
Submitted:
30 October 2024
Posted:
31 October 2024
You are already at the latest version
Abstract
This article reviews the transformative impact of single-cell whole genome sequencing (scWGS) on cancer biology research. ScWGS has revolutionized our understanding of tumor heterogeneity, clonal evolution, and the tumor microenvironment by enabling high-resolution analysis of individual cells' genomic, transcriptomic, and epigenomic profiles. The technology's unique capabilities, including detection of rare genomic events, simultaneous capture of multiple genomic features, and association with phenotypic data, have opened new avenues for cancer research and precision medicine. Integration of scWGS with other single-cell omics technologies has provided a multidimensional view of cellular states and regulatory mechanisms in cancer. Advanced data analysis tools, including machine learning and AI algorithms, have been crucial in interpreting the vast amounts of data generated, leading to the identification of new biomarkers and development of predictive models for patient stratification. The article also discusses emerging technologies like spatial transcriptomics and in situ sequencing, which promise to further enhance our understanding of tumor spatial organization and cellular interactions. As scWGS and related technologies continue to evolve, they are expected to drive significant advances in personalized cancer diagnostics, prognosis, and therapy, ultimately improving patient outcomes in the era of precision oncology.
Keywords:
single-cell whole genome sequencing
; tumor microenvironment
; tumor heterogeneity
; cancer research
; precision medicine
; artificial intelligence
1. Introduction
Human cancers are complex ecosystems composed of cells with distinct phenotypes, genotypes, and epigenetic states. Current cancer study models do not adequately reflect tumor composition in patients (Navin et al. 2011). Single-cell sequencing has revolutionized our understanding of cellular heterogeneity within cancer tissues, unlike traditional bulk RNA-seq analysis that provides an average transcriptional profile of a tissue sample, masking cellular diversity (Suvà and Tirosh 2019). Single cell RNA sequencing (scRNA-seq) enables the dissection of complex tumor ecosystems at single-cell resolution, revealing rare cell types, transition states, and intercellular interactions vital for cancer progression and therapeutic response (Patel et al. 2014; Tirosh et al. 2016; Puram et al. 2017).
Single-cell whole genome sequencing (scWGS) has revolutionized cancer research by providing a powerful platform to investigate clonal evolution, intratumoral heterogeneity, and treatment resistance (Navin et al. 2011). This technology enables the analysis of genomic and transcriptomic profiles at the individual cell level for the identification of specific cellular subpopulations within tumors associated with disease progression and treatment response (Han et al. 2022).
The advent of scWGS has transformed our understanding of cancer biology by providing an unprecedented view of the complex genetic landscapes of tumors at a single-cell level (Fu et al. 2020). This technique involves isolating individual cells, amplifying their DNA, and then sequencing the genomes to understand genetic variations at a single-cell level (Han et al. 2022). It has also revealed the remarkable heterogeneity present both within and between cancer cells (Loo et al. 2019). Meticulously analyzed genomic profiles of single cells, uncovered the trajectories driving tumor progression, the mechanisms underlying cancer cell metastasis, and the development of resistance to medical treatments (Tritschler et al. 2017; Rognoni and Watt 2018; Loo et al. 2019).
This review examines how single cell technologies have changed cancer biology research. It explores the technology's key contributions to understanding critical aspects of cancer, including tumor heterogeneity, evolution, and the tumor microenvironment through its ability to analyze genomic, transcriptomic, and epigenomic profiles at the individual cell level. The review covers the essential capabilities of single cell technology, its integration with other single-cell omics approaches, and the crucial role of advanced computational methods in data analysis. Additionally, it discusses the emergence of complementary technologies like spatial transcriptomics and in situ sequencing, while also addressing the potential future impact of these advancements on personalized cancer medicine. Throughout, this review emphasizes how these technological developments are driving progress in cancer diagnostics, prognostics, and therapeutic strategies, ultimately aiming to improve outcomes for cancer patients through more precise and personalized approaches.
2. Cancer Research
Within the conceptual framework of scWGS, three central capabilities stand out: fidelity, co-presence, and phenotypic association. These capabilities are essential for understanding the unique advantages of scWGS. The fidelity capability refers to its ability to detect DNA features such as mutations or modifications existing at low levels of mosaicism within a sample. Unlike bulk DNA sequencing (Evrony, Hinch, and Luo 2021)., the co-presence capability in single-cell DNA sequencing refers to its ability to capture multiple genomic features within a single cell (Evrony, Hinch, and Luo 2021). This capability allows the analysis of different aspects of the genome simultaneously, including copy number variations (CNVs), single nucleotide variants (SNVs), structural variations such as insertions and deletions, chromosomal rearrangements, and DNA methylation patterns.
Providing a more comprehensive understanding of the genetic landscape of individual cells. Lastly, the phenotypic association capability allows linking genomic information with cellular phenotypes (Macaulay et al. 2015; Evrony, Hinch, and Luo 2021). Connecting genetic data with specific cellular characteristics, researchers can gain insights into the functional implications of genomic variations at a single-cell level (Macaulay et al. 2015; Demaree et al. 2020; Evrony, Hinch, and Luo 2021). The co-presence and phenotypic association capabilities are crucial in heterogeneous samples, spanning from profiling cells within an organism to distinguishing between tumor and normal cells or analyzing cellular mixtures in complex environments like soil samples or the gut microbiome (Evrony, Hinch, and Luo 2021).
One of the fundamental applications of scWGS in cancer research is the characterization of circulating tumor cells (CTCs). These are defined as tumor cells that have been sloughed from the primary tumor into the circulatory or lymphatic systems (Ni et al. 2013; Dago et al. 2014; Lohr et al. 2014; D. Lin et al. 2021). Understanding their part in metastasis may contribute to better therapeutic management (Ju et al. 2022). In a study conducted by Polzer et al., scWGS technique was used to analyze the genomic profiles of CTCs in breast cancer patients, allowing the identification of fundamental principles guiding the evolution of individual tumors. These principles include the ability of tumor cells to generate genetic diversity through mutations and chromosomal rearrangements, as well as clonal selection of subpopulations with evolutionary advantages, such as resistance to therapy or increased metastatic capacity. These findings offer a new perspective on CTC dynamics, highlighting the coexistence of genetically distinct subpopulations, each with different metastatic potentials and therapeutic vulnerabilities (Polzer et al. 2014).
In addition to its application in studying CTCs, scWGS has enabled researchers to unravel the complex clonal architecture of solid tumors. This has revealed the hierarchical organization of genetically distinct subclones within a single tumor mass (Saadatpour et al. 2015). This approach has unveiled rare subpopulations of cells such as cancer stem cells or therapy-resistant clones, which can drive disease progression and treatment failure (Kharchenko, Silberstein, and Scadden 2014; Shekhar et al. 2014; Saadatpour et al. 2015). By reconstructing the evolutionary trajectories of these subclones, scWGS has provided valuable insights into the mechanisms underlying tumor heterogeneity and the emergence of resistant phenotypes (Y. Wang et al. 2014).
By comparing single cell data across multiple patients, researchers can identify common mutational patterns and chromosomal mutations that may contribute to the development and progression of specific cancer types (Wills and Mead 2015; S. Chen et al. 2023). This knowledge can inform the development of targeted therapies and guide personalized treatment strategies tailored to each patient's unique genetic profiles (Thiele et al. 2019; Reza et al. 2021; S. Chen et al. 2023).
Furthermore, single cell sequencing has allowed exploration of the intricate interaction between cancer cells and their microenvironment. By analyzing the genomic profiles of individual cells within the tumor environment, researchers can elucidate the complex dynamics between cancer cells, immune cells, and stromal components components (Figure 1) (Saadatpour et al. 2015). These recent developments have improved the understanding of complex interactions between tumors and their immune microenvironment in various human cancers, which would allow the development of effective immune therapies (Chew, Toh, and Abastado 2012; Bai and Cui 2022).
As the field of single cell technologies advances, the integration of multi omic data at a single-cell level, including transcriptomics, epigenomics, and proteomics, holds great promise for unraveling the complex regulatory networks driving cancer cell behavior (Evrony, Hinch, and Luo 2021). By combining these complementary datasets, researchers can gain a comprehensive understanding of the molecular mechanisms underlying tumor heterogeneity, paving the way for more effective and personalized cancer therapies.
3. Applications of Single-Cell Sequencing For Genomic Profiling in Human Cancer Cells
Single cell sequencing (SCS), especially single Cell DNA sequencing (scDNA-seq) technologies have impacted cancer analysis through the genomic profiling of single cells in the tumor and cancer. However, unlike the traditional genomics, single-cell genomics targets intratumoral heterogeneity and microenvironmental elements involved in response and resistance to treatment, providing a broader picture of cancer-related processes (Kim, Eum, and Lee 2021). Here we describe in detail some of the applications of scDNA-seq related to cancer:
- (a)
- Tumor heterogeneity and clonal evolution
Single-cell DNA sequencing has proven to be useful for interrogation of intratumoral heterogeneity and clonal evolution in a plethora of malignancies. Studies pointed out that tumor heterogeneity is due to clonal evolution, creating a fractal pattern of the primary and secondary main clones, sub-clones and single cells, raising issues on the best sampling strategy for tumor sequencing for clinical use (Munkácsy et al. , 2020). A great example is the use of single-cell DNA amplicon sequencing to track clonal heterogeneities of B-Cell Acute Lymphoblastic Leukemia (B-ALL) at diagnosis and during the course of chemotherapy treatment to investigate clonal evolution profile in response to therapy (S. Meyers et al. 2022).
The ability of scDNA-seq method has recently been demonstrated to track clonal evolution and identify dynamics of oncogenic cells in cancer progression and treatment (L. Xu et al. 2019). It is important to emphasize the applicability of single-cell DNA sequencing, where it is possible to directly obtain clonal genotypes at the cellular level and detect branches in clonal evolution, demonstrating that this approach is superior to other massive sequencing methods, such as bulk sequencing, for study clonal evolution (Borgsmüller et al. 2020). Rajan et al. (2023) analyzed, on 10 tumor samples and 12 019 tumor cells, intratumoral genomic heterogeneity and the evolution of somatic copy number alteration (SCNA) in structurally complex osteosarcoma genomes. In this study they describe genomic homogeneity on these, with a surprising remarkably conserved profiles of SCNA with a limited variability among the subclones; also they find a whole genome duplication (WGD) in many of tumors, these appear to be a mechanism to mitigate the effects of genetic deletions. The most genomic alterations in tumors were acquired early in the oncogenic process and remained stable over time, both in tumor progression and in response to therapy; this suggests an early catastrophic event, rather than sustained genomic instability as the primary cause of the tumor structural complexity. And these tumors exhibited a remarkable stability in their SCNA profiles from diagnosis to relapse, indicating that there is no significant evolution in response to treatments (Rajan et al. 2023).
- (b) Identification of Rare Mutations
ScDNA-seq has become a powerful tool in the identification of rare mutations in cancer, offering a high-resolution approach to uncover genomic complexity and compound mutations within individual cells (A. Y. Huang and Lee 2021). Examining cells individually provides the ability to reveal diverse evolutionary trajectories between primary and metastatic tumor cells, yielding insights into genetic heterogeneity within cancer populations (J. Tang et al. 2021). scDNA-seq has been instrumental in revealing the depth of genomic complexity and compound mutations within tumors, providing insights into the unique mutational profiles of individual cells (A. Y. Huang and Lee 2021). This technique has proven to be valuable in the study of somatic mutations allowing the detection of rare mutations that may go undetected with traditional massive sequencing methods (Jaberi et al. 2020). Gráf et al. (2021) illustrates how single-cell analysis can be used in conjunction with next-generation sequencing to identify mosaicism of BRCA2 mutations and decode the cellular processes underlying murine tumorigenesis (Gráf et al. 2021). Furthermore, this strategy improves the sensitivity of rare mutation detection of limited tumor cells by integrating laser capture microdissection (LCM) and NGS technologies, which shed light on BRCA1/2 mutations and the process of tumorigenesis.
- (c) Drugs Resistance and Mechanisms
ScDNA-seq is a powerful tool that helps understand the resistance that occurs against cancer drugs. The analysis of individual cells offers information about the heterogeneity within tumors and reveals which subpopulations can generate resistance to treatment (L. Liu et al. 2023). It has been shown that cancer stem cells, a small fraction of the tumor cell population, can cause progression, metastasis, and drug resistance (Pang et al. 2019). Therefore, it is extremely important to identify and target these specific cell populations to improve treatment results.
Another exemplary case is the study by Lee et al. (2022). In this study they employed a scDNA-seq approach to study clonal heterogeneity and clonal evolution in two patients with Myelodysplastic syndrome (MDS) refractory to hypomethylating agents (HMAs). Importantly, HMAs are the mainstay of treatment for MDS, however, in most patients, resistance to treatment and transformation of the disease into acute myeloid leukemia (AML) was observed. Two patients with HMA resistance and progression to AML were studied. By bulk sequencing, different single nucleotide variations (SNV) or insertions and deletions (INDEL) are detected in these patients, but using the scDNA-seq approach, rare cell clones with mutations that cannot be detected by bulk sequencing was detected, identifying pathogenic copy number variation (CNV) of GATA2, DNMT3A and TET2 which are associated with resistance against HMA but these CNVs could be coupled with small SNVs or INDELs of FLT3 and IDH2, all of this mutations works together, having an impact on disease progression and drug resistance (P. Lee et al. 2022).
Likewise, scDNA-seq also helps us discover and elucidate the mechanisms involved in these resistances, as in the case of quizartinib where on- and off-target mechanisms of resistance which can preexist therapy were identified. In this study they analyzed over 103 000 cells from 16 timepoints across 8 patients, identifying pathogens variants undetected by bulks. In this work they identify FLT3 tyrosine kinase domain (TKD) as a primary mechanism of resistance to quizartinib. The on-target resistance mechanism are: Kinase Domain (KD) Mutation, seven of eight patients developed at least one additional mutation in the FLT3 Kinase domain, most commonly at the D3895 locus, these KD mutations can occur on the native a (FLT3-ITD (internal tandem duplication) negative), in cis with the FLT3-ITD (Peretz et al. 2021).
- (d) Detection and Diagnosis the presence of Cancer
scDNA-seq has become a powerful tool to aid and complement cancer diagnosis by providing information on the genetic heterogeneity and molecular characteristics of tumors at the single cell level. Circulating tumor DNA (ctDNA) has been recognized as a valuable biomarker for molecular diagnosis and monitoring of cancer progression through blood samples (Grant et al. 2024). Single-cell sequencing techniques have enabled the detection of circulating tumor cells (CTCs) for liquid biopsy-based diagnosis of multiple cancers, allowing identification of genuine CTCs through concordant copy number alteration profiles (Shen et al. 2024).
Wang et al., (2022) presented a molecular algorithm based on single-cell genomics for early cancer detection (Z. Wang et al. 2022). The study uses single-cell sequencing to examine somatic copy number alterations (CNA) or oncogenic driver mutation profiles at the single-cell level to confirm cellular malignancy. The application of these technologies has also been seen in pancreatic cancer, significantly improving the sensitivity and specificity of cancer cell detection, allowing the analysis of rare cancer cells, circulating tumor cells and metastatic cells (Y. Liu et al. 2022; K. Zhang et al. 2023).
4. Applications of Single-Cell Sequencing for Transcriptomic Profiling in Human Cancer Cells
SCS has emerged as a pivotal method for exploring gene regulatory networks (GRN) and cellular dynamics (S. Chen et al. 2023). Among these technologies, single-cell RNA sequencing (scRNA-seq) provides a detailed view of transcript expression levels and patterns within individual cells across different subpopulations (Kuksin et al. 2021; Lähnemann et al. 2020; F. Tang et al. 2009). Here, we describe applications to sc-RNA related to cancer.
- (a)
- Identifying cancer stem cells and cells rare populations
CSCs (Cancer Stem Cells) are primitive, undifferentiated cells with characteristics like normal stem cells (Ren et al. 2021; Boesch et al. 2016). However, CSCs are biologically characterized by self-renewal, multi-directional differentiation and infinite proliferation, inducing anti-tumor drug resistance and metastasis (Pan et al. 2020). Pan et al., characterized and identified primary and metastatic duct renal cell carcinoma (CDRCC) and observed CSC specific markers to be correlated with poor prognosis of CDRCC and pinpointed inhibitors for effectively targeting CSCs as potential therapeutic strategies for CDRCC (Pan et al. 2020).
Other CSC malignancies such as pancreatic cancer have been studied by Ren et al., (2021), specifically pancreatic duct adenocarcinoma (PDAC) which is an aggressive and lethal malignancy (Ren et al. 2021). This study revealed the heterogeneity of ductal cells and identified nine main clusters: endocrine, acinar, endothelial, ductal, myeloid, fibro- blast, pericyte, T, and B cells and then were reclustered identifying 6 different clusters. Most enriched clusters in tumor tissue exhibited remarkably high copy number variations (CNV) levels. The expression of specific marker genes found to be particular from each cluster type for example on cluster 1 acinar epithelial related genes expressed PRSS1, CLPS (Segerstolpe et al. 2016); heat shock protein-related genes were expressed in cluster 6 are involved in protein transport and folding of ductal cells (Rosas et al. 2016). Clusters 3, 4, and 5 primarily expressed genes related to the proliferation and invasion potential of cells. single-cell trajectory analysis showed pancreatic duct cells originated from clusters 1 and 6, transitioned to cluster 2, and finally evolved into clusters 3, 4, and 5. In this study there could also be identified 202 cancer related genes (CRGs), including 140 upregulated CRGs and 62 downregulated CRGs and establish a relationship between key genes and patient survival. Frequency of mutation events in the high-risk cohort was significantly higher. Kras mutation indicated poor survival in patients. LY6D and MET were significantly more highly expressed in tumor tissues than in normal pancreatic tissues.
- (b) Determining heterogeneity within a cell population
Heterogeneity between different malignant cells is one of the fundamental characteristics of almost all human cancers (El-Sayes, Vito, and Mossman 2021; Lenz et al. 2022; Wen et al. 2022). By measuring transcriptomes at the single-cell level, scRNA-seq enables identification of cellular heterogeneity in far greater detail than conventional methods (Haide Chen, Ye, and Guo 2019). Furthermore, scRNA-seq can be used to identify specific modes of gene expression authorized for the elucidation of molecular mechanisms underlying tumor migration and invasion (Y. Zhang et al. 2021; Wen et al. 2022). Also, constructing gene regulatory networks (GRNs) from scRNA-seq data is used to explore intratumoral heterogeneity and can elucidate the critical genes involved in cancer development (Wen et al. 2022). For instance, Wouters et al. (2020) used single-cell transcriptomics in combination with GRNs and trajectory inference to study 10 melanoma cultures that have mapped the gene regulatory landscape of relapsed melanoma (Wouters et al. 2020; Wen et al. 2022).
- (c) Tumor immunology
The immune system is composed of a complex hierarchy of cell types that protect the organism against disease and maintain homeostasis (Haide Chen, Ye, and Guo 2019). Understanding how the immune system affects cancer development and progression has been one of the most challenging questions in immunology (Schreiber, Old, and Smyth 2011). Identifying heterogeneity of immune cells is the key to understanding the immune system. Advanced scRNA-seq technologies are revolutionizing our ability to study immunology. Recent studies from Hui et al. used scRNA-seq to reveal how neoadjuvant PD-1 blockade combined with chemotherapy remodels the tumor microenvironment in non-small cell lung cancer. They identified significant remodeling of immune cells within the tumor, including the expansion, activation, and phenotypic alterations of cytotoxic T cells, CD16+ natural killer (NK) cells, and regulatory T cells (Tregs) following therapy (Hui et al. 2023).
- (d) Cancer progression, drug development and cancer treatment
There is an increased interest in potential clinical application of single-cell techniques reflected in collaborative initiatives like LifeTime. LifeTime aims to understand the complex behavior of human cells during disease progression and analyze their response to the therapy, all at single-cell resolution (Rajewsky et al. 2020; Stein et al. 2021). To achieve that, further development and integration of multiomics methods is urgently needed (Stein et al. 2021).
ScRNA-seq has enabled identification of molecular pathways that allow prediction of survival (Sade-Feldman et al. 2018; Van de Sande et al. 2023), response to therapy (Jang et al. 2019; Van de Sande et al. 2023), likelihood of resistance (Tanaka et al. 2018; Jerby-Arnon et al. 2018; Van de Sande et al. 2023) and candidacy for alternative intervention (Cohen et al. 2021; Van de Sande et al. 2023).
Recently, an increasing number of clinical trials have been integrating RNA-seq in their design with various objectives; either biological description of the effect of treatments or with the intent to treat patients (Kuksin et al. 2021). Single-cell genome-plus-transcriptome sequencing is also a valuable tool to study the efficacy and safety of genome editing in germline therapy. Genome editing of human embryos or germ cells provides the means for introducing heritable genetic alterations, which may reduce the burden of genetic disease in specific familial situations (Vandereyken et al. 2023; Bekaert et al. 2022). For instance, Vishnubalaji and Alajez (2023) used computational algorithms to decipher the cellular composition of various Breast Cancer (BC) subtypes, including estrogen receptor-positive (ER+), HER2+, ER+HER2+, and triple-negative (TNBC) (Vishnubalaji and Alajez 2023). They analyzed transcriptomic data from 49,899 single-cells, derived from 26 BC patients, and integrated differentially expressed genes with CRISPR-Cas9 perturbational gene effects data from the Achilles project (R. M. Meyers et al. 2017). The key discoveries include ER+ BC: 13 targets, with RPS4X, RPL34, and VMP1 being more effective than ESR1. HER2+ BC: 44 targets, some stronger than ERBB2, with enriched processes in mRNA decay and protein targeting. TNBC: 29 targets, enriched in processes like protein targeting and mRNA processing. Overall, these findings point to new potential targets for treating each subtype.
Other studies are using tumor model systems exposed to treatment, or direct longitudinal sampling of patient tumor specimens before and during treatment to analyze single-cell genome-plus-transcriptome sequencing to understand the genetic subclones resistant to drug selection. Additionally, it will allow the study of how cells within these genetic subclones putatively apply cell plasticity to change their gene expression repertoire and accommodate different phenotypic cancer cell states able to withstand drug treatment and, eventually, acquire resistance (Rambow et al. 2018; Vandereyken et al. 2023). In turn, these approaches might enable the identification of potential cancer cell vulnerabilities, such as druggable molecular players involved in the acquisition of drug tolerance (Vandereyken et al. 2023).
Single-cell DNA sequencing enhanced our understanding of cancer in human cells, this technology allows tracking clonal evolution, detecting rare mutations, understanding drug resistances, improving cancer diagnosis with the implementation of single-cell RNA sequencing, to get an integral understanding of treatment in cancer (Figure 2).
5. ‘Co-Presence’ and ‘Phenotypic Association’ Capability of scSeq Technology
The 'co-presence' functionality of SCS technology refers to its ability to identify the various genomic features or variations concurrently present within each cell analyzed(Evrony, Hinch, and Luo 2021). For example, it can detect the simultaneous presence of multiple or single mutations(Petti et al. 2019; X. Xu et al. 2012), copy number alterations(Yan Gao et al. 2017; Wu et al. 2021), and structural variants(Aganezov et al. 2020) within the same single-cell. This capability provides insights into the interplay between different genomic aberrations and their potential combined effects on cellular processes or disease development(Funnell et al. 2022). By capturing the co-occurring genomic variations within individual cells, researchers can better understand the heterogeneity and complexity of genomic landscapes, particularly in diseases like cancer, where cells can harbor diverse combinations of mutations and other alterations. Furthermore, the ability to detect co-presence in SCS enables scientists to concurrently identify the manifestation of both genetic alterations and epigenetic changes within each cell analyzed(Shema, Bernstein, and Buenrostro 2019). As well as uncovered widespread variation among malignant cells concerning their cellular identity and developmental staging(Schuster 2022). Therefore, co-presence is a crucial capability of SCS that enables many of its unique applications, revolutionizing our understanding of genomics and disease development.
Conventional sequencing methods, which analyze entire cell populations, often face significant limitations. One such limitation is their inability to detect the simultaneous presence of multiple mutations within individual cells. This drawback results in a loss of information about the cellular diversity within the population(Misra, Jadhav, and Bapat 2022). In contrast, single-cell omics offer a superior approach to unraveling cellular diversity. SCS, in particular, preserves this information, allowing for a more holistic study of cells. Interestingly, studies have begun to use both, single-cell sequencing and multi-omics (bulked sequencing) datasets to analyze the molecular diversity of hepatocellular carcinoma (HCC) across inter- and intra-tumor levels(T. Wang et al. 2022). This approach exemplifies how we can enhance the understanding of co-presence in cell studies by integrating cutting-edge and traditional technologies.
The capability of SCS to associate phenotypes is a significant advancement in genomics. It allows for integrating genomic data from individual cells with simultaneous phenotypic profiling as a crucial link between a cell's genotype and phenotype(Evrony, Hinch, and Luo 2021). This feature aids in identifying cellular identities or states characterized by specific genomic features. However, it's important to note that existing single-cell data cannot directly associate cell clusters with phenotypic characteristics. Unfortunately, due to feasibility and sample size limitations, single-cell technology is not yet suitable for extensive cohort studies, with most experiments involving fewer than twenty patient samples, thereby lacking the statistical power needed to pinpoint the cell subpopulations driving the phenotype of interest(Redavid et al. 2022).
The complexities surrounding co-presence and phenotypic association analysis encompass the widely recognized technical noise, intricacies of data analysis, and the necessity for integrative approaches. Regardless of these challenges, SCS persists as a potent methodology for comprehensively investigating co-presence and phenotypic association in cellular entities.
6. Immune Cells Response in Tumor Microenvironment Using scWGS
Beyond the important roles immune cells play in determining the immune responsiveness of tumors, they are also important direct targets of immunotherapeutic strategies. It is thus pivotal to identify molecular mechanisms that modulate immune responsiveness and therapeutic performance of the tumor microenvironment (TME) immune cells. These mechanisms involve non-genetic events that pose significant regulatory elements to be characterized. For instance, Huang et al., highlighted the role of microvascular invasion-related malignant cells, which are driven by MYC pathway activation and MIF signaling within the TME of hepatocellular carcinoma (HCC). Their findings underscore how specific malignant subpopulations interact with immune cells, shaping prognosis and immune responsiveness(H. Huang et al. 2024). At the same time, cellular transition trajectory analysis in single-cell epigenomes further demonstrates that epigenetic features do not always directly predict future cell-fates or gene activities but are reciprocal to transcriptional dynamics. Identification and decoding of the epigenetic features associated with unique TME states and functions in vivo would thus require extensive studies of the single-cell TME epigenomes and their interplay with the transcriptional landscape(M. Kumar 2023). Because of these important roles, TME immune cells play in predicting the prognosis and therapeutic performance of distinct human cancer types, their molecular properties are urgently needed to be characterized with a personalized precision medicine, so that therapeutic strategies to modulate, within TME immune cells efficiently, can be designed(Sierant and Choi 2018).
It has been documented that various immune cells that are part of the TME can play important roles in promoting or inhibiting tumor development and metastasis. These cells, collectively called tumor-infiltrating immune cells 1 (TIICs), are also significantly associated with clinically relevant features, such as patient survival and response to immunotherapy. Efforts aiming to understand the molecular characteristics of immune cells, especially those within the TME have thus gained significant traction. To date, single-cell genomics has emerged as a widely used tool to study TIICs, and has identified various subpopulations of T cells and myeloid cells and shown spatial, differentiation, and functional heterogeneity within the TME(A. W. Zhang and Campbell 2020). For instance, Zhang et al. (2021) demonstrated age-related differences in the immune landscape of melanoma-bearing mice, where older mice exhibited a higher proportion of cytotoxic CD8+ T cells and fewer exhausted cells, contributing to enhanced antitumor immunity. This highlights the importance of dissecting immune cell subpopulations at the single-cell level to better understand their role in tumor progression and response to immunotherapies(C. Zhang et al. 2021).
Similarly, Chen et al. (2024) demonstrated that the upregulation of HMGB2 shapes the immunosuppressive microenvironment in HCC, correlating with exhausted T cells and poorer clinical outcomes. This underscores how epigenetic and transcriptional regulation of factors like HMGB2 can hinder immune cell functionality and drive resistance to immunotherapy, making it a key target for therapeutic intervention (Y.-Z. Chen, Meng, and Xiang 2024). Multi-omics technologies that provide single-cell data at the transcriptional/genomic, epigenetic, proteomic, and other levels have been pivotal in exploring the states and functions of TME immune cells. These have shown that epigenetic dynamics have a prominent influence on the cellular status of TME immune cells. Furthermore, parallel profiling of chromatin state and gene expression in single-cells revealed that chromatin modifications could be predictive of cell state dynamics and that cellular transcriptomes might not always reflect their epigenetic status. Myeloid-lineage immune cells are also influenced by epigenetic chromatin modifications, which can directly control their TME functions.
- (a)
- Tumor microenvironment
Tumors are highly complex entities composed of heterogeneous tumor cells, stroma cells and immune cells(Wolfe et al. 2022). Intertwined biological processes between different types of cells facilitate tumorigenesis, tumor progression and resistance to therapy, as well as, contributing to tissue homeostasis maintenance of normal physiological processes. Consequently, for effective therapeutic intervention, it is crucial to understand the underlying cellular and molecular knowledge and their roles in orchestrating the tumor microenvironment(S. Wang et al. 2023). The constituents of the TME and their role in tumorigenesis and in response to therapy are a rapidly evolving field, thanks to cutting-edge methodological advances. single-cell analysis has facilitated the understanding of immune cell responses in the TME and has allowed researchers to study the functional and molecular aspects of cancer immunologists in greater detail. For instance, Pires et al. (2020) demonstrated how T cells drive ECM remodeling and CSC reduction in fibrosarcomas, revealing how immune activity in the TME directly impacts tumor rejection and progression(Pires et al. 2020). A thorough understanding of immune cell activity in the TME is essential in developing newer drugs and treatment options in cancers.
- (b) Cellular Components of Tumor Microenvironment
These immune cells interact with each other and with cancer cells, secreting a variety of soluble factors, contributing to tumor initiation, progression, and invasion. The function of these cellular components and the tissue-specific microenvironment are deregulated in TME, with pro-tumorigenic and anti-tumorigenic activities being intertwined.
TME consists of non-cancerous cells, their associated cell products, and extracellular matrix within and around the tumor mass. The cellular components of TME are macrophages, granulocytes, mast cells, myeloid-derived suppressor cells, dendritic cells, T cells, B cells, natural killer cells, cancer-associated fibroblasts, mesenchymal stromal cells, endothelial cells, pericytes, and nerve cells. These cells perform various functions such as immune response, cell signaling, and extracellular matrix remodeling, crucial for tumor growth and progression. For instance, Anderson and highlighted that macrophage, especially M2 macrophages, along with myeloid-derived suppressor cells (MDSCs), contribute to immunosuppression by secreting cytokines such as IL-10 and TGF-β. These cytokines activate key signaling pathways like NF-κB and STAT3, facilitating tumor growth and immune evasion. Additionally, cancer-associated fibroblasts play a significant role in ECM remodeling, supporting metastasis and tumor invasion(Anderson and Simon 2020). Alcantara et al. (2023) further demonstrated that targeting STAT3 activation in MDSCs through CpG-STAT3 antisense oligonucleotide (ASO) therapy significantly reduces immunosuppression and enhances T-cell activity in renal and bladder cancer. This combination therapy, when used with anti-PD-1, improved tumor control by reducing MDSC-driven immune resistance and promoting tumor regression(Alcantara et al. 2023). Li et al. (2020), emphasize that tumor-associated macrophages (TAMs) and MDSCs, along with cytokines like IL-6 and TGF-β, contribute to chronic inflammation and immunosuppression within the TME. Targeting these cells and pathways could improve cancer therapies by disrupting the immunosuppressive TME(L. Li et al. 2020).
- (c) Overview of tumor microenvironment at single-cell resolution
The accessibility of single-cell sequencing to mixed cell populations in a non-perturbing manner has made it an attractive technique to rationalize the dynamics of cell heterogeneity in the TME. Due to their relatively low assay cost and high-throughput performance compared with imaging technologies, single-cell sequencing has been widely applied to study various types of tumors and mouse models, demonstrating that single-cell sequencing enables research of tumor cells andTME's cellularity and spatial distribution. Moreover, SCS reveals signaling interactions among different cell types, potentially uncovering novel factors driving the initiation and progression of cancers besides uncovering novel cell subsets with unknown functions.(V. Kumar et al. 2022; Nofech-Mozes et al. 2023)
In a translational perspective, single-cell sequencing provides vast advantages over the traditional bulk RNA-seq assay since the former could identify the complex heterogeneity in not only the cancer cells themselves but also historically stroma cells, opportunities, as well as challenges for developing novel cancer treatments. Single-cell sequencing provides comprehensive views on statics and dynamics of the cell composition in the TME along with their spatial distributions, which not only facilitate the discovery of potential biomarkers in a clinical routine but also advances researchers from bench to bedside, which would subsequently benefit cancer patients in the future.(J. J. Lee et al. 2021; Qian et al. 2020; Jianhong Zhang et al. 2021).
A central question addressed by single-cell sequencing of the tumor microenvironment is the cellular heterogeneity among stromal and immune cell types. Importantly, single-cell sequencing allows defining cell populations at higher resolution than possible by bulk sequencing, offering potential identification of biomarkers that would be otherwise diluted when bulk material is used. To this end, single-cell sequencing of dendritic cell subsets is a perfect test case. Dendritic cells are generally a rare cell type in the whole blood (0.36%) and single-cell technology could offer a better representation of the subsets such as plasmacytoid dendritic cells (pDC). The ability to define these dendritic cell subsets would have great implications in allowing personalized treatment of cancer through the application of active immunotherapies. Recently, single-cell RNA sequencing has been used to define dendritic cells, monocyte, and other diverse cell populations in triple-negative breast cancer (TNBC) and murine melanoma tumors. For instance, Gao et al. (2021) utilized single-cell RNA sequencing (scRNA-seq) to reveal the heterogeneity of monocyte-derived dendritic cells (moDCs) and type-2 conventional dendritic cells (cDC2s). Their study identified seven distinct DC subtypes, with functional differences in antigen processing and immune regulation, especially during maturation. This demonstrates the utility of scRNA-seq in uncovering novel immune cell subsets and their potential roles in shaping tumor immunity, offering insights into how these cell populations could be targeted for immunotherapy(Yuehan Gao et al. 2021). scRNA-seq allows to gain insight into spatial localization of tumor and stromal cells in the tumor microenvironment over bulk RNA sequencing technology. Distinct cellular localization within different parts of the tumor microenvironment, for example, within the tumor, invasive margin, and the center of the tumor, or between epithelial and stromal compartments, may allow a dissection of the distinct functions and mechanisms of tumor-associated cells.(Sun et al. 2022; Jian Liu et al. 2020; Ke et al. 2022).
7. Role of Single-Cell Data Analysis Technologies in Cancer Therapy
Cancer is a complex and heterogeneous disease, characterized by diverse cellular populations within a tumor and dynamic interactions between cancer cells and their microenvironment. This heterogeneity poses significant challenges in understanding disease mechanisms and developing effective treatments. The advent of single-cell data analysis technologies has revolutionized our ability to dissect the complexities of cancer at an unprecedented resolution, unveiling new insights into tumor biology and paving the way for personalized cancer therapy. The increase in SCS data and methodologies for wet-lab applications has been paralleled by advancements in data analysis tools to interpret this data(Svensson, Vento-Tormo, and Teichmann 2018; Zappia and Theis 2021).
The data analysis options available for a particular biological sample are largely dictated by the sequencing technique used to generate the raw data and depend on the research question and study objectives. The type of cell isolation employed in a study is closely aligned with its performance regarding capture efficiency and purity of target cells, which directly affects the output data and consequently the data analysis approach(G. Chen, Ning, and Shi 2019; Gross et al. 2015). For example, a droplet-based method is particularly effective for characterizing tissue composition, as it enables the capture of a substantial number of cells(Wolfien, David, and Galow 2021).
Single-cell data analysis generally involves distinct preprocessing and integration stages. Preprocessing focuses on preparing individual datasets through demultiplexing, quality control, filtering data and normalization. Integration then combines these datasets, using methods like data alignment and correction to account for technical variations (e.g., batch effects) and/or integrate data from different omics (Figure 3). Feature selection and dimensionality reduction are often performed after integration to facilitate downstream analyzes like clustering and trajectory inference. The pipeline choice can have a comparable or even greater impact on the ability to detect biological signals in scRNA-seq data than increasing the cell count from 96 to 384 cells analyzed(Vieth et al. 2019).
- (a)
- Preprocessing and Integration
The analysis of single-cell data begins with robust and effective data handling and integration. CellRanger is a key pipeline that primarily does the initial step of preprocessing raw single-cell RNA-seq by performing demultiplexing, alignment, and quantifying gene expression(Zheng et al. 2017). It uses these barcodes and UMIs to find low-quality cells and identify reads originating from the same cell(Tjoonk 2023). It is also known that it can cluster similar or the same cells into non-overlapping groups. Some tools merge data handling and integration with the subsequent analysis for a multimodal approach(Wolf, Angerer, and Theis 2018). Seurat is a great example of a multimodal analysis tool that enables researchers to do further analysis from data handling and integration(Hao et al. 2024). However, there are tools dedicated just to data handling experiments, like SingleCellExperiment(Risso et al. 2018; Amezquita et al. 2020) that include specialized methods to store and retrieve spike-in information, dimensionality reduction coordinates, and size factors for each cell, along with the usual metadata for genes and libraries. This flexible data representation is compatible with the Bioconductor ecosystem, allowing seamless integration with other analysis tools and pipelines. Additionally, tidySingleCellExperiment bridges the gap between SingleCellExperiment objects and the Tidyverse ecosystem(Wickham et al. 2019), promoting reproducible and efficient data handling and integration workflows.
Due to the difference in experimental methods and computational analysis in SCS, comparing directly cell identities between different experiments can be challenging. For this issue, tools like scmap(Kiselev, Yiu, and Hemberg 2017) address this by allowing users to integrate cell-type or individual cells in different experiments. Similarly, LIGER (Welch et al. 2019) can be used to compare and contrast across experimental batches, individuals, sex, tissues, species, and is particularly useful for handling different modalities like RNA-seq and ATAC-seq.
Unlike the single-cell Experiment, scmap offers further data exploration (downstream analysis) to identify clusters, find shared gene markers, compare clusters, and visualize clusters and gene expression, foir. Similarly, scMerge(Y. Lin et al. 2019) offers a robust method for merging multiple batches of single-cell RNA-seq data, accounting for potential batch effects and enabling integrated analyzes across diverse datasets. MAESTRO(C. Wang et al. 2020) besides doing quality control, normalization, and filtering of single-cell data, specializes in the integration of single-cell transcriptome and regulome for evaluating scATAC-seq clustering, automatic cell-type annotation, integration between scRNA-seq and scATAC-seq.
Moreover, many specialized tools analyze specific modalities. DropletUtils(A. Lun, Griffiths, and McCarthy 2021) facilitates the analysis of droplet-based single-cell data, while zellkonverter(Zappia L 2024) enables interoperability by converting between Python and R environments, allowing seamless data transfer between these platforms. Furthermore, data normalization and transformation methods, such as Linnorm(Yip et al. 2017), play a crucial role in ensuring data quality and comparability across samples. Linnorm provides robust normalization and transformation of data based on linear models and normality assumptions, enabling accurate downstream analyses. It preserves biological variations in scRNA-seq data and removes technical noises simultaneously.
Many studies utilize a multi-omics approach for cancer research, highlighting the importance of accurately integrating diverse data types for effective analysis. By combining transcriptomics, epigenomics, and proteomics, researchers can gain a comprehensive understanding of cellular states and the regulatory mechanisms that drive them(Ruan et al. 2024; Xing et al. 2023; Warfvinge et al. 2023). Tools like MOFA+(Argelaguet et al. 2020) and scArches(Lotfollahi et al. 2022) enable the integration of diverse omics data, revealing shared and unique features across different molecular layers. Spatial analysis techniques, such as spatialHeatmap(Jianhai Zhang et al. 2024), spicyR(Canete et al. 2021), and SpatialExperiment(Righelli et al. 2022), facilitate the exploration of spatial patterns and heterogeneity within tumor samples, providing insights into the tumor microenvironment and its influence on cancer progression and treatment response.
- (b) Clustering and downstream analysis
Identifying distinct cell populations and subgroups within heterogeneous tumor samples is a critical step in understanding cancer biology. Clustering algorithms like bluster (Aaron Lun 2024), celda (Campbell et al. 2021), and SC3 (Kiselev et al. 2017) group cells based on their transcriptome or genomic profiles, enabling the identification of distinct cell populations. Differential analysis methods, such as DEsingle (Z. Miao et al. 2018), and distinct (Tiberi et al. 2020) identify genes, pathways, or molecular features that are differentially expressed or regulated between cell populations or experimental conditions, providing insights into the molecular drivers of cellular heterogeneity.
In the other hand there a well-known multimodal tool like Seurat (Hao et al., 2024) and Scanpy (Wolf et al., 2018) that enable quality control, analysis, clustering and differential expression capabilities for scRNA-seq data, facilitating the identification and interpretation of cellular heterogeneity. Tools like SCENIC (Aibar et al., 2017) infer active transcription factors and gene regulatory networks, providing insights into gene regulation within tumor cells. CopyKAT (Gao et al., 2021) and InferCNV (inferCNV of the Trinity CTAT Project) specialize in inferring genome-wide copy number variation profiles from scRNA-seq data, a critical aspect of cancer genomics. Other recently used tools such as non-linear dimensional reduction (UMAP) cluster cells with a graph-based clustering approach (Khayatan, Hussain, and Tebyaniyan 2023; Y. Miao et al. 2024) and SingleR does cluster definition plus annotation (Aran et al. 2019).
- (c) Single-cell DNA and Whole Genome Sequencing (scWGS)
Single-cell DNA-sequencing (scDNA-seq) and whole genome sequencing (scWGS) techniques offer unprecedented insights into genomic alterations, such as copy number alterations, copy number variations, and clonal heterogeneity within tumors. Here, we present some tools for the downstream data analysis of scDNA-seq. The tool Ginkgo (Garvin et al. 2015) identifies copy number variations, CHISEL (Zaccaria and Raphael 2021) infer copy number alterations including whole-genome duplications (WGDs), while PyClone (Roth et al. 2014) and SCCNV (Dong et al. 2020) elucidate clonal populations and their evolutionary trajectories. SCcaller (Dong et al. 2017) and SCCNV identify single-nucleotide variations and copy number variations, respectively, and SCITE (Jahn, Kuipers, and Beerenwinkel 2016) designed tool to infer the evolutionary history of tumors using noisy and incomplete mutation profiles of single-cells from scDNA-seq data.
The integration of single-cell data analysis technologies has revolutionized our understanding of cancer biology, enabling the exploration of tumor heterogeneity, clonal evolution, and cellular interactions within the tumor microenvironment. As single-cell technologies continue to advance, data analysis tools will play a crucial role in leveraging the wealth of information contained within these datasets, ultimately paving the way for personalized and targeted cancer therapies tailored to individual patients' tumor profiles.
8. Emerging Technologies and Future Directions in scWGS in Cancer Biology
The rise of single-cell whole-genome sequencing (scWGS) has transformed cancer biology, offering detailed insights into the molecular landscapes of cancer cells. Deep learning techniques have proven highly effective in analyzing the vast and complex data generated by scWGS (Erfanian et al. 2023; Molho et al. 2024). These methods have outperformed traditional computational approaches in various domains (Premkumar et al. 2024; Zhu et al. 2024). In cancer biology, deep learning has facilitated the study of tumor cell heterogeneity, uncovering gene expression patterns and regulatory networks governing cell behavior (Halawani, Buchert, and Chen 2023; Molho et al. 2024).
Artificial intelligence (AI) has emerged as a powerful tool in scWGS analysis, with machine learning (ML) algorithms excelling at extracting valuable insights from high-dimensional and complex data (Qi and Zou 2023). In cancer research, AI has contributed to the development of prognostic models, biomarker identification, and characterization of the tumor microenvironment (Jianlan Liu et al. 2023; Mou et al. 2023). For example, Chen et al. (2023) used AI to construct a communication signature derived from cancer-associated fibroblasts (CAFs), enabling the stratification of clear cell renal cell carcinoma patients based on their immune profiles and potential response to immunotherapy (Hualin Chen et al. 2023). Similarly, Kang et al. (2024) employed AI techniques to identify tumor-associated macrophage subpopulations in prostate cancer, revealing subpopulations that may facilitate tumor progression by enhancing immune evasion and altering the tumor microenvironment, which can lead to therapeutic challenges in tumor progression and drug resistance (Kang et al. 2024).
Deep learning, a branch of AI, has emerged as a transformative force in scWGS, offering algorithms that can integrate multi-omics data, including genomics, transcriptomics, and proteomics, to unravel the complexities of tumor heterogeneity and identify cancer subtypes (He et al. 2023; J. Li et al. 2023). Danishuddin et al. (2024) explored the applications of deep learning in cancer genomic and proteomic studies, highlighting its potential to improve patient diagnosis, prognosis, and treatment strategies (Danishuddin, Khan, and Kim 2024). In glioma research, Luo et al. (2023) demonstrated the utility of deep learning in tumor segmentation, diagnosis, grading, and characterizing the tumor microenvironment, paving the way for personalized treatment approaches (Luo et al. 2023). As scWGS continues to evolve, deep learning will play a role in extracting meaningful insights from multi-omics data, driving the development of precision oncology.
In the future, developing technologies for single-cell analysis in cancer is crucial. Techniques in addition to DNA-based and RNA-based methods such as scEpigenetics, sc proteomics, sc metabolomics, sc CRISPR technologies and sc multi omics technologies, may be particularly helpful in advancing this field. Additionally, the advancements in machine learning, AI and Data Analysis will further enhance our understanding and capabilities in cancer biology and its implications. Consequently, more studies focusing on areas such as prostate cancer, breast cancer, cervical cancer, and solid tumors especially for complex matrices that are difficult to understand would be beneficial for future at the level of diagnosis, microenvironment, understanding of existing pre and post resistance, better targeted and efficient therapies, all supported by a robust bioinformatics analysis that will enhance these benefits through the incorporation of cutting-edge technologies such as artificial intelligence.
The integration of these tools into cancer profiling has greatly enhanced our ability to understand tumor complexity, genomic instability and clonal evolution. This comprehensive understanding of tumor complexity allows for more accurate predictions for treatment responses, identification of resistance mechanisms, and development of more personalized and effective therapeutic strategies, taking us to the way for precision oncology.
9. Conclusions
The introduction of single-cell whole genome sequencing (scWGS) has revolutionized our understanding of cancer biology, providing unprecedented insights into tumor heterogeneity, clonal evolution, and the complex interactions within the tumor microenvironment. This technological breakthrough has enabled researchers to analyze the genomic, transcriptomic, and epigenomic landscapes of single-cells and uncover the complex cellular dynamics that drive cancer progression, metastasis, and treatment resistance.
The unique capabilities of scWGS, particularly its accuracy in detecting rare genomic events, co-presence functionality in capturing multiple genomic features simultaneously, and phenotypic association potential, have opened new avenues for cancer research and precision medicine. These advances have facilitated the identification of rare cell populations such as cancer stem cells and elucidated the heterogeneous nature of tumors with unprecedented resolution.
Furthermore, the integration of scWGS with other single-cell omics technologies, including scRNA-seq, scATAC-seq and single-cell proteomics, has enabled a multidimensional view of cellular states and regulatory mechanisms in cancer. This integrative approach has enhanced our ability to decipher the complex interplay between genetic alterations, gene expression patterns and epigenetic modifications that contribute to tumor initiation, progression and therapeutic response.
The rapid development of data analysis tools and computational methods, including advanced machine learning and artificial intelligence algorithms, has been critical to harnessing the vast amounts of data generated by single-cell technologies. These sophisticated analytical approaches have enabled the identification of new biomarkers, the creation of predictive models for patient stratification and the development of targeted therapeutic strategies.
As this field continues to evolve, new technologies such as spatial transcriptomics and in situ sequencing will further improve our understanding of the spatial organization and cellular interactions within tumors. In addition, the integration of single-cell multiomics data with clinical information and longitudinal studies will be critical to translate these insights into actionable clinical strategies.
In summary, scWGS and related single-cell technologies have ushered in a new era of cancer research, offering unprecedented opportunities to unravel the complexity of tumor biology. As these technologies continue to evolve and become more accessible, they promise tremendous advances in advancing personalized cancer diagnostics, prognosis and therapy, ultimately improving patient outcomes in the era of precision oncology.
The challenges regarding co-presence and phenotypic association analysis include the well-known technical noise, data analysis complexities, and the need for integrative approaches. Dropout events, amplification bias, and sequencing errors are common technical noises that compromise the data quality. Data analysis complexities stem from difficulties in accurate variant calling, interpreting cellular heterogeneity, and integrating genomic data with phenotypic traits, which will be discussed further in the subsequent section. Moreover, integrative approaches such as multi-omics integration, longitudinal studies, and functional validation add complexity but are essential to creating robust studies. Other issues like sample preparation, scalability, cost, statistical power in cohorts, and the need for standardized protocols also present significant hurdles. Despite these challenges, SCS remains a powerful methodology for delving deeper into the analysis of co-presence and phenotypic association in cells.
Author Contributions
A.O.M wrote the first draft of the manuscript with the assistance of Y.J.A, D. L.M, N.G. and G.G who also contributed to preparing specific sections. E.N.G directed the project, provided expert insights, prepared the final manuscript and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
The research in the authors´s laboratories is supported by the grant FID23-105, from the National Secretariat of Science and Technology of Panamá (SENACYT) and the National System of Investigators of the SENACYT (SNI-SENACYT).
Acknowledgments
We thank Floris Foijer for various discussions. This review is focused on the impact of single cell sequencing for the advancement in cancer cell biology understanding. This review is not intended to provide comprehensive literature on the subject; therefore many appropriate citations may not be included for which we apologize.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Aganezov, Sergey, Sara Goodwin, Rachel M. Sherman, Fritz J. Sedlazeck, Gayatri Arun, Sonam Bhatia, Isac Lee, et al. 2020. “Comprehensive Analysis of Structural Variants in Breast Cancer Genomes Using Single-Molecule Sequencing.” Genome Research 30 (9): 1258–73. [CrossRef]
- Alcantara, Marice B., Wilson S. Tang, Dongfang Wang, Damian Kaniowski, Elaine Kang, Nazli Dizman, Alexander Chehrazi-Raffle, et al. 2023. “Targeting STAT3 in Tumor-Associated Antigen-Presenting Cells as a Strategy for Kidney and Bladder Cancer Immunotherapy.” Frontiers in Immunology 14:1274781. [CrossRef]
- Amezquita, Robert A., Aaron T. L. Lun, Etienne Becht, Vince J. Carey, Lindsay N. Carpp, Ludwig Geistlinger, Federico Marini, et al. 2020. “Orchestrating Single-Cell Analysis with Bioconductor.” Nature Methods 17 (2): 137–45. [CrossRef]
- Anderson, Nicole M., and M. Celeste Simon. 2020. “The Tumor Microenvironment.” Current Biology: CB 30 (16): R921–25.
- Aran, Dvir, Agnieszka P. Looney, Leqian Liu, Esther Wu, Valerie Fong, Austin Hsu, Suzanna Chak, et al. 2019. “Reference-Based Analysis of Lung Single-Cell Sequencing Reveals a Transitional Profibrotic Macrophage.” Nature Immunology 20 (2): 163–72. [CrossRef]
- Argelaguet, Ricard, Damien Arnol, Danila Bredikhin, Yonatan Deloro, Britta Velten, John C. Marioni, and Oliver Stegle. 2020. “MOFA+: A Statistical Framework for Comprehensive Integration of Multi-Modal Single-Cell Data.” Genome Biology 21 (1): 111. [CrossRef]
- Bai, Rilan, and Jiuwei Cui. 2022. “Development of Immunotherapy Strategies Targeting Tumor Microenvironment Is Fiercely Ongoing.” Frontiers in Immunology 13 (June):890166. [CrossRef]
- Bekaert, B., A. Boel, G. Cosemans, L. De Witte, B. Menten, and B. Heindryckx. 2022. “CRISPR/Cas Gene Editing in the Human Germline.” Seminars in Cell & Developmental Biology 131 (November):93–107.
- Boesch, Maximilian, Sieghart Sopper, Alain G. Zeimet, Daniel Reimer, Guenther Gastl, Burkhard Ludewig, and Dominik Wolf. 2016. “Heterogeneity of Cancer Stem Cells: Rationale for Targeting the Stem Cell Niche.” Biochimica et Biophysica Acta 1866 (2): 276–89. [CrossRef]
- Borgsmüller, Nico, Jose Bonet, Francesco Marass, Abel Gonzalez-Perez, Nuria Lopez-Bigas, and Niko Beerenwinkel. 2020. “BnpC: Bayesian Non-Parametric Clustering of Single-Cell Mutation Profiles.” Bioinformatics 36 (19): 4854–59. [CrossRef]
- Campbell, J., S. Corbett, Y. Koga, S. Yang, E. Reed, and Z. Wang. 2021. “Celda: CEllular Latent Dirichlet Allocation.” R Package Version 181.
- Canete, Nicolas P., Sourish S. Iyengar, James S. Wilmott, John T. Ormerod, Andrew N. Harman, and Ellis Patrick. 2021. “spicyR: Spatial Analysis of in Situ Cytometry Data in R.” bioRxiv. bioRxiv. [CrossRef]
- Chen, Geng, Baitang Ning, and Tieliu Shi. 2019. “Single-Cell RNA-Seq Technologies and Related Computational Data Analysis.” Frontiers in Genetics 10 (April):317. [CrossRef]
- Chen, Haide, Fang Ye, and Guoji Guo. 2019. “Revolutionizing Immunology with Single-Cell RNA Sequencing.” Cellular & Molecular Immunology 16 (3): 242–49. [CrossRef]
- Chen, Hualin, Wenjie Yang, Lin Ma, Yingjie Li, and Zhigang Ji. 2023. “Machine-Learning Based Integrating Bulk and Single-Cell RNA Sequencing Reveals the SLC38A5-CCL5 Signaling as a Promising Target for Clear Cell Renal Cell Carcinoma Treatment.” Translational Oncology 38 (101790): 101790. [CrossRef]
- Chen, Siyuan, Weibo Jiang, Yanhui Du, Manshi Yang, Yihan Pan, Huan Li, and Mengying Cui. 2023. “Single-Cell Analysis Technologies for Cancer Research: From Tumor-Specific Single Cell Discovery to Cancer Therapy.” Frontiers in Genetics 14 (October):1276959. [CrossRef]
- Chen, Yan-Zhu, Zhi-Shang Meng, and Zuo-Lin Xiang. 2024. “HMGB2 Drives Tumor Progression and Shapes the Immunosuppressive Microenvironment in Hepatocellular Carcinoma: Insights from Multi-Omics Analysis.” Frontiers in Immunology 15 (August):1415435. [CrossRef]
- Chew, Valerie, Han Chong Toh, and Jean-Pierre Abastado. 2012. “Immune Microenvironment in Tumor Progression: Characteristics and Challenges for Therapy.” Journal of Oncology 2012 (August):608406. [CrossRef]
- Cohen, Yael C., Mor Zada, Shuang-Yin Wang, Chamutal Bornstein, Eyal David, Adi Moshe, Baoguo Li, et al. 2021. “Identification of Resistance Pathways and Therapeutic Targets in Relapsed Multiple Myeloma Patients through Single-Cell Sequencing.” Nature Medicine 27 (3): 491–503. [CrossRef]
- Dago, Angel E., Asya Stepansky, Anders Carlsson, Madelyn Luttgen, Jude Kendall, Timour Baslan, Anand Kolatkar, et al. 2014. “Rapid Phenotypic and Genomic Change in Response to Therapeutic Pressure in Prostate Cancer Inferred by High Content Analysis of Single Circulating Tumor Cells.” PloS One 9 (8): e101777.
- Danishuddin, Shawez Khan, and Jong Joo Kim. 2024. “From Cancer Big Data to Treatment: Artificial Intelligence in Cancer Research.” The Journal of Gene Medicine 26 (1): e3629.
- Demaree, Benjamin, Cyrille L. Delley, Harish N. Vasudevan, Cheryl A. C. Peretz, David Ruff, Catherine C. Smith, and Adam R. Abate. 2020. “Joint Profiling of DNA and Proteins in Single Cells to Dissect Genotype-Phenotype Associations in Leukemia.” bioRxiv. bioRxiv. https://doi.org/10.1101/2020.02.26.967133.
- Dong, Xiao, Lei Zhang, Xiaoxiao Hao, Tao Wang, and Jan Vijg. 2020. “SCCNV: A Software Tool for Identifying Copy Number Variation from Single-Cell Whole-Genome Sequencing.” Frontiers in Genetics 11 (November):505441.
- Dong, Xiao, Lei Zhang, Brandon Milholland, Moonsook Lee, Alexander Y. Maslov, Tao Wang, and Jan Vijg. 2017. “Accurate Identification of Single-Nucleotide Variants in Whole-Genome-Amplified Single Cells.” Nature Methods 14 (5): 491–93.
- El-Sayes, Nader, Alyssa Vito, and Karen Mossman. 2021. “Tumor Heterogeneity: A Great Barrier in the Age of Cancer Immunotherapy.” Cancers 13 (4). https://doi.org/10.3390/cancers13040806.
- Erfanian, Nafiseh, A. Ali Heydari, Adib Miraki Feriz, Pablo Iañez, Afshin Derakhshani, Mohammad Ghasemigol, Mohsen Farahpour, et al. 2023. “Deep Learning Applications in Single-Cell Genomics and Transcriptomics Data Analysis.” Biomedecine & Pharmacotherapie [Biomedicine & Pharmacotherapy] 165 (115077): 115077.
- Evrony, Gilad D., Anjali Gupta Hinch, and Chongyuan Luo. 2021. “Applications of Single-Cell DNA Sequencing.” Annual Review of Genomics and Human Genetics 22 (1): 171–97.
- Fu, Luo-Qin, Wen-Lin Du, Mao-Hua Cai, Jia-Yu Yao, Yuan-Yuan Zhao, and Xiao-Zhou Mou. 2020. “The Roles of Tumor-Associated Macrophages in Tumor Angiogenesis and Metastasis.” Cellular Immunology 353 (104119): 104119.
- Funnell, Tyler, Ciara H. O’Flanagan, Marc J. Williams, Andrew McPherson, Steven McKinney, Farhia Kabeer, Hakwoo Lee, et al. 2022. “Single-Cell Genomic Variation Induced by Mutational Processes in Cancer.” Nature 612 (7938): 106–15.
- Gao, Yan, Xiaohui Ni, Hua Guo, Zhe Su, Yi Ba, Zhongsheng Tong, Zhi Guo, et al. 2017. “Single-Cell Sequencing Deciphers a Convergent Evolution of Copy Number Alterations from Primary to Circulating Tumor Cells.” Genome Research 27 (8): 1312–22.
- Gao, Yuehan, He Li, Zhaohuai Li, Lihui Xie, Xiuxing Liu, Zhaohao Huang, Binyao Chen, et al. 2021. “Single-Cell Analysis Reveals the Heterogeneity of Monocyte-Derived and Peripheral Type-2 Conventional Dendritic Cells.” The Journal of Immunology 207 (3): 837–48.
- Garvin, Tyler, Robert Aboukhalil, Jude Kendall, Timour Baslan, Gurinder S. Atwal, James Hicks, Michael Wigler, and Michael C. Schatz. 2015. “Interactive Analysis and Assessment of Single-Cell Copy-Number Variations.” Nature Methods 12 (11): 1058–60.
- Gráf, Alexandra, Márton Zsolt Enyedi, Lajos Pintér, Éva Kriston-Pál, Gábor Jaksa, Árpád Bálind, Éva Ezer, et al. 2021. “The Combination of Single-Cell and Next-Generation Sequencing Can Reveal Mosaicism for BRCA2 Mutations and the Fine Molecular Details of Tumorigenesis.” Cancers 13 (10). https://doi.org/10.3390/cancers13102354.
- Grant, Christopher Ryan, David J. Benjamin, Scott Cramer, and Arash Rezazadeh Kalebasty. 2024. “A Remnant Never Forgotten: The Utility of Circulating Tumor DNA in Treatment Guidance of Urachal Cancer.” Therapeutic Advances in Medical Oncology 16 (February):17588359241230743.
- Gross, Andre, Jonas Schoendube, Stefan Zimmermann, Maximilian Steeb, Roland Zengerle, and Peter Koltay. 2015. “Technologies for Single-Cell Isolation.” International Journal of Molecular Sciences 16 (8): 16897–919.
- Halawani, Raid, Michael Buchert, and Yi-Ping Phoebe Chen. 2023. “Deep Learning Exploration of Single-Cell and Spatially Resolved Cancer Transcriptomics to Unravel Tumour Heterogeneity.” Computers in Biology and Medicine 164 (107274): 107274.
- Han, Yingying, Dan Wang, Lushan Peng, Tao Huang, Xiaoyun He, Junpu Wang, and Chunlin Ou. 2022. “Single-Cell Sequencing: A Promising Approach for Uncovering the Mechanisms of Tumor Metastasis.” Journal of Hematology & Oncology 15 (1): 59.
- Hao, Yuhan, Tim Stuart, Madeline H. Kowalski, Saket Choudhary, Paul Hoffman, Austin Hartman, Avi Srivastava, et al. 2024. “Dictionary Learning for Integrative, Multimodal and Scalable Single-Cell Analysis.” Nature Biotechnology 42 (2): 293–304.
- He, Xiujing, Xiaowei Liu, Fengli Zuo, Hubing Shi, and Jing Jing. 2023. “Artificial Intelligence-Based Multi-Omics Analysis Fuels Cancer Precision Medicine.” Seminars in Cancer Biology 88 (January):187–200.
- Huang, August Yue, and Eunjung Alice Lee. 2021. “Identification of Somatic Mutations From Bulk and Single-Cell Sequencing Data.” Frontiers in Aging 2:800380.
- Huang, Haoran, Feifeng Wu, Yang Yu, Borui Xu, Dehua Chen, Yuwei Huo, and Shaoqiang Li. 2024. “Multi-Transcriptomics Analysis of Microvascular Invasion-Related Malignant Cells and Development of a Machine Learning-Based Prognostic Model in Hepatocellular Carcinoma.” Frontiers in Immunology 15 (August):1436131.
- Hui, Zhenzhen, Yulin Ren, Dong Zhang, Yulong Chen, Wenwen Yu, Jie Cao, Liang Liu, et al. 2023. “PD-1 Blockade Potentiates Neoadjuvant Chemotherapy in NSCLC via Increasing CD127+ and KLRG1+ CD8 T Cells.” NPJ Precision Oncology 7 (1): 48.
- Jaberi, Elham, Emilie Tresse, Kirsten Grønbæk, Joachim Weischenfeldt, and Shohreh Issazadeh-Navikas. 2020. “Identification of Unique and Shared Mitochondrial DNA Mutations in Neurodegeneration and Cancer by Single-Cell Mitochondrial DNA Structural Variation Sequencing (MitoSV-Seq).” EBioMedicine 57 (July):102868.
- Jahn, Katharina, Jack Kuipers, and Niko Beerenwinkel. 2016. “Tree Inference for Single-Cell Data.” Genome Biology 17 (1): 86.
- Jang, Jin Sung, Ying Li, Amit Kumar Mitra, Lintao Bi, Alexej Abyzov, Andre J. van Wijnen, Linda B. Baughn, et al. 2019. “Molecular Signatures of Multiple Myeloma Progression through Single Cell RNA-Seq.” Blood Cancer Journal 9 (1): 2.
- Jerby-Arnon, Livnat, Parin Shah, Michael S. Cuoco, Christopher Rodman, Mei-Ju Su, Johannes C. Melms, Rachel Leeson, et al. 2018. “A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade.” Cell 175 (4): 984–97.e24.
- Ju, Siwei, Cong Chen, Jiahang Zhang, Lin Xu, Xun Zhang, Zhaoqing Li, Yongxia Chen, Jichun Zhou, Feiyang Ji, and Linbo Wang. 2022. “Detection of Circulating Tumor Cells: Opportunities and Challenges.” Biomarker Research 10 (1): 58.
- Kang, Zhen, Yu-Xuan Zhao, Ren Shun Qian Qiu, Dong-Ning Chen, Qing-Shui Zheng, Xue-Yi Xue, Ning Xu, and Yong Wei. 2024. “Identification Macrophage Signatures in Prostate Cancer by Single-Cell Sequencing and Machine Learning.” Cancer Immunology, Immunotherapy: CII 73 (3): 41.
- Ke, May, Badran Elshenawy, Helen Sheldon, Anjali Arora, and Francesca M. Buffa. 2022. “Single Cell RNA-Sequencing: A Powerful yet Still Challenging Technology to Study Cellular Heterogeneity.” BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology 44 (11): e2200084.
- Kharchenko, Peter V., Lev Silberstein, and David T. Scadden. 2014. “Bayesian Approach to Single-Cell Differential Expression Analysis.” Nature Methods 11 (7): 740–42.
- Khayatan, Danial, Ahmed Hussain, and Hamid Tebyaniyan. 2023. “Exploring Animal Models in Oral Cancer Research and Clinical Intervention: A Critical Review.” Veterinary Medicine and Science 9 (4): 1833–47.
- Kim, Nayoung, Hye Hyeon Eum, and Hae-Ock Lee. 2021. “Clinical Perspectives of Single-Cell RNA Sequencing.” Biomolecules 11 (8). https://doi.org/10.3390/biom11081161.
- Kiselev, Vladimir Yu, Kristina Kirschner, Michael T. Schaub, Tallulah Andrews, Andrew Yiu, Tamir Chandra, Kedar N. Natarajan, et al. 2017. “SC3: Consensus Clustering of Single-Cell RNA-Seq Data.” Nature Methods 14 (5): 483–86.
- Kiselev, Vladimir Yu, Andrew Yiu, and Martin Hemberg. 2017. “Scmap - A Tool for Unsupervised Projection of Single Cell RNA-Seq Data.” bioRxiv. bioRxiv. https://doi.org/10.1101/150292.
- Kuksin, Maria, Daphné Morel, Marine Aglave, François-Xavier Danlos, Aurélien Marabelle, Andrei Zinovyev, Daniel Gautheret, and Loïc Verlingue. 2021. “Applications of Single-Cell and Bulk RNA Sequencing in Onco-Immunology.” European Journal of Cancer 149 (May):193–210.
- Kumar, Manish. 2023. “The Precision Oncology Approach to Molecular Cancer Therapeutics Targeting Oncogenic Signaling Pathways Is a Means to an End.” arXiv [q-bio.OT]. arXiv. http://arxiv.org/abs/2304.05411v11.
- Kumar, Vikrant, Kalpana Ramnarayanan, Raghav Sundar, Nisha Padmanabhan, Supriya Srivastava, Mayu Koiwa, Tadahito Yasuda, et al. 2022. “Single-Cell Atlas of Lineage States, Tumor Microenvironment, and Subtype-Specific Expression Programs in Gastric Cancer.” Cancer Discovery 12 (3): 670–91.
- Lähnemann, David, Johannes Köster, Ewa Szczurek, Davis J. McCarthy, Stephanie C. Hicks, Mark D. Robinson, and Catalina Vallejos. 2020. “Eleven Grand Challenges in Single-Cell Data Science.” Genome Biology. https://doi.org/10.1186/s13059-020-1926-6.
- Lee, Jaewon J., Vincent Bernard, Alexander Semaan, Maria E. Monberg, Jonathan Huang, Bret M. Stephens, Daniel Lin, et al. 2021. “Elucidation of Tumor-Stromal Heterogeneity and the Ligand-Receptor Interactome by Single-Cell Transcriptomics in Real-World Pancreatic Cancer Biopsies.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 27 (21): 5912–21.
- Lee, Paul, Rita Yim, Sin-Hang Fung, Kai-Kei Miu, Zhangting Wang, Ka-Chun Wu, Lester Au, Garret Man-Kit Leung, Victor Ho-Fun Lee, and Harinder Gill. 2022. “Single-Nucleotide Variations, Insertions/Deletions and Copy Number Variations in Myelodysplastic Syndrome during Disease Progression Revealed by a Single-Cell DNA Sequencing Platform.” International Journal of Molecular Sciences 23 (9). https://doi.org/10.3390/ijms23094647.
- Lenz, Guido, Giovana R. Onzi, Luana S. Lenz, Julieti H. Buss, Jephesson A. Dos Santos, and Karine R. Begnini. 2022. “The Origins of Phenotypic Heterogeneity in Cancer.” Cancer Research 82 (1): 3–11.
- Li, Junyu, Lin Li, Peimeng You, Yiping Wei, and Bin Xu. 2023. “Towards Artificial Intelligence to Multi-Omics Characterization of Tumor Heterogeneity in Esophageal Cancer.” Seminars in Cancer Biology 91 (June):35–49.
- Li, Lihong, Rui Yu, Tiange Cai, Zhen Chen, Meng Lan, Tengteng Zou, Bingyue Wang, Qi Wang, Yiye Zhao, and Yu Cai. 2020. “Effects of Immune Cells and Cytokines on Inflammation and Immunosuppression in the Tumor Microenvironment.” International Immunopharmacology 88 (November):106939.
- Lin, Danfeng, Lesang Shen, Meng Luo, Kun Zhang, Jinfan Li, Qi Yang, Fangfang Zhu, et al. 2021. “Circulating Tumor Cells: Biology and Clinical Significance.” Signal Transduction and Targeted Therapy 6 (1): 404.
- Lin, Yingxin, Shila Ghazanfar, Kevin Y. X. Wang, Johann A. Gagnon-Bartsch, Kitty K. Lo, Xianbin Su, Ze-Guang Han, et al. 2019. “scMerge Leverages Factor Analysis, Stable Expression, and Pseudoreplication to Merge Multiple Single-Cell RNA-Seq Datasets.” Proceedings of the National Academy of Sciences of the United States of America 116 (20): 9775–84.
- Liu, Jianlan, Pengpeng Zhang, Fang Yang, Keyu Jiang, Shiyi Sun, Zhijia Xia, Gang Yao, and Jian Tang. 2023. “Integrating Single-Cell Analysis and Machine Learning to Create Glycosylation-Based Gene Signature for Prognostic Prediction of Uveal Melanoma.” Frontiers in Endocrinology 14 (March):1163046.
- Liu, Jian, Tianmin Xu, Yuemei Jin, Bingyu Huang, and Yan Zhang. 2020. “Progress and Clinical Application of Single-Cell Transcriptional Sequencing Technology in Cancer Research.” Frontiers in Oncology 10:593085.
- Liu, Lingshan, Qiurui Zhang, Chenglong Wang, Heze Guo, Vincent Mukwaya, Rong Chen, Yichun Xu, et al. 2023. “Single-Cell Diagnosis of Cancer Drug Resistance through the Differential Endocytosis of Nanoparticles between Drug-Resistant and Drug-Sensitive Cancer Cells.” ACS Nano 17 (19): 19372–86.
- Liu, Ying, Gengqiu Luo, Yuanliang Yan, and Jinwu Peng. 2022. “A Pan-Cancer Analysis of Copper Homeostasis-Related Gene Lipoyltransferase 1: Its Potential Biological Functions and Prognosis Values.” Frontiers in Genetics 13 (October):1038174.
- Lohr, Jens G., Viktor A. Adalsteinsson, Kristian Cibulskis, Atish D. Choudhury, Mara Rosenberg, Peter Cruz-Gordillo, Joshua M. Francis, et al. 2014. “Whole-Exome Sequencing of Circulating Tumor Cells Provides a Window into Metastatic Prostate Cancer.” Nature Biotechnology 32 (5): 479–84.
- Loo, Jacky Fong-Chuen, Ho Pui Ho, Siu Kai Kong, Tza-Huei Wang, and Yi-Ping Ho. 2019. “Technological Advances in Multiscale Analysis of Single Cells in Biomedicine.” Advanced Biosystems 3 (11): e1900138.
- Lotfollahi, Mohammad, Mohsen Naghipourfar, Malte D. Luecken, Matin Khajavi, Maren Büttner, Marco Wagenstetter, Žiga Avsec, et al. 2022. “Mapping Single-Cell Data to Reference Atlases by Transfer Learning.” Nature Biotechnology 40 (1): 121–30.
- Lun, Aaron. 2024. “bluster: Clustering Algorithms for Bioconductor.” http://bioconductor.org/packages/bluster/.
- Lun, A., J. Griffiths, and D. McCarthy. 2021. “DropletUtils: Utilities for Handling Single-Cell Droplet data.(Bioconductor Version: Release (3.13)).”.
- Luo, Jiefeng, Mika Pan, Ke Mo, Yingwei Mao, and Donghua Zou. 2023. “Emerging Role of Artificial Intelligence in Diagnosis, Classification and Clinical Management of Glioma.” Seminars in Cancer Biology 91 (June):110–23.
- Macaulay, Iain C., Wilfried Haerty, Parveen Kumar, Yang I. Li, Tim Xiaoming Hu, Mabel J. Teng, Mubeen Goolam, et al. 2015. “G&T-Seq: Parallel Sequencing of Single-Cell Genomes and Transcriptomes.” Nature Methods 12 (6): 519–22.
- Meyers, Robin M., Jordan G. Bryan, James M. McFarland, Barbara A. Weir, Ann E. Sizemore, Han Xu, Neekesh V. Dharia, et al. 2017. “Computational Correction of Copy Number Effect Improves Specificity of CRISPR-Cas9 Essentiality Screens in Cancer Cells.” Nature Genetics 49 (12): 1779–84.
- Meyers, Sarah, Llucia Alberti-Servera, Olga Gielen, Margot Erard, Toon Swings, Jolien De Bie, Lucienne Michaux, et al. 2022. “Monitoring of Leukemia Clones in B-Cell Acute Lymphoblastic Leukemia at Diagnosis and During Treatment by Single-Cell DNA Amplicon Sequencing.” HemaSphere 6 (4): e700.
- Miao, Yuwen, Pan Wang, Jinyan Huang, Xin Qi, Yingjiqiong Liang, Wenquan Zhao, Huiming Wang, Jiong Lyu, and Huiyong Zhu. 2024. “Metabolomics, Transcriptome and Single-Cell RNA Sequencing Analysis of the Metabolic Heterogeneity between Oral Cancer Stem Cells and Differentiated Cancer Cells.” Cancers 16 (2). https://doi.org/10.3390/cancers16020237.
- Miao, Zhun, Ke Deng, Xiaowo Wang, and Xuegong Zhang. 2018. “DEsingle for Detecting Three Types of Differential Expression in Single-Cell RNA-Seq Data.” Bioinformatics (Oxford, England) 34 (18): 3223–24.
- Misra, Pratibha, Amruta R. Jadhav, and Sharmila A. Bapat. 2022. “Single-Cell Sequencing: A Cutting Edge Tool in Molecular Medical Research.” Armed Forces Medical Journal, India 78 (Suppl 1): S7–13.
- Molho, Dylan, Jiayuan Ding, Wenzhuo Tang, Zhaoheng Li, Hongzhi Wen, Yixin Wang, Julian Venegas, et al. 2024. “Deep Learning in Single-Cell Analysis.” ACM Transactions on Intelligent Systems and Technology 15 (3): 1–62.
- Mou, Lisha, Zuhui Pu, Yongxiang Luo, Ryan Quan, Yunhu So, and Hui Jiang. 2023. “Construction of a Lipid Metabolism-Related Risk Model for Hepatocellular Carcinoma by Single Cell and Machine Learning Analysis.” Frontiers in Immunology 14 (March):1036562.
- Navin, Nicholas, Jude Kendall, Jennifer Troge, Peter Andrews, Linda Rodgers, Jeanne McIndoo, Kerry Cook, et al. 2011. “Tumour Evolution Inferred by Single-Cell Sequencing.” Nature 472 (7341): 90–94.
- Ni, Xiaohui, Minglei Zhuo, Zhe Su, Jianchun Duan, Yan Gao, Zhijie Wang, Chenghang Zong, et al. 2013. “Reproducible Copy Number Variation Patterns among Single Circulating Tumor Cells of Lung Cancer Patients.” Proceedings of the National Academy of Sciences of the United States of America 110 (52): 21083–88.
- Nofech-Mozes, Ido, David Soave, Philip Awadalla, and Sagi Abelson. 2023. “Pan-Cancer Classification of Single Cells in the Tumour Microenvironment.” Nature Communications 14 (1): 1615.
- Pang, Long, Jing Ding, Yuxin Ge, Jianglin Fan, and Shih-Kang Fan. 2019. “Single-Cell-Derived Tumor-Sphere Formation and Drug-Resistance Assay Using an Integrated Microfluidics.” Analytical Chemistry 91 (13): 8318–25.
- Pan, Xiu-Wu, Hao Zhang, Da Xu, Jia-Xin Chen, Wen-Jin Chen, Si-Shun Gan, Fa-Jun Qu, et al. 2020. “Identification of a Novel Cancer Stem Cell Subpopulation That Promotes Progression of Human Fatal Renal Cell Carcinoma by Single-Cell RNA-Seq Analysis.” International Journal of Biological Sciences 16 (16): 3149–62.
- Patel, Anoop P., Itay Tirosh, John J. Trombetta, Alex K. Shalek, Shawn M. Gillespie, Hiroaki Wakimoto, Daniel P. Cahill, et al. 2014. “Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma.” Science (New York, N.Y.) 344 (6190): 1396–1401.
- Peretz, Cheryl A. C., Lisa H. F. McGary, Tanya Kumar, Hunter Jackson, Jose Jacob, Robert Durruthy-Durruthy, Mark J. Levis, Alexander Perl, Benjamin J. Huang, and Catherine C. Smith. 2021. “Single-Cell DNA Sequencing Reveals Complex Mechanisms of Resistance to Quizartinib.” Blood Advances 5 (5): 1437–41.
- Petti, Allegra A., Stephen R. Williams, Christopher A. Miller, Ian T. Fiddes, Sridhar N. Srivatsan, David Y. Chen, Catrina C. Fronick, Robert S. Fulton, Deanna M. Church, and Timothy J. Ley. 2019. “A General Approach for Detecting Expressed Mutations in AML Cells Using Single Cell RNA-Sequencing.” Nature Communications 10 (1): 3660.
- Pires, Ana, Alexander Greenshields-Watson, Emma Jones, Kathryn Smart, Sarah N. Lauder, Michelle Somerville, Stefan Milutinovic, et al. 2020. “Immune Remodeling of the Extracellular Matrix Drives Loss of Cancer Stem Cells and Tumor Rejection.” Cancer Immunology Research 8 (12): 1520–31.
- Polzer, Bernhard, Gianni Medoro, Sophie Pasch, Francesca Fontana, Laura Zorzino, Aurelia Pestka, Ulrich Andergassen, et al. 2014. “Molecular Profiling of Single Circulating Tumor Cells with Diagnostic Intention.” EMBO Molecular Medicine 6 (11): 1371–86.
- Premkumar, R., Arthi Srinivasan, K. G. Harini Devi, Deepika M, Gaayathry E, Pramod Jadhav, Abhishek Futane, and Vigneswaran Narayanamurthy. 2024. “Single-Cell Classification, Analysis, and Its Application Using Deep Learning Techniques.” Bio Systems 237 (105142): 105142.
- Puram, Sidharth V., Itay Tirosh, Anuraag S. Parikh, Anoop P. Patel, Keren Yizhak, Shawn Gillespie, Christopher Rodman, et al. 2017. “Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer.” Cell 171 (7): 1611–24.e24.
- Qian, Junbin, Siel Olbrecht, Bram Boeckx, Hanne Vos, Damya Laoui, Emre Etlioglu, Els Wauters, et al. 2020. “A Pan-Cancer Blueprint of the Heterogeneous Tumor Microenvironment Revealed by Single-Cell Profiling.” Cell Research 30 (9): 745–62.
- Qi, Ren, and Quan Zou. 2023. “Trends and Potential of Machine Learning and Deep Learning in Drug Study at Single-Cell Level.” Research (Washington, D.C.) 6 (March):0050.
- Rajan, Sanjana, Simone Zaccaria, Matthew V. Cannon, Maren Cam, Amy C. Gross, Benjamin J. Raphael, and Ryan D. Roberts. 2023. “Structurally Complex Osteosarcoma Genomes Exhibit Limited Heterogeneity within Individual Tumors and across Evolutionary Time.” Cancer Research Communications 3 (4): 564–75.
- Rajewsky, Nikolaus, Geneviève Almouzni, Stanislaw A. Gorski, Stein Aerts, Ido Amit, Michela G. Bertero, Christoph Bock, et al. 2020. “LifeTime and Improving European Healthcare through Cell-Based Interceptive Medicine.” Nature 587 (7834): 377–86.
- Rambow, Florian, Aljosja Rogiers, Oskar Marin-Bejar, Sara Aibar, Julia Femel, Michael Dewaele, Panagiotis Karras, et al. 2018. “Toward Minimal Residual Disease-Directed Therapy in Melanoma.” Cell 174 (4): 843–55.e19.
- Redavid, Immacolata, Maria Rosa Conserva, Luisa Anelli, Antonella Zagaria, Giorgina Specchia, Pellegrino Musto, and Francesco Albano. 2022. “Single-Cell Sequencing: Ariadne’s Thread in the Maze of Acute Myeloid Leukemia.” Diagnostics 12 (4): 996.
- Ren, Xuechen, Chengliang Zhou, Yu Lu, Fulin Ma, Yong Fan, and Chen Wang. 2021. “Single-Cell RNA-Seq Reveals Invasive Trajectory and Determines Cancer Stem Cell-Related Prognostic Genes in Pancreatic Cancer.” Bioengineered 12 (1): 5056–68.
- Reza, K. Kamil, Shuvashis Dey, Alain Wuethrich, Jing Wang, Andreas Behren, Fiach Antaw, Yuling Wang, Abu Ali Ibn Sina, and Matt Trau. 2021. “In Situ Single Cell Proteomics Reveals Circulating Tumor Cell Heterogeneity during Treatment.” ACS Nano 15 (7): 11231–43.
- Righelli, Dario, Lukas M. Weber, Helena L. Crowell, Brenda Pardo, Leonardo Collado-Torres, Shila Ghazanfar, Aaron T. L. Lun, Stephanie C. Hicks, and Davide Risso. 2022. “SpatialExperiment: Infrastructure for Spatially-Resolved Transcriptomics Data in R Using Bioconductor.” Bioinformatics (Oxford, England) 38 (11): 3128–31.
- Risso, Davide, Liam Purvis, Russell B. Fletcher, Diya Das, John Ngai, Sandrine Dudoit, and Elizabeth Purdom. 2018. “clusterExperiment and RSEC: A Bioconductor Package and Framework for Clustering of Single-Cell and Other Large Gene Expression Datasets.” PLoS Computational Biology 14 (9): e1006378.
- Rognoni, Emanuel, and Fiona M. Watt. 2018. “Skin Cell Heterogeneity in Development, Wound Healing, and Cancer.” Trends in Cell Biology 28 (9): 709–22.
- Rosas, Paola C., Ganachari M. Nagaraja, Punit Kaur, Alexander Panossian, Georg Wickman, L. Rene Garcia, Fahd A. Al-Khamis, and Alexzander A. A. Asea. 2016. “Hsp72 (HSPA1A) Prevents Human Islet Amyloid Polypeptide Aggregation and Toxicity: A New Approach for Type 2 Diabetes Treatment.” PloS One 11 (3): e0149409.
- Roth, Andrew, Jaswinder Khattra, Damian Yap, Adrian Wan, Emma Laks, Justina Biele, Gavin Ha, Samuel Aparicio, Alexandre Bouchard-Côté, and Sohrab P. Shah. 2014. “PyClone: Statistical Inference of Clonal Population Structure in Cancer.” Nature Methods 11 (4): 396–98.
- Ruan, Xinjia, Chong Lai, Leqi Li, Bei Wang, Xiaofan Lu, Dandan Zhang, Jingya Fang, Maode Lai, and Fangrong Yan. 2024. “Integrative Analysis of Single-Cell and Bulk Multi-Omics Data to Reveal Subtype-Specific Characteristics and Therapeutic Strategies in Clear Cell Renal Cell Carcinoma Patients.” Journal of Cancer 15 (19): 6420–33.
- Saadatpour, Assieh, Shujing Lai, Guoji Guo, and Guo-Cheng Yuan. 2015. “Single-Cell Analysis in Cancer Genomics.” Trends in Genetics: TIG 31 (10): 576–86.
- Sade-Feldman, Moshe, Keren Yizhak, Stacey L. Bjorgaard, John P. Ray, Carl G. de Boer, Russell W. Jenkins, David J. Lieb, et al. 2018. “Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma.” Cell 175 (4): 998–1013.e20.
- Schreiber, Robert D., Lloyd J. Old, and Mark J. Smyth. 2011. “Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion.” Science 331 (6024): 1565–70.
- Schuster, Linda Christina. 2022. “Dissecting Heterogeneity, Clonal Evolution, and Epigenetic Changes in Distinct and Molecularly Defined AML Subsets by Multi Omics Single-Cell Sequencing.” Heidelberg: archiv.ub.uni-heidelberg.de. https://doi.org/10.11588/heidok.00032274.
- Segerstolpe, Åsa, Athanasia Palasantza, Pernilla Eliasson, Eva-Marie Andersson, Anne-Christine Andréasson, Xiaoyan Sun, Simone Picelli, et al. 2016. “Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes.” Cell Metabolism 24 (4): 593–607.
- Shekhar, Karthik, Petter Brodin, Mark M. Davis, and Arup K. Chakraborty. 2014. “Automatic Classification of Cellular Expression by Nonlinear Stochastic Embedding (ACCENSE).” Proceedings of the National Academy of Sciences of the United States of America 111 (1): 202–7.
- Shema, Efrat, Bradley E. Bernstein, and Jason D. Buenrostro. 2019. “Single-Cell and Single-Molecule Epigenomics to Uncover Genome Regulation at Unprecedented Resolution.” Nature Genetics 51 (1): 19–25.
- Shen, Xiaohan, Jiao Dai, Lingchuan Guo, Zhigang Liu, Liu Yang, Dongmei Gu, Yinghong Xie, et al. 2024. “Single-Cell Low-Pass Whole Genome Sequencing Accurately Detects Circulating Tumor Cells for Liquid Biopsy-Based Multi-Cancer Diagnosis.” NPJ Precision Oncology 8 (1): 30.
- Sierant, Michael C., and Jungmin Choi. 2018. “Single-Cell Ssequencing in Cancer: Recent Applications to Immunogenomics and Multi-Omics Tools.” Genomics & Informatics 16 (4): e17.
- Stein, Catarina M., Ralf Weiskirchen, Frederik Damm, and Paulina M. Strzelecka. 2021. “Single-Cell Omics: Overview, Analysis, and Application in Biomedical Science.” Journal of Cellular Biochemistry 122 (11): 1571–78.
- Sun, Duanchen, Xiangnan Guan, Amy E. Moran, Ling-Yun Wu, David Z. Qian, Pepper Schedin, Mu-Shui Dai, et al. 2022. “Identifying Phenotype-Associated Subpopulations by Integrating Bulk and Single-Cell Sequencing Data.” Nature Biotechnology 40 (4): 527–38.
- Suvà, Mario L., and Itay Tirosh. 2019. “Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges.” Molecular Cell 75 (1): 7–12.
- Svensson, Valentine, Roser Vento-Tormo, and Sarah A. Teichmann. 2018. “Exponential Scaling of Single-Cell RNA-Seq in the Past Decade.” Nature Protocols 13 (4): 599–604. [CrossRef]
- Tanaka, Nobuyuki, Shintaro Katayama, Aparna Reddy, Kaneyasu Nishimura, Naoya Niwa, Hiroshi Hongo, Koichiro Ogihara, et al. 2018. “Single-Cell RNA-Seq Analysis Reveals the Platinum Resistance Gene COX7B and the Surrogate Marker CD63.” Cancer Medicine 7 (12): 6193–6204. [CrossRef]
- Tang, Fuchou, Catalin Barbacioru, Yangzhou Wang, Ellen Nordman, Clarence Lee, Nanlan Xu, Xiaohui Wang, et al. 2009. “mRNA-Seq Whole-Transcriptome Analysis of a Single Cell.” Nature Methods 6 (5): 377–82.
- Tang, Jie, Kailing Tu, Keying Lu, Jiaxun Zhang, Kai Luo, Haoxuan Jin, Lei Wang, et al. 2021. “Single-Cell Exome Sequencing Reveals Multiple Subclones in Metastatic Colorectal Carcinoma.” Genome Medicine 13 (1): 148. [CrossRef]
- Thiele, Jana- A., Pavel Pitule, James Hicks, and Peter Kuhn. 2019. “Single-Cell Analysis of Circulating Tumor Cells.” Methods in Molecular Biology (Clifton, N.J.) 1908:243–64. [CrossRef]
- Tiberi, Simone, Helena L. Crowell, Pantelis Samartsidis, Lukas M. Weber, and Mark D. Robinson. 2020. “distinct: A Novel Approach to Differential Distribution Analyses.” bioRxiv. bioRxiv. [CrossRef]
- Tirosh, Itay, Benjamin Izar, Sanjay M. Prakadan, Marc H. Wadsworth 2nd, Daniel Treacy, John J. Trombetta, Asaf Rotem, et al. 2016. “Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq.” Science (New York, N.Y.) 352 (6282): 189–96.
- Tjoonk, Niels. 2023. “What Is CellRanger and How Do You Use It?” Single Cell Discoveries. August 10, 2023. https://www.scdiscoveries.com/blog/knowledge/cellranger/.
- Tritschler, Sophie, Fabian J. Theis, Heiko Lickert, and Anika Böttcher. 2017. “Systematic Single-Cell Analysis Provides New Insights into Heterogeneity and Plasticity of the Pancreas.” Molecular Metabolism 6 (9): 974–90. [CrossRef]
- Vandereyken, Katy, Alejandro Sifrim, Bernard Thienpont, and Thierry Voet. 2023. “Methods and Applications for Single-Cell and Spatial Multi-Omics.” Nature Reviews. Genetics 24 (8): 494–515. [CrossRef]
- Van de Sande, Bram, Joon Sang Lee, Euphemia Mutasa-Gottgens, Bart Naughton, Wendi Bacon, Jonathan Manning, Yong Wang, et al. 2023. “Applications of Single-Cell RNA Sequencing in Drug Discovery and Development.” Nature Reviews. Drug Discovery 22 (6): 496–520. [CrossRef]
- Vieth, Beate, Swati Parekh, Christoph Ziegenhain, Wolfgang Enard, and Ines Hellmann. 2019. “A Systematic Evaluation of Single Cell RNA-Seq Analysis Pipelines.” Nature Communications 10 (1): 4667. [CrossRef]
- Vishnubalaji, Radhakrishnan, and Nehad M. Alajez. 2023. “Single-Cell Transcriptome Analysis Revealed Heterogeneity and Identified Novel Therapeutic Targets for Breast Cancer Subtypes.” Cells 12 (8). [CrossRef]
- Wang, Chenfei, Dongqing Sun, Xin Huang, Changxin Wan, Ziyi Li, Ya Han, Qian Qin, et al. 2020. “Integrative Analyses of Single-Cell Transcriptome and Regulome Using MAESTRO.” Genome Biology 21 (1): 198. [CrossRef]
- Wang, Shuo, Si-Tong Sun, Xin-Yue Zhang, Hao-Ran Ding, Yu Yuan, Jun-Jie He, Man-Shu Wang, Bin Yang, and Yu-Bo Li. 2023. “The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives.” International Journal of Molecular Sciences 24 (3). https://doi.org/10.3390/ijms24032943. [CrossRef]
- Wang, Tingjie, Ningxin Dang, Guangbo Tang, Zihang Li, Xiujuan Li, Bingyin Shi, Zhong Xu, et al. 2022. “Integrating Bulk and Single-Cell RNA Sequencing Reveals Cellular Heterogeneity and Immune Infiltration in Hepatocellular Carcinoma.” Molecular Oncology 16 (11): 2195–2213.
- Wang, Yong, Jill Waters, Marco L. Leung, Anna Unruh, Whijae Roh, Xiuqing Shi, Ken Chen, et al. 2014. “Clonal Evolution in Breast Cancer Revealed by Single Nucleus Genome Sequencing.” Nature 512 (7513): 155–60. [CrossRef]
- Wang, Zhuo, Yuyang Zhao, Xiaohan Shen, Yichun Zhao, Ziyuan Zhang, Huming Yin, Xiaojun Zhao, Haitao Liu, and Qihui Shi. 2022. “Single-Cell Genomics-Based Molecular Algorithm for Early Cancer Detection.” Analytical Chemistry 94 (5): 2607–14. [CrossRef]
- Warfvinge, Rebecca, Linda Geironson Ulfsson, Parashar Dhapola, Fatemeh Safi, Mikael N. E. Sommarin, Shamit Soneji, Henrik Hjorth-Hansen, et al. 2023. “Single Cell Multi-Omics Analysis of Chronic Myeloid Leukemia Links Cellular Heterogeneity to Therapy Response.” bioRxiv. https://doi.org/10.1101/2023.08.16.553504. [CrossRef]
- Welch, Joshua D., Velina Kozareva, Ashley Ferreira, Charles Vanderburg, Carly Martin, and Evan Z. Macosko. 2019. “Single-Cell Multi-Omic Integration Compares and Contrasts Features of Brain Cell Identity.” Cell 177 (7): 1873–87.e17. [CrossRef]
- Wen, Lu, Guoqiang Li, Tao Huang, Wei Geng, Hao Pei, Jialiang Yang, Miao Zhu, et al. 2022. “Single-Cell Technologies: From Research to Application.” Innovation (Cambridge (Mass.)) 3 (6): 100342. [CrossRef]
- Wickham, Hadley, Mara Averick, Jennifer Bryan, Winston Chang, Lucy McGowan, Romain François, Garrett Grolemund, et al. 2019. “Welcome to the Tidyverse.” Journal of Open Source Software 4 (43): 1686.
- Wills, Quin F., and Adam J. Mead. 2015. “Application of Single-Cell Genomics in Cancer: Promise and Challenges.” Human Molecular Genetics 24 (R1): R74–84. [CrossRef]
- Wolfe, Cecily, Yayi Feng, David Chen, Edwin Purcell, Anne Talkington, Sepideh Dolatshahi, and Heman Shakeri. 2022. “GeoTyper: Automated Pipeline from Raw scRNA-Seq Data to Cell Type Identification.” In 2022 Systems and Information Engineering Design Symposium (SIEDS). IEEE. https://doi.org/10.1109/sieds55548.2022.9799321.
- Wolf, F. Alexander, Philipp Angerer, and Fabian J. Theis. 2018. “SCANPY: Large-Scale Single-Cell Gene Expression Data Analysis.” Genome Biology 19 (1): 15. [CrossRef]
- Wolfien, Markus, Robert David, and Anne-Marie Galow. 2021. “Single-Cell RNA Sequencing Procedures and Data Analysis.” In Bioinformatics, 19–35. Exon Publications.
- Wouters, Jasper, Zeynep Kalender-Atak, Liesbeth Minnoye, Katina I. Spanier, Maxime De Waegeneer, Carmen Bravo González-Blas, David Mauduit, et al. 2020. “Robust Gene Expression Programs Underlie Recurrent Cell States and Phenotype Switching in Melanoma.” Nature Cell Biology 22 (8): 986–98. [CrossRef]
- Wu, Chi-Yun, Billy T. Lau, Heon Seok Kim, Anuja Sathe, Susan M. Grimes, Hanlee P. Ji, and Nancy R. Zhang. 2021. “Integrative Single-Cell Analysis of Allele-Specific Copy Number Alterations and Chromatin Accessibility in Cancer.” Nature Biotechnology 39 (10): 1259–69. [CrossRef]
- Xing, Zeyu, Dongcai Lin, Yuting Hong, Zihuan Ma, Hongnan Jiang, Ye Lu, Jiale Sun, et al. 2023. “Construction of a Prognostic 6-Gene Signature for Breast Cancer Based on Multi-Omics and Single-Cell Data.” Frontiers in Oncology 13 (November):1186858. [CrossRef]
- Xu, Liwen, Robert Durruthy-Durruthy, Dennis J. Eastburn, Maurizio Pellegrino, Omid Shah, Everett Meyer, and James Zehnder. 2019. “Clonal Evolution and Changes in Two AML Patients Detected with A Novel Single-Cell DNA Sequencing Platform.” Scientific Reports 9 (1): 11119. [CrossRef]
- Xu, Xun, Yong Hou, Xuyang Yin, Li Bao, Aifa Tang, Luting Song, Fuqiang Li, et al. 2012. “Single-Cell Exome Sequencing Reveals Single-Nucleotide Mutation Characteristics of a Kidney Tumor.” Cell 148 (5): 886–95. [CrossRef]
- Yip, Shun H., Panwen Wang, Jean-Pierre A. Kocher, Pak Chung Sham, and Junwen Wang. 2017. “Linnorm: Improved Statistical Analysis for Single Cell RNA-Seq Expression Data.” Nucleic Acids Research 45 (22): e179.
- Zaccaria, Simone, and Benjamin J. Raphael. 2021. “Characterizing Allele- and Haplotype-Specific Copy Numbers in Single Cells with CHISEL.” Nature Biotechnology 39 (2): 207–14. [CrossRef]
- Zappia L, Lun A. 2024. zellkonverter: Conversion Between scRNA-Seq Objects (version R package version 1.14.0). http://bioconductor.org/packages/zellkonverter/.
- Zappia, Luke, and Fabian J. Theis. 2021. “Over 1000 Tools Reveal Trends in the Single-Cell RNA-Seq Analysis Landscape.” Genome Biology 22 (1): 301. [CrossRef]
- Zhang, Allen W., and Kieran R. Campbell. 2020. “Computational Modelling in Single-Cell Cancer Genomics: Methods and Future Directions.” Physical Biology 17 (6): 061001. [CrossRef]
- Zhang, Cangang, Lei Lei, Xiaofeng Yang, Kaili Ma, Huiqiang Zheng, Yanhong Su, Anjun Jiao, et al. 2021. “Single-Cell Sequencing Reveals Antitumor Characteristics of Intratumoral Immune Cells in Old Mice.” Journal for Immunotherapy of Cancer 9 (10): e002809. [CrossRef]
- Zhang, Jianhai, Le Zhang, Brendan Gongol, Jordan Hayes, Alexander T. Borowsky, Julia Bailey-Serres, and Thomas Girke. 2024. “spatialHeatmap: Visualizing Spatial Bulk and Single-Cell Assays in Anatomical Images.” NAR Genomics and Bioinformatics 6 (1): lqae006. [CrossRef]
- Zhang, Jianhong, Chengyang Song, Ye Tian, and Xueying Yang. 2021. “Single-Cell RNA Sequencing in Lung Cancer: Revealing Phenotype Shaping of Stromal Cells in the Microenvironment.” Frontiers in Immunology 12:802080. [CrossRef]
- Zhang, Ke, Yuan Chen, Jie Zhu, Xinyu Ge, Junqing Wu, Peng Xu, and Jie Yao. 2023. “Advancement of Single-Cell Sequencing for Clinical Diagnosis and Treatment of Pancreatic Cancer.” Frontiers of Medicine 10 (August):1213136. [CrossRef]
- Zhang, Yijie, Dan Wang, Miao Peng, Le Tang, Jiawei Ouyang, Fang Xiong, Can Guo, et al. 2021. “Single-cell RNA Sequencing in Cancer Research.” Journal of Experimental & Clinical Cancer Research: CR 40 (1): 81.
- Zheng, Grace X. Y., Jessica M. Terry, Phillip Belgrader, Paul Ryvkin, Zachary W. Bent, Ryan Wilson, Solongo B. Ziraldo, et al. 2017. “Massively Parallel Digital Transcriptional Profiling of Single Cells.” Nature Communications 8 (January):14049.
- Zhu, Yuan, Litai Bai, Zilin Ning, Wenfei Fu, Jie Liu, Linfeng Jiang, Shihuang Fei, et al. 2024. “Deep Learning for Clustering Single-Cell RNA-Seq Data.” Current Bioinformatics 19 (3): 193–210. [CrossRef]
Figure 1.
Workflow for single-cell RNA sequencing (scRNA-seq) and bulk RNA sequencing. scRNA-seq reveals the distinct transcriptome of individual cells (top), while bulk RNA sequencing captures the average gene expression profile across all cells combined (bottom). Created in BioRender. Lab, G. (2024) https://BioRender.com/u93e754.
Figure 1.
Workflow for single-cell RNA sequencing (scRNA-seq) and bulk RNA sequencing. scRNA-seq reveals the distinct transcriptome of individual cells (top), while bulk RNA sequencing captures the average gene expression profile across all cells combined (bottom). Created in BioRender. Lab, G. (2024) https://BioRender.com/u93e754.
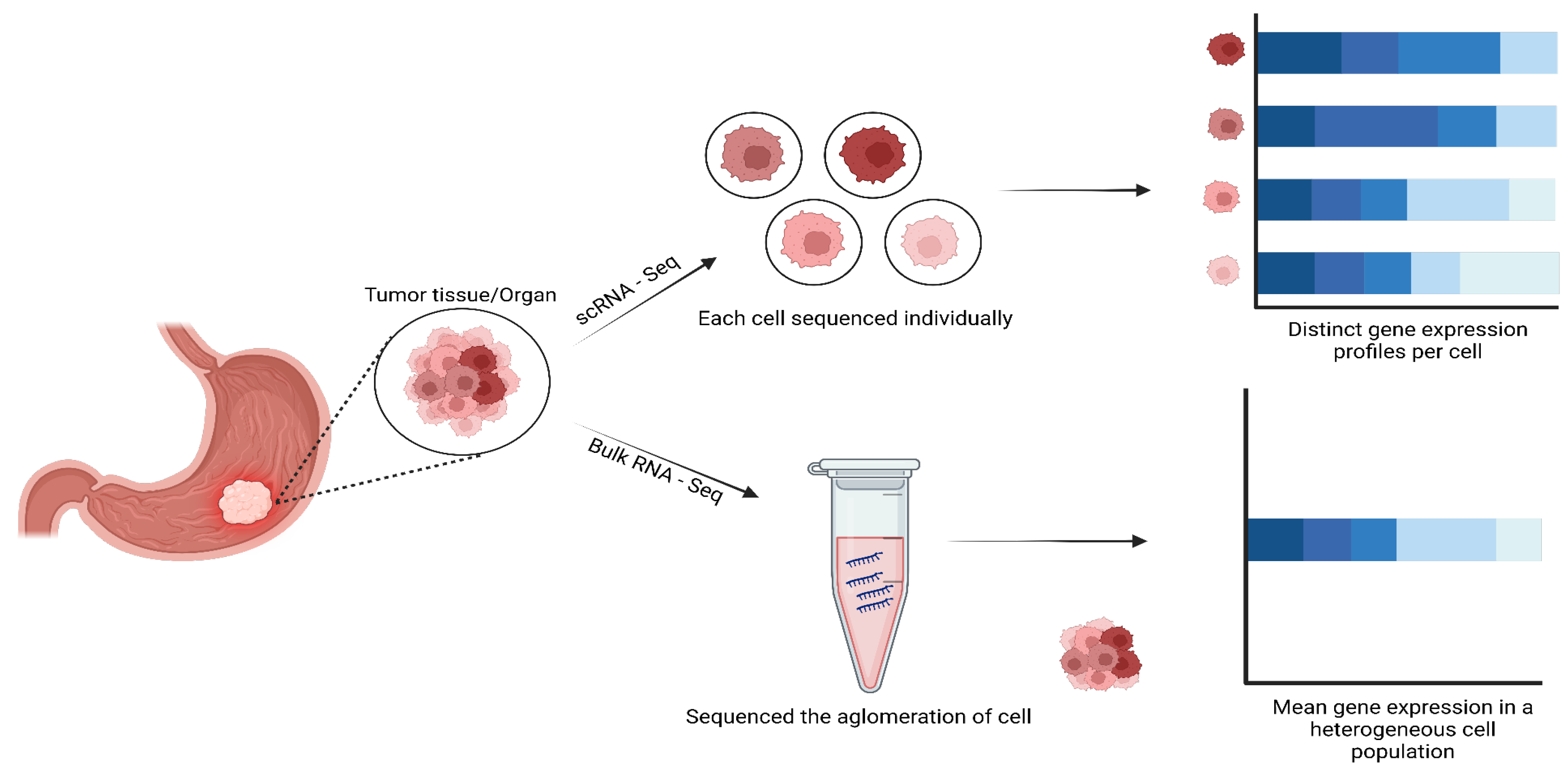
Figure 2.
Applications of single-cell sequencing for genomic and transcriptomic profiling in human cancer cells. Applications that are carried out by both technologies, independently or integrated, are found in the middle. Created in BioRender. Lab, G. (2024) https://BioRender.com/y81r919.
Figure 2.
Applications of single-cell sequencing for genomic and transcriptomic profiling in human cancer cells. Applications that are carried out by both technologies, independently or integrated, are found in the middle. Created in BioRender. Lab, G. (2024) https://BioRender.com/y81r919.
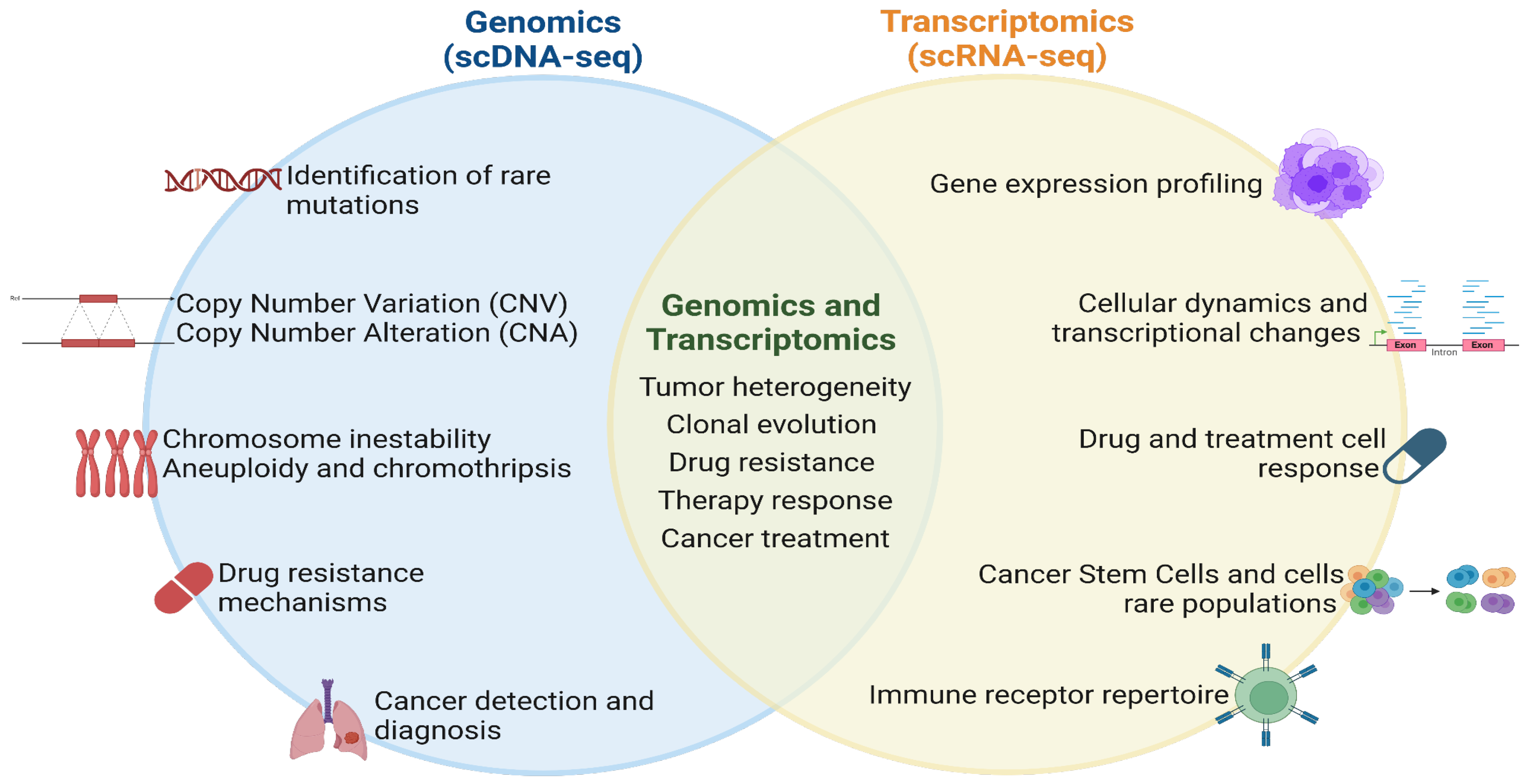
Figure 3.
Overview of common tools used in single-cell RNA sequencing (scRNA-seq) data handling workflow. The workflow begins with raw data acquisition in FastQ format, followed by demultiplexing and quality control to ensure reliability. It then transitions to preprocessing and integration stages. In preprocessing, data undergo filtering to eliminate low-quality cells, normalization to address technical variations, and correction for batch effects. The integration phase combines data from different samples and/or incorporates multi omics data for a comprehensive analysis of cell populations, depending on the study. Finally, the workflow culminates in downstream analysis, encompassing clustering to identify distinct cell populations, trajectory analysis to infer developmental pathways, differential expression analysis for gene variation, annotation for cell type identification, and interactive data visualization to interpret complex datasets. This systematic approach provides a simplified overview of the steps used for processing and analyzing scRNA-seq data. Created in BioRender. Lab, G. (2024) https://BioRender.com/r42z271.
Figure 3.
Overview of common tools used in single-cell RNA sequencing (scRNA-seq) data handling workflow. The workflow begins with raw data acquisition in FastQ format, followed by demultiplexing and quality control to ensure reliability. It then transitions to preprocessing and integration stages. In preprocessing, data undergo filtering to eliminate low-quality cells, normalization to address technical variations, and correction for batch effects. The integration phase combines data from different samples and/or incorporates multi omics data for a comprehensive analysis of cell populations, depending on the study. Finally, the workflow culminates in downstream analysis, encompassing clustering to identify distinct cell populations, trajectory analysis to infer developmental pathways, differential expression analysis for gene variation, annotation for cell type identification, and interactive data visualization to interpret complex datasets. This systematic approach provides a simplified overview of the steps used for processing and analyzing scRNA-seq data. Created in BioRender. Lab, G. (2024) https://BioRender.com/r42z271.
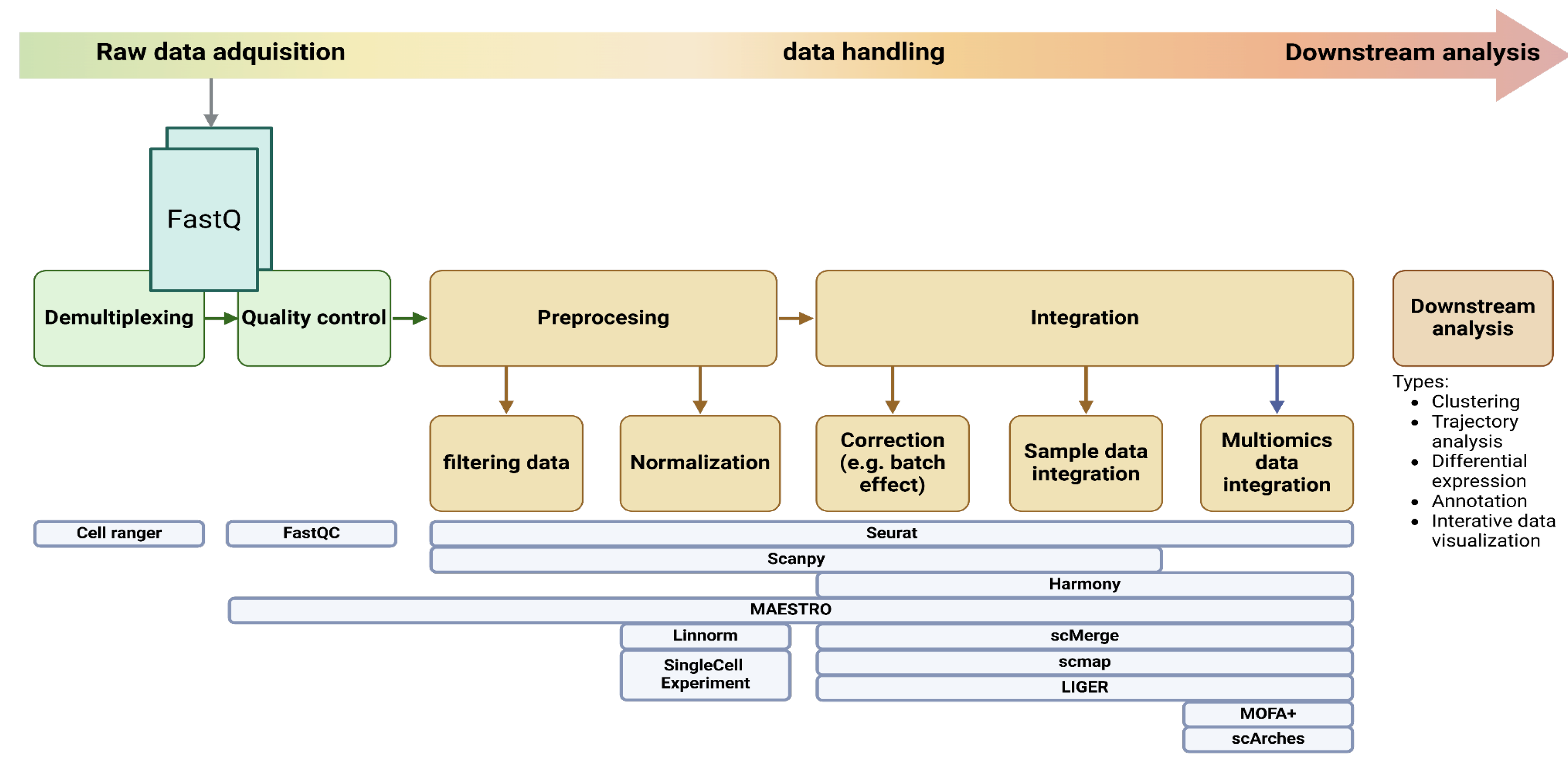
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



