
Submitted:
03 November 2024
Posted:
04 November 2024
You are already at the latest version
Abstract
Duchenne muscular dystrophy (DMD) is a severe genetic muscle disease due to mutations of dystrophin gene. There is no cure for DMD. Using a dystrophin-/-utrophin-/- (DKO-Hom) mouse model, we investigated the PGE2/EP2 pathway in the pathogenesis of dystrophic muscle and its potential as a therapeutic target. We found that Ep2, Ep4, Cox-2, 15-Pgdh mRNA, and PGE2 are significantly increased in DKO-Hom mice compared to wild-type (WT) mice. The EP2 and EP4 receptors are mainly expressed in CD68+ macrophages and are significantly increased in the muscle tissues of both dystrophin-/- (mdx) and DKO-Hom mice compared to WT mice. Osteogenic and osteoclastogenic gene expression in skeletal muscle also increased in DKO-Hom mice, which correlates with severe muscle heterotopic ossification (HO). Treatment of DKO-Hom mice with the EP2 antagonist PF04418948 for 2 weeks, increased body weight, reduced HO and muscle pathology by decreasing both total macrophages (CD68+) and senescent macrophages (CD68+P21+), while increasing endothelial cells (CD31+). PF04418948 also increased bone volume/total volume (BV/TV), trabecular thickness (Tb.Th) of the tibia trabecular bone and the cortical bone thickness of both the femur and tibia without affecting spine trabecular bone microarchitecture. In summary, our results indicate that targeting EP2 improves muscle pathology and maintains bone mass.
Keywords:
muscular dystrophy
; EP2
; EP4
; PGE2
; bone microarchitecture
; heterotopic ossification
; PF04418948
1. Introduction
Duchenne muscular dystrophy (DMD) is a severe genetic muscle disease due to the mutations of dystrophin gene and loss of dystrophin expression. It affects 1 in 3000 boys [1,2]. The manifestations of the DMD are severe, and patients died in the third or fourth decades and currently without cure. Dystrophin-/-/utrophin-/- (DKO-Hom) is a mouse model that recapitulates the disease’s clinical manifestation more closely than dystrophin -/- mice (Mdx) with more severe muscle histopathology including muscle necrosis, fibrosis, fat infiltration, heterotopic ossification (HO), kyphosis and short life span [3,4,5,6,7]. Previously, we have shown that DKO-Hom mice also exhibit a spectrum of musculoskeletal abnormalities, including bone osteopenia [8]. Bone osteopenia was determined to be a secondary consequence of the muscle disease, with mice demonstrating declines in osteoblasts, osteoclasts, and osteocytes [9]. Furthermore, we also found impaired fracture healing in DKO-Hom mice [10].
Restoring functional dystrophin is the fundamental solution for facilitating the cure, but few gene therapy approaches have been translated into clinical treatments due to varieties of mutations of the dystrophin gene. An early study using adeno-mini-utrophin intramuscularly (IM) injected into the tibialis anterior (TA) muscle of DKO-Hom mice found that utrophin was expressed in 95% of the myofibers after 30 days, while central nuclear myofibers decreased from 80% in the non-injected mice to 12% in the injected mice with a significant decrease in muscle necrosis [11]. Injection of adenoviral vector carrying full-length dystrophin gene into multiple muscles of DKO-Hom mice, widespread expression of exogenous dystrophin, significant reductions in centrally nucleated myofibers, and restoration of the dystrophin-associated proteins: beta-dystroglycan (beta-DG) and alpha-sarcoglycan (alpha-SG), as well as neuronal nitric oxide synthase (nNOS) were achieved. The treated DKO-Hom mice also demonstrated increased body weight, improved motor performance, and a prolonged life span [12]. Treatment with adeno-associated viral vector 6 (AAV-6)-microdystrophin alleviated most of the pathophysiological abnormalities associated with muscular dystrophy in the DKO-Hom mice [13]. Systemically injection of AAV1- mini-dystrophin (DysD3990) into DKO-Hom mice led to highly efficient mini-dystrophin gene expression in the muscle, ameliorated muscle pathologies, improvement in growth and motility, inhibition of spine and limb deformation, and extended life spans[14]. The combination of AAV-VEGFa and AAV-mini-dystrophin further improved muscle pathology as demonstrated by decreased inflammatory cell infiltration, central nuclear fibers, fibrosis, and increased dystrophin and nNOS expression [15]. Intraperitoneal injection (IP) of peptide-conjugated phosphorodiamidate morpholino oligomers (PPMOs) targeting exon 23 of dystrophin pre-mRNA in DKO-Hom mice induced near-normal level of dystrophin expression in all muscles examined, except for cardiac muscle, and a significant improvement in muscle function and dystrophic pathology [16]. By using the AAV-mediated exon skipping approach utilizing small nuclear RNA (snRNA) as a shuttle sequence, it was demonstrated that a single intravenous (IV) injection of scAAV9-U7ex23 in DKO-Hom mice induced near-normal level of dystrophin expression in all muscles examined, including the cardiac muscle. This approach resulted in a dramatic improvement of muscle function and dystrophic pathology as well as a remarkable extension of the lifespans of DKO-Hom mice [17]. Finally, ELEVIDYS, an AAV-based gene therapy (microdystrophin) is approved by FDA for the treatment of ambulatory pediatric patients aged 4 through 5 years with DMD with a confirmed mutation in the DMD gene [18].
CRISPR/Cas9 technology rendered new strategies to restore functional endogenous dystrophin by guided RNA-mediated excision of mutant dystrophin genes including exon 23. In 2016, it was first reported using AAV9 to deliver CRISPR/cas 9 and gene-editing components (gRNA) to postnatal mdx mice, a commonly used model of DMD. Different modes of AAV9 delivery were tested, including IP injection at postnatal day 1 (P1), IM injection at P12, and retro-orbital injection at P18. Each of these methods restored dystrophin protein expression in cardiac and skeletal muscle to varying degrees, and expression increased from 3 to 12 weeks after the injections. Postnatal gene editing also enhanced skeletal muscle function, as measured by grip strength tests 4 weeks after injection [19]. Duchêne BL et al. used pairs of sgRNAs targeting exons 47 and 58 and a normal reading frame was restored in myoblasts derived from muscle biopsies of 4 DMD patients with different exon deletions. Restoration of the DMD reading frame and dystrophin expression was also achieved in vivo in the heart of the del52hDMD/mdx [20]. Using a non-homologous end joining (NHEJ) CRISPR/cas 9 editing technology to remove mutant exon 23 in DKO-Hom mice was also reported. It was found that dystrophin restoration was most effective in the diaphragm, where a maximum of 5.7% of wild-type dystrophin expression was observed. However, CRISPR/Cas9-mediated gene editing did not extend lifespan in the DKO-Hom mice, and dystrophin was expressed in a within-fiber patchy manner in skeletal muscle tissues. Further analysis revealed many non-productive DNA repair events, including side effects such as AAV viral genome integration at the CRISPR cut sites. This study highlighted potential challenges for the successful development of CRISPR/cas9 therapies in the context of DMD [21].
More recently, base and prime editing was also used to edit the mutant dystrophin gene to restore its function and demonstrated high efficacy [22]. Using an adenine base editor (ABE) to modify splice donor sites of the dystrophin gene led to skipping of a common DMD deletion mutation of exon 51 (∆Ex51) in cardiomyocytes derived from human-induced pluripotent stem cells, and restored dystrophin expression. Prime editing was also capable of reframing the dystrophin open reading frame in these cardiomyocytes. IM injection of ∆Ex51 mice with AAV-9 encoding ABE components as a split-intein trans-splicing system resulted in successful gene editing and disease correction in vivo. This study demonstrated the effectiveness of nucleotide editing for the correction of diverse DMD mutations with minimal modification of the genome [23].
Cell therapy has also been explored for the treatment of DMD. Early studies showed that tail vein injection of bone marrow stem cells from C57BL/6 mice into irradiated DKO-Hom mice improved locomotive function and cardiograph activity with 7% of muscle fibers expressing dystrophin [24]. Transplantation of rat MSCs to DKO-Hom mice via the tail vein also improved motor function and muscle pathology and increased life span of the mice compared to DKO-Hom mice without MSC transplantation [24]. Rat MSC transplantation also improved locomotive function, increased dystrophin/utrophin expression, and increased life span from 22 weeks to 35 weeks for MSC-treated mice [25]. Transplantation of Pax3-induced ES-derived skeletal myogenic progenitors resulted in significant engraftment as evidenced by the presence of dystrophin+ myofibers and restoration of β-dystroglycan and eNOS within the sarcolemma, as well as enhanced strength of treated muscles [26]. Tail vein injection of embryonic like stem cells (ELSCs) isolated from human bone marrow improved motor function and decreased serum creatine kinase activity at 2 months after cell transplantation. In addition, dystrophin protein and messenger RNA were up-regulated and the skeletal muscle histology improved in these DKO-Hom mice transplanted with ELSCs [27].
Targeting abnormal expressed genes in DKO-Hom mice have also been shown to improve muscle histopathology. Sarcolipin (SLN) is an inhibitor of the sarco/endoplasmic reticulum (SR) Ca2+ ATPase (SERCA) and is abnormally elevated in the muscle of DMD patients and animal models. Either knocking down the SLN using genetic approaches or AAV-SLN siRNA attenuates muscle pathology and improves diaphragm, skeletal muscle, and cardiac function [28]. IL6 was also found to be significantly increased in DKO-Hom mice, IP injection of IL6 antibody MRL6-1 significantly increased embryonic myosin heavy chain and muscle diameter and reduced fibrosis via decreasing phosphorylated signal transducer and activator of transcription 3 (pSTAT3) [29].
Despite all the above research advancements, the current treatment of DMD patients is still limited and no cure is available. Inflammation remains a profound pathological change in the muscle of muscular dystrophy. Hence, anti-inflammation is equally important in DMD treatment in addition to the restoration of the dystrophin gene. Steroids that targeting inflammation are the only palliative therapy but have side effects including increasing fracture risk after long-term treatment [30,31,32]. The aim of this study was to investigate the role of the prostaglandin E 2/prostaglandin E 2 receptor 2 (PGE2/EP2) signaling pathway in the development of HO and muscle pathology in DKO-Hom mice and determine if targeting PGE2/EP2 signaling pathway can improve the pathology of muscular dystrophic muscle and bone health.
2. Materials and Methods
2.1. Mice Breeding
Dystrophin-/-Utrophin+/- (DKO-Het) mice were provided by our collaborator [14] and bred to generate Dystrophin-/- (Mdx) and Dystrophin-/-Utrophin-/- (DKO-Hom) mice for the experiments. Genotyping was performed at the time of weaning at 3 weeks after birth. All the experiments were performed according to the Institutional Animal Care and Use Committee by the University of Texas Health Science Center at Houston (Protocol # AWC-15-0073) and Colorado State University (#1234).
2.2. PGE2-EP2/4 Signaling and Inflammatory Gene Expression Detection Using RT-Q-PCR for 4-Week-Old Mice
Four-week-old C57BL/10J (WT) were purchased from Jackson Laboratories. Mdx and DKO-Hom mice were generated by breeding DKO-Het mice and were sacrificed at 4 weeks old to harvest muscle tissues. Gastrocnemius muscles were dissected and frozen in 2-methylbutane in liquid nitrogen on the cork using Cryogel for cryosectioning. Thigh muscle tissues were dissected and immediately placed in the -800C for later RNA extraction using Trizol reagent (Invitrogen). cDNA synthesis was performed using Iscript Reverse Transcription Supermix (1708841, BioRad). Q-PCR was performed using the SsoAdvanced Universal SYBR®® Green Supermix Q-PCR kit (1725271, BioRad). The targets analyzed included Cox-1, Cox-2,15-Pgdh, prostaglandin-E2 (PGE2) receptor 2 (Ep2), prostaglandin-E2 (PGE2) receptor 4 (Ep4), cluster differentiation 68(CD68), runt related transcription factor 2 (Runx2), osterix (Osx), cathepsin k (Ctsk), tartrate resistant acid phosphatase (Trap), follistatin (Fst), myostatin (Mstn), fibronectin type III domain containing 5 (Fndc5). All primers were designed using Primer 3 Input [33,34,35]. Primers used in this study are listed in Table 1.
2.3. Measurement of Prostaglandin 2 (PGE2) Using Enzyme Linked Immunosorbent Assay (ELISA)
A portion of the thigh muscle tissues were homogenized using buffer as suggested by Cayman Chemical to measure PGE2 using Cayman ELISA kit (Item No. 514010, Cayman Chemical, Ann Harbor, Michigan) following the manufacturer’s protocol and normalized to protein concentration. Protein concentration was measured with Pierce™ BCA Protein Assay Kits (Catalog number: 23225 ThermoFisher Scientific) following the manufacturer’s protocol.
2.4. Immunofluorescence Staining
We performed CD68/EP2 and CD68/EP4 double staining for gastrocnemius muscle to detect macrophage, EP2 and EP4 expression. Briefly, cryosections were cut into 8µm thickness using Leica CryoStat (Model CM 1950) and stored in -80°C. Section slides were taken out from -80 °C and dried for 10 minutes and then fixed with 4% paraformaldehyde for 8 minutes and then washed three times with phosphate-buffered saline (PBS) for further immunofluorescence staining, as previous described [36]. Briefly, after blocking with 5% donkey serum (017-000-121, Jackson ImmunoResearch Laboratories Inc.) in PBS, the slides were incubated with primary antibodies overnight at 4°C. The dilution of primary antibodies used for this study included rat anti-CD68 (AB53444, 1:100 dilution, Abcam), rabbit anti-EP4 Receptor (C-Term) Polyclonal Antibody (Item No. 101775, 1:50 dilution, Cayman Chemical), Recombinant Anti-Prostaglandin E Receptor EP2/PTGER2 antibody [EPR8030(B)] (ab167171, 1:250 dilution, Abcam). After washing with PBS three times with 5 minutes each, the slides were incubated with secondary antibodies based on primary antibodies used for each stain for 2 hours at ambient temperature. Secondary antibodies used were donkey anti-rat-488 (712-745-153, Jackson ImmunoResearch, Laboratories Inc 1:200) and donkey anti-rabbit-594 (711-585-152, 1:200 dilution, Jackson ImmunoResearch Laboratories Inc). After the secondary antibody incubation and another 3 washes, 4′,6-diamidino-2-phenylindole (DAPI) was used to reveal the nuclei. Immunofluorescence images were captured using Nikon-Ni microscope using 200X magnification and positive cell numbers were counted using Image J.
2.5. Muscle Histology
H&E staining was performed using AnaTech Hematoxylin Extra Strenth and Eosin Y according to the manufacturer’s protocol. Von Kossa staining was performed using an IHC World protocol for the detection of HO formation in the muscle tissues (https://www.ihcworld.com/_protocols/special_stains/von_kossa.htm). All chemical reagents were purchased from Sigma.
2.6. Mice Treatment with EP2 Antagonist PF04418948
Four-week-old DKO-Hom (male and female) mice were treated with vehicle (N=6)( 0.5% w/v methylcellulose + 0.1% v/v Tween 80 in purified water) according to literature [37] , or 10mg/kg/day PF04418948 in vehicle (0.5% w/v methylcellulose + 0.1% v/v Tween 80 in purified water) (n=8) (Cat. No. 4818, Tocris, R&D system) for 2 weeks by oral gavage daily. Mice were then sacrificed. Left side gastrocnemius muscle tissues were dissected and frozen in 2-methylbutane in liquid nitrogen on the cork for cryosection and staining. Left side thigh muscle were dissected and immediately frozen in -800C for RNA isolation. The right tibia and femur bone tissues together with attached intact muscle tissues as well as the skull and lumbar spine were dissected and fixed with neutral buffered formalin (NBF) for 72 hours for subsequent Micro-CT scanning and histology analysis.
2.7. Micro-CT Scanning and Analysis
Micro-CT scanning was performed after NBF fixation of the skull, lumbar spine, tibia and femur using Viva-CT 80 using scan parameters of 15µm voxel size, 70kVP and 114µA (Scanco Medical LLC) to detect HO and characterize bone microarchitecture. Spine L5 trabecular bone was quantified by contouring the entire vertebral trabecular bone while excluding the cortical bone portion using hand contouring and the morph function in the software. Proximal tibia cancellous bone was defined by contouring the trabecular bone right below the growth plate and extended to 50 slices (750um total). Both spine L5 and proximal tibia trabecular bone were defined with Gauss Sigma=0.8, Gauss support = 1 and threshold at 163. The cortical bone microarchitecture of the femur was quantified by defining 50 slices at the midshaft, using Gauss sigma =0.8, Gauss support = 1, and a threshold of 200, as previously described [9]. To quantify the HO in the surrounding muscle of the femur, we contoured the entire femur surrounding muscle by excluding femur cortical bone, and then defining HO using Gauss Sigma = 0.8, Gauss Support =1, and threshold of 163. In this case, total volume (TV) represented total muscle volume, and BV represented HO in the muscle. BV/TV represented HO volume ratio of total muscle volume.
2.8. Bone Histology Analysis and Immunohistochemistry
After Micro-CT scans, the bone tissues were decalcified using 10% ethylenediaminetetraacetic acid disodium salt dihydrate (E4884, Sigma-Aldrich) plus 1% sodium hydroxide for 4 weeks. Bone tissues were processed with gradient alcohol, cleared with xylene, and infiltrated in paraffin 9 (Fisher Scientific) and paraffin embedded using KD-TS3D1, Automatic Tissue Processor, and KD-BM & BL Tissue Embedding & Cooling System. Sections were cut into 5µm slices using a microtome. Herovici’s staining was performed as previously described [38,39]. H&E staining was performed, as detailed as above. TRAP staining was performed using Leukocyte Acid Phosphatase (TRAP) Kit (387-A Kit, Sigma) following the manufacturer’s protocol. Immunohistochemistry of osterix (OSX) using rabbit anti-OSX (ab 22552,1:1000, Abcam) was also performed according to previously described protocol [9] The secondary antibody used was Goat Anti-Rabbit IgG Antibody (H+L), Biotinylated (BA-1000-1.5, 1:300, Vector Laboratories). After secondary antibody incubation and washing, slides were incubated with ABC reagents for 2 hours at ambient temperature (VECTASTAIN®® Elite®® ABC-HRP Kit, Peroxidase (Standard) (PK-6100, Vector Laboratories). After another 3 times of washing with PBS, DAB Substrate Kit, Peroxidase (HRP), with Nickel, (3,3′-diaminobenzidine) (SK-4100) was used to reveal positive cells as brown color. Slides were washed with tap water and then counterstained with Hematoxylin QS (H-3404-100, Vector Laboratories) for 30 seconds, rinsed with tap water, and blue nuclei with running tap water for 10 minutes. Slides were then dehydrated in gradient alcohol, cleared with xylene, and mounted with Cytoseal (Fisher Scientific). Microscopic images were taken with a Nikon Ni bright field microscope. The number of TRAP+, OC+ and OSX+ cells were counted using Image J by normalized to 1 mm of bone surface.
2.9. Muscle Histology Analysis
Left side gastrocnemius tissues were harvested, and cryosection blocks were made as described in section 2.2. Immunofluorescence staining was performed as described in section 2.4. The primary antibodies used were rabbit anti-P21 (ab188224, 1:400 dilution, Abcam), rat anti-Gr-1 (BD557445,1:100, BD), and rat anti-CD31 (BD553370, BD 1:300); all the other steps are the same as section 2.5.
2.10. Q-PCR Analysis of the Thigh Muscle Tissues for PF04418948 Treated and Vehicle Treated DKO-Hom Mice
Thigh muscles were collected and immediately frozen on dry ice, and then stored at -80°C for later RNA extraction, CDNA synthesis and Q-PCR as detailed as section 2.2.
2.11. Statistical Analysis
All data were analyzed using Graphpad Prism 9.4. Student T tests were used for two group comparisons. Analysis of variance (ANOVA) was used for three or more group comparisons followed by Tukey’s post hoc multiple comparisons. For data with higher variation, Wilcoxon Rank Sum Test was used. P<0.05 was considered statistically significant.
3. Results
3.1. PGE2/EP2/4signaling Pathway Components all Significantly Up-Regulated in DKO-Hom Mice
We first performed reverse transcriptive quantitative polymerase chain reaction (Q-PCR) analysis of thigh muscle from wild type (WT), Mdx and DKO-Hom mice. We found a trend of increase of Ep2 mRNA in Mdx mice compared to WT mice, while Ep2 mRNA in DKO-Hom increased 30-fold compared to WT mice (Figure 1A). Ep4 mRNA also significantly increased in Mdx mice but increased 10-fold in DKO-Hom mice thigh muscle compared to WT mice (Figure 1B). Cyclooxygenase 1 (Cox-1) mRNA expression in the thigh muscle of Mdx mice showed no significant change compared to WT mice but demonstrated an increasing trend in DKO-Hom mice (P=0.0835) (Figure 1C). Cyclooxygenase 2 (Cox-2) mRNA expression significantly increased in both Mdx and DKO-Hom mice compared to WT mice (P=0.045 and 0.0096, respectively) (Figure 1D). 15-hydroxyprostaglandin dehydrogenase (15-Pgdh), the enzyme that catabolizes the degradation of PGE2, also increased in Mdx mice and significantly increased 6-fold in DKO-Hom mice compared to WT mice (Figure 1E). Further, cell differentiation 68 (CD68), a M1 macrophage marker, significantly increased in the thigh muscle of DKO-Hom mice compared to the WT mice (Figure 1f). ELISA results showed PGE2 was significantly elevated in the thigh muscle tissues of Mdx and DKO-Hom mice. PGE2 was also significantly higher in DKO-Hom mice compared to Mdx mice (Figure 1g).
3.2. Increased Osteogenic-Related Genes Correlated with HO in the Muscle Tissues of DKO-Hom Mice
Since we previously demonstrated that both Mdx and DKO-Hom muscle have severe HO formation as revealed by microCT [9]. We further tested osteogenesis and osteoclastogenesis gene expression in the thigh muscle of Mdx and DKO-Hom mice. The Q-PCR results showed that Runt related transcription factor 2 (Runx2) was significantly increased in both Mdx and DKO-Hom mice compared to WT mice (P=0.0347 and 0.0037, respectively) (Figure 2A). Osterix (Osx) mRNA also showed a trend of increase in DKO-Hom mice when compared to WT control mice (P=0.0919) (Figure 2B). Furthermore, cathepsin K (Ctsk) was found to be significantly elevated in DKO-Hom mice when compared to Mdx and WT mice while no significant increases were observed in Mdx mice compared to WT mice (Figure 2C). Further, tartrate resistant acid phosphatase (Trap) mRNA expression was also significantly increased in DKO-Hom mice compared to Mdx and WT mice (P=0.0218 and 0.0064, respectively) (Figure 2D). These results indicated that osteogenesis and osteoclastogenesis were both active in the muscle tissues of DKO-Hom mice and likely contributed to HO. We further investigated the myokine expression in the thigh muscle tissues. We found that follistatin (Fst) was significantly increased in the DKO-Hom mice compared to Mdx and WT mice (P=0.0117 and 0.0032, respectively) (Figure 2E). In addition, irisin (Fndc5) was also significantly up-regulated in the DKO-Hom mice compared to Mdx and WT mice (P=0.0303 and 0.0226, respectively) (Figure 2F). In addition, myostatin (Mstn) was significantly decreased in Mdx and DKO-Hom mice compared to WT mice (P=0.0004 and <0.0001, respectively) (Figure 2G). These myokine changes favor muscle regeneration and are likely compensatory for the muscle damage due to lack of dystrophin and utrophin. Finally, Von Kossa staining of the gastrocnemius muscle tissues revealed brown-black mineralization in the muscle tissues of both the Mdx and DKO-Hom mice, which further proved HO formation (Figure 2H).
3.3. EP2/EP4 Expression in the Muscle Tissues Are Predominately in the Macrophages
Next, we performed immunofluorescence staining for the gastrocnemius muscle tissues to detect which cells express EP2/EP4. We colocalized EP2 with CD68, a M1 macrophage marker. We found a significant increase of CD68+ macrophages in Mdx and DKO-Hom mice, compared to WT control mice (P<0.0001 for both) (Figure 3A,B). CD68+ EP2+ double positive cells also significantly increased in Mdx and DKO-Hom mice as compared to WT mice (P=0.0191 and 0.0019, respectively) (Figure 3A, C). There was also a significant increase of CD68- EP2+ cells in the muscle tissues of Mdx and DKO-Hom (Figure 3A,D). These results indicated EP2 expression was significantly increased in both macrophages and non-macrophages in muscle tissues of Mdx and DKO-Hom mice. CD68 and EP4 double staining was performed and revealed a significant increase of CD68+ cells in the muscle tissues of DKO-Hom mice compared to WT mice (P=0.0171) (Figure 3E,F). Furthermore, CD68+EP4+ cells were also significantly elevated in the muscle tissues of both Mdx and DKO-Hom mice compared to WT mice (P=0.0008 and <0.0001, respectively) (Figure 3E,G). CD68-EP4+ cells were also significantly increased in the DKO-Hom mice (P=0.0123) (Figure 3E,H). These results demonstrated that EP4 is significantly increased in both macrophage and non-macrophages in the gastrocnemius muscle tissues of DKO-Hom mice.
3.4. Treatment with EP2 Antagonist PF04418948 Decreased HO in the Muscle Tissues
To further investigate the role of EP2/PGE2 signaling pathway in the HO formation of muscle, we treated DKO-Hom mice at 4-week-old with the EP2 antagonist PF04418948 for 2 weeks by oral gavage at the dose of 10mg/Kg/day. Mice were sacrificed 2 weeks after PF04418948 treatment. Micro-Computer Tomography (Micro-CT) overviews of the skull (ventral view, Skull-V) (Figure 4A), lumbar spine (ventral view, Spine L) (Figure 4B) and femur (sagittal view, Femur-V) (Figure 4C) showed obvious decrease of HO formation in the muscle tissues in PF04418948 treated mice compared to the vehicle-treated mice. To quantify HO, we segmentalized entire femur muscle HO by drawing contour to exclude bone tissues. The segmental HO formation showed obvious decreases in the PF04418948 treated group compared to the vehicle treated group (Figure 4D). Quantification of the total muscle volume (TV) showed trend of increase in PF04418948 treated mice than vehicle treated mice(P=0.1576) (Figure 4E). Total femur muscle HO volume (BV) showed a decrease trend (P=0.1163) in PF04418948 treated versus the vehicle treated muscle (Figure 4F). Furthermore, BV/TV also trended to decrease in PF04418948 treated mice compared to the vehicle treated mice (P=0.1168) (Figure 4G). Additionally, mice body weight also was significantly higher in the PF04418948 treated group compared to the vehicle treated group at days 10 and 14 after treatment (Figure 4H).
3.5. PF04418948 Treatment Improved Muscle Pathology
To further investigate if PF04418948 treatment improves muscle pathology, Von Kossa staining was used to detect HO formation. The overall results showed that PF04418948 treatment decreased Von Kossa positive area, a finding consistent with results from the Micro-CT (Figure 5A). Furthermore, Hematoxylin and Eosin (H&E) staining demonstrated a large number of inflammatory cell patches (yellow arrows) in the vehicle treated group, while muscle from the PF04418948 treated group showed decreased inflammation and increased regeneration of long myofibers (multiple nucleated cells, green arrows) (Figure 5B). To investigate if PF04418948 treatment decreased inflammation and senescent cells, double immunofluorescent staining of CD68 (M1 macrophage) and P21 (senescent cells marker) was performed and revealed fewer green stained macrophage in the PF04418948-treated group compared to vehicle treated group. Furthermore, more central nuclear myofibers were identified in the PF04418948 treated group (Figure 5C). Quantification indicated that total CD68+ cells/200X field and CD68+P21+ senescent macrophages were significantly decreased in the PF04418948 treated group compared to the vehicle treated group (P=0.0316 and 0.0008, respectively) (Figure 5D,E). PF04418948 treatment also showed a trend of decrease in CD68-P21+(non-macrophage) senescent cells (P=0.1397) (Figure 5F). We also performed Gr-1 (neutrophil) and P21 double immunofluorescent staining and revealed that PF04418948 treatment did not change the total Gr-1+positive cells which were very few (Figure 5H). But Gr-1-P21+ cells (non-neutrophil senescent cells) were reduced by PF04418948 treatment (Figure 5I).
Finally, we performed CD31/P21 double immunofluorescence staining and revealed no significant differences in CD31+P21+ cells (senescent endothelial cells) between PF04418948 treated and the vehicle treated groups (Figure 5J-K). A trend of increased total CD31+ cells was detected in the PF04418948-treated group (Figure 5I,L), but no differences were detected in CD31-P21+ cells between the PF04418948-treated group and the control groups (Figure 5.M).
3.6. PF04418948 Treatment Changed HO Related Genes, Myokine, and Inflammatory Genes
We further performed Q-PCR analysis of the thigh muscle tissues for PF04418948 and the vehicle treated mice. We found a decreasing trend of Runx2 mRNA expression in the PF04418948 treated group compared to the vehicle treated group (Figure 6A). PF04418948 treatment significantly decreased Ctsk and Trap mRNA expression compared to the vehicle treatment (Figure 6B-C). Furthermore, PF04418948 treatment also significantly decreased Pax-7 expression (Figure 6D) and increased Mstn expression (Figure 6E). However, PF04418948 treatment demonstrated an increasing trend of IL-6 and IL1-β mRNA expression compared to the vehicle treated group (P=0.0767 and 0.0603, respectively).
3.7. PF04418948 Treatment Improved Trabecular Bone and Cortical Bone Microarchitecture of Long Bones
Micro-CT 3D images and quantification demonstrated that PF04418948 treatment did not change any trabecular bone parameters of spine L5 vertebrate trabecular bone including BV/TV, trabecular number (Tb.N), trabecular thickness (Tb.Th) and trabecular separation (Tb.Sp) (Figure 7A-E). Furthermore, Micro-CT 3D images and quantification revealed that PF04418948 treatment significantly increased BV/TV, Tb.Th, and demonstrated an increasing trend of BV density ( 613.74 vs 559.28 mgHA/cm3, P=0.116) of proximal tibia trabecular bone compared to vehicle treat group. No differences were found for Tb.N and Tb.Sp of the proximal tibia trabecular bone (Figure 7F-J). PF04418948 treatment also significantly increased the cortical thickness (Ct.Th) of femur cortical bone (Figure 7K-L) while did not change BV density (Figure 7M). In addition, PF04418948 treatment led to an increasing trend of tibia Ct.Th (0.1362 mm vs 0.1303mm, P=0.07) when compared to the vehicle treated group, and did not change BV density of tibia cortical bone)(data not shown). These results taken together demonstrated that PF04418948 treatment improved microarchitecture of long bones while not impacted spine trabecular bone.
3.8. Effects of PF04418948 Treatment on General Bone Morphology and Bone Matrix Collagen Type 1 (COL1)
H&E staining of the spine L5 vertebrate revealed no significant difference of trabecular bone structure between PF04418948 treated and the vehicle treated group (Figure 8A). Relatively more trabecular bone was found in proximal tibia of the PF04418948 treated group than the vehicle treated group and extended to the distal end of proximal tibia (Figure 8B). However, H&E staining of femoral cortical bone showed increased thickness in the PF04418948 group compared to the vehicle treated group (Figure 8C-D). Herovici’s staining, which differentiates COL 1 (pink red) and collagen type 3 (COL3) (dark blue), demonstrated spine L5 vertebrate trabecular bone in pink-red color and bone marrow in light blue color. No differences of trabecular bone density or thickness were found between PF04418948 treated group and the vehicle treat group (Figure 8E). Herovici’s staining of the proximal tibia demonstrated more pink-red collagen 1 positive trabecular bone in the PF04418948 treated group extending to distal side of proximal tibia compared to the vehicle treated group (Figure 8E). More strikingly, in the femur cortical bone, pink-red COL1 was obviously thicker in the PF04418948 treated group than the vehicle treated group (Figure 8G-H).
3.9. PF04418948 Treatment Increased Osteoclasts Without Changing Number of Osteoprogenitors in Trabecular Bone of Proximal Tibia
TRAP staining for osteoclasts was performed and quantified for spine L5 trabecular bone. No significant differences of TRAP+ cells were observed on the bone surface for the spine trabecular bone in the PF04418948 treated group when compared to the vehicle treated group (Figure 9A-B). Immunohistochemistry staining of OSX for osteoprogenitor cells showed a trend of increasing OSX+ cells (P=0.1461) in the PF04418948 treated group compared to the vehicle treated group (Figure 9 C-D). For the proximal tibia trabecular bone, PF04418948 treatment significantly increased TRAP+ osteoclasts than vehicle treated group (Figure 9E-F). However, PF004418948 treatment did significantly increase the OSX+ osteoprogenitor cells compared to vehicle treated group (Figure 9G-H).
4. Discussion
This study demonstrates that components of COX-2/PGE2/EP2/4 are all significantly upregulated in Mdx mice with more striking elevations in DKO-Hom mice, when compared to WT mice. Although 15-Pgdh, the enzyme which degrades PGE2, was also increased, the net result was a significant elevation of PGE2 in muscle tissues of the DKO-Hom mice. Both EP2 and EP4 were expressed in both CD68+macrophage and non-macrophage cells. PF04418948 treatment improved muscle pathology and decreased HO formation at a 10mg/Kg dosage potentially by decreasing the total CD68+ macrophages and senescent macrophages (CD68+P21+) without affecting neutrophils while increasing CD31+ endothelial cells. PF04418948 treatment also increased tibia trabecular bone BV/TV, Tb.Th, and femoral and tibia Ct.Th without negatively affecting the trabecular bone microarchitecture of spine L5. Our major findings are summarized in Figure 10.
Inflammation is one of the most profound clinical manifestations of DMD. Glucocorticoids (Deflazacort or prednisone) are currently considered the gold standard care for muscular dystrophy which improves the overall health of DMD patients and extend ambulatory time [40,41,42]. Further, Deflazacort treated boys have longer duration of ambulation than prednisone-treated patients (15.6 vs 13.5 years). Furthermore, Deflazacort was also associated with a lower risk of scoliosis, improved ambulatory function, a greater percentage of lean body mass, shorter statue, and lower body weight, after adjusting for age and steroid duration [43]. However, glucocorticoids treatment, including Deflazacort, increased vertebrate fracture risks for patients with DMD (with some reported 16-fold increase), and boys with a history of fracture(s) had a steep rate of functional decline [31,32,44,45]. Therefore, developing approaches targeting inflammation without negatively affecting bone is needed in conjunction with gene therapy to restore dystrophin.
This study investigated the role of the PGE2-EP2/4 pathway in pathogenesis of the DKO-Hom mice with the goal of developing a potential therapy for treatment of DMD patients. The Q-PCR results showed that Cox-2, Ep2 and Ep4 as well as 15-Pgdh are all significantly increased in the thigh muscle tissues of both Mdx and DKO-Hom mice, while COX-1 was not significantly changed. Furthermore, ELISA results demonstrated a significant elevation of PGE2 in both Mdx and DKO-Hom mice, although its degradation enzyme, 15-Pgdh, was also significantly increased. CD68 (M1 macrophage marker) mRNA is also dramatically increased in DKO-Hom mice (Figure 1). Immunofluorescent staining demonstrated that EP2 and EP4 receptors are present at significantly higher levels, in both macrophages and non-macrophages, in DKO-Hom mice compared to WT (Figure 2).
Thus far, very few studies have investigated PGE2 pathways in muscular dystrophy. Previous studies showed Mdx mice muscle tissues released more PGE2 in response to contraction [46,47] and followed similar pattern of creatine kinase release. In myotonic dystrophy type 1 (DM1), PGE2 was found to be increased through up-regulation of Cox-2, microsomal prostaglandin E synthase-1 (mPGES-1) and prostaglandin EP2/EP4 receptors. Up-regulation of PGE2 suppressed myogenic differentiation by decreasing the intracellular levels of calcium. Exogenous addition of acetylsalicylic acid, an inhibitor of Cox enzymes, abolished PGE2 abnormal secretion and restores the differentiation of DM1 muscle cells [48]. PGE2/EP2 acted downstream of notch signaling by inhibiting myogenic differentiation and promoting the self-renewal of human muscle progenitor cells [49]. Other study also demonstrated that COX2-KO muscled-derived stem cells are more easily differentiated into myofibers in calvarial bone defects even when they were transduced with retro-viral BMP4 [50]. However, no study, has investigated PGE2 pathways in the DKO-Hom mice which closely recapitulates the manifestations of DMD patients.
Since HO is one of the major muscle pathology changes in DKO-Hom mice [9,51], the osteogenic related genes were further investigated and found that Runx2, Ctsk and Trap mRNA are all significantly increased in Mdx mice and DKO-Hom mice, while OSX also shows a trend of upregulation in the DKO-Hom mice compared to WT mice. These results indicated that osteogenic-related genes and osteoclastogenic-related genes are up-regulated in the muscle tissues of DKO-Hom mice likely due to HO formation. However, Fst and Fndc5 (irisin) was significantly increased while Mstn was significantly decreased in Mdx and DKO-Hom mice compared to WT mice (Figure 2). These myokine gene expression changes favor muscle regeneration and likely did not contribute to the functional decline of DKO-Hom mice.
To further elucidate the role of PGE2/EP2 in the development of muscle pathology and HO formation, we treated DKO-Hom mice at 4 weeks old (a time when significant muscle damage and osteopenia were observed) with the EP2 antagonist PF04418948 for two weeks. Treatment with PF04418948 decreased HO as demonstrated by Micro-CT (skull, spine, and long bone surrounding muscle) and showed a decreasing trend of BV and BV/TV in the thigh muscle tissues. Von-Kossa staining also revealed decreased HO formation in the muscle tissues. H&E staining showed improved muscle pathology as revealed by a decrease in inflammatory cells with increased regenerated myofibers. PF04418948 treatment decreased total macrophages (CD68+) and senescent microphages (CD68+/P21+) but did not change Gr-1+ cells as well as Gr-1+/P21+ cells which are very few, and increased total CD31+ cells with no changes on CD31+/P21+ cells which are also very sparse in number (Figure 5). The Q-PCR results further showed that Runx-2, Ctsk and Trap expression were down-regulated which likely correlated with the decreased formation of HO as demonstrated by Micro-CT and Von Kossa staining. Interestingly, Pax-7 was down-regulated while Mstn expression up-regulated following treatment with PF04418948. Previously, it has been reported that Pax-7 and myogenin expression are mutually exclusive during myogenic differentiation; Pax-7 appears to be up-regulated in cells that escape differentiation and exit the cell cycle, suggesting a regulatory relationship between these two transcription factors. Indeed, overexpression of Pax-7 down-regulates MyoD, prevents myogenin induction, and blocks MyoD-induced myogenic conversion of 10T1/2 cells. Overexpression of Pax-7 also promotes cell cycle exit, even under proliferation conditions [52]. Pax-7 is also highly expressed during early myogenic differentiation of iPSCs and down-regulated when myogenin is induced [53]. A previous study showed Pax-7 is increased in the muscle tissues at 1 and 4 weeks in DKO-Hom mice and is decreased at 8 weeks compared to Mdx mice [5], while showing no difference compared to WT and Mdx mice at 4 weeks and decreased compared to WT and Mdx mice at 8 weeks [4]. These changes observed in the PF04428948 treated group are likely due to decreased muscle inflammation and increased regenerating fibers differentiated from the PAX-7 + satellite cells. The increase in Mstn is likely due to the improvement of muscle pathology, and its expression resumes to normal level because DKO-Hom mice have significantly decreased Mstn (Figure 2). The observation of no significant changes of Fst and Fndc5 indicate that the improvement of the muscle pathology was not due to regulation of these positive myokines. The insignificant changes in IL6 and IL1-β indicate that the beneficial effects of PF04418948 are not mediated by regulation of IL16 and IL1-β. It has been shown RhoA activation in macrophages also contributes to muscle HO formation [51] and that RhoA/Rock inhibition improves the beneficial effects of glucocorticoids in DKO-Hom mice [54]. The current study added a new mechanism of HO formation in DKO-Hom mice.
Previously, we have shown DKO-Hom mice developed osteopenia at 4 weeks of age [9]. Importantly, in this study, we demonstrated PF04418948 treatment increased BV/TV and Tb.Th of the proximal tibia. Strikingly, PF04418948 treatment increased the Ct.Th of the midshaft femur in addition to a trend of increasing of the tibia cortical bone thickness. This was further verified by COL1 staining (Herovici’s staining) (Figure 8). Increased TRAP+ osteoclasts were found in the proximal tibia, though no changes in the OSX+ osteogenic progenitor cells of the tibia trabecular bone were observed. Our previous study demonstrated that osteoblasts, osteoclasts, and osteocytes were all decreased in 4-week-old DKO-Hom mice trabecular bone [9] , but that osteoclasts increased at 6 weeks [9]. The increased osteoclasts in the proximal tibia were also likely caused by decreased inflammation (macrophages) in the muscle because monocyte-macrophages are also the progenitor cells of osteomacs which differentiate into osteoclasts on bone surface [55,56]. When muscle inflammation is improved, more monocyte-macrophage lineage cells will undergo osteoclast differentiation and resume normal balance of osteogenesis and osteoclastogenesis in the bone tissues.
The limitation of this study is that the treatment duration was only 2 weeks due to the rapid decline in overall health of the DKO-Hom mice. Whether long-term treatment would further improve muscle pathology and maintain bone microarchitecture requires additional studies. A previous study showed that COX-2KO mice had delayed fracture healing and can be largely rescued by EP4 agonist while EP2 agonist only marginally rescue the fracture healing phenotype of COX-2KO mice. These results indicate that EP2 plays a less important role in PGE2-mediated bone formation [57]. Therefore, targeting EP2 using antagonists may be relative safer for bone homeostasis. Periodical treatment may be more optimal, without the side effects observed in the intermittent glucocorticoid treatment.
5. Conclusion
In summary, this study elucidated the role of the PGE2/EP2 pathway in the development of muscle and bone pathology in a DKO-Hom mice model. EP2 antagonist PF04418948 improved muscle histopathology and the overall health of the DKO-Hom mice. PF044418948 treatment also increased proximal tibia trabecular bone BV/TV and Tb.Th while increased femoral and tibial cortical bone thickness without affecting spine L5 trabecular bone microarchitecture. Therefore, targeting EP2 is a potential palliative therapy for the treatment of DMD.
Acknowledgement: We thank Nicole Ehrhart and Laura Chubb from Colorado State University for their help with animal protocol.
Author Contributions
XG and JH conceived the project, XG designed experiments and wrote the original manuscript. XG, GZ performed the experiments. YC performed mice colony maintenance and genotyping. JJR and JEL revised the manuscript. BW provided DKO-Het mice and revised the manuscript. XX made the mechanism Graph (Figure 10). JH provided funding, project administration, revised and final approval of the manuscript.
Funding
This project is funded by NIH grant 1R01AR065445 and Shannon Philanthropy gifts.
Institutional Review Board Statement
This study was approved by IACUC of University of Texas Health Science Center at Houston (Protocol # AWC-15-0073) and Colorado State University (Protocol #1234).
Informed Consent Statement
Not Applicable.
Data Availability Statement
All original data will be made available upon query or request from corresponding authors.
Conflicts of Interest
JH received royalties annually from Cook Myosite Inc. All other authors have no conflict of interest.
References
- Koenig, M.; Hoffman, E.; Bertelson, C.; Monaco, A.; Feener, C.; Kunkel, L. Complete cloning of the duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Grady, R.; Teng, H.; Nichol, M.C.; Cunningham, J.C.; Wilkinson, R.S.; Sanes, J.R. Skeletal and Cardiac Myopathies in Mice Lacking Utrophin and Dystrophin: A Model for Duchenne Muscular Dystrophy. Cell 1997, 90, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Poddar, M.; Tang, Y.; Proto, J. D.; Sohn, J.; Mu, X.; Oyster, N.; Wang, B.; Huard, J. , Rapid depletion of muscle progenitor cells in dystrophic mdx/utrophin-/- mice. Hum Mol Genet 2014, 23, (18), 4786–800. [Google Scholar] [CrossRef]
- Mu, X.; Tang, Y.; Lu, A.; Takayama, K.; Usas, A.; Wang, B.; Weiss, K.; Huard, J. The role of Notch signaling in muscle progenitor cell depletion and the rapid onset of histopathology in muscular dystrophy. Hum. Mol. Genet. 2015, 24, 2923–2937. [Google Scholar] [CrossRef]
- Sohn, J.; Lu, A.; Tang, Y.; Wang, B.; Huard, J. Activation of non-myogenic mesenchymal stem cells during the disease progression in dystrophic dystrophin/utrophin knockout mice. Hum. Mol. Genet. 2015, 24, 3814–3829. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, C.; Liu, X.; Gao, L.; Zhang, W.; Huang, W.; Lu, X.; Wang, Z. [Study on the gene knockout model mice of Duchenne muscular dystrophy]. . 2003, 34, 210–3. [Google Scholar]
- Isaac, C.; Wright, A.; Usas, A.; Li, H.; Tang, Y.; Mu, X.; Greco, N.; Dong, Q.; Vo, N.; Kang, J.; et al. Dystrophin and utrophin “double knockout” dystrophic mice exhibit a spectrum of degenerative musculoskeletal abnormalities. J. Orthop. Res. 2012, 31, 343–349. [Google Scholar] [CrossRef]
- Gao, X.; Tang, Y.; Amra, S.; Sun, X.; Cui, Y.; Cheng, H.; Wang, B.; Huard, J. Systemic investigation of bone and muscle abnormalities in dystrophin/utrophin double knockout mice during postnatal development and the mechanisms. Hum. Mol. Genet. 2019, 28, 1738–1751. [Google Scholar] [CrossRef]
- Gao, X.; Sun, X.; Amra, S.; Cui, Y.; Deng, Z.; Cheng, H.; Zhang, G. W.; Huard, C. A.; Wang, B.; Huard, J. , Impaired bone defect and fracture healing in dystrophin/utrophin double-knockout mice and the mechanism. Am J Transl Res 2020, 12, (9), 5269–5282. [Google Scholar]
- Wakefield, P.M.; Tinsley, J.M.; A Wood, M.J.; Gilbert, R.; Karpati, G.; E Davies, K. Prevention of the dystrophic phenotype in dystrophin/utrophin-deficient muscle following adenovirus-mediated transfer of a utrophin minigene. Gene Ther. 2000, 7, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Kawano, R.; Ishizaki, M.; Maeda, Y.; Uchida, Y.; Kimura, E.; Uchino, M. Transduction of Full-length Dystrophin to Multiple Skeletal Muscles Improves Motor Performance and Life Span in Utrophin/Dystrophin Double Knockout Mice. Mol. Ther. 2008, 16, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Odom, G.L.; Gregorevic, P.; Allen, J.M.; Finn, E.; Chamberlain, J.S. Microutrophin Delivery Through rAAV6 Increases Lifespan and Improves Muscle Function in Dystrophic Dystrophin/Utrophin-deficient Mice. Mol. Ther. 2008, 16, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, J.; Fu, F.H.; Xiao, X. Systemic human minidystrophin gene transfer improves functions and life span of dystrophin and dystrophin/utrophin-deficient mice. J. Orthop. Res. 2009, 27, 421–426. [Google Scholar] [CrossRef]
- Xin, C.; Chu, X.; Wei, W.; Kuang, B.; Wang, Y.; Tang, Y.; Chen, J.; You, H.; Li, C.; Wang, B. Combined gene therapy via VEGF and mini-dystrophin synergistically improves pathologies in temporalis muscle of dystrophin/utrophin double knockout mice. Hum. Mol. Genet. 2021, 30, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Babbs, A.; Powell, D.; Kole, R.; Fletcher, S.; Wilton, S.D.; E Davies, K. Prevention of Dystrophic Pathology in Severely Affected Dystrophin/Utrophin-deficient Mice by Morpholino-oligomer-mediated Exon-skipping. Mol. Ther. 2010, 18, 198–205. [Google Scholar] [CrossRef]
- Goyenvalle, A.; Babbs, A.; Wright, J.; Wilkins, V.; Powell, D.; Garcia, L.; Davies, K. E. , Rescue of severely affected dystrophin/utrophin-deficient mice through scAAV-U7snRNA-mediated exon skipping. Hum Mol Genet 2012, 21, (11), 2559–71. [Google Scholar] [CrossRef]
- Delandistrogene moxeparvovec (Elevidys) for Duchenne muscular dystrophy. Med Lett Drugs Ther 2023, 65, (1686), 159–160.
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef]
- Duchêne, B.L.; Cherif, K.; Iyombe-Engembe, J.-P.; Guyon, A.; Rousseau, J.; Ouellet, D.L.; Barbeau, X.; Lague, P.; Tremblay, J.P. CRISPR-Induced Deletion with SaCas9 Restores Dystrophin Expression in Dystrophic Models In Vitro and In Vivo. Mol. Ther. 2018, 26, 2604–2616. [Google Scholar] [CrossRef]
- Hanson, B.; Stenler, S.; Ahlskog, N.; Chwalenia, K.; Svrzikapa, N.; Coenen-Stass, A.M.; Weinberg, M.S.; Wood, M.J.; Roberts, T.C. Non-uniform dystrophin re-expression after CRISPR-mediated exon excision in the dystrophin/utrophin double-knockout mouse model of DMD. Mol. Ther. - Nucleic Acids 2022, 30, 379–397. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.-M.; Koo, T.; Kim, K.; Lim, K.; Baek, G.; Kim, S.-T.; Kim, H.S.; Kim, D.-E.; Lee, H.; Chung, E.; et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018, 36, 536–539. [Google Scholar] [CrossRef]
- Chemello, F.; Chai, A.C.; Li, H.; Rodriguez-Caycedo, C.; Sanchez-Ortiz, E.; Atmanli, A.; Mireault, A.A.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci. Adv. 2021, 7, eabg4910. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, S.-L.; Liu, X.-R.; Huang, W.; Zhang, W.-X.; Lu, X.-L. [Bone marrow stem cells transplantation improve locomotive function of dystrophin/utrophin gene double knock-out mice]. . 2003, 25, 160–3. [Google Scholar] [PubMed]
- Li, Z.; Liu, H.-Y.; Lei, Q.-F.; Zhang, C.; Li, S.-N. Improved motor function in dko mice by intravenous transplantation of bone marrow-derived mesenchymal stromal cells. Cytotherapy 2011, 13, 69–77. [Google Scholar] [CrossRef]
- Filareto, A.; Darabi, R.; Perlingeiro, R. C. , Engraftment of ES-Derived Myogenic Progenitors in a Severe Mouse Model of Muscular Dystrophy. J Stem Cell Res Ther 2012, 10, (1). [Google Scholar] [CrossRef] [PubMed]
- Pang, R.-Q.; He, J.; Zhang, Y.-Y.; Xiong, F.; Ruan, G.-P.; Zhu, X.-Q.; Wang, Q.; Wang, J.-X.; Zhu, G.-X.; Zhao, J.; et al. Systemic delivery of human bone marrow embryonic-like stem cells improves motor function of severely affected dystrophin/utrophin–deficient mice. Cytotherapy 2014, 16, 1739–1749. [Google Scholar] [CrossRef]
- Voit, A.; Patel, V.; Pachon, R.; Shah, V.; Bakhutma, M.; Kohlbrenner, E.; McArdle, J.J.; Dell’italia, L.J.; Mendell, J.R.; Xie, L.-H.; et al. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Wada, E.; Tanihata, J.; Iwamura, A.; Takeda, S.; Hayashi, Y. K.; Matsuda, R. , Treatment with the anti-IL-6 receptor antibody attenuates muscular dystrophy via promoting skeletal muscle regeneration in dystrophin-/utrophin-deficient mice. Skelet Muscle 2017, 7, (1), 23. [Google Scholar] [CrossRef]
- Mayo, A.; Craven, B.; McAdam, L.; Biggar, W. Bone health in boys with Duchenne Muscular Dystrophy on long-term daily deflazacort therapy. Neuromuscul. Disord. 2012, 22, 1040–1045. [Google Scholar] [CrossRef]
- Singh, A.; Schaeffer, E.K.; Reilly, C.W. Vertebral Fractures in Duchenne Muscular Dystrophy Patients Managed With Deflazacort. J. Pediatr. Orthop. 2018, 38, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.; Wang, C.; Bushby, K.; Guglieri, M.; Horrocks, I.; Straub, V.; Ahmed, S. F.; Wong, S. C.; Network, U. K. N. C. , Fractures and Linear Growth in a Nationwide Cohort of Boys With Duchenne Muscular Dystrophy With and Without Glucocorticoid Treatment: Results From the UK NorthStar Database. JAMA Neurol 2019, 76, (6), 701–709. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B. C.; Remm, M.; Rozen, S. G. , Primer3--new capabilities and interfaces. Nucleic Acids Res 2012, 40, (15), e115. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Kõressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Usas, A.; Proto, J. D.; Lu, A.; Cummins, J. H.; Proctor, A.; Chen, C. W.; Huard, J. , Role of donor and host cells in muscle-derived stem cell-mediated bone repair: differentiation vs. paracrine effects. FASEB J 2014, 28, (8), 3792–809. [Google Scholar] [CrossRef]
- af Forselles, K. J.; Root, J.; Clarke, T.; Davey, D.; Aughton, K.; Dack, K.; Pullen, N. , In vitro and in vivo characterization of PF-04418948, a novel, potent and selective prostaglandin EP(2) receptor antagonist. Br J Pharmacol 2011, 164, (7), 1847–56. [Google Scholar] [CrossRef]
- Turner, N.J.; Pezzone, M.A.; Brown, B.N.; Badylak, S.F. Quantitative multispectral imaging of Herovici's polychrome for the assessment of collagen content and tissue remodelling. J. Tissue Eng. Regen. Med. 2011, 7, 139–148. [Google Scholar] [CrossRef]
- Gao, X.; Usas, A.; Tang, Y.; Lu, A.; Tan, J.; Schneppendahl, J.; Kozemchak, A.M.; Wang, B.; Cummins, J.H.; Tuan, R.S.; et al. A comparison of bone regeneration with human mesenchymal stem cells and muscle-derived stem cells and the critical role of BMP. Biomaterials 2014, 35, 6859–6870. [Google Scholar] [CrossRef]
- Guglieri, M.; Bushby, K.; McDermott, M. P.; Hart, K. A.; Tawil, R.; Martens, W. B.; Herr, B. E.; McColl, E.; Speed, C.; Wilkinson, J.; Kirschner, J.; King, W. M.; Eagle, M.; Brown, M. W.; Willis, T.; Griggs, R. C.; Group, F.-D. I. o. t. M. S.; Straub, V.; van Ruiten, H.; Childs, A. M.; Ciafaloni, E.; Shieh, P. B.; Spinty, S.; Maggi, L.; Baranello, G.; Butterfield, R. J.; Horrocks, I. A.; Roper, H.; Alhaswani, Z.; Flanigan, K. M.; Kuntz, N. L.; Manzur, A.; Darras, B. T.; Kang, P. B.; Morrison, L.; Krzesniak-Swinarska, M.; Mah, J. K.; Mongini, T. E.; Ricci, F.; von der Hagen, M.; Finkel, R. S.; O’Reardon, K.; Wicklund, M.; Kumar, A.; McDonald, C. M.; Han, J. J.; Joyce, N.; Henricson, E. K.; Schara-Schmidt, U.; Gangfuss, A.; Wilichowski, E.; Barohn, R. J.; Statland, J. M.; Campbell, C.; Vita, G.; Vita, G. L.; Howard, J. F., Jr.; Hughes, I.; McMillan, H. J.; Pegoraro, E.; Bello, L.; Burnette, W. B.; Thangarajh, M.; Chang, T. , Effect of Different Corticosteroid Dosing Regimens on Clinical Outcomes in Boys With Duchenne Muscular Dystrophy: A Randomized Clinical Trial. JAMA 2022, 327, (15), 1456–1468. [Google Scholar] [CrossRef]
- Wong, B.L.; Cook, T.; Miller, H. Prednisone and deflazacort in Duchenne muscular dystrophy: a patient perspective and plain language summary publication of the Cincinnati study. J. Comp. Eff. Res. 2022, 11, 779–786. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Sajeev, G.; Yao, Z.; McDonnell, E.; Elfring, G.; Souza, M.; Peltz, S.W.; Darras, B.T.; Shieh, P.B.; Cox, D.A.; et al. Deflazacort vs prednisone treatment for Duchenne muscular dystrophy: A meta-analysis of disease progression rates in recent multicenter clinical trials. Muscle Nerve 2019, 61, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Marden, J.R.; Freimark, J.; Yao, Z.; Signorovitch, J.; Tian, C.; Wong, B.L. Real-world outcomes of long-term prednisone and deflazacort use in patients with Duchenne muscular dystrophy: experience at a single, large care center. J. Comp. Eff. Res. 2020, 9, 177–189. [Google Scholar] [CrossRef]
- Phung, K.; McAdam, L.; Ma, J.; McMillan, H.J.; Jackowski, S.; Scharke, M.; Matzinger, M.-A.; Shenouda, N.; Koujok, K.; Jaremko, J.L.; et al. Risk factors associated with prevalent vertebral fractures in Duchenne muscular dystrophy. Osteoporos. Int. 2022, 34, 147–160. [Google Scholar] [CrossRef]
- Liaw, J.; Billich, N.; Carroll, K.; Adams, J.; Ryan, M.M.; Yiu, E.M.; Zacharin, M.; Simm, P.; Davidson, Z.E. Fracture risk and impact in boys with Duchenne muscular dystrophy: A retrospective cohort study. Muscle Nerve 2023, 67, 489–496. [Google Scholar] [CrossRef]
- McArdle, A.; Edwards, R.H.T.; Jackson, M.J. Effects of contractile activity on muscle damage in the dystrophin-deficient mdx mouse. Clin. Sci. 1991, 80, 367–371. [Google Scholar] [CrossRef]
- Jackson, M.J.; Brooke, M.H.; Kaiser, K.; Edwards, R. Creatine kinase and prostaglandin E, release from isolated Duchenne muscle. Neurology 1991, 41, 101–101. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, D.; Thebault, P.; Pelletier, R.; Chapdelaine, P.; Tarnopolsky, M.; Furling, D.; Puymirat, J. Abnormal prostaglandin E2 production blocks myogenic differentiation in myotonic dystrophy. Neurobiol. Dis. 2012, 45, 122–129. [Google Scholar] [CrossRef]
- Sakai-Takemura, F.; Nogami, K.; Elhussieny, A.; Kawabata, K.; Maruyama, Y.; Hashimoto, N.; Takeda, S.; Miyagoe-Suzuki, Y. Prostaglandin EP2 receptor downstream of Notch signaling inhibits differentiation of human skeletal muscle progenitors in differentiation conditions. Commun. Biol. 2020, 3, 1–13. [Google Scholar] [CrossRef]
- Gao, X.; Usas, A.; Lu, A.; Kozemchak, A.; Tang, Y.; Poddar, M.; Sun, X.; Cummins, J.H.; Huard, J. Cyclooxygenase-2 deficiency impairs muscle-derived stem cell-mediated bone regeneration via cellular autonomous and non-autonomous mechanisms. Hum. Mol. Genet. 2016, 25, 3216–3231. [Google Scholar] [CrossRef]
- Mu, X.; Lin, C.-Y.; Hambright, W.S.; Tang, Y.; Ravuri, S.; Lu, A.; Matre, P.; Chen, W.; Gao, X.; Cui, Y.; et al. Aberrant RhoA activation in macrophages increases senescence-associated secretory phenotypes and ectopic calcification in muscular dystrophic mice. Aging 2020, 12, 24853–24871. [Google Scholar] [CrossRef] [PubMed]
- Olguin, H.C.; Olwin, B.B. Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: a potential mechanism for self-renewal. Dev. Biol. 2004, 275, 375–388. [Google Scholar] [CrossRef] [PubMed]
- van der Wal, E.; Herrero-Hernandez, P.; Wan, R.; Broeders, M.; In ‘t Groen, S. L. M.; van Gestel, T. J. M.; van, I. W. F. J.; Cheung, T. H.; van der Ploeg, A. T.; Schaaf, G. J.; Pijnappel, W. , Large-Scale Expansion of Human iPSC-Derived Skeletal Muscle Cells for Disease Modeling and Cell-Based Therapeutic Strategies. Stem Cell Reports 2018, 10, (6), 1975–1990. [Google Scholar] [CrossRef]
- Mu, X.; Tang, Y.; Takayama, K.; Chen, W.; Lu, A.; Wang, B.; Weiss, K.; Huard, J. RhoA/ROCK inhibition improves the beneficial effects of glucocorticoid treatment in dystrophic muscle: implications for stem cell depletion. Hum. Mol. Genet. 2017, 26, 2813–2824. [Google Scholar] [CrossRef] [PubMed]
- Weivoda, M.M.; Bradley, E.W. Macrophages and Bone Remodeling. J. Bone Miner. Res. 2023, 38, 359–369. [Google Scholar] [CrossRef]
- Batoon, L.; Millard, S.M.; Wullschleger, M.E.; Preda, C.; Wu, A.C.-K.; Kaur, S.; Tseng, H.-W.; Hume, D.A.; Levesque, J.-P.; Raggatt, L.J.; et al. CD169+ macrophages are critical for osteoblast maintenance and promote intramembranous and endochondral ossification during bone repair. Biomaterials 2019, 196, 51–66. [Google Scholar] [CrossRef]
- Xie, C.; Liang, B.; Xue, M.; Lin, A. S.; Loiselle, A.; Schwarz, E. M.; Guldberg, R. E.; O’Keefe, R. J.; Zhang, X. , Rescue of impaired fracture healing in COX-2-/- mice via activation of prostaglandin E2 receptor subtype 4. Am J Pathol 2009, 175, (2), 772–85. [Google Scholar] [CrossRef]
Figure 1.
PGE2/EP2/4 pathway mRNA expressions in the thigh muscle of WT, Mdx and DKO-Hom mice by Q-PCR. (A) Ep2 mRNA expression. (B) Ep4 mRNA. (C) Cox-1 mRNA expression. (D) Cox-2 mRNA expression. (E)15Pgdh mRNA expression. (F) CD68 mRNA expression. (G) PGE2 level in muscle tissue homogenates of the 4-week-old mice. Exact P values are indicated between group bars.
Figure 1.
PGE2/EP2/4 pathway mRNA expressions in the thigh muscle of WT, Mdx and DKO-Hom mice by Q-PCR. (A) Ep2 mRNA expression. (B) Ep4 mRNA. (C) Cox-1 mRNA expression. (D) Cox-2 mRNA expression. (E)15Pgdh mRNA expression. (F) CD68 mRNA expression. (G) PGE2 level in muscle tissue homogenates of the 4-week-old mice. Exact P values are indicated between group bars.

Figure 2.
Osteogenesis, osteoclastogenesis and myokine mRNA expression in 4-week-old mice and Von-Kossa staining. (A) Runx2 mRNA expression. (B) Osx mRNA expression. (C) Ctsk mRNA expression. (D) Trap mRNA expression. (E) Fst mRNA expression. (F) Fndc5 mRNA expression. (G) Mstn mRNA expression. (H) Von Kossa staining for gastrocnemius muscle tissues. Large amount of brown-black color staining was found in the gastrocnemius muscle of Mdx and DKO-Hom mice which indicates heterotopic bone formation (mineralization). Scale bar =100µm. Exact P values are indicated between group bars.
Figure 2.
Osteogenesis, osteoclastogenesis and myokine mRNA expression in 4-week-old mice and Von-Kossa staining. (A) Runx2 mRNA expression. (B) Osx mRNA expression. (C) Ctsk mRNA expression. (D) Trap mRNA expression. (E) Fst mRNA expression. (F) Fndc5 mRNA expression. (G) Mstn mRNA expression. (H) Von Kossa staining for gastrocnemius muscle tissues. Large amount of brown-black color staining was found in the gastrocnemius muscle of Mdx and DKO-Hom mice which indicates heterotopic bone formation (mineralization). Scale bar =100µm. Exact P values are indicated between group bars.
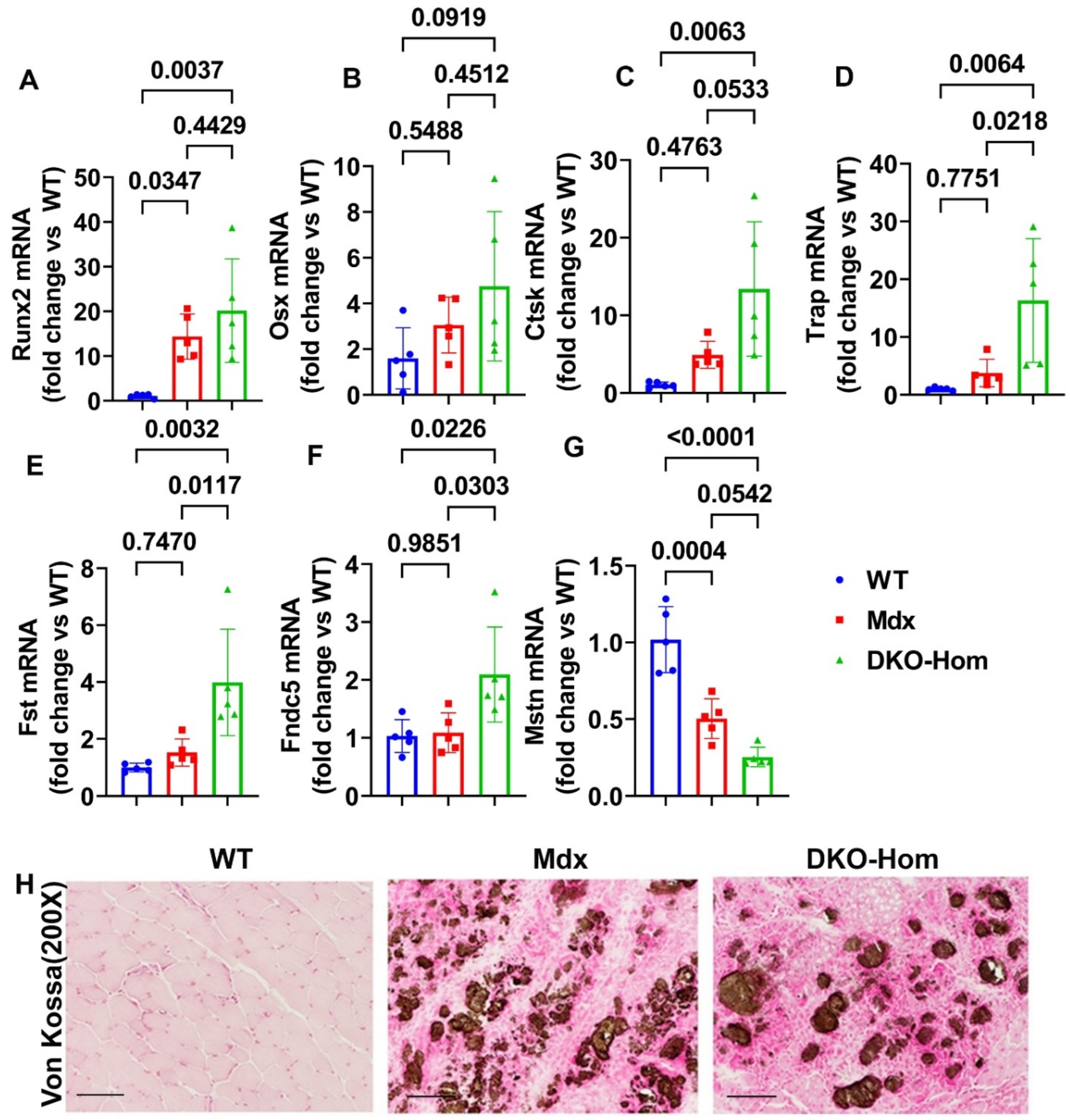
Figure 3.
Immunofluorescence staining of EP2 and EP4 receptors and colocalization with CD68 macrophage in 4-week-old muscle tissues. (A) EP2/CD68 double staining. CD68 stained with green, EP2 stained red, colocalized cells showed yellow or orange color. (B-D) Quantification of CD68+macrophage, CD68+EP2+ and CD68-EP2+ cells. (E) EP4/CD68 double staining. EP4 stained red, CD68 stained green. Colocalized cells stained yellow or orange. (F-H) Quantification of CD68+ macrophage, CD68+EP4+ and EP4+ cells. Exact P values are indicated between group bars. Scale bars=100µm.
Figure 3.
Immunofluorescence staining of EP2 and EP4 receptors and colocalization with CD68 macrophage in 4-week-old muscle tissues. (A) EP2/CD68 double staining. CD68 stained with green, EP2 stained red, colocalized cells showed yellow or orange color. (B-D) Quantification of CD68+macrophage, CD68+EP2+ and CD68-EP2+ cells. (E) EP4/CD68 double staining. EP4 stained red, CD68 stained green. Colocalized cells stained yellow or orange. (F-H) Quantification of CD68+ macrophage, CD68+EP4+ and EP4+ cells. Exact P values are indicated between group bars. Scale bars=100µm.
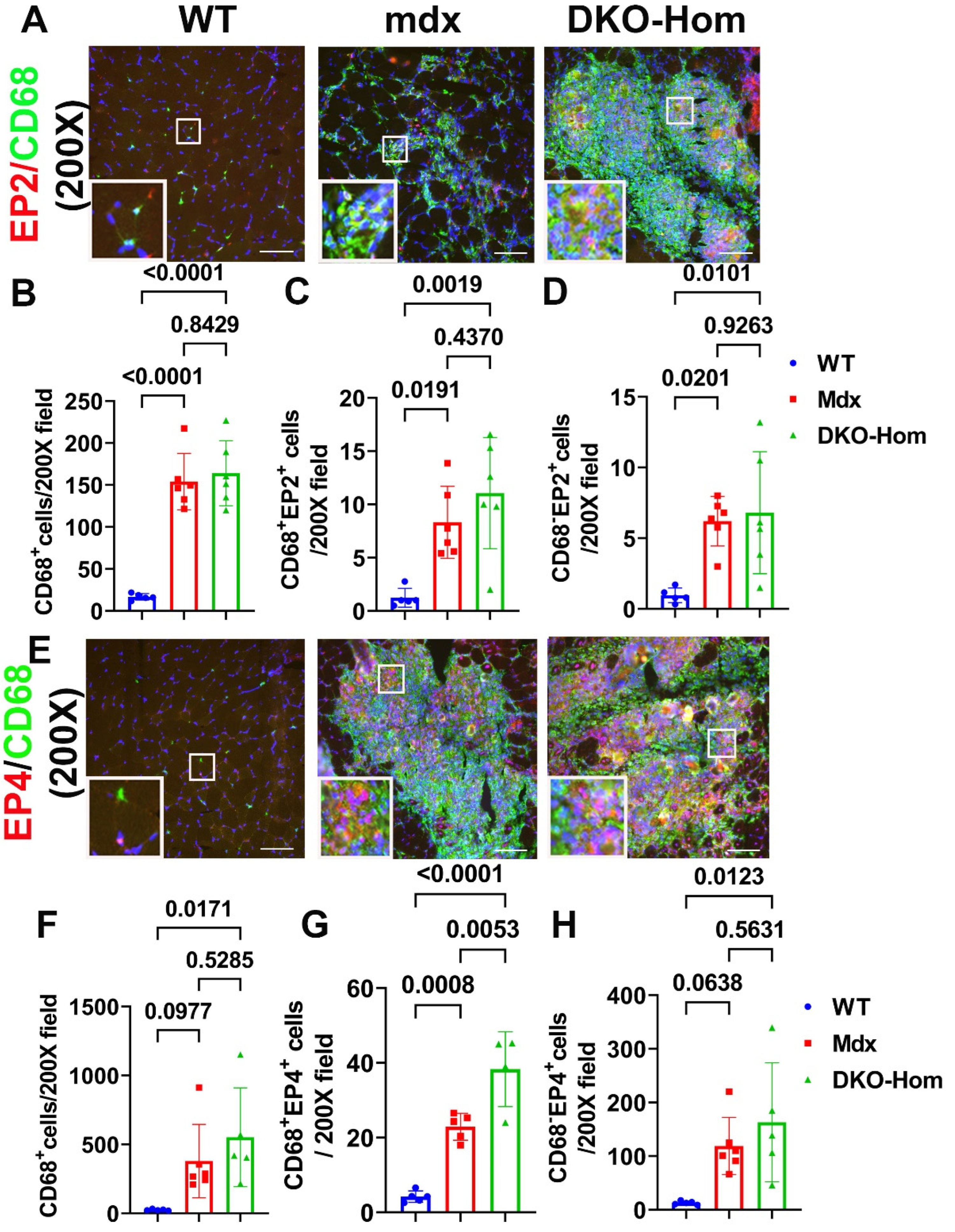
Figure 4.
Effect of EP2 antagonist PF04418948 on muscle HO formation. (A) Micro-CT 3D images of entire skull. (B) Lumbar spine overview for HO formation in spine surrounding muscles. (C) Micro-CT sagittal view of femur surrounding muscle tissues. (D) Micro-CT 3D segmental view of HO in thigh muscles. Red arrows indicate HO in all images. (E) Quantification of TV of the entire thigh muscle. (F) Micro-CT BV of entire thigh muscle. (G) BV/TV of HO in the thigh muscle. (H) Body weight at different time points of treatment. Scale bars for micro-CT images =1mm.
Figure 4.
Effect of EP2 antagonist PF04418948 on muscle HO formation. (A) Micro-CT 3D images of entire skull. (B) Lumbar spine overview for HO formation in spine surrounding muscles. (C) Micro-CT sagittal view of femur surrounding muscle tissues. (D) Micro-CT 3D segmental view of HO in thigh muscles. Red arrows indicate HO in all images. (E) Quantification of TV of the entire thigh muscle. (F) Micro-CT BV of entire thigh muscle. (G) BV/TV of HO in the thigh muscle. (H) Body weight at different time points of treatment. Scale bars for micro-CT images =1mm.
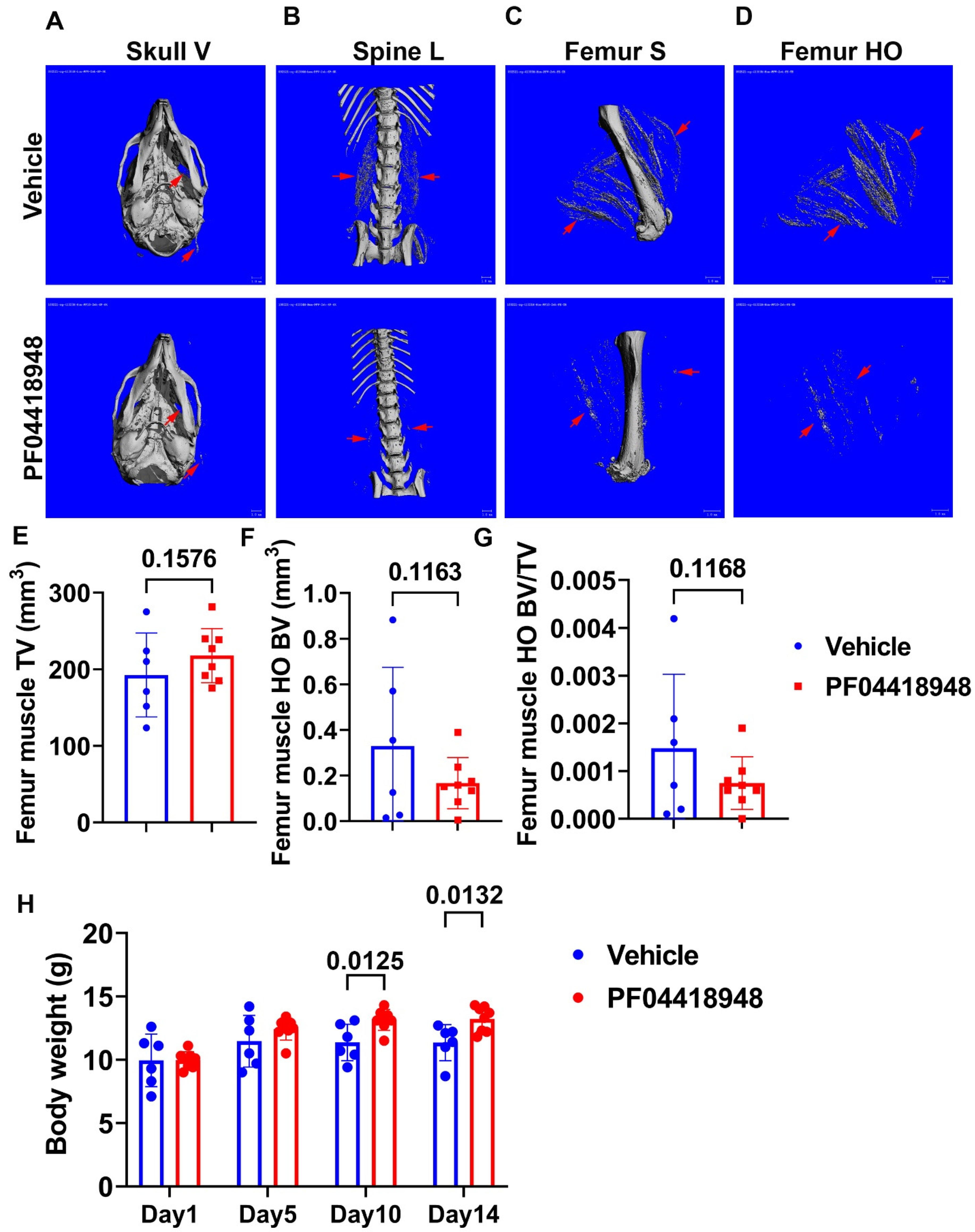
Figure 5.
Muscle histology and inflammation after treatment with PF04418948. (A) Von Kossa staining for HO. Brown-black color indicates HO. Scale bar=200µm. (B) H&E staining. A large number of inflammatory cells in the vehicle treated group were identified, while more regenerated muscle fibers were identified in the PF04418948 group. Yellow arrows indicate inflammatory cells, green arrows indicate regenerating multiple nucleated cells. Scale bars = 100µm. (C) CD68/P21 double staining. Insets highlighted positive cells, CD68 stained green, P21 stained red. Scale bars = 100µm. (D-F) Quantification of CD68+, CD68+P21+ and CD68-P21+cells. (G) Gr-1/P21 double staining. Insets highlighted positive cells, Gr-1 stained green, P21 stained red. Scale bars = 100µm. (H-I) Gr-1+, Gr-1-P21+ cell quantification. (J) CD31/P21 double staining. Insets highlighted positive cells. CD31 stain in green, P21 stain in red. Scale bars = 100µm. (K-M) CD31+P21+, Total CD31+ and CD31-P21+ cell quantification. Exact P values are shown between group bars.
Figure 5.
Muscle histology and inflammation after treatment with PF04418948. (A) Von Kossa staining for HO. Brown-black color indicates HO. Scale bar=200µm. (B) H&E staining. A large number of inflammatory cells in the vehicle treated group were identified, while more regenerated muscle fibers were identified in the PF04418948 group. Yellow arrows indicate inflammatory cells, green arrows indicate regenerating multiple nucleated cells. Scale bars = 100µm. (C) CD68/P21 double staining. Insets highlighted positive cells, CD68 stained green, P21 stained red. Scale bars = 100µm. (D-F) Quantification of CD68+, CD68+P21+ and CD68-P21+cells. (G) Gr-1/P21 double staining. Insets highlighted positive cells, Gr-1 stained green, P21 stained red. Scale bars = 100µm. (H-I) Gr-1+, Gr-1-P21+ cell quantification. (J) CD31/P21 double staining. Insets highlighted positive cells. CD31 stain in green, P21 stain in red. Scale bars = 100µm. (K-M) CD31+P21+, Total CD31+ and CD31-P21+ cell quantification. Exact P values are shown between group bars.
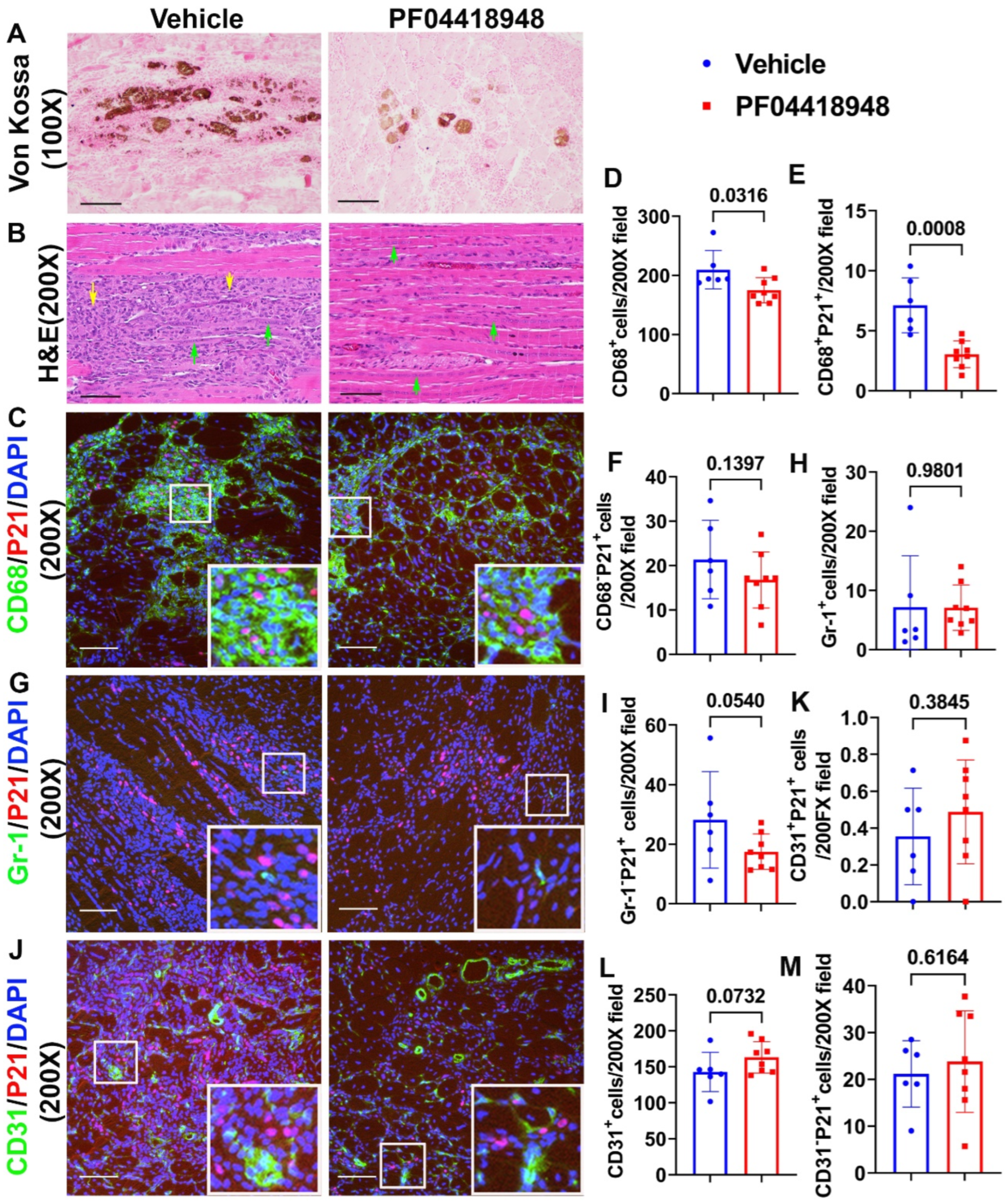
Figure 6.
Muscle gene expression after treatment with PF04418948 detected by Q-PCR. (A) Runx2 mRNA expression. (B) Ctsk mRNA expression. (C) Trap mRNA expression. (D) Pax-7 mRNA expression. (E) Mstn mRNA expression. (F) Il6 mRNA expression. (G) Il-1β mRNA expression.
Figure 6.
Muscle gene expression after treatment with PF04418948 detected by Q-PCR. (A) Runx2 mRNA expression. (B) Ctsk mRNA expression. (C) Trap mRNA expression. (D) Pax-7 mRNA expression. (E) Mstn mRNA expression. (F) Il6 mRNA expression. (G) Il-1β mRNA expression.
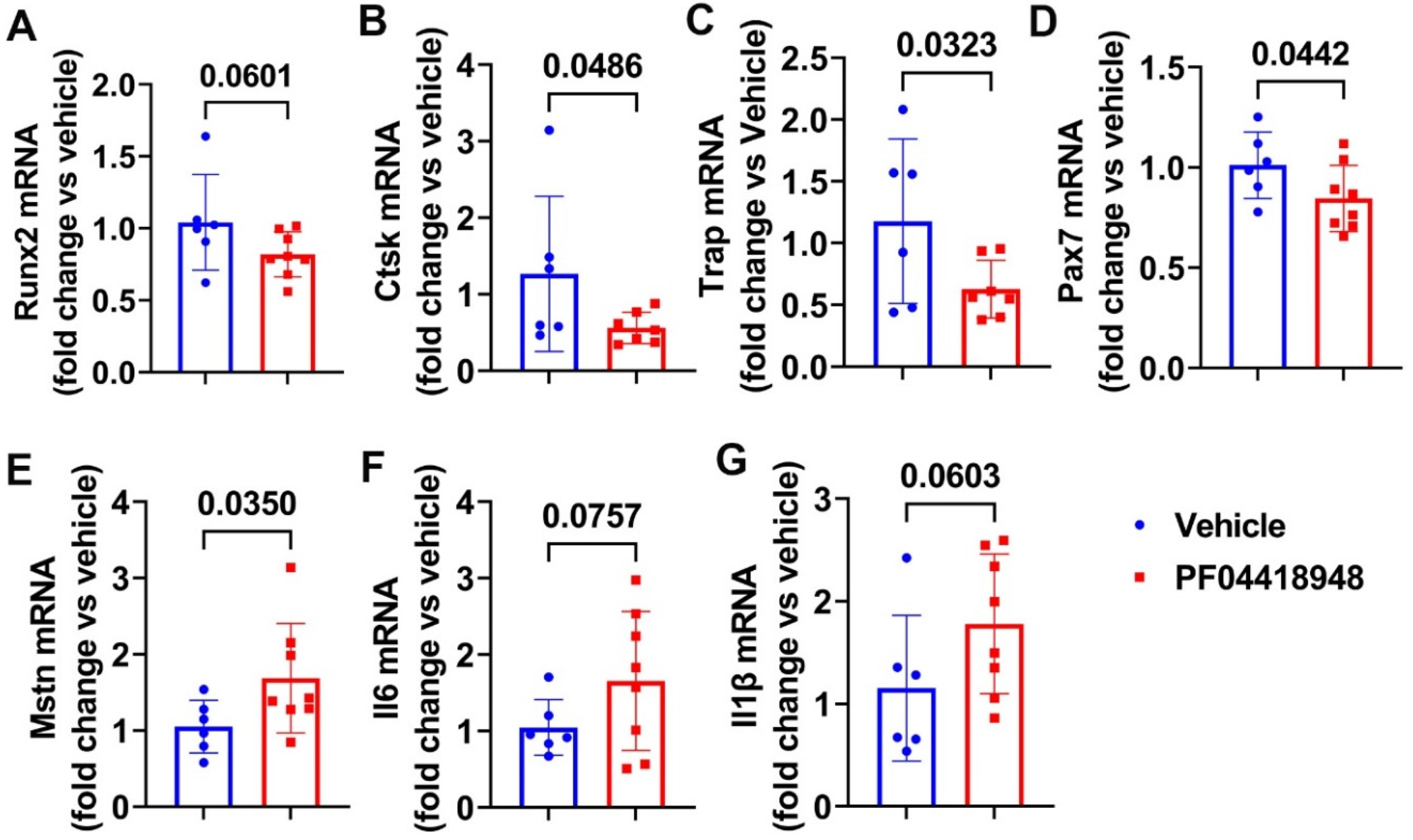
Figure 7.
Micro-CT analysis of bone tissues. (A) Micro-CT 3D images of spine L5 with ventral and dorsal views. (B-E) BV/TV, Tb.N, Tb.Th and Tb.Sp of spine L5. (F) Micro-CT 3D images of top and sagittal views of the proximal tibia trabecular bone. (G-H) BV/TV, Tb.N, Tb.Th, Tb.Sp of proximal tibia trabecular bone. (K) Micro-CT 3D view of femur cortical bone. (L-M) Ct.Th and BV density of femur cortical bone. Scale bars = 100µm. For proximal tibia BV/TV, Wilcoxon Rank Sum test was used due to higher variation. All other comparison used the Student T test.
Figure 7.
Micro-CT analysis of bone tissues. (A) Micro-CT 3D images of spine L5 with ventral and dorsal views. (B-E) BV/TV, Tb.N, Tb.Th and Tb.Sp of spine L5. (F) Micro-CT 3D images of top and sagittal views of the proximal tibia trabecular bone. (G-H) BV/TV, Tb.N, Tb.Th, Tb.Sp of proximal tibia trabecular bone. (K) Micro-CT 3D view of femur cortical bone. (L-M) Ct.Th and BV density of femur cortical bone. Scale bars = 100µm. For proximal tibia BV/TV, Wilcoxon Rank Sum test was used due to higher variation. All other comparison used the Student T test.
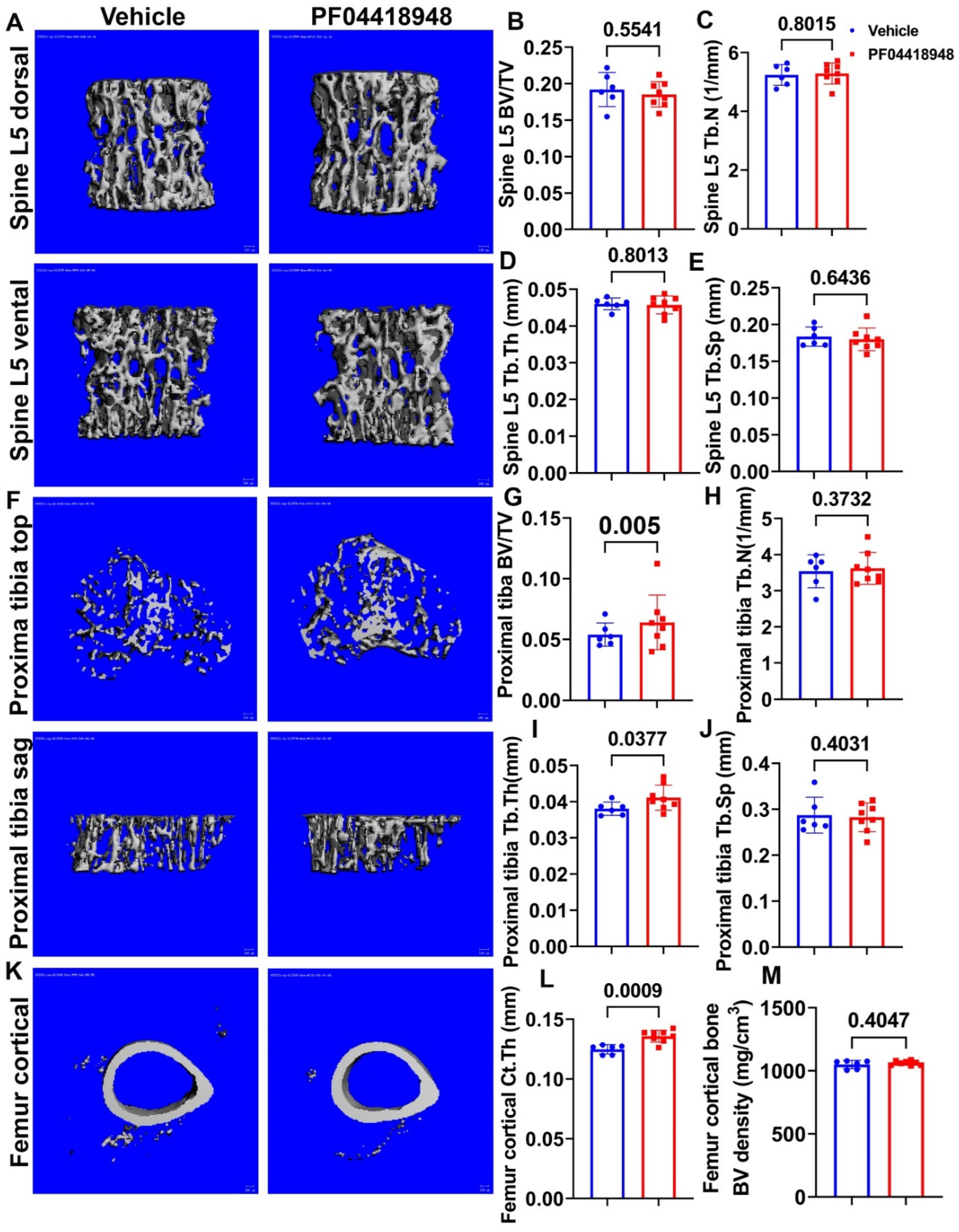
Figure 8.
H&E staining and Herovici’s staining of bone tissues of DKO-Hom mice treated with PF04418948 or vehicle. (A) H&E staining of spine L5 trabecular bone. No significant difference between PF04418948 treated and vehicle treated group was identified. (B) H&E staining of proximal tibia. More trabecular bone extended to the distal side of proximal tibia in the PF04418948 treated group compared to the vehicle treated group. (C-D) H&E staining of femur cortical bone at 40X and 200X. PF04418948 treated group showed thicker femur cortical bone than vehicle treated group. (E) Herovici’s staining of Spine L5 trabecular bone. COL1 stained a pink-red color, COL3 stained a dark blue color. No difference was found between the PF04418948 treated group and vehicle treated group. (F) Herovici’s staining of proximal tibia. The PF04418948-treated group showed more trabecular bone than vehicle treated group extending to the distal side of the proximal tibia. (G-H). Herovici’s staining of femur cortical bone at 40X and 200X. Femur cortical collagen 1 (pink-red) positive matrix is thicker in the PF04418948-treated than the vehicle-treated group. Scale bars = 500µm for 40X and 100µm for 200X.
Figure 8.
H&E staining and Herovici’s staining of bone tissues of DKO-Hom mice treated with PF04418948 or vehicle. (A) H&E staining of spine L5 trabecular bone. No significant difference between PF04418948 treated and vehicle treated group was identified. (B) H&E staining of proximal tibia. More trabecular bone extended to the distal side of proximal tibia in the PF04418948 treated group compared to the vehicle treated group. (C-D) H&E staining of femur cortical bone at 40X and 200X. PF04418948 treated group showed thicker femur cortical bone than vehicle treated group. (E) Herovici’s staining of Spine L5 trabecular bone. COL1 stained a pink-red color, COL3 stained a dark blue color. No difference was found between the PF04418948 treated group and vehicle treated group. (F) Herovici’s staining of proximal tibia. The PF04418948-treated group showed more trabecular bone than vehicle treated group extending to the distal side of the proximal tibia. (G-H). Herovici’s staining of femur cortical bone at 40X and 200X. Femur cortical collagen 1 (pink-red) positive matrix is thicker in the PF04418948-treated than the vehicle-treated group. Scale bars = 500µm for 40X and 100µm for 200X.
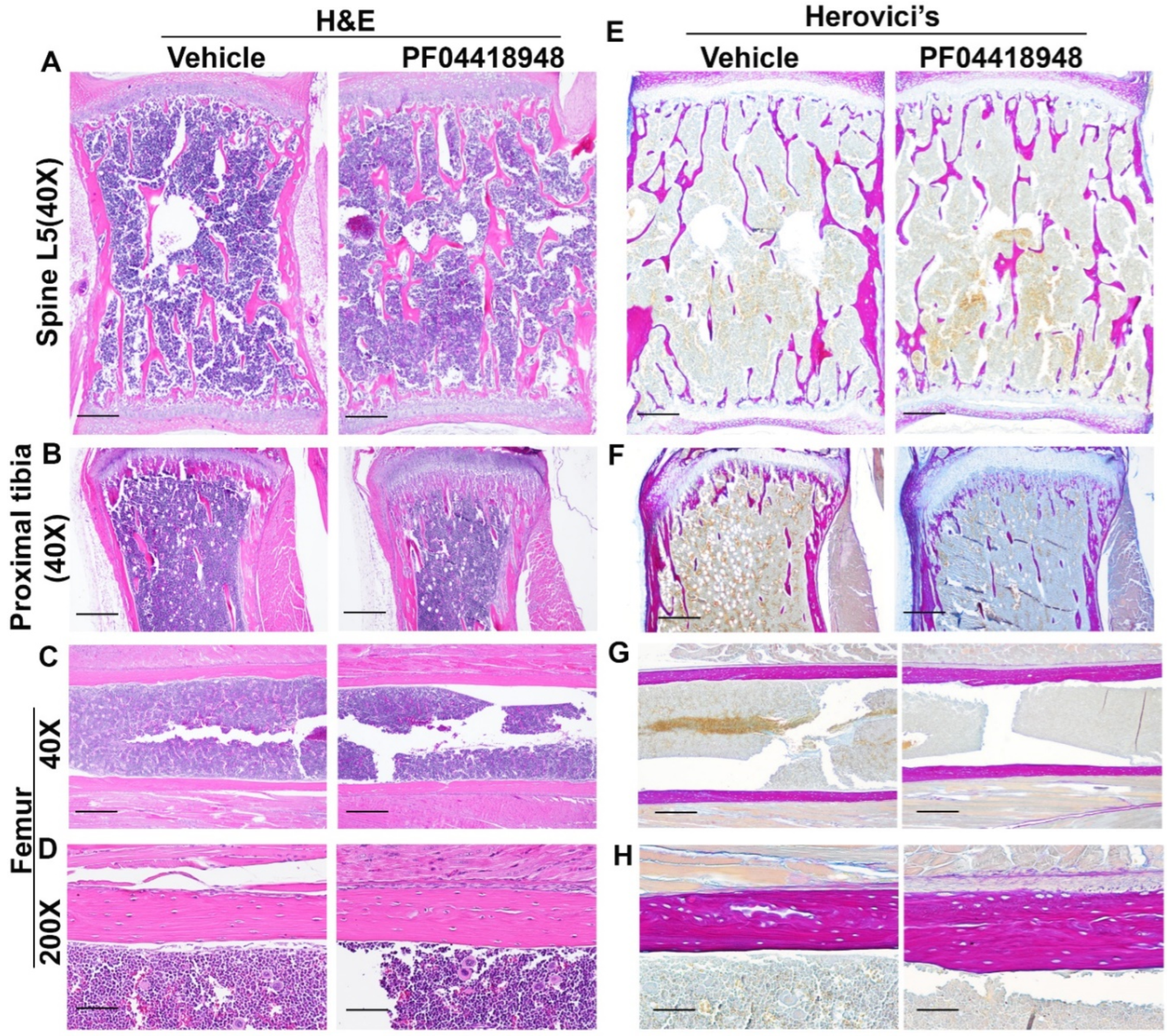
Figure 9.
Bone osteoclasts and osteoblast changes after PF04418948 treatment for DKO-Hom mice. (A-B) TRAP staining of spine L5 vertebrate trabecular bone. TRAP+ osteoclasts stained violet-red. No significant difference for TRAP+ cells/bone surface between the PF04418948 and the vehicle treated groups. (C-D) Immunohistochemistry staining of OSX for spine L5 vertebrate trabecular bone. OSX+ cells stained brown color in the nuclei. OSX+ positive cells/bone surface showed trend of increasing in the PF04418948 treated group. P=0.1461. (E-F) TRAP staining of proximal tibia and quantification. PF04418948 treatment significantly increased TRAP+ cells compared to the vehicle treated group. (G-H) Immunohistochemistry staining of OSX of the proximal tibia. No difference between PF04418948 treated and the vehicle treated mice. Scale bars =100µm for all the images.
Figure 9.
Bone osteoclasts and osteoblast changes after PF04418948 treatment for DKO-Hom mice. (A-B) TRAP staining of spine L5 vertebrate trabecular bone. TRAP+ osteoclasts stained violet-red. No significant difference for TRAP+ cells/bone surface between the PF04418948 and the vehicle treated groups. (C-D) Immunohistochemistry staining of OSX for spine L5 vertebrate trabecular bone. OSX+ cells stained brown color in the nuclei. OSX+ positive cells/bone surface showed trend of increasing in the PF04418948 treated group. P=0.1461. (E-F) TRAP staining of proximal tibia and quantification. PF04418948 treatment significantly increased TRAP+ cells compared to the vehicle treated group. (G-H) Immunohistochemistry staining of OSX of the proximal tibia. No difference between PF04418948 treated and the vehicle treated mice. Scale bars =100µm for all the images.
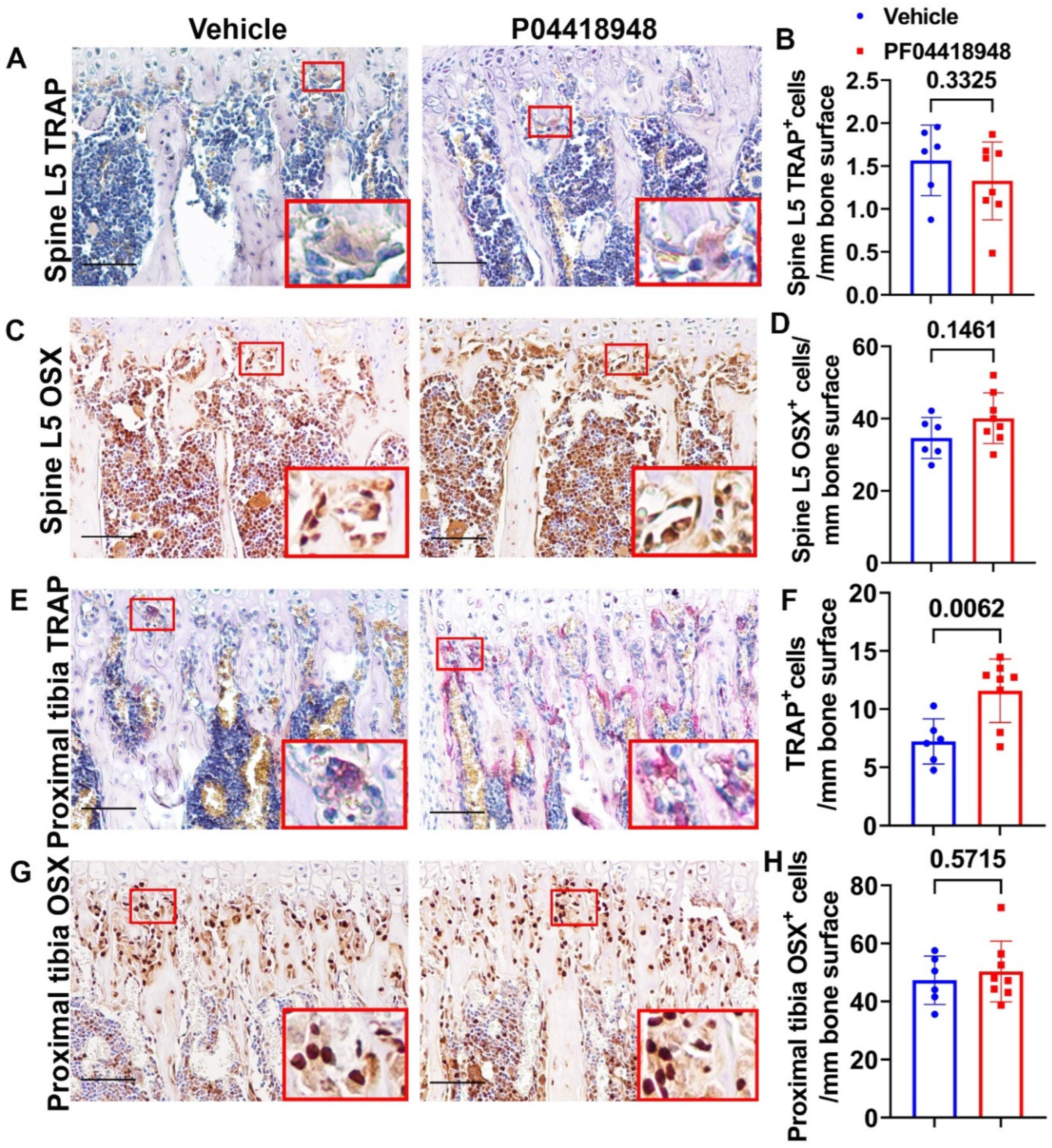
Figure 10.
Schematic mechanism summary of the results. This graph was created by Xiang Xiao using Figma.
Figure 10.
Schematic mechanism summary of the results. This graph was created by Xiang Xiao using Figma.
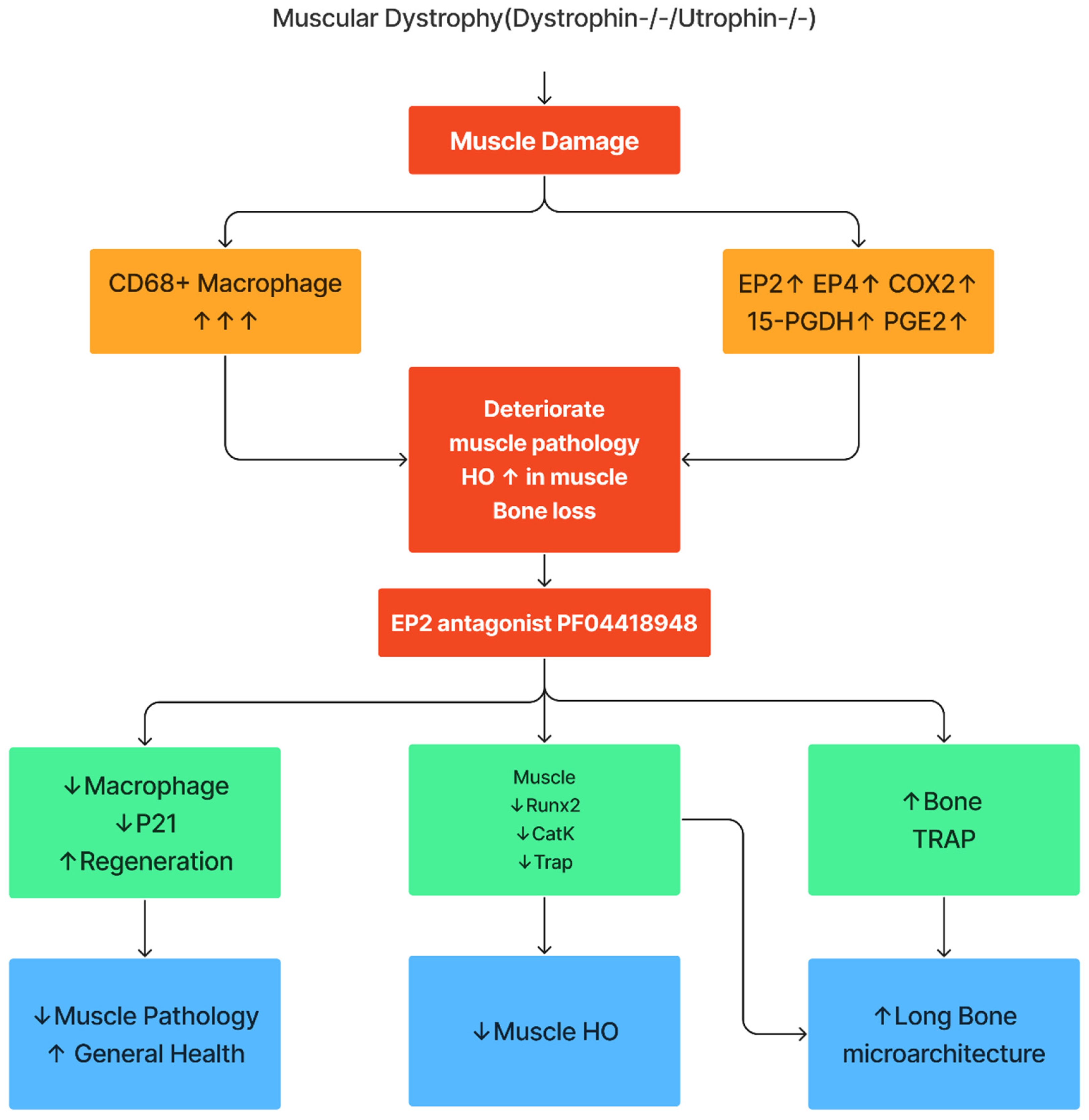

Table 1.
Primer information.
| Gene name | Forward primers (5′-3′) | Reverse primers (5′-3′) | Product size(bp) |
|---|---|---|---|
| Ep2 | ATGCTCCTGCTGCTTATCGT | AGGGCCTCTTAGGCTACTGC | 126 |
| Ep4 | TGGCTGTCACTGACCTTCTG | TGCATAGATGGCGAAGAGTG | 254 |
| Cox-1 | GTGGCTATTTCCTGCAGCTC | CAGTGCCTCAACCCCATAGT | 209 |
| Cox-2 | GGGCCCTTCCTCCCGTAGCA | CCATGGCCCAGTCCTCGGGT | 232 |
| 15-Pgdh | AGGTAGCATTGGTGGATTGG | CCACATCACACTGGACGAAC | 105 |
| CD68 | TTCTGCTGTGGAAATGCAAG | AGAGGGGCTGGTAGGTTGAT | 241 |
| Fst | TGACAATGCCACATACGCCA | CCTCCTCCTCCTCTTCCTCC | 131 |
| Mstn | TCAGACCCGTCAAGACTCCT | GGTCCTGGGAAGGTTACAGC | 253 |
| Il1β | ACTCATTGTGGCTGTGGAGA | TTGTTCATCTCGGAGCCTGT | 199 |
| Il-6 | CCGGAGAGGAGACTTCACAG | CAGAATTGCCATTGCACAAC | 134 |
| Runx2 | CCCAGCCACCTTTACCTACA | TATGGAGTGCTGCTGGTCTG | 150 |
| Osx | ACTCATCCCTATGGCTCGTG | GGTAGGGAGCTGGGTTAAGG | 238 |
| Ctsk | CCAGTGGGAGCTATGGAAGA | TGGTTCATGGCCAGTTCATA | 159 |
| Trap | CAGCAGCCAAGGAGGACTAC | ACATAGCCCACACCGTTCTC | 190 |
| Gapdh | CCGGGGCTGGCATTGCTCTC | GTGTTGGGGGCCGAGTTGGG | 190 |
| Pax7 | GACTCCGGATGTGGAGAAAA | GAGCACTCGGCTAATCGAAC | 145 |
| Fndc5 | CACGCGAGGCTGAAAAGATG | ACACCTGCCCACATGAAGAG | 130 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



