 465–471. [Google Scholar] [CrossRef]
- Katsinelos, P.; Kountouras, J.; Chatzis, J.; Christodoulou, K.; Paroutoglou, G.; Mimidis, K.; Beltsis, A.; Zavos, C. High-dose allopurinol for prevention of post-ERCP pancreatitis: a prospective randomized double-blind controlled trial. Gastrointest. Endosc. 2005, 61, 407–415. [Google Scholar] [CrossRef]
- Martinez-Torres, H.; Rodriguez-Lomeli, X.; Davalos-Cobian, C.; Garcia-Correa, J.; Maldonado-Martinez, J.M.; Medrano-Muñoz, F.; Fuentes-Orozco, C.; Gonzalez-Ojeda, A. Oral allopurinol to prevent hyperamylasemia and acute pancreatitis after endoscopic retrograde cholangiopancreatography. World J. Gastroenterol. 2009, 15, 1600–1606. [Google Scholar] [CrossRef]
Figure 1.
Surface representation of the XO crystal structure homodimer (PDB:3NRZ). Monomer A is in dark blue, while monomer B in light blue. The different protein cofactors (FAD, FeSII, FeSI, and Mo-pt), with the shortest distances between them marked, are represented on the right. The figure was created using PyMOL version 2.6.0.
Figure 1.
Surface representation of the XO crystal structure homodimer (PDB:3NRZ). Monomer A is in dark blue, while monomer B in light blue. The different protein cofactors (FAD, FeSII, FeSI, and Mo-pt), with the shortest distances between them marked, are represented on the right. The figure was created using PyMOL version 2.6.0.
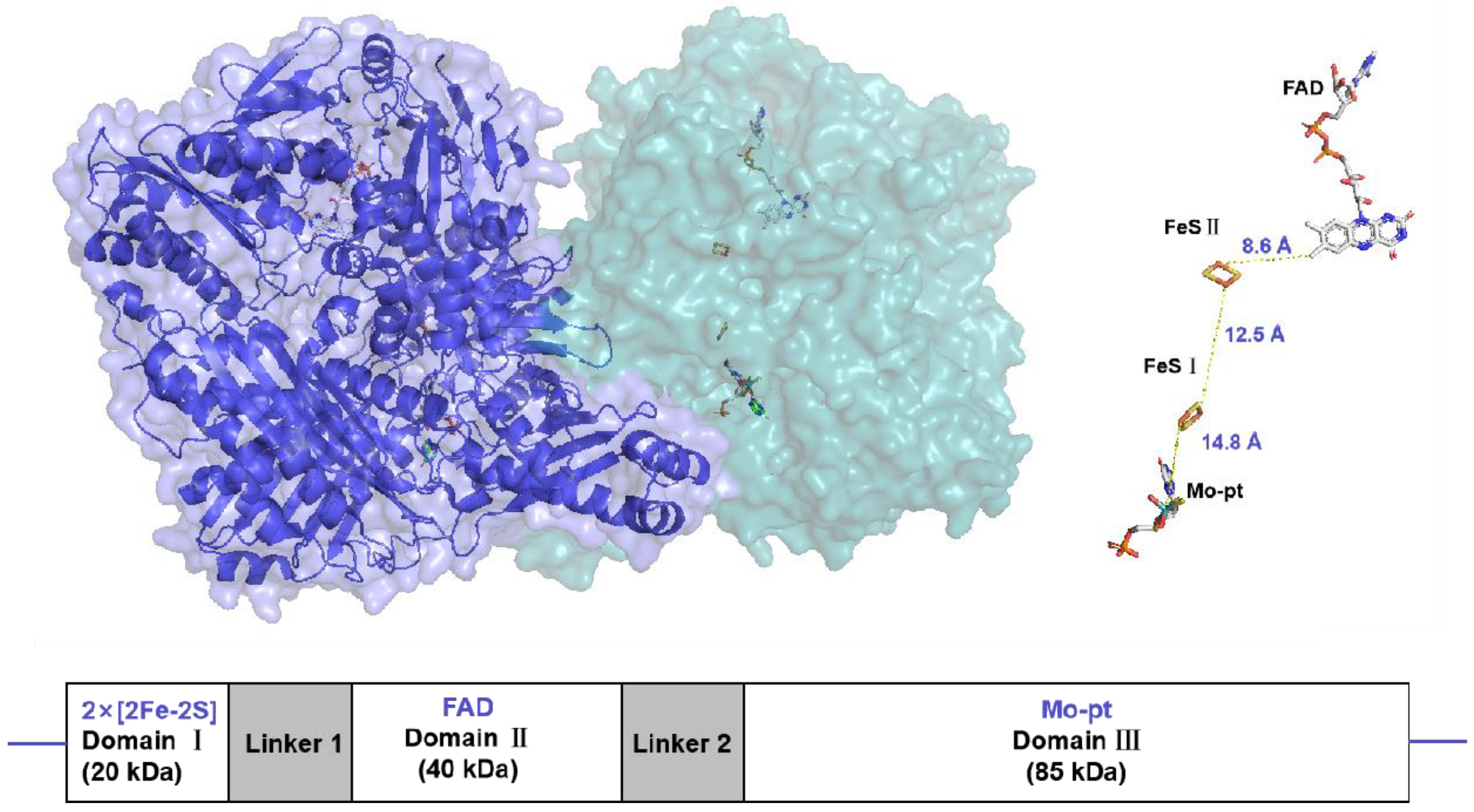
Figure 2.
Schematic diagram of the oxidative hydroxylation of hypoxanthine through xanthine to uric acid, and reactive oxygen species generation, in the metabolism of purines.
Figure 2.
Schematic diagram of the oxidative hydroxylation of hypoxanthine through xanthine to uric acid, and reactive oxygen species generation, in the metabolism of purines.
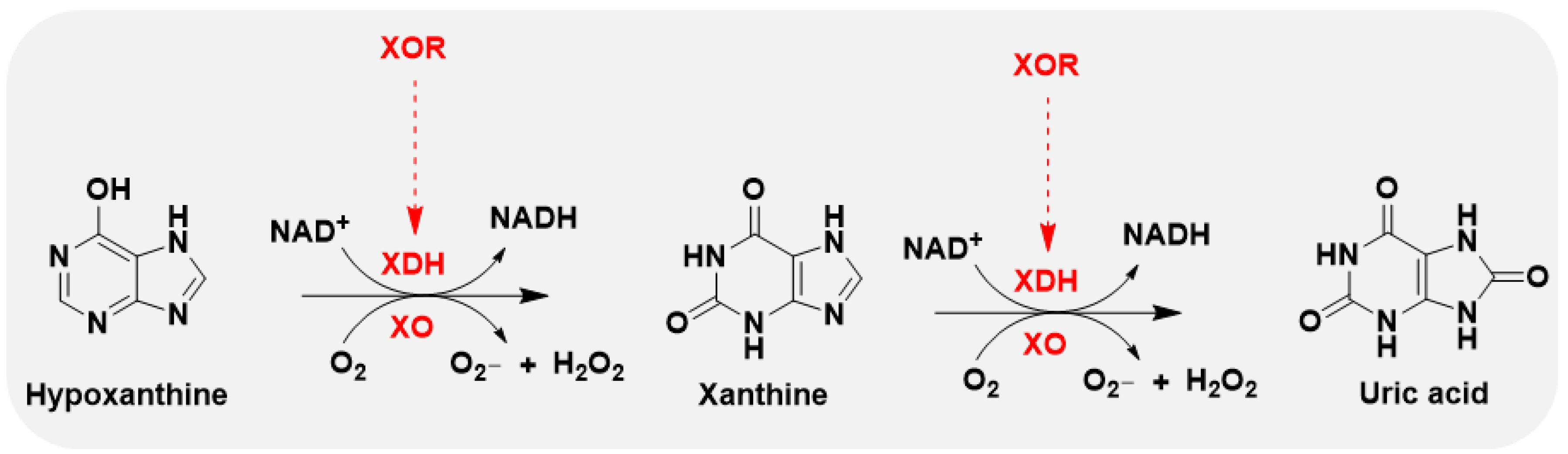

Table 1.
General structural formulae of recently reported XO inhibitors.
| Structure | Class | IC50 Value | Ref. |
|---|---|---|---|
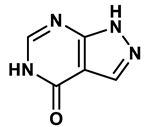 1 |
Purine derivatives | 0.2-50 μM | 19 |
 2 |
Purine derivatives | 0.115 μM | 21 |
 3 |
Purine derivatives | 0.45 μM | 22 |
 4 |
Purine derivatives | 0.065 μM | 23 |
 5 |
Thiazole derivative | 1.8 μM | 25 |
 6 |
1,2,3-triazoles derivative | 0.0053 μM | 26 |
 7 |
Pyrimidine derivative | 18 μM | 28 |
 8 |
Pyrazole derivative | 30 μM | 28 |
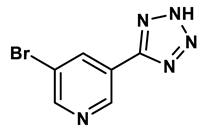 9 |
Isonicotinamide derivatives | 64 μM | 28 |
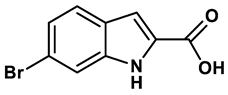 10 |
Imidazole derivatives | 82 μM | 28 |
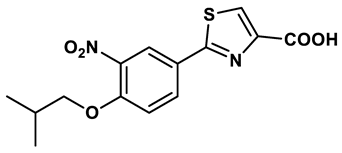 11 |
Thiazole derivative | 0.0486 μM | 29 |
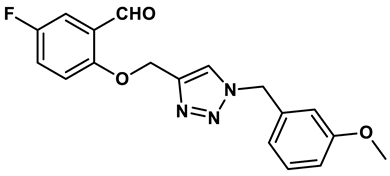 12 |
1,2,3-triazoles | 0.70 μM | 30 |
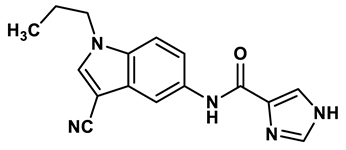 13 |
Imidazole derivatives | 0.018 μM | 31 |
 14 |
Isonicotinamide derivatives | 0.022 μM | 32 |
 15 |
Benzo[b]furan derivatives | 4.45 μM | 33 |
 16 |
3-Phenylcoumarins derivatives | 8.4 μM | 18 |
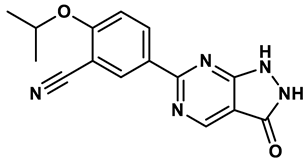 17 |
Pyrimidine derivatives | 0.039 μM | 34 |
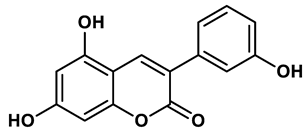 18 |
Coumarin analogues | 2.13 μM | 35 |
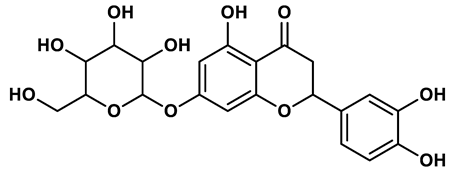 19 |
Flavonoid analogues | 10.6 μM | 41 |
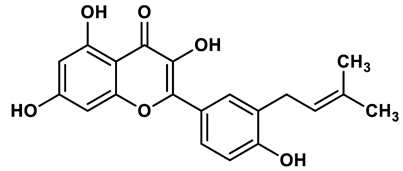 20 |
Flavonoid analogues | 8.45 μM | 42 |
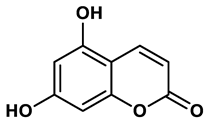 21 |
Coumarin analogues | 10.91 μM | 42 |
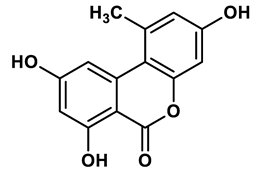 22 |
Ellagic acid analogues | 0.23 μM | 43 |
Table 2.
experimental studies on Inhibitors of XO.
| Year | AP Model | Species | Drug | Dose | Administration | Effects | Drug effect | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1985 | CDE | Mice | Allopurinol | 24 mg/kg | Therapeutic | ↓Serum amylase, pancreatic histological damage; survival rate | Positive | 59 |
| 1985 | FFA POSS ISCH |
Dog | Allopurinol | 100 mg |
Prophylactic | ↓Serum amylase, pancreas edema; pancreatic weight | Positive | 60 |
| 1987 | Cerulein | Mice | Allopurinol | 0.7-7 mg/kg/h | Prophylactic | No effect on pancreas edema; no change in serum amylase level | Negative | 45 |
| 1988 | Cerulein | Rat | Allopurinol | 20 mg/kg/h | Therapeutic | ↓Serum amylase; pancreatic weight | Positive | 69 |
| 1989 | NaTC; CDE |
Rat; Mice | Allopurinol | 100 mg/kg for rats; 50 mg/kg for mice |
Prophylactic | No change in serum amylase/lipase; pancreatic trypsin levels; no effect on histological damage, inflammation and survival | Negative | 68 |
| 1991 | ISCH+AA | Dog | Allopurinol | 100 mg/kg | Therapeutic | ↓Serum amylase; pancreas edema; pancreatic weight; hemorrhage | Positive | 61 |
| 1992 | PBDO + ISCH | Rat | Allopurinol | 20 mg/kg | Prophylactic | ↓Serum amylase; histological changes | Positive | 58 |
| 1996 | Cerulein | Rat | Allopurinol | 50 mg/kg | Prophylactic | No change in serum amylase, lipase and trypsin level; no effect on pancreatic histological damage | Negative | 71 |
| 1998 | L-arginine | Rat | Allopurinol | 100 or 200 mg/kg | Prophylactic | ↓Serum amylase; pancreatic MDA;histological damage; catalase activity ↑SOD; GPx |
Positive | 56 |
| 1998 | NaTC | Rat | Oxypurinol | 10 mM; 0.066 ml/min | Therapeutic | ↓Pancreatic MPO, MOD ↑GSH |
Positive | 50 |
| 1998 | ERCP | Dog | Allopurinol | 5 mg/kg | Prophylactic | ↓Serum amylase and lipase; histologic changes | Positive | 63 |
| 1999 | NaTC | Rat | Oxypurinol | 10 mM; 0.066 ml/min | Therapeutic | ↓Plasma lipase, pancreatic MPO and MOD | Positive | 98 |
| 2000 | L-arginine | Rat | Allopurinol | 200 mg/kg | Prophylactic | ↓Serum amylase; pancreatic MDA; plasma CCK-like bioactivity; serum TNF-α and IL-6 levels; histological damage; ↑SOD; GPx; |
Positive | 57 |
| 2000 | L-arginine | Rat | Allopurinol | 200 mg/kg | Prophylactic | ↓Serum amylase; histologic changes; | Positive | 55 |
| 2000 | NaTC | Rat | Oxypurinol | 10 mM; 0.066 ml/min | Therapeutic | ↓Plasma lipase; pancreatic MPO | Positive | 100 |
| 2002 | Ethyl alchohol | Dog | Allopurinol | 10 mg/kg | Prophylactic | ↓Pancreatic MPO | Positive | 54 |
| 2004 | NaTC | Rat | Oxypurinol | 5 mM; 0.066 mL/min for 30 minutes | Therapeutic | ↓Serum TNF-α; lung MPO activity | Positive | 64 |
| 2006 | NaTC | Rat | Allopurinol | 200 mg/kg | Therapeutic | ↓Serum amylase; plasma MDA; histological damage ↑ Plasma SOD and GSH-Px |
Positive | 67 |
| 2007 | NaTC | Rat | Allopurinol | 200 mg/kg | Therapeutic | ↓Serum amylase; tissue MDA; histological damage ↑Tissue SOD and GSH-Px |
Positive | 65 |
| 2012 | NaTC | Rat | Oxypurinol | 5 mM, 0.066 ml/min for 30 min | Therapeutic | ↓GSSH/GSH ratio; serum lipase ↑GSSH |
Positive | 66 |
CDE: cholinedeficient ethionine-supplemented diet. FFA: free fatty acid. POSS: partial duct obstruction combined with secretin stimulation. ISCH: ischemia. AA: acetaldehyde. PBDO: pancreatico-biliary duct obstruction. NaTC: intraductal infusion of 5% sodium taurocholate. MDA: Malondialdehyde. SOD: Superoxide dismutase. GSHPx: Glutathione peroxidase. MLNs: Mesenteric lymph nodes. MPO: myeloperoxidase.
Table 3.
Randomized placebo-controlled trials of allopurinol for acute pancreatitis.
| Year | Sample size | Study design | Intervention | Clinical outcome | Ref |
|---|---|---|---|---|---|
| 2001 | 300 | RCT | Allopurinol, 40 mg, 15 h and 3 h before ERCP or prednisone, 200 mg, 15 h and 3 h before ERCP vs placebo, 15 h and 3 h before ERCP | Neither prednisone nor allopurinol showed a beneficial influence on the incidence and severity of post-ERCP pancreatitis | 104 |
| 2005 | 243 | RCT | Allopurinol, 600 mg, 15 h and 3 h before ERCP vs placebo, 15 h and 3 h before ERCP | Pretreatment with high-dose, orally administered allopurinol decreases the frequency of post-ERCP pancreatitis | 109 |
| 2005 | 701 | RCT | Allopurinol, 600 and 300 mg, 4 h and 1 h before ERCP vs placebo | Prophylactic oral allopurinol did not reduce the frequency or the severity of post-ERCP pancreatitis | 105 |
| 2008 | 586 | RCT | Allopurinol,300 mg, 1 h before ERCP vs placebo | Allopurinol does not appear to reduce the overall risk of post-ERCP pancreatitis | 108 |
| 2009 | 170 | RCT | Allopurinol, 300 mg, 15 h and 3 h before ERCP vs placebo | Oral allopurinol before ERCP decreased the incidences of hyperamylasemia and pancreatitis in patients submitted to high-risk procedures | 110 |
| 2011 | 74 | RCT | Allopurinol,300 mg, 3 h and just before ERCP vs placebo | Allopurinol does not reduce the occurrence and amylase rise of post ERCP pancreatitis | 107 |
RCT: Randomized controlled trial. ERCP: Endoscopic retrograde cholangiopancreatography.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



