
Submitted:
04 November 2024
Posted:
05 November 2024
You are already at the latest version
Abstract
Natural products play a crucial role in drug discovery, primarily due to their structural complexity. The prenylated flavanone, glabranin ((S)-5,7-dihydroxy-8-(3-methylbut-2-en-1-yl)-2-phenylchroman-4-one), isolated plant Dalea elegans, has demonstrated neuroprotective effects, attributed to its inhibition of GSK-3β as per our previous in silico studies. Given the enzyme's diverse functions and its potential as a target for neurodegenerative diseases, our group synthesized and evaluated an 11C-labeled derivative of glabranin. We present its in vitro biological activity, including IC50, neuronal uptake in Alzheimer’s-affected brain regions, key physicochemical properties, and animal studies. This study confirms [11C]FLA's interaction with GSK-3β in vitro, highlighting the potential of radiotracers in bioactive compound research.
Keywords:
GSK-3β
; Alzheimer’s disease
; Glabranin
1. Introduction
Dementia is among the greatest global health crises of the 21st century. Presently, more than 50 million people are living with dementia worldwide, with this number estimated to triple to 152 million by 2050 as the world’s population grows older [1]. Alzheimer’s disease (AD) is the most common cause of dementia and is thought to account for 60–80 % of the cases [2].
AD is indeed a neurodegenerative disorder primarily characterized by cognitive decline and progressive memory loss. At the molecular level, are two key pathological, the presence of neurofibrillary tangles (NFTs) and insoluble β-amyloid (Aβ) plaques [3]. Aβ is derived from the proteolytic cleavage of β-amyloid precursor protein, whereas NFTs comprise hyper-phosphorylated forms of the microtubule-associated protein tau.
Despite the progress made, the efficacy of current drugs to treat, delay, or block the progression of neurodegenerative diseases remains limited [4,5,6,7]. Therefore, the discovery and development of new molecules that can improve current treatments are of great importance.
Natural products play a crucial role in the discovery of new drugs, essentially due to their structural complexity and ability to activate various intracellular signaling pathways through different mechanisms of action, while generally presenting fewer side effects [8]. Nature is a rich reservoir of structurally active metabolites with a broad spectrum of biological activities, including antibacterial, antifouling, antifungal, antioxidant, anticancer, anti-inflammatory, and neuroprotective properties among others [9]. Different molecules have been isolated from marine and terrestrial organisms, such as amino acid-derived alkaloids, aromatic compounds, fatty acids, sphingolipids, lactones, alcohols, peptides, polyacetylenes, quinones, quinolones, sterols, terpenes and terpenoids, among others.
The study of native species from different parts of the planet is of vital importance, since these metabolites are produced in response to physical and ecological pressures to ensure the survival of organisms and have evolved for billions of years in close association with biological systems, and therefore exhibit high specificity and high affinity to interact with target biological structures, making them excellent candidates to inspire the development of new drugs, with minimal side effects and significant health benefits [10,11]. Dalea L. (Fabaceae) is a uniquely American genus with more than 172 species. Its habitat extends from central Argentina and Chile to the southwestern United States. Different flavonoids have been isolated from Dalea Elegans, mostly belonging to the group of prenylated flavanones, with diverse biological activities, such as antioxidants, toxic effects on mitochondria isolated from rat liver and human tumor cells, antimicrobials and tyrosinase inhibitors [12].
In the study by Santi D., and colleagues [13], the neuroprotective activity of five prenylated flavanones isolated from Dalea species was evaluated in models of neurodegeneration in vitro. The flavanone (S)-5,7-dihydroxy-8-(3-methylbut-2-en-1-yl)-2-phenylchroman-4-one (Glabranin) (Figure 1) showed neuroprotective effects against oxidative stress-induced death in primary cultures of cerebellar granule neurons. In addition, an in-silico screening of the most potent compounds was performed, looking for neuroprotective-related protein targets responsible for the effects of these flavanones, based on ligand similarity search, pharmacophore comparison, and reverse docking.
These in-silico tests suggest that their neuroprotective potential could potentially be mediated by interaction with glycogen synthase kinase-3 beta (GSK-3β), a serine/threonine kinase that plays a leading role in the cascade of events that culminate in AD. GSK-3β plays a central role in a wide variety of cellular processes concerned with coordinating catabolic and anabolic pathways [14,15,16,17]. Particular interest has been focused on GSK-3β in the context of neuronal functions since its activity is essential for normal neurodevelopment and its over-activity is implicated in psychiatric conditions, cognitive dysfunction, and neurodegeneration [15,18,19,20,21,22,23]. Furthermore, its over-activity accounts for protein tau hyper-phosphorylation, increased β-amyloid production, and local plaque-associated microglial-mediated inflammatory responses, all of which are hallmark characteristics of AD.
Advances in imaging modalities such as positron emission tomography (PET) have encouraged its use for the evaluation of functional changes in brain pathologies. PET images provide information on pathological markers as well as pharmacokinetic properties to evaluate the absorption, distribution, metabolism, and excretion profiles of candidate drugs. Thus, PET imaging has become crucial for the characterization of new therapeutic agents.
Given the wide range of functions associated with GSK-3β, this enzyme has emerged as an interesting target for therapy and imaging in diverse diseases especially, AD. Considering all this information, our goal was to demonstrate the interaction between glabranin and GSK-3β as a putative target molecule involved in the neuroprotective effects of prenylated flavanones and to explore its potential as a GSK-3β marker. To accomplish these objectives, a radiolabeled glabranin derivative was prepared and its physicochemical properties were determined to explore its suitability to cross biological membranes, in particular the blood-brain barrier. In vivo and ex vivo animal biodistribution and imaging experiments were also performed to corroborate this capacity. In addition, this radiolabeled product was evaluated in vitro to determine its cellular and target specificity. Finally, the inhibitory activity towards GSK-3β, (IC50) was determined.
2. Results
2.1 Chemistry
The glabranin derivative (S)-7-hidroxy-5-methoxy-8-(3-methylbut-2-en-1-yl)-2-phenylcroman-4-ona (FLA) was synthesized using methyl iodide in alkaline medium. In the HPLC analysis, two species were detected. The product with a retention time (rt) of 13.2 minutes corresponds to an unreacted glabranin, while the product with 15.2 minutes rt corresponds to the methylated glabranin derivative (FLA) as confirmed by spectroscopic methods. The methylated derivative was used to verify the identity of the radiolabeled analog through coinjection. In addition, the synthesized compound was used to determine the IC50 of FLA to assess if methylation affects the inhibitory activity towards GSK3 (see biological evaluation).
2.2. Radiochemistry
11C labeling of glabranin was performed by O-methylation on the OH group at position 7 of the A ring as shown in Figure 2. The methylation position was selected due to the high reactivity of the above mentioned hydroxyl group reported in the literature [24,25].
The use of methyl iodide as a methylating agent (conditions I and II) did not yield the desired product and consequently was replaced by methyl triflate (condition III), due to its greater reactivity [26,27].
The radio-HPLC analytical chromatogram of the reaction mixture showed the presence of 11C-methanol (rt 5.2 min) generated by the hydrolysis of the 11C-methyl triflate, and a main peak with an rt 8-9 min corresponding to the desired product [11C]FLA.
Purification was successfully achieved by reverse phase semi-preparative HPLC. An example of the chromatographic profile of unpurified [11C]FLA is shown in Figure 3.
The highest yield (16 ± 3% decay-corrected yield) was obtained using [11C]MeOTf as a methylating agent and 2 µL de NaOH 0.5 M in anhydrous acetone at 25 °C for 4 min (entry III, Table 1).
After purification, the product was diluted with water, retained on a solid phase extraction cartridge (Sep-Pak(R) C18 Ligth), and finally diluted in aqueous 0.9 % NaCl with 10 % ethanol. Analytical radio-HPLC chromatograms showed the presence of the desired 11C-methylated product as a unique product with a retention time of 15.5 min. Coincidence with the nonradioactive analogous upon coinjection confirmed the identity of the radiolabeled product (Figure 4).
The potential methylation of the OH group in position 3 was not observed, thus confirming the literature reports about the higher reactivity of OH in position 7 in flavonones.
2.3. Physicochemical Evaluation
Characterization of [11C]FLA was performed by the following physicochemical studies: i) stability at the final formulation; ii) stability in plasma; iii) plasma protein binding, and iv) lipophilicity. These studies are predictive and could be then translated to the in vivo behavior of the radiotracer.
[11C]FLA was stable both in its final formulation and in human plasma for at least 150 min, with radiochemical purity (RCP) > 90%. This time frame is adequate for a 11C-labeled compound and sufficient to perform biological evaluations. Binding to plasmatic proteins was determined by size exclusion chromatography. PPB is a very important parameter that determines not only the pharmacokinetics of the drug but also its ability to penetrate the biological membranes [28]. The PPB of [11C]FLA, determined by molecular exclusion, was 72 ± 3% (n=3) and remained constant over time (for up to 60 min). The lipophilicity, determined as the partition coefficient between n-octanol and the phosphate buffer (pH = 7.4), was LogP 7.4 = 1.22 ± 0.05 n=3. This value is in accordance with the chemical structure of FLA and is within the optimal range to cross the BBB LogP at pH 7.4 between 0 to 3 [29].
The physicochemical studies corroborated that the molecule obtained is stable and presents adequate lipophilicity to cross membranes, which supports the proposal of this radiotracer for the experimental validation of in silico results about the interaction of these compounds with GSK3.
2.4. Ex Vivo Biodistribution Studies
Biodistribution studies with [11C]FLA were carried out in C57BL6J black mice at different times post-injection. Figure 5 shows the results expressed as % act/organ in the most significant fluids and organs as a function of time.
Biodistribution studies show that [11C]FLA is highly cleared from the blood (13.9 ± 1.7 % at 10 minutes post inject, 2.8 ± 1.7 % at 50 minutes post-inj.). Liver uptake was very high, even after 50 minutes of biodistribution time (12.2 ± 2.4 %) and excretion occurred mainly through the hepatobiliary system (% in intestines 44 ± 10 at 50 minutes post-inj.) with only a minor fraction excreted in the urine.
The tracer uptake in the blood and brain at different injection times of [11C]FLA are shown in Figure 6. At 20 minutes, when maximum uptake in the brain is reached (2.5 ± 0.29), clearance from the blood had already begun (5.58 ± 0.81 % at 10 minutes post inject, 1.84 ± 0.62 % at 20 minutes post-inj), suggesting some retention mechanism within the brain.
2.5. In Vivo Imagining Studies:
Static PET images were acquired in healthy C57BL/6J male mice, to confirm that the compound FLA is capable of crossing the blood-brain barrier.
Figure 7.
Static images (axial, sagittal, and coronal slices) at 20 min post-administration of [11C]FLA showing the uptake in a normal animal, in the body (upper images) and brain (lower images).
Figure 7.
Static images (axial, sagittal, and coronal slices) at 20 min post-administration of [11C]FLA showing the uptake in a normal animal, in the body (upper images) and brain (lower images).
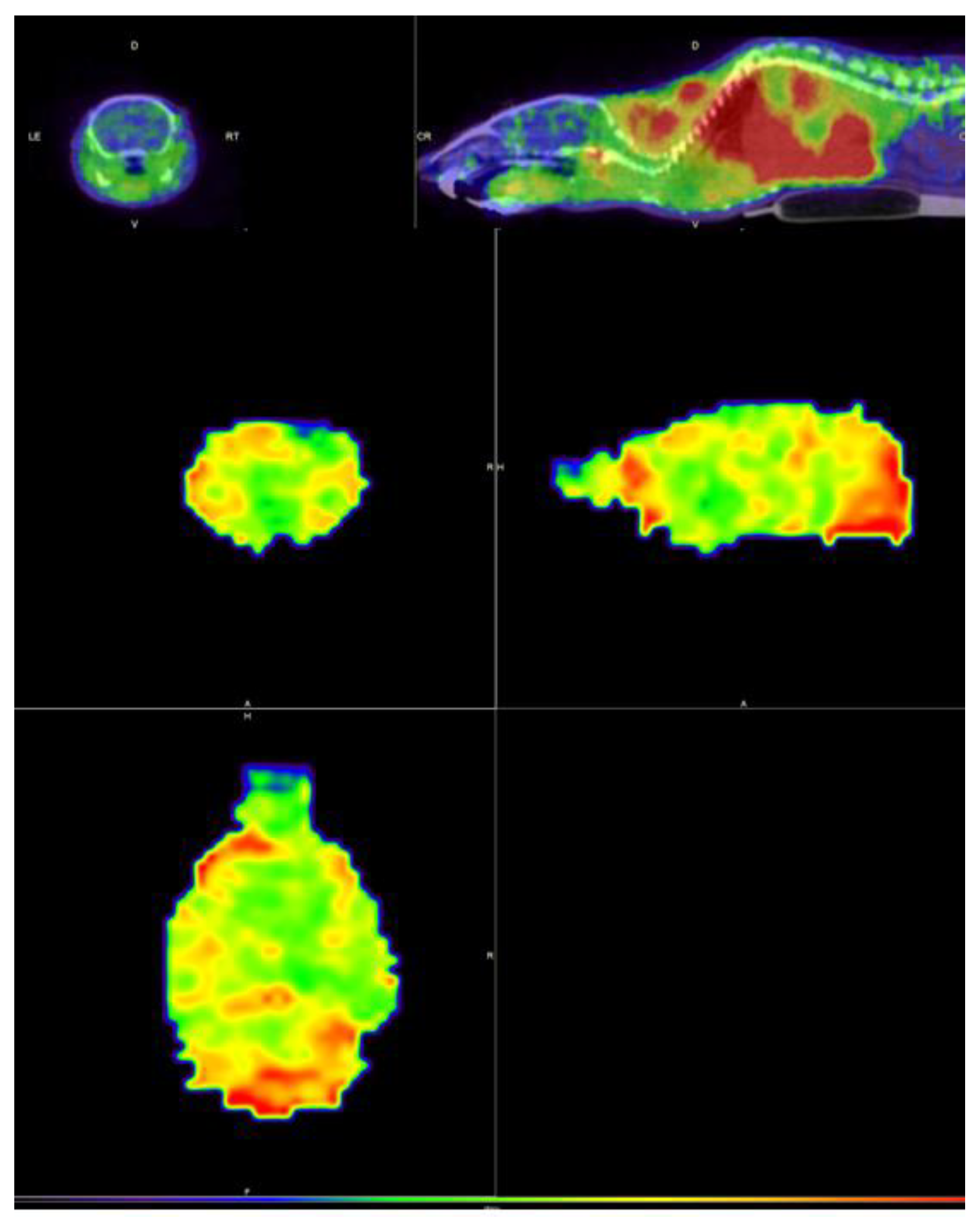
Uptake was seen in the brain, as expected by the logP at pH 7.4 value of the compound, and the result is consistent with the results obtained in the bi-distributions, which are shown previously.
2.6. In Vitro Evaluation of GSK-3β Inhibition
A luminescence method based on the quantification of the ATP amount present after the kinase reaction was employed to assess the GSK-3β inhibitory effect by glabranin and FLA. Compounds were first tested at a fixed concentration of 10 μM (in buffer), and a low GSK-3β inhibitory effect for both was observed. Then, the IC50 value was determined by performing a dose-response curve using different concentrations (1-100 μM). Nonlinear regression analysis showed an IC50 value in the micromolar range, being 44.8 ± 4.0 µM for glabranin, and 16.14 ± 0.6 µM for FLA (Figure 8).
This study confirmed literature reported inhibitory activity of GSK3 by glabranin and demonstrated that this activity was not eliminated by methylation. Furthermore, a lower IC50 of FLA indicated that methylation slightly potentiated the inhibition of the enzyme.
2.7. In Vitro Evaluation of [11C]FLA Uptake by Neurons
A primary culture of neurons from the hippocampus and cortex was used to test [11C]FLA uptake in vitro since these are the brain regions where GSK-3β is abundant [30]. Likewise, these are neuronal populations that are particularly affected by AD [31].
The uptake of [11C]FLA in cultured neurons was determined at three incubation times (5, 10, and 20 minutes). The uptake percentage varied significantly within the incubation time. 20 minutes was the time in which the highest percentage (15 %) of uptake was observed (Table 2).
[11C]FLA uptake by neurons was also evaluated at different activity loadings of the radiolabeled product. The variation of uptake percentage with the activity added to the neuron culture is shown in Table 3. A higher binding was obtained when the cultures were incubated with a lower activity.
These results were also used to optimize the conditions to test [11C]FLA uptake in other cell types such as astroglial cells. [11C]FLA uptake by primary cultures of astrocytes is used as a negative control since GSK-3β is primarily expressed in neurons [32].
2.8. In Vitro Evaluation of [11C]FLA Uptake by Astrocytes
The uptake of [11C]FLA by primary cultures of astrocytes is shown in Table 4. Compared with the uptake in neurons (Figure 2) the uptake by astrocytes was significantly lower at all studied times (5, 10, and 20 minutes). Furthermore, the uptake at 20 minutes was two-fold greater in neurons (14 %) than in astrocytes (7 %), suggesting the specificity of [11C]FLA uptake.
2.9. Competition Assay
2.9.1. [11C]FLA Interaction with GSK-3β Enzyme Was Evaluated
A competition assay between [11C]FLA and a recognized GSK-3β inhibitor, the 3-acetyl-4- (1-methyl-1H- indol-3-yl)-1H-pyrrole-2,5-dione (VP.36) was performed to further evaluate selectivity towards GSK-3β. Perez et al., previously reported a IC50 value for this maleimide derivative of 0.89 ± 0.19 µM [33].
The uptake of [11C]FLA by neurons varied significantly with VP.36 concentration, for example, 49.3 ± 8.2 % with 1.6 x 10-8 M of v.p 36 and 3.32 ± 0.4 % with 1.6 x 10-5 M as shown in Figure 9. This result strongly suggests that [11C]FLA interacts with GSK-3β in neurons.
3. Discussion
Historically, natural products have played a very important role in drug discovery due to many advantages, especially the higher chemical diversity and structural complexity in comparison to synthetic molecules [34]. The main areas of development have been cancer and infectious diseases [35,36,37], but some successful examples can also be found in cardiology (for example, statins) [38], and some neurodegenerative diseases (for example, fingolimod) in multiple sclerosis [39].
However, there are some important concerns regarding their use, mainly the lack of scientific evidence for their efficacy and safety [40].
Positron emission tomography can contribute to overcoming this drawback, providing information about target engagement, proof of mechanism, pharmacokinetic and pharmacodynamic profiles, etc [41]. PET is a molecular imaging technique that allows for the monitoring of a drug treatment’s effectiveness in vivo by studying the functionality of organs and biochemical processes. PET technology is very useful in the discovery and development of potential new drugs, as it allows determining whether a potential drug reaches its pharmacological target and obtaining information on whether it presents specific binding to a receptor, etc.
In the particular case of molecules targeting the central nervous system, as glaranin, a crucial point is the determination of penetration through the blood-brain barrier and access to the site of action. These points can easily be determined qualitatively and quantitatively by using PET technology by marking the molecule to be studied with a positron-emitting radionuclide [42,43]. The advantage of positron-emitting radionuclides such as 11C and 18F is that they can be easily incorporated into a wide variety of organic compounds without altering their structure, as well as their physical, chemical and biological properties [44].
In this context our goal was to demonstrate the interaction between glabranin, a potentially bioactive molecule derived from a native plant from our region, and GSK-3β as the molecular target described in the literature.
Our experimental design consisted of preparing an 11C derivative of glabranin through methylation. The experimental conditions were optimized, and the expected product could be isolated with an adequate yield and purity. Furthermore, [11C]FLA was stable both in the formulation medium (ethanol in NaCl 0,9%) and in human plasma for at least 150 min, thus allowing its used in in vitro and in vivo assays.
The structural verification through comparison with the non-radioactive analog allowed us to demonstrate that methylation occurred in the OH group in position 7, which was described in the literature as the most reactive position.
The physicochemical evaluation demonstrated that the lipophilicity of [11C]FLA was within the optimal range for crossing the membrane the blood-brain barrier and is, consequently, potentially able to reach its cellular target. The ability of [11C]FLA to cross the BBB was confirmed by biodistribution results and the PET image, in accordance to previous work with different flavonoids [45,46,47].
The biodistribution profile characterized by high hepatobiliary elimination and low uptake in non-target tissues, such as the heart, spleen, and muscles, at all analyzed times, is consistent with the high lipophilicity and PPB demonstrated by physicochemical evaluation and also. and reflects the absence of non-specific binding except in the lungs as reported for other flavonoids [48]. It is also important to note that the initial uptake of [11C]FLA in the blood was low, and decreased significantly throughout the denoting the tracer’s rapid blood clearance.
Additionally, we were able to verify that the compound crosses the blood-brain barrier, achieving its maximum uptake at 20 minutes. It is also important to note that when the maximum uptake in the brain is reached, the clearance from blood is already a significant indication that some retention mechanism inside the brain is in place.
To validate the use of the methylated analogue in the biological in vitro studies the IC50 of glabranin and the methylated derivative were determined, obtaining similar values in the micromolar range for both compounds (44.8 ± 4.0 µM and 16.1 ± 0.6 µM, respectively). Although these values are higher than the ones reported in the literature for other synthetic GSK-3β inhibitors (example 11C-Tideglusib IC50=60nM [49], these are, to the best of our knowledge, the first reported IC50 values for these type of compounds. Furthermore, the objective of elucidating whether glabranin and similar prenylated flavonones effectively interact with GSK-3β was fulfilled as reported by in silico studies.
Additional in vitro studies were included to further substantiate our hypothesis. Uptake of 11C-FLA by neurons from the hippocampus and cortex of mice (areas most affected in AD [31], and in which GSK-3β is most abundant) was compared with uptake in astrocytes (negative control). A high binding was observed in neuronal cultures, increasing with lower seeding activity and longer incubation time. It was also significantly higher than in the astrocyte cultures at all times tested (5, 10, and 20), being twice as high at 20 minutes, an expected result since GSK-3β is more abundant in neurons than in astrocytes.
The competition assay using a GSK-3 β inhibitor of proven activity (VP.36, IC50 0.89 ± 0.19 µM [33], showed a significant decrease in uptake, suggesting the specificity of the interaction with the enzyme. This assay corroborated experimentally the predictions of the in silico studies in the complex cellular environment where multiple factors are intervening.
4. Materials and Methods
4.1. Chemistry
Synthesis of (s)-7-hidroxy-5-methoxy-8-(3-methybut-2-en-1-yl)-2-pheenylcroman-4-ona (FLA)
(S)-7-hidroxy-5-methoxy-8-(3-methybut-2-en-1-yl)-2-phenylcroman-4-ona (FLA) was obtained by the direct methylation of glabranin (50 mg, 0.15mmol) with methyl iodide (0.200 ml, 3.22mmol) in basic conditions (K2CO3 (0.432 g, 3.13mmol) in acetone (5 ml) at room temperature and N2 atmosphere. The reaction progress was followed by analytical HPLC using a reverse phase Agilent ZORBAX Eclipse Plus column (Agilent) C18 4.6 × 150 mm 5 μm, a flow rate of 1.0 mL/min and the following solvent gradient: (A) acetic acid 0.1% (v/v) in water, (B) acetic acid 0.01% (v/v) in acetonitrile; 0 min: 10% (B), 0–10 min: 10 a 100% (B), 10–20 min: 100% (B). After 60 min of reaction, the chromatogram showed two products with retention times (rt) of 13.2 and 15.2 min, respectively. The products were isolated by analytical HPLC and identified using NMR spectroscopy. FLA: 1H NMR (CDCl3) δ 9.13 (s,1H), 8.45(s, 1H), 7.38-7.40 (m, 2H), 6.88-6.93 (m, 2H), 6.21 (s, 1H), 5.34 (dd, 1H), 5.18 (m, 1H), 3.73 (s, 3H), 3.25 (d, 2H), 1.60 (d, 6H).
4.2. Radiochemistry
Radiosynthesis of (S)-5-hydroxy-7-11C-methoxy-8-(3-methylbut-2-en-1-yl)-2-phenylchroman-4-one ([11C]FLA).
Three different labeling conditions were tested, which differ in methylating agent (methyl iodide [11C]CH3I or methyl triflate [11C]CH3OTf), base (NaOH, TBAF), time (1, 4 and 5 minutes), temperature (25 and 80°C) and solvent (DMSO and acetone) were assayed for the preparation of [11C]FLA.
[11C]CH3I or [11C]CH3OTf were prepared according to previously described methods. 1.
The optimized procedure was performed as follows: [11C]CH3OTf was bubbled into the reactor loaded with the precursor(S)-5,7-dihidroxy-8-(3-methylbut-2-en-1-yl)-2-phenylcroman-4-ona (0.2 mg, 0.745nmol) in anhydrous acetone (300 µL) and 2µL de NaOH 0.5 M. After the complete transfer of radioactivity, the reaction vial was incubated at 25 °C for 4 min. The reaction mixture was diluted with the acetonitrile: water (30:70) (1.5 mL) and purified onto a reverse-phase semipreparative VP 125/ 10 NUCLEODUR C18 HTec (Macherey-Nagel) 5 μm column with an isocratic mixture of deionized water and acetonitrile (70:30) and a flow rate of 8 mL/min. The radioactive product was collected, diluted with 50 mL of deionized water, and loaded in a solid phase extraction cartridge (C18 SepPak Light - Waters) to trap the radiotracer. Elution from the cartridge was achieved with absolute ethanol (1.0 mL) and the product was diluted with saline (10% ethanol in saline). The final product [11C]FLA was collected into a sterile vial through a sterile 0.22 μm pyrogen-free filter (Millex SA). The radiochemical purity of [11C]FLA was assessed using the above described HPLC system. Confirmation of the product identity was performed by the coinjection of the nonradioactive FLA.
4.3. Physicochemical Evaluation
Stability in Formulation Milieu (10% ethanol in NaCl 0.9%). Isolated [11C]FLA was incubated at room temperature for 3 h after labeling. The RCP purity was determined by HPLC using the above-described conditions.
Stability in Plasma. [11C]FLA (100 μL) was incubated in human plasma (900 μL) at 37°C for 2 h. After 60 and 120 min incubation time, samples (200 μL) were precipitated with ethanol (200 μL) and centrifuged (1500 rpm, 5 min), and the RCP purity was determined by HPLC using the above-described conditions.
Lipophilicity. Lipophilicity was studied through the partition coefficient (P) between n-octanol and phosphate buffer 0.125 M, pH 7.4. To a tube containing 2 mL of n-octanol and 2 mL of buffer, 1 mL of [11C]FLA was added. The mixture was shaken on a vortex mixer and centrifuged at 5000 rpm for 5 min. Four samples (0.2 mL) from each layer were counted in a γ-counter 3” × 3” well-type NaI solid scintillation detector coupled to a multichannel analyzer (Ametek-ORTEC). Lipophilicity was expressed as LogP, with P calculated as the mean value (cpm/mL) of the n-octanol layer divided by that of the buffer.
Protein Binding (PPB) [11C]FLA (25 μL) was incubated with human plasma (475 μL) at 37°C for up to 2 h. 50 μL samples taken at 30 and 60 min of incubation were added to Microspin G-50 columns (Pharmacia Biotech). Columns were centrifuged at 3300 for 2 min and the activity in the eluate and on the column were measured. The amount of the protein-bound radiotracer was calculated as the percentage of activity eluted from the column.
4.4. Biological Evaluation
C57BL/6J male mice, 13-14 weeks old were used for ex vivo and in vivo studies. The animals were housed under 12:12-h light/dark cycles; food and water were given ad libitum in rooms with controlled temperature and humidity at the Uruguayan Centre of Molecular Imaging (CUDIM) animal facility. The research protocol was carried out in accordance with the National Bioethics Committee requirements and under the current ethical regulations of the national law on animal experimentation N° 24042301. (National Commission of Animal Experimentation, CNEA, Uruguay). CUDIM’s Animal Bioethics Committee approved these protocols.
4.5. Ex Vivo Biodistribution Studies
Biodistributions studies were performed in C57BL/6J male mice, weighing between (36 ± 3) g (n=8). The animals were injected i.v. with 0.8–12.6 MBq (200–250 µL) of [11C]FLA into the caudal tail vein and sacrificed by cervical dislocation at 10, 20, and 50 min after injection (n =3 animals in each injection time). Organs (intestine, liver, kidneys, bladder, and brain), and blood samples were extracted, weighed and assayed for radioactivity in a gamma counter (a 300 × 3 00 well-type NaI [Tl] solid scintillation detector coupled to a multichannel analyser, ORTEC). The total urine volume was collected during the biodistribution period, and its activity was measured whit bladder. The percentage of the injected dose in the whole organ (%ID) and the percentage of the injected dose per gram of tissue (%ID/g). Corrections by different sample geometry were applied when necessary.
4.6. In Vivo Imaging Studies
Small animal PET-CT imaging was performed in a nanoScan® PET/CT Mediso Preclinical Imaging, based on LYSO cintillators. The spatial resolution of the scanner is 0.9 mm and the transaxial field of view (FOV) is 10.0 cm. The data were acquired in list mode in a 212 × 212 × 235 matrix with a pixel size of 0.4 × 0.4 × 0.4 mm and a coincidence window width of 1.0 nsec. The animals were anaesthetised with 2% isofluorane in an oxygen flow of 2 L/min, placed in prone position on the scanner bed and injected i.v. via the caudal tail vein with 200-300 μL of [11C]FLA (10.1 ± 2.3 MBq). PET images (static studies) acquisition started 15 min after radiotracer administration and performed over 20 min. Sinograms were reconstructed using 3D maximum likelihood expectation maximisation (3D-MLEM) with 4 iterations and 6 subsets.
4.7. In Vitro GSK-3β Inhibition Assay
Human recombinant GSK-3β and GSK3 Substrate (YRRAAVPPSPSLSRHSSPHQ(pS)EDEEE) were purchased from Promega (GSK-3β Kinase Enzyme System, Ref. V1991). Kinase-Glo luminescent kinase assay was obtained from Promega (Promega Biotech Iberica, SL, Ref. V6711). ATP and all other reagents were from Sigma-Aldrich (St. Louis, MO). The assay buffer contained 50 mM HEPES (pH 7.5), 1 mM EDTA, 1 mM EGTA, and 15 mM magnesium acetate. The GSK-3β activity assay was performed following the method published by Baki et al 2. The enzymatic reaction was performed in assay buffer (total volume, 40 μL) in white 96-well plates. In a typical assay, 10 μL of FLA or glabranin (dissolved in DMSO at 10mM concentration and then diluted in assay buffer to the desired concentration), and 10 μL (22 ng) of enzyme were added to each well followed by 20 μL of assay buffer containing substrate (GS-2 peptide) and ATP being the final concentration in the well 25 μM and 1 μM, respectively. The final DMSO concentration in the reaction mixture did not exceed 1%. After a 30-minute incubation period at room temperature, the enzymatic reaction was stopped with 40 μL of Kinase-Glo reagent. Glow-type luminescence was recorded after 10 minutes using a GloMax® Discover Microplate Reader. The activity is proportional to the difference between the total and consumed ATP. The inhibitory activities were calculated based on maximal activities measured in the absence of an inhibitor. The IC50 was defined as the concentration of each compound that reduces a 50 % the enzymatic activity concerning that without inhibitors. The IC50 value was determined by performing a nonlinear regression analysis using five different concentrations. Curve fitting was performed using the sigmoidal dose-response function on GraphPad Prism 6.0 software. The data shown are the mean of two different experiments.
4.8. Neurons and Astrocytes Primary Cultures
A primary culture of hippocampal and cortical neurons was prepared from 16-18-day-old fetuses of C57BL/6J. mice as previously described 3.
Cells were cultured in monolayer in 6-well plates at a cell density of 5x105 cells/well and were maintained in a neurobasal medium supplemented with 2% B27, 1% Glutamax, and penicillin/streptomycin (1%) at 37 °C and 5% CO2. The culture medium was partially changed every other day until the moment of the trials.
A culture of astrocytes from the hippocampus and cortex of neonatal C57BL/6J mice (1-2 days old) was prepared according to the method described by Diaz-Amarilla et al 3. Cells were cultured in a monolayer on individual plates P6 and maintained in DMEM medium supplemented with penicillin/streptomycin (1%) in a humidified incubator, at 37°C and 5% CO2.
4.9. Uptake Studies in Neurons and Astrocytes
After 12 days in vitro, the neurons or astrocytes were incubated with 14µCi (200µL) of [11C]FLA for 5, 10, and 20 minutes; and with 14µCi, 27µCi and 54µCi of the radiolabelled product during 20 min (assays were performed in triplicate) at 37ºC and 5% CO2, After the incubation time, the culture medium was preserved, and cells were washed with 1mL phosphate buffered saline (PBS). The cells were then detached with 500 µL trypsin-EDTA followed by 5 minutes of incubation at 37ºC and 5% CO2, the radioactivity of the culture medium and the radioactivity bound to the cells were measured in a counter solid scintillation. The uptake percentage was expressed as the percentage of radioactivity bound to cells relative to total activity (medium radioactivity + cell-bound radioactivity), as is shown in the following equation:
4.10. Competition Assay
The neurons were incubated at 37ºC and 5% CO2 for 20 min, with 14µCi (200µL) of [11C]FLA and increasing concentrations (16-1600nM) of the commercially available GSK3β inhibitor, 3-acetyl-4- (1-methyl-1H- indol-3-yl)-1H-pyrrole-2,5-dione (VP.36). After the incubation time, the culture medium was preserved, and the cells were washed with PBS and detached with trypsin-EDTA, as described above for uptake studies. The activity of the culture medium and the activity bound to the cells were measured and the percentage of uptake was calculated as described for uptake studies.
5. Conclusions
[11C]FLA, a methylated derivative from the natural compound glabranin, whose GSK-3β inhibitory activity was previously assessed by in silico studies, was synthesized and evaluated for the first time. Furthermore, the IC50 of glabranin was determined by our group, confirming the in silico prediction. Labeling, carried out with 11C-methylation was optimized. The purification process developed rendered a high radiochemical purity product with a reasonable yield.
[11C]FLA was stable over time. Lipophilic as expected with an adequate value to cross biological membranes, including BBB, a capacity confirmed in animal studies since uptake of [11C]FLA was seen in the brain.
In vitro studies showed a significant uptake of the radiotracer by neurons in culture, being greater than in astrocytes. This behavior reinforces the hypothesis of the specific interaction of [11C]FLA with GSK-3β since this enzyme is expressed mainly in neurons.
The methodology employed, obtaining and characterizing an 11C-labeled methylated derivative, allowed to easily perform the in vitro evaluation of a molecule with biological activity obtained from a native South American species. These results demonstrate the utility of PET technology in confirming the biological activity of natural products and its potential for drug discovery and evaluation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, M.Z. (Maia Zeni).; methodology, M.Z. (Maia Zeni), L.R. (Laura Reyes) MB (Manuela Bentura), L.M. Loreto Martinez-Gonzalez.; software, M.Z (Maia Zeni), F.A (Florencia Arredondo), .; formal analysis, M.Z.(Maia Zeni).; investigation, M.Z.(Maia Zeni).; resources M.Z.(Maia Zeni).; data curation, M.Z.(Maia Zeni).; writing—original draft preparation, M.Z.(Maia Zeni).; writing—review and editing, M.Z.(Maia Zeni).; visualization, M.Z.(Maia Zeni) F.A(Florencia Arredondo) and M.D. (Maria Daniela Santi), Diego Carvallo, J.A.(Juan A. Abin-Carriquiry) J.P (Juan Pablo Gambini ) and P.D. (Pablo Duarte); supervision, J.G. (Javier Giglio) A.R. (Ana Rey), (A.M.)Ana Martinez.; project administration, J.G. (Javier Giglio) and A.R. (Ana Rey). All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board (or Ethics Committee) of NAME OF INSTITUTE (protocol code XXX and date of approval).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are available upon reasonable request.
Acknowledgments
PEDECIBA Química and Agencia Nacional de Investigación e Inovación (ANII) MOV_CA_2021_1_171461, POS_NAC_D_2020_1_164001 and POS_NAC_2018_1_151615.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Singh DB, Gupta MK, Kesharwani RK, Sagar M, Dwivedi S, Misra K. Molecular drug targets and therapies for Alzheimer’s disease. Transl Neurosci. 2014;5(3):203-217. [CrossRef]
- Alzheimer Disease International. Informe Mundial sobre el Alzheimer 2019 Actitudes hacia la demencia. Glob voice Dement. Published online 2019:13.
- Hardy J. Has the Amyloid Cascade Hypothesis for Alzheimers Disease been Proved? Curr Alzheimer Res. 2006;3(1):71-73. [CrossRef]
- Durães F, Pinto M, Sousa E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals. 2018;11(2):44. [CrossRef]
- Mahase E. Three FDA advisory panel members resign over approval of Alzheimer’s drug. BMJ. Published online June 11, 2021:n1503. [CrossRef]
- Day GS, Scarmeas N, Dubinsky R, et al. Aducanumab Use in Symptomatic Alzheimer Disease Evidence in Focus. Neurology. 2022;98(15):619-631. [CrossRef]
- Reardon S. FDA approves Alzheimer’s drug lecanemab amid safety concerns. Nature. 2023;613(7943):227-228. [CrossRef]
- Alghamdi SS, Suliman RS, Aljammaz NA, Kahtani KM, Aljatli DA, Albadrani GM. Natural Products as Novel Neuroprotective Agents; Computational Predictions of the Molecular Targets, ADME Properties, and Safety Profile. Plants. 2022;11(4):549. [CrossRef]
- Newman DJ, Cragg GM. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J Nat Prod. 2020;83(3):770-803. [CrossRef]
- Hong J. Role of natural product diversity in chemical biology. Curr Opin Chem Biol. 2011;15(3):350-354. [CrossRef]
- Dzobo K. The Role of Natural Products as Sources of Therapeutic Agents for Innovative Drug Discovery. In: Comprehensive Pharmacology. Elsevier; 2022:408-422. [CrossRef]
- Santi MD, Peralta MA, Mendoza CS, Cabrera JL, Ortega MG. Chemical and bioactivity of flavanones obtained from roots of Dalea pazensis Rusby. Bioorg Med Chem Lett. 2017;27(8):1789-1794. [CrossRef]
- Santi MD, Arredondo F, Carvalho D, et al. Neuroprotective effects of prenylated flavanones isolated from Dalea species, in vitro and in silico studies. Eur J Med Chem. 2020;206:112718. [CrossRef]
- Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med. 2002;8(3):126-132. [CrossRef]
- Hur E-M, Zhou F-Q. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11(8):539-551. [CrossRef]
- Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol Ther. 2015;148:114-131. [CrossRef]
- Cormier KW, Woodgett JR. Recent advances in understanding the cellular roles of GSK-3. F1000Research. 2017;6:167. [CrossRef]
- Eldar-Finkelman H, Martinez A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front Mol Neurosci. 2011;4. [CrossRef]
- Kim YT, Hur E-M, Snider WD, Zhou F-Q. Role of GSK3 Signaling in Neuronal Morphogenesis. Front Mol Neurosci. 2011;4. [CrossRef]
- Kaidanovich-Beilin O, Beaulieu J-M, Jope RS, Woodgett JR. Neurological Functions of the Masterswitch Protein Kinase – Gsk-3. Front Mol Neurosci. 2012;5. [CrossRef]
- Llorens-Martà n M, Blazquez-Llorca L, Benavides-Piccione R, et al. Selective alterations of neurons and circuits related to early memory loss in Alzheimerâ€TMs disease. Front Neuroanat. 2014;8. [CrossRef]
- Pandey MK, DeGrado TR. Glycogen Synthase Kinase-3 (GSK-3)-Targeted Therapy and Imaging. Theranostics. 2016;6(4):571-593. [CrossRef]
- Sayas CL, Ávila J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells. 2021;10(4):721. [CrossRef]
- Panche AN, Diwan AD, Chandra SR. Flavonoids: an overview. J Nutr Sci. 2016;5:e47. [CrossRef]
- Cassidy A, Kay C. Phytochemicals: Classification and Occurrence. In: Encyclopedia of Human Nutrition. Elsevier; 2013:39-46. [CrossRef]
- Ferrieri RA. Production and Application of Synthetic Precursors Labeled with Carbon-l 1 and Fluorine-18.
- Wolf AP, Redvanly CS. Carbon-11 and radiopharmaceuticals. Int J Appl Radiat Isot. 1977;28(1-2):29-48. [CrossRef]
- Schmidt S, Gonzalez D, Derendorf H. Significance of Protein Binding in Pharmacokinetics and Pharmacodynamics. J Pharm Sci. 2010;99(3):1107-1122. [CrossRef]
- Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRX. 2005;2(4):541-553. [CrossRef]
- Yao HB, Shaw PC, Wong CC, Wan DCC. Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J Chem Neuroanat. 2002;23(4):291-297. [CrossRef]
- Wang X, L. Michaelis M, K. Michaelis E. Functional Genomics of Brain Aging and Alzheimers Disease: Focus on Selective Neuronal Vulnerability. Curr Genomics. 2010;11(8):618-633. [CrossRef]
- Muyllaert D, Kremer A, Jaworski T, et al. Glycogen synthase kinase-3β, or a link between amyloid and tau pathology? Genes, Brain Behav. 2008;7(SUPPL. 1):57-66. [CrossRef]
- Perez DI, Palomo V, Pérez C, et al. Switching Reversibility to Irreversibility in Glycogen Synthase Kinase 3 Inhibitors: Clues for Specific Design of New Compounds. J Med Chem. 2011;54(12):4042-4056. [CrossRef]
- Atanasov AG, Zotchev SB, Dirsch VM, Supuran CT. Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov. 2021;20(3):200-216. [CrossRef]
- Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov. 2015;14(2):111-129. [CrossRef]
- Newman DJ, Cragg GM. Natural Products as Sources of New Drugs from 1981 to 2014. J Nat Prod. 2016;79(3):629-661. [CrossRef]
- Atanasov AG, Waltenberger B, Pferschy-Wenzig E-M, et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol Adv. 2015;33(8):1582-1614. [CrossRef]
- Waltenberger B, Mocan A, Šmejkal K, Heiss E, Atanasov A. Natural Products to Counteract the Epidemic of Cardiovascular and Metabolic Disorders. Molecules. 2016;21(6):807. [CrossRef]
- Tintore M, Vidal-Jordana A, Sastre-Garriga J. Treatment of multiple sclerosis — success from bench to bedside. Nat Rev Neurol. 2019;15(1):53-58. [CrossRef]
- Di Paolo M, Papi L, Gori F, Turillazzi E. Natural Products in Neurodegenerative Diseases: A Great Promise but an Ethical Challenge. Int J Mol Sci. 2019;20(20):5170. [CrossRef]
- Nerella SG, Singh P, Sanam T, Digwal CS. PET Molecular Imaging in Drug Development: The Imaging and Chemistry Perspective. Front Med. 2022;9. [CrossRef]
- Brooks DJ. Positron emission tomography and single-photon emission computed tomography in central nervous system drug development. NeuroRX. 2005;2(2):226-236. [CrossRef]
- Nicolazzo JA, Charman SA, Charman WN. Methods to assess drug permeability across the blood-brain barrier. J Pharm Pharmacol. 2010;58(3):281-293. [CrossRef]
- Phelps ME. Positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci. 2000;97(16):9226-9233. [CrossRef]
- Youdim KA, Qaiser MZ, Begley DJ, Rice-Evans CA, Abbott NJ. Flavonoid permeability across an in situ model of the blood–brain barrier. Free Radic Biol Med. 2004;36(5):592-604. [CrossRef]
- Youdim KA, Dobbie MS, Kuhnle G, Proteggente AR, Abbott NJ, Rice-Evans C. Interaction between flavonoids and the blood–brain barrier: in vitro studies. J Neurochem. 2003;85(1):180-192. [CrossRef]
- Rangel-Ordóñez L, Nöldner M, Schubert-Zsilavecz M, Wurglics M. Plasma Levels and Distribution of Flavonoids in Rat Brain after Single and Repeated Doses of Standardized Ginkgo biloba Extract EGb 761®. Planta Med. 2010;76(15):1683-1690. [CrossRef]
- Lambrecht FY, Yilmaz O, Bayrak E, Kocagozoglu G, Durkan K. Could be radiolabeled flavonoid used to evaluate infection? J Radioanal Nucl Chem. 2010;283(2):503-506. [CrossRef]
- Domínguez JM, Fuertes A, Orozco L, del Monte-Millán M, Delgado E, Medina M. Evidence for Irreversible Inhibition of Glycogen Synthase Kinase-3β by Tideglusib. J Biol Chem. 2012;287(2):893-904. [CrossRef]
- Yao H-B, Shaw P-C, Wong C-C, Wan DC-C. Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J Chem Neuroanat. 2002;23(4):291-297. [CrossRef]
Figure 1.
Chemical structure of (S)-5,7-dihydroxy-8-(3-methylbut-2-en-1-yl)-2-phenylchroman-4-one (glabranin).
Figure 1.
Chemical structure of (S)-5,7-dihydroxy-8-(3-methylbut-2-en-1-yl)-2-phenylchroman-4-one (glabranin).
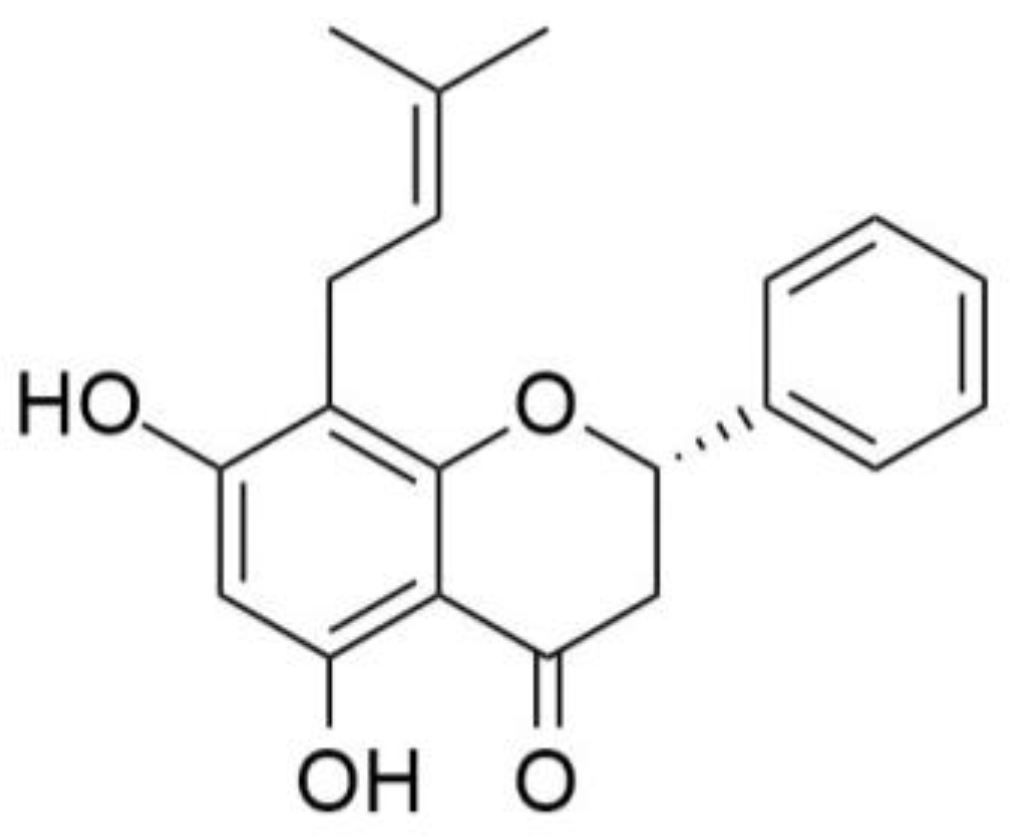
Figure 2.
Radiosynthesis of [11C]FLA Different reaction conditions were assayed to maximize labeling yield as shown in Table 1.
Figure 2.
Radiosynthesis of [11C]FLA Different reaction conditions were assayed to maximize labeling yield as shown in Table 1.
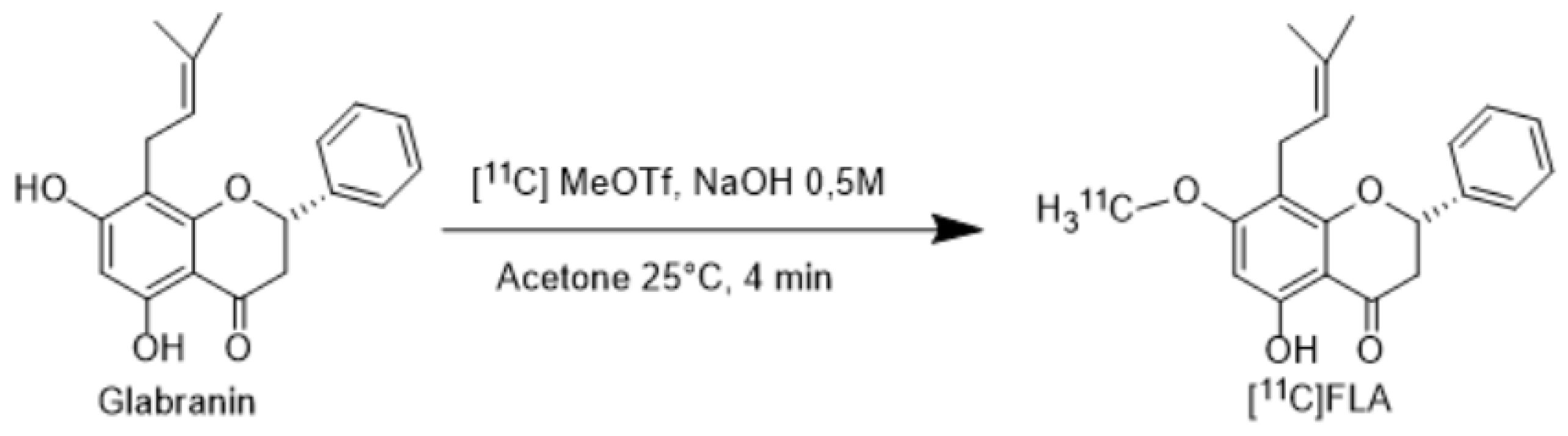
Figure 3.
Semipreparative chromatographic profile of radiosynthesis [11C]FLA.
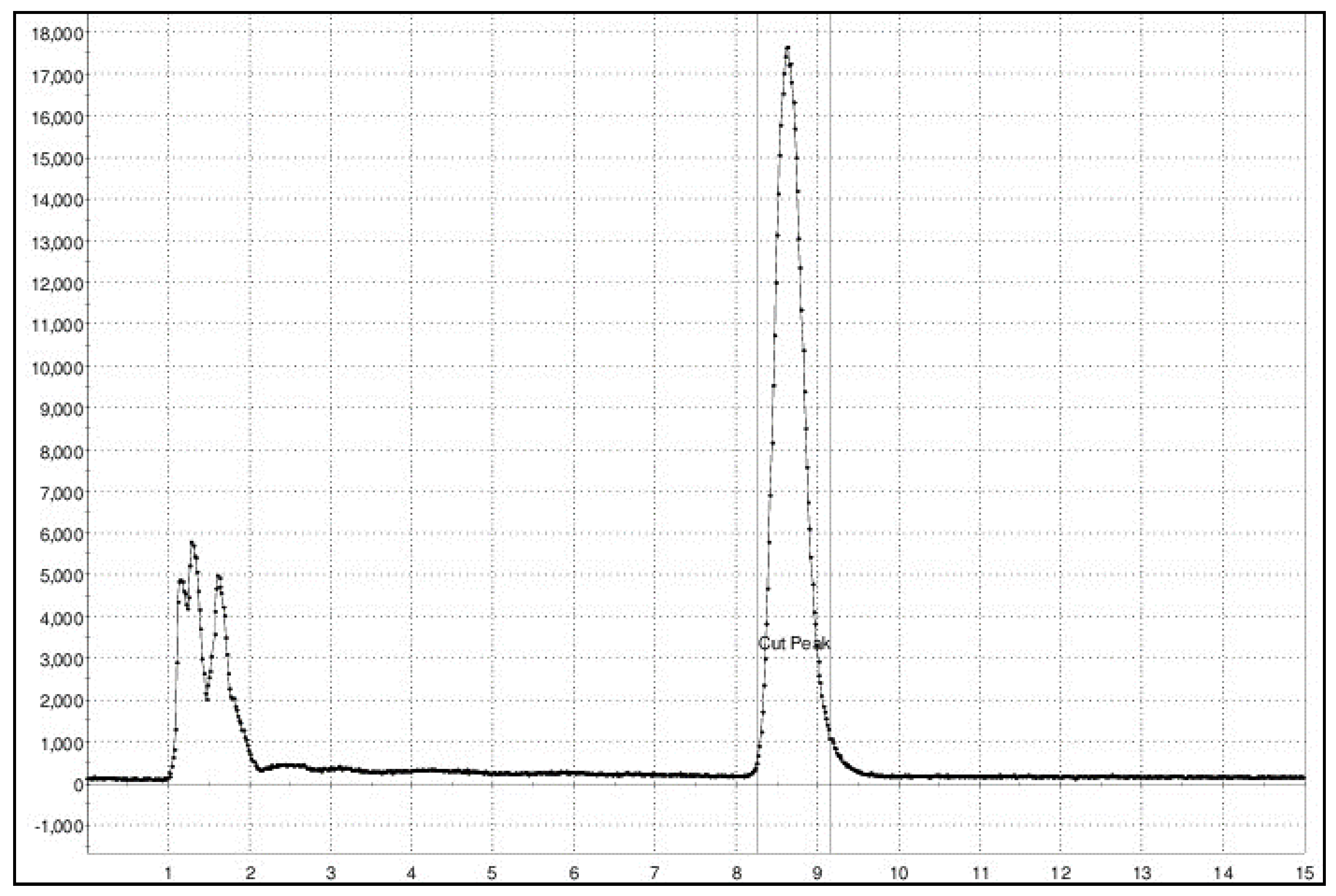
Figure 4.
Analytical radio-chromatographic profile of [11C]FLA, upper- UV detection a 290 nm; lower- gamma detection.
Figure 4.
Analytical radio-chromatographic profile of [11C]FLA, upper- UV detection a 290 nm; lower- gamma detection.
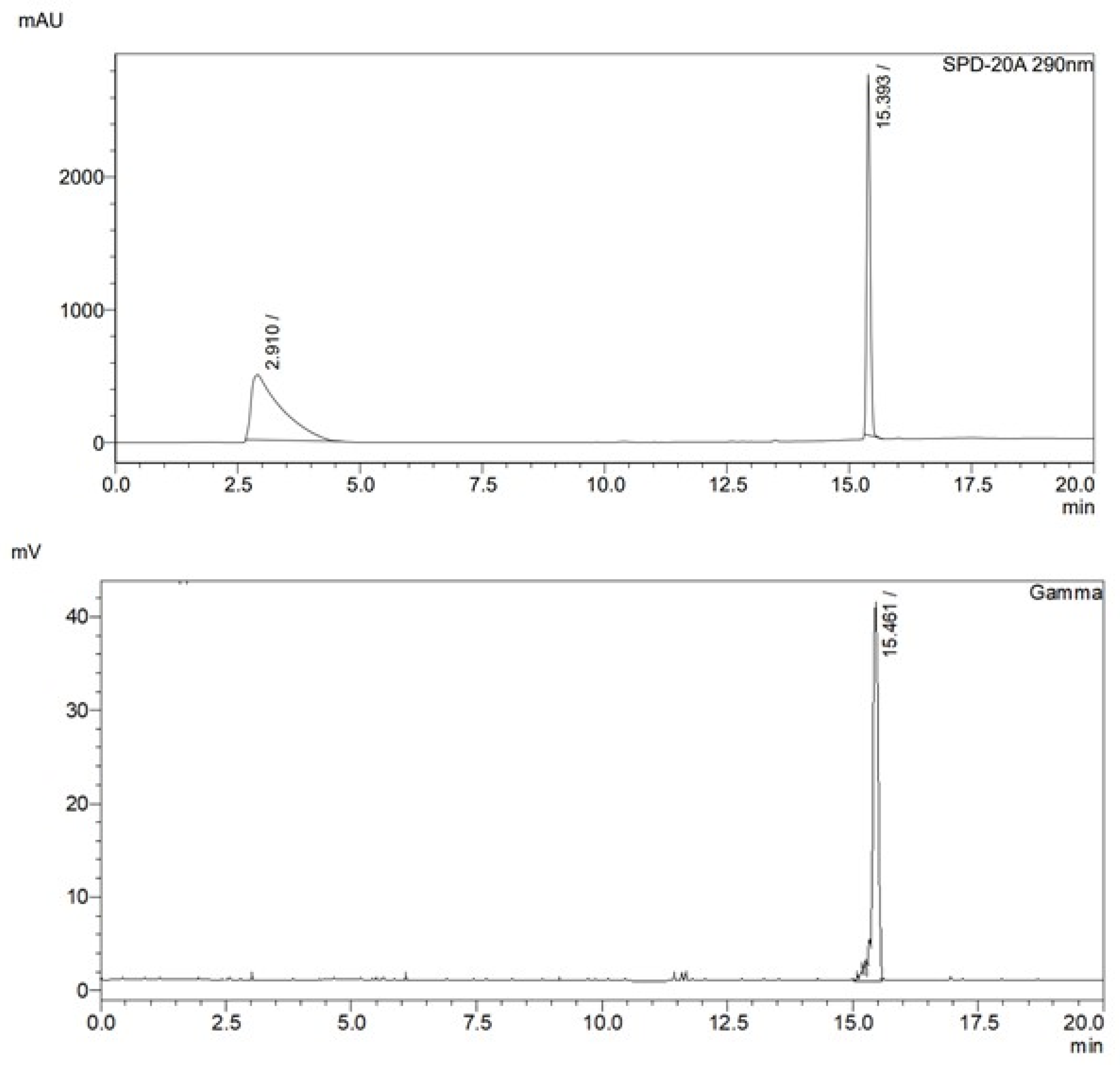
Figure 5.
Biodistribution of radioactivity after intravenous (i.v.) injection of [11C]FLA in C57BL6J black mice.
Figure 5.
Biodistribution of radioactivity after intravenous (i.v.) injection of [11C]FLA in C57BL6J black mice.
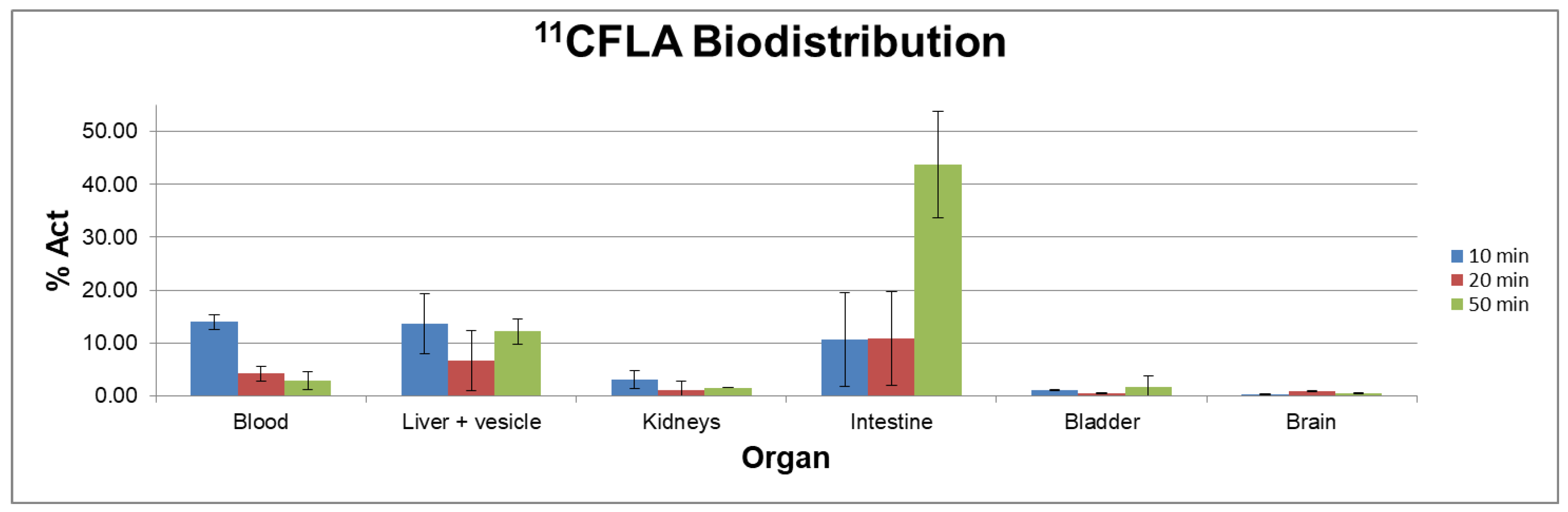
Figure 6.
% Activity/g vs t in blood and brain as a function of time after injection of [11C]FLA.
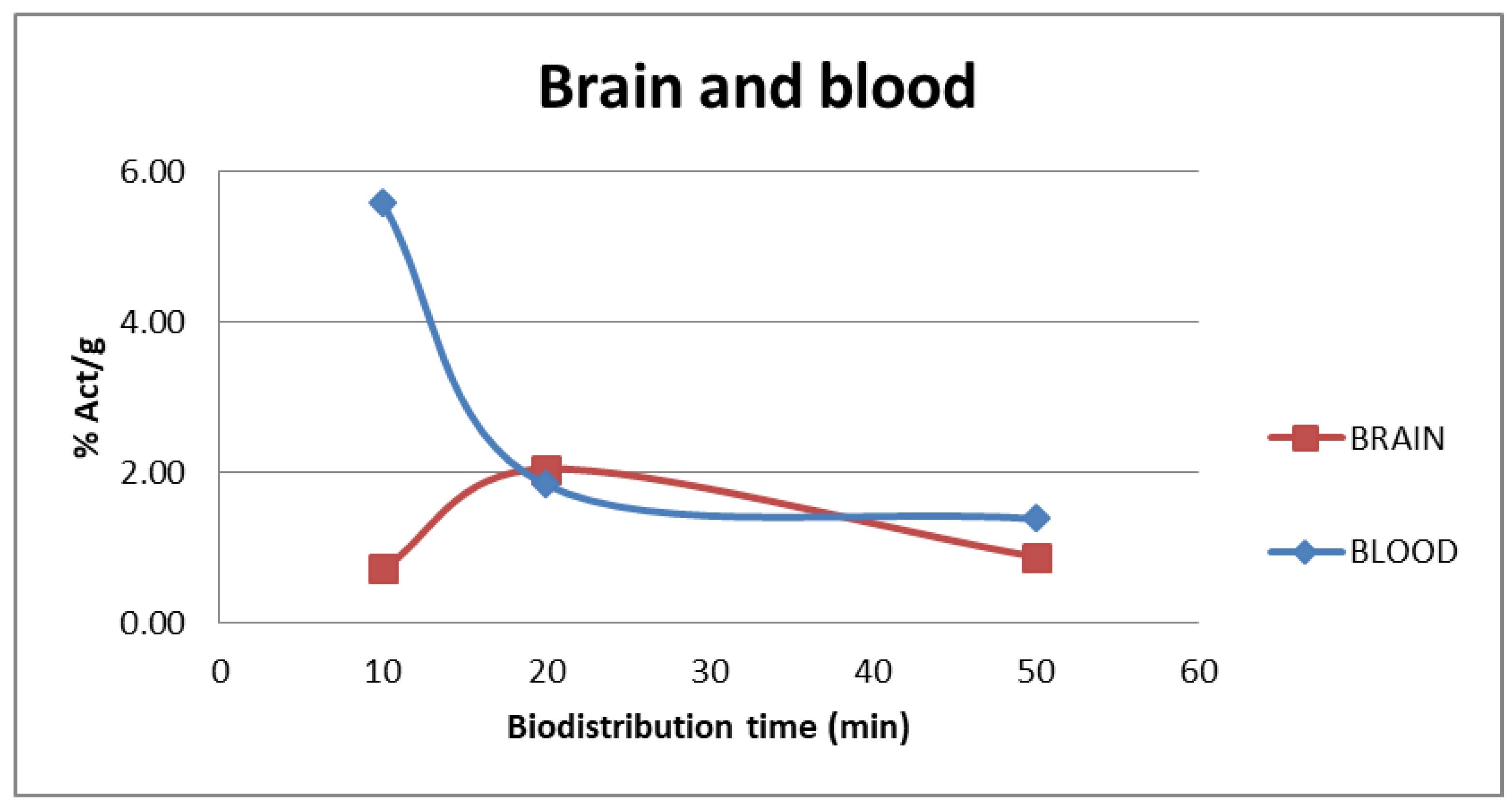
Figure 8.
Dose-response curve, by determination IC50 of glabranin and FLA.
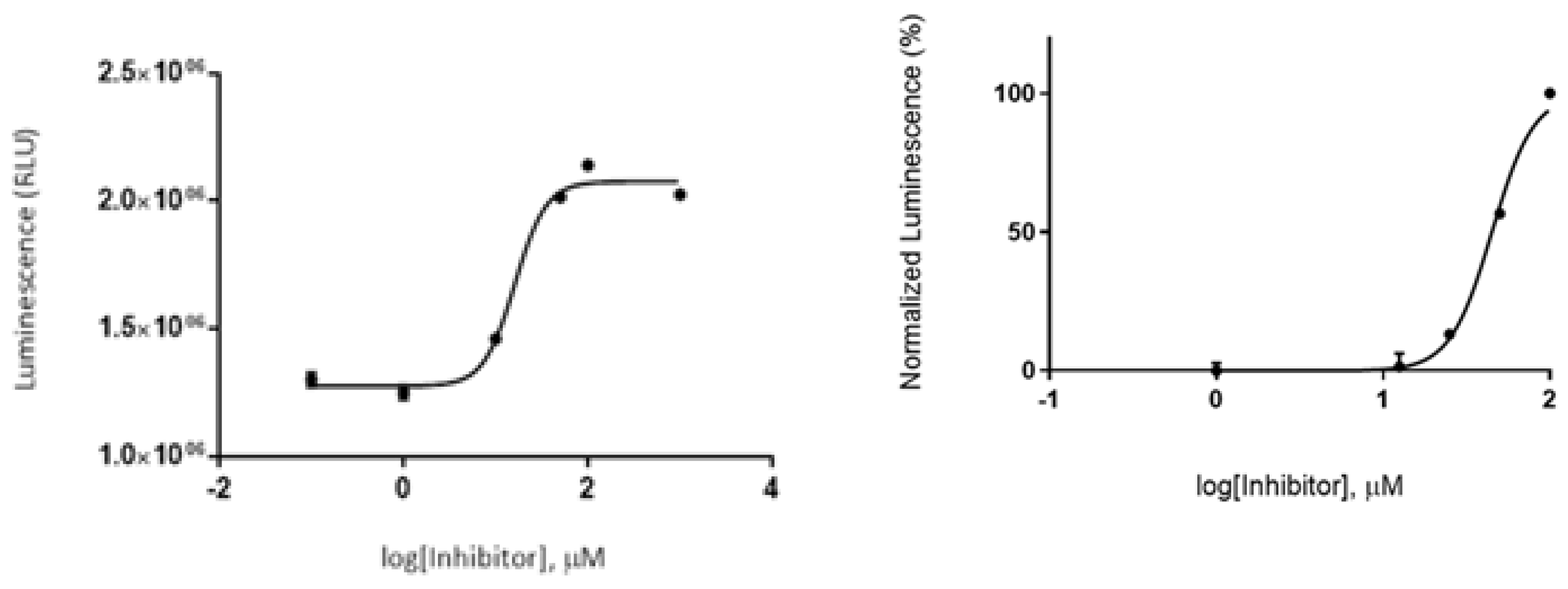
Figure 9.
[11C]FLA uptake by neurons when challenged against the commercially available GSK-3β inhibitor VP.36.
Figure 9.
[11C]FLA uptake by neurons when challenged against the commercially available GSK-3β inhibitor VP.36.
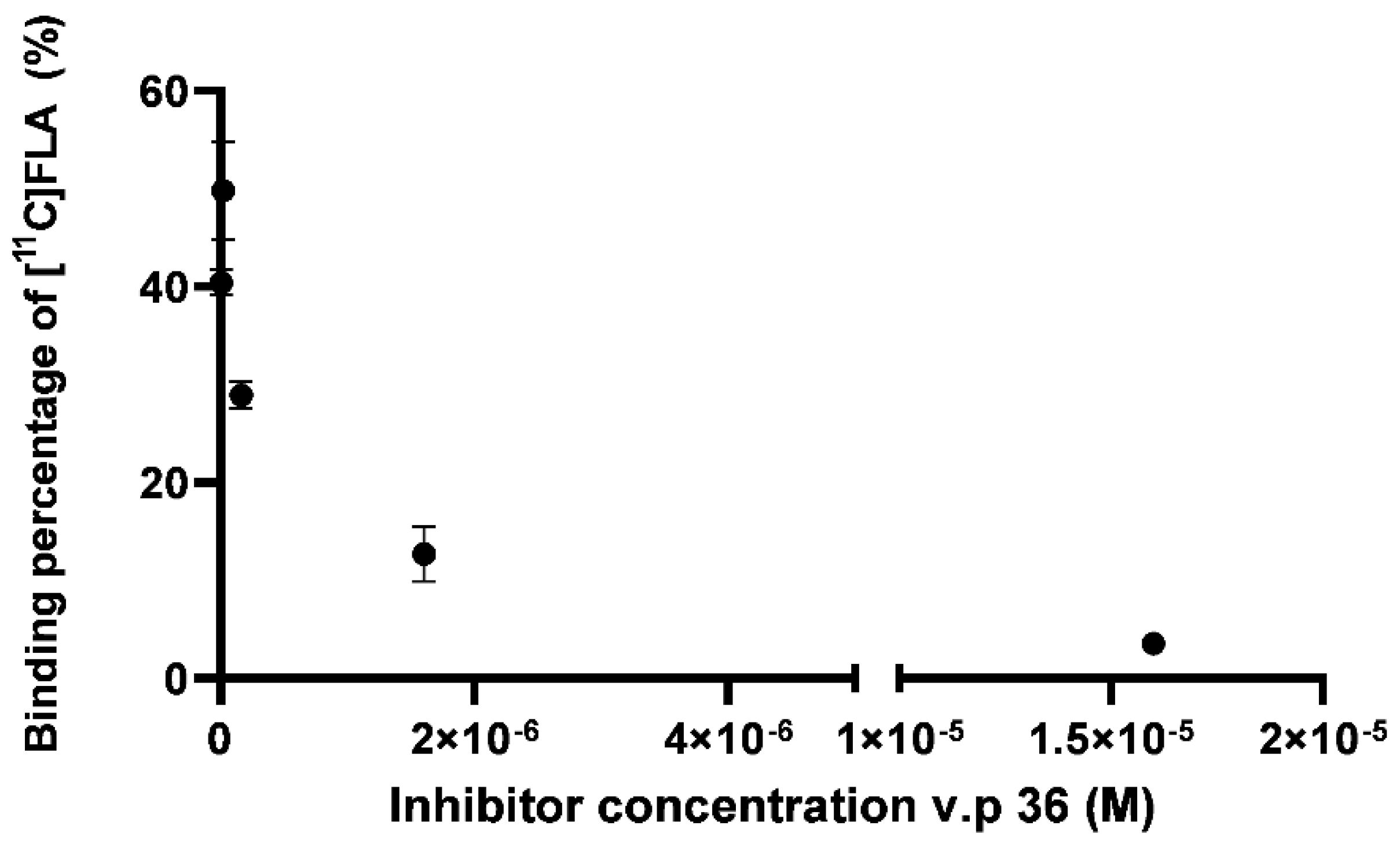

Table 1.
Summary of glabranin labeling conditions.
| Methylating agent | Base | Solvent | Temperature (°C) |
Time (min) |
Yield (%) |
|
|---|---|---|---|---|---|---|
| I | [11C]CH3I | NaOH 5M 5µL | DMSO 0.5 mL | 80 | 1 | 0 |
| II | [11C]CH3I | TBAF 1M 100µL | Acetone 0.4 mL and THF 0.15 mL | 80 | 5 | 0 |
| III | [11C]CH3OTf | NaOH 0.5M 2µL | Acetone 0.3 mL | 25 | 4 | 16 ± 3 (n=5) |
Table 2.
Uptake of [11C]FLA by neurons vs incubation time.
| Time (min) | Binding percentage (%) |
|---|---|
| 5 | 4.2 ± 0.7 |
| 10 | 8.4 ± 1.1 |
| 20 | 13.7 ± 1.5 |
Table 3.
Uptake 11C-FLA in neurons vs activity loaded.
| Activity (MBq) | Binding percentage (%) |
|---|---|
| 0.5 | 8.3 ± 0.1 |
| 1 | 6.5 ± 0.3 |
| 2 | 5.4 ± 0.1 |
Table 4.
Uptake [11C]FLA by astrocytes vs incubation time.
| Time (min) | Binding percentage (%) |
|---|---|
| 5 | 0.8 ± 0.1 |
| 10 | 1.0 ± 0.2 |
| 20 | 6.4 ± 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



