
Submitted:
05 November 2024
Posted:
05 November 2024
You are already at the latest version
Abstract
Advanced triple-negative breast cancer (TNBC) has poorer outcomes due to its aggressive behaviour and limited treatment options. While therapies like checkpoint inhibitors and PARP inhibitors (PARPi) offer some benefit, chemotherapy remains ineffective beyond the first line of treatment. Antibody-drug conjugates (ADCs) like sacituzumab govitecan-hziy (SG) represent a significant advancement. SG combines SN-38, an irinotecan derivative, with a TROP-2-targeting antibody via a pH-sensitive linking moiety, achieving an good drug:antibody ratio. Infused on Days 1 and 8 over 21 days, SG utilizes a moderately stable linker to ensure effective tumor targeting while minimizing side effects like diarrhea. By delivering SN-38 into the tumor niche, it induces double-stranded DNA damage, promoting cancer apoptosis. In a phase I-II study involving 108 mTNBC patients, SG achieved an overall response rate (ORR) of 33.3% and a median response period of 7.7 months. Common side effects included neutropenia, nausea, and fatigue. This review highlights clinical potential, pharmacokinetics, safety profile, resistance mechanisms of SG along with key ongoing clinical trials, emphasizing its role in managing refractory mTNBC, especially in third-line therapy.
Keywords:
Advanced triple-negative breast cancer (TNBC)
; Antibody-drug conjugates
1. Introduction
Breast cancer (BC) stands as the most prevalent neoplasm affecting females globally, with over 2.3 million freshly diagnosed cases documented in 2022 [1,2]. Due to its heterogeneous nature, it remains the second largest contributor to cancer-associated fatalities [3]. The St. Gallen International Breast Cancer Conference defined BC subgroups depending on either the presence or lack of hormone receptors: luminal A (ER/PR+, HER2−, Ki67+ < 20%, based on immuno-histochemistry staining pattern), luminal B (ER/PR+ < 20%, HER2−, Ki67+ ≥ 20%); HER2+ B2 (ER/PR+, HER2 upregulation), HER2 over expression (ER−, PR−, HER2 upregulation), basal-like TNBC (ER−, PR−, HER2−), and other unusual subtypes [4].
Triple-negative breast cancer or TNBC as it is commonly known is a notoriously malignant and recurrent class of BC with limited targeted therapies, relying mainly on cytotoxic chemotherapy. Median survival for advanced or metastatic TNBC (mTNBC) is around 12-14 months, with significant symptoms impacting quality of life [5]. Current single-agent chemotherapies for recurrent locally advanced TNBC and mTNBC show low response rates (5-10%) and progression-free survival (PFS) rate of mere 2-3 months [6]. Although immunotherapy and PARP inhibitors (PARPi) like olaparib can elicit responses in some TNBC patients, their survival benefits are uncertain.
Trophoblast cell surface antigen 2 (Trop-2), a surface protein up regulated in plethora of malignancies, presents a potential candidate for novel therapeutics. Sacituzumab govitecan (SG), an antibody-drug conjugate (ADC) directed against Trop-2, has shown efficacy in advanced TNBC. Administered at 10 mg/kg, SG delivers chemotherapy with manageable side effects. On April 22, 2020, SG was given accelerated authorization by the Food and Drug Administration (FDA) for treating mTNBC [7]. Despite promising Phase II/III results, ongoing trials are needed to fully assess SG's efficacy, tolerability, and Trop-2's function in malignancies. This review examines the development, mechanism of action, pharmacokinetics, adverse events (AEs), safety profile and clinical efficacy of SG, while emphasizing the need for further trials and clinical practice.
2. Triple-Negative Breast Cancer
Representing ~15-20% of total invasive BCs, TNBC differs significantly from other forms due to its distinct molecular and clinical hallmarks [8,9]. TNBC lacks estrogen, progesterone, and HER2 (human epidermal growth factor 2) receptors [10]. This receptor-negative status excludes TNBC patients from benefiting from hormonal or HER2-targeted therapeutic agents. As a result, TNBC is managed with chemotherapy before surgery, often referred to as neoadjuvant chemotherapy (NAC) [11,12], which remains the standard care, especially in early-stage disease. The overall response of TNBC to NAC is strongly related to long-term clinical efficacy, as NAC lowers the tumor load and nodal involvement [13].
2.1. Clinical Characteristics of TNBC
TNBC, typically diagnosed in younger women, is renowned for its extremely aggressive nature, greater metastatic potential, poorer overall outcomes, higher recurrence pattern and mortality [14]. It is well known fact that the probability of secondary relapse during the first five years of diagnosis is roughly three times greater in TNBC patients than in non-TNBC individuals [15]. However, after five years, the risk of late recurrence is considerably lower, at less than 3% [16]. The lungs, lymph nodes, and brain are the most common metastatic sites in TNBC [17], with survival rate less than 2 years [18].
Histologically, TNBC tumors are typically Grade 3, exhibit a fast-paced growth rate and abnormal cell cycle regulation, as evidenced by elevated Ki-67 levels [19]. Genetic predisposition plays a central function in TNBC progress, with BRCA1 and BRCA2 mutations being prevalent in TNBC patients across various populations, with a carrier rate of 10.6–30.9% [20,21]. International variations in BRCA mutation prevalence highlight the need for population-specific risk assessments. For instance, 23% of TNBC population in Mexico City had a BRCA1 founder mutation (BRCA1 ex9-12del) [22], while BRCA risk calculators designed for Caucasians often underestimate risk in Asian patients [23]
2.2. Molecular Subtypes and Heterogeneity of TNBC
The molecular heterogeneity of TNBC complicates its treatment, as it exhibits various subtypes with distinct genetic and biological behaviors. These subtypes differ in their gene expression profiles, further complicating the identification of universal therapeutic targets. This heterogeneity also contributes to the variability in clinical outcomes among TNBC patients, with some responding well to treatments, while others experience rapid disease progression.
Molecular heterogeneity of been progressively explored through advanced transcriptomics. Lehmann and coworkers initially conducted a comprehensive analysis of 21 publicly available microarray datasets, identifying seven distinct TNBC subtypes: basal-like 1 (BL1), basal-like 2 (BL2), immunomodulatory (IM), mesenchymal (M), mesenchymal-stem-like (MSL), luminal androgen receptor (LAR), and an unstable subtype (UNS) [24]. These classifications relied on the manifestation of key markers like ESR1, PGR, and ERBB2 and are defined by unique molecular signatures, including patterns of gene expression, mutations, and polymorphism in copy-numbers [25,26].
The BL1 subclass exhibit a substantial abundance of genes associated with DNA damage repair mechanisms, with a strikingly high frequency of TP53 mutations (up to 92%) and amplifications in genes like MYC, CDK6, and CCNE1 as well as deletions in BRCA2, PTEN, MDM2, and RB1. Meanwhile, the BL2 subclass focuses more on growth factor signaling and metabolic cascades. Together, these subtypes are characterized by high proliferation and show favorable responses to chemotherapy, particularly with mitotic inhibitors like taxanes. In contrast, the IM subclass is defined by up regulation of immune-related genes, including JAK/STAT and TNF signaling cascades, suggesting that immunotherapy might be a potential treatment avenue for this group [24].
The mesenchymal (M) and mesenchymal-stem-like (MSL) subtypes, while both associated with features such as cell motility and epithelial-mesenchymal transition (EMT), can be distinguished based on their gene expression profiles. MSL is particularly enriched in genes associated with angiogenesis and stem cell-like characteristics with under-expressed claudin. Despite being triple-negative, the LAR subclass shows a luminal pattern of RNA expression and is notably enriched for androgen receptor (AR) expression. Around 82% of LAR tumors express AR and have high levels of luminal markers such as FOXA1 and GATA3. This subtype also carries frequent mutations in key signaling genes like PIK3CA (55%), KMT2C (19%), and AKT1 (13%). Notably, LAR tumors have the lowest response rates to chemotherapy, as reflected by a poor pathological complete response (pCR) rate, underscoring the need for alternative therapeutic approaches targeting AR signaling or PI3K/AKT pathways [24]. Remarkably, these 7 classes were absent in a separate investigation using IHC-identified TNBC rather than gene expression-defined TNBC. Additionally, the reproducible nature of the BL2 and UNS clusters was inconsistent in studies diagnosing TNBC through mRNA expression [27].
In 2016, Lehmann and colleagues revisited the classification system (Figure 1), finding that some of the earlier subtypes, particularly IM and MSL, were prejudiced by the prevalence of cancer-associated stromal cells and tumor-infiltrating lymphocytes (TILs), which complicated their initial definitions. This led to a refined four-subtype classification—BL1, BL2, M, and LAR—commonly known as TNBCtype4 [28]. Each of these subtypes exhibits distinct clinical outcomes, including variations in response to chemotherapy and patterns of recurrence. For instance, BL1 tends to have the highest response rates (65.6%) to neoadjuvant chemotherapy, while LAR tumors often involve local lymph nodes and are highly prone to bone metastases [29].
Single-cell RNA sequencing studies have further complicated this picture, showing that most TNBC tumors contain multiple subtypes within a single mass, revealing the intratumoral heterogeneity that bulk sequencing methods fail to capture. This finding suggests that more nuanced and granular approaches, like single-cell analyses, are required to fully understand the biological processes at play and improve treatment strategies [30]. Further, variations in the intra- and inter-tumoral diversity between TNBC and ER+ BC may partially clarify the pitfalls in applying commercially available gene-expression assays for prognosis in TNBC.
2.3. Treatment Challenges
As TNBC cells lack hormone receptors, standard endocrine therapies and targeted treatments are ineffective in. Drug chemotherapy is typically the stronghold of managing TNBC, particularly in early-stage disease, where it is often administered as neoadjuvant chemotherapy (NAC), given before surgery to shrink tumors and reduce nodal involvement. While NAC can be effective in reducing tumor burden, approximately 50% of TNBC patients still exhibit residual disease after treatment, meaning that not all cancer cells are eradicated. New treatments for TNBC have emerged in recent years (Figure 2), focusing on overcoming the challenges associated with its aggressive behavior, lack of hormone receptors, and resistance to conventional therapies [31]. These novel approaches aim to enhance clinical outcomes by targeting molecular cascades specific to TNBC, as well as leveraging advancements in immunotherapy.
In the past decade, novel therapeutic interventions for TNBC have emerged, including PARPi that target DNA repair deficits in BRCA1/2-mutated TNBC, marking a substantial breakthrough for patients with these mutations. Immune checkpoint inhibitors (ICI), particularly PD-1 and PD-L1 inhibitors, show promise in TNBC treatment. Pembrolizumab, an anti-PD-1 inhibitor, enhances overall survival (OS) rate in PD-L1+ TNBC patients in the KEYNOTE-522 study, culminating in its FDA endorsement for use with chemotherapy as a neoadjuvant cure [32]. Other emerging therapeutics for TNBC includes PI3K/AKT/mTOR inhibitors and trastuzumab deruxtecan targeting HER2-low expression [33]. While advances have been made, current therapeutic options are limited to specific TNBC subgroups, and identifying new targets and designing effective ADCs remain challenging. Additionally, significant obstacles persist in developing accurate predictive biomarkers and overcoming immunotherapy resistance in TNBC.
2.3.1. Neoadjuvant Treatments
Neoadjuvant chemotherapy (NAC) is the most widely accepted course of therapy for stage II–III TNBC [34,35]. In chemotherapy, pCR, indicative of lack of metastasis in breast tissue and lymph nodes altogether, is linked to a fewer risk of relapse and improved OS [36]. Achieving pCR after NAC has been proven to indicate long-term health advantages [37] and serves as an intermediate marker for enhanced survival outcomes [38]. Residual disease post NAC is linked with a greater hazard of untimely relapse and distant metastasis, resulting in poorer long-term survival outcomes [39]. Traditionally, NAC involves treatment with anthracyclines, cyclophosphamides, and paclitaxel, resulting in a pCR rate of 35–45% [40,41,42]. Additionally, clinical trial designs like I-SPY 2 (NCT01042379) [43] and ARTEMIS (ISRCTN15384496) [44,45] aim to expedite the development of effective targeted treatments for NAC. Researchers are now testing new combinations of therapies, including the addition of PARPi and ICI to NAC, to improve response rates.
2.3.2. . Poly (ADP-ribose) polymerase (PARP) Inhibitors (PARPi)
PARPi have established substantial potential in treating TNBC, especially in subjects with BRCA mutations by targeting tumor cells with DNA repair deficiencies; specifically exploiting the inability of BRCA-mutated cells to repair DNA damage effectively [32]. By inhibiting PARP, the cancer cells accumulate more DNA damage, leading to cell death [46]. PARPi are now approved for use in metastatic or advanced TNBC with BRCA mutations [47]. Olaparib (Lynparza) [48] and Talazoparib (Talzenna) [49] have demonstrated superior progression-free survival (PFS) in clinical studies for BRCA-mutated BC, notably TNBC, and represent a targeted therapy option beyond traditional chemotherapy. While primarily used alone, ongoing trials are exploring its effectiveness when combined with other therapies.
2.3.3. AKT Inhibitors
The AKT signal transduction cascade is typically dysregulated in TNBC, contributing to tumor growth and survival [50]. Capivasertib and ipatasertib, both AKT inhibitors, have shown potential in treating TNBC when combined with chemotherapy. Capivasertib, evaluated in the PAKT trial, increased PFS from 4.2-5.9 months, with better outcomes in individuals having PIK3CA/AKT1/PTEN mutations [51,52]. An ongoing trial is exploring its use with other therapies like durvalumab (NCT03742102), while its tolerability and effectiveness are under evaluation in the phase III clinical study, CAPitello290 (NCT03997123). Similarly, ipatasertib was tested in the LOTUS trial, where combining it with paclitaxel boosted PFS, especially among subjects with low PTEN or specific mutations [53,54]. However, a phase III trial (IPATunity130) did not confirm these benefits [55]. Both drugs are under continued investigation in combination therapies, especially with immunotherapy, to enhance treatment outcomes for TNBC.
2.3.4. Cancer Vaccines
Cancer vaccines aim to augment the inherent capability of immunological cells to detect and eliminate malignant lesions. HER2-targeted vaccines, such as E75, GP2, and AE37, have been developed using HER2-derived peptides to stimulate immune responses in TNBC. E75 (nelipepimut-S) is a peptide vaccine targeting the HLA-A2-restricted nonapeptide from HER2's extracellular domain [56], while GP2 target its transmembrane domain [57]. AE37, a 12-mer peptide, targets the modified intracellular domain of HER2 [58]. E75 and GP2 activate CD8+ cytotoxic T cells via MHC class I, while AE37, presented by MHC class II, stimulates CD4+ T cells. These vaccines have proven to be efficacious for treating low HER2-expressing TNBC [59,60].
Additionally, dendritic cell (DC) vaccines, loaded with antigens such as MUC1 and P32, have demonstrated immense potential in preclinical studies, enhancing immune responses and inhibiting tumor growth [61]. Researchers are also developing vaccines targeting non-HER2 antigens like NY-ESO-1, WT1, MAGE-12, and MUC1. Peptide vaccines like PVX-410 and TAC vaccines such as P10s-PADRE have recently been investigated in clinical studies for TNBC [62].
2.3.5. Immunotherapy
The deficiency of endocrine receptors in TNBC complicates the development of targeted treatments [63]. Given the limitations of chemotherapy and the urgent need for more effective treatments, immunotherapy has surfaced to be a promising option for TNBC. This approach leverages the immune cells to identify and eliminate cancerous ones, with concurrent strategies designed to enhance immune responses.
ICI targeting PD-1 or PD-L1 can re-engage the immune system to attack tumors. Pembrolizumab (Keytruda), a PD-1 inhibitor, has been permitted for usage in PD-L1+ mTNBC patients when combined with chemotherapy, with clinical trials like KEYNOTE-355 showing significant improvements in PFS [64]. In contrast, atezolizumab (Tecentriq), another checkpoint inhibitor, was withdrawn from the market after inconsistent results in improving OS when combined with nab-paclitaxel [65]. Despite these challenges, ICI continue to be a vital area of research in TNBC, predominantly in conjunction with other strategies. Further, adding immune checkpoint inhibitors like pembrolizumab to NAC has been shown to increase pCR rates [66,67].
Additionally, therapeutic approaches such as adoptive cell transfer (ACT), including TILs and chimeric antigen receptor (CAR) T-cell therapy, are being explored due to their encouraging clinical efficacy as well as enhanced cancer detection and eradication [68]. Efforts to enhance functioning of cytotoxic T lymphocyte (CTL) [69] and tumor associated macrophages (TAMs)[70] as well as counter tumor immunosuppression [71] are also underway to improve treatment efficacy.
2.3.6. Antibody-Drug Conjugates (ADCs)
ADCs are an exclusively unique group of therapeutic interventions that attach an antibody specifically targeting cancer cells with a cytotoxic drug. A recently developed ADC against TNBC is Sacituzumab Govitecan, which targets Trop-2—an protein frequently up regulated in TNBC cells—and delivers a potent chemotherapy agent directly at the tumour sites, thereby minimizing damage to normal cells. SG was approved in 2020 for the treating mTNBC in individuals who had received two prior therapies. It has shown significant clinical benefits, including improved OS and PFS in heavily pretreated subjects. Table 1 summarizes the essential features of SG.
3. Sacituzumab govitecan, a Trop-2–directed ADC
In order to develop a targeted therapeutic intervention, a humanized monoclonal immunoglobulin (hRS7, Sacituzumab) was linked with a potent chemotherapeutic molecule, SN-38 (govitecan), a derivative of irinotecan (CPT-11), topoisomerase-I inhibitor, with the help of a proprietary hydrolyzable linking agent to form Sacituzumab govitecan (SG, IMMU-132, TrodelvyTM), a first-in-class third-generation ADC [72,73]. Because of its unique design, the medication can be precisely delivered to breast cancer cells without endangering healthy tissues.
3.1. The Discovery and Development of Sacituzumab Govitecan
3.1.1. Trop-2 and Its Expression Pattern
Initially identified in trophoblast cells [74], Trop-2, a transmembrane glycoprotein receptor expressed by TACSTD2 gene loci on chr 1p32, has been classified under various names including tumor-associated calcium signal transducer 2 (TACSTD2), membrane component chromosome 1 surface marker 1 (M1S1), gastrointestinal antigen 733-1 (GA733-1), and epithelial glycoprotein-1 (EGP-1) [75,76].
Trop-2 plays an essential part in tumorigenesis, especially in TNBC, by promoting epithelial-mesenchymal transition (EMT), distant metastasis, and resistance to programmed cell death. Trop-2 is related with various aggressive tumor characteristics, including lymph node inflammation, metastasis, and poor OS [77]. Its overexpression is observed in 95% of TNBC cases and 88% of metastatic TNBC [78,79], making it a significant therapeutic target [80,81]. Furthermore, circulating tumor cells that constitutively express Trop-2 on their surface serve as excellent biomarkers for EMT as well as distant metastasis [82].
ADCs based targeted therapies have emerged for Trop-2-expressing TNBC, although their effectiveness is limited, with response rates not exceeding 35% [80,83,84]. The ASCENT trial demonstrated improved PFS and OS in individuals over-expressing Trop-2, emphasizing the importance of this receptor as a therapeutic marker [85,86,87,88]. Trop-2's role in TNBC therapy is complex, with ADC trials largely targeting patients lacking other treatment options, rather than those with high Trop-2 expression [33]. Further, its expression in normal tissues raises concerns about side effects [75,89], since just 54.4% of TNBC patients express Trop-2 [90]. Fluctuations in Trop-2 expression induced by EMT and related to tumor plasticity, complicate its role as a biomarker [91].
Recent studies suggest that the loss of Trop-2 drives carcinogenesis in certain cancers, highlighting its context-dependent function [92,93]. While Trop-2 overexpression is associated with resistance to therapies like PD-L1 inhibitors [94], its under-expression may increase sensitivity to anti-HER3 ADCs [95]. The multifaceted functioning of Trop-2 and its tumor-specific pathways necessitates further to better understanding its therapeutic potential and its impact on cancer progression as well as reasons behind suboptimal treatment outcomes.
3.1.2. RS7 Monoclonal Antibody
The RS7 murine monoclonal antibody, initially generated by Stein et al. [96] to target human non-small-cell lung cancer (NSCLC), also binds to a wide array of neoplasms, notably BC cells, with some binding observed in non-cancerous cells [97,98]. It targets a 46 kDa glycoprotein receptor, phosphorylated at serine-303 by protein kinase C, which gets reduced to 35 kDa upon deglycosylatation. Later identified as epithelial glycoprotein-1 (EGP-1) [97,99], it is an essential component of intracellular ERK/MAPK signaling cascade [100]. RS7, known for its rapid internalization by cells [96], shows promise as a vehicle for radiometabolic therapy. Studies using radioiodine-labeled RS7 in a prostate tumor model via SPECT support its potential for targeting cancer cells in both imaging and treatment [101].
3.1.3. SN-38, a Camptothecin Derivative
SN-38 (7-ethyl-10-hydroxycamptothecin), a semi-synthetic analogue of camptothecin, and the bioactive metabolite of irinotecan, was selected for its well-established clinical efficacy. Camptothecins target cancer cells by engaging with topoisomerase I (TOPO-I), resulting in formation of double-stranded DNA lesions in the S-phase of cell cycle [102,103]. Irinotecan, a precursor drug of SN-38, is highly active against solid cancers, particularly mTNBC [104]. The cytochrome P450 3A4 (CYP3A4) enzyme converts it into two inert compounds, 7-ethyl-10- [4-N-(5-amino-pentanoic acid)-1-piperidino] carbonyl oxycamptothecin (APC) and 7-ethyl-10-(4-amino- 1-piperidino) carbonyl oxycamptothecin (NPC) [105].
SN-38 has an IC50 of 1.0–6.0 nM, but its hydrophobicity and limited coupling sites make it challenging to formulate and contributes to poor tolerability [106]. Modifications in the structure, like glucuronidation and lactone ring opening, reduce its potency. To improve solubility, a dipiperidino side chain was added at C10, creating irinotecan. Once this carbamate side chain cleaved in the liver carboxylesterases, SN-38, up to 1000 times more potent than irinotecan, is released but quickly gets glucuronidated by uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1), affecting its efficacy [107]. SN-38 and its glucuronide (SN-38G) are subsequently lost into the bile and intestines. Since UGT1A1 is associated with the metabolism of SN-38, medications that influence UGT1A1 enzyme function might boost or shorten the intake of SN-38 [108].
3.1.4. Conjugation of RS7 with SN-38
Researchers optimized SN-38’s performance by attaching linkers to the C10 and C20 positions, using polyethylene glycol (PEG) to enhance solubility and reduce aggregation [72,73,109]. A maleimide moeity was introduced to link SN-38 with sulfhydryl side chain on the IgG, creating a durable thioether linkage. Some linkers included a cathepsin B cleavage site (Phe-Lys) for tumor-specific release and a pH-sensitive benzyl carbonate bond to release the drug in acidic environments [72]. The original linker (CL2) exhibited intermediate serum stability (1–2 days) [109], and was later modified to a CL2A derivative by removing phenylalanine at the cathepsin B cleavage site, improving product quality and yield without affecting SN-38 release [73,110]. The CL2A linking agent, which contains a short PEG chain enhances solubility and couples SN-38 at the 20th position of the lactone ring, stabilizing it from converting to its less active carboxylate form (Figure 3). Furthermore, the 10th position of SN-38 gets shielded from glucuronidation after its attachment to RS7, allowing SN-38 to remain in its most active state until release.
The conjugation process involved linking the SN-38 complex to mildly reduced IgG, targeting eight specific thiol sites (two per heavy chain in hinge region and four in CH1-CL region) without disrupting the antibody's antigen-binding ability [73]. Liquid chromatography-Mass spectroscopy confirmed site-specific coupling away from the antigen-binding regions. Pharmacokinetic studies indicated the conjugate cleared similarly to native IgG, with stable chain association despite disulfide bond disruption. The conjugate maintained its immunoreactivity, and in mouse studies, the IgG component cleared from the body at a rate similar to that of the native IgG. SN-38 was released as expected, with none found after 72 hours, suggesting a minimal risk of off-target toxicity [106].
3.2. Mode of Action of Sacituzumab Govitecan
After administration, the hRS7 antibody binds to the Trop-2 receptor, facilitating internalization of the conjugate via endocytosis [111]. The acidic endosomal environment triggers hydrolysis of the pH-sensitive linker, releasing SN-38 into the cytosol when fused with the lysosomal compartment [112]. SN-38 binds and stabilizes topoisomerase I (TOPIB), leading to the formation a stable SN-38-TOPIB-DNA complex, which prevents the repair of DNA lesions [108,113]. As DNA replication proceeds, this results in irreversible single-stranded breaks, leading to cell death [87]. In addition, the membrane-permeable SN-38 is released into the tumor niche, inducing cytotoxic activity in nearby Trop-2+ and Trop-2- tumor cells through the bystander effect [114] (Figure 4), thus overcoming the heterogeneity of Trop-2 expression [73,115,116].
The pH-sensitive linker further facilitates SN-38 release in the acidic tumor microenvironment (TME), enabling targeted cell death of adjacent cells. This mechanism contrasts with anthracyclines, which targets topoisomerase II enzyme that repairs double-stranded DNA lesions. Nonetheless, both SN-38 and anthracyclines induce cell death through p53-mediated apoptosis, despite targeting different topoisomerases [117].
3.3. Dosing and Administration
Initially, clinical studies examined three SG dosages, viz. 8, 10, 12, and 18 mg/kg. While 12 mg/kg was recorded as the maximum tolerated dose (MTD), it necessitated repeated dosage adjustments and interruptions. As a result, 8- and 10-mg/kg dosages were utilized for future investigations. The most appropriate dosage for mTNBC, in terms of increased efficacy with manageable toxic reactions, was 10 mg/kg [118], which has now become the FDA-recommended dose. SG, supplied as a 180 mg single-dose injection, is reconstituted in 20 mL of sterile normal saline and diluted in an infusion bag, protected against sunlight. The final concentration must be maintained around 1.1-3.4 mg/mL, not exceeding a total volume of 500 mL. SG is infused intravenously on day 1 and day 8 over 21 days period. Dosage adjustments may be implemented if a patient's weight changes by more than 10%. For patients weighing over 178 kg, the dose is divided into two 500 mL administrations [119]. The first dosage is administered over a 3 hour period, with the patient continuously monitored during the treatment and for 30 minutes afterward. Subsequently, the drug can be given over 1–2 hour period, with a similar observation period. Infusion rates can be increased gradually if no Grade 1 hypersensitivity reactions occur [83,84]. The starting infusion rates vary but may begin at 50 mg/h and increasing to a peak of up to 1000 mg/h in later doses [119,120].
3.3. Pharmacokinetics (PK) profile of Sacituzumab govitecan
In a Phase I/II basket trial, SG was assessed for metastatic epithelial cancers without the need for Trop-2 expression testing. Phase I trial evaluated 25 individuals with three different doses of SG injections on days 1 and 8 over 21-day period. Based on the encouraging efficacy results, a dosage of 10 mg/kg was chosen for Phase II. This promising response led to further exploration of PK profile of SG across multiple studies using concentration-time statistics from the phase I/II IMMU-132-01, phase II TROPHY-U-01, and phase III ASCENT studies. These trials demonstrated no accumulation of SG or its active metabolite SN-38 after repeated treatment cycles, and exposure levels were consistent across the three studies [121].
Compared to hRS7 (Kd: 0.51 ± 0.04 nM), SG demonstrates enhanced binding strength for Trop-2 (Kd: 0.26 ± 0.14 nM) and supplies significantly higher levels of the active drug than irinotecan, with reduced systemic toxicity [119]. Clearance rates did not vary considerably between patients. Similar to results from preclinical studies, about 50% of the drug gets dissociated from the conjugate every day, and ~90% of the cargo was released from the ADC over three days [118]. Nevertheless, xenograft experiments revealed that SG leads to prolonged intratumoral retention of SN-38, with elevated drug levels persisting for 3 days after administration, against just 8 hrs for irinotecan [106].
Interestingly, population PK (popPK) modeling revealed that UGT1A1 polymorphisms, which affect the metabolism of SN-38, had no significant impact on PK of the ADC [122]. Furthermore, no notable changes in SG exposure were found in individuals with mild to moderate renal/hepatic impairment, nor were there differences related to age, sex, race, albumin levels, tumor type, UGT1A1 GT, use of UGT1A1 inducers or inhibitors or Trop-2 expression [123]. Exposure-response models showed that higher SG exposure was linked to improved outcomes, including longer PFS, OS, and better response rates. However, increased SG levels also raised the risk of side effects like diarrhea, neutropenia, nausea, vomiting, and hypersensitivity [124]. PopPK analysis illustrated a steady-state volume of distribution (Vss) of 3.6 L for SG, with a clearance rate of 0.13 L/h. The median elimination time of SG was recorded to be 23.4 hrs vs 17.6 hrs for SN-38.
In accordance with preclinical models, ~50% of the drug was released by the conjugate every 24 hrs [110]. In terms of PK parameters, SG and free SN-38 had a half-life (t½) of 16 and 18 hrs, respectively (Table 2); while the naked antibody, RS7, had a longer t½ of 103–114 hrs [125]. The t½ of SG corresponds to the rate of release of SN-38 from the ADC. SG attains elevated plasma levels than free SN-38, indicating that a significant portion of drug remains attached to the ADC during circulation [118].
SG's maximum concentration (Cmax) reached 239,000 ng/mL, with an area under the curve (AUC0–168) of 5,640,000 ng·h/mL. For SN-38, the Cmax was calculated to be 98 ng/mL, and the AUC0–168 was 3696 ng·h/mL. Notably, free SN-38 represented <5% of the total serum drug concentration, reflecting the strong binding between SN-38 and the conjugate. SG clearance was unaffected by cancer type, while free SN-38 levels had no correlation with neutropenia severity, and no antibody responses to the conjugate were reported [118].
UGT1A1 metabolizes SN-38, and its glucuronide product (SN-38G) is detected in blood. However, conjugation of SN-38 with RS7 protects it from glucuronidation, leading to a lower plasma SN-38G:SN-38 ratio for SG (1:5) vs irinotecan (>4:1). This suggests the need for further research to alleviate AEs in subjects exhibiting diminished UGT1A1 afunction [83,84,106]. No significant drug-drug interactions have been reported for SG; however, UGT1A1 inducers, inhibitors, or genetic variants may influence systemic SN-38 exposure as well as patient outcome [119]. In a phase I/II study, individuals with mutant UGT1A1*28 alleles, linked with reduced enzyme function, experience significantly more toxic effects, particularly neutropenia [83,84].
3.4. Clinical Efficacy of Sacituzumab Govitecan
The efficacy of SG was first evaluated in the phase I/II multicenter basket trial (IMMU-132-01) in 108 mTNBC individuals who had undergone prior treatments, at a dose of 10 mg/kg administered intravenously on days 1 and 8 of each 21-day treatment cycle. The median duration of treatment and median time to response was 5.1 and 2.0 months, respectively. The trial reported a 33% objective response rate (ORR), with a median PFS and OS of 5.5 and 13.0 months, respectively. Durable responses were noted, with median response duration of 7.7 months. After six and twelve months of therapy, the calculated probability of a response is 27.0% and 59.7%, respectively. Subgroup analysis in ER+HER2- cancer individuals showed a 31.5% response rate, with median PFS (mPFS) and median OS (mOS) of 5.5 and 12 months, respectively. The median duration of response was 8.7 months [83,84,114]. Based on the clinical outcomes of the trial, sacituzumab govitecan-hziy (Immunomedics, Inc.) received FDA authorization for use in individuals with mTNBC who had previously undergone no less than two treatments in metastatic setting [126].
In heavily pretreated mTNBC individuals, SG exhibited a 30% ORR, 8.9-month median response duration, and 46% clinical benefit rate. The mPFS and mOS was 6.0 and 16.6 months, respectively. Common grade ≥ 3 AEs were neutropenia, leukopenia, anemia, and febrile neutropenia. Trop-2 expression was strong in 88% of tissue samples, and no neutralizing antibodies were noticed following repeated therapy [127].
The phase III ASCENT trial involving 468 patients verified the effectiveness of SG in population with relapsed or refractory TNBC, leading to its FDA approval (Table 3). SG significantly improved mPFS to 5.6 months vs 1.7 months with physician's choice of single-agent treatment (TPC), and mOS was 12.1 months vs 6.7 months. Radiological responses were reported in 35% of individuals administered with SG in comparison with 5% in the chemotherapy group. However, SG was linked to greater frequencies of grade 3 or higher adverse reactions, particularly neutropenia. A subanalysis of ASCENT trial specifically evaluated outcomes based on whether patients had TNBC at their initial diagnosis. SG demonstrated notable benefits, with a mPFS of 4.6 months vs 2.3 months for TPC, and mOS of 12.4 months vs 6.7 months. The ORR was 31% for SG compared to 4% for TPC. The efficacy and safety profiles appeared comparable between populations with and without TNBC at first diagnosis; emphasizing the need for subtype reanalysis in mTNBC for optimal therapeutic interventions [85].
Biomarker analyses indicated that individuals with high or medium Trop-2 expression benefited better from the conjugate independent of their BRCA mutation level, although conclusions about low Trop-2 expression in patients remain uncertain. A subgroup examination revealed that SG may also be beneficial for TNBC individuals with brain metastases, although the findings were limited by sample size [86].
A phase II TROPiCS-02 trial (Table 3) demonstrated significant benefits for HR+HER2- mBC individuals treated with the conjugate. At the July 1, 2022, data cutoff, the study involving 543 patients found that SG improved OS (14.4 months) vs chemotherapy (11.2 months) with a higher ORR (21% vs 14%). Additionally, patients receiving SG experienced longer times to deterioration in global health status (4.3 months vs 3.0 months) and fatigue (2.2 months vs 1.4 months). The drug tolerability remained consistent with earlier investigations, albeit with one fatal event [128].
The active, not recruiting EVER-132-001 study (NCT04454437), evaluated the efficiency and safety of SG in 80 Chinese individuals with relapsing mTNBC who failed at least two previous drug regimens. The study found an ORR of 38.8% and a clinical benefit rate (CBR) of 43.8%, with a median PFS of 5.55 months. Overall, SG showed significant efficacy and a relatively controllable safety profile in this extensively pretreated group [129].
In an ongoing study of 50 participants with stage I-III TNBC (NCT04230109), 98% had already received 4 cycles of the ADC. Although baseline TROP2 expression could not predict pCR, higher Ki-67 levels and the presence of TILs were prognostic. While the pCR rate was 30% with an ORR of 64%, the 2-year event-free survival (EFS) rate was 95% for all participants and 100% for those with a pCR after a median follow-up of 18.9 months [130]. These findings support SG as a viable intervention for TNBC and BC regardless of initial subtype, highlighting the importance of reassessing molecular classification of mTNBC for best possible treatment strategies [131].
3.5. Safety Profile of Sacituzumab Govitecan
In the ASCENT trials (NCT02574455), the main AEs of any grade were: nausea (67%), neutropenia (64%), diarrhea (62%), fatigue (55%), anemia (50%), and vomiting (49%). Grade 3 and 4 AEs occured in 66% and 19% of mTNBC patients, respectively. Neutropenia was the major high-grade AE (grade 3: 26%; grade 4: 16%); however, mere 8% of population experienced grade ≥3 febrile neutropenia (grade 3: 6%; grade 4: 2%) [114,127].
The safety data of SG in phase II TROPiCS-02 study (NCT03901339) exhibited similarity to earlier investigations, with the exception of one fatality due to shock from neutropenic colitis [128]. In EVER-132-001 study (NCT04454437), 71.3% of population experienced high grade AEs, and 6.3% withdraw from therapy due to AEs [129]. In an ongoing phase II (NeoSTAR) trial (NCT04230109) frequently observed AEs included nausea, fatigue, alopecia, neutropenia, and rash.
An interruption in the therapy due to neutropenia was observed in 44% of cases, while the procedure was terminated in 3% of population. A decrease in SG dose was observed in 34% of individuals, with no therapy-associated mortality reported [132]. For high grade AEs, the dosage of ADC must be lowered by 25% for the first AE, 50% for the second effect and stopped after 3 AEs [133]. Upon detection of grade 3-4 neutropenia during scheduled therapy, treatment should be temporarily halted for 2-3 weeks to permit recovery to grade 1 before resuming the dosage [119].
3.6. Cost evaluation of Sacituzumab Govitecan
A 50-mL, 180 mg injection of SG is available in the market for $1,478.00/injection, while the estimated cost per 21-day treatment cycle is $12,478, based on the assumption of 94% dosage strength and a mean body weight of 71.1 kg [134]. Univariate analyses showed that outcomes particularly responsive to the pharmacological cost of SG and the utilities of PFS and advanced malignancy. Base-case calculations reveal that the incremental cost-effectiveness ratio (ICER) of SG in contrast to conventional drug therapy surpasses the US willingness-to-pay (WTP) benchmark of $150,000/QALY, rendering it cost-ineffective. In the overall mTNBC population, SG costs an additional $293,037 and provides 0.2340 more QALYs, resulting in an ICER of $1,252,295 per QALY (quality-adjusted life year) [135]. Based on Canadian Agency for Drugs and Technologies in Health (CADTH) reevaluation, the ICER for SG vs. TPC is $375,333 per QALY gained. SG must reduce its price by at least 87% to be affordable as per WTP criterion of $50,000 per QALY threshold [134].
3.6. Drug resistance to Sacituzumab Govitecan
Due to the complexity and variability of target antigens and payloads, ADC resistance mechanisms are diverse and may require individualized study for each ADC. Emerging issues following the approval of SG for HR+/HER2− and mTNBC include the need for a deeper understanding of newly developed resistance to the SG conjugate and strategies to overcome it [136]. A key factor in resistance to ADCs like SG arises from either downregulation or mutation of the receptor targets [137]. Lower Trop-2 levels has been associated with SG resistance, as demonstrated in a genomic analysis of a TNBC individual with underexpreed Trop-2 [138]. Individuals with reduced TROP-2 expression had shorter median PFS when treated with SG versus those with elevated expression (2.7 months vs. 6.9 months) [86]. Additionally, research showed that tamoxifen treatment drastically improved Trop-2 levels [139]. One strategy to overcome ADC resistance is upregulating target antigen expression [140]. Until now, temporal variations in Trop-2 expression during malignancy and therapy are not fully clear, indicating a potential arena for future research.
A recent study highlights the potential for cross-resistance between ADCs in mBC, especially when the same antibody target is used in consecutive treatments. While switching targets showed some improvement in PFS, the results were not statistically significant, suggesting the need for further investigation. Mechanisms like altered antigen expression or drug resistance may explain the reduced efficacy of subsequent ADCs. This study emphasizes the importance of optimizing ADC sequencing and exploring biomarker-driven approaches to improve patient outcomes and overcome resistance [141]. As such, success in addressing resistance will depend on a deeper understanding and evaluation of these mechanisms in diseased population.
4. Conclusions and Future Directions
SG is constantly under evaluation in clinical trials, both as a monotherapy and in conjunction with PARPi, chemotherapeutic agents, immunotherapies, across multiple cancer types, indicating that its usage may extend beyond third-line treatment for mTNBC and HR+ BC, suggesting broader future applications. The outcomes of these trials will offer significant insights into SG's potential across various clinical settings.
Combinations of SG with other cytotoxic drugs show promising antitumor potential. For example, using ATP binding cassette subfamily G member 2 (ABCG2) inhibitors with SG has been effective in overcoming resistance to SN-38 in various neoplasms [142]. The rationale for combining SG with immunotherapy is strong, as ADCs can induce immunogenic cell death and T cell infiltration, while ICIs restore exhausted T cells, leading to synergistic effects. A still recruiting phase I/II trial, Morpheus-panBC (NCT03424005), have shown some initial promising outcomes for ADC and ICI combinations, such as atezolizumab with SG, in breast cancer treatment [143].
Preclinical studies have already supported the synergy between SG and PARPi in TNBC (Table 4), and further clinical trials are anticipated [144]. A phase 1b trial (NCT04039230) tested a staggered dosing schedule of SG with the PARPi talazoparib in mTNBC to minimize AEs. This combination exhibited favourable tolerability with encouraging clinical efficacy, resulting in enhanced DNA damage. This combination strategy underscores the potential of revisiting previously rejected drug combinations for mTNBC [145].
Further, new strategies beyond traditional ADCs, such as immune-conjugates and nano-structures, are still in infancy. Liu et al. created a Trop-2-directed tetrakis-ranpirnase conjugate (IgG-Rap immunoRNase) using the hRS7 mAb, which demonstrated significant survival benefits in TNBC mouse models [146]. Next-generation anti-Trop-2 therapies are being developed to leverage different ADC linker structures, conjugation chemistries, and more potent drug payloads [147].
Follow-up research will likely concentrate on identifying and validating predictive biomarkers, with a particular emphasis on investigating the interplay between Trop-2 expression levels, UGT1A128 homozygosity, and clinical outcomes in cancer treatment [148]. Understanding how variations in Trop-2 expression correlate with UGT1A128 genetic polymorphisms may provide insights into individual patient responses to therapies targeting Trop-2, such as SG. Ultimately, such investigation could elucidate mechanisms of treatment efficacy and toxicity, enabling the development of more personalized treatment strategies that enhance therapeutic effectiveness while minimizing AEs and contribute to improved clinical outcomes in cancer management.
Future cancer therapies targeting the Trop-2 protein will likely prioritize strategies that selectively target the "cleaved" or activated form of Trop-2 present in neoplasms [149,150], while sparing the intact form expressed in normal tissues. This approach has the potential to enhance treatment efficiency whilst reducing off-target binding and diminishing toxicity, thus increasing the therapeutic window of Trop-2-targeted interventions.
In conclusion, the successful application of SG in mTNBC demonstrates that Trop-2-directed therapeutics is a promising avenue for exploration. However, challenges associated with existing targeted therapies illustrate the need for continued refinement and optimization of treatment strategies. Advances in the structural and functional analysis of Trop-2 and its expression patterns could guide the development of next-generation ADCs, enhancing treatment efficacy. Moreover, integrating SG with ICIs or PARPi aligns with the principles of precision medicine by combining therapies that target different aspects of tumor biology, thus aiming to improve clinical outcomes through a multifaceted approach. This strategic combination not only holds promise for improving efficacy but also emphasizes the necessity of personalized treatment regimens that consider individual patient characteristics and tumor profiles.
Author Contributions
SK: Writing – Original Draft Preparation, Writing – review & editing; Funding Acquisition; SB: Writing – review & editing; NZ: Writing – review & editing; RD: Writing – review & editing; KS: Writing – review & editing; RV: Writing – review & editing; SR: Writing – review & editing. AK: Writing – review & editing; ML: Conceptualization, Writing – review & editing.
Funding
This research has been funded by Scientific Research Deanship at the University of Ha’il, Saudi Arabia, through Project Number RG-24 063.
Data Availability Statement
All data is available within the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bray, F.; Aversano, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Kong, D.; Liu, J.; et al. Breast cancer heterogeneity and its implication in personalized precision therapy. Exp Hematol Oncol 2023, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Goldhirsch, A.; et al. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef]
- Huppert, L.A.; Gumusay, O.; Rugo, H.S. Emerging treatment strategies for metastatic triple-negative breast cancer. Ther Adv Med Oncol. 2022, 14, 17588359221086916. [Google Scholar] [CrossRef]
- Li, C.H.; Karantza, V.; Aktan, G.; Lala, M. Current treatment landscape for patients with locally recurrent inoperable or metastatic triple-negative breast cancer: a systematic literature review. Breast Cancer Res. 2019, 21, 143. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sacituzumab Govitecan: First Approval. Drugs. 2020, 80, 1019–1025. [Google Scholar] [CrossRef]
- Won, K.A.; Spruck, C. Triple-negative breast cancer therapy: Current and future perspectives. Int J Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; et al. Response to Neoadjuvant Therapy and Long-Term Survival in Patients With Triple-Negative Breast Cancer. J Clin Oncol. 2023, 41, 1809–1815. [Google Scholar] [CrossRef]
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-negative breast cancer: is there a treatment on the horizon? Oncotarget. 2017, 8, 1913–1924. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Kalari, K.R.; Suman, V.J.; Moyer, A.M.; Yu, J.; Visscher, D.W.; Dockter, T.J.; Vedell, P.T.; Sinnwell, J.P.; Tang, X.; et al. Tumor sequencing and patient-derived xenografts in the neoadjuvant treatment of breast cancer. J Natl Cancer Inst 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.A.; Chen, Y.H.; Solzak, J.P.; Ahmad, M.N.; Wedge, D.C.; Brinza, D.; Scafe, C.; Veitch, J.; Gottimukkala, R.; Short, W.; et al. Profiling molecular regulators of recurrence in chemorefractory triple-negative breast cancers. Breast Cancer Res. 2019, 21, 87. [Google Scholar] [CrossRef]
- Wu, Q.; Siddharth, S.; Sharma, D. Triple Negative Breast Cancer: A Mountain Yet to Be Scaled Despite the Triumphs. Cancers (Basel). 2021, 13, 3697. [Google Scholar] [CrossRef]
- Dent, R.; et al. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Reddy, S.M.; et al. Long-term survival outcomes of triple-receptor negative breast cancer survivors who are disease-free at 5 years and relationship with low hormone receptor positivity. Br. J. Cancer 2018, 118, 17–23. [Google Scholar] [CrossRef]
- Lin, N.U.; et al. Clinicopathologic features, patterns of recurrence, and survival among women with triple-negative breast cancer in the National Comprehensive. Cancer Netw. Cancer 2012, 118, 5463–5472. [Google Scholar] [CrossRef]
- Derakhshan, F.; Reis-Filho, J.S. Pathogenesis of triple-negative breast cancer. Annu Rev Pathol. 2022, 17, 181. [Google Scholar] [CrossRef]
- Keam, B.; Im, S.A.; Lee, K.H.; Han, S.W.; Oh, D.Y.; Kim, J.H.; Lee, S.H.; Han, W.; Kim, D.W.; Kim, T.Y.; et al. Ki-67 can be used for further classification of triple negative breast cancer into two subtypes with different response and prognosis. Breast Cancer Res. 2011, 13, R22. [Google Scholar] [CrossRef]
- Sharma, P.; Klemp, J.R.; Kimler, B.F.; et al. Germline BRCA mutation evaluation in a prospective triple-negative breast cancer registry: Implications for hereditary breast and/or ovarian cancer syndrome testing. Breast Cancer Res Treatment 2014, 145, 707–714. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Timms, K.; Liu, M.S.; et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res 2011, 17, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Villarreal-Garza, C.; Weitzel, J.N.; Llacuachaqui, M.; et al. The prevalence of BRCA1 and BRCA2 mutations among young Mexican women with triple-negative breast cancer. Breast Cancer Res Treatment 2015, 150, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, D.H. Distribution of BRCA1 and BRCA2 mutations in asian patients with breast cancer. J Breast Cancer 2013, 16, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Espinosa Fernandez, J.R.; Eckhardt, B.L.; Lee, J.; et al. Identification of triple-negative breast cancer cell lines classified under the same molecular subtype using different molecular characterization techniques: Implications for translational research. PLoS ONE. 2020, 15, e0231953. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; et al. Refinement of triple-negative breast cancer molecular subtypes: Implications for neoadjuvant chemotherapy selection. PLoS One. 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef]
- Manjunath, M.; Choudhary, B. Triple-negative breast cancer: A run-through of features, classification and current therapies. Oncol Lett. 2021, 22, 1–21. [Google Scholar] [CrossRef]
- So, J.Y.; Ohm, J.; Lipkowitz, S.; Yang, L. Triple negative breast cancer (TNBC): Non-genetic tumor heterogeneity and immune microenvironment: Emerging treatment options. Pharmacol Ther. 2022, 237, 108253. [Google Scholar] [CrossRef] [PubMed]
- Xiong, N.; Wu, H.; Yu, Z. Advancements and challenges in triple-negative breast cancer: a comprehensive review of therapeutic and diagnostic strategies. Front Oncol. 2024, 14, 1405491. [Google Scholar] [CrossRef] [PubMed]
- Rossi, V.; Turati, A.; Rosato, A.; Carpanese, D. Sacituzumab govitecan in triple-negative breast cancer: from bench to bedside, and back. Front. Immunol. 2024, 15, 1447280. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Curigliano, G.; Loibl, S.; Dubsky, P.; Gnant, M.; Poortmans, P.; Colleoni, M.; Denkert, C.; Piccart-Gebhart, M.; Regan, M.; et al. Estimating the Benefits of Therapy for Early-Stage Breast Cancer: The St. Gallen International Consensus Guidelines for the Primary Therapy of Early Breast Cancer 2019. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1541–1557. [Google Scholar] [CrossRef]
- Pusztai, L.; Foldi, J.; Dhawan, A.; DiGiovanna, M.P.; Mamounas, E.P. Changing Frameworks in Treatment Sequencing of Triple-Negative and HER2-Positive, Early-Stage Breast Cancers. Lancet Oncol. 2019, 20, e390–e396. [Google Scholar] [CrossRef]
- Holanek, M.; Selingerova, I.; Bilek, O.; Kazda, T.; Fabian, P.; Foretova, L.; et al. Neoadjuvant Chemotherapy of Triple-Negative Breast Cancer: Evaluation of Early Clinical Response, Pathological Complete Response Rates, and Addition of Platinum Salts Benefit Based on Real-World Evidence. Cancers (Basel). 2021, 13, 1586. [Google Scholar] [CrossRef]
- LeVasseur, N.; Sun, J.; Gondara, L.; Diocee, R.; Speers, C.; Lohrisch, C.; Chia, S. Impact of pathologic complete response on survival after neoadjuvant chemotherapy in early-stage breast cancer: a population-based analysis. J Cancer Res Clin Oncol. 2020, 146, 529–536. [Google Scholar] [CrossRef]
- Huang, M.; Qi, C.Z.; Ramsey, S.; Briggs, A.; Zhao, J.; Haiderali, A.; et al. Evaluation of pathological complete response as a trial-level surrogate for long-term survival outcomes among triple-negative breast cancer patients receiving neoadjuvant therapy. Ann Oncol. 2019, 30, iii34–iii35. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Untch, M.; Blohmer, J.-U.; Costa, S.D.; Eidtmann, H.; Fasching, P.A.; et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J. Clin. Oncol. 2012, 30, 1796–1804. [Google Scholar] [CrossRef]
- Balic, M.; Thomssen, C.; Würstlein, R.; Gnant, M.; Harbeck, N. St. Gallen/Vienna 2019: A Brief Summary of the Consensus Discussion on the Optimal Primary Breast Cancer Treatment. Breast Care. 2019, 14, 103–110. [Google Scholar] [CrossRef]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E. Early Breast Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2019, 30, 1194–1220. [Google Scholar] [CrossRef]
- Lee, J.S.; Yost, S.E.; Yuan, Y. Neoadjuvant Treatment for Triple Negative Breast Cancer: Recent Progresses and Challenges. Cancers (Basel). 2020, 12, 1404. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yee, D. I-SPY 2: a Neoadjuvant Adaptive Clinical Trial Designed to Improve Outcomes in High-Risk Breast Cancer. Curr Breast Cancer Rep. 2019, 11, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Yam, C.; Abuhadra, N.; Sun, R.; Adrada, B.E.; Ding, Q.Q.; White, J.B.; et al. Molecular Characterization and Prospective Evaluation of Pathologic Response and Outcomes with Neoadjuvant Therapy in Metaplastic Triple-Negative Breast Cancer. Clin Cancer Res. 2022, 28, 2878–2889. [Google Scholar] [CrossRef]
- Yam, C.; Yen, E.Y.; Chang, J.T.; Bassett, R.L.; Alatrash, G.; Garber, H.; et al. Immune Phenotype and Response to Neoadjuvant Therapy in Triple-Negative Breast Cancer. Clin Cancer Res. 2021, 27, 5365–5375. [Google Scholar] [CrossRef]
- Mandapati, A.; Lukong, K.E. Triple negative breast cancer: approved treatment options and their mechanisms of action. J Cancer Res Clin Oncol 2023, 149, 3701–3719. [Google Scholar] [CrossRef]
- McCrea, C.; Hettle, R.; Gulati, P.; Taneja, A.; Rajora, P. Indirect treatment comparison of olaparib and talazoparib in germline BRCA-mutated HER2-negative metastatic breast cancer. J Comp Eff Res. 2021, 10, 1021–1030. [Google Scholar] [CrossRef]
- Eikesdal, H.P.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann Oncol 2021, 32, 240–249. [Google Scholar] [CrossRef]
- McCann, K.E. Advances in the use of PARP inhibitors for BRCA1/2-associated breast cancer: talazoparib. Future Oncol 2019, 15, 1707–1715. [Google Scholar] [CrossRef]
- Zhang, Hp. , Jiang, Ry. , Zhu, Jy. et al. PI3K/AKT/mTOR signaling pathway: an important driver and therapeutic target in triple-negative breast cancer. Breast Cancer 2024, 31, 539–551. [Google Scholar] [CrossRef]
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Brunt, A.M.; Nemsadze, G.; et al. Capivasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer: the PAKT trial. JCO. 2020, 38, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Abraham, J.; Chan, S.; Brunt, A.M.; Nemsadze, G.; Baird, R.D.; et al. Abstract PD1-11: mature survival update of the double-blind placebo-controlled randomised phase II PAKT trial of first-line capivasertib plus paclitaxel for metastatic triple-negative breast cancer. Cancer Res. 2021, 81, D1–D11. [Google Scholar] [CrossRef]
- Kim, S.-B.; Dent, R.; Im, S.-A.; Espié, M.; Blau, S.; Tan, A.R.; et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017, 18, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Oliveira, M.; Isakoff, S.J.; Im, S.-A.; Espié, M.; Blau, S.; et al. Final results of the double-blind placebo-controlled randomized phase 2 LOTUS trial of first-line ipatasertib plus paclitaxel for inoperable locally advanced/metastatic triple-negative breast cancer. Breast Cancer Res Treat. 2021, 189, 377–386. [Google Scholar] [CrossRef]
- Dent, R.; Kim, S.-B.; Oliveira, M.; Barrios, C.; O’Shaughnessy, J.; Isakoff, S.J.; et al. Abstract GS3-04: double-blind placebo (PBO)-controlled randomized phase III trial evaluating first-line ipatasertib (IPAT) combined with paclitaxel (PAC) for PIK3CA/AKT1/PTEN-altered locally advanced unresectable or metastatic triple-negative breast cancer (aTNBC): primary results from IPATunity130 Cohort, A. Cancer Res. 2021, 81, GS3–04. [Google Scholar]
- Clifton, G.T.; Peoples, G.E.; Mittendorf, E.A. The development and use of the E75 (HER2 369–377) peptide vaccine. Future Oncol. 2016, 12, 1321–1329. [Google Scholar] [CrossRef]
- Clive, K.S.; Tyler, J.A.; Clifton, G.T.; Holmes, J.P.; Ponniah, S.; Peoples, G.E.; Mittendorf, EA. The GP2 peptide: a HER2/neu-based breast cancer vaccine. J Surg Oncol. 2012, 105, 452–458. [Google Scholar] [CrossRef]
- Brown, T.A.; Mittendorf, E.A.; Hale, D.F.; Myers, J.W.; Peace, K.M.; Jackson, D.O.; et al. Prospective, randomized, single-blinded, multi-center phase II trial of two HER2 peptide vaccines, GP2 and AE37, in breast cancer patients to prevent recurrence. Breast Cancer Res Treat. 2020, 181, 391–401. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Ardavanis, A.; Symanowski, J.; Murray, J.L.; Shumway, N.M.; Litton, J.K.; et al. Primary analysis of a prospective, randomized, single-blinded phase II trial evaluating the HER2 peptide AE37 vaccine in breast cancer patients to prevent recurrence. Ann Oncol. 2016, 27, 1241–1248. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Clifton, G.T.; Holmes, J.P.; Schneble, E.; Van Echo, D.; Ponniah, S.; et al. Final report of the phase I/II clinical trial of the E75 (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann Oncol. 2014, 25, 1735–1742. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Zhou, J.; Wang, J.; Jin, G.; Wang, X. Breast Cancer Vaccine containing a Novel toll-like receptor 7 agonist and an aluminum adjuvant exerts Antitumor effects. IJMS. 2022, 23, 15130. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M. , Butti, R., Panda, V.K. et al. Modulation of the tumor microenvironment and mechanism of immunotherapy-based drug resistance in breast cancer. Mol Cancer 2024, 23, 92. [Google Scholar] [CrossRef]
- Katz, H.; Alsharedi, M. Immunotherapy in triple-negative breast cancer. Med Oncol 2017, 35, 13. [Google Scholar] [CrossRef] [PubMed]
- Pembrolizumab (Keytruda): CADTH Reimbursement Review: Therapeutic area: Triple-negative breast cancer [Internet]. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2023. Clinical Review. Available from: https://www.ncbi.nlm.nih.gov/books/NBK596330.
- Preziosi, A.J.; Priefer, R. Oncology's trial and error: Analysis of the FDA withdrawn accelerated approvals. Life Sci. 2024, 346, 122615. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P. , Cortes, J., Pusztai, L., McArthur, H., Kümmel, S., Bergh, J., et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Schmid, P. , Salgado, R., Park, Y.H., Muñoz-Couselo, E., Kim, S.B., Sohn, J., Im, S.-A., Foukakis, T., Kuemmel, S., Dent, R., et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: Results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann. Oncol. [CrossRef]
- Lan, H.R.; Chen, M.; Yao, S.Y.; Chen, J.X.; Jin, K.T. Novel immunotherapies for breast cancer: Focus on 2023 findings. Int Immunopharmacol. 2024, 128, 111549. [Google Scholar] [CrossRef]
- Liu, Y. , Hu, Y. , Xue, J. et al. Advances in immunotherapy for triple-negative breast cancer. Mol Cancer 2023, 22, 145. [Google Scholar] [CrossRef]
- Padzińska-Pruszyńska, I.; Kucharzewska, P.; Matejuk, A.; Górczak, M.; Kubiak, M.; Taciak, B.; Król, M. Macrophages: Key Players in the Battle against Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2024, 25, 10781. [Google Scholar] [CrossRef]
- Ren, X. , Cheng, Z., He, J. et al. Inhibition of glycolysis-driven immunosuppression with a nano-assembly enhances response to immune checkpoint blockade therapy in triple negative breast cancer. Nat Commun 2023, 14, 7021. [Google Scholar] [CrossRef]
- Moon, S.-J. , Govindan, S.V.; Cardillo, T.M.; D’Souza, C.A.; Hansen, H.J.; Goldenberg, D.M. Antibody conjugates of 7-ethyl-10-hydroxycamptothecin (SN-38) for targeted cancer chemotherapy. J Med Chem. 2008, 51, 6916–6926. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Cardillo, T.M.; Govindan, S.V.; Rossi, E.A.; Sharkey, R.M. Trop-2 is a novel target for solid cancer therapy with sacituzumabgovitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget 2015, 6, 22496–22512. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.; Parks, D.R.; Rouse, R.V.; Herzenberg, L.A. Human trophoblast cell-surface antigens defined by monoclonal antibodies. Proc Natl Acad Sci USA. 1981, 78, 5147–5150. [Google Scholar] [CrossRef] [PubMed]
- Cubas, R.; Li, M.; Chen, C.; Yao, Q. Trop2: A possible therapeutic target for late stage epithelial carcinomas. Biochim Biophys Acta. 2009, 1796, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Shvartsur, A.; Bonavida, B. Trop2 and its overexpression in cancers: regulation and clinical/therapeutic implications. Genes Cancer. 2015, 6, 84–105. [Google Scholar] [CrossRef]
- Lin, H.; Huang, J.F.; Qiu, J.R.; Zhang, H.L.; Tang, X.J.; Li, H.; et al. Significantly upregulated TACSTD2 and Cyclin D1 correlate with poor prognosis of invasiveductal breast cancer. Exp Mol Pathol. 2013, 94, 73–78. [Google Scholar] [CrossRef]
- Zaman, S.; Jadid, H.; Denson, A.C.; Gray, J.E. Targeting Trop-2 in solid tumors: future prospects. Onco Targets Ther 2019, 12, 1781–1790. [Google Scholar] [CrossRef]
- Jeon, Y.; Jo, U.; Hong, J.; Gong, G.; Lee, H.J. Trophoblast cell-surface antigen 2 (TROP2) expression in triple-negative breast cancer. BMC Cancer. 2022, 22, 1014. [Google Scholar] [CrossRef]
- Okajima, D.; Yasuda, S.; Maejima, T.; Karibe, T.; Sakurai, K.; Aida, T.; et al. Datopotamab deruxtecan, a novel TROP2-directed antibody-drug conjugate, demonstrates potent antitumor activity by efficient drug delivery to tumor cells. Mol Cancer Ther. 2021, 20, 2329–2340. [Google Scholar] [CrossRef]
- Bardia, A.; Messersmith, W.A.; Kio, E.A.; Berlin, J.D.; Vahdat, L.; Masters, GA; et al. Sacituzumab govitecan, a Trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann Oncol. 2021, 32, 746–756. [Google Scholar] [CrossRef]
- Liao, Q.; Zhang, R.; Ou, Z.; Ye, Y.; Zeng, Q.; Wang, Y.; et al. TROP2 is highly expressed in triple-negative breast cancer CTCs and is a potential marker for epithelial mesenchymal CTCs. Mol Therapy: Oncol. 2024, 32, 200762. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Kalinsky, K. Sacituzumab Govitecan-hziy in Triple-Negative Breast Cancer. Reply. N Engl J Med. 2019, 380, 2382. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N Engl J Med. 2019, 380, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; et al.; ASCENT Clinical Trial Investigators Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N Engl J Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Tolaney, S.M.; Punie, K.; Loirat, D.; Oliveira, M.; Kalinsky, K.; et al. Biomarker analyses in the phase III ASCENT study of sacituzumab govitecan versus chemotherapy in patients with metastatic triple-negative breast cancer. Ann Oncol. 2021, 32, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A. , Hurvitz, S.A., Rugo, H.S., Brufsky, A., Cortes, J., Loibl, S., et al (2021d). A Plain Language Summary of the ASCENT Study: Sacituzumab Govitecan for Metastatic Triple-Negative Breast Cancer. Future Oncology, 17(30), 3911–3924. [CrossRef]
- Bardia, A.; Rugo, H.S.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; et al. Final results from the randomized phase III ASCENT clinical trial in metastatic triple-negative breast cancer and association of outcomes by human epidermal growth factor receptor 2 and trophoblast cell surface antigen 2 expression. J Clin Oncol. (2024)42:1738–44. [CrossRef]
- D’Arienzo, A.; Verrazzo, A.; Pagliuca, M.; Napolitano, F.; Parola, S.; Viggiani, M.; et al. Toxicity profile of antibody-drug conjugates in breast cancer: practical considerations. eClinical Medicine. (2023) 62:102113. [CrossRef]
- Dum, D.; Taherpour, N.; Menz, A.; Höflmayer, D.; Völkel, C.; Hinsch, A.; et al. Trophoblast cell surface antigen 2 expression in human tumors: A tissue microarray study on 18,563 tumors. Pathobiology. (2022) 89:245–58. [CrossRef]
- Kong, D.; Hughes, C.J.; Ford, H.L. Cellular plasticity in breast cancer progression and therapy. Front Mol Biosci. (2020) 7. [CrossRef]
- Hsu, E.C.; Rice, M.A.; Bermudez, A.; Jose, F.; Marques, G.; Aslan, M.; et al. Trop2 is a driver of metastatic prostate cancer with neuroendocrine phenotype via PARP1. Proc Natl Acad Sci USA. (2020) 117(4):2032–42. [CrossRef]
- Liu, X.; Deng, J.; Yuan, Y.; Chen, W.; Sun, W.; Wang, Y.; et al. Advances in Trop2- targeted therapy: Novel agents and opportunities beyond breast cancer. Pharmacol Ther. (2022) 239:108296. [CrossRef]
- Bessede, A.; Peyraud, F.; Besse, B.; Cousin, S.; Cabart, M.; Chomy, F.; et al. TROP2 is associated with primary resistance to immune checkpoint inhibition in patients with advanced non-small cell lung cancer. Clin Cancer Res. (2024) 30:779–85. [CrossRef]
- Redlich, N.; Robinson, A.M.; Nickel, K.P.; Stein, A.P.; Wheeler, D.L.; Adkins, D.R.; et al. Anti-Trop2 blockade enhances the therapeutic efficacy of ErbB3 inhibition in head and neck squamous cell carcinoma. Cell Death Dis. (2018) 9(1):5. [CrossRef]
- Stein, R.; Basu, A.; Chen, S.; Shih, L.B.; Goldenberg, D.M. Specificity and properties of MAb RS7-3G11 and the antigen defined by this pancarcinoma monoclonal antibody. Int J Cancer. 1993, 55, 938–946. [Google Scholar] [CrossRef]
- Stein, R.; Basu, A.; Goldenberg, D.M.; Lloyd, K.O.; Mattes, M.J. Characterization of cluster 13: the epithelial/carcinoma antigen recognized by MAb RS7. Int J Cancer Suppl. 1994, 8, 98–102. [Google Scholar] [CrossRef]
- Raji, R. , et al. , Uterine and ovarian carcinosarcomas overexpressing Trop-2 are sensitive to hRS7, a humanized anti-Trop-2 antibody, J. Exp. Clin. Cancer Res. 2011, 30, 106. [Google Scholar]
- De Leij, L.; Helrich, W.; Stein, R.; Mattes, M.J. SCLC-cluster-2 antibodies detect the pancarcinoma/epithelial glycoprotein EGP-2. Int J Cancer Suppl. 1994, 8, 60–63. [Google Scholar] [CrossRef]
- Basu, A.; Goldenberg, D.M.; Stein, R. The epithelial/carcinoma antigen EGP-1, recognized by monoclonal antibody RS7-3G11, is phosphorylated on serine 303. Int J Cancer. 1995, 62, 472–479. [Google Scholar] [CrossRef]
- Leung, K. 111In-Diethylenetriamine pentaacetic acid-anti-epithelial glycoprotein-1 hRS7 humanized monoclonal antibody. 2012 Jan 12 [Updated 2012 Apr 19]. In: Molecular Imaging and Contrast Agent Database (MICAD) [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2004-2013. Available from: https://www.ncbi.nlm.nih.gov/books/NBK92276/.
- Rothenberg, M.L. Topoisomerase I inhibitors: Review and update. Ann Oncol. 1997, 8, 837–855. [Google Scholar] [CrossRef]
- Hayashi, H.; Tsurutani, J.; Satoh, T.; et al. Phase II study of bi-weekly irinotecan for patients with previously treated HER2-negative metastatic breast cancer: KMBOG0610B. Breast Cancer. 2013, 20, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Suo, J.; Zhong, X.; He, P.; Zheng, H.; Tian, T. ; Yan X and Luo T (2021) A Retrospective Analysis of the Effect of Irinotecan-Based Regimens in Patients With Metastatic Breast Cancer Previously Treated With Anthracyclines and Taxanes. Front. Oncol. 11:654974. [CrossRef]
- Innocenti, F.; Iyer, L.; Ratain, M.J. Pharmacogenetics of anticancer agents: Lessons from amonafide and irinotecan. Drug Metab Dispos. 2001, 29, 596–600. [Google Scholar]
- Goldenberg, D.M.; Sharkey, R.M. Antibody-drug conjugates targeting TROP-2 and incorporating SN-38: a case study of anti-TROP-2 sacituzumabgovitecan. MAbs 2019, 11, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Mathijssen, R.H.; van Alphen, R.J.; Verweij, J.; et al. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin Cancer Res 2001, 7, 2182–2194. [Google Scholar]
- Gilead Sciences, Inc. 2023. Trodelvy® (sacituzumab govitecan-hziy) Impact of UGT1A1 Status on Safety Profile in Patients With mUC.
- Govindan, S.V.; Cardillo, T.M.; Moon, S.J.; Hansen, H.J.; Goldenberg, D.M. CEACAM5-targeted therapy of human colonic and pancreatic cancer xenografts with potent labetuzumab-SN-38 immunoconjugates. Clin Cancer Res. 2009, 15, 6052–6061. [Google Scholar] [CrossRef]
- Cardillo, T.M.; Govindan, S.V.; Sharkey, R.M.; Trisal, P.; Goldenberg, D.M. Humanized anti-Trop-2 IgG-SN-38 conjugate for effective treatment of diverse epithelial cancers: preclinical studies in human cancer xenograft models and monkeys. Clin Cancer Res. 2011, 17, 3157–3169. [Google Scholar] [CrossRef]
- Bravaccini, S.; Maltoni, R. Trop-2 therapy in metastatic triple negative breast cancer in Italy: clinical opportunity and regulatory pitfalls. J Pers Med 2021, 11, 1211. [Google Scholar] [CrossRef]
- Nagayama A et al (2020) Novel antibody–drug conjugates for triple negative breast cancer. Thera Adv Med Oncol 12:1758835920915980.
- Gilead Sciences Inc. Trodelvy 200 mg powder for concentrate for solution for infusion: EU summary of product characteristics. 2023. https:// www. ema. europa. eu. Accessed 18 Oct 2024.
- Kalinsky, K.; Diamond, J.R.; Vahdat, L.T.; Tolaney, S.M.; Juric, D.; O'Shaughnessy, J.; et al. Sacituzumab govitecan in previously treated hormone receptor-positive/HER2-negative metastatic breast cancer: final results from a phase I/II, single-arm, basket trial. Ann Oncol. 2020, 31, 1709–1718. [Google Scholar] [CrossRef]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer. 2017, 117, 1736–1742. [Google Scholar] [CrossRef]
- Sharkey, R.M.; McBride, W.J.; Cardillo, T.M.; et al. Enhanced delivery of SN-38 to human tumor xenografts with an anti-Trop-2-SN-38 antibody conjugate (sacituzumabgovitecan). Clin Cancer Res 2015, 21, 5131–5138. [Google Scholar] [CrossRef]
- Takeba, Y.; et al. Irinotecan activates p53 with its active metabolite, resulting in hepatocellular carcinoma apoptosis. J Pharmacol Sci 2007, 104, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Ocean, A.J.; Starodub, A.N.; Bardia, A.; et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2- SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: Safety and pharmacokinetics. Cancer. 2017, 123, 3843–3854. [Google Scholar] [CrossRef] [PubMed]
- Immunomedics Inc. (2021). Trodelvy (sacituzumab govitecan-hziy) package insert. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761115s000lbl.
- Cardillo, T.M.; Govindan, S.V.; Sharkey, R.M.; et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2/SN-38 antibody-drug conjugate: characterization and efficacy in pancreatic, gastric, and other cancers. Bioconjug Chem 2015, 26, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Sathe, A.G.; Singh, P.; et al. Pharmacokinetics of Sacituzumab Govitecan, a Trop-2-Directed Antibody-Topoisomerase I Inhibitor SN-38 Drug Conjugate, in Patients With Advanced Solid Tumors [Poster Abstract P-161]. Paper presented at: American Society for Clinical Pharmacology and Therapeutics (ASCPT) 2022 Annual Meeting; 16-18 March, 2022; Virtual.
- Sathe, A.G.; Singh, P.; Singh, I.; et al. Impact of UGT1A1 Polymorphisms on the Pharmacokinetics of Sacituzumab Govitecan [Poster Abstract P-151]. Paper presented at: American Society for Clinical Pharmacology and Therapeutics (ASCPT) 2022 Annual Meeting; 16-18 March, 2022; Virtual.
- Sathe, A.G.; Singh, I.; Jones, A.; et al. Population Pharmacokinetics of Sacituzumab Govitecan in Patients With Locally Advanced or Metastatic Breast Cancer or Other Solid Tumors [Poster PII-082]. Paper presented at: American Society for Clinical Pharmacology & Therapeutics,; March 22-24, 2023; Atlanta, GA.
- Singh, I.; Sathe, G.K.; Diderichsen, P.M.; et al. Exposure-Response Analyses of Sacituzumab Govitecan Efficacy and Safety in Patients With Metastatic Breast Cancer [Poster PO1-04-06]. Paper Paper Presented at: San Antonio Breast Cancer Symposium (SABCS); December 5-9, 2023; San Antonio, TX, USA.
- Seligson, J.M.; Patron, A.M.; Berger, M.J.; Harvey, R.D.; Seligson, N.D. Sacituzumab Govitecan-hziy: An Antibody-Drug Conjugate for the Treatment of Refractory, Metastatic, Triple-Negative Breast Cancer. Ann Pharmacother. 2021, 55, 921–931. [Google Scholar] [CrossRef]
- Wahby, S.; Fashoyin-Aje, L.; Osgood, C.L.; Cheng, J.; Fiero, M.H.; Zhang, L.; et al. FDA Approval Summary: Accelerated Approval of Sacituzumab Govitecan-hziy for Third-line Treatment of Metastatic Triple-negative Breast Cancer. Clin Cancer Res. 2021, 27, 1850–1854. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Diamond, J.R.; Moroose, R.L.; Isakoff, S.J.; Starodub, A.N.; et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients With Metastatic Triple-Negative Breast Cancer. J Clin Oncol. 2017, 35, 2141–2148. [Google Scholar] [CrossRef]
- Rugo, H.S.; Bardia, A.; Marmé, F.; Cortés, J.; Schmid, P.; Loirat, D.; et al. Overall survival with sacituzumab govitecan in hormone receptor-positive and human epidermal growth factor receptor 2-negative metastatic breast cancer (TROPiCS-02): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2023, 402, 1423–1433. [Google Scholar] [CrossRef]
- Xu, B.; Ma, F.; Wang, T.; Wang, S.; Tong, Z.; Li, W.; et al. A Phase IIb, single arm, multicenter trial of sacituzumab govitecan in Chinese patients with metastatic triple-negative breast cancer who received at least two prior treatments. Int J Cancer. 2023, 152, 2134–2144. [Google Scholar] [CrossRef]
- Spring, L.M.; Tolaney, S.M.; Fell, G.; Bossuyt, V.; Abelman, R.O.; et al. Response-guided neoadjuvant sacituzumab govitecan for localized triple-negative breast cancer: results from the NeoSTAR trial. Ann Oncol. 2024, 35, 293–301. [Google Scholar] [CrossRef]
- O'Shaughnessy, J.; Brufsky, A.; Rugo, H.S.; Tolaney, S.M.; Punie, K.; Sardesai, S.; et al. Analysis of patients without and with an initial triple-negative breast cancer diagnosis in the phase 3 randomized ASCENT study of sacituzumab govitecan in metastatic triple-negative breast cancer. Breast Cancer Res Treat. 2022, 195, 127–139. [Google Scholar] [CrossRef]
- Fenn, K.M.; Kalinsky, K. Sacituzumab govitecan: antibody-drug conjugate in triple-negative breast cancer and other solid tumors. Drugs Today (Barc). 2019, 55, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Spring, L.M.; Nakajima, E.; Hutchinson, J.; Viscosi, E.; Blouin, G.; Weekes, C.; Rugo, H.; Moy, B.; Bardia, A. Sacituzumab Govitecan for Metastatic Triple-Negative Breast Cancer: Clinical Overview and Management of Potential Toxicities. Oncologist. 2021, 26, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Sacituzumab Govitecan (Trodelvy): CADTH Reimbursement Recommendation: Indication: For the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer who have received 2 or more prior therapies, with at least 1 of them for metastatic disease [Internet]. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2022 Feb. Table 4, Cost and Cost-Effectiveness. Available from: https://www.ncbi.nlm.nih.gov/books/NBK595400/table/t04/.
- Xie, J., Li, S., Li, Y. et al. Cost-effectiveness of sacituzumab govitecan versus chemotherapy in patients with relapsed or refractory metastatic triple-negative breast cancer. BMC Health Serv Res 2023, 23, 706. [CrossRef]
- Khoury, R.; Saleh, K.; Khalife, N.; Saleh, M.; Chahine, C.; Ibrahim, R.; Lecesne, A. Mechanisms of Resistance to Antibody-Drug Conjugates. Int J Mol Sci. 2023, 24, 9674. [Google Scholar] [CrossRef]
- Hafeez, U. , Parakh, S., Gan, H.K., Scott, A.M., Antibody-drug conjugates for cancer therapy, Molecules (2020). 25, 4764. [CrossRef]
- Coates, J.T. , Sun, S., Leshchiner, I., Thimmiah, N., Martin, E.E., McLoughlin, D., et al. Parallel Genomic Alterations of Antigen and Payload Targets Mediate Polyclonal Acquired Clinical Resistance to Sacituzumab Govitecan in Triple-Negative Breast Cancer. Cancer Discov. 2021, 11, 2436–2445. [Google Scholar] [CrossRef]
- Zhu, J. , Wu, W., Togashi, Y., Nihira, N.T., Johmura, Y., Zhu, D., Nakanishi, M., Miyoshi, Y., Ohta, T. Alteration of Trop-2 expression in breast cancer cells by clinically used therapeutic agents and acquired tamoxifen resistance. Breast Cancer. 2022, 29, 1076–1087. [Google Scholar] [CrossRef]
- Lussier, D.M., et al., Radiation-induced neoantigens broaden the immunotherapeutic window of cancers with low mutational loads, Proc. Natl. Acad. Sci. USA. (2021). 118(24):e2102611118. [CrossRef]
- Abelman, R.O. , Spring, L., Fell, G.G., Ryan, P., Vidula, N., Medford, A.J., Shin, J., Abraham, E., Wander, S.A., Isakoff, S.J., et al. Sequential use of antibody-drug conjugate after antibody-drug conjugate for patients with metastatic breast cancer: ADC after ADC (A3) study. J. Clin. Oncol. 2023, 41, 1022. [Google Scholar] [CrossRef]
- Chang, C.H.; Wang, Y.; Zalath, M.; Liu, D.; Cardillo, T.M.; Goldenberg, D.M. Combining ABCG2 Inhibitors with IMMU-132, an Anti-Trop-2 Antibody Conjugate of SN-38, Overcomes Resistance to SN-38 in Breast and Gastric Cancers. Mol Cancer Ther. 2016, 15, 1910–1919. [Google Scholar] [CrossRef]
- Schmid, P. , Loi, S., De la Cruz Merino, L. et al. 181O Interim analysis (IA) of the atezolizumab (atezo) + sacituzumab govitecan (SG) arm in patients (pts) with triple-negative breast cancer (TNBC) in MORPHEUS-pan BC: A phase Ib/II study of multiple treatment (tx) combinations in pts with locally advanced/metastatic BC (LA/mBC). ESMO Open, (2024) 9, 103203. [CrossRef]
- Cardillo, T.M.; Sharkey, R.M.; Rossi, D.L.; et al. Synthetic lethality exploitation by an Anti-Trop-2-SN-38 antibody-drug conjugate, IMMU-132, plus PARP inhibitors in BRCA1/2-wild-type triple-negative breast cancer. Clin Cancer Res. 2017, 23, 3405–3415. [Google Scholar] [CrossRef]
- Bardia, A.; Coates, J.T., Spring L et al; Abstract 2638: Sacituzumab Govitecan, combination with PARP inhibitor, Talazoparib, in metastatic triple-negative breast cancer (TNBC): Translational investigation. Cancer Res 2022; 82 (12_Supplement): 2638. [CrossRef]
- Liu, D.; Cardillo, T.M.; Wang, Y.; Rossi, E.A.; Goldenberg, D.M.; Chang, C.H. Trop-2-targeting tetrakis-ranpirnase has potent antitumor activity against triple-negative breast cancer. Mol Cancer. 2014, 13, 53. [Google Scholar] [CrossRef]
- Liu, X.; Ma, L.; Li, J.; Sun, L.; Yang, Y.; Liu, T.; Xing, D.; Yan, S.; Zhang, M. Trop2-targeted therapies in solid tumors: advances and future directions. Theranostics. 2024, 14, 3674–3692. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.S.; Seligson, N.D.; Bottiglieri, S.; Carballido, E.; Cueto, A.D.; Imanirad, I.; Levine, R.; Parker, A.S.; Swain, S.M.; Tillman, E.M.; Hicks, J.K. UGT1A1 Guided Cancer Therapy: Review of the Evidence and Considerations for Clinical Implementation. Cancers (Basel). 2021, 13, 1566. [Google Scholar] [CrossRef] [PubMed]
- Trerotola, M.; Guerra, E.; Ali, Z.; et al. Trop-2 cleavage by ADAM10 is an activator switch for cancer growth and metastasis. Neoplasia 2021, 23, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Guerra, E.; Trerotola, M.; Relli, V.; et al. Trop-2 induces ADAM10-mediated cleavage of E-cadherin and drives EMT-less metastasis in colon cancer. Neoplasia 2021, 23, 898–911. [Google Scholar] [CrossRef]
Figure 1.
Molecular classification of Triple-negative breast cancer (TNBC) as reported by [28].
Figure 1.
Molecular classification of Triple-negative breast cancer (TNBC) as reported by [28].
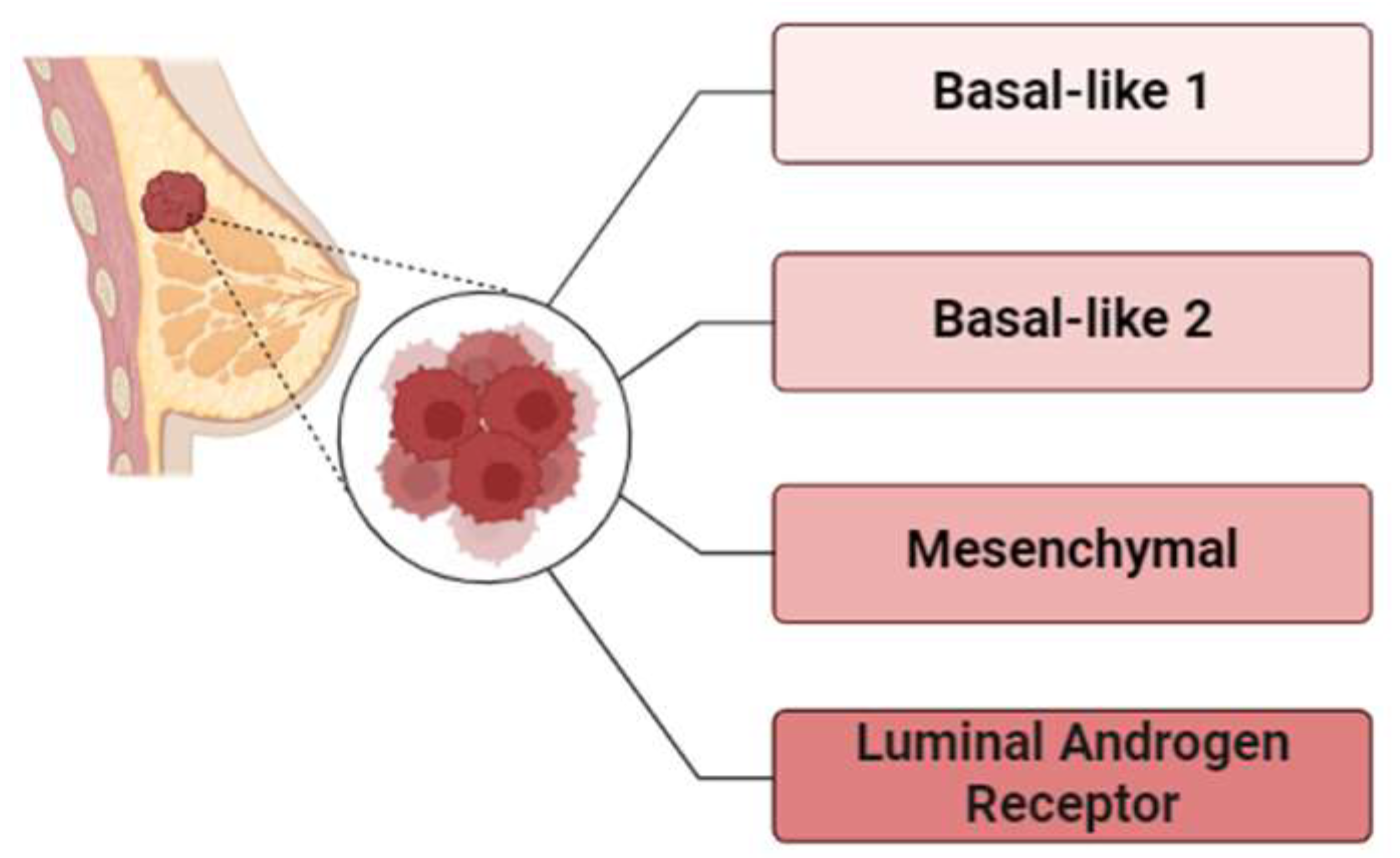
Figure 2.
Treatment options for triple-negative breast cancer (TNBC).
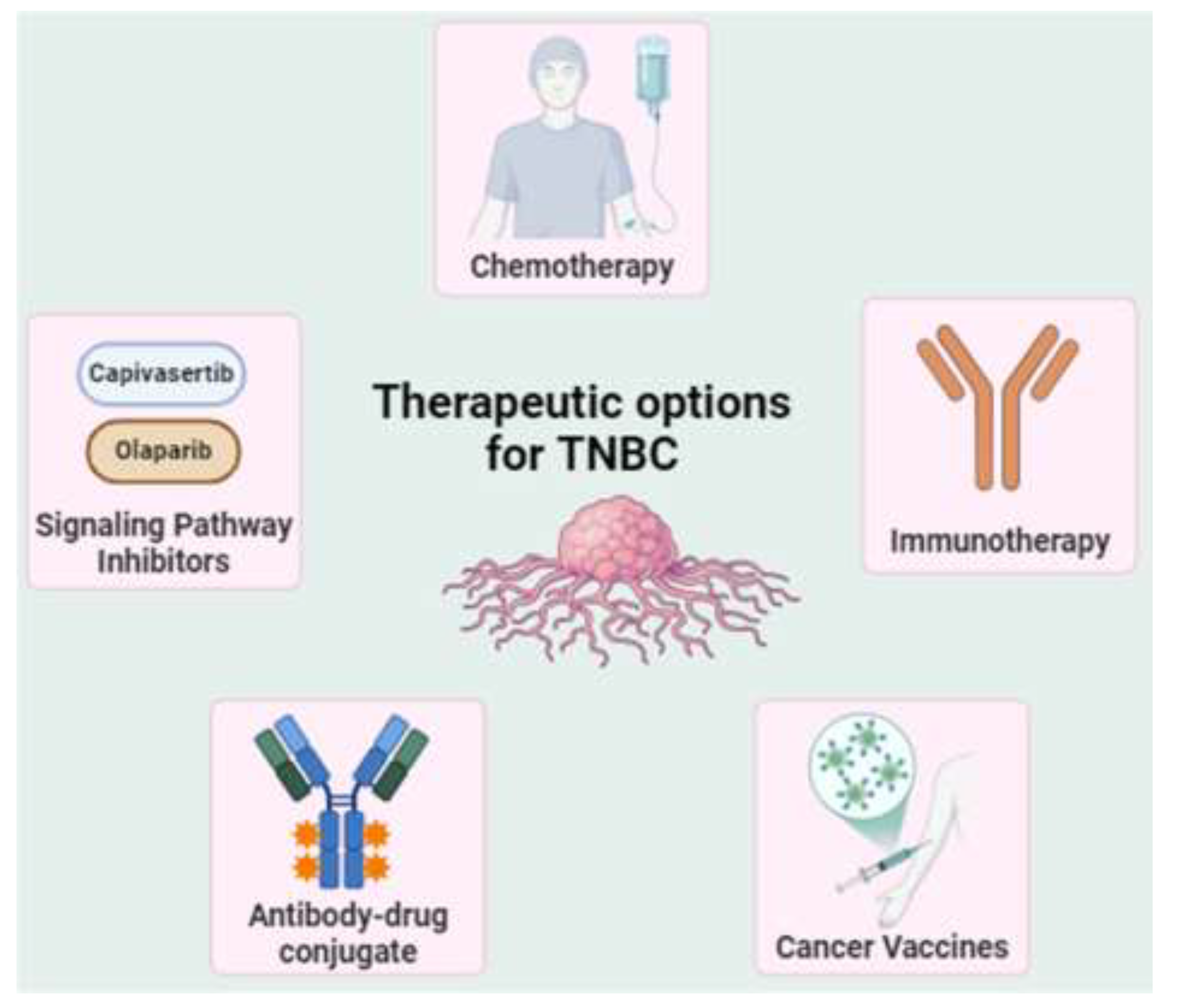
Figure 3.
Structure of SG. A scheme highlighting the chemical arrangement of the link between SN-38 (orange) and the hRS7 antibody (pink) with the CL2A linker (blue). The CL2A linker is specifically attached to the 20th position of SN-38, stabilizing its lactone ring while creating a pH-sensitive carbonate bond. A short polyethylene glycol (PEG) moeity is incorporated into the linker to enhance the water solubility of the SN-38 conjugate. Additionally, the maleimide at the linker terminus facilitates the formation of a stable thioether bond with sulfhydryl groups generated after mild reduction of the hRS7 antibody. The average drug-to-antibody ratio (DAR) is 7.6. (Republished with permission from Rossi et al. 2024, under Creative Commons Attribution License, CC BY). © 2024 Rossi, Turati, Rosato and Carpanese. doi: 10.3389/fimmu.2024.1447280.
Figure 3.
Structure of SG. A scheme highlighting the chemical arrangement of the link between SN-38 (orange) and the hRS7 antibody (pink) with the CL2A linker (blue). The CL2A linker is specifically attached to the 20th position of SN-38, stabilizing its lactone ring while creating a pH-sensitive carbonate bond. A short polyethylene glycol (PEG) moeity is incorporated into the linker to enhance the water solubility of the SN-38 conjugate. Additionally, the maleimide at the linker terminus facilitates the formation of a stable thioether bond with sulfhydryl groups generated after mild reduction of the hRS7 antibody. The average drug-to-antibody ratio (DAR) is 7.6. (Republished with permission from Rossi et al. 2024, under Creative Commons Attribution License, CC BY). © 2024 Rossi, Turati, Rosato and Carpanese. doi: 10.3389/fimmu.2024.1447280.
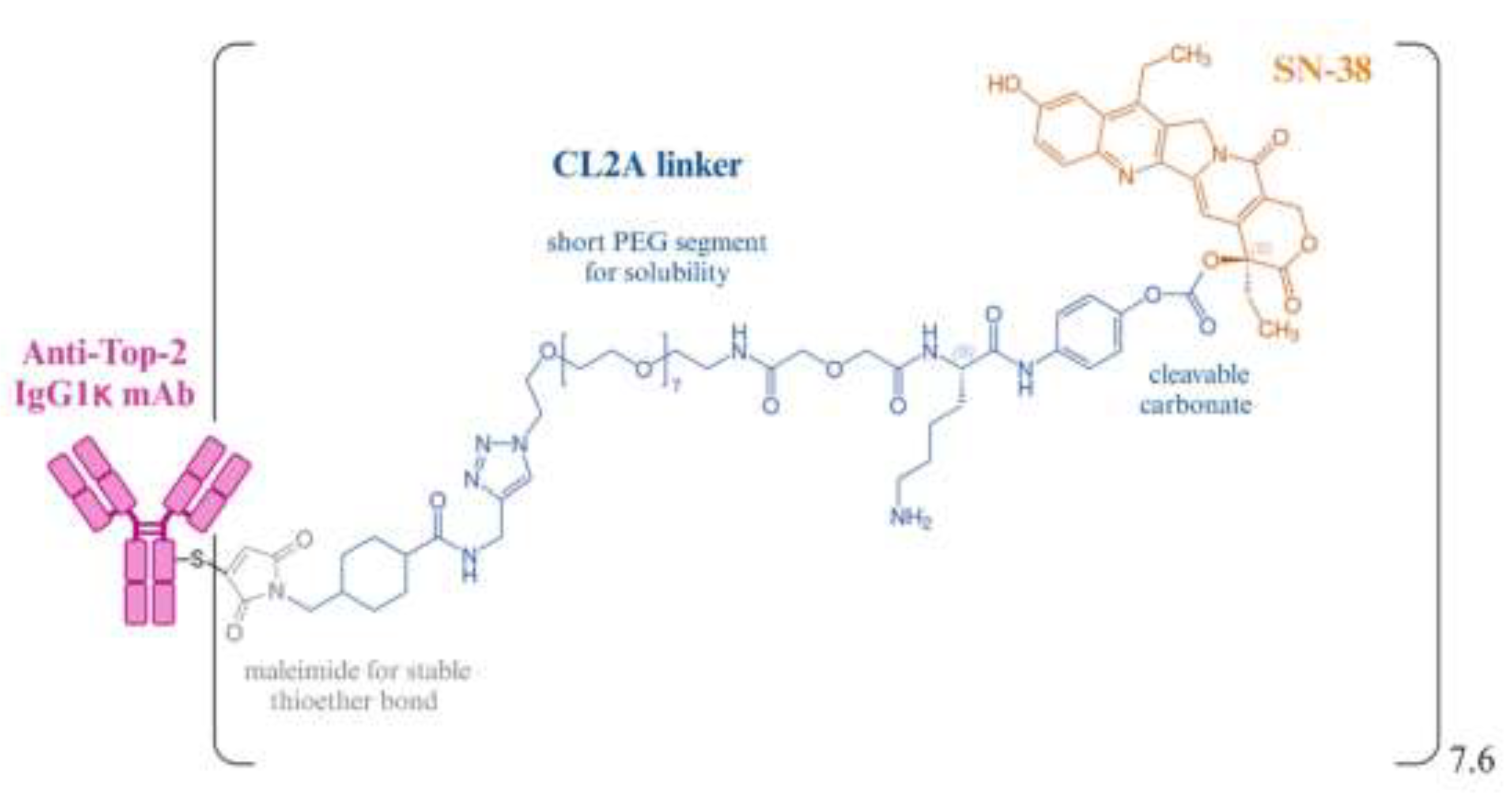
Figure 4.
Mechanism of action of Sacituzumab govitecan (Reproduced from Bardia, et al 2021c. Future Oncology under the Attribution-NoDerivatives 4.0 Unported License. doi: 10.2217/fon-2021-0868). © 2021 The Authors.
Figure 4.
Mechanism of action of Sacituzumab govitecan (Reproduced from Bardia, et al 2021c. Future Oncology under the Attribution-NoDerivatives 4.0 Unported License. doi: 10.2217/fon-2021-0868). © 2021 The Authors.
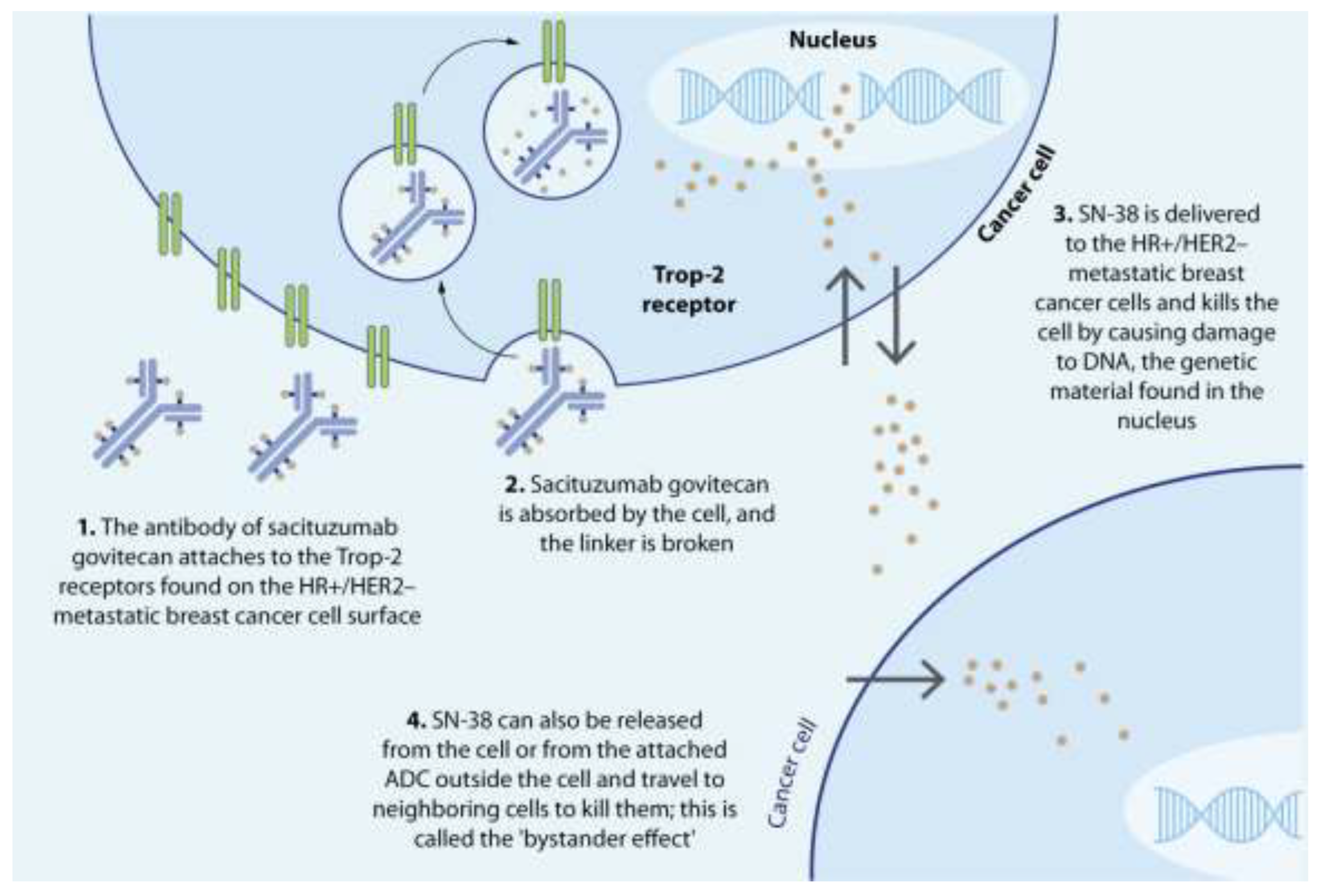

Table 1.
Key characteristics of sacituzumab govitecan-hziy (Trodelvy™).
| Parameters | Key features |
|---|---|
| Drug class | Antibody drug conjugate |
| Brand name | Trodelvy |
| Generic name | Sacituzumab govitecan-hziy |
| Company | Gilead Sciences Inc. |
| Dosage form | Injection |
| Color | Off-white powder |
| Reconstitution | Sterile normal saline |
| Mode of action | Antibody targets the Trop2 receptor on the cancer cell membrane SN-38 is released intracellularly as well as extracellularly SN-38 binds to and stabilizes topoisomerase IB |
| Indication | Advanced TNBC, after at least two lines of systemic treatment (including taxane), one in the metastatic setting Advanced HR+/HER2– breast cancer resistant to endocrine therapy, with at least two prior lines of treatments in the metastatic setting |
| FDA Approval date | April 22, 2020 for metastatic TNBC with at least two prior therapies February 3, 2023 for HR+/HER2– breast cancer |
| Dosage | 10 mg/kg intravenously |
| Maximum tolerated dose | 10 mg/kg/day, if weight changes by more than 10% from baseline, recalculate the dose |
| Administration | Premedicate with antipyretics and histamine antagonists Antiemetics for moderately emetogenic drugs on days 1, 8 every 21 days. Continue till progressive disease or dose-limiting toxicity |
| Toxicities % | Neutropenia (boxed warning): 61% Diarrhea (boxed warning): 65% Hypersensitivity: 37% |
| Molecular predictors of response |
No molecular predictors have been validated Testing for Trop2 expression is not needed |
| Special considerations | To consider UGT1A1 polymorphisms in patients with unusually severe or rapid onset adverse effects |
| Cost | $12,478 for a single 50-mL, 180 mg vial |
Table 2.
Pharmacokinetic Parameters for SG, SN38 and hRS7.
| Parameter | SG | SN-38 |
|---|---|---|
| Cmax, mean (CV%), (ng/mL) | 239,000 (11%) | 98 (45%) |
| AUC 0–168, mean (CV%), (ng.h/mL) | 5,640,000 (22%) | 3696 (56%) |
| Vd (L) | 3.6 | - |
| t½ (hours) | 16 | 18 |
| CLa, L/h | 149 | 270,000 |
| Vssa,L | 2820 | 6900 |
| Systemic clearance time (hrs) | 11-14 | 10-20 |
| Mean clearance rate, L/h | 0.13 | 11.2 |
| Median elimination time (hrs) | 23.4 | 17.6 |
Cmax, maximum serum concentration; AUC 0–168, area under the serum concentration curve from time zero to 168 h; Vd, volume of distribution; t½, half-life; CL, total body clearance, Vss, apparent volume of distribution at steady state following IV administration.
Table 3.
Landmark clinical trials of sacituzumab govitecan that led to FDA approval.
| Clinical Trial | Design | Intervention | Patient population |
No. of Pts. |
Response rates | Median PFS (months) | Median OS (months) | Grade ≥ 3 AEs | FDA approval |
|---|---|---|---|---|---|---|---|---|---|
| ASCENT (NCT02574455) [85] |
Phase III RCT |
SG vs. single agent CT | Metastatic TNBC refractory to two prior lines (including taxanes) | 529 | 31.1 % vs 4.2 % | 4.8 vs. 1.7 | 11.8 vs 6.9 |
Neutropenia (51%) Leukopenia (10%) Diarrhea (10%) Anemia (8%) |
Regulatory approval (April 7, 2021) |
| TROPiCS-02 (NCT03901339) [128] |
Phase III RCT | SG vs. single-agent CT | HR+/HER2- mBC prior taxane, CDK4/6 inhibitor and 2-4 prior CTs | 543 | 57 % vs 38 % |
5.5 vs. 4.0 | 13.9 vs 12.3 | Neutropenia (51%) Diarrhea (10%) |
Regulatory approval (February 3, 2023) |
RCT, randomized controlled trial; CT, chemotherapy; CDK 4/6, Cyclin-dependent kinase 4/6; OS, Overall survival; PFS, Progression-free survival; TNBC, Triple-negative breast cancer; FDA, Food and Drug Administration; AE, Adverse events.
Table 4.
Ongoing clinical trials of sacituzumab govitecan.
| Clinical Trial | Sponsor | Drugs | Phase | Enrollment | Status | Population | Primary Endpoints |
|---|---|---|---|---|---|---|---|
| NCT04039230 (ASSET) |
Massachusetts General Hospital |
SG + talazoparib | Ib/II | 75 | Recruiting | Metastatic TNBC | DLT, PFS, ORR |
| NCT04434040 (ASPRIA) |
Dana-Farber Cancer Institute | SG + atezolizumab | II | 40 | Active, not recruiting | Residual invasive disease in TNBC following NAC | Undetectable ctDNA |
| NCT04468061 (Saci-IO TNBC) | Dana-Farber Cancer Institute | SG with or without pembrolizumab | II | 110 | Recruiting | First-line PD-L1- mTNBC | PFS |
| NCT05633654 (ASCENT-05/AFT-65 OptimICE-RD/NSABP B-63) |
Gilead Sciences | SG + pembrolizumab vs. capecitabine | III | 1514 | Recruiting | TNBC with residual invasive disease after surgery and NAC | IDFS |
| NCT05382286 (ASCENT-04) |
Gilead Sciences | SG + pembrolizumab vs. TPC | III | 443 | Active, not recruiting | First-line metastatic TNBC whose tumors express PD-L1 | PFS |
| NCT03971409 (InCITe) | University of California, San Francisco |
SG + immunotherapy | II | 150 | Recruiting | Metastatic TNBC | ORR |
| NCT05143229 (ASSET) | University of Kansas Medical Center | SG + aleplisib | I | 18 | Recruiting | 2+L HER2+ mBC | RP2D |
| NCT03424005 (Morpheus-panBC) |
Hoffmann-La Roche | SG + Atezolizumab | I/II | 580 | Recruiting | Metastatic or Locally Advanced BC | ORR |
TPC: treatment of physician’s choice; BC: breast cancer; PFS: progression-free survival; ORR: overall response rate; DLTs: dose-limiting toxicities; IDFS: invasive disease-free survival; TNBC: triple-negative breast cancer; SG: sacituzumab govitecan; PD-L1: programmed death ligand-1; OS: overall survival; ctDNA: circulating tumor DNA; NAC: neoadjuvant chemotherapy; RP2D, recommended phase II dose; mBC: metastatic breast cancer.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



