
Submitted:
20 November 2024
Posted:
20 November 2024
You are already at the latest version
Abstract
The vascular endothelial growth factor (VEGF) family includes key mediators of vasculogenesis and angiogenesis. VEGFs are secreted by various cells of epithelial and mesenchymal origin and by some immune cells in response to physiological and pathological stimuli. In addition, immune cells express VEGF receptors and/or coreceptors and can respond to VEGFs in an autocrine or paracrine manner. This immunological role of VEGFs has opened the possibility to use the VEGF inhibitors already developed to inhibit tumor angiogenesis also in combination approaches with different immunotherapies to enhance the action of effector T lymphocytes against tumor cells. This review aims at analyzing the current knowledge on the crosstalk between VEGFs and the immune system and at highlighting those aspects that still need to be further investigated.
Keywords:
1. Introduction
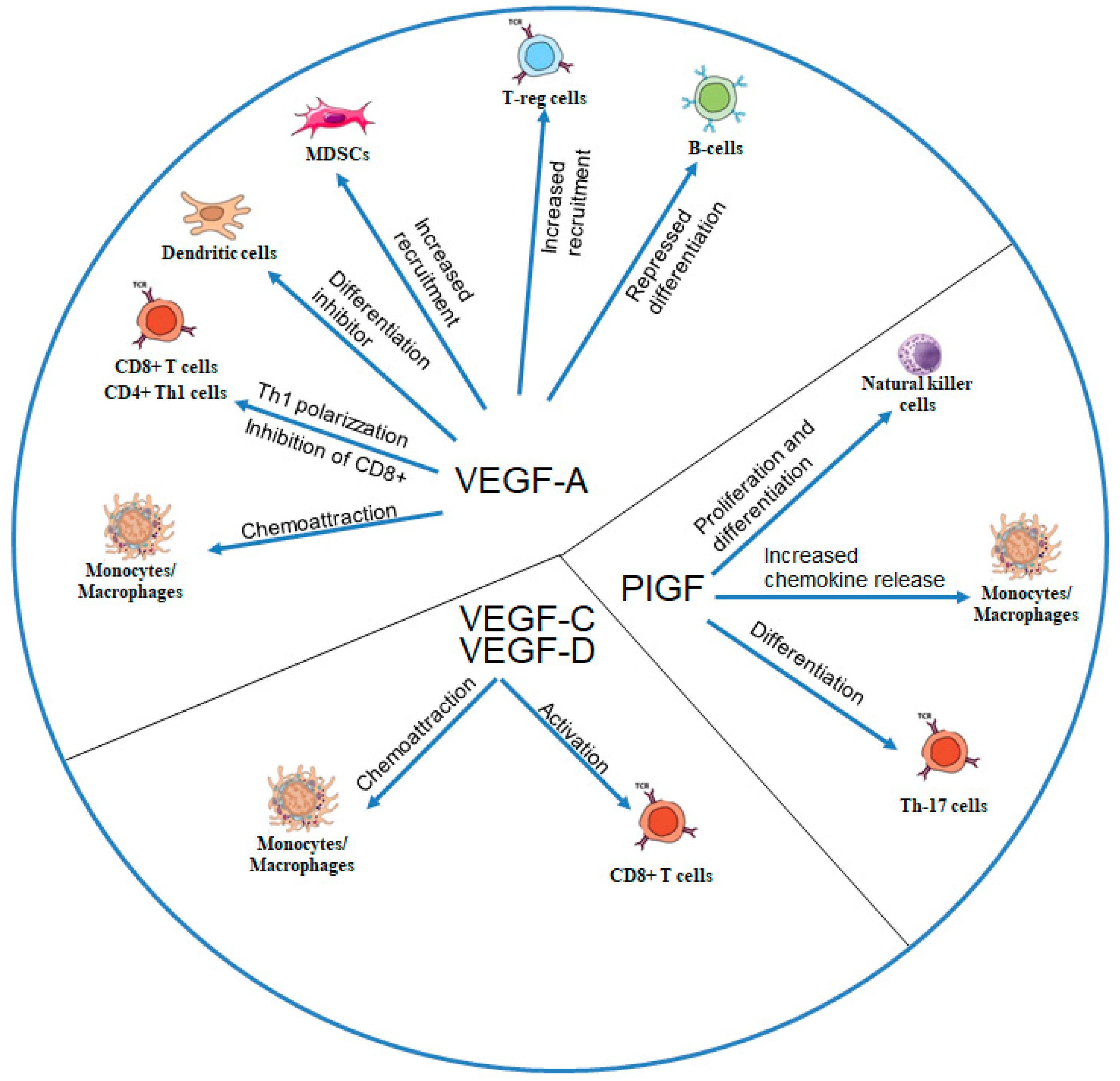
2. VEGF Family Members and Immune Cells
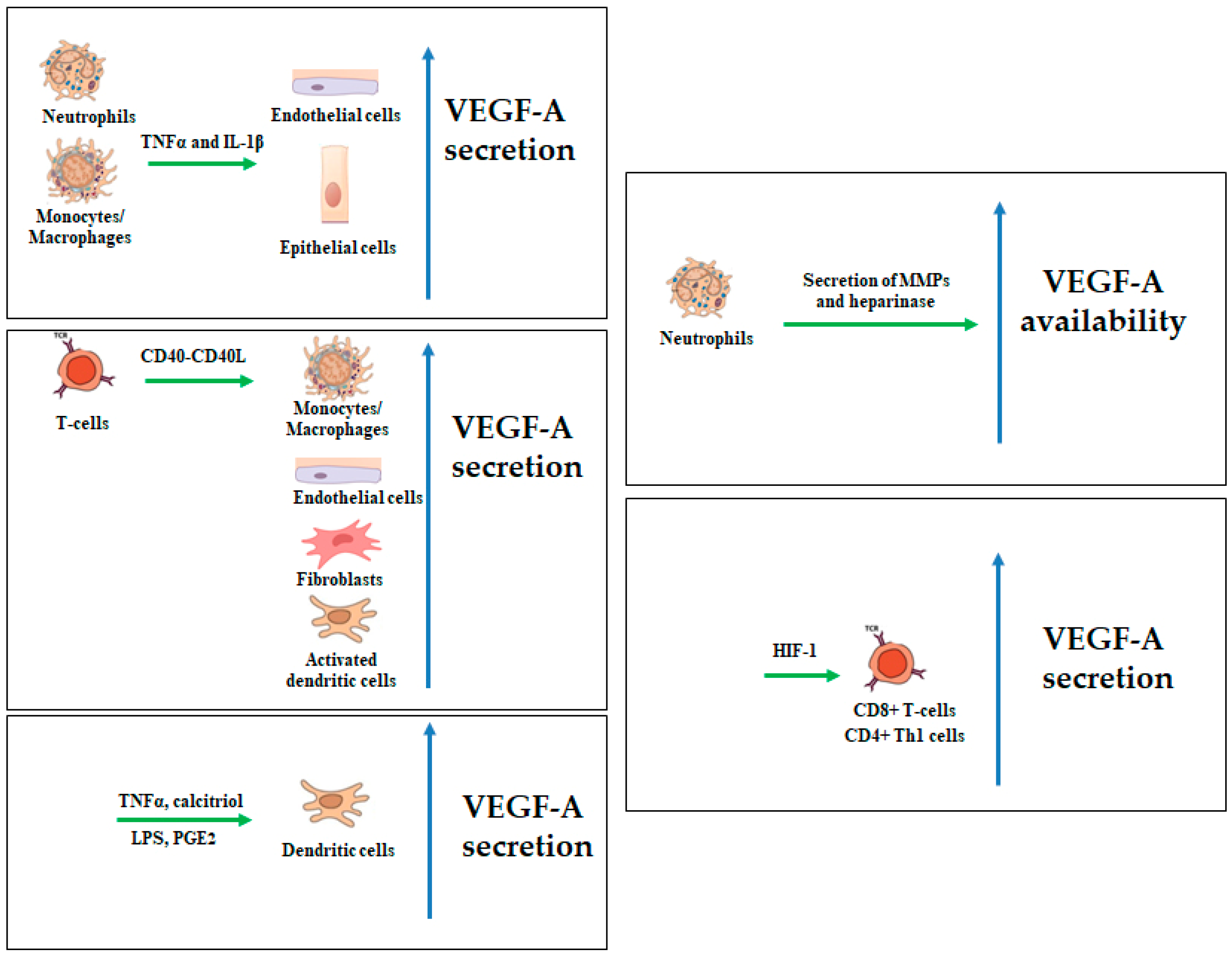
2.1. VEGF-A
2.2. VEGF-B and PlGF
2.3. VEGF-C and VEGF-D
3. Receptors of the VEGF Family and Their Expression on IMMUNE cells
3.1. VEGFR-1
3.2. VEGFR-2.
3.3. VEGFR-3
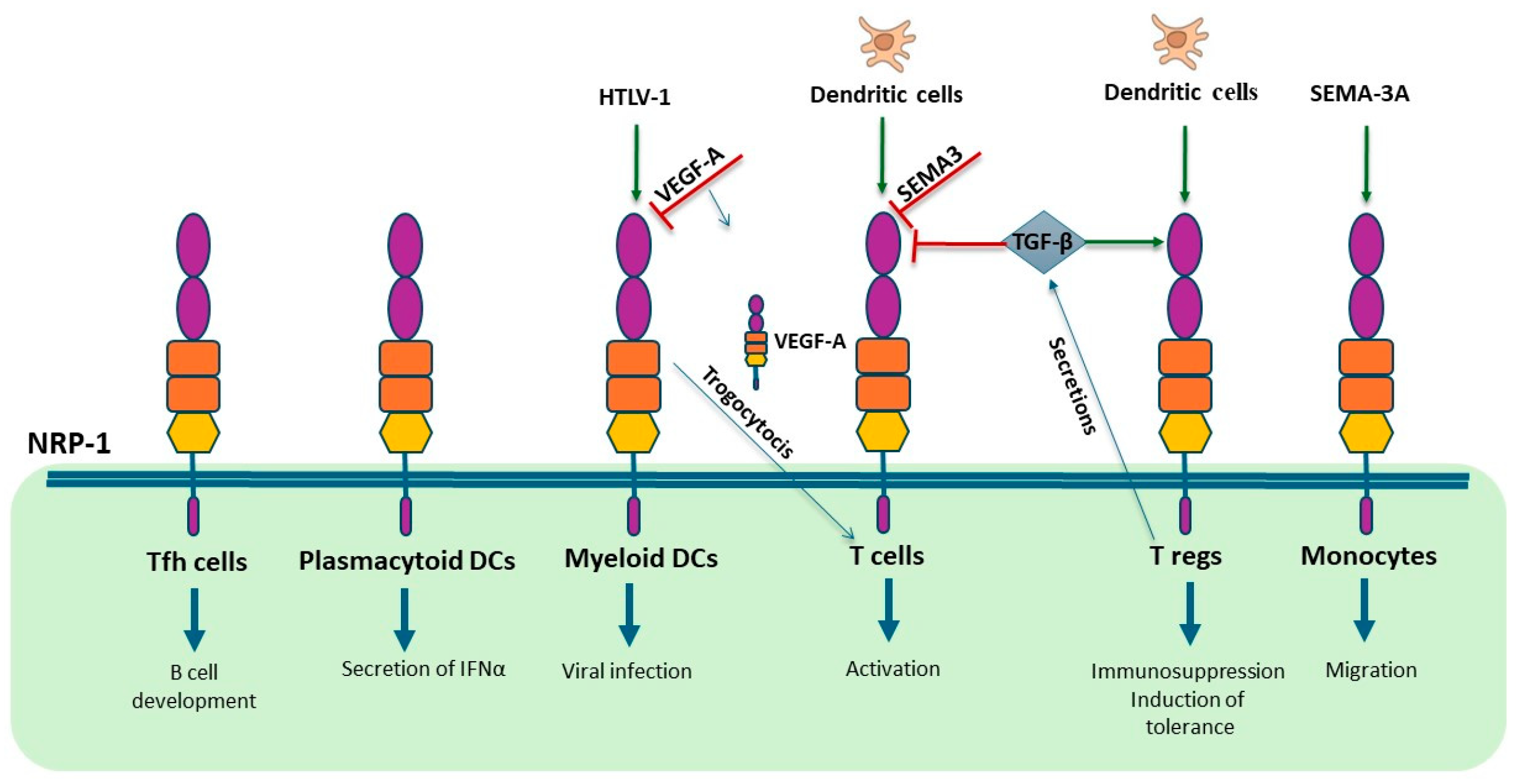
3.4. NRP-1 and NRP-2
4. Clinical Advancements and Challenges in Combining VEGF-Targeted Anti-Angiogenic Therapy with Checkpoint Inhibitor Immunotherapy

| Clinical trial ID | Study phase | Agent(s) | Anti-Angiogenic target | Anti-tumor immunity target | Cancer types | Ref. |
|---|---|---|---|---|---|---|
| NCT00790010 | I | Bevacizumab + Ipilimumab | VEGF-A | CTLA-4 | Melanoma | [80] |
| NCT01950390 | II | Bevacizumab + Ipilimumab | VEGF-A | CTLA-4 | Melanoma | [80] |
| NCT01633970 | I | Bevacizumab + Atezolizumab | VEGF-A | PD-L1 | RCC | [75] |
| NCT01984242 | II | Bevacizumab + Atezolizumab vs Sunitinib | VEGF-A, VEGFR | PD-L1 | RCC | [13,76] |
| NCT02420821 | III | Bevacizumab + Atezolizumab vs Sunitinib | VEGF-A, VEGFR | PD-L1 | RCC | [81,82] |
| NCT02231749 | III | Nivolumab + Ipilimumab vs Sunitinib | VEGFR, PDGFR | PD-1, CTLA-4 | RCC | [83,84] |
| NCT02493751 | I | Axitinib + Avelumab | VEGFR | PD-L1 | RCC | [85,86] |
| NCT02684006 | III | Axitinib + Avelumab vs Sunitinib | VEGFR, PDGFR | PD-L1 | RCC | [77,78] |
| NCT02724878 | II | Bevacizumab + Atezolizumab | VEGF-A | PD-L1 | RCC | [87] |
| NCT02853331 | III | Pembrolizumab + Axitinib vs Sunitinib | VEGFR, PDGFR | PD-1 | RCC | [81,88] |
| NCT02811861 | III | Pembrolizumab + Lenvatinib vs Sunitinib | VEGFR, PDGFR | PD-1 | RCC | [79] |
| NCT03721653 | II | Bevacizumab + Atezolizumab + FOLFOXIRI | VEGF-A | PD-L1 | CRC | [89,90] |
| NCT03434379 | II | Bevacizumab + Atezolizumab | VEGF-A | PD-L1 | HCC | [91,92] |
| NCT03006926 | I | Lenvatinib + Pembrolizumab | VEGFR, PDGFR | PD-1 | HCC | [93,94] |
| NCT03713593 | III | Lenvatinib + Pembrolizumab vs Lenvatinib | VEGFR, PDGFR | PD-1 | HCC | [95] |
| NCT02873962 | II | Bevacizumab + Nivolumab | VEGF-A | PD-1 | OC | [96] |
| NCT03038100 | III | Bevacizumab + Atezolizumab and Chemiotherapy | VEGF-A | PD-L1 | OC | [97] |
| NCT03170960 | I | Cabozantinib + Atezolizumab | VEGFR | PD-L1 | UC, RCC, NSCLC, HCC, | [98,99] |
| NCT02366143 | III | Atezolizumab + Bevacizumab + Paclitaxel/Carboplatin | VEGF-A | PD-L1 | NSCLC | [100] |
| NCT02443324 | I | Ramucirumab + Pembrolizumab | VEGFR | PD-1 | G/GEJ, NSCLC, UC, BTC | [101] |
| NCT02572687 | I | Ramucirumab + Durvalumab | VEGFR | PD-L1 | NSCLC, G/GEJ, HCC | [102] |
| NCT02856425 | I | Nintedanib + Pembrolizumab | PDGFR, VEGFR | PD-1 | Advanced solid tumors | [103] |
| NCT03377023 | I/II | Nintedanib + Nivolumab + Ipilimumab | PDGFR, VEGFR | PD-1, CTLA-4 | NSCLC | [104] |
5. Innovative Approaches Targeting VEGFs and Tumor-Mediated Immune Suppression
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Venkatakrishnan, G.; Parvathi, V.D. Decoding the mechanism of vascular morphogenesis to explore future prospects in targeted tumor therapy. Medical Oncology 2022, 39, 178. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Pezzella, F. Overview on the Different Patterns of Tumor Vascularization. Cells 2021, 10, 639. [Google Scholar] [CrossRef]
- Lorenc, P.; Sikorska, A.; Molenda, S.; Guzniczak, N.; Dams-Kozlowska, H.; Florczak, A. Physiological and tumor-associated angiogenesis: Key factors and therapy targeting VEGF/VEGFR pathway. Biomed Pharmacother 2024, 180, 117585. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.; LeCourter, J. The biology of VEGF and its receptors. Nat Med 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Reconciling VEGF With VPF: The Importance of Increased Vascular Permeability for Stroma Formation in Tumors, Healing Wounds, and Chronic Inflammation. Front Cell Dev Biol 2021, 9, 660609. [Google Scholar] [CrossRef]
- Pérez-Gutiérrez, L.; Ferrara, N. Biology and therapeutic targeting of vascular endothelial growth factor A. Nat Rev Mol Cell Biol 2023, 24, 816–834. [Google Scholar] [CrossRef]
- Ahmad, A.; Nawaz, M.I. Molecular mechanism of VEGF and its role in pathological angiogenesis. J Cell Biochem 2022, 123, 1938–1965. [Google Scholar] [CrossRef]
- Deng, J.; Qin, Y. Advancements and emerging trends in ophthalmic anti-VEGF therapy: a bibliometric analysis. Int Ophthalmol 2024, 44, 368. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Yang, F.; Lee, G.; Fan, Y. Navigating tumor angiogenesis: therapeutic perspectives and myeloid cell regulation mechanism. Angiogenesis 2024, 27, 333–349. [Google Scholar] [CrossRef]
- Voron, T.; Marcheteau, E.; Pernot, S.; Colussi, O.; Tartour, E.; Taieb, J.; Terme, M. Control of the immune response by pro-angiogenic factors. Front Oncol 2014, 4, 70. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol 2018, 9, 978. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Zeng, H.; Xu, Q.; Wang, J.; Xu, X.; Luo, J.; Zhang, L.; Luo, C.; Ying, J.; Li, J. The effect of anti-PD-1/PD-L1 antibodies combined with VEGF receptor tyrosine kinase inhibitors versus bevacizumab in unresectable hepatocellular carcinoma. Front Immunol 2023, 14, 1073133. [Google Scholar] [CrossRef]
- Cébe-Suarez, S.; Zehnder-Fjallman, A.; Ballmer-Hofer, K. The role of VEGF receptors in angiogenesis; complex partnerships. Cell Mol Life Sci 2006, 63, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. VEGF-VEGFR System as a Target for Suppressing Inflammation and other Diseases. Endocr Metab Immune Disord Drug Targets 2015, 15, 135–144. [Google Scholar] [CrossRef]
- Barleon, B.; Sozzani, S.; Zhou, D.; Weich, H.A.; Mantovani, A.; Marmé, D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 1996, 87, 3336–3343. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, M.; Palle, J.; Galy-Fauroux, I.; Terme, M. Direct and Indirect Modulation of T Cells by VEGF-A Counteracted by Anti-Angiogenic Treatment. Frontiers in Immunology 2021, 12. [Google Scholar] [CrossRef]
- Mor, F.; Quintana, F.J.; Cohen, I.R. Angiogenesis-inflammation cross-talk: vascular endothelial growth factor is secreted by activated T cells and induces Th1 polarization. J Immunol 2004, 172, 4618–4623. [Google Scholar] [CrossRef]
- Sozzani, S.; Rusnati, M.; Riboldi, E.; Mitola, S.; Presta, M. Dendritic cell-endothelial cell cross-talk in angiogenesis. Trends Immunol 2007, 28, 385–392. [Google Scholar] [CrossRef]
- Kong, X.; Bu, J.; Chen, J.; Ni, B.; Fu, B.; Zhou, F.; Pang, S.; Zhang, J.; Xu, S.; He, C. PIGF and Flt-1 on the surface of macrophages induces the production of TGF-β1 by polarized tumor-associated macrophages to promote lung cancer angiogenesis. Eur J Pharmacol 2021, 912, 174550. [Google Scholar] [CrossRef]
- Tayade, C.; Hilchie, D.; He, H.; Fang, Y.; Moons, L.; Carmeliet, P.; Foster, R.A.; Croy, B.A. Genetic depletion of placenta growth factor in mice alters uterine NK cells. J Immunol 2007, 178, 4267–4275. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.-A.; Kim, M.; Kang, M.-C.; Kong, J.-S.; Kim, K.-M.; Lee, S.; Hong, B.-K.; Jeong, G.H.; Lee, J.; Shin, M.-G.; et al. Placental growth factor regulates the generation of T H 17 cells to link angiogenesis with autoimmunity. Nat Immunol 2019, 20, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.; Zhang, T.; Chen, X.; Cao, W.; Lin, L.; Du, L.; Wang, Y.; Zhou, F.; He, X.; He, Y.; et al. Steroids Enable Mesenchymal Stromal Cells to Promote CD8(+) T Cell Proliferation Via VEGF-C. Adv Sci (Weinh) 2021, 8, 2003712. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Esumi, H. Reciprocal regulation between nitric oxide and vascular endothelial growth factor in angiogenesis. Acta Biochim Pol 2003, 50, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Li, H.; Liu, Y.; Li, Z. Regulation of VEGF-A expression and VEGF-A-targeted therapy in malignant tumors. J Cancer Res Clin Oncol 2024, 150, 221. [Google Scholar] [CrossRef]
- Melter, M.; Reinders, M.E.; Sho, M.; Pal, S.; Geehan, C.; Denton, M.D.; Mukhopadhyay, D.; Briscoe, D.M. Ligation of CD40 induces the expression of vascular endothelial growth factor by endothelial cells and monocytes and promotes angiogenesis in vivo. Blood 2000, 96, 3801–3808. [Google Scholar] [CrossRef]
- Cho, C.S.; Cho, M.L.; Min, S.Y.; Kim, W.U.; Min, D.J.; Lee, S.S.; Park, S.H.; Choe, J.; Kim, H.Y. CD40 engagement on synovial fibroblast up-regulates production of vascular endothelial growth factor. J Immunol 2000, 164, 5055–5061. [Google Scholar] [CrossRef]
- Tan, K.W.; Chong, S.Z.; Wong, F.H.; Evrard, M.; Tan, S.M.; Keeble, J.; Kemeny, D.M.; Ng, L.G.; Abastado, J.P.; Angeli, V. Neutrophils contribute to inflammatory lymphangiogenesis by increasing VEGF-A bioavailability and secreting VEGF-D. Blood 2013, 122, 3666–3677. [Google Scholar] [CrossRef]
- Perelman, N.; Selvaraj, S.K.; Batra, S.; Luck, L.R.; Erdreich-Epstein, A.; Coates, T.D.; Kalra, V.K.; Malik, P. Placenta growth factor activates monocytes and correlates with sickle cell disease severity. Blood 2003, 102, 1506–1514. [Google Scholar] [CrossRef]
- Selvaraj, S.K.; Giri, R.K.; Perelman, N.; Johnson, C.; Malik, P.; Kalra, V.K. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood 2003, 102, 1515–1524. [Google Scholar] [CrossRef]
- Parker, M.W.; Linkugel, A.D.; Goel, H.L.; Wu, T.; Mercurio, A.M.; Vander Kooi, C.W. Structural basis for VEGF-C binding to neuropilin-2 and sequestration by a soluble splice form. Structure 2015, 23, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.; Rossant, J.; Gertsenstein, M.; Breitman, M. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.; Zhang, L.; Bryce, D.; Peng, J. Increased hemangioblast commitment, not vascular disorganization, is the primary defect in flt-1 knock-out mice. Development 1999, 126, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Failla, C.M.; Carbo, M.; Morea, V. Positive and Negative Regulation of Angiogenesis by Soluble Vascular Endothelial Growth Factor Receptor-1. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Orecchia, A.; Lacal, P.M.; Schietroma, C.; Morea, V.; Zambruno, G.; Failla, C.M. Vascular endothelial growth factor receptor-1 is deposited in the extracellular matrix by endothelial cells and is a ligand for the integrin. J Cell Sci 2003, 116, 3479–3489. [Google Scholar] [CrossRef]
- Orecchia, A.; Mettouchi, A.; Uva, P.; Simon, G.C.; Arcelli, D.; Avitabile, S.; Ragone, G.; Meneguzzi, G.; Pfenninger, K.H.; Zambruno, G.; et al. Endothelial cell adhesion to soluble vascular endothelial growth factor receptor-1 triggers a cell dynamic and angiogenic phenotype. Faseb J 2014, 28, 692–704. [Google Scholar] [CrossRef]
- Osada, M.; Yamashita, A.; Akinaga, S.; Hosono, K.; Ito, Y.; Shibuya, M.; Asari, Y.; Amano, H. VEGFR1 TK signaling protects the lungs against LPS-induced injury by suppressing the activity of alveolar macrophages and enhancing the anti-inflammatory function of monocyte-derived macrophages. Toxicol Appl Pharmacol 2024, 492, 117083. [Google Scholar] [CrossRef]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.; Donners, M.M. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef]
- Lasser, S.A.; Ozbay Kurt, F.G.; Arkhypov, I.; Utikal, J.; Umansky, V. Myeloid-derived suppressor cells in cancer and cancer therapy. Nat Rev Clin Oncol 2024, 21, 147–164. [Google Scholar] [CrossRef]
- Meng, D.; Meng, M.; Luo, A.; Jing, X.; Wang, G.; Huang, S.; Luo, M.; Shao, S.; Zhao, X.; Liu, R. Effects of VEGFR1(+) hematopoietic progenitor cells on pre-metastatic niche formation and in vivo metastasis of breast cancer cells. J Cancer Res Clin Oncol 2019, 145, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Dalpati, N.; Rai, S.K.; Dash, S.P.; Kumar, P.; Singh, D.; Sarangi, P.P. Integrins α5β1 and αvβ3 Differentially Participate in the Recruitment and Reprogramming of Tumor-associated Macrophages in the In Vitro and In Vivo Models of Breast Tumor. J Immunol 2024. [Google Scholar] [CrossRef] [PubMed]
- Mimura, K.; Kono, K.; Takahashi, A.; Kawaguchi, Y.; Fujii, H. Vascular endothelial growth factor inhibits the function of human mature dendritic cells mediated by VEGF receptor-2. Cancer Immunol Immunother 2007, 56, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Dikov, M.M.; Ohm, J.E.; Ray, N.; Tchekneva, E.E.; Burlison, J.; Moghanaki, D.; Nadaf, S.; Carbone, D.P. Differential roles of vascular endothelial growth factor receptors 1 and 2 in dendritic cell differentiation. J Immunol 2005, 174, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Geindreau, M.; Ghiringhelli, F.; Bruchard, M. Vascular Endothelial Growth Factor, a Key Modulator of the Anti-Tumor Immune Response. International Journal of Molecular Sciences 2021, 22, 4871. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Meder, L.; Schuldt, P.; Thelen, M.; Schmitt, A.; Dietlein, F.; Klein, S.; Borchmann, S.; Wennhold, K.; Vlasic, I.; Oberbeck, S.; et al. Combined VEGF and PD-L1 Blockade Displays Synergistic Treatment Effects in an Autochthonous Mouse Model of Small Cell Lung Cancer. Cancer Res 2018, 78, 4270–4281. [Google Scholar] [CrossRef]
- Kim, C.G.; Jang, M.; Kim, Y.; Leem, G.; Kim, K.H.; Lee, H.; Kim, T.S.; Choi, S.J.; Kim, H.D.; Han, J.W.; et al. VEGF-A drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci Immunol 2019, 4. [Google Scholar] [CrossRef]
- Pavlakovic, H.; Becker, J.; Albuquerque, R.; Wilting, J.; Ambati, J. Soluble VEGFR-2: an antilymphangiogenic variant of VEGF receptors. Ann N Y Acad Sci 2010, 1207 Suppl 1, E7–15. [Google Scholar] [CrossRef]
- Kannan, S.; Rutkowski, J.M. VEGFR-3 signaling in macrophages: friend or foe in disease? Front Immunol 2024, 15, 1349500. [Google Scholar] [CrossRef]
- Maisel, K.; Hrusch, C.L.; Medellin, J.E.G.; Potin, L.; Chapel, D.B.; Nurmi, H.; Camacho, D.F.; Gleyzer, R.; Alitalo, K.; Sperling, A.I.; et al. Pro-lymphangiogenic VEGFR-3 signaling modulates memory T cell responses in allergic airway inflammation. Mucosal Immunol 2021, 14, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Ruhrberg, C. Neuropilin signalling in vessels, neurons and tumours. Semin Cell Dev Biol 2013, 24, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Valdembri, D.; Caswell, P.T.; Anderson, K.I.; Schwarz, J.P.; Konig, I.; Astanina, E.; Caccavari, F.; Norman, J.C.; Humphries, M.J.; Bussolino, F.; et al. Neuropilin-1/GIPC1 signaling regulates integrin traffic and function in endothelial cells. PLOS Biol 2009, 7, 115–132. [Google Scholar] [CrossRef]
- Prud'homme, G.J.; Glinka, Y. Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget 2012, 3, 921–939. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, M.; Gagnon, M.L.; Klagsbrun, M. Genomic organization of human neuropilin-1 and neuropilin-2 genes: identification and distribution of splice variants and soluble isoforms. Genomics 2000, 70, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Colotti, G.; Failla, C.M.; Lacal, P.M.; Ungarelli, M.; Ruffini, F.; Di Micco, P.; Orecchia, A.; Morea, V. Neuropilin-1 is required for endothelial cell adhesion to soluble vascular endothelial growth factor receptor 1. Febs j 2022, 289, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Bag, A.K.; Singh, R.K.; Talmadge, J.E.; Batra, S.K.; Datta, K. Multifaceted role of neuropilins in the immune system: potential targets for immunotherapy. Front Immunol 2017, 8, 1228. [Google Scholar] [CrossRef]
- Romeo, P.H.; Lemarchandel, V.; Tordjman, R. Neuropilin-1 in the immune system. Adv Exp Med Biol 2002, 515, 49–54. [Google Scholar] [CrossRef]
- Mendes-da-Cruz, D.A.; Lepelletier, Y.; Brignier, A.C.; Smaniotto, S.; Renand, A.; Milpied, P.; Dardenne, M.; Hermine, O.; Savino, W. Neuropilins, semaphorins, and their role in thymocyte development. Ann N Y Acad Sci 2009, 1153, 20–28. [Google Scholar] [CrossRef]
- Tordjman, R.; Lepelletier, Y.; Lemarchandel, V.; Cambot, M.; Gaulard, P.; Hermine, O.; Roméo, P.H. A neuronal receptor, neuropilin-1, is essential for the initiation of the primary immune response. Nat Immunol 2002, 3, 477–482. [Google Scholar] [CrossRef]
- Catalano, A.; Caprari, P.; Moretti, S.; Faronato, M.; Tamagnone, L.; Procopio, A. Semaphorin-3A is expressed by tumor cells and alters T-cell signal transduction and function. Blood 2006, 107, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.D.; Mueller, C.; Chae, W.J.; Alabanza, L.M.; Bynoe, M.S. Neuropilin-1 attenuates autoreactivity in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 2011, 108, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Bruder, D.; Probst-Kepper, M.; Westendorf, A.M.; Geffers, R.; Beissert, S.; Loser, K.; von Boehmer, H.; Buer, J.; Hansen, W. Neuropilin-1: a surface marker of regulatory T cells. Eur J Immunol 2004, 34, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Sarris, M.; Andersen, K.G.; Randox, F.; Mayr, L.; Betz, A.G. Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity 2008, 28, 402–413. [Google Scholar] [CrossRef]
- Renand, A.; Milpied, P.; Rossignol, J.; Bruneau, J.; Lemonnier, F.; Dussiot, M.; Coulon, S.; Hermine, O. Neuropilin-1 expression characterizes T follicular helper (Tfh) cells activated during B cell differentiation in human secondary lymphoid organs. PLoS One 2013, 8, e85589. [Google Scholar] [CrossRef]
- Grage-Griebenow, E.; Löseke, S.; Kauth, M.; Gehlhar, K.; Zawatzky, R.; Bufe, A. Anti-BDCA-4 (neuropilin-1) antibody can suppress virus-induced IFN-alpha production of plasmacytoid dendritic cells. Immunol Cell Biol 2007, 85, 383–390. [Google Scholar] [CrossRef]
- Bourbié-Vaudaine, S.; Blanchard, N.; Hivroz, C.; Roméo, P.H. Dendritic cells can turn CD4+ T lymphocytes into vascular endothelial growth factor-carrying cells by intercellular neuropilin-1 transfer. J Immunol 2006, 177, 1460–1469. [Google Scholar] [CrossRef]
- Lambert, S.; Bouttier, M.; Vassy, R.; Seigneuret, M.; Petrow-Sadowski, C.; Janvier, S.; Heveker, N.; Ruscetti, F.W.; Perret, G.; Jones, K.S.; et al. HTLV-1 uses HSPG and neuropilin-1 for entry by molecular mimicry of VEGF165. Blood 2009, 113, 5176–5185. [Google Scholar] [CrossRef]
- Carrer, A.; Moimas, S.; Zacchigna, S.; Pattarini, L.; Zentilin, L.; Ruozi, G.; Mano, M.; Sinigaglia, M.; Maione, F.; Serini, G.; et al. Neuropilin-1 identifies a subset of bone marrow Gr1- monocytes that can induce tumor vessel normalization and inhibit tumor growth. Cancer Res 2012, 72, 6371–6381. [Google Scholar] [CrossRef]
- Miyauchi, J.T.; Chen, D.; Choi, M.; Nissen, J.C.; Shroyer, K.R.; Djordevic, S.; Zachary, I.C.; Selwood, D.; Tsirka, S.E. Ablation of Neuropilin 1 from glioma-associated microglia and macrophages slows tumor progression. Oncotarget 2016, 7, 9801–9814. [Google Scholar] [CrossRef]
- Schellenburg, S.; Schulz, A.; Poitz, D.M.; Muders, M.H. Role of neuropilin-2 in the immune system. Mol Immunol 2017, 90, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Immunosuppressive effects of vascular endothelial growth factor. Oncol Lett 2022, 24, 369. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res 2014, 2, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hu, L.; Zhang, F.; Fang, M.; Xu, J.; Li, M.; Chen, Z. An investigative meta-analysis on the effectiveness and safety of integrating VEGF/VEGFR inhibitors with PD-1/PD-L1 inhibitors in cases with R/M HNSCC. Oral Oncol 2024, 153, 106814. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun 2016, 7, 12624. [Google Scholar] [CrossRef]
- Pal, S.K.; McDermott, D.F.; Atkins, M.B.; Escudier, B.; Rini, B.I.; Motzer, R.J.; Fong, L.; Joseph, R.W.; Oudard, S.; Ravaud, A.; et al. Patient-reported outcomes in a phase 2 study comparing atezolizumab alone or with bevacizumab vs sunitinib in previously untreated metastatic renal cell carcinoma. BJU Int 2020, 126, 73–82. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Motzer, R.J.; Rini, B.I.; Haanen, J.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Gravis-Mescam, G.; Uemura, M.; Lee, J.L.; et al. Updated efficacy results from the JAVELIN Renal 101 trial: first-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann Oncol 2020, 31, 1030–1039. [Google Scholar] [CrossRef]
- Motzer, R.J.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Alekseev, B.; Rha, S.Y.; Merchan, J.; Goh, J.C.; et al. Lenvatinib Plus Pembrolizumab Versus Sunitinib in First-Line Treatment of Advanced Renal Cell Carcinoma: Final Prespecified Overall Survival Analysis of CLEAR, a Phase III Study. J Clin Oncol 2024, 42, 1222–1228. [Google Scholar] [CrossRef]
- Wu, X.; Li, J.; Connolly, E.M.; Liao, X.; Ouyang, J.; Giobbie-Hurder, A.; Lawrence, D.; McDermott, D.; Murphy, G.; Zhou, J.; et al. Combined Anti-VEGF and Anti-CTLA-4 Therapy Elicits Humoral Immunity to Galectin-1 Which Is Associated with Favorable Clinical Outcomes. Cancer Immunol Res 2017, 5, 446–454. [Google Scholar] [CrossRef]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Alekseev, B.Y.; Lee, J.L.; Suarez, C.; Stroyakovskiy, D.; De Giorgi, U.; et al. Final Overall Survival and Molecular Analysis in IMmotion151, a Phase 3 Trial Comparing Atezolizumab Plus Bevacizumab vs Sunitinib in Patients With Previously Untreated Metastatic Renal Cell Carcinoma. JAMA Oncol 2022, 8, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Arén Frontera, O.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol 2019, 20, 1370–1385. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; McDermott, D.F.; Escudier, B.; Burotto, M.; Choueiri, T.K.; Hammers, H.J.; Barthélémy, P.; Plimack, E.R.; Porta, C.; George, S.; et al. Conditional survival and long-term efficacy with nivolumab plus ipilimumab versus sunitinib in patients with advanced renal cell carcinoma. Cancer 2022, 128, 2085–2097. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Larkin, J.; Oya, M.; Thistlethwaite, F.; Martignoni, M.; Nathan, P.; Powles, T.; McDermott, D.; Robbins, P.B.; Chism, D.D.; et al. Preliminary results for avelumab plus axitinib as first-line therapy in patients with advanced clear-cell renal-cell carcinoma (JAVELIN Renal 100): an open-label, dose-finding and dose-expansion, phase 1b trial. Lancet Oncol 2018, 19, 451–460. [Google Scholar] [CrossRef]
- Larkin, J.; Oya, M.; Martignoni, M.; Thistlethwaite, F.; Nathan, P.; Ornstein, M.C.; Powles, T.; Beckermann, K.E.; Balar, A.V.; McDermott, D.; et al. Avelumab Plus Axitinib as First-Line Therapy for Advanced Renal Cell Carcinoma: Long-Term Results from the JAVELIN Renal 100 Phase Ib Trial. Oncologist 2023, 28, 333–340. [Google Scholar] [CrossRef]
- McGregor, B.A.; McKay, R.R.; Braun, D.A.; Werner, L.; Gray, K.; Flaifel, A.; Signoretti, S.; Hirsch, M.S.; Steinharter, J.A.; Bakouny, Z.; et al. Results of a Multicenter Phase II Study of Atezolizumab and Bevacizumab for Patients With Metastatic Renal Cell Carcinoma With Variant Histology and/or Sarcomatoid Features. J Clin Oncol 2020, 38, 63–70. [Google Scholar] [CrossRef]
- Plimack, E.R.; Powles, T.; Stus, V.; Gafanov, R.; Nosov, D.; Waddell, T.; Alekseev, B.; Pouliot, F.; Melichar, B.; Soulières, D.; et al. Pembrolizumab Plus Axitinib Versus Sunitinib as First-line Treatment of Advanced Renal Cell Carcinoma: 43-month Follow-up of the Phase 3 KEYNOTE-426 Study. Eur Urol 2023, 84, 449–454. [Google Scholar] [CrossRef]
- Antoniotti, C.; Rossini, D.; Pietrantonio, F.; Catteau, A.; Salvatore, L.; Lonardi, S.; Boquet, I.; Tamberi, S.; Marmorino, F.; Moretto, R.; et al. Upfront FOLFOXIRI plus bevacizumab with or without atezolizumab in the treatment of patients with metastatic colorectal cancer (AtezoTRIBE): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol 2022, 23, 876–887. [Google Scholar] [CrossRef]
- Antoniotti, C.; Rossini, D.; Pietrantonio, F.; Salvatore, L.; Lonardi, S.; Tamberi, S.; Marmorino, F.; Moretto, R.; Prisciandaro, M.; Tamburini, E.; et al. Upfront Fluorouracil, Leucovorin, Oxaliplatin, and Irinotecan Plus Bevacizumab With or Without Atezolizumab for Patients With Metastatic Colorectal Cancer: Updated and Overall Survival Results of the ATEZOTRIBE Study. J Clin Oncol 2024, 42, 2637–2644. [Google Scholar] [CrossRef]
- Galle, P.R.; Finn, R.S.; Qin, S.; Ikeda, M.; Zhu, A.X.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.; et al. Patient-reported outcomes with atezolizumab plus bevacizumab versus sorafenib in patients with unresectable hepatocellular carcinoma (IMbrave150): an open-label, randomised, phase 3 trial. Lancet Oncol 2021, 22, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol 2022, 76, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase Ib Study of Lenvatinib Plus Pembrolizumab in Patients With Unresectable Hepatocellular Carcinoma. J Clin Oncol 2020, 38, 2960–2970. [Google Scholar] [CrossRef]
- Lee, M.M.P.; Chan, L.L.; Chan, S.L. The role of lenvatinib in the era of immunotherapy of hepatocellular carcinoma. J Liver Cancer 2023, 23, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kudo, M.; Merle, P.; Meyer, T.; Qin, S.; Ikeda, M.; Xu, R.; Edeline, J.; Ryoo, B.Y.; Ren, Z.; et al. Lenvatinib plus pembrolizumab versus lenvatinib plus placebo for advanced hepatocellular carcinoma (LEAP-002): a randomised, double-blind, phase 3 trial. Lancet Oncol 2023, 24, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Herold, C.; Gray, K.P.; Penson, R.T.; Horowitz, N.; Konstantinopoulos, P.A.; Castro, C.M.; Hill, S.J.; Curtis, J.; Luo, W.; et al. Assessment of Combined Nivolumab and Bevacizumab in Relapsed Ovarian Cancer: A Phase 2 Clinical Trial. JAMA Oncol 2019, 5, 1731–1738. [Google Scholar] [CrossRef]
- Moore, K.N.; Bookman, M.; Sehouli, J.; Miller, A.; Anderson, C.; Scambia, G.; Myers, T.; Taskiran, C.; Robison, K.; Mäenpää, J.; et al. Atezolizumab, Bevacizumab, and Chemotherapy for Newly Diagnosed Stage III or IV Ovarian Cancer: Placebo-Controlled Randomized Phase III Trial (IMagyn050/GOG 3015/ENGOT-OV39). J Clin Oncol 2021, 39, 1842–1855. [Google Scholar] [CrossRef]
- Pal, S.K.; McGregor, B.; Suárez, C.; Tsao, C.K.; Kelly, W.; Vaishampayan, U.; Pagliaro, L.; Maughan, B.L.; Loriot, Y.; Castellano, D.; et al. Cabozantinib in Combination With Atezolizumab for Advanced Renal Cell Carcinoma: Results From the COSMIC-021 Study. J Clin Oncol 2021, 39, 3725–3736. [Google Scholar] [CrossRef]
- Li, D.; Loriot, Y.; Burgoyne, A.M.; Cleary, J.M.; Santoro, A.; Lin, D.; Aix, S.P.; Garrido-Laguna, I.; Sudhagoni, R.; Guo, X.; et al. Cabozantinib plus atezolizumab in previously untreated advanced hepatocellular carcinoma and previously treated gastric cancer and gastroesophageal junction adenocarcinoma: results from two expansion cohorts of a multicentre, open-label, phase 1b trial (COSMIC-021). EClinicalMedicine 2024, 67, 102376. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Herbst, R.S.; Arkenau, H.T.; Santana-Davila, R.; Calvo, E.; Paz-Ares, L.; Cassier, P.A.; Bendell, J.; Penel, N.; Krebs, M.G.; Martin-Liberal, J.; et al. Ramucirumab plus pembrolizumab in patients with previously treated advanced non-small-cell lung cancer, gastro-oesophageal cancer, or urothelial carcinomas (JVDF): a multicohort, non-randomised, open-label, phase 1a/b trial. Lancet Oncol 2019, 20, 1109–1123. [Google Scholar] [CrossRef]
- Bang, Y.J.; Golan, T.; Dahan, L.; Fu, S.; Moreno, V.; Park, K.; Geva, R.; De Braud, F.; Wainberg, Z.A.; Reck, M.; et al. Ramucirumab and durvalumab for previously treated, advanced non-small-cell lung cancer, gastric/gastro-oesophageal junction adenocarcinoma, or hepatocellular carcinoma: An open-label, phase Ia/b study (JVDJ). Eur J Cancer 2020, 137, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Baldini, C.; Danlos, F.X.; Varga, A.; Texier, M.; Halse, H.; Mouraud, S.; Cassard, L.; Champiat, S.; Signolle, N.; Vuagnat, P.; et al. Safety, recommended dose, efficacy and immune correlates for nintedanib in combination with pembrolizumab in patients with advanced cancers. J Exp Clin Cancer Res 2022, 41, 217. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Wang, M. Prospect of immunotherapy combined with anti-angiogenic agents in patients with advanced non-small cell lung cancer. Cancer Manag Res 2019, 11, 7707–7719. [Google Scholar] [CrossRef]
- Lacal, P.M.; Atzori, M.G.; Ruffini, F.; Scimeca, M.; Bonanno, E.; Cicconi, R.; Mattei, M.; Bernardini, R.; D'Atri, S.; Tentori, L.; et al. Targeting the vascular endothelial growth factor receptor-1 by the monoclonal antibody D16F7 to increase the activity of immune checkpoint inhibitors against cutaneous melanoma. Pharmacol Res 2020, 159, 104957. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Gaucher, J.F.; Vidal, M.; Broussy, S. A Structural Overview of Vascular Endothelial Growth Factors Pharmacological Ligands: From Macromolecules to Designed Peptidomimetics. Molecules 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Deng, Q.; Chen, Z.; Yan, S.; Dong, Q.; Zhang, Y.; Cui, Y.; Li, L.; He, Y.; Shi, J. Recent development of VEGFR small molecule inhibitors as anticancer agents: A patent review (2021-2023). Bioorg Chem 2024, 146, 107278. [Google Scholar] [CrossRef] [PubMed]
- Mougel, A.; Méjean, F.; Tran, T.; Adimi, Y.; Galy-Fauroux, I.; Kaboré, C.; Mercier, E.; Urquia, P.; Terme, M.; Tartour, E.; et al. Synergistic effect of combining sunitinib with a peptide-based vaccine in cancer treatment after microenvironment remodeling. Oncoimmunology 2022, 11, 2110218. [Google Scholar] [CrossRef]
- Sasso, M.S.; Mitrousis, N.; Wang, Y.; Briquez, P.S.; Hauert, S.; Ishihara, J.; Hubbell, J.A.; Swartz, M.A. Lymphangiogenesis-inducing vaccines elicit potent and long-lasting T cell immunity against melanomas. Sci Adv 2021, 7. [Google Scholar] [CrossRef]
- Shamshiripour, P.; Hajiahmadi, F.; Lotfi, S.; Esmaeili, N.R.; Zare, A.; Akbarpour, M.; Ahmadvand, D. Next-Generation Anti-Angiogenic Therapies as a Future Prospect for Glioma Immunotherapy; From Bench to Bedside. Front Immunol 2022, 13, 859633. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



