
Submitted:
12 November 2024
Posted:
13 November 2024
You are already at the latest version
Abstract
A concise, transition metal free four step synthetic pathway has been developed for the synthesis of tetracyclic heterosteroidal compounds, 14-aza-12-oxasteroids, starting from readily available 2-naphthol analogues. After conversion of 2-naphthols to 2-naphtylamines by the Bucherer reaction, subsequent selective C-acetylation was done via the Sugasawa reaction and reduction of the acetyl group using borohydride, resulted into corresponding amino-alcohols. The naphthalene based amino-alcohols underwent double dehydrations and double intramolecular cyclisation with oxo-acids leading to one-pot formation of a C-N bond, a C-O bond and an amide bond in tandem, to generate two additional rings completing the steroidal framework. A series of 14-aza-12-oxasteroids were synthesized by our developed synthetic strategy, in moderate yields and structure of one of the final products, 12a-Methyl-11-phenyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one, was further confirmed by single crystal X-ray crystallography.
Keywords:
heterosteroids
; 14-aza-12-oxosteroid
; Sugasawa reaction
; Dean-Stark condensation
; double dehydration
1. Introduction
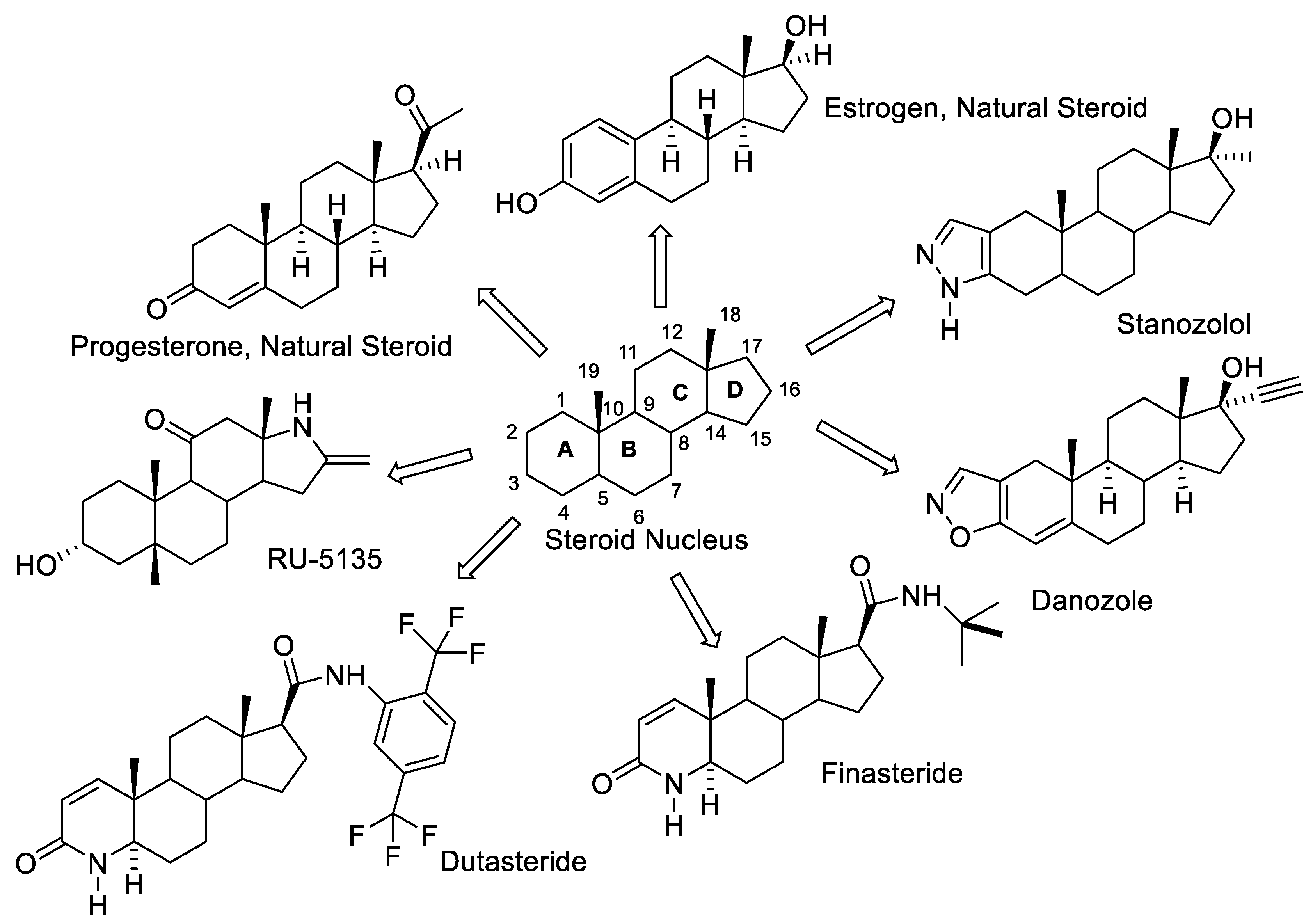
Steroids serve as an important class of natural products by virtue of their ability to penetrate cells and bind to membrane and nuclear receptors [1]. Steroidal hormones are classified as glucocorticoids, mineralocorticoids, estrogen, progestogens and androgens because of their binding to a particular receptor to express their biological response [2]. Estrogens (Figure 1), an aromatic ring containing steroidal hormone, binds with estrogen receptors (ER) and trigger them in promoting a variety of physiological responses such as bone maturation, neuroprotective effects, reproductive functions, modulation of blood lipid profile, and breast cell proliferation [3]. Progesterone is a pro-gestational steroid hormone secreted by the female reproductive system. It is associated to the menstrual cycle, pregnancy, and development of an embryo (Figure 1) [4]. Medicinal chemists are intrigued with the biological activities of various modified steroids for drug design and development process. While retaining the steroidal framework, the structural modifications allow for modulation of the biological activities for the resulting molecules. The naturally occurring steroidal frameworks have been modified in various ways with the aim of identifying active compounds with improved efficacy and reduced toxicity. Heterosteroids is one such modification of the natural steroids, where one or more carbons are replaced by heteroatom(s). Medicinal chemistry of heterosteroids have been extensively studied [5]. A few of heterosteroids have already been culminated into successful drugs, such as stanozolol (hereditary angioedema) [6], danazol (antiestrogen) [7], RU-5135 (antagonist of GABA in the CNS) [8], finasteride (antihyperplastics) [9], and dutasteride (benign prostatic hyperplasia) [10] (Figure 1).
Although, synthesis of these structurally complex molecules in a short route from readily available starting materials is a challenge in the field of organic chemistry, several aza and oxa-steroids have been synthesized by various research groups in past by following different synthetic protocols. Oumzil et al. [11], reported total synthesis of 11-oxasteroid having pyridine ring as the A ring, using intramolecular Diels-Alder reaction. Singh and Panda [12] described an efficient approach for the synthesis of 14-azasteroids using L-proline via intramolecular SN2′ cyclization reaction as a key step for the construction of nitrogen containing ring C. Bernath et al. [13], reported the stereospecific synthesis of 8-aza-12-oxasteroids. In continuation of our efforts of developing complex heterosteroids [14], mainly 14-azasterogen analogues, we herein report a concise synthesis of 14-aza-12-oxosteroids in four simple steps. We have previously reported the synthesis of tricyclic 7-oxa-2-azatricyclo[7.4.0.02,6]trideca-1(9),10,12-trien-3-ones and their homologs, by the reaction of keto acids with methyl chloroformate and variously substituted o-aminobenzyl alcohols using triethylamine as a base in toluene at room temperature [15], a similar type of synthesis with a different approach is reported here.
Figure 1.
Representative natural steroids and clinically used heterosteroids.
2. Results
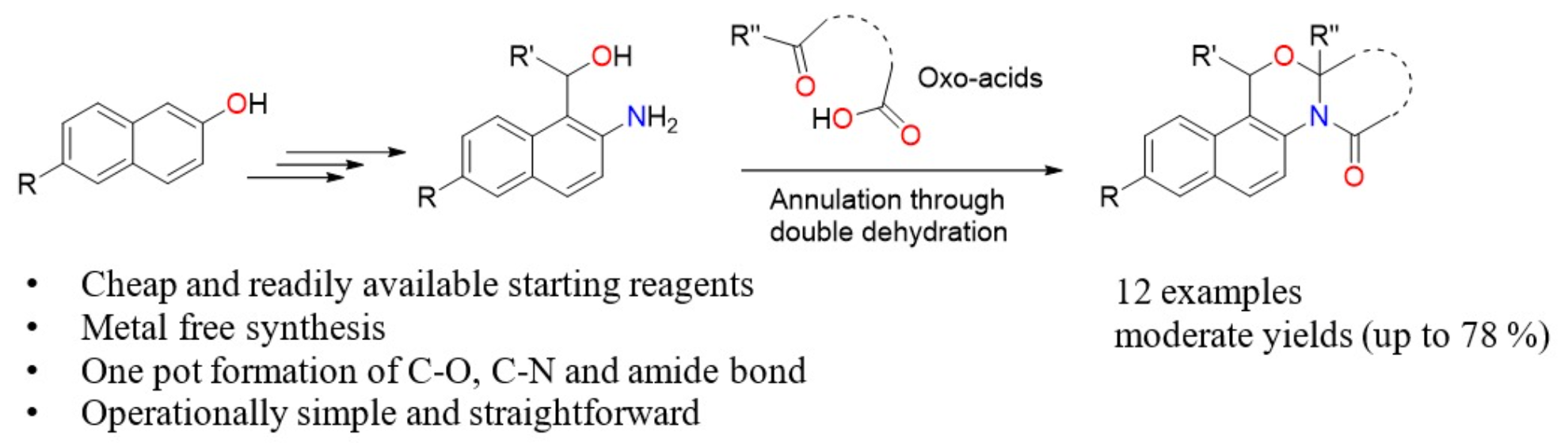
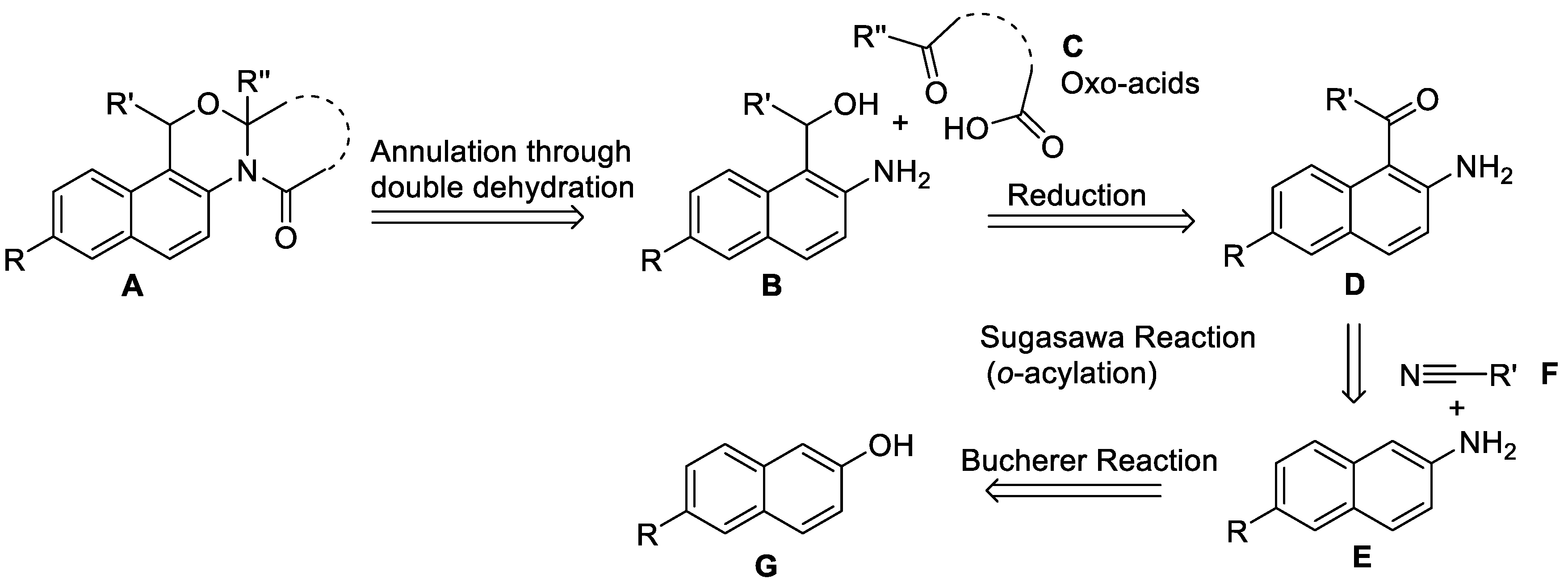
We envisioned a synthetic strategy to obtain our designed 14-aza-12-oxasteroids with general structure A, retrosynthetic analysis of which is presented in Scheme 1. Double dehydration and cyclization between B and oxo acids C would yield the target 14-aza-12-oxasteroids A. Naphthalene-based amino-alcohols B can be prepared by the reduction of 1-acyl-2-aminonaphthalenes D, which in turn can be synthesized via Sugasawa reaction [16] of E with various aryl and alkyl nitriles F. 2-Aminonapthalenes E can be obtained from commercially available 2-naphthols G via Bucherer reaction [17].
Our synthetic scheme started from commercially available 2-naphthol and 6-bromo-2-naphthol (Scheme 2), which were converted into 2-napthylamine (1a, 78% yield) and 6-bromo-2-napthylamine (1b, 85% yield), respectively by using Bucherer reaction [14a, 17]. The Friedel-Craft acylation is a common technique to attach acetyl groups on aromatic rings [18]. However, in an aniline system, coordination of the amino group with the Lewis acid results in a deactivated aromatic system. Sugasawa [16] took a unique approach to achieve ortho-acylation of anilines by using nitriles and two Lewis acids for the reaction. We successfully achieved ortho-acylation of 2-napthylamines (1a & 1b) by using various nitriles (benzyl nitrile, benzonitrile, acetonitrile and butyronitrile) by the Sugasawa protocol employing the BCl3 and AlCl3 system to obtain our desired amino-ketone compounds 2a-h (Scheme 2). The reactions were performed under inert atmosphere since moisture in the air would react with the Lewis acids to form respective hydroxides. During the work-up of this reaction, dilute acid was used to convert imine intermediate to the desired ketones. Although in case of compounds 2b and 2f, extensive amounts of conc. acids and significantly longer heating times were required. Requirement of the forcing conditions for imine hydrolysis was probably due to greater stability of these imines having conjugation with naphthalene and benzene rings. The ortho-acylated compounds 2a-h were obtained in 50-95% yields after purification by column chromatography. Further, these acylated 2-aminonaphthalenes 2a-h were reduced to corresponding amino alcohol analogues 3a-h, by borohydride reduction [19]. The crude compounds 3a-h were obtained in 93-98% yields, these compounds were used for the next reaction without further purification. A greener approach without involving any reagent, with recoverable solvent and lesser reaction time was explored to induce the double dehydration reaction between keto acids and the amino alcohols 3a-h utilising a Dean-Stark apparatus as compared to our previous report for the synthesis of similar tricyclic compounds [15]. It was gratifying that the attempt to achieve the double dehydration and subsequent formation of three bonds in tandem between naphthalene-based amino alcohols 3a-h and various keto acids (levulinic acid / 2-carboxybenzaldehyde / 4-acetylbutyric acid / (2-oxopropyl sulfanyl)-acetic acid) were successful. Formation of new C-N, C-O and amide bond along with double cyclization reaction leads to the synthesis of our desired 14-aza-12-oxasteroid compounds 4a-l, in moderate yields (30-78%, Scheme 2, Table 1). The structures of all the synthesized compounds 1a-b, 2a-2h, 3a-3h and 4a-4l were unambiguously established based on their spectral (1H-, 13C NMR and HRMS) data analysis. The structures of known compounds 1a and 1b were further confirmed on the basis of comparison of their physical and spectral data with those reported in the literature [14a, 17].
3. Discussion
The most plausible mechanism for the cyclization and dehydration reaction is shown in Scheme 3, taking 4c as a representative example. The amino alcohol 3b contains two nucleophiles with varying nucleophilicity. The amine group is more nucleophilic than the alcohol group, therefore it attacks the electrophilic ketone carbonyl on levulinic acid first, leading to dehydration and imine formation (I and II). Subsequent nucleophilic attack from the hydroxy group to the imine center resulted in intramolecular cyclisation, forming the amino-acetal C-ring in III. After proton transfer, the last nucleophilic attack from the secondary amino group to the carboxylic acid center carried out another intramolecular dehydration, forming the lactam as the D-ring in 4c.
The heterosteroidal products 4a-l obtained following this synthetic pathway were expected to give a mixture of up-to four stereoisomeric products: the (R,R) and (S,S) enantiomeric pair and the (R,S) and (S,R) enantiomeric pair, as we did not incorporated any means for enantioselection. In most cases (except in 4i and 4j) there was no evidence for the formation of mixtures of diastereomers in the NMR data. The final compounds 4a-h and 4k-l showed only one set of peaks in their NMR spectra (see Supporting Information), which indicates that these molecules were synthesized as racemic enantiomeric pairs. The analysis of the X-ray crystallographic data of compound 4c reveals that the unit cell has non-centrosymmetric space group and thus it cannot be a racemic mixture. This observation can be possibly due to the preferential crystallization of the C6-R, C4-S enantiomer which was randomly selected for crystallography. Also, from the crystal structure of compound 4c (Figure 2), it can be visualized that the methyl group of C4 and the phenyl group of C6 are in a trans geometry indicating the most thermodynamically stable product. This, along with NMR spectra, confirm that only one enantiomeric pair (R,S and S,R) was diastereoselectively formed in the reaction. In case of compounds 4i and 4j, where R′′ is H (instead of methyl group), mixture of diastereomers was obtained as evident from the NMR data. It appears that higher amount of steric hinderance caused by the presence of angular methyl group led to exclusive diastereoselectivity in case of compounds 4a-h and 4k-l. However, lower amount of steric hinderance in case of 4i and 4j, allowed for the formation of mixture of diastereomers albeit with some selectivity (see Experimental). The ratio of formation of these diastereomers was established by the proton NMR spectroscopy. The crystallographic data of compound 4c was deposited in the Cambridge Crystallographic Data Centre with CCDC no. 2006680. The ORTEP diagram of compound 4c is shown in Figure 2.
4. Materials and Methods
General: All the chemicals and solvents used for these syntheses were obtained from commercial sources and were used without modification, except for toluene and methanol. Toluene was dried with sodium metal overnight and distilled before reactions. Methanol was dried with molecular sieves overnight before use. Among the various keto acids used for our synthesis, levulinic acid, 2-carboxybenzaldehyde and 4-acetylbutyric acids were obtained from A.K. Scientific, Inc. USA. (2-Oxopropyl sulfanyl)-acetic acid was synthesized by following a literature procedure [20] starting from mercaptoacetic acid (obtained from Sigma Aldrich). TLCs were performed on pre-coated Merck silica gel 60F254 plates with the spots detected under UV light. The silica gel used in column chromatography was 230-400 mesh. NMR spectra were recorded using a Bruker AC-300 Avance spectrometer at 300 MHz for 1H NMR and 75 MHz for 13C{1H} NMR. The 1H and 13C{1H} NMR spectra were recorded in either CDCl 3 or DMSO-d6 solvents. 1H NMR spectra were reported relative to CHCl3 (δ: 7.26) or DMSO (δ: 2.50). 13C{1H} NMR spectra were reported relative to CDCl3 (δ: 77.16) or DMSO-d6 (δ: 39.52). HRMS (ESI) spectra were recorded using Orbitrap Q-exactive analyser. In X-ray crystallography, the crystal chosen was attached to the tip of a MicroLoop with Paratone-N oil. Measurements were made on a Bruker D8 VENTURE diffractometer equipped with a PHOTON III CMOS detector using monochromated Cu Kα radiation (λ = 1.54178 Å) from an Incoatec micro-focus sealed tube at 100 K. Among the various keto acids used for our synthesis, levulinic acid, 2-carboxybenzaldehyde and 4-acetylbutyric acids were obtained from a commercial supplier, A.K. Scientific, Inc. USA. (2-Oxopropyl sulfanyl)-acetic acid was synthesized by following a literature procedure starting from mercaptoacetic acid [20], which in turn was obtained from Sigma Aldrich, USA.
General procedure for the synthesis of 1-acyl-2-aminonaphthalenes (2a-h): In a N2-flushed 250 mL round bottom flask, 2-naphthylamines (2.00 g, 1a: 13.96 mmol; 1b: 9.01 mmol, 1 eq.) and AlCl3 (1.2 eq.) were added. The flask was flushed again with N2. Toluene was added to the reaction mixture followed by BCl3 heptane solution (1.0 M, 1.2 eq.). The R′CN (acetonitrile/butyronitrile/benzonitrile/benzyl nitrile, 6.5 eq) was then added to the reaction mixture. The mixture was refluxed and was monitored with TLC (2a-b: 3.5 h, 2c-d: 2 h, 2e-f: 4 h, 2g-h: 2.5 h). After cooling to room temperature, aq. HCl (1.0 M, 1.2 eq., 2b and 2f: 12.0 M, 500 eq.) was added to the resultant mixture forming a yellow-orange mixture, which was heated at 80 °C for 30 mins (4 hrs for 2b and 2f). During the work up, the reaction mixture was cooled to room temperature and the yellow-orange mixture was poured into ice-water (ca. 50 mL). This was followed by the addition of aq. NaOH (5.0 M) until pH > 13. More ice and NaOH pellets were used as needed with 2b and 2f, to bring the pH >13. The mixture was extracted with EtOAc (3 × 50 mL). The organic layers were added together and washed with brine (50 mL) and dried with anhydrous Na2SO4. The solvent was removed in vacuo to form the crude product. The crude product was purified by column chromatography to yield 2a-h.
1-(2-Aminonaphthalen-1-yl)-2-phenylethanone (2a): Dark orange solid; 50% yield (1.81 g); m.p. 72-73 °C; 1H NMR (CDCl3, 300 MHz): δ 7.85 (d, J = 9.0 Hz, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 9.0 Hz, 1H), 7.51 (t, J = 7.5 Hz, 1H), 7.39-7.28 (m, 6H), 6.83 (d, J = 9.0 Hz, 1H), 5.48 (brs, 2H), 4.36 (s, 2H); 13C{1H} NMR (CDCl3, 75 MHz): δ 203.6, 146.0, 135.6, 133.4, 129.4, 128.7, 128.4, 127.4, 126.6, 124.1, 122.6, 122.6, 119.2, 115.4, 50.0; ESI-HRMS: calcd. C18H16NO: 262.1226 [M+H]+; found 262.1227.
(2-Aminonaphthalen-1-yl)(phenyl)methanone (2b): Yellow crystals; 60% yield (2.06 g); m.p. 166-167 °C. 1H NMR (DMSO, 300 MHz): δ 7.78 (d, J = 9.0 Hz, 1H), 7.71 (d, J = 6.0 Hz, 1H), 7.66 (d, J = 6.0 Hz, 2H), 7.59 (t, J = 6.0 Hz, 1H), 7.46 (t, J = 7.5 Hz, 2H), 7.17-7.06 (m, 4H), 5.93 (brs, 2H); 13C{1H} NMR (DMSO, 75 MHz): δ 197.8, 145.7, 138.6, 132.8, 132.2, 131.4, 129.0, 128.5, 128.0, 126.3, 125.9, 123.3, 121.2, 118.9, 112.4; ESI-HRMS: calcd. C17H14NO: 248.1070 [M+H]; found 248.1070.
1-Acetyl-2-aminonaphthalene (2c): Orange solid; 63% yield (1.62 g); m.p. 107-108 °C. 1H NMR (CDCl3, 300 MHz): δ 7.82 (d, J = 9.0 Hz, 1H), 7.68 (d, J = 6.0 Hz, 1H), 7.65 (d, J = 6.0 Hz, 1H), 7.46 (t, J = 9.0 Hz, 1H), 7.27 (t, J = 9.0 Hz, 1H), 6.85 (d, J = 9.0 Hz, 1H), 5.80 (brs, 2H), 2.70 (s, 3H); 13C{1H} NMR (CDCl3, 75 MHz): δ 202.9, 146.4, 133.8, 132.6, 128.7, 127.7, 127.4, 124.4, 122.6, 119.5, 115.2, 32.3.
1-(2-Aminonaphalen-1-yl)butan-1-one (2d): Orange oil; 50% yield (1.33 g); 1H NMR (CDCl3, 300 MHz): δ 7.74-7.68 (m, 2H), 7.66 (d, J = 9.0 Hz, 1H), 7.46 (t, J = 9.0 Hz, 1H), 7.28 (t, J = 7.5 Hz, 1H), 6.87 (d, J = 9.0 Hz, 1H), 5.17 (brs, 2H), 2.99 (t, J = 7.5 Hz, 2H), 1.86 (sx, J = 7.8 Hz, 2H), 0.97 (t, J = 6.0 Hz, 3H); 13C{1H} NMR (CDCl3, 75 MHz): δ 207.7, 144.8, 132.8, 131.2, 128.6, 127.6, 127.2, 124.0, 122.5, 119.6, 116.4, 46.2, 19.3, 13.8; ESI-HRMS: calcd. C14H16NO: 214.1226 [M+H]+; found 214.1231.
1-(2-Amino-6-bromonaphthalen-1-yl)-2-phenylethanone (2e): Dark purple solid; 75% yield (2.29 g); m.p. 101-102 °C; 1H NMR (CDCl3, 300 MHz): δ 7.84 (s, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.55 (m, 2H), 7.34-7.26 (m, 5H), 6.83 (d, J = 12.0 Hz, 1H), 5.46 (brs, 2H), 4.26 (s, 2H); 13C{1H} NMR (CDCl3, 75 MHz): δ 203.2, 146.0, 135.3, 132.3, 130.6, 130.4, 129.3, 128.7, 128.5, 126.8, 125.7, 120.3, 115.9, 50.1; ESI-HRMS: calcd. C18H15BrNO: 340.0332 [M+H]+; found 340.0335.
(2-Amino-6-bromonaphthalen-1-yl)(phenyl)methanone (2f): Yellow crystals; 65% yield (1.90 g); m.p. 120-121 °C; 1H NMR (DMSO, 300 MHz): δ 7.97 (d, J = 1.5 Hz, 1H), 7.77 (d, J = 12.0 Hz, 1H), 7.64 (d, J = 6.0 Hz, 2H), 7.60 (d, J = 6.0 Hz, 1H), 7.46 (t, J = 7.5 Hz, 2H), 7.27 (d, J = 6.0 Hz, 1H), 7.16 (d, J = 9.0 Hz, 1H), 7.00 (d, J = 9.0 Hz, 1H), 6.03 (s, 2H); 13C{1H} NMR (DMSO, 75 MHz): δ 197.6, 146.4, 138.6, 133.3, 131.1, 130.9, 129.9, 129.3, 128.9, 127.4, 125.6, 120.4, 113.9, 112.3; ESI-HRMS: calcd. C17H13BrNO: 326.0175 [M+H]+; found 326.0184.
1-Acetyl-2-amino-6-bromonaphthalene (2g): Orange solid; 95% yield (2.25 g); m.p. 135-137 °C. 1H NMR (CDCl3, 300 MHz): δ 7.80 (s, 1H), 7.68 (d, J = 9.0 Hz, 1H), 7.55 (d, J = 9.0 Hz, 2H), 6.84 (d, J = 9.0 Hz, 1H), 5.94 (brs, 2H), 2.66 (s, 3H); 13C{1H} NMR (CDCl3, 75 MHz): δ 202.3, 147.0, 132.7, 131.2, 130.6, 130.4, 130.0, 128.8, 128.2, 125.9, 120.6, 32.2.
1-(2-Amino-6-bromonaphalen-1-yl)butan-1-one (2h): Brown solid; 60% yield (1.57 g); m.p. 42-43 °C; 1H NMR (CDCl3, 300 MHz): δ 7.81 (s, 1H), 7.59-7.47 (m, 3H), 6.86 (d, J = 9.0 Hz, 1H), 5.02 (brs, 2H), 2.92 (t, J = 7.5 Hz, 2H), 1.79 (q, J = 9.0 Hz, 2H), 0.95 (t, J = 7.5 Hz, 3H); 13C{1H} NMR (CDCl3, 75 MHz): δ 207.1, 145.1, 131.8, 130.7, 130.5, 130.2, 128.7, 125.6, 120.3, 116.1, 115.7, 46.2, 19.3, 13.8; ESI-HRMS: calcd. C14H15BrNO: 292.0332 [M+H]; found 292.0337.
General procedure for the synthesis of 1-(1-hydroxyalkyl)-2-aminonaphthalenes (3a-h): In a 50 mL flask, 2a-h (500 mg, 1 eq.) were dissolved in a 1:1 mixture of methanol and THF solution (10 mL). The reaction flask was flushed with N2 gas and cooled to 0 °C in an ice bath. While stirring, NaBH4 powder (6 eq., except 2b & 2f: 12 eq) was added in small batches. The reaction was stirred in a round bottom flask with a N2 balloon for 5 mins in the ice bath, and further stirred at room temperature until no starting material was observed on the TLC (ca. 1 hr). The solvent was removed in vacuo leaving a solid. Distilled water (ca. 5 mL) was added and the products were extracted with EtOAc (2 × 25 mL) and washed with distilled water (2 × 10 mL). The organic layer was dried with anhydrous Na2SO4 and the solvent was removed in vacuo yielding the products in pure form.
1-(2-Amimonaphthalen-1-yl)-2-phenylethanol (3a): Orange solid; 93% yield (467 mg); m.p. 129-131 °C; 1H NMR (CDCl3, 300 MHz): δ 7.82 (d, J = 6.0 Hz, 1H), 7.73 (d, J = 6.0 Hz, 1H), 7.61 (d, J = 9.0 Hz, 1H), 7.44 (t, J = 7.5 Hz, 1H), 7.39-7.25 (m, 6H), 6.89 (d, J = 9.0, 1H), 5.85 (dd, J = 9.0 Hz, 3.0 Hz, 1H), 3.81 (brs, 3H), 3.39 (dd, J = 15.0 Hz, 9.0 Hz,1H), 3.12 (dd, J = 12.0 Hz, 3.0 Hz, 1H); 13C{1H} NMR (CDCl3, 75 MHz): δ 143.1, 138.6, 131.8, 129.5, 129.1, 128.8, 128.5, 128.1, 126.6, 126.6, 121.8, 120.6, 120.3, 115.9, 72.0, 40.7; ESI-HRMS: calcd. C18H18NO: 264.1383 [M+H]; found 264.1383.
(2-Aminonaphthalen-1-yl)(phenyl)methanol (3b): Pale orange solid; 94% yield (474 mg); m.p. 131-133 °C; 1H NMR (DMSO, 300 MHz): δ 7.97 (d, J = 9.0 Hz, 1H), 7.67 (d, J = 9.0 Hz, 1H), 7.59 (d, J = 9.0 Hz, 1H), 7.38 (d, J = 6.0 Hz, 2H), 7.31-7.23 (m, 3H), 7.19-7.09 (m, 2H), 7.02 (d, J = 9.0 Hz, 1H), 6.62 (s, 1H), 6.16 (s, 1H), 5.61 (brs, 2H); 13C{1H} NMR (DMSO, 75 MHz): δ 144.7, 144.4, 132.8, 128.4, 128.3, 127.8, 127.0, 126.3, 126.0, 125.9, 122.1, 120.6, 119.9, 116.0, 68.5; ESI-HRMS: calcd. C17H16NO: 250.1226 [M+H]; found 250.1214.
1-(1-Hydroxyethyl)-2-aminonaphthalene (3c): Colourless solid; 98% yield (499 mg); m.p. 89.9-91.2 °C. 1H NMR (CDCl3, 300 MHz): δ 7.78 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 9.0 Hz, 1H), 7.58 (d, J = 9.0 Hz, 1H), 7.41 (t, J = 6.0 Hz, 1H), 7.23 (t, J = 6.0 Hz, 1H), 6.87 (d, J = 9.0 Hz, 1H), 5.91 (q, J = 7.5 Hz, 1H), 4.82 (brs, 1H), 3.33 (brs, 2H), 1.65 (d, J = 7.5 Hz, 3H); 13C{1H} NMR (CDCl3, 75 MHz): δ 142.8, 131.6, 128.8, 128.7, 127.9, 126.6, 121.8, 120.6, 120.2, 117.4, 67.0, 20.2. ESI-HRMS: calcd. for C12H14NO: 188.1070 [M+H]+; found 188.1068.
1-(2-Aminonaphthalen-1-yl)butan-1-ol (3d): Orange solid; 95% yield (480 mg); m.p. 86-88 °C; 1H NMR (DMSO, 300 MHz): δ 7.87 (d, J = 6.0 Hz, 1H), 7.64 (d, J = 9.0 Hz, 1H), 7.52 (d, J = 9.0 Hz, 1H), 7.32 (t, J = 7.5 Hz, 1H), 7.10 (t, J = 7.5 Hz, 1H), 6.97 (d, J = 9.0, 1H), 5.64 (brs, 2H), 5.53 (brs, 2H), 2.01-1.89 (m, 1H), 1.73-1.62 (m, 1H), 1.57-1.46 (m, 1H), 1.34-1.23 (m, 1H), 0.89 (t, J = 7.5 Hz, 3H); 13C{1H} NMR (DMSO, 75 MHz): δ 144.5, 132.2, 128.4, 127.7, 126.9, 121.2, 120.5, 120.0, 116.5, 68.1, 37.8, 19.1, 14.1; ESI-HRMS: calcd. C14H18NO: 216.1383 [M+H]+; found 216.1386.
1-(2-Amimo-6-bromonaphthalen-1-yl)-2-phenylethanol (3e): Purple solid; 93% yield (468 mg); m.p. 144-146 °C; 1H NMR (DMSO, 300 MHz): 7.86 (s, 1H), 7.54 (d, J = 9.0 Hz, 1H), 7.35 (d, J = 9.0 Hz, 1H), 7.25-7.21 (m, 4H), 7.18-7.12 (m, 2H), 7.06 (d, J = 9.0, 1H), 5.80 (brs, 2H), 5.61 (brs, 1H), δ 5.69 (s, 1H), 3.20 (dd, J = 15.0 Hz, 9.0 Hz, 1H), 2.96 (dd, J = 12.0 Hz, 6.0 Hz, 1H); 13C{1H} NMR (DMSO, 75 MHz): δ 145.1, 140.5, 139.3, 130.7, 129.9, 129.5, 128.4, 128.2, 127.9, 127.3, 125.8, 121.1, 115.9, 113.0, 69.6, 40.4; ESI-HRMS: calcd. C18H17BrNO: 342.0488 [M+H]+; found 342.0495.
(2-Amino-6-bromonaphthalen-1-yl)(phenyl)methanol (3f): Very pale orange solid; 94% yield (474 mg); m.p. 171-172 °C; 1H NMR (DMSO, 300 MHz): δ 7.94-7.89 (m, 2H), 7.58 (d, J = 9.0 Hz, 1H), 7.34 (d, J = 9.0 Hz, 3H), 7.25 (t, J = 7.5 Hz, 2H), 7.16 (t, J = 6.0 Hz, 1H), 7.06 (d, J = 9.0 Hz, 1H), 6.54 (s, 1H), 6.17 (s, 1H), 5.72 (brs, 2H); 13C{1H} NMR (DMSO, 75 MHz): δ 145.2, 144.2, 131.4, 129.8, 128.4, 127.8, 127.7, 126.3, 125.9, 124.9, 120.9, 116.1, 113.1, 68.2; ESI-HRMS: calcd. C17H15BrNO: 328.0332 [M+H]; found 328.0319.
1-(1-Hydroxyethyl)-2-amino-6-bromonaphthalene (3g): Colourless solid; 98% yield (495 mg); m.p. 120-123 °C. 1H NMR (DMSO-d6, 300 MHz): δ 7.90 (s, 1H), 7.87 (d, J = 3.0 Hz, 1H), 7.50 (d, J = 9.0 Hz, 1H), 7.39 (d, J = 3.0 Hz, 1H), 6.99 (d, J = 9.0 Hz, 1H), 5.74 (brs, 2H), 5.59 (q, J = 6.0 Hz, 1H), 5.52 (brs, 1H), 1.41 (d, J = 6.0 Hz, 3H); 13C{1H} NMR (DMSO-d6, 75 MHz): δ 144.6, 130.3, 129.9, 128.4, 128.2, 126.9, 123.7, 121.0, 117.1, 112.9, 64.4, 21.1; ESI-HRMS: calcd. for C12H13BrNO: 266.0175 [M+H]+; found 266.0173.
1-(2-Amino-6-bromonaphthalen-1-yl)butan-1-ol (3h): Brown solid; 98% yield (493 mg); m.p. 138-140 °C; 1H NMR (DMSO, 300 MHz): δ 7.87 (d, J = 1.5 Hz, 2H), 7.52 (d, J = 9.0 Hz, 1H), 7.40 (d, J = 9.0 Hz, 1H), 7.01 (d, J = 9.0, 1H), 5.80 (brs, 2H), 5.61 (brs, 1H), 5.42 (t, J = 6.0 Hz, 1H), 1.96-1.84 (m, 1H), 1.69-1.56 (m, 1H), 1.54-1.42 (m, 1H), 1.30-1.18 (m, 1H), 0.87 (t, J = 7.5 Hz, 3H); 13C{1H} NMR (DMSO, 75 MHz): δ 144.7, 130.8, 129.9, 128.4, 127.0, 121.1, 116.9, 113.0, 68.0, 36.9, 19.1, 14.1; ESI-HRMS: calcd. C14H17BrNO: 294.0488 [M+H]+; found 294.0490.
General procedure for the synthesis of 14-aza-12-oxasteroid analogs (4a-l): In a 50 mL round bottom flask with a stir bar, 3a-h (200 mg, 1 eq.) were dissolved in toluene (ca. 5 mL), and then appropriate keto acid [levulinic acid/2-carboxybenzaldehyde/4-acetylbutyric acid/(2-oxopropyl sulfanyl)-acetic acid, 1.5 eq.] was added. The reaction mixture was refluxed for 2 hrs with a Dean–Stark apparatus, with dry toluene filled in the vertical column. The solvent was removed from the reaction mixture in vacuo and the crude product was purified using column chromatography to yield respective 14-aza-12-oxasteroid analogs.
11-Benzyl-12a-methyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4a): Orange solid; 40% yield (104 mg); m.p. 124-126 °C; 1H NMR (CDCl3, 300 MHz): δ 8.16 (d, J = 9.0 Hz, 1H), 7.93 (t, J = 7.5 Hz, 2H), 7.84 (d, J = 9.0 Hz, 1H), 7.62 (t, J = 7.5 Hz, 1H), 7.52 (t, J = 7.5 Hz, 1H), 7.27-7.22 (m, 3H), 7.07 (d, J = 3.0, 2H), 5.85 (d, J = 3.0 Hz, 1H), 3.43 (d, J = 12.0 Hz, 1H), 3.06 (dd, J = 15.0 Hz, 6.0 Hz, 1H), 2.58 (t, J = 7.5 Hz, 2H), 2.37-2.20 (m, 2H), 1.42 (s, 3H); 13C{1H} NMR (DMSO, 75 MHz): δ 172.1, 137.7, 131.4, 129.5, 129.1, 128.4, 127.8, 126.5, 126.5, 124.9, 122.6, 121.8, 120.7, 89.3, 72.1, 42.8, 32.6, 29.8, 22.4; ESI-HRMS: calcd. C23H22NO2: 344.1645 [M+H]+; found 344.1644.
11-Benzyl-8-bromo-12a-methyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4b): Purple solid; 33% yield (82 mg); m.p. 106-108 °C; 1H NMR (CDCl3, 300 MHz): δ 8.16 (d, J = 9.0 Hz, 1H), 8.05 (d, J = 1.5 Hz, 1H), 7.79 (d, J = 9.0 Hz, 1H), 7.72 (d, J = 9.0 Hz, 1H), 7.66 (d, J = 9.0 Hz, 1H), 7.23-7.19 (m, 3H), 7.00-6.99 (m, 2H), 5.79 (dd, J =6.0 Hz, 3.0 Hz, 1H), 3.36 (dd, J = 15.0 Hz, 3.0 Hz, 1H), 3.04 (dd, J = 15.0 Hz, 6.0 Hz, 1H), 2.56 (t, J = 7.5 Hz, 2H), 2.18-2.34 (m, 2H), 1.40 (s, 3H); 13C{1H} NMR (DMSO, 75 MHz): δ 172.0, 137.2, 132.3, 131.7, 131.0, 129.8, 129.4, 127.8, 127.5, 126.6, 124.4, 122.0, 121.8, 118.8, 71.9, 89.3, 42.8, 32.6, 29.8, 22.3; ESI-HRMS: calcd. C23H21BrNO2: 421.0677 [M+H]+; found 421.0740.
12a-Methyl-11-phenyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4c): Yellow-orange powder; 45% yield (119 mg); m.p. 189-191 °C; 1H NMR (CDCl3, 300 MHz): δ 8.46 (d, J = 9.0 Hz, 1H), 7.90 (d, J = 9.0 Hz, 1H), 7.83 (d, J = 9.0 Hz, 1H), 7.48 (d, J = 9.0 Hz, 1H), 7.36 (t, J = 6.0 Hz, 1H), 7.32-7.26 (m, 6H), 6.45 (s, 1H), 2.71-2.66 (m, 2H), 2.29 (t, J = 9.0 Hz, 2H), 1.64 (s, 3H); 13C{1H} NMR (DMSO, 75 MHz): δ 172.6, 141.6, 132.3, 131.5, 129.7, 129.5, 129.2, 129.0, 129.0, 128.8, 126.6, 125.2, 124.6, 120.7, 120.6, 90.6, 75.8, 33.1, 30.6, 22.0; ESI-HRMS: calcd. C22H20NO2: 330.1489 [M+H]+; found 330.1489.
8-Bromo-12a-methyl-11-phenyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4d): Purple-brown solid; 30% yield (75 mg); m.p. 203-205 °C; 1H NMR (CDCl3, 300 MHz): δ 8.49 (d, J = 9.0 Hz, 1H), 7.97 (s, 1H), 7.79 (d, J = 9.0 Hz, 1H), 7.31-7.27 (m, 5H), 7.23-7.21 (m, 2H), 6.39 (s, 1H), 2.70-2.65 (m, 2H), 2.29 (t, J = 6.0 Hz, 2H), 1.64 (s, 3H); 13C{1H} NMR (DMSO, 75 MHz): δ 172.2, 140.9, 132.3, 132.2, 130.6, 129.5, 128.9, 128.5, 128.1, 127.8, 125.9, 121.4, 120.3, 118.8, 90.2, 75.2, 32.7, 30.1, 21.6; ESI-HRMS: calcd. C22H19BrNO2: 408.0594 [M+H]+; found 408.0584.
11,12a-Dimethyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4e): Viscous orange liquid; 34% yield (96 mg); 1H NMR (CDCl3, 300 MHz): δ 8.35 (d, J = 1.5 Hz, 1H), 7.87-7.78 (m, 3H), 7.53-7.43 (m, 2H), 5.66 (q, J = 6.0 Hz, 1H), 2.67 (t, J = 9.0 Hz, 2H), 2.30 (t, J = 9.0 Hz, 2H), 1.65 (d, J = 6.0 Hz, 3H), 1.47 (s, 3H); 13C{1H} NMR (CDCl3, 75 MHz): δ 172.1, 131.2, 130.2, 129.2, 128.9, 128.2, 126.2, 124.8, 123.3, 123.1, 120.4, 89.3, 68.0, 32.8, 30.1, 23.1, 21.5; ESI-HRMS: calcd. for C17H18NO2: 268.1332 [M+H]+; found 268.1328.
8-Bromo-11,12a-dimethyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]-oxazin-3(2H)-one (4f): Viscous orange liquid; 35% yield (92 mg); 1H NMR (CDCl3, 300 MHz): δ 8.35 (d, J =1.5 Hz, 1H), 7.98 (s, 1H), 7.69-7.39 (m, 3H), 5.58 (q, J = 6.0 Hz, 1H), 2.65 (t, J = 7.5 Hz, 2H), 2.27 (t, J = 7.5 Hz, 2H), 1.59 (d, J = 6.0 Hz, 3H), 1.43 (s, 3H); 13C{1H} NMR (CDCl3, 75 MHz): δ 172.2, 132.3, 130.7, 129.4, 127.7, 127.2, 124.8, 123.5, 121.4, 119.0, 118.7, 89.3, 67.7, 32.7, 30.0, 23.1, 21.4; ESI-HRMS: calcd. for C17H17BrNO2: 346.0437 [M+H]+; found 346.0433.
12a-Methyl-11-propyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4g): Orange solid; yield 45% (123 mg); m.p. 123-125 °C; 1H NMR (CDCl3, 300 MHz): δ 8.27 (d, J = 9.0 Hz, 1H), 7.87-7.76 (m, 3H), 7.52 (t, J = 9.0 Hz, 1H), 7.45 (t, J = 9.0 Hz, 1H), 5.58 (dd, J = 6.0 Hz, 1.5 Hz, 1H), 2.67 (t, J = 7.5 Hz, 2H), 2.30 (t, J = 7.5 Hz, 2H), 2.12-2.01 (m, 1H), 1.87-1.75 (m, 1H), 1.55-1.44 (m, 1H), 1.44 (s, 3H), 1.32-1.21 (m, 1H), 0.87 (t, J = 6.0 Hz, 3H). 13C{1H} NMR (DMSO, 75 MHz): δ 172.3, 131.2, 131.0, 129.2, 128.9, 128.1, 126.2, 124.8, 122.9, 122.6, 120.5, 89.3, 71.3, 38.8, 32.8, 30.1, 21.9, 18.0, 13.9; ESI-HRMS: calcd. C19H22NO2: 296.1645 [M+H]+; found 296.1631.
8-Bromo-12a-methyl-11-propyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]-oxazin-3(2H)-one (4h): Purple oil; 40% yield (102 mg); 1H NMR (CDCl3, 300 MHz): δ 8.29 (d, J = 9.0 Hz, 1H), 7.99 (d, J = 1.5 Hz, 1H), 7.70-7.62 (m, 2H), 7.56 (d, J = 9.0 Hz, 1H), 5.53 (dd, J = 6.0 Hz, 3.0 Hz, 1H), 2.66 (t, J = 7.5 Hz, 2H), 2.29 (t, J = 7.5 Hz, 2H), 2.06-1.95 (m, 1H), 1.82-1.70 (m, 1H), 1.51-1.39 (m, 1H), 1.43 (s, 3H), 1.23-1.15 (m, 1H), 0.85 (t, J = 7.5 Hz, 3H); 13C{1H} NMR (DMSO, 75 MHz): δ 172.3, 132.4, 131.3, 130.8, 129.4, 127.6, 127.2, 124.6, 122.7, 121.5, 118.7, 89.3, 71.1, 38.8, 32.8, 30.0, 21.8, 17.9, 13.8; ESI-HRMS: calcd. C19H21BrNO2: 374.0750 [M+H]+; found 374.0739.
7-Methyl-7H-naphtho[2',1':4,5][1,3]oxazino[2,3-a]isoindol-13(8aH)-one (4i): Viscous orange liquid; 78% yield (249 mg). Mixture of two diastereomers (6:4). Data for the major diastereomer are presented here; 1H NMR (CDCl3, 300 MHz): δ 8.56 (d, J = 1.5 Hz, 1H), 7.94-7.43 (m, 9H), 5.95 (q, J = 9.0 Hz, 1H), 5.79 (s, 1H), 1.72 (d, J = 9.0 Hz, 3H); 13C{1H} NMR (CDCl3, 75 MHz): δ 164.8 140.2, 132.7, 130.2, 128.9, 128.5, 126.6, 126.4, 124.7, 124.6, 124.0, 123.5, 123.2, 123.1, 122.4, 120.8, 119.0, 83.1, 72.4, 23.3; ESI-HRMS: calcd. for C20H16NO2: 302.1176 [M+H]+; found 302.1173.
4-Bromo-7-methyl-7H-naphtho[2',1':4,5][1,3]oxazino[2,3-a]isoindol-13(8aH)-one (4j): Colourless solid; 40% yield (112 mg); m.p. 230-233 °C. Mixture of two diastereomers (8:2). Data for the major diastereomer are presented here; 1H NMR (CDCl3, 300 MHz): δ 8.58 (d, J = 9.0 Hz, 1H), 8.03 (s, 1H), 7.95 (d, J = 9.0 Hz, 1H), 8.03-7.59 (m, 6H), 5.94 (q, J = 6.0 Hz, 1H), 5.87 (s, 1H), 1.70 (d, J = 6.0 Hz, 3H); 13C{1H} NMR (CDCl3, 75 MHz): δ 164.9, 140.2, 132.9, 132.2, 131.0, 130.5, 130.0, 129.7, 128.2, 127.7, 124.9, 124.2, 123.6, 122.5, 120.3, 118.6, 83.2, 72.3, 23.4; ESI-HRMS: calcd. for C20H15BrNO2: 380.0281 [M+H]+; found 380.0284.
12,13a-Dimethyl-1,2,3,13a-tetrahydronaphtho[2,1-d]pyrido[2,1-b][1,3]oxazin-4(12H)-one (4k): Viscous orange liquid; 30% yield (91 mg); 1H NMR (CDCl3, 300 MHz): δ 7.87-7.70 (m, 4H), 7.55-7.46 (m, 2H), 5.65 (q, J = 6.0 Hz, 1H), 2.63 (t, J = 9.0 Hz, 2H), 2.13 (t, J = 9.0 Hz, 2H), 1.63 (d, J = 6.0 Hz, 3H), 1.38 (s, 3H), 0.83 (t, J = 9.0 Hz, 2H); 13C{1H} NMR (CDCl3, 75 MHz): δ 170.1, 132.8, 131.6, 128.9, 128.8, 127.6, 127.1, 126.2, 125.2, 122.5, 86.1, 67.3, 37.0, 34.1, 25.0, 23.5, 17.0.
12,13a-Dimethyl-12,13a-dihydro-1H-naphtho[2,1-d][1,4]thiazino[3,4-b][1,3]oxazin-4(3H)-one (4l): Viscous orange liquid; 30% yield (92 mg); 1H NMR (CDCl3, 300 MHz): δ 7.87-7.66 (m, 4H), 7.56-7.47 (m, 2H), 5.65 (q, J = 7.5 Hz, 1H), 3.60 (m, 2H), 3.06 (s, 2H), 1.68 (d, J = 7.5 Hz, 3H), 1.54 (s, 3H); 13C{1H} NMR (CDCl3, 75 MHz): δ 165.6, 132.1, 131.6, 128.8, 128.7, 127.2, 126.3, 125.5, 124.6, 122.5, 87.7, 67.5, 39.0, 33.8, 23.4, 23.3; ESI-HRMS: calcd. for C17H18NO2S: 300.1053 [M+H]+; found 300.1047.
5. Conclusions
We have devised a concise synthesis of 14-aza-12-oxasteroids, 4a-l in four simple steps, starting from cheaply available 2-naphthol analogues. Bucherer reaction was employed to convert naphthol analogues to their corresponding naphthylamines, which in turn were ortho-acylated using Sugasawa reaction. Further, the 1-acylated-2-aminonaphthylenes were reduced to corresponding amino-alcohols. Double dehydrations and double intramolecular cyclisation of the synthesized amino-alcohols with four different oxo-acids resulted into one-pot formation of a C-N bond, a C-O bond and an amide bond in tandem. A series of twelve, 14-aza-12-oxasteroid analogs were synthesized in moderate yields. The structure, stereochemistry and connectivity of the bonds was further confirmed by X-ray crystal structure determination of one of the analogue i.e., 12a-methyl-11-phenyl-11,12a-dihydro-1H-naphtho[2,1-d]pyrrolo[2,1-b][1,3]oxazin-3(2H)-one (4c).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, A.J.; methodology, J.L. and D.W.; X-ray crystallography, K.R.; writing—original draft preparation, S.S., J.L. and D.W.; writing—review and editing, A.J.; supervision, A.J. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Natural Sciences and Engineering Research Council (NSERC) of Canada (salary support to JL and DW) and Beatrice Hunter Cancer Research Institute (BHCRI), Halifax (salary support to SS).
Data Availability Statement
All spectroscopic and x-ray data obtained during this research are included in the Supplementary Materials (see above).
Acknowledgments
Paraza Pharma, Inc. is thanked for their support in recording HRMS data of reported final products.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ibrahim-Ouali, M. Recent advances in oxasteroids chemistry. Steroids 2007, 72, 475–508. [Google Scholar] [CrossRef] [PubMed]
- (a) White, R.; Parker, M.G. Molecular mechanisms of steroid hormone action. Endocr-Relat. Cancer 1998, 5, 1–14; (b) Frye, C.A. Steroids, reproductive endocrine function, and affect. A review. Minerva Ginecol. 2009, 61, 541–562.
- Wall, E.H.; Hewitt, S.C.; Case, L.K.; Lin, C.-Y.; Korach, K.S.; Teuscher, C. The role of genetics in estrogen responses: a critical piece of an intricate puzzle. FASEB J 2014, 12, 5042–5054. [Google Scholar] [CrossRef] [PubMed]
- Taraborrelli, S. Physiology, production and action of progesterone. Acta Obstet. Gynecol. Scand. 2015, 94(S161), 8–16. [Google Scholar] [CrossRef] [PubMed]
- (a) Singh, H.; Jindal, D.P.; Yadav, M.R.; Kumar, M. Heterosteroids and drug research. In Progress in Medicinal Chemistry; Ellis, G.P., West, G.B., Eds.; Elsevier: 1991; pp. 233–300; (b) Ibrahim-Ouali, M.; Rocheblave, L. Recent advances in azasteroids chemistry. Steroids 2008, 73, 375–407; (c) Ibrahim-Ouali, M.; Santelli, M. Recent advances in thiasteroids chemistry. Steroids 2006, 71, 1025–1044; (d) Burbiel, J.; Bracher, F. Azasteroids as antifungals. Steroids 2003, 68, 587–594.
- Helfman, T.; Falanga, V. Stanozolol as a novel therapeutic agent in dermatology. JAAD 1995, 33, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Cottreau, C.M.; Ness, R.B.; Modugno, F.; Allen, G.O.; Goodman, M.T. Endometriosis and its treatment with danazol or lupron in relation to ovarian cancer. Clin. Cancer Res. 2003, 9, 51424. [Google Scholar]
- Curtis, D.R.; Malik, R. Glycine antagonism by RU 5135. Eur. J. Pharmacol. 1985, 110, 383. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Moore, R.A. Finasteride in the treatment of clinical benign prostatic hyperplasia: a systematic review of randomised trials. BMC Urol. 2002, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pohlman, G.D.; Pohlman, E.A.; Crawford, E.D. Dutasteride: A review of its use in the management of prostate disorders. Clin. Med. Insights Ther. 2011, 3, 172–177. [Google Scholar] [CrossRef]
- Oumzil, K.; Ibrahim-Ouali, M.; Santelli, M. First total synthesis of (±)-3-aza-11-oxa-1,3,5(10)-trieno steroids. Steroids 2006, 71, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Panda, G. L-Proline derived nitrogenous steroidal systems: An asymmetric approach to 14-azasteroids. RSC Advances 2013, 3(42), 19533–19544. [Google Scholar] [CrossRef]
- Bernath, G.; Fueloep, F.; Argay, G.; Kalman, A.; Sohar, P. Stereochemical studies. Saturated heterocycles. A simple stereospecific synthesis of oxazasteroids. Tetrahedron Lett. 1981, 22, 3797–3800. [Google Scholar] [CrossRef]
- (a) Abdelkhalik, A.M.; Paul, N.K.; Jha, A. Concise synthesis of 12a-methul-11-aryl-1,2-dihydrobenzo[f]pyrrolo[1,2-a]quinolin-3(12aH)-ones as racemic 14-azaestrongen analogues. Steroids 2015, 98, 107–113; (b) Jha, A.; Chou, T.-Y.; Al Jaroudi, Z.; Ellis, B.D.; Cameron, T.S. Aza-Diels–Alder reaction between N-aryl-1-oxo-1H-isoindolium ions and tert-enamides: Steric effects on reaction outcome. Beilstein J. Org. Chem. 2014, 10, 848–857.
- Jha, A.; Naidu, A.B.; Abdelkhalik, A.M. Transition metal-free one-pot cascade synthesis of 7-oxa-2-azatricyclo [7.4. 0.0 2, 6] trideca-1 (9), 10, 12-trien-3-ones from biomass-derived levulinic acid under mild conditions. Org. Biomol. Chem. 2013, 11, 7559–7565. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, T.; Toyoda, T.; Adachi, M.; Sasakura, K. Aminohaloborane in organic synthesis. 1. Specific ortho substitution reaction of anilines. J. Am. Chem. Soc. 1978, 100, 4842–4852. [Google Scholar] [CrossRef]
- Canete, A.; Melendrez, M.X.; Saitz, C.; Zanocco, A.L. Synthesis of aminonaphthalene derivatives using the Bucherer reaction under microwave irradiation. Synth. Commun. 2001, 31, 2143–2148. [Google Scholar] [CrossRef]
- Sartori, G.; Maggi, R. Use of solid catalysts in Friedel−Crafts acylation reactions. Chem. Rev. 2006, 106, 1077–1104. [Google Scholar] [CrossRef] [PubMed]
- (a) González-Morales, A.; Díaz-Coutiño, D.; Fernánez-Zertuche, M.; García-Barradas, O.; Ordóñez, M. Preparation of dimethyl (R)- and (S)-2-(2-aminophenyl)-2-hydroxyethylphosphonate from anthranilic acid. Tetrahedron: Asymmetry 2004, 15, 457–463. (b) Zhao, Y.; Huang, B.; Yang, C.; Chen, Q.; Xia, W. Sunlight-driven forging of amide/ester bonds from three independent components: an approach to carbamates. Org. Lett. 2016, 18(21), 5572–5575.
- Brink, M. “Long range”—kopplungen in einigen acetonylderivaten. Tetrahedron Lett. 1971, 29, 2753–2756. [Google Scholar] [CrossRef]
Scheme 1.
Retrosynthetic analysis for the synthesis of 14-aza-12-oxasteroids A.
Scheme 2.
Reagents and conditions: (i) (NH4)2SO3, aq. NH3, 100 psi, 150 °C (ii) (a) R'CN, BCl3, AlCl3, inert, toluene; (b) 1.0 M HCl; (iii) NaBH4, MeOH, THF, inert condition; (iv) Keto-acids [levulinic acid / 2-carboxybenzaldehyde / 4-acetylbutyric acid / (2-oxopropyl sulfanyl)-acetic acid], toluene, reflux, Dean-Stark distillation.
Scheme 2.
Reagents and conditions: (i) (NH4)2SO3, aq. NH3, 100 psi, 150 °C (ii) (a) R'CN, BCl3, AlCl3, inert, toluene; (b) 1.0 M HCl; (iii) NaBH4, MeOH, THF, inert condition; (iv) Keto-acids [levulinic acid / 2-carboxybenzaldehyde / 4-acetylbutyric acid / (2-oxopropyl sulfanyl)-acetic acid], toluene, reflux, Dean-Stark distillation.
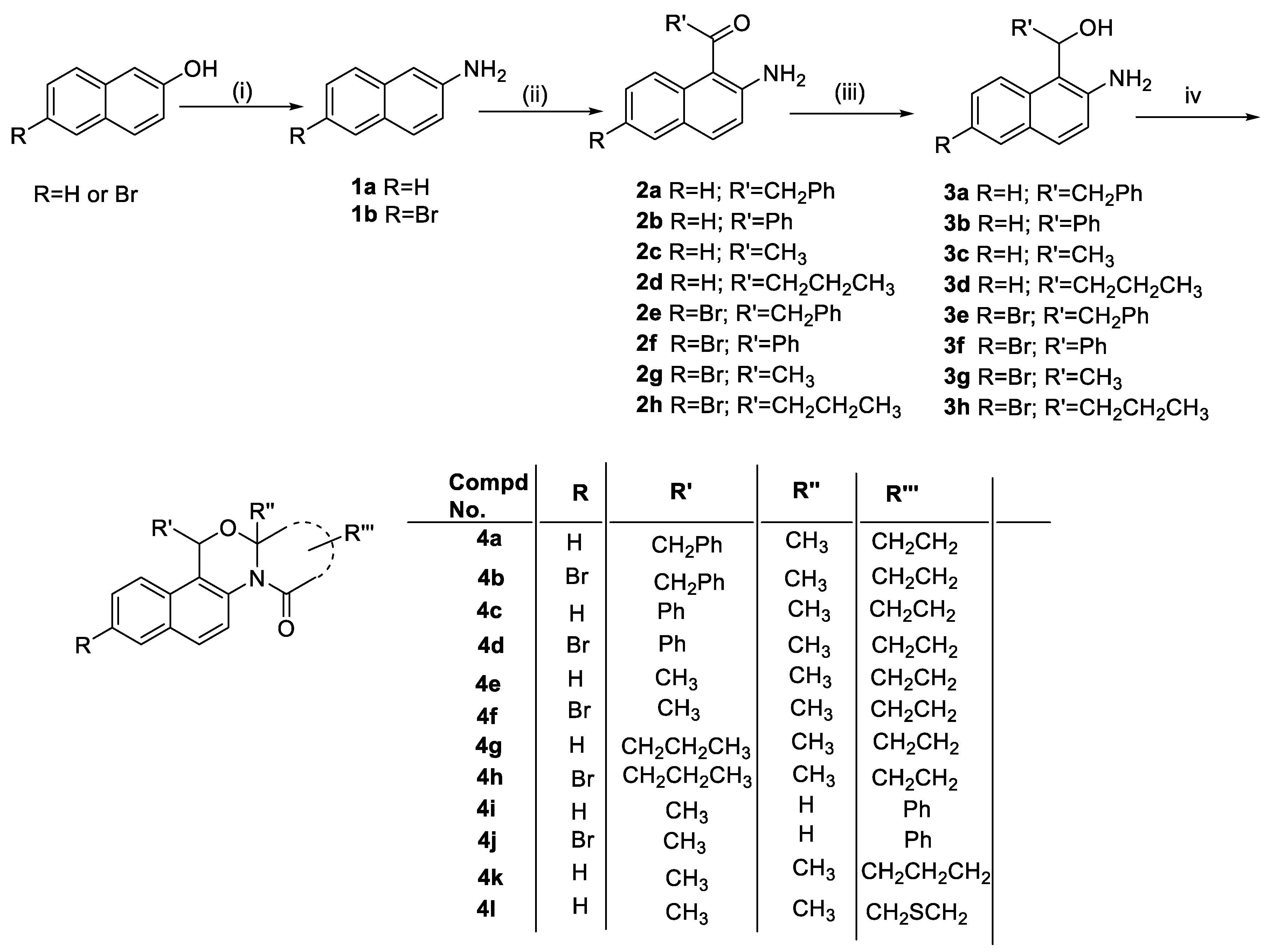
Scheme 3.
A plausible mechanism for synthesis of 14-aza-12-oxasteroids considering synthesis of 4c as the representative example.
Scheme 3.
A plausible mechanism for synthesis of 14-aza-12-oxasteroids considering synthesis of 4c as the representative example.
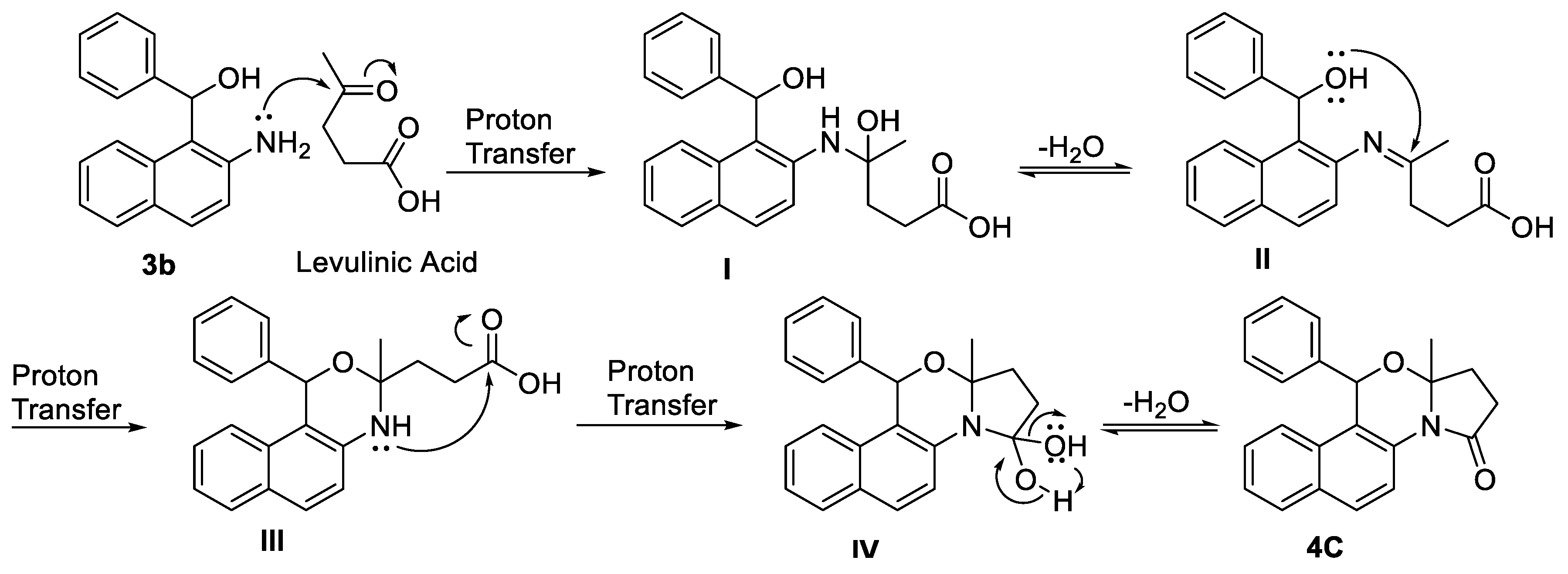
Figure 2.
(a) Ortep diagram, of compound 4c drawn in 50% thermal probability ellipsoids; (b) chemical structure of 4c with similar numbering pattern as X-ray crystal structure.
Figure 2.
(a) Ortep diagram, of compound 4c drawn in 50% thermal probability ellipsoids; (b) chemical structure of 4c with similar numbering pattern as X-ray crystal structure.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



