
Submitted:
20 November 2024
Posted:
22 November 2024
You are already at the latest version
Abstract
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR), an exceptionally potent genome-editing technique developed in 2012, is the ideal tool of the future for treating diseases by permanently correcting deleterious base mutations or disrupting disease-causing genes with great precision and efficiency. However, it is prone to cleaving double-stranded DNA in off-target genes and generating random mutations in the process. These drawbacks restrict its application in fundamental research and agriculture, and raises safety concerns in the field of medicine. Fortunately, the new gene editing technology derived from CRISPR/Cas9, known as prime editing, has the potential to provide targeted sequence insertion, deletion, and transversion, all while avoiding the formation of double-strand breaks, thus minimizing adverse effects. Meanwhile, the rapid development of this technology makes its application wider and broader. This review summarizes the current developments and optimizations of the prime editing (PE) system with improved editing efficiency and precision. Along with discussing the most recent delivery techniques and outlining the PE applications that are being used both in vitro and in vivo.
Keywords:
Prime editing
; Gene editing
; Gene therapy
1. Introduction
Programmable nucleases consisting of sequence-tailored guide RNAs (gRNAs) and Cas9 endonucleases are powerful tools for genome editing. Yet, the repair of double-strand DNA breaks (DSB) by error-prone end-joining processes confers an intrinsically high mutagenic character to nuclease-based genome editing[1]. In contrast, Prime editing (PE) is referred to as a ‘search and replace’ technology which can install any single base-pair change and precise insertions or deletions (indels) at specific genomic sequences without DSB formation[2]. The PE complex consists of a fusion protein comprising a catalytically impaired Cas9 endonuclease (often referred to as "Cas9 nickase") and an engineered Moloney murine leukemia virus (M-MLV) reverse transcriptase enzyme with a specially designed RNA molecule, which guides PE, named the prime editing guide RNA (pegRNA). The fusion protein is capable of both recognizing the target DNA site and copying new genetic information and the desired genetic changes into the genome. The pegRNAs are comprised of a 20 nucleotides spacer sequence which informs the Cas9 which DNA locus to target, a primer binding site (PBS), and a reverse transcriptase template (RTT). The PBS sequence binds to the genomic ssDNA after it has been cleaved and allows the latter to act as a primer to begin the synthesis of a complementary DNA strand. The RTT functions as the template for the reverse transcriptase to synthesize the required complementary strand with the desired alterations. A precise substitution of any nucleotide with any other nucleotide is theoretically possible by pegRNA. This makes it possible to permanently alter the genome by correcting point mutations and adding, removing, or changing a few nucleotides. The broad editing spectrum of prime editing technology has the potential to address a wide range of genetic diseases. According to research and computational analyses, prime editing's precision and versatility could theoretically correct up to 89% of known human genetic diseases[2]. Collectively, PE offers precise and versatile editing capabilities with reduced reliance on double-strand breaks (DSBs) and donor DNA templates compared to traditional CRISPR-Cas9-based methods[2].
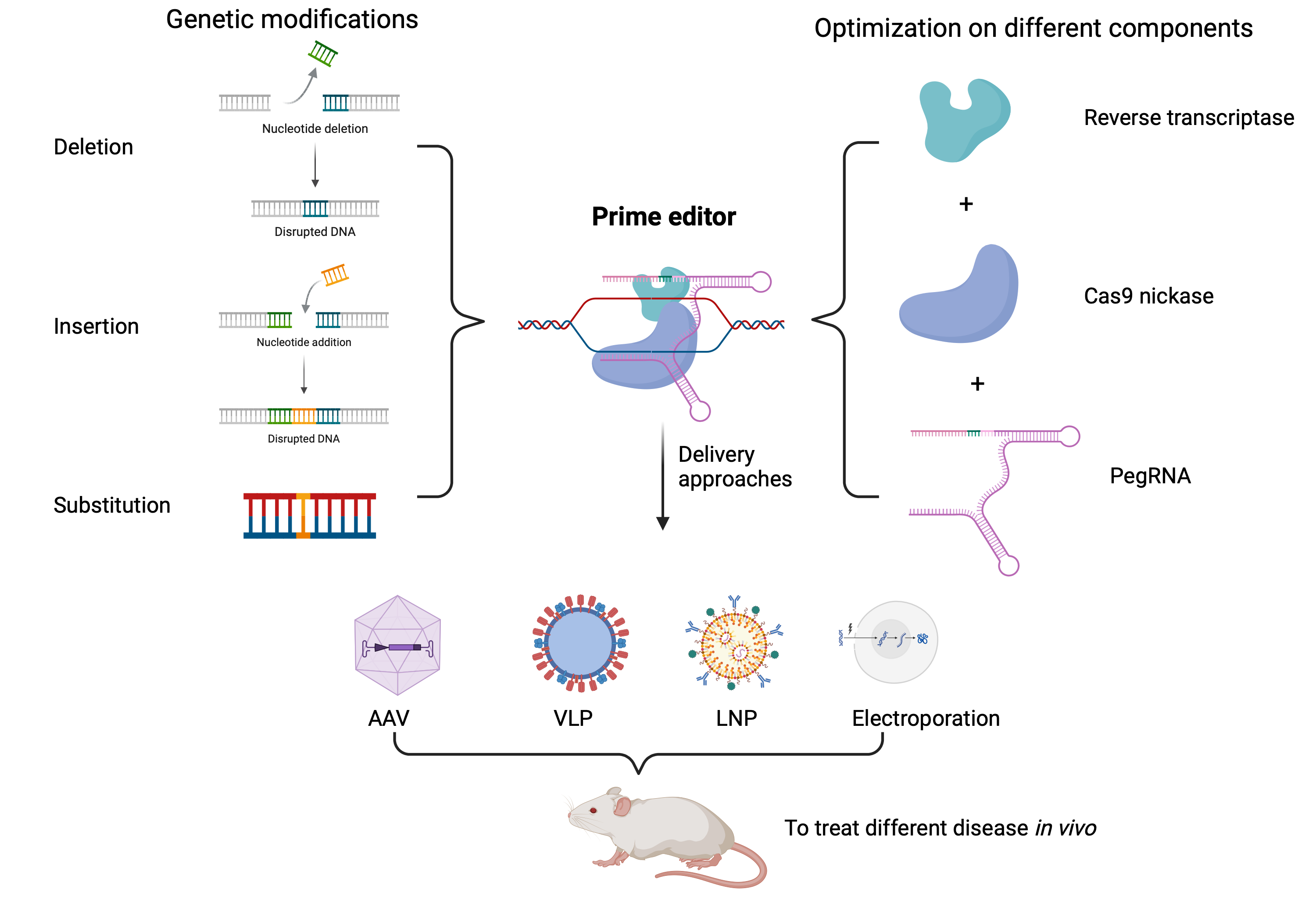
2. The Iteration of the Prime Editor from PE1 to PE7
PE1 system consists of three main elements: 1) a Cas9 nickase, which cuts a single DNA strand at a specific location, 2) a Reverse transcriptase (RT), which prepares the DNA containing the desired edition and 3) a pegRNA: an RNA which combines Cas9’s sgRNA sequence with the PBS and RTT. The PE2 system is identical to the PE1 system, with the exception that 5 mutations were added to the RT protein (D200N/L603W/T330P/T306K/W313F) to improve its activity, binding and thermostability [2]. The PE3 system is PE2 with an additional sgRNA to cause a ssDNA cut on the unedited strand. This favors the conservation of the edited strand compared to the PE2 system[2]. PE4 is based on PE2 but adds the MLH1dn gene, which encodes a mismatch repair inhibitory protein to increase the editing efficiency. However, it appears to be less successful with longer edits. The PE5 system is a combination of the PE3 system with MLH1dn to increase the gene editing efficiency. This is accomplished by protecting the edited sequence from the second round of modification[3]. Despite the various methods employed to achieve the appropriate editing efficiency in vitro, the large size of this system results in low in vivo efficiency. To solve this problem, PE6 was developed by using phage-assisted evolution to choose advantageous mutations in the Cas9 and RT domains. This led to the development of different RT-based prime editors, which were named PE6a to PE6d and which offered improvements in editor size (PE6a and b) and RT activity (PE6c and d). Furthermore, to gain a higher editing efficiency in mammalian cells using the PE system, several single mutations were added to the Cas9. Thus, PE6e-g were based on using various evolved and engineered Cas9 variants[4]. PE7 is the PEmax system (an optimized version of PE2: Cas9 mutations, nuclear localization, linker composition, and codon usage) fused to a truncated La protein, a small RNA-binding exonuclease protection factor, which is a 408-residue protein that consists of a highly conserved La motif, two RNA recognition motifs (RRM1 and RRM2) and a flexible region with a nuclear localization signal (NLS) at the C terminus. Fusing La protein to PEmax in multiple positions (that is the N terminus, the C terminus or between Cas9 nickase and MMLV-RT) improved intended editing efficiencies. The one which fuses the La N-terminal protein (1–194), containing the La motif and RRM1, into the C terminus of the PEmax system, is the most efficient and is named PE7 [5]. The modifications of all above prime editors are shown in Figure 1. At the same time, the summary of prime editor is shown in Table 1.
3. Optimization of the Editing Range of Prime Editing
3.1. Insertion and Integration
Prime editing was first developed to precisely rewrite short sequences of the genome. As a result, it experienced difficulties with longer edits. To explore what was contributing to this difficulty, Koeppel et al. [6] created a library of 3,604 sequences of varied lengths and analyzed their insertion frequency into four genomic locations belonging to three different human cell lines. This was done using different prime editor systems in varying DNA repair site situations. The results demonstrated that the length of the insertion sequence, nucleotide composition and secondary structure affected insertion rates. Moreover, they also discovered that the 3′ flap nucleases TREX1 and TREX2 suppress the insertion of longer sequences. They theorized that the nucleases degraded the 3’ flap intermediates (containing the insertion) before they could be integrated into the genome. Based on those modifications, they conclude that the pegRNA sequences between 15 and 21 nt could insert at higher rates than shorter ones in mismatch repair (MMR) proficient cells, and for the longer sequence over 30 nt, the 3′ flap nucleases TREX1 and TREX2 are the important factors.
As many hereditary diseases result from genes which contain mutations that are greater than one nucleotide in scope, the capacity to insert large DNA fragments is a requirement for the fields of gene-based therapies and genome engineering. GRAND editing, based on the PE3 system, uses two pegRNAs to insert DNA fragments larger than 400 bp, but the efficiency remained low [7]. Twin-PE, based on a similar methodology and developed by David Liu's group successfully inserted 5.6 kb attP-containing DNA donor plasmid into the genomes of human cells at AAVS1, CCR5 and ALB gene with 6.8%, 6.1% and 2.6% efficiency, respectively [8]. Afterwards, Zheng et al. [9] developed a Retrotransposon-like template jump prime editing (TJ) PE, which enabled them to insert the GFP gene (~800 bp) into recipient cells. The principle of this technology was motivated by the effective genomic insertion process of retrotransposons, which can replicate their genetic material into chromosomal DNA using target-primed reverse transcription (TPRT). In this study, the authors designed TJ-pegRNA harboring the insertion sequence as well as two primer binding sites (PBSs), with one PBS matching a nicking sgRNA site. Once the first template was inserted into the DNA, the second PBS on the DNA flap binds with the nick site generated by nicking sgRNA. Thus, the first inserted sequence will be recognized as the template for the second prime editing reaction to make the large insertion exon rewriting happen. Until now, all the above methods have not yet been applied to in vivo experiments. More recently, David Liu’s lab created Prime Assisted Site-Specific Integrase Gene Editing (PASSIGE)[10], a method for site-specific integration of large DNA genes (exceeding 10 kb), and its optimized version, eePASSIGE. This method shared the same underlying ideas of the technology named Programmable Addition via Site-Specific Targeting Elements (PASTE)[11]. Both PASSIGE and PASTE use the PE system and large serine integrases (LSRs), also called recombinases, to efficiently integrate foreign DNA into the genomes of eukaryotic cells. These two tools are based on using the programmability of the PE system to first write a short DNA sequence (< 50 bp) at a specific location in the genome, a recognition sequence (attB or attP) for a particular integrase, which generates the integration by delivering a template DNA that contains the matching recognition sequence (attP or attB) as well as the integrase.
Besides the above experiments conducted on human cells, Sun et al. [12] developed the Third-generation PrimeRoot editors which enable precise DNA insertion of 11.1 kb into plant genomes. In this system, they rely on the use of site-specific recombinases (SSRs) to produce massive insertions. During the recombination process, SSRs must first identify certain sequences called recombinase sites (RSs). Based on this mechanism, they link the DNA fragment onto the RSs so that it can be inserted into the plant genome by the RSs link to SSRs.
3.2. Deletion
To generate deletions using PE systems, two pegRNAs are used to target different strands. Choi et al. [13] reported that PRIME-Del can remove big fragments up to 10 kb with an editing efficiency of 1–30%. Put simply, they used PE2 with two pegRNAs to target different sites instead of delivering the PE3 system. Once the PE2 created a nick in the two desired sites, the two RTTs eliminated the portion that lay between the two sites. This result showed that in contrast with the CRISPR-Cas9/double sgRNAs technology, PRIME Del was more accurate and flexible in targeted genomic sequence deletion, marker labelling, and genomic rearrangement. Similarly, TwinPE,a method that uses a prime editor protein (PE2) and two prime editing guide RNAs (pegRNAs) for the programmable replacement or excision of DNA sequences at endogenous human genomic sites [8], can be used for deletion. The Single-anchor TwinPE was used to remove exon 51 (780 bp) of Duchenne muscular dystrophy (DMD) in HEK293T cells, with an average editing efficiency of 28% [8]. Furthermore, Jiang et al. [14] also developed a PE-Cas9-based deletion and repair (PEDAR) method for large fragment deletion (up to 10 kb). In this strategy, instead of using Cas9 nickase, they chose to link active Cas9 with RT to create PE-Cas9. Thus, under the direction of two pegRNAs that recognize both complementary DNA strands, PE-Cas9 can create two DSBs and remove the DNA fragment between the two cut locations.
3.3. Transversion
For transversion, base editing and prime editing are feasible for generating precise transversion in the genome DNA. However, base editing requires different enzymes fused with spCas9 for the different amino transversion, which is less precise than prime editing. Fortunately, with prime editing, it is possible to do all 12 types of transversion and any other types of substitution by providing the desired template sequence. However, the efficiency is highly dependent on the lengths of the PBS and RTT, and the position of the nicking sgRNA [2]. For each target site, it is necessary to test several pegRNAs with various combinations of RTT and PBS sequence lengths. The PBS and RTT often fall between 6-16 nt and 8-23 nt, respectively. There is no rule for designing the pegRNA to generate the highest efficiency for each gene. Thus, for each transversion modification, the different combinations of pegRNA need to be validated before further modifications. However, inverting large sequences is required to correct many human pathogenic alleles which contain multiple mutation points or large structure variants. Twin-PE mentioned above was shown to inverse a target sequence about 40 kb long which caused hunter disease by combining it with recombinase Bxb1 attB and attP[8].
4. Optimization of Prime Editor
4.1. Optimization of Cas Nickase
The optimization of Cas nickase has been accomplished by following four different paths: 1) Cas9 PAM requirements have been altered to expand the scope of sequences which can be targeted by using variants of SpCas9, 2) different sources of Cas9 have been used such as Staphylococcus aureus Cas9 (saCas9) to change both PAM requirements and packaging size, 3) different classes of Cas proteins (Cas12) have been used for similar goals and 4) mutation in Cas9 to increase the editing activity and reduce the indels. For the first aspect, as Lu et al. [15] summarized before, depending on the different variants of Cas9 protein, the required sequence of PAM is variable. Indeed, Kweon et al. [16] created PE2 variations utilizing several SpCas9 variants, including PE2-VQR, PE2-VRQR, PE2-NG, PE2-SpG, and PE2-SpRY. The PE2-SpRY can target 94.4% of pathogenic variants, thus, it is also named PAM-less prime editor.
The Cas9 nickase which is most frequently used for prime editing is the SpCas9 which comes from Streptococcus pyogenes. However, Cas9 can also be isolated from different species of bacteria. Thus, there are many others, such as Staphylococcus aureus (SaCas9), Streptococcus thermophilus (StCas9), Neisseria meningitidis (NmCas9), Francisella novicida (FnCas9), Campylobacter jejuni (CjCas9), and Streptococcus canis (ScCas9). More recently, a Cas9 variant was identified in Staphylococcus auricularis (SauriCas9) (3.3kb) which recognizes a 5’-NNGG-3’ PAM sequence, like CjCas9, has high editing activity, and is small enough for delivery by a single AAV[17]. For prime editing, Lan et al. [18] reported Mini-PE, which combines truncated MMLV RT with Campylobacter jejuni Cas9 (CjCas9; H559A). The truncated MMLV RT has 21 N-terminal amino acids (AA) and 200 C-terminal AA deleted, leaving 467 AA (two-thirds of the original RT). The total size of the mini-PE was lowered to 4.5 kb, which is suitable for recombinant AAV packaging[18]. However, the editing efficiency was low and should be the factor to be considered. David Liu’s lab[19] compared using SaCas9 and SpCas9 nickase based PE system, and they found that the one based on SpCas9 had higher efficiency than SaCas9.
Moreover, there are different types of nucleases based CRISPR/Cas systems, such as Cas12 and Cas13, and each one contains their own subtypes [20,21]. In combination with the prime editor, Liang et al [22] developed circular RNA-mediated prime editor (CPE) systems based on using Cas12 instead of using Cas9. This system preferentially detects T-rich genomic areas and has the potential for multiplexing.
Furthermore, some studies [23,24] showed that the nCas9 (H840 and D10A) occasionally generated DSB. Lee et al[23] tested 14 mutation combinations in nCas9 in 15 loci of HEK293T cells to avoid the formation of DSB, increase the activity of nCas9, and reduce unwanted editing. By combining H840A and N863A mutations, they abolished the function of the HNH domain, leading to single-strand breaks exclusively in the non-target strand. This decreased the frequency of indels while maintaining the same prime editing efficiency as the PE3 system.
Aside from that, Chauhan et al [25] developed the vPE system, which inserted four mutations in Cas9 nickase (R221K-K848A-H982A-N1317R) into a recently efficiency-boosting PE7 architecture. Among those modifications, K848A-H982A intended to extend the length of the 5' released from the nicking site, increasing its resection and reducing competition with the RTT template. R221K and N1317R are designed to boost the activity of Cas9 nickase.
4.2. Optimization of RT
The M-MLV RT employed in PE1 to PE5 is one of the most studied RTs. It consists of five domains. The palm (1-40, 125-159, 193-275 AA), finger (41-124, 160-192 AA) and thumb (276-361 AA) domains make up the DNA polymerase domain (1-361 AA). These domains play different roles in substrate binding, stability, and polymerase activity. The DNA polymerase domain is linked by a connection domain (362-496) to the RNase H domain (497-671) that degrades the RNA in RNA/DNA heteroduplex during DNA synthesis[26].
Several mutations have been found to improve the processivity, thermostability and substrate affinity of the M-MLV RT [27]. However, their combination in the PE needs to be tested and optimized for the targeted cells [26,28]. Anzalone et al.[2] screened multiple mutation combinations to create the pentamutant RT that was tested in the PE2 to PE5 systems. Chen et al. [29] codon-optimized this pentamutant for human cells in the process of developing the PEmax architecture. Similarly, Gao et al. [30] increased the expression level and editing efficiency by human codon-optimizing the RT using different algorithms.
Furthermore, since the loading capacity of the vector limits the delivery of the PE systems in vivo, many attempts have been made to minimize the size of the RT by trying different truncations of the M-MLV RT or using smaller orthologs that would yield equivalent editing efficiency. Although prior research showed that mutations in the RNase H domain can negatively affect the RT processivity and accuracy [27,31], its removal in the PE can lead to equivalent editing. Moreover, this domain interacts with peptidyl release factor 1 (eRF1), which recognizes a stop codon and recruits other termination factors [26,32], which prevents the binding of peptidyl release factor 3 (eRF3), promoting the stop codon readthrough and blocking the mRNA degradation by the non-sense mediated decay. Thus, Zheng et al.[33] suggested that prime editing could be safer if the RNase H domain is removed. After experimenting with several truncations, they discovered that the Δ497 truncation, which eliminates the RNase H domain, preserves an editing efficiency comparable to PE3 for substitutions and small insertions or deletions. Similarly, Böck et al.[34] observed similar editing efficiency between PE2 and PE2ΔRNase H in HEK293T for different substitutions in HEK293T cells. Damon et al.[28] also obtained similar editing with PE2 and PE2ΔRNaseH. However, when tested on complex editing types, such as long insertions or deletions that require TwinPE, the editing efficiency was, on average, 1.4-fold lower compared to the full-length PE2. Likewise, Lan et al.[35] found different truncations that produce editing comparable to PE2 while trying to make a mini-PE small enough to fit in a single AAV. The smallest one, which removes the first 21 and last 200 amino acids of the RT, combined with a compact CjCas9, can be delivered in vivo with a single AAV but with low editing efficiency. It has been previously described that the 23 first AA of the M-MLV RT are dispensable for its enzymatic function. Additionally, different truncations were tried using the plant prime editor (PPE) by Zong et al. [36]. The removal of the RNase H domain increased the editing by 2 folds for nucleotide substitutions at various sites in rice and wheat protoplast[36]. Overall, these truncations significantly reduce the PE size and offer additional splitting options for delivery in small vectors like AAV[28,30,33].
The connection domain links the RNase H to the polymerase, and some residues on its surface interact with the RNA template. Zheng et al.[33] removed part of the connection domain with Δ474 truncation, and this reduced the editing efficiency at some sites. The Δ418 truncation or the total removal of this domain resulted in low editing. Similarly, Zong et al.[36] eliminated the connection domain in the PPE and observed no editing. Gao et al.[30] had comparable editing with the Δ471 truncation and PE2 human codon-optimized in HEK293T cells for A to T substitution or CTT insertions. Examining further truncation, they discovered that they could eliminate an extra 60 bp at the N-terminal and 90 bp at the C-terminal without compromising the editing efficiency at those sites.
While working on different configurations for the PE, Grunewald et al.[37] discovered that an untethered RT and nSpCas9 had comparable editing efficiency to PE2. They hypothesized that instead of acting in cis on the fusion protein, the RT might act in trans from a molecule in solution. Their group used the split PE to screen smaller RT orthologues rapidly. They optimized the Marathon-RT from Eubacterium rectal but had 3 folds less editing for various edits in HEK293T cells compared to the full-length pentamutant M-MLV RT. Comparable editing between PE2 and a split PE2 was also reported by Liu et al.[38]. They co-transfected cells with PE2 mutants that were either defective for the M-MLV RT or nCas9 activity and had no editing. These results suggest that the RT in the PE, in opposition to a split PE, do not function in trans, probably due to local molecular clashes. They also tried 2 human codon-optimized bacterial RTs but had lower editing and higher indels compared to PE3.
To compare different RT orthologues, Doman et al.[28] tested 20 different RT orthologues. The results showed that the wild-type forms and all structure-based engineering ones had lower editing compared to PE2, especially the smallest ones. After that, they used the phage-assisted continuous evolution (PACE) to optimize the Escherichia coli Ec48 retrotransposon (1.2 kb) and Schizosaccharomyces pombe Tf1 retrotransposon (1.5kb) RT, named PE6a and PE6b, generated the editing efficiency averaging 80% of PEmax and giving comparable or better editing compared to PEmax depending on the edit tested respectively. However, both PE6a and PE6b are less efficient for long, complex edits and with structured RTT or PBS. To solve this problem, they added rational mutations to the evolved Tf1 to make PE6c. Also, they optimized the M-MLV RT with PACE to generate an RT of the same size as PEmax ΔRNaseH called PE6d. Both optimizations are more efficient with structured RTT or PBS and long edits that require twinPE. However, PE6d is not well suited for small prime edits.
4.3. Optimization of pegRNA
The design, stability, editing position of the pegRNA from the Cas9 nicking site, and auto-inhibitory interaction between PBS and the spacer sequence of pegRNA limited the use of PE systems. Thus, the most straightforward optimization method for improving the editing capability of the PE system is pegRNA modification.
Many methods have been developed to redesign the pegRNA, taking into account the assessment of off-target mutations, the support of various Cas variants, and the optimization variables for sequence design. The majority of these tools have been summarized in the reference review article[39]. This year, Park et al [40] reported a new web-based tool for pegRNA design and evaluation, which is used for saturation genome editing named SynDesign (https://deepcrispr.info/SynDesign). Besides, Mathis et al [41] developed PRIDICT 2.0, which evaluates the effectiveness of pegRNAs for all edit types up to 15 bp in length in primary cells in vivo as well as in cell lines with and without mismatch repair (https://www.pridict.it/epridict).
The pegRNA consists of a sgRNA and the addition of an unstructured RNA sequence to the 3'end of the sgRNA, which makes it unstable. To avoid degradation, David Liu's lab developed the engineered pegRNA (epegRNA), which has a structured RNA motif (two stable pseudoknots: prequeosine1-1 riboswitch aptamer (evopreQ1) or the frameshifting pseudoknot from Moloney murine leukemia virus (MMLV) hereafter named "mpknot") 3′ of the PBS that stops the pegRNA extension from degrading and editing-incompetent PE complexes from forming that fight for access to the desired genomic location. This method increased prime editing efficiency by 3-4-fold in many cell lines compared to unmodified pegRNA. At the same time, the authors tried to remove the linker between pseudoknots and pegRNA, and the result showed the linker is necessary to maintain the increased the editing efficiency. To further reduce the misfolding of the pegRNA, they trimmed the unnecessary sequence about 5nt from the evopreQ1 (TevopreQ1). The efficiency of different modifications (3nt insertion, nucleotide transversion) on different genes was increased from 3.8 to 6.8-fold [42]. Similarly, to reduce the degradation of the 3′-extended portion of pegRNAs, Zhang et al. [43] added a viral exoribonuclease-resistant RNAs (xrRNAs) motif, a group of conserved structures found in flaviviruses to the 3'end of pegRNA to protect the stability and activity. In this study, five different xrRNAs (Murray Valley encephalitis (MVE), West Nile virus (WNV), Zika, Dengue (Dengue), and Yellow Fever (YF) were tested to alter the pegRNA stability for base conversion in 293T cells. Among them, the 3' Zika xrRNA motif yields the optimal improvement. Instead of using those xrRNAs, another study added different RNA aptamer like stem-loops M2, PP7, Csy4, and BoxB to the 3'end of pegRNA to avoid its degradation. The editing efficiency of that modified pegRNA was compared to the ePE-Mpknot, ePE-EvopreQ1 at RUNX1 gene site (+5 G·C to T·A) in HEK293FT cells. The results demonstrated that none of the aptamer modifications generated higher efficiency than ePE-Mpknot and ePE-EvopreQ1. The aptamer-modified pegRNAs were split into two parts, i.e., the sgRNA and the Prime RNA (pRNA: PBS, RTT, aptamer), to increase the flexibility of the PE system, named split pegRNA prime editors (SnPEs). A Tornado circRNA system expression system was used to construct the circular prime RNA to stabilize the pRNA. However, the editing efficiency was lower than that of the classical PE system in most cases[44].
Furthermore, the editing efficiency was affected by the type of editing and the editing distance from the Cas9 nicking site. As the previous findings showed, modifications made to the invariant GG nucleotides of the PAM sequence for Cas9 (positions +5–6 relative to the dsDNA break) typically produced stronger phenotypes than other positions, with positions +1-4 remaining somewhat effective and positions +7–20 generally less effective, especially after the +16 position [45]. Meanwhile, the author also proposed that the location of the target sequence in a gene alters the editing efficiency. For example, the editing efficiency is higher if the editing site is located at the beginning of a gene due to it being more disruptive than other positions.
In addition, to decreasing the effect of the self-inhibition caused by the circularization structure of pegRNA, Ponnienselvan et al.[46] demonstrated that pegRNA binding and target recognition efficiency are impacted by the auto-inhibitory interaction that occurs between the spacer sequence and PBS. Thus, they demonstrated that reducing the complementarity between the PBS and spacer sequences could increase gene editing efficiency, which was achieved by reducing the length of PBS and conducting a transient cold shock before treating the cells. Similarly, to solve the same problem, Zhang et al.[47] applied three-point mutations to the 3' PBS region of the pegRNAs to prevent the misfolding of pegRNA and used a heat denaturation followed by slow cooling to refold the pegRNA structure. By conducting those strategies in zebrafish for generating the base substitution, the gene editing efficiency was increased up to 24.7 folds compared to unmodified pegRNA. In addition, Li et al [48] introduced same-sense mutations at the appropriate locations in the RTT to increase the PE-editing efficiency on average 353-fold. They also modified the secondary structure of the pegRNA to increase the stability of small hairpin by inserting the G/C pair. This modification was validated by using the PE3 system to generate a small indel and the efficiency was increased up to 10.6 folds.
5. PE Delivery Strategies
Ensuring the safe and effective delivery of gene editing components into the cell nucleus remains a challenge due to the presence of negatively charged proteins and nucleic acids, particularly the macro-protein prime editor. In general, delivery techniques can be divided into viral and non-viral delivery. While viral vectors were extensively employed in labs and clinical trials to administer genome editing treatments, the translation of these therapies was severely impeded by the possible immunogenicity associated with viral vectors and their potential random integration. Non-viral delivery methods, however, are offered as a potential solution. These vectors can be natural or artificially synthesized and have fewer safety concerns, although they have a somewhat lower delivery efficiency than viral delivery systems. However, the PE is 6.3 kb long for the coding sequence, and the pegRNA is negatively charged, which increases the delivery challenge to generate an efficient gene alteration in vivo.
5.1. Viral Vectors
5.1.1. Adeno-Associated Viruses (AAV)
AAV is a non-enveloped, replication-defective virus that does not cause disease in humans. It has a capacity of around 4.7 kb, which is much smaller than the size of the PE system. To address this limitation, the most well-known delivery method for PE is the dual AAV system, which separates the PE system into two parts and modifies the PE component with an intein, a segment protein that allows the fusion of two separated parts to form a complete protein by forming a peptide bond, and subsequently generating the gene editing function. Therefore, the studies related to the use of dual AAV systems focused on three parts: 1) comparing the splicing activity of inteins, 2) choosing the split site of the PE system, and 3) using a smaller size RT. To choose an intein to generate a higher efficiency, Pinto et al.[49] compared the splicing activity of 34 inteins to induce the fusion of two parts of the mCherry protein. Ten inteins out of all those orthologs were successfully used in vivo and in vitro. The results proposed that the reaction condition, the junction sequence, and protein expression level affect the intein efficiency. She et al. [50] compared the inteins Rhodothermus marinus (Rma) and Nostoc punctiform (Npu) to split the PE system at 11 different split sites. The Rma generated higher gene editing efficiency in most split sites.
Except for inteins, the split sites within SpCas9 are an important factor that alters the gene editing efficiency. She et al. [50] reported that the best on-target editing efficiency was obtained with a PE split located before amino acid 1105 (Ser) of SpCas9 and the use of the Rma intein. Zhi et al.[51] suggested that the PE split sites can only be within a small region from 990 (Asn) to 1050 (Ile). The split-PE-1005 and 1024 have higher editing efficiency with the Rma intein, especially Split-PE-1024 shows a higher editing efficiency with plasmid transfection in HEK293T cells. Davis et al.[19] proposed a similar split at the 1024 site of SpCas9, which allowed for therapeutically relevant editing in the liver (up to 46%), heart (up to 11%), and brain (up to 42% efficiency in the cortex) of mice. However, dual-vector designs are less efficient because the intein recombination system is only partially controlled. To prevent split issues that could impact Cas9 activity, Grünewald et al.[37] transfected the entire SpCas9 nickase (Cas9n) and RT independently. Gao et al.[52] reported a truncated reverse transcriptase which removed the RnaseH domain to insert 3 nt into the PCSK9 gene in mice, which were packaged into AAV8 with a total of 2 × 1012 vector genome (vg) injected by tail vein. The insertion efficiency reached 13.5% after 4 weeks. Meanwhile, Doman [4] developed a PE6 system with a smaller RT, the size is 270 bp smaller than the RnaseH version with several mutations to increase RT activity. This system was used to mediate large single-flap insertions in vivo, which generated an average of 40% and 62% loxP insertion in bulk and transduced cells.
5.1.2. Adenovector Particles (AdVPs)
Wang et al.[53] successfully used adenovector particles (AdVPs) to deliver the full length of prime editors (PE2 and PE3) to correct the Duchenne muscular dystrophy (DMD) gene in vitro. AdVPs containing the PE system were used to treat myoblasts and mesenchymal stem cells and had editing efficiencies of 80% and 64%, respectively. They also compared the delivery of PE2 and PE3 by AdVPs. The results demonstrated that the PE3 generated higher gene editing efficiency with fewer indels than PE2 delivery. Moreover, this strategy was further used to delete a DMD exon in myotubes produced by the fusion of human myoblasts by delivering PE2 and two pegRNAs. Dystrophin expression was detected in the AdVPs treated group. Collectively, this study provides a new method to deliver the full-length PE system. However, the immunogenicity and toxicity caused by AdVPs need to be considered when using them to deliver an in vivo PE system.
5.1.3. Helper-Dependent Adenovirus (HDAd)
Delivering the multicomponent structure of a PE system requires that the gene transfer vector has a big capacity. To deliver PEmax, a viral vector which could deliver over 16 kb is needed. However, the current lentiviral and recombinant adeno-associated virus (AAV) vectors can only carry payloads of 8 kb and 4.5 kb. Li [54] reported that they use the helper-dependent adenoviral (HDAd), a vector devoid of all viral coding sequences, which has a payload capacity of 35 kb to effectively transduce a broad range of cell types from different species irrespective of the cell cycle, and this led to long-term transgene expression. This strategy was used to deliver the prime editor into a sickle mouse model with corrections of over 40% for hemoglobin S alleles in vivo [55].
5.2. Non-Viral Vectors
5.2.1. Lipid Nanoparticles
Chen et al.[56] successfully reached 13% editing of the Pcsk9 gene, which encodes proprotein convertase subtilisin/kexin type 9, an enzyme that destroys low-density lipoprotein receptors (LDLRs). The immunodeficient mice were treated with lipid nanoparticles (LNP) containing PE components by retro-orbital intravenous injection. The authors sought to insert a 4 bp (TTAC) sequence into the Pcsk9 gene to introduce a premature termination codon and thereby shift the reading frame, thereby inactivating the gene. Inactivating the Pcsk9 gene is a preferred method for treating hypercholesterolemia since it lowers LDL-cholesterol (LDL-C) levels without causing negative side effects.
5.2.2. Virus-like Particles (VLPs)
More recently, David Liu’s lab [57] has applied a new method, virus-like particles (VLP), to deliver the full-length PE. Instead of using PE fused with Gag protein (which permitted the insertion of 26 PE molecules per VLP), they optimized the system by adding a P3–P4 coiled-coil peptide pair to the Gag protein and to the PE protein. Separating the GAG and PE allowed them to avoid the large size of the fusion protein (309 kDa) which was thought to hinder the VLP production. This strategy increased the gene editing efficiency for different mutations and reached 47% average editing among GFP+ nuclei in mouse brains by neonatal Intracerebroventricular injection.
6. Prime Editing in Therapeutic Applications
6.1. Creation of Pathogenic Cell Lines and Organoids
To study diseases in vitro, it is helpful to have a cell line or an organoid with a pathogenic mutation. Prime editing can install this mutation, allowing researchers to study the impact of the disease or of the treatment. For example, Schene et al. created a liver organoid containing the D482G, A>G mutation in the ABCB11 gene in 20% of cells to study bile salt export pump deficiency [58]. The Cerebrofrontofacial Baraitser-Winter syndrome and coloboma (a congenital malformation of one or more ocular structures) are caused by mutations in the ACTB gene, encoding β-actin. Using the prime editing technique, Binder et al. succeeded in creating pluripotent stem cells with the c.640G > T PTC (premature termination codons) mutation in exon 4 of the ACTB gene, making it possible to examine the molecular and functional consequences of the deficiency[59]. Geurts et al. used PE to generate intestinal organoids presenting either the F508del mutation or the R785* mutation in the CFTR (cystic fibrosis transmembrane conductance regulator) gene, two mutations responsible for cystic fibrosis [60]. Pancreatic adenocarcinoma was also studied by installing the G12V mutation in the KRAS gene of the zebrafish genome. However, off-target mutations were observed due to sequence homology [61]. In some patients with liver cancer, the CTNNB1 gene, which encodes β-catenin, has a deletion of 6 nucleotides. The introduction of this pathogenic mutation into a liver organoid was carried out by prime editing with success in 30% of the cells [62]. Hou et al. worked on X-linked severe combined immunodeficiency (X-SCID). This disease is caused by mutations in the interleukin-2 receptor gamma (IL2RG) gene. Prime editing was used to generate an in vitro disease model by introducing the c.458T>C mutation of the IL2RG gene, responsible for the disease, into T lymphocytes from healthy donors. Using this model, the authors were then able to correct 26% of the mutated cells [63].
6.2. Creation of Pathogenic Animal Models
After studying the edition or disease mechanisms in vitro, animal models recreate in vivo conditions to evaluate the effect of a mutation or the impact of a treatment on the phenotype. Lin et al. [60] generated a mouse model suffering from cataracts by deleting a nucleotide (G) in exon 3 of the CRYGC (crystallin gamma C) gene. This was accomplished through the microinjection of plasmids encoding the prime editing components into mouse embryos and obtained 38.2% nucleotide deletion as well as a nuclear cataract phenotype[64]. The P302L mutation in the TYR gene, encoding tyrosinase which controls melanin synthesis, is responsible for oculocutaneous albinism. This mutation was inserted by prime editing into zebrafish with a frequency of 3.33%, and the mutation was transmitted to 8.3% of the next generation[61]. Salem et al. installed in mice the S50A mutation in the CAPN2 gene, which encodes calpain 2, a ubiquitous protein with functions in the central nervous system and in the heart. In patients with pulmonary hypertension, serine at position 50 of the protein is hyperphosphorylated. The authors, therefore, developed this model to be able to study the impact of the mutation on the disease [65]. Qian et al. generated a rabbit model of Tay-Sachs disease, characterized by an accumulation of GM2 gangliosides in the nervous system. In 80% of cases, it is due to an insertion of four bases (TATC) in exon 11 of the HEXA gene encoding hexosaminidase A, causing a deficiency of the enzyme[66,67].
6.3. Correcting Mutations In Vitro
Prime editing is also used to engineer a treatment that would allow the exact correction of pathogenic mutations. This was first tested in vitro or ex vivo in cell lines. Alpha thalassemia is often caused by the CD142 mutation (UAA > CAA) in the HBA2 gene encoding the alpha 2 subunit of hemoglobin. Shao et al. used prime editing to correct 39% of the HUDEP2 (human umbilical cord-derived erythroid progenitor) in Hb CS (hemoglobin constant spring) cells [68] Chronic septic granulomatosis is caused by mutations in the NCF1 (neutrophil cytosol factor 1) gene, encoding the protein p47phox, a subunit of NADPH oxidase. The majority of patients have a homozygous deletion of 2 nucleotides (delGT) in exon 2 of the gene. Heath et al. corrected at least one allele in 75% of CD34+ myeloid cells carrying the delGT deletion[69]. Duchenne muscular dystrophy is caused by mutations in the DMD gene encoding dystrophin. Mbakam et al.[70] corrected the c.428 G>A mutation (with a rate of 28%) and the c.8713C>T mutation (22%) in myoblasts from patients in culture. Zhao et al. corrected the c.7893delC mutation of the DMD gene in patient fibroblasts with an edition rate of 31% [71]. The S210del mutation in the DGAT1 gene, encoding diacylglycerol O-acyltransferase 1, an enzyme required to metabolize lipids, causes an enteropathy characterized by diarrhea and malnutrition in infants, obtained 21% correction in intestinal cells isolated from patients [72]. Peterkova et al. corrected the c.1 A>G mutation in the FANCA gene, responsible for Fanconi anemia and reached 15% correction in patient fibroblasts[73]. Hypogonadism caused by the W495X mutation in the LHCGR (luteinizing hormone/choriogonadotropin receptor) gene results in insufficient testosterone and sperm production. Xia et al.[74] corrected 23.42% of Leydig stem cells from LhcgrW495X mutated mice, by delivering the N- and C-terminal parts of the PE using lentiviruses. Mutations in the CRB1 (crumbs homolog-1) gene can cause different phenotypes, including Leber congenital amaurosis and retinitis pigmentosa, which lead to vision loss. p.(C948Y) and p.(G1103R) are common mutations in this gene and they were corrected in induced pluripotent stem cells (IPSCs) from patients with a success rate of 72% [75]. Mutations in the HPRT1 (hypoxanthine phosphoribosyltransferase 1) gene cause Lesch-Nyhan syndrome, a disease characterized by hypotonia and developmental delay. Jang et al. [76] corrected up to 14% of the c.333_334ins(A) mutation in patient fibroblasts using PE. RYR1-related diseases are caused by mutations in the gene coding for the ryanodine receptor protein. Godbout et al.[77] generated a myoblast cell line presenting the T4709M mutation in the RYR1 gene in the homozygous state and then corrected the mutation with a rate of 59% by delivering the prime editing components under the RNA form.
6.4. Correcting Mutations In Vivo
Other studies reached the in vivo point, where they tested the edition in animal models. Mutations in the SERPINA1 gene (serpin peptidase inhibitor family A member 1) cause alpha-1-antitrypsin deficiency. The E342K G>A mutation of the SERPINA1 gene (PiZ allele) was corrected in 6.7% of the genes in mice by intravenous hydrodynamic injection of plasmid DNA encoding the different components of prime editing[78]. In a mouse model carrying the R207W (c.619>T) mutation of the Kcnq2 (potassium voltage-gated channel subfamily Q member 2) gene, responsible for epilepsy, Cao et al.[79] used prime editing and corrected 14% of mutated alleles. To treat hypercholesterolemia, Chen et al. [80] inserted in 13% of the genes 4 bp (TTAC) in the pcsk9 gene to introduce a premature termination codon and to inactivate the gene which encodes proprotein convertase subtilisin/kexin type 9, an enzyme that degrades low-density lipoprotein receptors (LDLR). Böck et al [81]. corrected the F263S (T>C) mutation in the Pahenu2 gene which causes phenylketonuria, a disease which impairs the assimilation of phenylalanine. By delivering the PE by an adenovirus into newborn mice, these authors achieved a gene modification of 11.1%. The most common variant in patients is c.1222C>T (p.R408W) and was corrected with an editing rate of 41% in liver cells of the R408W mice by Brooks et al. [82] using dual-AAV delivery[82]. The rd6 mice represent a model of recessive retinal degeneration caused by a 4 bp deletion in the mfrp (membrane frizzled-related protein) gene in a splice donor site. David R. Liu’s group successfully corrected this mutation at 15% in the eyes of rd6 mice through the subretinal injection of VLPs. This group also used PE delivered by subretinal injection of pseudoviral particles to correct the R44X (c.130 C > T) mutation of the rpe5 (retinal pigment epithelium) gene in the rd12 mouse, a model of retinal degeneration affected by Leber Congenital Amaurosis. An average correction of 7.2% of the retinal pigment epithelium was obtained along with a significant improvement in their visual function assessed by electroretinography [83]. Hong et al [84]. corrected the mutations c.3631C > T at 10.5% and c.2005C > T at 5.2% of the COL7A1 gene (collagen type VII alpha 1 chain), responsible for recessive dystrophic epidermolysis bullosa (RDEB). Prime editing treatment was carried out ex vivo on patient-derived fibroblasts then transplanted into immunodeficient athymic nude mice. Sickle cell disease is caused by an A>T mutation in the HBB gene encoding hemoglobin β. Prime editing was used ex vivo on patients' hematopoietic stem cells and corrected between 15% and 41% of the cells. These cells were grafted into mice which, 17 weeks after the transplant, presented between 28% and 43% healthy hemoglobin [85]. Tyrosinemia type I (HT-1) is caused by mutations, such as c.706G>A, in the fah gene, coding for fumarylacetoacetate hydrolase (FAAH), an enzyme responsible for tyrosine catabolism. Jang et al. corrected the in vivo deficit to 11.5% [86]. After treatment, 61% of hepatocytes expressed the fah gene. This percentage is higher than the editing rate of genomic DNA because most hepatocytes are polyploid[87]. DNA contamination from other cells also explains this difference [86,88,89].
7. Conclusion and Perspective
The prime editing system has advanced at an incredible rate from its discovery to characterization and potential applications in biological and agricultural sciences. This remarkable method has a high specificity and possibility for all kinds of gene manipulation compared to other nucleases and nickase-based methods. The optimization of each component of the PE system increases the editing range and editing efficiency for correcting different mutations. However, several concerns remain that need to be addressed. Most importantly, the editing efficiency on different cell lines needs to be further enhanced. To solve this problem, a better understanding of the structure and the molecular mechanism of the PE system needs to be elucidated, as well as the factors that may affect the PegRNA synthesis need to be considered if the plasmid is used. Furthermore, determining the guidelines for developing the appropriate length of pegRNA is crucial to minimize the wasteful effort involved in experimenting with various lengths of pegRNA PBS and RTT for every target site. Secondly, the factors affecting the editing efficiency in primary cells should be analyzed. For example, effective polymerization in MMLV-RT requires high intracellular dNTP levels. The enzymatic properties in primary cells can be enhanced by adding mutation in MMLV-RT to increase the gene editing efficiency. Most importantly, based on the goal of clinical gene therapy, investigations should be continued to find efficient and safe delivery vectors for effective in vivo treatments. Collectively, the PE system is a promising gene therapy technology, but it is still in its early years and must overcome various constraints to reach its potential.
Author Contributions
Y.Y.L conceived this review article. The review was written by Y.Y.L, C.B, N.S, A.S, and K.G., G.L. and J.P.T. All authors have read and agreed with the published version of the manuscript.
Funding
This research was funded by the Canadian Institutes of Health Research (CIHR) (OGB- 185747) and the Foundation for Cell and Gene Therapy. Y.Y.L is supported by the China Scholarship Council (CSC) Grant.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat Protoc 2013, 8, 2281–2308. [CrossRef]
- Anzalone, A. V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [CrossRef]
- Chen, P.J.; Hussmann, J.A.; Yan, J.; Knipping, F.; Ravisankar, P.; Chen, P.-F.; Chen, C.; Nelson, J.W.; Newby, G.A.; Sahin, M.; et al. Enhanced Prime Editing Systems by Manipulating Cellular Determinants of Editing Outcomes. Cell 2021, 184, 5635-5652.e29. [CrossRef]
- Doman, J.L.; Pandey, S.; Neugebauer, M.E.; An, M.; Davis, J.R.; Randolph, P.B.; McElroy, A.; Gao, X.D.; Raguram, A.; Richter, M.F.; et al. Phage-Assisted Evolution and Protein Engineering Yield Compact, Efficient Prime Editors. Cell 2023, 186, 3983-4002.e26. [CrossRef]
- Yan, J.; Oyler-Castrillo, P.; Ravisankar, P.; Ward, C.C.; Levesque, S.; Jing, Y.; Simpson, D.; Zhao, A.; Li, H.; Yan, W.; et al. Improving Prime Editing with an Endogenous Small RNA-Binding Protein. Nature 2024, 628, 639–647. [CrossRef]
- Koeppel, J.; Weller, J.; Peets, E.M.; Pallaseni, A.; Kuzmin, I.; Raudvere, U.; Peterson, H.; Liberante, F.G.; Parts, L. Prediction of Prime Editing Insertion Efficiencies Using Sequence Features and DNA Repair Determinants. Nat Biotechnol 2023, 41, 1446–1456. [CrossRef]
- Wang, J.; He, Z.; Wang, G.; Zhang, R.; Duan, J.; Gao, P.; Lei, X.; Qiu, H.; Zhang, C.; Zhang, Y.; et al. Efficient Targeted Insertion of Large DNA Fragments without DNA Donors. Nat Methods 2022, 19, 331–340. [CrossRef]
- Anzalone, A. V.; Gao, X.D.; Podracky, C.J.; Nelson, A.T.; Koblan, L.W.; Raguram, A.; Levy, J.M.; Mercer, J.A.M.; Liu, D.R. Programmable Deletion, Replacement, Integration and Inversion of Large DNA Sequences with Twin Prime Editing. Nat Biotechnol 2022, 40, 731–740. [CrossRef]
- Zheng, C.; Liu, B.; Dong, X.; Gaston, N.; Sontheimer, E.J.; Xue, W. Template-Jumping Prime Editing Enables Large Insertion and Exon Rewriting in Vivo. Nat Commun 2023, 14, 3369. [CrossRef]
- Pandey, S.; Gao, X.D.; Krasnow, N.A.; McElroy, A.; Tao, Y.A.; Duby, J.E.; Steinbeck, B.J.; McCreary, J.; Pierce, S.E.; Tolar, J.; et al. Efficient Site-Specific Integration of Large Genes in Mammalian Cells via Continuously Evolved Recombinases and Prime Editing. Nat Biomed Eng 2024. [CrossRef]
- Yarnall, M.T.N.; Ioannidi, E.I.; Schmitt-Ulms, C.; Krajeski, R.N.; Lim, J.; Villiger, L.; Zhou, W.; Jiang, K.; Garushyants, S.K.; Roberts, N.; et al. Drag-and-Drop Genome Insertion of Large Sequences without Double-Strand DNA Cleavage Using CRISPR-Directed Integrases. Nat Biotechnol 2023, 41, 500–512. [CrossRef]
- Sun, C.; Lei, Y.; Li, B.; Gao, Q.; Li, Y.; Cao, W.; Yang, C.; Li, H.; Wang, Z.; Li, Y.; et al. Precise Integration of Large DNA Sequences in Plant Genomes Using PrimeRoot Editors. Nat Biotechnol 2024, 42, 316–327. [CrossRef]
- Choi, J.; Chen, W.; Suiter, C.C.; Lee, C.; Chardon, F.M.; Yang, W.; Leith, A.; Daza, R.M.; Martin, B.; Shendure, J. Precise Genomic Deletions Using Paired Prime Editing. Nat Biotechnol 2022, 40, 218–226. [CrossRef]
- Jiang, T.; Zhang, X.-O.; Weng, Z.; Xue, W. Deletion and Replacement of Long Genomic Sequences Using Prime Editing. Nat Biotechnol 2022, 40, 227–234. [CrossRef]
- Lu, Y.; Happi Mbakam, C.; Song, B.; Bendavid, E.; Tremblay, J.-P. Improvements of Nuclease and Nickase Gene Modification Techniques for the Treatment of Genetic Diseases. Front Genome Ed 2022, 4. [CrossRef]
- Kweon, J.; Yoon, J.K.; Jang, A.H.; Shin, H.R.; See, J.E.; Jang, G.; Kim, J.I.; Kim, Y. Engineered Prime Editors with PAM Flexibility. Mol Ther 2021, 29, 2001–2007. [CrossRef]
- Hu, Z.; Wang, S.; Zhang, C.; Gao, N.; Li, M.; Wang, D.; Wang, D.; Liu, D.; Liu, H.; Ong, S.-G.; et al. A Compact Cas9 Ortholog from Staphylococcus Auricularis (SauriCas9) Expands the DNA Targeting Scope. PLoS Biol 2020, 18, e3000686. [CrossRef]
- Lan, T.; Chen, H.; Tang, C.; Wei, Y.; Liu, Y.; Zhou, J.; Zhuang, Z.; Zhang, Q.; Chen, M.; Zhou, X.; et al. Mini-PE, a Prime Editor with Compact Cas9 and Truncated Reverse Transcriptase. Mol Ther Nucleic Acids 2023, 33, 890–897. [CrossRef]
- Davis, J.R.; Banskota, S.; Levy, J.M.; Newby, G.A.; Wang, X.; Anzalone, A. V.; Nelson, A.T.; Chen, P.J.; Hennes, A.D.; An, M.; et al. Efficient Prime Editing in Mouse Brain, Liver and Heart with Dual AAVs. Nat Biotechnol 2024, 42, 253–264. [CrossRef]
- Makarova, K.S.; Koonin, E. V. Annotation and Classification of CRISPR-Cas Systems. In; 2015; pp. 47–75.
- Chen, P.J.; Liu, D.R. Prime Editing for Precise and Highly Versatile Genome Manipulation. Nat Rev Genet 2023, 24, 161–177. [CrossRef]
- Liang, R.; He, Z.; Zhao, K.T.; Zhu, H.; Hu, J.; Liu, G.; Gao, Q.; Liu, M.; Zhang, R.; Qiu, J.-L.; et al. Prime Editing Using CRISPR-Cas12a and Circular RNAs in Human Cells. Nat Biotechnol 2024. [CrossRef]
- Lee, J.; Lim, K.; Kim, A.; Mok, Y.G.; Chung, E.; Cho, S.-I.; Lee, J.M.; Kim, J.-S. Prime Editing with Genuine Cas9 Nickases Minimizes Unwanted Indels. Nat Commun 2023, 14, 1786. [CrossRef]
- Gopalappa, R.; Suresh, B.; Ramakrishna, S.; Kim, H. (Henry) Paired D10A Cas9 Nickases Are Sometimes More Efficient than Individual Nucleases for Gene Disruption. Nucleic Acids Res 2018, 46, e71–e71. [CrossRef]
- Chauhan, V.P.; Sharp, P.A.; Langer, R. Engineered Prime Editors with Minimal Genomic Errors 2024.
- Oscorbin, I.P.; Filipenko, M.L. M-MuLV Reverse Transcriptase: Selected Properties and Improved Mutants. Comput Struct Biotechnol J 2021, 19, 6315–6327. [CrossRef]
- Telesnitsky, A.; Goff, S.P. RNase H Domain Mutations Affect the Interaction between Moloney Murine Leukemia Virus Reverse Transcriptase and Its Primer-Template. Proceedings of the National Academy of Sciences 1993, 90, 1276–1280. [CrossRef]
- Doman, J.L.; Pandey, S.; Neugebauer, M.E.; An, M.; Davis, J.R.; Randolph, P.B.; McElroy, A.; Gao, X.D.; Raguram, A.; Richter, M.F.; et al. Phage-Assisted Evolution and Protein Engineering Yield Compact, Efficient Prime Editors. Cell 2023, 186, 3983-4002.e26. [CrossRef]
- Chen, P.J.; Hussmann, J.A.; Yan, J.; Knipping, F.; Ravisankar, P.; Chen, P.-F.; Chen, C.; Nelson, J.W.; Newby, G.A.; Sahin, M.; et al. Enhanced Prime Editing Systems by Manipulating Cellular Determinants of Editing Outcomes. Cell 2021, 184, 5635-5652.e29. [CrossRef]
- Gao, Z.; Ravendran, S.; Mikkelsen, N.S.; Haldrup, J.; Cai, H.; Ding, X.; Paludan, S.R.; Thomsen, M.K.; Mikkelsen, J.G.; Bak, R.O. A Truncated Reverse Transcriptase Enhances Prime Editing by Split AAV Vectors. Molecular Therapy 2022, 30, 2942–2951. [CrossRef]
- Mbisa, J.L.; Nikolenko, G.N.; Pathak, V.K. Mutations in the RNase H Primer Grip Domain of Murine Leukemia Virus Reverse Transcriptase Decrease Efficiency and Accuracy of Plus-Strand DNA Transfer. J Virol 2005, 79, 419–427. [CrossRef]
- Tang, X.; Zhu, Y.; Baker, S.L.; Bowler, M.W.; Chen, B.J.; Chen, C.; Hogg, J.R.; Goff, S.P.; Song, H. Structural Basis of Suppression of Host Translation Termination by Moloney Murine Leukemia Virus. Nat Commun 2016, 7, 12070. [CrossRef]
- Zheng, C.; Liang, S.-Q.; Liu, B.; Liu, P.; Kwan, S.-Y.; Wolfe, S.A.; Xue, W. A Flexible Split Prime Editor Using Truncated Reverse Transcriptase Improves Dual-AAV Delivery in Mouse Liver. Molecular Therapy 2022, 30, 1343–1351. [CrossRef]
- Böck, D.; Rothgangl, T.; Villiger, L.; Schmidheini, L.; Matsushita, M.; Mathis, N.; Ioannidi, E.; Rimann, N.; Grisch-Chan, H.M.; Kreutzer, S.; et al. In Vivo Prime Editing of a Metabolic Liver Disease in Mice. Sci Transl Med 2022, 14. [CrossRef]
- Lan, T.; Chen, H.; Tang, C.; Wei, Y.; Liu, Y.; Zhou, J.; Zhuang, Z.; Zhang, Q.; Chen, M.; Zhou, X.; et al. Mini-PE, a Prime Editor with Compact Cas9 and Truncated Reverse Transcriptase. Mol Ther Nucleic Acids 2023, 33, 890–897. [CrossRef]
- Zong, Y.; Liu, Y.; Xue, C.; Li, B.; Li, X.; Wang, Y.; Li, J.; Liu, G.; Huang, X.; Cao, X.; et al. An Engineered Prime Editor with Enhanced Editing Efficiency in Plants. Nat Biotechnol 2022, 40, 1394–1402. [CrossRef]
- Grünewald, J.; Miller, B.R.; Szalay, R.N.; Cabeceiras, P.K.; Woodilla, C.J.; Holtz, E.J.B.; Petri, K.; Joung, J.K. Engineered CRISPR Prime Editors with Compact, Untethered Reverse Transcriptases. Nat Biotechnol 2023, 41, 337–343. [CrossRef]
- Liu, B.; Dong, X.; Cheng, H.; Zheng, C.; Chen, Z.; Rodríguez, T.C.; Liang, S.-Q.; Xue, W.; Sontheimer, E.J. A Split Prime Editor with Untethered Reverse Transcriptase and Circular RNA Template. Nat Biotechnol 2022, 40, 1388–1393. [CrossRef]
- Petrova, I.O.; Smirnikhina, S.A. The Development, Optimization and Future of Prime Editing. Int J Mol Sci 2023, 24, 17045. [CrossRef]
- Park, J.; Yu, G.; Seo, S.-Y.; Yang, J.; Kim, H.H. SynDesign: Web-Based Prime Editing Guide RNA Design and Evaluation Tool for Saturation Genome Editing. Nucleic Acids Res 2024, 52, W121–W125. [CrossRef]
- Mathis, N.; Allam, A.; Tálas, A.; Kissling, L.; Benvenuto, E.; Schmidheini, L.; Schep, R.; Damodharan, T.; Balázs, Z.; Janjuha, S.; et al. Machine Learning Prediction of Prime Editing Efficiency across Diverse Chromatin Contexts. Nat Biotechnol 2024. [CrossRef]
- Nelson, J.W.; Randolph, P.B.; Shen, S.P.; Everette, K.A.; Chen, P.J.; Anzalone, A. V.; An, M.; Newby, G.A.; Chen, J.C.; Hsu, A.; et al. Engineered PegRNAs Improve Prime Editing Efficiency. Nat Biotechnol 2022, 40, 402–410. [CrossRef]
- Zhang, G.; Liu, Y.; Huang, S.; Qu, S.; Cheng, D.; Yao, Y.; Ji, Q.; Wang, X.; Huang, X.; Liu, J. Enhancement of Prime Editing via XrRNA Motif-Joined PegRNA. Nat Commun 2022, 13, 1856. [CrossRef]
- Feng, Y.; Liu, S.; Mo, Q.; Xiao, X.; Liu, P.; Ma, H. Enhancing Prime Editing Efficiency and Flexibility with Tethered and Split PegRNAs. Protein Cell 2022. [CrossRef]
- Cirincione, A.; Simpson, D.; Ravisankar, P.; Solley, S.C.; Yan, J.; Singh, M.; Adamson, B. A Benchmarked, High-Efficiency Prime Editing Platform for Multiplexed Dropout Screening. bioRxiv 2024. [CrossRef]
- Ponnienselvan, K.; Liu, P.; Nyalile, T.; Oikemus, S.; Maitland, S.A.; Lawson, N.D.; Luban, J.; Wolfe, S.A. Reducing the Inherent Auto-Inhibitory Interaction within the PegRNA Enhances Prime Editing Efficiency. Nucleic Acids Res 2023, 51, 6966–6980. [CrossRef]
- Zhang, W.; Petri, K.; Ma, J.; Lee, H.; Tsai, C.-L.; Joung, J.K.; Yeh, J.-R.J. Enhancing CRISPR Prime Editing by Reducing Misfolded PegRNA Interactions. Elife 2024, 12. [CrossRef]
- Li, X.; Zhou, L.; Gao, B.-Q.; Li, G.; Wang, X.; Wang, Y.; Wei, J.; Han, W.; Wang, Z.; Li, J.; et al. Highly Efficient Prime Editing by Introducing Same-Sense Mutations in PegRNA or Stabilizing Its Structure. Nat Commun 2022, 13, 1669. [CrossRef]
- Pinto, F.; Thornton, E.L.; Wang, B. An Expanded Library of Orthogonal Split Inteins Enables Modular Multi-Peptide Assemblies. Nat Commun 2020, 11, 1529. [CrossRef]
- She, K.; Liu, Y.; Zhao, Q.; Jin, X.; Yang, Y.; Su, J.; Li, R.; Song, L.; Xiao, J.; Yao, S.; et al. Dual-AAV Split Prime Editor Corrects the Mutation and Phenotype in Mice with Inherited Retinal Degeneration. Signal Transduct Target Ther 2023, 8, 57. [CrossRef]
- Zhi, S.; Chen, Y.; Wu, G.; Wen, J.; Wu, J.; Liu, Q.; Li, Y.; Kang, R.; Hu, S.; Wang, J.; et al. Dual-AAV Delivering Split Prime Editor System for in Vivo Genome Editing. Molecular Therapy 2022, 30, 283–294. [CrossRef]
- Gao, Z.; Ravendran, S.; Mikkelsen, N.S.; Haldrup, J.; Cai, H.; Ding, X.; Paludan, S.R.; Thomsen, M.K.; Mikkelsen, J.G.; Bak, R.O. A Truncated Reverse Transcriptase Enhances Prime Editing by Split AAV Vectors. Molecular Therapy 2022, 30, 2942–2951. [CrossRef]
- Wang, Q.; Capelletti, S.; Liu, J.; Janssen, J.M.; Gonçalves, M.A.F. V Selection-Free Precise Gene Repair Using High-Capacity Adenovector Delivery of Advanced Prime Editing Systems Rescues Dystrophin Synthesis in DMD Muscle Cells. Nucleic Acids Res 2024, 52, 2740–2757. [CrossRef]
- Li, C.; Georgakopoulou, A.; Newby, G.A.; Chen, P.J.; Everette, K.A.; Paschoudi, K.; Vlachaki, E.; Gil, S.; Anderson, A.K.; Koob, T.; et al. In Vivo HSC Prime Editing Rescues Sickle Cell Disease in a Mouse Model. Blood 2023. [CrossRef]
- Rosewell, A.; Vetrini, F. Helper-Dependent Adenoviral Vectors. J Genet Syndr Gene Ther 2011, 2. [CrossRef]
- Chen, Z.; Kelly, K.; Cheng, H.; Dong, X.; Hedger, A.K.; Li, L.; Sontheimer, E.J.; Watts, J.K. In Vivo Prime Editing by Lipid Nanoparticle Co-Delivery of Chemically Modified PegRNA and Prime Editor MRNA. GEN Biotechnology 2023, 2, 490–502. [CrossRef]
- An, M.; Raguram, A.; Du, S.W.; Banskota, S.; Davis, J.R.; Newby, G.A.; Chen, P.Z.; Palczewski, K.; Liu, D.R. Engineered Virus-like Particles for Transient Delivery of Prime Editor Ribonucleoprotein Complexes in Vivo. Nat Biotechnol 2024. [CrossRef]
- Schene, I.F.; Joore, I.P.; Oka, R.; Mokry, M.; van Vugt, A.H.M.; van Boxtel, R.; van der Doef, H.P.J.; van der Laan, L.J.W.; Verstegen, M.M.A.; van Hasselt, P.M.; et al. Prime Editing for Functional Repair in Patient-Derived Disease Models. Nat Commun 2020, 11, 5352. [CrossRef]
- Binder, S.; Ramachandran, H.; Hildebrandt, B.; Dobner, J.; Rossi, A. Prime-Editing of Human ACTB in Induced Pluripotent Stem Cells to Model Human ACTB Loss-of-Function Diseases and Compensatory Mechanisms. Stem Cell Res 2024, 75, 103304. [CrossRef]
- Geurts, M.H.; de Poel, E.; Pleguezuelos-Manzano, C.; Oka, R.; Carrillo, L.; Andersson-Rolf, A.; Boretto, M.; Brunsveld, J.E.; van Boxtel, R.; Beekman, J.M.; et al. Evaluating CRISPR-Based Prime Editing for Cancer Modeling and CFTR Repair in Organoids. Life Sci Alliance 2021, 4, e202000940. [CrossRef]
- Petri, K.; Zhang, W.; Ma, J.; Schmidts, A.; Lee, H.; Horng, J.E.; Kim, D.Y.; Kurt, I.C.; Clement, K.; Hsu, J.Y.; et al. CRISPR Prime Editing with Ribonucleoprotein Complexes in Zebrafish and Primary Human Cells. Nat Biotechnol 2022, 40, 189–193. [CrossRef]
- Abuhamad, A.Y.; Mohamad Zamberi, N.N.; Sheen, L.; Naes, S.M.; Mohd Yusuf, S.N.H.; Ahmad Tajudin, A.; Mohtar, M.A.; Amir Hamzah, A.S.; Syafruddin, S.E. Reverting TP53 Mutation in Breast Cancer Cells: Prime Editing Workflow and Technical Considerations. Cells 2022, 11, 1612. [CrossRef]
- Hou, Y.; Ureña-Bailén, G.; Mohammadian Gol, T.; Gratz, P.G.; Gratz, H.P.; Roig-Merino, A.; Antony, J.S.; Lamsfus-Calle, A.; Daniel-Moreno, A.; Handgretinger, R.; et al. Challenges in Gene Therapy for Somatic Reverted Mosaicism in X-Linked Combined Immunodeficiency by CRISPR/Cas9 and Prime Editing. Genes (Basel) 2022, 13, 2348. [CrossRef]
- Lin, J.; Liu, X.; Lu, Z.; Huang, S.; Wu, S.; Yu, W.; Liu, Y.; Zheng, X.; Huang, X.; Sun, Q.; et al. Modeling a Cataract Disorder in Mice with Prime Editing. Mol Ther Nucleic Acids 2021, 25, 494–501. [CrossRef]
- Salem, A.R.; Bryant, W.B.; Doja, J.; Griffin, S.H.; Shi, X.; Han, W.; Su, Y.; Verin, A.D.; Miano, J.M. Prime Editing in Mice with an Engineered PegRNA. Vascul Pharmacol 2024, 154, 107269. [CrossRef]
- Qian, Y.; Zhao, D.; Sui, T.; Chen, M.; Liu, Z.; Liu, H.; Zhang, T.; Chen, S.; Lai, L.; Li, Z. Efficient and Precise Generation of Tay–Sachs Disease Model in Rabbit by Prime Editing System. Cell Discov 2021, 7, 50. [CrossRef]
- Frisch, A.; Colombo, R.; Michaelovsky, E.; Karpati, M.; Goldman, B.; Peleg, L. Origin and Spread of the 1278insTATC Mutation Causing Tay-Sachs Disease in Ashkenazi Jews: Genetic Drift as a Robust and Parsimonious Hypothesis. Hum Genet 2004, 114, 366–376. [CrossRef]
- Shao, C.; Liu, Q.; Xu, J.; Xin, Y.; Zhang, J.; Ye, Y.; Luo, H.; Zhang, X.; Xu, X.; Xu, P. Prime Editing of the α-Thalassemia Hb Constant Spring Mutation. Blood 2023, 142, 5001–5001. [CrossRef]
- Heath, J.M.; Stuart Orenstein, J.; Tedeschi, J.G.; Ng, A.; Collier, M.D.; Kushakji, J.; Wilhelm, A.J.; Taylor, A.; Waterman, D.P.; De Ravin, S.S.; et al. Prime Editing Efficiently and Precisely Corrects Causative Mutation in Chronic Granulomatous Disease, Restoring Myeloid Function: Toward Development of a Prime Edited Autologous Hematopoietic Stem Cell Therapy. Blood 2023, 142, 7129–7129. [CrossRef]
- Happi Mbakam, C.; Rousseau, J.; Lu, Y.; Bigot, A.; Mamchaoui, K.; Mouly, V.; Tremblay, J.P. Prime Editing Optimized RTT Permits the Correction of the c.8713C>T Mutation in DMD Gene. Mol Ther Nucleic Acids 2022, 30, 272–285. [CrossRef]
- Zhao, X.; Qu, K.; Curci, B.; Yang, H.; Bolund, L.; Lin, L.; Luo, Y. Comparison of In-Frame Deletion, Homology-Directed Repair, and Prime Editing-Based Correction of Duchenne Muscular Dystrophy Mutations. Biomolecules 2023, 13, 870. [CrossRef]
- Bijker, L.E.; Uyttebroeck, S.; Hauser, B.; Vandenplas, Y.; Huysentruyt, K. Variants in DGAT1 Causing Enteropathy: A Case Report and Review of the Literature. Belgian Journal of Paediatrics 2023, 23, 275–279.
- Peterkova, L.; Racková, M.; Svaton, M.; Riha, P.; Novotna, D.; Sedlacek, P.; Sramkova, L.; Skvarova, K. P754: PRIME EDITING AS A NOVEL TOOL FOR PRECISE CORRECTION OF CAUSAL MUTATIONS IN FANCONI ANAEMIA GROUP A PATIENT-DERIVED CELLS. Hemasphere 2023, 7, e27248b5. [CrossRef]
- Xia, K.; Wang, F.; Tan, Z.; Zhang, S.; Lai, X.; Ou, W.; Yang, C.; Chen, H.; Peng, H.; Luo, P.; et al. Precise Correction of Lhcgr Mutation in Stem Leydig Cells by Prime Editing Rescues Hereditary Primary Hypogonadism in Mice. Advanced Science 2023, 10. [CrossRef]
- da Costa, B.L.; Jenny, L.A.; Maumenee, I.H.; Tsang, S.H.; Quinn, P.M.J. Analysis of CRB1 Pathogenic Variants Correctable with CRISPR Base and Prime Editing. In; 2023; pp. 103–107.
- Jang, G.; Shin, H.R.; Do, H.-S.; Kweon, J.; Hwang, S.; Kim, S.; Heo, S.H.; Kim, Y.; Lee, B.H. Therapeutic Gene Correction for Lesch-Nyhan Syndrome Using CRISPR-Mediated Base and Prime Editing. Mol Ther Nucleic Acids 2023, 31, 586–595. [CrossRef]
- Godbout, K.; Rousseau, J.; Tremblay, J.P. Successful Correction by Prime Editing of a Mutation in the RYR1 Gene Responsible for a Myopathy. Cells 2023, 13, 31. [CrossRef]
- de Serres, F.J.; Blanco, I.; Fernández-Bustillo, E. Health Implications of A1-Antitrypsin Deficiency in Sub-Sahara African Countries and Their Emigrants in Europe and the New World. Genetics in Medicine 2005, 7, 175–184. [CrossRef]
- Cao, B.; Huang, Y.; Tian, F.; Li, J.; Xu, C.; Wei, Y.; Liu, J.; Guo, Q.; Xu, H.; Zhan, L.; et al. Prime Editing-Based Gene Correction Alleviates the Hyperexcitable Phenotype and Seizures of a Genetic Epilepsy Mouse Model. Acta Pharmacol Sin 2023, 44, 2342–2345. [CrossRef]
- Chen, Z.; Kelly, K.; Cheng, H.; Dong, X.; Hedger, A.K.; Li, L.; Sontheimer, E.J.; Watts, J.K. In Vivo Prime Editing by Lipid Nanoparticle Co-Delivery of Chemically Modified PegRNA and Prime Editor MRNA. GEN Biotechnology 2023, 2, 490–502. [CrossRef]
- Böck, D.; Rothgangl, T.; Villiger, L.; Schmidheini, L.; Matsushita, M.; Mathis, N.; Ioannidi, E.; Rimann, N.; Grisch-Chan, H.M.; Kreutzer, S.; et al. In Vivo Prime Editing of a Metabolic Liver Disease in Mice. Sci Transl Med 2022, 14. [CrossRef]
- Brooks, D.L.; Whittaker, M.N.; Qu, P.; Musunuru, K.; Ahrens-Nicklas, R.C.; Wang, X. Efficient in Vivo Prime Editing Corrects the Most Frequent Phenylketonuria Variant, Associated with High Unmet Medical Need. The American Journal of Human Genetics 2023, 110, 2003–2014. [CrossRef]
- An, M.; Raguram, A.; Du, S.W.; Banskota, S.; Davis, J.R.; Newby, G.A.; Chen, P.Z.; Palczewski, K.; Liu, D.R. Engineered Virus-like Particles for Transient Delivery of Prime Editor Ribonucleoprotein Complexes in Vivo. Nat Biotechnol 2024. [CrossRef]
- Hong, S.-A.; Kim, S.-E.; Lee, A.-Y.; Hwang, G.-H.; Kim, J.H.; Iwata, H.; Kim, S.-C.; Bae, S.; Lee, S.E. Therapeutic Base Editing and Prime Editing of COL7A1 Mutations in Recessive Dystrophic Epidermolysis Bullosa. Molecular Therapy 2022, 30, 2664–2679. [CrossRef]
- Everette, K.A.; Newby, G.A.; Levine, R.M.; Mayberry, K.; Jang, Y.; Mayuranathan, T.; Nimmagadda, N.; Dempsey, E.; Li, Y.; Bhoopalan, S.V.; et al. Ex Vivo Prime Editing of Patient Haematopoietic Stem Cells Rescues Sickle-Cell Disease Phenotypes after Engraftment in Mice. Nat Biomed Eng 2023, 7, 616–628. [CrossRef]
- Jang, H.; Jo, D.H.; Cho, C.S.; Shin, J.H.; Seo, J.H.; Yu, G.; Gopalappa, R.; Kim, D.; Cho, S.-R.; Kim, J.H.; et al. Application of Prime Editing to the Correction of Mutations and Phenotypes in Adult Mice with Liver and Eye Diseases. Nat Biomed Eng 2021, 6, 181–194. [CrossRef]
- Wilkinson, P.D.; Delgado, E.R.; Alencastro, F.; Leek, M.P.; Roy, N.; Weirich, M.P.; Stahl, E.C.; Otero, P.A.; Chen, M.I.; Brown, W.K.; et al. The Polyploid State Restricts Hepatocyte Proliferation and Liver Regeneration in Mice. Hepatology 2019, 69, 1242–1258. [CrossRef]
- Kim, Y.; Hong, S.-A.; Yu, J.; Eom, J.; Jang, K.; Yoon, S.; Hong, D.H.; Seo, D.; Lee, S.-N.; Woo, J.-S.; et al. Adenine Base Editing and Prime Editing of Chemically Derived Hepatic Progenitors Rescue Genetic Liver Disease. Cell Stem Cell 2021, 28, 1614-1624.e5. [CrossRef]
- Jiang, T.; Zhang, X.-O.; Weng, Z.; Xue, W. Deletion and Replacement of Long Genomic Sequences Using Prime Editing. Nat Biotechnol 2022, 40, 227–234. [CrossRef]
Figure 1.
The iteration of the prime editor from PE1 to PE7.
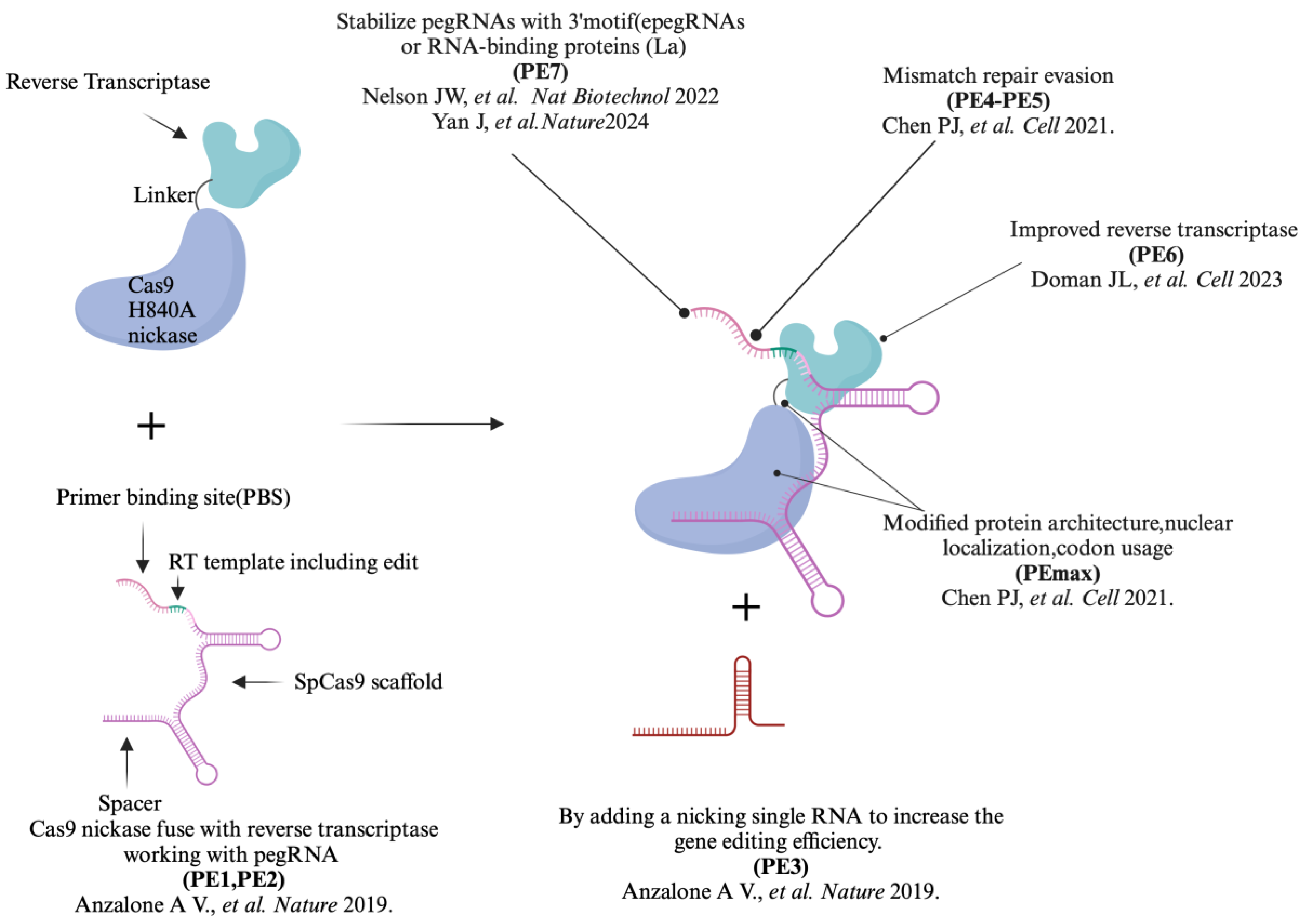

Table 1.
Summary of Prime editor iterations.
| Prime Editors |
Description | Efficiency | Key Features |
| PE1 | The original prime editor, utilizing a Cas9 nickase (H840A) fuse with reverse transcriptas (RT) and a pegRNA | 0.7-5.5% | Lower efficiency; foundational design for subsequent improvements. |
| PE2 | An improved version of PE1, incorporating 5 mutation points into RT changes | 1.6 to 5.1- fold compared to PE1 | Efficiency improved significantly and reduced off-target effects |
| PE3 | Further enhances PE2 by using additional sgRNA to achieve more precise editing | 3 - fold compare to PE2 | Increased targeting range, higher efficiency but with higher indels. |
| PE4 | A more advanced version of PE2 by adding a mismatch repair (MMR)-inhibiting protein | 7.7 - fold compared to PE2 | Enhance editing outcomes through co-expression of dominant negative MLH1 based on PE2 |
| PE5 | Advanced version of PE3 by adding a mismatch repair (MMR)-inhibiting protein | 2.0 - fold compared to PE3 | Enhance editing outcomes through co-expression of dominant negative MLH1 based on PE3 |
| PEmax | Advanced version of PE2, varying RT codon usage, SpCas9 nickase mutations, NLS sequences and the length and composition of peptide linkers between nCas9 and RT |
Higher than PE3 and PE5 | Further improvements in editing capabilities and versatility |
| PE6 | Optimization on Cas9 nickase and RT based on PEmax | 23 - fold compared to PEmax△RNaseH | PE6a to PE6d, which offered improvements in editor size (PE6a and b) and RT activity (PE6c and d); PE6e-g were based on using various evolved and engineered Cas9 variants |
| PE7 | PE7 is the PEmax system fused to a truncated La protein. | 5.2-fold improvement compared to PEmax | Stabilizing exogenous small RNAs therein, which avoid the pegRNA degradation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



