
Submitted:
21 November 2024
Posted:
22 November 2024
You are already at the latest version
Abstract
Within mammalian cells, diverse endocytic mechanisms, including phagocytosis, pinocytosis, and receptor-mediated endocytosis, serve as gateways exploited by many bacterial pathogens and toxins. Among these, caveolae-mediated endocytosis is characterized by lipid-rich caveolae and dimeric caveolin proteins. Caveolae are specialized microdomains on cell surfaces that impact cell signaling. Caveolin proteins facilitate the creation of caveolae and have three members in vertebrates: caveolin-1, caveolin-2, and caveolin-3. Many bacterial pathogens hijack caveolin machinery to invade host cells. For example, the Gram-positive facultative model intracellular bacterial pathogen Listeria monocytogenes exploits caveolin-mediated endocytosis for efficient cellular entry, translocation across the intestinal barrier, and cell-cell spread. Caveolin facilitates the internalization of group A Streptococci by promoting the formation of invaginations in the plasma membrane and avoiding fusion with lysosomes, thereby aiding intracellular survival. Caveolin plays a crucial role in internalizing and modulation of host immune responses by Gram-negative bacterial pathogens, such as Escherichia coli K1, Klebsiella pneumoniae, Pseudomonas aeruginosa, and Salmonella enterica Serovar Typhimurium. Here, we summarize how bacterial pathogens manipulate the host's caveolin system to facilitate bacterial entry and movement within and between host cells, support intracellular survival, evade immune responses, and trigger inflammation. The knowledge enhances the intervention of new therapeutic targets against caveolin in microbial invasion and immune evasion processes.
Keywords:
Caveolae
; Endocytosis
; Host-pathogen interaction
; Caveolin-1
; Caveolin-2
; Caveolin-3
; Listeria monocytogenes
; Microbial invasion
; Immune evasion
1. Introduction
The plasma membrane is not just a boundary for the cell; it is an active, multi-functional system involved in many cellular processes. In various types of cells, the plasma membrane is covered with small pits, 50–80 nm in size, known as caveolae, which form specialized regions within the membrane and were initially identified by Palade and Yamada in the 1950s, [1]. These plasma membrane domains are principally formed by caveolins, which are essential for the biogenesis of caveolae [2,3,4]. The first caveolin protein was discovered in 1989 by Glenney and colleagues [5,6,7]. Caveolins are oligomeric, cholesterol-binding integral plasma membrane proteins, ranging from 17-24 kDa, and are crucial in the invagination process of caveolae from the plasma membrane. To form caveolae, a core set of structural components is required, including Caveolin-1 (Cav-1), Caveolin-2 (Cav-2), Caveolin-3 (Cav-3), Cavin-1, and Pacsin/Syndapin proteins [8]. Following the discovery of Cav-2 and Cav-3, the original caveolin was renamed Cav-1 [9,10]. Cav-2 co-localizes with Cav-1 and is expressed in a variety of tissues, with the highest levels found in endothelial cells, adipocytes, fibroblasts, and smooth muscle cells, whereas Cav-3 expression is restricted to skeletal and cardiac muscle [7]. Recent cryo-electron microscopy studies suggest that the human Cav-1 complex is composed of 11 protomers organized into a tightly packed disc with a flat membrane-embedded surface [11].
The stability of caveolae also depends significantly on Eps15 homology domain (EHD) proteins, which associate with the neck region of caveolae and are essential for both caveolae formation and trafficking [8,12,13,14]. Cav-1 induces membrane curvature, an essential feature for creating caveolae [8], while cavins, which are peripheral membrane proteins with lipid-binding capabilities, form oligomeric complexes to stabilize caveolae structure [8,11,15]. Cavin1, for instance, binds both phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) and phosphatidylserine (PtdSer), further supporting the stability of Cav-1-rich areas [16,17]. Knockdown of Pacsin/Syndapin proteins revealed that the absence of these proteins led to the loss of caveolae morphology, suggesting their crucial role in membrane deformation required for caveola generation [8]. Beyond caveolae formation, Pacsins also contribute to caveolae function by recruiting Dynamin II to caveolae and interacting with EHD proteins [12].
Caveolins are thought to adopt a hairpin-like shape within the membrane, with their C- and N-termini facing the cytoplasm (Parton 2018). They are synthesized as integral membrane proteins in the endoplasmic reticulum (ER) via a signal recognition particle (SRP)--dependent pathway. Caveolins are transported through the Golgi apparatus to the plasma membrane [18]. They combine with Cavin-1 to form caveolae [19,20,21]. At the plasma membrane, caveolae represent a unique type of lipid raft with caveolin proteins, diverse lipid species, an invaginated structure, and enriched lipid leaflets. Cholesterol is essential for assembling these caveolae, along with high concentrations of negatively charged phospholipids, such as PtdSer and PtdIns(4,5)P2, which contribute to a distinct lipid environment within the caveolae [16].
Endocytosis is a cellular uptake process by which the cell regulates the translocation of molecules from outside. Based on the mechanism of endocytosis, it is subdivided into various categories. At the whole cell level, it is sub-divided into phagocytosis and pinocytosis. Phagocytosis is the engulfment and elimination of large particles by phagocytic cells such as macrophages and monocytes. On the other hand, pinocytosis is the uptake of small particles by non-phagocytic cells. Based on the dependency of the tetrameric GTPase dynamin (specifically Dynamin II), pinocytosis is again subdivided into two groups. The pinocytosis requiring Dynamin II is subdivided into clathrin-mediated endocytosis and caveolin-dependent endocytosis. Whereas dynamin-independent pathways are sub-divided into lipid-raft-dependent endocytosis, flotillin-dependent endocytosis, and macropinocytosis [22].
Caveolin-mediated endocytosis begins with the internalization of cargo through caveolae, which are specialized membrane invaginations rich in cholesterol and sphingolipids. Dynamin-2, a crucial GTPase, is essential for the fission of these caveolae from the plasma membrane. Following internalization, caveolin-positive vesicles may interact with early endosomes, where initial sorting occurs. Depending on the specific cargo and cellular needs, the vesicles can diverge to various intracellular compartments, including the Golgi apparatus or signaling endosomes. Unlike clathrin-mediated endocytosis, caveolin-mediated pathways often bypass lysosomes, avoiding degradation and instead supporting roles in signaling, lipid regulation, and transcytosis [8,23].
Caveolae is crucial in endocytosis, cholesterol and lipid metabolism, mechanosensation, and cellular signaling [7]. Besides these major functions, several bacterial pathogens hijack caveolin and caveolae-mediated pathways to bypass traditional host defenses, allowing them to establish infections and evade the immune system [24,25,26]. Over the past 70 years since its discovery, caveolin-mediated endocytosis and caveolin proteins have emerged as a central theme for their role in bacterial pathogenesis. They are now increasingly recognized as an essential factor in how bacteria invade, survive, and manipulate host cells [25,27,28,29]. In this review, we comprehensively summarize past and recent discoveries on how various bacterial pathogens exploit the host’s caveolin machinery for bacterial internalization, translocation into and across host cells, intracellular survival, immune evasion, and inflammation.
2. Gram-Positive Pathogens That Hijack the Caveolin-Mediated Endocytosis for Cellular Entry, Survival and Immune Evasion
2.1. Listeria monocytogenes
L. monocytogenes is a model Gram-positive facultative intracellular bacterial foodborne pathogen that causes listeriosis. Among the foodborne pathogens, it ranks third in the deaths caused yearly due to a high hospitalization (94%) and a high mortality rate (20-30%) [30]. The bacterium exploits its ability to cross critical host barriers, such as the intestinal, blood-brain, and placental barriers, which is crucial in its infection strategy [31,32]. Invasion and translocation across the gut epithelial cells are dependent on two essential proteins: Internalin A (InlA) and the Listeria adhesion protein (LAP) [33,34,35]. While the entry of L. monocytogenes via Peyer’s patches is mediated by the surface protein InlB [36]. After entry, the bacterium becomes enclosed in a phagosome, which is degraded by listeriolysin O (LLO), a protein encoded by the hly gene, allowing it to access the cytosol [37]. Once in the cytosol, L. monocytogenes employs the ActA protein to recruit the Arp2/3 complex, driving actin polymerization to enable its spread from one cell to another [38]. The formation of protrusions by L. monocytogenes also requires the secreted bacterial protein InlC, which interacts with the SH3 domain at the carboxyl terminus of the human scaffolding protein Tuba [39,40].
To cross the gut intestinal epithelial cells and traverse the intestinal barrier, L. monocytogenes utilizes the coordinated action of LAP-dependent paracellular translocation and InlA-mediated transcytosis [35]. The bacterial surface protein LAP binds to its host cell surface receptor heat shock protein 60 (Hsp60), leading to the internalization of tight junction proteins, Claudin-1, Occludin, and the adherens junction protein E-cadherin via Cav-1 and MLCK-mediated endocytosis [33,35,41,42] (Figure 1). Recent studies by our group have shown that pharmacological inhibition of caveolin in cell lines (Caco-2) and genetic knockout of Cav-1 in mice blocks LAP-induced increases in intestinal permeability, junctional endocytosis, and L. monocytogenes translocation [43] (Figure 2A,B). Once internalized, these junctional proteins are processed in early and recycling endosomes, resulting in the opening of cell junctions that permit L. monocytogenes to navigate through the intercellular spaces [43]. Following this initial junction opening by LAP, InlA subsequently binds directly to its receptor E-cadherin, facilitating InlA-mediated transcytosis across the gut epithelial barrier [43] (Figure 1).
While pioneering studies by Veiga et al. highlighted the essential role of clathrin in the initial actin-based invasion of L. monocytogenes into host cells, thereby establishing “bacterial-induced invasion” as the function of this nanoscale endocytic pathway, the role of caveolae in L. monocytogenes uptake could not be ruled out [44]. In cultured non-phagocytic cells, L. monocytogenes utilizes its surface proteins, InlA and InlB, to interact with human E-cadherin (hEcad) and c-Met, respectively, leading to cytoskeletal rearrangement via a zipper mechanism. Depleting cholesterol with methyl-β-cyclodextrin (MβCD) reduced the internalization of L. monocytogenes via both internalin's, indicating the role of lipid rafts in this entry mechanism [45]. Various lipid raft markers, including glycosylphosphatidylinositol-linked proteins and the ganglioside GM1, were observed at the entry sites, supporting the involvement of lipid rafts in internalization. While the interaction between InlA and E-cadherin is cholesterol-dependent, cholesterol depletion does not affect the InlB-c-Met interaction or its downstream PI3K signaling, although it impaired InlB-mediated actin polymerization [45,46].
The requirement of lipid rafts for InlA-mediated entry was further established by the recruitment of caveolin to the entry sites, which was necessary for bacterial internalization. InlB is essential for InlA-dependent entry into cells lacking constitutive phosphoinositide-3 kinase (PI3-K) activity, while it is not required in cells with activated PI3-K[46]. Overall, it can be concluded that InlA is an adhesion molecule within cholesterol-rich lipid rafts, triggering L. monocytogenes internalization through PI3-K activation and caveolin-mediated endocytosis [47].
The bacterium also employs caveolae to facilitate intercellular movement from one cell to another [48,49]. In non-pagocytic cells, when actin filament-rich protrusions containing the bacteria extend from one cell, they engage with ubiquitinated E-cadherin on adjacent cells. Caveolae play a crucial role by forming flattened invaginations that wrap around these bacterial protrusions, effectively engulfing them (Figure 3). The core proteins of caveolae, such as Cav-1, Cav-2, and subset of the caveolin-associated proteins (Cavin-2 and EHD2) are integral to this process, while additional clathrin-interacting proteins like Epsin-1 assist in bending the membrane to create these invaginations. This mechanism significantly enhances the bacterium’s ability to spread from one cell to another, as the actin filament-driven protrusions push the bacteria toward the host cell, deforming its surface [49] (Figure 3).
Unlike the E-cadherin-mediated uptake in non-phagocytic cells, in phagocytic cells, L. monocytogenes secrete listeriolysin O (LLO) that damages the plasma membrane of the infected cells and induces PtdSer inversion in the lipid rafts (Figure3). The bacterial protrusions having PtdSer bind to TIM4 which serves as the PtdSer receptor on the host cell membrane and initiates caveolin-mediated endocytosis to internalize the bacterium [47]. More recently, it was shown that the formin mDia1 is necessary for recruiting Filamin A, an F-actin cross-linking protein associated with caveolae, as well as Cav-1 to membrane invaginations [48].
2.2. Mycobacterium tuberculosis
M. tuberculosis is the causative agent of tuberculosis, a global threat affecting one-quarter of the world’s population. It utilizes cholesterol-dependent raft microdomains to invade and proliferate within phagocytic and non-phagocytic cells, such as epithelial cells, mast cells, macrophages, dendritic cells, fibroblasts, type II pneumocytes, and endothelial cells. Once internalized, the bacteria remain localized within caveolae, effectively circumventing the lytic activity of lysosomal enzymes and avoiding destruction by reactive oxygen and nitrogen species [50]. To invade myeloid-derived suppressor cells, M. tuberculosis interacts with a receptor embedded in lipid rafts on the host cell surface. Caveolae and associated proteins bind to the intracellular face of the lipid raft through cholesterol-binding interactions. This binding induces spontaneous invagination of the host cell's plasma membrane, activating dynamin at the neck of the invagination site, which facilitates the formation of vesicles that bud off into the cytosol. This mechanism allows M. tuberculosis to evade lysosomal fusion, promoting its survival within the host and enabling it to hijack the host's immune defenses [51].
Due to genetic similarity to M. tuberculosis, Mycobacterium bovis Bacillus Calmette-Guérin (BCG), is commonly used as a model organism for studying tuberculosis infection. Recent studies in mice show that Cav-1 regulates apoptosis and the inflammatory response in macrophages infected with BCG. Macrophages in Cav-1 deficient mice exhibit increased bacterial loads in the liver, suggesting that Cav-1 is essential for early elimination and protection against BCG. This process involves Cav-1’s regulation of acid sphingomyelinase (Asm)-dependent ceramide formation, apoptosis, and inflammatory cytokine production following BCG infection [52].
2.3. Staphylococcus aureus
S. aureus invades lung epithelial cells through lipid raft-mediated endocytosis, enabling it to evade immune defenses. A recent study identified α5β1 integrin and fibronectin-binding proteins (FnBPs) as initial adhesion points, with lipid rafts facilitating bacterial entry [53]. Disruption of lipid rafts reduced S. aureus internalization, and colocalization with the lipid raft marker ganglioside GM1 and Cav-1 confirmed this entry route. Alpha-hemolysin (Hla) was crucial for internalization, as Hla-deficient strains showed reduced internalization, implicating Hla’s interaction with Cav-1 in the internalization process [53].
2.4. Streptococcus species
Streptococci are a diverse group of Gram-positive pathogens classified into various groups based on their hemolytic properties and serological characteristics. The most medically relevant pathogenic species in humans are grouped into Group A, B, and other non-grouped streptococci. Streptococcus pyogenes (Group A Streptococcus) causes infections like strep throat, scarlet fever, and more severe conditions like necrotizing fasciitis and toxic shock syndrome. Most of the species of Streptococcus utilize caveolae to invade host cells. In Group A Streptococcus, the M protein and streptococcal fibronectin-binding protein I (SfbI) are the key virulence factors evading antibiotics and host immune defenses. Sfbl is pivotal in S. pyogenes invasion through a caveolae-dependent pathway [54]. SfbI, located on the bacterial surface, interacts with host fibronectin and α5β1 integrins, triggering the clustering of these integrins on the host cell surface [55]. This event promotes the aggregation of caveolae, leading to the formation of specialized membrane-bound compartments known as "caveosomes." The recruitment and invagination of caveolae around the bacterium facilitates its internalization into the host cell. Disrupting lipid rafts and cholesterol with MβCD and filipin prevented the invasion of S. pyogenes into HEp-2 cells [54]. Crucially, this process enables the bacteria to bypass lysosomal fusion, supporting S. pyogenes survival within epithelial and endothelial cells [54,55]. A recent study has revealed that the GAS effector protein NAD-glycohydrolase (Nga) acts as a negative regulator in Cav-1 mediated internalization of GAS in human epithelial cells [56].
Streptococcus agalactiae (Group B Streptococcus (GBS)) commonly colonizes the gastrointestinal and genitourinary tracts in humans. However, in newborns with compromised immune systems, it is a major cause of neonatal infections. GBS predominantly invade polarized cells from their lateral surfaces using α3β1 and α2β1 integrins, which trigger a cellular response involving tyrosine-kinase-dependent endocytosis and extensive actin remodeling [57]. Recent findings demonstrate that intact lipid rafts, Flotillin-1 and Cav-1, and the PI3K/AKT pathway are crucial for S. agalactiae to invade endothelial cells [58]. Disrupting cholesterol by pre-treating cells with MβCD or inhibiting the PI3K/AKT signaling pathway with LY294002 blocked bacterial invasion [58].
Streptococcus pneumoniae (non-grouped streptococci) causes invasive infections such as otitis media, sinusitis, lobar pneumonia, bacteremia, and meningitis. S. pneumoniae, invasion, and intracellular trafficking within respiratory epithelial cells involve caveolin-mediated and clathrin-mediated endocytosis (CME). The pneumococcal surface protein C (PspC) plays a crucial role in interacting with the polymeric immunoglobulin receptor (pIgR) on the host cell surface, facilitating bacterial invasion [59]. S. pneumoniae uptake via the PspC–pIgR pathway involves active dynamin-dependent caveolae and clathrin-coated vesicles. Cholesterol depletion from host cell membranes and disruption of lipid microdomains hindered pneumococcal internalization. Additionally, chemical inhibition of clathrin, or the functional inactivation of dynamin, caveolae, or clathrin via RNA interference (RNAi), significantly reduced pneumococcal uptake, indicating the involvement of both CME and caveolae in the invasion process [59].
3. Gram-Negative Bacterial Pathogens Manipulate Caveolin-Mediated Endocytosis to Infiltrate Host Cells, Secure a Niche for Survival and Evade the Immune System
3.1. Brucella spp.
Brucella suis, a Gram-negative facultative intracellular bacterium that survives and replicates within a membrane-bound compartment inside professional and nonprofessional phagocytic cells. By using cholesterol-sequestering drugs (filipin and- MβCD) and GM1binding (cholera toxin B) molecules and manipulating the lipid raft components—cholesterol and ganglioside GM1, it was found that lipid rafts, specifically under nonopsonic conditions, serve as entry points that allow the bacteria to prevent phagosome-lysosome fusion [60]. This suggests lipid rafts promote a stable environment within the host cell by restricting phagosome maturation at the membrane, supporting the short-term survival of B. suis. Moreover, lipid raft components like GPI-anchored proteins, GM1 gangliosides, and cholesterol are incorporated into these macropinosomes, while proteins like LAMP-1 and CD44 were excluded [61]. Disrupting lipid raft elements reduced VirB-dependent macropinocytosis and replication, implicating lipid raft-mediated entry in the pathogen's intracellular survival [61]. In contrast, the phagocytic trafficking and the intracellular survival of Brucella abortus are primarily mediated by clathrin and Rab5-mediated-mediated endocytosis [62].
3.2. Campylobacter jejuni
C. jejuni is a Gram-negative foodborne pathogen that causes human diarrheal diseases and is the leading cause of Guillain-Barre’s paralysis. Caveolae plays an essential role in the 81-176 strain of C. jejuni internalization in intestinal epithelial cells by serving as a special microdomain and facilitating the bacterium's interaction with the host membrane receptor located within caveolae. Disruption of membrane caveolae by pretreatment of intestinal cell monolayers with filipin III reduced pathogen entry by 95%, suggesting that caveolae-mediated interaction triggers signal transduction events that lead to cytoskeleton rearrangements, which are necessary for bacterial internalization [63]. Moreover, after internalizing epithelial cells via a caveolae-dependent pathway, C. jejuni-containing vacuole avoids lysosomal degradation and resides in a unique intracellular pathway that deviates from the canonical endocytic pathway [64]. Conversely, in macrophages, the bacteria are sent to lysosomes and swiftly eliminated, suggesting evolved strategies for survival in various host cell environments [64].
3.3. Chlamydia trachomatis
C. trachomatis is an obligate intracellular bacterial pathogen and a major causative agent of sexually transmitted infections [65]. The involvement of caveolae in the entry of C. trachomatis is an ongoing debate. Prior studies demonstrated that C. trachomatis serovar K invades the non-phagocytic human epithelial cells (HEp-2 and HeLa 229) and the phagocytic mouse macrophage cells (J-774A.1) by using cholesterol- and sphingolipid-rich lipid rafts containing caveolin [66]. Drugs that disrupt these lipid rafts by depleting cholesterol hindered chlamydial entry. The bacteria then form vesicles marked by caveolin, bypassing lysosomal fusion and acidification. Instead, they travel to the Golgi region, acquiring lipids and cholesterol. They maintain high levels of caveolin, typically recycled back to the plasma membrane, which supports their survival and replication.
Consistent with the above observations, another study reported that in HeLa cells infected with different Chlamydia species, both Cav-1 and Cav-2 are found at the inclusion membranes of C. pneumoniae, C. caviae, and C. trachomatis serovars E, F, and K [67]. Only Cav-2 colocalized with C. trachomatis serovars A, B, and C. This suggests a specific or indirect role for Cav-2 associated with these pathogens, possibly supporting their survival or replication at the inclusion membranes within host cells. However, another study using RNA interference (RNAi), immunoblotting, immunofluorescence, and RT-PCR showed that key structural elements of the clathrin-mediated endocytic pathway, such as clathrin heavy chain, Dynamin-2, heat shock 70-kDa protein 8, Arp2, and cortactin, but not caveolae, are not involved in C. trachomatis’ entry into the host cells [68].
3.4. Edwardsiella tarda
E. tarda is a Gram-negative bacterium known to cause bacteremia. As an intracellular pathogen, it can invade phagocytic and non-phagocytic cells, replicating within host cells. E. tarda primarily utilizes clathrin-mediated endocytosis for invasion, though it has also been shown to use caveolin-mediated endocytosis as an alternative, mainly when clathrin-dependent pathways are limited [69]. The invasion of E. tarda into macrophages was significantly reduced when clathrin- and caveolin-mediated endocytic pathways and endosome acidification were inhibited. However, blocking macropinocytosis did not affect bacterial invasion. A related species, Edwardsiella piscicida, a common fish pathogen, relies on caveolin-mediated pathways to enter non-phagocytic cells. Inhibitory drugs and shRNA-mediated downregulation to block specific endocytic pathways showed that the bacterium enters non-phagocytic cells through micropinocytosis and caveolin-mediated endocytosis, requiring cholesterol and dynamin for successful invasion [70].
3.5. Ehrlichia caffeensis and Anaplasma phagocytophilum
E. caffeensis and A. phagocytophilum are obligatory intracellular bacteria that are agents of human monocytic ehrlichiosis (HME) and human granulocytic ehrlichiosis (HGE), respectively [71]. These emerging tickborne zoonoses cause fever, headache, anorexia, and chills and are frequently accompanied by anemia and elevations in serum hepatic aminotransferases. To enter the host cell, E. caffeensis and A. phagocytophilum bind to their specific receptor located in the host cell surface caveolae that trigger activation of downstream signaling and enter the host cell by caveolae-mediated endocytosis that does not fuse with lysosomes [72]. Using fluorescence microscopy, Cav-1 was shown to co-localize with both early-stage and replicative bacterial inclusions. Additionally, proteins phosphorylated by tyrosine and PLC-γ2 were detected within the early inclusions. Clathrin was absent from all inclusions, suggesting a non-clathrin-mediated entry mechanism.
Furthermore, bacterial proteins from both E. chaffeensis and A. phagocytophilum were found to co-fractionate with Triton X-100-insoluble raft fractions. This suggests that caveolae encapsulating the bacteria retain essential signaling molecules required for entry and facilitate interactions with the recycling endosome pathway, which may contribute to the survival of these obligate intracellular pathogens within the host's immune response [72]. Recently, using yeast two-hybrid screening, co-immunoprecipitation, antibody blocking, and enzymatic inhibition, it was demonstrated that caveolae-dependent uptake of A. phagocytophilum via the invasion AipA9–21 and its interaction with host CD13 induces Src kinase signaling that mediates uptake into host cells [73].
3.6. Escherichia coli
Uropathogenic Escherichia coli (UPEC) is associated with a wide range of infections in humans beyond Urinary Tract Infections (UTIs) that include myositis, skin structure infection, osteomyelitis, epididymal-orchitis, meningitis. Severe E. coli infections lead to Systemic Inflammatory Response Syndrome (SIRS), which causes significant mortality [74]. UPECs that express type 1 pili are the primary causative agent of UTIs. To attach and invade host urinary bladder epithelial cells at the distal tip of each type 1 pilus, an adhesion protein known as FimH (Fimbrial Adhesin H) interacts with mannosylated glycoproteins, triggering bacterial entry [75].
CD48 is a GPI-anchored protein localized in plasmalemma caveolae in host cells, facilitating binding in FimH-expressing E. coli [76]. It forms a large bacteria-encapsulating compartment by recruiting intracellular vesicular caveolae to the bacterial attachment site. The involvement of caveolae in bacterial uptake was confirmed by immunoelectron microscopy, and several caveolin-specific markers, such as caveolin, GM1, and cholesterol, were observed to mobilize and encapsulate the bacteria. Cell fractionation studies suggested that the disruption of caveolae through cholesterol depletion inhibits bacterial uptake, confirming their involvement in the endocytic process that avoids classical lysosomal degradation pathways., thereby allowing the bacteria to remain viable intracellularly [28,76].
Escherichia coli K1 strain can cause meningitis by crossing the human blood-brain barrier (BBB). In the human brain, microvascular endothelial cells (HBMEC) that form the BBB, E. coli K1, bind to Gp96-like receptors, which cluster within caveolae. The bacterium triggers the activation of PKCα, which interacts with Cav-1. Overexpression of a dominant-negative version of Cav-1 with mutations in its scaffolding domain inhibited the interaction between phospho-PKCα and Cav-1 and prevented E. coli from invading HBMEC [77]. This interaction helps reorganize the actin cytoskeleton at the bacterial entry site and enwrap the bacteria that form caveolae. When bacteria internalize into caveolae, they avoid lysosomal fusion, enabling transcytosis across the BBB without being degraded, which aids their survival and spread within the host [77].
In addition to the Gp96-like receptor, the interaction of the virulence factor Invasion of Brain Endothelial cells A (IbeA) and its primary receptor Vimentin is the upstream signaling event that is required for the caveolae/lipid raft (LR)-dependent entry of E. coli K1 into HBMECs [78]. Vimentin is within a unique lipid raft containing cholesterol, sphingolipids, and Cav-1. After binding of IbeA with vimentin, caveolae on the host cell membrane are actively involved in clustering around the binding site and initiate a downstream signaling cascade, finally recruiting other proteins such as Cav-1, a7 nicotinic acetylcholine receptor (a7 nAChR) and polypyrimidine tract-binding protein-associated factor (PSF) into lipid raft. Subsequently, the NF-κB signaling pathway is activated through TAK1 and ERK. This process enhances bacterial penetration and leucocyte transmigration across the BBB and causes subsequent inflammation [79].
In E. coli K12-infected monocytes, the pattern-recognition receptor, Toll-like receptor 4 (TLR4), enhances caveolae-mediated endocytosis and bacterial uptake. Blocking TLR4, Src signaling, or the caveolae-mediated endocytosis pathway in transgenic sheep monocytes results in reduced bacterial uptake, a diminished capacity to clear bacteria, and increased endosomal pH. This indicates that caveolae-mediated endocytosis is crucial for effective bacterial internalization, destruction, and maintenance of an acidic endosomal environment required for the antimicrobial activity of monocytes. [80].
3.7. Francisella tularensis
F. tularensis is a Gram-negative, highly infectious bacterium that causes tularemia. Francisella relies on Cav-1 during its invasion of phagocytic cells but is dispensable in non-phagocytic cells [81]. Consistent with these observations, Tamilselvam et al. show that in murine macrophages during entry and the early stages of intracellular transport within the host cell, components associated with lipid rafts, such as cholesterol and Cav-1, were incorporated into vesicles containing Francisella [82]. Interfering with lipid rafts by depleting plasma membrane cholesterol, inducing raft internalization with cholera toxin, or removing raft-associated GPI-anchored proteins using phosphatidylinositol phospholipase C significantly reduced the entry of Francisella and its ability to multiply intracellularly [82].
3.8. Klebsiella pneumonia
K. pneumoniae is the third most commonly isolated bacterium in the blood of patients with sepsis. Using fluorescent microscopy, Huang Weaver et al. demonstrated that Cav-1 plays a role in the internalization of the pathogen into alveolar epithelial cells [83]. Although the transfection of dominant-negative Cav-1 significantly reduced bacterial internalization into lung cells, Cav-1 in the lipid raft also influenced DNA damage and repair responses by regulating reactive oxygen species, cell death, and inflammatory reactions, which is critical to host defense against K. pneumonia [83]. Along similar lines, recent studies using Cav-1 knockout mice showed that these mice exhibited significantly poorer outcomes following K. pneumoniae infections, with reduced survival, increased bacterial load, exacerbated tissue damage, heightened proinflammatory cytokine responses, and extensive systemic bacterial dissemination [84].
3.9. Leptospira
Leptospira is an emerging Gram-negative bacterium responsible for leptospirosis, a zoonotic disease that can spread through the bloodstream to organs such as the lungs, liver, kidneys, and even cerebrospinal fluid, exacerbating the illness [85]. To invade human and mouse blood vessel endothelial cells, renal tubule epithelial cells, and fibroblasts, Leptospira species employ a caveolae/integrin-β1-PI3K/FAK-microfilament endocytosis pathway (Figure 4). Once internalized, Leptospira forms specialized vesicles to avoid lysosome fusion; these vesicles utilize Rab5/Rab11 and Sec/Exo-SNARE proteins to assist in recycling and transport within the cell. Eventually, Leptospira exits the host cell through a FAK/microfilament/microtubule pathway mediated by the SNARE complex [85] (Figure 4). When entering vascular endothelial cells, Leptospira interacts with integrins and Cav-1 on the cell surface, promoting its internalization. In human umbilical vein endothelial cells (HUVECs), integrin β1 and Cav-1 aid bacterial entry by activating PI3K signaling, leading to cytoskeletal rearrangement through actin polymerization—an essential process for bacterial internalization and transcytosis (Zhao, Guo et al., 2022).
3.10. Neisseria gonorrhea
N. gonorrhea, a Gram-negative obligate human pathogen, that causes the sexually transmitted infection of gonorrhea. The bacterium invades host cells through a complex mechanism that involves caveolae. The PorBIA outer membrane protein on the bacterial surface binds to the host scavenger receptor SREC-I, leading to the recruitment of Cav-1 [86]. The presence of pili modulates the role of Cav-1 in bacterial invasion. When pili are absent, Cav-1 becomes phosphorylated and recruits PI3-K to facilitate downstream signaling required for bacterial uptake that promotes bacterial uptake by activating protein kinase D1 (PKD1). This uptake process is regulated by signaling pathways involving phospholipase C gamma 1 (PLCγ1), PI3-K, and PKD1, which work together to remodel the actin cytoskeleton, facilitating N. gonorrhea invasion [87]. Cav-1 also plays a role in the formation of ceramide-enriched membrane domains that compartmentalize receptors and signaling molecules, which is necessary for PorBIA-mediated bacterial invasion by concentrating SREC-I and organizing the signaling components involved in bacterial invasion [87].
3.11. Porphyromonas gingivalis
P. gingivalis is a bacterial pathogen responsible for human chronic periodontal disease. This pathogen employs multiple pathways to invade host cells, where caveolae play a crucial role. The bacterial fimbriae bind to ICAM-1 to invade host oral epithelium cells. Through this interaction, Cav-1 within caveolae vesicles internalizes P. gingivalis, enabling the bacterium to progress to early endosomes and eventually reach autophagosomes (Figure 4). The attachment of fimbriae also activates TLR2, which subsequently increases ICAM-1 expression and promotes clustering on the host cell surface [88].
Recent elaborate studies by Lei et al. demonstrated that P. gingivalis infected brain microvascular endothelial cells (BMECs) in vitro and in vivo showed increased caveolae and higher Cav-1 expression [89]. Downregulation of Cav-1 levels reduced P. gingivalis – mediated BBB permeability. P. gingivalis-arginine-specific gingipain (RgpA) colocalized with Cav-1. Additionally, P. gingivalis significantly lowered the expression of an essential host protein, the major facilitator superfamily domain containing 2a (Mfsd2a) critical for maintaining BBB integrity (Figure 4). Overexpression of Mfsd2a reduced both Cav-1 expression and P. gingivalis-mediated BBB permeability. The authors implicated that the Mfsd2a/Cav-1 transcytosis pathway is central to P. gingivalis-induced BBB permeability and pathogen entry, leading to neurological damage [89].
3.12. Pseudomonas aeruginosa
Pseudomonas is a Gram-negative bacterium that mainly causes pneumonia. The pathogen enters the host cells by lipid raft-mediated endocytosis. The activated Cav-2 forms a lipid raft on the host cell membrane that promotes P. aeruginosa entry, allowing bacteria to evade host immune defense and replicate intracellularly [90]. In epithelial cell internalization, Cav-1 facilitates the formation of an internalization platform with the cystic fibrosis transmembrane conductance regulator (CFTR) that promotes endocytosis [91].
P. aeruginosa employs lipid rafts, primarily intact lipid raft platforms and Cav-2, to invade type I pneumocytes in the lungs. Phosphorylation of Cav-2 is a critical step for regulating lipid raft-mediated endocytosis of bacterium. This mechanism assists P. aeruginosa invasion of the alveolar cells, leading to severe conditions like pneumonia and sepsis. When P. aeruginosa enters these cells, it colonizes with lipid raft components, which protect the bacteria from being cleared by immune cells [25].
By MS analysis, Thuenauer et al. recently demonstrated that LecB on the bacterial cells binds multiple apical receptors on the host cells (such as CEACAM1, MUCIN-1, ICAM1, and podocalyxin). This receptor clustering triggers the Src-PI3K-Rac signaling cascade, which leads to actin rearrangement and membrane protrusion formation that promotes bacterial uptake. On the apical plasma membrane, LecB also recruits Cav-1 to the apical plasma membrane, which is also critical in PI3-K activation and further facilitates endocytosis and bacterial uptake [92]. However, in-vivo studies with Cav-1 knockout mice revealed that the absence of Cav-1 in the lungs and spleen increased sensitivity to P. aeruginosa infection, increased mortality rate, elevated bacterial burdens, and elevated inflammatory responses via the Cav-1/STAT3/NF-κB axis [93,94].
3.13. Rickettsia spp.
Rickettsia species are Gram-negative obligate intracellular bacteria, with R. conorii and R. rickettsii being the primary species responsible for the Mediterranean and rocky mountain spotted fever, respectively [95]. These bacteria primarily infect the microvascular endothelium that lines blood vessels. They enter host cells using Fibroblast Growth Factor Receptor 1 (FGFR1) and Cav-1 mediated endocytosis. Sahni et al. demonstrated that pathogenic spotted fever group (SFG) rickettsiae can interact with heparan sulfate proteoglycans (HSPG) and the FGFR1 complex on the host cell surface, facilitating their internalization through FGFR1/Cav-1-mediated endocytosis [96]. Proteomic analysis revealed that the β-peptide of the rickettsial outer membrane protein A (OmpA) interacts with FGFR1, aiding in host cell invasion. Additionally, independent silencing of Cav-1 and Cav-2 suggests that rickettsiae may also use the Ku70 receptor for entry via Cav-2-dependent endocytosis [96]. Martinez et al. further demonstrated that inhibiting Ku70 impairs the internalization of R. conorii into host cells, indicating the involvement of Ku70-associated Cav-2-mediated endocytosis [97].
3.14. Salmonella enterica Serovar typhimurium
The Gram-negative facultative intracellular bacterium belonging to the non-typhoidal Salmonella family is a significant cause of foodborne illnesses globally. In the US alone, it causes as many as 1.35 million infections and 420 deaths annually [30]. The pathogen primarily spreads through contaminated food sources like poultry, eggs, and produce, leading to symptoms like diarrhea, fever, and abdominal cramps within 6 to 48 hours of infection.
S. enterica invade nonphagocytic cells by delivering the effector proteins SopE, SopE2, and SopB into the host cell through a type III secretion system (TTSS), encoded by pathogenicity island I (SPI-I). These effectors activate signaling pathways within the host cell, leading to various responses, notably the formation of membrane ruffles rich in actin that enable bacterial entry by a “trigger” like mechanism. At the bacterial attachment site, caveolae are present in the host cell membrane as an invagination cluster at the site of interaction and help recruit other core proteins like Cav-1 and Rac1 to the interaction site [98]. Bacterial effector protein SopE, with these core proteins, work together to rearrange the actin cytoskeleton, forming a membrane ruffle that allows bacteria to be engulfed by the host cells. However, unlike E. coli, S. enterica only manipulates caveolae to attach and invade the host cell; they do not use caveolae to survive inside the host cell. Once S. enterica is fully internalized, SopE levels decline in the caveolae and host cell cytoplasm, accompanied by a decrease in Rac1 activity. Reducing Cav-1 expression through siRNA treatment led to a lower Salmonella invasion rate than cells treated with control siRNA.
While S. enterica Serovar Typhimurium utilizes Cav-1 to enter non-phagocytic host cells through the apical plasma or basolateral membrane, Cav-2 expressed on the basolateral membrane inhibits Cdc42 activation and decreases pathogen uptake as Cdc42 promotes bacterial uptake and membrane ruffling. Knockdown of Cav-2 retards intestinal epithelial cell proliferation and increases bacterial uptake [99]. Interestingly, to counteract the decreased Cav-2 expression, the bacterium induces miR-29a transcription, relieving Cdc24 inhibition and increasing bacterial invasion [99,100].
In addition to classical enterocytes, S. enterica Serovar Typhimurium crosses the intestinal barrier through the M-cells of Peyer’s patches, the specialized immune cells in the epithelium covering Peyer’s patches of the small intestine. siRNA-mediated downregulation of Cav-1 in an in-vitro model of the M-cell-like model showed a significant reduction in bacterial transcytosis and uptake [101]. Moreover, in aged mice, Cav-1 showed high expression levels in the Peyer’s patches and spleen, and disruption of Cav-1 either by MβCD or siRNA in senescent non-phagocytic cells showed marked reduction of bacterial invasion [101]. The findings imply that elevated levels of Cav-1 in aging cells may contribute to the susceptibility of aged populations to microbial infections.
Interestingly, mice lacking Cav-1 (Cav-1⁻/⁻) had significantly higher bacterial loads in their spleen and other tissues when infected with Salmonella enterica serovar Typhimurium, compared to WT mice when challenged intravenously [102]. Although Cav-1-deficient mice had increased production of inflammatory signals, including chemokines and nitric oxide, they still suffered from a more severe infection and lower survival rates. In these mice, neutrophil infiltration into granulomas and liver damage increased, as indicated by higher levels of necrosis. Cav-1-deficient macrophages showed an exaggerated inflammatory response and produced more nitric oxide in response to bacterial LPS in laboratory conditions. These findings suggest that Cav-1 plays a crucial role in controlling the immune response, particularly in macrophages, and that the lack of Cav-1 leads to an overproduction of inflammatory mediators. This excessive production of toxic mediators from macrophages lacking Cav-1 contributes to the heightened susceptibility of Cav-1-deficient mice to S. enterica serovar Typhimurium [102].
3.15. Shigella flexneri
S. flexneri causes bacillary dysentery and, like L. monocytogenes, manipulates the host’s actin cytoskeleton, forming actin-rich "comet tails" for intracellular movement and then creating actin-based protrusions at the plasma membrane to move directly between neighboring host cells using caveolae. When a bacterial membrane protrusion encounters the membrane of the host cell, caveolin aids in physically distorting the plasma membrane around the bacterium providing a source of the membrane to release into the large invagination [103]. Like L. monocytogenes in S. flexneri endocytic invaginations, Cav-1 in colocalized along with other proteins and lipids, such as Cavin-2, EHD2, Dynamin-2, Epsin-1, PtdSer, and PtdIns as well as actin and the actin-associated proteins VASP, α-actinin-1, except α-actinin-4 which is only observed in case of L. monocytogenes [103]. Moreover, in contrast to L. monocytogenes, caveolin, Cavin-2, and EHD2 localize to clathrin-rich membrane invaginations formed during S. flexneri infections suggesting a co-occurrence of clathrin and caveolin pathways at the single-endocytic site and a rare case of these typically distinct pathways collaborating. The combined presence of both pathways may enhance structural stability, cargo specificity, or adaptability in response to infection.
4. Conclusions and Future Directions
Caveolae are small invaginations on the cell surface primarily formed by caveolins, which serve as the core structural components. Caveolin synthesis begins in the endoplasmic reticulum, followed by oligomerization in the Golgi apparatus. Once oligomers bind to Cavin-1, caveolae are formed at the plasma membrane, where cholesterol and phospholipids are essential in assembling distinct lipid pools. Caveolae have multiple functions, including roles in endocytosis, cholesterol and lipid metabolism, mechanosensation, and cellular signaling. Among these, caveolae-mediated endocytosis has garnered significant attention due to its utilization by various microorganisms to invade host cells and evade immune responses (Table 1). In this review, we explored the different roles of caveolae in microbial invasion and immune system evasion.
Among Gram-positive organisms, L. monocytogenes employ caveolae to invade non-phagocytic cells, cross the intestinal barrier, and spread cell-to-cell (Table 1). Similarly, M. tuberculosis, GAS, and S. pneumoniae use caveolae for invasion and survival within host cells after entry (Table 1). Different Gram-negative bacteria use slightly different mechanisms to enter host cells (Table 1). Among all the bacterial entry mechanisms, S. enterica Serovar Typhimurium demonstrates a unique and intriguing entry mechanism into host cells compared to other Gram-negative pathogens. In this case, Cav-1 and Cav-2 play opposing roles in bacterial entry into host cells. S. enterica Serovar Typhimurium utilizes Cav-1 to facilitate its entry through the apical or basolateral plasma membrane. In contrast, Cav-2, located on the basolateral membrane, inhibits bacterial uptake by suppressing the activation of Cdc42. Thus, Cav-1 and Cav-2 function antagonistically in regulating S. enterica Serovar Typhimurium entry into host cells. On the other hand, P. aeruginosa and Rickettsia use both Cav-1 and Cav-2 for internalization into host cells. A few Gram-negative bacterial pathogens like K. pneumoniae, E. coli K1, P. aeruginosa, and S. enterica Serovar Typhimurium have evolved sophisticated mechanisms to hijack caveolin for modulation of immune response and immune evasion (Table 1).
In addition to these bacterial pathogens, many bacterial toxins exploit the Cav-1-mediated endocytic route for entry [28]. To internalize into the host cell, the cholera toxin (CT) binds the GM1 receptor in caveolar membrane domains on the host cell membrane. This activates adenyl cyclase on the cytosolic surface of the basolateral membrane, leading to an increase in intracellular cAMP, which in intestinal crypt cells induces electrogenic Cl- secretion that leads to massive secretory diarrhea seen in cholera. Like cholera toxin, Aerolysin, the pore-forming toxin produced by Aeromonas hydrophila, and VacA toxin, produced by Helicobacter pylori that damages gastric-epithelial cells, exploit the Caveolin-dependent pathway to facilitate their entry and modulate its downstream effects on cellular signaling and immune response [28].
Many bacterial pathogens can cross the blood-brain barrier (BBB) and infect the brain, yet the role of caveolae in the invasion or translocation process is still unclear [104]. For example, Streptococcus, E. coli, and L. monocytogenes can all cause meningitis by crossing the BBB. While the role of caveolae in E. coli invasion is well established, the involvement of caveolae in Streptococcus and L. monocytogenes across the BBB is still unclear. This gap in knowledge presents an exciting opportunity for future research. Recent reports of unusual co-occurrence of clathrin and caveolin pathways utilized by S. flexneri at a single endocytic site suggest collaboration of these pathways [103]. Investigating the potential synergistic functions of these pathways may provide new insights into pathogen-host cell interactions and broader endocytic mechanisms.
Given its role in bacterial pathogenesis, Cav-1 has been proposed as a potential target for therapeutic interventions. By targeting the caveolae-mediated entry pathways, it might be possible to block bacterial invasion or disrupt the intracellular niches that protect bacteria from host defenses. For example, inhibitors of caveolae formation or Cav-1 function could prevent certain pathogens from gaining access to host cells. Along similar lines, a recent study showed that understanding the internalization mechanisms through Cav-1-dependent pathways was crucial for optimizing the delivery of the Human Papillomavirus HPV16E7 Affibody that enters cells to target intracellular proteins and neutralizes the HPV16 E7 oncoprotein, potentially stopping the progression of cancer at an early stage [105]. Modulating Cav-1 activity may also alter the immune response to enhance the host’s ability to clear infections. For example, Cav-1 has been suggested as a target for treating acne [106].
Caveolin, particularly Cav-1, is a critical factor in bacterial pathogenesis (Table 1). It provides pathogens with a means to invade host cells, evade immune detection, and establish chronic infections. By manipulating caveolae-mediated pathways, bacterial pathogens invade host cells, create favorable intracellular environments, modulate host signaling processes, and evade immune defenses. Further research into understanding the function of caveolin in bacterial infections deepens our understanding of host-pathogen interactions and opens new avenues for therapeutic strategies to disrupt these processes.
Author Contributions
Conceptualization, D.B., and R.D.; writing—original draft preparation, D.B., and R.D.; writing—review and editing, R.D.; funding acquisition, R.D. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by funds from KY-INBRE (part of award # P20GM103436-22 from the National Institute of General Medical Sciences (R.D.), the start-up funds at ODU (R.D.), and USDA-NIFA (Award # 2023-67017-40051; R.D.).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in collecting, analyzing, or interpreting the data, writing the manuscript, or deciding to publish.
References
- Yamada , E. The fine structure of the gall bladder epithelium of the mouse. The Journal of Biophysical and Biochemical Cytology 1955, 1, 445-458. [CrossRef]
- Fra, A.M.; Williamson, E.; Simons, K.; Parton, R.G. De novo formation of caveolae in lymphocytes by expression of VIP21-caveolin. Proceedings of the National Academy of Sciences 1995, 92, 8655-8659.
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001, 293, 2449-2452.
- Razani, B.; Engelman, J.A.; Wang, X.B.; Schubert, W.; Zhang, X.L.; Marks, C.B.; Macaluso, F.; Russell, R.G.; Li, M.; Pestell, R.G. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. Journal of Biological Chemistry 2001, 276, 38121-38138.
- Glenney, J.R. Tyrosine phosphorylation of a 22-kDa protein is correlated with transformation by Rous sarcoma virus. Journal of Biological Chemistry 1989, 264, 20163-20166.
- Glenney Jr, J.R.; Zokas, L. Novel tyrosine kinase substrates from Rous sarcoma virus-transformed cells are present in the membrane skeleton. The Journal of cell biology 1989, 108, 2401-2408.
- Bastiani, M.; Parton, R.G. Caveolae at a glance. Journal of cell science 2010, 123, 3831-3836.
- Parton, R.G. Caveolae: structure, function, and relationship to disease. Annual review of cell and developmental biology 2018, 34, 111-136.
- Scherer, P.E.; Okamoto, T.; Chun, M.; Nishimoto, I.; Lodish, H.F.; Lisanti, M.P. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proceedings of the National Academy of Sciences 1996, 93, 131-135.
- Song, K.S.; Scherer, P.E.; Tang, Z.; Okamoto, T.; Li, S.; Chafel, M.; Chu, C.; Kohtz, D.S.; Lisanti, M.P. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells: caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. Journal of Biological Chemistry 1996, 271, 15160-15165.
- Porta, J.C.; Han, B.; Gulsevin, A.; Chung, J.M.; Peskova, Y.; Connolly, S.; Mchaourab, H.S.; Meiler, J.; Karakas, E.; Kenworthy, A.K. Molecular architecture of the human caveolin-1 complex. Science advances 2022, 8, eabn7232.
- Morén, B.; Shah, C.; Howes, M.T.; Schieber, N.L.; McMahon, H.T.; Parton, R.G.; Daumke, O.; Lundmark, R. EHD2 regulates caveolar dynamics via ATP-driven targeting and oligomerization. Molecular biology of the cell 2012, 23, 1316-1329.
- Ariotti, N.; Hall, T.E.; Rae, J.; Ferguson, C.; McMahon, K.-A.; Martel, N.; Webb, R.E.; Webb, R.I.; Teasdale, R.D.; Parton, R.G. Modular detection of GFP-labeled proteins for rapid screening by electron microscopy in cells and organisms. Developmental cell 2015, 35, 513-525.
- Yeow, I.; Howard, G.; Chadwick, J.; Mendoza-Topaz, C.; Hansen, C.G.; Nichols, B.J.; Shvets, E. EHD proteins cooperate to generate caveolar clusters and to maintain caveolae during repeated mechanical stress. Current Biology 2017, 27, 2951-2962. e2955.
- Kovtun, O.; Tillu, V.A.; Ariotti, N.; Parton, R.G.; Collins, B.M. Cavin family proteins and the assembly of caveolae. Journal of cell science 2015, 128, 1269-1278.
- Kovtun, O.; Tillu, V.A.; Jung, W.; Leneva, N.; Ariotti, N.; Chaudhary, N.; Mandyam, R.A.; Ferguson, C.; Morgan, G.P.; Johnston, W.A. Structural insights into the organization of the cavin membrane coat complex. Developmental cell 2014, 31, 405-419.
- Hill, M.M.; Bastiani, M.; Luetterforst, R.; Kirkham, M.; Kirkham, A.; Nixon, S.J.; Walser, P.; Abankwa, D.; Oorschot, V.M.; Martin, S. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 2008, 132, 113-124.
- Monier, S.; Parton, R.G.; Vogel, F.; Behlke, J.; Henske, A.; Kurzchalia, T.V. VIP21-caveolin, a membrane protein constituent of the caveolar coat, oligomerizes in vivo and in vitro. Molecular biology of the cell 1995, 6, 911-927.
- Bastiani, M.; Liu, L.; Hill, M.M.; Jedrychowski, M.P.; Nixon, S.J.; Lo, H.P.; Abankwa, D.; Luetterforst, R.; Fernandez-Rojo, M.; Breen, M.R. MURC/Cavin-4 and cavin family members form tissue-specific caveolar complexes. Journal of Cell Biology 2009, 185, 1259-1273.
- Lajoie, P.; Goetz, J.G.; Dennis, J.W.; Nabi, I.R. Lattices, rafts, and scaffolds: domain regulation of receptor signaling at the plasma membrane. Journal of Cell Biology 2009, 185, 381-385.
- Hayer, A.; Stoeber, M.; Bissig, C.; Helenius, A. Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic 2010, 11, 361-382.
- Lin, A.E.-J.; Guttman, J.A. Hijacking the endocytic machinery by microbial pathogens. Protoplasma 2010, 244, 75-90.
- Cheng, Z.-J.; Deep Singh, R.; Marks, D.L.; Pagano, R.E. Membrane microdomains, caveolae, and caveolar endocytosis of sphingolipids. Molecular membrane biology 2006, 23, 101-110.
- Shin, J.-S.; Abraham, S.N. Caveolae as portals of entry for microbes. Microbes and infection 2001, 3, 755-761.
- Zaas, D.W.; Duncan, M.J.; Li, G.; Wright, J.R.; Abraham, S.N. Pseudomonas invasion of type I pneumocytes is dependent on the expression and phosphorylation of caveolin-2. Journal of Biological Chemistry 2005, 280, 4864-4872.
- Feng, H.; Guo, W.; Han, J.; Li, X.-A. Role of caveolin-1 and caveolae signaling in endotoxemia and sepsis. Life sciences 2013, 93, 1-6.
- Duncan, M.J.; Shin, J.S.; Abraham, S.N. Microbial entry through caveolae: variations on a theme. Cellular microbiology 2002, 4, 783-791.
- Shin, J.S.; Abraham, S. Co-option of endocytic functions of cellular caveolae by pathogens. Immunology 2001, 102, 2-7.
- Zaas, D.W.; Swan, Z.; Brown, B.J.; Wright, J.R.; Abraham, S.N. The expanding roles of caveolin proteins in microbial pathogenesis. Communicative & integrative biology 2009, 2, 535-537.
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.-A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States—major pathogens. Emerging infectious diseases 2011, 17, 7.
- Radoshevich, L.; Cossart, P. Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nature Reviews Microbiology 2018, 16, 32-46.
- Eallonardo, S.J.; Freitag, N.E. Crossing the Barrier: A Comparative Study of Listeria monocytogenes and Treponema pallidum in Placental Invasion. Cells 2023, 13, 88.
- Drolia, R.; Bhunia, A.K. Crossing the intestinal barrier via Listeria adhesion protein and internalin A. Trends in microbiology 2019, 27, 408-425.
- Lecuit, M.; Vandormael-Pournin, S.; Lefort, J.; Huerre, M.; Gounon, P.; Dupuy, C.; Babinet, C.; Cossart, P. A transgenic model for listeriosis: role of internalin in crossing the intestinal barrier. Science 2001, 292, 1722-1725.
- Drolia, R.; Tenguria, S.; Durkes, A.C.; Turner, J.R.; Bhunia, A.K. Listeria adhesion protein induces intestinal epithelial barrier dysfunction for bacterial translocation. Cell host & microbe 2018, 23, 470-484. e477.
- Chiba, S.; Nagai, T.; Hayashi, T.; Baba, Y.; Nagai, S.; Koyasu, S. Listerial invasion protein internalin B promotes entry into ileal Peyer's patches in vivo. Microbiology and immunology 2011, 55, 123-129.
- Portnoy, D.A.; Jacks, P.S.; Hinrichs, D. Role of hemolysin for the intracellular growth of Listeria monocytogenes. The Journal of experimental medicine 1988, 167, 1459-1471.
- Kocks, C.; Gouin, E.; Tabouret, M.; Berche, P.; Ohayon, H.; Cossart, P. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 1992, 68, 521-531.
- Tijoriwalla, S.; Liyanage, T.; Herath, T.U.; Lee, N.; Rehman, A.; Gianfelice, A.; Ireton, K. The host GTPase Dynamin 2 modulates apical junction structure to control cell-to-cell spread of Listeria monocytogenes. Infection and immunity 2024, 92, e00136-00124.
- Rajabian, T.; Gavicherla, B.; Heisig, M.; Müller-Altrock, S.; Goebel, W.; Gray-Owen, S.D.; Ireton, K. The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nature cell biology 2009, 11, 1212-1218.
- Drolia, R.; Amalaradjou, M.A.R.; Ryan, V.; Tenguria, S.; Liu, D.; Bai, X.; Xu, L.; Singh, A.K.; Cox, A.D.; Bernal-Crespo, V. Receptor-targeted engineered probiotics mitigate lethal Listeria infection. Nature communications 2020, 11, 6344.
- Liu, D.; Bai, X.; Helmick, H.D.; Samaddar, M.; Amalaradjou, M.A.R.; Li, X.; Tenguria, S.; Gallina, N.L.; Xu, L.; Drolia, R. Cell-surface anchoring of Listeria adhesion protein on L. monocytogenes is fastened by internalin B for pathogenesis. Cell Reports 2023, 42.
- Drolia, R.; Bryant, D.B.; Tenguria, S.; Jules-Culver, Z.A.; Thind, J.; Amelunke, B.; Liu, D.; Gallina, N.L.; Mishra, K.K.; Samaddar, M. Listeria adhesion protein orchestrates caveolae-mediated apical junctional remodeling of epithelial barrier for Listeria monocytogenes translocation. Mbio 2024, 15, e02821-02823.
- Veiga, E.; Cossart, P. Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nature cell biology 2005, 7, 894-900.
- Seveau, S.; Bierne, H.; Giroux, S.; Prévost, M.-C.; Cossart, P. Role of lipid rafts in E-cadherin–and HGF-R/Met–mediated entry of Listeria monocytogenes into host cells. The Journal of cell biology 2004, 166, 743-753.
- Seveau, S.; Tham, T.N.; Payrastre, B.; Hoppe, A.D.; Swanson, J.A.; Cossart, P. A FRET analysis to unravel the role of cholesterol in Rac1 and PI 3-kinase activation in the InlB/Met signalling pathway. Cellular microbiology 2007, 9, 790-803.
- Tsai, Y.-H.; Chen, W.-L. Host lipid rafts as the gates for Listeria monocytogenes infection: A Mini-Review. Frontiers in Immunology 2020, 11, 1666.
- Dhanda, A.S.; Vogl, A.W.; Ness, F.; Innocenti, M.; Guttman, J.A. mDia1 assembles a linear F-actin coat at membrane invaginations to drive Listeria monocytogenes cell-to-cell spreading. Mbio 2021, 12, e02939-02921.
- Dhanda, A.S.; Yu, C.; Lulic, K.T.; Vogl, A.W.; Rausch, V.; Yang, D.; Nichols, B.J.; Kim, S.H.; Polo, S.; Hansen, C.G. Listeria monocytogenes exploits host caveolin for cell-to-cell spreading. Mbio 2020, 11, 10.1128/mbio. 02857-02819.
- Muñoz, S.; Rivas-Santiago, B.; Enciso, J. Mycobacterium tuberculosis entry into mast cells through cholesterol-rich membrane microdomains. Scandinavian journal of immunology 2009, 70, 256-263.
- Kotzé, L.A.; Young, C.; Leukes, V.N.; John, V.; Fang, Z.; Walzl, G.; Lutz, M.B.; du Plessis, N. Mycobacterium tuberculosis and myeloid-derived suppressor cells: Insights into caveolin rich lipid rafts. EBioMedicine 2020, 53.
- Wu, Y.; Riehle, A.; Pollmeier, B.; Kadow, S.; Schumacher, F.; Drab, M.; Kleuser, B.; Gulbins, E.; Grassmé, H. Caveolin-1 affects early mycobacterial infection and apoptosis in macrophages and mice. Tuberculosis 2024, 147, 102493.
- Goldmann, O.; Lang, J.C.; Rohde, M.; May, T.; Molinari, G.; Medina, E. Alpha-hemolysin promotes internalization of Staphylococcus aureus into human lung epithelial cells via caveolin-1-and cholesterol-rich lipid rafts. Cellular and Molecular Life Sciences 2024, 81, 435.
- Rohde, M.; Müller, E.; Chhatwal, G.S.; Talay, S.R. Host cell caveolae act as an entry-port for group A streptococci. Cellular microbiology 2003, 5, 323-342.
- Jiang, Q.; Zhou, X.; Cheng, L.; Li, M. The adhesion and invasion mechanisms of Streptococci. Current Issues in Molecular Biology 2019, 32, 521-560.
- Toh, H.; Lin, C.-Y.; Nakajima, S.; Aikawa, C.; Nozawa, T.; Nakagawa, I. Group A Streptococcus NAD-glycohydrolase inhibits caveolin 1-mediated internalization into human epithelial cells. Frontiers in Cellular and Infection Microbiology 2019, 9, 398.
- De Gaetano, G.V.; Lentini, G.; Coppolino, F.; Famà, A.; Pietrocola, G.; Beninati, C. Engagement of α3β1 and α2β1 integrins by hypervirulent Streptococcus agalactiae in invasion of polarized enterocytes. Frontiers in Microbiology 2024, 15, 1367898.
- Ferreira, B.J.; Lannes-Costa, P.S.; Santos, G.d.S.; Mermelstein, C.; Einicker-Lamas, M.; Nagao, P.E. Involvement of lipid microdomains in human endothelial cells infected by Streptococcus agalactiae type III belonging to the hypervirulent ST-17. Memórias do Instituto Oswaldo Cruz 2020, 115, e190398.
- Asmat, T.M.; Agarwal, V.; Saleh, M.; Hammerschmidt, S. Endocytosis of Streptococcus pneumoniae via the polymeric immunoglobulin receptor of epithelial cells relies on clathrin and caveolin dependent mechanisms. International Journal of Medical Microbiology 2014, 304, 1233-1246.
- Naroeni, A.; Porte, F. Role of cholesterol and the ganglioside GM1 in entry and short-term survival of Brucella suis in murine macrophages. Infection and immunity 2002, 70, 1640-1644.
- Watarai, M.; Makino, S.i.; Fujii, Y.; Okamoto, K.; Shirahata, T. Modulation of Brucella-induced macropinocytosis by lipid rafts mediates intracellular replication. Cellular microbiology 2002, 4, 341-355.
- Lee, J.J.; Kim, D.G.; Kim, D.H.; Simborio, H.L.; Min, W.; Lee, H.J.; Her, M.; Jung, S.C.; Watarai, M.; Kim, S. Interplay between clathrin and Rab5 controls the early phagocytic trafficking and intracellular survival of Brucella abortus within HeLa cells. Journal of Biological Chemistry 2013, 288, 28049-28057.
- Hu, L.; McDaniel, J.P.; Kopecko, D.J. Signal transduction events involved in human epithelial cell invasion by Campylobacter jejuni 81-176. Microbial pathogenesis 2006, 40, 91-100.
- Watson, R.O.; Galán, J.E. Campylobacter jejuni survives within epithelial cells by avoiding delivery to lysosomes. PLoS pathogens 2008, 4, e14.
- Stelzner, K.; Vollmuth, N.; Rudel, T. Intracellular lifestyle of Chlamydia trachomatis and host–pathogen interactions. Nature Reviews Microbiology 2023, 21, 448-462.
- Norkin, L.C.; Wolfrom, S.A.; Stuart, E.S. Association of caveolin with Chlamydia trachomatis inclusions at early and late stages of infection. Experimental cell research 2001, 266, 229-238.
- Webley, W.C.; Norkin, L.C.; Stuart, E.S. Caveolin-2 associates with intracellular chlamydial inclusions independently of caveolin-1. BMC infectious diseases 2004, 4, 1-12.
- Hybiske, K.; Stephens, R.S. Mechanisms of Chlamydia trachomatis entry into nonphagocytic cells. Infection and immunity 2007, 75, 3925-3934.
- Sui, Z.-h.; Xu, H.; Wang, H.; Jiang, S.; Chi, H.; Sun, L. Intracellular trafficking pathways of Edwardsiella tarda: from clathrin-and caveolin-mediated endocytosis to endosome and lysosome. Frontiers in cellular and infection microbiology 2017, 7, 400.
- Hu, T.; Zhang, L.; Wang, W.; Yang, D.; Xiao, J.; Zhang, Y.; Liu, X.; Liu, Q. Edwardsiella piscicida enters nonphagocytic cells via a macropinocytosis-involved hybrid mechanism. Journal of Bacteriology 2019, 201, 10.1128/jb. 00548-00518.
- Rikihisa, Y. Anaplasma phagocytophilum and Ehrlichia chaffeensis: subversive manipulators of host cells. Nature Reviews Microbiology 2010, 8, 328-339.
- Lin, M.; Rikihisa, Y. Obligatory intracellular parasitism by Ehrlichia chaffeensis and Anaplasma phagocytophilum involves caveolae and glycosylphosphatidylinositol-anchored proteins. Cellular microbiology 2003, 5, 809-820.
- Lind, M.C.H.; Naimi, W.A.; Chiarelli, T.J.; Sparrer, T.; Ghosh, M.; Shapiro, L.; Carlyon, J.A. Anaplasma phagocytophilum invasin AipA interacts with CD13 to elicit Src kinase signaling that promotes infection. Mbio 2024, 15, e01561-01524.
- Vila, J.; Sáez-López, E.; Johnson, J.R.; Römling, U.; Dobrindt, U.; Cantón, R.; Giske, C.; Naas, T.; Carattoli, A.; Martínez-Medina, M. Escherichia coli: an old friend with new tidings. FEMS microbiology reviews 2016, 40, 437-463.
- Eto, D.S.; Gordon, H.B.; Dhakal, B.K.; Jones, T.A.; Mulvey, M.A. Clathrin, AP-2, and the NPXY-binding subset of alternate endocytic adaptors facilitate FimH-mediated bacterial invasion of host cells. Cellular microbiology 2008, 10, 2553-2567.
- Shin, J.-S.; Gao, Z.; Abraham, S.N. Involvement of cellular caveolae in bacterial entry into mast cells. Science 2000, 289, 785-788.
- Sukumaran, S.K.; Quon, M.J.; Prasadarao, N.V. Escherichia coli K1 internalization via caveolae requires caveolin-1 and protein kinase Cα interaction in human brain microvascular endothelial cells. Journal of Biological Chemistry 2002, 277, 50716-50724.
- Chi, F.; Jong, T.D.; Wang, L.; Ouyang, Y.; Wu, C.; Li, W.; Huang, S.H. Vimentin-mediated signalling is required for IbeA+ E. coli K1 invasion of human brain microvascular endothelial cells. Biochem J 2010, 427, 79-90. [CrossRef]
- Huang, S.-H.; Chi, F.; Peng, L.; Bo, T.; Zhang, B.; Liu, L.-Q.; Wu, X.; Mor-Vaknin, N.; Markovitz, D.M.; Cao, H. Vimentin, a novel NF-κB regulator, is required for meningitic Escherichia coli K1-induced pathogen invasion and PMN transmigration across the blood-brain barrier. PLoS One 2016, 11, e0162641.
- Li, Y.; Zhao, Y.; Xu, X.; Zhang, R.; Zhang, J.; Zhang, X.; Li, Y.; Deng, S.; Lian, Z. Overexpression of Toll-like receptor 4 contributes to the internalization and elimination of Escherichia coli in sheep by enhancing caveolae-dependent endocytosis. Journal of Animal Science and Biotechnology 2021, 12, 1-16.
- Law, H.; Lin, A.E.-J.; Kim, Y.; Quach, B.; Nano, F.E.; Guttman, J.A. Francisella tularensis uses cholesterol and clathrin-based endocytic mechanisms to invade hepatocytes. Scientific Reports 2011, 1, 192.
- Tamilselvam, B.; Daefler, S. Francisella targets cholesterol-rich host cell membrane domains for entry into macrophages. The Journal of Immunology 2008, 180, 8262-8271.
- Huang, H.; Weaver, A.; Wu, E.; Li, Y.; Gao, H.; Fan, W.; Wu, M. Lipid-based signaling modulates DNA repair response and survival against Klebsiella pneumoniae infection in host cells and in mice. American journal of respiratory cell and molecular biology 2013, 49, 798-807.
- Guo, Q.; Shen, N.; Yuan, K.; Li, J.; Wu, H.; Zeng, Y.; Fox III, J.; Bansal, A.K.; Singh, B.B.; Gao, H. Caveolin-1 plays a critical role in host immunity against Klebsiella pneumoniae by regulating STAT 5 and A kt activity. European journal of immunology 2012, 42, 1500-1511.
- Sun, A.-H.; Liu, X.-X.; Yan, J. Leptospirosis is an invasive infectious and systemic inflammatory disease. Biomedical Journal 2020, 43, 24-31.
- Faulstich, M.; Hagen, F.; Avota, E.; Kozjak-Pavlovic, V.; Winkler, A.C.; Xian, Y.; Schneider-Schaulies, S.; Rudel, T. Neutral sphingomyelinase 2 is a key factor for P or B-dependent invasion of Neisseria gonorrhoeae. Cellular microbiology 2015, 17, 241-253.
- Faulstich, M.; Böttcher, J.-P.; Meyer, T.F.; Fraunholz, M.; Rudel, T. Pilus phase variation switches gonococcal adherence to invasion by caveolin-1-dependent host cell signaling. PLoS pathogens 2013, 9, e1003373.
- Tamai, R.; Asai, Y.; Ogawa, T. Requirement for intercellular adhesion molecule 1 and caveolae in invasion of human oral epithelial cells by Porphyromonas gingivalis. Infection and immunity 2005, 73, 6290-6298.
- Lei, S.; Li, J.; Yu, J.; Li, F.; Pan, Y.; Chen, X.; Ma, C.; Zhao, W.; Tang, X. Porphyromonas gingivalis bacteremia increases the permeability of the blood-brain barrier via the Mfsd2a/Caveolin-1 mediated transcytosis pathway. International journal of oral science 2023, 15, 3.
- Zaas, D.W.; Swan, Z.D.; Brown, B.J.; Li, G.; Randell, S.H.; Degan, S.; Sunday, M.E.; Wright, J.R.; Abraham, S.N. Counteracting signaling activities in lipid rafts associated with the invasion of lung epithelial cells by Pseudomonas aeruginosa. Journal of Biological Chemistry 2009, 284, 9955-9964.
- Bajmoczi, M.; Gadjeva, M.; Alper, S.L.; Pier, G.B.; Golan, D.E. Cystic fibrosis transmembrane conductance regulator and caveolin-1 regulate epithelial cell internalization of Pseudomonas aeruginosa. American Journal of Physiology-Cell Physiology 2009, 297, C263-C277.
- Thuenauer, R.; Kühn, K.; Guo, Y.; Kotsis, F.; Xu, M.; Trefzer, A.; Altmann, S.; Wehrum, S.; Heshmatpour, N.; Faust, B.; et al. The Lectin LecB Induces Patches with Basolateral Characteristics at the Apical Membrane to Promote Pseudomonas aeruginosa Host Cell Invasion. mBio 2022, 13, e0081922. [CrossRef]
- Yuan, K.; Huang, C.; Fox, J.; Gaid, M.; Weaver, A.; Li, G.; Singh, B.B.; Gao, H.; Wu, M. Elevated inflammatory response in caveolin-1-deficient mice with Pseudomonas aeruginosa infection is mediated by STAT3 protein and nuclear factor κB (NF-κB). Journal of Biological Chemistry 2011, 286, 21814-21825.
- Gadjeva, M.; Paradis-Bleau, C.; Priebe, G.P.; Fichorova, R.; Pier, G.B. Caveolin-1 modifies the immunity to Pseudomonas aeruginosa. The journal of immunology 2010, 184, 296-302.
- Salje, J. Cells within cells: Rickettsiales and the obligate intracellular bacterial lifestyle. Nature Reviews Microbiology 2021, 19, 375-390.
- Sahni, A.; Patel, J.; Narra, H.P.; Schroeder, C.L.; Walker, D.H.; Sahni, S.K. Fibroblast growth factor receptor-1 mediates internalization of pathogenic spotted fever rickettsiae into host endothelium. PloS one 2017, 12, e0183181.
- Martinez, J.J.; Seveau, S.; Veiga, E.; Matsuyama, S.; Cossart, P. Ku70, a component of DNA-dependent protein kinase, is a mammalian receptor for Rickettsia conorii. Cell 2005, 123, 1013-1023.
- Lim, J.S.; Shin, M.; Kim, H.-J.; Kim, K.S.; Choy, H.E.; Cho, K.A. Caveolin-1 mediates Salmonella invasion via the regulation of SopE-dependent Rac1 activation and actin reorganization. The Journal of infectious diseases 2014, 210, 793-802.
- Hoeke, L.; Sharbati, J.; Pawar, K.; Keller, A.; Einspanier, R.; Sharbati, S. Intestinal Salmonella typhimurium infection leads to miR-29a induced caveolin 2 regulation. PLoS One 2013, 8, e67300. [CrossRef]
- De Almeida, C.J.G. Caveolin-1 and caveolin-2 can be antagonistic partners in inflammation and beyond. Frontiers in immunology 2017, 8, 1530.
- Lim, J.S.; Na, H.S.; Lee, H.C.; Choy, H.E.; Park, S.C.; Han, J.M.; Cho, K.A. Caveolae-mediated entry of Salmonella typhimurium in a human M-cell model. Biochemical and biophysical research communications 2009, 390, 1322-1327.
- Medina, F.A.; De Almeida, C.J.; Dew, E.; Li, J.; Bonuccelli, G.; Williams, T.M.; Cohen, A.W.; Pestell, R.G.; Frank, P.G.; Tanowitz, H.B. Caveolin-1-deficient mice show defects in innate immunity and inflammatory immune response during Salmonella enterica serovar Typhimurium infection. Infection and immunity 2006, 74, 6665-6674.
- Dhanda, A.S.; Guttman, J.A. Localization of host endocytic and actin-associated proteins during Shigella flexneri intracellular motility and intercellular spreading. The Anatomical Record 2023, 306, 1088-1110.
- Badaut, J.; Blochet, C.; Obenaus, A.; Hirt, L. Physiological and pathological roles of caveolins in the central nervous system. Trends in neurosciences 2024.
- Zhang, Q.; Zhu, H.; Cui, Z.; Li, Y.; Zhuo, J.; Ye, J.; Zhang, Z.; Lian, Z.; Du, Q.; Zhao, K.-N. The HPV16E7 Affibody as a Novel Potential Therapeutic Agent for Treating Cervical Cancer Is Likely Internalized through Dynamin and Caveolin-1 Dependent Endocytosis. Biomolecules 2022, 12, 1114.
- Kruglikov, I.L.; Scherer, P.E. Caveolin-1 as a possible target in the treatment for acne. Experimental dermatology 2020, 29, 177-183.
- Zhao, X.; Guo, J.; Jia, X.; Yang, Y.; Liu, L.; Nie, W.; Fang, Z. Internalization of Leptospira interrogans via diverse endocytosis mechanisms in human macrophages and vascular endothelial cells. PLOS Neglected Tropical Diseases 2022, 16, e0010778.
Figure 1.
Schematic depicting the mechanism of L. monocytogenes LAP and Caveolin-mediated translocation across the intestinal epithelial barrier and subsequent InlA-mediated internalization across non-phagocytic cells. LAP on L. monocytogenes binds to its host cell surface receptor heat shock protein 60 (Hsp60), inducing endocytosis of tight junction proteins, claudin-1, occludin, and the adherens junction protein E-cadherin via Caveolin-1 and MLCK-mediated endocytosis. This disrupts cell junctions, allowing L. monocytogenes to pass through the paracellular spaces. InlA subsequently binds to its receptor E-cadherin at the adherens junctions to mediate transcytosis across the epithelial barrier. In non-phagocytic cells, the bacterial surface protein InlA and InlB interact with E-cadherin and c-met, leading to the cytoskeletal rearrangement via a zipper mechanism that triggers L. monocytogenes internalization through PI3-K activation and Caveolin-mediated endocytosis. Figure created using Biorender and adapted from [43].
Figure 1.
Schematic depicting the mechanism of L. monocytogenes LAP and Caveolin-mediated translocation across the intestinal epithelial barrier and subsequent InlA-mediated internalization across non-phagocytic cells. LAP on L. monocytogenes binds to its host cell surface receptor heat shock protein 60 (Hsp60), inducing endocytosis of tight junction proteins, claudin-1, occludin, and the adherens junction protein E-cadherin via Caveolin-1 and MLCK-mediated endocytosis. This disrupts cell junctions, allowing L. monocytogenes to pass through the paracellular spaces. InlA subsequently binds to its receptor E-cadherin at the adherens junctions to mediate transcytosis across the epithelial barrier. In non-phagocytic cells, the bacterial surface protein InlA and InlB interact with E-cadherin and c-met, leading to the cytoskeletal rearrangement via a zipper mechanism that triggers L. monocytogenes internalization through PI3-K activation and Caveolin-mediated endocytosis. Figure created using Biorender and adapted from [43].
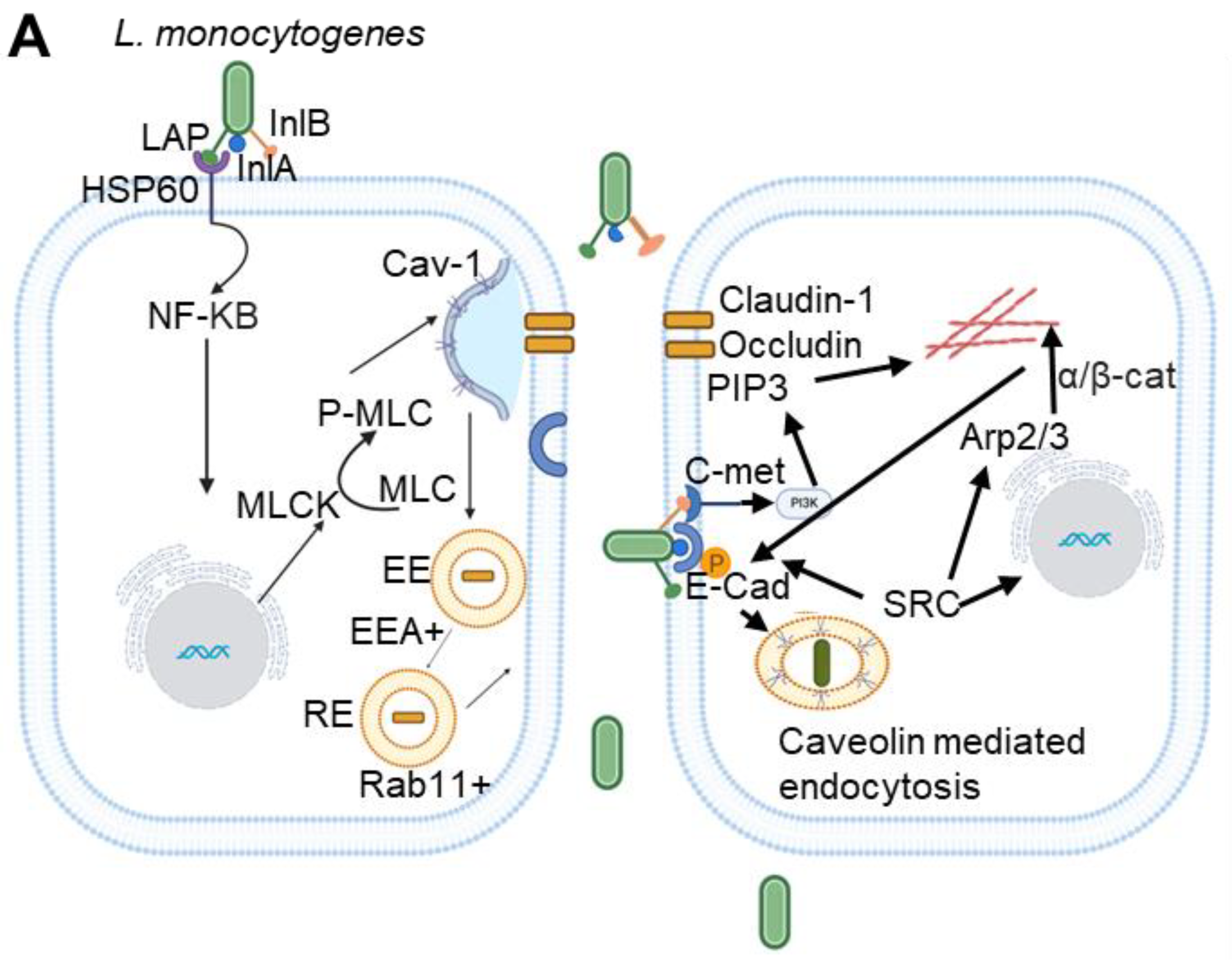
Figure 2.
L. monocytogenes translocation and junctional protein expression are affected in caveolin-1 knockout mice. (A) Figure 2A represents immunostaining data from ileum and colon tissues collected from wild-type (WT) and Cav-1 knockout (Cav-1−/−) mice, which were orally administered 5 × 108 CFU of InlAm or InlAm lap− (murinized Listeria strains). The findings, from a recent study by our group [43], show that WT mice challenged with InlAm strains display increased L. monocytogenes translocation into the lamina propria of ileum and colon tissues. In contrast, Cav-1−/− mice challenged with the InlAm strain exhibit bacterial restriction to the intestinal lumen. (B) Figure 2B represents confocal immunofluorescence micrographs of ileal tissues harvested from infected mice. These images illustrate structural changes in the intestinal cell junctions, marked by increased P-MLC and mislocation of junctional proteins, facilitating bacterial translocation. The InlAm-challenged WT mice show increased expression of P-MLC (green; arrows) and mislocalization (intracellular puncta, endocytosis) of Claudin-1, Occludin, and E-cadherin (arrows). The figure is taken from [43], and permission will be acquired before publication. .
Figure 2.
L. monocytogenes translocation and junctional protein expression are affected in caveolin-1 knockout mice. (A) Figure 2A represents immunostaining data from ileum and colon tissues collected from wild-type (WT) and Cav-1 knockout (Cav-1−/−) mice, which were orally administered 5 × 108 CFU of InlAm or InlAm lap− (murinized Listeria strains). The findings, from a recent study by our group [43], show that WT mice challenged with InlAm strains display increased L. monocytogenes translocation into the lamina propria of ileum and colon tissues. In contrast, Cav-1−/− mice challenged with the InlAm strain exhibit bacterial restriction to the intestinal lumen. (B) Figure 2B represents confocal immunofluorescence micrographs of ileal tissues harvested from infected mice. These images illustrate structural changes in the intestinal cell junctions, marked by increased P-MLC and mislocation of junctional proteins, facilitating bacterial translocation. The InlAm-challenged WT mice show increased expression of P-MLC (green; arrows) and mislocalization (intracellular puncta, endocytosis) of Claudin-1, Occludin, and E-cadherin (arrows). The figure is taken from [43], and permission will be acquired before publication. .
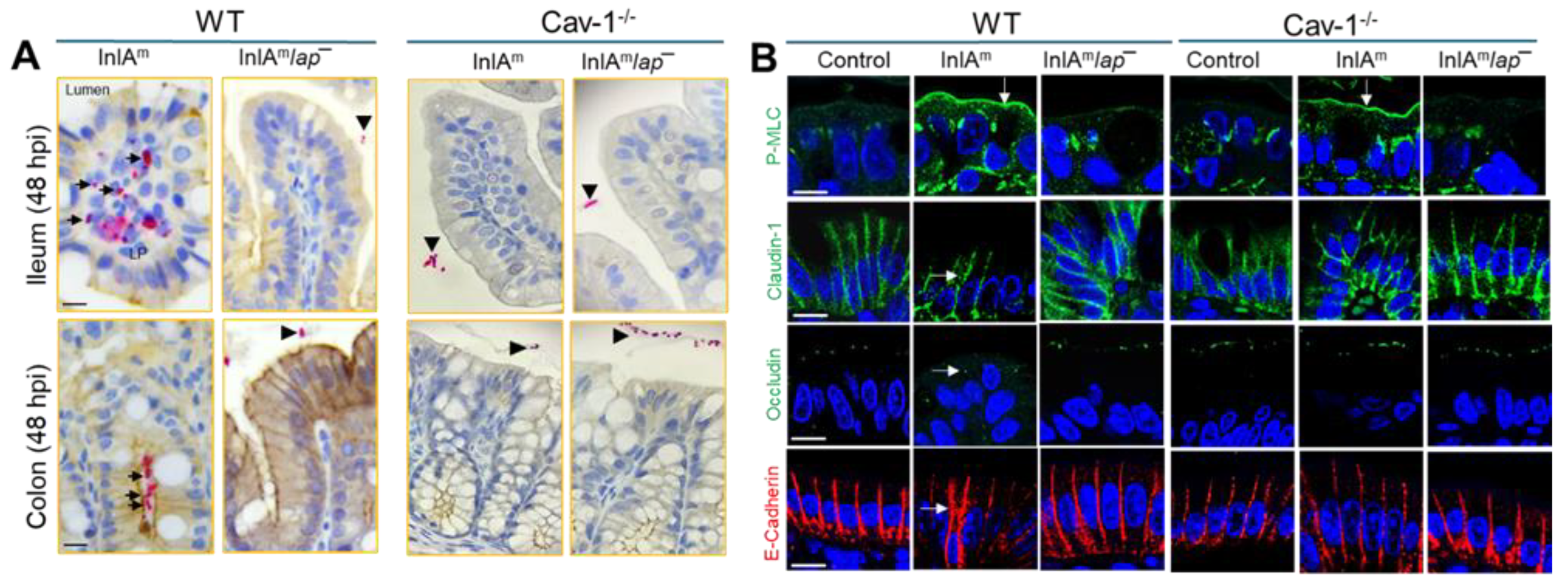
Figure 3.
Schematic representation of the cell-to-cell spread mechanism of L. monocytogenes in phagocytic and non-phagocytic cells. In non-phagocytic cells, when actin filament-rich protrusions containing the bacteria extend from one cell, they bind to ubiquitinated E-cadherin in adjacent cells. This binding triggers caveolae to form a flattened invagination that wraps around these bacterial protrusions, effectively engulfing them with the help of some core proteins of caveolae, such as Cav-1, Cav-2, a subset of the Caveolin-associated proteins (Cavin-2 and EHD2), and clathrin-interacting Epsin that assists in bending the membrane to create these invaginations.In phagocytic cells, internalized actin protrusions containing L. monocytogenes secrete LLO, which disrupts phosphatidyl serine on the plasma membrane. Both actin protrusions and phosphatidyl serine-positive L. monocytogenes bind to the TIM4 receptor on the host cell surface, which causes internalization of L. monocytogenes via Caveolin-mediated endocytosis. Figure created using Biorender.
Figure 3.
Schematic representation of the cell-to-cell spread mechanism of L. monocytogenes in phagocytic and non-phagocytic cells. In non-phagocytic cells, when actin filament-rich protrusions containing the bacteria extend from one cell, they bind to ubiquitinated E-cadherin in adjacent cells. This binding triggers caveolae to form a flattened invagination that wraps around these bacterial protrusions, effectively engulfing them with the help of some core proteins of caveolae, such as Cav-1, Cav-2, a subset of the Caveolin-associated proteins (Cavin-2 and EHD2), and clathrin-interacting Epsin that assists in bending the membrane to create these invaginations.In phagocytic cells, internalized actin protrusions containing L. monocytogenes secrete LLO, which disrupts phosphatidyl serine on the plasma membrane. Both actin protrusions and phosphatidyl serine-positive L. monocytogenes bind to the TIM4 receptor on the host cell surface, which causes internalization of L. monocytogenes via Caveolin-mediated endocytosis. Figure created using Biorender.
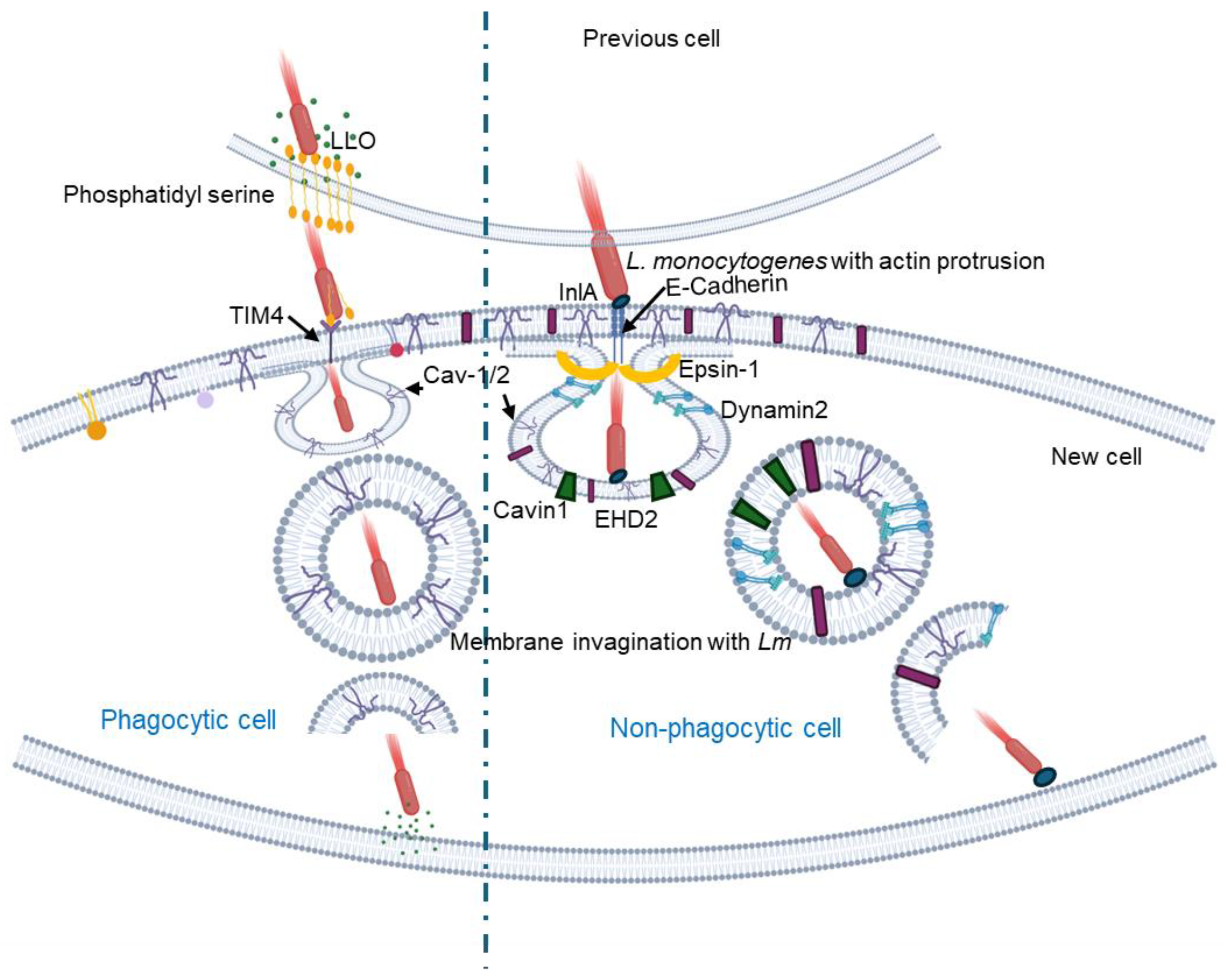
Figure 4.
Schematics depicting the internalization mechanism of P. gingivalis and Leptospiral via Caveolin-mediated endocytosis. (A). The interaction of the virulent factor RgpA of P. gingivalis with Cav-1 in the host cell facilitates the internalization of P. gingivalis via caveolae. P. gingivalis inhibits the integrity of Mfsd2a, leading to enhanced transcytosis across the blood-brain barrier and increased Cav-1 expression, which induces albumin uptake to the cell. (Adapted from [89]) .(B) LeptospiraI species interacts with Integrin-β-1 on host cells, triggers caveolin to form an invagination, and through caveolae/integrin-b1-PI3K/FAK-microfilament endocytosis pathway, it enters the host cell. To avoid fusion with lysosomes, it forms leptospiral vesicles inside the host cell, and these vesicles recruit Rab5/Rab11 and Sec/Exo-SNARE proteins in endocytic recycling and vesicular transport systems for intracellular migration and finally release from the cells through a SNARE complex-mediated FAK/microfilament/microtubule endocytosis pathway. Figure created using Biorender.
Figure 4.
Schematics depicting the internalization mechanism of P. gingivalis and Leptospiral via Caveolin-mediated endocytosis. (A). The interaction of the virulent factor RgpA of P. gingivalis with Cav-1 in the host cell facilitates the internalization of P. gingivalis via caveolae. P. gingivalis inhibits the integrity of Mfsd2a, leading to enhanced transcytosis across the blood-brain barrier and increased Cav-1 expression, which induces albumin uptake to the cell. (Adapted from [89]) .(B) LeptospiraI species interacts with Integrin-β-1 on host cells, triggers caveolin to form an invagination, and through caveolae/integrin-b1-PI3K/FAK-microfilament endocytosis pathway, it enters the host cell. To avoid fusion with lysosomes, it forms leptospiral vesicles inside the host cell, and these vesicles recruit Rab5/Rab11 and Sec/Exo-SNARE proteins in endocytic recycling and vesicular transport systems for intracellular migration and finally release from the cells through a SNARE complex-mediated FAK/microfilament/microtubule endocytosis pathway. Figure created using Biorender.
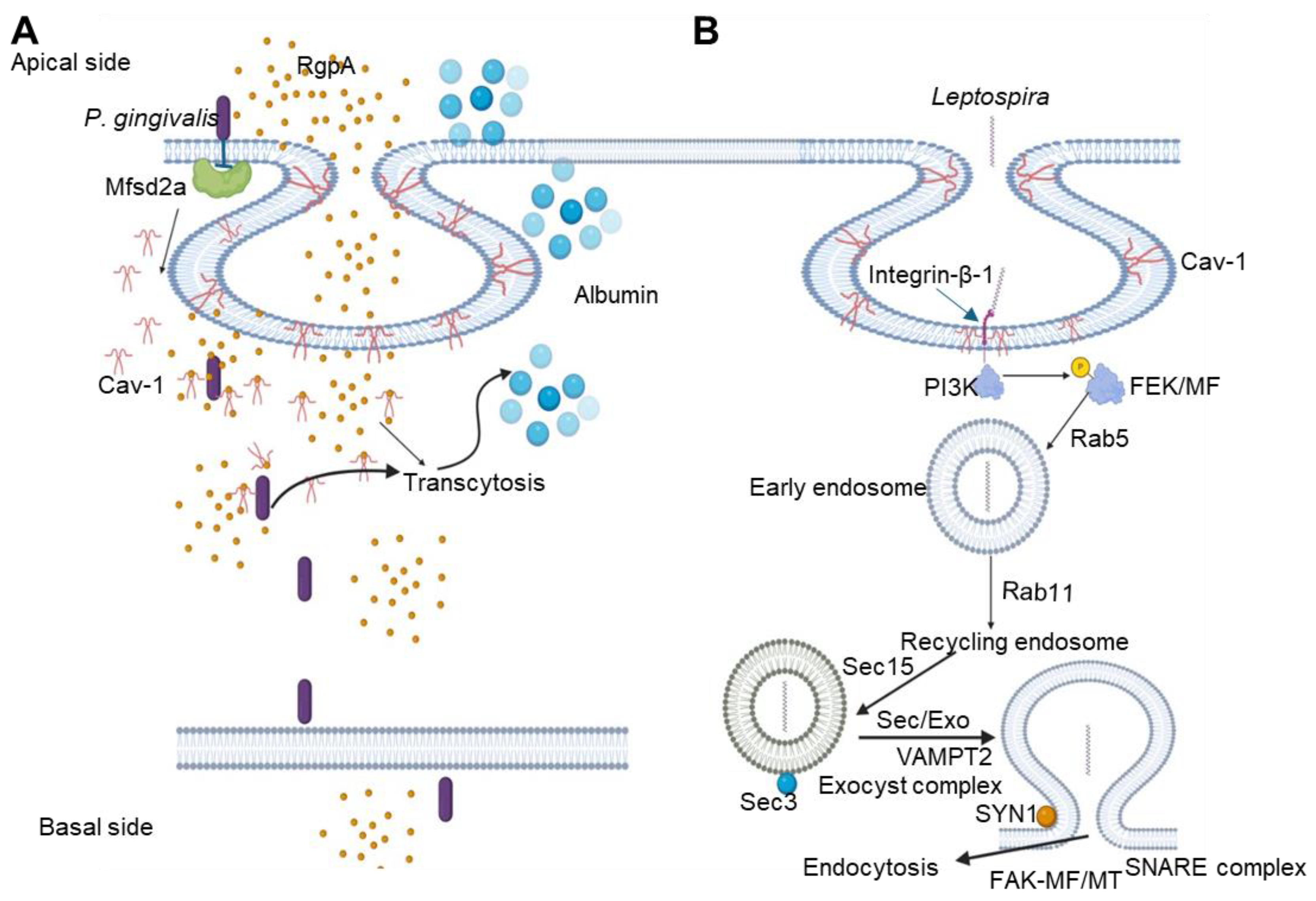

Table 1.
A comparative table of bacterial pathogens exploitation of Caveolin-mediated endocytosis for invasion, intracellular survival, cell-cell spread, and modulation of immune responses.
Table 1.
A comparative table of bacterial pathogens exploitation of Caveolin-mediated endocytosis for invasion, intracellular survival, cell-cell spread, and modulation of immune responses.
| Pathogen | Pathway | Caveolin involved | Reference |
|---|---|---|---|
| Bacterial invasion and intracellular survival | |||
| Anaplasma phagocytophilum | Bacterial internalization and intracellular survival within caveosome. | Cav-1 | [72] |
| Brucella spp. | Caveolin-mediated entry. | Cav-1 | [60,61] |
| Campylobacter jejuni | Helps in bacterial internalization and intracellular survival. | Cav-1 | [63,64] |
| Edwardsiella tarda | Caveolin mediated invasion and intracellular survival. | Cav-1 | [69,70] |
| Ehrlichia chaffeensis | Bacterial internalization and intracellular survival within caveosome. | Cav-1 | [72] |
| Escherichia coli | Caveolin mediated invasion and intracellular survival. | Cav-1 | [77,79] |
| Fransicella tularensis | Caveolin-mediated entry into macrophages and hepatocytes. Proliferation inside macrophages. | Cav-1 | [81,82] |
| Klebsiella pneumonae | Caveolin-mediated internalization. | Cav-1 | [83] |
| Leptospira | Caveolin-mediated entry. | Cav-1 | [107] |
| Listeria monocytogenes | Apical junctional remodeling for bacterial translocation and internalization. | Cav-1 | [43,45] |
| Neisseria gonorrhoeae | Caveolin-mediated invasion. | Cav-1 | [86,87]. |
| Porphyromonas gingivalis | Caveolin-mediated internalization. | Cav-1 | [88,89] |
| Pseudomonas aeruginosa | Lipid raft mediated endocytosis. | Cav-1 and Cav-2 | [25,91,92] |
| Rickettsia spp. | Caveolin mediated endocytic pathway for bacterial entry. | Cav-1 and Cav-2 | [92,95,96] |
| Salmonella enterica Serovar Typhimurium | Caveolin-mediated internalization and transcytosis. | Cav-1 and Cav-2. | [99,101] |
| Streptococcus spp. | Invasion and intracellular survival. Caveosome-mediated internalization. | Cav-1 | [55] |
| Intracellular survival and cell-cell spread | |||
| Leptospira | Intracellular migration through the vesicular transport system initiated by Caveolin. | Cav-1 | [85] |
| Listeria monocytogenes | Cell-to-cell spreading. | Cav-1 | [48,49] |
| Shigella flexneri | Cell-to-cell spreading. | Cav-1 | [103] |
| Modulation of host immune responses | |||
| Escherichia coli K1 | Increase inflammation in brain cells. | Cav-1 | [79] |
| Klebsiella pneumoniae | Modulation of host immunity through STAT5-Akt signaling pathway. | Cav-1 | [84] |
| Mycobacterium bovis Bacillus Calmette-Guérin (BCG) | Cav-1 regulates apoptosis and the inflammatory response in macrophages infected with BCG. | Cav-1 | [52] |
| Pseudomonas aeruginosa | Downregulates inflammatory response in host cells. | Cav-1 | [91,93,94] |
| Salmonella enterica Serovar Typhimurium | Regulate anti-inflammatory responses in macrophages. | Cav-1 | [102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



