
Submitted:
30 October 2024
Posted:
31 October 2024
You are already at the latest version
Abstract
Cellular reprogramming of somatic cells towards induced pluripotency is a multistep stochastic process mediated by the transcription factors Oct4, Sox2, Klf4 and c-Myc (OSKM), which orchestrate global epigenetic and transcriptional changes. We performed a large-scale analysis of integrated ChIP-seq, ATAC-seq and RNA-seq data and revealed the spatiotemporal highly dynamic pattern of OSKM DNA binding during reprogramming. We found that OSKM show distinct temporal patterns of binding to different classes of pluripotency-related enhancers. Genes involved in reprogramming are regulated by the coordinated activity of multiple enhancers, which are sequentially bound by OSKM for strict transcriptional control. Based on these findings, we developed an unbiased approach to identify Reprogramming Inducible Enhancers (RIEs), constructed enhancer-traps and isolated cells undergoing reprogramming in real-time. We used a representative RIE taken from the Upp1 gene fused to GFP and isolated cells at different time points during reprogramming and found that they have unique developmental capacities as they are reprogrammed with high efficiency due to their distinct molecular signatures. In conclusion, our experiments have led to the development of an unbiased method to identify and isolate reprogrammable cells in real-time by exploiting the functional dynamics of OSKM, which can be used as efficient reprogramming biomarkers.
Keywords:
transcriptional regulation
; transcription factors
; enhancers
; chromatin structure
; genomics
; cellular reprogramming
; OSKM
; iPSCs
1. Introduction
Cellular reprogramming of somatic cells towards induced Pluripotent Stem Cells (iPSCs) via the over-expression of Oct4, Sox2, Klf4 and c-Myc (OSKM) provides an excellent model system to study the mechanisms by which a small set of transcription factors (TFs) could reactivate the pluripotency gene network to drive cell fate specification [1,2]. Reprogramming is a stochastic process, achieved only in a few cells, which manage to alter their chromatin structure to efficiently and appropriately regulate the necessary gene modules [3,4,5]. During the early stages of reprogramming, somatic cells need to undergo a series of morphological and functional changes, including among others an increase in the proliferation rate, a transition towards glycolytic metabolic pathways and a decrease in size [4,6]. In addition, mesenchymal cells undergo the Mesenchymal to Epithelial Transition process (MET) between day 2 to day 4 of reprogramming [7,8,9]. We have previously identified a gene regulatory network reconstructed from nine transcriptional regulators (TRs), the 9TR-GRN, which is required for the establishment of pluripotency [10]. The 9TR-GRN consists of Taf1c, Tead4, Tfap4, Rcan1, Cbfa2t3, Gli2, Irf6, Ovol1 and the Nanog TFs, all of which are direct OSKM targets. The 9TR-GRN is assembled in a stochastic and stepwise manner between days 1 and 6 of reprogramming only in a small number of cells expressing all factors, and is required for the gradual establishment of pluripotency [10]. These and other studies have suggested that OSKM-induced cellular reprogramming is highly plastic, as it is accompanied by the transient expression of genes from unrelated lineages, regardless of the starting cell type or species [11].
Previous landmark studies have provided insights for the OSKM-driven epigenetic and transcriptional changes occurring in the cells undergoing reprogramming [12,13,14,15,16,17,18,19,20,21]. Oct4, Sox2 and Klf4 function as pioneer transcription factors by binding to MEF-closed chromatin during the first days of reprogramming with or without c-Myc [14,18,19,22,23]. OSKM trigger chromatin opening in these sites leading to the stepwise induction of pluripotency genes either alone or in combination with other transcriptional regulators [12,13,14,15,17,18,19,20,21,24,25]. In parallel, OSKM participate in the deactivation of somatic and mesenchymal gene networks regulated by the presence of macroH2A-containing nucleosomes [26]. In addition, OSKM bind accessible and active somatic enhancers causing repression of their associated genes through various complex mechanisms including interactions with the somatic factors, whereas their concomitant binding to pluripotency enhancers activates the expression of the linked genes. OSKM-driven somatic gene repression involves chromatin deacetylation, displacement and/or down-regulation of the somatic TFs and up-regulation of negative transcriptional regulators [17,18,19,24].
Given that cellular reprogramming is a purely stochastic process, predictions regarding which cells will be reprogrammed and the timing for completion of the process are currently impossible. However, various gene markers have been proposed for the identification of pure and efficiently reprogrammable cell populations in vivo [27]. Among them, the best markers for mouse cells involve the early down-regulation of the mesenchymal surface antigen Thy1, the activated expression of alkaline phosphatase (Alpl), the upregulation of the embryonic antigen SSEA1, and the late activation of the Oct4 and/or the Nanog regulatory regions and the reconstruction of the 9TR-GRN [10,28,29,30,31,32,33]. Despite the existing methods to study cell populations entering the efficient reprogramming path(s), for identifying the actual mechanisms responsible for the gradual acquisition of pluripotency remain still elusive. Given the molecular complexity of the reprogramming process and the development of various reprogramming systems (different TF combinations, chemical molecules, genetically modified cells, etc) [27,34,35,36,37] it will be beneficial to establish new efficient ways to isolate cells during their transition to pluripotency in real-time, in order to illuminate the shady processes controlling reprogramming.
In this study, we have performed a large-scale analysis of integrated ChIP-seq, ATAC-seq and RNA-seq data and revealed the spatiotemporal highly dynamic and complex pattern of OSKM binding to the mouse genome during the course of cellular reprogramming. The combinatorial integration of multiple datasets ensures a more reliable and complete picture of the genomic OSKM binding activity, free of putative system-dependent discrepancies. We found that OSKM show distinct temporal patterns of binding at different groups of ESC enhancers. These highly dynamic and complex DNA binding events are associated with the function and expression of the neighboring genes by affecting the local chromatin dynamics. We found that genes essential for reprogramming (MET/EMT and pluripotency genes) are regulated by the coordinated activity of multiple enhancers, which are sequentially bound by OSKM to warrant strict transcriptional control. Based on these findings we developed an unbiased approach to identify Reprogramming Inducible Enhancers (RIEs) aiming to generate enhancer-traps for isolating in real-time cells undergoing reprogramming. We used a construct bearing the RIE taken from the Upp1 gene fused to GFP as a marker to isolate cells undergoing reprogramming in real-time. We demonstrated that these cells have acquired unique developmental capacities since they are reprogrammed with high efficiency due to their distinct molecular signatures, such as the high-level expression of 9TR GRN components, which are co-expressed in pre-iPSCs cells. In conclusion, our experiments have led to the development of an unbiased method to identify and isolate reprogrammable cells in real-time by exploiting the functional dynamics of OSKM that controls the transcriptional potential of RIEs, which can be used as efficient reprogramming biomarkers.
2. Results
2.1. Creation of an Integrated ChIP-Seq Dataset to Monitor Global Binding of OSKM During Cellular Reprogramming
To investigate the complex molecular mechanisms involved in TF-induced cellular reprogramming, we examined the binding of the OSKM factors to the mouse genome by integrating publicly available time-course ChIP-seq datasets [10,16,17,18,38] (Figure 1A, Table S1, Table S2) followed by computational analysis. We anticipated that a large-scale analysis of such integrated data acquired from a number of different experiments using the OSKM MEFs reprogramming system is an important parameter for obtaining a representative picture of the OSKM binding activity. Furthermore, the OSKM ChIP-seq time-course dataset (day 0, day 1, day 3, day 6 of reprogramming and in ESCs) was integrated with ATAC-seq and RNA-seq experiments in order to monitor both the requirements and the functional consequences of OSKM genomic binding. The ESCs RNA-seq data used in our analysis were obtained from our previously published dataset [26] (Table S1).
2.2. An Unprecedented Highly Dynamic OSKM Binding During Cellular Reprogramming
Our large scale analysis revealed that OSK tend to co-bind in a gradually increasing mode from day 1 to day 6 of reprogramming (Figure 1B, Oct4, Sox2 and Klf4 rows), while Myc preferentially co-binds with Klf4 only (Figure 1B, Myc rows) [14,17]. Interestingly, we discovered that the highly preferential Oct4-Sox2 co-binding in ESCs represents a stable state established late during reprogramming, because during the long reprogramming process, Sox2 shares more binding sites with Klf4 than with Oct4 (Figure 1B, compare Sox2 row with Oct4 and Klf4 columns at the different time points). Furthermore, comparison of the common Oct4 (“Oct4 to Oct4”), Sox2 (“Sox2 to Sox2”), Klf4 (‘Klf4 to Klf4”) and Myc (“Myc to Myc”) binding sites between days 1 and 3 and between days 3 and 6, revealed that ~50% of their total binding sites on day 1 are lost on day 3 and similarly, their binding sites were lost from day 3 to day 6 (Figure 1C, “D1→D3” and “D3→D6” columns). Importantly, with the exception of Klf4, OSM, especially Oct4 and Sox2, are highly mobile preserving only 10–30% of their day 6 sites in ESCs (Figure 1C, 1D, “D6→ES” column). These observations strongly suggest that the highly dynamic OSKM binding generates transient chromatin landscapes bearing unique functional potentials via the sequential occupation and abandonment of sites, which altogether, are not similar to the landscape found at the terminal and stable ESC state (Figure 1C,D). Overall, our data suggest that on the road to achieve pluripotency, OSKM are in a constant mobility phase onto chromatin rewiring various enhancer landscapes, thus providing a reasonable molecular explanation for the generation of cellular plasticity in reprogramming [39].
2.3. The Combinatorial Binding of OSKM to Early and Late Elements Prefigures the Induction of Pluripotency-Related Genes During Reprogramming
To investigate how the continuously changing OSKM DNA binding landscape during reprogramming culminates in the establishment of the unique stable DNA binding pattern in ESCs, we grouped the total sites occupied by OSKM from the beginning of reprogramming until the acquisition of pluripotency (ESCs) according to their temporal DNA binding pattern. We identified four major classes of sites: the early bound ESC sites (ESCs sites occupied from day 1), late-bound ESCs sites (ECS sites occupied after day 3 of reprogramming), transient sites (sites that are occupied transiently during reprogramming), and the MEF sites (sites bound by KM in MEFs and OSKM during reprogramming, but not in ESCs) (Figure 1E). Collectively, during reprogramming the majority of the total sites (>200,000 sites) bound by OSKM are occupied by these factors transiently only. This finding further underscores the dynamic and the stochastic nature of reprogramming.
More specifically, we found that of the total 83,674 OSKM binding sites in ESCs (Figure 2A, top panel), 31,188 (37.3%) sites are bound by OSKM from day 1 (early-bound sites) (Figure 2A, left panel), while the remaining 52,486 sites are sequentially occupied at days 3, 6, and later (late-bound sites) (Figure 2A, right panel). Furthermore, we found that the early-bound sites are preferentially located at ±0-5kb from the TSSs of the closest genes (more proximal regions), whereas the late-bound sites are primarily located between 5 and 500kb from the TSSs (distal regions) (Figure 2B,E). OSKM early binding events coincide with the rapid down-regulation and up-regulation of neighboring genes mainly involved in the somatic state and developmental and housekeeping processes, respectively, whereas the late-bound sites correlate to a more delayed activation (after day 3) of genes involved in developmental and differentiation processes (compare Figure 2C-D with Figure 2F-G).
Intriguingly, we found that the vast majority of the OSKM-bound genes in ESCs (9,992 genes) bear both early- and late-bound OSKM sites (Figure S1A, Table S3), thus suggesting that these genes could be regulated in a combinatorial manner by OSKM via widely separated cis-regulatory elements occupied at different times during reprogramming. Some of these genes are induced during reprogramming, such as the critical pluripotent regulators Pou5f1 (Oct4), Sox2 and Nanog (OSN) and various other reprogramming TFs and cofactors, like Bhlhe40, Ehf, Elf3, Esrrb, Etv5, Lin28a/b, Nr5a2, Sall1/4, Tbx3, Tfcp2l1, Utf1 and Zic3 (Table S3, Figure S1C,F). Importantly, 6 out of the 9 TRs (i.e. Rcan1, Tead4, Tfap4, Gli2, Ovol1 and Nanog) previously shown to reconstruct the 9TR gene regulatory network required for cellular reprogramming [10], are also regulated by the combinatorial action of early- and late- OSKM binding at proximal and distal regulatory elements, respectively. Of note, this group of genes includes also TFs which are down-regulated during reprogramming, like Snai1, Twist1/2 and Zeb1/2, all of which are involved in maintaining the mesenchyme phenotype [26,40], and thus inhibit reprogramming. Interestingly, although the OSKM ESC early-bound genes display a relatively stable expression pattern during the first 6-8 days of reprogramming, their expression is dramatically decreased towards ESCs, even though OSKM are still bound at their respective DNA elements (Figure S1B), whereas the late-bound OSKM ESC genes are induced at the latest phases of reprogramming (compare Figure S1C and Figure S1D) including among others genes involved in extracellular matrix (ECM) organization (Figure S1G and Table S3).
As predicted from our analysis, a significant fraction of the early-bound OSKM sites (37.4%) lie in open chromatin in MEFs (Figure 2A, left panel heatmap and Figure 2H, top panel) bearing also motifs for somatic TFs (e.g. Fosl, Jun and Tead families) (Figure S2A), whereas the OSKM late-bound elements are in a closed chromatin configuration in MEFs (Figure 2A, right panel heatmap and Figure 2H, lower panel), but contain various motifs for pluripotent TFs, such as Nanog and are linked to genes involved exclusively in cell fate decision processes (Figure 2G, Figure S2B). The vast majority of the late-bound sites (90%) remain closed throughout reprogramming (low ATAC-seq signal, Figure 2A right panel heatmap, and Figure 2H lower panel). We speculate that since these genes are bound by the master pluripotency factors Oct4, Sox2 and Nanog are in a “poised” state in ESCs, that is, ready to be expressed during differentiation upon receiving the right developmental signals [41].
The pioneering activity of OSKM in initiating reprogramming begins with their binding to the early-bound sites, a significant fraction of which is constitutively open in MEFs and remain open throughout reprogramming (23,282 sites), and a fraction of these lie within ESC-specific super-enhancers [17,42] (Figure 2I, Table S4). We also note that stable binding of OSKM in these open sites is also marked by the co-presence of somatic TF motifs, suggesting that these regions could play a binary role in both the mesenchymal and the pluripotent state (Figure 2I, Venn diagram, Figure S2C). We assume that OSKM binding to these sites eventually replaces pre-bound somatic TFs (Figure 2M, see a list of genes in Table S3). Intriguingly, a small percentage of early-bound ESCs sites, closes and re-opens at various stages of reprogramming (Figure 2J-L), thus causing dynamic changes in the gene expression programs of the associated genes (Figure 2N-P). This finding further underscores the continuously changing chromatin landscape controlled by OSKM.
2.4. Identification of Reprogramming-Inducible Enhancers in the Mouse Genome
So far, we have provided a comprehensive compilation of putative genomic regulatory regions bound by OSKM during different stages of cellular reprogramming by performing genome-wide occupation analytical studies integrated with gene expression and open chromatin signatures (Figure 1E, Figure 2). Previous studies have identified a large number of biochemical and functional interactions between O/S/K/M, thus implying that they collaboratively initiate cellular reprogramming [17,22]. This unprecedented and carefully-coordinated OSKM dynamic binding along with the binding of other TFs, including the 9TR network, lead to a sequential occupation of markedly different genomic regions between early and later stages of reprogramming, thus driving cells to abandon their mesenchymal phenotype and induce their transition towards pluripotency. For example, only the small percentage of cells in which the 9TR network is successfully reconstructed and activated by the direct action of OSKM will be reprogrammed into iPSCs [10] (Figure S3A-I). For an initial assessment of the existence and the biological relevance of putative Reprogramming Inducible Enhancers (RIEs), we searched the genome for such enhancers with in vivo reprogramming activity. Furthermore, the identification of RIEs will allow the isolation and study of the precious rare cell populations undergoing successful transition from the somatic to the pluripotent phenotype in real time. Our previous analysis identified early-bound ESC genomic sites (Figure 2A, left panel) implicated in the up-regulation of genes related to the transition to pluripotency (Figure 2C-D, Figure S1C,F). Importantly, 19,226 of these sites are bound by OSKM only after the initiation of reprogramming (Figure 3A, right panel de novo sites), and therefore we assumed that they could be contained within putative RIEs.
Based on the above, we carefully inspected these specific genomic sites to assess the value of these elements in functioning as true RIEs. More specifically, we searched the de novo acquired OSKM early-bound sites focusing on regions residing up to ±2.5kb from the TSS of up-regulated genes during reprogramming (Figure 3B). We did not take into account the biological function of the genes close to these elements, or any data regarding genomic regions previously characterized as regulatory elements. Thus, our RIEs identification approach was based on an unbiased logic. The resulting 702 sites were further filtered to remove loci bearing 10 or less Oct4, Sox2, Klf4 or Myc peaks in total. Based on this distribution, we defined 66 co-bound regions of adjacent OSKM peaks that are no more than 800bp apart on average, which we assumed that could function as putative RIEs (Table S5). Of these, three test enhancers were selected as representative examples for further functional validation analyses: a 600bp-region located ~700 bp upstream of the Lefty1 gene (“Lefty1700” element, Figure 3C), a 300bp region located ~1800 bp upstream of the Pou5f1 gene (“Pou5f11800” element, Figure 3D) and a 300bp region residing ~800 bp upstream of the Upp1 gene (“Upp1800” element, Figure 3E). Lefty1 encodes for a protein belonging to the TGF-β family of ligands which plays a role in the determination of the left-right symmetry in the developing embryo and it is a stemness marker [43,44]. Pou5f1 encodes for the Oct4 transcription factor protein. Oct4 has a vital role in regulation of pluripotency by forming the core of the pluripotency transcription factor network along with Sox2 and Nanog [45]. Finally, Upp1 encodes for a uridine phosphorylase, an enzyme catalyzing the reversible phosphorolysis of uridine, often found up-regulated in rapidly dividing malignant cells [46,47]. All three elements are stably-bound by OSK from day 1 throughout reprogramming to ESCs, with the exception of Upp1800, where Klf4 binding occurs from day 3 and afterwards. Importantly, all three elements are bound by Nanog in ESCs (Figure 3C-E). Furthermore, OSK early binding (day 1) correlates with an increase of the corresponding gene expression within the first 24 hours (Figure 3F-H). Taken together, these observations support the idea that the above genomic elements could indeed function as RIEs. In addition, ATAC-seq analysis revealed that these sites are occupied by nucleosomes in MEFs, but become accessible early in reprogramming (Figure S4A-C). More specifically, OSK binding at Lefty1700 correlates with an immediate opening of the local chromatin, while the Pou5f11800 and Upp1800 become accessible after day 3. Accordingly, although at the beginning of reprogramming all three putative RIEs are marked by the repressive histone modification H3K27me3, during reprogramming there is a gradual replacement of the repressive mark by the H3K27ac modification, a transcriptionally active promoter/enhancer marker, a finding consistent with their function as RIEs (Figure S4D-F) [17]. Interestingly, all three RIEs coincide with ESC-related Multiple Transcription-factor binding Loci (MTLs) - regions short in size, bound by multiple TFs, which are believed to act as sites of enhanceosome assembly in ESCs (Figure 3C-E, bottom). The elements have been also annotated as ES-specific Super-enhancer regions (Figure 3C-E, bottom) [42,48]. Taken together, the Lefty1700, Pou5f11800 and Upp1800 identified by our unbiased approach (Figure 3B), share many structural characteristics with functional enhancer elements and they are part of previously known regulatory elements in ESCs and therefore, they could indeed function as true RIEs.
2.5. The Upp1800 Element Functions as a Reprogramming Inducible Enhancer Marking Cells Undergoing Reprogramming to Pluripotency
To investigate whether the three putative regulatory elements act as true RIEs, we generated reporter constructs in which a minimal promoter driving the expression of the GFP gene was fused to the test enhancers immediately upstream, that is, in a position similar to their natural genomic location (Figure 4A, left panel). The resulting constructs were cloned into a vector suitable for the generation of lenti-viral particles. Next, the produced lenti-viruses bearing the putative RIE reporters were used to transduce MEFs with the OKSM transgene incorporated into the Col1a1 locus and the rtTA*M2 transactivator under the ROSA 26 promoter [49,50]. Interestingly, we found that initiation of cellular reprogramming with the addition of DOX is accompanied by the activation of the putative RIEs. We observed a robust GFP expression driven by all three putative RIEs during reprogramming (Figure 4B, Figure S5A-B). More specifically, during the first 3 days of reprogramming, a gradually increasing number of GFP-expressing cells (GFP(+) cells) appeared within the population of cells undergoing reprogramming. Morphological examination revealed that the majority of the GFP(+) cells had abandoned their mesenchymal phenotype as they appeared smaller, round, with an increased number of intercellular contacts (especially since day 3) (arrows in Figure 4B and Figure S5A-B). This is a profound alteration characterizing cells that undergo MET [8]. Importantly, many of the GFP(+) cells cluster within the early iPSC colonies, especially those cells expressing the Lefty1700 and Upp1800 reporters (see day 6 in Figure 4B and Figure S5A). In contrast, expression of the Pou5f11800 reporter appears to be less restricted to the early iPSC colonies. These experiments strongly suggest that the Lefty1700, Pou5f11800 and Upp1800 function as ESC RIEs and they mark cells undergoing phenotypical transitions during reprogramming. Significantly, the Lefty1700 and Upp1800 reporters, not only specifically mark activation of GFP gene expression after initiation of reprogramming, but they also mark the cells that will form the early iPSCs colonies (Figure 4B, Figure S5A). On the other hand, the Pou5f11800 RIE suffices to warrantee transcriptional activation in the context of reprogramming only, without marking specifically the early iPSCs formations (Figure S5B).
Next, we asked whether these putative RIEs suffice to regulate the expression of their adjacent genes during reprogramming. Thus, we compared the expression pattern of the endogenous genes (Lefty1, Pou5f1 and Upp1) with the expression of the exogenous GFP controlled by the respective regulatory elements (Figure 4C, Figure S5C-D). Remarkably, we found that both the Lefty1 and Upp1 GFP reporters display expression patterns that are similar to their endogenous counterparts, thus implying that these elements are the main regulatory enhancers controlling the expression of these genes. Of note, the small temporal difference (1-2 days) observed between the GFP and the endogenous gene expression pattern could be explained by the fact that as the exogenous RIE-GFP construct can be incorporated in various random open chromatin genomic sites, it may lack an appropriate epigenetic landscape, which normally could delay the endogenous gene expression. Since the Lefty1700 element simulates the expression pattern of the endogenous gene during the first 6 days only, followed by a gradual decrease in its activity, it implies that this element is not sufficient to support Lefty1 gene expression throughout the entire course of reprogramming, but it suffices only for the initial stages of the process (Figure S5C). On the other hand, the Pou5f11800 element did not mimic the expression program of the endogenous gene (Figure S5D), thus suggesting that Pou5f1 expression is controlled by additional regulatory elements during reprogramming.
2.6. The Cells “Marked” by the Reprogramming Inducible Enhancer Upp1800 Element Achieve Earlier and More Robust Induction of the 9TR Network Leading to Efficient Reprogramming
We described above the integration of multiple features that led us to conclude that the Upp1800 and Lefty1700 enhancers function as autonomous RIEs activated early during reprogramming marking the cells undergoing MET in early iPSC colonies (Figure 4B, Figure S5A). Therefore, we used the RIE Upp1800 GFP reporter as a tool to isolate the cells undergoing reprogramming by monitoring GFP expression in order to explore their transcriptional and phenotypical properties (Figure 4A, right panel). We chose the Upp1800 GFP reporter because it accurately recapitulates the expression of the endogenous Upp1 gene throughout reprogramming (Figure 4C). FACS-isolated Upp1800-GFP(+) cells and GFP(-) cells on day 4 of reprogramming were re-plated in parallel and their reprogramming efficiency was calculated. We found that the GFP(+) cells produced two times more AP-positive iPSC colonies as compared to the GFP(-) cells (Figure 4D). This finding is in agreement with the reporter assays, where we observed high level expression of Upp1800 driven GFP expression in early iPSC colonies (Figure 4B, Figure S5E).
To investigate the molecular signature of the Upp1800-GFP(+) cells that enables them for efficient reprogramming, as opposed to the GFP(-) cells, we performed RNA-seq experiments side by side with the GFP(-) cells used as a control to “capture” the underlying molecular pathways supporting highly efficient reprogramming. Comparison of the RNA-seq data between GFP(+) and GFP(-) cells revealed that these two subpopulations are characterized by distinct transcriptiome profiles, since we identified a significant number of up- and down-regulated Differentially Expressed Genes (DEGs) (Figure 4E-F). More specifically, the GFP(+) cells, where the Upp1800 RIE is activated, bear 718 up-regulated and 394 down-regulated DEGs as compared to the GFP(-) cells (Figure 4F, Table S6). Functional enrichment analysis (GSEA) revealed that the up-regulated genes are related to intercellular adhesion and various signaling processes, whereas down-regulated genes are involved in chemotaxis and response to chemokines, processes indicative of the complex cell-cell interactions and the phenotypic alterations occurring during the early days of reprogramming (Figure 4G).
To further investigate the reconstruction of molecular networks marking the reprogrammable cells, we used the MCODE algorithm [51] to cluster groups of DEGs involved in the various biological processes described above (Figure S6). This detailed analysis verified that indeed in the GFP(+) cells, clusters of down-regulated genes are formed which are involved in chemotaxis, in responses to extracellular signals (Figure S6, Clusters 1-2), in cell migration and EMT (Figure S6, Clusters 1-2, 4-5, 9). For example, Bmp4, an EMT-promoting gene functioning as a reprogramming inhibitor [52,53] is down-regulated in GFP(+) cells, thus relieving the cells from a reprogramming barrier (Figure S6, Cluster 5). On the contrary, genes involved in the establishment of cell adhesion and epithelial functions are up-regulated, including among others Dcn and various basement membrane components like laminin and collagen IV genes (Figure S6, Clusters 5, 9). These genes function by facilitating MET, a requirement for reprogramming. Furthermore, genes involved in embryonic development and morphogenesis are also up-regulated in Upp1800-GFP(+) cells (Figure S6, Clusters 5, 7-8). Of these, an important example includes the telomerase reverse transcriptase gene (Tert) (Figure S6, Cluster 5). This finding further validates our experimental approach since regulation of telomere length by the induced Tert is critical for the achievement of the high pluripotency potential of ES and iPS cells [54,55,56,57]. In this context, we also identified that Tpp1, a gene regulating Tert binding to telomeres during MEF reprogramming [58], is also up-regulated in the Upp1800-GFP(+) cells (Table S6). As expected, we showed that a number of Hox TFs, previously implicated in high efficiency reprogramming [59], are also up-regulated in the RIE-GFP(+) cells (Figure S6, Cluster 7). Taken together these data further validate our previous results indicating that the RIEs could mark specific cells in a population, which undergo global morphological and molecular alterations in the beginning of reprogramming. More specifically, we have been able to “tag” cells that undergo MET in which specific sets of genes involved in the transition have been activated.
We have previously shown that the small percentage of cells completing reprogramming reconstruct a dynamic gene regulatory network known as 9TR-GRN [10]. We found that the TFs Gli2 and Irf6, which are direct targets of OSKM and components of the 9TR-GRN are up-regulated in the Upp1800 GFP(+) cells (Figure 4H, Figure S5F, S6I). Next, we sorted Upp1800-GFP(+) cells and GFP(-) cells on day 4 of reprogramming followed by replating to evaluate their molecular characteristics at later time points. First, we examined the EMT/mesenchymal markers Snail1 and Vimentin (Vim) along with the 9TR-GRN members Irf6 and Nanog, 12 days after cell sorting and re-plating (Figure 4I, Figure S5G). We found that the GFP(+) cells bear decreased levels of the EMT master regulator Snail1, despite the fact that Snail1 expression was nearly equal between the two cell populations on day 4 before re-plating (Figure S5G, left panel). This observation suggests that the GFP(+) cells, which over-express Gli2 and Irf6 early after OSKM induction, abandon the mesenchymal phenotype more efficiently. Interestingly, the classic EMT marker Vim is down-regulated in the GFP(+) cells as early as from day 4 (when the sorting is performed) (Figure S5G, right panel), in agreement with our previous data indicating that the Upp1-GFP(+) cells have epithelial characteristics (Figure 4B, 4G, Figure S6E,I). Intriguingly, Irf6 is expressed at 4 times higher levels in Upp1800-GFP(+) than in GFP(-) cells on day 4, but later these expression levels are similar (Figure 4I). This implies that the RIE Upp1800 marks cells acquiring a temporal “advantage” over the rest of the population in reconstructing the 9TR-GRN network. As a result, these cells could acquire earlier and more efficiently various pluripotent molecular components. Consistent with this is our observation that 12 days after sorting, the GFP(+) cells express more Nanog transcripts than the GFP(-) cells (Figure 4I). Taken together, the data described above strongly suggest that the RIE Upp1800 is an efficient enhancer-trap for the early identification and isolation of cells entering the reprogramming trajectory.
3. Discussion
Cellular reprogramming is a stepwise stochastic process characterized by the emergence of multiple intermediate cell states towards MET from which a small percentage of cells continues to be reprogrammed to achieve pluripotency [3,4,5,10]. Reactivation of the pluripotency gene regulatory network by OSKM requires the rewiring of many gene regulatory elements to permanently inactivate somatic enhancers, to transiently activate enhancers of genes required for initiation of the process and finally to activate the pluripotency enhancers. In this study, we have highlighted the complexity of OSKM action by creating a composite OSKM ChIP-seq dataset [10,16,17,18,38] used to map in detail the association of the reprogramming factors with the genome to reveal how they orchestrate the gradual acquisition of pluripotency. Contrary to many previous studies, we focused on the temporal and permanent OSKM binding patterns to different genomic elements aiming to examine how the OSKM ESC binding pattern is achieved and established during reprogramming. Within this context, we identified novel putative regulatory elements bound by OSKM in a complex and dynamic fashion, some of which are capable of functioning as RIEs. We have exploited the transcriptional regulatory property of these elements and isolated cells undergoing reprogramming in real-time. Their characterization not only independently validated our previously identified 9TR-GRN as a critical player of reprogramming, but importantly, it also revealed the simultaneous reconstruction of additional reprogramming-specific gene networks controlling the global morphological and molecular alterations required for achieving pluripotency.
We discovered that of the 83,674 genomic sites occupied by OSKM in ESCs, 11,962 sites are pre-bound in MEFs by the endogenously expressed KM, and then they are immediately co-occupied, at least some of them, by OS from day 1 (Early ESC sites). These results suggest that pre-bound KM recruit OS via cooperative interactions. This is an important observation because the role of Myc is usually overlooked in reprogramming and it is often omitted from the transcription-factor “cocktails” [60,61]. However, even in the absence of Myc co-expression with OSK, it is still present in MEFs and its inhibition has dramatic effects in the reprogramming efficiency [21,60].
In contrast to the stably bound OSKM throughout reprogramming, a large number of genomic sites are occupied by OSKM (individually or as a group) in a temporal manner. Apparently, during reprogramming, OSKM become highly mobile by constantly binding to new sites and/or abandoning others inducing alterations in chromatin structure and the expression of the nearby genes. Thus, the dynamic binding of OSKM is directly related to the re-organization of chromatin and the gradual establishment of the pluripotent epigenetic landscape. It is conceivable to assume that at least in part, the inherent stochasticity of cellular reprogramming is due to the low percentage of cells that will be able to establish the correct pattern of dynamic OSKM binding suitable for orchestrating the correct transcriptional programs towards pluripotency.
An interesting implication of the dynamic OSKM binding patterns described herein stems from the fact that Klf4 appears to regulate the formation of “hubs” composed of pluripotency genes and ESC Super-enhancers [38]. Thus, we propose that the temporal pattern of Klf4 binding during reprogramming shown here may regulate the instruction of different hubs consisting of different enhancers associated transiently with the same or different genes during reprogramming. Based on the above, we propose that single genes may “use” multiple OSKM-bound enhancer elements, loaded at different time-points, to ensure the execution of an accurate reprogramming expression pattern.
Our extensive characterization of the various classes of OSKM-bound elements and their association with pluripotency genes prompted us to test whether some of these elements can be used as enhancer traps to isolate and study the small percentage of cells that are stochastically “chosen” to be reprogrammed. We developed and applied an unbiased method based on the OSKM global binding pattern for the identification of putative RIEs among the early-bound regions. We identified 66 putative RIEs, including the Lefty1700, Pou5f11800 and Upp1800 elements. We showed that all three elements function as true reprogramming inducible enhancers. Interestingly, these elements map within ESC-specific Super-enhancers [42] and within loci that have been previously implicated in regulation of gene expression [62,63,64], thus validating our unbiased approach used to identify RIEs.
We found that, at least one of these RIEs, the Upp1800 element when fused to a minimal promoter-GFP cassette can accurately reproduce the expression of the endogenous gene specifically in the cells undergoing reprogramming. Therefore, we used the Upp1800-GFP reporter to isolate GFP(+) and GFP(-) cells during the first days of reprogramming. Importantly, the GFP(+) isolated cells have twice as much the capacity to complete their reprogramming process as compared to the control GFP(-) cells, since we showed that they acquire more efficiently the epithelial phenotype. This observation is also supported by our finding that genes involved in MET are significantly enriched in the Upp1800-GFP(+) cells (e.g. Dcn, Col4a1/2, Lama2, Pdgfrb, Serpine1/2, Slit3, Tagln, Thbs2 etc.). Importantly, we found that the GFP(+) cells express at high levels the 9TR-GRN members Gli2 and Irf6, as early as on day 2 of reprogramming, leading to a more robust activation of the 9TR network. As Nanog induction is the final component required for reconstructing the 9TR-GRN network its efficient up-regulation paves the route to pluripotency (Figure 4J). In summary, by using the OSKM complex DNA binding patterns during reprogramming together with gene expression data as guides, we were able to accurately identify functional enhancers induced in reprogramming (RIEs). We provided evidence that RIEs can be used as effective biomarkers for the identification and isolation of rare cell populations that are being appropriately “prepared” to enter reprogramming successfully very early in the process. This may have important implications in the development of methods to achieve high reprogramming efficiencies for medical applications.
4. Materials and Methods
4.1. Experimental Protocols
4.1.1. MEFs Isolation from Mouse Embryos
For our cellular reprogramming experiments we used mouse embryonic fibroblasts (MEFs) from cross-bred homogygous transgenic mice originating from the B6;129S4-Col1a1tm1(tetO-Pou5f1,-Klf4,-Sox2,-Myc)Hoch/J (RRID:IMSR_JAX:011001) and B6.Cg-Gt(ROSA)26Sortm1(rtTA*M2)Jae/J (RRID:IMSR_JAX:006965) strains (“4F-MEFs”) [49,50]. MEFs were isolated as described before [10]. In brief, mouse embryos E13.5-14.5 were harvested from euthanized pregnant female mice. After removing the head, the heart and the liver, the emrbyonic tissue was chopped up using a razor blade and turned into single-cell suspension by incubation in 0.25% Trypsin solution in PBS (Gibco™, #15090046) and multiple passes through 18G and 21G syringes. MEFs were cultured at 37οC and 5% CO2 concentration, in high-glucose DMEM (Sigma-Aldrich, D6429) supplemented with heat-inactivated FBS at 10% final concentration (Pan Biotech, P30-1985), 1x GlutaMAX™ Supplement (Gibco™, #35050-061), 1x MEM Non-Essential Amino Acids Solution (Gibco™, #11140-050) and Penicillin-Streptomycin mix diluted at 1/100 (Gibco™, #15140-122).
4.1.2. Cellular Reprogramming Protocol
An appropriate number of “4F-MEFs” (depending on the protocol) was placed in plates (day 0) and cellular reprogramming (OKSM over-expression) was induced with the addition of doxycycline (DOX) at 2ug/ml (Sigma-Aldrich, D9891) in MEF medium as described above, but supplemented with 15% FBS. The medium was refreshed every two days. Upon the emergence of the first early iPSC colonies (around day 8) the medium was replaced with ESC medium: KnockOut™ DMEM (Gibco, #10829018), 20% Pansera FBS designed for ESCs (Pan Biotech, P30-2602), 1x GlutaMAX™ Supplement (Gibco™, #35050-061), 1x MEM Non-Essential Amino Acids Solution (Gibco™, #11140-050), Penicillin-Streptomycin mix diluted at 1/100 (Gibco™, #15140-122) and 20ng/mL mLIF (Santa Cruz Biotechnology, sc-4378).
4.1.3. Nanog Chromatin Immunoprecipitation Followed by High-Throughput Sequencing
ChIP was performed as described before [10]. Cells were cross-linked with 1% formaldehyde at room temperature for 10 min, and then quenched with 0.125M glycine for 5 min. Cells were washed with PBS and harvested with scraper. Lysis was performed in Lysis buffer [50mM Hepes (pH 7.9), 140mM NaCl, 1mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100], washed in Wash buffer [10mM Tris-HCL (pH 8.1), 200mM NaCl, 1mM EDTA (pH 8.0), 0.5mM EGTA (pH 8.0)] and then resuspended in Sonication buffer [0.1% SDS, 1mM EDTA, 10mM Tris (pH 8.1)]. Chromatin was sheared to 200-500bp fragments in the Covaris S2 sonicator using TC12×12mm tubes (Tube AFA Fiber and Cap, Covaris) for 12 min (200 cycles per burst). After the sonication, we added Triton X-100 and NaCl to reach a final concentration of 1% and 150mM, respectively. The chromatin was then centrifuged and filtered via a 0.2um filter syringe. 10ug chromatin was incubated with 3,6ug rabbit IgG (crude serum) and 10ug anti-Nanog (Cell Signaling Technology Cat# 8785, RRID:AB_11220438) at 4oC overnight. The next day pre-equilibrated protein G DynabeadsTM (Thermo Fisher Scientific, 10004D) in IP buffer [0.1% SDS, 1mM EDTA, 10mM Tris (pH 8.1), 1% Triton X-100, 150mM NaCl] were mixed with the chromatin and antibody solutions. The mix was left to incubate for 1 hour at room temperature. Next, the beads were washed with Low salt [0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Hepes-KOH (pH 7.9), 150mM NaCl], High salt [0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Hepes-KOH (pH 7.9), 500mM NaCl] and LiCl buffer [100mM Tris-HCl (pH 7.5), 0.5M LiCl, 1% NP-40, 1% Sodium deoxycholate]. The immunoprecipitated chromatin was digested with Proteinase K (Roche Life Sciences, 03115828001) at 50οC for 15 min, followed by incubation with RNase A at 65°C overnight and an extra step of Proteinase K for 1 hour at 50οC. The released DNA was cleaned-up using NucleoMag clean-up and selection kit (Macherey-Nagel, 744970) and the elution was performed in TE/10 buffer. Eluted DNA was then used for downstream analyses.
The Nanog ChIP-seq library construction and sequencing were carried out at the Greek Genome Center (GGC) of BRFAA. Libraries were generated with NEBNext® Ultra II™ DNA Library prep Kit for Illumina® (E7645L, NEB), as indicated by the manufacturer’s guidelines using 10ng of DNA (chromatin that was not incubated with the antibody was used as input DNA). The quality of the generated libraries was validated with the Agilent Bioanalyzer DNA 1000 chip. Subsequently, the libraries were quantitated with the Qubit™ High Sensitivity (HS) spectrophotometric method and pooled in equimolar amounts for sequencing. Approximately, 25 million, 101bp-long, single-end reads were generated with Illumina NovaSeq 6000 sequencer. The data of the Nanog ChIP-seq experiment are deposited in the Gene Expression Omnibus data repository under the GEO accession ID GSE274131.
4.1.4. ATAC-Seq
ATAC-seq libraries from different time-points were prepared starting from 50,000 cells, following previously published protocols [65,66,67,68]. Briefly, cell pellets were lysed in Lysis Buffer [10mM Tris-HCl, pH 7.5, 10mM NaCl, 3mM MgCl2, 0.1% NP-40, 0.1% Tween-20, 0.01% Digitonin (Promega, G9441)] for 3 min on ice, washed with Wash Buffer [10mM Tris-HCl, pH 7.5, 10mM NaCl, 3mM MgCl2, 0.1% Tween-20] and centrifuged at 500g for 10 min at 4oC. Each pellet (nuclei) was resuspended in 50ul Transposition reaction mix [1x Tagment DNA Buffer (Illumina, 15027866), 16.5ul 1x PBS, 0.1% Tween-20, 0.01% Digitonin (Promega, G9441), 2.5ul Tn5 Transposase (Tagment DNA Enzyme 1, Illumina, 15027865)] and incubated at 37oC for 30 min on a thermomixer at 1000rpm. DNA purification was performed using the Qiagen MinElute Reaction Cleanup Kit (Qiagen, 28204). Library purification was performed with the AMPure XP beads (Beckman Coulter, A63880). The quality of libraries was assessed using an Agilent 2100 Bioanalyzer and libraries were quantified with the Qubit™ High Sensitivity (HS) spectrophotometric method (Invitrogen). Libraries were sequenced paired-end using a NovaSeq 6000 sequencer at the Greek Genome Center at BRFAA. The data of the ATAC-seq experiment are deposited in the Gene Expression Omnibus data repository under the GEO accession ID GSE274130.
4.1.5. Construction of Enhancer Reporters and Production of Lenti-Viral Particles
Cloning of regulatory elements and construction of enhancer reporters
We isolated the three identified enhancer elements (Lefty1700, Pou5f11800, Upp1800) from the mouse genome via PCR using specific primer pairs (Table S7). The primers were designed to enclose the identified OSKM peaks of the respective RIE element in the smallest amplicon size possible. For the construction of our enhancer reporters we utilized the plasmidic vector LeGO-G2 [69]. LeGO-G2 was a gift from Boris Fehse (Addgene plasmid #25917; http://n2t.net/addgene:25917; RRID: Addgene_25917). The strong regulatory element SFFV upstream of the EGFP gene was removed and replaced each time by one of the three isolated enhancer elements and a minimal promoter sequence. As minimal promoter was used either the endogenous proximal promoter sequences of each of the three genes (Table S7), or the SCP1 element [70] which was isolated from the pSTARR-seq_human vector [71]. pSTARR-seq_human was a gift from Alexander Stark (Addgene plasmid #71509; http://n2t.net/addgene:71509; RRID: Addgene_71509). The two types of promoters (SCP1 and endogenous) did not affect the pattern of EGFP expression (data not shown) and thus, were used interchangeably in our reporter assays. LeGO-G2 was used as a positive control in our experiments since it showed stable GFP expression over-time.
4.1.6. Generation of Lenti-Viral Particles
To introduce our constructs into the MEF genome we produced lenti-viral particles via transfection in HEK293T cells (Human Embryonic Kidney 293T; RRID:CVCL_0063). The HEK293T cells were transfected with a combination of the plasmids pMD2.G, psPAX2 and each one of our constructs (separately) using the classic calcium phosphate method. The day after the transfection the supernatant was replaced with fresh medium and 3 days after the medium change, the lenti-viral particles were harvested. HEK293T were cultured at 37οC and 5% CO2 concentration, in high-glucose DMEM (Sigma-Aldrich, D6429) supplemented with heat-inactivated FBS at 10% final concentration (Pan Biotech, P30-1985), 1x GlutaMAX™ Supplement (Gibco™, #35050-061), 1x MEM Non-Essential Amino Acids Solution (Gibco™, #11140-050) and Penicillin-Streptomycin mix diluted at 1/100 (Gibco™, #15140-122). Plasmids pMD2.G (Addgene plasmid #12259; http://n2t.net/addgene:12259; RRID:Addgene_12259) and psPAX2 (Addgene plasmid #12260; http://n2t.net/addgene:12260; RRID:Addgene_12260) were a gift from Didier Trono.
4.1.7. Reporter Assays
- MEFs transduction and cellular reprogramming
For the reporter assays we transduced ~0.8-1x106 early-passage “4F-MEFs” with the above described lenti-viral particles in the presence of polybrene at a final concentration of 6ug/ml (Sigma-Aldrich, H9268) overnight, followed by change of medium. After 2-3 days, an appropriate number of transduced-MEFs (depending on the assay) was re-plated and cellular reprogramming was induced with the addition of DOX (day 0), as described above.
- 2.
- Microscopy
~600.000 “4F-MEFs” transduced with the enhancer reporters were plated in 10cm plates and cellular reprogramming was induced after 24 hours by the addition of DOX, as described above. Cells were observed regularly under the microscope for the monitoring of the reprogramming process and the detection of GFP(+) cells. A Leica DM IRE2 inverted microscope was used (phase contrast and fluorescence microscopy) with an ORCA-Flash 4.0 LT digital camera (Hamamatsu, C11440-42U, SN 001432). The images were captured with the HCImage Live software (Hamamatsu) and processed using FIJI/ImageJ [72]. Brightness adjustments were made uniformly to all the images of a single experiment in order to correctly depict the increase/decrease of GFP intensity between the different time-points of each experiment. The pixel intensities of the images in Figure S5E were transformed using the LOG function to be able to depict both time-points with very low and very high GFP intensity.
- 3.
- Reporter assay for the comparison of exogenous-GFP and endogenous gene expression pattern (Figure 4C, Figure S5C-D)
~70.000 “4F-MEFs” transduced with the enhancer reporters were plated in 6-well plates and cellular reprogramming was induced the next day by the addition of DOX, as already described. For each enhancer-reporter two biological replicates were prepared. Cells were collected using 1x Trypsin solution (Gibco™, 15090046) at different time-points for RNA extraction.
- 4.
- Sorting of Upp1800-GFP(+) and GFP(-) cells for transcriptome analysis and reprogramming efficiency calculation
1.5-3x106 transduced “4F-MEFs” were plated in 10cm plates and reprogramming was initiated after 24 hours (day 0). At least two biological replicates were prepared for each experiment. At day 2 (for day 2 Upp1800 RNA-seq) or day 4 (for efficiency calculation and transcriptome analysis in Figure 4I and Figure S5G) cells were harvested using 1x Trypsin solution (Gibco™, 15090046), washed with PBS and stained with 0.3uM DAPI for 5-10 min (Roche, 236276). Next, cells were washed again with PBS and resuspended in cold Sorting buffer [1x PBS, 5% FBS, 1mM EDTA] to a final concentration of 5-10x106 cells/ml. Cell aggregates were removed prior to sorting with 50um strainer cap filters (BD Biosciences, 340629). GFP(+) and GFP(-) sorted cells aimed for day 2/day 4 transcriptome analyses (RNA-seq and analysis in Figure 4I and S5G, respectively) were used directly for RNA extraction (100-200.000 cells per sample). For reprogramming efficiency calculation alive GFP(+) and GFP(-) cells were counted using Trypan blue and plated in equal numbers on 12-well plates (70-80.000 cells per well). After letting cells to overcome stress and expand for almost 10-12 days, they formed healthy iPSC colonies. To compensate for inequalities in cell duplication rates caused by the stress of the sorting process, before estimating the reprogramming efficiency we re-plated equal numbers of GFP(+) and GFP(-) cells in 6-well plates (100-200.000 cells per well) covered with gelatin (Sigma-Aldrich, G1393) and feeders (C57Bl/6 MEFs treated with 10ug/ml Mitomycin C for 2.5-3 hours). iPSC colonies were left to grow and were cultured 1 week without DOX, prior to AP staining. GFP(+) and GFP(-) sorted cells aimed to be used for transcriptome analysis at a later time-point (“12 days after sorting” in Figure 4I and S5G) were re-plated on 12-well plates (100.000 cells per well) and used for RNA-extraction 12 days later.
- 5.
- Alkaline Phosphatase (AP) staining for reprogramming efficiency calculation
Reprogramming cell cultures were subjected to Alkaline Phosphatase (AP) staining for the identification of iPSC colonies as described before [10]. In brief, cells growing on plates were washed with PBS and fixed with 4% formaldehyde at 4oC for 10 min. The cells were washed with NTMT buffer [100mM Tris-HCl, 100mM NaCl, 50mM MgCl2 and 0.1% Tween-20, pH 9.5] and then stained for AP activity, using NBT/BCIP substrate solution (Roche, 11681451001) diluted 1/100 in NTMT buffer. Reprogramming efficiency was calculated by dividing the number of AP(+) iPSC colonies with the total number of cells which were seeded on the feeder-covered plates and then, by multiplying by 100. Reprogramming efficiency evaluation was performed in biological triplicates.
- 6.
- RNA isolation and real-time PCR
Cells were lysed with NucleoZOL (740404, Macherey-Nagel) and their RNA was extracted following the manufacturer’s instructions. Reverse transcription followed to produce cDNA with the PrimeScript™ RT Reagent Kit (Perfect Real Time) (TaKaRa, RR037A) using oligodT primers and random 6mers. The cDNA quantification was performed in the CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad) using the 2x KAPA SYBR® FAST Universal mix (KAPA, KK4618) and the appropriate primer pair (Table S7). Calculation of expression values was performed with the 2^(-ΔCt) method and Gapdh served as the reference gene. GFP expression values of the enhancer-reporters in Figure 4C and Figure S5C-D were normalized to the GFP values of LeGO-G2 at the respective time-points, to compensate for putative fluctuations in GFP expression caused by the intense epigenetic alterations occuring in reprogramming at the random chromatin integration sites of the reporters.
- 7.
- RNA sequencing
- Reprogramming time-course
“4F-MEFs” (day 0) and cells undergoing reprogramming were isolated at different time-points (day 1, day 2, day 3, day 4, day 6, day 8) and their RNA was extracted using NucleoZOL (740404, Macherey-Nagel), as by the manufacturer’s instructions. Two biological replicates were prepared for each sample. The RNAs were DNAseI-treated and cleaned-up using the classic phenol-chloroform procedure. RNA-seq experiments were carried out in the Greek Genome Center (GGC) of the Biomedical Research Foundation of the Academy of Athens (BRFAA). RNA-seq libraries were prepared using the NEBNext® Ultra II™ Directional RNA Library prep Kit for Illumina® (E7760, NEB) with 1ug of total RNA input. Library QC was performed with the Agilent Bioanalyzer DNA 1000 kit and quantitation with the Qubit™ High Sensitivity (HS) spectrophotometric method (Invitrogen). Sequencing was performed in the Illumina NovaSeq 6000 sequencer and we obtained on average 22x106 single-end, reverse-stranded, 101bp-long reads per sample. The data of the RNA-seq experiment are deposited in the Gene Expression Omnibus data repository under the GEO accession ID GSE274132.
- Upp1800-GFP sorted cells
GFP(+) and GFP(-) cells collected after sorting (as described above) were subjected to RNA extraction using the NucleoSpin RNA kit (740955, Macherey-Nagel). The DNA was removed by a DNAseI on-column step, as instructed by the kit’s manual. The library preparation and sequencing was performed as described above. Two biological replicates were sequenced per sample. The data of the RNA-seq experiment are deposited in the Gene Expression Omnibus data repository under the GEO accession ID GSE279976.
4.2. Bioinformatics Analyses
4.2.1. ChIP-Seq Data Analysis
- 8.
- Selection and primary analysis of published O/S/K/M and Nanog ChIP-seq datasets
For our O/S/K/M ChIP-seq analysis we utilized multiple datasets derived either from our laboratory (current publication and [10]), or by others [16,17,18,38], deposited under the GEO IDs GSE114581, GSE274131 GSE90893, GSE67520, GSE101905 and GSE113429 (Table S1). We selected datasets which use the 4-factor reprogramming system (OKSM) and MEFs as the starting population. Prior to merging with other datasests, we re-analyzed our O/S/K/M ChIP-seq data under the GEO ID GSE114581 [10], wishing to take advantage of the latest adjustments to the existing algorithms. In more detail, Fastq files were aligned against the mm10 mouse genome with Bowtie2 aligner [73] and the output files were filtered for low-quality reads, duplicates, blacklist regions and mitochondrial genome entries with custom-made pipeline from BEDtools [74] and SAMtools [75]. Peak calling was performed with MACS2 callpeak command [76]. Bigwig files were constructed with the deepTools bamCompare tool [77], using the “subtract“ option to remove Input signal from the respective IP signal, and choosing RPKM for normalization. The same pipeline was performed for our Nanog ChIP-seq experiment analysis, but first, the quality of the Fastq files was examined with the FastQC software [78]. Finally, for the production of bigwig files for Figure 3 and Figure S3, raw data of the O/S/K/M published datasets which were used in this study (GSE90893, GSE67520, GSE101905, GSE113429) were downloaded from the SRA database and analyzed as described above.
- 9.
- Merging of datasets
Our O/S/K/M ChIP-seq dataset (GSE114581) was re-analyzed as described above, while the other ChIP-seq peaks were used as published (GSE90893, GSE67520, GSE101905, GSE113429) (see Table S1 for more details). The peaks (bed files) were combined per time-point for each individual transcription factor (using cat and sort functions) and overlaps between peaks from different datasets were combined using the BEDtools mergeBed tool (option “-d 300”) [74]. For the day 1 time-point we combined the 18 hours and day 1 data and for the day 6 time-point we combined the day 5 and day 6 data. We also combined data from both high- (SSEA1+) and low- (Thy1+) efficiency cells -when available, since we are interested in studying the bulk population of cells undergoing reprogramming. For the analysis of OSKM binding as a group (Figure 1E and Figure 2), we merged the Oct4, Sox2, Klf4 and Myc peaks per time-point, as we did above (cat, sort, mergeBed -d 300). The peaks of the combined dataset are presented in Table S2.
For the visualization of the combined dataset in the Integrative Genomics Viewer (IGV) [79] (Figure 3 and S3) we used the normalized bigwig files from each dataset, as produced by the analysis described above. Then, the different bigwig files were merged and averaged for each factor per time-point, using the WiggleTools mean tool [80].
- 10.
- ATAC-seq analysis
Bioinformatics analyses were performed using The Galaxy Suite [81]. The quality of the sequencing reads was evaluated using the FastQC algorithm [78]. Reads were trimmed to 50bp using the Trimmomatic tool [82]. Sequencing reads were mapped to the mm10 version of the mouse genome using the Bowtie2 algorithm with the “very sensitive end to end” option [73]. The Samtools Fixmate command was used [75] prior to duplicate removal with the MarkDuplicates command from Picard tools using the option “do not write duplicates to the output file”. Reads from all samples were downsampled to the same sequencing depth using the Downsample BAM option from Picard tools. Peaks were called using the MACS2 algorithm with the options “no model”, “extension-200”, “shift-100” and a q-value cut-off of 0.05 [76]. Bigwig files were constructed using the bamCoverage command from the Deeptools suite [83].
4.2.2. Investigation of OSKM Binding Sites
- 11.
- Calculation of common binding sites between Oct4, Sox2, Klf4 and Myc
For the analysis presented in Figure 1B-D we utilized our combined dataset after downsampling the Oct4 day 1, day 3 and day 6 datasets. This was necessary in order to avoid a bias over Oct4 overlaps, due to the higher number of Oct4 peaks in our combined dataset. We downsampled randomly the number of Oct4 peaks to match the average number of the Sox2 and Klf4 peaks of the respective time-point, using a custom-made pipeline (sed command). For the calculation of the common sites between each pair of the OSKM factors we used BEDTools intersect (-wa -u options) [74].
- 12.
- Identification of ESC, transient and MEF sites
For this analysis (Figure 1E and Figure 2) we used the merged OSKM peaks and not the individual Oct4, Sox2, Klf4 and Myc sites (see ChIP-seq analysis above). All sites where OSKM were present at the ESC stage were considered as “ESC OSKM binding sites”. For the identification of the “early-bound” and “late-bound” categories of ESC sites presented in Figure 1E and 2A we performed intersection of all the ESC sites with the day 1 OSKM binding sites using the BEDTools intersect tool (-wa -u and -v options, respectively) [74]. For the early-bound ESC subcategories in Figure 2I-L, we performed successive intersections as described above (-wa -u or -v options), with the day 3 and day 6 OSKM binding sites, accordingly. The same was done for the identification of MEF “pre-bound” and “de novo sites” presented in Figure 3A.
For the OSKM “Transient sites” (Figure 1E) we followed a similar logic of successive intersections, as described above, in order to combine all the different categories of transiently-bound sites (sites where OSKM bind at least at one intermediate time-point, but neither in MEFs, nor in ESCs). In more detail, for the transient sites where OSKM bind only at one time-point (i.e. day 1, day 3, or day 6) we removed (-v option) from the respective bed files (i.e. the total day 1, day 3, or day 6 OSKM peaks) the overlaps with the ESC OSKM sites and subsequently, the overlaps with the other two remaining intermediate time-points (i.e. we removed the day 3-day 6, day 1-day 6 and day 1-day 3 OSKM peaks, respectively). Finally, we removed the common peaks of the resulting files with the KM MEF binding sites. The remaining sites were considered the day 1-, day 3- and day 6-only transient OSKM sites (data not shown). The transient sites where OSKM bind at two intermediate time-points (i.e. day 1-day 3, day 1-day 6, and day 3-day 6 sites, data not shown) were calculated by pairwise intersections of the respective time-point OSKM peaks. In each case, for every overlap we kept the original sites of both intersected files and merged them (cat, sort and mergeBed with default options). Then, we removed the overlaps with the remaining intermediate time-point (i.e. day 6, day 3 or day 1, respectively), with ESCs and subsequently with MEFs. Finally, for the transient sites where OSKM bind at all intermediate time-points (day 1-day 3-day 6 sites, data not shown), we intersected in a stepwise manner the three respective OSKM files and for every overlap we kept and merged the original sites of all intersected files. From the resulting file we kept only the sites where OSKM were not also present in ESCs and MEFs. The final file containing all “Transient sites” (shown in Figure 1E) was produced by merging all the above categories of sites (cat, sort and mergeBed with default options).
For the “MEF sites, lost at ESCs” (Figure 1E) we combined the different categories of sites where (OS)KM bind at MEFs but not in ESCs. In more detail, the sites bound only at MEFs (data not shown), were calculated by keeping the KM MEF peaks which did not overlap with ESCs and neither with any of the other time-points in reprogramming. The rest of the MEF sites, which are lost progressively in reprogramming (data not shown) were calculated by merging all the transient site files before the removal of the overlaps to the MEF peaks and intersecting them with the MEFs KM peaks. The final file containing all “MEF sites, lost at ESCs” (shown in Figure 1E) was produced by merging the two above categories of sites (cat, sort and mergeBed with default options).
Using multiIntersect tool, we ensured that the identified (sub)categories of the ESC, transient and MEF OSKM-bound sites did not overlap with each other. Thus, all OSKM sites presented in this study belong to a single (sub)category of sites.
- 13.
- Overlaps of OSKM sites with ESC Super-enhancers
For the calculation of overlaps between the ESC Super-enhancers [42] and the various types of OSKM ESC sites presented in Table S4 we used the BEDTools intersect tool, as above (-wa -u and -c options) [74].
- 14.
- Epigenetic characterization of OSKM binding sites
The percentage of statistically significant ATAC-open and ATAC-closed chromatin regions (heatmaps and Venn diagrams in Figure 2A,I-L) was calculated using the BEDTools intersect tool for each category of sites and for all ATAC-seq examined time-points. The tornado plots were constructed with the plotHeatmap tool of deepTools (v3.5.2) [77]. The matrices that were used as input for the plotHeatmap were calculated with computeMatrix reference-point mode and the options “--referencePoint center” “--averageTypeBins mean”, “--missingDataAsZero”, “-bs 5”.
- 15.
- Gene assignment to OSKM binding sites
Assignment of the various OSKM binding sites to genes was performed with GREAT on-line tool (v4.0.4) [84,85] using the whole genome as background and the “Single nearest gene” option for 1000kb maximum distance. For the calculation of common gene sets between the different OSKM ESC site subcategories (Figure S1, Table S3) we used the Venny tool (v2.1) [86].
- 16.
- Motif analysis
Our de novo motif analysis was performed with HOMER [87]. We used the options “-size given” and “-preparse”. The motifs presented in this paper have a final enrichment p-value<1e-10, appear at least at 5% of the target sequences and have a score ≥0.9.
4.3. Method for the Identification of Putative Reprogramming Inducible Enhancers
To identify Reprogramming Inducible Enhancers (RIEs) in the mouse genome we used the de novo acquired early-bound ESC sites (Figure 3A, right panel). Using GREAT (v4.0.4) [84,85] and the “Single nearest gene” option for 2.5kb maximum distance, we found that 2865 genes are associated with the above regions. Among them, we chose only the sites close to the 610 genes which are up-regulated between MEFs and ESCs, with Log2FC>2 and p-adjusted<0.05 (702 sites). Then, we intersected the resulting sites with all the individual Oct4, Sox2, Klf4 and Myc peaks in reprogramming using BEDTools (-c option) [74] to identify and remove regions with a low number of binding events (=<10 peaks). The remaining 66 sites were considered as putative RIE elemements (Table S5).
4.4. RNA-Seq Analysis
The quality of the sequenced samples was evaluated by the FastQC tool [78]. Then, the FASTQ files were aligned to the UCSC mm10 genome version with the HISAT2 tool (v2.1.0) using the options “-5 4 -3 4” to trim the 5’ and 3’ ends of the reads and “--rna-strandness R” [88]. Annotation of the SAM files to produce the raw counts was performed with the HTSeq htseq-count tool (v0.6.1p1 and 2.0.3) [89] and the options “-s reverse” “-m intersection-nonempty”, using the UCSC mm10refGene.gtf genome annotation file. Normalized counts were calculated using DESeq2 (v1.30.1, estimateSizeFactors) [90]. To calculate the normalized counts for the reprogramming time-course we used also the raw counts of the sample “ESCs wild type” as they are uploaded in the GEO database (GSE215883, Table S1). Genes with zero normalized counts at all time-points were excluded when calculating the median expression value of multiple genes. DEGs were considered the genes with Log2FC>0.58 or <-0.58 and p-adjusted<0.05.
4.5. Functional Enrichment of Gene Sets
For the functional enrichment analyses presented in Figure 2D, 2G and 4G we used Webgestalt [91], while the analyses in Figure S1 were performed with EnrichR [92,93,94]. Finally, for the networks analysis in Figure S6 we utilized the STRING Enrichment tool of the stringApp [95] in Cytoscape [96]. We utilized the libraries GO Biological Process 2023 and GO Molecular Function 2023 [97,98], KEGG Pathways [99,100,101], Reactome Pathways [102], MSigDB Hallmark 2020 [103,104] and PanglaoDB Augmented 2021 [105].
4.6. Network Analysis
For the network analysis of the day 2 Upp1800-GFP(+)/(-) RNA-seq dataset presented in Figure S6 we inserted all the DEGs (Log2FC>0.58 or <-0.58 and p-adjusted<0.05) in Cytoscape STRING protein query [96] to create a “full STRING network” with confidence cut-off 0.4. Then, the network was clustered with the MCODE application [51] using the default parameters and without the “haircut” option. The Log2FC value of each DEG node was assigned to a blue-red color gradient (blue for down-regulated genes, red for up-regulated genes). The color scale has the same minimum and maximum values for all networks appearing in Figure S6.
4.7. Other Graphical Representations
Ggplot2 package [106] was used in R (v4.0.5) for the construction of the gene expression line-plots, the functional enrichment and motif analyses dot-plots, the PCA and volcano plots. All heatmaps were designed using the pheatmap package in R. Statistical analyses and plots in Figure 4 and S5 were performed using GraphPad Prism version 8.0.2 for Windows, GraphPad Software, Boston.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: OSKM early- and late-bound ESC genes; Figure S2: Motif analysis of the ESC OSKM genomic sites; Figure S3: OSKM and Nanog binding to the 9TR-GRN loci; Figure S4: Chromatin structure at the Lefty1700, Pou5f11800 and Upp1800 elements during cellular reprogramming; Figure S5: The Lefty1700 and Pou5f11800 identified putative reprogramming inducible enhancers (RIEs) and additional data for the Upp1800-GFP(+) and GFP(-) cells during cellular reprogramming; Figure S6: Dense subnetworks of the DEGs from the day 2 Upp1800-GFP(+) and GFP(-) cells RNA-seq; Table S1: The datasets used in the present study; Table S2: Combined dataset ChIP-seq peaks for OSKM and Nanog; Table S3: Genes associated with the identified OSKM-bound sites using the GREAT algorithm; Table S4: Overlap of the ESC Super-enhancers with the identified OSKM-bound sites; Table S5: List of the identified putative Reprogramming Inducible Enhancers (RIEs); Table S6: Day 2 Upp1800-GFP(+) and GFP(-) RNA-seq dataset (normalized counts and DEGs list); Table S7: Primers used in the present study and DNA sequences of the cloned RIEs.
Author Contributions
Conceptualization, E.K., D.V., A.Po. and D.T.; methodology, E.K., D.V., S.F., A.Po. and G.V.; validation, E.K. and D.V.; formal analysis, E.K., D.V. and S.F.; investigation, E.K., D.V., S.F., A.Po., A.Pa. and G.V.; data curation, E.K., D.V. and S.F.; writing—original draft preparation, E.K.; writing—review and editing, E.K., D.V., S.F. and D.T; visualization, E.K. and D.V.; supervision, D.V.; project administration, D.T.; funding acquisition, D.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants to DT from the Greek General Secretariat for Research and Innovation (GSRI) (Cooperative Grants Synergasia I #969, Excellence Award Aristeia I #1567), Emblematic Action Against COVID19, Human Foundation for Research and Innovation (015999), BIOIMAGING.GR (MIS-5002755, European Committee FP7 projects (STPredicta, and Biofos), from the European Economic Area (EL0084), and from the KMW offsets program,. DV was also supported by a scholarship from the Bodossaki Foundation. EK was also supported from the State Scholarships Foundation (Operational Programme MIS-5000432) and from).
Institutional Review Board Statement
The animal study protocol was approved by the Biomedical Research Foundation of Academy of Athens Aanimal welfare and Ethics committee.
Data Availability Statement
The new data produced and presented in the study (raw and analyzed files) are included in the Supplementary Material and/or are openly available in the Gene Expression Omnibus data repository under the GEO accession IDs GSE274130, GSE274131, GSE274132 and GSE279976. The data remain in private status and reviewers can access the records with the following secure tokens: for GSE274130, please use qzodmkcehnshzmv; for GSE274131, please use glepeyqmtzwrla; for GSE274132, please use klcbgawwhbaxhqd; for GSE279976, please use kribkkaqxnuthir. Already published data used in the study (either analyzed again, or used as published) are available in the Gene Expression Omnibus data repository under the GEO accession IDs GSE114581, GSE90893, GSE67520, GSE101905, GSE113429 and GSE215883. A detailed list of all the datasets used in this study is also available in the supplementary material.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–72. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Elite and stochastic models for induced pluripotent stem cell generation. Nature 2009, 460, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Buganim Y, Faddah DA, Jaenisch R. Mechanisms and models of somatic cell reprogramming. Nat Rev Genet. 2013, 14, 427–39. [Google Scholar] [CrossRef]
- Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol. 2016, 17, 183–93. [Google Scholar] [CrossRef]
- David L, Polo JM. Phases of reprogramming. Stem Cell Res. 2014, 12, 754–61. [Google Scholar] [CrossRef]
- Samavarchi-Tehrani P, Golipour A, David L, Sung H, Beyer TA, Datti A, et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010, 7, 64–77. [Google Scholar] [CrossRef]
- Li R, Liang J, Ni S, Zhou T, Qing X, Li H, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010, 7, 51–63. [Google Scholar] [CrossRef]
- Shu X, Pei D. The function and regulation of mesenchymal-to-epithelial transition in somatic cell reprogramming. Curr Opin Genet Dev. 2014, 28, 32–7. [Google Scholar] [CrossRef]
- Papathanasiou M, Tsiftsoglou SA, Polyzos AP, Papadopoulou D, Valakos D, Klagkou E, et al. Identification of a dynamic gene regulatory network required for pluripotency factor-induced reprogramming of mouse fibroblasts and hepatocytes. EMBO J. 2021, 40, e102236. [Google Scholar] [CrossRef]
- Deng W, Jacobson EC, Collier AJ, Plath K. The transcription factor code in iPSC reprogramming. Curr Opin Genet Dev. 2021, 70, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Sridharan R, Tchieu J, Mason MJ, Yachechko R, Kuoy E, Horvath S, et al. Role of the murine reprogramming factors in the induction of pluripotency. Cell 2009, 136, 364–77. [Google Scholar] [CrossRef] [PubMed]
- Polo JM, Anderssen E, Walsh RM, Schwarz BA, Nefzger CM, Lim SM, et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 2012, 151, 1617–32. [Google Scholar] [CrossRef] [PubMed]
- Soufi A, Donahue G, Zaret KS. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 2012, 151, 994–1004. [Google Scholar] [CrossRef]
- Cacchiarelli D, Trapnell C, Ziller MJ, Soumillon M, Cesana M, Karnik R, et al. Integrative Analyses of Human Reprogramming Reveal Dynamic Nature of Induced Pluripotency. Cell 2015, 162, 412–424. [Google Scholar] [CrossRef]
- Chen J, Chen X, Li M, Liu X, Gao Y, Kou X, et al. Hierarchical Oct4 Binding in Concert with Primed Epigenetic Rearrangements during Somatic Cell Reprogramming. Cell Rep. 2016, 14, 1540–1554. [Google Scholar] [CrossRef]
- Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, et al. Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell. 2017, 168, 442–459e20. [Google Scholar] [CrossRef]
- Knaupp AS, Buckberry S, Pflueger J, Lim SM, Ford E, Larcombe MR, et al. Transient and Permanent Reconfiguration of Chromatin and Transcription Factor Occupancy Drive Reprogramming. Cell Stem Cell. 2017, 21, 834–845e6. [Google Scholar] [CrossRef]
- Li D, Liu J, Yang X, Zhou C, Guo J, Wu C, et al. Chromatin Accessibility Dynamics during iPSC Reprogramming. Cell Stem Cell. 2017, 21, 819–833e6. [Google Scholar] [CrossRef]
- Schwarz BA, Cetinbas M, Clement K, Walsh RM, Cheloufi S, Gu H, et al. Prospective Isolation of Poised iPSC Intermediates Reveals Principles of Cellular Reprogramming. Cell Stem Cell. 2018, 23, 289–305e5. [Google Scholar] [CrossRef]
- Zviran A, Mor N, Rais Y, Gingold H, Peles S, Chomsky E, et al. Deterministic Somatic Cell Reprogramming Involves Continuous Transcriptional Changes Governed by Myc and Epigenetic-Driven Modules. Cell Stem Cell. 2019, 24, 328–341e9. [Google Scholar] [CrossRef] [PubMed]
- Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, Zaret KS. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 2015, 161, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Roberts GA, Ozkan B, Gachulincová I, O’Dwyer MR, Hall-Ponsele E, Saxena M, et al. Dissecting OCT4 defines the role of nucleosome binding in pluripotency. Nat Cell Biol. 2021, 23, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Li D, Shu X, Zhu P, Pei D. Chromatin accessibility dynamics during cell fate reprogramming. EMBO Rep. 2021, 22, e51644. [Google Scholar] [CrossRef]
- King HW, Klose RJ. The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. Elife 2017, 6, 1–24. [Google Scholar] [CrossRef]
- Valakos D, Klagkou E, Kokkalis A, Polyzos A, Kyrilis FL, Banos A, et al. Combinatorial targeting of a specific EMT/MET network by macroH2A variants safeguards mesenchymal identity. PLoS One 2023, 18, e0288005. [Google Scholar] [CrossRef]
- Theunissen TW, Jaenisch R. Molecular control of induced pluripotency. Cell Stem Cell 2014, 14, 720–34. [Google Scholar] [CrossRef]
- Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 2007, 1, 55–70. [Google Scholar] [CrossRef]
- Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 2007, 448, 318–24. [Google Scholar] [CrossRef]
- Lengner CJ, Camargo FD, Hochedlinger K, Welstead GG, Zaidi S, Gokhale S, et al. Oct4 expression is not required for mouse somatic stem cell self-renewal. Cell Stem Cell 2007, 1, 403–15. [Google Scholar] [CrossRef]
- Stadtfeld M, Maherali N, Breault DT, Hochedlinger K. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell. 2008, 2, 230–40. [Google Scholar] [CrossRef] [PubMed]
- Brambrink T, Foreman R, Welstead GG, Lengner CJ, Wernig M, Suh H, et al. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell. 2008, 2, 151–9. [Google Scholar] [CrossRef] [PubMed]
- Hotta A, Cheung AYLL, Farra N, Vijayaragavan K, Séguin CA, Draper JS, et al. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat Methods 2009, 6, 370–6. [Google Scholar] [CrossRef] [PubMed]
- Rais Y, Zviran A, Geula S, Gafni O, Chomsky E, Viukov S, et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nature 2013, 502, 65–70. [Google Scholar] [CrossRef]
- Chen J, Liu J, Chen Y, Yang J, Chen J, Liu H, et al. Rational optimization of reprogramming culture conditions for the generation of induced pluripotent stem cells with ultra-high efficiency and fast kinetics. Cell Res. 2011, 21, 884–94. [Google Scholar] [CrossRef]
- Bar-Nur O, Brumbaugh J, Verheul C, Apostolou E, Pruteanu-Malinici I, Walsh RM, et al. Small molecules facilitate rapid and synchronous iPSC generation. Nat Methods 2014, 11, 1170–6. [Google Scholar] [CrossRef]
- Vidal SE, Amlani B, Chen T, Tsirigos A, Stadtfeld M. Combinatorial modulation of signaling pathways reveals cell-type-specific requirements for highly efficient and synchronous iPSC reprogramming. Stem cell reports. 2014, 3, 574–84. [Google Scholar] [CrossRef]
- Di Giammartino DC, Kloetgen A, Polyzos A, Liu Y, Kim D, Murphy D, et al. KLF4 is involved in the organization and regulation of pluripotency-associated three-dimensional enhancer networks. Nat Cell Biol. 2019, 21, 1179–1190. [Google Scholar] [CrossRef]
- Huyghe A, Trajkova A, Lavial F. Cellular plasticity in reprogramming, rejuvenation and tumorigenesis: a pioneer TF perspective. Trends Cell Biol. 2024, 34, 255–267. [Google Scholar] [CrossRef]
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014, 15, 178–96. [Google Scholar] [CrossRef]
- Young, RA. Control of the embryonic stem cell state. Cell. 2011, 144, 940–54. [Google Scholar] [CrossRef] [PubMed]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013, 153, 307–19. [Google Scholar] [CrossRef] [PubMed]
- Meno C, Saijoh Y, Fujii H, Ikeda M, Yokoyama T, Yokoyama M, et al. Left-right asymmetric expression of the TGF beta-family member lefty in mouse embryos. Nature 1996, 381, 151–5. [Google Scholar] [CrossRef] [PubMed]
- Tabibzadeh S, Hemmati-Brivanlou A. Lefty at the crossroads of “stemness” and differentiative events. Stem Cells. 2006, 24, 1998–2006. [Google Scholar] [CrossRef]
- Jerabek S, Merino F, Schöler HR, Cojocaru V. OCT4: dynamic DNA binding pioneers stem cell pluripotency. Biochim Biophys Acta. 2014, 1839, 138–54. [Google Scholar] [CrossRef]
- Watanabe S-I, Hino A, Wada K, Eliason JF, Uchida T. Purification, cloning, and expression of murine uridine phosphorylase. J Biol Chem. 1995, 270, 12191–6. [Google Scholar] [CrossRef]
- Li Y, Jiang M, Aye L, Luo L, Zhang Y, Xu F, et al. UPP1 promotes lung adenocarcinoma progression through the induction of an immunosuppressive microenvironment. Nat Commun. 2024, 15, 1200. [Google Scholar] [CrossRef]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008, 133, 1106–17. [Google Scholar] [CrossRef]
- Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005, 121, 465–77. [Google Scholar] [CrossRef]
- Stadtfeld M, Maherali N, Borkent M, Hochedlinger K. A reprogrammable mouse strain from gene-targeted embryonic stem cells. Nat Methods. 2010, 7, 53–5. [Google Scholar] [CrossRef]
- Bader GD, Hogue CW V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003, 4, 2. [Google Scholar] [CrossRef]
- Chen J, Liu H, Liu J, Qi J, Wei B, Yang J, et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat Genet. 2013, 45, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Zeng S, Zhang Y, Ma J, Deng G, Qu Y, Guo C, et al. BMP4 promotes metastasis of hepatocellular carcinoma by an induction of epithelial-mesenchymal transition via upregulating ID2. Cancer Lett. 2017, 390, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Huang J, Wang F, Okuka M, Liu N, Ji G, Ye X, et al. Association of telomere length with authentic pluripotency of ES/iPS cells. Cell Res. 2011, 21, 779–92. [Google Scholar] [CrossRef]
- Wang F, Yin Y, Ye X, Liu K, Zhu H, Wang L, et al. Molecular insights into the heterogeneity of telomere reprogramming in induced pluripotent stem cells. Cell Res. 2012, 22, 757–68. [Google Scholar] [CrossRef]
- Kinoshita T, Nagamatsu G, Saito S, Takubo K, Horimoto K, Suda T. Telomerase reverse transcriptase has an extratelomeric function in somatic cell reprogramming. J Biol Chem. 2014, 289, 15776–87. [Google Scholar] [CrossRef]
- Hidema S, Fukuda T, Date S, Tokitake Y, Matsui Y, Sasaki H, et al. Transgenic expression of Telomerase reverse transcriptase (Tert) improves cell proliferation of primary cells and enhances reprogramming efficiency into the induced pluripotent stem cell. Biosci Biotechnol Biochem. 2016, 80, 1925–33. [Google Scholar] [CrossRef]
- Tejera AM, Stagno d’Alcontres M, Thanasoula M, Marion RM, Martinez P, Liao C, et al. TPP1 is required for TERT recruitment, telomere elongation during nuclear reprogramming, and normal skin development in mice. Dev Cell. 2010, 18, 775–89. [Google Scholar] [CrossRef]
- Nagamatsu G, Saito S, Kosaka T, Takubo K, Kinoshita T, Oya M, et al. Optimal ratio of transcription factors for somatic cell reprogramming. J Biol Chem. 2012, 287, 36273–36282. [Google Scholar] [CrossRef]
- Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008, 26, 101–6. [Google Scholar] [CrossRef]
- Wernig M, Meissner A, Cassady JP, Jaenisch R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell. 2008, 2, 10–2. [Google Scholar] [CrossRef] [PubMed]
- Cao D, Nimmakayalu MA, Wang F, Zhang D, Handschumacher RE, Bray-Ward P, et al. Genomic structure, chromosomal mapping, and promoter region analysis of murine uridine phosphorylase gene. Cancer Res. 1999, 59, 4997–5001. Available online: http://www.ncbi.nlm.nih.gov/pubmed/10519414.
- Okumura-Nakanishi S, Saito M, Niwa H, Ishikawa F. Oct-3/4 and Sox2 regulate Oct-3/4 gene in embryonic stem cells. J Biol Chem. 2005, 280, 5307–17. [Google Scholar] [CrossRef] [PubMed]
- Nakatake Y, Fukui N, Iwamatsu Y, Masui S, Takahashi K, Yagi R, et al. Klf4 cooperates with Oct3/4 and Sox2 to activate the Lefty1 core promoter in embryonic stem cells. Mol Cell Biol. 2006, 26, 7772–82. [Google Scholar] [CrossRef]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013, 10, 1213–8. [Google Scholar] [CrossRef]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015, 109, 21.29.1–21299. [Google Scholar] [CrossRef]
- Ackermann AM, Wang Z, Schug J, Naji A, Kaestner KH. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Mol Metab. 2016, 5, 233–244. [Google Scholar] [CrossRef]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017, 14, 959–962. [Google Scholar] [CrossRef]
- Weber K, Bartsch U, Stocking C, Fehse B. A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Mol Ther. 2008, 16, 698–706. [Google Scholar] [CrossRef]
- Juven-Gershon T, Cheng S, Kadonaga JT. Rational design of a super core promoter that enhances gene expression. Nat Methods. 2006, 3, 917–22. [Google Scholar] [CrossRef]
- Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 2013, 339, 1074–7. [Google Scholar] [CrossRef] [PubMed]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012, 9, 676–82. [Google Scholar] [CrossRef] [PubMed]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012, 9, 357–9. [Google Scholar] [CrossRef] [PubMed]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010, 26, 841–2. [Google Scholar] [CrossRef]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009, 25, 2078–9. [Google Scholar] [CrossRef]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–5. [Google Scholar] [CrossRef]
- Andrews S (Babraham, B. FastQC: a quality control tool for high throughput sequence data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011, 29, 24–6. [Google Scholar] [CrossRef]
- Zerbino DR, Johnson N, Juettemann T, Wilder SP, Flicek P. WiggleTools: parallel processing of large collections of genome-wide datasets for visualization and statistical analysis. Bioinformatics. 2014, 30, 1008–9. [Google Scholar] [CrossRef]
- Galaxy Community. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014, 30, 2114–20. [Google Scholar] [CrossRef] [PubMed]
- Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–91. [Google Scholar] [CrossRef] [PubMed]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa Y, Dyer ES, Bejerano G. WhichTF is functionally important in your open chromatin data? Ay F, editor. PLoS Comput Biol. 2022, 18, e1010378. [Google Scholar] [CrossRef]
- Oliveros, JC. VENNY. An interactive tool for comparing lists with Venn Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010, 38, 576–89. [Google Scholar] [CrossRef]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015, 31, 166–9. [Google Scholar] [CrossRef]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov M V., Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–7. [Google Scholar] [CrossRef] [PubMed]
- Xie Z, Bailey A, Kuleshov M V., Clarke DJB, Evangelista JE, Jenkins SL, et al. Gene Set Knowledge Discovery with Enrichr. Curr Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Doncheva NT, Morris JH, Gorodkin J, Jensen LJ. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25, 25–9. [Google Scholar] [CrossRef]
- Gene Ontology Consortium, Aleksander SA, Balhoff J, Carbon S, Cherry JM, Drabkin HJ, et al. The Gene Ontology knowledgebase in 2023. Baryshnikova A, editor. Genetics. 2023, 224, 1–14. [Google Scholar] [CrossRef]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011, 27, 1739–40. [Google Scholar] [CrossRef] [PubMed]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Franzén O, Gan L-M, Björkegren JLM. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford). 2019, 2019, 1–9. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, 2016. [Google Scholar]
Figure 1.
OSKM bind to the genome in a dynamic fashion during cellular reprogramming. (A) Graphical representation of the experimental approach used to map the OSKM binding sites during cellular reprogramming and their functional output. We integrated published O/S/K/M ChIP-seq experiments to construct a composite ChIP-seq dataset representing the day 0 (D0), day 1 (D1), day 3 (D3), day 6 (D6) time-points together with the ESC stage. These data were further integrated with results obtained from ATAC-seq at the same time-points and with RNA-seq experiments performed at day 0 (D0), day 1 (D1), day 2 (D2), day 3 (D3), day 4 (D4), day 6 (D6), day 8 (D8) and ESCs.; (B) Heatmaps depicting the extent of the combinatorial binding of O/S/K/M throughout reprogramming (Day 1, Day 3, Day 6, and ESC). Values are calculated as the percentage of binding sites occupied by the Oct4 (O), Sox2 (S), Klf4 (K) and Myc (M) transcription factors (rows) in combination with each of the other three factors (columns), per time-point. The value 100% depicts the total binding sites occupied by each factor. Compare row name to column name.; (C) Heatmap depicting the percentage of DNA binding sites occupied by Oct4, Sox2, Klf4 and Myc, which have been preserved between two sequential time-points. For example, the value 38.87% (intersection of Oct4 to Oct4 row with D1→D3 column) represents the percentage of the Oct4 binding sites at day 1 which have been preserved at day 3.; (D) Line-graph depicting the mobility of O/S/K/M by plotting the relative number of new binding sites occupied by each of the O/S/K/M between two sequential time-points of reprogramming.; (E) Schematic representation of the different classes of OSKM binding sites occupied during cellular reprogramming. OSKM are depicted as multi-color circles bound to DNA per time-point (left). The absence of OSKM binding to a specific set of sites at a given time-point as compared to ESCs and vice-versa is marked with an “X”. Each class of OSKM sites is divided in regions either proximal (≤5kb) or distal (>5kb) relative to the neighboring genes’ TSS. The size of the circles in the table represents the number of OSKM binding sites of each of the corresponding class. The median expression fold between ESCs and MEFs (Day 0) for each class of genes is depicted as a heatmap in the figure.
Figure 1.
OSKM bind to the genome in a dynamic fashion during cellular reprogramming. (A) Graphical representation of the experimental approach used to map the OSKM binding sites during cellular reprogramming and their functional output. We integrated published O/S/K/M ChIP-seq experiments to construct a composite ChIP-seq dataset representing the day 0 (D0), day 1 (D1), day 3 (D3), day 6 (D6) time-points together with the ESC stage. These data were further integrated with results obtained from ATAC-seq at the same time-points and with RNA-seq experiments performed at day 0 (D0), day 1 (D1), day 2 (D2), day 3 (D3), day 4 (D4), day 6 (D6), day 8 (D8) and ESCs.; (B) Heatmaps depicting the extent of the combinatorial binding of O/S/K/M throughout reprogramming (Day 1, Day 3, Day 6, and ESC). Values are calculated as the percentage of binding sites occupied by the Oct4 (O), Sox2 (S), Klf4 (K) and Myc (M) transcription factors (rows) in combination with each of the other three factors (columns), per time-point. The value 100% depicts the total binding sites occupied by each factor. Compare row name to column name.; (C) Heatmap depicting the percentage of DNA binding sites occupied by Oct4, Sox2, Klf4 and Myc, which have been preserved between two sequential time-points. For example, the value 38.87% (intersection of Oct4 to Oct4 row with D1→D3 column) represents the percentage of the Oct4 binding sites at day 1 which have been preserved at day 3.; (D) Line-graph depicting the mobility of O/S/K/M by plotting the relative number of new binding sites occupied by each of the O/S/K/M between two sequential time-points of reprogramming.; (E) Schematic representation of the different classes of OSKM binding sites occupied during cellular reprogramming. OSKM are depicted as multi-color circles bound to DNA per time-point (left). The absence of OSKM binding to a specific set of sites at a given time-point as compared to ESCs and vice-versa is marked with an “X”. Each class of OSKM sites is divided in regions either proximal (≤5kb) or distal (>5kb) relative to the neighboring genes’ TSS. The size of the circles in the table represents the number of OSKM binding sites of each of the corresponding class. The median expression fold between ESCs and MEFs (Day 0) for each class of genes is depicted as a heatmap in the figure.
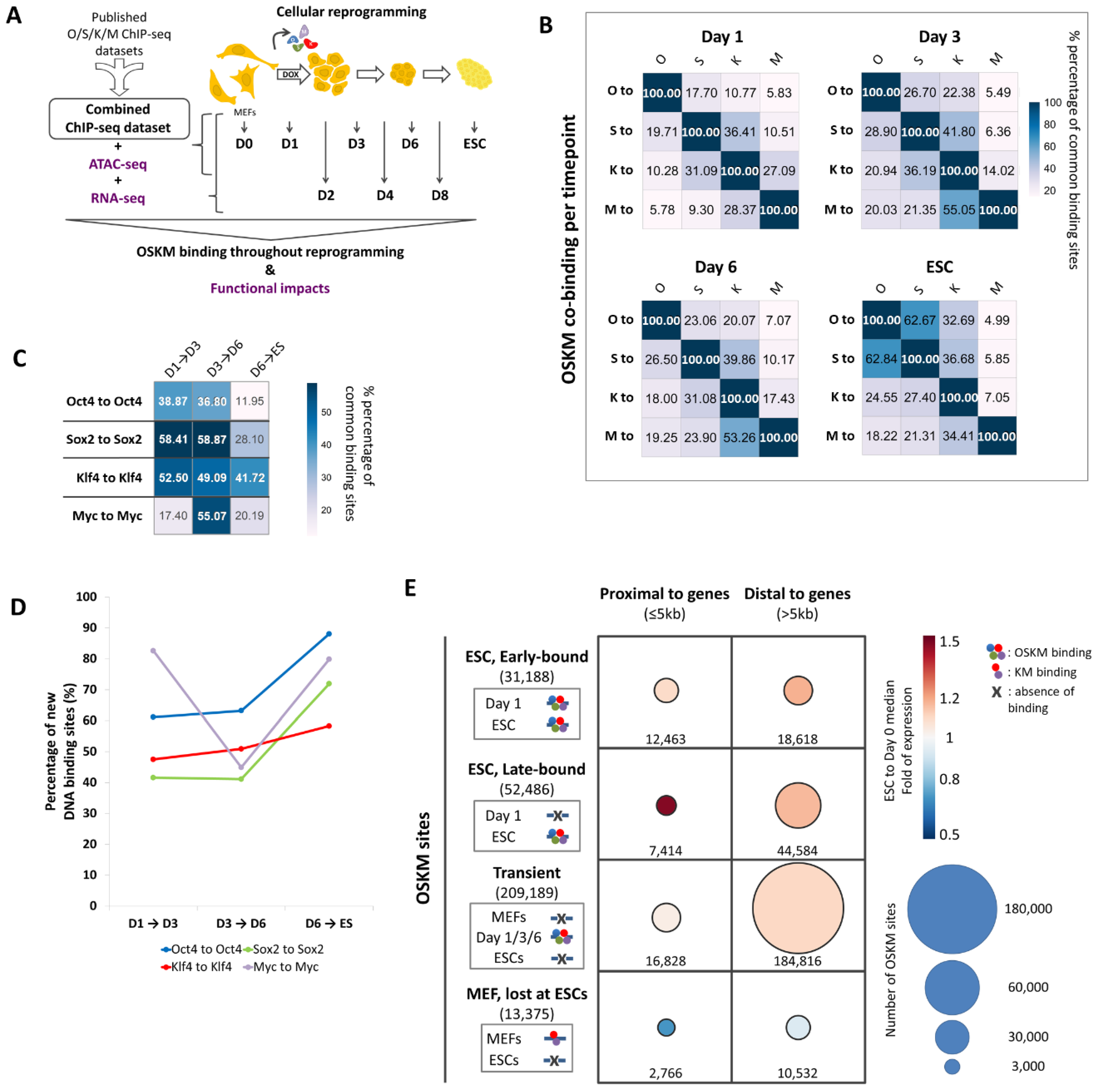
Figure 2.
Identification and characterization of the ESC OSKM binding sites acquired during cellular reprogramming. (A) Graphical representation summarizing the acquisition of the ESCs OSKM binding sites during reprogramming. OSKM are depicted as multi-color circles bound to DNA. The absence of OSKM binding to a specific set of sites on Day 1 as compared to ESCs is marked with an “X”. The heatmap shown at the bottom part of the figure depicts the percentage of each class of OSKM sites lying in open chromatin regions during reprogramming (MEFs, Day 1, Day 3, Day 6, ESCs). Green and purple colors correspond to the percentage of binding sites accessible per time-point (green for >30%, purple for <30%). The Venn diagrams depict the common chromatin open sites between MEFs and ESCs for each class of sites.; (B) Association of the Early-bound OSKM ESC sites with the neighboring genes using the GREAT algorithm. Depicted is the distribution of the OSKM sites relative to the nearest gene TSS within 1000kb distance.; (C) Shown is a line-graph depicting normalized counts for the expression of genes associated with the early-bound OSKM ESC sites during cellular reprogramming. The median expression value is shown for each time-point.; (D) Functional enrichment analysis (Over-representation analysis) of the genes located near the early-bound OSKM ESC sites. Depicted are the top 10 terms as sorted using FDR (FDR<0.01). The GO Biological Process library (non-redundant terms) was used.; (E) Same as in B, but for the late-bound OSKM sites.; (F) Same as in C, but for the late-bound OSKM sites.; (G) Same as in D, but for the late-bound OSKM sites.; (H) Summary and tornado plots depicting the ATAC-seq signal at the early- and late-bound OSKM ESC sites from -1kb to +1km from the center of the OSKM peaks during cellular reprogramming (Day 0, Day 1, Day 3, Day 6, ESCs). The OSKM sites were sorted in a descending order based on the ATAC-seq signal in ESCs.; (I) Graphical representation summarizing the acquisition of the stably-bound OSKM ESC binding sites during reprogramming. OSKM are depicted as multi-color circles bound to DNA. The absence of OSKM binding to a specific set of sites on Day 3, or Day 6, as compared to ESCs is marked with an “X”. The heatmap shown at the bottom part of the figure depicts the percentage of the stably-bound OSKM ESC sites lying in open chromatin regions during reprogramming (MEFs, Day 1, Day 3, Day 6, ESCs). Green and purple colors correspond to the percentage of binding sites accessible per each time-point (green for >30%, purple for <30%). The Venn diagrams depict the common chromatin open sites between MEFs and ESCs.; (J) As in I, but for the early OSKM ESC sites bound on Day 1, Day 3 and ESCs (Dynamically-bound Early ESC sites, where OSKM are absent at Day 6).; (K) As in I, but for the early OSKM ESC sites bound on Day 1, Day 6 and ESCs (Dynamically-bound Early ESC sites, where OSKM are absent at Day 3).; (L) As in I, but for the early OSKM ESC sites bound only on Day 1 and ESCs (Dynamically-bound Early ESC sites, where OSKM are absent at Day 3 and 6).; (M) As in C, but for the stably-bound OSKM ESC sites.; (N) As in C, but for the early OSKM ESC sites bound on Day 1, Day 3 and ESCs (Dynamically-bound Early ESC sites, where OSKM are absent at Day 6).; (O) As in C, but for the early OSKM ESC sites bound on Day 1, Day 6 and ESCs (Dynamically-bound Early ESC sites, where OSKM are absent at Day 3).; (P) As in C, but for the early OSKM ESC sites bound only on Day 1 and ESCs (Dynamically-bound Early ESC sites, where OSKM are absent at Day 3 and 6).
Figure 2.
Identification and characterization of the ESC OSKM binding sites acquired during cellular reprogramming. (A) Graphical representation summarizing the acquisition of the ESCs OSKM binding sites during reprogramming. OSKM are depicted as multi-color circles bound to DNA. The absence of OSKM binding to a specific set of sites on Day 1 as compared to ESCs is marked with an “X”. The heatmap shown at the bottom part of the figure depicts the percentage of each class of OSKM sites lying in open chromatin regions during reprogramming (MEFs, Day 1, Day 3, Day 6, ESCs). Green and purple colors correspond to the percentage of binding sites accessible per time-point (green for >30%, purple for <30%). The Venn diagrams depict the common chromatin open sites between MEFs and ESCs for each class of sites.; (B) Association of the Early-bound OSKM ESC sites with the neighboring genes using the GREAT algorithm. Depicted is the distribution of the OSKM sites relative to the nearest gene TSS within 1000kb distance.; (C) Shown is a line-graph depicting normalized counts for the expression of genes associated with the early-bound OSKM ESC sites during cellular reprogramming. The median expression value is shown for each time-point.; (D) Functional enrichment analysis (Over-representation analysis) of the genes located near the early-bound OSKM ESC sites. Depicted are the top 10 terms as sorted using FDR (FDR<0.01). The GO Biological Process library (non-redundant terms) was used.; (E) Same as in B, but for the late-bound OSKM sites.; (F) Same as in C, but for the late-bound OSKM sites.; (G) Same as in D, but for the late-bound OSKM sites.; (H) Summary and tornado plots depicting the ATAC-seq signal at the early- and late-bound OSKM ESC sites from -1kb to +1km from the center of the OSKM peaks during cellular reprogramming (Day 0, Day 1, Day 3, Day 6, ESCs). The OSKM sites were sorted in a descending order based on the ATAC-seq signal in ESCs.; (I) Graphical representation summarizing the acquisition of the stably-bound OSKM ESC binding sites during reprogramming. OSKM are depicted as multi-color circles bound to DNA. The absence of OSKM binding to a specific set of sites on Day 3, or Day 6, as compared to ESCs is marked with an “X”. The heatmap shown at the bottom part of the figure depicts the percentage of the stably-bound OSKM ESC sites lying in open chromatin regions during reprogramming (MEFs, Day 1, Day 3, Day 6, ESCs). Green and purple colors correspond to the percentage of binding sites accessible per each time-point (green for >30%, purple for <30%). The Venn diagrams depict the common chromatin open sites between MEFs and ESCs.; (J) As in I, but for the early OSKM ESC sites bound on Day 1, Day 3 and ESCs (Dynamically-bound Early ESC sites, where OSKM are absent at Day 6).; (K) As in I, but for the early OSKM ESC sites bound on Day 1, Day 6 and ESCs (Dynamically-bound Early ESC sites, where OSKM are absent at Day 3).; (L) As in I, but for the early OSKM ESC sites bound only on Day 1 and ESCs (Dynamically-bound Early ESC sites, where OSKM are absent at Day 3 and 6).; (M) As in C, but for the stably-bound OSKM ESC sites.; (N) As in C, but for the early OSKM ESC sites bound on Day 1, Day 3 and ESCs (Dynamically-bound Early ESC sites, where OSKM are absent at Day 6).; (O) As in C, but for the early OSKM ESC sites bound on Day 1, Day 6 and ESCs (Dynamically-bound Early ESC sites, where OSKM are absent at Day 3).; (P) As in C, but for the early OSKM ESC sites bound only on Day 1 and ESCs (Dynamically-bound Early ESC sites, where OSKM are absent at Day 3 and 6).
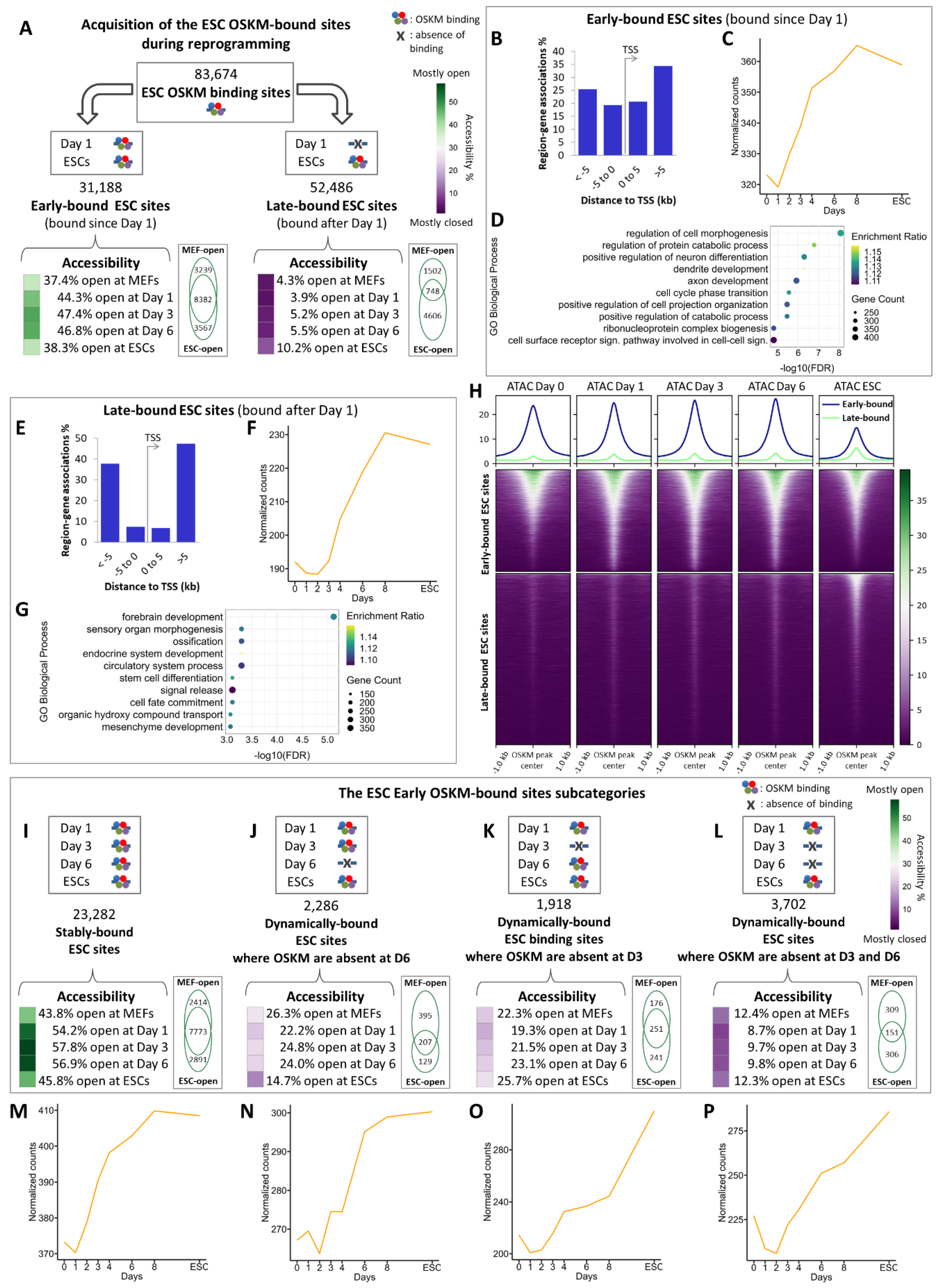
Figure 3.
Identification of putative reprogramming inducible enhancers (RIEs) in the mouse genome. (A) Graphical representation summarizing the acquisition of the early-bound OSKM ESC sites. The left panel shows sites that are pre-bound by KM in MEFs, whereas the right panel depicts the de novo sites occupied by OSKM after initiation of reprogramming. OSKM are depicted as multi-color circles bound to DNA. The absence of OSKM binding to a specific set of sites in MEFs as compared to ESCs is marked with an “X”.; (B) Graphical representation of the unbiased method used to identify putative RIE elements along with the sequential filtering of the OSKM-bound genomic elements.; (C) Shown are ChIP-seq bigwig files in the IGV browser depicting the binding of Oct4 (blue), Sox2 (green), Klf4 (red), Myc (purple) and Nanog (cyan) to Lefty1700 putative RIE in MEFs undergoing reprogramming (Day 1, Day 3 and Day 6) and in control MEFs (Day 0) and ESCs. The scale for each snapshot is shown on the right. The binding signal has been calculated as RPKM after subtraction of the Input signal from the respective IP signal. Statistically significant peaks are depicted with a black bar below the respective lane. The ESC Multiple Transcription Factor binding Loci (ESC-MTL) is depicted as a light green bar. The Lefty1 ESC-specific Super-enhancer (ESC-SE) is depicted as a petrol bar. The position of the Lefty1700 element is depicted as a dark grey bar at the bottom of the panel.; (D) As in C, but for the Pou5f11800 element.; (E) As in C, but for the Upp1800 element.; (F) Shown is a line-graph depicting normalized expression units of the Lefty1 gene during reprogramming. The bottom part of the panel summarizes the binding of OSKM at each time-point (data taken from Figure 3C). Oct4 is depicted in blue, Sox2 in green, Klf4 in red and c-Myc in purple.; (G) As in F, but for the Pou5f1 gene and the Pou5f11800 element. Data taken from Figure 3D.; (H) As in F, but for the Upp1 gene and the Upp1800 element. Data taken from Figure 3E.
Figure 3.
Identification of putative reprogramming inducible enhancers (RIEs) in the mouse genome. (A) Graphical representation summarizing the acquisition of the early-bound OSKM ESC sites. The left panel shows sites that are pre-bound by KM in MEFs, whereas the right panel depicts the de novo sites occupied by OSKM after initiation of reprogramming. OSKM are depicted as multi-color circles bound to DNA. The absence of OSKM binding to a specific set of sites in MEFs as compared to ESCs is marked with an “X”.; (B) Graphical representation of the unbiased method used to identify putative RIE elements along with the sequential filtering of the OSKM-bound genomic elements.; (C) Shown are ChIP-seq bigwig files in the IGV browser depicting the binding of Oct4 (blue), Sox2 (green), Klf4 (red), Myc (purple) and Nanog (cyan) to Lefty1700 putative RIE in MEFs undergoing reprogramming (Day 1, Day 3 and Day 6) and in control MEFs (Day 0) and ESCs. The scale for each snapshot is shown on the right. The binding signal has been calculated as RPKM after subtraction of the Input signal from the respective IP signal. Statistically significant peaks are depicted with a black bar below the respective lane. The ESC Multiple Transcription Factor binding Loci (ESC-MTL) is depicted as a light green bar. The Lefty1 ESC-specific Super-enhancer (ESC-SE) is depicted as a petrol bar. The position of the Lefty1700 element is depicted as a dark grey bar at the bottom of the panel.; (D) As in C, but for the Pou5f11800 element.; (E) As in C, but for the Upp1800 element.; (F) Shown is a line-graph depicting normalized expression units of the Lefty1 gene during reprogramming. The bottom part of the panel summarizes the binding of OSKM at each time-point (data taken from Figure 3C). Oct4 is depicted in blue, Sox2 in green, Klf4 in red and c-Myc in purple.; (G) As in F, but for the Pou5f1 gene and the Pou5f11800 element. Data taken from Figure 3D.; (H) As in F, but for the Upp1 gene and the Upp1800 element. Data taken from Figure 3E.

Figure 4.
The Upp1800 element is a reprogramming inducible enhancer (RIE) which marks cells undergoing reprogramming and the early iPSCs colonies. (A) Shown is the experimental outline used to test the transcriptional capacity of the putative RIEs.; (B) Representative fluorescence microscopy images taken from a time-course reprogramming experiment using MEFs transduced with the lenti-virus bearing the Upp1800-GFP reporter cassette. The brightfield (phase contrast) and fluorescence images were merged in each time-point. The white arrows point to cells abandoning the MEF phenotype (MET). Early iPSC formations are indicated with yellow dashed lines. Scale bar: 150μm.; (C) Line-graph depicting the expression of the endogenous Upp1 gene (orange line, right axis) in comparison with the expression of the Upp1800-GFP transgene (green line, left axis). Shown are the mean expression values from two biological replicates and the standard error.; (D) Bar-graphs showing the iPSCs generation efficiency (%) of the isolated (day 4) Upp1800-GFP(+) cells undergoing reprogramming, as compared to the Upp1800-GFP(-) cells isolated from the same experiment used as control. Three biological replicates are depicted. The mean and the standard error are also depicted. Unpaired two-tailed t-test was performed (t=3.525, df=4, p-value=0.024). Representative images of the AP-staining plates are also shown at the bottom. *: p-value < 0.05; (E) PCA analysis of the RNA-seq profile of Upp1800-GFP(+) (green) and GFP(-) cells (red) isolated on day 2 of reprogramming.; (F) Volcano plot depicting the number of DEGs identified between Upp1800-GFP(+) and GFP(-) cells. Cut-offs for Volcano plot: p adjusted<0.05 and Log2FC>0.58 or <-0.58.; (G) Functional enrichment analysis (GSEA) of the genes expressed in Upp1800-GFP(+) cells isolated on day 2 as compared to control Upp1800-GFP(-) cells. Log2FC was used for ranking. The GO Biological Process library (non-redundant terms) was used. Depicted are the top 5 terms with both positive and negative normalized enrichment scores.; (H) Dot-plots depicting normalized counts for the expression of the Gli2 and Irf6 genes in the Upp1800-GFP(+) and GFP(-) cells, as calculated by RNA-seq on day 2 of reprogramming. Two biological replicates are depicted. The mean and the standard error are also depicted.; (I) Bar-chart depicting the fold change of Irf6 and Nanog expression between the Upp1800-GFP(+) and GFP(-) cells on day 4 (day of isolation) and 12 days after isolation of the cells. The dashed line represents the fold of expression equal to 1. Two biological replicates are depicted for each cell type. The mean and the standard error are also indicated.; (J) Schematic representation of the 9TR-GRN network’s sequential assembly along with the network nodes’ expression status in cells bearing the Upp1800-GFP transgene on Day 1 (top panel, before sorting) and after sorting to Upp1800-GFP(+) and GFP(-) cells (Day 2 to Day 14 of reprogramming, middle and bottom panel, respectively). The representation is shown according to Papathanasiou et al., 2021 [10]. The green arrows represent the positive effect of one regulator to another. The red-lines represent inhibitory effects of one regulator to another.
Figure 4.
The Upp1800 element is a reprogramming inducible enhancer (RIE) which marks cells undergoing reprogramming and the early iPSCs colonies. (A) Shown is the experimental outline used to test the transcriptional capacity of the putative RIEs.; (B) Representative fluorescence microscopy images taken from a time-course reprogramming experiment using MEFs transduced with the lenti-virus bearing the Upp1800-GFP reporter cassette. The brightfield (phase contrast) and fluorescence images were merged in each time-point. The white arrows point to cells abandoning the MEF phenotype (MET). Early iPSC formations are indicated with yellow dashed lines. Scale bar: 150μm.; (C) Line-graph depicting the expression of the endogenous Upp1 gene (orange line, right axis) in comparison with the expression of the Upp1800-GFP transgene (green line, left axis). Shown are the mean expression values from two biological replicates and the standard error.; (D) Bar-graphs showing the iPSCs generation efficiency (%) of the isolated (day 4) Upp1800-GFP(+) cells undergoing reprogramming, as compared to the Upp1800-GFP(-) cells isolated from the same experiment used as control. Three biological replicates are depicted. The mean and the standard error are also depicted. Unpaired two-tailed t-test was performed (t=3.525, df=4, p-value=0.024). Representative images of the AP-staining plates are also shown at the bottom. *: p-value < 0.05; (E) PCA analysis of the RNA-seq profile of Upp1800-GFP(+) (green) and GFP(-) cells (red) isolated on day 2 of reprogramming.; (F) Volcano plot depicting the number of DEGs identified between Upp1800-GFP(+) and GFP(-) cells. Cut-offs for Volcano plot: p adjusted<0.05 and Log2FC>0.58 or <-0.58.; (G) Functional enrichment analysis (GSEA) of the genes expressed in Upp1800-GFP(+) cells isolated on day 2 as compared to control Upp1800-GFP(-) cells. Log2FC was used for ranking. The GO Biological Process library (non-redundant terms) was used. Depicted are the top 5 terms with both positive and negative normalized enrichment scores.; (H) Dot-plots depicting normalized counts for the expression of the Gli2 and Irf6 genes in the Upp1800-GFP(+) and GFP(-) cells, as calculated by RNA-seq on day 2 of reprogramming. Two biological replicates are depicted. The mean and the standard error are also depicted.; (I) Bar-chart depicting the fold change of Irf6 and Nanog expression between the Upp1800-GFP(+) and GFP(-) cells on day 4 (day of isolation) and 12 days after isolation of the cells. The dashed line represents the fold of expression equal to 1. Two biological replicates are depicted for each cell type. The mean and the standard error are also indicated.; (J) Schematic representation of the 9TR-GRN network’s sequential assembly along with the network nodes’ expression status in cells bearing the Upp1800-GFP transgene on Day 1 (top panel, before sorting) and after sorting to Upp1800-GFP(+) and GFP(-) cells (Day 2 to Day 14 of reprogramming, middle and bottom panel, respectively). The representation is shown according to Papathanasiou et al., 2021 [10]. The green arrows represent the positive effect of one regulator to another. The red-lines represent inhibitory effects of one regulator to another.
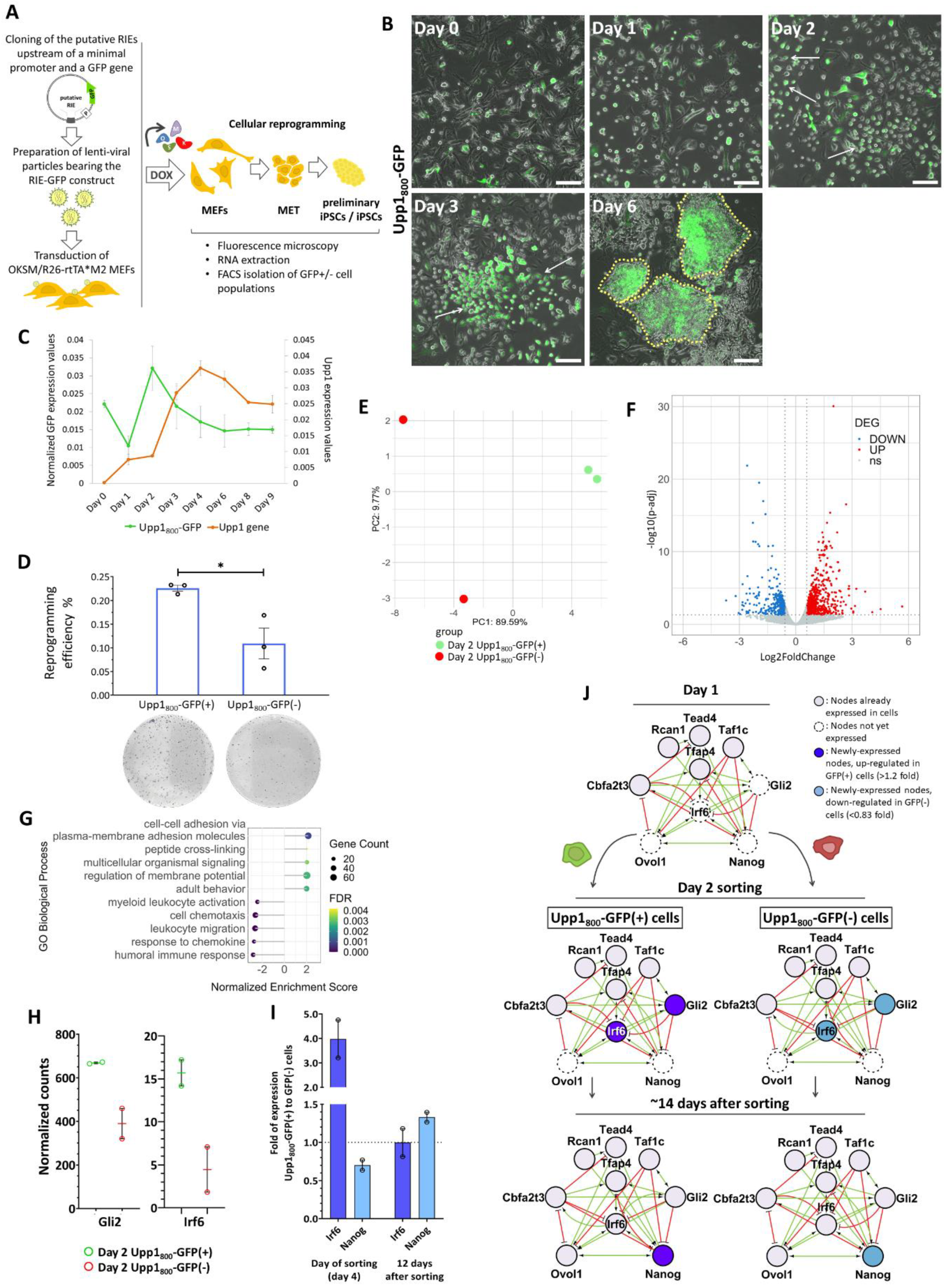
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



