
Submitted:
30 October 2024
Posted:
31 October 2024
You are already at the latest version
Abstract
GNAO1 encephalopathies are a group of neglected genetic disorders primarily occurring due to de-novo point mutations in the Gαo protein-encoding gene in human GNAO1. This gene is reported to be highly conserved among Caenorhabditis elegans (C. elegans) and humans, with a sequence similarity of nearly 80%. Signaling pathways involved in various neurotransmitters, including GPCR pathways can be easily studied in the C. elegans model system. Therefore, using this model system to delineate downstream effectors and clinical targets to Gαo can be highly advantageous. Mutations that cause GNAO1 encephalopathy can be easily replicated in transgenic C. elegans and validated by rescuing phenotypic defects, primarily locomotion and egg-laying defects in worms. Although there are recent technical advancements in understanding the interacting proteins, there are unclear and uncertain hypotheses that explain the effect of Gαo mutations in humans. Coming to the clinical aspect of this disorder, there are no available approved diagnostic procedures to detect GNAO1 encephalopathy in the early stages of life. The present diagnostic procedures reiterate symptoms and overlap with other neurological symptoms that record neglected data of cases. Therefore, here we provide an overview of past research and a perspective of future work, with the primary objective of focusing on GNAO1 encephalopathy.
Keywords:
Caenorhabditis elegans (C. elegans)
; GNAO1 encephalopathy
; Gαo
; G-proteins
; mutations
; disorders
; neurotransmitter
; phenotype
; signaling pathway
1. Gαo Signaling Pathway Is Associated with GNAO1 Encephalopathy
G-protein coupled receptors (GPCRs) are the largest class of surface receptors, constituting about 5% of the genome. To date, humans have approximately 1000 GPCRs classified, each with a unique and distinct function [1]. The major functions of GPCRs involve mediating most of the cellular responses to taste, olfaction, and vision [2]. The prominent role of GPCRs is implicated in the fact that they bind to a variety of ligands like neurotransmitters, ions, and hormones and are majorly involved in the transmission of extracellular signals into intracellular responses. The significance of GPCRs lies in their ubiquity and diversity as they are involved in a wide range of biological functions from regulation of blood pressure and heart rate to control of immune responses and release of hormones [3]. GPCRs are coupled with G-proteins (guanosine-binding) proteins, which act as molecular switches, transmitting signals from activated cell surface receptors to intracellular proteins. The activation of G-proteins and their subsequent interaction with effector proteins mediate a wide range of cellular responses, including changes in enzyme activity, ion channel opening or closing, and modulation of intracellular signaling pathways [4]. In its usual conformation, the G-proteins exist as heterotrimeric complexes involving the Gα, Gβ, and Gγ subunits. Neurotransmitter receptors stimulate Gα subunits to exchange bound GDP for GTP. Gα-GTP then separates from the Gβγ, and these activated G-protein subunits can evoke responses in the cell. Signaling is terminated when the Gα proteins hydrolyze their bound GTP, thus returning to the GDP-bound state and re-associating with Gα (Figure 1A) [5]. G-proteins are further classified into four main families: Gαs, Gαi/o, Gαq, and Gα12, each mediating different physiological responses depending on the tissue type and signal received [6]. Among these, the Gαi/o family, Gαo (encoded by the GNAO1 gene) is the most abundant G-protein found in brain tissue and controls both the development and adult physiology of the brain. It is particularly crucial for regulating neuronal signaling, specifically in neurodevelopment and synaptic transmission [7]. Recently, Gαo has been characterized as an inducer of neuronal differentiation [8]. In C. elegans, the homolog of the human Gαo protein is GOA-1. This protein plays a vital role in inhibiting neurotransmitter release by negatively regulating synaptic vesicle exocytosis [9]. GOA-1 acts via several signaling pathways, including those involving diacylglycerol (DAG) and protein kinase C (PKC), modulating locomotion and egg-laying behaviors in C. elegans [10]. Mutations in GOA-1 result in hyperactive neurotransmission that is reflected by hyperactive locomotion and increased egg-laying behavior due to dysregulated synaptic vesicle cycling [11]. For the other Gα subunits, the downstream effectors molecules are established using a combination of forward genetics and biochemical analysis. However, Gαo remains the only type of Gα protein in higher eukaryotes for which an effector has not been identified (Figure 1B). This is a remarkable gap in our knowledge since Gαo constitutes 1% of the total membrane protein in the brain, making it orders of magnitude more abundant than other neural G-proteins [12]. Over the past two decades, many unsuccessful attempts have been made to classify the effector molecules (signaling proteins) for Gαo. The challenge in identifying Gαo effectors through forward genetics could be attributed to several factors. First, the redundancy of multiple functionally similar effectors may mask the effects of mutations in single genes, making it difficult to observe changes in Gαo signaling [13]. Second, if a Gαo effector is vital for survival or reproduction, mutations could result in lethality or impair recovery of such mutants in genetic screens [14].
The human GNAO1 gene encodes Gαo, the α subunit of heterotrimeric G-proteins that play important neuro-modulatory functions by coupling with various GPCRs, including dopamine, serotonin, and opioid receptors. [15]. Although various signaling events in which Gαo is involved have been described, systematic actions of Gαo in the nervous system have not been fully understood. Numerous de-novo GNAO1 mutations are associated with neurodevelopmental disorders, collectively termed GNAO1 encephalopathy-related disorders or GNAO1 encephalopathy, which mainly include developmental and epileptic encephalopathy 17 (also called early infantile epileptic encephalopathy) (DEE) and neurodevelopmental disorder with involuntary movements (NEDIM) [16]. GNAO1 encephalopathy has a broad, emerging phenotypic spectrum. One core phenotype is weakened movement which can be a characteristic of chorea, dystonia, and dyskinesia, and other altered phenotypes can feature epilepsy and developmental delay. [17]. Evaluation of GNAO1 disorder-associated mutations in mice has reiterated some of the phenotypes of GNAO1 encephalopathy, including impaired movement and seizure susceptibility. Testing pathological GNAO1 mutations in other model organisms will be valuable in assessing the conserved functional effects of these genetic perturbations, and their influence on movement. Gαo is highly conserved in invertebrates including the nematode C. elegans where its ortholog, G-protein o-alpha subunit (GOA)-1, regulates locomotion and egg-laying circuits [14,18]. The extremely well-defined genetics of GOA-1 in C. elegans make this an ideal in-vivo system for evaluating the functional impacts of pathological GNAO1 mutations. To date, efforts to characterize pathological mutations of GNAO1 at the molecular level have yielded promising, but conflicting results in mammalian and rodent systems. An early study evaluated a pertussis toxin-insensitive version of Gαo using a heterologous cell-based assay and placed these pathological mutations in three categories: loss of function, gain of function, and normal function [14]. Recent evaluation of rescued Gαo indicated that GNAO1 mutations result in loss of function with several mutations reported to antagonize transduction of GPCR signals by acting as dominant negatives [15]. Particularly notable are differing in vitro results with G203R, R209C, and the less well-characterized G42R mutation. These were initially described as gain of function or normal function and were subsequently found to be loss of function and dominant negative. As a result of these differing conclusions, the functional effects, and mechanisms of GNAO1 pathological mutations remain unresolved. Intense interest has emerged in understanding Gαo function in the nervous system and developing intervention strategies for GNAO1 encephalopathy. Thus, there is a persuasive need to use in-vivo models to study the behavioral impact of GNAO1 disorder-associated mutations. Here, we highlight the importance of studying GNAO1 mutations and effectors of Gαo, in the context of GNAO1 encephalopathy using C. elegans as the model system.
2. Current Status of GNAO1 Encephalopathy
2.1. GNAO1 Encephalopathy: Current Treatments and Clinical Insights
GNAO1 encephalopathy remains considerably rare with only around 400 annual cases reported globally, according to the latest data [19]. However, the number is possibly an underrepresentation of cases, especially from developing countries due to a lack of awareness and diagnostic challenges on genetic diseases. There is no available cure for GNAO1 encephalopathy-related disorders [20]. Investigation on GNAO1-related disorders has identified that deep brain stimulation treatments are efficacious, yet they pose the risk of surgical procedures and failure of medical machinery [21]. A case study by Weihao and his colleagues in 2022 suggested the efficacy of the drug oxcarbazepine in treating GNAO1-related movement disorders. Oxcarbazepine is conventionally used for the treatment of epilepsy as oxcarbazepine is a sodium ion channel blocker. Administration of drugs such as tiapride hydrochloride, phenobarbital, benzodiazepines, and various hormones were found to be ineffective in treating GNAO1-associated movement disorders. Oxcarbazepine is a potent drug to treat movement disorders caused by GNAO1 mutation which can treat abnormalities like rigidity and twisting of limbs and trunk or chorea particularly in the variant p.Glu237Lys [20]. Clinical investigation performed by Ananth et al., identified the use of neuroleptics and tetrabenazine treatment (commonly used against symptoms of chorea) for 6 patients facing continuous recurring missense mutation of GNAO1, analyzed by whole exome sequencing, is only useful to a limited degree as the high dosage treatment only worsened the symptoms of chorea in these patients [22]. Another investigation carried out by Danti et al. 2017 on 7 patients suffering from de-novo missense and splice site GNAO1 mutations, identified by next-generation sequencing highlighted that tetrabenazine was moderately controlled dyskinesia for 2 patients. One patient experienced drug-resistant seizures, and the other five had adequately controlled epilepsy. One patient’s life was saved with emergency deep brain stimulation (DBS) [23]. The other emerging area of treatment for genetic mutations is gene therapy where the RNAi approach has been widely explored. RNA interference is the biological process that involves the silencing of gene expression mediated by the formation of double-stranded RNA in the system. This mechanism for the first time was discovered in the C. elegans model system [24]. RNAi as a potential approach to gene therapy was described way back in 2003 [25]. For the first time, evidence supporting the successful RNAi-based gene therapy has been shown by targeted nanoparticles in human cancerous cell lines [26]. These advancements have paved the way for RNAi-based therapeutic approaches for genetic diseases causing neuronal abnormalities like epilepsy and movement disorders. One such approach involves the strategy “Silence-and-replace” mechanism where shRNA mediated by Adeno-Associated Vector (AAV)-DJ serotype vectors in primary mouse neuronal cultures that resulted in suppression of endogenous Gαo [27] RNAi as a therapeutic approach to GNAO1 encephalopathies is an active area of research in GPCR biology.
2.2. GNAO1 Encephalopathy: Mutation Analysis and Phenotypic Defects
Recent studies suggest that silencing the pathogenic variant at the genetic level can prevent phenotypic defects [28]. All the major mutations occurring in GNAO1 have been represented in Figure 2A which fall under two categories of neurodevelopmental disorders, i.e., NEDIM and DEE17. Among these, studies indicate the occurrence of more frequent G203R, R209C, or E246K mutations in GNAO1 encephalopathies [29]. It has been shown in mice models that contain G203R pathogenic mutant of GNAO1 are neonatally lethal even in heterozygous conditions [30], but the same heterozygous variant has been successfully silenced using RNA inference in cell-based assays [28]. The phenotypic defects of GNAO1 encephalopathies in mouse models are evident in the later stages of their life while in humans, it is pediatric. The mutations are further classified as Loss of function and Gain of Function, affecting the levels of active Gαo. As discussed in Figure 2B, the defects in humans and mice are indistinguishable making it difficult to assess the outcomes of the mutations. In contrast, C. elegans model system, the phenotypic defects are evident with distinguishable locomotory and egg-laying defects. These findings collectively suggest the need for a simpler yet conserved model system to find the potential treatment for GNAO1 encephalopathy. C. elegans has proved to be an important in-vivo system for assessing the functional genetic effects of GNAO1 pathological mutations. Prior research has shown evidence that C. elegans is useful in investigating the molecular genetic basis of neurodevelopmental disorders [31,32,33,34]. Indeed, C. elegans could be an ideal tool for functionally evaluating the increasing number of GNAO1 pathological variants identified to date.
3. C. elegans as an Advanced Model System to Study GNAO1 Mutations
GNAO1 mutations remain highly understudied because of the non-availability of human subjects due to the extremely low number of reported cases and technical difficulties, permissions, and ethical concerns associated with obtaining human samples. To mitigate the mutations responsible for GNAO1 encephalopathy, researchers have traditionally been using rodent models, particularly mice [30]. There were reported studies of unsuccessful attempts in mice models to study GNAO1-related disorders [35]. Further, in the quest to find a simpler model system, an attempt was made to replace Drosophila melanogaster’s Gαo with human GNAO1 which led to the production of an inaccurate nucleotide sequence of the gene. While approximately 75% of human genes associated with diseases have counterparts in Drosophila, the proteins they produce differ, which may limit the translation of findings from the Drosophila model to human conditions, particularly in drug discovery [36]. These reports establish the need for the use of the alternative model system with the ease of genetic manipulation and evident phenotypic defects. C. elegans has been established as a valuable experimental model for understanding GNAO1-related disorders and exploring potential treatments [37]. The researchers created two genetically modified strains with mutations at key positions Glu246 and Arg209, known to be crucial in Gαo. These loss-of-function (LOF) mutations caused varied reductions in Gαo-mediated signaling, resulting in excessive neurotransmitter release from different neuron types. This led to hyperactive behaviors like increased egg laying and movement [38]. Notably, single-copy mutations demonstrated cell-specific dominant-negative effects, determined by the specific altered residue. Similar to earlier mutants S47G and A221D, caffeine effectively mitigated the excessive movement in animals with the R209H and E246K mutations, implying caffeine’s independent action from the mutation type. In contrast, the adenosine receptor antagonist Istradefylline worked in R209H animals but not in E246K worms, suggesting caffeine’s multifaceted mode of action [37]. Overall, these findings deepen our understanding of the disease mechanisms and provide more evidence for caffeine’s potential efficacy in managing dyskinesia linked to GNAO1 loss of function mutations. In contrast, models involving the gain-of-function (GOF) mutation of Gαo, [G203R] or Gαo [R209C] in striatal neurons led to impaired locomotor behavior in mice which provides insights into G protein functioning and its mutants [39], but its relevance as a disease model is still not clear. Interestingly, hyperactivity seen in C215Y/+ mice is associated with the motor cortex rather than anatomical defects in the striatum [30]. These findings underscore the complexity of modeling GNAO1 encephalopathy and the importance of accurately replicating the disease phenotype in animal models for meaningful research and therapeutic development. C. elegans provides an excellent opportunity to define the genetic mechanisms by which GNAO1 variants affect locomotor behavior and could be key for resolving outstanding mechanistic issues about the molecular pathology of GNAO1 encephalopathy. Moreover, C. elegans has the potential to be developed as an in-vivo platform capable of evaluating large numbers of GNAO1 variants and could be used for genetic and small molecule screens targeting GNAO1. In C. elegans mutations associated with disorders, tested by CRISPR editing of the native GOA-1/Gαo locus, lead to abnormal locomotor behavior also same result was found in previous research in rodents and suggests emerging common principles governing how GNAO1 pathological variants impact Gαo, function in live organisms. The similarities between locomotor behaviors in C. elegans and rodent models make this correspondence quite reasonable. Both are sensitive to dopaminergic modulation, the C. elegans motor circuit comprising excitatory cholinergic and inhibitory GABAergic motor neurons, and GABAergic striatal neurons (dMSNs and iMSNs) responsible for motor coordination in mice [40,41,42,43]. In both cases, loss of Gαo function results in hyperactive locomotion. Furthermore, Gαo, signaling serves to inhibit neuronal activity in both C. elegans and mammals [44,45]. These conserved features have proven advantageous in the profiling of GNAO1 variants.
The spectrum of GNAO1 encephalopathy is wide and continually evolving. As mentioned previously, the central aspect of this condition is compromised motor function, along with epilepsy and developmental delay which are prevalent features of this phenotype. Gαo being a major G-protein, is expressed across many brain regions including the cerebellum wherein it is cellularly localized within the Purkinje fibers, basket, and stellate cells which inhibit GABAergic interneurons and Golgi neuronal cells. It forms connections with various crucial G protein-coupled receptors (GPCRs) such as GABA-β, α2 adrenergic, adenosine A1 (A1R), and dopamine D2 (D2R) receptors. These receptors are essential for controlling neurotransmitter release, movement, and neural development functions. There are numerous postulated downstream targets in the signaling pathway of Gαo, as well as the other members of the Gα family. Many of the targets affected by Gαo signaling are also implicated in disorders related to movement. Mutations in other signaling molecules remain understudies but are of great importance. For example, genetic mutations in ADCY5, the gene responsible for encoding adenylyl cyclase type 5, have been identified in patients with dyskinesia and dystonia [46,47]. Acknowledging the significant diversity in clinical manifestations is crucial to comprehend the molecular mechanisms behind GNAO1-related disorders. This diversity encompasses early-onset epileptic encephalopathy, as indicated by [17], as well as patients with intricate movement disorders, some accompanied by epilepsy [22]. The prevailing characteristics among patients harboring GNAO1 mutations are hypotonia and developmental delay irrespective of their clinical presentation or biochemical traits. After these, choreoathetosis and dystonia are the subsequent most frequent observations [17,48,49]. While a notable proportion of individuals exhibit atypical EEG or MRI results, fewer than fifty percent of those with GNAO1 mutations displayed distinctly aberrant EEG patterns, primarily among patients with epilepsy and loss-of-function (LOF) mutations. This diversity, encompassing variations in clinical presentation and impact on brain structure/function, suggests an involvement of both neurodevelopmental changes and disruptions in functional signaling. The latter factor seems to be more prominent in patients with gain-of-function (GOF) mutations, who exhibit fewer indications of structural brain abnormalities and display partial positive responses to drug interventions. The potentially uncertain underlying factors contributing to GNAO1-related movement disorders can be elucidated by examining GNAO1 signaling. One potential pathway involves the canonical inhibition of cAMP by Gαo, which can be facilitated by Gαo or the liberated Gβγ [50,51]. Notably, mutations in ADCY5, responsible for encoding an AC protein that generates cAMP, also lead to movement abnormalities in human patients. The disruption of cAMP signaling has been linked to impaired brain function [52]. Consequently, disturbances in cAMP levels could perturb the delicately balanced neurodevelopmental system suggesting the operation of Gαo via the cAMP pathway might be important in movement disorders, yet the downstream effectors are not studied [48].
A second theoretical foundation for GNAO1-associated movement disorders pertains to Gαo’s involvement in governing neurotransmitter release. The deficiency of crucial neurotransmitters like catecholamines (such as dopamine, epinephrine, and norepinephrine) and serotonin has been extensively studied in the context of movement disorders or seizures [53]. Gαo’s presynaptic role in regulating neurotransmitter release presents another potential avenue in the understanding of movement disorder etiology.
A third conceivable perspective, centered on developmental considerations, involves potential alterations in the maturation of neurons, a process vital during appropriate stages of neurological development. Consequently, children with developmental abnormalities might display irregular behaviors. Notably, most individuals with GNAO1-linked movement disorders also suffer from significant developmental delays. Morphologically, MRI scans may reveal widespread atrophy and delayed myelination. In general, genetic factors account for around 40% of cases involving developmental delay, including intellectual disability [54]. Control over cAMP levels and neurotransmitter release can influence ongoing neural functions as well as neurological development. Within this framework, GNAO1-associated movement disorders could arise from disruptions in either or both processes. The former scenario would likely be more amenable to therapeutic interventions compared to the latter.
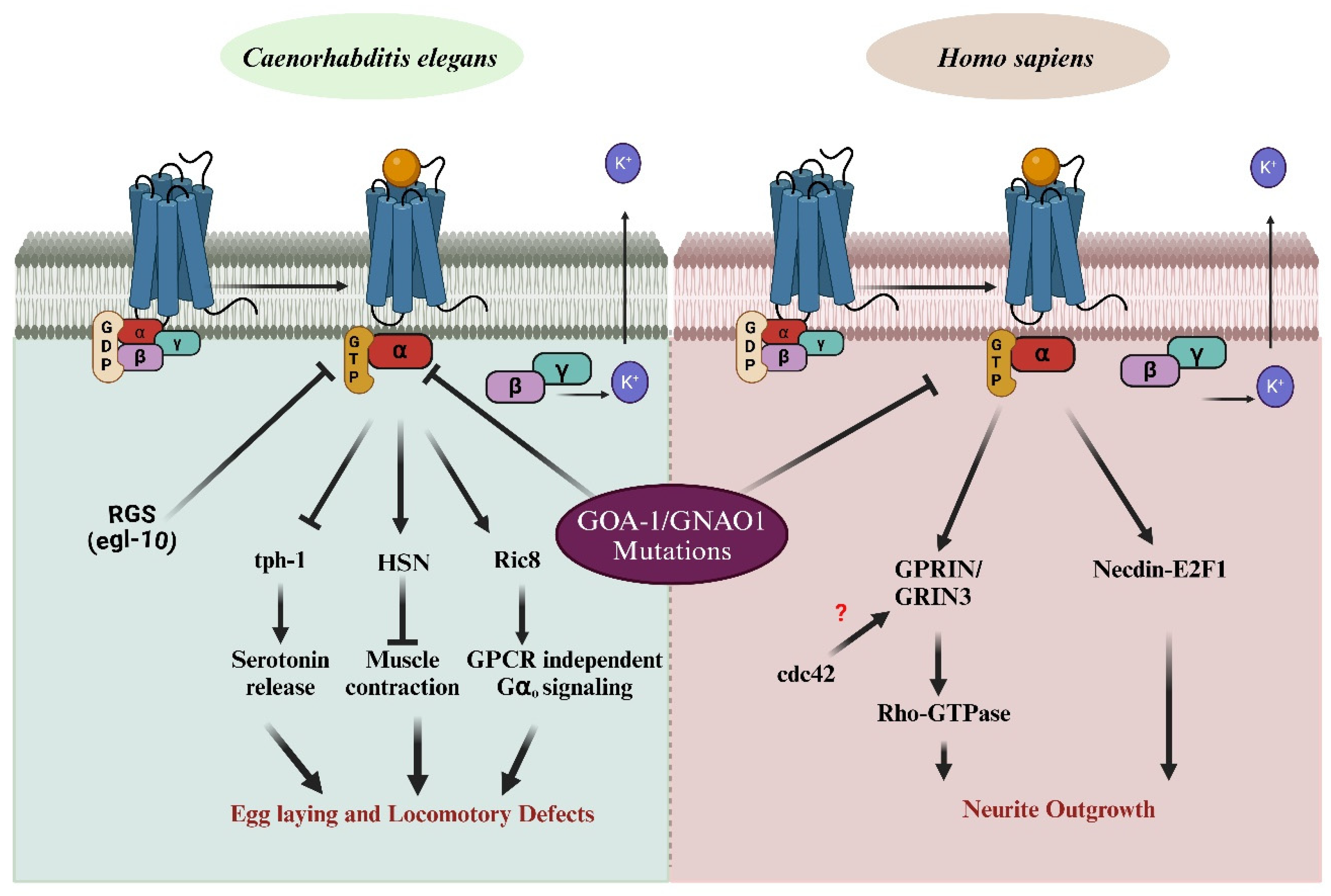
The conserved nature of G-protein signaling across species enables us to use the C. elegans model system, while also highlighting the specific downstream effectors unique to each organism. GNAO1 mutations significantly impair normal biological functions, manifesting as distinct phenotypes—locomotory defects in C. elegans and defective neuronal development in humans. Understanding these pathways can provide critical insights into the molecular mechanisms underlying GNAO1-related diseases and may offer therapeutic targets for treating associated disorders. In C. elegans, the Gαo protein is involved in several signaling cascades. Upon activation, Gα dissociates from the Gβγ subunits, initiating a downstream signaling cascade that regulates key biological processes. These include serotonin release via tph-1, muscle contraction through the hermaphrodite-specific motor neuron (HSN) [55], and GPCR-independent signaling through Ric8 [56], which mediates Gα signaling even in the absence of receptor activation. The regulator of G-protein signaling (RGS) protein, egl-10, modulates Gα activity by accelerating GTP hydrolysis [57], returning Gα to its inactive state bound to GDP. Mutations in GNAO1 disrupt these pathways, resulting in egg-laying and locomotory defects in C. elegans, highlighting the critical role of G-protein signaling in neural and muscular functions. In Homo sapiens, mutations in GNAO1 are linked to neurological disorders, including epilepsy and movement disorders. Similar to C. elegans, Gα dissociates from Gβγ upon activation by GPCRs. However, in humans, GNAO1 mutations impact neuronal signaling pathways involved in neurite outgrowth. Specifically, these mutations influence Rho-GTPase signaling, which is modulated by GPRIN (G-protein-regulated inducer of neurite outgrowth) [58]. These proteins possibly interact with GNAO1 and Rho-GTPase via the CDC-42 pathway [59], which regulates cytoskeletal dynamics essential for neurite outgrowth and neuronal morphology. Additionally, the interaction between GNAO1 and Necdin-E2F1 modulates cell cycle regulation, further affecting neuronal differentiation [60]. Defects in these pathways due to GNAO1 mutations result in impaired neurite development, contributing to the observed neurological phenotypes in patients. (Figure 3). A clear picture of all the events is still unknown, opening a plethora of opportunities in the field of GNAO1 biology.
4. Conclusion and Perspective
The study of GNAO1 encephalopathy, a genetic disorder caused by mutations in the GNAO1 gene, which encodes for the Gαo subunit of G-proteins, presents significant challenges due to the complexity of G-protein-coupled receptor (GPCR) signaling pathways in the nervous system [61]. Gαo, one of the most abundant membrane proteins in the brain, plays a crucial role in neuro-modulation by interacting with various GPCRs, such as dopamine, serotonin, and opioid receptors [39]. This genetic disorder manifests in patients as a broad spectrum of symptoms, including developmental delays, epilepsy, involuntary movements, and other motor dysfunctions [62]. The exact mechanisms underlying these phenotypes remain poorly understood, and while mammalian models have provided some insight into the effects of GNAO1 mutations, they often present conflicting results, making it difficult to derive conclusive findings. In this context, C. elegans a nematode with a well-conserved G-protein signaling pathway similar to that in humans, has emerged as an ideal in-vivo model for studying the functional impacts of GNAO1 mutations. C. elegans Gαo (goa-1) shares approximately 80% sequence similarity with the human GNAO1 gene [63], making it a valuable tool for assessing the consequences of mutations on locomotion, neurotransmitter release, and other nervous system functions.
Several advantages contribute to C. elegans being the preferred model system for GNAO1 research. The simplicity of its neural circuit, coupled with its highly conserved genetic pathways, allows researchers to effectively replicate the mutations observed in human GNAO1 encephalopathy. These mutations can be introduced into C. elegans using CRISPR editing, enabling precise study of their effects on behavior, such as locomotion and egg-laying, which mirror the impaired movement and motor dysfunctions seen in human patients [38,64]. Additionally, C. elegans’ rapid life cycle and ease of genetic manipulation facilitate large-scale screenings of GNAO1 variants, providing a high-throughput platform for studying the functional impact of these mutations. Previous studies in rodent models have confirmed the involvement of Gαo in movement disorders, but C. elegans offers the added benefit of being able to dissect the molecular mechanisms of these mutations with greater precision and at a faster pace. For example, recent research using C. elegans has demonstrated that mutations in key residues, such as Glu246 and Arg209, lead to hyperactive behaviors like excessive movement and egg-laying, reflecting the overactive neurotransmitter release seen in patients with GNAO1 mutations [38]. The ability to observe these phenotypic changes in real time makes C. elegans an invaluable model for understanding how specific mutations disrupt Gαo-mediated signaling pathways.
In terms of therapeutic research, C. elegans has proven instrumental in identifying potential treatments for GNAO1 encephalopathy. For instance, studies have shown that caffeine can mitigate the hyperactive phenotypes caused by certain GNAO1 mutations, offering a potential avenue for pharmacological intervention. This finding highlights the multifaceted nature of caffeine’s action, as it was effective across different mutations, suggesting that it may act on multiple pathways involved in Gαo signaling [21]. Similarly, drugs like oxcarbazepine, traditionally used to treat epilepsy, have shown promise in managing movement disorders associated with GNAO1 mutations, further emphasizing the utility of C. elegans in pre-clinical drug screening [20]. These findings underscore the potential of C. elegans not only as a model for understanding the genetic basis of GNAO1 encephalopathy but also as a platform for discovering novel therapeutic approaches. Beyond its use in studying movement disorders, C. elegans offers a unique opportunity to investigate the role of GNAO1 mutations in epilepsy, a common feature of GNAO1 encephalopathy. Although much of the research has focused on the motor dysfunctions caused by GNAO1 mutations, seizures are a significant and often life-threatening symptom of this disorder [65]. C. elegans’ well-characterized nervous system and responsiveness to various neurotransmitters, including GABA and dopamine, make it an ideal system for studying the mechanisms of seizure induction and progression. This model has already been used to study seizure-like activity in other genetic disorders, and its application to GNAO1 research could provide new insights into how these mutations increase seizure susceptibility. While C. elegans provides a powerful model for studying GNAO1 encephalopathy, there remain significant challenges in fully elucidating the downstream pathways through which Gαo mutations exert their effects. The pathways involving GNAO1 are intricate, with numerous interactions between GPCRs, G proteins, and their downstream effectors, many of which remain undefined. Moreover, the clinical diagnosis of GNAO1 encephalopathy is often delayed due to the overlap of symptoms with other neurological disorders, further complicating efforts to understand the full spectrum of this disease. Future research in C. elegans will need to focus on mapping these pathways and identifying specific molecular targets that could be used for early diagnosis and intervention. In conclusion, C. elegans serves as an invaluable model for studying the molecular and phenotypic consequences of GNAO1 mutations. Its conservation of Gαo signaling pathways, combined with its genetic tractability, provides a unique platform for both basic research and therapeutic development. By utilizing C. elegans researchers can gain deeper insights into the mechanisms underlying GNAO1 encephalopathy and develop targeted treatments that address the complex interplay of motor dysfunctions, seizures, and developmental delays characteristic of this disorder. As research continues to advance, C. elegans will undoubtedly play a crucial role in unraveling the mysteries of GNAO1-related neurological disorders and improving outcomes for affected individuals.
Author Contributions
Review Structuring- S.K, S.Y, S.S.V; Literature Searching- S.Y, S.S.V, S.B, S.G, S.N, S.N.B; Manuscript Draft- S.K. S.Y, S.S.V, S.B, S.G, S.N, S.N.B; Manuscript Critical Revision- S.K, Figure Preparation- S.S.V, S.Y.
Acknowledgments
We heartily thank our lab members Ajay Pradhan, Debolina Sarkar, Vandna Maurya, and Lavan Bansal for their critical evaluation and valuable feedback on the manuscript. S.K.’s laboratory is funded by the Department of Biotechnology, India (BT/PR38584/MED/122/247/202), Department of Science and Technology (DST), India (CRG/2021/000732) and DBT/Wellcome Trust India Alliance (IA/I/22/2/506480). S.Y is supported by the ICMR-Junior Research Fellowship.
Conflicts of Interest
The authors declare no competing interests.
Abbreviations
- ADCY5—Adenylyl cyclase type 5
- CRISPR—Clustered Regularly Interspaced Short Palindromic Repeats
- DAG—Diacylglycerol
- DBS—Deep brain stimulation
- DEE-17—Developmental and epileptic encephalopathy-17
- EEG—Electroencephalography
- GABA—Gamma-aminobutyric acid
- GDP- Guanosine diphosphate
- GNAO1—G-protein subunit alpha-O-1
- GPCR—G-protein-coupled receptor
- GTP- Guanosine triphosphate
- HSN—Hermaphrodite-specific motor neuron
- MRI—Magnetic resonance imaging
- NEDIM—Neurodevelopmental disorder with involuntary movements
- PKC—Protein kinase C
- RGS—Regulator of G-protein signaling
- RNAi—RNA interference
References
- Zhang M, Chen T, Lu X, Lan X, Chen Z, Lu S. G protein-coupled receptors (GPCRs): advances in structures, mechanisms, and drug discovery. Signal Transduct Target Ther. 2024 Apr 10;9(1):88. [CrossRef]
- Clapham DE, Neer EJ. G protein beta gamma subunits. Annu Rev Pharmacol Toxicol. 1997; 37:167-203. [CrossRef]
- Villaseca S, Romero G, Ruiz MJ, Pérez C, Leal JI, Tovar LM, Torrejón M. Gαi protein subunit: A step toward understanding its non-canonical mechanisms. Front Cell Dev Biol. 2022 Aug 24; 10:941870. [CrossRef]
- Wickman K, Clapham DE. Ion channel regulation by G proteins. Physiol Rev. 1995 Oct;75(4):865-85. [CrossRef]
- Jiang M, Bajpayee NS. Molecular mechanisms of Go signaling. Neurosignals. 2009;17(1):23-41. [CrossRef]
- Hepler JR, Gilman AG. G proteins. Trends Biochem Sci. 1992 Oct;17(10):383-7. [CrossRef]
- Wettschureck N, Offermanns S. Mammalian G proteins and their cell type-specific functions. Physiol Rev. 2005 Oct;85(4):1159-204. [CrossRef]
- Ju H, Lee S, Kang S, Kim SS, Ghil S. The alpha subunit of Go modulates cell proliferation and differentiation through interactions with Necdin. Cell Commun Signal. 2014 Jul 10; 12:39. [CrossRef]
- Mendel JE, Korswagen HC, Liu KS, Hajdu-Cronin YM, Simon MI, Plasterk RH, Sternberg PW. Participation of the protein Go in multiple aspects of behavior in C. elegans. Science. 1995 Mar 17;267(5204):1652-5. [CrossRef]
- Ségalat L, Elkes DA, Kaplan JM. Modulation of serotonin-controlled behaviors by Go in Caenorhabditis elegans. Science. 1995 Mar 17;267(5204):1648-51. [CrossRef]
- Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, Rand JB. A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci U S A. 1996 Oct 29;93(22):12593-8. [CrossRef]
- Collins KM, Koelle MR. Postsynaptic ERG potassium channels limit muscle excitability to allow distinct egg-laying behavior states in Caenorhabditis elegans. J Neurosci. 2013 Jan 9;33(2):761-75. [CrossRef]
- Cuppen E, van der Linden AM, Jansen G, Plasterk RH. Proteins interacting with Caenorhabditis elegans Galpha subunits. Comp Funct Genomics. 2003;4(5):479-91. [CrossRef]
- Koelle MR. Neurotransmitter signaling through heterotrimeric G proteins: insights from studies in C. elegans. WormBook. 2018 Dec 11;2018:1-52. [CrossRef]
- Wang D, Dao M, Muntean BS, Giles AC, Martemyanov KA, Grill B. Genetic modeling of GNAO1 disorder delineates mechanisms of Gαo dysfunction. Hum Mol Genet. 2022 Feb 21;31(4):510-522. [CrossRef]
- Briere L, Thiel M, Sweetser DA, Koy A, Axeen E. GNAO1-Related Disorder. 2023 Nov 9. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024. PMID: 37956232. DOI is not available. Available from https://pubmed.ncbi.nlm.nih.gov/37956232/.
- Nakamura K, Kodera H, Akita T, Shiina M, Kato M, Hoshino H, Terashima H, Osaka H, Nakamura S, Tohyama J, Kumada T, Furukawa T, Iwata S, Shiihara T, Kubota M, Miyatake S, Koshimizu E, Nishiyama K, Nakashima M, Tsurusaki Y, Miyake N, Hayasaka K, Ogata K, Fukuda A, Matsumoto N, Saitsu H. De Novo mutations in GNAO1, encoding a Gαo subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am J Hum Genet. 2013 Sep 5;93(3):496-505. [CrossRef]
- Collins KM, Bode A, Fernandez RW, Tanis JE, Brewer JC, Creamer MS, Koelle MR. Activity of the C. elegans egg-laying behavior circuit is controlled by competing activation and feedback inhibition. Elife. 2016 Nov 16;5:e21126. [CrossRef]
- Sáez González, M., Kloosterhuis, K., van de Pol, L., Baas, F., & Mikkers, H. (2023). Phenotypic Diversity in GNAO1 Patients: A Comprehensive Overview of Variants and Phenotypes. Human mutation, 2023, Article 6628283. [CrossRef]
- Ling W, Huang D, Yang F, Yang Z, Liu M, Zhu Q, Huang J, Zhou R, Chen X. Treating GNAO1 mutation-related severe movement disorders with oxcarbazepine: a case report. Transl Pediatr. 2022 Sep;11(9):1577-1587. [CrossRef]
- Liu Y, Zhang Q, Wang J, Liu J, Yang W, Yan X, Ouyang Y, Yang H. Both subthalamic and pallidal deep brain stimulation are effective for GNAO1-associated dystonia: three case reports and a literature review. Ther Adv Neurol Disord. 2022 Apr 29; 15:17562864221093507. [CrossRef]
- Ananth AL, Robichaux-Viehoever A, Kim YM, Hanson-Kahn A, Cox R, Enns GM, Strober J, Willing M, Schlaggar BL, Wu YW, Bernstein JA. Clinical Course of Six Children With GNAO1 Mutations Causing a Severe and Distinctive Movement Disorder. Pediatr Neurol. 2016 Jun; 59:81-4. [CrossRef]
- Danti FR, Galosi S, Romani M, Montomoli M, Carss KJ, Raymond FL, Parrini E, Bianchini C, McShane T, Dale RC, Mohammad SS, Shah U, Mahant N, Ng J, McTague A, Samanta R, Vadlamani G, Valente EM, Leuzzi V, Kurian MA, Guerrini R. GNAO1 encephalopathy: Broadening the phenotype and evaluating treatment and outcome. Neurol Genet. 2017 Mar 21;3(2):e143. [CrossRef]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998 Feb 19;391(6669):806-11. [CrossRef]
- Caplen NJ. RNAi as a gene therapy approach. Expert Opin Biol Ther. 2003 Jul;3(4):575-86. [CrossRef]
- Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010 Apr 15;464(7291):1067-70. [CrossRef]
- Lunev EA, Shmidt AA, Vassilieva SG, Savchenko IM, Loginov VA, Marina VI, Egorova TV, Bardina MV. [Effective Viral Delivery of Genetic Constructs to Neuronal Culture for Modeling and Gene Therapy of GNAO1 Encephalopathy]. Mol Biol (Mosk). 2022 Jul-Aug;56(4):604-618. Russian. [CrossRef]
- Klementieva NV, Lunev EA, Shmidt AA, Loseva EM, Savchenko IM, Svetlova EA, Galkin II, Polikarpova AV, Usachev EV, Vassilieva SG, Marina VI, Dzhenkova MA, Romanova AD, Agutin AV, Timakova AA, Reshetov DA, Egorova TV, Bardina MV. RNA Interference Effectors Selectively Silence the Pathogenic Variant GNAO1 c.607 G > A In Vitro. Nucleic Acid Ther. 2024 Apr;34(2):90-99. [CrossRef]
- Koval A, Larasati YA, Savitsky M, Solis GP, Good JM, Quinodoz M, Rivolta C, Superti-Furga A, Katanaev VL. In-depth molecular profiling of an intronic GNAO1 mutant as the basis for personalized high-throughput drug screening. Med. 2023 May 12;4(5):311-325.e7. [CrossRef]
- Silachev D, Koval A, Savitsky M, Padmasola G, Quairiaux C, Thorel F, Katanaev VL. Mouse models characterize GNAO1 encephalopathy as a neurodevelopmental disorder leading to motor anomalies: from a severe G203R to a milder C215Y mutation. Acta Neuropathol Commun. 2022 Jan 28;10(1):9. [CrossRef]
- Locke CJ, Williams SN, Schwarz EM, Caldwell GA, Caldwell KA. Genetic interactions among cortical malformation genes that influence susceptibility to convulsions in C. elegans. Brain Res. 2006 Nov 20;1120(1):23-34. [CrossRef]
- Dexter PM, Caldwell KA, Caldwell GA. A predictable worm: application of Caenorhabditis elegans for mechanistic investigation of movement disorders. Neurotherapeutics. 2012 Apr;9(2):393-404. [CrossRef]
- Lee TY, Yoon KH, Lee JI. NGT-3D: a simple nematode cultivation system to study Caenorhabditis elegans biology in 3D. Biol Open. 2016 Apr 15;5(4):529-34. [CrossRef]
- Opperman KJ, Mulcahy B, Giles AC, Risley MG, Birnbaum RL, Tulgren ED, Dawson-Scully K, Zhen M, Grill B. The HECT Family Ubiquitin Ligase EEL-1 Regulates Neuronal Function and Development. Cell Rep. 2017 Apr 25;19(4):822-835. [CrossRef]
- Polikarpova AV, Egorova TV, Lunev EA, Tsitrina AA, Vassilieva SG, Savchenko IM, Silaeva YY, Deykin AV, Bardina MV. CRISPR/Cas9-generated mouse model with humanizing single-base substitution in the Gnao1 for safety studies of RNA therapeutics. Front Genome Ed. 2023 Apr 3; 5:1034720. [CrossRef]
- Savitsky M, Solis GP, Kryuchkov M, Katanaev VL. Humanization of Drosophila Gαo to Model GNAO1 Paediatric Encephalopathies. Biomedicines. 2020 Oct 6;8(10):395. [CrossRef]
- Di Rocco M, Galosi S, Lanza E, Tosato F, Caprini D, Folli V, Friedman J, Bocchinfuso G, Martire A, Di Schiavi E, Leuzzi V, Martinelli S. Caenorhabditis elegans provides an efficient drug screening platform for GNAO1-related disorders and highlights the potential role of caffeine in controlling dyskinesia. Hum Mol Genet. 2022 Mar 21;31(6):929-941. [CrossRef]
- Ravi B, Zhao J, Chaudhry SI, Signorelli R, Bartole M, Kopchock RJ, Guijarro C, Kaplan JM, Kang L, Collins KM. Presynaptic Gαo (GOA-1) signals to depress command neuron excitability and allow stretch-dependent modulation of egg laying in Caenorhabditis elegans. Genetics. 2021 Aug 9;218(4):iyab080. [CrossRef]
- Muntean BS, Masuho I, Dao M, Sutton LP, Zucca S, Iwamoto H, Patil DN, Wang D, Birnbaumer L, Blakely RD, Grill B, Martemyanov KA. Gαo is a major determinant of cAMP signaling in the pathophysiology of movement disorders. Cell Rep. 2021 Feb 2;34(5):108718. [CrossRef]
- Baik JH, Picetti R, Saiardi A, Thiriet G, Dierich A, Depaulis A, Le Meur M, Borrelli E. Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature. 1995 Oct 5;377(6548):424-8. [CrossRef]
- Xu, H., Wu, XR., Wewer, U. et al. Murine muscular dystrophy caused by a mutation in the laminin α2 (Lama2) gene. Nat Genet 8, 297–302 (1994). [CrossRef]
- Chase DL, Pepper JS, Koelle MR. Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans. Nat Neurosci. 2004 Oct;7(10):1096-103. [CrossRef]
- Allen NE, Sherrington C, Paul SS, Canning CG. Balance and falls in Parkinson’s disease: a meta-analysis of the effect of exercise and motor training. Mov Disord. 2011 Aug 1;26(9):1605-15. [CrossRef]
- Ségalat L, Elkes DA, Kaplan JM. Modulation of serotonin-controlled behaviors by Go in Caenorhabditis elegans. Science. 1995 Mar 17;267(5204):1648-51. [CrossRef]
- Jiang M, Bajpayee NS. Molecular mechanisms of go signaling. Neurosignals. 2009;17(1):23-41. [CrossRef]
- Meijer, I. A., Miravite, J., Kopell, B. H., & Lubarr, N. (2017). Deep Brain Stimulation in an Additional Patient with ADCY5 -Related Movement Disorder. Journal of Child Neurology, 32(4), 438-439. [CrossRef]
- Mencacci NE, Erro R, Wiethoff S, Hersheson J, Ryten M, Balint B, Ganos C, Stamelou M, Quinn N, Houlden H, Wood NW, Bhatia KP. ADCY5 mutations are another cause of benign hereditary chorea. Neurology. 2015 Jul 7;85(1):80-8. [CrossRef]
- Kelly M, Park M, Mihalek I, Rochtus A, Gramm M, Pérez-Palma E, Axeen ET, Hung CY, Olson H, Swanson L, Anselm I, Briere LC, High FA, Sweetser DA; Undiagnosed Diseases Network; Kayani S, Snyder M, Calvert S, Scheffer IE, Yang E, Waugh JL, Lal D, Bodamer O, Poduri A. Spectrum of neurodevelopmental disease associated with the GNAO1 guanosine triphosphate-binding region. Epilepsia. 2019 Mar;60(3):406-418. [CrossRef]
- Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., Dlugos, D., Eichler, E. E., Epstein, M. P., Glauser, T., Goldstein, D. B., Han, Y., Heinzen, E. L., Hitomi, Y., Howell, K. B., Johnson, M. R., Kuzniecky, R., Lowenstein, D. H., Lu, Y. F., Madou, M. R. Z., Marson, A. G., ... Winawer, M. R. (2013). De novo mutations in epileptic encephalopathies. Nature, 501(7466), 217-221. [CrossRef]
- Dortch-Carnes J, Potter DE. Delta-opioid agonist-stimulated inositol phosphate formation in isolated, rabbit iris-ciliary bodies: role of G(i/o) proteins and Gbetagamma-subunits. Exp Eye Res. 2003 Dec;77(6):647-52. [CrossRef]
- Gill A, Hammes SR. G beta gamma signaling reduces intracellular cAMP to promote meiotic progression in mouse oocytes. Steroids. 2007 Feb;72(2):117-23. [CrossRef]
- Borlikova G, Endo S. Inducible cAMP early repressor (ICER) and brain functions. Mol Neurobiol. 2009 Aug;40(1):73-86. [CrossRef]
- Mercimek-Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, Hahn CD, Kannu P, Kobayashi J, Minassian BA, Moharir M, Siriwardena K, Weiss SK, Weksberg R, Snead OC 3rd. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia. 2015 May;56(5):707-16. [CrossRef]
- Miclea D, Peca L, Cuzmici Z, Pop IV. Genetic testing in patients with global developmental delay / intellectual disabilities. A review. Clujul Med. 2015;88(3):288-92. [CrossRef]
- Tanis, J. E., Moresco, J. J., Lindquist, R. A., & Koelle, M. R. (2008). Regulation of serotonin biosynthesis by the G proteins Gαo and Gαq controls serotonin signaling in Caenorhabditis elegans. Genetics, 178(1), 157-169. [CrossRef]
- Solis GP, Koval A, Valnohova J, Kazemzadeh A, Savitsky M, Katanaev VL. Neomorphic Gαo mutations gain interaction with Ric8 proteins in GNAO1 encephalopathies. J Clin Invest. 2024 Jun 14;134(15):e172057. [CrossRef]
- Koelle MR. A new family of G-protein regulators—the RGS proteins. Curr Opin Cell Biol. 1997 Apr;9(2):143-7. [CrossRef]
- Savoia Claudia, Pujol Julien B, Vaucher Angelique, Marchi Umberto De, Rieker Claus, Heikkilä, Eija, Núñez Galindo, Antonio, Dayon Loïc, Dioum, Elhadji M. GPRIN1 modulates neuronal signal transduction and affects mouse-learning behavior,” Mar. 30, 2018. [CrossRef]
- Taira R, Akamine S, Okuzono S, Fujii F, Hatai E, Yonemoto K, Takemoto R, Kato H, Masuda K, Kato TA, Kira R, Tsujimura K, Yamamura K, Ozaki N, Ohga S, Sakai Y. Gnao1 is a molecular switch that regulates the Rho signaling pathway in differentiating neurons. Sci Rep. 2024 Jul 24;14(1):17097. [CrossRef]
- Ju H, Lee S, Kang S, Kim SS, Ghil S. The alpha subunit of Go modulates cell proliferation and differentiation through interactions with Necdin. Cell Commun Signal. 2014 Jul 10; 12:39. [CrossRef]
- Solis GP, Kozhanova TV, Koval A, Zhilina SS, Mescheryakova TI, Abramov AA, Ishmuratov EV, Bolshakova ES, Osipova KV, Ayvazyan SO, Lebon S, Kanivets IV, Pyankov DV, Troccaz S, Silachev DN, Zavadenko NN, Prityko AG, Katanaev VL. Pediatric Encephalopathy: Clinical, Biochemical and Cellular Insights into the Role of Gln52 of GNAO1 and GNAI1 for the Dominant Disease. Cells. 2021 Oct 14;10(10):2749. [CrossRef]
- JoJo Yang QZ, Porter BE, Axeen ET. GNAO1-related neurodevelopmental disorder: Literature review and caregiver survey. Epilepsy Behav Rep. 2022 Dec 31; 21:100582. [CrossRef]
- Wang D, Dao M, Muntean BS, Giles AC, Martemyanov KA, Grill B. Genetic modeling of GNAO1 disorder delineates mechanisms of Gαo dysfunction. Hum Mol Genet. 2022 Feb 21;31(4):510-522. [CrossRef]
- Miller KG, Emerson MD, Rand JB. Goalpha and diacylglycerol kinase negatively regulate the Gqalpha pathway in C. elegans. Neuron. 1999 Oct;24(2):323-33. [CrossRef]
- Saitsu H, Fukai R, Ben-Zeev B, Sakai Y, Mimaki M, Okamoto N, Suzuki Y, Monden Y, Saito H, Tziperman B, Torio M, Akamine S, Takahashi N, Osaka H, Yamagata T, Nakamura K, Tsurusaki Y, Nakashima M, Miyake N, Shiina M, Ogata K, Matsumoto N. Phenotypic spectrum of GNAO1 variants: epileptic encephalopathy to involuntary movements with severe developmental delay. Eur J Hum Genet. 2016 Jan;24(1):129-34. [CrossRef]
Figure 1.
Overview of G-protein-coupled receptor (GPCR) signaling and known G-protein effectors. A) The diagram illustrates the cyclic process of GPCR activation and G-protein signaling. (Top, left) In the inactive state, the GPCR is bound to an intracellular heterotrimeric G-protein, composed of Gα, Gβ, and Gγ subunits, with GDP bound to Gα. (Top, right) Ligand binding to the extracellular domain of the GPCR induces a conformational change that facilitates GDP release and GTP binding to Gα, activating the G-protein complex. (Bottom, right) The Gα subunit dissociates from the Gβγ dimer, and both subunits can interact with downstream effectors, initiating intracellular signaling pathways. (Bottom, left) GTP hydrolysis by the RGS proteins (Regulators of G-protein Signaling) leads to the reformation of the inactive heterotrimeric complex and terminates the signaling. This cycle repeats as long as the ligand remains bound to the GPCR. B) Four distinct Gα subtypes (Gαs, Gαq, Gα12, Gαo) are shown, each modulating different downstream effectors: adenylate cyclase (AC) for Gαs, phospholipase C β (PLCβ) and TRIO (Trio Rho Guanine Nucleotide Exchange Factor) for Gαq, and Rho GEF (Guanine Exchange Factor) for Gα12. The effector for Gαo is currently unknown (denoted by “?”). Mutations in the GNAO1 gene, which encodes the Gαo protein, are associated with pediatric GNAO1 encephalopathy, as highlighted on the right side of the diagram. These mutations can lead to severe neurological conditions, underscoring the clinical relevance of Gαo in brain development and function. (Figures created with BioRender.com).
Figure 1.
Overview of G-protein-coupled receptor (GPCR) signaling and known G-protein effectors. A) The diagram illustrates the cyclic process of GPCR activation and G-protein signaling. (Top, left) In the inactive state, the GPCR is bound to an intracellular heterotrimeric G-protein, composed of Gα, Gβ, and Gγ subunits, with GDP bound to Gα. (Top, right) Ligand binding to the extracellular domain of the GPCR induces a conformational change that facilitates GDP release and GTP binding to Gα, activating the G-protein complex. (Bottom, right) The Gα subunit dissociates from the Gβγ dimer, and both subunits can interact with downstream effectors, initiating intracellular signaling pathways. (Bottom, left) GTP hydrolysis by the RGS proteins (Regulators of G-protein Signaling) leads to the reformation of the inactive heterotrimeric complex and terminates the signaling. This cycle repeats as long as the ligand remains bound to the GPCR. B) Four distinct Gα subtypes (Gαs, Gαq, Gα12, Gαo) are shown, each modulating different downstream effectors: adenylate cyclase (AC) for Gαs, phospholipase C β (PLCβ) and TRIO (Trio Rho Guanine Nucleotide Exchange Factor) for Gαq, and Rho GEF (Guanine Exchange Factor) for Gα12. The effector for Gαo is currently unknown (denoted by “?”). Mutations in the GNAO1 gene, which encodes the Gαo protein, are associated with pediatric GNAO1 encephalopathy, as highlighted on the right side of the diagram. These mutations can lead to severe neurological conditions, underscoring the clinical relevance of Gαo in brain development and function. (Figures created with BioRender.com).
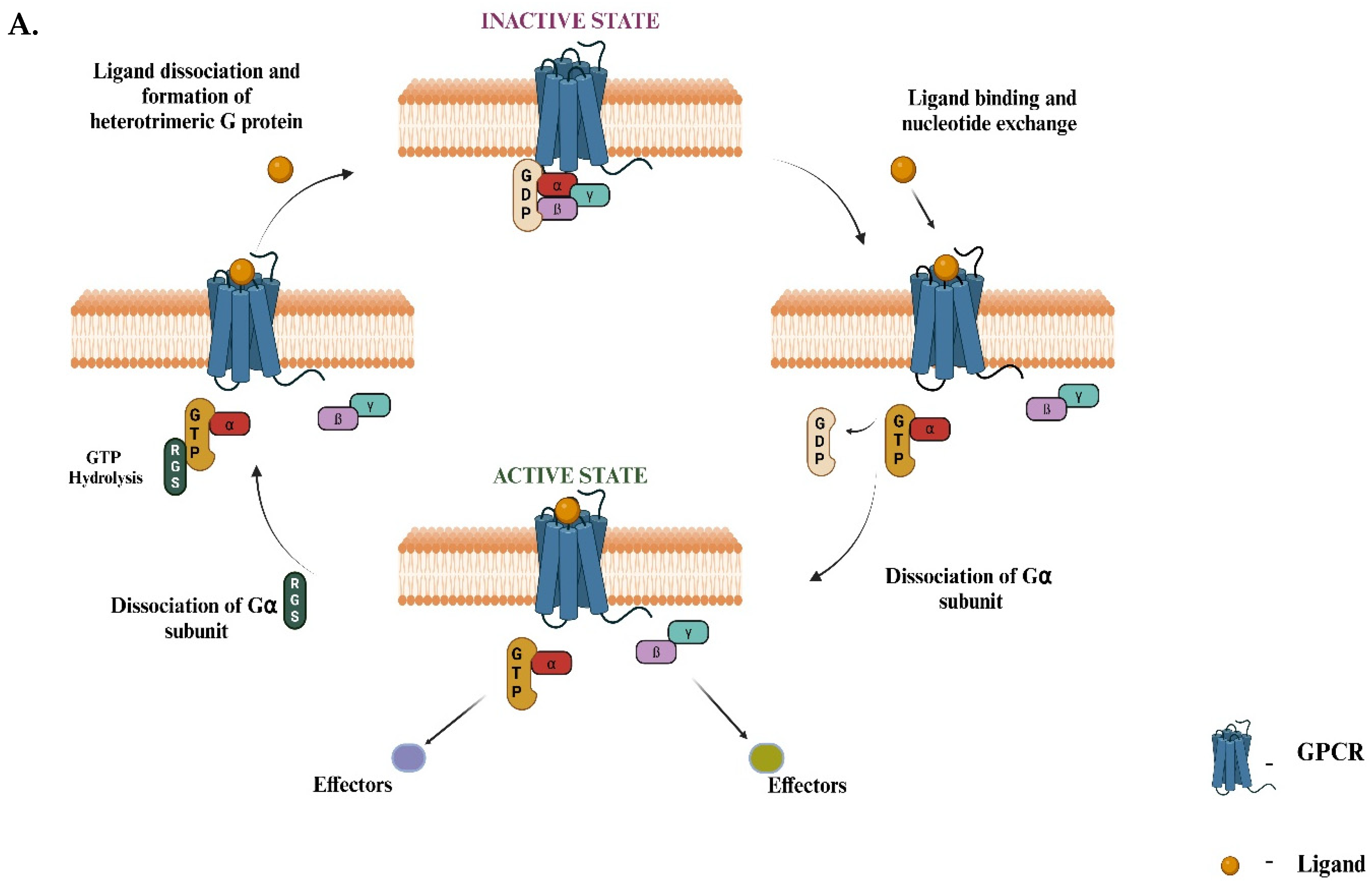
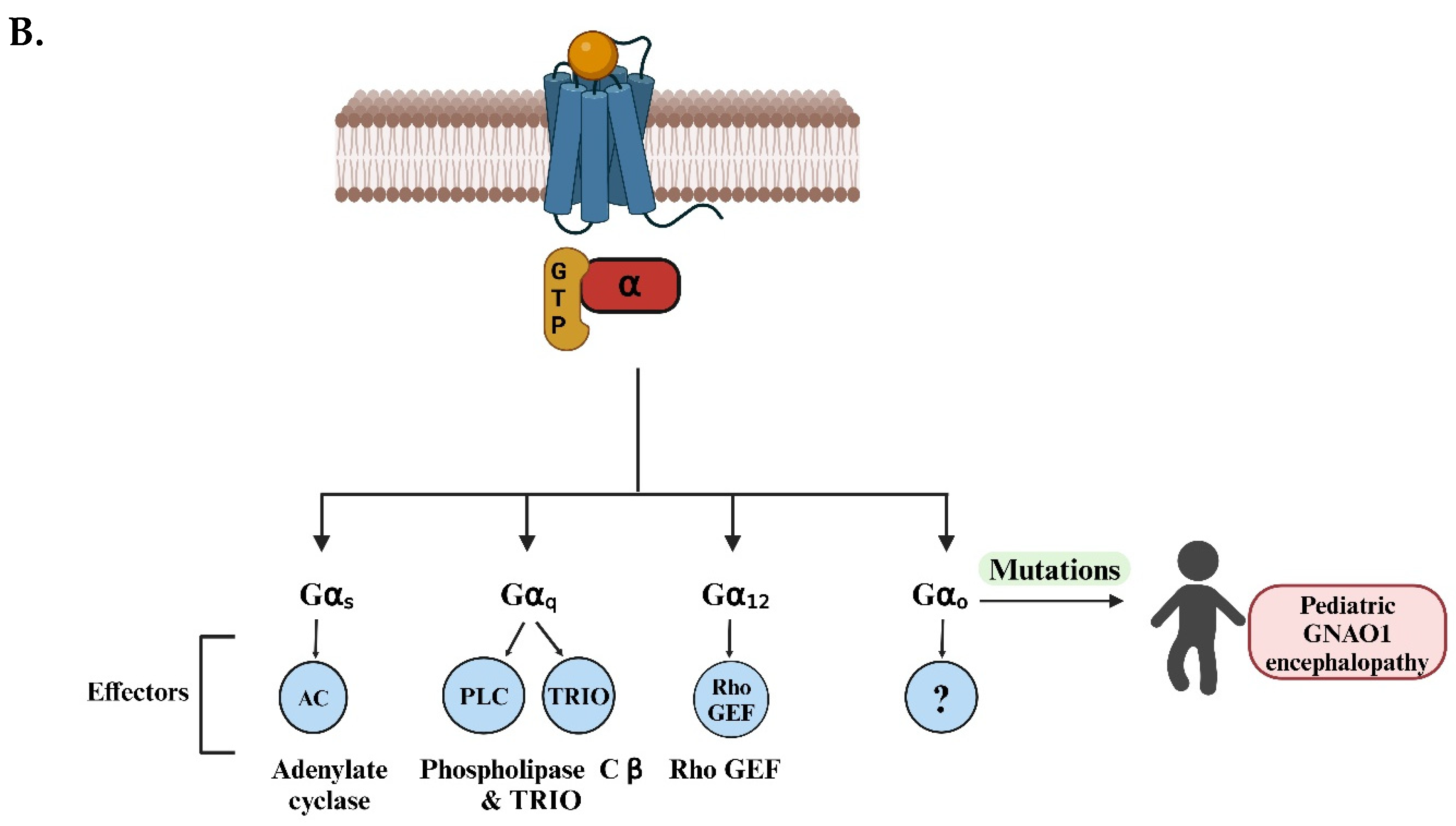
Figure 2.
Mutational landscape of GNAO1 mutations associated with NEDIM and DEE17 syndromes and overview of their phenotypic effects in Homo sapiens, Mus musculus, and Caenorhabditis elegans A) The linear schematic represents the hGNA01 protein sequence (1-354 amino acids), highlighting the locations of disease-related mutations. Mutations identified in patients with Neurodevelopmental Disorder with Involuntary Movements (NEDIM) are shown above the protein, while mutations associated with Developmental and Epileptic Encephalopathy 17 (DEE17) are indicated below the protein. Key mutations include G40R, S47G, I56T, and A227V among others, with multiple variations at residue R209. B) Mutations in the GNAO1 gene are categorized into three groups: loss-of-function mutations (left, blue), neutral mutations (center, black), and gain-of-function mutations (right, red). Each category lists relevant mutations based on their functional impact. Loss-of-function mutations include G40R, D174G, L199P, R209H, A227V, N270H, Y231C, F275S, and I279N, while gain-of-function mutations include G184S, G203R, G42R, and E246K. R209C and R209G represent neutral mutations. The bottom section highlights the phenotypic changes associated with these mutations across different species, including Homo sapiens(humans), Mus musculus (mice), and C. elegans (nematodes). Loss-of-function mutations are associated with movement disorders and developmental delays in humans, whereas gain-of-function mutations cause epileptic encephalopathies. In Mus musculus, loss-of-function mutations result in hyperlocomotion, but no phenotypic data is available for gain-of-function mutations. In C. elegans, loss-of-function mutations lead to hyperlocomotion and premature egg-laying, while gain-of-function mutations cause lethargic movement and delayed egg-laying. (Figures created with BioRender.com).
Figure 2.
Mutational landscape of GNAO1 mutations associated with NEDIM and DEE17 syndromes and overview of their phenotypic effects in Homo sapiens, Mus musculus, and Caenorhabditis elegans A) The linear schematic represents the hGNA01 protein sequence (1-354 amino acids), highlighting the locations of disease-related mutations. Mutations identified in patients with Neurodevelopmental Disorder with Involuntary Movements (NEDIM) are shown above the protein, while mutations associated with Developmental and Epileptic Encephalopathy 17 (DEE17) are indicated below the protein. Key mutations include G40R, S47G, I56T, and A227V among others, with multiple variations at residue R209. B) Mutations in the GNAO1 gene are categorized into three groups: loss-of-function mutations (left, blue), neutral mutations (center, black), and gain-of-function mutations (right, red). Each category lists relevant mutations based on their functional impact. Loss-of-function mutations include G40R, D174G, L199P, R209H, A227V, N270H, Y231C, F275S, and I279N, while gain-of-function mutations include G184S, G203R, G42R, and E246K. R209C and R209G represent neutral mutations. The bottom section highlights the phenotypic changes associated with these mutations across different species, including Homo sapiens(humans), Mus musculus (mice), and C. elegans (nematodes). Loss-of-function mutations are associated with movement disorders and developmental delays in humans, whereas gain-of-function mutations cause epileptic encephalopathies. In Mus musculus, loss-of-function mutations result in hyperlocomotion, but no phenotypic data is available for gain-of-function mutations. In C. elegans, loss-of-function mutations lead to hyperlocomotion and premature egg-laying, while gain-of-function mutations cause lethargic movement and delayed egg-laying. (Figures created with BioRender.com).
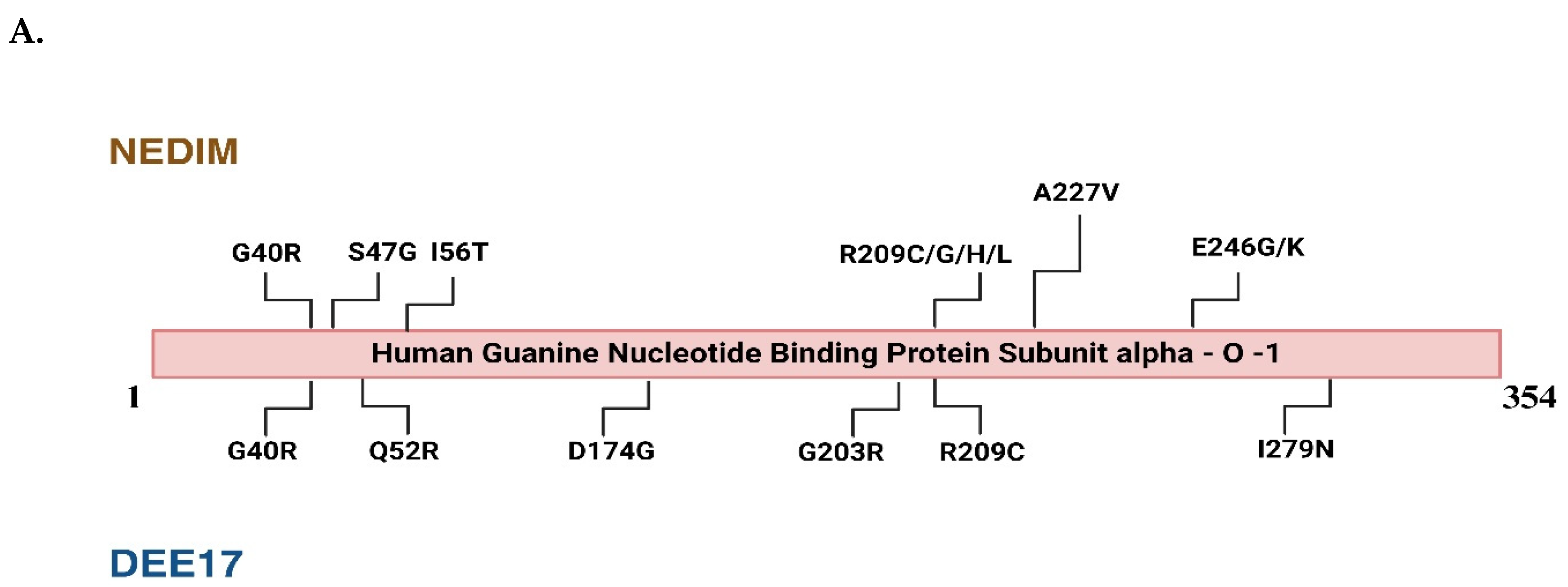
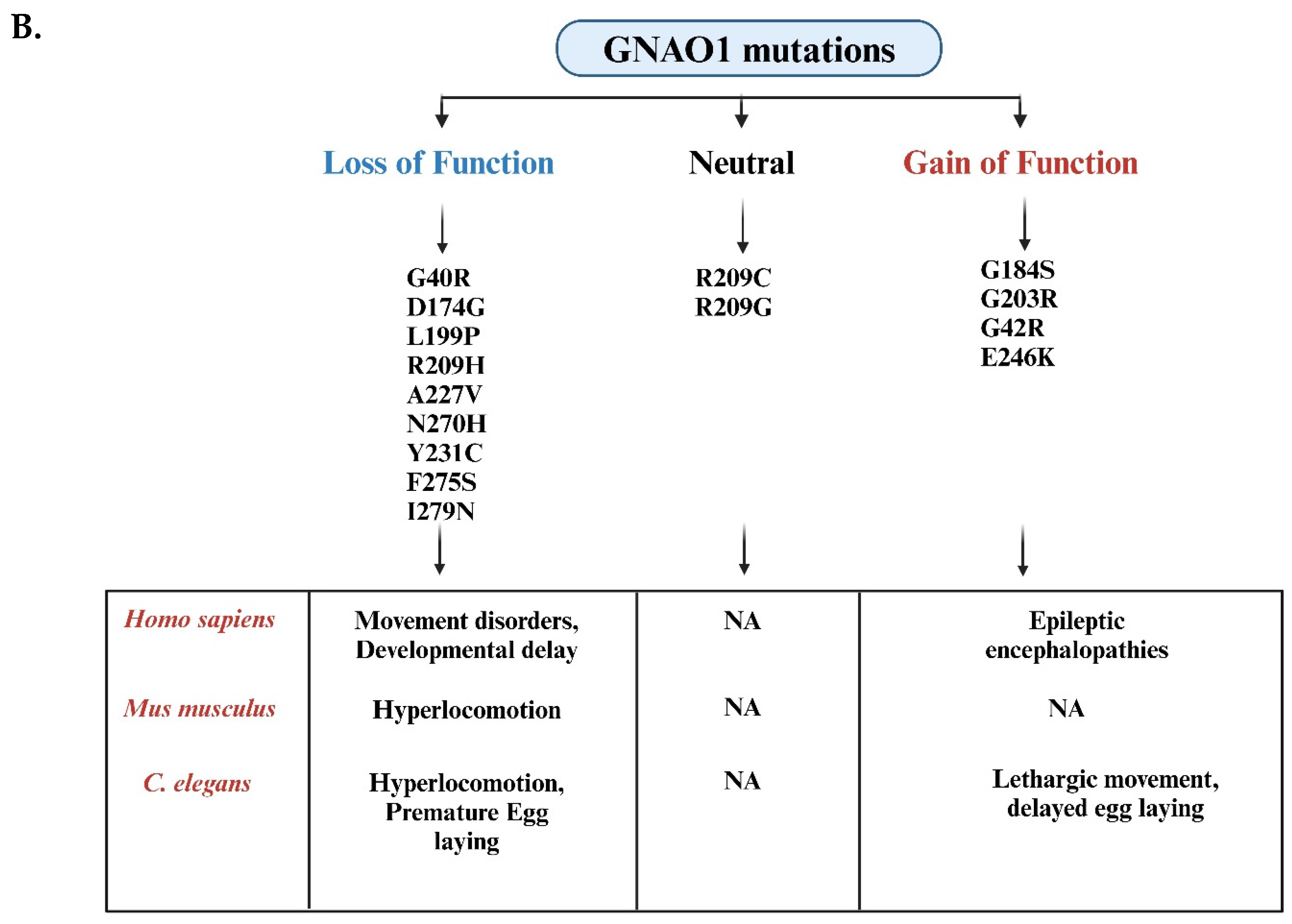
Figure 3.
Impact of GNAO1 mutations on G protein signaling pathways in C. elegans and Homo sapiens. The left panel illustrates normal G-protein signaling pathways in C. elegans, showing the interaction of Gα (bound to GDP) with Gβγ upon GPCR activation. GNAO1 (Gα) regulates processes such as serotonin release, muscle contraction, and egg-laying behavior via downstream effectors such as tbh-1 and Ric-8. Mutations in GNAO1 lead to defects in egg laying and locomotion. The right panel demonstrates the G protein signaling in Homo sapiens. GNAO1 mutations disrupt neurite outgrowth by affecting the interaction with Rho-GTPase signaling, Necdin-EF1, and the GRIN/GRPN complex, with potential involvement of cdc-42, which remains unclear. Arrows indicate pathways affected by GNAO1 mutations, contributing to abnormal neuronal growth and function. RGS—Regulator of G-protein Signaling; tph-1—Tryptophan Hydroxylase; Ric8—Resistance to inhibitors of Cholinesterase; GRIN-1—Glutamate Inotropic Receptor NMDA Subunit -1; cdc-42—Cell Division Control protein–42 (Figures created with BioRender.com).
Figure 3.
Impact of GNAO1 mutations on G protein signaling pathways in C. elegans and Homo sapiens. The left panel illustrates normal G-protein signaling pathways in C. elegans, showing the interaction of Gα (bound to GDP) with Gβγ upon GPCR activation. GNAO1 (Gα) regulates processes such as serotonin release, muscle contraction, and egg-laying behavior via downstream effectors such as tbh-1 and Ric-8. Mutations in GNAO1 lead to defects in egg laying and locomotion. The right panel demonstrates the G protein signaling in Homo sapiens. GNAO1 mutations disrupt neurite outgrowth by affecting the interaction with Rho-GTPase signaling, Necdin-EF1, and the GRIN/GRPN complex, with potential involvement of cdc-42, which remains unclear. Arrows indicate pathways affected by GNAO1 mutations, contributing to abnormal neuronal growth and function. RGS—Regulator of G-protein Signaling; tph-1—Tryptophan Hydroxylase; Ric8—Resistance to inhibitors of Cholinesterase; GRIN-1—Glutamate Inotropic Receptor NMDA Subunit -1; cdc-42—Cell Division Control protein–42 (Figures created with BioRender.com).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



