
Submitted:
30 October 2024
Posted:
31 October 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Coxsackievirus A6 (CVA6) has become increasingly clinically relevant as a cause of Hand, Foot and Mouth Disease (HFMD) globally since 2008. However, most laboratories do not routinely determine the enteroviral type of positive samples. The non-pharmaceutical measures introduced to curb transmission during the SARS-CoV-2 pandemic may also have been perturbed CVA6 epidemiology. We thus aimed to determine the prevalence, clinical presentation and genetic relationship of CVA6 across three complete epidemic seasons: one pre- and two post-SARS-CoV-2-emergence in our regional healthcare setting. Surplus diagnostic nucleic acid from diagnosed enteroviral positives diagnosed between September to December 2018 and May 2021 to April 2023 was subject to VP1 gene sequencing to determine CVA6 cases and interrogate their phylogenetic relationship. Confirmed CVA6 cases were also retrospectively clinically audited. CVA6 infections were identified in 33 and 69 individuals pre- and post-pandemic respectively, with cases peaking in November of 2018 and 2022, but in October of 2021. HFMD was the primary diagnosis in 85.5% of post-pandemic cases, but only 69.7% pre-pandemic, where respiratory and neurological symptoms (45.5% and 12.1% respectively) were significantly elevated. Complete VP1 sequence was retrieved for 94% of CVA6 cases, revealing study infections were genetically diverse and suggestive of multiple local and international transmission chains. CVA6 presented a significant clinical burden in our regional UK hospital setting both pre- and post-pandemic and was subject to dynamic clinical and genetic epidemiology.
Keywords:
Coxsackievirus A6
; Enterovirus
; CVA6
; genetic epidemiology
; Hand foot and mouth disease
; HFMD
1. Introduction
Human enteroviral infections are contracted predominantly via the faecal-oral route and exhibit a broad spectrum of disease ranging from asymptomatic or self-limiting illness through to high clinical consequence and associated mortality [1,2]. The causative Enteroviruses (EV) can be subcategorised into four species, EV-A to EV-D comprising over 100 types and are more prevalent within younger age groups, with severe outcomes including acute flaccid myelitis, endocarditis, and meningitis [3,4]. Less severe and more frequent enteroviral disease manifestations include hand, foot and mouth disease (HFMD), presenting classically with short-lived combinations of low-grade fever, malaise, a maculopapular rash or blisters on the hands, soles and buttocks and ulcerative lesions of the throat, mouth and tongue [5].
EV-A types are the leading cause of HFMD, principally EV-A71 and several members of the genetically diverse Coxsackievirus (CV) grouping CVA6, CVA10 and CVA16 [6]. Since 2008, CVA6 has been increasingly diagnosed in HFMD outbreaks across the world [7,8,9,10,11]. These recent epidemics have featured an atypical HFMD presentation of a more disseminated rash or principally herpangina [12,13,14,15] and can include a significant number of affected adults [16,17]. Onychomadesis - the separation of the nail from its matrix due to cessation in growth - can also be observed in atypical HFMD [18,19], occurring circa 8 weeks post infection in the majority of cases [20]. In rarer instances, CVA6 infection can lead to more severe complications like aseptic meningitis, encephalitis, acute flaccid myelitis and sometimes fatality [17,21,22,23]. Even though the majority of CVA6 infections occur without sequalae, higher mortality rates are often observed amongst the immunosuppressed due to the development of neurological complications [24]. The high prevalence of CVA6 infection combined with the potential for severity has prompted interest in the development of a vaccine [25].
HFMD is routinely diagnosed by clinical presentation, however, the causative virus can be confirmed via molecular methods involving the amplification and detection of the viral genome. Specific molecular assays targeting the 5’ UTR have previously been successful at identifying the presence of enteroviruses [26], however due to genetic drift occurring mainly within the capsid coding region and recombination [27,28], sequencing the VP1 gene is used predominantly to determine EV types [29,30]. CVA6 detection can be accomplished via analysis of skin and nasopharyngeal swabs, stool samples and / or fluid extracted from cutaneous lesions and vesicles. The European Union and the USA currently have no mandatory legislations with regards to notifying incidences of HFMD, causing difficulties in surveillance and identifying transmission trends of this enterovirus [31,32]. In contrast HFMD is a notifiable disease in China, allowing national surveillance and enterovirus prevalence to be identified [33].
CVA6 has a single stranded positive sense RNA genome comprised of approximately 7400 bases and can be distinguished into four genotypes A-D, further subcategorised into sub-genotypes including B1, B2, C1, C2 and D1-3 [34]. Sub-genotype D3 has recently been evidenced as the most prevalent sub-genotype [35,36,37], however the non-pharmaceutical interventions of the SARS-CoV-2 pandemic may have inadvertently affected circulation of enteroviruses including CVA6 [38]. We thus aimed to investigate the genetic and clinical epidemiology of CVA6 diagnosed in our regional UK hospital in epidemic seasons before and after the emergence of SARS-CoV-2.
2. Materials and Methods
The study was conducted in two periods: initially between 1st September to 17th December 2018 when a significant Enterovirus D68 epidemic was experienced and previously reported [39]. A second period followed between 11th May 2021 and 26th April 2023, when our diagnostic laboratory re-commenced enteroviral testing post-pandemic and sought to evaluate routine enteroviral typing. Whilst the precise start and end of the SARS-CoV-2 pandemic are open to debate, to simplify the narrative in this study, all 2021-2023 samples are referred to as ‘post-pandemic’.
Samples were screened for enteroviral infection using both commercial and in-house assays as described previously [30,39]. Briefly, samples were determined EV-positive by the commercial clinical assay (AusDiagnostics, Australia) as part of the routine care diagnostic pathway. Where available, surplus nucleic acid was re-screened using in house assays targeting partial VP1 and 5’ NCR genomic regions and candidate positives were confirmed by Sanger sequencing [29,30]. Some patients had more than one sample taken and were thus sequenced more than once also. Additionally, a minority of diagnostic samples were submitted for typing at the National Reference Laboratory, either confirming CVA6 infection or independently identifying further CVA6 in 4 cases without residual material available.
Confirmed CVA6 positive samples were then further amplified with two novel PCR assays targeting overlapping genomic regions covering the entire VP1 gene, based on all CVA6 lineage exemplars as presented by Bian et al 2015 [40] using MEGA7 [41] and Primer3 (https://primer3.ut.ee/). This pilot amplification returned inconsistent results, with approximately 50% amplification success (data not shown), but Sanger sequencing and phylogenetic analysis of successfully amplified samples indicated all were of the globally most common lineage D3 [37]. Therefore, the primers were modified and / or redesigned to specifically target CVA6 lineage D3 based on lineage D3 reference sequences and a single contig generated from a study sample in the preliminary VP1 amplification.
Final primer pairs employed to amplify and sequence full length VP1 were set A: CVA6D3_VP1Af AGACTGGAGGGGTATACGACT (positions 2258 – 2278 on CVA6 Kyoto5 D3 strain, GenBank accession number AB779618.1) and CVA6D3_VP1Ar GGTATGATTTYCTGCTATCCGG (2935 – 2914) generating a product size 678bp; and set B: CVA6D3_VP1Bf GCGCTTTGAYGCCGAGTT (2814 – 2831) and CVA6D3_VP1Br TCTCGTGAGCTACTTTCCCA (3473 – 3454) generating a product size of 660bp. Both products were amplified in 20µl reactions with HotStarTaq (QIAGEN) as per manufacturer’s recommendations, 1-2 µl hexamer-primed cDNA template and 200 µM forward and reverse primers, thermocycled as follows: 95oC for 15 min then 55 cycles of 95oC for 20 s, 55oC 20s and 72oC for 60s. PCR products of the expected size when visualised by gel electrophoresis were sequenced with primers CVA6D3_VP1Ar and CVA6D3_VP1Bf as appropriate. Sequences were basecalled using FinchTV and identity was preliminarily assessed using NCBI Standard Nucleotide BLAST (BLASTn) and the Genome Detective online Enterovirus Genotyping Tool (Version 2.9) [42].
Sequences were compared to a global reference dataset generated by retrieving all available full length VP1 lineage D sequences CVA6 (n = 4358) from GenBank in December 2023, as well as a smaller cohort to identify lineage [35]. These were aligned with the study sequences using Geneious software, and a phylogenetic tree was assembled in IQ tree 2 [43] using the SYM+I+R6 model and 1000 SH-aLRT bootstraps, then annotated using FigTree (http://tree.bio.ed.ac.uk/software/figtree/). Complete CVA6 sequences from this study are deposited on GenBank under accession numbers MZ576353 - MZ576381 and ABXXXXXX – ABXXXXXX.
Available clinical data was audited under local study number 23-005C, as approved by the Nottingham University Hospitals NHS Trust. As part of the anonymisation process of clinical data export, ages were only recorded in discrete months or years. Without formal prospective categorisation, rash type reporting was highly varied from multiple clinical sources and degrees of experience, including: Vesicular (n = 30), eczema coxsackium (n = 18), HFMD (n = 8), crusted lesions (n = 2) impetigo (n = 2), as well as macular, maculopapular, nappy rash, papular, combinations of the aforementioned or type not stated (n = 16). Therefore, this data category was reduced to the binary presence or absence of a rash for statistical investigation, and accordingly no distinction could be made between classical and atypical HFMD.
Swabs taken from any external anatomy were designated as skin swabs and those involving any sampling of the throat e.g. nose and throat as oral swabs. Any non-skin or oral cavity specimen, including whole blood, faeces, nasopharyngeal aspirates (all n = 2) and ocular swabs, bronchoalveolar lavage and sputum (all n = 1) were grouped together as ‘other’ for analyses.
Statistical analyses were performed on the available clinical data segregated by the time of sample acquisition. Samples from 2018 were compared against those sampled in 2021-2023. Contingency analyses of categorical data were performed by χ2 test, as implemented in GraphPad Prism 10.3.1.
3. Results
3.1. Epidemiology
CVA6 was identified in 33 of 143 cases (23.1%) of sequence-confirmed enteroviral diagnosis between 1st September and 17th December 2018, the second most prevalent type behind EV-D68 as we previously reported [39]. The second study period commenced on 11th May 2021 and continued to 26th April 2023, indicated CVA6 infection in 69 of 185 successfully typed cases (37.3%, the most common in this period). 19 of the study-typed CVA6 cases were independently referred for typing at the National Enterovirus Reference Laboratory and all returned a CVA6 result. Both study-identified EV-D68 & CVA6 co-infections were also confirmed by the reference lab, however 79 of the 102 total CVA6-positive cases (77.45%) were not referred for typing.
CVA6 infections were seen predominantly between August and December, but in the sole fully screened year of 2022, sporadic incidences were observed throughout. The CVA6 epidemic season of 2018 peaked in November, whereas the first post-pandemic season sampled in 2021, saw an earlier peak in October, followed by a return to a November peak in 2022. Seasonal peak and breadth were also broadly similar in each year, particularly so between 2018 and 2021, peaking at 18 and 16 cases per month respectively, with more cases in the month preceding the peak than after (Figure 1). Overall respiratory illness testing information is presented in Figure S1, indicating a pre-pandemic peak in November of 2018 coinciding with CVA6 epidemiology. Post-pandemic testing by the respiratory 16-plex capable of enteroviral detection was at a more consistent but lower level between September and December, due to an increased local focus on point-of-care-testing for SARS-CoV-2, Influenza A & B and Respiratory Syncytial virus at admission [44].
3.2. Clinical and Laboratory Characteristics
CVA6 positive patients’ clinical presentations were audited retrospectively, comparing cases recorded before and after the emergence of SARS-CoV-2 (Table 1). The retrospective clinical audit was unable to accurately distinguish between rash and according classical and atypical HFMD presentation based on non-uniform reporting, so these varied presentations were grouped together (see methods).
Strikingly, CVA6 presented much more frequently in the clinical audit with a principal diagnosis of HFMD post-pandemic (p = 0.0057, Table 1). This was further supported by the increased frequency of a symptomatic rash post-pandemic, noted in 89.9% of cases (p = 0.0106, Table 1), whereas the higher level of respiratory involvement pre-pandemic (45.5% compared to 18.8% post-pandemic, p = 0.073) tallied with the accordingly lower principal HFMD diagnosis (Table 1). Indeed, 7 of 11 non-HFMD primary diagnoses were classified as respiratory and two more involved EV-D68 co-infection.
Age at presentation was overwhelmingly in the young (84 of 102 cases, 82.35%, Table 1), but not neonatal, with just a single case under 6 months of age, whilst 12 adults were also diagnosed. There was a relatively lower proportion of cases in the under 1 year olds in conjunction with a greater proportion of 2 year olds in the post-pandemic group (Table 1 and Figure S2), which may be suggestive of a later median age of infection, but this was far from statistical significance in our dataset limited by delineation in years to further anonymise, rather than more precisely by months (p = 0.5569, table 1). Alongside the principal differences in CVA6 presentation, co-morbidities were also significantly elevated post-Pandemic (p = 0.0018, Table 1). However, this is likely to be strongly linked to the greater proportion of primary HFMD diagnoses, with 20 of 29 post-pandemic instances of co-morbidities noting eczema, a key factor for exacerbation of HFMD, all but one of which was indeed recorded principally as an HFMD presentation.
Neurological symptoms were also highlighted to be significantly more prevalent pre-pandemic, but only four patients presented as such, one with an EV-D68 co-infection diagnosed with Acute Flaccid Paralysis [39]. Other cases were in young children suffering febrile convulsions, one requiring intensive care, the other not, despite an episode of respiratory arrest. The final neurological case was less severe in a young adult experiencing parathesis in addition to a more typical CVA6 picture of respiratory symptoms, with chest pain, shortness of breath and fever. Additional unusual presentations included single instances of SIDS and also conjunctivitis (all additional clinical data not shown).
In general, the cohort indicated an imbalance towards male gender with near identical ratios of approximately 6 males to 4 females in both Pandemic eras (Table 1). Similarly, proportions of hospital admission, fever and gastrointestinal symptoms were also not significantly different pre- and post-pandemic (Table 1). Collectively, these indicated more than half of cases were assessed as outpatients, fever was only reported in around a third of patients, whilst gastrointestinal symptoms were even less frequent, circa 1 in 10 cases (Table 1). Co-infection by other routinely screened-for viruses [45] was rare (data not shown) and thus not further assessed.
3.3. Phylogenetic Analysis of CVA6 VP1 Sequences
To interrogate the genetic relationship between the CVA6 viruses causing infections during our study period, we conducted a phylogenetic analysis of the complete VP1 sequence from both our study samples and the complete available global dataset as of December 2023. The PCR amplification strategy generated full length VP1 sequence for all but 6 samples (92 of 98 CVA6 infections determined locally, with 4 determined exclusively by the National Enteroviral Reference Laboratory). Of these, three failed to amplify only with primer set A (bases 2258 - 2935), one with primer set B only (bases 2814 - 3473) and two with both primer sets (but had been confirmed CVA6 by preliminary sequencing [30]). Using available sequence data, a third base position mismatch was identified in the reverse priming site of primer set A (data not shown), however this was also common in successfully amplified samples, suggesting a further undetected mismatch in the forward primer of set A was responsible for PCR failure. Similarly, no mismatches could be identified in the forward priming region of set B, suggesting an isolated mismatch in the reverse set B primer. Two of the A amplicon failures were of low template input, as was one of the two samples that failed for both, whilst the other dual failure was the sputum sample (data not shown). Partial VP1 sequences were also of common D3 lineage (data not shown).
As expected, due to preliminary investigation and final amplification strategy (see methods), all samples were of lineage D3 (Figure S3)- accordingly, only lineage D samples were included in the full phylogenetic tree. However, within this lineage there was considerable diversity not only amongst reference sequences but also within our local contemporary sequences.
The study sequences distributed across genotype D3, were located in 11 different well-supported subgroups (Figure 2), with pre-pandemic 2018 sequences found exclusively in subgroups 4-6 and 8-11. The majority of these 2018 season sequences (21 of 29) are presented in Figure 3, alongside contemporary 2017-18 European isolates from elsewhere in the UK [46], France and Spain [13], as well as Russia, China and Venezuela. UK study sequences were not always discretely grouped with each other, in some cases showing a closer genetic relationship to geographically distant samples than each other e.g. NUH_CVA6_18 presents in a well-supported branch of three with MK252985 and MK252987 from Russia (Figure 3, subgroup 11). Another well-supported branch contains only a single 2018 study sequence, but also those from October 2021 and November 2022, alongside contemporary French and Russian samples. This subgroup is itself most closely related to an exclusively Indian subgroup (Figure 3, subgroup 10). Similarly, subgroup 10 presents an isolated 2018 study sequence (NUH_CVA6_2) on a well-supported branch with exclusively Japanese sequences from the year before, as well as two sequences (NUH_CVA6_16 and 27) which group amongst isolates from France and India [47]. Subgroup 8 (Figure S4) contains only a single 2018 study sequence on a long branch in a well-supported grouping with a 2016 Australian sample [48].
Whilst these isolated study sequences are suggestive of intercontinental transmission but lack supporting epidemiological evidence, sample NUH_CVA6_14 (Figure S5) unusually appeared on a relatively long single branch length and presented with a recent travel history to several South East Asian countries but not including China, sequences from which comprehensively outgroup the study sample. Surprisingly this patient was not only co-infected with an Enterovirus D68 strain, but this also stood distinct from contemporary UK EV-D68 genome sequences, as we have previously reported [39].
The post-SARS-CoV-2 emergence years and peak seasons of 2021 and 2022 exhibited similar patterns of diversity, with 2021 sequences predominating in subgroup 3 and 2022 in subgroup 2 (Figure 4). Each sub-group contained a vice-versa minority i.e. five 2022 sequences amongst the twenty-five 2021 sequences (subgroup 3) and a sole 2021 sequence amongst the seventeen 2022 sequences (subgroup 2). This may be suggestive of local endemic circulation and persistence of CVA6 strains across the core epidemic seasons as well as lineage displacement (August to December, Figure 1). Subgroup 2 further persisted into 2023 with 4 of the 5 infections investigated that year, albeit before the main epidemic season after the study’s end. In contrast, the dwindling subgroup 3 was not detected. Interestingly subgroups 2 and 3 were homogenously represented by our UK study sequences and out grouped at well supported nodes by 2018 Chinese strains, as were the sparser study sequences in subgroup 1 (Figure S6) in contrast to the aforementioned interspersal of reference sequences seen in subgroup 11 that contained the bulk of the 2018 infections. The smaller 2022 infection-sequenced subgroup 6 was similarly homogenous, but out grouped by a French isolate from 2017 (Figure S7). These samples were more closely related to an isolated 2018 sequence than other post-pandemic infections.
Identical sequences were observed in all cases of serially sampled patients, for example study samples 91 & 92, 104 & 105 (Figure 4). Identical sequences were also derived from different cases, for example samples 1, 4 & 8 (Figure S8) and 15 & 17 (Figure 3). However, the clinical audit indicated these were unrelated by family or immediate UK Postcode level geography and thus unlikely by direct contact.
4. Discussion
Our study covered the two sampling periods, both before and after the emergence of SARS-CoV-2 where we undertook universal in-house enteroviral typing, rather than selective referral to a reference laboratory service [30,39]. Whilst a key objective in the pre-pandemic era was to assess the impact of EV-D68 in a hospital setting [39,49], it became apparent that CVA6 was the second-most prevalent enterovirus detected and thus demanded further investigation. This finding was re-enforced when we undertook a post-pandemic pilot project to determine the genotype of all diagnosed infections. High prevalence of CVA6 amongst clinically diagnosed enteroviral infections was in agreement with other universal screening studies, most significantly and relevantly in the recent and largest study of enteroviral infections in Europe to date [4] but also in the US [50] and worldwide in general [40].
The seasonality of enterovirus infections can sometimes be oversimplified as a broadly summer infection [51], which may be true for some types but not others [4]. However, here we demonstrate that, in agreement with others, CVA6 undoubtedly peaks in the Autumn in the UK [52]. A subtle shift in transmission peak was observed in 2021, potentially due to the unprecedented non-pharmaceutical intervention measures introduced to limit SARS-CoV-2 transmission in the UK and elsewhere globally [44]. Diminished pandemic transmission of other enteroviruses has also been recorded [49] with an accompanying uncertainty about infection rates and severity as interventional measures were lifted with a generally older susceptible population [53].
Age at CVA6 diagnosis broadly followed trends reported elsewhere, with a large UK-based cohort also finding most (52%) to be 1–5 years of age and a further quarter (23%) to be between 4 and 12 months old [52], as well as a similar demographic more broadly in Europe [4]. Interestingly our hypothesized but not statistically significant study trend of children acquiring CVA6 slightly later in life in the immediately post-pandemic era was in contradiction to a similar study in the Netherlands [38].
By comparison, 2021 Respiratory Syncytial Virus infections were recorded earlier than expected and also with increased severity [54,55]. Perturbance of typical viral transmission patterns has been assessed most intensively with Influenza epidemiology [56] and observed most significantly with the lack of detection of Influenza B Yamagata lineage since March 2020 [57]. Although influenza epidemics were observed to be of lower intensity and shorter in a study of nine Asian countries in 2021 [58], our data indicated a similar size CVA6 infection burden in 2021 compared to 2018. The greater proportion of males presenting at circa 60% in each group appears to be a general feature of HFMD as this was in close agreement with a predominantly EV-A71- and CVA16-causal HFMD cohort in China [59].
Whilst our limited CVA6 cohort split across three epidemic seasons with non-uniformly recorded clinical details was unable to fully probe disease manifestations, particularly in terms of rash appearance, others have done so comprehensively in contemporary European pre-pandemic cohorts, finding an approximately 3 to 1 caseload of atypical rather than typical HFMD presentation [37]. Instead we focused on more universal parameters of infection and their variability pre- and post-pandemic, finding an elevated respiratory tract involvement in 2018, supported by findings in linked parameters with more respiratory tract sampling and lesser rash observation and HFMD primary diagnosis in this year. The pre-pandemic sample profile allies more closely with results from the broad European cohort of Bubba and colleagues, with CVA6 detected by skin swab in approximately half of all cases and secondarily in the respiratory tract in approximately a quarter [4]. Others have found CVA6 to be the most commonly detected enterovirus of the respiratory tract [60]. In another extensive, but more closely matched study set exclusively in the UK from 2006 to 2017 (>1000 CVA6 cases), the sample set comprised 73.5% skin (and vesicle) swabs and just 13.2% respiratory specimens, similar to our more disparate post-pandemic sampling [52].
The limited sampling from non-skin/oral sites and associated non-respiratory/HFMD principal diagnosis seen in our results, was reflected elsewhere [4,52], although the circa one-third febrile cases recorded in both pre- and post-pandemic groups was approximately three-fold greater than the European average [4]. However, neurological manifestations such as the febrile convulsions we saw in the smaller 2018 group have been significant proportions of other CVA6 cohorts [60]. The severe neurological complication Acute Flaccid Paralysis (AFP) was also seen in CVA6 cases in China earlier in the mid-2000s [35], although our sole case was co-infected with EV-D68, which may have been the principal causative agent [39,61]. EV-D68 and CVA6 co-infection has also been observed previously presenting classical symptomology of both enteroviruses [62]. Given that these viruses were the most prevalent in our recent surveillance pre- and post-pandemic here and elsewhere [39], occasional co-infections should accordingly be expected, as should co-infection by intra-typic strains of CVA6 that must have facilitated the extensive genomic recombination characterised in whole genome sequencing studies [27,28,37].
We confirmed the recent dominance of CVA6 genotype D3 in the UK as reported both elsewhere in continental Europe [13,37] and China [35], although our exclusively VP1 sequencing approach lacked the ability to further characterise the aforementioned genomic recombinants hypothesised to enhance its post-millennium emergence and pathogenicity [27,37,63]. However, whole genome sequencing only further exacerbates the apparent complexity of strains beyond the genotype D3 designation that was apparent in our phylogenetic analyses. Whilst we observed less intermingling between study and non-study sequences post-pandemic, this is likely to be at least in part an artefactual result of limited available reference material to date as many viral sequencing laboratories have diverted time and resources to prioritise SARS-CoV-2 studies.
In the better-sampled pre-pandemic era, considerable sub-genotype diversity and by implication labyrinthine transmission chains were indicated. Closely sampled CVA6 genomes from our regional UK centre were more closely related to those in other countries or even continents than each other in many instances, as was also the case in our related EV-D68 study [39]. The intercontinental viral transmission illustrated by groups such as ours during the emergence of SARS-CoV-2 [64], was again demonstrated with the likely import of an East Asian CVA6 sub-lineage alongside an East-Asian EV-D68 sub-lineage [39], most surprisingly in the same co-infected returning traveller. Where pre-pandemic study sequences were most closely related, or even identical in the VP1 region covered, clinical auditing indicated no obvious clues to epidemiological linkage, further emphasising hidden subclinical transmission events and the unsampled burden of infection. A study sampled in the preceding years of 2016-17 in Australia also showed a similarly mixed epidemiological picture of genetically isolated viruses, alongside clusters suggestive of endemic transmission chains across seasons [48].
Others have attempted to compare enteroviral genetic diversity pre- and post-pandemic, finding a reduction in genetic variability may have occurred with the lockdown measures introduced, although not notably in CVA6 [38]. Whilst suitably cautious, waiting for additional evidence from further complementary studies such as presented here, they equally note that genetic change arising from transmission bottlenecks may indeed become clinically relevant in the future.
In conclusion, the global epidemiology of viral infections is experiencing an unprecedented era, following a phase of restrictive measures and their subsequent relaxation, leaving an atypical global immune landscape within which to renew transmission [65]. Our study indicates this phenomenon has indeed perturbed CVA6 in the UK, with evidence of an effect on circulating lineages, their peak epidemic season and their associated clinical manifestation. The global capability in virological surveillance demonstrated during the SARS-CoV-2 pandemic could be increasingly applied to monitor the ever-changing epidemiology of enteroviral infections also [66].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Proportion of respiratory diagnostic test results in pre- and post- pandemic eras; Figure S2: CVA6 case proportion by age grouping in pre- and post-pandemic eras; Figure S3: Lineage analysis of CVA6 complete VP1 gene; Figure S4: Phylogenetic analysis of CVA6 complete VP1 subgroup 8; Figure S5: Phylogenetic analysis of CVA6 complete VP1 subgroup 4; Figure S6: Phylogenetic analysis of CVA6 complete VP1 subgroup 1; Figure S7: Phylogenetic analysis of CVA6 complete VP1 subgroup 6; Figure S8: Phylogenetic analysis of CVA6 complete VP1 subgroup 5; Figure S9: Phylogenetic analysis of CVA6 complete VP1 subgroup 7.
Author Contributions
Conceptualization, C.P.M.; Methodology, C.P.M.; Validation, C.P.M., A.J. and J.H.; Formal Analysis, C.P.M., T.T., A.T. and J.H.; Investigation, C.P.M., A.J., L.B., H.H-W., N.A., B.C., G.C. and W.I.; Resources, C.P.M. and G.C.; Data Curation, C.P.M., A.J. and S.A.; Writing – Original Draft Preparation, C.P.M., A.J. and C.J.; Writing – Review & Editing, All authors; Visualization, C.P.M., A.T., J.H. and S.A.; Supervision, C.P.M.; Project Administration, C.P.M.; Funding Acquisition, C.P.M.
Funding
This work was funded in part by a grant from the UK Clinical Virology Network.
Institutional Review Board Statement
Ethical review and approval were waived for this study, as only targeted genomic investigations on surplus acellular nucleic acids specimens of a clinically diagnosed virus were undertaken. Available clinical data was retrospectively audited by members of the pathway of clinical care team only, under study number 23-005C, as approved by the Nottingham University Hospitals NHS Trust.
Informed Consent Statement
Patient consent was waived as study audited clinical and virological parameters obtained during routine consented healthcare.
Data Availability Statement
The original sequencing data presented in the study is openly available in GenBank as described in the Materials and Methods section. The additional raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Abzug MJ. Presentation, Diagnosis, and Management of Enterovirus Infections in Neonates. Pediatric Drugs 2004; 6:1 - 10.
- Glaser C, Wilson MR. Enteroviruses: the elephants in the room. Lancet Infect Dis 2020; 20:153-5.
- Lugo D, Krogstad P. Enteroviruses in the early 21st century: new manifestations and challenges. Curr Opin Pediatr 2016; 28:107-13.
- Bubba L, Broberg EK, Jasir A, Simmonds P, Harvala H, Enterovirus study c. Circulation of non-polio enteroviruses in 24 EU and EEA countries between 2015 and 2017: a retrospective surveillance study. Lancet Infect Dis 2020; 20:350-61.
- Esposito S, Principi N. Hand, foot and mouth disease: current knowledge on clinical manifestations, epidemiology, aetiology and prevention. Eur J Clin Microbiol Infect Dis 2018; 37:391-8.
- Gonzalez G, Carr MJ, Kobayashi M, Hanaoka N, Fujimoto T. Enterovirus-Associated Hand-Foot and Mouth Disease and Neurological Complications in Japan and the Rest of the World. Int J Mol Sci 2019; 20.
- Anh NT, Nhu LNT, Van HMT, et al. Emerging Coxsackievirus A6 Causing Hand, Foot and Mouth Disease, Vietnam. Emerg Infect Dis 2018; 24:654-62.
- Li J, Zhu R, Huo D, et al. An outbreak of Coxsackievirus A6-associated hand, foot, and mouth disease in a kindergarten in Beijing in 2015. BMC Pediatr 2018; 18:277.
- Luchs A, Azevedo LS, Souza EV, et al. Coxsackievirus A6 strains causing an outbreak of hand-foot-and-mouth disease in Northeastern Brazil in 2018. Rev Inst Med Trop Sao Paulo 2022; 64:e16.
- Sinclair C, Gaunt E, Simmonds P, et al. Atypical hand, foot, and mouth disease associated with coxsackievirus A6 infection, Edinburgh, United Kingdom, January to February 2014. Euro Surveill 2014; 19:20745.
- Osterback R, Vuorinen T, Linna M, Susi P, Hyypia T, Waris M. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis 2009; 15:1485-8.
- Huang WC, Huang LM, Lu CY, Cheng AL, Chang LY. Atypical hand-foot-mouth disease in children: a hospital-based prospective cohort study. Virol J 2013; 10:209.
- Andres C, Guasch E, Pinana M, et al. Recombinant CV-A6 strains related to hand-foot-mouth disease and herpangina at primary care centers (Barcelona, Spain). Future Microbiol 2019; 14:499-507.
- Li W, Gao HH, Zhang Q, et al. Large outbreak of herpangina in children caused by enterovirus in summer of 2015 in Hangzhou, China. Sci Rep 2016; 6:35388.
- Zhao TS, Du J, Li HJ, et al. Molecular epidemiology and clinical characteristics of herpangina children in Beijing, China: a surveillance study. PeerJ 2020; 8:e9991.
- Drago F, Ciccarese G, Broccolo F, Rebora A, Parodi A. Atypical hand, foot, and mouth disease in adults. J Am Acad Dermatol 2017; 77:e51-e6.
- Broccolo F, Drago F, Ciccarese G, et al. Severe atypical hand-foot-and-mouth disease in adults due to coxsackievirus A6: Clinical presentation and phylogenesis of CV-A6 strains. J Clin Virol 2019; 110:1-6.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol 2010; 48:49-54.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis 2011; 11:346.
- Miyamoto A, Hirata R, Ishimoto K, et al. An outbreak of hand-foot-and-mouth disease mimicking chicken pox, with a frequent association of onychomadesis in Japan in 2009: a new phenotype caused by coxsackievirus A6. Eur J Dermatol 2014; 24:103-4.
- Fujimoto T, Iizuka S, Enomoto M, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg Infect Dis 2012; 18:337-9.
- Yang F, Yuan J, Wang X, et al. Severe hand, foot, and mouth disease and coxsackievirus A6-Shenzhen, China. Clin Infect Dis 2014; 59:1504-5.
- Yang X, Li Y, Zhang C, et al. Clinical features and phylogenetic analysis of severe hand-foot-and-mouth disease caused by Coxsackievirus A6. Infect Genet Evol 2020; 77:104054.
- S A, Sabeena S, Bhat KG, Bharani KC, Ramachandran S, Arunkumar G. Coxsackievirus A6 (CV-A6) Encephalomyelitis in an Immunocompromised Child: a Case Report and Brief Review of the Literature. Jpn J Infect Dis 2018; 71:388-9.
- Qian SS, Wei ZN, Jin WP, et al. Efficacy of a coxsackievirus A6 vaccine candidate in an actively immunized mouse model. Emerg Microbes Infect 2021; 10:763-73.
- Oberste MS, Maher K, Flemister MR, Marchetti G, Kilpatrick DR, Pallansch MA. Comparison of classic and molecular approaches for the identification of untypeable enteroviruses. J Clin Microbiol 2000; 38:1170-4.
- Gaunt E, Harvala H, Osterback R, et al. Genetic characterization of human coxsackievirus A6 variants associated with atypical hand, foot and mouth disease: a potential role of recombination in emergence and pathogenicity. J Gen Virol 2015; 96:1067-79.
- Simmonds P, Welch J. Frequency and dynamics of recombination within different species of human enteroviruses. J Virol 2006; 80:483-93.
- Nix WA, Oberste MS, Pallansch MA. Sensitive, seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J Clin Microbiol 2006; 44:2698-704.
- Howson-Wells HC, Winckles S, Aliker C, et al. Enterovirus subtyping in a routine UK laboratory setting between 2013 and 2017. J Clin Virol 2020; 132:104646.
- Hassel C, Mirand A, Lukashev A, et al. Transmission patterns of human enterovirus 71 to, from and among European countries, 2003 to 2013. Euro Surveill 2015; 20:30005.
- Lott JP, Liu K, Landry ML, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol 2013; 69:736-41.
- Wang J, Zhang S. Epidemiological characteristics and trends of hand-foot-mouth disease in Shanghai, China from 2011 to 2021. Front Public Health 2023; 11:1162209.
- Liu H, Zhang M, Feng C, et al. Characterization of Coxsackievirus A6 Strains Isolated From Children With Hand, Foot, and Mouth Disease. Front Cell Infect Microbiol 2021; 11:700191.
- Song Y, Zhang Y, Ji T, et al. Persistent circulation of Coxsackievirus A6 of genotype D3 in mainland of China between 2008 and 2015. Sci Rep 2017; 7:5491.
- Mirand A, Cohen R, Bisseux M, et al. A large-scale outbreak of hand, foot and mouth disease, France, as at 28 September 2021. Euro Surveill 2021; 26.
- Tomba Ngangas S, Bisseux M, Jugie G, et al. Coxsackievirus A6 Recombinant Subclades D3/A and D3/H Were Predominant in Hand-Foot-And-Mouth Disease Outbreaks in the Paediatric Population, France, 2010-2018. Viruses 2022; 14.
- Forero EL, Knoester M, Gard L, et al. Changes in enterovirus epidemiology after easing of lockdown measures. J Clin Virol 2023; 169:105617.
- Howson-Wells HC, Tsoleridis T, Zainuddin I, et al. Enterovirus D68 epidemic, UK, 2018, was caused by subclades B3 and D1, predominantly in children and adults, respectively, with both subclades exhibiting extensive genetic diversity. Microb Genom 2022; 8.
- Bian L, Wang Y, Yao X, Mao Q, Xu M, Liang Z. Coxsackievirus A6: a new emerging pathogen causing hand, foot and mouth disease outbreaks worldwide. Expert Rev Anti Infect Ther 2015; 13:1061-71.
- Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 2016; 33:1870-4.
- Kroneman A, Vennema H, Deforche K, et al. An automated genotyping tool for enteroviruses and noroviruses. J Clin Virol 2011; 51:121-5.
- Minh BQ, Schmidt HA, Chernomor O, et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 2020; 37:1530-4.
- Francis RV, Billam H, Clarke M, et al. The Impact of Real-Time Whole-Genome Sequencing in Controlling Healthcare-Associated SARS-CoV-2 Outbreaks. J Infect Dis 2022; 225:10-8.
- Bagasi AA, Howson-Wells HC, Clark G, et al. Human Bocavirus infection and respiratory tract disease identified in a UK patient cohort. J Clin Virol 2020; 129:104453.
- Majumdar M, Celma C, Pegg E, Polra K, Dunning J, Martin J. Detection and Typing of Human Enteroviruses from Clinical Samples by Entire-Capsid Next Generation Sequencing. Viruses 2021; 13.
- Yoshitomi H, Ashizuka Y, Ichihara S, et al. Molecular epidemiology of coxsackievirus A6 derived from hand, foot, and mouth disease in Fukuoka between 2013 and 2017. J Med Virol 2018; 90:1712-9.
- Cobbin JCA, Britton PN, Burrell R, et al. A complex mosaic of enteroviruses shapes community-acquired hand, foot and mouth disease transmission and evolution within a single hospital. Virus Evol 2018; 4:vey020.
- Simoes MP, Hodcroft EB, Simmonds P, et al. Epidemiological and clinical insights into the enterovirus D68 upsurge in Europe 2021/22 and the emergence of novel B3-derived lineages, ENPEN multicentre study. J Infect Dis 2024.
- Abedi GR, Watson JT, Pham H, Nix WA, Oberste MS, Gerber SI. Enterovirus and Human Parechovirus Surveillance - United States, 2009-2013. MMWR Morb Mortal Wkly Rep 2015; 64:940-3.
- Moriyama M, Hugentobler WJ, Iwasaki A. Seasonality of Respiratory Viral Infections. Annu Rev Virol 2020; 7:83-101.
- Kamau E, Nguyen D, Celma C, et al. Seroprevalence and Virologic Surveillance of Enterovirus 71 and Coxsackievirus A6, United Kingdom, 2006-2017. Emerg Infect Dis 2021; 27:2261-8.
- Messacar K, Baker RE, Park SW, Nguyen-Tran H, Cataldi JR, Grenfell B. Preparing for uncertainty: endemic paediatric viral illnesses after COVID-19 pandemic disruption. Lancet 2022; 400:1663-5.
- Cai W, Kondgen S, Tolksdorf K, et al. Atypical age distribution and high disease severity in children with RSV infections during two irregular epidemic seasons throughout the COVID-19 pandemic, Germany, 2021 to 2023. Euro Surveill 2024; 29.
- Honemann M, Thiem S, Bergs S, et al. In-Depth Analysis of the Re-Emergence of Respiratory Syncytial Virus at a Tertiary Care Hospital in Germany in the Summer of 2021 after the Alleviation of Non-Pharmaceutical Interventions Due to the SARS-CoV-2 Pandemic. Viruses 2023; 15.
- Fricke LM, Glockner S, Dreier M, Lange B. Impact of non-pharmaceutical interventions targeted at COVID-19 pandemic on influenza burden - a systematic review. J Infect 2021; 82:1-35.
- Paget J, Caini S, Del Riccio M, van Waarden W, Meijer A. Has influenza B/Yamagata become extinct and what implications might this have for quadrivalent influenza vaccines? Euro Surveill 2022; 27.
- Davis WW, Mott JA, Olsen SJ. The role of non-pharmaceutical interventions on influenza circulation during the COVID-19 pandemic in nine tropical Asian countries. Influenza Other Respir Viruses 2022; 16:568-76.
- Liu B, Luo L, Yan S, et al. Clinical Features for Mild Hand, Foot and Mouth Disease in China. PLoS One 2015; 10:e0135503.
- Lau SKP, Zhao PSH, Sridhar S, et al. Molecular epidemiology of coxsackievirus A6 circulating in Hong Kong reveals common neurological manifestations and emergence of novel recombinant groups. J Clin Virol 2018; 108:43-9.
- Messacar K, Asturias EJ, Hixon AM, et al. Enterovirus D68 and acute flaccid myelitis-evaluating the evidence for causality. Lancet Infect Dis 2018; 18:e239-e47.
- de Sousa IP, Jr. , Giamberardino HI, Raboni SM, et al. Simultaneous enterovirus EV-D68 and CVA6 infections causing acute respiratory distress syndrome and hand, foot and mouth disease. Virol J 2021; 18:88.
- Yu F, Zhu R, Jia L, et al. Sub-genotype change and recombination of coxsackievirus A6s may be the cause of it being the predominant pathogen for HFMD in children in Beijing, as revealed by analysis of complete genome sequences. Int J Infect Dis 2020; 99:156-62.
- Chappell JG, Tsoleridis T, Clark G, et al. Retrospective screening of routine respiratory samples revealed undetected community transmission and missed intervention opportunities for SARS-CoV-2 in the United Kingdom. J Gen Virol 2021; 102.
- Gomez GB, Mahe C, Chaves SS. Uncertain effects of the pandemic on respiratory viruses. Science 2021; 372:1043-4.
- Fischer TK, Simmonds P, Harvala H. The importance of enterovirus surveillance in a post-polio world. Lancet Infect Dis 2022; 22:e35-e40.
Figure 1.
Sequence-confirmed CVA6 prevalence by month. 2018 samples (red line) were only assessed between September and December, then enteroviral typing recommenced in May of 2021 (blue) and was continued through the entirety of 2022 (yellow) to the end of April in 2023 (grey).
Figure 1.
Sequence-confirmed CVA6 prevalence by month. 2018 samples (red line) were only assessed between September and December, then enteroviral typing recommenced in May of 2021 (blue) and was continued through the entirety of 2022 (yellow) to the end of April in 2023 (grey).
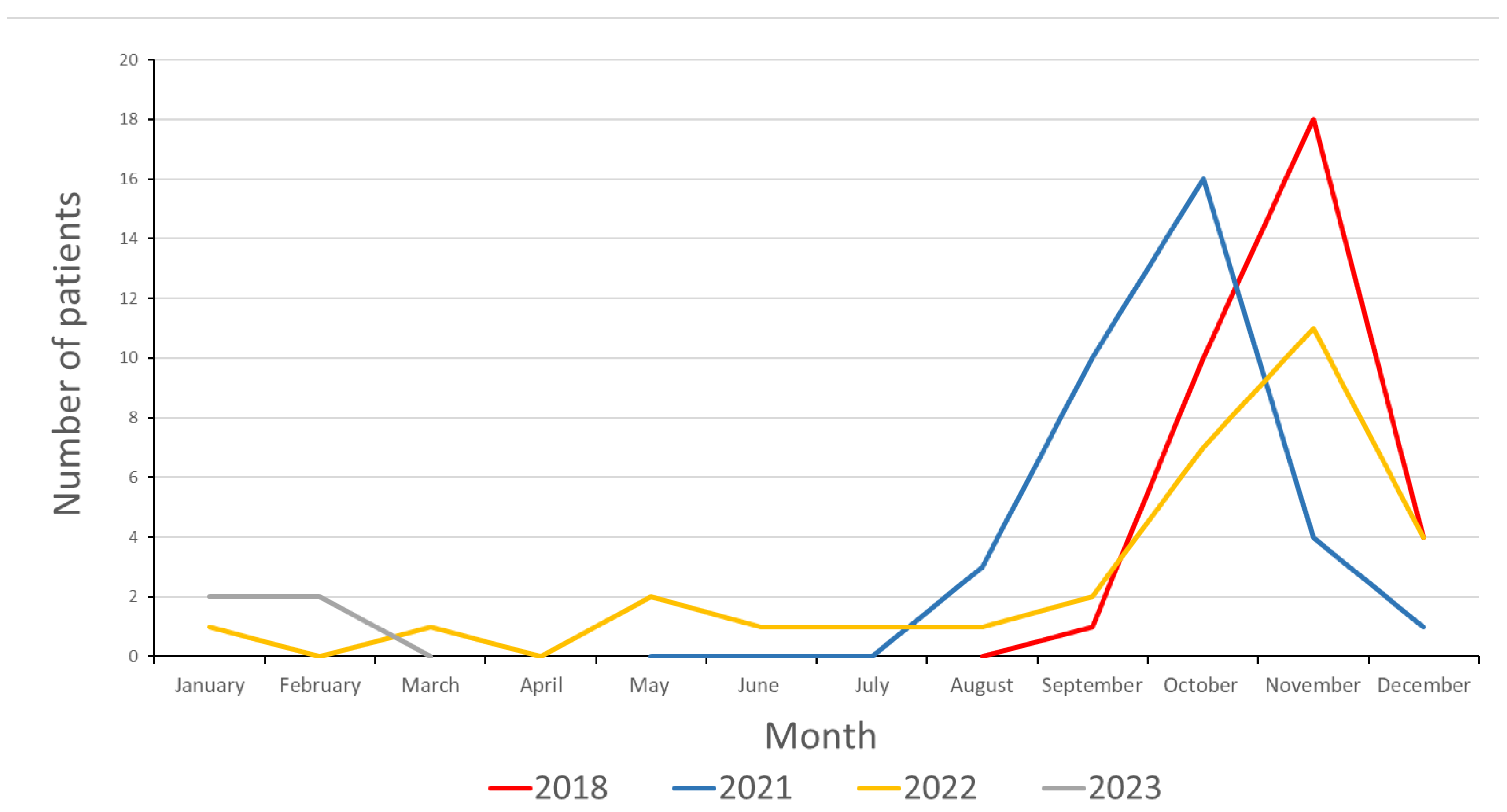
Figure 2.
Molecular phylogenetic analysis of complete CVA6 VP1 gene of study and available reference sequences. The phylogenetic tree includes 27 novel sequences from 2018 (red), 28 novel sequences from 2021 (green), 31 novel sequences from 2022 (blue), 5 novel sequences from 2023 (purple) and 4358 publicly available sequences from GenBank, representing all available lineage D sequences. The tree was constructed using the maximum likelihood method with SYM+I+R6 model in IQ-TREE2. Subgroups have been illustrated for clarity, and highlighted in later subtrees (Figure 3, Figure 4, Figures S4–S9). Bootstrapping based on 1000 replications was performed but were omitted for clarity. Tree is midpoint rooted, with a scale of 0.002.
Figure 2.
Molecular phylogenetic analysis of complete CVA6 VP1 gene of study and available reference sequences. The phylogenetic tree includes 27 novel sequences from 2018 (red), 28 novel sequences from 2021 (green), 31 novel sequences from 2022 (blue), 5 novel sequences from 2023 (purple) and 4358 publicly available sequences from GenBank, representing all available lineage D sequences. The tree was constructed using the maximum likelihood method with SYM+I+R6 model in IQ-TREE2. Subgroups have been illustrated for clarity, and highlighted in later subtrees (Figure 3, Figure 4, Figures S4–S9). Bootstrapping based on 1000 replications was performed but were omitted for clarity. Tree is midpoint rooted, with a scale of 0.002.
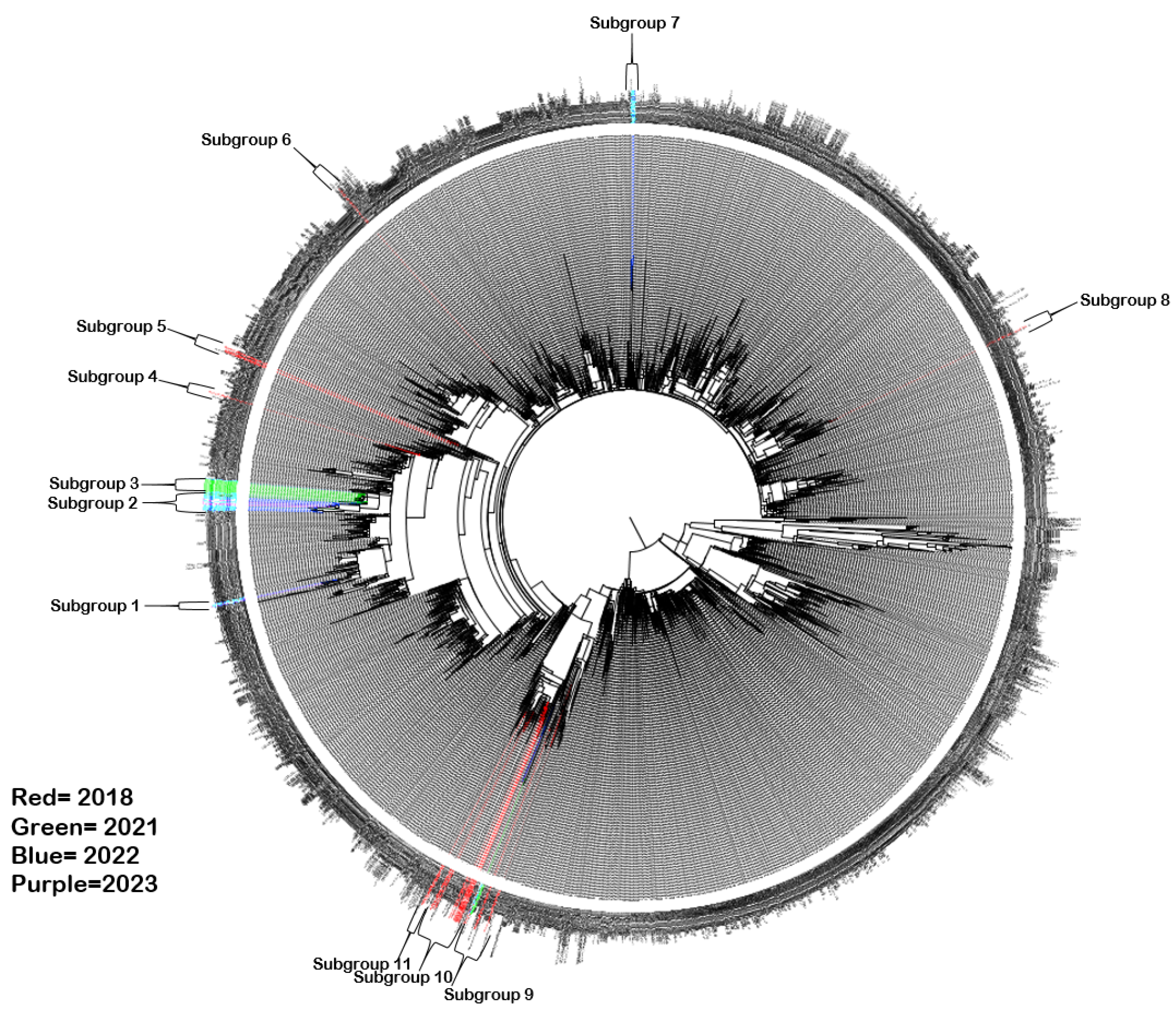
Figure 3.
Phylogenetic analysis of CVA6 complete VP1 subgroups 9-11. Molecular phylogenetic analysis of full VP1 CVA6 gene using the maximum likelihood method with SYM+I+R6 model in IQ-TREE2. Analysis was completed to include the subgroup 9, 10 and 11 sequences highlighted in Figure 2, including 21 novel sequences from 2018 (red), 2 novel sequences from 2021 (green), a novel sequence from 2022 (blue) and 133 publicly available sequences from GenBank. Where branches have been collapsed for clarity (represented by triangles), country of origin and year for all sequences has been annotated. Bootstrapping based on 1000 replications is shown next to each branch at a scale of 0.002, with bootstrap values below 70 omitted. Tree is midpoint rooted.
Figure 3.
Phylogenetic analysis of CVA6 complete VP1 subgroups 9-11. Molecular phylogenetic analysis of full VP1 CVA6 gene using the maximum likelihood method with SYM+I+R6 model in IQ-TREE2. Analysis was completed to include the subgroup 9, 10 and 11 sequences highlighted in Figure 2, including 21 novel sequences from 2018 (red), 2 novel sequences from 2021 (green), a novel sequence from 2022 (blue) and 133 publicly available sequences from GenBank. Where branches have been collapsed for clarity (represented by triangles), country of origin and year for all sequences has been annotated. Bootstrapping based on 1000 replications is shown next to each branch at a scale of 0.002, with bootstrap values below 70 omitted. Tree is midpoint rooted.
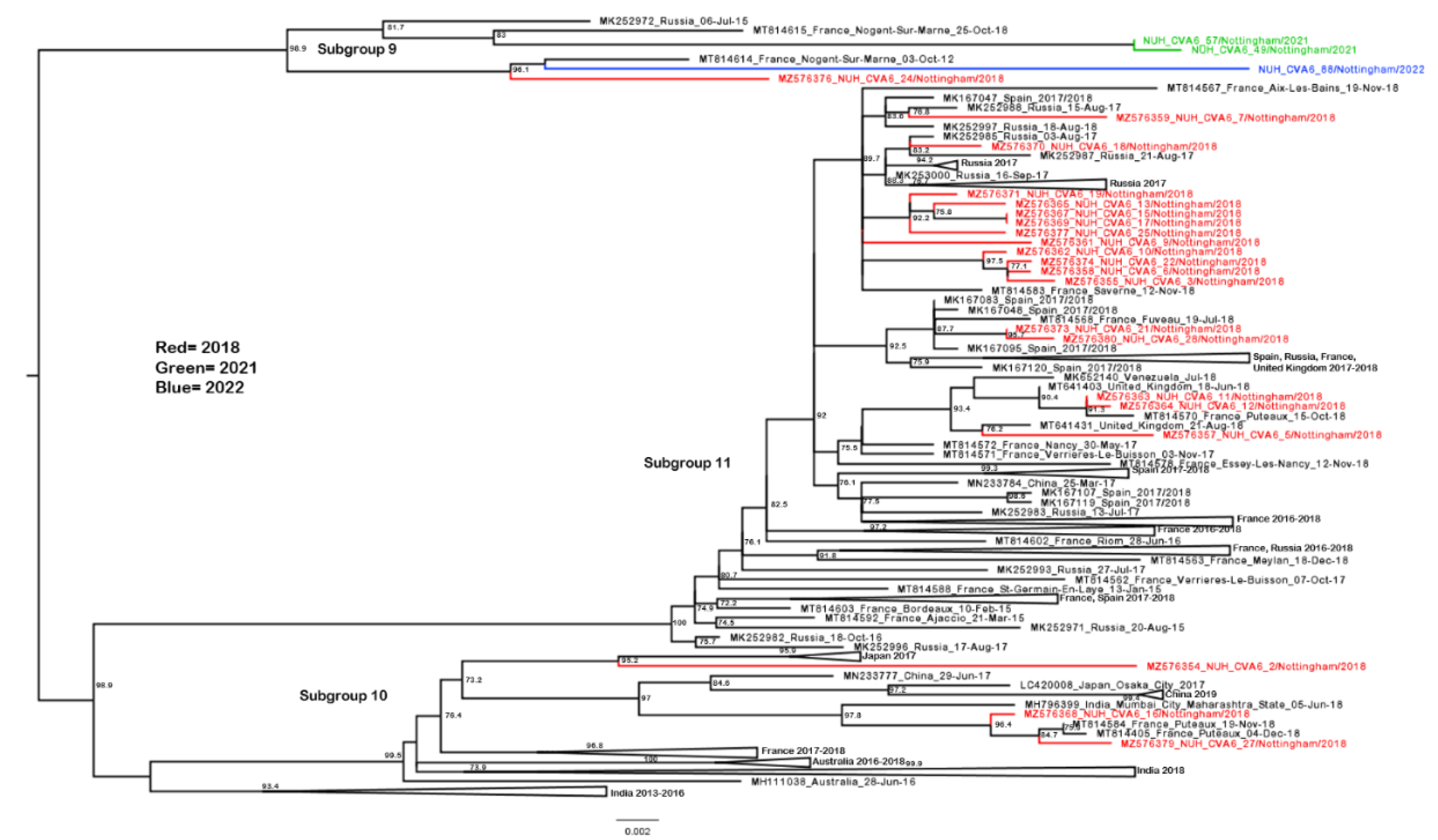
Figure 4.
Phylogenetic analysis of CVA6 complete VP1 subgroups 2 and 3. Molecular phylogenetic analysis of full VP1 CVA6 gene using the maximum likelihood method with SYM+I+R6 model in IQ-TREE2. Analysis was completed to include the subgroup 2 and 3 sequences highlighted in Figure 2, including 26 novel sequences from 2021 (green), 22 novel sequences from 2022 (blue), 4 novel sequences from 2023 (purple), and 6 publicly available sequences from GenBank. Bootstrapping based on 1000 replications is shown next to each branch at a scale of 0.002, with bootstrap values below 70 omitted. Tree is midpoint rooted.
Figure 4.
Phylogenetic analysis of CVA6 complete VP1 subgroups 2 and 3. Molecular phylogenetic analysis of full VP1 CVA6 gene using the maximum likelihood method with SYM+I+R6 model in IQ-TREE2. Analysis was completed to include the subgroup 2 and 3 sequences highlighted in Figure 2, including 26 novel sequences from 2021 (green), 22 novel sequences from 2022 (blue), 4 novel sequences from 2023 (purple), and 6 publicly available sequences from GenBank. Bootstrapping based on 1000 replications is shown next to each branch at a scale of 0.002, with bootstrap values below 70 omitted. Tree is midpoint rooted.
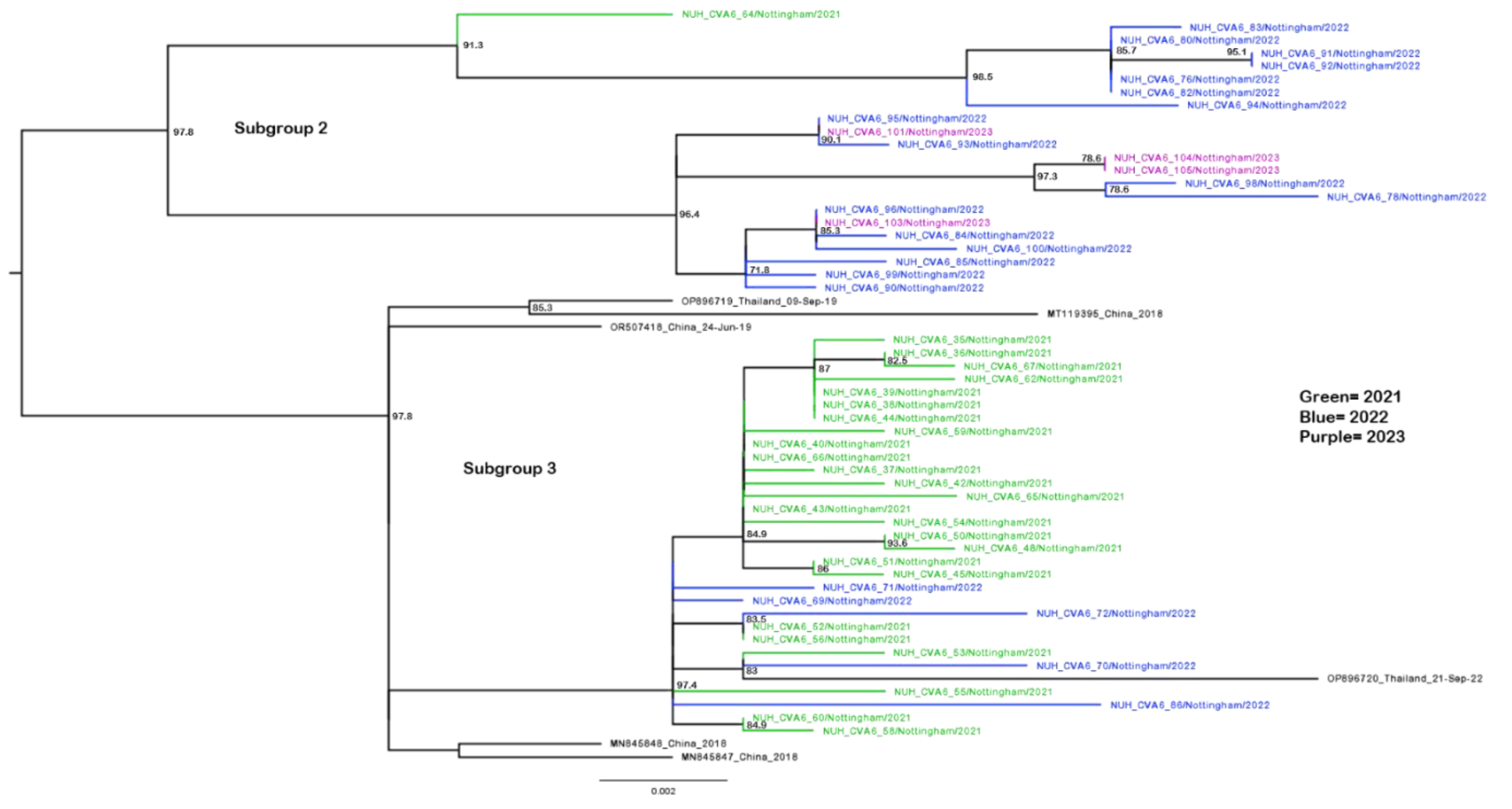

Table 1.
Clinical and laboratory data from the 102 CVA6 positive study cases.
| Pandemic era | |||
| Pre- | Post- | ||
| Cases | 33 | 69 | |
| Clinical feature | N (%) | N (%) | p value |
| Male gender | 20 (60.6) | 43 (62.3) | 0.866 |
| Hospital admission | 14 (42.4) | 24 (34.8) | 0.4552 |
| Comorbidities | 3 (9.1) | 26 (37.7) | 0.0018 |
| Neurological symptoms | 4 (12.1) | 0 (0.0) | 0.0039 |
| Fever | 13 (39.4) | 21 (30.4) | 0.4543 |
| Gastro | 4 (12.1) | 7 (10.1) | 0.8211 |
| Rash | 23 (69.7) | 62 (89.9) | 0.0106 |
| Respiratory symptoms | 15 (45.5) | 13 (18.8) | 0.073 |
| HFMD primary diagnosis | 22 (66.7) | 59 (85.5) | 0.0057 |
| Sample site | |||
| Skin swab | 15 (45.5) | 59 (85.5) | <0.0001 |
| Oral swab | 13 (39.4) | 6 (8.7) | |
| Other sample | 5 (15.2) | 4 (5.8) | |
| Age group | 0.5569 | ||
| Under 1 year | 9 (27.3) | 13 (18.8) | |
| 1 year | 15 (45.5) | 27 (39.1) | |
| 2 years | 3 (9.1) | 11 (15.9) | |
| 3 years | 0 (0.0) | 6 (8.7) | |
| 4 to 17 years of age | 1 (3.0) | 5 (7.2) | |
| Adult | 5 (15.2) | 7 (10.1) | |
* Patients were grouped into pre- (1st September and 17th December 2018, n =33) and post-pandemic (11th May 2021 to 26th April 2023, n = 69) cohorts for analysis. Categorical analyses were performed using a χ2 test, comparing each clinical feature of infection, the site of sampling, and age at time of sampling.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



