
Submitted:
30 October 2024
Posted:
31 October 2024
You are already at the latest version
Abstract
One of the major challenges in gene therapy (GT) is the efficient and safe introduction of nucleic acids (NA) into eukaryotic cells. This process requires overcoming various biological barriers and navigating complex pathways to reach target cells and achieve their biological function. To address this obstacle, numerous transfection methods have been developed, including physical techniques and the use of genetic vectors, both viral and non-viral. However, until now, no transfection method is 100% safe and efficient. Within the spectrum of non-viral genetic vectors, cationic liposomes formed by cationic lipids (CL), stand out for their ability to protect and deliver therapeutic NA. These liposomes offer greater biocompatibility and lower immunogenicity compared to viral vectors, although they still do not match the efficiency of viral delivery systems. Consequently, ongoing research focuses on synthesizing a wide variety of cationic lipids in the search for compounds that provide high transfection efficiency with minimal cytotoxicity. This study aimed to design and synthesize a novel CL (CholCadLys) derived from natural cellular molecules for transferring genetic material to eukaryotic cells. The lipid was synthesized using: cholesteryl chloroformate for the hydrophobic region, cadaverine as a linker, and lysine for the polar region, connected by carbamate and amide bonds, respectively. Identification was confirmed through thin-layer chromatography, purification by preparative chromatography, and characterization via infrared spectroscopy and mass spectrometry. The synthesis yielded a 60% success rate, with stable nanoliposomes averaging 76 nm in diameter. Liposomes were formed using this CL and commercial neutral lipids, characterized by transmission electron microscopy and Nanoparticle Tracking Analysis. These liposomes, combined with plasmid DNA, formed lipoplexes used to transfect Hek-293 FT cells, achieving up to 40% transfection efficiency without cytotoxicity in the mixture of CholCadLys and CholCad. This novel CL demonstrates potential as an efficient, safe, and cost-effective gene transfer system, facilitating further development in gene therapy.
Keywords:
cationic lipid
; lipoplexes
; gene therapy
; lipofection
1. Introduction
Modern therapeutics have sought to modify the natural course of diseases through symptomatic, pathophysiological, and etiological approaches, with the latter being the most desirable. In diseases with a genetic component, the etiology lies in one or more defective genes that can be repaired or replaced by normal genes, a process known as GT [1,2,3]. GT has garnered significant interest in the scientific community due to its potential to treat genetic diseases by introducing therapeutic NA, such as DNA, inhibitory RNAs (miRNAs or siRNAs), aptamers, and targeted editing systems like CRISPR/Cas [4,5,6].
A key challenge in GT is the efficient and safe introduction of NAs into eukaryotic cells, which must navigate various biological barriers to reach target cells. Various transfection methods have been developed, including physical methods and the use of genetic vectors, both viral and non-viral [7,8,9,10]. Physical methods, although operationally simple, are invasive and limited to ex vivo applications [11]. Viral genetic vectors, while efficient, present immunogenicity issues and limitations in the size of the transported NAs [12]. On the other hand, non-viral genetic vectors, such as liposomes, polymers, and nanoparticles, offer greater biocompatibility and lower immunogenicity, although they have yet to achieve the efficiency of viral vectors [8,13].
Among non-viral genetic vectors, liposomes are notable for their ability to protect and deliver therapeutic NAs. Classified as either unilamellar or multilamellar based on their structure, liposomes can be composed of anionic, neutral, or cationic lipids, each with its own advantages and limitations regarding stability, biocompatibility, and transfection efficiency [7,14]. CL-based liposomes exhibit acceptably high transfection efficiencies, minimal to no immunogenicity, unrestricted NA cargo capacity, excellent storage stability, and ease of handling [15,16,17,18]. In 1987, Felgner and colleagues designed and synthesized the first CL used as a transfection agent, N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA), which forms stable lipoplexes with NAs, facilitating the delivery and bioavailability of therapeutic genetic material in eukaryotic cells [19].
CLs are synthetic amphipathic lipids characterized by one or more positive charges on their hydrophilic head, which is connected to a hydrophobic tail via a linker [20,21,22]. The most commonly observed hydrophilic groups are primary, secondary, tertiary amines, or ammonium salts. The hydrophobic tails are typically composed of aliphatic chains, cholesterol, or other types of steroids. Finally, the linkers between the hydrophobic and hydrophilic regions consist of molecules that form ether, ester, carbamate, or amide bonds, and their nature affects the biodegradation rate [22,23]. Therefore, the structure of CLs is one of the key factors in promoting the transfection process and determining their cytotoxicity [24]. This is because the effectiveness and safety of these lipids depend not only on a single domain but on the combination of their entire structure. Consequently, a wide variety of CLs continue to be synthesized in the ongoing search for those that offer high transfection efficiency and without cytotoxicity [9].
Accordingly, a wide variety of CLs have been designed and synthesized to form liposomes and used as vectors to transfer NA to eukaryotic cells in clinical studies, some of which are commercially available. Among the most popular are DOTMA (Lipofectin) [19], DOTAP [25], DMRIE [26], DOGS (Transfectam) [27], DOSPA (Lipofectamine) [28], and DC-Chol [29]. Although some of these lipids are already employed in in vitro studies, these have shown that they can exhibit cytotoxic effects or induce apoptosis in certain cell lines [15,30]. Consequently, research has focused on synthesizing CLs with low or no cytotoxicity and high transfection efficiency. Therefore, the primary objective of this study is to design and synthesize a novel CL derived from a basic amino acid, intended for use in the transfer of genetic material. This will contribute to the development of gene delivery vectors that meet the necessary safety and efficacy standards for GT to become a reliable and competitive alternative to traditional therapies.
2. Materials and Methods
2.1. Design of the Novel CL: Cholesteryl-Cadaverine-Lysine (CholCadLys)
For the design of the CholCadLys lipid, two primary factors were taken into consideration:
- Structural Conformity: The CholCadLys lipid should conform to the structural scheme consisting of a hydrophobic region, a linker, and a hydrophilic region; main components of an amphipathic lipid.
- Natural Compatibility: The components used to create the CholCadLys lipid should be molecules that are naturally found as constituents and/or metabolites within eukaryotic cells.
Taking these factors into account, the lipid needed to have a molecular structure capable of forming liposomes, associating with DNA, and being non-cytotoxic. Based on these premises, the components selected for lipid synthesis were:
- A Steroid for the Lipid Bilayer: Cholesteryl chloroformate (Sigma-Aldrich, St. Louis, MO, USA) was chosen for its ability to integrate into lipid bilayers.
- A Small Primary Amine as a Linker: Cadaverine (Sigma-Aldrich) was selected to function as a connector between the hydrophobic body and the hydrophilic head due to its small size and primary amine group.
- A Basic Amino Acid for the Polar Head: L-lysine (Sigma-Aldrich) was selected to confer two positive charges to the hydrophilic region, enhancing its ability to associate with DNA.
This design ensures that the amphipathic lipid integrates well within biological membranes, provides effective DNA binding and delivery capabilities, and minimizes potential cytotoxic effects. The components chosen were carefully selected to fulfill the necessary criteria for forming stable and efficient liposomal structures for gene delivery in therapeutic applications.
2.2. Synthesis and Purification of CholCadLys Lipid
The synthesis of the CholCadLys lipid was carried out as follows: In a round-bottom flask, 1 mmol of cholesteryl chloroformate (activated cholesterol) dissolved in 15 ml of chloroform ACS (Fermont, Monterrey, México) was placed with 2 mmol of pyridine (Sigma-Aldrich) as a catalyst for the reaction. Then, 2 mmol of cadaverine dissolved in chloroform was added dropwise. Subsequently, 2 mmol of L-lysine dissolved in chloroform was added dropwise. The reaction was conducted with constant magnetic stirring at room temperature for 2 hours. Next, the organic phase was separated using a fraction collector and washed with bidistilled water (v/v) to extract the excess cadaverine, pyridine and L-lysine. Finally, the organic phase was completely evaporated to obtain a yellow powder. The synthesized CholCadLys lipid was identified using thin-layer chromatography (TLC) Silica gel 60 F254 (Merck, Darmstadt, Germany). The development system employed was a mixture of chloroform:methanol:acetic acid:water (Sigma-Aldrich) (65:25:8:4 v/v). The chromatoplates were revealed using two methods: 1) spraying with 1% ninhydrin in 96% ethanol and heating to 37°C until a purple coloration appeared, indicating the presence of free amino groups, and 2) immersing the chromatoplate in 5% sulfuric acid (H2SO4) in absolute ethanol, allowing it to dry for 45 minutes, and then exposing the chromatoplate to a flame, revealing the presence of organic matter.
To purify the synthesized lipid, a Preparative Layer Chromatography (PLC) Silica gel 60 (Merck) was developed on 20 x 20 cm plates with a thickness of 500 μm. The sample was dissolved in chloroform-methanol (JT Baker, México) (2:1 v/v) and applied across the entire width of the plate. The development system employed was mentioned above. The chromatography was developed for 3 hours, after which the plate was dried at room temperature using a fan. To locate the CholCadLys lipid on the chromatoplate, the leftmost centimeter was developed with 1.0% ninhydrin (Sigma-Aldrich) in ethanol (Merck). The entire row of the chromatoplate at the height where the CholCadLys lipid was revealed and then scraped off. The silica gel containing the lipid was washed four times with the chloroform-methanol mixture (2:1 v/v). The mixture was centrifuged at 2,500 x g for 5 minutes to remove the silica gel, and the supernatants from the washes were collected. The organic solvents were then evaporated to dryness, yielding the purified CholCadLys lipid in solid form.
2.3. Characterization of CholCadLys Lipid
2.3.1. Infrared Spectroscopy
To confirm the presence of the expected functional groups in the CholCadLys lipid, an IR spectroscopy test was conducted to obtain and analyze the corresponding spectrum. In a potassium chloride cell, 50 µL of the test sample was added and the solvent was allowed to evaporate to dryness. The cell was then placed in the Perkin Elmer FT-IR Spectrum GX2000 spectrophotometer and scanned over the range of 4000 to 400 cm⁻¹.
2.3.2. Mass Spectrometry
To determine the exact mass of the CholCadLys lipid and evaluate whether it corresponds to the calculated theoretical mass, a mass spectrometry test was performed. Additionally, this allowed for an assessment of the sample's purity. The sample was directly introduced into the Bruker micrOTOF-Q II 10392 mass spectrometer, using the following program: ESI source, positive ion polarity, nebulizer at 0.4 Bar, active focus, drying temperature at 180ºC, scan range from 5 m/z to 3000 m/z, and capillary voltage at 4500 V. This instrumental determination confirms the existence of the desired chemical structure and indicates the sample's purity.
2.4. Isolation and Purification of the Plasmid DNA (pDNA)
The strain Escherichia coli Top10, transformed with the pIRES2-EGFP plasmid (BD Clontech, Mountain View, CA, USA), was used to inoculate a 1 L flask of Luria-Bertani (LB) broth supplemented with kanamycin (60 μg/mL) and incubated for 12 hours at 37°C with agitation at 150 rpm using a Lab-Line 3590 rotary shaker. Following this, chloramphenicol was added to a final concentration of 50 μg/mL, and the culture was incubated for another 12 hours at 37°C with agitation. The bacterial cells were harvested by centrifugation at 4,000 x g for 20 minutes at 4°C. The extraction and purification of the pDNA were carried out using the alkaline lysis method with the UltraClean® Maxi Plasmid Prep Kit (MO BIO Labs, Carlsbad, CA, USA). Following purification, the concentration of the pDNA was measured using a NanoDrop series UV/visible spectrophotometer (Thermo Scientific).
2.5. Cationic Liposome Formation
To determine the ability of the CholCadLys lipid to form liposomes, a liposome formation assay via sonication was performed. The procedure followed the method reported by Oseguera (2015). In sterile, screw-capped 13 x 100 mm tubes, the CholCadLys and a helper lipid, both dissolved in chloroform, were added. The helper lipids used were: Cholesterol or Dioleylphosphatidylethanolamine (DOPE) in different molar proportions. Chloroform was evaporated under reduced pressure with rotation until complete dryness, forming a lipid film at the bottom of the tubes. Based on the total amount of lipids, a specific volume of sterile PBS was added to achieve a final concentration of 0.3 µg of total lipids/300µL. The mixture then underwent three cycles of sonication, each lasting 20 seconds, with 45 seconds of rest between cycles, using a Laboratory Supplies Co. bath sonicator. The formation of liposomes was evidenced by obtaining a homogeneous turbid suspension without clumps. The liposomes were considered stable if the suspension remained milky and free of clumps after 3 hours of formation.
2.6. Formation of Lipoplexes
Lipoplexes were formed using stable cationic liposomes at a ratio of 1 μg total lipids (in a volume of 30 μL D-MEM without serum) using varying amounts of pDNA (BD Clontech, Mountain View, CA, USA), different total lipids/DNA (w/w) ratios: 1:0.66, 1:0.4, 1:0.28, 1:0.22, and 1:0.18, in 30 μL of the same medium. The mixtures were incubated for 30 minutes at 37°C. This process resulted in suspensions of lipoplexes in 60 μL of serum-free medium, ready for transfection into cultured cells. For comparison of transfection efficiency, a concentration of 0.5 μg of Lipofectamine TM 2000 (Thermo Fisher Scientific) was used according to the manufacturer's instructions.
2.7. Characterization of Liposomes and Lipoplexes
2.7.1. Characterization of Lipoplexes by Agarose Gel Electrophoresis
The interaction of DNA with cationic liposomes was characterized by electrophoresis to verify the electrostatic interaction between these molecules. Agarose gel electrophoresis was performed using 0.8% agarose gels in 1X Tris-Borate-EDTA (TBE) buffer (89 mM Tris, 89 mM boric acid, 2 mM EDTA), pH 8.2, at 80 V for 1.5 hours. The gel was stained with ethidium bromide (5 μg/mL) and visualized using a High Performance Ultraviolet Transilluminator. Images were captured with a Kodak Gel Logic 100 gel documentation system.
2.7.2. Transmission Electron Microscopy (TEM)
The structural characteristics of cationic liposomes and lipoplexes were analyzed using TEM with negative staining technique. Aliquots of 20 µL of cationic liposomes or lipoplexes were placed on 200-mesh copper grids previously coated with formvar. The samples were incubated for 5 minutes at room temperature, and excess liquid was removed by blotting with filter paper. The grids were then stained with a 1% aqueous solution of phosphotungstic acid for 45 seconds. Excess stain was removed by blotting with filter paper, and the samples were examined using a JEOL JEM 1010 transmission electron microscope at an operating voltage of 60 kV and a magnification of 100,000x.
2.7.3. Nanoparticle Tracking Analysis (NTA)
To determine particle count per mL and verify particle size, cationic liposomes and lipoplexes were analyzed using the NanoSight NS300. This instrument utilizes NTA to characterize nanoparticles ranging from 10 nm to 2000 nm in solution. Each particle was analyzed individually yet simultaneously through direct observation and laser diffraction measurement.
2.8. Transfection Into the Eukaryotic Cell Line
The Hek-293 FT cell line (human embryonic kidney cells) was cultured in 25 cm³ Nunc flasks with DMEM supplemented with 10% fetal bovine serum (FBS, Sigma–Aldrich). Cell cultures at 80% confluence were used in 96-well plates. The culture medium was removed, and the cells were washed twice with 1X PBS. Subsequently, 40 μL of the lipoplex preparation was added. For the transfection process, the cells were incubated with the lipoplexes for 2 hours at 37°C in a 5% CO2 atmosphere. The medium was removed, and the samples were washed twice with PBS. Then, fresh medium supplemented with 10% SFB was added to continue incubation for another 36 hours The transfection efficiency was evaluated by fluorescence microscopy 24 hours post-transfection.
2.9. Determination of Cell Viability Post-Transfection
Eukaryotic cell cultures at 80% confluence were used 24 hours post-passage. The culture medium was removed, and the cells were washed twice with 1X PBS. Subsequently, 40 μL of the lipoplex preparation was added. The cells were incubated with the lipoplexes for 2 hours at 37°C in a 5% CO2 atmosphere. Following incubation, the lipoplexes were removed, and the cells were washed twice with 50 µL of 1X PBS. Then, 20 µL of 0.2% trypan blue dye (Sigma-Aldrich) in 1X PBS was added, and the cells were incubated for 10 minutes at 37°C in a 5% CO2 atmosphere. The vital dye, which stains only damaged or dead cells, was then removed, and the cells were washed twice with 50 µL of 1X PBS. Cell viability was determined by observing the cells under the Axio Vert.A1 microscope (Carl Zeiss, Oberkochen, Germany) and calculating the percentage of dead or damaged cells relative to the total number of viable cells.
3. Results
3.1. The Synthesis and Purification of CholCadLys Lipid Enables a High-Yield Formation of Cationic Lipids
The synthesis of the CholCadLys lipid involved the formation of carbamate and amide bonds between the activated cholesterol, the cadaverine as linker, and the basic amino acid Lysine. The final CholCadLys lipid obtained is depicted in Figure 1.
The synthesis was carried out using activated cholesterol:cadaverine:lysine molar ratio 1:2:2 to promote the production of the desired product with a higher yield. The synthesized and purified CholCadLys lipid exhibited a semi-solid consistency, a light brown color, and a waxy appearance. The identification of the lipid (unpurified and purified) was performed through analytical chromatography and it was revealed with 1% ninhydrin in ethanol, where 2 spots were observed, of which the intermediate one correspond to the cationic lipid, being an amphipathic molecule, while the amino acid that did not react, being polar, remained at the point of application (Figure 2 (I.A and II.A)) and with 5% sulfuric acid in ethanol, the steroid used as raw material moved in front of the solvent, being a highly hydrophobic molecule (Figure 2 (I.B and II.B)), in both revelation techniques an Rf of 0.8 was detected (Figure 2). In each chromatogram, the raw materials were applied at different spots as follows: activated cholesterol (1), the unpurified or purified lipid obtained directly from the synthesis (2), and the basic amino acid (3) (Figure 2 (I. and II.)). The yield of the reaction was on average 60%.
3.2. Elongation of the Carbonyl (C=O) bond of the Amide and Carbamate Bond are Structural Features of the Cholcadlys Lipid
Infrared spectroscopy was conducted on the purified synthesized CholCadLys lipid, resulting in the corresponding spectrum of the molecule. Signals representing characteristic bonds were observed in all obtained spectra: a signal ranging between 1688.59 and 1641.63 cm-1 corresponding to the elongation of the carbonyl (C=O) bond of the amide and carbamate bond. Similarly, signals corresponding to the elongation of the –NH groups were observed at 3298.84 cm-1, while signals corresponding to the C-H elongations of the methyl and methylene groups of the hydrocarbon chains were observed between 2918 and 2849 cm-1. Signals corresponding to the CH3 elongations were observed at 1451 - 1413 cm-1 (Figure 3). Thus, these signals confirm the presence of a molecule, comprising the hydrophobic portion linked to the basic amino acid through an amide bond. This validates the presence of the expected functional groups of the designed CholCadLys lipid.
The synthesized CholCadLys lipid underwent mass spectrometry, from which the corresponding spectrum was obtained. In the spectrum, the exact mass of the synthesized lipid was determined, which coincided with the theoretical mass obtained using the Chem Draw Ultra 8.0.3 program (Informer Technologies, Inc., Los Ángeles, CA, USA), amounting to 643.5 g/mol (Figure 4). Therefore, the identity of this new lipid could be confidently confirmed.
3.3. The Extraction and Characterization of pIRES2-EGFP plasmid DNA Confirm its Molecular Integrity
The pIRES2-EGFP plasmid, sized at 5308 bp, served as the genetic material. It comprises the Cytomegalovirus CMV promoter for expression in eukaryotic cells, the IRES2-EGFP gene encoding the green fluorescent protein as a reporter gene, and the kanamycin resistance gene (Kan) (Figure 5).
The integrity of the pDNA was determined by electrophoretic mobility in a 1% agarose gel. Two of the three possible bands for a plasmid (form I or supercoiled circular, form II or relaxed circular, and form III or linear) were observed. Molecular form I (supercoiled) was obtained at 98%, while molecular form II (relaxed circular) was obtained at 2% (figure 6). Therefore, the plasmid DNA exhibited a high degree of integrity, ensuring a high level of confidence in genetic expression.
3.4. Formation of Stable Liposomes with Different Molar Ratios of Cholesterol and DOPE as Helper Lipids
Cationic liposomes were formed using the Felgner sonication method, modified by Ibáñez et al. (1996), incorporating two helper lipids DOPE or cholesterol (Sigma-Aldrich) at different molar ratios. During the preparation of liposomes with all planned lipid mixtures, it was observed that not all of them were stable. The stability of liposomes depends on the interaction between lipids, both the CholCadLys and the helper lipid, to form a stable lipid bilayer in the aqueous medium. Preparations yielding stable liposomes exhibited a milky appearance characteristic of vesicle suspensions due to light refraction. The lipid mixtures forming stable liposomes included CholCadLys:cholesterol in molar proportions of 1:1, 2:1, and the CholCadLys:DOPE in molar ratios of 1:1 and 1:2.
3.5. Electrophoresis Reveals pDNA Interactions with Liposomes
Electrophoresis was performed on lipoplexes formed with the stable liposomes to determine the electrostatic interaction between liposomes and pDNA. This assay provides insight into the interaction between liposomes and genetic material in lipoplexes. Three different behaviors were observed in the electropherograms: pDNA present in the loading well (lipoplex), free pDNA without delay, and free pDNA with delay. The behavior of pDNA in different selected lipoplexes was compared, using varying amounts of pDNA, i.e., different total lipids/DNA (w/w) ratios: 1:0.66, 1:0.4, 1:0.28, 1:0.22, and 1:0.18. The quantity of pDNA molecules was gradually decreased, while maintaining a fixed amount of liposomes (1 μg total lipids). For this test, the same volume was loaded into the wells of the gel.
In the case of free pDNA, the lipoplexes exhibited different behaviors. The CholCadLys lipid mixed with cholesterol-cadaverine (CholCad) IUPAC name ([10,13-dimethyl-17-(6-methylheptan-2-yl)-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-yl] N-(5 aminopentyl)carbamate) [32] and DOPE as a helper lipid showed pDNA without delay only in the ratio 1:0.66 total lipids/DNA, while a slight delay was observed in the ratio 1:0.40 total lipids/DNA. However, there was always free pDNA, indicating two possibilities: not all pDNA interacted with liposomes, and the ratio of total lipids to pDNA was not appropriate, suggesting an excess of added pDNA (Figure 7).
3.6. TEM Analysis Reveals Size and Morphology for Transfection
The total lipids/pDNA ratio 1:0.4 demonstrated optimal electrostatic interaction, which is ideal for transfection, as it strikes a balance that is neither too strong nor too weak, ensuring efficient delivery of the genetic material, which was used for the subsequent assays. To characterize the liposomes, those that were stable were selected for future transfection, including those containing cholesterol in a molar ratio of 1:1 and 2:1, as well as those containing DOPE in a molar ratio of 1:1 and 1:2. The liposomes appeared spherical and ovoid with a heterogeneous size ranging from 26.4 to 126 nm in diameter in all mixtures (Figure 8). Similarly, lipoplexes were analyzed by TEM, revealing complexes of various sizes ranging from 110 to 420 nm that formed associations with tubular-shaped (CholCadLys:Cholesterol 2:1) (Figure 8D) junctions (CholCadLys:Cholesterol 1:1) (Figure 8B) in all lipid mixtures (Figure 8).
3.7. Comparative Analysis of Size and Concentration of Liposomes and Lipoplexes for Transfection Applications
The liposomes were observed to be spherical with a heterogeneous size ranging from 102 to 136.6 nm in diameter, and the approximate particle count was 2 particle / mL (Table 1). Similarly, lipoplexes total lipids/pDNA ratio 1:0.4 were analyzed, revealing associations among them in multiple forms, with a heterogeneous size ranging from 92.3 to 142 nm in diameter, and the approximate particle count was 2 particles / mL. The quantity of liposomes per ml is crucial for transfection (Table 1).
3.8. Lipid Mixtures Containing DOPE as a Helper Lipid Increase Transfection Efficiency
The highest transfection efficiencies were achieved when using lipoplexes with w/w ratios total lipids/pDNA 1:0.4. In general, transfections were carried out with lipoplexes containing the CholCadLys and CholCad lipids, in combination with cholesterol or DOPE in different molar proportions. Very few transfected cells were observed with the CholCadLys lipid containing cholesterol, resulting in a transfection efficiency of 2% (Figure 9 C, D). However, when transfection was performed with lipoplexes formed with the CholCad and DOPE (Figure 9 E, F, G), and the CholCadLys lipid and DOPE (Figure 9 I, J), a transfection efficiency of up to 10% was achieved.
Given that the best transfections were obtained with the CholCadLys and the CholCad lipids, and these lipids mixed with DOPE, a mixture of these lipids was prepared to observe the effect on transfection efficiency. Stable liposomes were formed with the lipid mixture as follows: CholCadLys - CholCad in a molar ratio of 1:2; CholCadLys - CholCad in 2:1; CholCadLys - CholCad - DOPE in 1:1:2; CholCadLys - CholCad - DOPE in 2:1:1. Lipoplexes were then formed, and the Hek-293 FT cell line was transfected.
An increase in transfection efficiency was observed with the mixture of CLs in different proportions. Using the CholCadLys - CholCad in 1:2 and the CholCadLys - CholCad in 2:1, a transfection efficiency of up to 30% was achieved (Figure 9 K, L). Meanwhile, mixing the CholCadLys - CholCad - DOPE in 1:1:2 and the CholCadLys - CholCad - DOPE in 2:1:1 resulted in a transfection efficiency of up to 40% (Figure 9 M, N), which was superior to the LipofectamineTM 2000 (Thermo Fisher Scientific) used as comparison method for this transfection system in this cell line.
3.9. Lipid mixtures containing CholCadLys do not exhibit cytotoxic effects in cell culture
The cytotoxic effect of lipoplexes formed with the helper lipids cholesterol or DOPE in different molar ratios was evaluated using the trypan blue exclusion dye. The test was conducted after completing the transfection period. No blue-stained cells (dead or damaged cells) were observed in the cultures of cells treated with various CholCadLys lipoplexes. In contrast, when cells were transfected with LipofectamineTM 2000 (Thermo Fisher Scientific), approximately 50% of cells were observed to be non-viable (stained). Untreated cells were used as a negative control (Figure 10).
4. Discussion
Genetic carriers enable the transport and protection of genetic material for transfection, as the uptake of naked genetic material by eukaryotic cells is challenging and inefficient [33,34]. Therefore, genetic carriers that facilitate the transfection process are commonly required. Currently, the most common and safe genetic carriers are cationic liposomes; however, their main drawback is low transfection efficiency [35,36,37]. In this study, a CL derived from cholesterol, cadaverine linked by a carbamate bond, and lysine linked by an amide bond were synthesized to enhance transfection efficiency and avoid cytotoxicity. These molecules were chosen as they are normally part of cells, making them potentially innocuous for in vitro and in vivo applications.
It's important to note that CLs derived from cholesterol have been previously used for gene delivery to eukaryotic cells with acceptable transfection efficiencies. However, most of them exhibited some degree of cytotoxicity due to the polar region they contain, preventing their establishment as effective non-viral gene delivery vectors [38,39]. Studies have shown that cells can hydrolyze CLs used for transfection, with units linked by carbamate bonds, without causing cytotoxicity [39,40,41]. Additionally, the amide bond formed in this newly synthesized CL is highly recognized by the cell, as it links the amino acids constituting peptides and proteins, making its hydrolysis non-problematic for the cell.
The purification of the CholCadLys lipid was achieved through differential solubility using liquid-liquid extractions with chloroform and water. In the aqueous phase, excess polar molecules (pyridine, cadaverine, and lysine) were separated, while the complete lipids and cholesterol chloroformate remained in the organic phase. Although, preparative chromatography was also employed to obtain a purer lipid, separating it completely from excess raw material. The yield of the reaction was around 60%, which is considered acceptable [42]. However, this could be improved by modifying certain parameters such as temperature and the amount of raw material used, favoring the formation of the final product. Increasing the amount of raw material is a viable option due to the low synthesis cost, using economical reagents and not requiring any sophisticated equipment, both for synthesis and purification, considering it as an advantage.
Monitoring the synthesis and identifying the CholCadLys lipid was carried out through thin-layer chromatography on silica gel (TLC), which is essential for the analysis of lipids and other small molecules like amino acids, nucleotides, and vitamins, among others [43]. TLC is a sensitive and precise identification tool, reliable for initial correct identification. It does not require complex instrumentation, making it simple to use, providing advantages for its application. For the elution of the chromatographic plate, a solvent mixture suitable for amphipathic molecules was used as the mobile phase. Therefore, an Rf value between the point of application and the solvent front (0.8 > Rf > 0.3) indicates the presence of amphipathic molecules, such as cationic lipids. It was observed that cholesteryl chloroformate, which is highly soluble in organic solvents, migrates with the mobile phase on the chromatographic plate. In contrast, L-lysine, due to its two functional groups capable of forming hydrogen bonds (the amino and hydroxyl groups, both acting as hydrogen bond donors and acceptors) and its short hydrocarbon chain, remained at the application point. Furthermore, the majority of lysine was eliminated in the aqueous phase. The presence of an intermediate spot with an Rf of 0.8 confirmed the presence of an amphipathic compound, corresponding to the cationic lipid. This chromatography also allowed a rough assessment of the lipid's purity after chromatographic elution. Note that two developing reactions were used to identify the molecules, as they indicate two different characteristics: the ninhydrin development indicated the presence of free amino groups, which provide the positive charge to the cationic lipid and form its hydrophilic part; the sulfuric acid development indicated the presence of organic matter in the hydrophobic region and the general presence of organic contaminants, both in the crude reaction mixture and after purification.
To confirm the presence of the CholCadLys lipid, other techniques such as infrared spectroscopy (IR) and mass spectrometry were necessary. IR spectroscopy identified the functional groups of the molecules, confirming the nucleophilic substitution between the cholesterol and the cadaverine and the amidation between the cadaverine and the lysine. Mass spectrometry identified the exact mass of the compounds and indirectly indicated their purity, showing the signal of the synthesized lipid along with other signals that could correspond to unreacted compounds or secondary compounds formed during the synthesis such as a lysine molecule attached to a cholesteryl chloroformate molecule, a cadaverine molecule attached to a cholesteryl chloroformate molecule, and two lysine molecules (polymerized) attached to a cholesteryl chloroformate molecule. This suggests that the synthesized lipid is not completely pure. However, it is also known that the same method used for analysis can fragment the evaluated molecule, which could account for the signals detected prior to those of the molecule of interest.
Cationic liposomes were formed from amphipathic CLs mixed with colipids, mainly providing stability to the liposome's lipid bilayer. The most used helper lipids are DOPE, cholesterol, or dioleoylphosphatidylcholine (DOPC). In this study, liposome stability was tested using the synthesized CL and helper lipids. Not all lipid mixtures tested formed stable liposomes. Liposome stability depends on the molecular structure of the CLs and helper lipids for the lipid bilayer to form properly. The study found that the CL formed liposomes with both cholesterol and DOPE. However, not all molar ratios used formed stable liposomes, yet all liposomes were tested in the transfection process. The lipid size, determined by the size of the hydrophobic and hydrophilic parts, is the major parameter for liposome stability. The geometry of the synthesized CL appears to be a thin cylinder, and DOPE's molecular geometry is conical [44,45]. Despite DOPE being a neutral lipid, its interaction with the synthesized CL could provide stabilization through electrostatic interactions, making liposomes more stable when formed with DOPE as a helper lipid [46,47].
With stable cationic liposomes, lipoplexes were prepared for transfecting the Hek-293 FT cell line. To perform transfection, the first step involved obtaining pDNA with high integrity and purity to ensure proper gene expression and avoid cytotoxicity due to bacterial contaminants (RNA and proteins), focusing solely on liposome-induced cytotoxicity. Lipoplexes result from the electrostatic interaction of the positive charges of the hydrophilic region of CLs with the negative charges of the phosphate groups in the genetic material (pDNA). The DNA-liposome binding is crucial for efficient GT, as electrostatic interaction determines the release and intracellular delivery of genetic material [48,49]. Additionally, liposomes, besides acting as genetic carriers, allow the condensation of genetic material on their surface, preventing degradation by nucleases [50,51], and participate in the interaction with target cells for efficient internalization or delivery of genetic material [52,53].
Using the lipoplexes formed from stable liposomes, the Hek-293 FT cell line was transfected. Clear differences in gene transfer efficiency were observed based on the helper lipid used, with higher transfection efficiency achieved when DOPE was employed. In vitro studies have shown that DOPE is more efficient for gene transfer [54,55,56], a finding confirmed by the results of transfections with CholCadLys lipid.
Furthermore, in vitro transfection assays have shown that cholesterol is the most effective helper lipid for forming lipoplexes, likely due to its role in stabilizing lipid bilayers within cellular membranes [57,58], however, in this work transfection efficiency significantly decreased when lipoplexes were formed with cholesterol as a helper lipid. This may be since, as the hydrophobic portion of the synthesized CholCadLys lipid is a cholesterol molecule, it cannot interact effectively with cholesterol. Cholesterol stabilizes the phospholipids and glycolipids in the plasma membrane, but two cholesterol molecules cannot associate correctly in the lipid bilayer [59,60], resulting in less stable and less efficient lipoplexes for genetic material transfer.
Since both the CholCadLys and the CholCad lipids showed good transfection efficiency, a mixture of these two CLs with DOPE was prepared to assess if efficiency could be increased. The transfection efficiency was found to be up to 40%, which was very similar to that obtained with commercial liposomes LipofectamineTM 2000 (Thermo Fisher Scientific) (used as a comparison method). This outcome may be attributed to stabilizing the liposome bilayer surface through electrostatic interactions between DOPE and the synthesized CLs, leaving two positive charges on the surface, enhancing electrostatic interaction with DNA, and improving the transfection process.
Viability assays revealed that cells showed no damage with any of the mixtures containing CholCadLys lipid. Therefore, it can be asserted that the synthesized CholCadLys lipid, being composed of molecules naturally found in the cell, is not cytotoxic. Once the lipoplex is taken up by the cell, and the pDNA can migrate to the cell nucleus, the next step would be the hydrolysis of the amide and carbamate bonds by nonspecific enzymes [61,62,63], generating cell-native molecules (lysine,cadeverine, and cholesterol). Lysine can be used for protein synthesis, or the cell can utilize it to oxidize it to CO2 and water plus ATP. The primary amine can be used in the cell for polyamine production, and the activated cholesterol, which can serve as a precursor for bile salts, steroid hormones, vitamin D, and as a structural component of cell membranes [64].Therefore, these CLs, lacking cytotoxicity, can be considered safe for gene transfer, both in vitro.
Characterizing the lipoplexes allowed the comparison of their structural and physicochemical characteristics with TEM using negative staining. All liposomes were observed to have spherical or ovoid shapes, with heterogeneous sizes ranging from 26.4 to 126 nm. This size is considered suitable for gene transfer, as studies have shown that small unilamellar liposomes (~70 nm) are more efficient for gene transfer compared to larger ones (~135 nm) [65,66]. In the case of lipoplexes, there was no variation in size when genetic material was added. However, lipoplexes formed with the mixture of lipids containing DOPE exhibited associations between them with tubular connections, resembling a beaded necklace, which has been described with other CLs [67].
Agarose gel electrophoresis was performed to determine the interaction between liposomes and pDNA, using a lipid/DNA (w/w) ratio of 1:0.4 as a reference, as this ratio resulted in the best transfection efficiency with the tested CLs. Increasing the amount of pDNA relative to the amount of liposomes resulted in free or weakly associated pDNA on the liposome surface (retarded pDNA), saturating the positive charges on the liposome surface and hindering interaction with the cell membrane, consequently reducing transfection [68,69]. Conversely, decreasing the amount of DNA relative to the amount of liposomes allowed better interaction and uptake by the cell, as surplus positive charges remained for interaction during transfection. The lipid/DNA ratio of 1:0.4, which showed the best transfection efficiencies, implies that this ratio is ideal to allow the amount of pDNA used to associate with the positive charges on the liposome's surface without saturating it completely. This ensures there are remaining positive charges on the lipoplex surface for interaction with the cell membrane during transfection. Modifying this ratio altered transfection efficiency accordingly.
This study demonstrated the effectiveness of the CholCadLys mixed with different helper lipid ratios for gene transfer in Hek-293 FT cells. However, further studies are needed to enhance its effectiveness in transfection across different cell lines, primary cultures, and experimental animals, with the goal of eventually using them as safe and efficient genetic carriers in humans.
5. Conclusions
The CL amphipathic nature was confirmed through the Rf value. When the CholCadLys lipid was mixed with helper lipids in various molar ratios, it formed stable liposomes and lipoplexes with heterogeneous sizes, depending on the lipid mixture used. Additionally, it was demonstrated that the CholCadLys lipid is capable of transfecting eukaryotic cells in culture with low cytotoxicity. The transfection efficiency was found to depend on the lipid mixtures used, with the most efficient transfections occurring with mixtures containing DOPE as a helper lipid. Therefore, the mixture of CholCadLys and CholCad enhances the transfection efficiency lipid shows potential as a genetic vector for the delivery of NA to cultured cells, offering new possibilities for developing more compatible and efficient gene delivery systems for future in vivo gene therapies. However, further testing with different cell lines is recommended to confirm its efficiency and cytotoxicity.
Author Contributions
Conceptualization, D.M.B.-E., M.M.-S. and M.I.-H.; methodology, D.M.B.-E., validation, M.M.-S., D.M.B.-E.; formal analysis, M.M.-S.; investigation, D.M.B.-E.; resources, D.M.B.-E., M.M.-S and M.I.-H.; writing—original draft preparation, D.M.B.-E.; writing—review and editing, M.M.-S., R.A.M-R; visualization, M.M.-S.; supervision, M.I.-H.; project administration, D.M.B.-E. and M.I.-H.; funding acquisition, M.I.-H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Instituto Politécnico Nacional (IPN), through Secretaría de Investigación y Posgrado (SIP), linked to project numbers 20171406, 20181251, 20195226, 20200448, 20221571 and 20231845.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
Diana Marcela Bravo Estupiñan is a doctoral student thanks to the support of the Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCyT) at Instituto Politécnico Nacional (IPN). Hek-293 FT cell line was provided by Daniel Romero Trejo (CINVESTAV).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- High, K.A.; Roncarolo, M.G. Gene Therapy. N. Engl. J. Med. 2019, 381, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.C.; Wang, Z.L.; Xu, T.; He, Z.Y.; Wei, Y.Q. The approved gene therapy drugs worldwide: from 1998 to 2019. Biotechnol. Adv. 2020, 40, 107502. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Rivas-garcía, L.; Baptista, P.V.; Fernandes, A.R. Gene therapy in cancer treatment: Why go nano? Pharmaceutics 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Afonin, K.A.; Chandler, M. Aptamers as Modular Components of Therapeutic Nucleic Acid Nanotechnology. 2021, pp. 825–882. Available online: https://www.taylorfrancis.com/chapters/edit/10.1201/9781003122005-34/aptamers-modular-components-therapeutic-nucleic-acid-nanotechnology-martin-panigaj-brittany-johnson-weina-ke-jessica-mcmillan-ekaterina-goncharova-morgan-chandler-kirill-afonin.
- Gupta, A.; Andresen, J.L.; Manan, R.S.; Langer, R. Nucleic acid delivery for therapeutic applications. Adv. Drug Deliv. Rev. 2021, 178, 113834. [Google Scholar] [CrossRef]
- Luthra, R.; Kaur, S.; Bhandari, K. Applications of CRISPR as a potential therapeutic. Life Sci. 2021, 284. [Google Scholar] [CrossRef]
- Ibáñez, M.; Oseguera, B. Nanoliposomas catiónicos como vehículos para terapia génica. In Nanobiotecnología: Fundamentos y perspectivas Tecnología de frontera para resolver los problemas del mañana; Ciudad de México, 2016; pp. 380–910 ISBN 3841752705.
- Zu, H.; Gao, D. Non-viral Vectors in Gene Therapy: Recent Development, Challenges, and Prospects. AAPS J. 2021, 23. [Google Scholar] [CrossRef]
- Cui, S.; Wang, Y.; Gong, Y.; Lin, X.; Zhao, Y.; Zhi, D.; Zhou, Q.; Zhang, S. Correlation of the cytotoxic effects of cationic lipids with their headgroups. Toxicol. Res. (Camb). 2018, 7, 473–479. [Google Scholar] [CrossRef]
- Yin, H.; Kauffman, K.J.; Anderson, D.G. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 2017, 16, 387–399. [Google Scholar] [CrossRef]
- Sayed, N.; Allawadhi, P.; Khurana, A.; Singh, V.; Navik, U.; Pasumarthi, S.K.; Khurana, I.; Banothu, A.K.; Weiskirchen, R.; Bharani, K.K. Gene therapy: Comprehensive overview and therapeutic applications. Life Sci. 2022, 294, 120375. [Google Scholar] [CrossRef]
- Butt, M.H.; Zaman, M.; Ahmad, A.; Khan, R.; Mallhi, T.H.; Hasan, M.M.; Khan, Y.H.; Hafeez, S.; Massoud, E.E.S.; Rahman, M.H.; et al. Appraisal for the Potential of Viral and Nonviral Vectors in Gene Therapy: A Review. Genes 2022, Vol. 13, Page 1370 2022, 13, 1370. [Google Scholar] [CrossRef]
- Sainz-ramos, M.; Gallego, I.; Villate-beitia, I.; Zarate, J.; Maldonado, I.; Puras, G.; Pedraz, J.L. How Far Are Non-Viral Vectors to Come of Age and Reach Clinical Translation in Gene Therapy? Int. J. Mol. Sci. 2021, Vol. 22, Page 7545 2021, 22, 7545. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Agrawal, M.K. A historical perspective of liposomes-a bio nanomaterial. Mater. Today Proc. 2021, 45, 2963–2966. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, M.C.P.; Radaic, A.; Garcia-Fossa, F.; da Cruz-Höfling, M.A.; Vinolo, M.A.R.; de Jesus, M.B. The in vivo toxicological profile of cationic solid lipid nanoparticles. Drug Deliv. Transl. Res. 2020, 10, 34–42. [Google Scholar] [CrossRef]
- Ewert, K.K.; Scodeller, P.; Simón-Gracia, L.; Steffes, V.M.; Wonder, E.A.; Teesalu, T.; Safinya, C.R. Cationic Liposomes as Vectors for Nucleic Acid and Hydrophobic Drug Therapeutics. Pharm. 2021, Vol. 13, Page 1365 2021, 13, 1365. [Google Scholar] [CrossRef]
- Thongbamrer, C.; Roobsoong, W.; Sattabongkot, J.; Opanasopit, P.; Yingyongnarongkul, B. ek Serum Compatible Spermine-Based Cationic Lipids with Nonidentical Hydrocarbon Tails Mediate High Transfection Efficiency. ChemBioChem 2022, 23, e202100672. [Google Scholar] [CrossRef]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 7413–7417. [Google Scholar] [CrossRef]
- Koynova, R.; Tenchov, B. Cationic Lipids: Molecular Structure/Transfection Activity Relationships and Interactions with Biomembranes. Top. Curr. Chem. 2010, 296, 51–93. [Google Scholar] [CrossRef]
- Marshall, J.; Nietupski, J.B.; Lee, E.R.; Siegel, C.S.; Rafter, P.W.; Rudginsky, S.A.; Chang, C.D.; Eastman, S.J.; Harris, D.J.; Scheule, R.K.; et al. Cationic Lipid Structure and Formulation Considerations for Optimal Gene Transfection of the Lung. J. Drug Target. 2000, 7, 453–469. [Google Scholar] [CrossRef]
- Zhi, D.; Bai, Y.; Yang, J.; Cui, S.; Zhao, Y.; Chen, H.; Zhang, S. A review on cationic lipids with different linkers for gene delivery. Adv. Colloid Interface Sci. 2018, 253, 117–140. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhi, D.; Zhang, S. Cationic Liposomes in Different Structural Levels for Gene Delivery. Non-Viral Gene Ther. 2011. [Google Scholar] [CrossRef]
- Paecharoenchai, O.; Niyomtham, N.; Apirakaramwong, A.; Ngawhirunpat, T.; Rojanarata, T.; Yingyongnarongkul, B.E.; Opanasopit, P. Structure relationship of cationic lipids on gene transfection mediated by cationic liposomes. AAPS PharmSciTech 2012, 13, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.K.; Hwang, G.B.; Seu, Y.B.; Choi, J.S.; Jin, K.S.; Doh, K.O. DOTAP/DOPE ratio and cell type determine transfection efficiency with DOTAP-liposomes. Biochim. Biophys. Acta - Biomembr. 2015, 1848, 1996–2001. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, V.; Anderson, D.; Lan, J.; Schifferli, K.; Jessee, J. DMRIE-C reagent for transfection of suspension cells and for RNA transfection. Focus (Madison). 1995, 17, 84–86. [Google Scholar]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. DNA Transfection Mediated by Cationic Lipid Reagents. Cold Spring Harb. Protoc. 2019, 2019, pdb–prot095414. [Google Scholar] [CrossRef]
- Zhen, S.; Li, X. Liposomal delivery of CRISPR/Cas9. Cancer Gene Ther. 2019 277 2019, 27, 515–527. [Google Scholar] [CrossRef]
- Yang, S. ye; Zheng, Y.; Chen, J. yin; Zhang, Q. yang; Zhao, D.; Han, D. en; Chen, X. jing Comprehensive study of cationic liposomes composed of DC-Chol and cholesterol with different mole ratios for gene transfection. Colloids Surfaces B Biointerfaces 2013, 101, 6–13. [Google Scholar] [CrossRef]
- Soenen, S.J.H.; Brisson, A.R.; De Cuyper, M. Addressing the problem of cationic lipid-mediated toxicity: The magnetoliposome model. Biomaterials 2009, 30, 3691–3701. [Google Scholar] [CrossRef]
- Latulippe DR, Zydney AL. Separation of plasmid DNA isoforms by highly converging flow through small membrane pores. J Colloid Interface Sci. 2011, 357, 548–553. [Google Scholar] [CrossRef]
- Cui, K.; Liang, D.; Sweedler, D. Compositions comprising cationic amphiphiles and colipids for delivering therapeutic molecules 2012, 1–48.
- Jiang, T.; Gonzalez, K.M.; Cordova, L.E.; Lu, J. Nanotechnology-enabled gene delivery for cancer and other genetic diseases. Expert Opin. Drug Deliv. 2023, 20, 523. [Google Scholar] [CrossRef]
- Wang, C.; Pan, C.; Yong, H.; Wang, F.; Bo, T.; Zhao, Y.; Ma, B.; He, W.; Li, M. Emerging non-viral vectors for gene delivery. J. Nanobiotechnology 2023, 21, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Simões, S.; Filipe, A.; Faneca, H.; Mano, M.; Penacho, N.; Düzgünes, N.; de Lima, M.P. Cationic liposomes for gene delivery. Expert Opin. Drug Deliv. 2005, 2, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Jang, W.Y.; Ko, Y.T. The Effect of Surface Charges on the Cellular Uptake of Liposomes Investigated by Live Cell Imaging. Pharm. Res. 2017, 34, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Tseu, G.Y.W.; Kamaruzaman, K.A. A Review of Different Types of Liposomes and Their Advancements as a Form of Gene Therapy Treatment for Breast Cancer. Molecules 2023, 28, 1498. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.; Huan, M.L.; Wan, N.; Qiu, H.; Zhou, S.Y.; Zhang, B. Le Novel Cholesterol-Based Cationic Lipids as Transfecting Agents of DNA for Efficient Gene Delivery. Int. J. Mol. Sci. 2015, 16, 5666. [Google Scholar] [CrossRef]
- Shi, J.; Yu, S.; Zhu, J.; Zhi, D.; Zhao, Y.; Cui, S.; Zhang, S. Carbamate-linked cationic lipids with different hydrocarbon chains for gene delivery. Colloids Surf. B. Biointerfaces 2016, 141, 417–422. [Google Scholar] [CrossRef]
- Zhi, D.; Zhang, S.; Zhao, Y.; Cui, S.; Wang, B.; Chen, H.; Zhi, D.; Zhao, D.; Zhi, D.; Zhang, S.; et al. In Vitro Study of Carbamate-Linked Cationic Lipid for Gene Delivery Against Cervical Cancer Cells. Adv. Mater. Phys. Chem. 2013, 2, 229–232. [Google Scholar] [CrossRef]
- Liu, D.; Qiao, W.; Li, Z.; Cui, X.; Li, K.; Yu, L.; Yan, K.; Zhu, L.; Cheng, L. Carbamate-linked cationic lipids for gene delivery. Bioorg. Med. Chem. 2008, 16, 995–1005. [Google Scholar] [CrossRef]
- Vogel, A.; Tatchell, A.; Furnis, B.; Hannaford, A.; Smith, P. Vogel’s Textbook of Practical Organic Chemistry; 1996; ISBN 9780582462366; 0582462363.
- Coskun, O. Separation techniques: Chromatography. North. Clin. Istanbul 2016, 3, 156. [Google Scholar] [CrossRef]
- Cullis, P.R.; de Kruijff, B.; Hope, M.J.; Nayar, R.; Schmid, S.L. Phospholipids and membrane transport. Can. J. Biochem. 1980, 58, 1091–1100. [Google Scholar] [CrossRef]
- Karanth, H.; Murthy, R.S.R. pH-Sensitive liposomes-principle and application in cancer therapy. J. Pharm. Pharmacol. 2010, 59, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Hirsch-Lerner, D.; Zhang, M.; Eliyahu, H.; Ferrari, M.E.; Wheeler, C.J.; Barenholz, Y. Effect of “helper lipid” on lipoplex electrostatics. Biochim. Biophys. Acta - Biomembr. 2005, 1714, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Ermilova, I.; Swenson, J. DOPC versus DOPE as a helper lipid for gene-therapies: molecular dynamics simulations with DLin-MC3-DMA. Phys. Chem. Chem. Phys. 2020, 22, 28256–28268. [Google Scholar] [CrossRef] [PubMed]
- Nsairat, H.; Alshaer, W.; Odeh, F.; Esawi, E.; Khater, D.; Bawab, A. Al; El-Tanani, M.; Awidi, A.; Mubarak, M.S. Recent advances in using liposomes for delivery of nucleic acid-based therapeutics. OpenNano 2023, 11, 100132. [Google Scholar] [CrossRef]
- Dan, N.; Danino, D. Structure and kinetics of lipid–nucleic acid complexes. Adv. Colloid Interface Sci. 2014, 205, 230–239. [Google Scholar] [CrossRef]
- Lebrón, J.A.; Ostos, F.J.; López-lópez, M.; Moyá, M.L.; Sales, C.; García, E.; García-calderón, C.B.; García-calderón, M.; Peña-gómez, M.J.; Rosado, I.V.; et al. Metallo-Liposomes of Ruthenium Used as Promising Vectors of Genetic Material. Pharmaceutics 2020, 12, 482. [Google Scholar] [CrossRef]
- Crook, K.; McLachlan, G.; Stevenson, B.J.; Porteous, D.J. Plasmid DNA molecules complexed with cationic liposomes are protected from degradation by nucleases and shearing by aerosolisation. Gene Ther. 1996, 3, 834–839. [Google Scholar]
- dos Santos Rodrigues, B.; Banerjee, A.; Kanekiyo, T.; Singh, J. Functionalized Liposomal Nanoparticles for Efficient Gene Delivery System to Neuronal Cell Transfection. Int. J. Pharm. 2019, 566, 717. [Google Scholar] [CrossRef]
- Durymanov, M.; Reineke, J. Non-viral Delivery of Nucleic Acids: Insight Into Mechanisms of Overcoming Intracellular Barriers. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Du, Z.; Munye, M.M.; Tagalakis, A.D.; Manunta, M.D.I.; Hart, S.L. The Role of the Helper Lipid on the DNA Transfection Efficiency of Lipopolyplex Formulations. Sci. Rep. 2014, 4, 7107. [Google Scholar] [CrossRef]
- Colosimo, A.; Serafino, A.; Sangiuolo, F.; Di Sario, S.; Bruscia, E.; Amicucci, P.; Novelli, G.; Dallapiccola, B.; Mossa, G. Gene transfection efficiency of tracheal epithelial cells by DC-Chol–DOPE/DNA complexes. Biochim. Biophys. Acta - Biomembr. 1999, 1419, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, S.; Kanegae, N.; Nishina, K.; Kamikawa, Y.; Koiwai, K.; Masunaga, H.; Sakurai, K. The role of the helper lipid dioleoylphosphatidylethanolamine (DOPE) for DNA transfection cooperating with a cationic lipid bearing ethylenediamine. Biochim. Biophys. Acta - Biomembr. 2013, 1828, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Betker, J.L.; Kullberg, M.; Gomez, J.; Anchordoquy, T.J. Cholesterol Domains Enhance Transfection. Ther. Deliv. 2013, 4, 453. [Google Scholar] [CrossRef]
- Hosseini, E.S.; Nikkhah, M.; Hosseinkhani, S. Cholesterol-rich lipid-mediated nanoparticles boost of transfection efficiency, utilized for gene editing by CRISPR-Cas9. Int. J. Nanomedicine 2019, 14, 4353–4366. [Google Scholar] [CrossRef]
- de Santis, A.; Scoppola, E.; Ottaviani, M.F.; Koutsioubas, A.; Barnsley, L.C.; Paduano, L.; D’Errico, G.; Krauss, I.R. Order vs. Disorder: Cholesterol and Omega-3 Phospholipids Determine Biomembrane Organization. Int. J. Mol. Sci. 2022, 23, 5322. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef]
- Zhu, L.; Mahato, R.I. Lipid and polymeric carrier-mediated nucleic acid delivery. Expert Opin. Drug Deliv. 2010, 7, 1209. [Google Scholar] [CrossRef]
- Mahesh, S.; Tang, K.C.; Raj, M. Amide Bond Activation of Biological Molecules. Mol. 2018, Vol. 23, Page 2615 2018, 23, 2615. [Google Scholar] [CrossRef]
- D’Souza, A.J.M.; Topp, E.M. Release from polymeric prodrugs: linkages and their degradation. J. Pharm. Sci. 2004, 93, 1962–1979. [Google Scholar] [CrossRef]
- Craig, M.; Yarrarapu, S.N.S.; Dimri, M.; Biochemistry, Cholesterol. StatPearls. 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK513326/.
- Liu, C.; Zhang, L.; Zhu, W.; Guo, R.; Sun, H.; Chen, X.; Deng, N. Barriers and Strategies of Cationic Liposomes for Cancer Gene Therapy. Mol. Ther. Methods Clin. Dev. 2020, 18, 751–764. [Google Scholar] [CrossRef]
- Elsana, H.; Olusanya, T.O.B.; Carr-wilkinson, J.; Darby, S.; Faheem, A.; Elkordy, A.A. Evaluation of novel cationic gene based liposomes with cyclodextrin prepared by thin film hydration and microfluidic systems. Sci. Reports 2019 91 2019, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Zhang, S.; Jiang, H.; Zhao, B.; Lv, H. Lipoplex morphologies and their influences on transfection efficiency in gene delivery. J. Control. Release 2007, 123, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, J.; Gao, Y.; Zhu, J.; Wientjes, M.G.; Au, J.L.S. Relationships between Liposome Properties, Cell Membrane Binding, Intracellular Processing, and Intracellular Bioavailability. AAPS J. 2011, 13, 585. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, R.; Coelho, F.; Silva, B.F.B. Lipid-Nucleic Acid Complexes: Physicochemical Aspects and Prospects for Cancer Treatment. Mol. 2020, Vol. 25, Page 5006 2020, 25, 5006. [Google Scholar] [CrossRef]
Figure 1.
Molecular structure of synthesized CholCadLys lipid. The formation of the carbamate bond between cholesteryl chloroformate and cadaverine and the amide bond between cadaverine and lysine is observed. UIPAC name: 10,13-dimethyl-17-(6-methylheptan-2-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl (5-(2,6-diaminohexanamido)pentyl)carbamate.
Figure 1.
Molecular structure of synthesized CholCadLys lipid. The formation of the carbamate bond between cholesteryl chloroformate and cadaverine and the amide bond between cadaverine and lysine is observed. UIPAC name: 10,13-dimethyl-17-(6-methylheptan-2-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl (5-(2,6-diaminohexanamido)pentyl)carbamate.
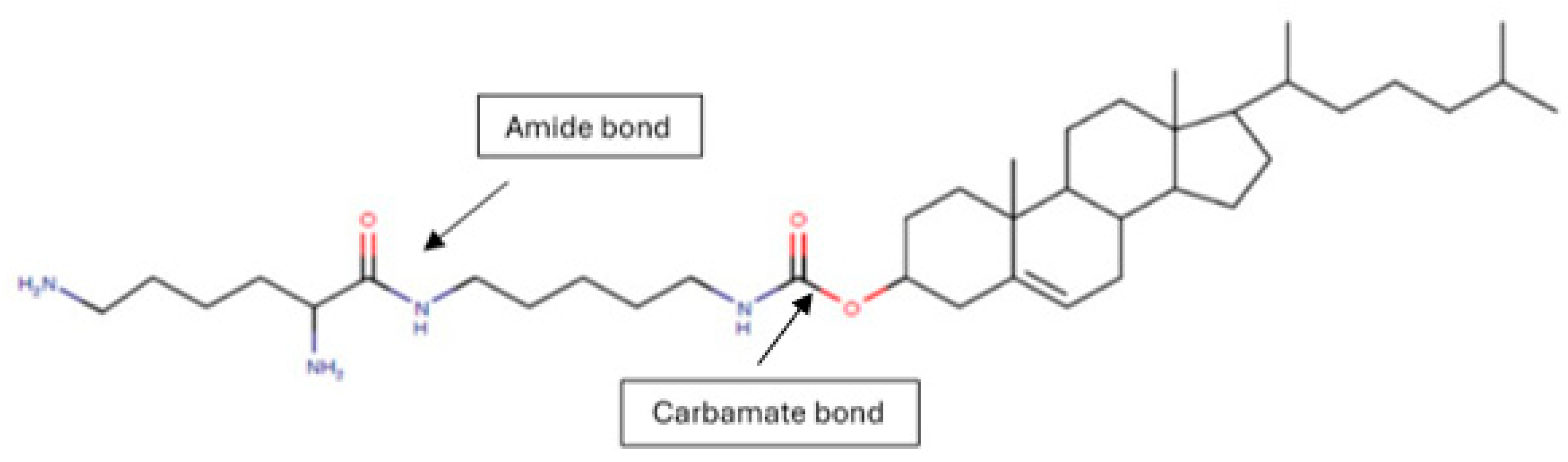
Figure 2.
(I.) Identification of the unpurified synthesized CholCadLys lipid. The raw material was placed in the chromatogram (1: cholesteryl chloroformate, 2: A mixture of raw materials, reaction intermediates, and the unpurified synthesized cationic lipid can be observed and 3: L-lysine). Chromatogram A of each lipid was revealed with ninhydrin (presence of free amino groups), while chromatogram B was revealed with sulfuric acid (presence of organic matter). (II.) Identification of the purified synthesized CholCadLys lipid. The raw material was placed in the chromatogram (1: cholesteryl chloroformate, 2: purified synthesized lipid and 3: L-lysine). Chromatogram A of each lipid was revealed with ninhydrin (presence of free amino groups), while chromatogram B was revealed with sulfuric acid (presence of organic matter). .
Figure 2.
(I.) Identification of the unpurified synthesized CholCadLys lipid. The raw material was placed in the chromatogram (1: cholesteryl chloroformate, 2: A mixture of raw materials, reaction intermediates, and the unpurified synthesized cationic lipid can be observed and 3: L-lysine). Chromatogram A of each lipid was revealed with ninhydrin (presence of free amino groups), while chromatogram B was revealed with sulfuric acid (presence of organic matter). (II.) Identification of the purified synthesized CholCadLys lipid. The raw material was placed in the chromatogram (1: cholesteryl chloroformate, 2: purified synthesized lipid and 3: L-lysine). Chromatogram A of each lipid was revealed with ninhydrin (presence of free amino groups), while chromatogram B was revealed with sulfuric acid (presence of organic matter). .

Figure 3.
Infrared spectrum of the synthesized CholCadLys lipid. The signal at 1688.59 – 1641.63 cm⁻¹ corresponds to the C=O stretching of the carbonyl of the amide bond. The signal at 3298.84 cm-1 corresponds to the elongation of the -NH groups, the signals at 2918.33 cm-1 and 2849.56 cm⁻¹ correspond to the C-H elongations of the methyl and methylene groups of the hydrocarbon chains, and the signals 1451 – 1413 cm⁻¹, corresponding to the CH3 elongations.
Figure 3.
Infrared spectrum of the synthesized CholCadLys lipid. The signal at 1688.59 – 1641.63 cm⁻¹ corresponds to the C=O stretching of the carbonyl of the amide bond. The signal at 3298.84 cm-1 corresponds to the elongation of the -NH groups, the signals at 2918.33 cm-1 and 2849.56 cm⁻¹ correspond to the C-H elongations of the methyl and methylene groups of the hydrocarbon chains, and the signals 1451 – 1413 cm⁻¹, corresponding to the CH3 elongations.
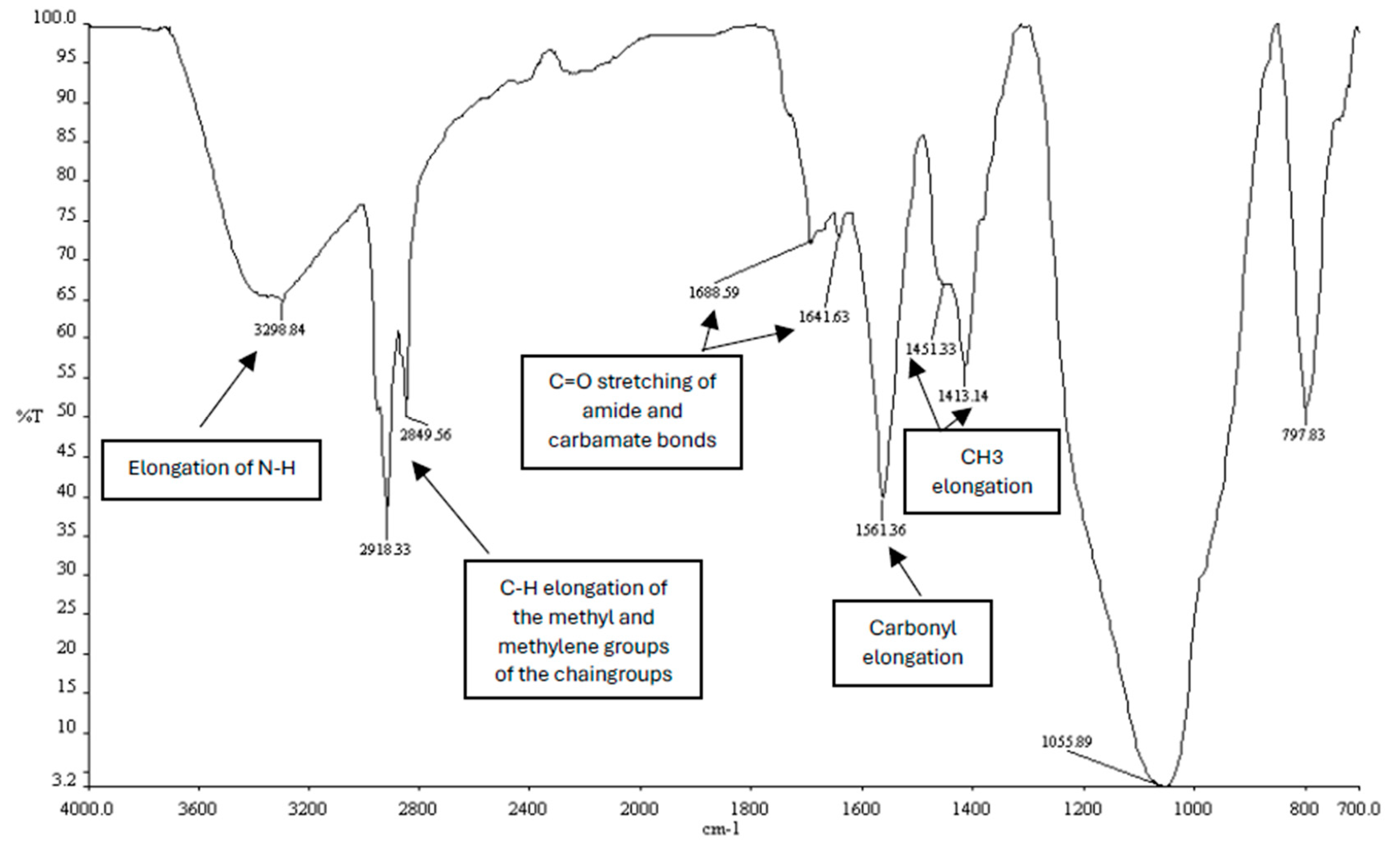
Figure 4.
Mass spectrum of the CholCadLys lipid. A signal with an exact mass of 643.5439 Da, corresponding to the CholCadLys lipid was observed.
Figure 4.
Mass spectrum of the CholCadLys lipid. A signal with an exact mass of 643.5439 Da, corresponding to the CholCadLys lipid was observed.
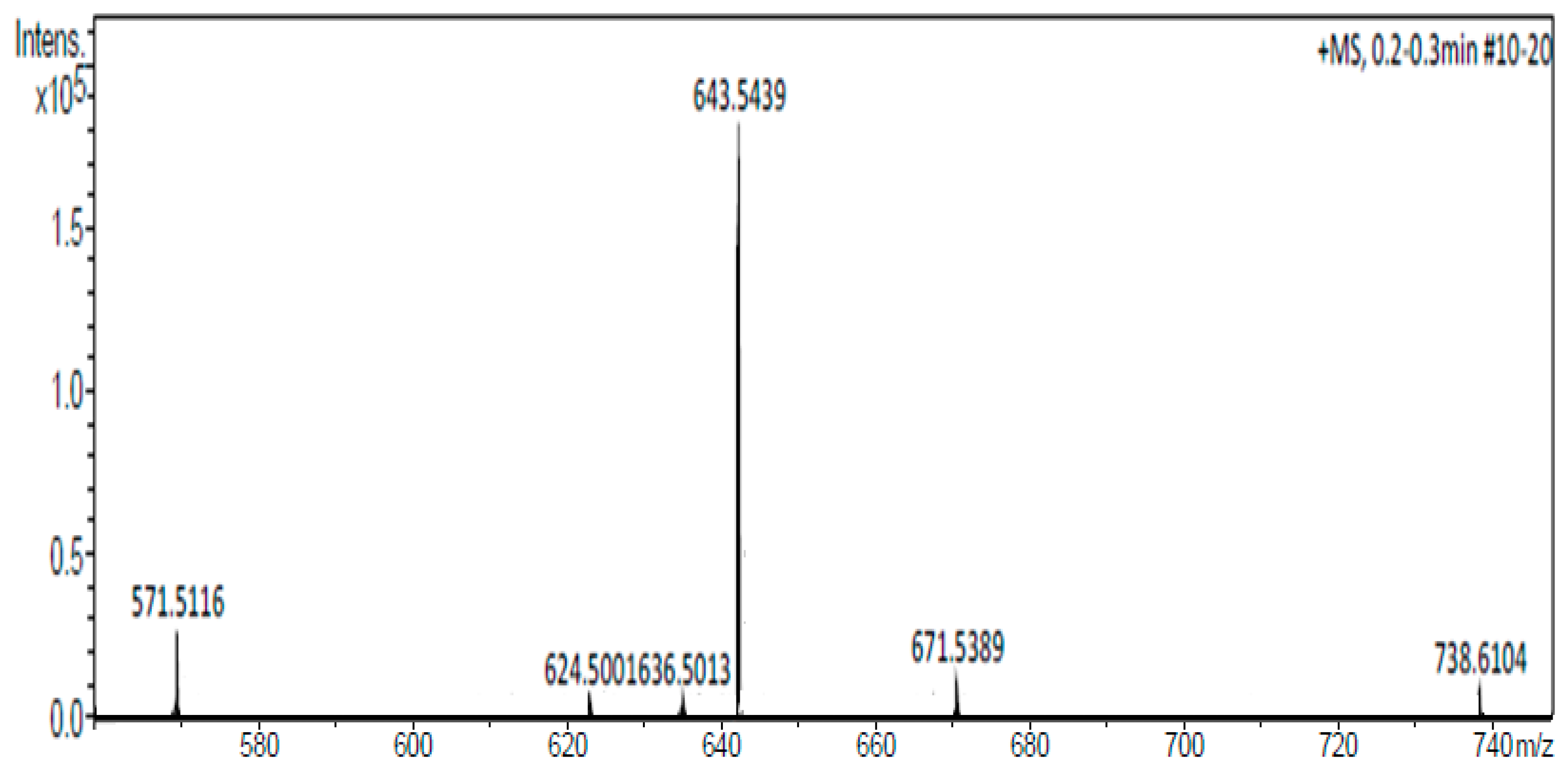
Figure 5.
Genetic map of the pIRES2-EGFP plasmid. The plasmid components and specific restriction enzyme cleavage sites are depicted.
Figure 5.
Genetic map of the pIRES2-EGFP plasmid. The plasmid components and specific restriction enzyme cleavage sites are depicted.
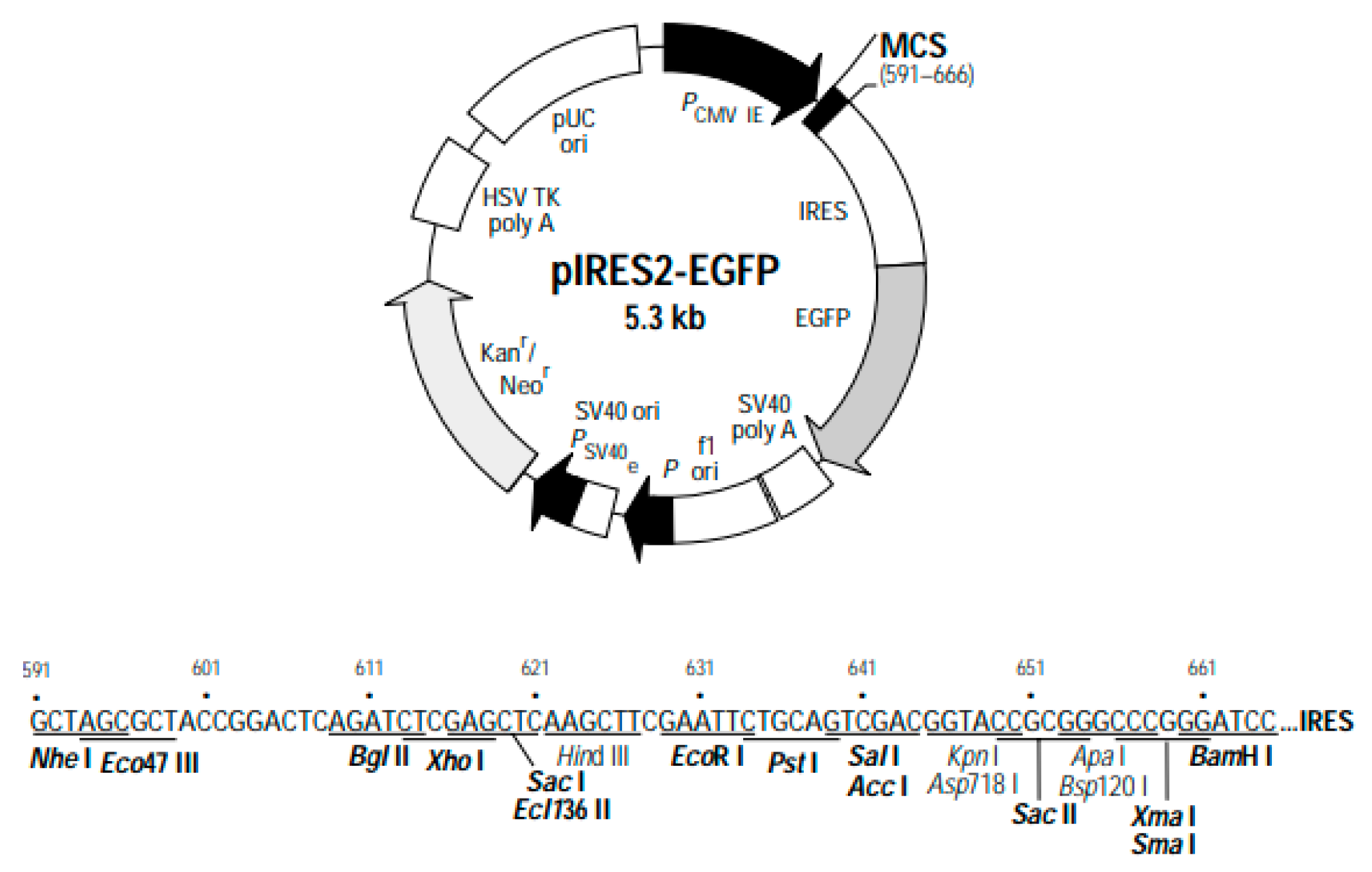
Figure 6.
Electropherogram of the purified pIRES2-EGFP plasmid integrity. Lane 1: Quick-Load 1Kb DNA Ladder molecular size marker; Lane 2: pIRES2-EGFP control pDNA; Lanes 3-4: 1 and 2 μL of plasmid DNA extraction and purification.
Figure 6.
Electropherogram of the purified pIRES2-EGFP plasmid integrity. Lane 1: Quick-Load 1Kb DNA Ladder molecular size marker; Lane 2: pIRES2-EGFP control pDNA; Lanes 3-4: 1 and 2 μL of plasmid DNA extraction and purification.
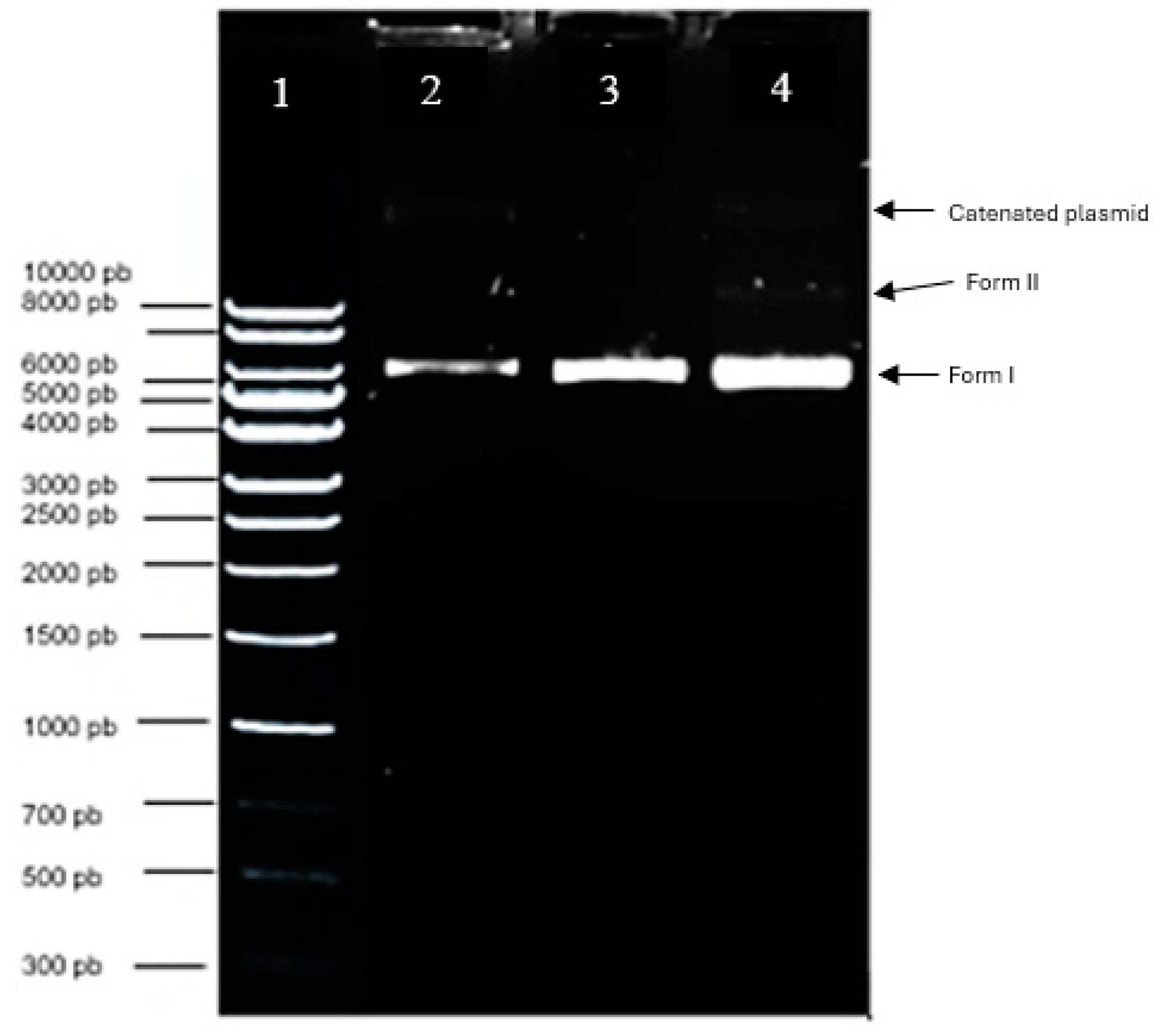
Figure 7.
Electropherograms of lipoplexes formed with CholCadLys and helper lipids at different w/w ratios (1 μg total lipids/different amounts of pDNA). A) CholCadLys - DOPE 2:1; B) CholCadLys - CholCad 1:2; C) CholCadLys - CholCad 2:1; D) CholCadLys - CholCad - DOPE 1:1:2; E) CholCadLys - CholCad - DOPE 2:1:1. 1. pDNA control. 2. Ratio 1:0.66 μg DNA. 3. Ratio 1:0.40 μg DNA. 4. Ratio 1:0.28 μg DNA. 5. Ratio 1:0.22 μg DNA. 6. Ratio 1:0.18 μg DNA.
Figure 7.
Electropherograms of lipoplexes formed with CholCadLys and helper lipids at different w/w ratios (1 μg total lipids/different amounts of pDNA). A) CholCadLys - DOPE 2:1; B) CholCadLys - CholCad 1:2; C) CholCadLys - CholCad 2:1; D) CholCadLys - CholCad - DOPE 1:1:2; E) CholCadLys - CholCad - DOPE 2:1:1. 1. pDNA control. 2. Ratio 1:0.66 μg DNA. 3. Ratio 1:0.40 μg DNA. 4. Ratio 1:0.28 μg DNA. 5. Ratio 1:0.22 μg DNA. 6. Ratio 1:0.18 μg DNA.
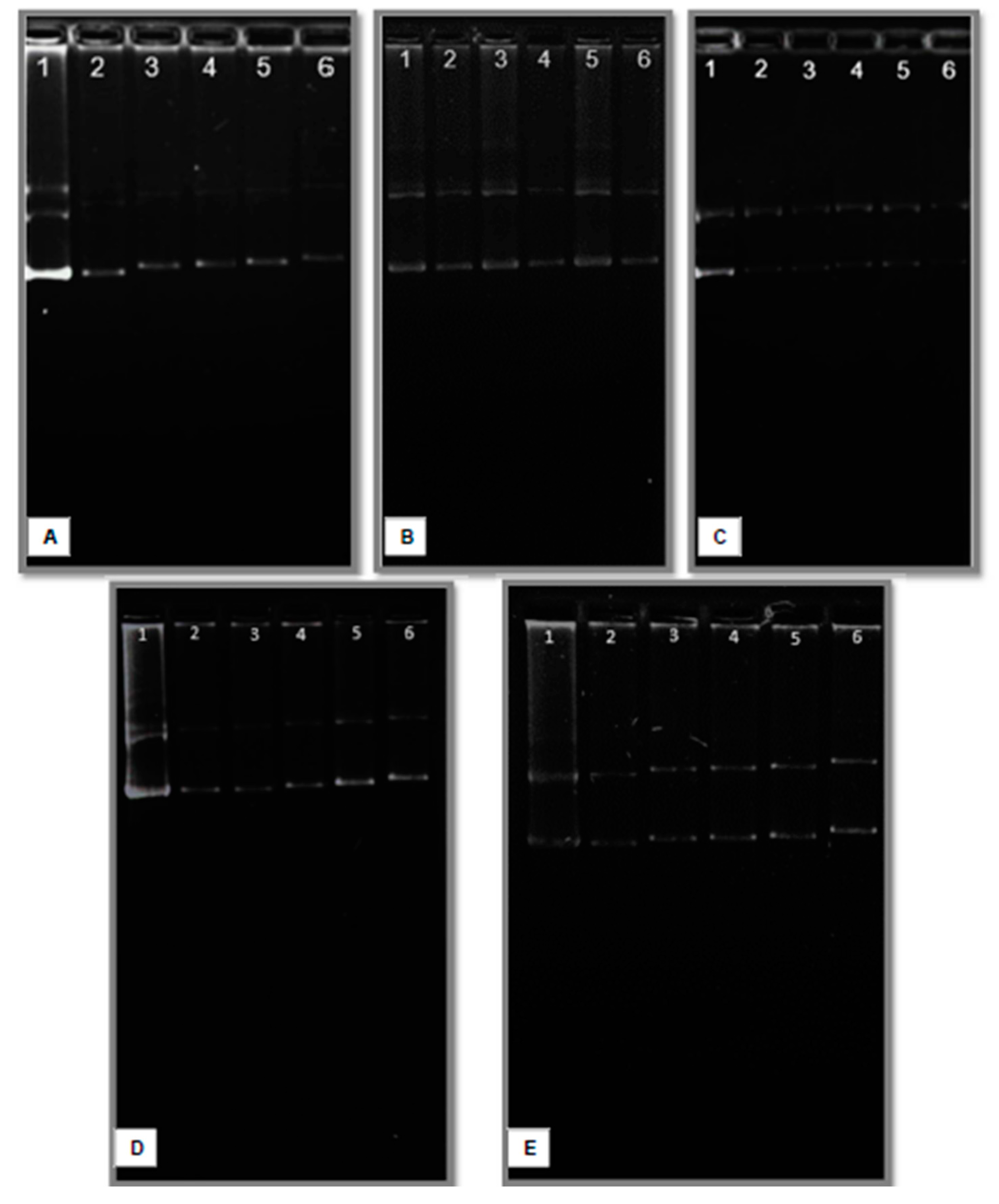
Figure 8.
TEM micrographs of liposomes and lipoplexes (total lipids/pDNA ratio 1:0.4). A) Liposomes formed with CholCadLys:cholesterol at a molar ratio of 1:1. B) Lipoplexes formed with CholCadLys:cholesterol at a molar ratio of 1:1. C) Liposomes formed with CholCadLys:cholesterol at a molar ratio of 2:1. D) Lipoplexes formed with CholCadLys:cholesterol at a molar ratio of 2:1. E) Liposomes formed with CholCadLys:DOPE at a molar ratio of 1:1. F) Lipoplexes formed with CholCadLys:DOPE at a molar ratio of 1:1. G) Liposomes formed with CholCadLys:DOPE at a molar ratio of 1:2. H) Lipoplexes formed with CholCadLys:DOPE at a molar ratio of 1:2.
Figure 8.
TEM micrographs of liposomes and lipoplexes (total lipids/pDNA ratio 1:0.4). A) Liposomes formed with CholCadLys:cholesterol at a molar ratio of 1:1. B) Lipoplexes formed with CholCadLys:cholesterol at a molar ratio of 1:1. C) Liposomes formed with CholCadLys:cholesterol at a molar ratio of 2:1. D) Lipoplexes formed with CholCadLys:cholesterol at a molar ratio of 2:1. E) Liposomes formed with CholCadLys:DOPE at a molar ratio of 1:1. F) Lipoplexes formed with CholCadLys:DOPE at a molar ratio of 1:1. G) Liposomes formed with CholCadLys:DOPE at a molar ratio of 1:2. H) Lipoplexes formed with CholCadLys:DOPE at a molar ratio of 1:2.
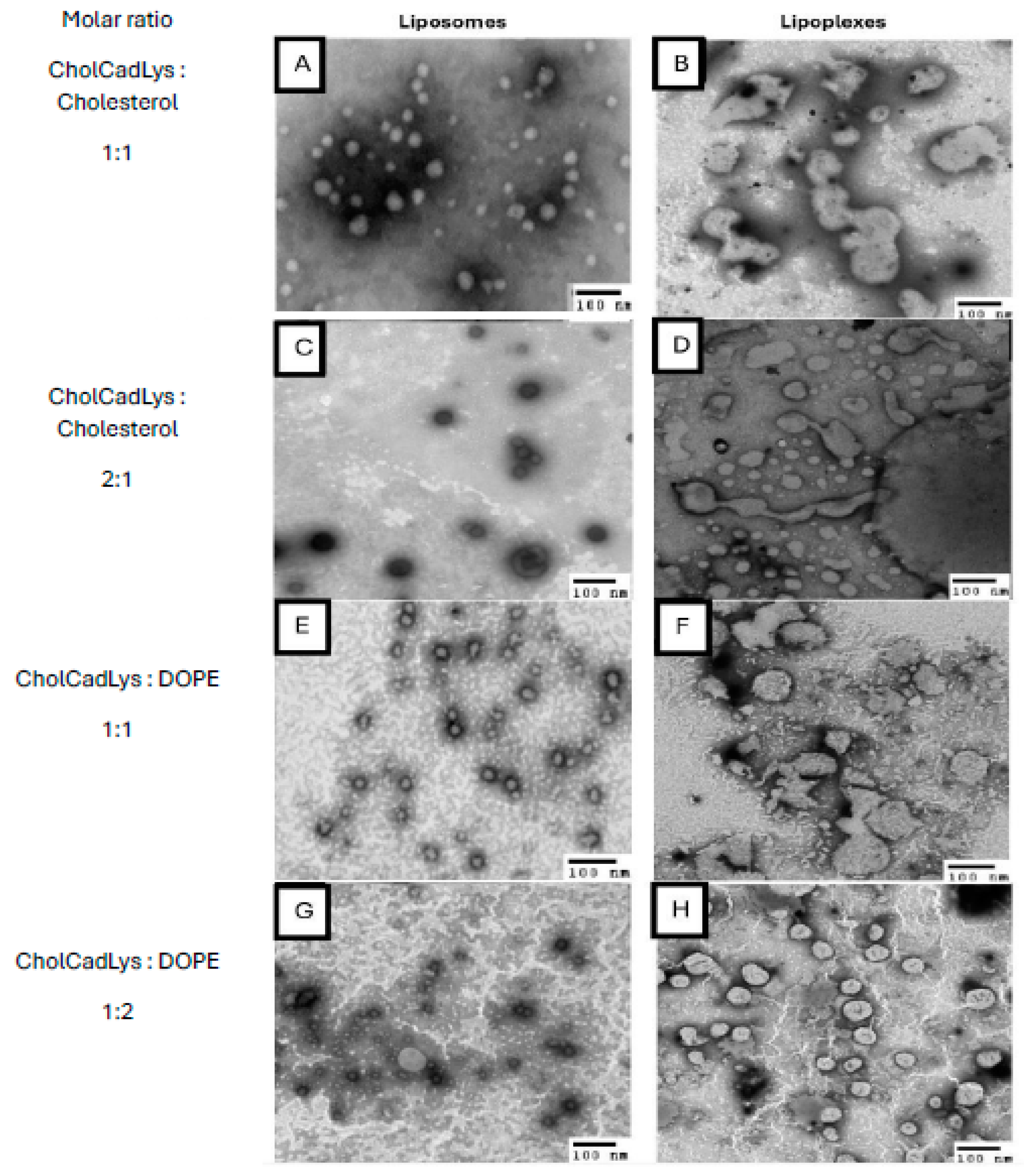
Figure 9.
Transfection in Hek-293 FT Cell Line. Different lipoplex mixtures were prepared using the synthesized lipid and a helper lipid in various ratios. A) Untransfected control cells; B) CholCadLys; C) CholCadLys - Cholesterol 1:1; D) CholCadLys - Cholesterol 2:1; E) CholCad - DOPE 1:1; F) CholCad - DOPE 1:2; G) CholCad - DOPE 2:1; I) CholCadLys - DOPE 1:1; J) CholCadLys - DOPE 1:2; K) CholCadLys - CholCad 1:2; L) CholCadLys - CholCad 2:1; M) CholCadLys - CholCad - DOPE 1:1:2; N) CholCadLys - CholCad - DOPE 2:1:1; Ñ) Lipofectamine 0.5 μg - 0.400 μg DNA.
Figure 9.
Transfection in Hek-293 FT Cell Line. Different lipoplex mixtures were prepared using the synthesized lipid and a helper lipid in various ratios. A) Untransfected control cells; B) CholCadLys; C) CholCadLys - Cholesterol 1:1; D) CholCadLys - Cholesterol 2:1; E) CholCad - DOPE 1:1; F) CholCad - DOPE 1:2; G) CholCad - DOPE 2:1; I) CholCadLys - DOPE 1:1; J) CholCadLys - DOPE 1:2; K) CholCadLys - CholCad 1:2; L) CholCadLys - CholCad 2:1; M) CholCadLys - CholCad - DOPE 1:1:2; N) CholCadLys - CholCad - DOPE 2:1:1; Ñ) Lipofectamine 0.5 μg - 0.400 μg DNA.
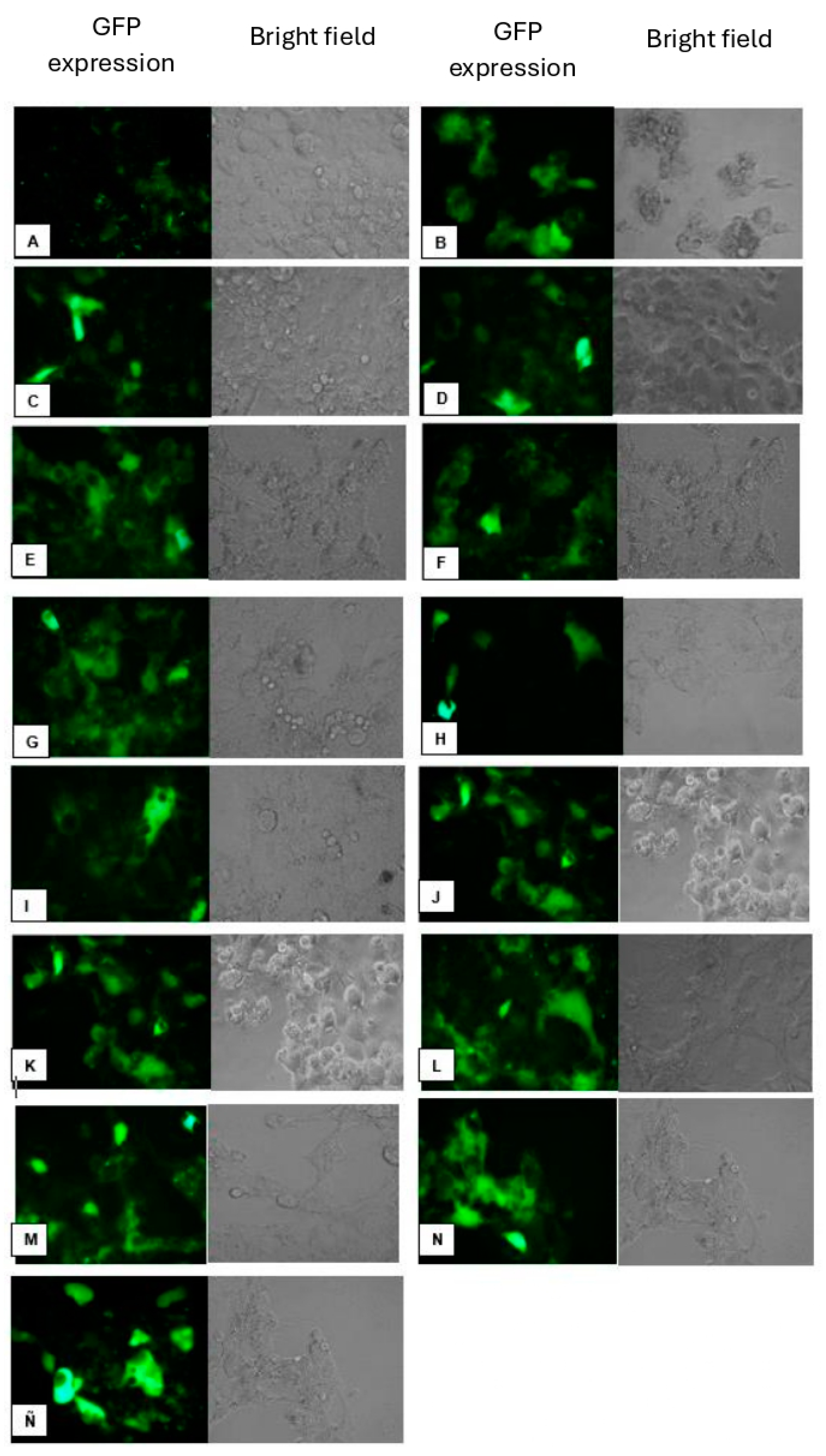
Figure 10.
Viability Effect on Hek-293 FT Cell Line. A) Negative control cells; B) Lipoplexes formed with cholesterol as a helper lipid; C) Lipoplexes formed with DOPE as a helper lipid; D) Lipoplexes formed with: CholCadLys - CholCad - DOPE 1:1:2; E) Lipoplexes formed with: CholCadLys - CholCad - DOPE 2:1:1; F) Lipofectamine 0.5 μg.
Figure 10.
Viability Effect on Hek-293 FT Cell Line. A) Negative control cells; B) Lipoplexes formed with cholesterol as a helper lipid; C) Lipoplexes formed with DOPE as a helper lipid; D) Lipoplexes formed with: CholCadLys - CholCad - DOPE 1:1:2; E) Lipoplexes formed with: CholCadLys - CholCad - DOPE 2:1:1; F) Lipofectamine 0.5 μg.
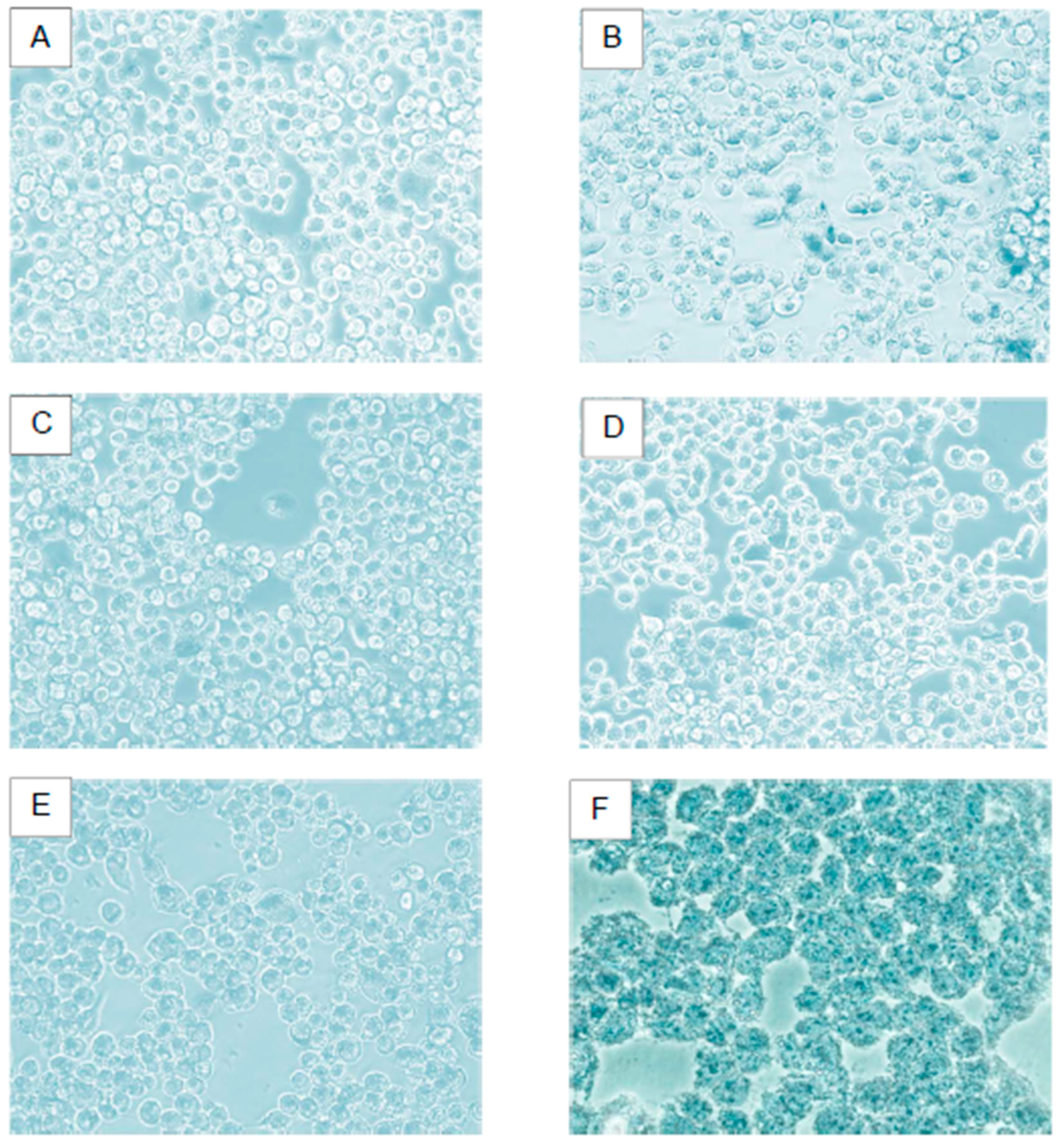

Table 1.
Size and Count of Liposomes and Lipoplexes.
| Molar Ratios (CholCadLys : Helper Lipid) |
Average Size (nm) | Concentration (particles/mL) |
|||
| Liposomes | Lipoplexes | Liposomes | Lipoplexes | ||
| Cholesterol | 1:1 | 136.6 | 132 | ||
| 2:1 | 114.8 | 92.3 | |||
| DOPE | 1:1 | 102.4 | 142 | ||
| 1:2 | 116.7 | 107.2 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



