
Submitted:
04 November 2024
Posted:
04 November 2024
You are already at the latest version
Abstract
The effect of the Mn3+ ion on the local distortions of the FeO6 octahedra in orthoferrite samples was investigated. Mössbauer spectroscopy measurements for a series of the HoFe1−xMnxO3 (x=0.0−0.7) orthoferrite samples with space group Pnma was carried out at temperatures above the Néel point (700 K). The electric field gradient (EFG) tensor on Fe ions for these compounds was found using the first-principle calculations. The concentration dependence of the quadrupole splitting was resulted from the experimental and theoretical data. The absence of an electron contribution to the quadrupole splitting at all the manganese concentrations was demonstrated. It was shown by the theoretical calculations that the values of all the electric field gradient components increase significantly with the manganese concentration in the system, and the eigenvectors Vxx and Vyy of the electric field gradient tensor sharply change their direction at concentrations of x > 0.1.
Keywords:
orthoferrites
; electric field gradient
; Mössbauer spectroscopy
; first-principle calculations
1. Introduction
To date, crystals of rare-earth orthoferrites (R = La, Pr, ..., Lu, and Y) of almost all available compositions have been synthesized and studied [1,2,3]. The crystal structure is described by space group of the orthorhombic system and the unit cell contains four formula units (Z = 4). This is the distorted structure of a perfect perovskite, in which distortions are mainly caused by oxygen ion displacements leading to rotations of FeO octahedra (Figure 1). Briefly, this structure can be described as a frame of vertex-sharing FeO octahedral with rare-earth element atoms in voids between transition ion octahedra.
Orthoferrites undergo magnetic phase transitions of several types, including the spontaneous spin-reorientation transitions [4]. The complex magnetic behavior of the compounds still evokes keen interest of both experimentalist and theoreticians [3]. Over the past few decades, rare-earth orthoferrites have been in focus of researchers [5,6]. The magnetic structure of the high-temperature orthoferrite phase is a canted antiferromagnet. The relatively small canting of the magnetic moments in the subsystem causes a weak ferromagnetic moment in these compounds [7]. Orthoferrite crystals have high Néel temperature , which decreases monotonically as the atomic number of a rare-earth element grows [8].
Below the Néel temperature, the orthoferrites undergo a spin-reorientation transition, during which the weak ferromagnetic moment changes its direction by 90[9,10]. The transition temperature varies over a wide range, from units to hundreds of Kelvin, depending on the rare-earth ion type. The highest temperature of the spin-reorientation transition is observed in the crystal and amounts to 480 K [11,12]. Ultrafast switching of the magnetization of domain walls opens up broad prospects for use of orthoferrites in technology [3]. Therefore, the task of tuning the temperature of this transition is crucial. As was shown previously, a way to control the transition temperature is isovalent substitution in the subsystem [13,14]. This substitution, among other things, affects the Néel temperature. As has been shown recently, changes in the angles and distances in (Fe, Mn)O oxygen octahedra play an important role, leading to changes in the electric field gradient (EFG) on ions [14,15]. A similar behavior is observed when a rare-earth element in the subsystem is replaced [16,17]. In [14], it was shown that substitution of Jahn–Teller Mn ions for iron ions in the crystals induces additional distortions of the (Fe,Mn)O oxygen octahedron and, consequently, a strong increase in the temperature of the spin-reorientation transition, which can be indicative of the impact of the oxygen octahedron distortions on the magnetic properties of orthoferrites.
In this study, the Mössbauer effect measurements a series of the orthoferrites and theoretical calculations of the EFG tensor were carried out for establishing the effect of substituting manganese ions on the degree of distortion of the immediate environment of an iron ion. For this purpose, the concentration dependences of the quadrupole splitting, EFG tensor components, field asymmetry parameter, and EFG tensor eigenvector directions were found.
2. Materials and Methods
A series of the ( single crystals was synthesized by optical floating zone melting. At the first stage, to obtain the (x = 0, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, and 0.7) samples, powders of the initial , , and oxides (99.9%, Alfa Aesar) were mixed in the desired ratio and annealed at a temperature of 925 C for 18 h. The annealed powders were poured into a rubber mold and pressed in a hydrostatic press under a pressure of ≈100 MPa. The obtained cylindrical samples were then annealed in a vertical furnace at a temperature of 1400 C for 16 h. After annealing, the synthesized polycrystalline ( samples were placed in an FZ-T-4000-H-VIII-VPO-PC optical floating zone furnace (Crystal Systems Corp.) to grow single crystals. The growth occurred in air under normal pressure at a relative rod rotation speed of 30 rpm. The growth rates varied from 3 to 1 mm/h, depending on the manganese concentration in the samples.
The synthesized rod-shaped ( single crystals had a diameter of up to 7 mm and a length of up to 10 cm. The quality of the samples of the entire series and their orientation along the three crystallographic axes were tested by X-ray Laue diffraction. As an example, Figure 2a presents a typical view of a single crystal and Laue patterns for the sample with a manganese concentration of x = 0.3 along different crystallographic directions (Figure 2b–d). For all the samples, one can see narrow symmetrical reflections corresponding to space group Pnma (♯62). Our investigations showed that the crystallographic axis direction makes an angle of about 23 with the single crystal growth direction.
Samples for the Mössbauer study were prepared by grinding the ( single crystals to a powder. For the samples with manganese concentrations above 0.5, the high-temperature Mössbauer measurements require too much time because of the low concentration and strong absorption by a rare-earth atom. The resulting powder with a mg/cm sample by iron content was pressed in aluminum foil 20 mm in diameter. Mössbauer spectra of the investigated samples were obtained on an MS-1104Em spectrometer equipped with an MRP-750K furnace (Research Institute of Physics, Southern Federal University) with a radioactive source in the transmission geometry at a temperature T = 700 K, since the measurements in the paramagnetic state yield information about distortions in the close vicinity of the Mössbauer isotope caused by structural factors [18].
The spectra were processed in two stages. At the first stage, possible nonequivalent states of iron were determined by calculating the quadrupole splitting probability distributions [19]. Using the results obtained, a preliminary model spectrum was formed. At the next stage, the model spectrum was adjusted to the experimental spectrum by varying the entire set of hyperfine parameters using the least squares method in the linear approximation. The chemical shift values are given relative to metallic iron (Fe).
The calculations were carried out within the density functional theory using the Perdew–Burke–Ernzerhof exchange-correlation functionals with the generalized gradient approximation (PBE–GGA) implemented in the VASP package [20,21]. The number of plane waves was limited by energy of 600 eV. The Monkhorst–Pack grid [22] was chosen to be . The calculation used the GGA + U method in the Dudarev approximation [23], in which the parameter U for the iron ion was chosen to be 2 eV. Configurations of valence electrons were , Fe: for Ho ions and for O ions. The values of the EFG tensor components were calculated using the technique described in [24] implemented in the VASP package.
3. Results and Discussion
The room-temperature Mössbauer spectra were described in detail in [14]. It should be noted that, with increasing concentration of cations, an increase in the chemical shift of the spectra is observed. In addition, an interplay was suggested between the structural distortions and the magnetic reorientation transition temperature as a result of substitution of the Jahn–Teller cation for the cation [14]. Therefore, to thoroughly examine the direct effect of substitution of the cation on the degree of distortion of the nearest environment of iron ions, Mössbauer spectra were recorded above the temperature of magnetic ordering of the subsystem at 700 K. The obtained spectra are shown in Figure 3.
At a temperature of 700 K, the spectra for all the samples have the form of quadrupole doublets. Their hyperfine parameters are given in Table 1. The obtained hyperfine parameters of the compound at 700 K correspond to those reported previously in [17]. According to the experimental data obtained here, the quadrupole splitting has the smallest value for this composition. With an increase in the concentration, the value increases in the series (). The results obtained show an almost threefold increase in the quadrupole splitting on iron atoms already at with a tendency for further growth.
A similar situation was observed in the system [25], where the observed growth was attributed to an increase in the degree of distortion of octahedra. As a result, the crystal field acting on the nucleus of the atom is significantly changed. Although the nucleus is only sensitive to the second derivative of the crystal field (EFG, ), there is a direct correlation between the parameters of the second-order crystal field splitting and the lattice contribution to the quadrupole splitting [26]. At the same time, when replacing by cations with a spherical shell in the series of orthoferrites, the value barely grows even at a significant degree of substitution (), despite the difference between ionic radii of the cations. Thus, one can speak about the decisive role of the cation type in changing the degree of distortion of the nearest environment [15].
The Mössbauer spectra of orthoferrites in the paramagnetic state allow direct estimation of the effect of cation substitution on the degree of distortions of the FeO octahedron of the orthoferrite structure under the interaction of the quadrupole moment of a nucleus with the EFG on Fe nuclei in the crystal. In the paramagnetic state, the value is only determined by the quadrupole transitions ( and ) of the nucleus of the Mössbauer atom. In general, the quadrupole splitting of the Mössbauer spectrum has two contributions: (i) from charges of the ions surrounding the Mössbauer nuclei and (ii) from intrinsic electrons, in particular, resulting from the covalent admixture.
To identify the main contribution, first-principle calculations of the electric field gradient tensor within the framework of the method [24] implemented in the VASP package were performed for the iron ion in the compound (x = 0, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7). All calculations were performed using the lattice parameters and relative coordinates of atoms obtained from the X-ray diffraction experiment [14]. Relaxation of the crystal structure of the studied crystals was not carried out. At the same time, calculations for samples with different concentrations of manganese were performed without accounting the manganese ion. That is, in fact, calculations were performed for the crystal using lattice parameters and atomic coordinates corresponding to crystals with specific concentrations of manganese. This approach can be justified for the following reasons: 1) this greatly simplifies the calculation procedure and, as a result, significantly saves calculation time; 2) the experiment demonstrates the effect on iron atoms; 3) such an approach allows one to evaluate the contribution of manganese valence electrons to the experimentally observed value of the quadrupole shift by directly comparing the concentration dependences of obtained from theoretical calculations, where manganese ions were not taken into account, with those obtained from experimental Mössbauer data, where electrons are implemented from both iron ions and manganese ions.
Basing on the data of the theoretical calculation using formula (1) [27], the calculated Mössbauer quadrupole splitting values were obtained as a function of the manganese concentration.
where Q is a nuclear quadrupole moment (Q = 0.16 b for 57Fe), is an asymmetry parameter (will be defined below). The results are presented in Figure 4.
It can be clearly seen that the calculated data are in good agreement with the experiment and the approximation straight lines have the same slope for both the experimental and theoretical values. At all the concentrations, there is a difference only by a constant value. This means that there is no additional contributions depending on the Mn concentration to the shift value.
Good qualitative and quantitative agreement of the values confirms the appropriateness of the approach used in the calculations described above. Thus, the most important conclusion can be made that Mn cations affect the Mössbauer spectra mainly due to distortions of the crystal lattice, while there is no admixture of electron density to the iron cations, which is somewhat different from the results of the study [13].
Using the calculated data, concentration dependences of the components , , and (||>||>||) of the diagonalized EFG tensors and the EFG asymmetry parameter (Eq. (2)) were plotted [27]. The plots are shown in Figure 5. The linear growth of the field components with increasing concentration is seen. The calculation yielded a value of V/m for the principal component of the EFG tensor of the compound, which is consistent with the data reported in [16] ( V/m), where the lattice contribution to the EFG in unsubstituted orthoferrite crystals was thoroughly studied by measuring the perturbed angular correlation spectra.
The asymmetry parameter abruptly drops in the low-concentration region and has a constant value at . This follows from the change in the EFG symmetry at , which becomes closer to axial. The EFG in the unsubstituted sample has no axial symmetry, contrary to the assumption of the previous work [16].
The relation between and should be closely inspected. The first one is only 1.5 times smaller than the second one for all the considered concentrations of manganese ion. This is a sufficiently large value. Thus, the gradient of the electric field for this series of crystals does not have a pronounced axial symmetry. Therefore, it is necessary to monitor the behavior of the both components and of the field gradient.
The coordinates of the eigenvectors of the components of the electric field gradient were calculated for structures corresponding to concentrations of manganese ion . It should be noted that in the initial compound, due to the orthorhombic structural symmetry, the oxygen octahedron FeO is non-perfect and has some distortions. All the Fe–O bond have different lengths. When manganese is substituted for the iron ion, the degree of distortion of the (Fe,Mn)O octahedron increases, the two axes of the octahedron are decreasing.
Figure 6 shows the directions of the calculated EFG eigenvectors at concentrations of x = 0, 0.1, 0.3, and 0.7. The vector barely change its direction at all the concentrations, which passes close to the longest axis of the FeO octahedron. A bright feature is the behavior of the vector . In the initial compound (x = 0), this vector lies in a plane close to the base of the octahedron, perpendicular to its longest axis, and passes near the middle of the octahedron base edge. At , the vector of the field component rotates by 27 in the plane close to the octahedron base, shifting closer to one of oxygen atoms (see Figure 6). At x = 0.3, the angle of rotation of the vector increases to 39. At , the angle of rotation of the vector is 53. In this case, its direction almost coincides with the shortest axis of the FeO oxygen octahedron. The direction of the vector behaves similarly (the field vectors , and are mutually perpendicular).
The obtained features of the behavior of EFG on the Fe ion (a significant increase in the values of the field components and a sharp change in the direction of the field vectors and due to the Mn doping), apparently, should lead to a change in the balance of exchange constants in the 3d subsystem of HoFeMnO compounds. This is consistent with the results of [14],where a change in the ground magnetic state at concentrations , as well as the disappearance of the orientation phase transition at concentrations were found.
4. Conclusions
A series of single crystals of the () orthoferrites were grown. The Mössbauer effect was measured at temperatures above the Neel point (700 K) on the () orthoferrite samples with sp. gr. Pnma. A monotonic increase in the quadrupole splitting with increasing manganese concentration in the crystal lattice was found. The density functional theory calculation of the EFG tensor on iron cations for the entire series of the samples () showed that the main reason for the observed growth is the crystal lattice distortion by the Jahn–Teller cation; the electron contribution to the quadrupole splitting caused by the covalent admixture of electrons was absent. At the same time, a significant increase in values of all the EFG tensor components was observed with an increase in the manganese concentration in the crystal lattice, while the crystal field symmetry in the subsystem approaches the axial type.
As the calculation of the EFG on the Fe ion demonstrated, a concentration change in the directions of the EFG eigenvectors and was detected at , while the direction of the vector remains unchanged. In our opinion, such a motion of the EFG components with an increase in the concentration of cations can lead to an increase in the temperature of the spin-reorientation transition in this system due to a change in the balance of the exchange constants in the subsystem of the compounds.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, K.Sh.; methodology, S.S., S.S.; validation, K.Sh.; formal analysis, M.P., Y.K., K.Sh.; investigation, T.B. S.S., S.S., Y.K., M.P.; resources, A.S., D.G.; data curation, M.P., Y.K.; writing—original draft preparation, Y.K., M.P., D.G., K.Sh.; writing—review and editing, Y.K., K.Sh.; visualization, Y.K., M.P.; supervision, K.Sh.; project administration, K.Sh., S.S.; funding acquisition, K.Sh.
Funding
This study was supported by the Russian Science Foundation and the Krasnoyarsk Territorial Foundation for Support of Scientific and R
Acknowledgments
The Mössbauer measurements were carried out on the equipment of the Center for Collective Use, Krasnoyarsk Scientific Center, Siberian Branch of the Russian Academy of Sciences.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kamal Warshi, M.; Mishra, V.; Sagdeo, A.; Mishra, V.; Kumar, R.; Sagdeo, P. Synthesis and characterization of RFeO3: experimental results and theoretical prediction. Advances in Materials and Processing Technologies 2018, 4, 558–572. [Google Scholar] [CrossRef]
- Akbashev, A.; Semisalova, A.; Perov, N.; Kaul, A. Weak ferromagnetism in hexagonal orthoferrites RFeO3 (R= Lu, Er-Tb). Applied Physics Letters 2011, 99. [Google Scholar] [CrossRef]
- Tang, J.; Ke, Y.; He, W.; Zhang, X.; Zhang, W.; Li, N.; Zhang, Y.; Li, Y.; Cheng, Z. Ultrafast Photoinduced Multimode Antiferromagnetic Spin Dynamics in Exchange-Coupled Fe/RFeO3 (R= Er or Dy) Heterostructures. Advanced Materials 2018, 30, 1706439. [Google Scholar] [CrossRef] [PubMed]
- Belov, K.; Zvezdin, K.; Kadomtseva, A. (in Russian). Journal of Experimental and Theoretical Physics 1974, 67, 90–99. [Google Scholar]
- Gareeva, Z.; Sharafullin, I.; Zvezdin, A. 2D-Perovskite Multiferroics: Interface-Induced Magnetoelectric Effect in Perovskite-Based Multiferroic Superlattices. Crystals 2023, 13, 1404. [Google Scholar] [CrossRef]
- Mai, V.Q.; Tien, N.A. low-temperature co-precipitation synthesis of HoFeO3 nanoparticles. Crystals 2021, 11, 238. [Google Scholar] [CrossRef]
- Eibschütz, M.; Shtrikman, S.; Treves, D. Mössbauer studies of Fe 57 in orthoferrites. Physical review 1967, 156, 562. [Google Scholar] [CrossRef]
- Sawatzky, G.; Van Der Woude, F. Covalency effects in hyperfine interactions. Le Journal de Physique Colloques 1974, 35, C6–47. [Google Scholar] [CrossRef]
- Ryan, D.; Stoyel, Q.; Veryha, L.; Xu, K.; Ren, W.; Cao, S.; Yamani, Z. A Single-Crystal Mössbauer Study of Spin Reorientations in the Multi-Ferroic HoFeO 3. IEEE Transactions on Magnetics 2017, 53, 1–5. [Google Scholar] [CrossRef]
- Nikolov, O.; Hall, I.; Godfrey, K. A Mossbauer study of temperature-driven spin reorientations in Dy1-xHoxFeO3. Journal of Physics: Condensed Matter 1995, 7, 4949. [Google Scholar] [CrossRef]
- White, R. Review of recent work on the magnetic and spectroscopic properties of the rare-earth orthoferrites. Journal of Applied Physics 1969, 40, 1061–1069. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, G.; Shen, J.; Gao, K.; Zhou, Y.; Lu, Z.; Wu, Y.; Liu, M. Study on the structure, Mössbauer and magnetic properties of polycrystalline Mn-doped SmFeO3. Physica B: Condensed Matter 2024, 674, 415552. [Google Scholar] [CrossRef]
- Kotnana, G.; Jammalamadaka, S.N. Band gap tuning and orbital mediated electron–phonon coupling in HoFe1- xCrxO3 (0? x? 1). Journal of Applied Physics 2015, 118. [Google Scholar] [CrossRef]
- Shaykhutdinov, K.; Skorobogatov, S.; Knyazev, Y.; Kamkova, T.; Vasiliev, A.; Semenov, S.; Pavlovsky, M.; Krasikov, A. CONTROL OF SPIN-REORIENTATION TRANSITION TEMPERATURE IN SINGLE CRYSTALS OF ORTHOFERRITES HoFe1- xMnxO3 (in Russian). JOURNAL OF EXPERIMENTAL AND THEORETICAL PHYSICS 2024, 165, 685–699. [Google Scholar]
- Kotnana, G.; Reddy, V.R.; Jammalamadaka, S.N. Magnetic and hyperfine interactions in HoFe1- xCrxO3 (0<x<1) compounds. Journal of Magnetism and Magnetic Materials 2017, 429, 353–358. [Google Scholar]
- Rearick, T.M.; Catchen, G.L.; Adams, J.M. Combined magnetic-dipole and electric-quadrupole hyperfine interactions in rare-earth orthoferrite ceramics. Physical Review B 1993, 48, 224. [Google Scholar] [CrossRef]
- Angadi, V.J.; Manjunatha, K.; Kubrin, S.; Kozakov, A.; Kochur, A.; Nikolskii, A.; Petrov, I.; Shevtsova, S.; Ayachit, N. Crystal structure, valence state of ions and magnetic properties of HoFeO3 and HoFe0.8Sc0.2O3 nanoparticles from X-ray diffraction, X-ray photoelectron, and Mössbauer spectroscopy data. Journal of Alloys and Compounds 2020, 842, 155805. [Google Scholar] [CrossRef]
- Bayukov, O.; Buznik, V.; Ikonnikov, V.; Petrov, M. Determination of the magnetic structure of the weak ferromagnet Fe3BO6 using the Mössbauer effect(in Russian). Solid State Physics 1976, 18, 2319. [Google Scholar]
- Knyazev, Y.V.; Tarasov, A.; Platunov, M.; Trigub, A.; Bayukov, O.; Boronin, A.; Solovyov, L.; Rabchevskii, E.; Shishkina, N.; Anshits, A. Structural and electron transport properties of CaFe2O4 synthesized in air and in helium atmosphere. Journal of Alloys and Compounds 2020, 820, 153073. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical review B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Physical review letters 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Physical review B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+ U study. Physical Review B 1998, 57, 1505. [Google Scholar] [CrossRef]
- Petrilli, H.M.; Blöchl, P.E.; Blaha, P.; Schwarz, K. Electric-field-gradient calculations using the projector augmented wave method. Physical Review B 1998, 57, 14690. [Google Scholar] [CrossRef]
- Usman, I.B. Synthesis and characterization of Sm-based orthoferrite compounds, SmFe1-xMnxO3. PhD thesis, 2010.
- Vermaas, A.; Groeneveld, W.; Reedijk, J. Ligand-Field Calculations on Pseudo-Tetragonal High-Spin Fe (II) Compounds: Part I: Calculations of paramagnetic susceptibility and electric quadrupole splitting. Zeitschrift für Naturforschung A 1977, 32, 1393–1403. [Google Scholar] [CrossRef]
- Gütlich, P.; Bill, E.; Trautwein, A.X. Mössbauer spectroscopy and transition metal chemistry: fundamentals and applications; Springer Science & Business Media, 2010.
Figure 1.
Unit cell of the (0 crystal. Atoms of the transition element are located inside oxygen octahedra.
Figure 1.
Unit cell of the (0 crystal. Atoms of the transition element are located inside oxygen octahedra.

Figure 2.
Typical single crystal of ( (a) with the Laue diffraction patterns (b-d).
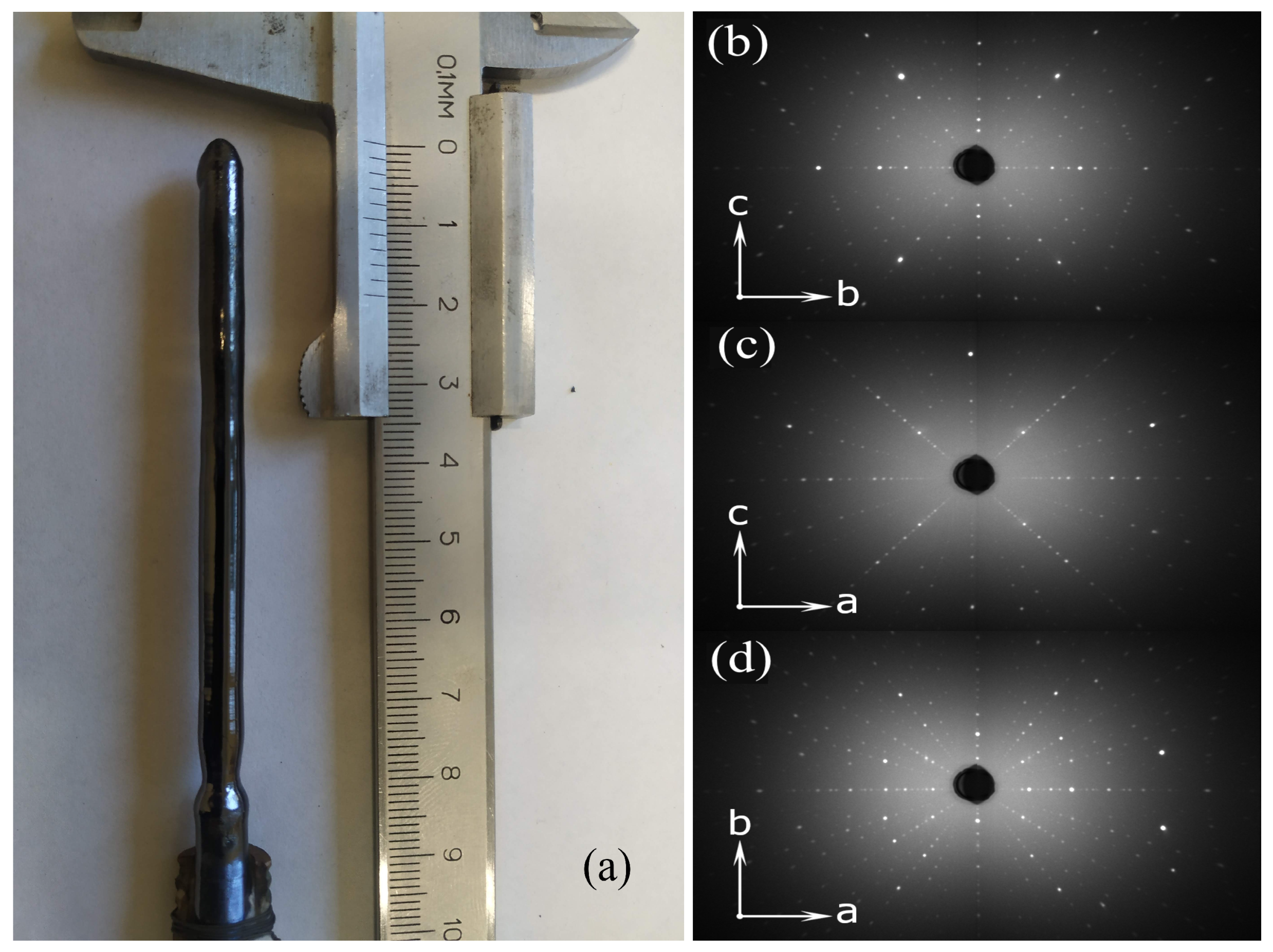
Figure 3.
Mössbauer spectra of the () samples at K. Dots show the experimental spectra and the solid line, the result of their processing. The value of the effect is given to the right of the spectra.
Figure 3.
Mössbauer spectra of the () samples at K. Dots show the experimental spectra and the solid line, the result of their processing. The value of the effect is given to the right of the spectra.
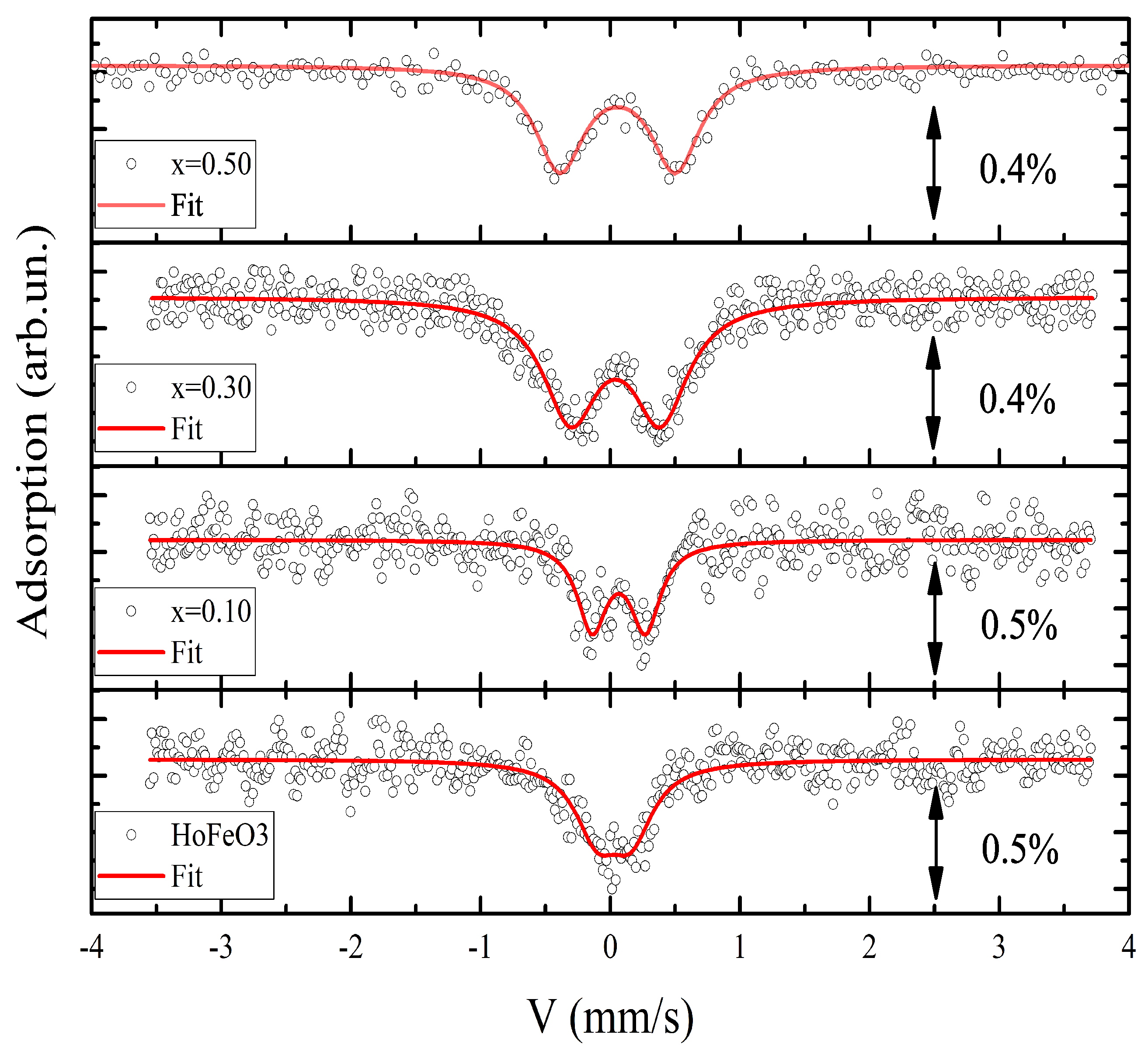
Figure 4.
Concentration dependence of quadrupole splitting in the orthoferrite series. • shows experimental values of the hyperfine parameter and ▪, its calculated values.
Figure 4.
Concentration dependence of quadrupole splitting in the orthoferrite series. • shows experimental values of the hyperfine parameter and ▪, its calculated values.
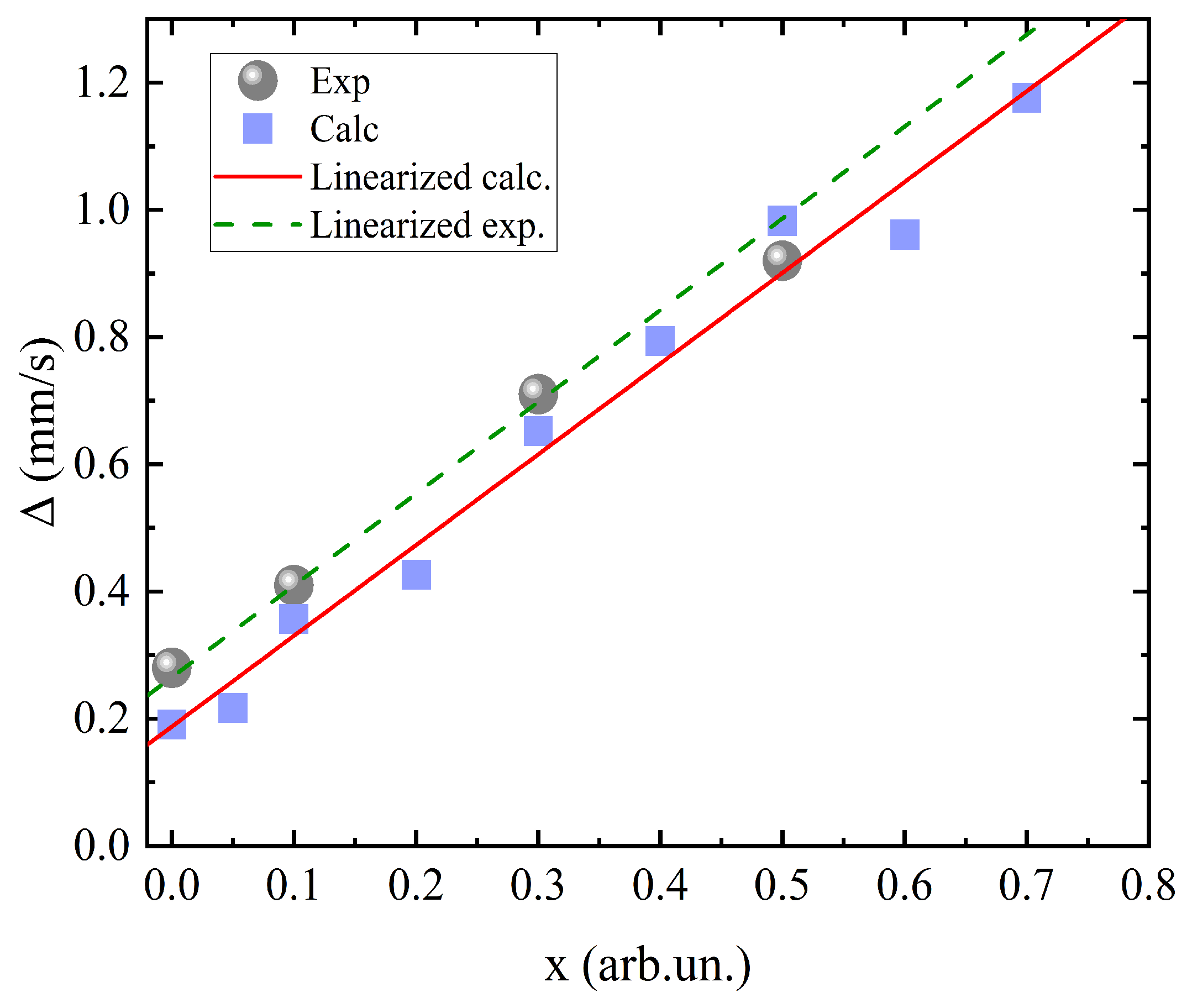
Figure 5.
Concentration dependence of the EFG asymmetry parameter for the () orthoferrite series.
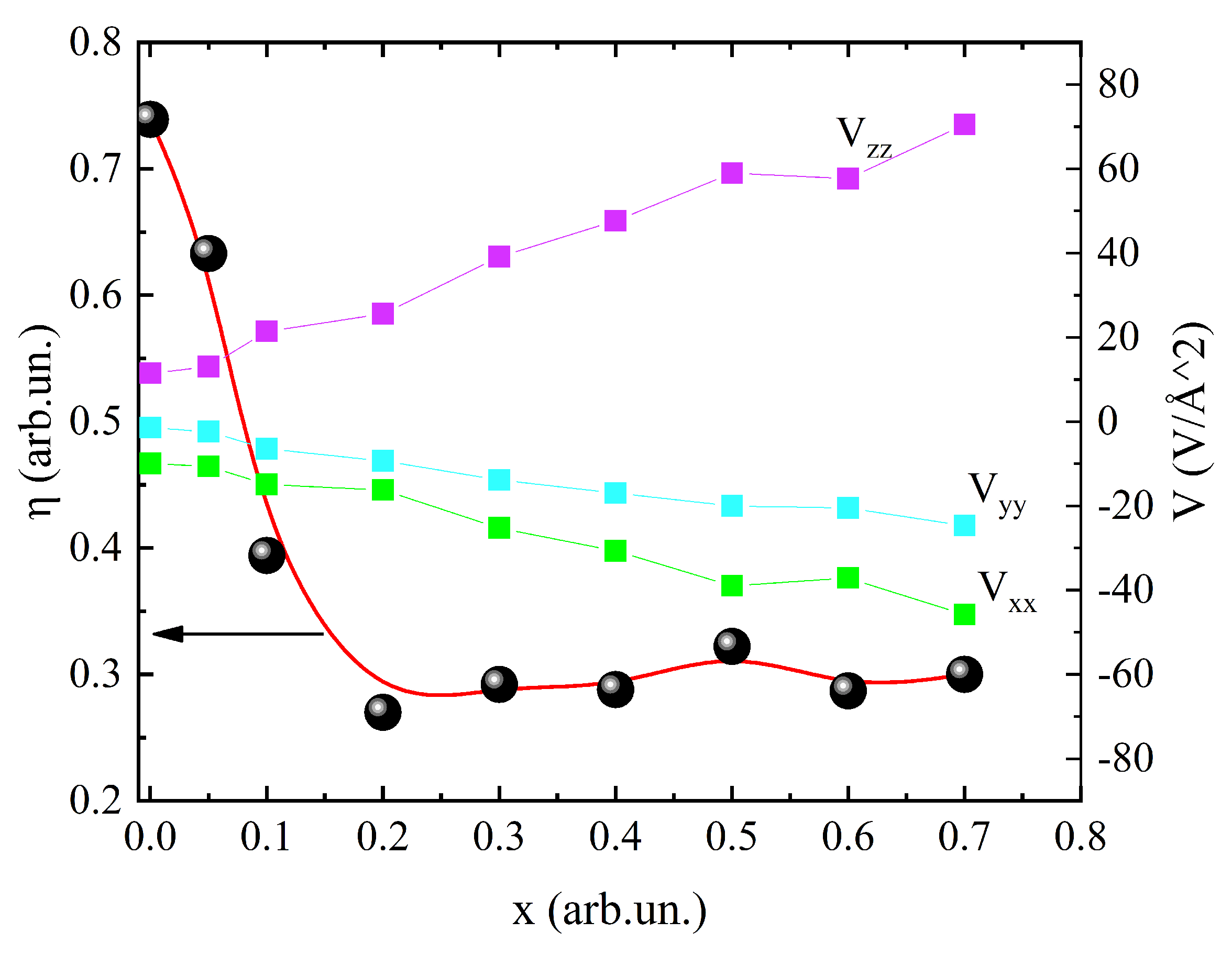
Figure 6.
Concentration dependence of the EFG component vectors in the () orthoferrite series in two projections.
Figure 6.
Concentration dependence of the EFG component vectors in the () orthoferrite series in two projections.
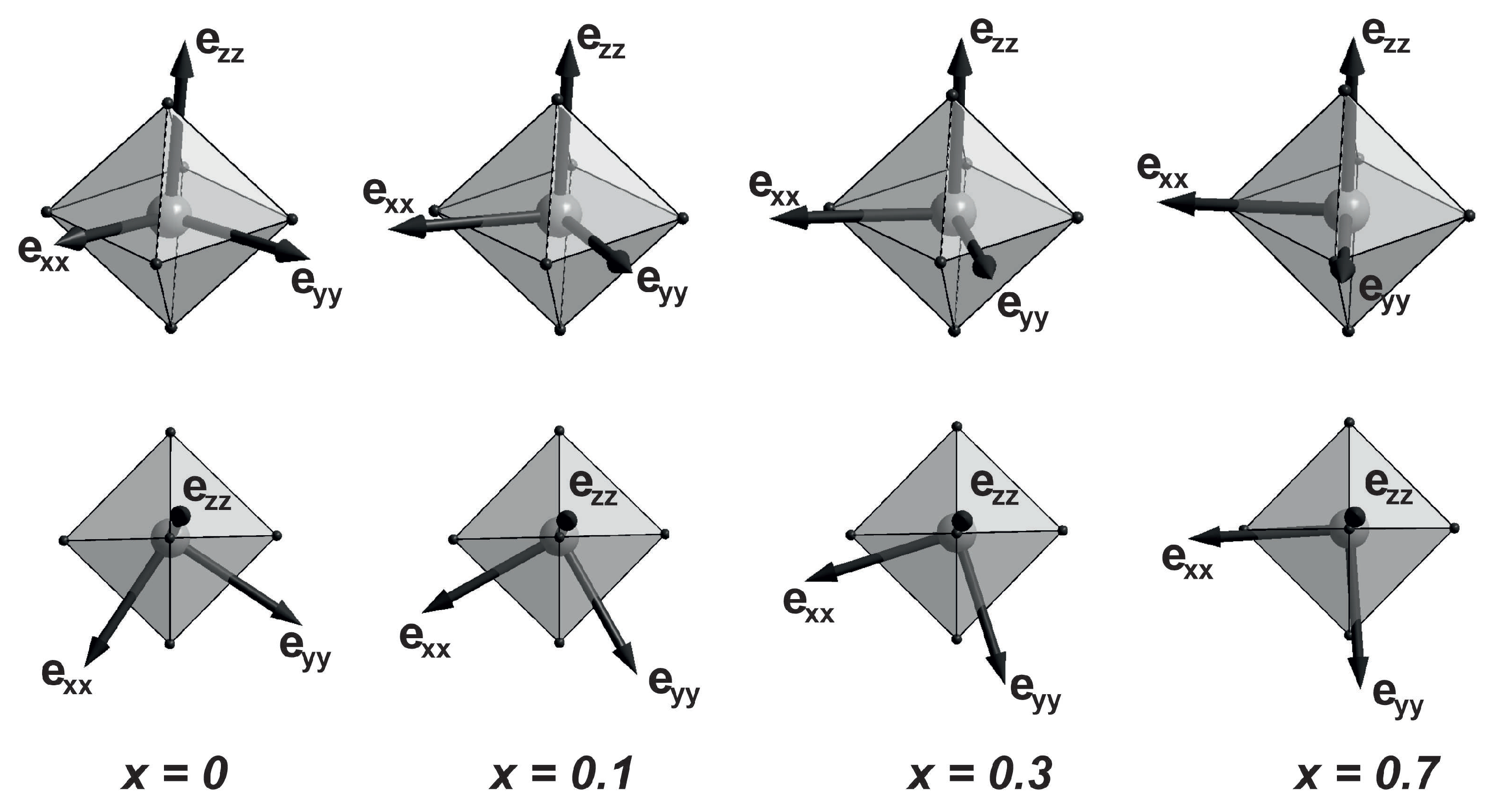

Table 1.
Mössbauer parameters of the () samples at a temperature of 700 K. is the chemical shift with respect to -Fe, is the quadrupole splitting, W is the full width at half maximum of the Mössbauer line, and A is the relative occupancy of sites
Table 1.
Mössbauer parameters of the () samples at a temperature of 700 K. is the chemical shift with respect to -Fe, is the quadrupole splitting, W is the full width at half maximum of the Mössbauer line, and A is the relative occupancy of sites
| x | , | , | W, | A, | Origin |
|---|---|---|---|---|---|
| ±0.005 mm/s | ±0.01 mm/s | ±0.01 mm/s | ±0.03 arb. un. | ||
| 0.0 | 0.067 | 0.28 | 0.41 | 1.0 | |
| 0.10 | 0.098 | 0.41 | 0.29 | 1.0 | |
| 0.30 | 0.093 | 0.71 | 0.31 | 1.0 | |
| 0.50 | 0.088 | 0.92 | 0.44 | 1.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 1996 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



