
Submitted:
30 October 2024
Posted:
31 October 2024
You are already at the latest version
Abstract
The neurobiology of depression establishes a close relationship between this psychiatric disorder and alterations in neuroplasticity associated with neuronal atrophy and a reduction in dendritic arborization in the prefrontal cortex and hippocampus. In this sense, the therapeutic effect of antidepressants is associated with changes in the brain associated with neuroplasticity, neurogenesis and synaptogenesis through the activation of intracellular signaling pathways associated with changes at the neurochemical and behavioral level in animal models used to study depression. Antidepressants increase the synaptic availability of monoamines (monoaminergic hypothesis) such as serotonin, noradrenaline and GABA by inhibiting their reuptake or degradation and activating intracellular signaling pathways such as the cAMP-CREB cascade, which regulates the expression of genes related to neuroplasticity and neurogenesis in various brain structures associated with depression. Although acute treatment alters the number of receptors, the therapeutic effect lasts 3-4 weeks and depends on the increase in the density of dendritic spines and the expression of proteins such as BDNF and GAP-43 in the hippocampus and cerebral cortex. This review focuses on the effects of acute and chronic treatment with monoaminergic antidepressants and new drugs and other pharmacological alternatives in preclinical studies with the aim of demonstrating their mechanism of action and relationship to neuroplasticity.
Keywords:
Neuroplasticity
; Antidepressants
; Acute and chronic treatment
; Depression
; Mechanisms of action
; Neurotrophic factors
1. Introduction
The therapeutic effect of antidepressants is based on complex mechanisms involving neuroplasticity that go beyond the traditional monoaminergic deficit hypothesis. Neuroplasticity is the adaptive ability of the brain to reorganise and form new connections under normal and pathological conditions [1]. Depression causes dendritic neuronal atrophy and a reduction in glial cells and dendritic arborization in neurons of the prefrontal cortex and hippocampus. In addition, an increase in dendritic branching of neurons of the cerebral amygdala was reported [2]. The neu-rotrophic hypothesis states that treatment with antidepressants promotes neu-roadaptive changes at the level of the brain, changes that counteract some of the characteristic symptoms of depression [3]. Chronic antidepressant treatment is known to promote complex mechanisms at the cellular, molecular and structural levels of neurons, via intracellular signalling pathways involved in survival and neuroplasticity. Chronic treatment with antidepressants stimulates the function of cAMP-responsive element binding protein (CREB), a transcription factor that regulates the expression of genes involved in neuroplasticity, cell survival and cognition [1].
It promotes neurogenesis, dendritic arborization and synaptogenesis in the hippocampus and prefrontal cortex and reverses the pathological effects of stress and depression [4]. The cAMP-MAPK-CREB-BDNF cascade is involved in dendritic restructuring, neurogenesis and cell survival [5]. Chronic stress has been reported to decrease neuroprotective factors such as brain-derived neurotrophic factor (BDNF), which negatively affects neuroplasticity and increases neuronal atrophy. This phenomenon leads to a reduction in synaptic contacts and brain volume [2,6]. In contrast, other subcortical regions such as the amygdala show hypertrophy in the context of depression, possibly contributing to the anxiety and altered emotions characteristic of this disorder [7]. These findings underscore the critical importance of understanding the neurobiological mechanisms underlying depression as a basis for developing more effective and targeted therapeutic approaches that can counteract the observed brain changes.
Treatment alternatives have been tested at an experimental level with the aim of improving the efficacy and safety of treatments for depression. This research is of crucial importance, especially considering that response rates to existing treatments are only 50% [8]. In this sense, the effect of chronic treatment with antidepressants has been confirmed in animal models such as forced swimming, where the effect of relatively low doses of fluoxetine on behavioural hopelessness has been observed [9,10]. In addition, an increase in BDNF specifically in the hippocampus and prefrontal cortex of mice [11] and an increase in the expression of 5-HT1A and 5-HT2A receptors in the raphe, hippocampus and cerebral cortex of the rat [12] have been observed. However, acute treatment, especially at high doses administered 24, 5 and 1 hour before the forced swim test, has been shown to alter behavioural indicators of hopelessness in rats. These effects appear to be related to an increase in levels of neurotransmitters such as gamma-aminobutyric acid (GABA), dopamine (DA), noradrenaline (NA) and serotonin (5-HT), without necessarily causing plastic changes in brain structures that control emotional processing [13,14]. These findings on antidepressants have led to the exploration of new pharmacological strategies targeting non-monoaminergic mechanisms.
The aim of this review is to provide a comprehensive understanding of the different antidepressants used experimentally, to elucidate the mechanisms of action of the different antidepressants and to determine the differences between acute and chronic antidepressant treatment on an experimental level on neuroplasticity in the rodent brain. To prepare this review, we conducted a comprehensive bibliographic search in PubMed, Scopus and Web of Science using specific keywords such as antidepressants, acute treatment, chronic treatment, forced swimming, neuroplasticity and experimental depression. The search was refined to include only studies published since 2000. Inclusion criteria focused on research articles, reviews, abstracts and position statements examining the effects of different types of antidepressants at an experimental level during chronic and acute treatment, as well as their mechanism of action and changes in neuroplasticity. Exclusion criteria excluded those studies that had no antidepressant effect, did not have access to full text, or were not rigorously peer-reviewed.
Data extraction followed a structured approach, capturing critical information on study design, key findings and the relevance of each study to understanding the differences in antidepressant effect between acute and chronic administration and the potential mechanism of action of each effect. We assessed the quality and validity of the studies based on methodological rigor, reproducibility and peer-reviewed status. This approach ensured that only high quality studies were included, allowing a comprehensive synthesis of the current state of knowledge.
2. Depression Overview
According to the World Health Organization, depression is a mental disorder characterized by a depressed mood or a lack of interest in activities for prolonged periods of time [15]. This debilitating emotional state is accompanied by symptoms such as cognitive impairment, hopelessness, despair, feelings of emptiness, changes in sleeping and eating patterns, difficulty concentrating and suicidal thoughts [16]. The effects of depression extend beyond the person affected and significantly affect their family, social and community relationships [15]. Depression is one of the leading causes of disability worldwide, affecting approximately 3.8 of the world's population, or around 280 million people [17,18,19,20]. Depression, particularly in patients with medical conditions, exacerbates the burden of disease and negatively impacts quality of life [21,22]. Building on this, the recent COVID-19 pandemic that emerged in 2020 has further exacerbated this situation, with factors such as childbirth and loss of loved ones contributing to increased rates of depression [23,24,25].
Neurobiological research on depression has shown a strong link between this psychiatric disorder and changes in neuroplasticity. These changes are particularly evident in neuronal atrophy and the reduction of dendritic branching in key brain structures such as the prefrontal cortex and hippocampus. A model has been proposed in which prolonged stress decreases neuroprotective factors such as BDNF, which negatively affects neuronal plasticity and increases neuronal atrophy. This process leads to a reduction in synaptic contacts and brain volume [2,26]. Conversely, other subcortical regions, such as the amygdala, exhibit hypertrophy associated with depression, which may contribute to the anxiety and altered emotions characteristic of this disorder [7]. These findings highlight the importance of understanding the neurobiological mechanisms underlying depression in order to develop more effective and targeted therapeutic approaches that can counteract the observed brain changes. The treatment of depression includes pharmacological and non-pharmacological options such as psychotherapy, but antidepressants can cause a number of adverse effects [27,28]. The low adherence to treatment and the occurrence of side effects have driven the search for therapeutic alternatives [29,30,31].
Preclinical research into treatment alternatives is being conducted with the aim of improving the efficacy and safety of depression treatments. This research is crucial, especially considering that response rates to existing treatments are only around 50% [32]. Therefore, research into antidepressant alternatives, such as herbal medicines, is crucial for improving treatment outcomes. Given the chronic nature of depression and the need to find both acute and long-term treatment solutions, the ongoing evaluation of these alternatives is critical to addressing the significant global burden of depression.
3. Mechanisms of Action of Antidepressants and Neuroplasticity
The main mechanism of action of antidepressants focuses on the synaptic availability of monoamines by inhibiting reuptake or degradation [33]. It is known that acute treatment has an effects on the number of receptors. The pharmacological response to chronic treatments in the clinic suggests neuroplasticity mechanisms necessary for the establishment of the therapeutic effect [34]. In addition, chronic administration of antidepressants leads to changes in the sensitivity of presynaptic and postsynaptic receptors [35]. These mechanisms usually focus on the restructuring of neuronal synapses, a process known as neuroplasticity of the neurotransmission systems involved, mainly 5-HT, NA and GABA. This process takes about 3 to 4 weeks [12], which explains the long latency period with which the therapeutic effects of drugs and substances with antidepressant activity can be observed.
The mechanism of action of drugs with antidepressant effects, such as tricyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs) and selective serotonin reuptake inhibitors (SSRIs), have in common the enhancement of serotonergic neurotransmission [35], but their therapeutic effects are based on complex mechanisms (Shelton, 2000). Some of the mechanisms activate the adenylate cyclase-protein kinase A cascade and inhibit the phospholipase C-protein kinase C mechanisms that regulate gene expression. They also alter the expression levels of mRNAs of various receptors, neurotrophic factors and neuropeptides [36]. Crystallographic studies of G-protein-coupled receptors and neurotransmitter transporters have enabled us to understand the mechanism of antidepressants and to develop new drugs [37].
The molecular and cellular mechanisms by which antidepressants exert their effects are complex, i.e. they go beyond increasing the availability of monoamines in the synaptic space [38]. Acute administration of antidepressants can modulate the neuronal ERK/MAPK (extracellular signal-regulated kinase/mitogen-activated protein kinase) signalling pathway in the prefrontal cortex, which contributes to the restoration of neurogenesis and neuroplasticity [39]. Some antidepressants such as fluoxetine and ketamine have the ability to bind to the trkB receptor, a mechanism by which they could induce plastic changes at the brain level [40]. Chronic treatment, in turn, causes greater activation of the cAMP system, thereby increases the expression of transcription factors such as CREB, which regulate target genes such as BDNF [41]. In addition, it has been reported to increase neurogenesis in the adult hippocampus [40]. These changes in neuroplasticity strengthen synaptic connectivity and remodelling of neuronal circuits in the emotional circuitry [38].
Several studies have reported that fluoxetine ingestion induces long-lasting behavioral changes and neuroplasticity in the hippocampus and cortex in intact adult rodents [42,43]. In this sense, young rats (9 weeks old) treated with 10 mg/kg fluoxetine twice daily for three weeks showed greater motivation to explore novel environments in the Y-maze, with no effects on anhedonia and anxiety, assessed respectively with the sucrose test and the Elevated Plus Maze. However, an increase in the number of 5-bromodeoxyuridine-positive (BrdU+) cells, the density of dendritic spines in layer II/III pyramidal neurons of the medial prefrontal cortex, and the expression levels of BDNF/tropomyosin receptor kinase B (TrkB) were observed. These changes can persist for up to 20 days after the last fluoxetine dose [43].
Chronic treatment with fluoxetine has been shown to induce neuroplasticity in several brain regions of rodents. In the somatosensory cortex of rats, it increases c-fos expression and dendritic spine density [44]. In mice, administration of 10 mg/kg fluoxetine over a two-week period led to a reduction in hopelessness, anhedonia and anxiety. In addition, fluoxetine treatment decreased the deterioration of hippocampal neurons and the number of dendritic spines tended to increase, which is attributed to synaptic plasticity [45]. Similarly, in the chronic social isolation model in rats, administration of 5 mg/kg fluoxetine for three weeks was shown to reverse the behavioral effects induced by stress. On the other hand, the synaptosomal polysialic acid-neuronal cell adhesion molecule (plasticity-related molecule in the hippocampus) (PSA-NCAM), a molecular marker of plasticity, increases in the hippocampus of chronically isolated rats, an effect that is reversed by fluoxetine treatment [46]. However, the effects of fluoxetine in young rats are reported to differ from those in adults. As PSA-NCAM expression increases in several brain regions, the number of proliferating cells in the subventricular zone decreases [47].
These results show that the effect of fluoxetine on neuroplasticity varies according to treatment duration, age and brain region, as the mechanism by which fluoxetine achieves the antidepressant effect is complex. Another study examined the effect of chronic treatment with fluoxetine on neurogenesis and the expression of growth-associated protein 43 (GAP43), a synaptic protein, in the hip pocampus of rats exposed to chronic unpredictable mild stress (CUMS), a behavioral model similar to depression. The results of this study showed that the treatment decreased immobility in the forced swim test and increased the expression of BrdU-positive cells and GAP-43 (a protein associated with neuronal plasticity). However, fluoxetine has a greater effect on neurite outgrowth than on neurogenesis [48].
On the other hand, the most commonly used tools in preclinical research to elucidate the mechanisms of action of drugs or molecules with antidepressant potential include the forced swim test, water uptake with sucrose or tail suspension, etc. These studies can be carried out with acute or chronic administration. Forced swimming, for example, is one of the most widely used and validated behavioral models for the study of drugs and substances with potential antidepressant effects, which assesses the hopelessness of the behavior. In this model, drugs or substances with antidepressant potential such as fluoxetine reduce immobility behavior and prolong the latency to the first immobility period, which is interpreted as an increase in the animal's motivation to escape from the stressful situation presented by the forced swim [9], a typical effect of clinically effective antidepressants. Furthermore, antidepressants that are ineffective when administered acutely have been shown to exert an antidepressant effects when administered chronically to experimental animals [49]. This chronic administration is necessary if the effects of stress on immobility and changes in neuroplasticity are to be reversed [45,49].
4. Pharmacological Treatment of Depression
The pharmacological treatment of major depressive disorder (MDD) is based on the use of serotonergic drugs, despite their limited efficacy. Several mechanistically novel drugs have been developed in recent years, but many fail in clinical trials. Several hypotheses have been proposed to explain the pathophysiology of MDD, suggesting that physio-logical processes such as neuroplasticity, circadian rhythms and metabolism are potential targets [50].
Despite their limited efficacy, these antidepressants, especially SSRIs, are still the most important drugs for the pharmacological treatment of MDD. Approximately 35–50% of patients do not respond to treatment [51,52]. In addition, lethargy, sedation and sexual dysfunction are side effects of serotonergic antidepressants, making it difficult for patients to adhere to the required treatment regimens [53,54]. The time interval of at least two weeks to see an improvement in depressive symptoms when using currently available medications and the lack of response in treatment-resistant depressed patients are also issues that need to be addressed [55].
The monoaminergic hypothesis for depression, formulated in part on the basis of the antidepressant efficacy of serotonergic drugs, has not found sufficient support in the studies conducted to date [56]. Therefore, there is an urgent need to find new non-serotonergic drug targets with greater pharmacological efficacy and fewer side effects.
Early studies in MDD patients showed decreased levels of the monoamines 5-HT, DA and NE in the brain, leading to the so-called “monoaminergic hypothesis”, that led to the development of several classes of antidepressants that primarily target the monoaminergic system [57,58]. MAOIs were the first compounds to show antidepressant activity by increasing the availability of monoamines in the synaptic cleft by preventing their degradation by monoamine oxidase (MAO) enzymes [50]. However, the use of MAOIs was restricted due to adverse effects and their reported toxicity [59,60].
Another class of drugs are the TCAs and tetracyclic antidepressants. TCAs such as imipramine cause an improvement in depressive state in some patients by inhibiting presynaptic NE and 5-HT reuptake transporters, leading to an increase in the concentration of NE and 5-HT in the synaptic cleft [50]. However, cyclic antidepressants can also act on other postsynaptic receptors (α-adrenergic, histaminergic and cholinergic). However, they are associated with adverse effects such as dizziness and memory impairment [60,61].
In the search for antidepressants with improved efficacy and effectiveness, pharmaceutical companies began researching ligands that selectively inhibit 5-HT reuptake. This led to the development of fluoxetine, the first SSRI approved by the Food and Drug Administration (FDA) [62]. Since then, other SSRIs have proven effective and are prescribed as first-line treatment for depression. SSRIs work by increasing the availability of 5-HT in the synaptic cleft, specifically by inhibiting 5-HT reuptake transporters. SSRIs have fewer side effects than TCAs, which typically include sexual dysfunction, insomnia and loss of appetite [58,60].
Another class of medications used to treat MDD are atypical antidepressants, which differ in their mechanism of action from other treatments and often act on multiple targets. 5-HT and NA reuptake inhibitors (SNRIs), such as venlafaxine and duloxetine, have been developed to selectively inhibit both the 5-HT and NA transporters [61]. There is evidence that SNRIs may be more effective than SSRIs in the treatment of MDD. Unfortunately, this effect seems to be specifically related to venlafaxine, which, however, has a higher discontinuation rate due to side effects [63,64,65].
Atypical or multimodal antidepressants act on other neurotransmitter systems. For example, bupropion is a DA and NA reuptake inhibitor that has a higher affinity for the DA reuptake transporter [66], while agomelatine is a melatonin receptor agonist and an inhibitor of certain serotonergic receptors [67]. Another class of antidepressants is referred to as “noradrenergic and serotonergic specific antidepressants” or NaSSAs. These drugs, represented mainly by mirtazapine, antagonize the a2-adrenergic receptor and inhibit certain serotonergic receptors [68]. Vortioxetine, one of the drugs recently approved by the FDA, inhibits 5-HT reuptake by inhibiting its transporter and has a high affinity for several types of 5-HT receptors [69]. The common side effects of atypical antidepressants are usually mild and include nausea, dry mouth, insomnia, nervousness, and low libido.
Recently, some antidepressants have been launched, such as es-ketamine (Spravato) [70], a glutamatergic antagonist, the combination of dextrom-thorphan, an N-methyl-D-aspartate (NMDA) receptor antagonist, with bupropion (Auvelity) [71] and brexanolone (Zulresso), a positive allosteric modulator of GABAA [72]. Although these are new drugs for the treatment of MDD, they have been developed for specific indications, making it difficult to use them for specific types of depression. For example, Zulresso is only prescribed for postpartum depression; Spravato is only indicated for patients who do not respond to drug treatment for depression and may have sedation and cognitive impairment as side effects; and Auvelity has addiction potential, as dextromethorphan is known to have this problem. These new drugs can be seen as significant advances in the pharmacotherapy of MDD. However, they have limitations due to side effects, so the search for antidepressants with better effectiveness and efficacy for the treatment of MDD should continue [50]. The most commonly used antidepressants for the treatment of depression are listed in Table 1.
5. Effect of Acute Antidepressant Treatment on an Experimental Level
In preclinical studies, it is possible to observe the effect of high doses of antidepressants during acute administration, that is impossible to replicate in the clinic. There is evidence that acute treatment with antidepressants can have rapid effects on a subset of symptoms [82]. However, chronic administrations of two to four weeks are necessary to achieve a desired pharmacological effect according to the Hamilton scale [83].
Several studies have shown that a dose of 20 mg/kg fluoxetine administered 23.5, 5 and 1 hour before the forced swim test reduced immobility behavior in male rats in the Porsolt pond through effects on the 5-HT2C receptor [84]. On the other hand, [85] showed that fluoxetine at doses of 5 and 10 mg/kg reduced immobility and increased swimming behavior in ovariectomized female rats during treatment for 23.5, 5 and 1 h before the forced swim test. On the other hand, [14] reported that a dose of 10 mg/kg fluoxetine reduced immobility in female and male rats. However, low doses of 5 mg/kg fluoxetine do not have the same effect in male rats under the same dosing regimen [86]. Furthermore, antidepressants that are ineffective when administered acutely have been shown to produce antidepressant-like effects when administered chronically [49,86,87]. Such chronic administration in experimental sub-trials tends to be necessary to reverse the effects of stress on immobility [49,87].
The most commonly used antidepressants in clinical practise are selective inhibitors such as 5-HT and NA inhibitors, MAOIs and TCAs, which represent the best pharmacological options. Nevertheless, a high percentage of patients do not achieve sustained remission due to adverse effects or the long time it takes for the therapeutic effects to kick in. It is therefore necessary to explore more effective and safer drugs with a faster onset of action. In this sense, two scenarios have been observed: i) the fact that in preclinical research, conventional antidepressants can produce different effects at effective or high acute doses, ranging from the emergence of anxiety-like behaviours or states [88] to antidepressant effects in preclinical models after 1 to 3 administrations within 24 hours, such as fluoxetine [13,14] and citalopram [88] in both sexes and different age groups [14].
These drugs have changed the paradigm of antidepressants, in which a long latency to therapeutic effect was considered an indispensable prerequisite, related to the establishment of slow-onset plastic changes in the various neural circuits that regulate motivated behavior and that are associated with changes in neurotrophin levels [89,90], which will be described later.
On the other hand, recent studies in both preclinical and clinical research have found new agents capable of producing antidepressant-like effects after a single administration and over a period of several hours [91,92,93]. These agents are substances previously known for other pharmacological properties, such as ketamine, which is used as an anesthetic and whose been discovered have been discovered at the clinical level [94], hallucinogens such as psilocybin and neurosteroids such as allopregnanolone, which have been shown to be effective in the treatment of depression over a period of 24 to 36 hours [95].
Other studies have reported that SSRI treatment increases synaptic 5-HT availability to a limited extent when administered acutely (2 to 4 administrations), due to inhibitory effects on release mediated by somatodendritic 5-HT1A receptors and terminal 5-HT1B autoreceptors [88], whose inhibitory effect on release is decreased after chronic treatment or by their antagonism [96]. It has been known for more than two decades that acute antidepressant treatment with 10 mg/kg fluoxetine induces changes in the expression of the c-fos gene that differ from the expression patterns of the same gene after chronic treatments and that help to explain the plastic changes of chronic treatment [97]. For example, acute intraperitoneal administration of 10 mg/kg imipramine can reverse immobility time in the forced swim test, with no observed effects on neurotrophin levels (NGF and BDNF) in the hippocampus and cerebral cortex [98]. Desipramine, reboxetine, bupropion and pagyline reduce immobility in the tail suspension model in intact mice in the same way as the SSRIs sertraline, fluoxetine and paroxetine, but not in NE- and epinephrine-deficient mice by disruption of the DO-beta-hydroxylase gene, in which only citalopram reduced immobility [99], emphasizing the importance of the noradrenergic system for the acute effects of the first group of drugs. While a single administration of citalopram (30 mg/kg) showed no effect, three administrations of citalopram (10 mg/kg) intraperitoneally within 24 hours increased CREB phosphorylation in the hippocampus and decreased immobility time in the tail suspension test in mice, but not in CREBαΔ mutants, indicating its importance for the establishment of antidepressant effects. In addition, these three administrations succeeded in blocking the hypothermia response induced by the serotonergic agonist 8-OHDPAT, suggesting that within 24 hours the 5 HT1A and possibly 5HT7 autoreceptors were desensitized [88]. However, these effects were not observed when animals were exposed to another model of depression such as forced swimming with a single dose of citalopram (10 mg/kg, i.p.), but with selective 5 HT4 receptor agonists: RS 67333 (1.5 mg/kg, i.p.) and prucalopride (2.5 mg/kg, i.p.), which induce changes in CREB phosphorylation after only 3 days of treatment (Lucas et al, 2007), which underlines the potential of 5 HT4 receptor agonists as potential fast-acting antidepressants [100].
Ketamine is a non-competitive NMDA receptor antagonist that is primarily used as an anesthetic but also has antidepressant effects when administered acutely. Acute administration of ketamine has shown antidepressant effects in rat [101] and mouse models [102,103,104].
For example, in the forced swim test, high doses of imipramine (20 and 30 mg/kg) and ketamine (10 and 15 mg/kg weight) reduce immobility without altering lo-motor activity and increase energy metabolism by increasing creatine kinase activity in the striatum, cerebral cortex, prefrontal cortex and cerebellum [105]; but only the highest dose of ketamine increases BDNF and target of rapamycin (m TOR) protein in the hippocampus of rats [106]. The increase in BDNF was also observed in the prefrontal cortex of 14-month-old rats [107]. In addition, ketamine can reduce the levels of the interleukins IL-6 and IL-1β and increase the 5-HT/tryptophan ratio in the hippocampus [108]. In mice, acute administration of ketamine was also observed to increase the expression of the neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) in the dentate gyrus of the hippocampus, while blocking the signalling of this peptide attenuated the acute effects of ketamine in the tail suspension test, forced swimming, and sucrose consumption [109]. However, the antidepressant effects of ketamine are not always observed and depend on the dose, route of administration [102] and treatment protocol [110].
Ketamine promotes a cascade of cellular events that begin with the release of glu-tamate, activation of AMPA receptors that activate mTOR-mediated plasticity, BDNF, and the synthesis of synaptic proteins that facilitate plasticity in structures such as the prefrontal cortex [111,112] or increase the activity of immature neurons without increasing neurogenesis [113].
In male Wistar-Kyoto rats, which exhibit a depressive-like phenotype, simultaneous administration of treatment psychedelic substances psilocybin (1 mg/kg) and LSD (0.15 mg/kg) resulted in antidepressant-like effects in the forced swim test that appeared to last longer than the effects of ketamine [114]. Although both psilocybin and ketamine induce changes in the dopaminergic, serotonergic and GABAergic neurotransmission systems [115], a single administration of psilocybin in mice produces antidepressant-like effects in the forced swim test that are related to changes in the neuroplasticity of the hippocampus and prefrontal cortex in mice [116]. These plastic changes seem to be related to the higher affinity of these psychedelics to the TrkB receptors for BDNF [117]. It should not be overlooked that psilocin and psilo-cybin have no effect in the forced swim test in other cases, perhaps due to factors such as the genetic characteristics of the animals or the time after treatment at which the swim tests were carried out [118,119].
In 2019, the FDA approved brexenolone in an intravenous dosage form, the active ingredient of which is the neurosteroid allopregnanolone, for the treatment of postpartum depression. The decline in progesterone and neurosteroid levels in the postpartum period, which is seen as a kind of withdrawal from the effects of these substances, is considered to be part of the etiology of postpartum depression [120], an idea that had previously been proposed for the depression observed in premenstrual syndrome [121]. The antidepressant effect of allopregnanolone and other neurosteroids [122] has already been documented in preclinical studies in the forced swim model in mice [123] and in male [124] and female rats without ovarian hormones [125] from the first acute administration by modulating the activity of the GABA-A receptor. Apparently, the mechanism of brexenolone involves activation of the GABA-A receptor at synaptic and extrasynaptic levels, resulting in inhibition of circuits necessary for the treatment of postpartum depression [126]. These mechanisms also involve the BDNF signaling pathway, but seem to be independent of AMPA receptor activation [127]. The mechanisms of this work and their similarity to other agents of plant origin point to the possibility of new substances such as flavonoids, among which chrysin stands out [128].
The discovery of substances with mechanisms of action that provide virtually immediate antidepressant effects has led to the search for new substances such as reelin [129,130] with rapid action profiles that act by mimicking the mechanism of action of ketamine [92] or by novel mechanisms of action [131].
6. Effect of Chronic Antidepressant Treatment on an Experimental Level
Depression is one of the most common mental disorders, characterized by persistent sadness and lack of interest or pleasure in previously rewarding or enjoyable activities [132]. Unfortunately, evidence suggests that approximately 300 million people worldwide will suffer from depression in 2021, according to the Institute for Health Metrics and Evaluation [133].
SSRIs are widely used for the treatment and management of various mood disorders, especially depression [134]. Three SSRIs, including fluoxetine, sertraline, and es-citalopram, produce modest improvements (approximately 5–10%) in standardized depression scores in adolescent patients with moderate major depression without significantly increasing the risk of suicidal ideation or behavior [135].
It seems that SSRIs in chronic PTSD reduce basal levels of diurnal cortisol and cortisol reactivity to stress, leading to an improvement in mood [136]. As we know, the main mechanism of action of SSRIs is the blockade of the 5-HT transporter (SERT), which causes an increase of 5-HT in the synaptic cleft [137]. However, the therapeutic effects of SSRIs cannot be fully summarized in a simple inhibition of SERT [138]. Several studies have shown that SSRIs can alter the level of BDNF. For example, long-term treatment with fluoxetine increases BDNF expression in the hippocampus of depressed rats [90] and in the olfactory bulb of depressed mice [139]. Sertraline and escitalopram also increase BDNF levels, which improves depression [140]. On the other hand, clinical studies have also shown this effect [141].
Several studies have shown that antidepressant treatment affects neuroplasticity and increases neurogenesis, especially with antidepressants that act on 5-HT and NA in regions such as the hippocampus and cerebral cortex. This increase in the formation of new neurons is crucial for the recovery of cognitive and emotional function in patients with MDD [142]. Antidepressants also act by modifying synaptogenesis, i.e. the formation of new synapses between neurons, and have effects on neuronal growth factors such as BDNF, which play an important role in promoting neuroplasticity and neuronal survival and reversing the reduction in the volume of the hippocampus, a key structure of the limbic system in MDD [142].
Other studies have shown that chronic treatment with fluoxetine increases synaptic plasticity through effects on BDNF in the hippocampus and cortex. In addition, fluoxetine increases the volume of the hippocampus in people with MDD [143]. Similarly, antidepressants have been shown to increase levels of vascular endothe-lial growth factor (VEGF). This factor is crucial for promoting neurogenesis, angiogenesis and neuronal survival, as it promotes the formation of new neurons and synaptic connectivity [143]. For example, VEGF has been reported to regulate the effects of antidepressants such as lamotrigine, suggesting that this factor could serve as a biomarker to monitor response to antidepressant treatment [144].
On the other hand, other studies have shown that the insulin-like growth factor (IGF-1) plays an important role in the effect of antidepressants. In patients with MDD, it has been observed that serum levels of this factor are elevated, while they decrease again after the onset of the effect of SSRIs. IGF-1 has neuroprotective properties in the CNS and also promotes neurogenesis. IGF-1 influences the effect of antidepressants because it activates the MAP/ERK and PI3K signaling pathways, which are crucial for neuronal survival and synaptic plasticity [145].
The relationship between brain-derived neurotrophic factor (BDNF) and its receptor, TrkB (tropomyosin receptor kinase B), and antidepressants is an important area of research in the field of neuropharmacology. In this sense, a wide range of antidepressants such as SSRIs, dual antidepressants and naturally occurring products such as chrysin (7,8-dihydroxyflavone) have been reported to increase BDNF levels, favoring the binding of this neurotrophin to its receptor TrkB, which activates several intracellular signaling pathways such as MAPK/ERK and PI3K, essential for promoting plastic changes in the brain and maintaining neuronal survival under stress conditions [146]. Figure 1 describes the mechanisms involved in acute and chronic antidepressant treatment that have been reported to date.
Similarly, conventional antidepressants, which generally focus on the monoaminergic system, may alter the function of NMDA receptors that are important for long-term potentiation, which is essential for the regulation of processes such as memory and for changes in emotional and affective state. Ketamine, an NMDA receptor antagonist, has shown a rapid and long-lasting antidepressant effect, while riluzole, an inhibitor of glutamate release that also increases the expression of the glutamate transporter in the glia, helps to eliminate depressive symptoms [147]. The effects of drugs with antidepressant activity are described in Table 2.
7. Pharmacological alternatives in the treatment of depression
One of the limitations of pharmacological treatment of depression is the adverse effects. Therefore, the search for alternatives through other pharmacological approaches such as natural products, modulation of the inflammatory process and the microbiota is promising [154]. In this sense, the presence of saponins, terpenes and fla-vonoids in several herbal remedies has been demonstrated to have antidepressant effects through the inhibition of SERT, NA and DA [155]. On the other hand, it has been reported that plant molecules can regulate mood due to their antioxidant capacity. In this sense, rutin (quercetin-3-rutinoside) administered for 14 days at a dose of 80 mg/kg to rats with reserpine-induced depression was reported to prolong swimming time compared to the control group [156]. This effect is due to the antioxidant capacity of rutin and the decrease in acetylcholinesterase activity.
The combination of plant extracts with antidepressant activity in rats has shown a better effect when they were combined than when they were administered individually. In this regard, the combination of Bupleurum Chinese DC (Chaihu) and Paeonia lactiflora Pall (Baishao) administered at a dose of 7.5 mg/kg to rats with unpredictable chronic stress for 28 days showed an antidepressant effect by shortening the time of immobility and increasing sucrose preference. The metabolomics results suggest that the antidepressant effect of the combination of Chaihui and Baishao is due to its ability to activate multiple signaling pathways and metabolites. The MAPK and arachidonic acid signaling pathways are critical for neuroplasticity as they can influence synaptogenesis, inflammation and growth factor expression, essential mechanisms for reducing the characteristic symptoms of depression [157].
Another natural product that has shown antidepressant properties in rats subjected to the CUMS model is crocin (Crocus sativus Linn), a compound derived from saffron. Administration of crocin at a dose of 25 mg/kg over a four-week period was shown to increase sucrose consumption and locomotor activity and decrease immobility time in the forced swim test. In addition, metabolomics tools and network pharmacology were used to identify the major pathways of action of crocin, which include biosynthesis of tryptophan, phenylalanine, histidine, glycerolipids and steroid hormones. These results demonstrate the mechanism of action of crocin, which is an alternative treatment for depression [158]. In another study using the CUMS model in rats, the suspension of Ziziphi spinosae 100 mg/kg/day for 4 weeks was shown to have an antidepressant effect in the forced swim test and sucrose consumption. There is also an increase in serum levels of 5-HT and its metabolite 5-hydroxyindoleacetic acid (5HIAA). Results that could modulate the serotonergic system as a possible mechanism of action [159].
Modulation of the inflammatory process represents an alternative to improving the symptoms of depression. In this sense, the antidepressant effect of the flavonoid quercetin was investigated in a model of LPS-induced depression in rats. Administration of 50 mg/kg quercetin over a 7-day period resulted in an increased in mobility time and grooming time and a decrease in proinflammatory mediators in the brain compared to the LPS-treated group. This suggests that quercetin has antidepressant properties via inhibition of neuroinflammation mediated by modulation of the microglial signaling pathway in the hippocampus and prefrontal cortex of the brain [160].
On the other hand, there are reports on the use of probiotics as a complementary alternative in the pharmacological treatment of depression. Probiotics can modulate the gut-brain axis, inflammation and the synthesis of various neurotransmitters. In this context, it was reported that 10-week treatment of adult male rats with a mixture of 8 strains of probiotics (a preparation called "Ecologic Barrier") at a dose of 4.5 grams, equivalent to 1.125 × 10^10 colony-forming units (CFU), reduced depressive behavior. This treatment reduced depressive behavior, improved the immune response, induced changes in the expression of genes related to HPA axis feedback, and increased the expression of genes related to neuroplasticity and neuroprotection, such as BDNF [161]. Similarly, treatment with probiotics in stressed rats restores gut flora, an effect that reduces behavioral variables indicative of depression and increases levels of NA and 5-HT and inhibits stress hormones such as adrenocorticotropic hormone (ACTH) and corticosterone [162]. In addition, probiotics have shown anxiolytic and antidepressant effects in stress-sensitive rat models that were also subjected to internal deprivation to induce additional stress. In addition, changes in the concentration of DA, 5-HT and their metabolites were detected in the hippocampus and striatum. These results suggest that treatment with probiotics alters gut flora and brain neurochemistry, an effect that is related to neuroplasticity and emotional state [163].
8. Conclusions
Depression is a complex mental disorder that affects millions of people worldwide. Its neurobiology is associated with structural and neurochemical changes in brain regions such as the hippocampus, the prefrontal cortex and the amygdala. Preclinical studies of antidepressants and their relationship to neuroplasticity are a very active area of research, reflecting the complexity of depression and contributing to understanding. These studies suggest that acute antidepressant treatments show immediate effects in behavioral models of depression such as forced swimming. However, the long-term effects of chronic treatment are crucial for the activation of intracellular signaling pathways related to neuroadaptive changes in the brain and thus for the establishment of therapeutic effect. These neuroadaptive changes are promoted by SSRIs and tricyclic antidepressants, which increase the bioavailability of various neurotransmitters at the synaptic level. In addition to traditional treatments, complementary alternatives should be considered, such as the use of probiotics and plant-based compounds, which have been shown to have an antidepressant effect and promote brain neuroplasticity. This review shows the effects of chronic antidepressant treatment at the preclinical level on neuroplasticity and its importance in establishing therapeutic efficacy against depression.
9. Future Directions
Research on the neurobiology of depression should continue to investigate non-monoaminergic mechanisms such as neuroinflammation, changes in the HPA axis and gut microbiome, and focus on mechanisms related to neuroplasticity. In this context, further preclinical research on the evaluation and efficacy of pharmacological alternatives will allow us to understand the mechanisms involved in the therapeutic effect of depression.
Acknowledgments
Rosas-Sánchez received support from the National Council of Humanities, Sciences and Technologies CONAHCYT Postdoctoral Stays for Mexico CVU 714866.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Tardito, D.; Perez, J.; Tiraboschi, E.; Musazzi, L.; Racagni, G.; Popoli, M. Signaling pathways regulating gene expression, neuroplasticity, and neurotrophic mechanisms in the action of antidepressants: a critical overview. Pharmrev. 2006, 58, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med. 2016, 22, 238–249. [Google Scholar] [CrossRef]
- Racagni, G.; Popoli, M. Cellular and molecular mechanisms in the long-term action of antidepressants. Dialogues Clin. Neurosci. 2008, 10, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Serafini, G.; Pompili, M.; Innamorati, M.; Giordano, G.; Montebovi, F.; Sher, L.; Girardi, P. The role of microRNAs in synaptic plasticity, major affective disorders and suicidal behavior. Neurosci. Res. 2012, 73, 179–190. [Google Scholar] [CrossRef]
- Arantes-Gonçalves, F.; Coelho, R. Depressão e tratamento. Apoptose, neuroplasticidade e antidepressivos. Acta Med. 2006, 19, 9–20. [Google Scholar]
- Gray, J.D.; Milner, T.A.; McEwen, B.S. Dynamic plasticity: the role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience. 2013, 239, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Anilkumar, S.; Chattarji, S.; Buwalda, B. Repeated social stress leads to contrasting patterns of structural plasticity in the amygdala and hippocampus. Behav. Brain Res. 2018, 347, 314–324. [Google Scholar] [CrossRef]
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major depressive disorder. Nat. Rev. Dis. Prim. 2016, 2, 16065. [Google Scholar] [CrossRef]
- Contreras, C.M.; Rodríguez-Landa, J.F.; Gutiérrez-García, A.G.; Bernal-Morales, B. The lowest effective dose of fluoxetine in the forced swim test significantly affects the firing rate of lateral septal nucleus neurones in the rat. J. Psychopharmacol. 2001, 15, 231–236. [Google Scholar] [CrossRef]
- Lozano-Hernandez, R.; Rodríguez-Landa, J.F.; Hernández-Figueroa, J.D.; Saavedra, M.; Ramos-Morales, F.R.; Cruz-Sánchez, J.S. Antidepressant-like effects of two commercially available products of Hypericum perforatum in the forced swim test: a long-term study. J. Med. Plants Res. 2010, 4, 131–137. [Google Scholar]
- Jesse, C.R.; Donato, F.; Giacomeli, R.; Del Fabbro, L.; da Silva Antunes, M.; De Gomes, M.G.; Souza, L.C. Chronic unpredictable mild stress decreases BDNF and NGF levels and Na+, K+-ATPase activity in the hippocampus and prefrontal cortex of mice: Antidepressant effect of chrysin. Neuroscience. 2015, 289, 367–380. [Google Scholar] [CrossRef]
- German-Ponciano, L.J.; Rosas-Sánchez, G.U.; Ortiz-Guerra, S.I.; Soria-Fregozo, C.; Rodríguez-Landa, J.F. Effects of chrysin on mRNA expression of 5-HT 1A and 5-HT 2A receptors in the raphe nuclei and hippocampus. Rev. Bras. Farmacogn. 2021, 31, 353–360. [Google Scholar] [CrossRef]
- Page, M.E.; Detke, M.J.; Dalvi, A.; Kirby, L.G.; Lucki, I. Serotonergic mediation of the effects of fluoxetine, but not desipramine, in the rat forced swimming test. Psychopharmacology. 1999, 147, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Guasti, A.; Olivares-Nazario, M.; Reyes, R.; Martínez-Mota, L. Sex and age differences in the antidepressant-like effect of fluoxetine in the forced swim test. Pharmacol. Biochem. Behav. 2017, 152, 81–89. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Depression. In WHO fact sheets. Available online: https://www.who.int/news-room/fact-sheets/detail/depression (accessed on 28 May 2024).
- Clack, S.; Ward, T. The classification and explanation of depression. Behav. Chang. 2019, 36, 41–55. [Google Scholar] [CrossRef]
- Institute for Health Metrics and Evaluation. Global Health Data Exchange (GHDx). Available online: https://vizhub.healthdata.org/gbd-results/ (accessed on 4 March 2023).
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Woody, C. A.; Ferrari, A. J.; Siskind, D. J.; Whiteford, H. A.; Harris, M. G. A systematic review and meta-regression of the prevalence and incidence of perinatal depression. J. Affect. Disord. 2017, 219, 86–92. [Google Scholar] [CrossRef]
- Ribeiro, J.D.; Huang, X.; Fox, K.R.; Franklin, J.C. Depression and hopelessness as risk factors for suicide ideation, attempts and death: meta-analysis of longitudinal studies. Br. J. Psychiatry. 2018, 212, 279–286. [Google Scholar] [CrossRef]
- Puyat, J.H.; Kazanjian, A.; Wong, H.; Goldner, E. Comorbid chronic general health conditions and depression care: a population-based analysis. Psychiatr. Serv. 2017, 68, 907–915. [Google Scholar] [CrossRef]
- Walker, J.; Burke, K.; Wanat, M.; Fisher, C.; Fielding, J.; Quinlan, R.; Coker, O.; Murray, G.; Sharpe, M. The prevalence of depression in general hospital inpatients: a systematic review and meta-analysis of interview-based studies. Psychol. Med. 2018, 48, 2285–2298. [Google Scholar] [CrossRef]
- Guo, Y.; Sims, O.T.; Qin, W.; Yang, F. Factors associated with symptoms of depression and psychological distress during the COVID-19 pandemic. Behav. Sci. 2021, 11, 13. [Google Scholar] [CrossRef]
- Ustun, G. Determining depression and related factors in a society affected by COVID-19 pandemic. Int. J. Soc. Psychiatry. 2021, 67, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Bueno-Notivol, J.; Gracia-García, P.; Olaya, B.; Lasheras, I.; López-Antón, R.; Santabárbara, J. Prevalence of depression during the COVID-19 outbreak: A meta-analysis of community-based studies. Int. J. Clin. Health Psychol. 2021, 21, 100196. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, C.G.; Sanacora, G.; Duman, R.S.; Krystal, J.H. Ketamine and rapid-acting antidepressants: A window into a new neurobiology for mood disorder therapeutics. Annu. Rev. Med. 2015, 66, 509–523. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [CrossRef]
- Sisay, T.; Wami, R. Adverse drug reactions among major depressive disorders: Patterns by age and gender. Heliyon. 2021, 7, e08655. [Google Scholar] [CrossRef]
- Evans-Lacko, S.; Aguilar-Gaxiola, S.; Al-Hamzawi, A.; Alonso, J.; Benjet, C.; Bruffaerts, R.; Chiu, W.T.; de Girolamo, G.; Florescu, S.; Gureje, O.; et al. Socio-economic variations in the mental health treatment gap for people with anxiety, mood, and substance use disorders: Results from the WHO World Mental Health (WMH) surveys. Psychol. Med. 2018, 48, 1560–1571. [Google Scholar] [CrossRef]
- Keyloun, K.R.; Hansen, R.N.; Hepp, Z.; Gillard, P.; Thase, M.E.; Devine, E.B. Adherence and persistence across antidepressant therapeutic classes: A retrospective claims analysis among insured US patients with major depressive disorder (MDD). CNS Drugs. 2017, 31, 421–432. [Google Scholar] [CrossRef]
- Marwaha, S.; Palmer, E.; Suppes, T.; Cons, E.; Young, A.H.; Upthegrove, R. Novel and emerging treatments for major depression. Lancet. 2023, 401, 141–153. [Google Scholar] [CrossRef]
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major depressive disorder. Nat. Rev. Dis. Prim. 2016, 2, 16065. [Google Scholar] [CrossRef]
- Elhwuegi, A. S. Central monoamines and their role in major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2004, 28, 435–451. [Google Scholar] [CrossRef]
- Kusmider, M.; Faron-Górecka, A.; Solich, J.; Pabian, P.; Dziedzicka-Wasylewska, M. Time-course of changes in key catecholaminergic receptors and trophic systems in rat brain after antidepressant administration. Neurochem. Int. 2020, 141, 104885. [Google Scholar] [CrossRef] [PubMed]
- Owens, M. J. Molecular and cellular mechanisms of antidepressant drugs. Depress. Anxiety. 1996, 4, 153–159. [Google Scholar] [CrossRef]
- Shelton, R. C. Cellular mechanisms in the vulnerability to depression and response to antidepressants. Psychiatr. Clin. North Am. 2000, 23, 713–729. [Google Scholar] [CrossRef]
- Jarończyk, M.; Walory, J. Novel molecular targets of antidepressants. Molecules. 2022, 27, 533. [Google Scholar] [CrossRef]
- Sharp, T. Molecular and cellular mechanisms of antidepressant action. Behav. Neurobiol. Depress. Treat. 2013, 309–325. [Google Scholar] [CrossRef]
- Di Benedetto, B.; Radecke, J.; Schmidt, M.V.; Rupprecht, R. Acute antidepressant treatment differently modulates ERK/MAPK activation in neurons and astrocytes of the adult mouse prefrontal cortex. Neuroscience. 2013, 232, 161–168. [Google Scholar] [CrossRef]
- Thompson, S. M. Plasticity of synapses and reward circuit function in the genesis and treatment of depression. Neuropsychopharmacology. 2023, 48, 90–103. [Google Scholar] [CrossRef]
- Duman, R. S.; Heninger, G. R.; Nestler, E. J. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 1997, 54, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Neumann, M.; Hansen, K.; Hong, S. M.; Kim, S.; Noble-Haeusslein, L. J.; Liu, J. Fluoxetine increases hippocampal neurogenesis and induces epigenetic factors but does not improve functional recovery after traumatic brain injury. J. Neurotrauma. 2011, 28, 259–268. [Google Scholar] [CrossRef]
- Song, T.; Wu, H.; Li, R.; Xu, H.; Rao, X.; Gao, L. ;... & Lei, H. Repeated fluoxetine treatment induces long-lasting neurotrophic changes in the medial prefrontal cortex of adult rats. Behav. Brain Res. 2019, 365, 114–124. [Google Scholar] [CrossRef]
- Guirado, R.; Varea, E.; Castillo-Gómez, E.; Gómez-Climent, M. A.; Rovira-Esteban, L.; Blasco-Ibáñez, J. M.; Nacher, J. Effects of chronic fluoxetine treatment on the rat somatosensory cortex: activation and induction of neuronal structural plasticity. Neurosci. Lett. 2009, 457, 12–15. [Google Scholar] [CrossRef]
- Qian, X.; Zhang, Y.; Tan, H. J. Chronic fluoxetine treatment reverses depressive-like behaviors in mice via enhancing neuroplasticity. J. Pharmacol. Pharmacother. 2023, 14, 259–267. [Google Scholar] [CrossRef]
- Djordjevic, A.; Djordjevic, J.; Elaković, I.; Adzic, M.; Matić, G.; Radojcic, M. B. Fluoxetine affects hippocampal plasticity, apoptosis and depressive-like behavior of chronically isolated rats. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 36, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Varea, E.; Guirado, R.; Gilabert-Juan, J.; Martí, U.; Castillo-Gómez, E.; Blasco-Ibáñez, J. M. ;... & Nacher, J. Expression of PSA-NCAM and synaptic proteins in the amygdala of psychiatric disorder patients. J. Psychiatr. Res. 2012, 46, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Zavvari, F.; Nahavandi, A.; Goudarzi, M. Fluoxetine attenuates stress-induced depressive-like behavior through modulation of hippocampal GAP43 and neurogenesis in male rats. J. Chem. Neuroanat. 2020, 103, 101711. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Page, M.E.; Lucki, I. Differential behavioral effects of the antidepressants reboxetine, fluoxetine, and moclobemide in a modified forced swim test following chronic treatment. Psychopharmacology. 2005, 182, 335–344. [Google Scholar] [CrossRef]
- Tsugiyama, L.E.; Moraes, R.C.M.; Moraes, Y.A.C.; Francis-Oliveira, J. Promising new pharmacological targets for depression: The search for efficacy. Drug Discov. Today. 2023, 28, 103804. [Google Scholar] [CrossRef]
- Trivedi, M.H.; Hollander, E.; Nutt, D.; Blier, P. Clinical evidence and potential neurobiological underpinnings of unresolved symptoms of depression. J. Clin. Psychiatry. 2008, 69, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, M.H.; Rush, A.J.; Wisniewski, S.R.; Nierenberg, A.A.; Warden, D.; Ritz, L.; StarD Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STARD: implications for clinical practice. Am. J. Psychiatry. 2006, 163, 28–40. [Google Scholar] [CrossRef]
- Sharpley, A.L.; Cowen, P.J. Effect of pharmacologic treatments on the sleep of depressed patients. Biol. Psychiatry. 1995, 37, 85–98. [Google Scholar] [CrossRef]
- Harrigan, R.A.; Brady, W.J. ECG abnormalities in tricyclic antidepressant ingestion. Am. J. Emerg. Med. 1999, 17, 387–393. [Google Scholar] [CrossRef]
- Machado-Vieira, R.; Baumann, J.; Wheeler-Castillo, C.; Latov, D.; Henter, I.D.; Salvadore, G.; Zarate Jr, C.A. The timing of antidepressant effects: A comparison of diverse pharmacological and somatic treatments. Pharmaceuticals. 2010, 3, 19–41. [Google Scholar] [CrossRef]
- Moncrieff, J.; Cooper, R.E.; Stockmann, T.; Amendola, S.; Hengartner, M.P.; Horowitz, M.A. The serotonin theory of depression: A systematic umbrella review of the evidence. Mol. Psychiatry. 2023, 28, 3243–3256. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, R.M. History and evolution of the monoamine hypothesis of depression. J. Clin. Psychiatry 2000, 61, 4–6. [Google Scholar] [PubMed]
- Berton, O.; Nestler, E.J. New approaches to antidepressant drug discovery: beyond monoamines. Nat. Rev. Neurosci. 2006, 7, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Shulman, K.I.; Herrmann, N.; Walker, S.E. Current place of monoamine oxidase inhibitors in the treatment of depression. CNS Drugs. 2013, 27, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Hillhouse, T.M.; Porter, J.H. A brief history of the development of antidepressant drugs: From monoamines to glutamate. Exp. Clin. Psychopharmacol. 2015, 23, 1. [Google Scholar] [CrossRef]
- Vaishnavi, S.N.; Nemeroff, C.B.; Plott, S.J.; Rao, S.G.; Kranzler, J.; Owens, M.J. Milnacipran: a comparative analysis of human monoamine uptake and transporter binding affinity. Biol. Psychiatry. 2004, 55, 320–322. [Google Scholar] [CrossRef]
- Wong, D.T.; Perry, K.W.; Bymaster, F.P. The discovery of fluoxetine hydrochloride (Prozac). Nat. Rev. Drug Discov. 2005, 4, 764–774. [Google Scholar] [CrossRef]
- Stahl, S.M.; Grady, M.M.; Moret, C.; Briley, M. SNRIs: the pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectr. 2005, 10, 732–747. [Google Scholar] [CrossRef]
- Papakostas, G.I.; Thase, M.E.; Fava, M.; Nelson, J.C.; Shelton, R.C. Are antidepressant drugs that combine serotonergic and noradrenergic mechanisms of action more effective than the selective serotonin reuptake inhibitors in treating major depressive disorder? A meta-analysis of studies of newer agents. Biol. Psychiatry. 2007, 62, 1217–1227. [Google Scholar] [CrossRef]
- de Silva, V.A.; Hanwella, R. Efficacy and tolerability of venlafaxine versus specific serotonin reuptake inhibitors in treatment of major depressive disorder: A meta-analysis of published studies. Int. Clin. Psychopharmacol. 2012, 27, 8–16. [Google Scholar] [CrossRef]
- Fava, M.; Rush, A.J.; Thase, M.E.; Clayton, A.; Stahl, S.M.; Pradko, J.F.; Johnston, J.A. Fifteen years of clinical experience with bupropion HCl: From bupropion to bupropion SR to bupropion XL. Prim. Care Companion J. Clin. Psychiatry. 2005, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Laudon, M.; Frydman-Marom, A. Therapeutic effects of melatonin receptor agonists on sleep and comorbid disorders. Int. J. Mol. Sci. 2014, 15, 15924–15950. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhu, X.; Gillespie, A.; Feng, Y.; Zhou, J.; Chen, X. ;... & Chen, R. Effectiveness of mirtazapine as add-on to paroxetine v. paroxetine or mirtazapine monotherapy in patients with major depressive disorder with early non-response to paroxetine: a two-phase, multicentre, randomized, double-blind clinical trial. Psychol. Med. 2021, 51, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Bang-Andersen, B.; Ruhland, T.; Jørgensen, M.; Smith, G.; Frederiksen, K.; Jensen, K.G.; Stensbøl, T.B. Discovery of 1-[2-(2,4-dimethylphenylsulfanyl) phenyl] piperazine (Lu AA21004): A novel multimodal compound for the treatment of major depressive disorder. J. Med. Chem. 2011, 54, 3206–3221. [Google Scholar] [CrossRef] [PubMed]
- Nikayin, S.; Murphy, E.; Krystal, J.H.; Wilkinson, S.T. Long-term safety of ketamine and esketamine in treatment of depression. Expert Opin. Drug Saf. 2022, 21, 777–787. [Google Scholar] [CrossRef]
- Khabir, Y.; Hashmi, M.R.; Asghar, A.A. Rapid-acting oral drug (Auvelity) for major depressive disorder. Ann. Med. Surg. 2022, 82. [Google Scholar] [CrossRef]
- Cornett, E.M.; Rando, L.; Labbé, A.M.; Perkins, W.; Kaye, A.M.; Kaye, A.D.; Urits, I. Brexanolone to treat postpartum depression in adult women. Psychopharmacol. Bull. 2021, 51, 115. [Google Scholar]
- Blier, P.; Ward, H.E.; Tremblay, P.; Laberge, L.; Hébert, C.; Bergeron, R. Combination of antidepressant medications from treatment initiation for major depressive disorder: A double-blind randomized study. Am. J. Psychiatry. 2010, 167, 281–288. [Google Scholar] [CrossRef]
- Kato, T.; Furukawa, T.A.; Mantani, A.; Kurata, K.I.; Kubouchi, H.; Hirota, S. ; SUN☺ D Investigators. Optimising first-and second-line treatment strategies for untreated major depressive disorder—the SUN☺ D study: A pragmatic, multi-centre, assessor-blinded randomised controlled trial. BMC Med. 2018, 16, 1–16. [Google Scholar] [CrossRef]
- Henssler, J.; Alexander, D.; Schwarzer, G.; Bschor, T.; Baethge, C. Combining antidepressants vs antidepressant monotherapy for treatment of patients with acute depression: A systematic review and meta-analysis. JAMA Psychiatry. 2022, 79, 300–312. [Google Scholar] [CrossRef]
- Fornaro, M.; Martino, M.; Mattei, C.; Prestia, D.; Vinciguerra, V.; De Berardis, D.; Fornaro, P. Duloxetine-bupropion combination for treatment-resistant atypical depression: A double-blind, randomized, placebo-controlled trial. Eur. Neuropsychopharmacol. 2014, 24, 1269–1278. [Google Scholar] [CrossRef]
- Mørk, A.; Pehrson, A.; Brennum, L.T.; Nielsen, S.M.; Zhong, H.; Lassen, A.B.; Stensbøl, T.B. Pharmacological effects of Lu AA21004: A novel multimodal compound for the treatment of major depressive disorder. J. Pharmacol. Exp. Ther. 2012, 340, 666–675. [Google Scholar] [CrossRef]
- Kanes, S.J.; Colquhoun, H.; Doherty, J.; Raines, S.; Hoffmann, E.; Rubinow, D.R.; Meltzer-Brody, S. Open-label, proof-of-concept study of brexanolone in the treatment of severe postpartum depression. Hum. Psychopharmacol. 2017, 32, e2576. [Google Scholar] [CrossRef] [PubMed]
- Melón, L.; Hammond, R.; Lewis, M.; Maguire, J. A novel, synthetic, neuroactive steroid is effective at decreasing depression-like behaviors and improving maternal care in preclinical models of postpartum depression. Front. Endocrinol. 2018, 9, 703. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Briefing document: New Drug Application 211371/New Drug Application, brexanolone for the treatment of postpartum depression. 2018. (accessed on 30 May 2024).
- Patterson, R.; Balan, I.; Morrow, A.L.; Meltzer-Brody, S. Novel neurosteroid therapeutics for post-partum depression: perspectives on clinical trials, program development, active research, and future directions. Neuropsychopharmacology. 2024, 49, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.M.; Bowden, C.L.; Frazer, A. Rethinking depression and the actions of antidepressants: uncovering the links between the neural and behavioral elements. J. Affect. Disord. 2010, 120, 16–23. [Google Scholar] [CrossRef]
- Feighner, J.P.; Entsuah, A.R.; McPherson, M.K. Efficacy of once-daily venlafaxine extended release (XR) for symptoms of anxiety in depressed outpatients. J. Affect. Disord. 1998, 47, 55–62. [Google Scholar] [CrossRef]
- Cryan, J.F.; Lucki, I. Antidepressant-like behavioral effects mediated by 5-hydroxytryptamine2C receptors. J. Pharmacol. Exp. Ther. 2000, 295, 1120–1126. [Google Scholar] [PubMed]
- Estrada-Camarena, E.; Fernández-Guasti, A.; López-Rubalcava, C. Antidepressant-like effect of different estrogenic compounds in the forced swimming test. Neuropsychopharmacology. 2003, 28, 830–838. [Google Scholar] [CrossRef]
- Detke, M.J.; Johnson, J.; Lucki, I. Acute and chronic antidepressant drug treatment in the rat forced swimming test model of depression. Exp. Clin. Psychopharmacol. 1997, 5, 107. [Google Scholar] [CrossRef]
- Rodríguez-Landa, J. F.; Cueto-Escobedo, J.; Flores-Aguilar, L. Á.; Rosas-Sánchez, G. U.; Rovirosa-Hernández, M. D. J.; García-Orduña, F.; Carro-Juárez, M. The aqueous crude extracts of Montanoa frutescens and Montanoa grandiflora reduce immobility faster than fluoxetine through GABAA receptors in rats forced to swim. J. Evid.-Based Integr. Med. 2018, 23, 2515690X18762953. [Google Scholar] [CrossRef]
- Mombereau, C.; Gur, T.L.; Onksen, J.; Blendy, J.A. Differential effects of acute and repeated citalopram in mouse models of anxiety and depression. Int. J. Neuropsychopharmacol. 2010, 13, 321–334. [Google Scholar] [CrossRef]
- Peng, Z.; Zhang, C.; Yan, L.; Zhang, Y.; Yang, Z.; Wang, J.; Song, C. EPA is more effective than DHA to improve depression-like behavior, glia cell dysfunction, and hippocampal apoptosis signaling in a chronic stress-induced rat model of depression. Int. J. Mol. Sci. 2020, 21, 1769. [Google Scholar] [CrossRef]
- Kang, Z.; Ye, H.; Chen, T.; Zhang, P. Effect of electroacupuncture at siguan acupoints on expression of BDNF and TrkB proteins in the hippocampus of post-stroke depression rats. J. Mol. Neurosci. 2021, 71, 2165–2171. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, G.M.; Aaronson, S.T.; Alvarez, O.; Atli, M.; Bennett, J.C.; Croal, M.; DeBattista, C.; Dunlop, B.W.; Feifel, D.; Hellerstein, D.J.; Husain, M.I.; Kelly, J.R.; Lennard-Jones, M.R.; Licht, R.W.; Marwood, L.; Mistry, S.; Páleníček, T.; Redjep, O.; Repantis, D.; Schoevers, R.A.; Malievskaia, E. Single-dose psilocybin for a treatment-resistant episode of major depression: Impact on patient-reported depression severity, anxiety, function, and quality of life. J. Affect. Disord. 2023, 327, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; Kavalali, E.T.; Monteggia, L.M. Ketamine and rapid antidepressant action: New treatments and novel synaptic signaling mechanisms. Neuropsychopharmacology. 2024, 49, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Lopes, E.I.T.C.; Cavalcanti-Ribeiro, P.; Palhano-Fontes, F.; Gonçalves, K.T.D.C.; Nunes, E.A.; Lima, N.B.M.; Santos, N.C.; Brito, A.J.C.; de Araujo, D.B.; Galvão-Coelho, N.L. Rapid and long-lasting effects of subcutaneous esketamine on suicidality: An open-label study in patients with treatment-resistant depression. J. Psychiatr. Res. 2024, 176, 254–258. [Google Scholar] [CrossRef]
- Chrenek, C.; Duong, B.; Khullar, A.; McRee, C.; Thomas, R.; Swainson, J. Use of ketamine for treatment resistant depression: Updated review of literature and practical applications to a community ketamine program in Edmonton, Alberta, Canada. Front. Psychiatry. 2024, 14, 1283733. [Google Scholar] [CrossRef]
- Epperson, C.N.; Rubinow, D.R.; Meltzer-Brody, S.; Deligiannidis, K.M.; Riesenberg, R.; Krystal, A.D.; Bankole, K.; Huang, M.Y.; Li, H.; Brown, C.; Kanes, S.J.; Lasser, R. Effect of brexanolone on depressive symptoms, anxiety, and insomnia in women with postpartum depression: Pooled analyses from 3 double-blind, randomized, placebo-controlled clinical trials in the HUMMINGBIRD clinical program. J. Affect. Disord. 2023, 320, 353–359. [Google Scholar] [CrossRef]
- Hjorth, S.; Bengtsson, H.J.; Kullberg, A.; Carlzon, D.; Peilot, H.; Auerbach, S.B. Serotonin autoreceptor function and antidepressant drug action. J. Psychopharmacol. 2000, 14, 177–185. [Google Scholar] [CrossRef]
- Lino-de-Oliveira, C.; Sales, A.J.; Del Bel, E.A.; Silveira, M.C.; Guimarães, F.S. Effects of acute and chronic fluoxetine treatments on restraint stress-induced Fos expression. Brain Res. Bull. 2001, 55, 747–754. [Google Scholar] [CrossRef]
- Possamai-Della, T.; Dal-Pont, G.C.; Resende, W.R.; Aguiar-Geraldo, J.M.; Peper-Nascimento, J.; Quevedo, J.; Valvassori, S.S. Imipramine can be effective on depressive-like behaviors, but not on neurotrophic factor levels in an animal model for bipolar disorder induced by ouabain. Mol. Neurobiol. 2022, 59, 7170–7181. [Google Scholar] [CrossRef]
- Cryan, J.F.; O'Leary, O.F.; Jin, S.H.; Friedland, J.C.; Ouyang, M.; Hirsch, B.R.; Page, M.E.; Dalvi, A.; Thomas, S.A.; Lucki, I. Norepinephrine-deficient mice lack responses to antidepressant drugs, including selective serotonin reuptake inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 8186–8191. [Google Scholar] [CrossRef]
- Sgambato, V. The Serotonin 4 Receptor Subtype: A Target of Particular Interest, Especially for Brain Disorders. Int. J. Mol. Sci. 2024, 25, 5245. [Google Scholar] [CrossRef] [PubMed]
- Koncz, S.; Papp, N.; Pothorszki, D.; Bagdy, G. (S)-Ketamine but Not (R)-Ketamine Shows Acute Effects on Depression-Like Behavior and Sleep-Wake Architecture in Rats. Int. J. Neuropsychopharmacol. 2023, 26, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Sałat, K.; Siwek, A.; Starowicz, G.; Librowski, T.; Nowak, G.; Drabik, U.; Gajdosz, R.; Popik, P. Antidepressant-like effects of ketamine, norketamine and dehydronorketamine in forced swim test: Role of activity at NMDA receptor. Neuropharmacology. 2015, 99, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, P.J.; Kounelis-Wuillaume, S.K.; Gheidi, A.; Morrow, J.D.; Spencer-Segal, J.L.; Watson, B.O. Sex- and stress-dependent effects of a single injection of ketamine on open field and forced swim behavior. Stress. 2021, 24, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, P.J.; Yen, J.Y.; Watson, B.O. Stress-sensitive antidepressant-like effects of ketamine in the mouse forced swim test. PLoS ONE. 2019, 14, e0215554. [Google Scholar] [CrossRef]
- Assis, L.C.; Rezin, G.T.; Comim, C.M.; Valvassori, S.S.; Jeremias, I.C.; Zugno, A.I.; Quevedo, J.; Streck, E.L. Effect of acute administration of ketamine and imipramine on creatine kinase activity in the brain of rats. Rev. Bras. Psiquiatr. 2009, 31, 247–252. [Google Scholar] [CrossRef]
- Garcia, L.S.; Comim, C.M.; Valvassori, S.S.; Réus, G.Z.; Barbosa, L.M.; Andreazza, A.C.; Stertz, L.; Fries, G.R.; Gavioli, E.C.; Kapczinski, F.; Quevedo, J. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2008, 32, 140–144. [Google Scholar] [CrossRef]
- Hernández-Hernández, E.; Ledesma-Corvi, S.; Jornet-Plaza, J.; García-Fuster, M.J. Fast-acting antidepressant-like effects of ketamine in aged male rats. Pharmacol. Rep. 2024. [Google Scholar] [CrossRef]
- Zhang, G.F.; Wang, J.; Han, J.F.; Guo, J.; Xie, Z. M.; Pan, W.; Yang, J.J.; Sun, K.J. Acute single dose of ketamine relieves mechanical allodynia and consequent depression-like behaviors in a rat model. Neurosci. Lett. 2016, 631, 7–12. [Google Scholar] [CrossRef]
- Zhang, H.L.; Sun, Y.; Wu, Z.J.; Yin, Y.; Liu, R.Y.; Zhang, J.C.; Zhang, Z.J.; Yau, S.Y.; Wu, H.X.; Yuan, T.F.; Zhang, L.; Adzic, M.; Chen, G. Hippocampal PACAP signaling activation triggers a rapid antidepressant response. Mil. Med. Res. 2024, 11, 49. [Google Scholar] [CrossRef]
- Acevedo, J.; Mugarura, N.E.; Welter, A.L.; Johnson, E.M.; Siegel, J.A. The Effects of Acute and Repeated Administration of Ketamine on Memory, Behavior, and Plasma Corticosterone Levels in Female Mice. Neuroscience. 2023, 512, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.; Gardier, A.M. Fast-acting antidepressant activity of ketamine: highlights on brain serotonin, glutamate, and GABA neurotransmission in preclinical studies. Pharmacol. Ther. 2019, 199, 58–90. [Google Scholar] [CrossRef] [PubMed]
- Guilloux, J.P.; Nguyen, T.M.L.; Gardier, A.M. Ketamine: A neuropsychotropic drug with an innovative mechanism of action. Biol. Aujourd'hui. 2023, 217, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Rawat, R.; Tunc-Ozcan, E.; Dunlop, S.; Tsai, Y.H.; Li, F.; Bertossi, R.; Peng, C.Y.; Kessler, J.A. Ketamine's rapid and sustained antidepressant effects are driven by distinct mechanisms. Cell. Mol. Life Sci. 2024, 81, 105. [Google Scholar] [CrossRef]
- Hibicke, M.; Landry, A.N.; Kramer, H.M.; Talman, Z.K.; Nichols, C.D. Psychedelics, but not ketamine, produce persistent antidepressant-like effects in a rodent experimental system for the study of depression. ACS Chem. Neurosci. 2020, 11, 864–871. [Google Scholar] [CrossRef]
- Wojtas, A.; Bysiek, A.; Wawrzczak-Bargiela, A.; Maćkowiak, M.; Gołembiowska, K. Limbic System Response to Psilocybin and Ketamine Administration in Rats: A Neurochemical and Behavioral Study. Int. J. Mol. Sci. 2023, 25, 100. [Google Scholar] [CrossRef]
- Zhao, X.; Du, Y.; Yao, Y.; Dai, W.; Yin, Y.; Wang, G.; Li, Y.; Zhang, L. Psilocybin promotes neuroplasticity and induces rapid and sustained antidepressant-like effects in mice. J. Psychopharmacol (Oxford, England) 2024, 38, 489–499. [Google Scholar] [CrossRef]
- Moliner, R.; Girych, M.; Brunello, C.A.; Kovaleva, V.; Biojone, C.; Enkavi, G.; Antenucci, L.; Kot, E.F.; Goncharuk, S.A.; Kaurinkoski, K.; Kuutti, M.; Fred, S.M.; Elsilä, L.V.; Sakson, S.; Cannarozzo, C.; Diniz, C.R.A.F.; Seiffert, N.; Rubiolo, A.; Haapaniemi, H.; Meshi, E.; Castrén, E. Psychedelics promote plasticity by directly binding to BDNF receptor TrkB. Nat. Neurosci. 2023, 26, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Jefsen, O.; Højgaard, K.; Christiansen, S.L.; Elfving, B.; Nutt, D.J.; Wegener, G.; Müller, H.K. Psilocybin lacks antidepressant-like effect in the Flinders Sensitive Line rat. Acta Neuropsychiatr. 2019, 31, 213–219. [Google Scholar] [CrossRef]
- Kolasa, M.; Nikiforuk, A.; Korlatowicz, A.; Solich, J.; Potasiewicz, A.; Dziedzicka-Wasylewska, M.; Bugno, R.; Hogendorf, A.; Bojarski, A.; Faron-Górecka, A. Unraveling psilocybin's therapeutic potential: Behavioral and neuroplasticity insights in Wistar-Kyoto and Wistar male rat models of treatment-resistant depression. Psychopharmacology. 2024. [Google Scholar] [CrossRef]
- Reddy, D.S. Neurosteroid replacement therapy for catamenial epilepsy, postpartum depression and neuroendocrine disorders in women. J. Neuroendocrinol. 2022, 34, e13028. [Google Scholar] [CrossRef]
- Contreras, C.M.; Azamar-Arizmendi, G.; Saavedra, M.; Hernández-Lozano, M. A five-day gradual reduction regimen of chlormadinone reduces premenstrual anxiety and depression: A pilot study. Arch. Med. Res. 2006, 37, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S.; Kaur, G.; Kulkarni, S.K. Sigma (sigma1) receptor mediated anti-depressant-like effects of neurosteroids in the Porsolt forced swim test. Neuroreport. 1998, 9, 3069–3073. [Google Scholar] [CrossRef]
- Khisti, R.T.; Chopde, C.T.; Jain, S.P. Antidepressant-like effect of the neurosteroid 3alpha-hydroxy-5alpha-pregnan-20-one in mice forced swim test. Pharmacol. Biochem. Behav. 2000, 67, 137–143. [Google Scholar] [CrossRef]
- Rodríguez-Landa, J.F.; Contreras, C.M.; García-Ríos, R.I. Allopregnanolone microinjected into the lateral septum or dorsal hippocampus reduces immobility in the forced swim test: participation of the GABAA receptor. Behav. Pharmacol. 2009, 20, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Landa, J.F.; Contreras, C.M.; Bernal-Morales, B.; Gutiérrez-García, A.G.; Saavedra, M. Allopregnanolone reduces immobility in the forced swimming test and increases the firing rate of lateral septal neurons through actions on the GABAA receptor in the rat. J. Psychopharmacol. (Oxf.) 2007, 21, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S.; Mbilinyi, R.H.; Estes, E. Preclinical and clinical pharmacology of brexanolone (allopregnanolone) for postpartum depression: a landmark journey from concept to clinic in neurosteroid replacement therapy. Psychopharmacology. 2023, 240, 1841–1863. [Google Scholar] [CrossRef]
- Shirayama, Y.; Fujita, Y.; Oda, Y.; Iwata, M.; Muneoka, K.; Hashimoto, K. Allopregnanolone induces antidepressant-like effects through BDNF-TrkB signaling independent from AMPA receptor activation in a rat learned helplessness model of depression. Behav. Brain Res. 2020, 390, 112670. [Google Scholar] [CrossRef] [PubMed]
- Cueto-Escobedo, J.; Andrade-Soto, J.; Lima-Maximino, M.; Maximino, C.; Hernández-López, F.; Rodríguez-Landa, J.F. Involvement of GABAergic system in the antidepressant-like effects of chrysin (5,7-dihydroxyflavone) in ovariectomized rats in the forced swim test: Comparison with neurosteroids. Behav. Brain Res. 2020, 386, 112590. [Google Scholar] [CrossRef]
- Machado-Vieira, R.; Salvadore, G.; Luckenbaugh, D.A.; Manji, H.K.; Zarate, C.A., Jr. Rapid onset of antidepressant action: A new paradigm in the research and treatment of major depressive disorder. J. Clin. Psychiatry. 2008, 69, 946–958. [Google Scholar] [CrossRef]
- Brymer, K.J.; Johnston, J.; Botterill, J.J.; Romay-Tallon, R.; Mitchell, M.A.; Allen, J.; Pinna, G.; Caruncho, H.J.; Kalynchuk, L.E. Fast-acting antidepressant-like effects of Reelin evaluated in the repeated-corticosterone chronic stress paradigm. Neuropsychopharmacology. 2020, 45, 1707–1716. [Google Scholar] [CrossRef]
- Cai, M.; Zhu, Y.; Shanley, M.R.; Morel, C.; Ku, S.M.; Zhang, H.; Shen, Y.; Friedman, A.K.; Han, M.H. HCN channel inhibitor induces ketamine-like rapid and sustained antidepressant effects in chronic social defeat stress model. Neurobiol. Stress. 2023, 26, 100565. [Google Scholar] [CrossRef]
- Malhi, G.S.; Mann, J.J. Depression. Lancet. 2018, 392, 2299–2312. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, H.; Nie, K.; Gao, Y.; Su, H.; Wang, Z.; Dong, H. Traditional herbal formula Jiao-tai-wan improves chronic restrain stress-induced depression-like behaviors in mice. Biomed. Pharmacother. 2022, 153, 113284. [Google Scholar] [CrossRef]
- Serebruany, V.L.; Suckow, R.F.; Cooper, T.B.; O’Connor, C.M.; Malinin, A.I.; Krishnan, K.R.R. Relationship between release of platelet/endothelial biomarkers and plasma levels of sertraline and N-desmethylsertraline in acute coronary syndrome patients receiving SSRI treatment for depression. Am. J. Psychiatry. 2005, 162, 1165–1170. [Google Scholar] [CrossRef]
- DeLucia, V.; Kelsberg, G.; Safranek, S. Which SSRIs most effectively treat depression in adolescents? J. Fam. Pract. 2016, 65, 632–634. [Google Scholar]
- Vermetten, E.; Vythilingam, M.; Schmahl, C.; De Kloet, C.; Southwick, S.M.; Charney, D.S.; Bremner, J.D. Alterations in stress reactivity after long-term treatment with paroxetine in women with posttraumatic stress disorder. Ann. N.Y. Acad. Sci. 2006, 1071, 184–202. [Google Scholar] [CrossRef]
- Chu, A.; Wadhwa, R. Selective serotonin reuptake inhibitors. In StatPearls [Internet]. StatPearls Publishing 2022.
- Edinoff, A.N.; Akuly, H.A.; Hanna, T.A.; Ochoa, C.O.; Patti, S.J.; Ghaffar, Y.A.; Kaye, A.M. Selective serotonin reuptake inhibitors and adverse effects: A narrative review. Neurol. Int. 2021, 13, 387–401. [Google Scholar] [CrossRef]
- Hu, B.; Doods, H.; Treede, R.D.; Ceci, A. Duloxetine and 8-OH-DPAT, but not fluoxetine, reduce depression-like behaviour in an animal model of chronic neuropathic pain. Neurosci. Lett. 2016, 619, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhu, X.; Yu, P.; Sheng, T.; Wang, Y.; Ye, Y. Crocin ameliorates depressive-like behaviors induced by chronic restraint stress via the NAMPT-NAD+-SIRT1 pathway in mice. Neurochem. Int. 2022, 157, 105343. [Google Scholar] [CrossRef]
- Abdallah, M.S.; Mosalam, E.M.; Zidan, A.A.A.; Elattar, K.S.; Zaki, S.A.; Ramadan, A.N.; Ebeid, A.M. RETRACTED ARTICLE: The Antidiabetic Metformin as an Adjunct to Antidepressants in Patients with Major Depressive Disorder: A Proof-of-Concept, Randomized, Double-Blind, Placebo-Controlled Trial. Neurotherapeutics. 2020, 17, 1897–1906. [Google Scholar] [CrossRef]
- Hayley, S.; Litteljohn, D. Neuroplasticity and the next wave of antidepressant strategies. Front. Cell. Neurosci. 2013, 7, 218. [Google Scholar] [CrossRef] [PubMed]
- Levy, M. J.; Boulle, F.; Steinbusch, H. W.; van den Hove, D. L.; Kenis, G.; Lanfumey, L. Neurotrophic factors and neuroplasticity pathways in the pathophysiology and treatment of depression. Psychopharmacology. 2018, 235, 2195–2220. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Li, N.; Li, T. VEGF regulates antidepressant effects of lamotrigine. Eur. Neuropsychopharmacol. 2012, 22, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E.; Dantzer, R.; Kelley, K.W.; McCusker, R.H. Central administration of insulin-like growth factor-I decreases depressive-like behavior and brain cytokine expression in mice. J. Neuroinflamm. 2011, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.C.; Wu, J.; Fujita, Y.; Yao, W.; Ren, Q.; Yang, C. ;... & Hashimoto, K. Antidepressant effects of TrkB ligands on depression-like behavior and dendritic changes in mice after inflammation. Int. J. Neuropsychopharmacol 2015, 18, pyu077. [Google Scholar] [CrossRef]
- Huang, Y.J.; Lane, H.Y.; Lin, C.H. New treatment strategies of depression: based on mechanisms related to neuroplasticity. Neural Plast. 2017, 2017, 4605971. [Google Scholar] [CrossRef] [PubMed]
- Ledesma-Corvi, S.; Jornet-Plaza, J.; García-Fuster, M.J. Aromatase inhibition and ketamine in rats: Sex-differences in antidepressant-like efficacy. Biol. Sex Differ. 2023, 14, 73. [Google Scholar] [CrossRef]
- Li, S.; Zhu, Z.; Lan, T.; Wu, Y.; Li, Y.; Wang, C.; Jian, W.; Yu, S.Y. Levomilnacipran ameliorates lipopolysaccharide-induced depression-like behaviors and suppressed the TLR4/Ras signaling pathway. Int. Immunopharmacol. 2023, 122, 110595. [Google Scholar] [CrossRef]
- Filipović, D.; Novak, B.; Xiao, J.; Yan, Y.; Yeoh, K.; Turck, C.W. Chronic fluoxetine treatment of socially isolated rats modulates prefrontal cortex proteome. Neuroscience. 2022, 501, 52–71. [Google Scholar] [CrossRef]
- Seo, M.K.; Lee, J.G.; Park, S.W. Effects of escitalopram and ibuprofen on a depression-like phenotype induced by chronic stress in rats. Neurosci. Lett. 2019, 696, 168–173. [Google Scholar] [CrossRef]
- Huang, D.; Zhang, L.; Yang, J.Q.; Luo, Y.; Cui, T.; Du, T.T.; Jiang, X.H. Evaluation on monoamine neurotransmitters changes in depression rats given with sertraline, meloxicam or/and caffeic acid. Genes Dis. 2018, 6, 167–175. [Google Scholar] [CrossRef]
- Alkon, D.L.; Hongpaisan, J.; Sun, M.K. Effects of chronic bryostatin-1 on treatment-resistant depression in rats. Eur. J. Pharmacol. 2017, 807, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Elias, E.; Zhang, A.Y.; Manners, M.T. Novel pharmacological approaches to the treatment of depression. Life. 2022, 12, 196. [Google Scholar] [CrossRef]
- Pedersen, M. E.; Szewczyk, B.; Stachowicz, K.; Wieronska, J.; Andersen, J.; Stafford, G. I.; van Staden, J.; Pilc, A.; Jäger, A. K. Effects of South African traditional medicine in animal models for depression. J. Ethnopharmacol. 2008, 119, 542–548. [Google Scholar] [CrossRef]
- Foudah, A.I.; Alqarni, M.H.; Alam, A.; Devi, S.; Salkini, M.A.; Alam, P. Rutin improves anxiety and reserpine-induced depression in rats. Molecules. 2022, 27, 7313. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qin, X.M.; Tian, J.S.; Gao, X.X.; Du, G.H.; Zhou, Y.Z. Integrated network pharmacology and metabolomics to dissect the combination mechanisms of Bupleurum chinense DC-Paeonia lactiflora Pall herb pair for treating depression. J. Ethnopharmacol. 2021, 264, 113281. [Google Scholar] [CrossRef]
- Luo, Y.; Zhong, Z.; Li, H.; Wang, L.; Guo, D.; Dong, X.; Liu, J.; Xie, M.; Wu, M.; Xiang, Y.; Zhang, X.; Meng, P. Integrating serum metabolomics and network analysis to explore the antidepressant activity of crocin in rats with chronic unexpected mild stress-induced depression. Pharm. Biol. 2023, 61, 1414–1430. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, M.; Lu, X.; Wei, R.; Xu, J. Ziziphi spinosae lily powder suspension in the treatment of depression-like behaviors in rats. BMC Complement. Altern. Med. 2017, 17, 238. [Google Scholar] [CrossRef]
- Adeoluwa, O.A.; Olayinka, J.N.; Adeoluwa, G.O.; Akinluyi, E.T.; Adeniyi, F.R.; Fafure, A.; Nebo, K.; Edem, E.E.; Eduviere, A.T.; Abubakar, B. Quercetin abrogates lipopolysaccharide-induced depressive-like symptoms by inhibiting neuroinflammation via microglial NLRP3/NFκB/iNOS signaling pathway. Behav. Brain Res. 2023, 450, 114503. [Google Scholar] [CrossRef]
- Abildgaard, A.; Elfving, B.; Hokland, M.; Wegener, G.; Lund, S. Probiotic treatment reduces depressive-like behaviour in rats independently of diet. Psychoneuroendocrinology. 2017, 79, 40–48. [Google Scholar] [CrossRef]
- Li, Q.; Li, L.; Niu, X.; Tang, C.; Wang, H.; Gao, J.; Hu, J. Probiotics alleviate depressive behavior in chronic unpredictable mild stress rat models by remodeling intestinal flora. NeuroReport. 2021, 32, 686–693. [Google Scholar] [CrossRef]
- Daugé, V.; Philippe, C.; Mariadassou, M.; Rué, O.; Martin, J.C.; Rossignol, M.N. ;... & Naudon, L. A probiotic mixture induces anxiolytic-and antidepressive-like effects in fischer and maternally deprived long evans rats. Front. Behav. Neurosci. 2020, 14, 581296. [Google Scholar] [CrossRef]
Figure 1.
Proposed mechanisms that mediate the antidepressant effect experimentally.
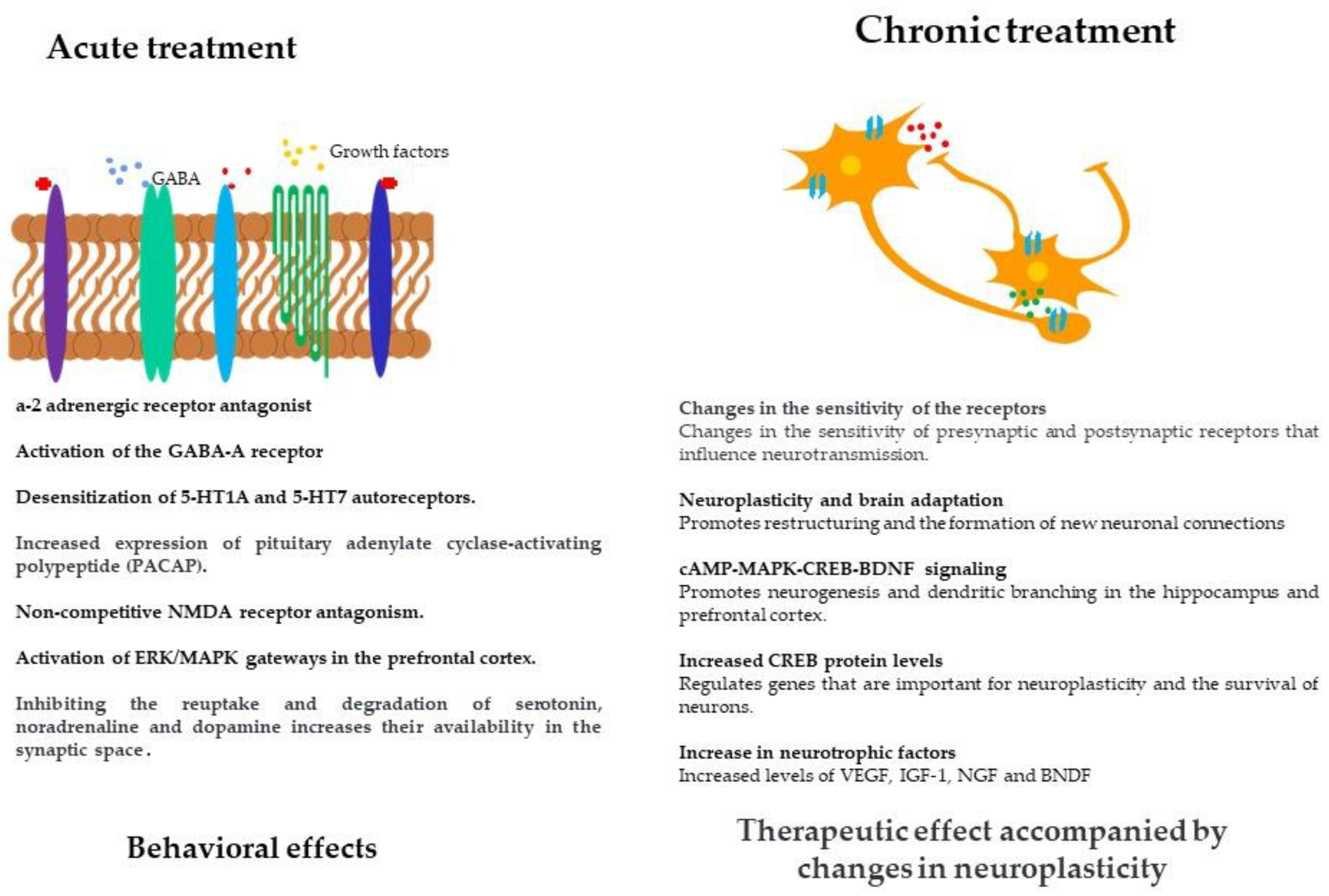

Table 1.
Classes of antidepressants that are used to treat depression.
| Class | Pharmacological target | Mechanism of action | Reference |
| Monoamine Oxidase Inhibitors (IMAO) | |||
| Selegiline | Monoamine Oxidase Enzyme Type A and B. | Prevents the degradation of 5-hydroxytryptamine (5-HT); increases the availability of 5-HT in the synapses. | [59,60]. |
| Fenelzina | |||
| Tricyclic antidepressants (TCA) | |||
| Amitriptyline | 5-HT reuptake transporter and norepinephrine (NE) reuptake transporter. | Inhibits NE and 5-HT reuptake transporters; increases the availability of 5-HT and NE at the synapses; binds to postsynaptic noradrenaline, histamine and acetylcholine receptors. | [58,59]. |
| Imipramine | |||
| Nortriptyline | |||
| Selective serotonin reuptake inhibitors (SSRIs) | |||
| Fluoxetine | 5-HT reuptake transporters | In particular, they inhibit the reuptake of 5-HT, which increases the availability of serotonin in the synapse. | [58,60,64]. |
| Paroxetine | |||
| Escitalopram | |||
| Sertraline | |||
| Specific noradrenergic and serotonergic antidepressants | |||
| Mirtazapine | α2 NE receptors and 5-HT receptors | α-2 NE receptor antagonists that cause an increased release of 5-HT and NE; they act as antagonists/agonists of several specific 5-HT receptors. | [68,73,74,75]. |
| Mianserin | |||
| Norepinephrine and serotonin reuptake inhibitors | |||
| Venlafaxine | 5-HT receptors and NE uptake transporters | Inhibits the reuptake of 5-HT and NE and increases their availability in the synapses. | [75,76]. |
| Duloxetine | |||
| Atypical antidepressants | |||
| Agomelatine | 5-HT receptors, NE receptors and melatonin receptors | Bupropion acts as a DA and NE reuptake inhibitor; vortioxetine acts as an agonist/antagonist of several 5-HT and NE receptors; agomelatine activates melatonin receptors and antagonizes some 5-HT receptors. | [76,77]. |
| Bupropion | |||
| Vortioxetine | |||
| New antidepressants | |||
| Ketamine | Antagonist of the ionotropic glutamate receptor, NMDA 3A. Potentiator of the 5-hydroxytryptamine receptor 3A. Antagonist of the neuronal acetylcholine receptor subunit alpha-7. Inhibitor of nitric oxide synthase brain. Agonist and partial agonist of the dopamine D2 receptor. Agonist of the kappa-type opioid receptor. Antagonist of 5-hydroxytryptamine receptor 2 and 5-hydroxytryptamine receptor 1. |
Ketamine interacts with N-methyl-D-aspartate (NMDA) receptors, opioid receptors, monoaminergic receptors, muscarinic receptors and voltage sensitive Ca ion channels. Unlike other general anaesthetic agents, ketamine does not interact with GABA receptors | [70,71]. |
| Brexenolone | Positive allosteric modulator GABA(A) Receptor | Brexanolone is a neuroactive steroid that occurs naturally in the body (as natural allopregnanolone) when the female sex hormone progesterone is metabolized. This steroid compound is also thought to act as a barbitu-like positive allosteric modulator of synaptic and extrasynaptic GABAA receptors. In this way, brexanolone may enhance the activity of GABA at these receptors by opening the calcium channels of GABAA receptors more frequently and for longer periods of time. It is also thought that brexanolone triggers this effect on GABAA receptors at a binding site that differs from those of benzodiazepines. |
[78,79,80,81]. |
Table 2.
Effect of chronic treatment with antidepressants.
| Drug | Dosage (route of administration, dose and duration of treatment) | Experimental subject | Experimental model | Identified effect | Mechanism | Reference |
|---|---|---|---|---|---|---|
| Ketamine Letrozole |
Ketamine 5 mg/kg. Letrozole 1 mg/kg. 7 days of treatment Intraperitoneal route of administration |
Adult female and male rats of the Sprague-Dawley strain. | Forced Swim Test (FST) | Reduction of FST immobility in men with ketamine and reduction of FST immobility in women with letrozole. The combination of ketamine and letrozole reduces FST immobility in men. | It is assumed that they act via NMDA receptors and could modulate neurotrophic factors in the prefrontal cortex. | [148] |
| Levomilnacipran | 30 mg/kg. 14 days of treatment Intraperitoneal administration route |
Male Wistar rats. | Depression induced with lipopolysaccharides (LPS) 0.5 mg/kg for 2 weeks. Sucrose preference. FST. Open Field Test. |
Levomilnacipran reduces immobility behavior in the FST. It reverses the increase in transcript levels of the proinflammatory cytokines IL-1β, INF-γ and TNF-α. | Levomilnacipran inhibits the activation of the TLR4/NF-κB and Ras/p38 signaling pathways and modulates the ERK/CREB/BDNF pathway. | [149] |
| Fluoxetine | 15 mg/kg/day. 6 weeks Intraperitoneal administration route |
Male Wistar Rats | Chronic social isolation (CSIS) (6 weeks) Sucrose preference FST |
Increased sucrose consumption. Reduction of immobility time in the FST | Expression of calcium/calmodulin-dependent protein kinase 1 (CaMKK1). Phosphorylation of the cAMP-responsive element-binding protein (CREB). Expression of BDNF. |
[150] |
| Escitalopram Ibuprofen |
Escitalopram 10 mg/kg Ibuprofen 40 mg/kg Combination of both 21 days of treatment Intraperitoneal administration route |
Adult male Sprague-Dawley rats | Stress from restriction FST |
Reduction of immobility in FST with individual and combined treatment. Reduction in corticosterone levels. Increase in BDNF and p11 levels. |
Positive regulation of BDNF and p11 | [151] |
| Meloxicam Caffeic acid Sertraline |
Meloxicam 3 mg/kg – 1mg/kg Caffeic acid 30 mg/kg – 10 mg/kg Sertraline 5mg/kg Meloxicam 1mg/kg + Caffeic acid 10mg/kg 21 days of treatment Intraperitoneal administration route |
Adult male Sprague-Dawley rats | CUMS 6 weeks Open Field Test FST |
All treatments reduced immobility in FST. Caffeic acid inhibits NA reduction and increases Trp and MHGP. Meloxicam inhibits NA reduction and increases Trp, MHGP and Tyr. |
Inhibition of COX-2 and reduction of 5-HIAA. | [152] |
| Bryostatin-1 Imipramine |
Bryostatin-1 (20 µg/m2) Intravenous administration by tail Imipramine (15 mg/kg) intraperitoneal administration 5.5 weeks of treatment |
Male Wistar Rats | Open space swimming test Morris water maze Visible platform test. |
Bryostatin-1 reduces immobility after 2 weeks of treatment. Bryostatin-1 restored the rats' ability in spatial learning and spatial memory recall. |
Protein kinase C (PKC)ε activation | [153] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



