
Submitted:
01 November 2024
Posted:
04 November 2024
You are already at the latest version
Abstract
Two novel lipids, acylglycerol and glycerophospholipid containing succinate-linked provitamin D3 at sn-2 position of glycerol backbone, have been synthesized from dihydroxyacetone (DHA) and sn-glycerophosphocholine (GPC), respectively. Three-step synthesis of 1,3-dipalmitoyl-2-(7-dehydrocholesterylsuccinoyl)glycerol involved esterification of DHA with palmitic acid, reduction of carbonyl group and conjugation of resulting 1,3-dipalmitoylglycerol with 7-dehydrocholesterol hemisuccinate (7-DHC HS). Using NaBH3CN as a reducing agent was crucial to avoid any acyl migration and obtain the final product with 100% regioisomeric purity. 1-Palmitoyl-2-(7-dehydrocholesterylsuccinoyl)-sn-glycero-3-phosphocholine was produced in two-step process by the esterification of GPC at sn-1 position with palmitic acid and subsequent conjugation of 1-palmitoyl-sn-glycero-3-phosphocholine with 7-DHC HS. Apart from the main product, some amount of its regioisomer containing provitamin D3 linked at sn-1 position and palmitic acid at sn-2 position was detected which indicated the acyl migration of palmitic acid from sn-1 to sn-2 position in the intermediate 1-palmitoyl-sn-glycerophosphocholine. Synthesized novel lipids were fully characterized by spectroscopic data. They can find application as novel lipid-based prodrugs as the additives to sunscreen creams, providing controlled release of provitamin D3 in the epidermis.
Keywords:
lipid prodrugs
; 7-dehydrocholesterol
; modified acylglycerols
; modified phospholipids
; succinate linker
; acyl migration
1. Introduction
Research indicates the importance of vitamin D not only in rickets [1] and musculoskeletal disorders [2] but also in affecting immune responses [3,4] as well as in anticancer processes [5,6]. Its deficiency is associated with serious health consequences, connected with diabetes [7], cardiovascular [8], autoimmune [9] and cancer diseases [10]. The basic source of vitamin D3 for humans is a production in the skin under the influence of ultraviolet radiation (UVB, 280-310 nm), which results in the isomerization of provitamin D3 (7-dehydrocholesterol, 7-DHC) to previtamin D3, which is further thermally isomerized to vitamin D3 [11].
Vitamin D deficiency is becoming more and more common due to low exposure to the sun in winter, as well as the use of sunscreen creams protecting the skin against UV radiation, which reduces the endogenous production of this compound. For that reason, there is a growing interests in the production of novel delivery systems of vitamin D for oral administration, food fortification or transdermal delivery [12].
As it was shown in the investigations on an animal model, over 80% of provitamin D3 and vitamin D3 are present in the skin in the storage form of esters with fatty acids, which are gradually hydrolyzed in epidermis in the presence of the transporting protein DBP. This protein binds the released vitamin D3 molecule in the blood serum and transports it to the liver, where it is hydroxylated at the C-25 carbon atom [13]. The described mechanism of controlled release of the free vitamin slows down its metabolism, limits its toxicity by controlling the concentration in the body, and through its successive transformation into the active 25-hydroxy derivative prevents vitamin D deficiency, also in the periods of low availability of sunlight.
Taking into account these findings, the aim of our research is to design and synthesize 7-DHC esterified with acylglycerols and phospholipids, which could serve as a storage form of esterified provitamin D3 in the skin. Such lipid-based prodrugs of 7-DHC prepared in the form of liposome formulations can be potentially used as the additives to sunscreen creams, providing controlled release of provitamin D3 in the epidermis. In this paper we present the synthesis and structural characterization of two 7-DHC derivatives: acylglycerol-based prodrug in which 7-DHC is linked through the sn-2 position of glycerol backbone and its glycerophospholipid-based analogue.
Production of symmetrical glyceride prodrugs is a common strategy to produce triglyceride-based prodrugs. Following this approach there were synthesized several acylglycerols containing the bioactive molecules at sn-2 position e.g., non-steroidal anti-inflammatory drugs (NSAIDs) [14,15,16], anti-HIV agent didanosine [17], antineoplastic agent chlorambucil [18], antibacterial drug norfloxacin [19] or testosterone [20]. Also, conjugation of bioactive molecule at sn-2 position is applied to obtain phospholipid-based prodrugs, i.a. containing NSAIDs like indomethacine [21] ibuprofen or naproxen [22], anticonvulsant valproic acid [23] or antibiotic fumagillin [24].
2. Results and Discussion
Our strategy for the design of target compounds involved the synthesis of 1,3-dipalmitoylglycerol and 1-palmitoyl-sn-glycero-3-phosphocholine followed by their conjugation with 7-dehydrocholesterol at sn-2 position. We decided to attach 7-DHC residue through succinate linker as it was successfully applied in our laboratory to the synthesis of acylglycerols containing stigmasterol [25,26,27]. Conjugation of bioactive molecule via succinate linker to obtain different sterol-modified phospholipid was reported earlier for modified phospholipids containing cholesterol [28], stigmasterol [29] or dehydroepiandrosterone (DHEA) [30] residues.
2.1. Synthesis of 7-Dehydrocholesterol Hemisuccinate
7-Dehydrocholesterol hemisuccinate (7-DHC HS) was obtained in the reaction of 7-dehydrocholesterol with succinic anhydride in anhydrous pyridine in the presence of DMAP. The procedure was generally in accordance to the one described earlier for stigmasterol hemisuccinate [25] but the purification step required modification. As the crystallization from methanol afforded product in only 32% yield, flash chromatography was used instead to significantly improve the yield to 76%. Tian et al. [31] reported synthesis of 7-DHC HS but no details of synthetic procedure were given and 1H NMR description was also incomplete. Therefore in this paper we decided to give full spectroscopic data including NMR, IR and HRMS (Section 4.3 in Materials and Methods).
2.2. Synthesis of 1,3-Dipalmitoyl-2-(7-dehydrocholestyrylsuccinoyl)glycerol
In most of the synthetic pathways leading to the modified symmetrical glycerides, including those developed in our research team for the synthesis of stigmasterol-modified acylglycerols [26,27], the starting material was dihydroxyacetone (DHA). Generally, it was esterified by the fatty acid, reduced to 1,3-diacylglycerol and conjugated with the appropiate drug. The similar procedure was applied for the synthesis of acylglycerol with 7-DHC residue described here (Scheme 1).
Contrary to the previous reports where DHA (1) was esterified with palmitoyl chloride in anhydrous pyridine [14,32], in our investigation we used palmitic acid in the presence of DCC and DMAP to obtain 1,3-dipalmitoyloxypropan-2-one (2) in 67% yield after purification by flash chromatography. This Steglich procedure we successfully applied earlier for the synthesis of 1,3-dimirystoyloxypropan-2-one [26] and 1,3-dioleoyloxypropan-2-one [27]. Detailed spectroscopic analysis (IR, NMR, HRMS) confirmed the structure of the obtained diester 2. Searching for the literature reports we did not find any spectroscopic data of 1,3-dipalmitoyloxypropan-2-one (2) therefore we decided to report them in this paper (Section 4.4.1 in Materials and methods).
The next step required the reduction of carbonyl group of 2 to obtain 1,3-dipalmitoylglycerol. Using sodium borohydride as the reducing agent, apart from the desired 1,3-diacylglycerols, we observed the formation of their 1,2-regioisomers as a result of migration of acyl group from sn-1 to sn-2 position. Similar phenomenon was observed earlier by Cockman et al. [33] as well as in our previous investigations during the reduction of 1,3-diacyloxypropan-2-ones [26,27]. To avoid the neccessity of regioisomeric diacylglycerols separation by flash chromatography and improve the yield of desired 1,3-isomer, we carried out the reduction of 2 with sodium cyanoborohydride in tetrahydrofuran at pH 4 following the procedure proposed by Cockman et al. [33]. Both TLC analysis of the reaction mixture and NMR analysis showed no acyl migration and 1,3-dipalmitoylglycerol was isolated as the only product in 86% yield without time-consuming chromatographic purification. The spectroscopic data of 2 were in fully accordance with those published earlier [27].
In the final step, 1,3-dipalmitoylglycerol (3) was esterified with 7-DHC HS in the typical Steglich procedure. After flash chromatography pure 1,3-dipalmitoyl-2-(7-dehydrocholesterylsuccinoyl)glycerol (4) was obtained in 78% yield.
2.3. Synthesis of 1-Palmitoyl-2-(7-dehydrocholesterylsuccinoyl)-sn-glycero-3-phosphocholine (7)
A two-step process was applied to obtain 1-palmitoyl-2-(7-dehydrocholesterylsuccinoyl)-sn-glycero-3-phosphocholine 7 (Scheme 2).
The first step involved the preparation of 1-palmitoyl-sn-glycero-3-phosphocholine (1-PA GPC), following the method previously described by Niezgoda [34]. In this process, sn-glycerophosphocholine (GPC) was first transformed into a cyclic acetal intermediate and then selectively acylated using palmitoyl chloride in the presence of triethylamine (TEA) to obtain the final product. The resulting 1-PA GPC was purified by flash chromatography and after drying procedure residual moisture was removed to afford final product in 71% yield.
The second step involved the Steglich esterification of 1-PA GPC with 7-DHC HS at 4°C to form the final product. Despite using relatively high molar ratios of DCC and DMAP in relation to 1-PA-GPC and long reaction time (48h), TLC analysis indicated only partial conversion of the substrate. Additional one equivalent of DCC and prolonged reaction time gave the product in an unsatisfactory 16% yield.
Diaryl carbodiimides, such as DCC, are prone to hydrolysis in the presence of water, which reduces the overall efficiency of the esterification [35]. Therefore we concluded that the low yield might have been caused by insufficient drying of the substrates. Therefore, in the next trial the 1-PA GPC and 7-DHC HS were rigorously dried by adding different anhydrous solvents, including acetone, ethanol and dichloromethane and evaporating the azeotropes using a rotary evaporator. This procedure was repeated several times and such prepared anhydrous substrates were subjected to Steglich esterification which significantly increased the isolated yield of phospholipid 7 to 51%.
2.4. Spectroscopic Identification of the Final Products 4 and 7
HRMS analysis of both compounds confirmed their molecular masses. The detailed NMR measurements, using 2D spectroscopy (correlation COSY, HMQC and HMBC), let us to identify the signals from glycerol skeleton, residue of 7-DHC and palmitic acid, succinyl linker and for the phospholipid 7 also the phosphocholine part of the molecule. All the spectra of compounds 4 and 7 are included in Supplementary Materials, and full spectroscopic data are given in Materials and Methods, Section 4.4.3 and 4.5.2.
On 1H NMR spectra the presence of palmitic acid residue was confirmed by the characterictic signals coming from one (phospholipid 7) or two (acylglycerol 4) chains. Among them, the particularly visible were two triplets, one from terminal methyl group at 0.87 ppm and one from methylene protons at the α carbons at 2.31 ppm (acylglycerol 4) or 2.27 ppm (phospholipid 7). On the spectra of both products multiplets of CH2 group at β position were observed in the range of 1.54 – 1.60 ppm and the signals from other CH2-protons of acyl chains were overlapped multiplets in the range of 1.18 – 1.31 ppm. On 13C NMR the most characteristic signals from palmitic acid residue were those of carbonyl carbons from ester group at 173.31 ppm (acylglycerol 4) or 173.60 ppm (phospholipid 7), carbons α and β (34.00 and 24.83 ppm for acylglycerol 4 and 34.05 and 24.86 ppm for phospholipid 7) as well as ω-2, ω-1 and ω carbons (31.91, 22.68, and 14.10 respectively for acylglycerol 4 and and analogously 31.93, 22.69, 14.12 ppm for phospholipid 7).
As the chemical shifts of signals from sterol part of the acylglycerol 4 observed on 13C NMR spectra were in fully accordance with those reported by Wilson et al. for 7-dehydrocholesterol acetate [36], we could uneqivocally assign them to the corresponding carbons. Consequently, using data from HMQC and HMBC spectra, we could also identify the signals from particular protons on the 1H NMR spectra. Among them the most distinctive was triplet of triplets from H-3 at 4.71 ppm and signals from olefinic protons: doublet of triplets at 5.38 ppm from H-7 and doublet of doublets at 5.56 ppm from H-6. Separate signals were also observed for both protons from CH2-4 group (2.36 and 2.49 ppm) H-12β (2.08 ppm), H-9 (1.98 ppm), H-25 (1.52 ppm) and H-22 (1.01 ppm). Moreover, we found singlets from methyl groups at C-18 and C-19 (0.61 and 0.94 ppm respectively) as well as doublets from methyl groups at C-26 and C-27 (0.86 and 0.87 ppm), and C-21 (0.93 ppm). The signals from remaining protons of sterol part were superimposed but using 2D spectroscopy analysis their location in particular multiplets was possible and consistent with the data obtained by Wilson for 7-dehydrocholesterol acetate [36]. The analogous assignment was made for the carbons and protons of sterol residue on the spectra of phospholipid 7.
In the case of both synthesized lipids the attachment of 7-DHC by succinate linker was confirmed by multiplets of two methylene groups in the range of 2.55 – 2.66 ppm on 1H NMR spectra and signals of carbonyl atoms on 13C NMR spectra, at 171.37 and 171.49 ppm for acylglycerol 4 as well as 171.64 an 171.89 ppm for phospholipid 7.
The presence of phosphoric acid residue in phospholipid 7 was confirmed by the signal at – 0.95 ppm on 31P NMR spectrum and the choline residue was identified by the singlet from three methyl group attached to the nitrogen atom at 3.33 ppm and two broad singlets from methylene groups CH2-α and CH2-β at 4.28 ppm and 3.76 ppm, respectively on 1H NMR spectrum. Apart from singlet at 54.33 ppm from the carbons of three methyl groups, 13C spectroscopy showed also signals from corresponding carbons of choline fragment, represented by characteristic doublets with 13C-31P coupling constants 2J=3.3 Hz for C-α atom and 3J=6.1 Hz for C-β.
Valuable information confirming the symmetrical structure of 4 were provided by the analysis of signals from the atoms of glycerol backbone. On 1H NMR spectrum, two doublets of doublets were found at 4.15 and 4.25 ppm (Figure 1A), each representing two magnetically equivalent protons: one from CH2 group at sn-1 and one from CH2 group at sn-3. On the 13C NMR spectra (Figure 1B) only one signal from carbons at sn-1 and sn-3 positions was found (61.93 ppm). Likewise, carbonyl atoms of palmitic acyl residues at sn-1 and sn-3 were also represented by only one signal at 173.31 ppm. All these data fully confirmed the presence of 7-DHC residue at sn-2 position and lack of any acyl migration during the reaction. The similar observations were done in our previous paper for the confirmation of symmetrical structure of 1,3-diacyl-2-stigmasterylsuccinoylglycerols [26,27].
In the case of phospholipid 7 analysis of the spectrum range characteristic for protons of glycerol skeleton as well as for carbonyl carbons of ester moieties was also very informative. On 1H NMR spectra of phospholipid 7 (Figure 2A) protons from CH2 group at sn-3 resonated in the range of 3.92-3.98 ppm whereas those from CH2 group at sn-1 were observed as two separate doublets of doublets at 4.14 ppm and 4.34 ppm. The multiplet from sn-2 proton was significantly shifted downfield to 5.20 ppm. The carbon-phosphorus coupling gave characteristic doublets on the 13C NMR spectrum for Csn-2 at 71.16 ppm (3J = 7.4 Hz) and Csn-3 at 63.53 ppm (2J = 4.0 Hz).
Careful spectral analysis of phospholipid 7 let us also identify some minor signals in the area of resonance of glycerol protons (Figure 2A). The most separated and visible was doublet of doublets at 4.40 ppm, shifted downfield in a relation to major signal from one of the CH2-1 protons by 0.06 ppm. On the other hand, minor signal at 5.17 ppm was slightly shifted towards higher field by 0.03 ppm compared with major signal from H-2. Another minor signals overlapped less or more with their neighbouring major signals. Similarly, on 13C NMR, apart from major signals of carbonyl atoms three minor signals were also observed in this area (Figure 2B).
The above-mentioned minor signals on NMR spectra let us to the assumption that they must have come from the corresponding protons of regioisomeric phospholipid, containing 7-DHC residue attached to sn-1 position and palmitic acid linked to sn-2 position. To support our hypothesis we compared the chemical shifts of minor and major signals detected on the spectra of phospholipid 7 with the chemical shifts of the corresponding signals reported for their analogs containing dehydroepiandrosterone (DHEA). (Table 1). We take into consideration two regiosomeric glycerophospholipids containing DHEA at sn-2 and oleic acid at sn-1 (1-OA-2-DHEA PChol) as well as DHEA at sn-1 and oleic acid at sn-2 (1-DHEA-2-OA PChol), synthesized by Kłobucki et al. [30].
One can see that comparing to its regiosomer, in the spectra of glycerophospholipid containing fatty acid residue at sn-2 position and sterol molecule at sn-1 position (1-DHEA-2-OL-PChol), one of CH2-1 protons resonates at lower field (4.39 ppm versus 4.36 ppm) and proton H-2 at higher field (5.18 ppm versus 5.20 ppm). On the other hand, compared to 1-sterol glycerophospholipid (1-DHEA-2-OL-PChol), in the spectra of regiosomer with sterol residue at sn-2 position (1-OA-2-DHEA-PChol) the difference in chemical shifts between signals from carbonyl carbons of succinate linker is significantly lower (Δδ = 0.06 versus 0.4) and signal from carbon atom of fatty acid residue is significantly shifted upfield (173.61 versus 173.16). Similar differences in chemical shifts between minor and major signals observed on the analyzed spectra of phospholipid 7 confirmed that apart from expected product (major signals), some amount of 2-palmitoyl-1-(7-dehydrocholesterylsuccinoyl)-sn-glycero-3-phosphocholine (minor signals) was formed.
Formation of this regiosomer can be explained by the partial transformation of intermediate 1-palmitoyl-sn-glycero-3-phosphocholine (1-PA GPC) (6) to 2-palmitoyl-sn-glycero-3-phosphocholine (2-PA GPC) through acyl migration of palmitic acid residue and the subsequent Steglich esterification of 2-PA GPC. Acyl migration in lysophospholipids between sn-1 and sn-2 positions is a common phenomenon observed during chemical and enzymatic modifications of phospholipids [34,37,38,39,40] which leads to a regioisomeric mixtures of products, reducing the yield and purity of final glycerophospholipids. This isomerization is is favoured by i.a. alkaline pH, temperature and solvent polarity [41]. Kiełbowicz et al. [42] observed the acyl migration in lysophospholipids containining palmitic acid in different organic solvents, including isopropanol and dichloromethane used in the synthesis described here. The migration can be also promoted by silica gel used in the chromatographic purification [43]. Sugasini and Subbaiah studied the effect of acyl group on the migration and found that saturated fatty acids, like palmitic acid, are more suspectible for migration than polyunsaturated fatty acids [44].
4. Materials and Methods
4.1. Chemicals
Dihydroxyacetone (≥98%), palmitic acid (≥99%), 7-dehydrocholesterol (≥95%), 4-(dimethylamino)pyridine (DMP, ≥99%), N,N’-dicyclohexylcarbodiimide (DCC, 99%), sodium cyanoborohydride, dibutyltin(IV) oxide (DBTO, 98%), triethylamine (TEA, ≥99,5%), palmitoyl chloride (98%), DOWEX® 50WX8 hydrogen form, ethanol-free chloroform (≥99%), anhydrous tetrahydrofurane (THF, ≥99.9%), anhydrous dichloromethane (DCM, ≥99.8%), anhydrous pyridine (99.8%), and hexane (ACS reagent, ≥99%) were purchased from Merck (Darmstadt, Germany). sn-Glycero-3-phosphocholine (GPC) was purchased from Bachem AG (Bubendorf, Switzerland). Acetic acid (99.5-99.9%) and isopropanol were purchased from POCH (Gliwice, Poland). Anhydrous isopropanol was prepared by dissolving sodium (8g/L) in alcohol dried previously by distillation from CaSO4. Subsequently, the solution was distilled once again and fresh distillate was kept with MS 3A molecular sieves beads for 96h to obtain isopropanol containing only 9 ppm of water. Other chemicals of analytical grade were purchased from Chempur (Piekary Śląskie, Poland).
4.2. Analytical Methods
The progress of reactions was reviewed by thin layer chromatography (TLC) using 0.2 mm aluminum plates coated with silica gel 60 F254 (Merck, Darmstadt, Germany). Chromatograms were examined by spraying the plates with a solution of 1% Ce(SO4)2 and 2% H3[P(Mo3O10)4] in 10% H2SO4 and heating the plates to 120 – 200 ◦C or 0.05% solution of primuline in acetone:water mixture (4:1, v/v) which made the spots visible under UV light (λ = 365 nm).
Reaction products were purified by flash chromatography was carried out using puriFlash® SX520 Plus Interchim system (Interchim, Montluçon, France) gradient pump, UV detector, and fraction collector. The samples were dry loaded on a pre-column puriFlash® F0012 and the products were separated on puriFlash® SIHP 30 μm columns using gradient elution with solvent mixtures as indicated in the respective method sections (flow 26 mL/min, pressure 15 mbar).
Nuclear Magnetic Resonance spectra (1H NMR, 13C NMR, COSY, HMQC, HMBC) were recorded on Bruker Avance II 600 MHz spectrometer (Bruker, Rheinstetten, Germany) or Jeol 400 MHz Year Hold Magnet spectrometer (Jeol Ltd., Tokyo, Japan). Chemical shifts were correlated to the residual solvent signal (CDCl3, δH = 7.26, δC = 77.00). Infrared spectroscopy (IR) was carried out on Nicolet iS10 FTIR Spectrometer (Thermo Scientific™, Waltham, MA, USA) equipped with monolithic diamond ATR crystal attachment. High Resolution Mass Spectra (HRMS) were recorded on Bruker Daltonics ESI-Q-TOF maXis impact mass spectrometer (Bruker, Billerica, MA, USA) or Waters Xevo G2 mass spectrometer (Waters, Milford, MA, USA) using positive electrospray ionization (ESI) techniques.
4.3. Preparation of 7-Dehydrocholesteryl Hemisuccinate (7-DHC HS)
7-Dehydrocholesterol (1.5 g, 3.9 mmol), succinic anhydride (1.37 g, 13.7 mmol) and DMAP (0.45 g, 3.9 mmol) were dissolved in 40 mL of anhydrous pyridine. The reaction was carried out on a magnetic stirrer (Heidolph, Schwabach, Germany) at 60 °C in a thermostated glycerol bath under reflux condenser for 24 h. When the substrate reacted completely, (TLC, chloroform/methanol/acetic acid 95:5:0.1) the reaction mixture was acidified with 1 M HCl to pH 2. The product was extracted with DCM and the organic layer was washed with brine until neutralization and dried over anhydrous MgSO4. After filtration and solvent evaporation under vacuum, the crude product was purified by flash chromatography using a gradient elution system (from hexane:AcMe:AcOH 100:0:0 to hexane:AcMe:AcOH, 4:1:0.01, v/v/v) to afford pure 7-dehydrocholesteryl hemisuccinate (1.14 g, yield 76%) with following physical and spectroscopic data:
White crystals, mp 155-160 °C, Rf = 0.18 (hexane:AcMe:AcOH, 4:1:0.01); 1H NMR (600 MHz, CDCl3) δ: 0.61 (s, 3H, CH3-18), 0.86 and 0.87 (two d, J=6.6 Hz, 6H, CH3-26 and CH3-27), 0.93 (d, J=6.7 Hz, 3H, CH3-21), 0.94 (s, 3H, CH3-19), 1.02 (m, 1H, one of CH2-22), 1.07-1.17 (m, 3H, CH2-24 and one of CH2-23), 1.18-1.27 (m, 2H, H-17 and H-12α), 1.28-1.42 (m, 6H, H-16β, one of CH2-23, one of CH2-22, H-1α, H-15β and H-20), 1.52 (m, 1H, H-25), 1.55-1.61 (m, 2H, H-2β and H-11α), 1.66-1.74 (m, 2H, H-15α and H-11β), 1.85-1.99 (m, 4H, H-14α, H-1β, H-2α and H-16α), 1.98 (m, 1H, H-9), 2.08 (m, 1H, H-12β), 2.36 (m, 1H, H-4β), 2.49 (ddd, J=14.5, 4.8 and 2.2 Hz, 1H, H-4α), 2.61 and 2.68 (two m, 4H, –O(O)C–CH2–CH2–COOH), 4.72 (m, 1H, H-3), 5.38 (dt, J=5.6 and 2.5 Hz, 1H, H-7), 5.56 dd, J=5.6 and 2.2 Hz, 1H, H-6); 13C NMR (150 MHz, CDCl3) δ: 11.82 (C-18), 16.16 (C-19) 18.83 (C-21), 21.01 (C-11), 22.54 and 22.80 (C-26, C-27), 23.00 (C-15), 23.87 (C-23), 27.99 (C-25), 28.07 (C-2, C-16), 29.00 and 29.22 (–O(O)C–CH2–CH2–COOH), 36.10 (C-22), 36.14 (C-20), 36.52 (C-4), 37.04 (C-10), 37.86 (C-1), 39.12 (C-12), 39.48 (C-24), 42.88 (C-13), 46.00 (C-9), 54.45 (C-14), 55.87 (C-17), 73.35 (C-3), 116.27 (C-7), 120.27 (C-6), 138.35 (C-5), 141.60 (C-8), 171.58 (–O(O)C–CH2–CH2–COOH), 177.93 –O(O)C–CH2–CH2–COOH); IR (ATR): νmax =2953, 1732, 1709, 1174, 1466 cm-1; HRMS (ESI): m/z calcd for C31H48O4: 507.3445 [M+Na]+; found: 507.3447.
4.4. Synthesis of Target Acylglycerol with 7-Dehydrocholesterol Residue
4.4.1. Preparation of 1,3-Dipalmitoyloxypropan-2-one (2)
Dihydroxyacetone (0.5 g, 5.6 mmol), N,N′-dicyclohexylcarbodiimide (DCC, 2.66 g, 12.88 mmol) and 4-(dimethylamino) pyridine (DMAP, 1.57 g, 12.88 mmol) were dissolved in ethanol-free chloroform. Palmitic acid (3.35 g, 13.07 mmol) in ethanol-free chloroform was added dropwise to the reaction mixture to reach the total volume of 100 mL. After 24 h of stirring at room temperature the total substrate conversion was observed (TLC: hexane/ethyl acetate 5:1, visualization by primuline test). White precipitate was removed by filtration through a funnel with a G4 sintered disc. Next, the filtrate was washed with 0.5 M HCl, saturated NaCl solution (until neutral), dried with anhydrous MgSO4, filtered, and concentrated in vacuo. Purified product (2.13 g, 67% yield) was obtained by flash chromatography using a gradient elution method (from hexane:DCM 100:0 to hexane:DCM 35:65). Its physical and spectroscopic data are given below:
White crystals, mp 73-78 °C, lit. 77-78°C [45] Rf = 0.45 (hexane: dichloromethane, 1:2), 1H NMR (400 MHz, CDCl3) δ: 0.88 (t, J=6.9 Hz, 6H, 2 × -OC(O)(CH2)14CH3), 1.20–1.38 (m, 48H, 2 × -OC(O)CH2CH2(CH2)12CH3), 1.61-1.70 (m, 4H, 2 × -OC(O)CH2CH2(CH2)12CH3), 2.42 (t, J=7.5 Hz, 4H, 2 × -OC(O)CH2(CH2)13CH3), 4.74 (s, 4H, CH2-1 and CH2-3); 13C NMR (100 MHz, CDCl3) δ: 14.11 (2 × -OC(O)(CH2)14CH3), 22.68 (2 × -OC(O)(CH2)13CH2CH3), 24.79 (2 × -OC(O)CH2CH2(CH2)12CH3), 29.05, 29.22, 29.35, 29.42, 29.58, 29.64, 29.67 (2 × -OC(O)CH2CH2(CH2)10CH2CH2CH3), 31.91 (2 × -OC(O)(CH2)10CH2CH2CH3), 33.72 (2 × -OC(O)CH2(CH2)11CH3), 66.13 (C-1, C-3), 172.93 (2 × -OC(O)(CH2)14CH3), 198.15 (C-2); IR (ATR): νmax =2916, 2850, 1746, 1732, 1709, 1175, 719 cm-1; HRMS (ESI): m/z calcd for C35H66O5: 567.4988 [M+H]+; found: 567.4982.
4.4.2. Preparation of 1,3-Dipalmitoylglycerol (3)
NaBH3CN (0.083 g, 1.3 mmol) was added portion wise to the stirring solution of 1,3-dipalmitoyloxypropan-2-one (2) (0.5 g, 0.88 mmol) in THF (40 mL). Subsequently, few mL of glacial acetic acid was added until the reaction mixture was acidified to pH 4. After 40 minutes, when no substrate was observed by TLC (hexane/ethyl acetate 5:1, primuline), the solvent was evaporated in vacuo, the residue was suspended in water and the product was extracted by dichloromethane. After standard work-up of the organic layer (washing with brine, drying with anhydrous MgSO4 and solvent evaporation), 0.43 g (86% yield) of pure 1,3-dipalmitoylglycerol were obtained.
4.4.3. Preparation of 1,3-Dipalmitoyl-2-(7-dehydrocholesterylsuccinoyl)glycerol (4)
7-DHC HS (0.192 g, 0.40 mmol) dissolved in ethanol-free chloroform was added in portions to the stirring mixture of 1,3-dipalmitoylglycerol (3) (0.15 g, 0.26 mmol), DCC (0.083 g, 0.40 mmol) and DMAP (0.053 g, 0.43 mmol) in the same solvent (total volume 20 mL). The solution was stirred at room temperature for 24 h and after total substrate consumption (TLC, hexane/ethyl acetate 5:1), the reaction was worked up as described for 1,3-dipalmitoylpropanone. Pure product (0.211 g, yield 78%) was obtained by flash chromatography using a gradient elution system (from hexane:DCM 100:0 to hexane:DCM 20:80). Its physical and spectroscopic data are as follows:
Waxy solid, Rf = 0.55 (hexane:ethyl acetate, 5:1); 1H NMR (600 MHz, CDCl3) δ: 0.61 (s, 3H, CH3-18), 0.86 and 0.87 (two d, J=6.6 Hz, 6H, CH3-26 and CH3-27), 0.87 (t, J=6.9 Hz, 6H, 2 × -OC(O)(CH2)14CH3), 0.93 (d, J=6.9 Hz, 3H, CH3-21), 0.94 (s, 3H, CH3-19), 1.00 (m, 1H, one of CH2-22), 1.06-1.17 (m, 3H, CH2-24 and one of CH2-23), 1.18-1.31 (m, 50H, H-17 and H-12α and 2 × -OC(O)CH2CH2(CH2)12CH3), 1.31-1.42 (m, 6H, H-16β, one of CH2-23, one of CH2-22, H-1α, H-15β and H-20), 1.52 (m, 1H, H-25), 1.55-1.64 (m, 6H, H-2β, H-11α and 2 × -OC(O)CH2CH2(CH2)12CH3), 1.66-1.74 (m, 2H, H-15α and H-11β), 1.85-1.95 (m, 4H, H-14α, H-1β, H-2α and H-16α), 1.98 (m, 1H, H-9), 2.08 (ddd, J=12.5, 4.3 and 2.5 Hz, 1H, H-12β), 2.31 (t, J=7.6 Hz, 4H, 2 × -OC(O)CH2(CH2)13CH3), 2.36 (m, 1H, H-4β), 2.49 (ddd, J=14.5, 4.5 and 2.3 Hz, 1H, H-4α), 2.58–2.66 (m, 4H, -OC(O)CH2CH2(O)CO-DChol), 4.15 (dd, J=11.9 and 5.9 Hz, 2H, one of CH2 at sn-1 and one of CH2 at sn-3), 4.29 (dd, J=11.9 and 3.8 Hz, 2H, one of CH2 at sn-1 and one of CH2 at sn-3), 4.71 (tt, J=11.5 and 4.5 Hz, 1H, H-3), 5.27 (tt, J=5.9 and 3.8 Hz, 1H, Hsn-2), 5.38 (dt, J=5.6 and 2.6 Hz, 1H, H-7), 5.56 (dd, J=5.6 and 2.3 Hz, 1H, H-6); 13C NMR (150 MHz, CDCl3) δ: 11.80 (C-18), 14.10 (2 × -OC(O)(CH2)14CH3), 16.14 (C-19) 18.82 (C-21), 21.00 (C-11), 22.53 and 22.80 (C-26, C-27), 22.68 (2 × -OC(O)(CH2)13CH2CH3), 22.99 (C-15), 23.87 (C-23), 24.83 (2 × -OC(O)CH2CH2(CH2)12CH3), 27.99 (C-25), 28.06 (C-2, C-16), 29.10, 29.12, 29.28, 29.36, 29.49, 29.63, 29.66 and 29.70 (-OC(O)CH2CH2(O)CO-DChol, 2 × -OC(O)CH2CH2(CH2)10CH2CH2CH3), 31.91 (2 × -OC(O)(CH2)12CH2CH2CH3), 34.00 (2 × -OC(O)CH2(CH2)13CH3), 36.09 (C-22), 36.13 (C-20), 36.57 (C-4), 37.04 (C-10), 37.88 (C-1), 39.11 (C-12), 39.48 (C-24), 42.87 (C-13), 46.01 (C-9), 54.44 (C-14), 55.86 (C-17), 61.93 (Csn-1 and Csn-3), 69.46 (Csn-2), 73.23 (C-3), 116.26 (C-7), 120.30 (C-6), 138.29 (C-5), 141.59 (C-8), 171.37 and 171.49 (-OC(O)CH2CH2(O)CO-Stig), 173.31 (2 × -OC(O)(CH2)14CH3); IR (ATR): νmax = 2919, 2850, 1735, 1468, 1155 cm-1; HRMS (ESI): m/z calcd for C66H114O8: 1057.8406 [M+Na]+; found: 1057.8398
4.5. Synthesis of Glycerophosholipid with 7-Dehydrocholesterol Residue
4.5.1. Preparation of 1-Palmitoyl-sn-glycero-3-phosphocholine (6)
The synthesis of 1-palmitoyl-sn-glycero-3-phosphocholine was carried out according to the procedure described by Niezgoda et al. [34]. sn-Glycero-3-phosphocholine (5, 0.95 g, 3.7 mmol) was dried by repeated (3 x) evaporation with an anhydrous CH2Cl2 and then suspended with DBTO (0.92 g, 3.7 mmol) in anhydrous isopropanol (15 mL) and refluxed for 1 h. The reaction was cooled to the room temperature and TEA (5.5 mmol) was added followed by palmitoyl chloride (1.52 g, 5.5 mmol). After 0.5 h of the reaction, the organic solvent was evaporated from the reaction mixture under vacuum. The crude residue was purified by flash chromatography using a gradient elution system (from DCM:MeOH:H2O 95:5:0 to DCM:MeOH:H2O 65:30:5, v/v/v) and dried by the evaporation from anhydrous ethanol followed by keeping in vacuum desiccator to obtain 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (1.29 g, yield 72%).
Physical data: white solid, Rf = 0.22 (DCM:MeOH:H2O 65:30:5), spectroscopic data consistent with literature data [43].
4.5.2. Preparation of 1-Palmitoyl-2-(7-dehydrocholesterylsuccinoyl)-sn-glycero-3-phosphocholine (7)
To the stirring mixture of rigorously dried 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (6, 0.15 g, 0.3 mmol), 7-DHC HS (0.308 g, 0.64 mmol) and DMAP (0.064 g, 0.3 mmol) dissolved in 9 mL of anhydrous CH2Cl2, a solution of DCC (0.081 g, 0.67 mmol) in 3 mL of CH2Cl2 was added. The reaction was carried out for 96 h at 4°C. The resulting precipitate was filtered off using a Shott funnel with G4 sintered disc and a Dowex® 50WX8 H+ form was added to the reaction mixture. After 30 min of stirring, the resin was filtered off and the solvent was evaporated under vacuum. The crude product was purified by flash chromatography using a gradient elution system (from DCM:MeOH:H2O 95:5:0 to DCM:MeOH:H2O 65:30:5, v/v/v) to afford pure 1-palmitoyl-2-(7-dehydrocholesteryl)succinoyl-sn-glycero-3-phosphocholine (0.147 g, yield 51%) with following physical and spectroscopic data:
Waxy white solid, Rf = 0.51 (DCM:MeOH:H2O 65:30:5); 1H NMR (600 MHz, CDCl3) δ: 0.61 (s, 3H, CH3-18), 0.85 and 0.86 (two d, J=6.6 Hz, 6H, CH3-26 and CH3-27), 0.87 (t, J=7.0 Hz, 3H, -OC(O)(CH2)14CH3), 0.93 (d, J=6.3 Hz, 3H, CH3-21), 0.94 (s, 3H, CH3-19),), 1.01 (m, 1H, one of CH2-22), 1.06-1.17 (m, 3H, CH2-24 and one of CH2-23), 1.18-1.31 (m, 26H, H-17 and H-12α and -OC(O)CH2CH2(CH2)12CH3), 1.31-1.41 (m, 6H, H-16β, one of CH2-23, one of CH2-22, H-1α, H-15β and H-20), 1.51 (m, 1H, H-25), 1.54-1.60 (m, 4H, H-2β, H-11α and -OC(O)CH2CH2(CH2)12CH3), 1.64-1.74 (m, 2H, H-15α and H-11β), 1.84-1.93 (m, 4H, H-14α, H-1β, H-2α and H-16α), 1.97 (m, 1H, H-9), 2.08 (m, 1H, H-12β), 2.27 (t, J=7.5 Hz, 2H, -OC(O)CH2(CH2)13CH3), 2.35 (m, 1H, H-4β), 2.47 (m, 1H, H-4α), 2.55–2.66 (m, 4H, -OC(O)CH2CH2(O)CO-DChol), 3.33 (s, 9H, -N(CH3)3), 3.76 (broad s, 2H, CH2-β), 3.92-3.98 (m, 2H, CH2 at sn-3), 4.14 (dd, J= 12.0 and 7.2 Hz, 1H, one of CH2 at sn-1), 4.28 (broad s, 2H, CH2-α), 4.34 (dd, J=12.0 and 2.9 Hz, 1H, one of CH2 at sn-1), 4.67 (tt, J=11.2 and 4.4 Hz, 1H, H-3), 5.20 (m, 1H, Hsn-2), 5.37 (m, 1H, H-7), 5.55 (m, 1H, H-6); 13C NMR (150 MHz, CDCl3) δ: 11.80 (C-18), 14.12 (-OC(O)(CH2)14CH3), 16.13 (C-19), 18.82 (C-21), 21.00 (C-11), 22.54 and 22.80 (C-26, C-27), 22.69 (-OC(O)(CH2)13CH2CH3), 23.01 (C-15), 23.91 (C-23), 24.86 (-OC(O)CH2CH2(CH2)12CH3), 27.99 (C-25), 28.07 (C-2, C-16), 29.13, 29.21, 29.26, 29.39, 29.64, 29.70, 29.77 (-OC(O)CH2CH2(O)CO-DChol, -OC(O)CH2CH2(CH2)10CH2CH2CH3), 31.93 (-OC(O)(CH2)12CH2CH2CH3), 34.05 (-OC(O)CH2(CH2)13CH3), 36.10 (C-22), 36.16 (C-20), 36.60 (C-4), 37.04 (C-10), 37.87 (C-1), 39.11 (C-12), 39.48 (C-24), 42.87 (C-13), 45.99 (C-9), 54.33 (-N(CH3)3, 54.43 (C-14), 55.89 (C-17), 59.32 (d, J=3.3 Hz, C-α), 62.74 (Csn-1), 63.53 (d, J=4.0 Hz, Csn-3), 66.28 (d, J=6.1 Hz, C-β), 71.16 (d, J=7.4 Hz, Csn-2), 73.16 (C-3), 116.27 (C-7), 120.32 (C-6), 138.28 (C-5), 141.57 (C-8), 171.64 (-OC(O)CH2CH2(O)CO-DChol), 171.89 (-OC(O)CH2CH2(O)CO-DChol), 173.60 (-OC(O)(CH2)14CH3); 31P NMR (243 MHz, CDCl3) δ: - 0.95; IR (ATR): νmax = 3388, 2923, 2851, 1733, 1467, 1247, 1161, 1092 cm-1; HRMS (ESI): m/z calcd for C55H96NO10P: 962.6850 [M+H]+; found: 962.6898.
5. Conclusions
In conclusion, two novel lipid derivatives of provitamin D3 have been successfully synthesized and characterized by spectroscopic analysis. Three-step synthesis of acylglycerol containing two palmitic acid residues at sn-1 and sn-3 position and succinate-linked DHC at sn-2 position was efficient. Using NaBH3CN as the reduction agent let to avoid the acyl migration in 1,3-dipalmitoylglycerol and formation of only the desired regioisomer of final acylglycerol 4. For the preparation of glycerophospholipid with the DHC linked to sn-2 position (7) improved synthetic methods should be used to prevent acyl migration, involving the control of reaction pH, limitation of chromatographic separation, or change of the solvent for the synthesis of intermediate 1-PA GPC and its further Steglich esterification.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, W.G.; investigation, W.G., S.O., N.N., A.C., methodology, N.N., S.O.; formal analysis, P.F., P.W.; writing—original draft, W.G., S.O., N.N., A.C.; writing—review and editing, W.G.; supervision, W.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the statutory activities of the Department of Food Chemistry
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bhattarai, H.K.; Shrestha, S.; Rokka, K.; Shakya, R. Vitamin D, Calcium, Parathyroid Hormone, and Sex Steroids in Bone Health and Effects of Aging. J. Osteoporos. 2020, 2020, 9324505. [Google Scholar] [CrossRef] [PubMed]
- Wintermeyer, E.; Ihle, C.; Ehnert, S.; Stöckle, U.; Ochs, G.; de Zwart, P.; Flesch, I.; Bahrs, C.; Nussler, A.K. Crucial role of vitamin D in the musculoskeletal system. Nutrients 2016, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Deluca, H.F.; Cantorna, M.T. Vitamin D: its role and uses in immunology 1. FASEB J. 2001, 15, 2579–2585. [Google Scholar] [CrossRef] [PubMed]
- Ghaseminejad-Raeini, A.; Ghaderi, A.; Sharafi, A.; Nematollahi-Sani, B.; Moossavi, M.; Derakhshani, A.; Sarab, G.A. Immunomodulatory actions of vitamin D in various immune-related disorders: a comprehensive review. Front. Immunol. 2023, 14, 1–16. [Google Scholar] [CrossRef]
- Chiang, K.-C.; C. Chen, T. The Anti-cancer Actions of Vitamin D. Anticancer. Agents Med. Chem. 2012, 13, 126–139. [CrossRef]
- Deeb, K.K.; Trump, D.L.; Johnson, C.S. Vitamin D signalling pathways in cancer: Potential for anticancer therapeutics. Nat. Rev. Cancer 2007, 7, 684–700. [Google Scholar] [CrossRef]
- Harinarayan, C.V. Vitamin D and diabetes mellitus. Hormones 2014, 13, 163–181. [Google Scholar] [CrossRef]
- Wang, T.J.; Pencina, M.J.; Booth, S.L.; Jacques, P.F.; Ingelsson, E.; Lanier, K.; Benjamin, E.J.; D’Agostino, R.B.; Wolf, M.; Vasan, R.S. Vitamin D deficiency and risk of cardiovascular disease. Circulation 2008, 117, 503–511. [Google Scholar] [CrossRef]
- Sîrbe, C.; Rednic, S.; Grama, A.; Pop, T.L. An Update on the Effects of Vitamin D on the Immune System and Autoimmune Diseases. Int. J. Mol. Sci. 2022, 23, 9784. [Google Scholar] [CrossRef]
- Seraphin, G.; Rieger, S.; Hewison, M.; Capobianco, E.; Lisse, T.S. The impact of vitamin D on cancer: A mini review. J. Steroid Biochem. Mol. Biol. 2023, 231, 106308. [Google Scholar] [CrossRef]
- Matthias Wacker and; Michaela F. Holick Sunlight and Vitamin D - A global perspective for health. Dermatoendocrinol. 2013, 5, 211–217. [CrossRef]
- Glowka, E.; Stasiak, J.; Lulek, J. Drug delivery systems for vitamin D supplementation and therapy. Pharmaceutics 2019, 11, 347. [Google Scholar] [CrossRef] [PubMed]
- Duchow, E.G.; Sibilska-Kaminski, I.K.; Plum, L.A.; DeLuca, H.F. Vitamin D esters are the major form of vitamin D produced by UV irradiation in mice. Photochem. Photobiol. Sci. 2022, 21, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Redasani, V.K.; Bari, S.B. Synthesis and evaluation of glyceride prodrugs of naproxen. Open J. Med. Chem. 2013, 3, 87–92. [Google Scholar] [CrossRef]
- Khan, M.S.Y.; Akhter, M. Synthesis, pharmacological activity and hydrolytic behavior of glyceride prodrugs of ibuprofen. Eur. J. Med. Chem. 2005, 40, 371–376. [Google Scholar] [CrossRef]
- Khan, M.S.Y.; Akhter, M. Glyceride derivatives as potential prodrugs: Synthesis, biological activity and kinetic studies of glyceride derivatives of mefenamic acid. Pharmazie 2005, 60, 110–114. [Google Scholar]
- Lalanne, M.; Paci, A.; Andrieux, K.; Dereuddre-Bosquet, N.; Clayette, P.; Deroussent, A.; Ré, M.; Vassal, G.; Couvreur, P.; Desmaële, D. Synthesis and biological evaluation of two glycerolipidic prodrugs of didanosine for direct lymphatic delivery against HIV. Bioorganic Med. Chem. Lett. 2007, 17, 2237–2240. [Google Scholar] [CrossRef]
- Garzon-Aburbeh, A.; Poupaert, J.H.; Claesen, M.; Dumont, P.; Atassi, G. 1,3-Dipalmitoylglycerol Ester of Chlorambucil as a Lymphotropic, Orally Administrable Antineoplastic Agent. J. Med. Chem. 1983, 26, 1200–1203. [Google Scholar] [CrossRef]
- Dhaneshwar, S.; Tewari, K.; Joshi, S.; Godbole, D.; Ghosh, P. Diglyceride prodrug strategy for enhancing the bioavailability of norfloxacin. Chem. Phys. Lipids 2011, 164, 307–313. [Google Scholar] [CrossRef]
- Hu, L.; Quach, T.; Han, S.; Lim, S.F.; Yadav, P.; Senyschyn, D.; Trevaskis, N.L.; Simpson, J.S.; Porter, C.J.H. Glyceride-Mimetic Prodrugs Incorporating Self-Immolative Spacers Promote Lymphatic Transport, Avoid First-Pass Metabolism, and Enhance Oral Bioavailability. Angew. Chemie - Int. Ed. 2016, 55, 13700–13705. [Google Scholar] [CrossRef]
- Dvir, E.; Friedman, J.E.; Lee, J.Y.; Koh, J.Y.; Younis, F.; Raz, S.; Shapiro, I.; Hoffman, A.; Dahan, A.; Rosenberg, G.; et al. A novel phospholipid derivative of indomethacin, DP-155 [mixture of 1-steroyl and 1-palmitoyl-2-{4-[1-(p-chlorobenzoyl)-5-methoxy-2-methyl-3- indolyl acetamido]butanoyl}-sn-glycero-3-phosophatidyl choline], shows superior safety and similar efficacy in re. J. Pharmacol. Exp. Ther. 2006, 318, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Kłobucki, M.; Urbaniak, A.; Grudniewska, A.; Kocbach, B.; Maciejewska, G.; Kiełbowicz, G.; Ugorski, M.; Wawrzeńczyk, C. Syntheses and cytotoxicity of phosphatidylcholines containing ibuprofen or naproxen moieties. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Duvdevani, R.; Shapiro, I.; Elmann, A.; Finkelstein, E.; Hoffman, A. The oral absorption of phospholipid prodrugs: In vivo and in vitro mechanistic investigation of trafficking of a lecithin-valproic acid conjugate following oral administration. J. Control. Release 2008, 126, 1–9. [Google Scholar] [CrossRef]
- Zhou, H.F.; Yan, H.; Senpan, A.; Wickline, S.A.; Pan, D.; Lanza, G.M.; Pham, C.T.N. Suppression of inflammation in a mouse model of rheumatoid arthritis using targeted lipase-labile fumagillin prodrug nanoparticles. Biomaterials 2012, 33, 8632–8640. [Google Scholar] [CrossRef]
- Rudzińska, M.; Grudniewska, A.; Chojnacka, A.; Gładkowski, W.; Maciejewska, G.; Olejnik, A.; Kowalska, K. Distigmasterol-Modified Acylglycerols as New Structured Lipids—Synthesis, Identification and Cytotoxicity. Molecules 2021, 26, 6837. [Google Scholar] [CrossRef]
- Gładkowski, W.; Włoch, A.; Pruchnik, H.; Chojnacka, A.; Grudniewska, A.; Wysota, A.; Dunal, A.; Castro, D.R.; Rudzińska, M. Acylglycerols of Myristic Acid as New Candidates for Effective Stigmasterol Delivery—Design, Synthesis, and the Influence on Physicochemical Properties of Liposomes. Molecules 2022, 27, 3406. [Google Scholar] [CrossRef]
- Gładkowski, W.; Chojnacka, A.; Włoch, A.; Pruchnik, H.; Grudniewska, A.; Dunal, A.; Dudek, A.; Maciejewska, G.; Rudzińska, M. Conjugates of 1,3- and 1,2-Acylglycerols with Stigmasterol: Synthesis, NMR Characterization, and Impact on Lipid Bilayers. Chempluschem 2023, 88, e202300161. [Google Scholar] [CrossRef]
- Huang, Z.; Szoka, F.C. Sterol-modified phospholipids: Cholesterol and phospholipid chimeras with improved biomembrane properties. J. Am. Chem. Soc. 2008, 130, 15702–15712. [Google Scholar] [CrossRef]
- Huang, Z.; Jaafari, M.R.; Szoka, F.C. Disterolphospholipids: Nonexchangeable lipids and their application to liposomal drug delivery. Angew. Chemie - Int. Ed. 2009, 48, 4146–4149. [Google Scholar] [CrossRef]
- Kłobucki, M.; Grudniewska, A.; Smuga, D.A.; Smuga, M.; Jarosz, J.; Wietrzyk, J.; Maciejewska, G.; Wawrzeńczyk, C. Syntheses and antiproliferative activities of novel phosphatidylcholines containing dehydroepiandrosterone moieties. Steroids 2017, 118, 109–118. [Google Scholar] [CrossRef]
- Tian, N.N.; Li, C.; Tian, N.; Zhou, Q.X.; Hou, Y.J.; Zhang, B.W.; Wang, X.S. Syntheses of 7-dehydrocholesterol peroxides and their improved anticancer activity and selectivity over ergosterol peroxide. New J. Chem. 2017, 41, 14843–14846. [Google Scholar] [CrossRef]
- Bentley, P.H.; McCrae, W. An Efficient Synthesis of Symmetrical 1,3-Diglycerides. J. Org. Chem. 1970, 35, 2082–2083. [Google Scholar] [CrossRef]
- Cockman, S.J.; Joll, C.A.; Mortimer, B.C.; Redgrave, T.G.; Stick, R.V. The Synthesis of Some Esters of Glycerol with Special Attention to the Problem of Acyl Migration. Aust. J. Chem. 1990, 43, 2093–2097. [Google Scholar] [CrossRef]
- Niezgoda, N.; Mituła, P.; Kempińska, K.; Wietrzyk, J.; Wawrzeńczyk, C. Synthesis of phosphatidylcholine with conjugated linoleic acid and studies on its cytotoxic activity. Aust. J. Chem. 2013, 66, 354–361. [Google Scholar] [CrossRef]
- Tordini, F.; Bencini, A.; Bruschi, M.; De Gioia, L.; Zampella, G.; Fantucci, P. Theoretical study of hydration of cyanamide and carbodiimide. J. Phys. Chem. A 2003, 107, 1188–1196. [Google Scholar] [CrossRef]
- Wilson, W.K.; Sumpter, R.M.; Warren, J.J.; Rogers, P.S.; Ruan, B.; Schroepfer, G.J. Analysis of unsaturated C27 sterols by nuclear magnetic resonance spectroscopy. J. Lipid Res. 1996, 37, 1529–1555. [Google Scholar] [CrossRef]
- Plückthun, A.; Dennis, E.A. Acyl and Phosphoryl Migration in Lysophospholipids: Importance in Phospholipid Synthesis and Phospholipase Specificity. Biochemistry 1982, 21, 1743–1750. [Google Scholar] [CrossRef]
- Niezgoda, N.; Gliszczy, A.; Gładkowski, W.; Chojnacka, A. Lipase-catalyzed enrichment of egg yolk phosphatidylcholine with conjugated linoleic acid. J. Mol. Catal. B Enzym. 2016, 123, 14–22. [Google Scholar] [CrossRef]
- Wang, T.; Cheng, J.; Wang, N.; Zhang, X.; Jiang, L.; Yu, D.; Wang, L. Study on the stability of intermediates in the process of enzymatic hydrolysis of phosphatidic acid by phospholipase A1. Lwt 2021, 142, 111015. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, N.; Wang, D.; Luo, S.; Zhang, H.; Yu, D.; Wang, L.; Elfalleh, W.; Liao, C. Preparation of vacuum-assisted conjugated linoleic acid phospholipids under nitrogen: Mechanism of acyl migration of lysophospholipids. Food Chem. 2024, 436, 137680. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, L.; Qu, L.; Wang, X.; Liu, Y. Mechanism and effective factors of acyl migration of lysophospholipid. Chinese J. Org. Chem. 2014, 34, 2529–2536. [Google Scholar] [CrossRef]
- Kiełbowicz, G.; Smuga, D.; Gładkowski, W.; Chojnacka, A.; Wawrzeńczyk, C. An LC method for the analysis of phosphatidylcholine hydrolysis products and its application to the monitoring of the acyl migration process. Talanta 2012, 94, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Papangelis, A.; Ulven, T. Synthesis of Lysophosphatidylcholine and Mixed Phosphatidylcholine. J. Org. Chem. 2022, 87, 8194–8197. [Google Scholar] [CrossRef] [PubMed]
- Sugasini, D.; Subbaiah, P.V. Rate of acyl migration in lysophosphatidylcholine (LPC) is dependent upon the nature of the acyl group. Greater stability of sn-2 docosahexaenoyl LPC compared to the more saturated LPC species. PLoS ONE 2017, 12, 1–13. [Google Scholar] [CrossRef]
- Schlenk, H.; Lamp, B.G.; Wallace DeHaas, B. Syntheses of Derivatives of Dihydroxyacetone and of Glycerides. J. Am. Chem. Soc. 1952, 74, 2550–2552. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of acylglycerol 4 containing succinyl-linked 7-DHC at sn-2 position. Reagents and conditions: (i) succinic anhydride, DMAP, anhydrous pyridine, 60°C, 24 h (ii) palmitic acid, DCC, DMAP, CHCl3, r.t., 24 h (iii) NaBH3CN, THF, glacial MeCOOH, r.t., 40 min. (iv) DCC, DMAP, CHCl3, r.t., 24 h.
Scheme 1.
Synthesis of acylglycerol 4 containing succinyl-linked 7-DHC at sn-2 position. Reagents and conditions: (i) succinic anhydride, DMAP, anhydrous pyridine, 60°C, 24 h (ii) palmitic acid, DCC, DMAP, CHCl3, r.t., 24 h (iii) NaBH3CN, THF, glacial MeCOOH, r.t., 40 min. (iv) DCC, DMAP, CHCl3, r.t., 24 h.
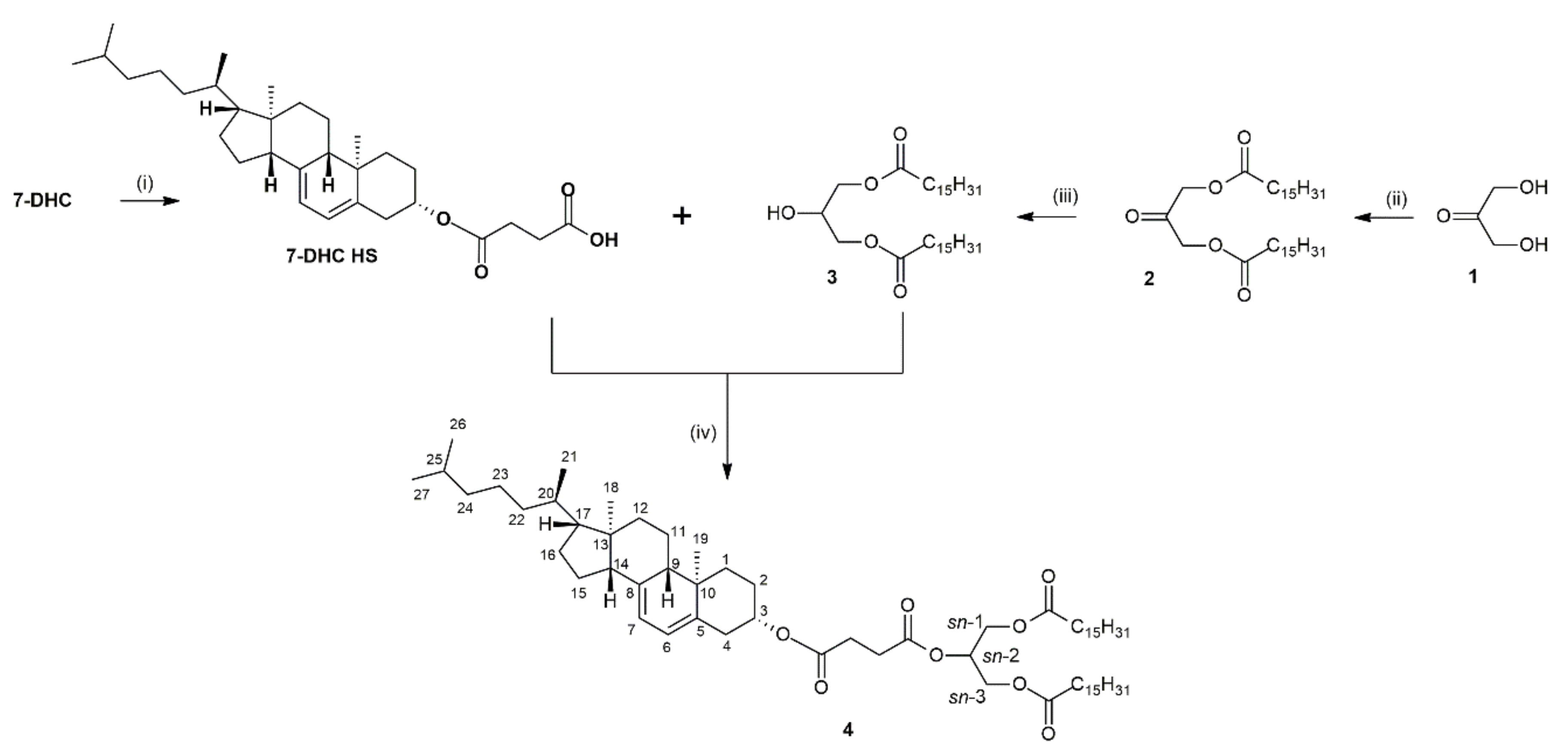
Scheme 2.
Synthesis of glycerophospholipid 7 containing succinyl-linked 7-DHC at sn-2 position. Reagents and conditions: (i) DBTO, iPrOH, reflux, 1h then TEA, pamitoyl chloride, r.t., 0.5 h (ii) DCC, DMAP, CH2Cl2, 4 °C, 96 h.
Scheme 2.
Synthesis of glycerophospholipid 7 containing succinyl-linked 7-DHC at sn-2 position. Reagents and conditions: (i) DBTO, iPrOH, reflux, 1h then TEA, pamitoyl chloride, r.t., 0.5 h (ii) DCC, DMAP, CH2Cl2, 4 °C, 96 h.
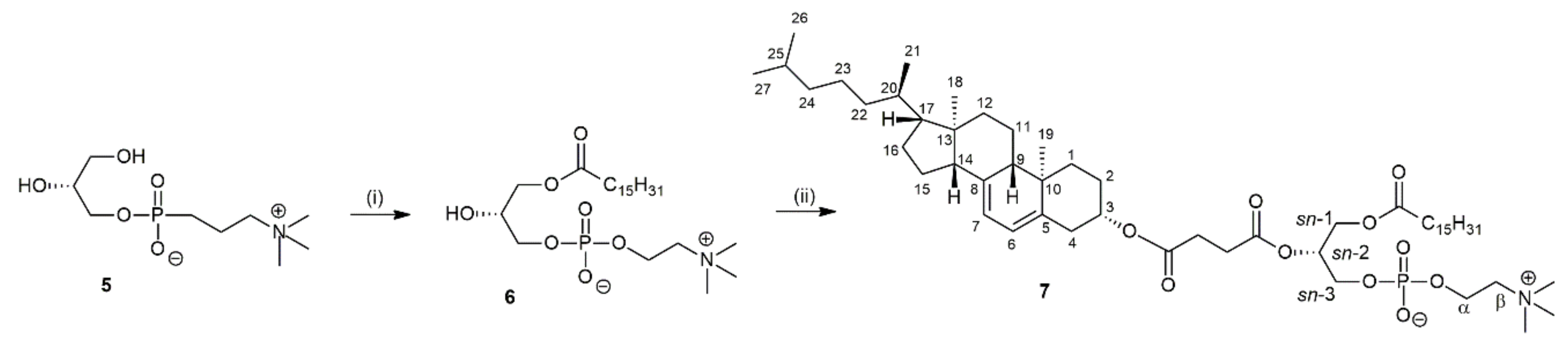
Figure 1.
Fragment of 1H NMR (A) and 13C NMR (B) spectrum of acylglycerol 4.
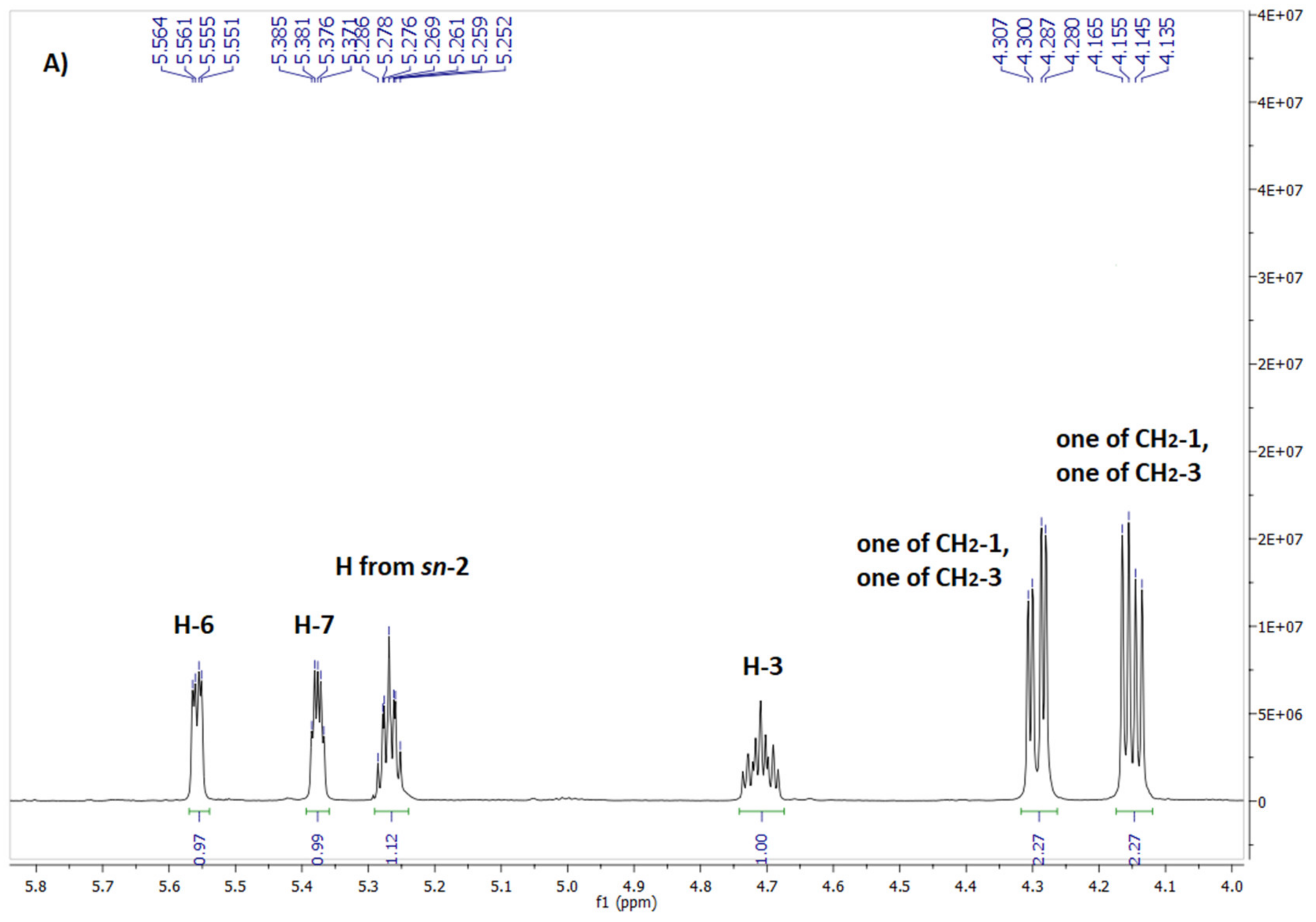
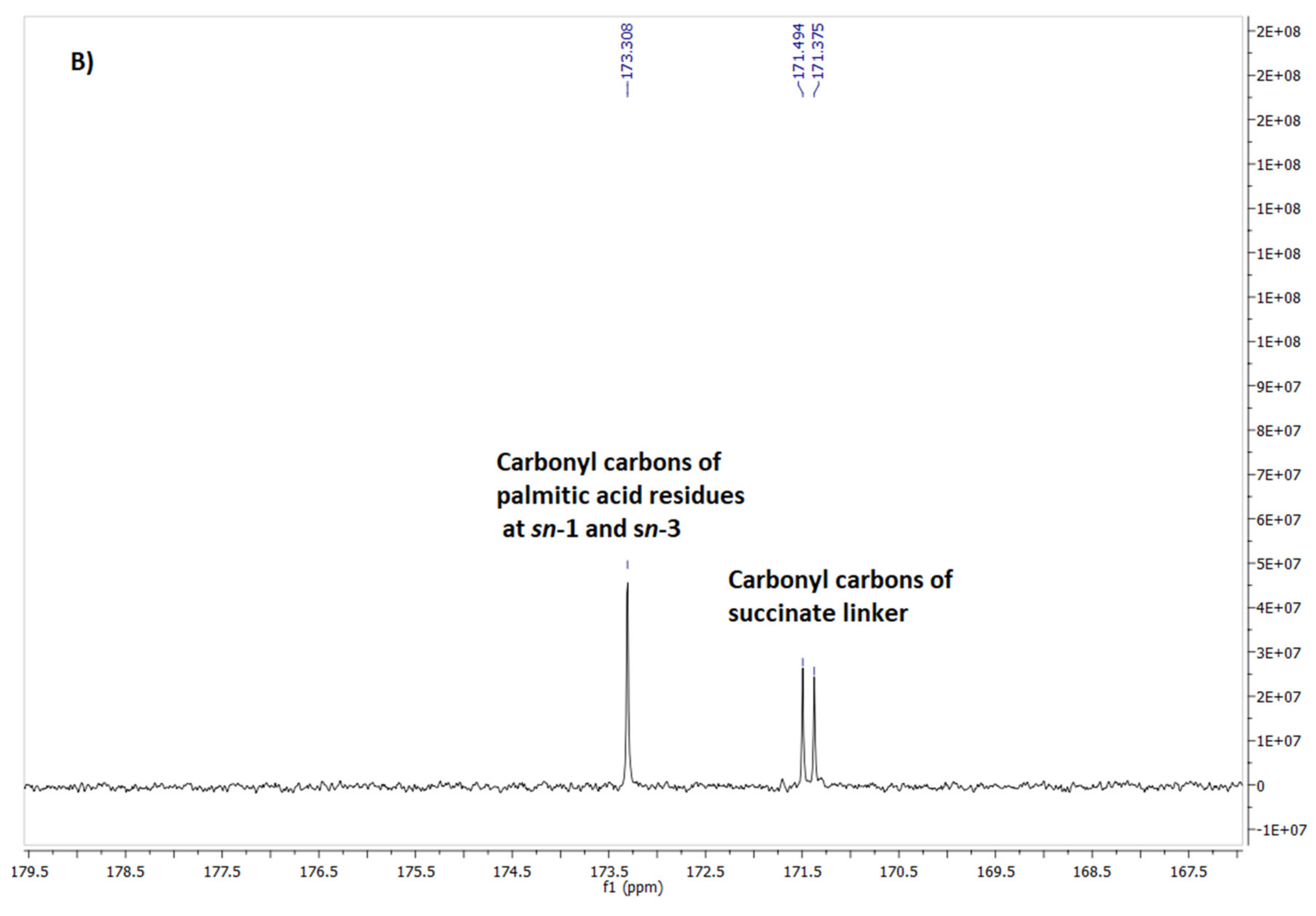
Figure 2.
Fragment of 1H NMR (A) and 13C NMR (B) spectrum of phospholipid 7.
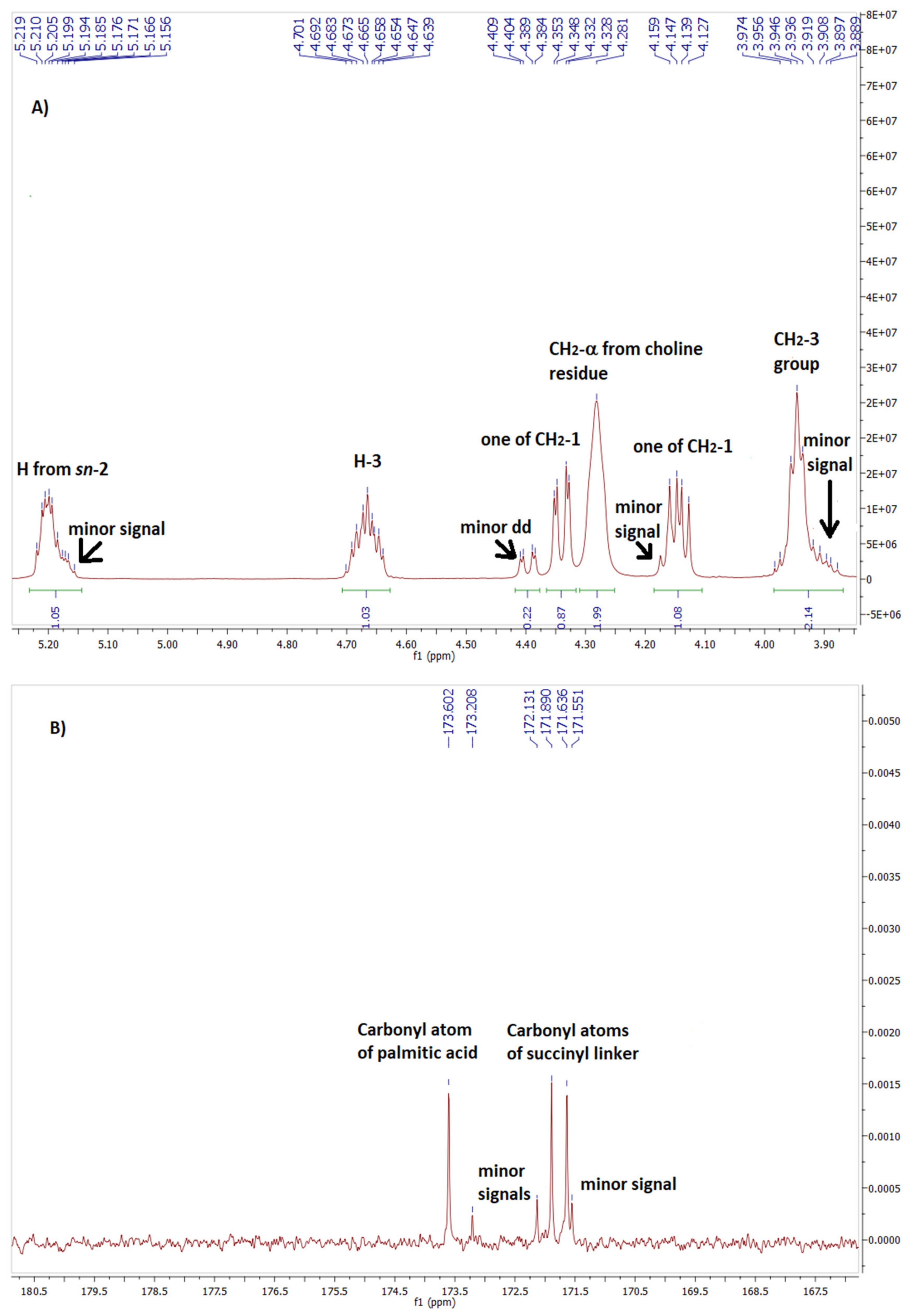

Table 1.
Chemical shifts of selected major and minor signals observed on the spectra of phospholipid 7 and two analogous glycerophospholipids containing hemisuccinate-attached DHEA and oleic acid residue (1-OA-2-DHEA-PChol1 and 1-DHEA-2-OL-PChol2).
Table 1.
Chemical shifts of selected major and minor signals observed on the spectra of phospholipid 7 and two analogous glycerophospholipids containing hemisuccinate-attached DHEA and oleic acid residue (1-OA-2-DHEA-PChol1 and 1-DHEA-2-OL-PChol2).
| Proton or carbon | Chemical shift [ppm] | |||
|---|---|---|---|---|
| Major signals on the spectrum of phospholipid 7 | 1-OA-2-DHEA- PChol3 |
Minor signals on the spectrum of phospholipid 7 | 1-DHEA-2-OA- PChol3 |
|
| one of the proton from CH2 at sn-1 | 4.14 | 4.14 | Overlapped with 4.14 | 4.14 |
| one of the proton from CH2 at sn-1 | 4.34 | 4.36 | 4.40 | 4.39 |
| H at sn-2 | 5.20 | 5.20 | 5.17 | 5.18 |
| carbonyl carbons from hemisuccinate linker | 171.64 and 171.89 | 171.63 and 171.69 | 171.55 and 172.13 | 171.55 and 171.95 |
| carbonyl carbon from fatty acid residue | 173.60 | 173.61 | 173.21 | 173.16 |
1 1-Oleoyl-2-(3β-O-succinylandrost-5-en-17-one)-sn-glycero-3-phosphocholine. 2 1-(3β-O-Succinylandrost-5-en-17-one)-2-oleoyl-sn-glycero-3-phosphocholine. 3 Data from Kłobucki et al. [30].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



